

Abstract
The ability to probe the membrane potential of multiple genetically defined neurons simultaneously would have a profound impact on neuroscience research. Genetically encoded voltage indicators are a promising tool for this purpose, and recent developments have achieved a high signal-to-noise ratio in vivo with 1-photon fluorescence imaging. However, these recordings exhibit several sources of noise and signal extraction remains a challenge. We present an improved signal extraction pipeline, spike-guided penalized matrix decomposition-nonnegative matrix factorization (SGPMD-NMF), which resolves supraand subthreshold voltages in vivo. The method incorporates biophysical and optical constraints. We validate the pipeline with simultaneous patch-clamp and optical recordings from mouse layer 1 in vivo and with simulated and composite datasets with realistic noise. We demonstrate applications to mouse hippocampus expressing paQuasAr3-s or SomArchon1, mouse cortex expressing SomArchon1 or Voltron, and zebrafish spines expressing zArchon1.
In brief
Xie et al. present a pipeline to extract accurate waveforms from in vivo voltage imaging recordings of neurons. The method preserves both subthreshold dynamics and spikes and rejects sources of noise, including correlated background, motion, and blood flow. This pipeline improves accessibility of voltage imaging technologies.
Graphical abstract
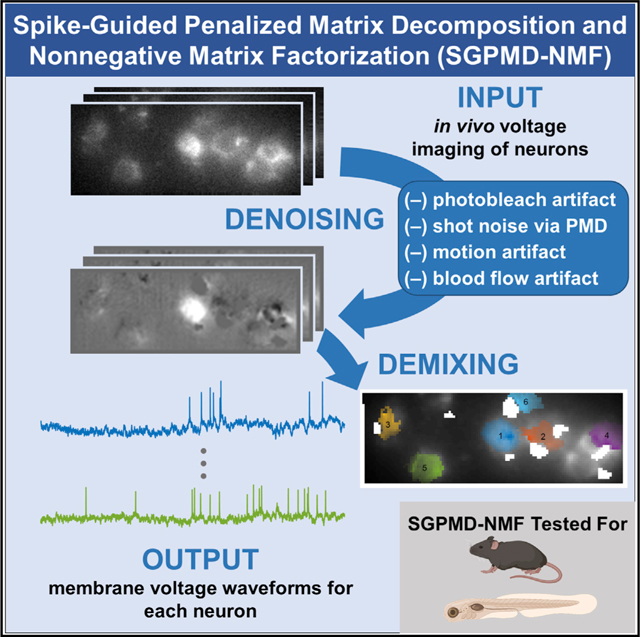
INTRODUCTION
A technology to measure the membrane potential of multiple neurons simultaneously in behaving animals would be a transformative tool for neuroscience research (Bando et al., 2019; Knöpfel and Song, 2019; Yang and St-Pierre, 2016). Although calcium imaging (Farhi et al., 2019; Sofroniew et al., 2016) and extracellular arrays (e.g., neuropixels) (Jun et al., 2017) can report the spiking outputs of hundreds or even thousands of neurons, subthreshold dynamics are invisible to these methods. Subthreshold voltages reflect synaptic and neuromodulatory inputs, as well as the activity of many endogenous ion channels. Thus, measurements of subthreshold voltages can be useful for functional connectivity mapping and for determining the input-output transformations that a neuron or microcircuit implements. By measuring subthreshold waveforms under different levels of optogenetic depolarization, one can resolve the distinct contributions of excitatory and inhibitory synaptic inputs to membrane voltage (Fan et al., 2020). Voltage imaging can also resolve spike waveforms and distinguish spike types, e.g., complex versus simple spikes. History-dependent and subcellular variations in spike waveform can be important in governing vesicle release probability in axons (Panzera and Hoppa, 2019) and in controlling synaptic plasticity in dendrites (Sjöström et al., 2008). These examples provide a strong motivation for measuring membrane voltage in vivo, despite substantial technical challenges.
Several recent efforts introduced improvements to genetically encoded voltage indicators (GEVIs): QuasAr3 (Adam et al., 2019), Archon (Piatkevich et al., 2019), Voltron (Abdelfattah et al., 2019), and ASAP3 (Villette et al., 2019). In combination with advanced optical instrumentation, these GEVIs achieved fluorescence voltage imaging from identified neurons in vivo (Bando et al., 2019). However, these recordings have substantial statistical and systematic noise sources that present a challenge for extraction of true neuronal voltage dynamics.
Voltage imaging in vivo presents far more stringent technical challenges than does calcium imaging. First, accurate detection of action potentials requires frame rates of ~1,000 frames/s (or even higher for fast-spiking interneurons), versus typically ~10 frames/s or slower for Ca2+ imaging. Photons from voltage reporters are thus divided into ~100-fold thinner time bins and consequently have ~10-fold higher shot noise relative to signal, all else being equal.
Second, GEVIs typically yield 10%–40% ΔF/F per spike, whereas the best Ca2+ indicators have severalfold increases in brightness for a single action potential but are insensitive to subthreshold potentials (Chen et al., 2013; Tian et al., 2009). For millivolt-level subthreshold events, GEVIs typically produce ΔF/F < 0.5%. Thus, the subthreshold signals in voltage imaging can be as much as 100-fold smaller than the spike signals in Ca2+ imaging.
Third, the most advanced voltage imaging schemes rely on 1-photon (1P) optics (although there has been progress in 2-photon (2P) voltage imaging; Villette et al., 2019). Consequently, 1P voltage imaging experiments are susceptible to optical crosstalk between distinct in-focus signal sources and from out-of-focus cells, blood flow, or background autofluorescence.
Fourth, subthreshold voltages often have strong correlations between cells, whereas spiking tends to be less correlated (Lampl et al., 1999). Thus, the true voltage signal of interest is often correlated with sources of crosstalk.
Fifth, voltage signals only originate at the cell membrane, a nanometers-thick 2D manifold, whereas Ca2+ signals come predominantly from the cytoplasm throughout the cell body. Voltage measurements are thus far more sensitive to motion artifacts or to misalignment of illumination, sample, and detection. During behavioral tasks, motion artifacts may also correlate with the true voltage signal.
Progress against these challenges requires combined efforts on three fronts: (1) molecular tools, through the development of brighter, faster, and more sensitive voltage reporters; (2) instrumentation, through the creation of microscopes with improved depth penetration, spatial resolution, sensitivity, and speed; and (3) algorithms, through the software to convert gigantic video data files into estimates of time-dependent membrane voltage from defined neurons. This paper deals with the third aspect.
In a voltage imaging movie, each pixel encodes a time-dependent signal that contains a sum of signals from one or more neurons and from various noise sources. The software challenge is to identify (potentially nonlinear) combinations of pixels that give optimal estimates of the voltages from individual neurons while rejecting noise sources and crosstalk from partially overlapping cells. This is called the demixing or source-separation problem. Efforts have focused on algorithms that facilitate identification of spikes within the single-neuron traces (Cai et al., 2020). Here we seek to identify both spikes and subthreshold voltages so as to learn about neuronal outputs and inputs.
Extracting subthreshold signals from voltage imaging datasets presents unique challenges. Correlated signal and noise dynamics confound well-established signal extraction techniques, such as joint principal-component analysis (PCA) and independent-component analysis (ICA) (Mukamel et al., 2009), that assume statistical independence between distinct sources. Advanced image demixing techniques for in vivo 2P Ca2+ imaging data are primarily focused on identifying spiking events and thus can safely neglect many sources of noise (Pnevmatikakis et al., 2016).
A joint penalized matrix decomposition (PMD) and nonnegative matrix factorization (NMF) approach has been proposed to denoise and demix voltage imaging data (Buchanan et al., 2019). This method can extract cell signals that have a high signal-to-noise ratio (SNR) from in vitro voltage imaging movies, in which motion artifacts, blood flow, light scattering, and temporally varying background can all be ignored.
Here, we build upon the PMD-NMF pipeline to account for fluctuating background dynamics that might be correlated with the desired in-focus neural signals. We call the augmented pipeline spike-guided PMD-NMF (SGPMD-NMF). Our procedure takes advantage of several robust statistical structures within voltage imaging data. First, action potentials have lower correlations between cells and to noise sources than do slower subthreshold signals. We thus extract the spatial footprints of cells from temporally high-pass-filtered movies, a process that preserves spikes but suppresses subthreshold fluctuations (spike-guided NMF). We then apply the same spatial footprints to extract the slower subthreshold voltage dynamics from the unfiltered movies and optimize pixel weights within the footprints for this setting. Spiking and subthreshold footprints coincide when cells are electrotonically compact, a reasonable assumption for studies performed with soma-localized GEVIs. The tradeoff is that the algorithm requires cells to spike. Cells that show purely subthreshold dynamics during the analyzed epoch are not detected, although in principle one could use regular PMD-NMF to find nonspiking sources after accounting for all spiking sources.
Second, the spatial profiles of out-of-focus background sources are more dispersed and smoother compared with the spatial profiles of in-focus cell signals. By optimizing the smoothness of the background, we determine how to apportion low-frequency fluorescence signals among signal and background, even when these two sources overlap in space and are correlated in time.
Using these constraints, we accurately resolve the subthreshold dynamics of neurons recorded in vivo. We validate our methods with (1) simulated data containing realistic noise and in which ground truth is known by construction, (2) simultaneous in vivo patch-clamp and voltage imaging recordings taken in cortical layer 1 of anesthetized mice, and (3) composite movies created by summing real in vivo single-cell recordings from mouse hippocampus. We compare SGPMD-NMF, conventional PMD-NMF, PCA-ICA, and simple region of interest (ROI)-based analysis. SGPMD-NMF produces traces that more closely resemble ground truth than the other methods. Finally, we apply our pipeline to datasets from different organisms (mouse and zebrafish) and using different indicators (paQuasAr3-s, SomArchon1, zArchon1, and Voltron) to show the broad applicability of SGPMD-NMF. Code and application to an example dataset are available here: https://github.com/adamcohenlab/invivo-imaging. Analysis was conducted using MATLAB and Python.
Signal extraction algorithm
The SGPMD-NMF algorithm comprises two steps: (1) denoising and (2) demixing. The substeps are shown in Figure 1.
Figure 1. Pipeline for denoising and demixing in vivo voltage imaging data.

The denoising steps (blue) comprise a set of distinct corrections for the sources of statistical noise and systematic artifacts that can arise in vivo. The demixing steps (green) use action potentials to identify the cell footprints and the differing spatial profiles of in-focus versus out-of-focus sources to apportion subthreshold dynamics between cells and background.
Denoising
The denoising steps address several distinct sources of noise: motion artifacts, photobleaching, shot noise, and blood autofluorescence. The relative contributions of these noise sources depend on the details of the preparation: density of expressing cells, whether the animal is awake or anesthetized, imaging depth, and microscopy modality. A user should evaluate the necessity of each type of correction individually.
The raw dataset is first corrected for in-plane motion using NoRMCorre (Pnevmatikakis and Giovannucci, 2017). This step aligns the neuron locations between frames of the movie. The function also returns the horizontal (x) and vertical (y) displacements of each frame relative to a reference frame.
Photobleaching often does not follow simple monoexponential kinetics, particularly when the illumination is nonuniform, such that different molecules experience different local illumination intensities. Moreover, variations in local microenvironment can cause heterogeneous bleaching rates even under uniform illumination. Rather than using an exponential fit, each pixel in the registered movie is corrected for photobleaching with a 3rd-order spline-based detrending fit. In selecting the detrending interval, one must select an interval short enough to correct for the fastest photobleaching transients, but not so short that subthreshold depolarizations (e.g., network up states) are removed. Typically, 5 s is appropriate.
Shot noise is removed from the movie via a spatially localized PMD approach (Buchanan et al., 2019). This approach uses the uncorrelation of shot noise between pixels, whereas true voltage signals typically extend over multiple contiguous pixels. The PMD approach calculates local filters that aim to preserve the correlated signal structure in the movie while rejecting uncorrelated noise. To achieve reasonable run time for long movies, we typically apply the PMD algorithm to a subset of contiguous frames selected from the full movie. PMD returns spatial filters, which are then applied to the full movie to obtain the denoised version of the full movie. This approach allows the PMD algorithm to run at a fixed time even as the movie duration increases.
NoRMCorre (Pnevmatikakis and Giovannucci, 2017) did not completely correct for two types of motion artifacts. First, it did not correct for small out-of-plane z motions that affected the focus of the image. Second, it did not correct for relative motion of the sample and spatially structured fluorescence excitation light. Structured illumination can dramatically improve SNR in stationary samples by minimizing out-of-focus fluorescence (Adam et al., 2019; Chien et al., 2017; Fan et al., 2020). However, brain motion shifts the signal sources relative to the illumination, leading to modulation of fluorescence that is not corrected by rigid-body image translations. To reduce the effects of these artifacts, we employed a generalized linear model to project motion-correlated signals out of each pixel in the denoised movie. We removed from each pixel any components of the signal correlated with the regressors [x(t), y(t), x2(t), y2(t), x(t)y(t)], where x(t) and y(t) represent the motion traces calculated by NoRMCorre.
In some movies, even after motion decorrelation, we observed an additional ~5–6 Hz periodic artifact because of the animal’s heartbeat. To remove this artifact, we first isolated the heartbeat signal by bandpass filtering the motion-decorrelated movie in a frequency window around the heartbeat frequency. We then conducted PCA on the filtered movie. The first principal spatial component constituted the spatial profile of the heartbeat artifact, which we kept for removing the heartbeat signal at a later step. Finally, to reduce contamination from blood flow, we added software functionality to manually select and mask regions containing blood vessels.
Demixing
After cleaning the noise from the movie, we approached the demixing problem of separating individual neurons and background spatial components, or footprints. We took advantage of the greater statistical independence of spiking compared with subthreshold voltages to identify the single-cell footprints. The PMD denoising step enabled us to isolate spiking signals at the single-pixel level with a temporal high-pass filter. Without PMD denoising, finding single-trial, single-pixel spikes would be impeded by temporally uncorrelated shot noise. To reduce analysis time for large fields of view (FOVs), we applied 2 × 2 binning to the denoised movie before demixing.
First, we applied a sliding-window temporal high-pass filter (cutoff of 10 ms) to each pixel in the denoised movie to remove the low-frequency subthreshold signals. The high-pass-filtered movie was then processed by a local nonnegative matrix factorization (localNMF) demixer, which detected cell profiles by identifying sets of highly correlated, spatially contiguous pixels (Buchanan et al., 2019). The initial demixer output, A0, consisted of the d × n matrix of cell footprints, where d is the number of pixels in a frame and n is the number of spiking cells. The value of n depends on a user-specified threshold for cross-correlations between different regions of the FOV. If the SNR is high, cells can be detected in principle from a single spike; if the SNR is low, more spikes may be necessary. Footprints from nearby cells were allowed to overlap. A threshold was applied to the initial estimates of the cell footprints, A0, to set small values to zero and thereby to delimit the spatial support of the cells. If there was a substantial heartbeat artifact in the movie, then we used linear regression to express each frame of the denoised movie (before high-pass filtering) as a linear combination of the cell footprints and the heartbeat spatial profile. We then removed the component apportioned to the heartbeat signal.
We then sought to identify the time course of the background dynamics. Background was defined as spatially extended signals that accounted for variance in the movie not accounted for by cell signals. PCA was applied to the original movie (after denoising but before high-pass filtering), with analysis restricted to pixels not in the thresholded A0: A user-specified number of background components was selected, typically ≤10. These components represent contributions to the fluorescence from out-of-focus cells, breathing artifacts, or residual motion artifacts. We assumed that the time course of the background contributions to each cell would be a linear combination of the off-cell background dynamics.
The next step was to apportion the on-cell dynamics between background and signal. Initially the background footprints were set to zero under all cells. We then used a fast hierarchical alternating least-squares (HALS) algorithm (Friedrich et al., 2017) to fit background weights and cell temporal components to the full denoised movie (including the low-frequency dynamics). By using cell spatial weights initialized from the spiking-only data, we ensured that the extracted low-frequency dynamics had the same spatial footprints as the spiking, a consequence of the electrotonic compactness of cell bodies. After iterating the least-squares fit, the output gave updated cell spatial footprints, A1, and a fit for the d × r matrix of background spatial footprints, B0, where r is the estimated rank of the background.
At this step, B0 typically contained contamination from A1; i.e., the spatial maps of the background had structures that resembled the in-focus cells. This crosstalk implied that subthreshold signals were not optimally apportioned between in-focus cells and background. To remedy this crosstalk, we sought to remove the spatial footprints of the cells from the images of the background. Here we used the fact that the out-of-focus background was spatially smoother than the in-focus signal.
We created updated background spatial components , where Wopt is a n × r matrix of weights selected to maximize a measure the spatial smoothness in each column of B1.
We chose each column, j, of the weight matrix by optimizing the following through gradient descent:
where is the nearest-neighbor discrete approximation to the gradient operator, and
where pixels (i, j) are in the edge set ε when the pixels are adjacent. The gradients at points where the objective was nondifferentiable were set to zero for the optimization procedure.
With the updated footprints, we implemented a least-squares estimate of membrane voltage by regressing the cell spatial components and updated background images on the full denoised and motion-corrected (but not high-pass-filtered) movie. This process gave the final output traces, which isolated cells and accurately separated their subthreshold signals from the background dynamics. A key feature of this algorithm is that it did not impose statistical independence of cell and background temporal sequences or spatial profiles.
Both the PMD denoising and the localNMF demixing steps scale linearly in computational cost with the number of movie frames, so to reduce the time for analysis, in our implementation, we ran the PMD denoising and localNMF demixing steps on only a subset of the frames of the movie (typically 6,000 frames for denoising and 10,000 frames for demixing) to obtain spatial filters (for denoising) or cell and background spatial footprints (for demixing), which were then applied to the whole movie. This approach reduced the total time of analysis for a 96 × 284 pixel movie of 60,000 frames to under 20 min on the Harvard Faculty of Arts and Sciences Research Computing (FASRC) Cannon cluster.
RESULTS
SGPMD-NMF pipeline reliably recovers cell signals in the presence of a correlated background
We verified the pipeline on simulated data containing two partially overlapping disk-shaped cells and a spatially heterogeneous time-varying rank-1 background (Figure 2A). To impose correlated subthreshold dynamics, we constructed sets of three subthreshold waveforms with a specified 3 × 3 cross-correlation matrix and then assigned one waveform to each of the cells and one to the background. The subthreshold oscillations had a root-meansquare (RMS) amplitude of 2 mV. We then added spikes (2-ms wide) atop the waveforms at independent Poisson-distributed intervals. Spike height and subthreshold power spectrum were selected to approximately correspond to in vivo recordings. To model the impact of shot noise, the overall brightness was scaled by an adjustable factor, and then the total photon counts at each pixel were selected from a Poisson random variable with a mean equal to the mean fluorescence for that pixel.
Figure 2. Validation of the SGPMD-NMF algorithm on simulated data.

(A) Example field of view comprising two simulated cells with spatial overlap and correlated subthreshold dynamics and a broad background whose dynamics were also correlated with each of the cells.
(B) Comparison of signal extraction via PCA-ICA versus SGPMD-NMF. The signals extracted via SGPMD-NMF were substantially closer to the ground truth than were the signals extracted via PCA-ICA.
(C) Quantification (mean ± SD) of SGPMD-NMF performance as a function of signal characteristics compared with PCA-ICA, ROI average, and PMD-NMF. Here is the cross-correlation of X and Y, C1 and C2 are the signals of the two cells, and B is the background. When subscripts are omitted, the calculation is averaged over both cells. Superscripts ‘‘in’’ and ‘‘out’’ refer to the ground truth voltage input and the extracted fluorescence output, respectively. ΔVrms is the root-meansquare voltage difference between extracted signal and ground truth voltage (lower values indicate better performance). In (i), (ii), (iv), (v), (vii), and (viii), (gray dashed line; closer proximity to this line indicates better performance). In (ii), (iii), (v), (vi), (viii), and (ix), movie brightness = 1. In a few trials, PMD-NMF failed to converge and the results were omitted. The number of such trials is listed in the first row in parentheses and applies to plots in the second and third rows.
We explored the ability of the algorithm to retrieve the input signals as a function of the overall movie brightness and the subthreshold correlations. The overall movie brightness (e.g., controlled by illumination intensity) determined the relative contributions of Poisson-distributed shot noise versus low-rank signal and background sources. First, we compared the performance of PCA-ICA and SGPMD-NMF (Figure 2B). In the displayed example, we analyzed a simulation in which the subthreshold correlations between cells and between each cell and the rank-1 background were all 0.5. When all traces were normalized (mean of 0 and standard deviation [SD] of 1), the RMS error of the traces extracted by PCA-ICA was 0.44, whereas for SGPMD-NMF, it was 0.11. Furthermore, the correlation between the extracted PCA-ICA traces for the two cells was 0.19, whereas the correlation between the extracted SGPMD-NMF traces was 0.53, close to the ground truth value of 0.5. This lower correlation from PCA-ICA compared with ground truth is expected, because ICA explicitly searches for independent components. Altogether, these results demonstrate that SGPMD-NMF offers superior performance compared with PCA-ICA in extracting subthreshold dynamics.
We then tested the ability of the SGPMD-NMF algorithmto preserve the correlations between cell 1 and cell 2, and between each cell and its respective ground truth, under different conditions of SNR and subthreshold correlations (Figure 2C, i–vi). Under all but the most stringent conditions, the algorithm extracted the input signals with high fidelity. A movie brightness of 1 represented the SNR of a typical in vivo recording from Adam et al. (2019). We also compared the performance of PCA-ICA, ROI average, and PMD-NMF against the results of SGPMD-NMF. In all settings, SGPMD-NMF displayed the best performance in terms of correspondence to ground truth and preservation of ground truth cell-to-cell subthreshold correlations.
To quantify the correspondence between the ground truth inputs and the SGPMD-NMF outputs, we calculated the RMS difference between input and output. Fluorescence recordings do not natively come with an absolute voltage scale, so to make the comparison, we scaled the fluorescence to match the 2 mV RMS amplitude of the subthreshold inputs. Under all conditions, except for the lowest movie brightness, the RMS difference between input and output was under 1 mV, indicating faithful extraction of ground truth voltages (Figure 2C, vii–ix). Compared against each of the other analysis pipelines, SGPMD-NMF had the lowest RMS difference between ground truth and extracted voltage.
SGPMD-NMF corresponds with electrophysiology ground truth in vivo
To assess the correspondence between SGPMD-NMF and electrophysiology ground truth in vivo, we compared in vivo fluorescence recordings of single neurons in mouse cortex L1 expressing Voltron with simultaneous whole-cell patch clamp (Figure 3). The patch-clamp data were smoothed and downsampled to match the 400 Hz sampling frequency of the voltage imaging data. Figure 3A shows that the spatial profile identified for the active cell was localized, and Figure 3B shows that the SGPMD-NMF extracted signal and spike profiles corresponded to the ground truth patch-clamp recording. Because of the sparse expression in these experiments, there was little background or signal contamination from other cells. As a result, all signal extraction approaches performed well, although across five cells, the average RMS difference between voltage imaging trace and ground truth was modestly lower for SGPMD-NMF (2.6 mV) compared with PMD-NMF (3.3 mV; p = 0.038, paired 1-sided t test, Figure 3C). The RMS difference was calculated in windows of 5,000 frames, the same window size used for detrending, and averaged across all windows in the recording.
Figure 3. Validation of SGPMD-NMF with in vivo electrophysiology.

(A) Spatial footprint (top) and signal (bottom) extracted from an in vivo voltage imaging recording (mouse cortex L1 expressing Voltron) with simultaneous patch clamp.
(B) Inset of signal shown in (A) with patch-clamp ground truth recording overlaid. Further insets are marked in green and magenta and shown to the right.
(C) RMS differences between electrophysiological ground truth and voltage imaging signal extracted by SGPMD-NMF (2.6 mV) and PMD-NMF (3.3 mV; mean displayed by bar, n = 5 independent recordings). p = 0.038, paired 1-sided t test.
SGPMD-NMF is robust to motion, background, and overlapping sources
To test the impact of the various steps of SGPMD-NMF pipeline, we extracted signals from a recording of a hippocampal neuron in an awake mouse expressing SomArchon1 using versions of SGPMD-NMF with individual steps omitted (Figures 4A–4D). Because it was not feasible to acquire simultaneous patch-clamp and voltage imaging recordings under these conditions, we qualitatively assessed whether the extracted signals were consistent with expected membrane voltage dynamics. Application of the full SGPMD-NMF pipeline identified a well-localized cell and clear spikes that rode atop subthreshold depolarizations (Figure 4A). When the motion regression step was omitted, nonphysiological negative-going transients contaminated the signal during periods of animal motion (Figure 4B). When the background smoothing step was omitted, the spike signals were substantially diminished relative to baseline noise because too much of the background crosstalk and noise was incorrectly ascribed to the cell (Figure 4C). With just PMD-NMF (Buchanan et al., 2019), the spatial footprint was poorly localized to the cell location, a consequence of the strong subthreshold correlation between in-focus cell and out-of-focus background (Figure 4D).
Figure 4. Validation of SGPMD-NMF algorithm against sources with motion, background, and overlapping cells.

(A–D) Four analyses of the same recording (mouse hippocampal CA1 pyramidal cells expressing SomArchon1). Top: image of the cell footprint. Middle: extracted fluorescence trace. Bottom: close-up of the fluorescence in the boxed region.
(A) Full SGPMD-NMF.
(B) SGPMD-NMF with the motion regression step omitted.
(C) SGPMD-NMF with the background smoothing step omitted.
(D) PMD-NMF.
(E) Left: composite movies were formed by adding two separately acquired single-cell movies (mouse hippocampal CA1 oriens interneurons expressing paQuasAr3-s) with a 20-pixel lateral offset between the cells. Right: signals were extracted from the source movies individually and from the composite movie jointly.
(F) Temporal cross-correlations of input and output traces showed good fidelity of extracted relative to input traces. Here is the cross-correlation of X and Y normalized by its value at a lag-1 time step (see STAR Methods for details). Shadings show mean ± SEM over 4 composite movies.
(G) Left: second set of composite movies was formed by adding two separately acquired single-cell movies of the same cell (mouse hippocampal CA1 oriens interneurons expressing paQuasAr3-s), one with the cell in focus and one with the cell 20 μm out of focus. The lateral offset between the two cells ranged from 0 pixels to 20 pixels. Right: signals were extracted from the source movies individually and from the composite movie jointly. In the composite movie, only the in-focus cell was extracted as a cell signal, and the out-of-focus cell was treated as background.
(H) Correlation of output traces with corresponding input traces as a function of overlap between the two cells. Error bars show mean ± SEM over 4 composite movies.
A challenge with validating the algorithm’s preservation of intercellular subthreshold correlations is that it is technically infeasible to make whole-cell patch-clamp recordings from multiple neurons in vivo while simultaneously performing voltage imaging. Thus, there was no ground truth data against which to compare the outputs of SGPMD-NMF when applied to multiple cells.
To address this challenge, we created composite movies by adding together real in vivo voltage imaging movies of well-isolated hippocampal neurons expressing paQuasAr3-s. By adjusting the lateral offset of the two cell images, we could control the degree of spatial overlap in the composite movie. This approach tested the algorithm beyond the worst case of in vivo crosstalk, because two real cells could never occupy the same volume.
We extracted the single-cell voltage traces from the individual recordings (giving input traces) and then applied SGPMD-NMF to the composite recordings (giving output traces) (Figure 4E). We performed the calculation for four pairs of cells (giving four trials). The correlation between the input traces was near zero, as expected for recordings taken at different times and different FOVs. In in vivo recordings, the distance of closest approach of in-focus cells is approximately 1 cell diameter, corresponding to 20 pixels in our movies. At this separation, the lag-1 cross-correlation (see STAR Methods for details) between the output traces of the two cells was (mean ± SEM), which was not significantly different from zero (see STAR Methods for rationale for using lag-1 correlations). The mean lag-1 correlation between each output trace and its corresponding input trace at this separation was . The temporal auto- and cross-correlation functions of the output traces closely matched the corresponding functions of the input traces (Figure 4F).
We next tested the algorithm performance in the presence of out-of-focus spatially overlapping sources. We added in vivo voltage imaging movies of well-isolated hippocampal neurons expressing paQuasAr3-s to subsequent recordings of the same cells taken with 20 μm defocus (Figure 4G). Addition of an out-of-focus background cell with a lateral offset of 20 pixels did not substantially perturb the extracted waveform of the in-focus cell, with (mean ± SEM, n = 4 pairs) (Figure 4H). Shifting the out-of-focus cell to directly beneath the in-focus cell lowered this correlation to .
SGPMD-NMF pipeline works for multiple species and reporters
After validating SGPMD-NMF in simulations and composite datasets, we then applied the pipeline to analyze multi-cell in vivo recordings acquired with different species, brain regions, cell types, reporters, and imaging modalities (Table 1). The reporters and imaging systems have all been published previously (Abdelfattah et al., 2019; Adam et al., 2019; Fan et al., 2020; Piatkevich et al., 2018). The zebrafish data were acquired on a previously unpublished transgenic line expressing zArchon1 under control of the Vglut2a enhancer (STAR Methods).
Table 1.
In vivo datasets analyzed via SGPMD-NMF
| Brain region | Cell type | Reporter | Imaging modality | Reference |
|---|---|---|---|---|
| Mouse cortex L1 (Figure 3) | interneurons (Ndnf+) | Voltron + patch clamp | wide-field epifluorescence | reporter: Abdelfattah et al., 2019 data: this work |
| Mouse hippocampus CA1 (Figure 4) |
oriens interneurons | paQuasAr3-s | micromirror-based, soma-targeted, structured illumination | data: Adam et al., 2019 |
| Mouse hippocampus CA1 (Figures 4 and 5; Video S1) |
pyramidal cells | SomArchon1 | micromirror-based, soma-targeted, structured illumination | reporter: Piatkevich et al., 2019
data from setup: Adam et al., 2019 |
| Mouse cortex L1 (Figure 6; Videos S2 and S5) |
interneurons (Ndnf+) | Voltron | wide-field epifluorescence | data: Abdelfattah et al., 2019 |
| Mouse cortex L1 (Figure S1; Video S3) |
interneurons (5-HT3AR+) | SomArchon1 | holographic peripheral membrane-targeted structured illumination | data: Fan et al., 2020 |
| Zebrafish spinal cord (Figure S2; Video S4) |
excitatory (Vglut2a+) | zArchon1 | light sheet | transgenics: this work reporter: Piatkevich et al., 2018 |
Diverse datasets included different species (mouse and zebrafish), regions of the central nervous system (hippocampus, cortex, and spinal cord), reporters (paQuasAr3-s, SomArchon1, zArchon1, and Voltron), and imaging modalities (structured illumination, holographic, wide-field epifluorescence, and light sheet). See also Figures S1 and S2 and Videos S1, S2, S3, S4, and S5.
Videos S1, S2, S3, and S4 show each step of the SGPMD-NMF pipeline for each of the preparations. SGPMD-NMF identified spiking cells and separately extracted time-dependent signals from in-focus cells and from multiple background components (Figures 5, 6, S1, and S2). The videos show that the initial denoising steps of SGPMD-NMF substantially reduced speckle, motion, and blood flow artifacts. The movies of the residuals (denoised movie minus signal and background) show that most sources of variation under spiking cells are accounted for in either signal or background and that the extracted waveforms of the spiking cells in the signal look biologically plausible (e.g., elevated spike rates during periods of subthreshold depolarization). The background movie is spatially smooth near the identified cells and shows low-frequency temporal dynamics that are highly correlated across many pixels.
Figure 5. Voltage imaging in the mouse hippocampus CA1 pyramidal cell layer (PCL) using SomArchon1.

Cells expressed SomArchon1 and were imaged via micromirror-based, soma-targeted, structured illumination.
(A) Top: image of the field of view, showing dense neurons as occurs in the pyramidal cell layer of CA1. The cell footprints are overlaid. Regions contaminated by blood flow are masked in white. Bottom: extracted single-cell traces. Subthreshold depolarizations clearly coincided with elevated spike rates, giving confidence that the subthreshold waveforms reflect membrane potential.
(B) Background components from SGPMD-NMF. The two components that explained the most variance in the movie are included, with each component’s spatial profile above the corresponding temporal trace.
(C) Average across pixels in cell 5 in the denoised movie (mean ROI), SGPMD-NMF reconstructed signal movie (SGPMD-NMF), reconstructed background movie (background), and residual movie (residual).
(D) Scatterplot of the relative variance of each cell background versus signal.
(E) Comparison of the pairwise cell-cell cross-correlations between SGPMD-NMF and PCA-ICA. Most (12 of 15) pairwise correlations had a smaller magnitude for PCA-ICA versus SGPMD-NMF.
Figure 6. Voltage imaging in mouse cortex L1 using Voltron.

Cells expressed Voltron and were imaged via wide-field epifluorescence.
(A) Top: image of the field of view. The cells are labeled at the centroid of their footprints. Bottom: extracted single-cell traces. Insets shown in (C) are marked in red and black.
(B) Background components from SGPMD-NMF. The two components that explained the most variance in the movie are included, with each component’s spatial profile above the corresponding temporal trace.
(C) Inset of eight single-cell traces over a window of approximately 3 s.
(D) Average over pixels in cell 6 in the denoised movie (mean ROI), SGPMD-NMF reconstructed signal movie (SGPMD-NMF), reconstructed background movie (background), and residual movie (residual).
(E) Scatterplot of the relative variance of each cell background versus signal. Anticorrelation between relative variance of background and signal results because together, background and signal account for >99% of the total variance. If background and signal were uncorrelated, they would fall along the line x + y = 1. Deviations below the line x + y = 1 indicate positive correlation between signal and background.
(F) Top: SD image of the reconstructed sum of background and residual. Magenta masks indicate 14 manually selected bright spots, or missed cells. Bottom: mean ROI (on denoised movie) of the 14 missed cells. These traces showed little or no spiking activity.
(G) Skewness of temporally high-pass-filtered mean ROI traces (on denoised movie) of the 14 missed cells and 59 detected cells. Skewness provides a measure of spiking activity relative to baseline noise. Error bars show mean ± SEM. The missed cells displayed substantially less spiking activity compared with the detected cells.
To assess the performance of SGPMD-NMF, we compared the SGPMD-NMF outputs to what one would obtain from a flat average across each cell footprint after denoising, i.e., the mean of the ROI (Figures 5C, 6D, S1C, and S2C). The ROI-based signals contained all sources of background, whereas SGPMD-NMF traces isolated cell signals from background. To quantify the comparison, we quantified the ratios of the variances of the SGPMD-NMF signal, background, and residual traces to the variance of the mean ROI trace (Table S1). Most (>99%) of the variance of the denoised movie within the cell ROIs was explained by a combination of signal and background, demonstrating that SGPMD-NMF accounted for almost all time-varying signals in the movies. Specifically, except for the zebrafish spinal cord dataset, the variance of the signal was on average larger than the variance of the background, indicating the cell regions had a strong signal. For the zebrafish spinal cord dataset, we found that the background had a larger variance than the signal because of strong contributions of blood flow to the background.
Using the mouse hippocampus data, we also conducted a comparison of SGPMD-NMF to PCA-ICA (Figure 5E). For the six cells identified in both methods, we calculated the 15 pairwise correlations between cell signals. For all but 3 cell pairs, which happened to have low cross-correlations, the magnitude of correlation of the PCA-ICA signals was lower than that of the SGPMD-NMF signals. Based on analysis with simulated data (Figure 2), we infer that PCA-ICA likely underestimated cell-cell cross-correlations in membrane voltage.
SGPMD-NMF analysis of the data from mouse cortex L1 using the Voltron reporter identified 59 spiking cells in a single FOV, including several overlapping footprints. However, the residual contained several objects that looked like bright cells but were not picked up by the algorithm. To determine the reason for this, we studied the signals from ROIs around these cells. These cells all had low SNR and lacked clearly discernible spikes (Figure 6F). We quantified the number and amplitude of spikes via skewness (Figure 6G). The mean skewness of the ROI signals of these cells after a 250 ms sliding-window temporal high-pass filter was 0.14 ± 0.03 (mean ± SEM). For cells identified by SGPMD-NMF, the same metric was 0.88 ± 0.09. We inferred that SGPMD-NMF did not pick up these cells because they did not pass the initial spike-finding step. To determine whether we could identify nonspiking cells, we analyzed a cropped portion of the dataset (Video S5). We employed a multi-pass strategy for localNMF that identified additional cells with lower SNR (Buchanan et al., 2019).
Finally, we applied the SGPMD-NMF pipeline to recordings in mouse cortical L1 cells expressing SomArchon1 (Figure S1) and to zebrafish spinal cord expressing the zebrafish-optimized variant, zArchon1 (Figure S2). In both cases, the pipeline extracted realistic-looking cell morphologies and spikes riding atop subthreshold depolarizations.
DISCUSSION
In vivo voltage imaging is a promising approach for studying the intercellular correlations in spiking and subthreshold dynamics. However, the data analysis must be performed with great care to avoid introducing spurious correlations or systematic artifacts. We have demonstrated a robust software pipeline for demixing voltage signals and background in complex, noisy tissues.
Parameter tuning is an important aspect of obtaining results from SGPMD-NMF. In the denoising steps, the temporal detrending filter window and block size for the block-wise PMD can be adjusted. We find that results are robust to small changes in both parameters. A filter window of 5,000 frames and a block size approximately the size of one neuron led to good results on movies sampled at 1 kHz and 400 Hz. The other parameters of the PMD denoiser are automatically set by simulation (Buchanan et al., 2019). For the demixing steps, the high-pass filter window for isolating spikes, number of background components, and localNMF parameters can be adjusted. A high-pass filter window of between 5 and 10 ms worked well across all tested datasets. A good number of background components can be found by the greatest number of components such that the resulting background traces do not have spikes. The localNMF parameters can be tuned by checking that the neuron spatial profiles are localized appropriately. These parameters typically do not need tuning across movies of similar SNR. The limited parameter tuning of SGPMD-NMF compares favorably with techniques in which, e.g., signal and background are defined manually. In such cases, there is often ambiguity about how to distinguish signal from background and how to avoid contamination from nearby or out-of-focus sources.
An outstanding challenge is to identify strategies for creating masks that optimize the SNR of the extracted traces. Both shot noise and systematic noise (e.g., artifacts from blood flow, uncorrected motion, and diffusing intracellular vesicles) vary over a cell. The many combinations of pixel weights give, on average, the correct voltage but differ in noise statistics. If the noise is independent across pixels, then the HALS approach optimizes the signal. When this assumption is not true, appropriate incorporation of a spatiotemporally correlated noise model could improve the quality of the extracted traces. Automated detection of blood vessels would also decrease manual labor in running the pipeline.
An independent voltage imaging spike detection pipeline, VolPy, was recently introduced by Cai et al. (2020). This pipeline is largely complementary to ours, because it focuses on extracting spike times, rather than accuracy of subthreshold dynamics. VolPy uses a supervised neural network to find neurons based on summary images of the datasets. A similar approach could be adopted here to use cell morphology to identify both spiking and nonspiking cells as initial footprint guesses in the SGPMD-NMF demixing step; this will be an interesting direction to explore in future work. Compared with VolPy, SGPMD-NMF offers more effective demixing in several respects. First, denoising of temporally uncorrelated noise via PMD allows improved accuracy in demixing (Buchanan et al., 2019). Second, SGPMD-NMF enables recovery of subthreshold signals and correlations with high fidelity by incorporating a background model that can have temporal dynamics correlated with cell signals. Without a proper method to account for the background, subthreshold signals of extracted cells will be inaccurate. Third, blood artifacts, which are not considered by VolPy, contaminate extracted cell signals, because regions of blood flow often overlap with cell footprints. In many cases, these blood artifacts are difficult to see in the raw data but become readily apparent after the denoising step used here. Although our method does not include a spike-finding step, such algorithms are available in the literature and can be appended to our pipeline. Specifically, the SpikePursuit algorithm (Abdelfattah et al., 2019) and the improved version incorporated in the VolPy package (Cai et al., 2020) have been optimized for fluorescent voltage recordings.
With progress in 2P voltage imaging (Villette et al., 2019), the challenges of optical crosstalk are expected to abate, but other challenges arise. Voltage signals only come from the intersection of the submicron 2P laser focus and the nanometers-thick cell membrane, leading to extreme sensitivity to motion artifacts. Furthermore, the stringent optical sectioning of 2P microscopy implies that all fluorescence must originate in a narrow equatorial belt of the cell, compared with 1P microscopy, which can average over the entire surface area of the cell. As a result, the requirements on per-molecule fluorescence are more demanding in 2P than in 1P microscopy, so photobleaching is more of a concern. In movies in which photobleaching is substantial, one could still apply SGPMD-NMF to identify cell and background footprints on the early part of the recording when SNR is high and then apply these footprints to later parts of the recording to get cell temporal signals for the full movie. Although photobleaching-induced loss of SNR is unavoidable, this approach will make the best use of the available data.
A fascinating application of GEVI technology would be to map dendritic voltages in vivo. This application would require broadly trafficked GEVIs, as opposed to soma-localized GEVIs; advances in imaging to sample fine dendritic processes; and novel signal extraction algorithms to accommodate that subthreshold and spiking waveforms may vary in different ways across the dendritic tree.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead contact, Adam E. Cohen (cohen@chemistry.harvard.edu).
Materials availability
Optogenetic constructs used in this study are available from Addgene as paQuasAr3-s (Addgene 107703, 107704), SomArchon1 (Addgene 126943, 126512), Voltron (Addgene 119036), zArchon1 (Addgene 108427).
Data and code availability
Code to run SGPMD-NMF on an example dataset is available on GitHub here: https://github.com/adamcohenlab/invivo-imaging. Instructions for installing and running the code are here: http://bit.ly/sgpmdnmf-instructions. All SGPMD-NMF analyses done in the paper were run on the Harvard FASRC Cannon cluster. The TreFiDe dependency package requires Linux; other aspects of the code do not have operating system prerequisites.
The simultaneous in vivo whole-cell patch-clamp recording and voltage imaging dataset is available at https://dx.doi.org/10.25378/janelia.c.5325254 (Rozsa et al., 2021). The zebrafish spinal cord voltage imaging dataset is available at https://doi.org/10.6084/m9.figshare.14153339 (Böhm et al., 2021). All other voltage imaging datasets analyzed have been previously published (Table 1).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Harvard University or of Janelia Farm Research Campus.
Simultaneous whole-cell patch-clamp recording and voltage imaging in vivo
In vivo whole-cell recordings were made in NDNF-Cre mice (JAX 28536) (4 females, 3 males; 54–160 days old at the time of the first surgery).
Zebrafish spinal cord
Recordings were done in 5 day-post-fertilization (dpf) transgenic zebrafish larvae expressing UAS:zArchon1-GFP under the control of vGlut2a:Gal4 (Satou et al., 2013). The sex of the larvae was not determined because it is not practical to determine sex at this developmental stage.
METHOD DETAILS
Simultaneous whole-cell patch-clamp recording and voltage imaging in vivo
AAV2/1-syn-FLEX-Voltron-ST (Abdelfattah et al., 2019) (titer: 2×1012 GC/ml) was injected at 6–10 injection sites 200 μm deep into the somatosensory cortex (30 nL each; injection rate, 1 nl/s) (Liu et al., 2020). Headbars and cranial windows (2.5 mm diameter) were implanted centered on 1.6 mm lateral, 1.2 mm posterior from lambda (Daie et al., 2021). To prepare the JF dye for injection, 100 nanomoles of lyophilized JF525 were dissolved in 20 μl of DMSO, 20 μl Pluronic F-127 (20% w/v in DMSO), and 60 μl of PBS (final dye concentration 1 μM). 33–71 days after the first surgery, mice were anesthetized with 2%–3% isoflurane and 100 μL of the dye solution was injected into the retro-orbital sinus of the right eye using a 30 gauge needle. One day later we removed the cranial window and performed durotomy (Goldey et al., 2014). The craniotomy was filled with 10–15 μL of 1% agarose, then a D-shaped coverslip was secured on top to suppress brain motion, but leaving access to the brain on the lateral side of the craniotomy.
Micropipettes (3–6 MΩ) were filled with (in mM) 126 K-gluconate, 4 KCl, 4 ATP-Mg, 0.3 GTP-Na2, 10 HEPES, 10 creatine phosphate (pH 7.25; 300 mOsm). Somatic whole-cell recordings were obtained from layer 1 interneurons (20–100 μm depth from brain surface) visualized with infrared oblique illumination (Szucs et al., 2009). The infrared LED (Osram SFH 4550, 850 nm) was attached to the imaging objective with a custom-printed holder and illuminated the craniotomy 30–45° from normal. Warm saline (35–37°C) was perfused in the craniotomy to keep the cortex at physiological temperature. We recorded in current clamp mode. Signals were filtered at 20 kHz (Multiclamp 700B, Axon Instruments) and digitized at 100 kHz using acq4 (Campagnola et al., 2014). A wide-field fluorescence microscope equipped with a water immersion objective (40X, NA 0.8, Olympus) was used for imaging. Illumination was delivered using a 525 nm LED (Mightex, LCS-0525–60-22); intensity at the sample, < 20 mW/mm2. A custom filter set (517/20 nm (excitation, Semrock FF01–517/20), 537LP nm (emission, Chroma RET537lp), and a 532LP dichroic mirror (Chroma, RT532rdc)) was used for fluorescence imaging of Voltron525. Images were collected using a sCMOS camera (Hamamatsu Orca Flash 4.0 v3) at frame rates of 400 Hz or 1000 Hz. The pixel size was 0.502 μm/pixel. Mice were under isoflurane anesthesia and imaged in darkness.
Zebrafish spinal cord
Larvae were paralyzed by immersion in 1 mg/ml α-bungarotoxin for ~1 min and mounted in a drop of 1.5% low melting point agarose. Imaging was done on a custom light sheet microscope using a 639 nm red laser (MLL-FN-639, CNI lasers) to illuminate a 480 μm wide region with ~300 mW of laser light. Images were collected through a low magnification high NA objective (XLPLN25XWMP2, Olympus) a 100 mm tube lens (TTL100-A, Thorlabs) and a 664 nm long pass filter. Images were recorded at 1 kHz on a sCMOS camera (Hamamatsu Flash 4.0). During the recording, larvae were presented with a forward moving grating at 15 mm/s to induce swimming. To ensure naturalistic behavior, the ventral nerve root signal was electrophysiologically recorded at the same time and the fictive swim signal fed back to control the speed of the backward motion on the grating as described in Ahrens et al. (2013).
QUANTIFICATION AND STATISTICAL ANALYSIS
Lag-1 cross-correlation calculation for composite movie analysis
We calculated , the lag-1 cross-correlation as follows. First, we denote the cross-correlation function of X and Y as , which is a discrete function centered at t = 0. t denotes the lag. Then,
which is a function of time. The lag-1 cross-correlation reduces the effect of uncorrelated shot noise on the correlation between noisy signals X and Y.
PCA-ICA analysis
All PCA-ICA analyses were conducted on full movies that had been denoised following SGPMD-NMF. The movies were then high-pass filtered in time with a 50 Hz high-pass filter. The high pass filtered movies were then segmented using PCA followed by time-domain ICA (Mukamel et al., 2009). The maximum number of sources from PCA-ICA was set to 20, and further components that did not correspond to cells were eliminated manually by inspection of the temporal and spatial components.
Supplementary Material
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
| Deposited data | ||
| Simultaneous in vivo
whole-cell patch-clamp recording and voltage imaging (Voltron in mouse L1) |
This paper | https://dx.doi.org/10.25378/janelia.c.5325254 |
| zebrafish spinal cord voltage
imaging (zArchon1) |
This paper | https://doi.org/10.6084/m9.figshare.14153339 |
| Experimental models: Organisms/strains | ||
| Mouse: NDNF-Cre: B6.Cg-Ndnftm1.1(folA/cre)Hze/J | Jackson Laboratory | Stock #028536 |
| Zebrafish: Tg(vGlut2a:Gal4), Tg(UAS:zArchon-GFP) | vGlut2a:Gal4: National BioResource Project, Zebrafish, Core Institution, Japan; UAS:zArchon-GFP: Cohen lab | N/A |
| Recombinant DNA | ||
| paQuasAr3-s | Addgene | 107703, 107704 |
| SomArchon1 | Addgene | 126943, 126512 |
| Voltron | Addgene | 119036 |
| zArchon1 | Addgene | 108427 |
| Software and algorithms | ||
| SGPMD-NMF | This paper | https://github.com/adamcohenlab/invivo-imaging |
| TreFiDe | Buchanan et al., 2019 | https://github.com/ikinsella/trefide |
| Python | Python Software Foundation | https://www.python.org/ |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
Highlights.
SGPMD-NMF is a signal extraction pipeline for in vivo voltage imaging recordings
Recovers supra-and subthreshold signals in the presence of correlated background
Extracted voltage signals correspond to patch-clamp electrophysiology in vivo
Effective across multiple species and voltage indicators
ACKNOWLEDGMENTS
M.E.X., Y.A., L.Z.F., U.L.B., and A.E.C. were supported by the Harvard Data Science Initiative, NIH grant R01MH117042, and the Howard Hughes Medical Institute. I.K., D.Z., and L.P. were supported by NIH grant R01EB22913. A.S., M.R., and K.S. were supported by the Howard Hughes Medical Institute. We thank C. Cai and A. Giovannuci for helpful discussions. We thank T. Kawashima, M. Ahrens, E. Jung, K. Piatkevich, and E. Boyden for zArchon1 zebrafish transgenics. The computations in this paper were run on the FASRC Cannon cluster supported by the FAS Division of the Science Research Computing Group at Harvard University.
Footnotes
DECLARATION OF INTERESTS
A.E.C. is a co-founder of Q-State Biosciences. The other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108954.
REFERENCES
- Abdelfattah AS, Kawashima T, Singh A, Novak O, Liu H, Shuai Y, Huang YC, Campagnola L, Seeman SC, Yu J, et al. (2019). Bright and photostable chemigenetic indicators for extended in vivo voltage imaging. Science 365, 699–704. [DOI] [PubMed] [Google Scholar]
- Adam Y, Kim JJ, Lou S, Zhao Y, Xie ME, Brinks D, Wu H, Mostajo-Radji MA, Kheifets S, Parot V, et al. (2019). Voltage imaging and optogenetics reveal behaviour-dependent changes in hippocampal dynamics. Nature 569, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrens MB, Orger MB, Robson DN, Li JM, and Keller PJ (2013). Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat. Methods 10, 413–420. [DOI] [PubMed] [Google Scholar]
- Bando Y, Grimm C, Cornejo VH, and Yuste R (2019). Genetic voltage indicators. BMC Biol. 17, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm UL, Xie ME, and Cohen AE (2021). Voltage imaging in zebrafish spinal cord with zArchon1. figshare. 10.6084/m9.figshare.14153339.v1. [DOI]
- Buchanan EK, Kinsella I, Zhou D, Zhu R, Zhou P, Gerhard F, Ferrante J, Ma Y, Kim SH, Shaik MA, et al. (2019). Penalized matrix decomposition for denoising, compression, and improved demixing of functional imaging data. bioRxiv. 10.1101/334706. [DOI]
- Cai C, Friedrich J, Pnevmatikakis EA, Podgorski K, and Giovannucci A (2020). VolPy: automated and scalable analysis pipelines for voltage imaging datasets. bioRxiv. 10.1101/2020.01.02.892323. [DOI] [PMC free article] [PubMed]
- Campagnola L, Kratz MB, and Manis PB (2014). ACQ4: an open-source software platform for data acquisition and analysis in neurophysiology research. Front. Neuroinform 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien MP, Brinks D, Adam Y, Bloxham W, Kheifets S, and Cohen AE (2017). Two-photon photoactivated voltage imaging in tissue with an Archaerhodopsin-derived reporter. bioRxiv. 10.1101/211946. [DOI] [PMC free article] [PubMed]
- Daie K, Wang T, Singh A, Finkelstein A, Kim JJ, Rozsa M, and Svoboda K (2021). ALM Window Surgery. protocolsio. 10.17504/protocols.io.zhgf33w. [DOI]
- Fan LZ, Kheifets S, Böhm UL, Wu H, Piatkevich KD, Xie ME, Parot V, Ha Y, Evans KE, Boyden ES, et al. (2020). All-optical electrophysiology reveals the role of lateral inhibition in sensory processing in cortical layer 1. Cell 180, 521–535.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhi SL, Parot VJ, Grama A, Yamagata M, Abdelfattah AS, Adam Y, Lou S, Kim JJ, Campbell RE, Cox DD, and Cohen AE (2019). Wide-Area All-Optical Neurophysiology in Acute Brain Slices. J. Neurosci 39, 4889–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich J, Yang W, Soudry D, Mu Y, Ahrens MB, Yuste R, Peterka DS, and Paninski L (2017). Multi-scale approaches for high-speed imaging and analysis of large neural populations. PLoS Comput. Biol 13, e1005685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldey GJ, Roumis DK, Glickfeld LL, Kerlin AM, Reid RC, Bonin V, Schafer DP, and Andermann ML (2014). Removable cranial windows for long-term imaging in awake mice. Nat. Protoc 9, 2515–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JJ, Steinmetz NA, Siegle JH, Denman DJ, Bauza M, Barbarits B, Lee AK, Anastassiou CA, Andrei A, Aydın ç., et al. (2017). Fully integrated silicon probes for high-density recording of neural activity. Nature 551, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöpfel T, and Song C (2019). Optical voltage imaging in neurons: moving from technology development to practical tool. Nat. Rev. Neurosci 20, 719–727. [DOI] [PubMed] [Google Scholar]
- Lampl I, Reichova I, and Ferster D (1999). Synchronous membrane potential fluctuations in neurons of the cat visual cortex. Neuron 22, 361–374. [DOI] [PubMed] [Google Scholar]
- Liu L, Chen S, Li N, and Svoboda K (2020). Virus injection. protocolsio. 10.17504/protocols.io.bctxiwpn. [DOI]
- Mukamel EA, Nimmerjahn A, and Schnitzer MJ (2009). Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63, 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzera LC, and Hoppa MB (2019). Genetically Encoded Voltage Indicators Are Illuminating Subcellular Physiology of the Axon. Front. Cell. Neurosci 13, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatkevich KD, Jung EE, Straub C, Linghu C, Park D, Suk HJ, Hochbaum DR, Goodwin D, Pnevmatikakis E, Pak N, et al. (2018). A robotic multidimensional directed evolution approach applied to fluorescent voltage reporters. Nat. Chem. Biol 14, 352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatkevich KD, Bensussen S, Tseng HA, Shroff SN, Lopez-Huerta VG, Park D, Jung EE, Shemesh OA, Straub C, Gritton HJ, et al. (2019). Population imaging of neural activity in awake behaving mice. Nature 574, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pnevmatikakis EA, and Giovannucci A (2017). NoRMCorre: An online algorithm for piecewise rigid motion correction of calcium imaging data. J. Neurosci. Methods 291, 83–94. [DOI] [PubMed] [Google Scholar]
- Pnevmatikakis EA, Soudry D, Gao Y, Machado TA, Merel J, Pfau D, Reardon T, Mu Y, Lacefield C, Yang W, et al. (2016). Simultaneous Denoising, Deconvolution, and Demixing of Calcium Imaging Data. Neuron 89, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozsa M, Singh A, and Svoboda K (2021). Voltron 1.0 simultaneous imaging and whole cell patch clamp recordings in NDNF-Cre mice. figshare. 10.25378/janelia.c.5325254. [DOI]
- Satou C, Kimura Y, Hirata H, Suster ML, Kawakami K, and Higashijima S (2013). Transgenic tools to characterize neuronal properties of discrete populations of zebrafish neurons. Development 140, 3927–3931. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Rancz EA, Roth A, and Häusser M (2008). Dendritic excitability and synaptic plasticity. Physiol. Rev 88, 769–840. [DOI] [PubMed] [Google Scholar]
- Sofroniew NJ, Flickinger D, King J, and Svoboda K (2016). A large field of view two-photon mesoscope with subcellular resolution for in vivo imaging. eLife 5, e14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szucs P, Pinto V, and Safronov BV (2009). Advanced technique of infrared LED imaging of unstained cells and intracellular structures in isolated spinal cord, brainstem, ganglia and cerebellum. J. Neurosci. Methods 177, 369–380. [DOI] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, et al. (2009). Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villette V, Chavarha M, Dimov IK, Bradley J, Pradhan L, Mathieu B, Evans SW, Chamberland S, Shi D, Yang R, et al. (2019). Ultrafast Two-Photon Imaging of a High-Gain Voltage Indicator in Awake Behaving Mice. Cell 179, 1590–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HH, and St-Pierre F (2016). Genetically Encoded Voltage Indicators: Opportunities and Challenges. J. Neurosci 36, 9977–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code to run SGPMD-NMF on an example dataset is available on GitHub here: https://github.com/adamcohenlab/invivo-imaging. Instructions for installing and running the code are here: http://bit.ly/sgpmdnmf-instructions. All SGPMD-NMF analyses done in the paper were run on the Harvard FASRC Cannon cluster. The TreFiDe dependency package requires Linux; other aspects of the code do not have operating system prerequisites.
The simultaneous in vivo whole-cell patch-clamp recording and voltage imaging dataset is available at https://dx.doi.org/10.25378/janelia.c.5325254 (Rozsa et al., 2021). The zebrafish spinal cord voltage imaging dataset is available at https://doi.org/10.6084/m9.figshare.14153339 (Böhm et al., 2021). All other voltage imaging datasets analyzed have been previously published (Table 1).