

Abstract
Our understanding of the potential role of sodium channels in multiple sclerosis (MS) has grown substantially in recent years. The channels have long had a recognized role in the symptomatology of the disease, but now also have suspected roles in causing permanent axonal destruction, and a potential role in modulating the intensity of immune activity. Sodium channels might also provide an avenue to achieve axonal and neuronal protection in MS, thereby impeding the otherwise relentless advance of permanent neurological deficit. The symptoms of MS are largely determined by the conduction properties of axons and these, in turn, are largely determined by sodium channels. The number, subtype and distribution of the sodium channels are all important, together with the way that channel function is modified by local factors, such as those resulting from inflammation (eg, nitric oxide). Suspicion is growing that sodium channels may also contribute to the axonal degeneration primarily responsible for permanent neurological deficits. The proposed mechanism involves intra‐axonal sodium accumulation which promotes reverse action of the sodium/calcium exchanger and thereby a lethal rise in intra‐axonal calcium. Partial blockade of sodium channels protects axons from degeneration in experimental models of MS, and therapy based on this approach is currently under investigation in clinical trials. Some recent findings suggest that such systemic inhibition of sodium channels may also promote axonal protection by suppressing inflammation within the brain.
INTRODUCTION
The course of multiple sclerosis (MS) is very variable between patients, but there is commonly an initial relapsing/remitting period characterized by “attacks” from which a full recovery may be made, followed after a decade or so by a more progressive course when there is a steady accumulation of permanent neurological deficit. The neurological deficits in the relapsing/remitting and progressive phases tend to have different underlying mechanisms, and sodium channels may contribute to each in different ways. Our knowledge of how sodium channels contribute to the production of neurological deficits far outweighs our knowledge of how (and indeed whether) they contribute to the immunological mechanisms of MS, but some recent findings suggest they may affect the properties of immunological cells.
To understand the role of sodium channels in MS it may be helpful briefly to summarize their elementary properties. The sodium channels of interest in MS are mainly voltage gated and located in the cell membranes of neurons and their axons, or in the membranes of immune cells. Voltage‐gated sodium channels are normally closed, but they open transiently in response to depolarization of the membrane in which they are embedded. A small depolarization may only open a small percentage of the channels but although the channels may often open for only less than 1 ms, while they are open a stream of sodium ions flows through the channels into the cell. Sodium ions are positively charged and so they carry a current across the membrane and this serves to depolarize the cell still further, promoting the opening of additional sodium channels. In this way many sodium channels can open almost simultaneously, and, in excitable cells such as neurons, this can create the “explosion” of inward current that characterizes an action potential. Once open, sodium channels close automatically, often within a millisecond, although, as we shall see later, under pathological conditions the behavior of the channels changes and then the sodium current may become more persistent.
SODIUM CHANNELS AND THE SYMPTOMATOLOGY OF MULTIPLE SCLEROSIS
Loss of function—relapses
Demyelination. Relapses in MS are typically characterized by a temporary (weeks or months) loss of function resulting in symptoms such as paralysis, blindness and numbness. Such deficits are primarily attributable to axonal conduction block, and probably the most important cause of this is demyelination. (Inflammation might also be able to block conduction in otherwise normal axons, as discussed below.) Demyelination is a potent cause of axonal dysfunction (135), especially if whole internodes of myelin are lost (ie, segmental demyelination), as commonly occurs in MS. Several factors play a role in the conduction block, but dominant is the fact that although sodium channels are aggregated in very high density (∼1000/µm2) precisely at the nodes of Ranvier, there is only a low density (<25/µm2) normally located beneath the myelin sheath (116, 143). This pattern is ideal for saltatory conduction, but if the myelin is removed by demyelination, as occurs in MS, it exposes a long length of axonal membrane that appears to be inexcitable. Some computer simulations predict that conduction may be possible along such demyelinated axolemma (23, 145, 149), but this has yet to be shown experimentally. The (relative) inexcitability of the freshly demyelinated membrane is devastating for conduction because the gap exposed by the loss of a whole myelinated internode is far too long for the current generated by the last active node, or hemi‐node (ie, the “node” at the start of the demyelinated stretch), to be able to excite the next hemi‐node, in the absence of myelin to guide the current and reduce capacitance. Conduction therefore fails. This conduction failure is arguably the most important cause of neurological deficit in MS. The failure was predicted by Charcot (37), and it was the first deficit to be detected experimentally, first in the peripheral nervous system (49, 92) and later in the central (94, 95, 96). The conduction block is not necessarily permanent however, as discussed below.
Conduction can also fail even when demyelination is less extensive and confined only to the paranodes resulting in nodal widening (63, 87, 139), a pathology known to occur in MS (153). Failure in this case arises from the increase in membrane capacitance at the widened node, which can require more current to depolarize it than is available. The widened nodal gap also means that the exciting local current flowing out at the node is no longer funneled to flow specifically across the nodal membrane, but rather it becomes dissipated over a larger area.
A potential cause of conduction failure in MS is the loss of sodium channels at nodes of Ranvier, as has been reported in animals with peripheral autoimmune inflammatory demyelinating disease (102). However, whether such loss occurs in MS is not known.
Inflammation/immunology. There is a growing realization that inflammation may be an important cause of temporary neurological deficits in MS (14, 155). The case can be made most clearly when the deficits are quite brief (days), because then they are not easily explained by demyelination. Perhaps the most convincing evidence has arisen from observations made during therapy with Campath‐1H, a humanized monoclonal antibody directed against the CD52 antigen expressed by lymphocytes and monocytes (99). Within hours of the intravenous administration of the antibody, patients with MS experienced a resurrection of symptoms that had been expressed earlier in the course of their disease, but from which they had made a recovery. As old symptoms were resurrected rather than new ones appearing, the deficits presumably arose from conduction block in axons that had been previously damaged by the disease process, but the mechanism remained unclear. The resurrected symptoms were expressed for less than a day, ruling out any mechanisms based on structural changes such as demyelination with repair by remyelination. Conduction block caused by pyrexia (Uhthoff’s phenomenon, see below) was also ruled out by other observations. The administration of the Campath‐1H provoked a surge in the circulating concentration of cytokines, particularly tumor necrosis factor‐α, interferon‐γ and interleukin‐6 (99, 152), suggesting that cytokines may have directly impaired function in (presumably) demyelinated axons, although subsequent observations (38) indicated that the prime suspect, tumor necrosis factor‐α, was not responsible. Consistent with this interpretation, pretreatment of the patients with intravenous methylprednisolone was found to prevent both the cytokine surge and the expression of symptoms, suggesting that the cytokine/inflammatory response was directly involved. However, experimental study did not provide confirmation that these cytokines had direct effects on conduction (113). It now seems that the best explanation for the puzzle involves nitric oxide. Although not affecting axons directly, the cytokine surge would be expected to provoke the production of nitric oxide by the microglial and inflammatory cells located in the existing lesions, and nitric oxide can block axonal conduction, particularly in demyelinated axons (see below). Thus, at low concentrations of nitric oxide demyelinated axons would be preferentially affected, providing a plausible explanation for the transient exacerbation provoked by Campath‐1H administration.
It is also possible that other factors associated with inflammation may cause more prolonged neurological deficits by affecting sodium channels, although the evidence is weak and confusing. The concept of circulating factors that could block neurological function was introduced in 1965 (22), but despite a number of studies inspired by these early observations the identity of the factors was, and has remained, obscure [see discussion in Smith (127)]. A pentapeptide named endocaine, or QYNAD (an acronym standing for the sequence of amino acids) was more recently reported to be present specifically in the cerebrospinal fluid of patients with the inflammatory neurological disorders MS or Guillain‐Barré syndrome and to have a local anesthetic‐like effect on sodium channels, like lignocaine (6, 30). The implication that QYNAD might be responsible for neurological deficits in MS was greeted with interest, particularly when QYNAD was reported to block conduction in sciatic nerve (151). However, initial excitement was dampened by the failure of several laboratories to reproduce the findings on sodium channels (44, 112) or to find effects on the electrical activity of a neuronal network (105). Disturbingly, some of the original observations have been retracted (31), but not the main effects on sodium channels, and these have also received some support from other studies (97, 106). At this time, QYNAD’s properties remain potentially interesting, but extraordinarily elusive.
It has long been conjectured that antibodies might affect axonal conduction in MS, perhaps by binding to sodium channels (146, 147), but until recently there was little convincing evidence in support. However, preliminary findings have now been presented that anti‐neurofascin antibodies are present in MS; and that monoclonal antibodies to neurofascin colocalize with sodium channels at central nodes of Ranvier (90). Moreover, administration of the monoclonal antibodies to animals with experimental autoimmune encephalomyelitis (EAE; a model of MS) exacerbated the severity of disease, raising the possibility that, in MS, anti‐neurofascin antibodies may have deleterious electrophysiological consequences promoting loss of function.
Another study has recently described conduction block in the rat optic nerve induced by cerebrospinal fluid from some patients with MS (36). Whether this effect is mediated by effects on sodium channels is not yet clear. Similarly, there is equivocal evidence that T cells can block axonal conduction (50, 154), but as the mechanism may not involve sodium channels it will not be discussed further here.
Other factors associated with the inflammatory response that can affect sodium channel function, and so conceivably the expression of symptoms, are some cytokines and prostaglandins. For example, interleukin‐2 can affect sodium currents, at least in muscle (77), and tumor necrosis factor‐α affects sodium conductance, at least in Aplysia neurons (122), although this evidence must be weighed against a preliminary study which found no clear effects of tumor necrosis factor‐α on demyelinated axons (113). Prostaglandin E2 affects the tetrodotoxin‐resistant sodium current in rat dorsal root ganglion, and colonic, neurons (55, 64) resulting in increased excitability (55). Whether these effects would occur within the central nervous system in MS is not clear.
Nitric oxide. There is abundant evidence that nitric oxide is produced within the lesions of MS (128) and so it may be important that nitric oxide is also capable of blocking axonal conduction (113, 126), especially in demyelinated axons (113) (Figure 1; see also Figure 7). Experimentally, the block can be imposed within minutes of exposure to nitric oxide, maintained for hours, and relieved within minutes of removing the nitric oxide (113). The mechanism(s) is not known, but nitric oxide can affect the function of sodium channels (3, 66, 114, 123) and the sodium pump (65, 121), in addition to the function of other ion channels, perhaps by acting on channel thiols (3, 85, 125). Nitric oxide may alternatively block conduction by inhibiting mitochondrial metabolism (21, 32) and causing membrane depolarization (61, 62)
Figure 1.
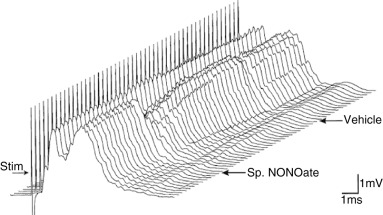
A series of compound action potentials showing conduction through a demyelinating ethidium bromide lesion induced in the rat dorsal columns 23 days previously. The records were obtained 2 minutes apart and are plotted in three‐dimensional perspective with the earliest records at the front. The compound action potential has two peaks: the first shows conduction in normal axons, and the second shows delayed conduction in demyelinated axons. After the first 12 records were obtained an injection of a nitric oxide donor, spermine NONOate, was made into the lesion. The reduced amplitude of the second peak alone shows that the nitric oxide selectively blocked conduction in the demyelinated axons. A later, control injection of vehicle alone at the same site had no appreciable effect on conduction. Reproduced from Redford et al (113).
Figure 7.

Four series of records showing compound action potentials obtained simultaneously from four dorsal spinal roots in an anesthetized rat. The individual records were taken 4 minutes apart and so span several hours: the earliest records are plotted at the front. Initially the roots were stimulated at 1 Hz (ie, once per second) and after 30 minutes the medium bathing the roots was exchanged either for fresh control medium (top plots) or for control medium with the addition of the sodium channel‐blocking agent flecainide (bottom plots). After an additional 15 minutes the stimulation rate for all the roots was increased to 100 Hz, and this frequency was maintained for 5 h, after which the rate was reduced back to 1 Hz. For 2 h (bar) during the 5 h period the roots were exposed to nitric oxide which promptly blocked conduction: a higher concentration of nitric oxide was used here in comparison with Figure 1. When the nitric oxide was washed away, conduction was restored to the roots treated with flecainide, but not in the control roots. The failure of conduction in the control roots is because the axons degenerated in response to the impulse activity in conjunction with exposure to nitric oxide, whereas the inclusion of flecainide provided almost complete protection. Modified from Kapoor et al (76).
SODIUM CHANNELS AND THE RESTORATION OF FUNCTION—REMISSION
Neurological function can be restored in MS by at least four mechanisms, any of which can contribute to the remissions that characterize relapsing/remitting disease. One mechanism is simply the resolution of any inflammation that may have held normal function in abeyance, and another involves “plastic” gray matter adaptations to compensate for functional loss, but the remaining two mechanisms involve the reorganization of axonal sodium channels and will be considered further.
Restoration of conduction to demyelinated axons. The low sodium channel density along freshly exposed demyelinated axolemma initially prevents conduction because the axolemma is inexcitable, as described above, but one of the adaptations of demyelinated axons to the demyelinated state is the acquisition of sodium channels distributed along the demyelinated axolemma. The first evidence for this was the observation in the peripheral nervous system that conduction could occur along demyelinated axons and that it adopted a continuous, rather than saltatory, mode of conduction across the demyelinated segment (26, 27). Later experiments revealed that conduction could also occur in peripheral axons by micro‐saltation between new node‐like foci of sodium channels, termed phi‐nodes, that form along the demyelinated axolemma every 100–400 (mean 255) µm (133). However, the techniques employed to prove successful conduction in demyelinated peripheral axons could not be applied in the central nervous system and so proof here awaited a study in which conduction was demonstrated by intra‐axonal recordings from individual dorsal column axons that were then labeled iontophoretically and reconstructed in three‐dimensions through the lesion at the electron microscope level (58). Conduction was proven to occur over demyelinated regions several internodes in length (2500 µm), and could be restored within 2–3 weeks of the induction of the demyelinating lesion. Although successful conduction is theoretically favored by a small axon diameter, it was observed in demyelinated axons as large as 5.5 µm in diameter (58), suggesting that most demyelinated central axons will be physically capable of conduction under ideal conditions (even though many fail to conduct in practice).
Initially it was not known whether the internodal sodium channels upon which conduction depended were original ones that had spread from the positions of the old nodes, but more recently it has become clear that at least some of the channels must be inserted de novo because the new channels can be of a different subtype than is usually expressed at adult nodes. During development, before ensheathment with glial cells, axons have a diffuse distribution of Nav1.2 along the axolemma (20, 74), and it is presumably these that support impulse conduction prior to myelination (150). As myelination proceeds, Nav1.2 channels become clustered at the new nodes, but they are gradually replaced by Nav1.6 channels so that adult nodes are populated by Nav1.6 channels (20, 35, 74). Interestingly, axons demyelinated caused by EAE show an increased expression of not only Nav1.6 along the demyelinated axolemma, but also of Nav1.2 (40). The Nav1.2 channels were presumably inserted in response to the demyelination. Sodium channel immunoreactivity could occur over regions of more than 30 µm in optic nerve axons, and was associated with up‐regulation of mRNA for Nav1.2 in retinal ganglion neurons. Notably, the same pattern of changes in Nav1.2 and Nav1.6 has also been observed along demyelinated axons within MS lesions (42) (Figure 2).
Figure 2.

Immunohistochemical labeling of Nav1.2 (B,D,F,H) and Nav1.6 (A,C,E,G) sodium channels in the spinal cord and optic nerve in multiple sclerosis (MS). Myelin basic protein (MBP; green) is plentiful in the normal tissue (A,B) but almost absent from the demyelinated lesions. In the normal tissue the sodium channel labeling is punctate (nodal) for Nav1.6, but absent for Nav1.2. In contrast, both types of channel show almost continuous labeling along demyelinated axons (eg, C,D,E,F; white arrows). Reproduced from Craner et al (42).
The central demyelinated axons studied by Felts and colleagues were found to conduct even when at least 88% of their surface area was entirely devoid of any glial contacts across several internodes (58). This last point is noted because, where present, astrocytes may play a role in the location of sodium channels as astrocytic processes make intimate contacts with demyelinated axons specifically at sites that exhibit a node‐like, electron‐dense undercoating deemed to indicate the location of high densities of sodium channels (15, 19, 118). The presence of conduction in the absence of substantial astroglial ensheathment may be related to islands of sodium channels that have been detected at the electron microscope level along naked demyelinated dorsal column axons in the rat, using well‐characterized anti‐sodium channel antibodies (Figure 3: T. Deerinck, M.H. Ellisman, P.A. Felts, S.R. Levinson and K.J. Smith, unpub. obs.). The islands have the longitudinal dimension of nodes of Ranvier, that is, approximately 1 µm, but they sometimes appear on just one side of the axon, rather than circumferentially as would be expected if they represented the locations of the old nodes. If the aggregations represent nascent nodes, it is interesting that they form in the apparent absence of the processes of astroglial, or any other, cells. The appearance of these axons is reminiscent of the clusters of sodium channels found in the absence of any ensheathing cells along peripheral, dystrophic axons (47).
Figure 3.
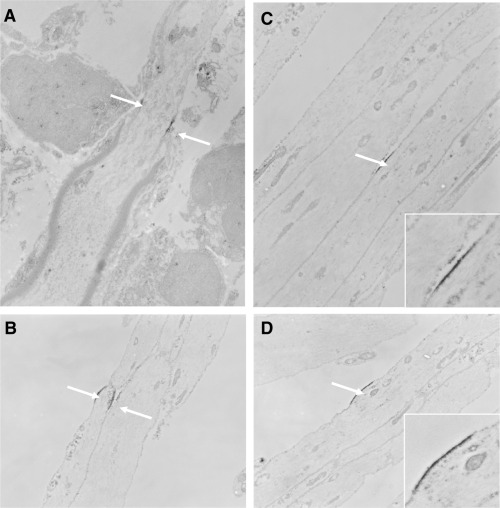
Electron micrographs showing longitudinal sections through central demyelinated axons in the rat dorsal columns 1 month after the intraspinal injection of ethidium bromide accompanied by β‐irradiation (to prevent repair by remyelination). The tissue is unstained but labeled with an anti‐sodium channel antibody. A. A hemi‐node at the edge of a demyelinated segment shows labeling along the axolemma at either side of the axon at the location of the old node. B. Node‐like labeling along a naked demyelinated axon. C,D. Unilateral labeling of sodium channels along naked demyelinated axons: insets show higher magnification. T. Deerinck, M.H. Ellisman, P.A. Felts, S.R. Levinson and K.J. Smith, unpublished observations.
The restoration of conduction in demyelinated axons can explain the clinical observations of apparently “silent” demyelinated lesions, namely large demyelinated lesions in pathways (eg, the optic nerves) where conduction block would have been expected to produce symptoms, but where no, or only minimal, deficit was detected (88, 103, 108, 141).
The conduction velocity along the demyelinated portion of central axons is very much reduced, as it is in peripheral axons (26), and it is associated with a refractory period for transmission prolonged from the normal range of 0.5–1.4 ms, to 1.0–6.0 ms, with one axon requiring a gap of 27 ms between impulses before the second was faithfully transmitted (58). This insecurity of conduction in demyelinated axons, coupled with the properties of sodium channels, results in a range of curious phenomena related to body temperature: in an extreme example, one patient temporarily went blind if she used a hair dryer (29). The exacerbation of symptoms upon body warming, known as Uhthoff’s sign (124, 136), is a common phenomenon in MS and it is attributed to the fact that the temperature coefficient for sodium inactivation is larger than that for activation (45). The consequence is that the duration of action potentials gets fractionally shorter upon warming and this reduces the current that is available to depolarize the demyelinated membrane to its firing threshold. In axons on the verge of conduction failure, as central demyelinated axons often are, this additional compromise can result in conduction block. Thus a patient with demyelination of the optic pathway, for example, can suffer temporary conduction block and blindness upon head warming resulting from the use of a hair dryer.
The low security of conduction at sites of demyelination also contributes to the early loss of function upon repeated use of demyelinated axons, which can result in progressive weakness upon exercise (93), or perhaps cause the fading or blurring of vision upon fixated gaze (91, 144). These deficits have been related to the fact that repeated conduction along demyelinated axons can result in intermittent periods (eg, 0.2–2.0 s) of complete conduction block, interspersed with periods when conduction through the lesion occurs faithfully (57, 134). The periods of conduction block result from membrane hyperpolarization in response to activity of the electrogenic Na+/K+ ATPase (sodium pump) induced by the raised intracellular sodium resulting from the preceding impulse activity (24, 26).
Abnormalities in channel expression, and their consequences, are perhaps best exemplified in the Purkinje neurons of the cerebellum. Black and colleagues (16) have found that the mRNA and protein of the Nav1.8 subtype of sodium channel are clearly expressed in the Purkinje neurons of animals with EAE and patients with progressive MS, but not at all in normal Purkinje neurons. The Nav1.8 channels are expressed with annexin II (39), a protein that facilitates insertion of the channel into the cell membrane, which is consistent with a belief that the channels are functional. The Nav1.8 channel is resistant to steady state inactivation (4) which means that it can open even when the membrane is depolarized: other subtypes inactivate under these conditions. The Nav1.8 channel also recovers rapidly from inactivation (54), and thus it will, for example, support repetitive firing even in depolarized neurons. Now the electrical behavior of a neuron is largely determined by the aggregate properties of its sodium channels, and so the atypical properties of Nav1.8 channels would be expected to confer atypical properties on the Purkinje neurons of MS patients. Consistent with this expectation, Purkinje neurons transfected to express Nav1.8 channels display atypical properties, including the production of sustained repetitive firing in response to a sustained depolarizing current (115). Purkinje neurons from mice with EAE display similar properties (120). Such unusual behavior is particularly interesting when considered with the fact that patients with MS commonly exhibit cerebellar deficits with characteristics consistent with a sodium channel abnormality (148). For example, the deficits are frequently persistent, rather than remitting, may have a paroxysmal expression (in common with inherited channelopathies), and they typically show a favorable response to therapy with sodium channel‐blocking agents such as carbamazepine.
Hyperexcitability. During the course of their disease, many patients with MS experience so‐called “positive” symptoms, namely the gain of phenomena such as tingling sensations and neuropathic pain. These effects can be attributed to changes in the pattern of expression and distribution of different types of sodium (and potassium) channels that can confer not only excitability to demyelinated axolemma, but also hyperexcitability. Affected axons can become spontaneously active, generating long trains of impulses that arise ectopically at the site of demyelination, conducting in both directions from it (129, 130). The impulses traveling to the brain are interpreted as tingling etc. sensations referred to the body part innervated by the axons (see also 100), or as flashes of light in the case of optic nerve axons (46). Hyperexcitable axons can generate either continuous discharges (at 10–50 Hz), or may fire in busts, with burst durations of 0.1–5 s, separated by silent periods of 0.1–100 s. Continuous discharges, in particular, have been related to the appearance of a slow, persistent inward sodium current that can appear at demyelinated membranes (75, 117, 138) (Figure 4), and this mechanism is consistent with the fact that bothersome tingling sensations respond well to sodium channel‐blocking agents such as carbamazepine. Sensory axons are more prone to hyperexcitability than their motor counterparts, perhaps because they have a greater expression of persistent sodium currents (25, 98), and so sensory manifestations of hyperexcitability are more common than motor, although phenomena such as myokymia have been reported (68).
Figure 4.

Portions from an intra‐axonal, micropipette recording from a location at or near the demyelinated portion of a central axon in an ethidium bromide lesion in the rat spinal cord. The records show oscillations in the membrane potential which either routinely (A) or periodically (B,C) initiate action potentials. The almost sinusoidal waveform is clearly observed where the depolarizations are subthreshold for initiating spikes (B,C). The oscillations are abolished by sodium channel‐blocking agents (not shown) indicating that they arise from currents flowing through sodium channels. Reproduced from Kapoor et al (75).
Hyperexcitability in demyelinated axons is normally accompanied by the acquisition of mechanosensitivity, which results in the generation of phasic and tonic bursts of ectopic impulses upon slight deformation (particularly stretching) of the site of demyelination (129, 130). Sensory axons are again more susceptible than motor axons, and demyelination affecting the sensory axons of the cervical dorsal columns can result in the perception of electric shock or tingling sensations upon stretching the cervical cord by bending the neck forwards (Lhermitte’s symptom) (73, 84).
Remyelination. Repair of demyelinated axons by remyelination is common in some patients with MS (107, 110, 111). Based on observations in experimental lesions there is little doubt that the remyelinated axons will conduct impulses, although this remains unproven in MS. Central axons in which conduction had been blocked by experimental demyelination were shown to conduct when repaired by spontaneous remyelination (131, 132), even when the new myelin internodes possessed as few as five lamellae (58). Furthermore, maturation of the new sheaths was accompanied by the restoration of near‐normal conduction velocity, refractory period for transmission and ability faithfully to conduct trains of impulses at high frequency (131, 132). The type of remyelinating cell does not seem to be very important, as remyelination is effective in restoring conduction irrespective of whether it is achieved primarily by oligodendrocytes (131, 132), or endogenous Schwann cells (56), or by transplanted cells of various types (69, 70, 79, 142). At least in the case of Schwann cells, the new nodes formed by remyelination regain the normal configuration of ion channels, including the presence of Nav1.6 channels in the nodal membrane (18) (Figure 5). Accordingly, remyelination has been associated with the restoration of function at the behavioral level in experimental lesions (72), and also probably in clinical studies (33), and where it occurs, remyelination probably makes an important contribution to remissions.
Figure 5.

Immunohistochemical labeling of mature (>1 year old) nodes of Ranvier formed on central axons in the rat dorsal columns during the process of remyelination by endogenous Schwann cells. The axons were formerly demyelinated by the intraspinal injection of ethidium bromide. Upper panel: green shows the location of Caspr, found on either side of nodes of Ranvier. The picture labeled 2A shows that Nav1.2 sodium channels are not present at the remyelinated nodes, although the antibody recognizes them at nodes in developing optic nerve (inset). The remyelinated nodes label clearly for the presence of Nav1.6 sodium channels bounded by Caspr labeling (B). Peripheral myelin is shown by P0 labeling (blue in C,D) and it is clear that Nav1.6 sodium channels, but not Nav1.2, are present at the nodes formed by remyelination. Scale bars 10 µm. Lower panel: green marks ezrin, a molecule expressed at the nodal microvilli of Schwann cells. Nav1.2 sodium channels are not present at the central nodes formed by remyelination by Schwann cells (pictures labeled 4A, B, C), but Nav1.6 channels are expressed at the nodal gap (D, E, F). Reproduced from Black et al (18).
AXONAL DEGENERATION AND INJURY—PROGRESSIVE DISEASE
Axonal degeneration is believed to be the main cause of the permanent neurological deficits that characterize the progressive forms of MS. A number of mechanisms have been proposed to account for the degeneration but here we focus only on the growing evidence that sodium channels may play an important role in a mechanism involving energy insufficiency.
Pathological evidence shows that axons degenerate in large numbers in acute, inflammatory demyelinating lesions where the magnitude of the damage is proportional to the intensity of inflammation (59, 140). Such studies indicate a role for factors associated with the inflammatory response, including nitric oxide. Nitric oxide is produced in large amounts within inflammatory MS lesions (128) and one of its most important potential effects is that it can potently inhibit mitochondrial function (21, 32, 51), reducing the production of ATP. Most ATP in the brain is destined to fuel the pumps that maintain ion gradients (5), and of these the most important is the Na+/K+ ATPase, or sodium pump. Thus axons passing through nitric oxide‐rich inflammatory lesions in MS may be relatively starved of the ATP required to maintain sodium homeostasis, in which case they could be particularly vulnerable to the energy demands resulting from sustained impulse activity. An experiment based on this reasoning found that, indeed, axons could be killed by physiological frequencies of impulse activity, if this occurred while the axons were exposed to nitric oxide (135). The axons will have suffered a raised entry of sodium ions because of the impulse activity, perhaps exacerbated by a nitric oxide‐induced modification of the sodium channels that makes them pass a persistent sodium current, namely a persistent drain of sodium ions into the axon (3, 66). By analogy with axons rendered experimentally ischemic, a high internal sodium ion concentration can result in a lethal influx of calcium ions via reverse action of the sodium/calcium exchanger (137). Thus impulse activity in the presence of nitric oxide could initiate a sequence of events that culminates in the calcium‐mediated digestion of the axon [described in more detail in Bechtold and Smith (8)]. In patients, impulse activity would, of course, be substantially increased in any axons that became hyperexcitable and spontaneously active, as they can generate continuous trains of impulses at frequencies up to 50 Hz (7, 75, 117, 129, 130). The realization that sodium loading may be an important cause of axonal degeneration has stimulated research into strategies for axonal protection, as discussed below.
There is evidence that it may be preferentially the Nav1.6 subtype of sodium channel that predisposes axons to damage. Thus Craner and colleagues labeled dorsal column axons from animals with EAE for myelin basic protein, Nav1.2, Nav1.6, the sodium/calcium exchanger, and β‐amyloid precursor protein as a marker for axonal damage (41) (Figure 6). Nearly all (92%) of the damaged axons were positive for Nav1.6, whereas only 38% were positive for Nav1.2, and, of these, 95% also expressed Nav1.6. Interestingly, most (73%) of the damaged axons coexpressed Nav1.6 with the sodium/calcium exchanger, but only 4% of apparently undamaged axons expressed both these markers. The authors concluded that co‐expression of Nav1.6 and the sodium/calcium exchanger is associated with axonal injury. Notably, similar findings were made in acute MS lesions (42). The association of Nav1.6 with axonal injury is potentially related to the fact that Nav1.6 channels produce a larger persistent sodium current, than, say, Nav1.2, and so would tend to promote sodium influx even in the absence of impulse activity (119).
Figure 6.
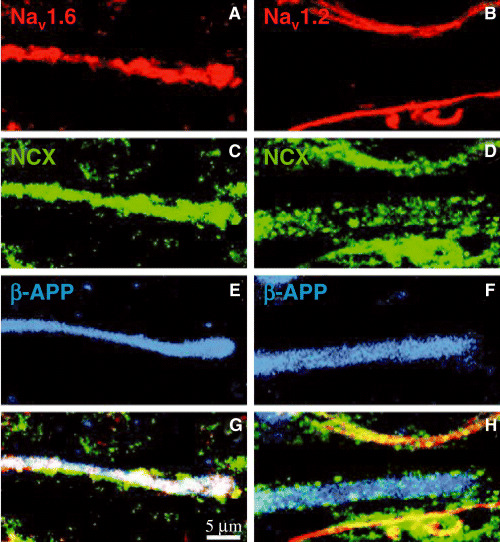
Images of tissue from multiple sclerosis immunohistochemically labeled for different molecules. The panels on the left show the same microscopic field, and a single, different field is shown on the right. A and B show labeling for Nav1.6 and Nav1.2 sodium channels respectively (red), C and D show labeling for the sodium/calcium exchanger (NCX, green), E and F show β‐amyloid precursor protein (β‐APP, blue) and G and H show the merged images. The same structure (which is presumably a damaged, ie, β‐APP+, axon) is labeled by all the antibodies in the left panel, but on the right the β‐APP+ structure is weakly positive for NCX, but it is not positive for Nav1.2. The data indicate that damaged axons are positive for Nav1.6 and NCX, but not for Nav1.2. Reproduced from Craner et al (42).
The damage resulting from sodium channel activity may be augmented by a calcium‐mediated modification of channel structure. It has recently been found that sodium‐mediated calcium entry in stretch‐injured axons can result in the activation of proteases that cleave part of the α‐subunit of the sodium channel responsible for channel inactivation. The resulting failure of inactivation results in a persistent inward sodium current that results in more calcium entry, in a positive feedback mechanism (71). Working by another mechanism, calcium may also increase the sodium current amplitude of Nav1.6 channels, and affect their inactivation kinetics, via calmodulin (67). It seems reasonable to propose that these mechanisms may occur in MS, and, if so, this would amplify the sodium and calcium entry, promoting the activation of local calcium‐dependent degradative enzymes, and resulting in lysis and degeneration of the axon.
Although, on the evidence above, demyelinated axons might appear to be especially vulnerable to sodium‐mediated degeneration, and although this seems intuitively logical, it is not inevitably true. Thus although it is true that conduction of a single impulse along a demyelinated axon will result in much more axonal sodium loading than if the axon were myelinated, it is also true that a demyelinated axon will not conduct a very heavy impulse load, that is, it will not conduct at very high frequency, and it will not conduct at moderate frequency for very long before conduction intermittently fails (134). In these ways demyelinated axons gain protection from sodium loading, whereas normal axons remain vulnerable. Indeed, it was normal axons that degenerated in response to impulse activity in the presence of nitric oxide in the study referred to earlier in which axons were stimulated in the presence of nitric oxide (135): demyelinated axons were not studied in this way, and might not, in any case, have conducted at all in the presence of nitric oxide. It is notable that a detailed pathological study of MS found little or no correlation between plaque load and axonal loss, indicating that demyelination may not be the primary cause of axonal loss (48).
The prediction of energy insufficiency in MS caused by nitric oxide, described above, is largely circumstantial, but direct evidence supporting a belief that an energy deficient state may exist in MS lesions, perhaps by a different mechanism, has recently been provided by a study of expression levels of 33 000 characterized genes in postmortem motor cortex (52). The investigators found that 26 nuclear‐encoded mitochondrial genes were decreased, as was the functional activity of mitochondrial respiratory chain complexes I and III, specifically in neurons. If, as the authors suggest, these observations mean that ATP supply is diminished in MS neurons, then these cells will be particularly vulnerable to sodium accumulation, and damage by reverse sodium/calcium exchange. It is thus possible that some observations of a hypoxia‐like state within MS tissue (1, 2) owe their origin not to true tissue hypoxia, but rather to an energy‐insufficient state arising from the inadequacy of mitochondrial function.
The mechanisms for degeneration and protection described above seem to apply most directly to the intensely inflammatory lesions of relapsing/remitting MS where expression of the inducible (high output) form of nitric oxide synthase (iNOS) is particularly prominent and axons degenerate in large numbers. However, there is also a “slow burning” axonal degeneration that continues in chronic inactive demyelinated plaques and that may contribute significantly to clinical progression of the disease (80). In this regard it is noteworthy that although intense inflammation may be lacking in such cases, the axonal degeneration nonetheless occurs on a background of residual chronic inflammation where many microglial cells are intensely positive for iNOS (83). It is therefore possible that axons in progressive MS, as well as those in relapsing/remitting disease, may be vulnerable to nitric oxide‐mediated energy insufficiency, and hence to sustained impulse activity.
The mechanisms underlying the symptomatology of MS, including those not dependent on sodium channels, have recently been reviewed in detail (136).
SODIUM CHANNELS AND IMMUNE CELLS
There is growing evidence that sodium channels may affect not only the properties of neuronal cells, but also the properties of some immune cells. Such effects could explain why Bechtold and colleagues found that flecainide therapy in EAE and experimental autoimmune neuritis (EAN; a model of Guillain‐Barré syndrome) reduced not only the number of degenerating axons, but also the intensity of the inflammation at the peak of disease (10, 11). Thus, in EAE, flecainide therapy significantly reduced the number of activated macrophages/microglia and also the number of cells positive for iNOS (10). Similarly, phenytoin reduced the spinal cord infiltrates in mice with EAE by 75% (43). In fact it is not entirely clear whether the protective effect of sodium channel blockade in EAE is caused by effects on axons, or on immune cells, or both, although the axonal protection achieved in models where inflammation is absent (76) indicates that at least some of the protection is mediated on axons directly.
If sodium channel inhibition truly has anti‐inflammatory effects within the CNS, there are several ways in which it could be mediated. For example, a role for sodium currents in T cell activation and co‐stimulation has been suggested (60, 78, 82), and fast transient sodium currents have been observed in several T cell and B cell lines (60). Furthermore, initial work in our laboratory has suggested that therapeutically relevant doses of lamotrigine and phenytoin can inhibit T cell activation and proliferation, for example, they reduce the proliferation of lymph node cells in response to phytohemagglutinin (89). With regard to B cells, both amiloride‐ and tetrodotoxin‐sensitive sodium channels have been reported on human and rat B cells (28, 34, 104, 109) and blockade of amiloride‐sensitive sodium channels inhibited antibody secretion by lymphocyte hybridomas (156). In this context it is notable that flecainide therapy of rats with EAN was found to cause a reduction in the serum titer of anti‐peripheral myelin antibodies (11), although it is possible that this reduction was related to the reduced axonal damage which may have limited the stimulus for B cells to produce antibodies.
Sodium channels are also present in macrophages and microglial cells, namely cells that perform effector and other functions in MS, EAE and EAN, and the expression of Nav1.6 channels is increased in activated microglia and macrophages in MS and EAE (43). Voltage‐gated sodium channels have been described in rat (81) and human (101) microglia, and it has been suggested that the activation of a sodium current might be necessary to trigger microglial activation and facilitate a subsequent immune response (53). In support of this suggestion, the sodium channel‐blocking agent lignocaine has been found to inhibit superoxide release from rabbit alveolar macrophages (13) and Craner and colleagues (43) found that treatment of activated rat microglia with the sodium channel‐blocking agent tetrodotoxin reduced microglial phagocytic activity by 40%.
THERAPY
It is now accepted that axonal degeneration is a major cause of permanent neurological deficit in MS, and this has sharpened the need to identify therapeutic strategies for axonal protection. The realization that electrical activity and sodium loading could be an important cause of axonal degeneration raised the possibility that axons might gain protection by partial blockade of their sodium channels, or blockade of the reverse mode of action of the sodium/calcium exchanger (8). This possibility was confirmed by the observation that axons survived exposure to nitric oxide if the sodium channel‐blocking agents flecainide or lidocaine (135) or tetrodotoxin or sipatrigine (62) were included in the incubation medium, or if the sodium/calcium exchange‐blocking agent bepridil was added (76). Furthermore, axons survived sustained impulse conduction in the presence of nitric oxide if flecainide was present, even though the dose was sufficiently low that conduction continued despite the presence of the drug (76) (Figure 7). The partial blockade of sodium channels mediated by the drugs presumably limited the rise in the sodium ion concentration within axons, keeping it below the level at which damaging calcium entry occurred via reverse action of the sodium/calcium exchanger (see above).
To determine whether the pharmacological blockade of sodium channels might form the basis of a therapy in MS, additional experiments examined whether such drugs could protect axons from degeneration in animal models of the disease, namely different forms of EAE. Flecainide and lamotrigine were found to be protective in a chronic relapsing rat model of EAE (9, 10, 12) and also in a model of Guillain‐Barré syndrome (11). In EAE, flecainide therapy, from 7 days post immunization to the end of the experiment at day 28, reduced axonal degeneration from the control value of 38% degeneration in severely affected animals, to only 2% in similarly severely affected animals (9, 10). Phenytoin was also found to provide significant protection in a progressive model of EAE in the mouse (86), even when therapy was extended over a 6‐month period (17). Importantly, therapy with sodium channel‐blocking agents improved the functional outcome (9, 10, 17, 86). Based on these encouraging observations, clinical trials of lamotrigine and phenytoin have been initiated in London and New Haven, respectively, treating patients with secondary and primary progressive MS. Success in these trials will indicate that the seemingly relentless accumulation of permanent neurological deficit in progressive MS can be subdued, with the hope that patients can retain their neurological function in the long term.
ACKNOWLEDGMENTS
Dr. Marija Sajic is thanked for her expert comments on the manuscript. Work in the author’s laboratory is supported by grants from the Medical Research Council (UK), European Union “NeuroMiSe”, and the Multiple Sclerosis Society of Great Britain and Northern Ireland.
REFERENCES
- 1. Aboul‐Enein F, Lassmann H (2005) Mitochondrial damage and histotoxic hypoxia: a pathway of tissue injury in inflammatory brain disease? Acta Neuropathol (Berl) 109:49–55. [DOI] [PubMed] [Google Scholar]
- 2. Aboul‐Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Bruck W, Lucchinetti C, Schmidbauer M, Jellinger K, Lassmann H (2003) Preferential loss of myelin‐associated glycoprotein reflects hypoxia‐like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol 62:25–33. [DOI] [PubMed] [Google Scholar]
- 3. Ahern GP, Hsu S‐F, Klyachko VA, Jackson MB (2000) Induction of persistent sodium current by exogenous and endogenous nitric oxide. J Biol Chem 275:28810–28815. [DOI] [PubMed] [Google Scholar]
- 4. Akopian AN, Sivilotti L, Wood JN (1996) A tetrodotoxin‐resistant voltage‐gated sodium channel expressed by sensory neurons. Nature 379:257–262. [DOI] [PubMed] [Google Scholar]
- 5. Ames A III (2000) CNS energy metabolism as related to function. Brain Res Brain Res Rev 34:42–68. [DOI] [PubMed] [Google Scholar]
- 6. Aulkemeyer P, Hausner G, Brinkmeier H, Weber F, Wurz A, Rudel R (2000) The small sodium‐channel blocking factor in the cerebrospinal fluid of multiple sclerosis patients is probably an oligopeptide. J Neurol Sci 172:49–54. [DOI] [PubMed] [Google Scholar]
- 7. Baker M, Bostock H (1992) Ectopic activity in demyelinated spinal root axons of the rat. J Physiol (Lond) 451:539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bechtold DA, Smith KJ (2005) Sodium‐mediated axonal degeneration in inflammatory demyelinating disease. J Neurol Sci 233:27–35. [DOI] [PubMed] [Google Scholar]
- 9. Bechtold DA, Kapoor R, Smith KJ (2002) Axonal protection mediated by flecainide therapy in experimental inflammatory demyelinating disease. J Neurol 249(Suppl. 1):204. [Google Scholar]
- 10. Bechtold DA, Kapoor R, Smith KJ (2004) Axonal protection using flecainide in experimental autoimmune encephalomyelitis. Ann Neurol 55:607–616. [DOI] [PubMed] [Google Scholar]
- 11. Bechtold DA, Yue X, Evans RM, Davies M, Gregson NA, Smith KJ (2005) Axonal protection in experimental autoimmune neuritis by the sodium channel blocking agent flecainide. Brain 128:18–28. [DOI] [PubMed] [Google Scholar]
- 12. Bechtold DA, Miller SJ, Dawson AC, Sun Y, Kapoor R, Berry D, Smith KJ (2006) Axonal protection achieved in a model of multiple sclerosis using lamotrigine. J Neurol 253:1542–1551. [DOI] [PubMed] [Google Scholar]
- 13. Bidani A, Heming TA (1997) Effects of lidocaine on cytosolic pH regulation and stimulus‐induced effector functions in alveolar macrophages. Lung 175:349–361. [DOI] [PubMed] [Google Scholar]
- 14. Bitsch A, Wegener C, Costa C, Bunkowski S, Reimers CD, Prange HW, Bruck W (1999) Lesion development in Marburg’s type of acute multiple sclerosis: from inflammation to demyelination. Mult Scler 5:138–146. [DOI] [PubMed] [Google Scholar]
- 15. Black JA, Felts P, Smith KJ, Kocsis JD, Waxman SG (1991) Distribution of sodium channels in chronically demyelinated spinal cord axons: immuno‐ultrastructural localization and electrophysiological observations. Brain Res 544:59–70. [DOI] [PubMed] [Google Scholar]
- 16. Black JA, Dib‐Hajj S, Baker D, Newcombe J, Cuzner ML, Waxman SG (2000) Sensory neuron‐specific sodium channel SNS is abnormally expressed in the brains of mice with experimental allergic encephalomyelitis and humans with multiple sclerosis. Proc Natl Acad Sci USA 97:11598–11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Black JA, Liu S, Hains BC, Saab CY, Waxman SG (2006) Long‐term protection of central axons with phenytoin in monophasic and chronic‐relapsing EAE. Brain 129:3196–3208. [DOI] [PubMed] [Google Scholar]
- 18. Black JA, Waxman SG, Smith KJ (2006) Remyelination of dorsal column axons by endogenous Schwann cells restores the normal pattern of Nav1.6 and Kv1.2 at nodes of Ranvier. Brain 129:1319–1329. [DOI] [PubMed] [Google Scholar]
- 19. Blakemore WF, Smith KJ (1983) Node‐like axonal specializations along demyelinated central nerve fibres: ultrastructural observations. Acta Neuropathol 60:291–296. [DOI] [PubMed] [Google Scholar]
- 20. Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G (2001) Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 30:91–104. [DOI] [PubMed] [Google Scholar]
- 21. Bolanos JP, Almeida A, Stewart V, Peuchen S, Land JM, Clark JB, Heales SJR (1997) Nitric oxide‐mediated mitochondrial damage in the brain: mechanisms and implications for neurodegenerative diseases. J Neurochem 68:2227–2240. [DOI] [PubMed] [Google Scholar]
- 22. Bornstein MB, Crain SM (1965) Functional studies of cultured brain tissues as related to “demyelinative disorders. Science 148:1242–1244. [DOI] [PubMed] [Google Scholar]
- 23. Bostock H (1994) The pathophysiology of demyelination. In: Multiple Sclerosis: Current Status of Research and Treatment. Herndon RM, Seil FJ (eds), pp. 89–112. Demos Publications, Inc.: New York. [Google Scholar]
- 24. Bostock H, Grafe P (1985) Activity‐dependent excitability changes in normal and demyelinated rat spinal root axons. J Physiol (Lond) 365:239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bostock H, Rothwell JC (1997) Latent addition in motor and sensory fibres of human peripheral nerve. J Physiol 498:277–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bostock H, Sears TA (1976) Continuous conduction in demyelinated mammalian nerve fibers. Nature 263:786–787. [DOI] [PubMed] [Google Scholar]
- 27. Bostock H, Sears TA (1978) The internodal axon membrane: electrical excitability and continuous conduction in segmental demyelination. J Physiol (Lond) 280:273–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bradford AL, Ismailov II, Achard JM, Warnock DG, Bubien JK, Benos DJ (1995) Immunopurification and functional reconstitution of a Na+ channel complex from rat lymphocytes. Am J Physiol 269:C601–C611. [DOI] [PubMed] [Google Scholar]
- 29. Brickner RM (1950) The significance of localised vasoconstrictions in multiple sclerosis. Res Publ Assoc Res Nerv Ment Dis 28:236–244. [PubMed] [Google Scholar]
- 30. Brinkmeier H, Aulkemeyer P, Wollinsky KH, Rudel R (2000) An endogenous pentapeptide acting as a sodium channel blocker in inflammatory autoimmune disorders of the central nervous system. Nat Med 6:808–811. [DOI] [PubMed] [Google Scholar]
- 31. Brinkmeier H, Weber F, Aulkemeyer P, Wollinsky KH, Rudel R (2003) The pentapeptide QYNAD does not block voltage‐gated sodium channels. Neurology 60:1871–1872. [DOI] [PubMed] [Google Scholar]
- 32. Brown GC, Borutaite V (2002) Nitric oxide inhibition of mitochondrial respiration and its role in cell death. Free Radic Biol Med 33:1440–1450. [DOI] [PubMed] [Google Scholar]
- 33. Brusa A, Jones SJ, Plant GT (2001) Long‐term remyelination after optic neuritis: a 2‐year visual evoked potential and psychophysical serial study. Brain 124:468–479. [DOI] [PubMed] [Google Scholar]
- 34. Bubien JK, Warnock DG (1993) Amiloride‐sensitive sodium conductance in human B lymphoid cells. Am J Physiol 265:C1175–C1183. [DOI] [PubMed] [Google Scholar]
- 35. Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR (2000) Sodium channel Na(v)1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc Natl Acad Sci USA 97:5616–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Centonze D, Rossi S, Boffa L, Versace V, Palmieri MG, Caramia MD, Bernardi G (2005) CSF from MS patients can induce acute conduction block in the isolated optic nerve. Eur J Neurol 12:45–48. [DOI] [PubMed] [Google Scholar]
- 37. Charcot JM (1868) Histologie de la sclerose en plaque. Gaz Hop (Paris) 41:554–566. [Google Scholar]
- 38. Coles AJ, Wing MG, Molyneux P, Paolillo A, Davie CM, Hale G, Waldmann H, Compston A (1999) Monoclonal antibody treatment exposes three mechanisms underlying the clinical course of multiple sclerosis. Ann Neurol 46:296–304. [DOI] [PubMed] [Google Scholar]
- 39. Craner MJ, Lo AC, Black JA, Baker D, Newcombe J, Cuzner ML, Waxman SG (2003) Annexin II/p11 is up‐regulated in Purkinje cells in EAE and MS. Neuroreport 14:555–558. [DOI] [PubMed] [Google Scholar]
- 40. Craner MJ, Lo AC, Black JA, Waxman SG (2003) Abnormal sodium channel distribution in optic nerve axons in a model of inflammatory demyelination. Brain 126:1552–1561. [DOI] [PubMed] [Google Scholar]
- 41. Craner MJ, Hains BC, Lo AC, Black JA, Waxman SG (2004) Co‐localization of sodium channel Nav1.6 and the sodium‐calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain 127:294–303. [DOI] [PubMed] [Google Scholar]
- 42. Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG (2004) Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci USA 101:8168–8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Craner MJ, Damarjian TG, Liu S, Hains BC, Lo AC, Black JA, Newcombe J, Cuzner ML, Waxman SG (2005) Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia 49:220–229. [DOI] [PubMed] [Google Scholar]
- 44. Cummins TR, Renganathan M, Stys PK, Herzog RI, Scarfo K, Horn R, Dib‐Hajj SD, Waxman SG (2003) The pentapeptide QYNAD does not block voltage‐gated sodium channels. Neurology 60:224–229. [DOI] [PubMed] [Google Scholar]
- 45. Davis FA, Schauf CL (1981) Approaches to the development of pharmacological interventions in multiple sclerosis. Adv Neurol 31:505–510. [PubMed] [Google Scholar]
- 46. Davis FA, Bergen D, Schauf C, McDonald I, Deutsch W (1976) Movement phosphenes in optic neuritis: a new clinical sign. Neurology 26:1100–1104. [DOI] [PubMed] [Google Scholar]
- 47. Deerinck TJ, Levinson SR, Bennett GV, Ellisman MH (1997) Clustering of voltage‐sensitive sodium channels on axons is independent of direct Schwann cell contact in the dystrophic mouse. J Neurosci 17:5080–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. DeLuca GC, Williams K, Evangelou N, Ebers GC, Esiri MM (2006) The contribution of demyelination to axonal loss in multiple sclerosis. Brain 129:1507–1516. [DOI] [PubMed] [Google Scholar]
- 49. Denny‐Brown D, Brenner C (1944) Lesion in peripheral nerve resulting from compression by spring clip. Arch Neurol Psychiatry 52:1–19. [Google Scholar]
- 50. Devaux J, Forni C, Beeton C, Barbaria J, Beraud E, Gola M, Crest M (2003) Myelin basic protein‐reactive T cells induce conduction failure in vivo but not in vitro. Neuroreport 14:317–320. [DOI] [PubMed] [Google Scholar]
- 51. Duchen MR (2004) Roles of mitochondria in health and disease. Diabetes 53(Suppl. 1):S96–S102. [DOI] [PubMed] [Google Scholar]
- 52. Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, Gudz T, Macklin WB, Lewis DA, Fox RJ, Rudick R, Mirnics K, Trapp BD (2006) Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol 59:478–489. [DOI] [PubMed] [Google Scholar]
- 53. Eder C (1998) Ion channels in microglia (brain macrophages). Am J Physiol 275:C327–C342. [DOI] [PubMed] [Google Scholar]
- 54. Elliott AA, Elliott JR (1993) Characterization of TTX‐sensitive and TT X‐resistant sodium currents in small cells from adult rat dorsal root ganglia. J Physiol (Lond) 463:39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. England S, Bevan S, Docherty RJ (1996) PGE2 modulates the tetrodotoxin‐resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP‐protein kinase a cascade. J Physiol 495:429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Felts PA, Smith KJ (1992) Conduction properties of central nerve fibers remyelinated by Schwann cells. Brain Res 574:178–192. [DOI] [PubMed] [Google Scholar]
- 57. Felts PA, Kapoor R, Smith KJ (1995) A mechanism for ectopic firing in central demyelinated axons. Brain 118:1225–1231. [DOI] [PubMed] [Google Scholar]
- 58. Felts PA, Baker TA, Smith KJ (1997) Conduction in segmentally demyelinated mammalian central axons. J Neurosci 17:7267–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ferguson B, Matyszak MK, Esiri MM, Perry VH (1997) Axonal damage in acute multiple sclerosis lesions. Brain 120:393–399. [DOI] [PubMed] [Google Scholar]
- 60. Gallin EK (1991) Ion channels in leukocytes. Physiol Rev 71:775–811. [DOI] [PubMed] [Google Scholar]
- 61. Garthwaite G, Goodwin DA, Garthwaite J (1999) Nitric oxide stimulates cGMP formation in rat optic nerve axons, providing a specific marker of axon viability. Eur J Neurosci 11:4367–4372. [DOI] [PubMed] [Google Scholar]
- 62. Garthwaite G, Goodwin DA, Batchelor AM, Leeming K, Garthwaite J (2002) Nitric oxide toxicity in CNS white matter: an in vitro study using rat optic nerve. Neuroscience 109:145–155. [DOI] [PubMed] [Google Scholar]
- 63. Gilliatt RW (1982) Electrophysiology of peripheral neuropathies—an overview. Muscle Nerve 5:S108–S116. [PubMed] [Google Scholar]
- 64. Gold MS, Zhang L, Wrigley DL, Traub RJ (2002) Prostaglandin E(2) modulates TTX‐R I(Na) in rat colonic sensory neurons. J Neurophysiol 88:1512–1522. [DOI] [PubMed] [Google Scholar]
- 65. Guzman NJ, Fang MZ, Tang SS, Ingelfinger JR, Garg LC (1995) Autocrine inhibition of Na+/K(+)‐ATPase by nitric oxide in mouse proximal tubule epithelial cells. J Clin Invest 95:2083–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hammarstrom AKM, Gage PW (1999) Nitric oxide increases persistent sodium current in rat hippocampal neurons. J Physiol (Lond) 520:451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Herzog RI, Liu C, Waxman SG, Cummins TR (2003) Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties. J Neurosci 23:8261–8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hjorth RJ, Willison RG (1973) The electromyogram in facial myokymia and hemifacial spasm. J Neurol Sci 20:117–126. [DOI] [PubMed] [Google Scholar]
- 69. Honmou O, Felts PA, Waxman SG, Kocsis JD (1996) Restoration of normal conduction properties in demyelinated spinal cord axons in the adult rat by transplantation of exogenous Schwann cells. J Neurosci 16:3199–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Imaizumi T, Lankford KL, Kocsis JD (2000) Transplantation of olfactory ensheathing cells or Schwann cells restores rapid and secure conduction across the transected spinal cord. Brain Res 854:70–78. [DOI] [PubMed] [Google Scholar]
- 71. Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH (2004) Traumatic axonal injury induces proteolytic cleavage of the voltage‐gated sodium channels modulated by tetrodotoxin and protease inhibitors. J Neurosci 24:4605–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jeffery ND, Blakemore WF (1997) Locomotor deficits induced by experimental spinal cord demyelination are abolished by spontaneous remyelination. Brain 120:27–37. [DOI] [PubMed] [Google Scholar]
- 73. Kanchandani R, Howe JG (1982) Lhermitte’s sign in multiple sclerosis: a clinical survey and review of the literature. J Neurol Neurosurg Psychiat 45:308–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kaplan MR, Cho MH, Ullian EM, Isom LL, Levinson SR, Barres BA (2001) Differential control of clustering of the sodium channels Na(v)1.2 and Na(v)1.6 at developing CNS nodes of Ranvier. Neuron 30:105–119. [DOI] [PubMed] [Google Scholar]
- 75. Kapoor R, Li YG, Smith KJ (1997) Slow sodium‐dependent potential oscillations contribute to ectopic firing in mammalian demyelinated axons. Brain 120:647–652. [DOI] [PubMed] [Google Scholar]
- 76. Kapoor R, Davies M, Blaker PA, Hall SM, Smith KJ (2003) Blockers of sodium and calcium entry protect axons from nitric oxide‐mediated degeneration. Ann Neurol 53:174–180. [DOI] [PubMed] [Google Scholar]
- 77. Kaspar A, Brinkmeier H, Rudel R (1994) Local anaesthetic‐like effect of interleukin‐2 on muscular Na+ channels: no evidence for involvement of the IL‐2 receptor. Pflugers Arch 426:61–67. [DOI] [PubMed] [Google Scholar]
- 78. Khan NA, Poisson JP (1999) 5‐HT3 receptor‐channels coupled with Na+ influx in human T cells: role in T cell activation. J Neuroimmunol 99:53–60. [DOI] [PubMed] [Google Scholar]
- 79. Kohama I, Lankford KL, Preiningerova J, White FA, Vollmer TL, Kocsis JD (2001) Transplantation of cryopreserved adult human Schwann cells enhances axonal conduction in demyelinated spinal cord. J Neurosci 21:944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, Linington C, Schmidbauer M, Lassmann H (2000) Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol 157:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Korotzer AR, Cotman CW (1992) Voltage‐gated currents expressed by rat microglia in culture. Glia 6:81–88. [DOI] [PubMed] [Google Scholar]
- 82. Lai ZF, Chen YZ, Nishimura Y, Nishi K (2000) An amiloride‐sensitive and voltage‐dependent Na+ channel in an HLA‐DR‐restricted human T cell clone. J Immunol 165:83–90. [DOI] [PubMed] [Google Scholar]
- 83. Lassmann H, Wekerle H (2006) The pathology of multiple sclerosis. In: McAlpine’s Multiple Sclerosis. Compston A, Confavreux C, Lassmann H, McDonald I, Miller D, Noseworthy J, Smith K, Wekerle H (eds), pp. 557–599. Churchill Livingstone: London. [Google Scholar]
- 84. Lhermitte J, Bollack J, Nicholas M (1924) Les douleurs à type de décharge électrique consécutives à la flexion céphalique dans la sclérose en plaques. Rev Neurol 2:56–62. [Google Scholar]
- 85. Li Z, Chapleau MW, Bates JN, Bielefeldt K, Lee H‐C, Abboud FM (1998) Nitric oxide as an autocrine regulator of sodium currents in baroreceptor neurons. Neuron 20:1039–1049. [DOI] [PubMed] [Google Scholar]
- 86. Lo AC, Saab CY, Black JA, Waxman SG (2003) Phenytoin protects spinal cord axons and P axonal conduction and neurological function in a model of neuroinflammation in vivo . J Neurophysiol 90:3566–3571. [DOI] [PubMed] [Google Scholar]
- 87. Lu JL, Sheikh KA, Wu HS, Zhang J, Jiang ZF, Cornblath DR, Asbury AK, Griffin JW, Ho TW (2000) Physiologic‐pathologic correlation in Guillain‐Barre syndrome in children. Neurology 54:33–39. [DOI] [PubMed] [Google Scholar]
- 88. Mackay RP, Hirano A (1967) Forms of benign multiple sclerosis with report of two “clinically silent” cases discovered at autopsy. Trans Am Neurol Assoc 92:143–149. [PubMed] [Google Scholar]
- 89. Makowska A, Bechtold DA, Sajic M, Gregson N, Hughes RAC, Smith KJ (2004) Effect of sodium channel blocking agents on T cell activation. J Immunol 154:88. [Google Scholar]
- 90. Mathey E, Derfuss T, Storch M, Hales K, Rasband M, Velhin S, Hohlfeld R, Meint E, Linington C (2007) Neurofascin: a novel target for antibody mediated axonal injury in multiple sclerosis. Eur J Neurol (in press). [Google Scholar]
- 91. McDonald I (1998) Pathophysiology of multiple sclerosis. In: McAlpine’s Multiple Sclerosis. Compston A, Ebers G, Lassmann H, McDonald I, Matthews B, Wekerle H (eds), pp. 359–378. Churchill Livingstone: London. [Google Scholar]
- 92. McDonald WI (1963) The effects of experimental demyelination on conduction in peripheral nerve: a histological and electrophysiological study. II. Electrophysiological observations. Brain 86:501–524. [DOI] [PubMed] [Google Scholar]
- 93. McDonald WI (1975) Mechanisms of functional loss and recovery in spinal cord damage. In: Outcome of Severe Damage to the Central Nervous System. 23–33. [DOI] [PubMed]
- 94. McDonald WI, Sears TA (1969) Effect of demyelination on conduction in the central nervous system. Nature 221:182–183. [DOI] [PubMed] [Google Scholar]
- 95. McDonald WI, Sears TA (1970) The effects of experimental demyelination on conduction in the central nervous system. Brain 93:583–598. [DOI] [PubMed] [Google Scholar]
- 96. McDonald WI, Sears TA (1970) Effect of a demyelinating lesion on conduction in the central nervous system studied in single nerve fibres. J Physiol (Lond) 207:53P–54P. [PubMed] [Google Scholar]
- 97. Meuth SG, Budde T, Duyar H, Landgraf P, Broicher T, Elbs M, Brock R, Weller M, Weissert R, Wiendl H (2003) Modulation of neuronal activity by the endogenous pentapeptide QYNAD. Eur J Neurosci 18:2697–2706. [DOI] [PubMed] [Google Scholar]
- 98. Mogyoros I, Bostock H, Burke D (2000) Mechanisms of paresthesias arising from healthy axons. Muscle Nerve 23:310–320. [DOI] [PubMed] [Google Scholar]
- 99. Moreau T, Coles A, Wing M, Isaacs J, Hale G, Waldmann H, Compston A (1996) Transient increase in symptoms associated with cytokine release in patients with multiple sclerosis. Brain 119:225–237. [DOI] [PubMed] [Google Scholar]
- 100. Nordin M, Nystrom B, Wallin U, Hagbarth KE (1984) Ectopic sensory discharges and paresthesiae in patients with disorders of peripheral nerves, dorsal roots and dorsal columns. Pain 20:231–245. [DOI] [PubMed] [Google Scholar]
- 101. Norenberg W, Illes P, Gebicke‐Haerter PJ (1994) Sodium channel in isolated human brain macrophages (microglia). Glia 10:165–172. [DOI] [PubMed] [Google Scholar]
- 102. Novakovic SD, Levinson SR, Schachner M, Shrager P (1998) Disruption and reorganization of sodium channels in experimental allergic neuritis. Muscle Nerve 21:1019–1032. [DOI] [PubMed] [Google Scholar]
- 103. O’Riordan JI, Losseff NA, Phatouros C, Thompson AJ, Moseley IF, Macmanus DG, McDonald WI, Miller DH (1998) Asymptomatic spinal cord lesions in clinically isolated optic nerve, brain stem, and spinal cord syndromes suggestive of demyelination. J Neurol Neurosurg Psychiat 64:353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Oh Y, Warnock DG (1997) Expression of the amiloride‐sensitive sodium channel beta subunit gene in human B lymphocytes. J Am Soc Nephrol 8:126–129. [DOI] [PubMed] [Google Scholar]
- 105. Otto F, Kieseier BC, Gortz P, Hartung HP, Siebler M (2005) The pentapeptide QYNAD does not inhibit neuronal network activity. Can J Neurol Sci 32:344–348. [DOI] [PubMed] [Google Scholar]
- 106. Padmashri R, Chakrabarti KS, Sahal D, Mahalakshmi R, Sarma SP, Sikdar SK (2004) Functional characterization of the pentapeptide QYNAD on rNav1.2 channels and its NMR structure. Pflugers Arch 447:895–907. [DOI] [PubMed] [Google Scholar]
- 107. Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H, Sorensen P, Brueck W, Lucchinetti C, Lassmann H (2006) Remyelination is extensive in a subset of multiple sclerosis patients. Brain 129:3165–3172. [DOI] [PubMed] [Google Scholar]
- 108. Phadke JG, Best PV (1983) Atypical and clinically silent multiple sclerosis: a report of 12 cases discovered unexpectedly at necropsy. J Neurol Neurosurg Psychiat 46:414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Pieri C, Recchioni R, Moroni F, Balkay L, Marian T, Tron L, Damjanovich S (1989) Ligand and voltage gated sodium channels may regulate electrogenic pump activity in human, mouse and rat lymphocytes. Biochem Biophys Res Commun 160:999–1002. [DOI] [PubMed] [Google Scholar]
- 110. Prineas JW, Connell F (1979) Remyelination in multiple sclerosis. Ann Neurol 5:22–31. [DOI] [PubMed] [Google Scholar]
- 111. Prineas JW, Kwon EE, Sharer LR, Cho E‐S (1987) Massive early remyelination in acute multiple sclerosis. Neurology 37(Suppl. 1):109. [Google Scholar]
- 112. Quasthoff S, Pojer C, Mori A, Hofer D, Liebmann P, Kieseier BC, Schreibmayer W (2003) No blocking effects of the pentapeptide QYNAD on Na+ channel subtypes expressed in Xenopus oocytes or action potential conduction in isolated rat sural nerve. Neurosci Lett 352:93–96. [DOI] [PubMed] [Google Scholar]
- 113. Redford EJ, Kapoor R, Smith KJ (1997) Nitric oxide donors reversibly block axonal conduction: demyelinated axons are especially susceptible. Brain 120:2149–2157. [DOI] [PubMed] [Google Scholar]
- 114. Renganathan M, Cummins TR, Hormuzdiar WN, Black JA, Waxman SG (2000) Nitric oxide is an autocrine regulator of Na+ currents in axotomized C‐type DRG neurons. J Neurophysiol 83:2431–2442. [DOI] [PubMed] [Google Scholar]
- 115. Renganathan M, Gelderblom M, Black JA, Waxman SG (2003) Expression of Nav1.8 sodium channels perturbs the firing patterns of cerebellar Purkinje cells. Brain Res 959:235–242. [DOI] [PubMed] [Google Scholar]
- 116. Ritchie JM, Rogart RB (1977) Density of sodium channels in mammalian myelinated nerve fibers and nature of the axonal membrane under the myelin sheath. Proc Natl Acad Sci USA 74:211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rizzo MA, Kocsis JD, Waxman SG (1996) Mechanisms of paresthesiae, dysesthesiae, and hyperesthesiae: role of Na+ channel heterogeneity. Eur Neurol 36:3–12. [DOI] [PubMed] [Google Scholar]
- 118. Rosenbluth J, Tao‐Cheng J‐H, Blakemore WF (1985) Dependence of axolemmal differentiation on contact with glial cells in chronically demyelinated lesions of cat spinal cord. Brain Res 358:287–302. [DOI] [PubMed] [Google Scholar]
- 119. Rush AM, Dib‐Hajj SD, Waxman SG (2005) Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J Physiol 564:803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Saab CY, Craner MJ, Kataoka Y, Waxman SG (2004) Abnormal Purkinje cell activity in vivo in experimental allergic encephalomyelitis. Exp Brain Res 158:1–8. [DOI] [PubMed] [Google Scholar]
- 121. Sato T, Kamata Y, Irifune M, Nishikawa T (1995) Inhibition of purified (Na+,K+)‐ATPase activity from porcine cerebral cortex by NO generating drugs. Brain Res 704:117–120. [DOI] [PubMed] [Google Scholar]
- 122. Sawada M, Hara N, Maeno T (1991) Analysis of a decreased Na+ conductance by tumor necrosis factor in identified neurons of Aplysia kurodai. J Neurosci Res 28:466–473. [DOI] [PubMed] [Google Scholar]
- 123. Sawada M, Ichinose M, Hara N (1995) Nitric oxide induces an increased Na+ conductance in identified neurons of Aplysia. Brain Res 670:248–256. [DOI] [PubMed] [Google Scholar]
- 124. Selhorst JB, Saul RF (1995) Uhthoff and his symptom. J Neuroophthalmol 15:63–69. [PubMed] [Google Scholar]
- 125. Shrager P (1977) Slow sodium inactivation in nerve after exposure to sulhydryl blocking reagents. J Gen Physiol 69:183–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Shrager P, Custer AW, Kazarinova K, Rasband MN, Mattson D (1998) Nerve conduction block by nitric oxide that is mediated by the axonal environment. J Neurophysiol 79:529–536. [DOI] [PubMed] [Google Scholar]
- 127. Smith KJ (1994) Conduction properties of central demyelinated and remyelinated axons, and their relation to symptom production in demyelinating disorders. Eye 8:224–237. [DOI] [PubMed] [Google Scholar]
- 128. Smith KJ, Lassmann H (2002) The role of nitric oxide in multiple sclerosis. Lancet Neurol 1:232–241. [DOI] [PubMed] [Google Scholar]
- 129. Smith KJ, McDonald WI (1980) Spontaneous and mechanically evoked activity due to central demyelinating lesion. Nature 286:154–155. [DOI] [PubMed] [Google Scholar]
- 130. Smith KJ, McDonald WI (1982) Spontaneous and evoked electrical discharges from a central demyelinating lesion. J Neurol Sci 55:39–47. [DOI] [PubMed] [Google Scholar]
- 131. Smith KJ, Blakemore WF, McDonald WI (1979) Central remyelination restores secure conduction. Nature 280:395–396. [DOI] [PubMed] [Google Scholar]
- 132. Smith KJ, Blakemore WF, McDonald WI (1981) The restoration of conduction by central remyelination. Brain 104:383–404. [DOI] [PubMed] [Google Scholar]
- 133. Smith KJ, Bostock H, Hall SM (1982) Saltatory conduction precedes remyelination in axons demyelinated with lysophosphatidyl choline. J Neurol Sci 54:13–31. [DOI] [PubMed] [Google Scholar]
- 134. Smith KJ, Felts PA, Kapoor R (1997) Axonal hyperexcitability: mechanisms and role in symptom production in demyelinating diseases. Neuroscientist 3:237–246. [Google Scholar]
- 135. Smith KJ, Kapoor R, Hall SM, Davies M (2001) Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol 49:470–476. [PubMed] [Google Scholar]
- 136. Smith KJ, McDonald I, Miller D, Lassmann H (2006) The pathophysiology of multiple sclerosis. In: McAlpine’s Multiple Sclerosis. Compston A, Confavreux C, Lassmann H, McDonald I, Miller D, Noseworthy J, Smith K, Wekerle H (eds), pp. 601–659. Churchill Livingstone: London. [Google Scholar]
- 137. Stys PK (1998) Anoxic and ischemic injury of myelinated axons in CNS white matter: from mechanistic concepts to therapeutics. J Cereb Blood Flow Metab 18:2–25. [DOI] [PubMed] [Google Scholar]
- 138. Stys PK, Sontheimer H, Ransom BR, Waxman SG (1993) Noninactivating, tetrodotoxin‐sensitive Na+ conductance in rat optic nerve axons. Proc Natl Acad Sci USA 90:6976–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Sumner AJ, Saida K, Saida T, Silberberg DH, Asbury AK (1982) Acute conduction block associated with experimental antiserum‐mediated demyelination of peripheral nerve. Ann Neurol 11:469–477. [DOI] [PubMed] [Google Scholar]
- 140. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L (1998) Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338:278–285. [DOI] [PubMed] [Google Scholar]
- 141. Ulrich J, Groebke‐Lorenz W (1983) The optic nerve in multiple sclerosis: a morphological study with retrospective clinico‐pathological correlations. Neuro-Ophthalmology 3:149–159. [Google Scholar]
- 142. Utzschneider DA, Archer DR, Kocsis JD, Waxman SG, Duncan ID (1994) Transplantation of glial cells enhances action potential conduction of amyelinated spinal cord axons in the myelin‐deficient rat. Proc Natl Acad Sci USA 91:53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Waxman SG (1977) Conduction in myelinated, unmyelinated, and demyelinated fibers. Arch Neurol 34:585–589. [DOI] [PubMed] [Google Scholar]
- 144. Waxman SG (1981) Clinicopathological correlations in multiple sclerosis and related diseases. Adv Neurol 31:169–182. [PubMed] [Google Scholar]
- 145. Waxman SG (1989) Demyelination in spinal cord injury. J Neurol Sci 91:1–14. [DOI] [PubMed] [Google Scholar]
- 146. Waxman SG (1995) Sodium channel blockade by antibodies: a new mechanism of neurological disease? Ann Neurol 37:421–423. [DOI] [PubMed] [Google Scholar]
- 147. Waxman SG (2000) Do “demyelinating” diseases involve more than myelin? Nat Med 6:738–739. [DOI] [PubMed] [Google Scholar]
- 148. Waxman SG (2005) Cerebellar dysfunction in multiple sclerosis: evidence for an acquired channelopathy. Prog Brain Res 148:353–365. [DOI] [PubMed] [Google Scholar]
- 149. Waxman SG, Ritchie JM (1993) Molecular dissection of the myelinated axon. Ann Neurol 33:121–136. [DOI] [PubMed] [Google Scholar]
- 150. Waxman SG, Black JA, Kocsis JD, Ritchie JM (1989) Low density of sodium channels supports action potential conduction in axons of neonatal rat optic nerve. Proc Natl Acad Sci USA 86:1406–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Weber F, Rudel R, Aulkemeyer P, Brinkmeier H (2002) The endogenous pentapeptide QYNAD induces acute conduction block in the isolated rat sciatic nerve. Neurosci Lett 317:33–36. [DOI] [PubMed] [Google Scholar]
- 152. Wing MG, Moreau T, Greenwood J, Smith RM, Hale G, Isaacs J, Lachmann PJ, Compston A (1996) Mechanism of first‐dose cytokine‐release syndrome by CAMPATH 1‐H: involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA‐1) on NK cells. J Clin Invest 98:2819–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wolswijk G, Balesar R (2003) Changes in the expression and localization of the paranodal protein Caspr on axons in chronic multiple sclerosis. Brain 126:1638–1649. [DOI] [PubMed] [Google Scholar]
- 154. Yarom Y, Naparstek Y, Lev‐Ram V, Holoshitz J, Ben‐Nun A, Cohen IR (1983) Immunospecific inhibition of nerve conduction by T lymphocytes reactive to basic protein of myelin. Nature 303:246–247. [DOI] [PubMed] [Google Scholar]
- 155. Youl BD, Turano G, Miller DH, Towell AD, Macmanus DG, Moore SG, Barrett G, Kendall BE, Moseley IF, Tofts PS, Halliday AM, McDonald WI (1991) The pathophysiology of acute optic neuritis. An association of gadolinium leakage with clinical and electrophysiological deficits. Brain 114:2437–2450. [DOI] [PubMed] [Google Scholar]
- 156. Zhou ZH, Bubien JK (2002) ENaC plays a role in regulated antibody secretion by hybridomas. Am J Physiol Cell Physiol 283:C1480–C1491. [DOI] [PubMed] [Google Scholar]