


Abstract
Targeted mRNA expression panels, measuring up to 800 genes, are used in academic and clinical settings due to low cost and high sensitivity for archived samples. Most samples assayed on targeted panels originate from bulk tissue comprised of many cell types, and cell-type heterogeneity confounds biological signals. Reference-free methods are used when cell-type-specific expression references are unavailable, but limited feature spaces render implementation challenging in targeted panels. Here, we present DeCompress, a semi-reference-free deconvolution method for targeted panels. DeCompress leverages a reference RNA-seq or microarray dataset from similar tissue to expand the feature space of targeted panels using compressed sensing. Ensemble reference-free deconvolution is performed on this artificially expanded dataset to estimate cell-type proportions and gene signatures. In simulated mixtures, four public cell line mixtures, and a targeted panel (1199 samples; 406 genes) from the Carolina Breast Cancer Study, DeCompress recapitulates cell-type proportions with less error than reference-free methods and finds biologically relevant compartments. We integrate compartment estimates into cis-eQTL mapping in breast cancer, identifying a tumor-specific cis-eQTL for CCR3 (C–C Motif Chemokine Receptor 3) at a risk locus. DeCompress improves upon reference-free methods without requiring expression profiles from pure cell populations, with applications in genomic analyses and clinical settings.
INTRODUCTION
Academic and clinical settings have prioritized the collection of tissue samples of mixed cell types for molecular profiling and biomarker studies (1–3). Bulk tissue, especially from cancerous tumors, is comprised of different cell types, many rare, and each contributing varied biological signal to an assay (e.g. mRNA expression) (4,5). This cell-type heterogeneity makes it difficult to distinguish variability that reflects shifts in cell populations from variability that reflects changes in cell-type-specific expression (6). Since RNA-seq technology was developed, cell-type deconvolution from mRNA expression has become important in genetic and genomic association studies: either using compositions in regression models as covariates to adjust for the association between cell type proportions and phenotype (7–10), or using them as inputs to solve for cell-type specific quantities (11,12). Cell-type deconvolution methods can be reference-based (supervised) (13–19) or reference-free (unsupervised) (20–25), depending on whether cell-type-specific expression profiles are available for the component cell-types. When reference panels are unavailable, as in understudied tissues or populations (26), reference-free deconvolution is the only viable option. Even in cases where reference expression profiles are available, reference-based methods may provide inaccurate proportion estimates if the mixed tissue and references represent different clinical settings or phenotypes (27).
Given the advent of single-cell technologies and studies into cell trajectories, the concept of cell types in bulk tissue has been debated (28). Especially in perturbed or diseased tissues, like cancerous tumors, individual cells may be present in different states or various cells of possibly different identities may contribute, in aggregate, to the same biological process and have similar molecular profiles (29–31). While previous reference-free methods rely on searching the feature space for compartment-specific molecular features from the entire transcriptome and thus require a large feature space (22,25,32), reference-free deconvolution methods can, with fewer assumptions, identify tissue compartments or isolated units of a tissue that represent either a biological process or a cell type (33). Thus, reference-free methods have important advantages over reference-based methods but may require a large number of features for optimal performance (32,34).
Many important datasets may have fewer expression targets than those required for existing reference-free deconvolution methods. Targeted mRNA expression assays are optimized for gene expression quantification in samples stored clinically and use a panel of up to 800 genes without requiring cDNA synthesis or amplification steps (35–37). These technologies offer key advantages in sensitivity, technical reproducibility, and strong robustness for profiling formalin-fixed, paraffin-embedded (FFPE) samples (35,38). Given these advantages, targeted expression profiling is increasingly being used for molecular studies (36,37,39–42), especially prospective studies involving FFPE samples stored over several years (43) and diagnostic assays in clinical settings (3,44). Due to its viability in diagnostics, it is important to identify reference-free deconvolution methods that overcome the need for searching for compartment-specific genes from the assay's feature space (22,25,32), given the limited feature space in targeted panels.
Previous groups have proposed methods for efficiently reconstructing full gene expression profiles from sparse measurements of the transcriptome, borrowing techniques from image reconstruction using compressed sensing (45,46) and machine learning (47–50). For example, Cleary et al. developed a blind compressed sensing method that recovers gene expression from multiple composite measurements of the transcriptome (up to 100 times fewer measurements than genes) by using modules of interrelated genes in an unsupervised manner. Another imputation method by Viñas et al. (51) used recent machine learning methodology (52) to provide efficient and accurate transcriptomic reconstruction in healthy, unperturbed tissue from the Genotype-Tissue Expression (GTEx) Project (53,54). These ideas provide a promising avenue to expand the feature space of targeted panels, rendering them more applicable for reference-free deconvolution methods.
Here, we present DeCompress, a semi-reference-free deconvolution method for targeted panels. DeCompress requires a reference RNA-seq or microarray dataset from similar bulk tissue assayed by the targeted expression panel to train a compressed sensing model to expand the feature space in a targeted panel. We show the advantages of using DeCompress over other reference-free methods with two simulation analyses and five real data applications. Lastly, we examine the impact of tissue compartment deconvolution on downstream analyses, such as cis-eQTL analysis using expression data from the Carolina Breast Cancer Study (CBCS) (55). DeCompress is the first deconvolution method to focus on targeted expression panels and is available freely as an R package on GitHub at https://github.com/bhattacharya-a-bt/DeCompress.
MATERIALS AND METHODS
The DeCompress algorithm
DeCompress takes in two expression matrices from similar bulk tissue as inputs: the target expression matrix from a targeted panel of gene expression with 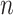 samples and
samples and 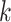 genes, and a reference expression matrix from an RNA-seq and microarray panel with
genes, and a reference expression matrix from an RNA-seq and microarray panel with 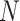 samples and
samples and 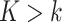 genes. Ideally, both the target and reference expression matrices should be on the raw expression scale (not log-transformed), as total RNA abundance of a gene in bulk tissue is a linear combination of that gene's compartment-specific RNA abundances. We refer to DeCompress as a semi-reference-free method, as it requires a reference expression matrix but not compartment-specific expression profiles (as in reference-based methods). For a user-defined number of compartments, DeCompress outputs compartment proportions for all samples in the target and the compartment-specific expression profiles for the genes used in deconvolution; these compartments reflect groups of cells that have similar biological processes or molecular profiles. The method follows three general steps, as detailed in Figure 1: (1) selection of the compartment-specific genes from the reference, (2) compressed sensing to expand the targeted panel to a DeCompressed expression matrix with these compartment-specific genes, and (3) ensemble deconvolution on the DeCompressed dataset. Full mathematical and algorithmic details for DeCompress are provided in Supplemental Methods. DeCompress is available as an R package on GitHub (https://github.com/bhattacharya-a-bt/DeCompress).
genes. Ideally, both the target and reference expression matrices should be on the raw expression scale (not log-transformed), as total RNA abundance of a gene in bulk tissue is a linear combination of that gene's compartment-specific RNA abundances. We refer to DeCompress as a semi-reference-free method, as it requires a reference expression matrix but not compartment-specific expression profiles (as in reference-based methods). For a user-defined number of compartments, DeCompress outputs compartment proportions for all samples in the target and the compartment-specific expression profiles for the genes used in deconvolution; these compartments reflect groups of cells that have similar biological processes or molecular profiles. The method follows three general steps, as detailed in Figure 1: (1) selection of the compartment-specific genes from the reference, (2) compressed sensing to expand the targeted panel to a DeCompressed expression matrix with these compartment-specific genes, and (3) ensemble deconvolution on the DeCompressed dataset. Full mathematical and algorithmic details for DeCompress are provided in Supplemental Methods. DeCompress is available as an R package on GitHub (https://github.com/bhattacharya-a-bt/DeCompress).
Figure 1.

Schematic for the DeCompress algorithm. DeCompress takes in a reference RNA-seq or microarray matrix with 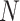 samples and
samples and 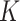 genes, and the target expression with
genes, and the target expression with 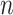 samples and
samples and 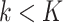 genes. The algorithm has three general steps: (1) finding the
genes. The algorithm has three general steps: (1) finding the 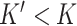 genes in the reference that are cell-type specific, (2) training the compressed sensing model that projects the feature space in the target from
genes in the reference that are cell-type specific, (2) training the compressed sensing model that projects the feature space in the target from 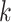 genes to the
genes to the 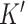 cell-type specific genes, and (3) decompressing the target to an expanded dataset and deconvolving this expanded dataset. DeCompress outputs cell-type proportions and cell-type specific profiles for the
cell-type specific genes, and (3) decompressing the target to an expanded dataset and deconvolving this expanded dataset. DeCompress outputs cell-type proportions and cell-type specific profiles for the 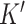 genes.
genes.
The first step of DeCompress is to use the reference dataset to find a set of 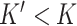 genes that are representative of different compartments that comprise the bulk tissue. These
genes that are representative of different compartments that comprise the bulk tissue. These 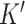 genes, called the compartment-specific genes, can be supplied by the user if prior gene signatures can be applied. If any such gene signatures are not available, DeCompress borrows from previous reference-free methods to determine this set of genes [Linseed (22) or TOAST (32)]. If the user cannot determine the total number of compartments, the number of compartments can be estimated from the reference by assessing the cumulative total variance explained by successive singular value decomposition modes or the number of columns in the basis of a non-negative matrix factorization of the reference matrix.
genes, called the compartment-specific genes, can be supplied by the user if prior gene signatures can be applied. If any such gene signatures are not available, DeCompress borrows from previous reference-free methods to determine this set of genes [Linseed (22) or TOAST (32)]. If the user cannot determine the total number of compartments, the number of compartments can be estimated from the reference by assessing the cumulative total variance explained by successive singular value decomposition modes or the number of columns in the basis of a non-negative matrix factorization of the reference matrix.
After a set of compartment-specific genes are determined, DeCompress uses the reference to infer a model that predicts the expression of each of these compartment-specific genes from the genes in the target. Predictive modeling procedures borrow ideas from compressed sensing (45,46,56), a technique that was developed to reconstruct a full image from sparse measurements of it: the estimation procedure can be broken down into solving a system of equations using either linear or non-linear regularized optimization, with options for parallelization when the sample size of the reference dataset is large. These optimization methods are detailed in Supplemental Methods. The predictive models are curated into a compression matrix, which is then used to expand the original target (with 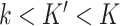 genes) into the artificially DeCompressed expression matrix (with the
genes) into the artificially DeCompressed expression matrix (with the 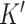 compartment-specific genes). This compressed sensing model and feature expansion helps recover expression of compartment-specific genes that are not assayed in the target but also those genes that have high variance in groups or subtypes present only in the target (Supplemental Results, Supplemental Figure S1). In practice, we observed that regularized linear regression (lasso, ridge, or elastic net regression (48)) provides the best prediction of gene expression (Supplemental Figure S2), and the user may either model the gene expression using the traditional Gaussian family or assume that the errors follow a Poisson distribution to account for the scale of the original data (not log-transformed).
compartment-specific genes). This compressed sensing model and feature expansion helps recover expression of compartment-specific genes that are not assayed in the target but also those genes that have high variance in groups or subtypes present only in the target (Supplemental Results, Supplemental Figure S1). In practice, we observed that regularized linear regression (lasso, ridge, or elastic net regression (48)) provides the best prediction of gene expression (Supplemental Figure S2), and the user may either model the gene expression using the traditional Gaussian family or assume that the errors follow a Poisson distribution to account for the scale of the original data (not log-transformed).
Lastly, ensemble deconvolution is performed on the DeCompressed expression matrix to estimate (i) compartment proportions on the samples in the target and (ii) the compartment-specific expression profiles for the 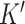 genes used in deconvolution. Several options for reference-free deconvolution are provided in DeCompress. We also provide options that uses a reference-based method, unmix from the DESeq2 package (57), based on compartment expression profiles estimated from the reference RNA-seq or microarray dataset (i.e. an approximate compartment expression profile is estimated from a non-negative matrix factorization of the reference dataset). Estimates from the method that best recovers the DeCompressed expression matrix are chosen. Supplemental Table S1 provides summaries of the methods employed in DeCompress. In practice, we recommend that users iterate the DeCompress process over a range of numbers of compartments and validate the estimated compartments and gene signatures against known biology. An example biological validation process is described in detail when we apply DeCompress to a large NanoString nCounter expression dataset from the CBCS (43).
genes used in deconvolution. Several options for reference-free deconvolution are provided in DeCompress. We also provide options that uses a reference-based method, unmix from the DESeq2 package (57), based on compartment expression profiles estimated from the reference RNA-seq or microarray dataset (i.e. an approximate compartment expression profile is estimated from a non-negative matrix factorization of the reference dataset). Estimates from the method that best recovers the DeCompressed expression matrix are chosen. Supplemental Table S1 provides summaries of the methods employed in DeCompress. In practice, we recommend that users iterate the DeCompress process over a range of numbers of compartments and validate the estimated compartments and gene signatures against known biology. An example biological validation process is described in detail when we apply DeCompress to a large NanoString nCounter expression dataset from the CBCS (43).
Benchmarking analysis
Using simulations and published datasets, we benchmarked DeCompress against six other reference-free methods: deconf (20), CellDistinguisher (25), Linseed (22), DeconICA (see Data Availability), CDSeq (23), and iterative non-negative matrix factorization with feature selection using TOAST (32) (see Supplemental Table S1). We implemented all methods with default settings provided by the respective software packages. All these datasets provide a matrix of known compartment proportions. To measure the performance of each method, we calculate the error between the estimated and true compartment proportions as the mean squared error (MSE) (i.e. the mean row-wise MSE between the two matrices). We also permute the columns in the estimated matrix (corresponding to compartments) to align compartments accordingly between the known and estimated proportions to minimize the MSE for each method. In all benchmarking analyses (in-silico and published mixing experiments and CBCS), we use default settings for DeCompress: TOAST to select compartment-specific genes in the reference, elastic net with mixing parameter 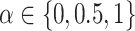 to train compressed sensing models, and ensemble deconvolution of the DeCompressed dataset using CellDistinguisher, Linseed, TOAST + NMF, and DeconICA; we did not use CDSeq on DeCompressed datasets due to running time, but it is a viable option for users of DeCompress. We set the number of compartments in the benchmarking analyses to the number of compartments in the true proportion matrices.
to train compressed sensing models, and ensemble deconvolution of the DeCompressed dataset using CellDistinguisher, Linseed, TOAST + NMF, and DeconICA; we did not use CDSeq on DeCompressed datasets due to running time, but it is a viable option for users of DeCompress. We set the number of compartments in the benchmarking analyses to the number of compartments in the true proportion matrices.
In-silico mixing experiments
We performed in-silico mixing experiments using single-cell RNA-seq expression data (GEO accession number: GSE136148) from single-cell suspensions of fresh frozen mouse mammary gland tissue (19). After processing and clustering (58,59), we identified expression profiles for four well-characterized sets of cells (>50 cells in each cluster): epithelial cells, fibroblasts, adipocytes, and immune cells (see Supplemental Methods). We randomly generated compartment proportions for each of these tissue types by generating a proportion matrix drawn from a half-normal distribution with variance 1 and dividing each row by the row sum. For a reference, we then simulated mixed RNA-seq expression data for 200 samples and scaled these mixed expression profiles with multiplicative noise randomly generated from a half-Normal distribution with 0 mean and standard deviations of 4 and 8. Across 25 simulations, we simulated another mixed RNA-seq dataset with the same noise parameters and selected 200, 500, and 800 of the genes with mean and standard deviations above the median mean and standard deviations of all genes to generate a targeted panel. We added more normally-distributed multiplicative noise with zero mean and unit variance to simulate a batch difference between the reference and target matrix.
We additionally performed in-silico mixing experiments using expression data from the Genotype-Tissue Expression (GTEx) Project (dbGAP accession number phs000424.v7.p2) (53,54). Here, we obtained median transcripts per million (TPM) data for four tissue types largely present in bulk mammary tissue: subareolar mammary cells, EBV-transformed lymphocytes, transformed fibroblasts, and subcutaneous adipose. Using these median profiles, we generated a reference RNA-seq dataset and targeted panel as in the single-cell experiment. For comparison to compartments with dissimilar expression profiles, we repeated these simulations for four other tissues: mammary tissue, pancreas, pituitary and whole blood. Full details for this simulation framework are provided in Supplemental Methods.
Existing mixing experiments
We also benchmarked DeCompress in four published mixing experiments: (i) microarray expression for mixed rat brain, liver, and lung biospecimens (GEO Accession Number: GSE19830), commonly used as a benchmarking dataset in deconvolution studies (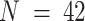 ) (11), (ii) RNA-seq expression (GSE123604) for a mixture of breast cancer cells, fibroblasts, normal mammary cells and Burkitt's lymphoma cells (
) (11), (ii) RNA-seq expression (GSE123604) for a mixture of breast cancer cells, fibroblasts, normal mammary cells and Burkitt's lymphoma cells (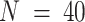 ) (23), (iii) microarray expression (GSE97284) for laser capture micro-dissected prostate tumors (
) (23), (iii) microarray expression (GSE97284) for laser capture micro-dissected prostate tumors (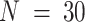 ) (60) and (iv) RNA-seq expression (GSE64098) for a mixture of two lung adenocarcinoma cell lines (
) (60) and (iv) RNA-seq expression (GSE64098) for a mixture of two lung adenocarcinoma cell lines (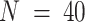 ) (61,62). As in the in-silico mixing using GTEx data, we generated pseudo-targeted panels by randomly selecting 200, 500, and 800 of the genes with mean and standard deviations above the median mean and standard deviations of all genes. For the rat mixture dataset, we used 30 of the 42 samples as a reference microarray matrix (with multiplicative noise, as in GTEx) and deconvolved with the remaining 12 samples in the target matrix. In the remaining three datasets, we obtained normalized RNA-seq reference matrices from The Cancer Genome Atlas: TCGA-BRCA breast tumor expression for the breast cancer cell line mixture, TCGA-PRAD prostate tumor expression for the prostate tumor microarray study, and TCGA-LUAD for the lung adenocarcinoma mixing study. These datasets are summarized in Supplemental Table S2.
) (61,62). As in the in-silico mixing using GTEx data, we generated pseudo-targeted panels by randomly selecting 200, 500, and 800 of the genes with mean and standard deviations above the median mean and standard deviations of all genes. For the rat mixture dataset, we used 30 of the 42 samples as a reference microarray matrix (with multiplicative noise, as in GTEx) and deconvolved with the remaining 12 samples in the target matrix. In the remaining three datasets, we obtained normalized RNA-seq reference matrices from The Cancer Genome Atlas: TCGA-BRCA breast tumor expression for the breast cancer cell line mixture, TCGA-PRAD prostate tumor expression for the prostate tumor microarray study, and TCGA-LUAD for the lung adenocarcinoma mixing study. These datasets are summarized in Supplemental Table S2.
Applications in Carolina Breast Cancer Study (CBCS) data
We lastly used expression data from the Carolina Breast Cancer Study for validation and analysis (55). Paraffin-embedded tumor blocks were requested from participating pathology laboratories for each sample, were reviewed, and were assayed for gene expression using the NanoString nCounter system, as discussed previously (43). As described before (10,63), the expression data (406 genes and 11 housekeeping genes) were pre-processed and normalized using quality control steps from the NanoStringQCPro package, median ratio normalization using DESeq2 (57,64), and estimation and removal of unwanted technical variation using the RUVSeq and limma packages (65,66). The resulting normalized dataset comprised of samples from 1199 patients, comprising of 628 women of African descent (AA) and 571 women of European descent (EA). A study pathologist analyzed tumor microarrays (TMAs) from 148 of the 1199 patients to estimate area of dissections originating from epithelial tumor, intratumoral stroma, immune infiltrate, and adipose tissue (10). These compartment proportions of the 148 samples were used for benchmarking of DeCompress against other reference-free methods.
Date of death and cause of death were identified by linkage to the National Death Index. All diagnosed with breast cancer have been followed for vital status from diagnosis until date of death or date of last contact. Breast cancer-related deaths were classified as those that listed breast cancer (International Statistical Classification of Disease codes 174.9 and C-50.9) as the underlying cause of death on the death certificate. Of the 1199 samples deconvolved, 1153 had associated survival data with 330 total deaths, 201 attributed to breast cancer.
Over-representation and gene set enrichment analysis
We conducted over-representation (ORA) and gene set enrichment analysis (GSEA) to identify significantly enriched gene ontologies using WebGestaltR (67). Specifically, we considered biological process ontologies categorized by The Gene Ontology Consortium (68,69) at FDR-adjusted 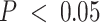 .
.
Survival analysis
Here, we defined a relevant event as a death due to breast cancer. We aggregated all deaths not due to breast cancer as a competing risk. Any subjects lost to follow-up were treated as right-censored observations. We built cause-specific Cox models (70) by modeling the hazard function of breast cancer-specific mortality with the following covariates: race, PAM50 molecular subtype (71), age, compartment-specific proportions and an interaction term between molecular subtype and compartment proportion. We compared these compartment-specific survival models with the nested baseline model that did not include compartment proportions using partial likelihood ratio tests. We tested for the statistical significance of parameter estimates using Wald-type tests, adjusting for multiple testing burden using the Benjamini-Hochberg procedure at a 10% false discovery rate (72).
eQTL analysis
CBCS genotype data is measured on the OncoArray. Approximately 50% of the SNPs for the OncoArray were selected as a ‘GWAS backbone’ (Illumina HumanCore), which aimed to provide high coverage for many common variants through imputation. The remaining SNPs were selected from lists supplied by six disease-based consortia, together with a seventh list of SNPs of interest to multiple disease-focused groups. Approximately 72 000 SNPs were selected specifically for their relevance to breast cancer. The sources for the SNPs included in this backbone, as well as backbone manufacturing, calling, and quality control, are discussed in depth by the OncoArray Consortium (73,74). All samples were imputed using the October 2014 (v.3) release of the 1000 Genomes Project (75) as a reference panel in the standard two-stage imputation approach, using SHAPEIT2 for phasing and IMPUTEv2 for imputation (76–78). All genotyping, genotype calling, quality control, and imputation was done at the DCEG Cancer Genomics Research Laboratory (73,74).
From the provided genotype data, we excluded variants (i) with a minor frequency less than 1% based on genotype dosage and (ii) that deviated significantly from Hardy-Weinberg equilibrium at P < 10−8 using the appropriate functions in PLINK v1.90b3 (79). Finally, we intersected genotyping panels for the AA and EA samples, resulting in 5 989 134 autosomal variants. We excluded 334,391 variants on the X chromosome. CBCS genotype data was coded as dosages, with reference and alternative allele coding as in the National Center for Biotechnology Information's Single Nucleotide Polymorphism Database (dbSNP) (80).
As previously described (10), using the 1199 samples (621 AA, 578 EA) with expression data, we assessed the additive relationship between the gene expression values and genotypes with linear regression analysis using MatrixeQTL (81). We consider a baseline linear model with log-transformed gene expression of a gene of interest as the dependent variable, SNP dosage as the primary predictor of interest, and the following covariates: age, BMI, post-menopausal status, and the first 5 principal components of the joint AA and EA genotype matrix. We also considered a compartment-specific interaction model that adds compartment proportion from DeCompress and an interaction term between the SNP dosage and compartment proportion (8,9). This interaction model subtly changes the interpretation of the main SNP dosage effect, representing an estimate of the eQTL effect size at 0% compartment-specific cells. Thus, we recover compartment-specific eQTLs by testing the interaction effect, which measures how the magnitude of an eQTL differs between the two cell types. The interaction model was fit using MatrixeQTL’s linear-cross implementation. It is important to note that we model the log-transformed expression here, as existing methods for modeling expression on genotype do not support interaction terms (82–84).
We compared eQTLs mapped in CBCS here with eQTLs in GTEx. We downloaded healthy tissue eQTLs from the Genotype-Tissue Expression (GTEx) Project and cross-referenced eGenes and corresponding eSNPs between CBCS and GTEx in healthy breast mammary tissue, EBV-transformed lymphocytes, transformed fibroblasts, and subcutaneous adipose tissue. We considered these tissues mainly due to their high relative composition in bulk breast tumor samples, as shown previously in many studies (23,85–87). The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 14 May 2020. We also downloaded iCOGs GWAS summary statistics for breast cancer risk (88–90) to assess any overlap between CBCS eQTLs and GWAS-detected risk variants.
RESULTS
Benchmarking DeCompress against other reference-free deconvolution methods
We benchmarked DeCompress performance across seven datasets (see Supplemental Table S2): (i) in-silico mixing experiments using single-cell expression profiles from mouse mammary tissue (19) and tissue-specific expression profiles from the Genotype-Tissue Expression (GTEx) Project (53,54), (ii) expression from four published datasets with known compartment proportions (11,23,60,61) and (iii) and tumor expression from the Carolina Breast Cancer Study (43,55). We compared the performance of DeCompress against five other reference-free deconvolution methods (summarized in Supplemental Table S1): deconf (20), Linseed (22), DeconICA, CDSeq (23), iterative non-negative matrix factorization with feature selection using TOAST (TOAST + NMF) (32) and CellDistinguisher (25). Estimated compartment proportions were compared to simulated or reported true compartment proportions with the mean squared error (MSE) between the two matrices (see Materials and Methods). In total, we observed that DeCompress recapitulates compartment proportions with the least error compared to reference-free deconvolution methods.
In-silico experiments
We first considered in-silico mixing experiments using single-cell expression profiles from mouse mammary gland data (19), specifically from four cell-types: fibroblasts, epithelial cells, adipocytes, and immune cells (see Materials and Methods). Figure 2A shows the performance of DeCompress compared to reference-free methods across 25 simulated targeted panels of increasing numbers of genes on the simulated targeted panels and 200 samples. In general, DeCompress estimated compartment proportions with the lowest MSE across datasets of different feature sizes and the two error settings, with CDSeq and DeconICA producing similarly low errors in estimation. In the high noise setting with 500 and 800 genes on the target, CDSeq shows lower errors than DeCompress. Spearman correlations between true and estimated proportions (element-wise correlation across the two matrices) were consistently largest across all simulation settings using DeCompress compared to other methods (Figure 2A); among the benchmarked reference-free methods, only CDSeq consistently showed positive correlations to the true compartment proportions.
Figure 2.

Benchmarking results for in-silico single cell RNA-seq mixing experiments. (A) Boxplots of mean squared error (MSE) and Spearman correlation (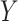 -axis; top and bottom panel, respectively) between true and estimated compartment proportions in in-silico scRNA-seq experiments across various methods (
-axis; top and bottom panel, respectively) between true and estimated compartment proportions in in-silico scRNA-seq experiments across various methods (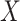 -axis), with 25 simulated datasets per number of genes. Boxplots are colored by the number of genes in each simulated dataset. (B) Scatter plots of MSE (X-axis) and Spearman correlation (Y-axis) across all simulation settings, colored and shaped by method. The Spearman correlation between MSE and correlation is also provided. (C) Boxplots of Spearman correlation (
-axis), with 25 simulated datasets per number of genes. Boxplots are colored by the number of genes in each simulated dataset. (B) Scatter plots of MSE (X-axis) and Spearman correlation (Y-axis) across all simulation settings, colored and shaped by method. The Spearman correlation between MSE and correlation is also provided. (C) Boxplots of Spearman correlation (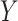 -axis) between true and estimated compartment-specific proportions in in-silico scRNA-seq experiments across various methods (
-axis) between true and estimated compartment-specific proportions in in-silico scRNA-seq experiments across various methods (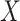 -axis), with 25 simulated datasets per number of genes. Boxplots are colored by the number of genes in each simulated dataset.
-axis), with 25 simulated datasets per number of genes. Boxplots are colored by the number of genes in each simulated dataset.
To put estimates of MSE and correlation in perspective, we considered two methods of generating a null distribution for these metrics: (i) randomly generating 10 000 random proportion matrices and (ii) permuting estimated proportion matrices across samples 10 000 times (Supplemental Figure S3). We found that DeCompress, CDSeq, and DeconICA estimates greatly outperform both nulls, whereas MSE and correlations for other methods overlap with these null distributions. In general, in subsequent analyses, we choose to compare with a randomly generated null (null distribution 1 from above) as it is a common basis of comparison across all methods and included this in Figure 2A. We also found a strong negative association (Spearman correlations of 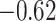 and
and 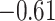 at high and low error settings, both with
at high and low error settings, both with 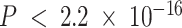 ) between MSE and correlation between the truth and estimates (Figure 2B). This inverse association between MSE and correlation has been reported in previous deconvolution analyses (17,19,32). Thus, to be consistent with previous analyses, we mainly consider MSE as the performance metric in subsequent analyses. Moreover, we found that compartment proportion estimates from DeCompress shows moderate positive correlation (Spearman correlation
) between MSE and correlation between the truth and estimates (Figure 2B). This inverse association between MSE and correlation has been reported in previous deconvolution analyses (17,19,32). Thus, to be consistent with previous analyses, we mainly consider MSE as the performance metric in subsequent analyses. Moreover, we found that compartment proportion estimates from DeCompress shows moderate positive correlation (Spearman correlation 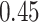 ;
; 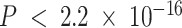 ) with the truth overall, in sharp contrast to other methods (Supplemental Figure S4).
) with the truth overall, in sharp contrast to other methods (Supplemental Figure S4).
We also assessed correlations between true and estimated compartment-specific expression profiles. Here, for the six benchmarked methods, we computed Spearman correlations between true and estimated compartment-specific expression profiles for the 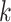 genes on the targeted panel. For DeCompress, we computed these correlations for the
genes on the targeted panel. For DeCompress, we computed these correlations for the 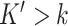 compartment-specific genes on the artificial DeCompressed expression matrix. Figure 2C shows that CDSeq showed largest correlations with the truth, with deconf, TOAST, CellDistinguisher and DeCompress with slightly lower correlations. High correlations of DeCompress estimates with true compartment-specific expression profiles suggests that the compression matrix generally preserves rank order of the compartment-specific genes.
compartment-specific genes on the artificial DeCompressed expression matrix. Figure 2C shows that CDSeq showed largest correlations with the truth, with deconf, TOAST, CellDistinguisher and DeCompress with slightly lower correlations. High correlations of DeCompress estimates with true compartment-specific expression profiles suggests that the compression matrix generally preserves rank order of the compartment-specific genes.
With these in-silico single cell RNA-seq mixtures, we performed sensitivity analysis with respect to the choice of the number of compartments (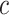 ) to assess how choosing fewer or more compartments might affect results. To orient the compartments across different
) to assess how choosing fewer or more compartments might affect results. To orient the compartments across different 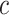 , we used canonical correlation analysis (CCA) on the estimated per-compartment expression profiles (91). As expected, we found that choosing lower values of c (
, we used canonical correlation analysis (CCA) on the estimated per-compartment expression profiles (91). As expected, we found that choosing lower values of c (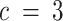 compared to
compared to 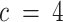 ) resulted in the merging of two compartments, while other compartments stayed relatively similar across runs. Likewise, choosing higher values of
) resulted in the merging of two compartments, while other compartments stayed relatively similar across runs. Likewise, choosing higher values of 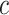 (
(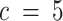 compared to
compared to 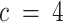 ) resulted in the splitting of a single compartment into two, while the other compartments stayed relatively similar. Contribution plots to the shared canonical variates for a specific instance of simulations (4 true compartments, 500 genes on the target, 100 samples in both the reference and target) are presented in Supplemental Figure S5.
) resulted in the splitting of a single compartment into two, while the other compartments stayed relatively similar. Contribution plots to the shared canonical variates for a specific instance of simulations (4 true compartments, 500 genes on the target, 100 samples in both the reference and target) are presented in Supplemental Figure S5.
Next, we conducted in-silico mixing experiments using median GTEx expression profiles of mammary tissue, lymphocytes, fibroblasts, and subcutaneous adipose (see Materials and Methods). Figure 3A shows the performance of DeCompress compared to other reference-free methods across 25 simulated targeted panels of increasing numbers of genes on the simulated targeted panels. In general, we found that DeCompress gives more accurate estimates of compartment proportions than the other methods at both settings for multiplicative noise. As the number of genes in the targeted panel increased, the difference in MSE between DeCompress and the other methods remains largely constant. Linseed and DeconICA, methods that search for mutually independent axes of variation that correspond to compartments, consistently perform poorly on these simulated datasets, possibly due to the relative similarity between the expression profiles for these compartments and the small number of genes on the targeted panels. deconf, TOAST + NMF (matrix factorization-based methods) and CellDistinguisher (topic modeling) perform similarly to one another and only moderately worse in comparison to DeCompress. CDSeq shows a trend of decreasing median MSE as the number of genes on the target increases, though the range in the MSE values is large.
Figure 3.

Benchmarking results for in-silico GTEx mixing experiments and real data examples. (A) Boxplots of mean squared error (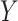 -axis) between true and estimated cell-type proportions in in-silico GTEx mixing experiments across various methods (
-axis) between true and estimated cell-type proportions in in-silico GTEx mixing experiments across various methods (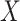 -axis), with 25 simulated datasets per number of genes. GTEx mixing was done at two levels of multiplicative noise, such that errors were drawn from a Normal distribution with zero mean and standard deviation 8 (left) and 4 (right). Boxplots are colored by the number of genes in each simulated dataset. (B) Boxplots of MSE (
-axis), with 25 simulated datasets per number of genes. GTEx mixing was done at two levels of multiplicative noise, such that errors were drawn from a Normal distribution with zero mean and standard deviation 8 (left) and 4 (right). Boxplots are colored by the number of genes in each simulated dataset. (B) Boxplots of MSE (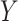 -axis) between true and estimated cell-type proportions over 25 simulated GTEx mixed expression datasets with 500 genes, multiplicative noise drawn from a Normal distribution with zero mean and standard deviation 10, and 2 (left), 3 (middle) and 4 (right) different cell-types. Boxplots are collected by the reference-free method tested. (C) Boxplots of mean squared error (
-axis) between true and estimated cell-type proportions over 25 simulated GTEx mixed expression datasets with 500 genes, multiplicative noise drawn from a Normal distribution with zero mean and standard deviation 10, and 2 (left), 3 (middle) and 4 (right) different cell-types. Boxplots are collected by the reference-free method tested. (C) Boxplots of mean squared error (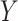 -axis) between true and estimated cell-type proportions in 25 simulated targeted panels of 200, 500, and 800 genes (
-axis) between true and estimated cell-type proportions in 25 simulated targeted panels of 200, 500, and 800 genes (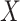 -axis), using four different datasets: breast cancer cell-line mixture (top-left) (23), rat brain, lung, and liver cell-line mixture (top-right) (11), prostate tumor samples (bottom-left) (60), and lung adenocarcinoma cell-line mixture (bottom-right) (61). Boxplots are colored by the benchmarked method. The red line indicates the median null MSE when generating cell-type proportions randomly. If a red line is not provided, then the median null MSE is above the scale provided on the
-axis), using four different datasets: breast cancer cell-line mixture (top-left) (23), rat brain, lung, and liver cell-line mixture (top-right) (11), prostate tumor samples (bottom-left) (60), and lung adenocarcinoma cell-line mixture (bottom-right) (61). Boxplots are colored by the benchmarked method. The red line indicates the median null MSE when generating cell-type proportions randomly. If a red line is not provided, then the median null MSE is above the scale provided on the 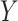 -axis.
-axis.
We also investigated how the number of compartments affects the performance of the seven reference-free methods. Using GTEx, we generated another set of in-silico mixed targeted panels (500 genes) using 2 (human mammary tissue and lymphocytes), 3 (mammary, lymphocytes, fibroblasts) and 4 (mammary, fibroblasts, lymphocytes, and adipose) compartments and applied all methods to estimate the compartment proportions. Figure 3B provides boxplots of the MSE across 25 simulated targeted panels using DeCompress and the other five benchmarked methods. For all seven methods, the median MSE for these datasets remained similar as the number of compartments increased, though the range in the MSE decreases considerably. In particular, the performance of DeconICA increases considerably as more compartments were used for mixing, as mentioned in its documentation. Here again, we found that DeCompress gave the smallest median MSE between the true and estimated cell proportions, with Linseed and CDSeq performing well in the four cell-type setting. In total, results from these in-silico mixing experiments show both the accuracy and precision of DeCompress in estimated compartment proportions.
The four cell types we used for the above analyses simulated bulk mammary tissue but contained compartments with highly correlated gene expression profiles (Supplemental Figure S6A). We recreated the in-silico mixing experiments with four compartments with minimal correlations: mammary tissue, pancreas, pituitary gland, and whole blood (Supplemental Figure S6A). In mixtures with these tissues, we found that DeCompress also outperformed the reference-free methods, with a clear decrease in median MSE as the number of genes on the simulated targeted panels are increased (Supplemental Figure S6B).
Publicly available datasets
Although in-silico mixing experiments showed strong performance of DeCompress, we sought to benchmark DeCompress with previously published datasets that have known compartment mixture proportions. We downloaded expression data from a breast cancer cell-line mixture (RNA-seq) (23), rat brain, lung, and liver cell-line mixture (microarray) (11), prostate tumor with compartment proportions estimated with laser-capture microdissection (microarray) (60), and lung adenocarcinoma cell-line mixture (RNA-seq) (61) and generated pseudo-targeted panels with 200, 500, and 800 genes (see Materials and Methods). For the rat mixture dataset, we trained the compression sensing model on a randomly selected training split with added noise to simulate a batch effect between the training and targeted panel; for the other three cancer-related datasets, reference RNA-seq data was downloaded from The Cancer Genome Atlas (TCGA) (2).
Overall, DeCompress showed the lowest MSE across all three datasets, in comparison to the other reference-free methods (Figure 3C, Supplemental Figure S7). The patterns observed in the GTEx results are evident in these real datasets, as well. Deconvolution using Linseed gave variable performance across datasets (high variability in model performance), with very small ranges in MSEs in the rat microarray and lung adenocarcinoma datasets while highly variable MSEs in the breast cancer and prostate cancer datasets. We investigated the performance of the methods (four reference-free methods and unmix from the DESeq2 package (57)) incorporated into the ensemble deconvolution step of DeCompress in deconvolving the DeCompressed expression, using the breast, prostate, and lung cancer datasets (Supplemental Figure S8). We found that unmix gives accurate estimates of compartment proportions in the breast cancer and prostate tumor datasets, where the compartments are like those in bulk tumors. However, in the case of the lung adenocarcinoma mixing dataset (mixture of two lung cancer cell lines), unmix does not consistently outperform the reference-free methods, perhaps owing to a dissimilarity between the lung adenocarcinoma mixture dataset and TCGA-LUAD reference dataset. We lastly investigated a scenario where the reference and target assays measure different bulk tissue. Using the breast cancer cell-line mixtures pseudo-targets and a TCGA-LUAD reference, DeCompress estimated compartment proportions with larger errors, such that the distribution of MSEs intersect with a null distribution of MSEs from randomly generated compartment proportion matrices (Supplemental Figure S9). In general, these results suggest that DeCompress performs best when using a reference from a tissue properly aligned to the target and iterating over deconvolution methods implemented in the ensemble deconvolution.
Carolina Breast Cancer Study (CBCS) expression
We finally benchmarked DeCompress against the other reference-free deconvolution methods in breast tumor expression data from the Carolina Breast Cancer Study (CBCS) (43,55) on 406 breast cancer-related genes on 1199 samples. We used RNA-seq breast tumor expression from TCGA to determine compartment-specific genes for four compartments and train the compression matrix for deconvolution in CBCS using DeCompress; 393 of the 406 genes on the CBCS panel were measured in TCGA-BRCA. For validation, a study pathologist trained a computational algorithm to estimate compartment proportions based on tissue areas using 148 tumor microarrays (TMAs) (92). We treat these estimated compartment proportions for epithelial tumor, adipose, stroma, and immune infiltrate as a ‘silver standard.’ However, it is important to note the distinction between the estimated proportions from gene expression deconvolution and the study pathologist: gene expression deconvolution methods output proportions based on RNA content, whereas the pathological algorithm is based on tissue areas. Previous methods have suggested that gene expression-based deconvolution over- and underestimates proportions of compartments that produce, respectively, high and low levels of RNA, but RNA content and tissue compartment areas are generally positively correlated (22,23).
First, to determine whether the DeCompressed expression matrix accurately represented expression for samples in the target, we split the 393 genes into 5 groups and trained TCGA-based predictive models of genes in each ‘test’ group using those in the other four ‘training’ groups. This scheme allowed for assessment of how well genes in the four training groups could predict expression of genes in the test group, both in-sample via cross-validation across samples in TCGA data and out-of-sample applied to CBCS data. Overall, in-sample cross-validation prediction of gene expression across 5 folds of samples in TCGA is strong (median adjusted 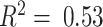 ), with a drop-off in out-sample performance in CBCS (median adjusted
), with a drop-off in out-sample performance in CBCS (median adjusted 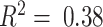 ), shown in Figure 4A. We also trained models stratified by estrogen-receptor (ER) status, a major, biologically-relevant classification in breast tumors (93,94). These ER-specific models showed slightly better out-sample performance compared to the overall models (median adjusted
), shown in Figure 4A. We also trained models stratified by estrogen-receptor (ER) status, a major, biologically-relevant classification in breast tumors (93,94). These ER-specific models showed slightly better out-sample performance compared to the overall models (median adjusted 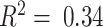 and
and 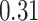 in ER-specific and overall models, respectively), though in-sample performance of the ER-specific models was similar to overall models with roughly the same median
in ER-specific and overall models, respectively), though in-sample performance of the ER-specific models was similar to overall models with roughly the same median 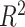 (Figure 4A). In a similar fashion, we predicted gene expression in CBCS using a miscast reference, TCGA lung adenocarcinoma data, observing a distribution of prediction
(Figure 4A). In a similar fashion, we predicted gene expression in CBCS using a miscast reference, TCGA lung adenocarcinoma data, observing a distribution of prediction 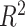 that is far shifted to the left with a median adjusted
that is far shifted to the left with a median adjusted 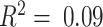 (Supplemental Figure S10). This observation, as seen in the mixing experiments, further reinforces the importance of selecting a proper reference for all steps of DeCompress.
(Supplemental Figure S10). This observation, as seen in the mixing experiments, further reinforces the importance of selecting a proper reference for all steps of DeCompress.
Figure 4.

Benchmarking results with Carolina Breast Cancer Study expression data. (A) Kernel density plots of predicted adjusted 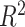 per-sample in in-sample TCGA prediction (left) through cross-validation and out-sample prediction in CBCS (right), colored by overall and ER-specific models. (B) MSE (Y-axis) between true and estimated cell-type proportions in CBCS across all methods (
per-sample in in-sample TCGA prediction (left) through cross-validation and out-sample prediction in CBCS (right), colored by overall and ER-specific models. (B) MSE (Y-axis) between true and estimated cell-type proportions in CBCS across all methods (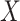 -axis). Random indicates the mean MSE over 10 000 randomly generated cell-type proportion matrices. (C) Spearman rank correlations (
-axis). Random indicates the mean MSE over 10 000 randomly generated cell-type proportion matrices. (C) Spearman rank correlations (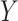 -axis) between compartment-wise true and estimated proportions across all benchmarked methods (
-axis) between compartment-wise true and estimated proportions across all benchmarked methods (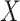 -axis). These correlations measure the association between estimated compartment proportions and the ‘silver standard’ pathologist-estimated tissue compartment areas across all samples for the four compartments. Correlations marked with a star are significantly different from 0 at
-axis). These correlations measure the association between estimated compartment proportions and the ‘silver standard’ pathologist-estimated tissue compartment areas across all samples for the four compartments. Correlations marked with a star are significantly different from 0 at 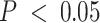 .
.
Next, as in the GTEx mixing simulations and the 4 published datasets, DeCompress recapitulated true compartment proportions with the minimum error (Figure 4B), ∼33% less error than TOAST + NMF, CDSeq, and Linseed, the second tier of methods with respect to MSE. To provide some context to the magnitude of these errors, we randomly generated 10 000 compartment proportion matrices for 148 samples and 4 compartments. The mean MSE is provided in Figure 4B, showing that two of the five benchmarked methods (CellDistinguisher and DeconICA) exceeded this randomly generated null MSE value. We also observed that correlations between tissue area compartment and DeCompress-estimated compartment proportions are positive and significantly non-zero for three of four compartments, computed as the Spearman rank correlation between the two estimates across all samples for the four proposed compartments (Figure 4C). Unlike those from TOAST + NMF, DeCompress estimates of compartment-specific compartment proportions were positively correlated with estimates of tissue compartment areas (Supplemental Figure S11). Though these correlations are not large in magnitude, we observed the DeCompress estimates are well-calibrated with the tissue area estimates, consistent with previous observations that RNA content-based estimates are generally positively associated with area-based estimates (22,23). We note here that CDSeq’s Bayesian machinery considers priors on read length and gene length to adjust for this discrepancy; however, the nCounter assay is not a sequencing technology (35).
Comparison of computational speed
The computational cost of DeCompress is high, owing primarily to training the compressed sensing models. Non-linear estimation of the columns of the compression matrix is particularly slow (Supplemental Figure S12). In practice, we recommend running an elastic net method (LASSO, elastic net, or ridge regression) which is both faster (Supplemental Figure S12) and generally gives larger cross-validation 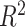 (Supplemental Figure S2). As we see that the cross-validation
(Supplemental Figure S2). As we see that the cross-validation 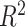 distributions of the non-linear methods overlap with that of the linear methods and no one method clearly outperforms others (Supplemental Figure S2), we include all methods in the DeCompress package but set the default methods as elastic net, ridge regression, and lasso. Given enough computational resources, iterating over all possible options provided (least angle regression, elastic net regression, and non-linear optimization) will provide the most accurate compressed sensing model. The median cross-validation
distributions of the non-linear methods overlap with that of the linear methods and no one method clearly outperforms others (Supplemental Figure S2), we include all methods in the DeCompress package but set the default methods as elastic net, ridge regression, and lasso. Given enough computational resources, iterating over all possible options provided (least angle regression, elastic net regression, and non-linear optimization) will provide the most accurate compressed sensing model. The median cross-validation 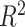 for elastic net and ridge regression is ∼16% larger than least angle regression and LASSO, and nearly 25% larger than the non-linear optimization methods. Using CBCS data with 1199 samples and 406 genes, we ran all benchmarked deconvolution methods 25 times and recorded the total runtimes (Supplemental Figure S13). For DeCompress, we used TCGA-BRCA data with 1212 samples as the reference. Running DeCompress in serial (∼7 min) takes around 5 times longer than the next slowest reference-free deconvolution method (TOAST + NMF, ∼1.5 min), though DeCompress can meet TOAST + NMF in runtime if implemented in parallel with enough workers (∼50 s). The computational cost of CDSeq is high, probably owing to its Markov chain Monte Carlo sampling scheme. These computations were conducted on a high-performance cluster (RedHat Linux operating system) with 25GB of RAM using the bigstatsr package to efficiently manage memory and fit models (95).
for elastic net and ridge regression is ∼16% larger than least angle regression and LASSO, and nearly 25% larger than the non-linear optimization methods. Using CBCS data with 1199 samples and 406 genes, we ran all benchmarked deconvolution methods 25 times and recorded the total runtimes (Supplemental Figure S13). For DeCompress, we used TCGA-BRCA data with 1212 samples as the reference. Running DeCompress in serial (∼7 min) takes around 5 times longer than the next slowest reference-free deconvolution method (TOAST + NMF, ∼1.5 min), though DeCompress can meet TOAST + NMF in runtime if implemented in parallel with enough workers (∼50 s). The computational cost of CDSeq is high, probably owing to its Markov chain Monte Carlo sampling scheme. These computations were conducted on a high-performance cluster (RedHat Linux operating system) with 25GB of RAM using the bigstatsr package to efficiently manage memory and fit models (95).
Applications of DeCompress in the Carolina Breast Cancer Study
Given the strong performance of DeCompress in benchmarking experiments, we estimated compartment proportions for 1199 subjects in CBCS with transcriptomic data assayed with NanoString nCounter. As reference-free methods output proportions for agnostic compartments, identifying approximate descriptors for compartments is often difficult. Here, we first outline a framework for assigning modular identifiers for compartments identified by DeCompress, guided by compartment-specific gene signatures. Then, we assess performance of using compartment-specific proportions in downstream analyses of breast cancer outcomes and gene regulation. Using TCGA breast cancer (TCGA-BRCA) expression as a training set, we iteratively searched for compartment-specific features using TOAST + NMF (32) (Step 1 in Figure 1) and included canonical compartment markers for guidance using a priori knowledge (29,96,97) (see Materials and Methods). After expanding the targeted CBCS expression to these genes using a compressed sensing model based on elastic net, lasso, or ridge regression, we estimated compartment proportions. We iterated across three to five possible tissue compartments, as per the recommendations in Materials and Methods. At an assumed five compartments, we observed that compartment-specific gene signatures that were enriched for clearly distinct sets of biological process ontologies consistent with compartments generally identified in bulk breast tumors.
Identifying approximate modules for DeCompress-estimated compartments
We leveraged compartment-specific gene signatures to annotate each compartment with modular identifiers. First, we conducted over-representation analysis (ORA) (67) of gene signatures for all five compartments, revealing cell cycle regulation ontologies (FDR-adjusted 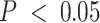 ) for compartment 4 (C4), shown in Figure 5A. Gene set enrichment analysis (GSEA) for the C4 gene signature (98) revealed significant enrichments for cell differentiation and development process ontologies (Supplemental Figure S14). ORA analysis also assigned immune-related ontologies to the C2 gene signatures at FDR-adjusted
) for compartment 4 (C4), shown in Figure 5A. Gene set enrichment analysis (GSEA) for the C4 gene signature (98) revealed significant enrichments for cell differentiation and development process ontologies (Supplemental Figure S14). ORA analysis also assigned immune-related ontologies to the C2 gene signatures at FDR-adjusted 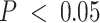 and ERBB signaling to C3, though this enrichment did not achieve statistical significance. The stem cell differentiation, extracellular matrix, and morphogenic ontologies in C5 suggest that this compartment may resemble stromal or tumor-adjacent normal mammary tissue (60,99,100). C1 gene signatures were not enriched for ontologies in that conclusive compartment assignment, though these catabolic, morphogenic, and extracellular process ontologies (Figure 5A) are often present in activated cells in the tumor microenvironment (101). From these results, we hypothesized that C3 resembled HER2-enriched tumor cells, C4 an epithelial tumor compartment, C2 an immune cell compartment and C1 and C5 presumptively stromal or tumor-adjacent mammary tissue found in the tumor microenvironment.
and ERBB signaling to C3, though this enrichment did not achieve statistical significance. The stem cell differentiation, extracellular matrix, and morphogenic ontologies in C5 suggest that this compartment may resemble stromal or tumor-adjacent normal mammary tissue (60,99,100). C1 gene signatures were not enriched for ontologies in that conclusive compartment assignment, though these catabolic, morphogenic, and extracellular process ontologies (Figure 5A) are often present in activated cells in the tumor microenvironment (101). From these results, we hypothesized that C3 resembled HER2-enriched tumor cells, C4 an epithelial tumor compartment, C2 an immune cell compartment and C1 and C5 presumptively stromal or tumor-adjacent mammary tissue found in the tumor microenvironment.
Figure 5.

Identification of DeCompress-estimated compartments. (A) Bar plot of 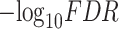 -adjusted
-adjusted 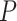 -values for top gene ontologies (
-values for top gene ontologies (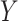 -axis) enriched in compartment-specific gene signatures. (B) Heatmap of Pearson correlations between compartment-specific gene signatures (
-axis) enriched in compartment-specific gene signatures. (B) Heatmap of Pearson correlations between compartment-specific gene signatures (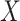 -axis) and GTEx median expression profiles and MCF7 single-cell profiles (Y-axis). Significant correlations at nominal
-axis) and GTEx median expression profiles and MCF7 single-cell profiles (Y-axis). Significant correlations at nominal 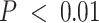 are indicated with an asterisk. (C) Boxplots of estimated immune (left) and tumor (C3 + C4 compartments, right) proportions (
are indicated with an asterisk. (C) Boxplots of estimated immune (left) and tumor (C3 + C4 compartments, right) proportions (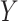 -axis) across PAM50 molecular subtypes (
-axis) across PAM50 molecular subtypes (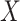 -axis).
-axis).
To investigate these hypotheses further, we computed Spearman correlations between the compartment-specific gene expression profiles and median tissue-specific expression profiles from GTEx (53,54) and single cell RNA-seq profiles of MCF7 breast cancer cells (102) (Figure 5B; Supplemental Figure S15). Here, we find that C4 shows strong positive correlations with fibroblasts, lymphocytes, multiple collagenous organs (such as blood vessels, skin, mucosal esophagus, vagina, and uterus (103–105)), and MCF7 cells, breast tumor cells that cannot have ERBB2 gene amplification (106). We hypothesize that strong correlation with lymphocytes reflects immune infiltrate. C1 showed strong positive associations with epithelial organs but also with adipose, lending more evidence to the hypothesis that C1 may represent tumor-adjacent normal mammary tissue. The C3 gene signature was significantly correlated with expression profiles of secretory organs (salivary glands, pancreas, liver) (107). C2 and C5, however, did not exhibit strong correlations with bulk organs and cell-types assayed in GTEx. In sum, these gene expression correlations support our hypotheses, especially for C1, C3, and C4.
Lastly, we subjected the C2, C3, and C4 compartments to further biological validation by investigating established associations between tumor tissue composition, breast cancer subtypes, and race. Distributions of the hypothesized immune (C2) and tumor (C3 + C4 proportions) compartment proportions revealed significant differences across PAM50 molecular subtypes (Figure 5C; Kruskal–Wallis test of differences with 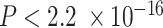 ) (71). These trends across subtypes were consistent with evidence that Basal-like and HER2-enriched subtypes, known to have high proliferation and high epithelial cellularity, had the largest proportions of estimated tumor and immune compartments, while Luminal A, Luminal B and Normal-like subtypes showed lower proportions (43,71,108). Though not statistically significant, we also noticed that the C1 was enriched in Normal-like and Luminal A tumor and C5 in Basal-like and HER2-enriched tumors (Supplemental Figure S16). Next, we found strong differences in C4 and total tumor compartment estimates across race (Supplemental Figure S17A). C3 and C4 also have strong correlations with ER (estrogen receptor) and HER2-scores, gene-expression based continuous variables that indicate clinical subtypes based on ESR1 and ERBB2 gene modules (Supplemental Figure S17B); however, none of the C3, C4, immune or tumor compartment estimates showed significant differences across clinical ER status determined by immunohistochemistry (Supplemental Figure S17C). We considered the incorporation of estimates of compartment proportions in building models of breast cancer survival (Supplemental Results and Supplemental Table S3).
) (71). These trends across subtypes were consistent with evidence that Basal-like and HER2-enriched subtypes, known to have high proliferation and high epithelial cellularity, had the largest proportions of estimated tumor and immune compartments, while Luminal A, Luminal B and Normal-like subtypes showed lower proportions (43,71,108). Though not statistically significant, we also noticed that the C1 was enriched in Normal-like and Luminal A tumor and C5 in Basal-like and HER2-enriched tumors (Supplemental Figure S16). Next, we found strong differences in C4 and total tumor compartment estimates across race (Supplemental Figure S17A). C3 and C4 also have strong correlations with ER (estrogen receptor) and HER2-scores, gene-expression based continuous variables that indicate clinical subtypes based on ESR1 and ERBB2 gene modules (Supplemental Figure S17B); however, none of the C3, C4, immune or tumor compartment estimates showed significant differences across clinical ER status determined by immunohistochemistry (Supplemental Figure S17C). We considered the incorporation of estimates of compartment proportions in building models of breast cancer survival (Supplemental Results and Supplemental Table S3).
Incorporating compartment proportions into eQTL models detects more tissue-specific gene regulators
We investigated how incorporating estimated compartment proportions affected cis-expression quantitative trait loci (cis-eQTL) mapping in breast tumors, a common application of deconvolution methods in assessing sources of variation in gene regulation (9,109). In previous eQTL studies using CBCS expression, several bulk breast tumor cis-eGenes (i.e. the gene of interest in an eQTL association between SNP and gene expression) were found in healthy mammary, subcutaneous adipose, or lymphocytes from GTEx (10). We included DeCompress proportion estimates for the tumor (C3 + C4 estimates) and immune (C2) compartments in a race-stratified, genetic ancestry-adjusted cis-eQTL interaction model (see Materials and Methods), as proposed by Geeleher et al. and Westra et al. (8,9). We found that sets of compartment-specific cis-eGenes generally had few intersections with bulk cis-eGenes (Figure 6A), though we detected more cis-eQTLs with the immune- and tumor-specific interaction models (Supplemental Figure S18). At FDR-adjusted 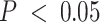 , of 209 immune-specific cis-eGenes identified in women of European ancestry (EA), 7 were also mapped in the bulk models (with no compartment proportion covariates), and no tumor-specific cis-eGenes were identified with the bulk models. Similarly, at FDR-adjusted
, of 209 immune-specific cis-eGenes identified in women of European ancestry (EA), 7 were also mapped in the bulk models (with no compartment proportion covariates), and no tumor-specific cis-eGenes were identified with the bulk models. Similarly, at FDR-adjusted 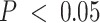 , in women of African ancestry (AA), 27 of 331 and 9 of 124 cis-eGenes identified with the immune- and tumor-compartment interaction models were also mapped with the bulk models, respectively. Manhattan plots for cis-eQTLs across the whole genome across bulk, tumor, and immune show the differences in eQTL architecture in these compartment-specific eQTL mappings in EA and AA samples (Supplemental Figures S19 and S20, respectively). Furthermore, we generally detected more cis-eQTLs at FDR-adjusted
, in women of African ancestry (AA), 27 of 331 and 9 of 124 cis-eGenes identified with the immune- and tumor-compartment interaction models were also mapped with the bulk models, respectively. Manhattan plots for cis-eQTLs across the whole genome across bulk, tumor, and immune show the differences in eQTL architecture in these compartment-specific eQTL mappings in EA and AA samples (Supplemental Figures S19 and S20, respectively). Furthermore, we generally detected more cis-eQTLs at FDR-adjusted 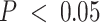 with the immune-specific interactions than the bulk and tumor-specific interactions (EA: 565 bulk cis-eQTLs, 65 tumor cis-eQTLs, 8927 immune cis-eQTLs; AA: 237 bulk cis-eQTLs, 449 tumor cis-eQTLs, 7676 immune cis-eQTLs; Supplemental Figure S18). All eQTLs with FDR-adjusted
with the immune-specific interactions than the bulk and tumor-specific interactions (EA: 565 bulk cis-eQTLs, 65 tumor cis-eQTLs, 8927 immune cis-eQTLs; AA: 237 bulk cis-eQTLs, 449 tumor cis-eQTLs, 7676 immune cis-eQTLs; Supplemental Figure S18). All eQTLs with FDR-adjusted 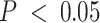 are provided in Supplemental Data.
are provided in Supplemental Data.
Figure 6.

Compartment-specific cis-eQTL mapping in the Carolina Breast Cancer Study. (A) Venn diagram of bulk, tumor-, and immune-specific cis-eGenes identified in European-ancestry (left) and African-ancestry samples (right) in CBCS. (B) Enrichment analysis of immune- (red) and tumor-specific (blue) cis-eGenes in CBCS, plotting the –log10 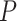 -value of enrichment (
-value of enrichment (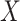 -axis) and description of gene ontologies (
-axis) and description of gene ontologies (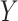 -axis). The size of the point represents the relative enrichment ratio for the given ontology. (C) Scatterplots of GTEx (
-axis). The size of the point represents the relative enrichment ratio for the given ontology. (C) Scatterplots of GTEx (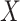 -axis) and CBCS effect size (
-axis) and CBCS effect size (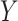 -axis) for significant CBCS cis-eQTLs that were mapped in GTEx. Each point is colored by the GTEx tissue in which the cis-eQTL has the lowest
-axis) for significant CBCS cis-eQTLs that were mapped in GTEx. Each point is colored by the GTEx tissue in which the cis-eQTL has the lowest 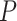 -value. Reference dotted lines for the
-value. Reference dotted lines for the 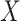 - and
- and 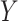 -axes are provided. (D) For risk variants from GWAS for breast cancer from iCOGs (88–90), scatterplot of –log10
-axes are provided. (D) For risk variants from GWAS for breast cancer from iCOGs (88–90), scatterplot of –log10 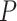 -values of bulk (
-values of bulk (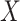 -axis) and compartment-specific cis-eQTLs (
-axis) and compartment-specific cis-eQTLs (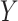 -axis), colored blue for tumor- and red for immune-specific models. A 45-degree reference line is provided. In the top right corner, three tumor-specific cis-eQTLs are labelled with the eGene CCR3 as they are significant at FDR-adjusted
-axis), colored blue for tumor- and red for immune-specific models. A 45-degree reference line is provided. In the top right corner, three tumor-specific cis-eQTLs are labelled with the eGene CCR3 as they are significant at FDR-adjusted 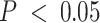 . (E) Tumor-specific eQTL effect sizes and 95% confidence intervals (
. (E) Tumor-specific eQTL effect sizes and 95% confidence intervals (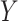 -axis) for rs56387622 on CCR3 expression across various estimates of tumor purity. The eQTL effect size from the bulk model is given in blue.
-axis) for rs56387622 on CCR3 expression across various estimates of tumor purity. The eQTL effect size from the bulk model is given in blue.
We analyzed the sets of EA and AA tumor- and immune-specific eGenes in CBCS with ORA analysis for biological processes (Figure 6B). We found that, in general, these sets of eGenes were concordant with the compartment in which they were mapped. All at FDR-adjusted 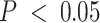 , AA tumor-specific eGenes showed enrichment for cell cycle and developmental ontologies, while immune-specific eGenes were enriched for leukocyte activation and migration and response to drug pathways. Similarly, EA tumor-specific eGenes showed enrichments for cell death and proliferation ontologies, and immune-specific eGenes showed cytokine and lymph vessel-associated processes. We then cross-referenced bulk and tumor-specific cis-eGenes found in the CBCS EA sample with cis-eGenes detected in healthy tissues from GTEx: mammary tissue, fibroblasts, lymphocytes, and adipose (see Materials and Methods), similar to previous pan-cancer germline eQTL analyses (10,110). We attributed several of the bulk cis-eGenes to healthy GTEx tissue (all but 2), but tumor-specific cis-eGenes were less enriched in healthy tissues (Supplemental Figure S21). We compared the cis-eQTL effect sizes for significant CBCS cis-eSNPs found in GTEx. As shown in Figure 6C, 98 of 220 bulk cis-eQTLs detected in CBCS that were also found in GTEx were mapped in healthy tissue, with strong positive correlation between effect sizes (Spearman
, AA tumor-specific eGenes showed enrichment for cell cycle and developmental ontologies, while immune-specific eGenes were enriched for leukocyte activation and migration and response to drug pathways. Similarly, EA tumor-specific eGenes showed enrichments for cell death and proliferation ontologies, and immune-specific eGenes showed cytokine and lymph vessel-associated processes. We then cross-referenced bulk and tumor-specific cis-eGenes found in the CBCS EA sample with cis-eGenes detected in healthy tissues from GTEx: mammary tissue, fibroblasts, lymphocytes, and adipose (see Materials and Methods), similar to previous pan-cancer germline eQTL analyses (10,110). We attributed several of the bulk cis-eGenes to healthy GTEx tissue (all but 2), but tumor-specific cis-eGenes were less enriched in healthy tissues (Supplemental Figure S21). We compared the cis-eQTL effect sizes for significant CBCS cis-eSNPs found in GTEx. As shown in Figure 6C, 98 of 220 bulk cis-eQTLs detected in CBCS that were also found in GTEx were mapped in healthy tissue, with strong positive correlation between effect sizes (Spearman 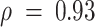 ). The remaining 122 eQTLs that could not be detected in healthy GTEx tissue contained some discordance in the direction of effects, though correlations between these effect sizes were also high (
). The remaining 122 eQTLs that could not be detected in healthy GTEx tissue contained some discordance in the direction of effects, though correlations between these effect sizes were also high (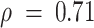 ). In contrast, we were unable to detect any of the CBCS tumor-specific cis-eQTLs as significant eQTLs in GTEx healthy tissue, and the correlation of these effect sizes across CBCS and GTEx was poor (Spearman
). In contrast, we were unable to detect any of the CBCS tumor-specific cis-eQTLs as significant eQTLs in GTEx healthy tissue, and the correlation of these effect sizes across CBCS and GTEx was poor (Spearman 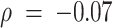 ). These results suggest that this compartment-specific eQTL mapping, especially those that are tumor-specific, identified eQTLs that are not enriched for eQTLs from healthy tissue.
). These results suggest that this compartment-specific eQTL mapping, especially those that are tumor-specific, identified eQTLs that are not enriched for eQTLs from healthy tissue.
To evaluate any overlap of compartment-specific eQTLs with SNPs implicated with breast cancer risk, we extracted 932 risk-associated SNPs in women of European ancestry from iCOGS (88–90) at FDR-adjusted 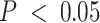 that were available on the CBCS OncoArray panel (73). Figure 6D shows the raw
that were available on the CBCS OncoArray panel (73). Figure 6D shows the raw 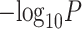 -values of the association of these SNPs with their top cis-eGenes in the bulk and tumor- and immune-specific interaction models. In large part, none of these eQTLs reached FDR-adjusted
-values of the association of these SNPs with their top cis-eGenes in the bulk and tumor- and immune-specific interaction models. In large part, none of these eQTLs reached FDR-adjusted 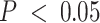 , except for three cis-eQTLs, with their strengths of association favoring the bulk eQTLs. However, we detected three tumor-specific EA cis-eQTLs in near-perfect linkage disequilibrium of
, except for three cis-eQTLs, with their strengths of association favoring the bulk eQTLs. However, we detected three tumor-specific EA cis-eQTLs in near-perfect linkage disequilibrium of 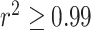 (strongest association with rs56387622) with chemokine receptor CCR3, a gene whose expression was previously found to be associated with breast cancer outcomes in luminal-like subtypes (111,112). As estimated tumor purity increases, the cancer risk allele C at rs56387622 has a consistently strong negative effect on CCR3 expression (Figure 6E). We find that CCR3 expression is not significantly different across tumor stage and ER status but is significantly different across PAM50 molecular subtype (Supplemental Figure S22). In sum, results from our cis-eQTL analysis show the advantage of including DeCompress-estimated compartment proportions in downstream genomic analyses to identify compartment-specific associations that may be relevant in disease pathways.
(strongest association with rs56387622) with chemokine receptor CCR3, a gene whose expression was previously found to be associated with breast cancer outcomes in luminal-like subtypes (111,112). As estimated tumor purity increases, the cancer risk allele C at rs56387622 has a consistently strong negative effect on CCR3 expression (Figure 6E). We find that CCR3 expression is not significantly different across tumor stage and ER status but is significantly different across PAM50 molecular subtype (Supplemental Figure S22). In sum, results from our cis-eQTL analysis show the advantage of including DeCompress-estimated compartment proportions in downstream genomic analyses to identify compartment-specific associations that may be relevant in disease pathways.
DISCUSSION
Here, we presented DeCompress, a semi-reference-free deconvolution method catered towards targeted expression panels that are commonly used for archived tissue in clinical and academic settings (3,35). Unlike traditional reference-based methods that require compartment-specific expression profiles, DeCompress requires only a reference RNA-seq or microarray dataset on similar bulk tissue to train a compressed sensing model that projects the targeted panel into a larger feature space for deconvolution. Such reference datasets are much more widely available than compartment-specific expression on the same targeted panel. We benchmarked DeCompress against reference-free methods (20,22,23,25,32) using in-silico single-cell (19) and GTEx mixing experiments (53,54), four published datasets with known compartment proportions (11,23,60,61), and a large, heterogeneous NanoString nCounter dataset from the CBCS (43,55). In these analyses, we showed that DeCompress efficiently and accurately expanded the feature space of the target and recapitulated true compartment proportions with the lowest error and the strongest compartment-specific positive correlations, especially when the reference dataset is properly aligned with the tissue assayed in the target and ensemble deconvolution is executed across all implemented deconvolution methods. We tested the performance of DeCompress by incorporating compartment estimates in eQTL mapping to reveal immune- and tumor-compartment-specific breast cancer eQTLs.
While DeCompress has several important strengths, it has some limitations. First, DeCompress, like other deconvolution methods (22,23,25,32), may over- or underestimate tissue compartment areas. Linseed and CDSeq, methods built for RNA-seq data, include scaling factors using ERCC spike-ins or read length (22,23), though these options are not available for targeted panels. Second, DeCompress has a high computational cost, owing mainly to feature selection and compressed sensing training steps. We recommend running mainly linear optimization methods in this step and have implemented parallelization options and efficient memory mapping (95) to bring computation time on par with the iterative framework proposed in TOAST (32). However, DeCompress estimates compartment proportions both accurately and precisely, compared to other reference-free methods, and provides a strong computational alternative that is much faster than costly lab-based measurement of composition. Third, DeCompress is a semi-reference-free method and shares the limitations of reference-based methods – namely concerns with the proper selection of a reference dataset. As seen in the lung adenocarcinoma example, where TCGA-LUAD data was not an accurate reflection of a mixture of adenocarcinoma cell-lines, DeCompress performance was slightly lower than with datasets properly matched to their references. Yet, in this setting, DeCompress performance was on par with that of the other reference-free methods that do not use a misaligned reference. Lastly, also in common with reference-free methods, the compression model may also be sensitive to phenotypic variation in the reference, as evidenced by the increase in out-sample prediction 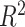 in ER-specific models compared to overall models in CBCS. This specificity may be leveraged to train more accurate models by using more than one reference dataset to reflect clinical or biological heterogeneity in the targeted panel. Researchers may employ more systematic methods of assessing the similarity of the reference and target datasets, like measuring the distance between the two matrices (i.e. norms based on the singular values of matrices) or comparing the correlation structure of overlapping genes in the feature spaces of the reference and target. These evaluations will help with selecting a proper reference for a targeted panel to be deconvolved using DeCompress.
in ER-specific models compared to overall models in CBCS. This specificity may be leveraged to train more accurate models by using more than one reference dataset to reflect clinical or biological heterogeneity in the targeted panel. Researchers may employ more systematic methods of assessing the similarity of the reference and target datasets, like measuring the distance between the two matrices (i.e. norms based on the singular values of matrices) or comparing the correlation structure of overlapping genes in the feature spaces of the reference and target. These evaluations will help with selecting a proper reference for a targeted panel to be deconvolved using DeCompress.
DeCompress also shares some challenges with reference-free deconvolution methods, such as the selection of an appropriate number of compartments. Previous groups have emphasized reliance on a priori knowledge for deconvolving well-studied tissues, such as blood and brain (113,114). However, diseased tissues, like bulk cancerous tumors, especially in understudied subtypes or populations, are more difficult to deconvolve due to the similarity between compartments, many of which may be rare or reflect transient cell states (29,94,115,116). For this reason, we included several data-driven approaches for estimating the number of compartments from variation in the gene expression and recommended applying prior domain knowledge about the tissue of interest. We also observed, through simulations, that selecting too many or too few compartments a priori lead to signal from true compartments splitting into compartments with smaller proportions or aggregating into compartments with larger proportions, respectively. Overestimating the number of compartments may lead to difficulties in assigning identities to the compartment, whereas underestimating the compartments may lead to ignoring important biological variation that is present in the DeCompress-ed expression. It is also important to carefully consider the gene module-based annotations for the unidentified estimated compartments, especially in bulk tissue where traditional ideas of compartments are inapplicable (28). Several previous reference-free methods have leveraged in vitro mixtures of highly distinct cell lines in training and testing (11,22), namely the rat cell line mixture (GSE19830) (11). Though this dataset is easy to deconvolve and thus useful in testing methodology, the extreme differences in gene expression between these three tissue types renders this dataset sub-optimal for methods benchmarking. Furthermore, assigning estimated compartments to known tissues in this dataset is straightforward and does not capture the difficulty of this task in typical deconvolution applications. Instead, our applications in breast cancer expression with CBCS provided such a difficult statistical challenge. Our outlined approach of first comparing compartment-specific gene signatures to known tissue profiles from GTEx or single-cell profiles, then analyzing these signatures with ORA or GSEA, and lastly checking hypotheses against known biological trends provides a structured framework for addressing the compartment identification problem.
Our downstream eQTL analysis in CBCS breast tumor expression also provided some insight into gene regulation, similar to recent work into deconvolving immune subpopulation eQTL signals from bulk blood eQTLs (109). In breast cancer, Geeleher et al. previously showed that a similarly implemented interaction eQTL model gave better mapping of compartment-specific eQTLs (8,9). Our results are consistent with this finding, especially since tumor- and immune-specific eGenes were enriched for commonly associated ontologies. However, unlike Geeleher et al, we generally detected a larger number of immune- and tumor-specific eQTLs and eGenes than in the bulk, unadjusted models. We believe that this larger number of compartment-specific eGenes may be due to the specificity of the genes assayed by the CBCS targeted panel. As the panel included 406 genes, all previously implicated in breast cancer pathogenesis, proliferation, or response (10,43,117), the interaction model will detect SNPs that have large effects on compartment-specific genes. The interaction term is interpreted as the difference in eQTL effect sizes between samples of 0% and 100% of the given compartment; accordingly, for genes implicated in specific breast cancer pathways, we expect to see large differences in compartment-specific eQTL effects (118–120). Though this interaction model is straight-forward in its interpretation for the tumor compartment (i.e. a sample of 100% tumor cells versus 100% tumor-associated normal cells), this interpretation may be tenuous for less well-defined compartments, like an immune compartment that includes several different immune cells. This interaction term's effect size may also be inflated for compartment estimates that have low mean and high variance across the samples. In addition, we did not consider trans-acting eQTLs that are often attributed to compartment heterogeneity, though we believe that methods employing mediation or cross-condition analysis can be integrated with compartment estimates to map compartment-specific trans-eQTLs relevant in breast cancer (121–123).
Relevant to risk and proliferation of breast cancer, we detected a locus of cis-eSNPs associated with expression of CCR3 (C–C chemokine receptor type s3) that were GWAS-identified risk SNPs (88–90) but were not significantly associated with CCR3 expression using the bulk models and were not detected in GTEx. If one or more causal SNPs in this genomic region affects CCR3 expression only in cancer cells and the effect on CCR3 expression is the main mechanism by which the locus predisposes individuals to breast cancer, we can hypothesize that an earlier perturbation in the development of cancer (e.g. transcription factor or microRNA activation) may cause this SNP’s tumorigenic effect. Given this perturbation in precancerous mammary cells, individuals with the risk allele would convey the tumorigenic effects of decreased CCR3 expression. It has been previously shown that increased peritumoral CCR3 expression is associated with improved survival times in luminal-like breast cancers (111,112). The CCR3 receptor has been shown to be the primary binding site of CCL11 (eotaxin-1), an eosinophil-selective chemoattractant cytokine (124,125), and accordingly CCR3 antagonism prohibited chemotaxis of basophils and eosinophils, a phenomenon observed in breast cancer activation and proliferation (126,127). Without DeCompress and the incorporation of estimated compartment proportions in the eQTL model, this association between eSNP and CCR3 expression would not have been detected in this dataset (128).
DeCompress, our semi-reference-free deconvolution method, provides a powerful method to estimate compartment-specific proportions for targeted expression panels that have a limited number of genes and only requires RNA-seq or microarray expression from a similar bulk tissue. Our method's estimates recapitulate known compartments with less error than reference-free methods and provide compartments that are biologically relevant, even in complex tissues like bulk breast tumors. We provide examples of using these estimated compartment proportions in downstream studies of outcomes and eQTL analysis. Given the wide applications of reference-free deconvolution, the popularity of targeted panels in both academic and clinical settings, and increasing need for analyzing heterogeneous and dynamic tissues, we anticipate creative implementations of DeCompress to give further insight into expression variation in complex diseases.
DATA AVAILABILITY
The DeCompress package is available as R software on GitHub: https://github.com/bhattacharya-a-bt/DeCompress. Sample code for replication and results from the eQTL analysis are provided: https://github.com/bhattacharya-a-bt/DeCompress_supplement. CBCS expression data is publicly available at GSE148426. CBCS genotype datasets analyzed in this study are not publicly available as many CBCS patients are still being followed and accordingly is considered sensitive; the data is available from M.A.T upon reasonable request. GTEx median expression profiles are available from dbGAP accession number phs000424.v7.p2. Single cell RNA-seq expression from Dong et al is available from GEO: GSE136148. Data from the published mixture experiments are available from GEO: GSE19830, GSE123604, GSE97284 and GSE64098. Single-cell expression profiles of MCF7 cells were obtained from GSE52716. Expression data from The Cancer Genome Atlas is available from the Broad GDAC Firehose repository (https://gdac.broadinstitute.org/) with accession number phs000178.v11.p8. Software for DeconICA is available from the following DOI: 10.5281/zenodo.1250070, with documentation and code at https://github.com/UrszulaCzerwinska/DeconICA/tree/v0.1.0.
Supplementary Material
ACKNOWLEDGEMENTS
We think the Carolina Breast Cancer Study participants and volunteers. We also thank Katie Hoadley, Yuchao Jiang, Linnea Olsson, Chuck Perou, Luca Pinello and Naim Rashid for valuable discussion during the research process. We thank Erin Kirk and Jessica Tse for their invaluable support during the research process. We thank the DCEG Cancer Genomics Research Laboratory and acknowledge the support from Stephen Chanock, Rose Yang, Meredith Yeager, Belynda Hicks, and Bin Zhu.
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from: the GTEx Portal (https://storage.googleapis.com/gtex_analysis_v7/rna_seq_data/GTEx_Analysis_2016–01-15_v7_RNASeQCv1.1.8_gene_median_tpm.gct.gz) with dbGaP accession number phs000424.v7.p2 on 05/14/20.
Notes
Present address: Michael I. Love, Department of Biostatistics, Department of Genetics, University of North Carolina-Chapel Hill, Chapel Hill, NC 27516, USA.
Contributor Information
Arjun Bhattacharya, Department of Pathology and Laboratory Medicine, David Geffen School of Medicine, University of California-Los Angeles, Los Angeles, CA 90095, USA.
Alina M Hamilton, Department of Pathology and Laboratory Medicine, University of North Carolina-Chapel Hill, Chapel Hill, NC 27516, USA.
Melissa A Troester, Department of Pathology and Laboratory Medicine, University of North Carolina-Chapel Hill, Chapel Hill, NC 27516, USA; Department of Epidemiology, University of North Carolina-Chapel Hill, Chapel Hill, NC 27516, USA.
Michael I Love, Department of Biostatistics, University of North Carolina-Chapel Hill, Chapel Hill, NC 27516, USA; Department of Genetics, University of North Carolina-Chapel Hill, Chapel Hill, NC 27516, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Susan G. Komen® provided financial support for CBCS study infrastructure; National Institutes of Health, National Cancer Institute [P01-CA151135, P50-CA05822, U01-CA179715 to M.A.T.]; M.I.L. is supported by R01-HG009937 (National Human Genome Research Institute) and P01-CA142538 (National Cancer Institute); A.B. and M.I.L are supported by P30-ES010126 (National Institute of Environmental Health Sciences); A.M.H. is supported by 1T32GM12274 (National Institute of General Medical Sciences); the Translational Genomics Laboratory is supported in part by grants from the National Cancer Institute [3P30CA016086]; University of North Carolina at Chapel Hill University Cancer Research Fund. The funders had no role in the design of the study, the collection, analysis, or interpretation of the data, the writing of the manuscript, or the decision to submit the manuscript for publication. Funding for open access charge: NHGRI (to M.I.L.).
Conflict of interest statement. The authors have no conflicts of interest to disclose. This study was approved by the Office of Human Research Ethics at the University of North Carolina at Chapel Hill, and written informed consent was obtained from each participant. All experimental methods abided by the Helsinki Declaration.
REFERENCES
- 1. Bennett A.D., Schneider A.J., Arvanitakis Z., Wilson S.R.. Overview and findings from the religious orders study. Curr. Alzheimer Res. 2013; 9:628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., Sander C., Stuart J.M., Chang K., Creighton C.J.et al.. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wallden B., Storhoff J., Nielsen T., Dowidar N., Schaper C., Ferree S., Liu S., Leung S., Geiss G., Snider J.et al.. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med. Genomics. 2015; 8:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tellez-Gabriel M., Ory B., Lamoureux F., Heymann M.F., Heymann D.. Tumour heterogeneity: the key advantages of single-cell analysis. Int. J. Mol. Sci. 2016; 17:2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McGregor K., Bernatsky S., Colmegna I., Hudson M., Pastinen T., Labbe A., Greenwood C.M.T.. An evaluation of methods correcting for cell-type heterogeneity in DNA methylation studies. Genome Biol. 2016; 17:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuhn A., Thu D., Waldvogel H.J., Faull R.L.M.M., Luthi-Carter R.. Population-specific expression analysis (PSEA) reveals molecular changes in diseased brain. Nat. Methods. 2011; 8:945–947. [DOI] [PubMed] [Google Scholar]
- 7. Guintivano J., Aryee M.J., Kaminsky Z.A.. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013; 8:290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. André G.;, Westra H.-J., Arends D., Esko T., Peters M.J., Schurmann C., Schramm K.. Cell specific eQTL analysis without sorting cells. PLoS Genet. (2015); 24:1005223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geeleher P., Nath A., Wang F., Zhang Z., Barbeira A.N., Fessler J., Grossman R.L., Seoighe C., Stephanie Huang R.. Cancer expression quantitative trait loci (eQTLs) can be determined from heterogeneous tumor gene expression data by modeling variation in tumor purity. Genome Biol. 2018; 19:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhattacharya A., García-Closas M., Olshan A.F., Perou C.M., Troester M.A., Love M.I.. A framework for transcriptome-wide association studies in breast cancer in diverse study populations. Genome Biol. 2020; 21:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shen-Orr S.S., Tibshirani R., Khatri P., Bodian D.L., Staedtler F., Perry N.M., Hastie T., Sarwal M.M., Davis M.M., Butte A.J.. Cell type-specific gene expression differences in complex tissues. Nat. Methods. 2010; 7:287–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim-Hellmuth S., Aguet F., Oliva M., Muñoz-Aguirre M., Kasela S., Wucher V., Castel S.E., Hamel A.R., Viñuela A., Roberts A.L.et al.. Cell type-specific genetic regulation of gene expression across human tissues. Science. 2020; 369:eaaz8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bertsekas D.P. Convex Optimization Algorithms Athena Scientific. 1999; Belmot, Massachusetts. [Google Scholar]
- 14. Zhong Y., Wan Y.-W., Pang K., Chow L.M.L., Liu Z.. Digital sorting of complex tissues for cell type-specific gene expression profiles. BMC Bioinformatics. 2013; 14:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Quon G., Haider S., Deshwar A.G., Cui A., Boutros P.C., Morris Q.. Computational purification of individual tumor gene expression profiles leads to significant improvements in prognostic prediction. Genome Med. 2013; 5:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Z., Cao S., Morris J.S., Ahn J., Liu R., Tyekucheva S., Gao F., Li B., Lu W., Tang X.et al.. Transcriptome Deconvolution of Heterogeneous Tumor Samples with Immune Infiltration. iScience. 2018; 9:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen B., Khodadoust M.S., Liu C.L., Newman A.M., Alizadeh A.A.. Profiling tumor infiltrating immune cells with CIBERSORT. Methods in Molecular Biology. 2018; 1711:Humana Press Inc; 243–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang J., Devlin B., Roeder K.. Using multiple measurements of tissue to estimate subject- and cell-type-specific gene expression. Bioinformatics. 2020; 36:782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dong M., Thennavan A., Urrutia E., Li Y., Perou C.M., Zou F., Jiang Y.. SCDC: bulk gene expression deconvolution by multiple single-cell RNA sequencing references. Brief. Bioinform. 2021; 22:416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Repsilber D., Kern S., Telaar A., Walzl G., Black G.F., Selbig J., Parida S.K., Kaufmann S.H., Jacobsen M.. Biomarker discovery in heterogeneous tissue samples -taking the in-silico deconfounding approach. BMC Bioinformatics. 2010; 11:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X., Park J., Susztak K., Zhang N.R., Li M.. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 2019; 10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zaitsev K., Bambouskova M., Swain A., Artyomov M.N.. Complete deconvolution of cellular mixtures based on linearity of transcriptional signatures. Nat. Commun. 2019; 10:12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang K., Meng Q., Shats I., Umbach D.M., Li M., Li Y., Li X., Li L.. CDSeq: A novel complete deconvolution method for dissecting heterogeneous samples using gene expression data. PLoS Comput. Biol. 2019; 15:e1007510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Sherf U., Speed T.P.. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249–264. [DOI] [PubMed] [Google Scholar]
- 25. Newberg L.A., Chen X., Kodira C.D., Zavodszky M.I.. Computational de novo discovery of distinguishing genes for biological processes and cell types in complex tissues. PLoS One. 2018; 13:e0193067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schelker M., Feau S., Du J., Ranu N., Klipp E., MacBeath G., Schoeberl B., Raue A.. Estimation of immune cell content in tumour tissue using single-cell RNA-seq data. Nat. Commun. 2017; 8:2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yousefi P., Huen K., Quach H., Motwani G., Hubbard A., Eskenazi B., Holland N.. Estimation of blood cellular heterogeneity in newborns and children for epigenome-wide association studies. Environ. Mol. Mutagen. 2015; 56:751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Clevers H. What is your conceptual definition of “cell type” in the context of a mature organism?. Cell Syst. 2017; 4:255–259. [DOI] [PubMed] [Google Scholar]
- 29. Wu S.Z., Roden D.L., Wang C., Holliday H., Harvey K., Cazet A.S., Murphy K.J., Pereira B., Al-Eryani G., Hou R.et al.. Single-cell analysis reveals diverse stromal subsets associated with immune evasion 1 in triple-negative breast cancer. 2020; bioRxiv doi:06 June 2020, preprint: not peer reviewed 10.1101/2020.06.04.135327. [DOI] [PMC free article] [PubMed]
- 30. Barkley D., Yanai I.. Plasticity and clonality of cancer cell states. Trends Cancer. 2019; 5:655–656. [DOI] [PubMed] [Google Scholar]
- 31. van der Leun A.M., Thommen D.S., Schumacher T.N.. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer. 2020; 20:218–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li Z., Wu H.. TOAST: improving reference-free cell composition estimation by cross-cell type differential analysis. Genome Biol. 2019; 20:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peng X.L., Moffitt R.A., Torphy R.J., Volmar K.E., Yeh J.J.. De novo compartment deconvolution and weight estimation of tumor samples using DECODER. Nat. Commun. 2019; 10:4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li Z., Wu Z., Jin P., Wu H.. Dissecting differential signals in high-throughput data from complex tissues. Bioinformatics. 2019; 35:3898–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Geiss G.K., Bumgarner R.E., Birditt B., Dahl T., Dowidar N., Dunaway D.L., Fell H.P., Ferree S., George R.D., Grogan T.et al.. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008; 26:317–325. [DOI] [PubMed] [Google Scholar]
- 36. Marczyk M., Fu C., Lau R., Du L., Trevarton A.J., Sinn B.V., Gould R.E., Pusztai L., Hatzis C., Symmans W.F.. The impact of RNA extraction method on accurate RNA sequencing from formalin-fixed paraffin-embedded tissues. BMC Cancer. 2019; 19:1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mercer T.R., Gerhardt D.J., Dinger M.E., Crawford J., Trapnell C., Jeddeloh J.A., Mattick J.S., Rinn J.L.. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol. 2012; 30:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Veldman-Jones M.H., Brant R., Rooney C., Geh C., Emery H., Harbron C.G., Wappett M., Sharpe A., Dymond M., Barrett J.C.et al.. Evaluating robustness and sensitivity of the NanoString Technologies nCounter Platform to Enable Multiplexed Gene Expression Analysis of clinical samples. Cancer Res. 2015; 75:2587–2593. [DOI] [PubMed] [Google Scholar]
- 39. Brasó-Maristany F., Filosto S., Catchpole S., Marlow R., Quist J., Francesch-Domenech E., Plumb D.A., Zakka L., Gazinska P., Liccardi G.et al.. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat. Med. 2016; 22:1303–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Urrutia A., Duffy D., Rouilly V., Posseme C., Djebali R., Illanes G., Libri V., Albaud B., Gentien D., Piasecka B.et al.. Standardized whole-blood transcriptional profiling enables the deconvolution of complex induced immune responses. Cell Rep. 2016; 16:2777–2791. [DOI] [PubMed] [Google Scholar]
- 41. Scott D.W., Wright G.W., Williams P.M., Lih C.-J., Walsh W., Jaffe E.S., Rosenwald A., Campo E., Chan W.C., Connors J.M.et al.. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood. 2014; 123:1214–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ng S.W.K., Mitchell A., Kennedy J.A., Chen W.C., McLeod J., Ibrahimova N., Arruda A., Popescu A., Gupta V., Schimmer A.D.et al.. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016; 540:433–437. [DOI] [PubMed] [Google Scholar]
- 43. Troester M.A., Sun X., Allott E.H., Geradts J., Cohen S.M., Tse C.-K., Kirk E.L., Thorne L.B., Mathews M., Li Y.et al.. Racial differences in PAM50 subtypes in the Carolina Breast Cancer Study. JNCI J. Natl. Cancer Inst. 2018; 110:176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vieira A.F., Schmitt F.. An update on breast cancer multigene prognostic tests—emergent clinical biomarkers. Front. Med. 2018; 5:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Candès E.J., Romberg J.. Quantitative robust uncertainty principles and optimally sparse decompositions. Found. Comput. Math. 2006; 6:227–254. [Google Scholar]
- 46. Candès E.J., Romberg J., Tao T.. Robust uncertainty principles: exact signal reconstruction from highly incomplete frequency information. IEEE Trans. Inf. Theory. 2006; 52:489–509. [Google Scholar]
- 47. Efron B., Hastie T., Johnstone I., Tibshirani R.. Least angle regression. Ann. Statist. 2004; 32:407–499. [Google Scholar]
- 48. Friedman J., Hastie T., Tibshirani R.. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010; 33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 49. Liao S.J. An explicit, totally analytic approximate solution for Blasius’ viscous flow problems. Int. J. Non. Linear. Mech. 1999; 34:759–778. [Google Scholar]
- 50. Goodfellow I.J., Pouget-Abadie J., Mirza M., Xu B., Warde-Farley D., Ozair S., Courville A., Bengio Y.. Generative adversarial nets. Adv. Neural Inform. Process. Syst. 2014; 27:2672–2680. [Google Scholar]
- 51. Viñas R., Azevedo T., Gamazon E.R., Liò P.. Gene expression imputation with generative adversarial imputation nets. 2020; bioRxiv doi:10 June 2020, preprint: not peer reviewed 10.1101/2020.06.09.141689. [DOI]
- 52. Yoon J., Jordon J., Van Der Schaar M.. GAIN: missing data imputation using generative adversarial nets. 2018; arXiv doi:07 Jun 2018, preprint: not peer reviewedhttps://arxiv.org/abs/1806.02920v1.
- 53. Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., Hasz R., Walters G., Garcia F., Young N.et al.. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ardlie K.G., DeLuca D.S., Segrè A.V., Sullivan T.J., Young T.R., Gelfand E.T., Trowbridge C.A., Maller J.B., Tukiainen T., Lek M.et al.. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Newman B., Moorman P.G., Millikan R., Qaqish B.F., Geradts J., Aldrich T.E., Liu E.T.. The Carolina Breast Cancer Study: integrating population-based epidemiology and molecular biology. Breast Cancer Res. Treat. 1995; 35:51–60. [DOI] [PubMed] [Google Scholar]
- 56. Donoho D.L. Compressed sensing. IEEE Trans. Inf. Theory. 2006; 52:1289–1306. [Google Scholar]
- 57. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R.et al.. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019; 20:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Benayoun B.A., Pollina E.A., Singh P.P., Mahmoudi S., Harel I., Casey K.M., Dulken B.W., Kundaje A., Brunet A.. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res. 2019; 29:697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tyekucheva S., Bowden M., Bango C., Giunchi F., Huang Y., Zhou C., Bondi A., Lis R., Van Hemelrijck M., Andrén O.et al.. Stromal and epithelial transcriptional map of initiation progression and metastatic potential of human prostate cancer. Nat. Commun. 2017; 8:doi:10.1038/s41467-017-00460-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Holik A.Z., Law C.W., Liu R., Wang Z., Wang W., Ahn J., Asselin-Labat M.-L., Smyth G.K., Ritchie M.E.. RNA-seq mixology: designing realistic control experiments to compare protocols and analysis methods. Nucleic. Acids. Res. 2016; 45:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu R., Holik A.Z., Su S., Jansz N., Chen K., Leong H.S., Blewitt M.E., Asselin-Labat M.-L., Smyth G.K., Ritchie M.E.. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic. Acids. Res. 2015; 43:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bhattacharya A., Hamilton A.M., Furberg H., Pietzak E., Purdue M.P., Troester M.A., Hoadley K.A., Love M.I.. An approach for normalization and quality control for NanoString RNA expression data. Brief Bioinform. 2020; doi:10.1093/bib/bbaa163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bullard J.H., Purdom E., Hansen K.D., Dudoit S.. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010; 11:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Risso D., Ngai J., Speed T.P., Dudoit S.. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014; 32:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liao Y., Wang J., Jaehnig E.J., Shi Z., Zhang B.. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic. Acids. Res. 2019; 47:199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Consortium T.G.O. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019; 47:D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T.et al.. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Austin P.C., Fine J.P.. Practical recommendations for reporting fine-gray model analyses for competing risk data. Stat. Med. 2017; 36:4391–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Parker J.S., Mullins M., Cheang M.C.U., Leung S., Voduc D., Vickery T., Davies S., Fauron C., He X., Hu Z.et al.. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009; 27:1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Benjamini Y., Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B. 1995; 57:289–300. [Google Scholar]
- 73. Amos C.I., Dennis J., Wang Z., Byun J., Schumacher F.R., Gayther S.A., Casey G., Hunter D.J., Sellers T.A., Gruber S.B.et al.. The OncoArray Consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol. Biomarkers Prev. 2017; 26:126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lilyquist J., Ruddy K.J., Vachon C.M., Couch F.J.. Common genetic variation and breast cancer risk—past, present, and future. Cancer Epidemiol. Biomarkers Prev. 2018; 27:380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Auton A., Abecasis G.R., Altshuler D.M., Durbin R.M., Bentley D.R., Chakravarti A., Clark A.G., Donnelly P., Eichler E.E., Flicek P.et al.. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. O’Connell J., Gurdasani D., Delaneau O., Pirastu N., Ulivi S., Cocca M., Traglia M., Huang J., Huffman J.E., Rudan I.et al.. A general approach for haplotype phasing across the full spectrum of relatedness. PLos Genet. 2014; 10:e1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Delaneau O., Marchini J., Zagury J.-F.. A linear complexity phasing method for thousands of genomes. Nat. Methods. 2012; 9:179–181. [DOI] [PubMed] [Google Scholar]
- 78. Howie B.N., Donnelly P., Marchini J.. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009; 5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., De Bakker P.I.W., Daly M.J.et al.. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007; 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K.. DbSNP: the NCBI database of genetic variation. Nucleic. Acids. Res. 2001; 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shabalin A.A. Gene expression Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012; 28:1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Palowitch J., Shabalin A., Zhou Y.H., Nobel A.B., Wright F.A.. Estimation of cis-eQTL effect sizes using a log of linear model. Biometrics. 2018; 74:616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sun W. A statistical framework for eQTL mapping using RNA-seq Data. Biometrics. 2012; 68:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mohammadi P., Castel S.E., Brown A.A., Lappalainen T.. Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res. 2017; 27:1872–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ellsworth R.E., Blackburn H.L., Shriver C.D., Soon-Shiong P., Ellsworth D.L.. Molecular heterogeneity in breast cancer: State of the science and implications for patient care. Semin. Cell Dev. Biol. 2017; 64:65–72. [DOI] [PubMed] [Google Scholar]
- 86. Turashvili G., Brogi E.. Tumor heterogeneity in breast cancer. Front. Med. 2017; doi:10.3389/fmed.2017.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wen Y., Wei Y., Zhang S., Li S., Liu H., Wang F., Zhao Y., Zhang D., Zhang Y.. Cell subpopulation deconvolution reveals breast cancer heterogeneity based on DNA methylation signature. Brief. Bioinform. 2016; 18:426–440. [DOI] [PubMed] [Google Scholar]
- 88. Michailidou K., Hall P., Gonzalez-Neira A., Ghoussaini M., Dennis J., Milne R.L., Schmidt M.K., Chang-Claude J., Bojesen S.E., Bolla M.K.et al.. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013; 45:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Michailidou K., Beesley J., Lindstrom S., Canisius S., Dennis J., Lush M.J., Maranian M.J., Bolla M.K., Wang Q., Shah M.et al.. Genome-wide association analysis of more than 120, 000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015; 47:373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Michailidou K., Lindström S., Dennis J., Beesley J., Hui S., Kar S., Lemaçon A., Soucy P., Glubb D., Rostamianfar A.et al.. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017; 551:92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. González I., Déjean S., Martin P.G.P., Baccini A.. CCA: An R package to extend canonical correlation analysis. J. Stat. Softw. 2008; 23:doi:10.18637/jss.v023.i12. [Google Scholar]
- 92. Sandhu R., Chollet-Hinton L., Kirk E.L., Midkiff B., Troester M.A.. Digital histologic analysis reveals morphometric patterns of age-related involution in breast epithelium and stroma. Hum. Pathol. 2016; 48:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sørlie T., Tibshirani R., Parker J., Hastie T., Marron J.S., Nobel A., Deng S., Johnsen H., Pesich R., Geisler S.et al.. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. U. S. A. 2003; 100:8418–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Perou C.M., Sørile T., Eisen M.B., Van De Rijn M., Jeffrey S.S., Ress C.A., Pollack J.R., Ross D.T., Johnsen H., Akslen L.A.et al.. Molecular portraits of human breast tumours. Nature. 2000; 406:747–752. [DOI] [PubMed] [Google Scholar]
- 95. Prive F., Aschard H., Ziyatdinov A., Blum M.G.B.. Efficient analysis of large-scale genome-wide data with two R packages: Bigstatsr and bigsnpr. Bioinformatics. 2018; 34:2781–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Azizi E., Carr A.J., Plitas G., Mazutis L., Rudensky A.Y., Pe’er D., Cornish A.E., Konopacki C., Prabhakaran S., Nainys J.et al.. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment resource single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018; 174:1293–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nguyen Q.H., Pervolarakis N., Blake K., Ma D., Davis R.T., James N., Phung A.T., Willey E., Kumar R., Jabart E.et al.. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat. Commun. 2018; 9:2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yu G., Wang L.G., Han Y., He Q.Y.. ClusterProfiler: an R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol. 2012; 16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Aran D., Sirota M., Butte A.J.. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015; 6:8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Aran D., Camarda R., Odegaard J., Paik H., Oskotsky B., Krings G., Goga A., Sirota M., Butte A.J.. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 2017; 8:doi:10.1038/s41467-017-01027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dakhova O., Ozen M., Creighton C.J., Li R., Ayala G., Rowley D., Ittmann M.. Global gene expression analysis of reactive stroma in prostate cancer. Clin. Cancer Res. 2009; 15:3979–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rothwell D.G., Li Y., Ayub M., Tate C., Newton G., Hey Y., Carter L., Faulkner S., Moro M., Pepper S.et al.. Evaluation and validation of a robust single cell RNA-amplification protocol through transcriptional profiling of enriched lung cancer initiating cells. BMC Genomics. 2014; 15:1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Smith B.A., Balanis N.G., Nanjundiah A., Sheu K.M., Tsai B.L., Zhang Q., Park J.W., Thompson M., Huang J., Witte O.N.et al.. A Human Adult Stem Cell Signature Marks Aggressive Variants across Epithelial Cancers. Cell Rep. 2018; 24:3353–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Uhlen M., Zhang C., Lee S., Sjöstedt E., Fagerberg L., Bidkhori G., Benfeitas R., Arif M., Liu Z., Edfors F.et al.. A pathology atlas of the human cancer transcriptome. Science. 2017; 357:eaan2507. [DOI] [PubMed] [Google Scholar]
- 105. Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A.et al.. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 106. Lee A., Oesterreich S., Davidson N.. MCF-7 cells—changing the course of breast cancer research and care for 45 years. J. Natl. Cancer Inst. 2015; 107:doi:10.1093/jnci/djv073. [DOI] [PubMed] [Google Scholar]
- 107. Prat A., As Pascual T., De Angelis C., Gutierrez C., Llombart-Cussac A., Wang T., Cort J., Rexer B., Par L., Forero A.et al.. HER2-enriched subtype and ERBB2 expression in HER2-positive breast cancer treated with dual HER2 blockade. J. Natl. Cancer Inst. 2020; 112:46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. D’Arcy M., Fleming J., Robinson W.R., Kirk E.L., Perou C.M., Troester M.A., D’Arcy M., Fleming J., Robinson W.R., Kirk E.L.et al.. Race-associated biological differences among Luminal A breast tumors. Breast Cancer Res. Treat. 2015; 152:437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Aguirre-Gamboa R., de Klein N., di Tommaso J., Claringbould A., van der Wijst M.G., de Vries D., Brugge H., Oelen R., Võsa U., Zorro M.M.et al.. Deconvolution of bulk blood eQTL effects into immune cell subpopulations. BMC Bioinformatics. 2020; 21:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Calabrese C., Lehmann K., Urban L., Liu F., Erkek S., Fonseca N.A., Kahles A., Kilpinen H., Markowski J.3, P.G. et al.. Assessing the Gene Regulatory Landscape in 1,188 Human Tumors. 2017; bioRxiv doi:29 November 2017, preprint: not peer reviewed 10.1101/225441. [DOI]
- 111. Gong D.H., Fan L., Chen H.Y., Ding K.F., Yu K.Da. Intratumoral expression of CCR3 in breast cancer is associated with improved relapse-free survival in luminal-like disease. Oncotarget. 2016; 7:28570–28578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Thomas J.K., Mir H., Kapur N., Bae S., Singh S.. CC chemokines are differentially expressed in Breast Cancer and are associated with disparity in overall survival. Sci. Rep. 2019; 9:4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Reinius L.E., Acevedo N., Joerink M., Pershagen G., Dahlén S.-E., Greco D., Söderhäll C., Scheynius A., Kere J.. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012; 7:e41361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Montaño C.M., Irizarry R.A., Kaufmann W.E., Talbot K., Gur R.E., Feinberg A.P., Taub M.A.. Measuring cell-type specific differential methylation in human brain tissue. Genome Biol. 2013; 14:R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Chen Y.P., Wang Y.Q., Lv J.W., Li Y.Q., Chua M.L.K.K., Le Q.T., Lee N., Dimitrios Colevas A., Seiwert T., Hayes D.N.et al.. Identification and validation of novel microenvironment-based immune molecular subgroups of head and neck squamous cell carcinoma: implications for immunotherapy. Ann. Oncol. 2019; 30:68–75. [DOI] [PubMed] [Google Scholar]
- 116. Hoadley K.A., Yau C., Hinoue T., Wolf D.M., Lazar A.J., Drill E., Shen R., Taylor A.M., Cherniack A.D., Thorsson V.et al.. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018; 173:291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. D’Arcy M., Fleming J., Robinson W.R., Kirk E.L., Perou C.M., Troester M.A.. Race-associated biological differences among Luminal A breast tumors. Breast Cancer Res. Treat. 2015; 152:437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wang F., Dohogne Z., Yang J., Liu Y., Soibam B.. Predictors of breast cancer cell types and their prognostic power in breast cancer patients. BMC Genomics. 2018; 19:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Troester M.A., Hoadley K.A., Sørlie T., Herbert B.S., Børresen-Dale A.L., Lønning P.E., Shay J.W., Kaufmann W.K., Perou C.M.. Cell-type-specific responses to chemotherapeutics in breast cancer. Cancer Res. 2004; 64:4218–4226. [DOI] [PubMed] [Google Scholar]
- 120. Schaefer M.H., Serrano L.. Cell type-specific properties and environment shape tissue specificity of cancer genes. Sci. Rep. 2016; 6:20707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Yang F., Gleason K.J., Wang J., Consortium T.G., Duan J., He X., Pierce B.L., Chen L.S.. CCmed: cross-condition mediation analysis for identifying robust trans-eQTLs and assessing their effects on human traits. 2019; bioRxiv doi:13 October 2019, preprint: not peer reviewed 10.1101/803106. [DOI]
- 122. Shan N., Wang Z., Hou L.. Identification of trans-eQTLs using mediation analysis with multiple mediators. BMC Bioinformatics. 2019; 20:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Pierce B.L., Tong L., Chen L.S., Rahaman R., Argos M., Jasmine F., Roy S., Paul-Brutus R., Westra H.J., Franke L.et al.. Mediation analysis demonstrates that trans-eQTLs are often explained by cis-mediation: a genome-wide analysis among 1,800 South Asians. PLoS Genet. 2014; 10:e1004818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Jöhrer K., Zelle-Rieser C., Perathoner A., Moser P., Hager M., Ramoner R., Gander H., Höltl L., Bartsch G., Greil R.et al.. Up-regulation of functional chemokine receptor CCR3 in human renal cell carcinoma. Clin. Cancer Res. 2005; 11:2459–2465. [DOI] [PubMed] [Google Scholar]
- 125. Miyagaki T., Sugaya M., Murakami T., Asano Y., Tada Y., Kadono T., Okochi H., Tamaki K., Sato S.. CCL11-CCR3 interactions promote survival of anaplastic large cell lymphoma cells via ERK1/2 activation. Cancer Res. 2011; 71:2056–2065. [DOI] [PubMed] [Google Scholar]
- 126. Bryan S.A., Jose P.J., Topping J.R., Wilhelm R., Soderberg C., Kertesz D., Barnes P.J., Williams T.J., Hansel T.T., Sabroe I.. Responses of leukocytes to chemokines in whole blood and their antagonism by novel CC-chemokine receptor 3 antagonists. Am. J. Respir. Crit. Care Med. 2002; 165:1602–1609. [DOI] [PubMed] [Google Scholar]
- 127. Samoszuk M.K., Nguyen V., Gluzman I., Pham J.H.. Occult deposition of eosinophil peroxidase in a subset of human breast carcinomas. Am. J. Pathol. 1996; 148:701–706. [PMC free article] [PubMed] [Google Scholar]
- 128. Alasoo K., Rodrigues J., Mukhopadhyay S., Knights A.J., Mann A.L., Kundu K., Hale C., Dougan G., Gaffney D.J.. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat. Genet. 2018; 50:424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The DeCompress package is available as R software on GitHub: https://github.com/bhattacharya-a-bt/DeCompress. Sample code for replication and results from the eQTL analysis are provided: https://github.com/bhattacharya-a-bt/DeCompress_supplement. CBCS expression data is publicly available at GSE148426. CBCS genotype datasets analyzed in this study are not publicly available as many CBCS patients are still being followed and accordingly is considered sensitive; the data is available from M.A.T upon reasonable request. GTEx median expression profiles are available from dbGAP accession number phs000424.v7.p2. Single cell RNA-seq expression from Dong et al is available from GEO: GSE136148. Data from the published mixture experiments are available from GEO: GSE19830, GSE123604, GSE97284 and GSE64098. Single-cell expression profiles of MCF7 cells were obtained from GSE52716. Expression data from The Cancer Genome Atlas is available from the Broad GDAC Firehose repository (https://gdac.broadinstitute.org/) with accession number phs000178.v11.p8. Software for DeconICA is available from the following DOI: 10.5281/zenodo.1250070, with documentation and code at https://github.com/UrszulaCzerwinska/DeconICA/tree/v0.1.0.