

Abstract
This paper presents the design and study of a first-in-class cyclic peptide inhibitor against the SARS-CoV-2 main protease (Mpro). The cyclic peptide inhibitor is designed to mimic the conformation of a substrate at a C-terminal autolytic cleavage site of Mpro. The cyclic peptide contains a [4-(2-aminoethyl)phenyl]-acetic acid (AEPA) linker that is designed to enforce a conformation that mimics a peptide substrate of Mpro. In vitro evaluation of the cyclic peptide inhibitor reveals that the inhibitor exhibits modest activity against Mpro and does not appear to be cleaved by the enzyme. Conformational searching predicts that the cyclic peptide inhibitor is fairly rigid, adopting a favorable conformation for binding to the active site of Mpro. Computational docking to the SARS-CoV-2 Mpro suggests that the cyclic peptide inhibitor can bind the active site of Mpro in the predicted manner. Molecular dynamics simulations provide further insights into how the cyclic peptide inhibitor may bind the active site of Mpro. Although the activity of the cyclic peptide inhibitor is modest, its design and study lays the groundwork for the development of additional cyclic peptide inhibitors against Mpro with improved activities.
Keywords: COVID-19, SARS-CoV-2, Main protease, Cyclic peptide inhibitor, Cyclophane
Graphical abstract
1. Introduction
Cyclic peptides are an important class of drugs. The binding affinities and specificities of cyclic peptides rival those of small molecule drugs and biomacromolecule drugs [[1], [2], [3], [4], [5], [6]]. Cyclic peptides often exhibit enhanced conformational stability and improved biological activity compared to linear analogues [7,8]. This conformational rigidity increases resistance to degradation by endogenous proteases, leading to increased plasma stability [9]. Peptide cyclization can also facilitate passage of the peptide through a cell membrane [10,11]. Although cyclic peptides often do not meet common physicochemical guidelines for drug-like cellular uptake and bioavailability, such as Lipinski's Rules of 5, a variety of cyclic peptide drugs are active against intracellular targets [[12], [13], [14], [15], [16], [17], [18]].
Secondary structure and backbone rigidity often impart a greater propensity for cyclic peptides to cross the plasma membrane [[19], [20], [21], [22], [23], [24], [25], [26]]. The cyclic undecapeptide immunosuppressant drug cyclosporine A is an archetypal example for cell-permeable cyclic peptides. Despite its large size (MW = 1202 Da), cyclosporine A is orally bioavailable and passively crosses the plasma membrane. Cyclosporine A achieves its remarkable permeability and bioavailability by adopting a rigid secondary structure comprising two β-strands connected by a β-turn to form an antiparallel β-sheet. The amide backbone of cyclosporine A is also highly methylated, and the remaining unmethylated NH groups are intramolecularly hydrogen-bonded.
Cyclic peptide inhibitors that target viral proteins, such as the SARS-CoV-2 main protease (Mpro), may offer promise as antiviral drugs with pharmacological properties similar to cyclosporine A. Mpro is one of the best-characterized drug targets for coronaviruses [[27], [28], [29], [30], [31], [32], [33], [34], [35], [36], [37], [38], [39], (b), (c), (d), [40]]. Mpro cleaves the initially translated viral polyprotein into its component proteins within cells infected by SARS-CoV-2. Cleavage generally occurs immediately after a Gln residue, and the Gln residue is typically preceded by a hydrophobic residue, most often Leu. The residue that follows the Gln is often a small amino acid, such as Ser, Ala, or Asn. Mpro autolytically cleaves itself from the polyprotein [41]. Inhibiting Mpro activity slows or halts viral replication, offering the promise of improved clinical outcomes for COVID-19 and other coronavirus diseases. Furthermore, there are no known human proteases with similar cleavage specificity to Mpro, suggesting that it should be possible to develop inhibitors that target Mpro without off-target toxicity.
Peptide-based inhibitors previously developed to target the SARS-CoV Mpro have effectively been repurposed and modified to target the SARS-CoV-2 Mpro — N3 from Jin et al., 13b from Zhang et al., and 11a and 11b from Dai et al. [16,42,43] These inhibitors block SARS-CoV-2 replication in cell-based studies, making them promising antiviral drug candidates. While the Mpro inhibitors N3, 13b, 11a, and 11b have shown promise against inhibiting SARS-CoV-2 replication, additional Mpro inhibitors will most likely be needed for their improved properties or to be used in combination therapies [44].
In this paper, we describe the design and in vitro evaluation of UCI-1 (University of California, Irvine Coronavirus Inhibitor-1), a first-in-class cyclic peptide that we hypothesized would inhibit the SARS-CoV-2 Mpro, which is required for viral replication (Fig. 1 ). UCI-1 is a cyclic pentapeptide containing four amino acids from a Mpro substrate constrained in a macrocycle linked together with a [4-(2-aminoethyl)phenyl]-acetic acid (AEPA) group to create a paracyclophane. This rigidified macrocycle is designed to mimic the conformation of a C-terminal autolytic cleavage site of a naturally occurring Mpro substrate. Evaluation of UCI-1 in an in vitro Mpro inhibition assay reveals that UCI-1 inhibits the SARS-CoV-2 Mpro at mid-micromolar concentrations. LC/MS analysis indicates that UCI-1 resists cleavage by Mpro, despite containing a scissile amide bond. Furthermore, UCI-1 is found to be non-toxic toward human embryonic kidney cells at concentrations that inhibit Mpro [45].
Fig. 1.

(A) Chemical structure of a general cyclic peptide inhibitor illustrating the arrangement of the P2, P1, P1′, and P2′ positions and [4-(2-aminoethyl)phenyl]-acetic acid (AEPA) and the envisioned binding interactions with the S2, S1, S1′, S2′, and S3′ pockets in the Mpro active site. (B) Chemical structure of UCI-1.
2. Results
Design of UCI-1. We designed the cyclic peptide inhibitor UCI-1 based on the crystal structure of an inactive SARS-CoV Mpro (C145A) variant with a 10 amino-acid C-terminal extension corresponding to the C-terminal prosequence of Mpro (PDB 5B6O) (“Mpro 316”, Fig. 2 ) [46]. The SARS-CoV Mpro amino acid sequence is 96% identical to the SARS-CoV-2 Mpro amino sequence, and the three-dimensional structure of the SARS-CoV-2 Mpro is highly similar to the structure of the SARS-CoV Mpro [47]. In the Mpro 316 crystal structure, C-terminal residues 301–310 (SGVTFQGKFK) extend into and complex with the active site of another Mpro 316 molecule in an adjacent asymmetric unit (Fig. 2 inset). This complex reveals how the P2–P1–P1′-P2′-P3′ positions (residues 305–309, FQGKF) of the C-terminal autolytic cleavage site fit into the active site of Mpro 316.
Fig. 2.

Crystal structure of Mpro316 showing two Mpro316 dimers in two adjacent asymmetric units (PDB 5B6O). One dimer is shown in grey surface view; the other dimer is shown in green cartoons. The inset shows a detailed view of residues 301–310 of the C-terminal autolytic cleavage site of one Mpro316 molecule in the active site of another Mpro316 molecule.
We designed UCI-1 to mimic the conformation that the P2–P1–P1′-P2′-P3′ residues adopt in the active site of Mpro 316. In the active site of Mpro 316, these residues adopt a “kinked” conformation in which the phenyl group of Phe309 at the P3′ position points toward the backbone of Phe305 at the P2 position (Fig. 2 inset). To mimic this conformation, we envisioned linking the phenyl group of Phe309 to the backbone of Phe305 to create a macrocycle. To realize this design, we used the molecular visualization software PyMOL (version 2.2.2, Schrödinger) to build a model of the envisioned cyclic peptide by modifying Phe305 and Phe309 in the active site of Mpro 316 (Fig. 3 ). In PyMOL, we deleted residues 301–304 to expose the amino group on Phe305; we also deleted residue 310 and the carbonyl of Phe309. We then connected the para position of Phe309 to the amino group of Phe305 with a CH2CO group to create a macrocycle. The newly created amino acid derived from Phe309 thus constitutes the amino acid AEPA.
Fig. 3.

Design process for creating the cyclic peptide inhibitor UCI-1 from the C-terminal autolytic substrate in the active site of Mpro: (1) Delete residues 301–304 and 310 as well as the carbonyl of Phe309. (2) Build a CH2CO group on the para position of the phenyl group on Phe309. (3) Create a bond between the carbonyl carbon of the newly created CH2CO group on Phe309 and the amino group of Phe305 and minimize (clean) the structure. (4) Mutate Gly307 to serine.
We recognized that upon linking Phe309 and Phe305 as described above, Phe305 and Gln306 were poised to form a β-turn in which the carbonyl group of AEPA hydrogen bonds with the amino group of Gly307. We envisioned that β-turn formation in the cyclic peptide inhibitor would promote rigidity of the cyclic scaffold. To introduce additional conformational rigidity to the macrocycle, we also mutated Gly307 to serine, which is the most common residue at the P1’ position among the 11 known SARS-CoV-2 Mpro cleavage sites. The resulting cyclic peptide inhibitor UCI-1 was then further studied as described below. In an accompanying paper, we present a tutorial on the design process using free software [48].
Synthesis of UCI-1. We synthesized UCI-1 by Fmoc-based solid-phase peptide synthesis of the protected linear peptide H2N-FQSK-AEPA-COOH on 2-chlorotrityl chloride resin, followed by cleavage of the linear protected peptide from the resin and subsequent solution-phase macrocyclization and global deprotection (Scheme 1 ). UCI-1 was purified using reverse-phase HPLC. The synthesis and purification proceeded smoothly on a 0.1 mmol scale and yielded 22 mg of purified UCI-1 as the TFA salt. Detailed procedures for synthesis of UCI-1 are given in the Materials and Methods section.
Scheme 1.

Synthesis of UCI-1.
Enzyme inhibition assay. To evaluate whether UCI-1 inhibits the SARS-CoV-2 Mpro, we used a fluorescence-based Mpro inhibition assay with native Mpro (Life Sensors) and the fluorogenic Mpro substrate K(Dabcyl)-TSAVLQSGFRKM-E(EDANS)-NH2 [28,29,49]. For the inhibition assay, we pre-incubated Mpro with varying concentrations of UCI-1 (23.4–750 μM) in assay buffer for 30 min. We then added the fluorogenic Mpro substrate and monitored Mpro activity in a continuous kinetic assay with 360 nm excitation and 460 nm emission. In each well, the concentration of Mpro was 0.1 μM and the concentration of the fluorogenic substrate was 50 μM. Fig. S1 shows the data from the continuous kinetic assay. Initial rates for Mpro activity in the presence or absence of UCI-1 were obtained by fitting a straight line to the linear portions of the curves from the continuous kinetic assay. The % activity was calculated by comparing the initial rate at each concentration to the initial rate in the absence of inhibitor. Non-linear regression was then used to fit the data and estimate an IC50 of ca. 160 μM for UCI-1 (Fig. 5A).
Fig. 5.

Enzyme inhibition assay of UCI-1 (A) and control peptides peptide-1a (B) and peptide-1b (C). The activity of Mpro was measured in the presence of increasing concentrations of UCI-1, peptide-1a, or peptide-1b. A dose-response curve was determined by non-linear regression and used to estimate the IC50. All data are shown as the mean of three technical replicates, with error bars representing the standard deviation. Panels D and E show the chemical structures of peptide-1a and peptide-1b.
To better understand the significance of inhibition of Mpro by UCI-1, we synthesized and tested two control peptides derived from UCI-1 — the acyclic analog of UCI-1 “peptide-1a” (Fig. 5D) and the diastereomer “peptide-1b” (Fig. 5E) — that were predicted to not inhibit Mpro. Peptide-1a exhibits little or no inhibition at concentrations at or below 750 μM (Fig. 5B). This finding indicates that the cyclic structure of UCI-1 is important for its activity. There is a slight reduction in rate of cleavage of the fluorogenic substrate upon addition of 1500 μM peptide-1a. This reduction in rate may reflect either slight inhibition of Mpro or that the acyclic peptide acts as a competitive substrate at high concentrations. Peptide-1b exhibits little or no inhibition at concentrations at or below 1500 μM (Fig. 5C). The inactivity of peptide-1b suggests that the shape and stereochemistry of UCI-1 are critical for fitting into and blocking the active site of Mpro, and that inhibition by UCI-1 does not simply result from its hydrophobicity and cyclic structure.
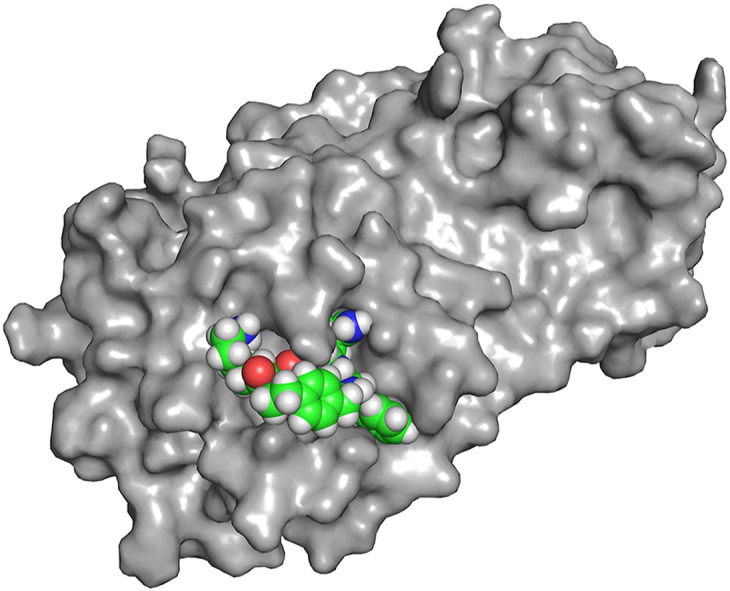
Conformational analysis, docking, and molecular dynamics simulations of UCI-1. To better understand the three-dimensional structure of UCI-1 and how it could interact with the active site of the SARS-CoV-2 Mpro, we performed conformational analysis and docking studies. Conformational searching of UCI-1 (MacroModel with the MMFFs force field and GB/SA water) revealed that UCI-1 adopts a global minimum energy conformation that resembles the kinked conformation that residues 305–309 adopt in the active site of Mpro 316 (Fig. 2, Fig. 4 A). In the global minimum energy conformation, the AEPA residue acts as a rigid spacer, with Phe, Gln, Ser, and Lys forming a bridge. As we had envisioned, the Phe and Gln residues adopt a β-turn conformation, with Phe at the i+1 position and Gln at the i+2 position. The Phe side chain is well situated to fit into the S2 pocket, and the Gln side chain is well situated to fit into the S1 pocket. The Ser, Lys, and AEPA residues, in turn, are poised to occupy the S1′, S2′, and S3’ pockets.
Fig. 4.

(A) Lowest energy conformer of UCI-1. (B) Superposition of the 20 lowest energy conformers from conformational searching. The difference in energy between the lowest and highest energy conformers among these 20 is 7.0 kJ/mol (C) UCI-1 in complex with the SARS-CoV-2 Mpro active site generated in Autodock Vina. The SARS-CoV-2 Mpro crystal structure with PDB accession number 6YB7 was used in the docking study.
The cyclic peptide appears to be particularly rigid. In the conformational search, the peptide backbones of most of the conformers adopt the conformation described above, differing only in side chain geometry and the type of β-turn formed by Phe305 and Gln306 (Fig. 4B). To evaluate how UCI-1 might fit in the active site of Mpro, we docked UCI-1 with the X-ray crystallographic structure of the SARS-CoV-2 Mpro PDB 6YB7 using Autodock Vina [50]. This initial docking study revealed that UCI-1 fits into the active site of the SARS-CoV-2 Mpro in the envisioned manner (Fig. 4C).
To further evaluate how UCI-1 may achieve inhibition of Mpro, we performed a complementary docking experiment on UCI-1, and then performed a molecular dynamics simulation on the docked structure. In this study, we first docked UCI-1 with an equilibrated version of the X-ray crystallographic structure of the SARS-CoV-2 Mpro dimer PDB 6Y2E, which has previously been simulated in explicit water [51]. Next, we chose a low-energy configuration of UCI-1 bound to an equilibrated crystal structure of the Mpro dimer PDB 6Y2E that best matched the docked structure observed in the initial docking experiment with PDB 6YB7. Finally, we performed 10 ns of molecular dynamics simulation of the docked structure using the CHARMM36 force field and TIP3P water at 310 K. This procedure was then repeated for peptide-1a and peptide-1b to gain insight into why these control peptides do not substantially inhibit Mpro.
In the UCI-1 docking experiment, the side chains of UCI-1 occupy the active site Mpro in the envisioned manner, where Phe occupies the S2 pocket, Gln occupies the S1 pocket, Ser occupies the S1′ pocket, Lys occupies the S2′ pocket, and the AEPA group occupies the S3′ pocket (Fig. S2). Over the course of the simulation, UCI-1 stays bound to the active site and the amino acids maintain their positions in the pockets of the active site—only the Lys side chain appears to leave its respective pocket over the 10 ns simulation (Fig. S3). In contrast, the control peptides explore more of the active site and their side chains do not remain in their respective pockets as frequently. Notably, the d-Phe side chain of peptide-1b does not stay in the S2 pocket, and at times travels towards the S3’ pocket, causing the peptide to lift away from the active site. The linear control, peptide-1a, does not appear to bind well in any of the pockets.
Stability of UCI-1 toward Mpro. The amide bond between residues at positions P1 and P1’ of UCI-1 has the potential to be cleaved by Mpro, because residues at these positions correspond to the cleavage site of Mpro substrates. To determine whether Mpro cleaves UCI-1, we used LC/MS to analyze UCI-1 in the well solution from the 96-well plate of the enzyme inhibition assay after 4 h. To aid in LC/MS identification of the cleavage product of UCI-1, we synthesized peptide-1c, which would correspond to the authentic UCI-1 Mpro cleavage product. We then spiked the well solution with peptide-1c to identify and determine the limits of detection of the putative cleavage product.
UCI-1 is not appreciably cleaved by Mpro (Fig. S2). LC/MS analysis of 5 μL of the 187.5 μM UCI-1 enzyme inhibition assay well solution spiked with 20 μM peptide-1c shows a peak at 2.2 min that corresponds to peptide-1c (Fig. S2B). The peak at 2.2 min is absent in the LC/MS trace acquired before spiking the well solution with peptide-1c (Fig. S2A), indicating that there is little to no cleavage of UCI-1 by Mpro. The ion current at 2.2 min in the LC/MS trace acquired before spiking with peptide-1c shows no significant evidence of the UCI-1 cleavage product (Fig. S2C), whereas the ion current at 2.2 min in the LC/MS trace acquired after spiking with peptide-1b shows the mass of peptide-1c (Fig. S2D), providing additional evidence that UCI-1 resists cleavage by Mpro. These findings confirm that UCI-1 acts as an inhibitor and not a competitive substrate. The resistance of UCI-1 to cleavage by Mpro is consistent with the observations by others that cyclic peptides often resist proteolytic cleavage [[52], [53], [54], [55], [56], [57]].
Cytotoxicity of UCI-1. To evaluate whether UCI-1 is cytotoxic, we exposed human embryonic kidney (HEK-293) cells to varying concentrations of UCI-1 (0–256 μM) for 72 h, and then assessed cell death using a lactate dehydrogenase (LDH) release assay. At the highest concentration evaluated, UCI-1 elicits little or no LDH release from HEK-293 cells, indicating that UCI-1 is not cytotoxic at concentrations up to 256 μM (Fig. S3).
3. Discussion
UCI-1 is noteworthy, because it represents a direct creation of a cyclic peptide inhibitor of Mpro from the crystal structure of a linear peptide substrate. UCI-1 is created from the linear peptide substrate by the addition of only a single methylene group to the para position of Phe309 that is then linked to the backbone carbonyl of Phe305 to create a rigid peptide macrocycle linked together by AEPA. In synthesizing UCI-1, the cyclization proceeds smoothly, without formation of detectable oligomers, uncyclized linear peptide, or side products.
The apparent rigidity of UCI-1 demonstrates an emergent property of the AEPA linker, whereby a short strand of amino acids can span across the AEPA linker to create a macrocycle that adopts a β-turn structure and a doughnut-like shape. To further evaluate the utility of the AEPA linker, we designed a homologue of UCI-1 in which we replaced the rigid AEPA linker with a flexible tri-alanine linker (tri-Ala-UCI-1) and performed conformational searching of this homologue (MacroModel with the MMFFs force field and GB/SA water). Like UCI-1, tri-Ala-UCI-1 is a 21-membered macrocycle. Conformational searching of tri-Ala-UCI-1 reveals that the global minimum energy conformation of tri-Ala-UCI-1 adopts a compact closed structure, rather than an open structure (Fig. 6 ). While UCI-1 contains an open ring with a stabilizing β-turn structure and a “kinked” conformation that resembles the conformation of the linear peptide Mpro substrate, tri-Ala-UCI-1 lacks these features. Because the AEPA amino acid replaces a tripeptide and enforces an open structure, it is a useful template for inducing a well-defined conformation in the spanning tetrapeptide [[58], [59],59].
Fig. 6.

Global minimum energy conformations of UCI-1 (A) and tri-Ala-UCI-1 (B).
UCI-1 is a paracyclophane containing four α-amino acids, in addition to AEPA. Conformationally constrained paracyclophane macrocycles containing AEPA and two, three, or five α-amino acids can also be envisioned. Molecular modeling of homologous AEPA macrocycles derived from UCI-1 with two, three, and five amino acids demonstrates that all the macrocycles adopt doughnut-like open structures in which the amino acids are extended above the AEPA linker (Fig. 7 ). In additional modeling studies, we have found that AEPA macrocycles that contain amino acids other than glycine adopt this open doughnut-like structure. Macrocycles containing only two α-amino acids are strained to the point of bending the AEPA unit out of planarity — a common property of smaller paracyclophanes (Fig. 7A) [60]. Nevertheless, we were able to synthesize cyclo(AEPA-FQ) and found that the cyclization proceeded smoothly, without the formation of detectable oligomers, uncyclized linear peptide, or side products.
Fig. 7.

Global minimum energy conformations of homologous AEPA macrocycles derived from UCI-1 that contain two (A), three (B) and five (C) amino acids spanned across the AEPA linker.
4. Conclusion
In spite of the rapid development of vaccines to combat COVID-19, antiviral drugs are still needed. Antiviral drugs that slow or halt viral replication can lead to a shortened time to recovery from COVID-19, offering the promise of improved mortality rates and alleviating the tremendous strain experienced by hospitals during the COVID-19 pandemic [61]. Antiviral drugs could also be used as first line of defense in a future coronavirus outbreak or pandemic, or even as a prevention.
The design of the UCI-1 demonstrates that cyclic peptides that mimic the conformation of linear peptide substrates of Mpro can be developed as inhibitors against Mpro. Almost all of the Mpro inhibitors that have been reported thus far are linear peptides that contain a “warhead” that forms a covalent bond with the active cysteine of Mpro. While the activity of UCI-1 is modest compared to other known Mpro inhibitors, UCI-1 lays the groundwork for developing additional cyclic peptide inhibitor analogues with improved activity against Mpro. Design and development of the next generation of UCI-1 analogues will likely produce better inhibitors, and we anticipate that the studies described here will pave the way for the design of more potent cyclic peptide inhibitors of Mpro with potential to be developed into drugs. We also anticipate that the AEPA unit will prove useful in the design of other conformationally constrained cyclic peptides with potential pharmacological applications.
5. Materials and methods
Synthesis of 2-(4-(2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)ethyl)phenyl)acetic acid (Fmoc-AEPA). A 50 mL round-bottom flask equipped with a magnetic stirring bar was charged with 100 mg (0.55 mmol, 1 equiv) of 2-(4-(2-aminoethyl)phenyl)acetic acid dissolved in 10 mL H2O. 0.156 mL (1.10 mmol, 2 equiv) of Et3N was added. 160 mg of Fmoc-OSu (0.50 mmol, 0.9 equiv) was dissolved in 10 mL CH3CN and added to the reaction mixture. The reaction was run for 2 h at room temperature. While it was running, the reaction was monitored by TLC (3:1 EtOAc/hexanes + 10% MeOH, Rf = 0.44) to determine the consumption of starting material (Rf = 0.77) and an appearance of fulvene (Rf = 0.81). 10 mL of EtOAc was then added to the reaction mixture and the organic layer was removed. The aqueous layer was acidified with 30 mL 1 M HCl, and then 10 mL of EtOAc was added. The organic layer was washed with water and brine, dried over MgSO4, and solvent was evaporated in vacuo to afford 0.140 g (70%) of Fmoc-AEPA-OH as a white powder. The Fmoc-AEPA-OH was used in solid-phase peptide synthesis without further purification. The product contains a minor contaminant (<10%) of Fmoc-AEPA-AEPA-OH, as detected by 1H NMR spectroscopy. HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C25H23NO4Na 424.1525 found 424.1507.
Syntheses of UCI-1 and peptide-1b. 2-Chlorotrityl chloride resin (300 mg, 1.6 mmol/g) was added to a Bio-Rad Poly-Prep chromatography column (10 mL). The resin was suspended in dry CH2Cl2 (10 mL) and allowed to swell for 30 min. The solution was drained from the resin and a solution of Fmoc-AEPA-OH (0.50 equiv, 64 mg, 0.16 mmol) in 6% (v/v) 2,4,6-collidine in dry CH2Cl2 (8 mL) was added immediately and the suspension was gently agitated for 12 h. The solution was then drained and a mixture of CH2Cl2/MeOH/N,N-diisopropylethylamine (DIPEA) (17:2:1, 10 mL) was added immediately. The mixture was gently agitated for 1 h to cap the unreacted 2-chlorotrityl chloride resin sites. The resin was then washed with dry CH2Cl2 (2x) and dried by passing nitrogen through the vessel. This procedure yielded 0.51 mmol/g of loaded resin.
The Fmoc-AEPA-2-chlorotrityl resin was transferred to a peptide synthesis coupling vessel and subjected to cycles of peptide coupling with Fmoc-protected amino acid building blocks. The linear peptide was synthesized from the C-terminus to the N-terminus. Each coupling cycle consisted of i. Fmoc-deprotection with 20% (v/v) piperidine in DMF for 5 min (2x), ii. washing with DMF (3x), iii. coupling of the amino acid (5 equiv) in the presence of HCTU (4.5 equiv) and 20% (v/v) N-methylmorpholine (2,4,6-collidine) in DMF for 10–20 min. iv. washing with DMF (3x). After coupling of the last amino acid, the terminal Fmoc group was removed with 20% (v/v) piperidine in DMF. The resin was transferred from the coupling vessel to a Bio-Rad Poly-Prep chromatography column.
The linear peptide was cleaved from the resin by agitating the resin for 1 h with a solution of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) in CH2Cl2 (1:4, 7 mL) [62]. The suspension was filtered and the filtrate was collected in a 250-mL round-bottomed flask. The resin was washed with additional HFIP in CH2Cl2 (1:4, 7 mL) and then with CH2Cl2 (2 × 10 mL). The combined filtrates were concentrated by rotary evaporation to give a white solid. The white solid was further dried by vacuum pump to afford the crude protected linear peptide, which was cyclized without further purification.
The crude protected linear peptide was dissolved in dry DMF (150 mL). HOBt (5 equiv) and HBTU (5 equiv) were added to the solution. NMM (12 equiv) was added to the solution and the mixture was stirred under nitrogen for 24 h. The mixture was then concentrated under reduced pressure to afford the crude protected cyclic peptide.
The protected cyclic peptide was dissolved in TFA/triisopropylsilane (TIPS)/H2O (18:1:1, 20 mL) in a 250-mL round-bottomed flask equipped with a nitrogen-inlet adaptor. The solution was stirred for 1.5 h. The reaction mixture was then concentrated by rotary evaporation under reduced pressure to afford the crude cyclic peptide as a thin yellow film on the side of the round-bottomed flask. The crude cyclic peptide was immediately subjected to purification by reverse-phase HPLC (RP-HPLC).
The peptide was dissolved in H2O and acetonitrile (7:3, 10 mL), and the solution was filtered through a 0.2 μm syringe filter and purified by RP-HPLC (gradient elution with 10–30% CH3CN over 50 min). Pure fractions were concentrated by rotary evaporation and lyophilized. The synthesis of UCI-1 yielded 22 mg (18.7% based on resin loading) of the peptide as the TFA salt. The synthesis of peptide-1b yielded 16 mg (13.6% based on resin loading) of the peptide as the TFA salt.
Synthesis of peptide-1a. Rink amide AM resin (300 mg, 0.68 mmol/g) was added to a peptide synthesis coupling vessel. The resin was suspended in dry DMF (10 mL) and allowed to swell for 30 min. The solution was drained from the resin and the Fmoc group was removed with 20% (v/v) piperidine in DMF for 20 min. The solution was drained and washed with DMF (5x). Fmoc-AEPA-OH (0.50 equiv, 40.8 mg, 0.102 mmol), HATU (0.5 equiv), and HOAt (0.5 equiv) in 20% (v/v) 2,4,6-collidine in dry DMF (8 mL) was then added to the resin mixed for 12 h. The solution was then drained, washed with DMF (3x) and a mixture of acetic anhydride/pyridine (3:2, 10 mL) was added. The resin was mixed for 15 min to cap the unreacted resin sites. The resin was then washed with DMF (3x). This procedure yielded 0.17 mmol/g of loaded resin.
The Fmoc-AEPA-Rink amide AM resin was subjected to cycles of peptide coupling with Fmoc-protected amino acid building blocks as described above, ending with cleavage of the terminal Fmoc. The resin was then washed with CH2Cl2 (3x) and then dried by pushing N2 gas through the Poly-Prep column. The peptide was cleaved from the resin and globally deprotected by mixing the dried resin with TFA/triisopropylsilane (TIPS)/H2O (18:1:1, 10 mL) and gently rocking for 2.5 h. The peptide was drained into a glass beaker, precipitated in cold ether, and subjected to purification by RP-HPLC as described above. The synthesis of peptide-1a yielded 17 mg (40.4% yield based on resin loading) of the peptide as the TFA salt.
Synthesis of peptide-1c. 2-Chlorotrityl chloride resin (300 mg, 1.6 mmol/g) was added to a Bio-Rad Poly-Prep chromatography column (10 mL). The resin was suspended in dry CH2Cl2 (10 mL) and allowed to swell for 30 min. The solution was drained from the resin and a solution of Fmoc-Gln(Trt)-OH (0.50 equiv, 146.57 mg, 0.24 mmol) in 6% (v/v) 2,4,6-collidine in dry CH2Cl2 (8 mL) was added immediately and the suspension was gently agitated for 12 h. The solution was then drained and a mixture of CH2Cl2/MeOH/N,N-diisopropylethylamine (DIPEA) (17:2:1, 10 mL) was added immediately. The mixture was gently agitated for 1 h to cap the unreacted 2-chlorotrityl chloride resin sites. The resin was then washed with dry CH2Cl2 (2x) and dried by passing nitrogen through the vessel. This procedure yielded 0.36 mmol/g of loaded resin.
The Fmoc-Gln(Trt)-2-chlorotrityl resin was subjected to cycles of peptide coupling with Fmoc-protected amino acid building blocks as described above, ending with cleavage of the terminal Fmoc. The resin was then washed with CH2Cl2 (3x) and then dried by pushing N2 gas through the Poly-Prep column. The peptide was cleaved from the resin and globally deprotected by mixing the dried resin with TFA/triisopropylsilane (TIPS)/H2O (18:1:1, 10 mL) and gently rocking for 2.5 h. The peptide was drained into a glass beaker, precipitated in cold ether, and subjected to purification by RP-HPLC as described above. The synthesis of peptide-1c yielded 13 mg (13.6% yield based on resin loading)of the peptide as the TFA salt.
Synthesis of H2N–K(Dabcyl)-TSAVLQSGFRKM-E(EDANS)-NH2. Rink amide AM resin (300 mg, 0.68 mmol/g) was added to a peptide synthesis coupling vessel. The resin was suspended in dry DMF (10 mL) and allowed to swell for 30 min. The solution was drained from the resin and the Fmoc group was removed with 20% (v/v) piperidine in DMF for 20 min. The solution was drained and washed with DMF (5x). Fmoc-Glu(EDANS)-OH (1.5 equiv, 189 mg, 0.306 mmol), HATU (1.5 equiv), and HOAt (1.5 equiv) in 20% (v/v) 2,4,6-collidine in dry DMF (8 mL) was then added to the resin mixed for 12 h. The solution was then drained, washed with DMF (3x) and a mixture of acetic anhydride/pyridine (3:2, 10 mL) was added. The resin was mixed for 15 min to cap the unreacted resin sites. The resin was then washed with DMF (3x). This procedure yielded 0.48 mmol/g of loaded resin.
Fmoc-Glu(EDANS)-Rink amide AM resin was subjected to cycles of peptide coupling with Fmoc-protected amino acid building blocks as described above. For coupling Fmoc-Lys(Dabcyl)-OH, Fmoc-Lys(Dabcyl)-OH (2 equiv, 180.3 mg, 0.408 mmol), HATU (2 equiv), and HOAt (2 equiv) in 20% (v/v) 2,4,6-collidine in dry DMF was added to the resin and mixed for 24 h. The resin was then washed with DMF (3x) and the terminal Fmoc was removed with 20% (v/v) piperidine in DMF for 20 min. The resin was then washed with DMF (3x) followed by CH2Cl2 (3x) and then dried by pushing N2 gas through the Poly-Prep column. The peptide was cleaved from the resin and globally deprotected by mixing the dried resin with TFA/triisopropylsilane (TIPS)/H2O (18:1:1, 20 mL) in a round bottom flask and stirring for 2.5 h. The peptide was drained into a glass beaker, precipitated in cold ether, and subjected to purification by RP-HPLC as described above. The synthesis of H2N–K(Dabcyl)-TSAVLQSGFRKM-E(EDANS)-NH2 yielded 42 mg (12.6% based on resin loading) of the peptide as the TFA salt.
Enzyme inhibition assay. A proprietary assay buffer containing detergent was used for the inhibition assays (BPS Bioscience, catalog # 79956). The substrate with the cleavage site of Mpro (indicated by the arrow, ↓), H2N–K(Dabcyl)-TSAVLQ↓SGFRKM-E(EDANS)-NH2, was used in the fluorescence resonance energy transfer (FRET) based continuous kinetic assay, using a black-walled 96-well plate. The emergence of EDANS fluorescence due to the cleavage of the substrate by Mpro was monitored at 460 nm, with excitation at 360 nm, using a Varioskan LUX fluorescence spectrophotometer (Thermo Fisher) using the top-read mode at 37 °C. Stock solutions (20 mg/mL) of UCI-1, peptide-1a, and peptide-1b were prepared gravimetrically by dissolving the peptides in deionized H2O and then diluted in assay buffer to create 10X solutions for each concentration in a serial dilution. For the determination of the IC50, 0.1 μM SARS-CoV-2 Mpro was incubated with UCI-1, peptide-1a, or peptide-1b at various concentrations (23.4–750 μM for UCI-1 and 187.5–1500 μM for peptide-1a and peptide-1b) in assay buffer at 37 °C for 30 min. Afterward, the reaction was initiated by adding the FRET peptide substrate at a 50 μM final concentration (final well volume: 50 μL). The IC50 value for UCI-1 was determined using the GraphPad Prism 8.4.3 software by plotting the initial rates. Measurements of enzymatic activity were performed in triplicate and are presented as the mean ± standard deviations (s.d.).
LC/MS analysis of enzyme inhibition assay well solution. LC/MS analysis of the pooled 187.5 μM UCI-1 well solutions from the enzyme inhibition assay was performed on a Waters Xevo Qtof G2XS equipped with a C4 column. For both LC/MS traces, 5 μL of the well solution (diluted 1:2 in deionized H2O) was injected on the column. For the spiking experiment, a 10 mg/mL stock solution of peptide-1c was prepared gravimetrically in deionized H2O, and an aliquot of the stock solution was diluted in deionized H2O to create a 1 mM working solution. A 1-μL aliquot of the 1 mM working solution of peptide-1c was added to 100 μL of the well solution to achieve a final peptide-1c concentration of 10 μM.
LDH release assay. The LDH release assay was performed using the Pierce LDH Cytotoxicity Assay Kit from Thermo Scientific. Experiments were performed in replicates of five, and an additional 10 wells were used for controls. Cells were cultured in the inner 60 wells (rows B–G, columns 2–11) of the 96-well plate. DMEM:F12 media (100 μL) was added to the outer wells (rows A and H and columns 1 and 12), in order to ensure the greatest reproducibility of data generated from the inner wells. A 10-mg/mL stock solution of UCI-1 was prepared gravimetrically in sterile deionized H2O that had been passed through a 0.2 μm nylon syringe filter. The stock solution was used to create a 2560 μM solution of UCI-1, which was serially diluted in sterile deionized H2O to create 10x working solutions of UCI-1.
HEK-293 cells were plated in a 96-well plate at 15,000 cells per well. Cells were incubated in 100 μL of a 1:1 mixture of DMEM:F12 media supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a 5% CO2 atmosphere and allowed to adhere to the bottom of the plate for 24 h. After 24 h, the culture media was removed and replaced with 90 μL of serum-free DMEM:F12 media. A 10-μL aliquot of the working solutions of UCI-1 was added to each well, for well concentrations of 256 μM–32 μM. Experiments were run in replicates of five. Five wells were used as controls and received 10-μL aliquots of sterile deionized water (vehicle). Another five wells were left untreated, to be subsequently used as controls with lysis buffer for the LDH release assay. Cells were incubated at 37 °C in a 5% CO2 atmosphere for 72 h.
After 72 h, 10 μL of 10x lysis buffer — included with the assay kit — was added to the five untreated wells, and the cells were incubated for an additional 45 min. After 45 min, a 50-μL aliquot of the supernatant media from each well was transferred to a new 96-well plate and 50 μL of LDH substrate solution, prepared according to manufacturer's protocol, was added to each well. The treated plates were stored in the dark for 30 min. The absorbance of each well was measured at 490 and 680 nm (A490 and A680). Data were processed by calculating the differential absorbance for each well (A490−A680) and comparing those values to those of the lysis buffer controls and the untreated controls:
| % cell death = [(A490−A680)compound − (A490−A680)vehicle] ⁄ [(A490−A680)lysis − (A490−A680)vehicle] |
Docking of UCI-1 to SARS-CoV-2 Mpro. The model of the SARS-CoV-2 Mpro was generated as follows: Starting coordinates of SARS-CoV-2 Mpro were generated from the SARS-CoV-2 Mpro crystallographic structure (PDB 6YB7) using PyMOL and were saved as a new PDB file. In PyMOL, the dimethyl sulfoxide molecule that sits in the active site of 6YB7 was deleted. A minimum energy (global minimum) structure of UCI-1 was generated by conformational searching in MacroModel (MMFFs force field in GB/SA water). iBabel was used to convert the PDB file generated in MacroModel into a PDBQT file prior to docking. Docking was performed using AutoDock Tools and AutoDock Vina. In AutoDock Tools, a grid was chosen to encompass the active site of SARS-CoV-2 Mpro in the size of 25x25 × 25 Å and with the coordinates x = 9.250, y = −5.944, z = 18.944. SARS CoV-2 Mpro was treated as a rigid receptor in these calculations. The lowest energy cluster, as determined by AutoDock Vina, was chosen to represent the docking model shown in Fig. 7C.
Molecular Dynamics Simulation. The initial SARS-CoV-2 Mpro model that was used for MD simulations was generated from the final frame of the published wild type Mpro trajectory [50], which was originally seeded with PDB structure 6Y2E (see reference for details). Initial peptide conformations were obtained by docking UCI-1, peptide-1a, and peptide-1b respectively to the A chain active site of the initial Mpro model using AutoDock Vina [49]; the search box in each case was given a radius equal to 10 times the peptide's radius of gyration, centered on residue M165 (chosen because of its central location in the conserved substrate binding region, as confirmed by both visual inspection of the structure and the analysis of Jin et al. (2020)) [31], and an exhaustiveness of 32 was employed. Out of the 25 minimum energy poses, the pose most closely resembling the conformation found when docking against PDB 6YB7 was retained for use as the initial condition. CHARMM representations of the peptide models were created using CGenFF version 3.01 [63,64,65,66]. The combined peptide/dimer structures were solvated in TIP3P water [67] using VMD version 1.9.3; [68] a cubic water box with minimum margin of 10 Å from the protein complex on each side was employed, with NaCl added to achieve neutral charge. Each complex was subsequently minimized for 10,000 iterations using namd 2.13 [69] with the CHARMM 36 m force field [70], followed by a 10 ps simulation period for box adjustment. (All simulations were performed using periodic boundary conditions with an NpT ensemble at 310 K and 1 atm, Langevin dynamics with a damping coefficient of 1/ps and a Nosé-Hoover Langevin piston71,72 were respectively employed for temperature and pressure control. Rigid bonds were used only for water molecules, and a 1 fs step size was employed.) Following adjustment, each complex was simulated for 10 ns, with 1,000 frames being retained for subsequent analysis (1/ps). Visualization and analysis of the resulting trajectories was performed with VMD.
Author contributions
A.G.K. and J.S.N. conceived and designed UCI-1 and the research. A.G.K., M.K., and M.A.M. synthesized Fmoc-AEPA-OH. A.G.K., C.M.T.P., and M.A.M. performed the peptide synthesis. A.G.K. performed the enzyme inhibition assays and the LC/MS experiments. G.G. performed the LDH release assays. A.G.K. and J.S.N. performed the conformational analysis. M.K. performed the initial docking study to PDB 6YB7. E.M.D and C.T.B performed the docking experiments and molecular dynamics simulations with PDB 6YE2. A.G.K. and J.S.N. wrote the manuscript. All authors read and approved the manuscript.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We thank Dr. Rachel Martin and Dr. Vy Dong for helpful advice and discussions, and the Mass Spectrometry Facility in the UCI Department of Chemistry for assistance with LC/MS experiments.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2021.113530.
Funding
C.T.B. and E.M.D. were supported by NASA award 19-EXO19-0070.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Schlippe Y.V., Hartman M.C., Josephson K., Szostak J.W. In vitro selection of highly modified cyclic peptides that act as tight binding inhibitors. J. Am. Chem. Soc. 2012, Jun 27;134(25):10469–10477. doi: 10.1021/ja301017y. Epub 2012 Mar 29. PMID: 22428867; PMCID: PMC3384292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vinogradov A.A., Yin Y., Suga H. Macrocyclic peptides as drug candidates: recent progress and remaining challenges. J. Am. Chem. Soc. 2019, Mar 13;141(10):4167–4181. doi: 10.1021/jacs.8b13178. Epub 2019 Feb 27. PMID: 30768253. [DOI] [PubMed] [Google Scholar]
- 3.Gang D., Kim D.W., Park H.S. Cyclic peptides: promising scaffolds for biopharmaceuticals. Genes. 2018, Nov 16;9(11):557. doi: 10.3390/genes9110557. PMID: 30453533; PMCID: PMC6267108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sawyer T. CHAPTER 1:renaissance in peptide drug discovery: the third wave. in Peptide-based Drug Discovery: Challenges and New Therapeutics. 2017:1–34. doi: 10.1039/9781788011532-00001. [DOI] [Google Scholar]
- 5.Morrison C. Constrained peptides' time to shine? Nat. Rev. Drug Discov. 2018, Jul 30;17(8):531–533. doi: 10.1038/nrd.2018.125. PMID: 30057410. [DOI] [PubMed] [Google Scholar]
- 6.Dougherty P.G., Sahni A., Pei D. Understanding cell penetration of cyclic peptides. Chem. Rev. 2019, Sep 11;119(17):10241–10287. doi: 10.1021/acs.chemrev.9b00008. Epub 2019, May 14. PMID: 31083977; PMCID: PMC6739158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katsara M., Tselios T., Deraos S., Deraos G., Matsoukas M.T., Lazoura E., Matsoukas J., Apostolopoulos V. Round and round we go: cyclic peptides in disease. Curr. Med. Chem. 2006;13(19):2221–2232. doi: 10.2174/092986706777935113. PMID: 16918350. [DOI] [PubMed] [Google Scholar]
- 8.Clardy J., Walsh C. Lessons from natural molecules. Nature. 2004, Dec 16;432:829–837. doi: 10.1038/nature03194. 7019. PMID: 15602548. [DOI] [PubMed] [Google Scholar]
- 9.Qian Z., Rhodes C.A., McCroskey L.C., Wen J., Appiah-Kubi G., Wang D.J., Guttridge D.C., Pei D. Enhancing the cell permeability and metabolic stability of peptidyl drugs by reversible bicyclization. Angew Chem. Int. Ed. Engl. 2017, Feb 1;56(6):1525–1529. doi: 10.1002/anie.201610888. Epub 2016 Dec 30. PMID: 28035784; PMCID: PMC5296932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rezai T., Yu B., Millhauser G.L., Jacobson M.P., Lokey R.S. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J. Am. Chem. Soc. 2006, Mar 1;128(8):2510–2511. doi: 10.1021/ja0563455. PMID: 16492015. [DOI] [PubMed] [Google Scholar]
- 11.Qian Z., Martyna A., Hard R.L., Wang J., Appiah-Kubi G., Coss C., Phelps M.A., Rossman J.S., Pei D. Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry. 2016, May 10;55(18):2601–2612. doi: 10.1021/acs.biochem.6b00226. Epub 2016 Apr 28. PMID: 27089101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gavenonis J., Sheneman B.A., Siegert T.R., Eshelman M.R., Kritzer J.A. Comprehensive analysis of loops at protein-protein interfaces for macrocycle design. Nat. Chem. Biol. 2014, Sep;10(9):716–722. doi: 10.1038/nchembio.1580. Epub 2014 Jul 20. PMID: 25038791; PMCID: PMC4138238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Driggers E.M., Hale S.P., Lee J., Terrett N.K. The exploration of macrocycles for drug discovery--an underexploited structural class. Nat. Rev. Drug Discov. 2008, Jul;7(7):608–624. doi: 10.1038/nrd2590. PMID: 18591981. [DOI] [PubMed] [Google Scholar]
- 14.Mallinson J., Collins I. Macrocycles in new drug discovery. Future Med. Chem. 2012, Jul;4(11):1409–1438. doi: 10.4155/fmc.12.93. PMID: 22857532. [DOI] [PubMed] [Google Scholar]
- 15.Heinis C. Drug discovery: tools and rules for macrocycles. Nat. Chem. Biol. 2014, Sep;10(9):696–698. doi: 10.1038/nchembio.1605. Epub 2014 Jul 20. PMID: 25038789. [DOI] [PubMed] [Google Scholar]
- 16.Giordanetto F., Kihlberg J. Macrocyclic drugs and clinical candidates: what can medicinal chemists learn from their properties? J. Med. Chem. 2014, Jan 23;57(2):278–295. doi: 10.1021/jm400887j. Epub 2013 Sep 17. PMID: 24044773. [DOI] [PubMed] [Google Scholar]
- 17.Bockus A.T., McEwen C.M., Lokey R.S. Form and function in cyclic peptide natural products: a pharmacokinetic perspective. Curr. Top. Med. Chem. 2013;13(7):821–836. doi: 10.2174/1568026611313070005. PMID: 23578026. [DOI] [PubMed] [Google Scholar]
- 18.Smolewski P., Robak T. The discovery and development of romidepsin for the treatment of T-cell lymphoma. Expet Opin. Drug Discov. 2017, Aug;12(8):859–873. doi: 10.1080/17460441.2017.1341487. Epub 2017 Jun 22. PMID: 28641053. [DOI] [PubMed] [Google Scholar]
- 19.Lättig-Tünnemann G., Prinz M., Hoffmann D., Behlke J., Palm-Apergi C., Morano I., Herce H.D., Cardoso M.C. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun. 2011, Aug 30;(2):453. doi: 10.1038/ncomms1459. PMID: 21878907; PMCID: PMC3265364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bird G.H., Mazzola E., Opoku-Nsiah K., Lammert M.A., Godes M., Neuberg D.S., Walensky L.D. Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat. Chem. Biol. 2016, Oct;12(10):845–852. doi: 10.1038/nchembio.2153. Epub 2016 Aug 22. PMID: 27547919; PMCID: PMC5055751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang H., Yin L., Kim K.H., Cheng J. Helical poly(arginine) mimics with superior cell-penetrating and molecular transporting properties. Chem. Sci. 2013, Oct;4(10):3839–3844. doi: 10.1039/C3SC51328A. PMID: 25400902; PMCID: PMC4232443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamashita H., Kato T., Oba M., Misawa T., Hattori T., Ohoka N., Tanaka M., Naito M., Kurihara M., Demizu Y. Development of a cell-penetrating peptide that exhibits responsive changes in its secondary structure in the cellular environment. Sci. Rep. 2016, Sep 9;6:33003. doi: 10.1038/srep33003. PMID: 27609319; PMCID: PMC5016780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qian Z., Martyna A., Hard R.L., Wang J., Appiah-Kubi G., Coss C., Phelps M.A., Rossman J.S., Pei D. Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry. 2016, May 10;55(18):2601–2612. doi: 10.1021/acs.biochem.6b00226. Epub 2016 Apr 28. PMID: 27089101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolfe J.M., Fadzen C.M., Holden R.L., Yao M., Hanson G.J., Pentelute B.L. Perfluoroaryl bicyclic cell-penetrating peptides for delivery of antisense oligonucleotides. Angew Chem. Int. Ed. Engl. 2018, Apr 16;57(17):4756–4759. doi: 10.1002/anie.201801167. Epub 2018 Mar 14. PMID: 29479836; PMCID: PMC6248909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frost J.R., Scully C.C., Yudin A.K. Oxadiazole grafts in peptide macrocycles. Nat. Chem. 2016, Dec;8(12):1105–1111. doi: 10.1038/nchem.2636. Epub 2016 Oct 24. PMID: 27874866. [DOI] [PubMed] [Google Scholar]
- 26.Matsui K., Kido Y., Watari R., Kashima Y., Yoshida Y., Shuto S. Highly conformationally restricted cyclopropane tethers with three-dimensional structural diversity drastically enhance the cell permeability of cyclic peptides. Chemistry. 2017, Mar 2;23(13):3034–3041. doi: 10.1002/chem.201604946. Epub 2016 Dec 27. PMID: 27878880. [DOI] [PubMed] [Google Scholar]
- 27.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003, Jun 13;300(5626):1763–1767. doi: 10.1126/science.1085658. Epub 2003 May 13. PMID: 12746549. [DOI] [PubMed] [Google Scholar]
- 28.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, Sep;281(18):4085–4096. doi: 10.1111/febs.12936. Epub 2014 Aug 11. PMID: 25039866; PMCID: PMC7163996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J., von Brunn A., Leyssen P., Lanko K., Neyts J., de Wilde A., Snijder E.J., Liu H., Hilgenfeld R. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020, May 14;63(9):4562–4578. doi: 10.1021/acs.jmedchem.9b01828. Epub 2020 Feb 24. PMID: 32045235; PMCID: PMC7098070. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020, Apr 24;368(6489):409–412. doi: 10.1126/science.abb3405. Epub 2020 Mar 20. PMID: 32198291; PMCID: PMC7164518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020, Apr 9 doi: 10.1038/s41586-020-2223-y. Epub ahead of print. PMID: 32272481. [DOI] [PubMed] [Google Scholar]
- 32.Dai W., Zhang B., Su H., Li J., Zhao Y., Xie X., Jin Z., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., Jiang H., Rao Z., Zhang L.K., Xu Y., Yang H., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020, Apr 22 doi: 10.1126/science.abb4489. Epub ahead of print. PMID: 32321856; PMCID: PMC7179937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xue X., Yu H., Yang H., Xue F., Wu Z., Shen W., Li J., Zhou Z., Ding Y., Zhao Q., Zhang X.C., Liao M., Bartlam M., Rao Z. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 2008, Mar;82(5):2515–2527. doi: 10.1128/JVI.02114-07. Epub 2007 Dec 19. PMID: 18094151; PMCID: PMC2258912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vuong W., Khan M.B., Fischer C., Arutyunova E., Lamer T., Shields J., Saffran H.A., McKay R.T., van Belkum M.J., Joyce M., Young H.S., Tyrrell D.L., Vederas J.C., Lemieux M.C. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. bioRxiv. 2020 doi: 10.1101/2020.05.03.073080. 05.03.073080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Douangamath A., Fearon D., Gehrtz P., Krojer T., Lukacik P., Owen C.D., Resnick E., Strain-Damerell C., Aimon A., Ábrányi-Balogh P., Brandaõ-Neto J., Carbery A., Davison G., Dias A., Downes T.D., Dunnett L., Fairhead M., Firth J.D., Jones S.P., Keely A., Keserü G.M., Klein H.F., Martin M.P., Noble M.E.M., O'Brien P., Powell A., Reddi R., Skyner R., Snee M., Waring M.J., Wild C., London N., von Delft F., Walsh M.A. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. bioRxiv. 2020;5(27):118117. doi: 10.1101/2020.05.27.118117. PPR:PPR168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dragovich P.S., Prins T.J., Zhou R., Webber S.E., Marakovits J.T., Fuhrman S.A., Patick A.K., Matthews D.A., Lee C.A., Ford C.E., Burke B.J., Rejto P.A., Hendrickson T.F., Tuntland T., Brown E.L., Meador J.W., 3rd, Ferre R.A., Harr J.E., Kosa M.B., Worland S.T. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as L-glutamine replacements. J. Med. Chem. 1999, Apr 8;42(7):1213–1224. doi: 10.1021/jm9805384. PMID: 10197965. [DOI] [PubMed] [Google Scholar]
- 37.Westberg M., Su Y., Zou X., Ning L., Hurst B., Tarbet B., Lin M.Z. Rational design of a new class of protease inhibitors for the potential treatment of coronavirus diseases. bioRxiv. 2020 doi: 10.1101/2020.09.15.275891. Preprint. [DOI] [Google Scholar]
- 38.Günther S., Reinke P.Y.A., Fernández-García Y., Lieske J., Lane T.J., Ginn H.M., Koua F.H.M., Ehrt C., Ewert W., Oberthuer D., Yefanov O., Meier S., Lorenzen K., Krichel B., Kopicki J.D., Gelisio L., Brehm W., Dunkel I., Seychell B., Gieseler H., Norton-Baker B., Escudero-Pérez B., Domaracky M., Saouane S., Tolstikova A., White T.A., Hänle A., Groessler M., Fleckenstein H., Trost F., Galchenkova M., Gevorkov Y., Li C., Awel S., Peck A., Barthelmess M., Schluenzen F., Lourdu Xavier P., Werner N., Andaleeb H., Ullah N., Falke S., Srinivasan V., França B.A., Schwinzer M., Brognaro H., Rogers C., Melo D., Zaitseva-Doyle J.J., Knoska J., Peña-Murillo G.E., Mashhour A.R., Hennicke V., Fischer P., Hakanpää J., Meyer J., Gribbon P., Ellinger B., Kuzikov M., Wolf M., Beccari A.R., Bourenkov G., von Stetten D., Pompidor G., Bento I., Panneerselvam S., Karpics I., Schneider T.R., Garcia-Alai M.M., Niebling S., Günther C., Schmidt C., Schubert R., Han H., Boger J., Monteiro D.C.F., Zhang L., Sun X., Pletzer-Zelgert J., Wollenhaupt J., Feiler C.G., Weiss M.S., Schulz E.C., Mehrabi P., Karničar K., Usenik A., Loboda J., Tidow H., Chari A., Hilgenfeld R., Uetrecht C., Cox R., Zaliani A., Beck T., Rarey M., Günther S., Turk D., Hinrichs W., Chapman H.N., Pearson A.R., Betzel C., Meents A. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science. 2021, Apr 2 doi: 10.1126/science.abf7945. Epub ahead of print. PMID: 33811162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vatansever E.C., Yang K.S., Drelich A.K., Kratch K.C., Cho C.C., Kempaiah K.R., Hsu J.C., Mellott D.M., Xu S., Tseng C.K., Liu W.R. Bepridil is potent against SARS-CoV-2 in vitro. Proc. Natl. Acad. Sci. U. S. A. 2021,Mar 9;118(10) doi: 10.1073/pnas.2012201118. PMID: 33597253; PMCID: PMC7958448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.For recent examples of in silico designed and discovered inhibitors see: (a) Porto W.F. Virtual screening of peptides with high affinity for SARS-CoV-2 main protease. Comput. Biol. Med. 2021 Apr 2;133:104363. doi: 10.1016/j.compbiomed.2021.104363. Epub ahead of print. PMID: 33862305; PMCID: PMC8018786. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Breidenbach J., Lemke C., Pillaiyar T., Schäkel L., Al Hamwi G., Diett M., Gedschold R., Geiger N., Lopez V., Mirza S., Namasivayam V., Schiedel A.C., Sylvester K., Thimm D., Vielmuth C., Phuong Vu L., Zyulina M., Bodem J., Gütschow M., Müller C.E. Targeting the main protease of SARS-CoV-2: from the establishment of high throughput screening to the design of tailored inhibitors. Angew Chem. Int. Ed. Engl. 2021 Apr 26;60(18):10423–10429. doi: 10.1002/anie.202016961. Epub 2021 Mar 24. PMID: 33655614; PMCID: PMC8014119. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang C.H., Stone E.A., Deshmukh M., Ippolito J.A., Ghahremanpour M.M., Tirado-Rives J., Spasov K.A., Zhang S., Takeo Y., Kudalkar S.N., Liang Z., Isaacs F., Lindenbach B., Miller S.J., Anderson K.S., Jorgensen W.L. Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent. Sci. 2021 Mar 24;7(3):467–475. doi: 10.1021/acscentsci.1c00039. Epub 2021 Feb 22. PMID: 33786375; PMCID: PMC7931627. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Verma N., Henderson J.A., Shen J. Proton-coupled conformational activation of SARS coronavirus main proteases and opportunity for designing small-molecule broad-spectrum targeted covalent inhibitors. J. Am. Chem. Soc. 2020 Dec 30;142(52):21883–21890. doi: 10.1021/jacs.0c10770. Epub 2020 Dec 15. PMID: 33320670; PMCID: PMC7754784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hegyi A., Ziebuhr J. Conservation of substrate specificities among coronavirus main proteases. J. Gen. Virol. 2002 Mar;83(Pt 3):595–599. doi: 10.1099/0022-1317-83-3-595. PMID: 11842254. [DOI] [PubMed] [Google Scholar]
- 42.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020 Apr 9 doi: 10.1038/s41586-020-2223-y. Epub ahead of print. PMID: 32272481. [DOI] [PubMed] [Google Scholar]
- 43.Dai W., Zhang B., Su H., Li J., Zhao Y., Xie X., Jin Z., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., Jiang H., Rao Z., Zhang L.K., Xu Y., Yang H., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020 Apr 22 doi: 10.1126/science.abb4489. Epub ahead of print. PMID: 32321856; PMCID: PMC7179937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen M.S., Corey L. Combination prevention for COVID-19. Science. 2020 May 08;368(6491):550. doi: 10.1126/science.abc5798. [DOI] [PubMed] [Google Scholar]
- 45.An initial version of this work was deposited in bioRxiv on August 3. 2020. Reference. [DOI] [Google Scholar]
- 46.Muramatsu T., Takemoto C., Kim Y.T., Wang H., Nishii W., Terada T., Shirouzu M., Yokoyama S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. U. S. A. 2016 Nov 15;113(46):12997–13002. doi: 10.1073/pnas.1601327113. Epub 2016 Oct 31. PMID: 27799534; PMCID: PMC5135343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020 Apr 24;368(6489):409–412. doi: 10.1126/science.abb3405. Epub 2020 Mar 20. PMID: 32198291; PMCID: PMC7164518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang S, Krumberger M, Morris MA, Parrocha CMT, Kreutzer AG, Nowick JS. Structure-based drug design of an inhibitor of the SARS-CoV-2 (COVID-19) main protease using free software: a tutorial for students and scientists. Submitted to Eur. J. Med. Chem. [DOI] [PMC free article] [PubMed]
- 49.The Fluorogenic Mpro Substrate We Have Synthesized and Used in Our Studies Contains a Dabcyl Group on the ε-amine of Lysine. Previous Studies Have Used an Isomer of This Substrate with the Dabcyl Group on the Terminal Amine.
- 50.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010 Jan 30;31(2):455–461. doi: 10.1002/jcc.21334. PMID: 19499576; PMCID: PMC3041641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cross T.J., Takahashi G.R., Diessner E.M., Crosby M.G., Farahmand V., Zhuang S., Butts C.T., Martin R.W. Sequence characterization and molecular modeling of clinically relevant variants of the SARS-CoV-2 main protease. Biochemistry. 2020 Oct 6;59(39):3741–3756. doi: 10.1021/acs.biochem.0c00462. Epub 2020 Sep 24. PMID: 32931703; PMCID: PMC7518256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schlippe Y.V., Hartman M.C., Josephson K., Szostak J.W. In vitro selection of highly modified cyclic peptides that act as tight binding inhibitors. J. Am. Chem. Soc. 2012 Jun 27;134(25):10469–10477. doi: 10.1021/ja301017y. Epub 2012 Mar 29. PMID: 22428867; PMCID: PMC3384292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vinogradov A.A., Yin Y., Suga H. Macrocyclic peptides as drug candidates: recent progress and remaining challenges. J. Am. Chem. Soc. 2019 Mar 13;141(10):4167–4181. doi: 10.1021/jacs.8b13178. Epub 2019 Feb 27. PMID: 30768253. [DOI] [PubMed] [Google Scholar]
- 54.Morrison C. Constrained peptides' time to shine? Nat. Rev. Drug Discov. 2018 Jul 30;17(8):531–533. doi: 10.1038/nrd.2018.125. PMID: 30057410. [DOI] [PubMed] [Google Scholar]
- 55.Dougherty P.G., Sahni A., Pei D. Understanding cell penetration of cyclic peptides. Chem. Rev. 2019 Sep 11;119(17):10241–10287. doi: 10.1021/acs.chemrev.9b00008. Epub 2019 May 14. PMID: 31083977; PMCID: PMC6739158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nowick J.S., Chung D.M., Maitra K., Maitra S., Stigers K.D., Sun Y. An unnatural amino acid that mimics a tripeptide β-strand and forms β-Sheet-like hydrogen-bonded dimers. J. Am. Chem. Soc. 2000;122:7654–7661. [Google Scholar]
- 57.Khasanova T.V., Khakshoor O., Nowick J.S. Functionalized analogues of an unnatural amino acid that mimics a tripeptide beta-strand. Org. Lett. 2008 Nov 20;10(22):5293–5296. doi: 10.1021/ol8021897. Epub 2008 Oct 21. PMID: 18937484; PMCID: PMC2659333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cram D.J., Cram J.M. Cyclophane chemistry: bent and battered benzene rings. Acc. Chem. Res. 1971;4:204–213. [Google Scholar]
- 59.Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., Hohmann E., Chu H.Y., Luetkemeyer A., Kline S., Lopez de Castilla D., Finberg R.W., Dierberg K., Tapson V., Hsieh L., Patterson T.F., Paredes R., Sweeney D.A., Short W.R., Touloumi G., Lye D.C., Ohmagari N., Oh M.D., Ruiz-Palacios G.M., Benfield T., Fätkenheuer G., Kortepeter M.G., Atmar R.L., Creech C.B., Lundgren J., Babiker A.G., Pett S., Neaton J.D., Burgess T.H., Bonnett T., Green M., Makowski M., Osinusi A., Nayak S., Lane H.C., ACTT-1 Study Group Members Remdesivir for the treatment of covid-19 - preliminary report. N. Engl. J. Med. 2020 May 22 doi: 10.1056/NEJMoa2007764. Epub ahead of print. PMID: 32445440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bollhagen R., Schmiedberger M., Barlosb K., Grell E. A new reagent for the cleavage of fully protected peptides synthesised on 2-chlorotrityl chloride resin. J. Chem. Soc., Chem. Commun. 1994:2559–2560. [Google Scholar]
- 61.Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., Mackerell A.D., Jr. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010 Mar;31(4):671–690. doi: 10.1002/jcc.21367. PMID: 19575467; PMCID: PMC2888302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu W., He X., Vanommeslaeghe K., MacKerell A.D., Jr. Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012 Dec 5;33(31):2451–2468. doi: 10.1002/jcc.23067. Epub 2012 Jul 23. PMID: 22821581; PMCID: PMC3477297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vanommeslaeghe K., MacKerell A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 2012 Dec 21;52(12):3144–3154. doi: 10.1021/ci300363c. Epub 2012 Nov 28. PMID: 23146088; PMCID: PMC3528824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vanommeslaeghe K., Raman E.P., MacKerell A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012 Dec 21;52(12):3155–3168. doi: 10.1021/ci3003649. Epub 2012 Nov 28. PMID: 23145473; PMCID: PMC3528813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 66.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996 Feb;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. 27-8. PMID: 8744570. [DOI] [PubMed] [Google Scholar]
- 67.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005 Dec;26(16):1781–1802. doi: 10.1002/jcc.20289. PMID: 16222654; PMCID: PMC2486339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B.L., Grubmüller H., MacKerell A.D., Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017 Jan;14(1):71–73. doi: 10.1038/nmeth.4067. Epub 2016 Nov 7. PMID: 27819658; PMCID: PMC5199616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martyna G.J., Tobias D.J., Klein M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 70.Feller S.E., Zhang Y., Pastor R.W., Brooks B.R. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.