

Abstract
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor with roles in inflammation and tumorigenicity. A fraction of STAT3 localizes in mitochondria, where it augments tumorigenesis via regulation of mitochondrial functions, including modulation of respiration and redox status. We show a novel mechanism for mitochondrial STAT3 regulation of redox homeostasis in triple‐negative breast cancer cells. Loss of STAT3 diminished complex I dehydrogenase activity and impaired NAD+ regeneration, leading to impaired expression of glutathione biosynthetic genes and other antioxidant genes. Expressing mitochondrially restricted STAT3 or replenishment of the cellular NAD pool restored antioxidant gene expression, as did complementation of the NADH dehydrogenase activity by expression of the STAT3‐independent yeast dehydrogenase, NDI1. These NAD‐regulated processes contributed to malignant phenotypes by promoting clonal cell growth and migration. Proximity interaction and protein pull‐down assays identified three components of complex I that associated with mitochondrial STAT3, providing a potential mechanistic basis for how mitochondrial STAT3 affects complex I activity. Our data document a novel mechanism through which mitochondrial STAT3 indirectly controls antioxidant gene regulation through a retrograde NAD+ signal that is modulated by complex I dehydrogenase activity.
Keywords: breast cancer, glutathione, mitochondria, oxidative stress, reactive oxygen species, STAT3
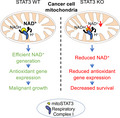
STAT3 interacts with respiratory complex I in mitochondria, leading to enhanced NADH dehydrogenase activity, facilitating efficient regeneration of NAD+ during respiration. NAD+ acts as a retrograde signal linking mitochondrial metabolism to changes in nuclear gene expression, leading to induction of antioxidant genes that contribute to the maintenance of redox balance and malignant cell growth, survival, and migration.
Abbreviations
- GSH
glutathione
- IDH
isocitrate dehydrogenase
- KO
knockout
- MTS
mitochondrial targeting sequence
- NAD(H)
nicotinamide adenine dinucleotide
- PGC1α
peroxisome proliferator‐activated receptor‐gamma co‐activator‐1alpha
- ROS
reactive oxygen species
- SIRT
sirtuin
- SOD
superoxide dismutase
- STAT3
signal transducer and activator of transcription 3
- TNBC
triple negative breast cancer
- WT
wild‐type
1. Introduction
Cellular redox homeostasis is an essential prerequisite for growth and survival of aerobic organisms and is regulated by numerous biochemical pathways that balance production and elimination of reactive oxygen species (ROS). ROS levels are elevated in cancer, where they contribute to DNA damage, proliferation, and metastasis [1]. Although elevated ROS levels are considered drivers of tumorigenesis, they can also trigger apoptosis, senescence, or catastrophic DNA damage; therefore, antioxidant pathways that buffer ROS are necessary for cell survival [2]. A major antioxidant pathway operating in many cancers involves enzymes that detoxify ROS, many of which are products of genes regulated by the NRF2 transcription factor [3], including enzymes involved in the synthesis of the major cellular redox buffer, glutathione (GSH) [4], and enzymes that directly detoxify oxidative environments [5, 6]. Mitochondria are a major source of ROS in tumor cells, with respiratory complex I and III being major contributors [7]. For instance, reduced complex I activity can increase cellular ROS. Mitochondrial complex I is also a major site for generating the cofactor nicotinamide adenine dinucleotide (NAD+) through NADH oxidation, and NAD+/NADH balance maintained by complex I plays a critical role in tumor progression [8]. The matrix arm of complex I, consisting of seven core subunits, binds multiple cofactors including a flavin mononucleotide (FMN) molecule. NADH donates a pair of electrons to this FMN molecule, which then flows through the respiratory chain to ubiquinone. This transfer of electrons at the Fe‐sulfur center N1 can generate ROS. In vitro characterization of purified mitochondria shows a strong correlation between complex I‐dependent superoxide production and NAD+/NADH ratio [9, 10]. For example, addition of NADH to purified bovine heart mitochondria generates superoxide, and this phenomenon can be quenched by addition of excess NAD+ [11]. Imbalance in the cellular NAD pool increases disease progression through ROS production and inflammation [12]. NAD+/NADH ratio can also affect gene transcription, largely through the action of the histone deacetylase silent mating type information regulation homolog 1 (SIRT1), which targets chromatin in a NAD+‐dependent manner [13]. SIRT1 target genes help maintain the cellular antioxidant capacity via synthesis of GSH [14] and can contribute to cell survival [15]. SIRT3 is a mitochondrially localized NAD+‐dependent deacetylase [16]. SIRT3 substrates include proteins involved in amino acid metabolism (GDH, AceCS2) and electron transport chain (SDH, NDUFA9), as well as the antioxidant proteins, manganese superoxide dismutase 2 (SOD2), and isocitrate dehydrogenase 2 (IDH2) [17, 18, 19, 20, 21, 22, 23], highlighting the role of SIRT3 in maintaining energy homeostasis. Loss of SIRT3, or reduced SIRT3 activity due to lower NAD+/NADH levels, accelerates tumor formation, accompanied by heightened ROS. This effect is attributed to reduced SOD2 activity, a ROS detoxifying enzyme that is deacetylated by SIRT3 [24, 25].
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that regulates numerous biological functions, including tumorigenicity and inflammation [26]. STAT3 is activated by phosphorylation of tyrosine 705 (Y705) and serine 727 (S727) by protein kinases regulated by cytokine and growth factor receptors and by some oncogenes. Tyrosine‐phosphorylated STAT3 is considered the major transcriptionally active form and can contribute to oncogenesis [27], although nonphosphorylated STAT3 has also been implicated in transcriptional regulation and cancer [28]. STAT3 is constitutively activated in many cancers, including breast cancer [29]. For instance, the IL‐6/STAT3 signaling axis contributes to the growth of stem cell‐like breast cancer growth [30].
In addition to its nuclear functions, a pool of STAT3 localizes to mitochondria [31, 32] where it contributes to a number of cellular functions, including mitochondrial respiration via modulation of complex I, II and V, maintenance of overall mitochondrial health through mPTP closure, and regulation of mitochondrial gene expression, and these functions contribute to Ras‐mediated cellular transformation [33, 34, 35, 36]. Moreover, mitochondrial STAT3 also regulates cellular redox homeostasis. Cells lacking STAT3 display increased ROS levels, both mitochondrial and cellular, and lower levels of GSH [37]. The increased ROS levels can be attributed to a dysfunctional electron transport chain, reduced GSH levels or both, and it remains unclear how mitochondrial STAT3 regulates the cellular GSH pool. While it is known that increased oxidative stress can activate nuclear STAT3 through phosphorylation on Y705, probably through transient inactivation of protein tyrosine phosphatases [38], it remains unclear whether STAT3 contributes to antioxidant gene expression, either through its nuclear or mitochondrial functions. Mechanisms through which metabolic changes in mitochondria are communicated to the nucleus to influence gene expression are complex [39], and it is unclear how mitochondrial STAT3‐regulated process can affect expression of GSH biosynthetic genes, which are nuclear encoded.
In this study, we show that the presence of STAT3 in mitochondria affected the basal expression of the GSH biosynthetic genes GCLC and GCLM, and of two major antioxidant genes, NQO1 and HMOX1. This regulation was independent of STAT3 nuclear action and did not appear to involve changes in NRF2. We found that compromised complex I activity in the absence of mitochondrial STAT3 affected NAD+ regeneration, resulting in reduced NAD+/NADH ratios, which correlated with impaired antioxidant gene expression and reduced malignant cell survival and migration. Moreover, mitochondrial STAT3 interacted with the dehydrogenase subunit of complex I.
2. Materials and methods
2.1. Materials
All chemicals were purchased from Sigma‐Aldrich (St. Louis, MO, USA) unless otherwise specified. Reagents with limited water solubility were initially dissolved in dimethylsulfoxide or ethanol before dilution in cell growth media (final solvent concentrations, < 0.1%).
2.2. Cell culture
Cells were cultured in Dulbecco's Modified Eagle Medium (DMEM; GE Healthcare, Piscataway, NJ, USA) supplemented with 10% Bovine Calf Serum (Sigma‐Aldrich) and Gentamicin (Cellgro, Corning Life Sciences, Tewksbury, MA, USA) in a 95% air/5% CO2 humidified atmosphere. The human triple‐negative breast cancer cell line MDA‐MB‐231 and human lung cancer cell line A549 were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells stably expressing Cas9 and guide RNA against STAT3 (gRNA sequence: AGATTGCCCGGATTGTGGCC) or a Cas9 alone (siEV) were maintained in the above media containing 5 μg·mL−1 of puromycin. MDA‐MB‐231 cells stably expressing STAT3 WT or mitochondrial targeting sequence (MTS) were maintained in the above media containing 200 µg·mL−1 of hygromycin. Cells were treated with SIRT1 inhibitor EX‐527 (Cayman Chemicals, Ann Arbor, MI, USA) or SIRT3 inhibitor 3‐TYP (TargetMol), where indicated in the figure legends.
2.3. Cell viability assay
Cell viability was measured in triplicate by crystal violet staining, as previously described [40]. In brief, cells seeded in 96‐well plates (Corning Incorporated, Corning, NY, USA) at a density of 104 cells/well were allowed to attach overnight (16 h at 37 °C), followed by addition of 0.375 mm hydrogen peroxide with or without supplementation with N‐Acetyl cysteine (NAC) (25 mm), Trolox (250 µm), or NAM (10 mm), and then cultured for an additional 8 h at 37 °C, followed by staining with 50 µL per well of crystal violet.
2.4. Measurements of ROS generation in living cells
Cells were cultured in 6‐well plates for 16 h and then exposed to NAC (25 mm), Trolox (250 µm), or NAM (10 mm) for 8 h. Cells were labeled with 2.5 µm MitoSox Red (Thermo Fisher, Waltham, MA, USA) for 30 min at 37 °C, according to the manufacturer's instructions, and fluorescent intensities were measured by flow cytometry.
2.5. Measurement of mitochondrial membrane potential
The mitochondrial membrane potential was measured in triplicate samples using the cationic, cell‐permeant dye tetramethylrhodamine ethyl ester (TMRE) from Thermo Fisher Scientific as previously described [31]. In brief, cells grown in DMEM overnight were washed with PBS, and incubated with 250 nm TMRE for 30 min at 37 °C for dye loading. The cells were washed with PBS prior to being collected for fluorescence analysis by flow cytometry.
2.6. Gene expression analyses
For quantitative reverse transcriptase‐polymerase chain reaction (RT‐PCR) analyses, total RNA was isolated using TriZol (Thermo Fisher) and reverse‐transcribed with Moloney murine leukemia virus (M‐MLV). The resulting cDNAs were amplified by qRT‐PCR with SYBR green (Molecular Probes, Eugene, OR, USA) using the primers shown in Table S1. Relative expression was determined by comparison to a standard curve generated from serial dilutions of cDNA containing abundant target sequences, and normalized to the expression of beta‐2‐microglobulin (B2M).
2.7. Mitochondrial preparation
Cells were harvested and washed once with PBS. The cell pellet was resuspended in mitochondria purification buffer (MPB: 10 mm Tris, 1 mm EGTA, 200 mm sucrose, pH 7.4) containing freshly added protease inhibitors (Thermo Fisher), 2 mm Na3VO4, 1 mm DTT, and 1 mm sodium‐β‐glycerophosphate, and incubated on ice for 10 min. The cell suspension was disrupted with 40 strokes in a Dounce homogenizer and centrifuged at 800 g for 5 min to pellet nuclei and unbroken cells. The supernatant was transferred to a fresh tube and spun at 10 000 g for 10 min to pellet mitochondria. Mitochondria were resuspended in MPB with 0.02% digitonin and incubated on ice for 5 min, and spun at 10 000 g for 10 min. Mitochondria were washed 2× with MPB to remove any digitonin, and protein content was quantified by using the Bio‐Rad Coomassie dye assay.
2.8. Western Blot analysis
For western blot analysis, mitochondria or cell pellets were lysed in RIPA buffer (50 mm Tris‐HCl, 150 mm NaCl, 0.2% SDS, 0.5% sodium deoxycholate, 1% Triton X‐100, 5% glycerol) containing freshly added protease inhibitors (Thermo Fisher), 2 mm Na3VO4, 1 mm DTT and 1 mm sodium‐β‐glycerophosphate, and incubated on ice for 10 min. Lysates were spun at 20 000 g for 10 min, and clarified lysates were resolved on SDS/PAGE, transferred to Polyvinylidene fluoride membranes, blocked with 5% milk, and probed with the antibodies. Unless otherwise specified, all antibodies and detection reagents were used at 1 : 2000 dilution prepared in 5% BSA in TBST: STAT3‐alpha (D1A5) (CST, Danvers, MA, USA, catalog no. 8768S), STAT3 (124H6) (CST, catalog no. 9139S), NDUFV2 (ABclonal, Woburn, MA, USA, catalog no. A7442), NDUFS2 (ABclonal, catalog no. A12858), NDUFAF2 (ABclonal, catalog no. A14296), SDHA (ABclonal, catalog no. A2594), Tubulin (Sigma, catalog no. 26628228), Streptavidin‐HRP (BD, Franklin Lakes, NJ, USA, catalog no. 554066). The blots were developed using a chemiluminescent detection kit (Advansta WesternBright ECL HRP substrate) on a LI‐COR Odyssey® Fc Imaging System.
2.9. BioID fusion protein and mass spectrometry analysis
STAT3‐BioID (SB) was constructed by fusing the sequence for MTS‐STAT3 [31] to BioID2 [41] in a lentiviral vector carrying puromycin resistance. MTS‐BioID was constructed by fusing the sequence for MTS [31] to BioID2 [41] in a lentiviral vector carrying puromycin resistance. MDA‐MB‐231 STAT3 WT cells were infected to stably express SB or MTS‐BioID and were maintained under 5 µg·mL−1 of puromycin.
MDA‐MB‐231 cells expressing SB or MTS‐BioID were grown in the presence of biotin, and mitochondria were prepared and lysed as described in Section 2.8, except the concentration of SDS was raised to 1%. Biotinylated proteins were collected from mitochondrial lysates on streptavidin Sepharose (Amersham Biosciences, Division of GE Healthcare, Piscataway, NJ, USA), and eluted proteins were identified by mass spectroscopy (MS) as described previously [42, 43].
In Brief, samples were reduced and alkylated with dithiothreitol (1 h at 57 °C) and iodoacetamide (45 min at room temperature). The samples were loaded on a NuPAGE 4–12% Bis‐Tris gel (Thermo Fisher Scientific) and run for 20 min at 200 V. The gel was stained with GelCode Blue Staining Reagent (Thermo Fisher). The gel bands were excised, cut into 1‐mm3 pieces, and destained for 15 min in a 1 : 1 (v/v) solution of methanol and 100 mm ammonium bicarbonate. The buffer was exchanged and the samples destained for another 15 min. This was repeated for another three cycles. The gel plugs were dehydrated by washing with acetonitrile and further dried by placing them in a SpeedVac for 20 min.
The samples were digested by adding 250 ng of trypsin (Thermo Fisher) onto the dried gel plugs followed by 300 μL of 100 mm ammonium bicarbonate. Digestion was carried out overnight at room temperature with gentle shaking. The digestion was halted by adding 300 μL of R2 50 μm Poros beads in 5% formic acid and 0.2% trifluoro acetic acid and agitated for 2 h at 4 °C. Beads were loaded onto equilibrated C18 ziptips. The Poros beads were washed with 0.5% acetic acid. The peptides were eluted with 40% acetonitrile in 0.5% acetic acid followed by 80% acetonitrile in 0.5% acetic acid. The organic solvent was removed using a SpeedVac concentrator and the samples reconstituted in 0.5% acetic acid.
An aliquot of each sample was loaded onto an Acclaim PepMap trap column (2 cm × 75 µm) in line with an EASY‐Spray analytical column (50 cm × 75 µm ID PepMap C18, 2 μm bead size) using the auto sampler of an EASY‐nLC 1000 HPLC (Thermo Fisher Scientific) with solvent A consisting of 2% acetonitrile in 0.5% acetic acid and solvent B consisting of 80% acetonitrile in 0.5% acetic acid. The peptides were gradient eluted into a Q Exactive HF‐X Mass Spectrometer (Thermo Fisher Scientific) using the following gradient: 5–35% in 60 min, 35–45% in 10 min, followed by 45–100% in 10 min. The gradient was held at 100% for another 10 min. MS1 spectra were recorded with a resolution of 45 000, an AGC target of 3e6, with a maximum ion time of 45 ms, and a scan range from 400 to 1500 m/z. The MS/MS spectra were collected with a resolution of 15 000, an AGC target of 1e5, maximum ion time of 120 ms, one microscan, 2 m/z isolation window, a Normalized Collision Energy of 27, and included charge states from +2 to +7.
2.9.1. MS data analysis
All acquired MS2 spectra were searched against the UniProt human database using Sequest within Proteome Discoverer 1.4. The search parameters were as follows: precursor mass tolerance ± 10 p.p.m., fragment mass tolerance ± 0.02 Da, digestion parameters trypsin allowing two missed cleavages, fixed modification of carbamidomethyl on cysteine, variable modification of oxidation on methionine, and variable modification of deamidation on glutamine and asparagine and a 1% peptide and protein FDR searched against a decoy database. The results were filtered to only include proteins identified by at least two unique peptides and likely contaminates were removed [44]. Known mitochondrial proteins were identified by comparison to the human MitoCarta3.0 database [45] and analyzed for pathway enrichment by using Enrichr [46] and by the Molecular Signatures Database v7.2 [47].
2.10. Complex I and complex II activity assay
Two microgram of purified mitochondria was incubated in a 96‐well plate with 100 µL of complex I assay buffer (25 mm KPO4, 2 mm KCN, 3.5 g·L−1 BSA, 60 µm DCIP, 70 µm DCU, 1 µm antimycin A) with or without 10 µm rotenone for 10 min at 37 °C. Five millimolar NADH was added to start the reaction, and absorbance was measured continuously at 600 nm at 30‐s intervals for 10 min at 37 °C. Complex I activity was defined as: 1 U = 1 µm DCIP reduced per min per µg of protein.
Two microgram of purified mitochondria was incubated in a 96‐well plate with 100 µL of Complex II assay buffer (80 mm KPO4, 1 g·L−1 BSA, 2 mm EDTA, 0.2 mm ATP, 80 µm DCIP, 50 µm DCU, 1 µm Antimycin A, 3 µm rotenone) for 10 min at 37 °C. Ten millimolar sodium succinate and 0.3 mm KCN were added to start the reaction, and absorbance was measured continuously at 600 nm at 30 s intervals for 10 min at 37 °C. Complex II activity was defined as: 1 U = 1 µm DCIP reduced per min per µg of protein.
2.11. Clonogenic assay
Cells grown in DMEM were plated in 6‐well dishes at 1000 cells/well. Colonies were allowed to grow for 10–14 days, with media change every 2 days. After large clones were visible, the colonies were washed with PBS and stained with crystal violet before being counted using ImageJ to determine colony number and size.
2.12. Wound healing scratch assay
Cells were plated at 105 cells per well in a 12‐well dish. Following attachment, the cell monolayer was scratched in a straight line using a 200‐µL pipette tip. Cell monolayers were imaged with a ZOE fluorescent cell imager immediately and after 24 h. Images were analyzed using imagej (NIH, Bethesda, MD, USA) to quantify wound closure.
2.13. NAD/NADH measurement
Cells were harvested, and NAD+/NADH content was measured by using the NAD/NADH‐Glo Assay kit (Promega, Madison, WI, USA), according to instructions.
2.14. GSH measurement
Cells were harvested and GSH content was measured by using the GSH Assay kit (Promega), according to instructions.
2.15. NRF2 luciferase reporter gene assay
MDA‐MB‐231 STAT3 WT and knockout (KO) cells were transfected with a luciferase reporter plasmid programmed by the antioxidant‐responsive element (ARE) sequence from the promoter of the human NAD(P)H quinone oxidoreductase gene, as previously described [48]. In brief, cells were transfected with the ARE‐luciferase plasmid and a TK‐renilla luciferase control plasmid using Lipofectamine 2000. Forty‐eight hours after transfection, both firefly and renilla luciferase activities were measured with the dual luciferase reporter assay kit (Promega), and expression data were calculated from the firefly/renilla luciferase ratios and reported as relative light units (RLU).
2.16. Statistical analysis
Experiments were performed in triplicate, and data were presented as the mean ± SD (n = 3) and analyzed by unpaired Student’s t‐test or one‐way analysis of variance (ANOVA). All data shown are representative of at least three independent biological replicates and were analyzed by using graphpad prism version 8.0 (GraphPad Software, San Diego CA, USA).
3. Results
3.1. Mitochondrial STAT3 positively regulates complex I function and cellular NAD+ concentration
To determine whether STAT3 is critical for mitochondrial functions in triple‐negative breast cancer (TNBC), we created STAT3 KO cell lines by CRISPR/Cas9 gene targeting in the TNBC cell line MDA‐MB‐231 [40] and compared them to wild‐type (WT) counterparts. We also generated STAT3 KO cell lines reconstituted with either WT STAT3 or with artificially mitochondrially restricted forms of STAT3 (MTS), including a version that encoded the phosphorylation‐deficient S727A mutation, previously shown to impair mitochondrial STAT3 functions [31]. STAT3 depletion and reconstitution were validated by western blot (Fig. S1A). STAT3 was expressed in reconstituted lines at ~ 25–50% of endogenous levels, with the WT protein expressed in both cytosolic and mitochondrial fractions, while MTS‐STAT3 accumulated exclusively in mitochondria.
Mitochondria are the major site for ROS production in the cell, and impaired electron transport chain activity generates increased ROS [49]. It has also been shown that loss of STAT3 leads to decreased oxidative phosphorylation and increased mitochondrial ROS (mROS) [37]. Therefore, we checked whether the absence of STAT3 induced oxidative stress in MDA‐MB‐231 cells. Cells lacking STAT3 had almost two times more mROS than WT cells (Fig. 1A). This increased mROS accumulation could be prevented by reconstitution with either WT or MTS‐STAT3, and could be quenched by treatment with the antioxidants NAC and Trolox. Since MTS‐STAT3 expression was sufficient to return mROS levels to WT levels, we concluded that mitochondrial STAT3 functions are critical for the regulation of ROS in TNBC.
Fig. 1.
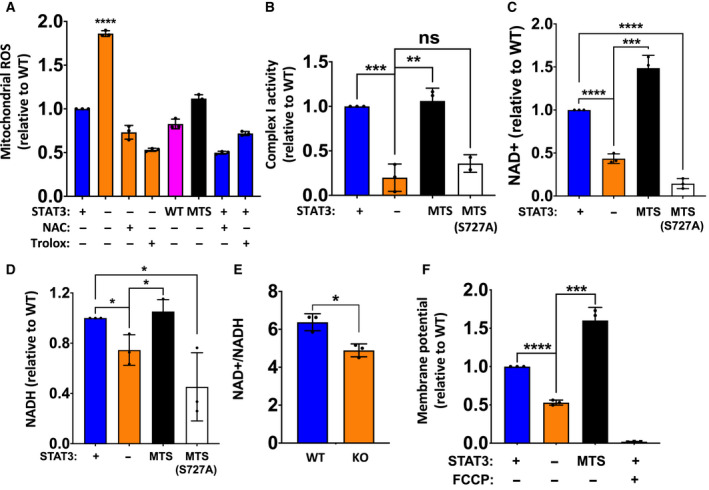
Mitochondrial STAT3 positively regulates complex I function and cellular NAD+ concentration. (A) Mitochondrial ROS measured for STAT3 WT, KO, KO + WT, and KO + MTS MDA‐MB‐231 cells. STAT3 WT and KO cells were treated with antioxidants (25 mm NAC or 250 μm Trolox) for 8 h where indicated before measurement. Only STAT3 KO was significantly different from others (P < 0.001, one‐way ANOVA). (B) Complex I activity for STAT3 WT, KO, KO + MTS (WT), and KO + MTS (S727A) cells. (C) NAD+ and (D) NADH levels in STAT3 WT, KO, KO + MTS (WT), and KO + MTS (S727A) cells. (E) NAD+/NADH ratio in MDA‐MB‐231 STAT3 WT and KO cells. (F) Mitochondrial membrane potential measured for STAT3 WT, KO, and KO + MTS cells using TMRE. Control cells were treated with 50 nm FCCP as indicated for 10 min before measurement. Student's t‐test: ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, ns = not significant, n = 3. Error bars represent ± SD.
Complex I is one of the main contributors to mitochondrial superoxide production [50], and has been shown to have reduced activity in the absence of STAT3 [31, 32]. Hence, we measured complex I activity in MDA‐MB‐231 STAT3 WT and KO cells. Complex I activity was significantly lower in STAT3 KO cells, reduced to ~ 20% the level in WT cells (Fig. 1B); however, there was no significant change in complex II activity in the absence of STAT3 (Fig. S1B). Importantly, reconstitution of STAT3 KO cells with MTS‐STAT3 fully restored complex I activity (Fig. 1B), indicating that enzyme activity was dependent on the mitochondrial pool of STAT3 protein. We also tested STAT3 KO cells reconstituted with the MTS‐STAT3‐S727A mutant, because phosphorylation of this residue has been implicated in mitochondrial STAT3 functions [31]. Reconstitution with MTS‐STAT3‐S727A did not restore complex I function (Fig. 1B), confirming a critical role for this residue and probably its phosphorylation in the regulation of complex I activity by STAT3 in TNBC. Although these results documented positive regulation of complex I activity by mitochondrial STAT3, no differences were observed in complex I protein levels (NDUFS2, NDUFV2, NDUFAF2) between STAT3 WT and KO mitochondria (Fig. S1C). This result suggests that mitochondrial STAT3 enhances complex I enzymatic activity, rather than its synthesis or assembly.
Mitochondrial complex I, through its NADH: quinone oxidoreductase activity, maintains the cellular pool of NAD by oxidizing NADH to NAD+, while coupling the translocation of protons across the mitochondrial membrane [51]. To determine how loss of STAT3 affected cellular dinucleotide pools, we measured NAD+ and NADH levels in STAT3 WT and KO cells. In the absence of STAT3, dinucleotide levels were reduced, with NAD+ levels reduced to ~ 40% of WT levels (Fig. 1C), while NADH levels were less affected (Fig. 1D). Reconstitution of KO cells with MTS‐STAT3 but not with MTS‐STAT3‐S727A restored NAD+ and NADH concentrations to endogenous levels (Fig. 1C,D). These results document a role for mitochondrial STAT3 in the maintenance of NAD abundance in TNBC cells, possibly through modulation of complex I activity.
Increased mROS due to inhibition of electron transport chain complexes leads to mitochondrial membrane depolarization [52]. Indeed, measurement of mitochondrial membrane potential revealed that STAT3 KO cells have about 50% lower membrane potential than WT cells (Fig. 1F). Normal membrane potential was restored in STAT3 KO cells by reconstitution with MTS‐STAT3. Specificity of the membrane potential measurements was confirmed by the reduced potential observed following treatment with the ETC decoupling agent, FCCP (Fig. 1F).
3.2. Mitochondrial STAT3 regulates expression of GSH biosynthetic and antioxidant genes
NAD+ abundance is critical for expression of antioxidant genes, including genes belonging to the GSH biosynthetic pathway [14]. Therefore, we investigated whether the observed dinucleotide reductions in MDA‐MB‐231 STAT3 KO cells impacted the expression of redox genes. To that end, we quantified basal mRNA levels of GCLC (glutamate‐cysteine ligase catalytic subunit), GCLM (glutamate‐cysteine ligase modifier subunit), HMOX1 (heme oxygenase 1), and NQO1 (NAD(P)H quinone dehydrogenase) in STAT3 WT and KO cells.
Given the heightened oxidative stress observed in STAT3 KO cells (Fig. 1), we anticipated an induction of antioxidant gene expression, as increased ROS burden can induce transcriptional activation of genes involved in antioxidant synthesis through activation of NRF2 [53]. In contrast, we observed a significant reduction (50–80%) in the expression of these genes in cells lacking STAT3 (Fig. 2A). We also measured cellular GSH and found that MDA‐MB‐231 STAT3 KO cells had ~ 50% lower amounts of cellular GSH (Fig. S2A), as observed previously in other cancer cells [37]. To confirm the direct involvement of mitochondrial STAT3 in gene regulation, antioxidant gene expression was measured in KO cells reconstituted with MTS‐STAT3. Expression of all four genes was restored to WT levels following expression of MTS‐STAT3 (Fig. 2B). Consistent with the restored mRNA levels, GSH concentration was also restored by reconstitution with MTS‐STAT3 (Fig. S2A). We confirmed the inability of MTS‐STAT3 to restore expression of conventional STAT3 target genes, when reconstituted in STAT3 KO cells. Loss of STAT3 led to a significant decrease in SOCS3 mRNA levels, which were restored by WT‐STAT3 but not by MTS‐STAT3 (Fig. S2B). This result demonstrated that mitochondrial STAT3 impacts the regulation of these nuclear‐encoded genes, in spite of its lack of direct nuclear transcriptional function [54].
Fig. 2.

Mitochondrial STAT3 regulates expression of GSH biosynthetic genes and antioxidant genes. (A) Basal mRNA levels of GSH biosynthesis pathway genes (GCLC, GCLM, NQO1, and HMOX1) in MDA‐MB‐231 STAT3 WT and KO cells. (B) mRNA levels of antioxidant genes in MDA‐MB‐231 STAT3 KO cells after reconstitution with MTS‐STAT3. (C) mRNA levels of antioxidant genes in MDA‐MB‐231 STAT3 WT and KO cells after treatment with 50 µm rotenone or 250 µm Trolox for 8 h. (D) mRNA levels of antioxidant genes in MDA‐MB‐231 cells after reconstitution with NDI1 or treatment with 10 mm nicotinamide (NAM) for 16 h. (E) NAD+/NADH ratio measured in STAT3 WT, KO, KO + NDI1, or KO cells treated with 10 mm NAM for 48 h. Student’s t‐test: ***P < 0.001, **P < 0.01, *P < 0.05, ns = not significant, n = 3. Error bars represent ± SD.
We wanted to understand whether the loss of antioxidant gene expression could be attributed to the reduced complex I activity observed in STAT3 KO cells. To that end, we measured the mRNA levels of these genes in STAT3 WT and KO cells treated with the complex I inhibitor, rotenone. Treatment of cells with 50 µm rotenone for 8 h caused a modest reduction (~ 20–30%) in the expression of these genes (Fig. 2C) that mirrored the reductions observed in the absence of STAT3. We also treated both STAT3 WT and KO cells with the cell‐permeable vitamin E analog and free radical scavenger, Trolox, to determine whether mitigating the increased ROS in STAT3 KO cells could rescue gene expression. Treating cells with 250 µm Trolox for 8 h caused a modest increase in the expression of GCLC in both STAT3 WT and KO cells, but it did not restore its level in KO cells to those seen in WT cells (Fig. 2A). Moreover, Trolox treatment had no impact on GCLM or NQO1 in either WT or KO cells (Fig. 2C).
Since ROS levels did not appear to be responsible for changes in antioxidant gene expression, we tested whether reduced levels of NAD+ were responsible for impaired gene expression. To check this, we augmented NADH dehydrogenase activity in STAT3 KO cells by ectopically expressing yeast NDI1. Yeast NDI1 is a rotenone‐resistant and STAT3‐independent NADH dehydrogenase that is capable of bypassing defects in complex I function when expressed in mammalian cells [40, 55, 56]. Interestingly, NDI1 expression restored antioxidant gene expression in STAT3 KO cells to WT levels (Fig. 2D). We also treated STAT3 KO cells with nicotinamide (NAM), a precursor capable of boosting cellular NAD+ levels [57]. Treatment of STAT3 KO cells with 10 mm NAM restored expression of antioxidant genes to WT levels (Fig. 2D). Treatment with NAM or expression of NDI1 in KO cells also normalized NAD+/NADH ratios (Fig. 2E).
3.3. STAT3‐dependent antioxidant gene expression is not mediated by changes in NRF2 function
Since the antioxidant genes observed to have lower expression in the absence of STAT3 are regulated by NRF2 during oxidative stress, we determined the status of the NRF2 pathway in cells lacking STAT3. First, we examined antioxidant gene expression in the non‐small‐cell lung carcinoma cell line, A549, which displays constitutive NRF2 activity due to a null mutation in the negative regulator, KEAP1 [58]. Similar to MDA‐MB‐431 cells, A549 cells expressed reduced levels of antioxidant genes GCLC and HO1 when the STAT3 gene was disrupted (Fig. 3A), in spite of the presence of constitutively active NRF2.
Fig. 3.
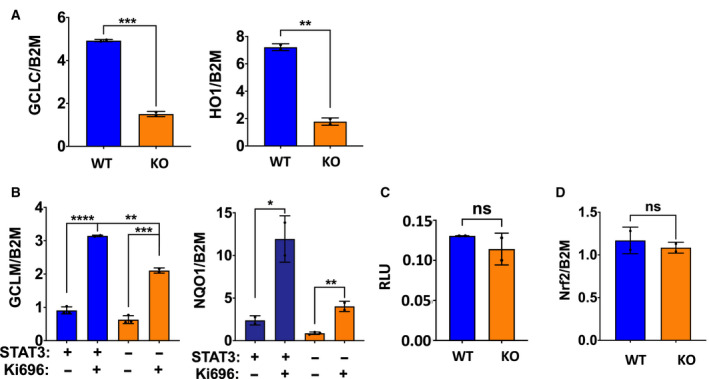
STAT3‐dependent antioxidant gene expression is not mediated by changes in NRF2 function. (A) Basal mRNA expression of antioxidant genes in NSCLC cell line A549 STAT3 WT and KO. (B) mRNA expression of antioxidant genes in MDA‐MB‐231 STAT3 WT and KO cells treated with 1 μm of the NRF2 activator, KI696 for 48 h. (C) Luminescence assay in MDA‐MB‐231 STAT3 WT and KO cells using a reporter plasmid responsive to NRF2. Luciferase data are expressed as RLU. (D) Basal mRNA levels of NRF2 in MDA‐MB‐231 STAT3 WT and KO cells. Student’s t‐test: ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, ns = not significant, n = 3. Error bars represent ± SD.
We also confirmed that the NRF2 pathway is not rendered defective by the absence of STAT3 by treating cells with a small molecule activator of NRF2, KI696 [59]. KI696 treatment induced robust expression of antioxidant genes in both WT and KO cells (Fig. 3B), consistent with the NRF2 pathway remaining functional in these cells regardless of the status of STAT3. However, while fold induction of GCLM and NQO1 in response to KI696 treatment was equivalent between WT and KO cells, their absolute levels were lower in KO cells reflecting impaired basal gene expression in the absence of STAT3.
We also measured the basal activity of NRF2 using a reporter assay. MDA‐MB‐231 STAT3 WT and KO cells were transfected with a luciferase reporter driven by NRF2‐responsive promoter elements [48]. Both WT and KO cells transfected with this reporter expressed similar levels of luciferase (Fig. 3C), demonstrating that basal NRF2 transcriptional activity was equivalent in the two cell lines. Finally, measurement of NRF2 mRNA levels in STAT3 WT and KO cells demonstrated no differences in expression between the two cell lines (Fig. 3D). Overall, these results exclude an impairment in the NRF2 pathway as a primary cause for reduced basal antioxidant gene expression in cells lacking STAT3 and indicate a requirement for STAT3 in addition to NRF2.
3.4. Mitochondrial STAT3 regulates clonogenic potential and sensitivity to oxidative stress
To explore the biological consequences of mitochondrial STAT3 function, we examined its requirement for cell growth and survival. To examine clonogenic potential, we plated WT and KO cells at limiting dilution and measured colony formation after 10 days. STAT3 KO cells showed reduced clonogenic potential, resulting in the growth of 50% fewer colonies (Fig. 4A). Importantly, WT levels of clonogenic growth were restored by expression of MTS‐STAT3. Moreover, the diminished clonal growth of STAT3 KO cells was also reverted by culturing the cells in the presence of NAM. Nonetheless, there were no significance differences in the proliferation rates of STAT3 WT and KO cells when cultured under standard growth conditions (Fig. S2C). We also found that STAT3 KO cells undergo 50% more cell death following oxidative stress, by exposing cells to H2O2 (Fig. 4B). Cell viability could be restored by countering the oxidative stress with antioxidants (25 mm NAC or 250 µm Trolox; Fig. 4B), consistent with the enhanced sensitivity to stress being due to the altered redox balance in STAT3 KO cells. We also treated STAT3 KO cells with NAM prior to exposure to the oxidative stress. Consistent with expectations, NAM pretreatment also protected STAT3 KO cells from peroxide‐induced cell death (Fig. 4B). Finally, we examined whether loss of STAT3 reduced cellular migration by performing a wound healing assay [60]. STAT3 KO cells migrated twofold less that WT cells, when migration potential was measured after 16 h (Fig. 4C). Similar to the role of mitochondrial STAT3 and NAD in clonal growth, the reduced migration of STAT3 KO cells was restored either by expression of MTS‐STAT3 or by supplementation with NAM. Together, these results show that mitochondrial STAT3, through its ability to maintain normal pools of NAD+, regulates clonal growth, oxidant resistance, and cell survival and migration.
Fig. 4.
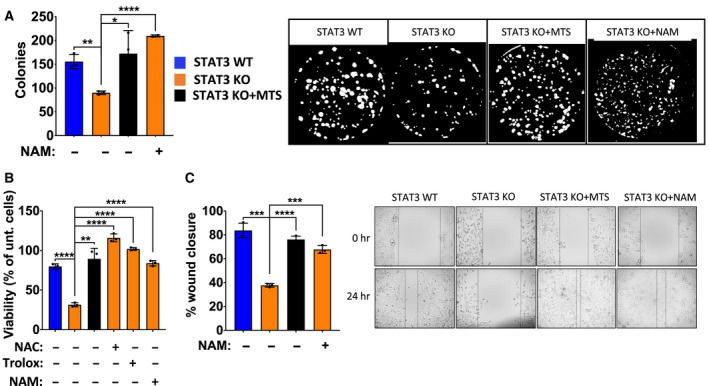
Mitochondrial STAT3 regulates clonogenic potential and sensitivity to oxidative stress. (A) Number of colonies formed by MDA‐MB‐231 STAT3 WT, KO, KO + MTS cells, and KO cells treated with 10 mm NAM for 10 d. Representative image from ImageJ is shown. (B) Cell viability measured by crystal violet of MDA‐MB‐231 STAT3 WT, KO, and KO + MTS cells treated with 0.375 mm H2O2 or 0.375 mm H2O2 + antioxidants (25 mm NAC, 250 μm Trolox, or 10 mm NAM) for 8 h. (C) Wound healing scratch assay measured for MDA‐MB‐231 STAT3 WT, KO, KO + MTS cells, and KO cells treated with 10 mm NAM for 24 h. Student’s t‐test: ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, ns = not significant, n = 3. Error bars represent ± SD.
3.5. Mitochondrial STAT3 interacts with complex I components
To explore how mitochondrial STAT3 could regulate complex I activity in TNBC, we examined protein partners of MTS‐STAT3. To this end, we fused MTS‐STAT3 to the BioID2 protein to create a proximity‐labeling probe, STAT3‐BioID (SB) (Fig. 5A), and expressed it in MDA‐MB‐231 cells. The fusion protein was confirmed to localize to mitochondria (Fig. 5B), and biotinylation of target proteins was mainly observed in the mitochondrial fraction (Fig. 5C). Total protein biotinylation increased in response to treatment time with biotin (Fig. 5C), and biotinylated proteins could be quantitatively recovered from mitochondrial extracts by streptavidin pull‐down (Fig. 5C).
Fig. 5.

Mitochondrial STAT3 interacts with complex I components. (A) Cartoon of the STAT3‐BioID construct (SB). (B) Western blot showing cytosolic and mitochondrial fractions from MDA‐MB‐231 WT cells expressing SB. The SB construct is larger in size than the endogenous protein (indicated in the figure) and is mainly located in the mitochondrial fraction. (C) Streptavidin‐HRP blot for cells expressing SB and treated with 50 μm biotin for 18 h showing that biotinylated proteins are mainly restricted to the mitochondrial fraction (left). Time course of biotin treatment showing time‐dependent increase in biotinylated proteins (middle). Pull‐down with streptavidin sepharose from mitochondrial lysates expressing SB and treated with 50 μm biotin for 18 h showing near quantitative recovery of biotinylated proteins (right). (D) Table showing list of complex I proteins biotinylated in the presence of SB. (E) Streptavidin sepharose was used to pull‐down biotinylated proteins from mitochondrial lysates of MDA‐MB‐231 STAT3 WT cells expressing SB treated with 50 μm biotin for 18 h, and complex I components (NDUFV2, NDUFS2, NDUFAF2) were identified by immunoblotting. (F) Left: Immunoprecipitation of endogenous STAT3 verifies interaction with complex I components (NDUFV2, NDUFS2, NDUFAF2). Right: Pull‐down with IgG as control. Mw are expressed in KDa.
To evaluate proteins in the vicinity of STAT3, SB‐expressing cells were treated with biotin, and the biotinylated proteins recovered by streptavidin pull‐down were identified by MS (Data S1). We used the Enrichr and Molecular Signatures enrichment analysis tools [46, 47, 61] to identify pathways defined by the proteins preferentially biotinylated by mitochondrial STAT3. Proteins associated with oxidative phosphorylation were detected, due in part to enrichment of a number of complex I subunit components. Of the 17 protein subunits of the oxidoreductase (N/Q) module of complex I involved in NADH oxidation and ubiquinone reduction [62], 11 were present in the biotinylated fraction (Fig. 5D).
To confirm these interactions, biotinylated proteins from mitochondrial lysates of cells treated with 50 µm biotin for 18 h were recovered on streptavidin beads and identified by western blotting. NDUFS2, NDUFV2, and NDUFAF2, three constituents or assembly factors of complex I identified by mass spectrometry, were independently confirmed to be proximity labeled by STAT3‐BioID (Fig. 5E). Interestingly, endogenous STAT3 was detected in the biotinylated fraction, indicating that the SB fusion protein interacted with endogenous STAT3, possibly due to dimerization of mitochondrial STAT3, as previously suggested [35]. In contrast, the complex II component SDHA was not recovered in the pull‐down fraction (Fig. 5E). As control, we used mitochondrial lysates from cells expressing a MTS‐BioID construct lacking STAT3. Mitochondrial lysates from these cells were biotinylated (Fig. S3, right panel), but pull‐down using streptavidin beads did not recover any complex I proteins or STAT3 (Fig. S3, left panel). To confirm these interactions with endogenous STAT3, mitochondrial lysates from MDA‐MB‐231 cells were immunoprecipitated using an antibody against STAT3. NDUFS2, NDUFV2, and NDUFAF2 were all co‐immunoprecipitated with STAT3, while SDHA was not (Fig. 5F). None of these proteins were recovered in IgG control immunoprecipitation experiments (Fig. 5F, right panel). Mitochondrial STAT3 interacted most strongly with NDUFV2, whereas the interaction with NDUFS2 and the complex I assembly factor NDUFAF2 appeared weaker (Fig. 5F). Notably, although STAT3 was quantitatively recovered by immunoprecipitation, a significant fraction of these endogenous complex I proteins were left in the supernatant. Overall, these results indicate that mitochondrial STAT3 interacts nonstoichiometrically with a portion of complex I. Interestingly, the complex I components identified as STAT3 interactors are associated with the dehydrogenase module, consistent with altered dehydrogenase activity in the absence of STAT3.
4. Discussion
It has become increasingly clear that cancer cells display increased ROS production and that elevated ROS contributes to malignancy by augmenting many of the characteristics of cancer cell behavior [1, 2]. However, ROS are also harmful, so an increased redox potential is essential for cancer cell survival [63]. One of the major cellular sources of ROS is mitochondrial metabolism, due to ROS generation through the activity of the respiratory chain, largely due to complexes I and III [49]. Several studies have demonstrated increased ROS in tumor cells following loss of STAT3, and this effect has been shown to be mediated, at least in part, by mitochondrial STAT3 [64], which regulates the activity of complex I [31, 32]. Inefficient complex I function due to the absence of STAT3 can therefore lead to increased mitochondrial ROS, and mitochondrial STAT3 has been proposed to act as a redox sensor [65, 66]. Another source of increased ROS in the absence of STAT3 are decreased GSH levels, due to reduced ROS buffering capacity [37]. However, the basis for the decreased GSH observed in the absence of STAT3 has remained unknown.
In this study, we found that mitochondrial STAT3 modulates GSH levels at least in part by regulating the expression of genes involved in GSH synthesis. However, STAT3 did not act directly as a transcription factor, since mitochondrial STAT3 was sufficient to maintain expression of these nuclear genes. Instead, regulation of gene expression must depend on a retrograde signal that facilitates communication between mitochondria and nuclei [39], a process that promotes adaptation to metabolic flux. Retrograde signaling can be direct, in which a mitochondrially localized transcription factor is sent to the nucleus under appropriate circumstances [67]. Retrograde signaling can also occur through a less direct route, in which a mitochondrial metabolite influences cellular responses, including changes in nuclear gene expression [68, 69]. Our data suggest that NAD+ serves this function to regulate GSH synthesis. STAT3 controlled the abundance of cellular NAD+ through maintenance of complex I dehydrogenase activity. Absence of STAT3 led to reduced complex I activity, resulting in impaired regeneration of NAD+ from NADH oxidation, leading to a reduced overall NAD+/NADH ratio. Importantly, normal NAD+/NADH ratios could be restored by expression of mitochondrially localized STAT3, increased NAD synthesis due to nutritional supplementation with the NAD precursor NAM, or by complementing impaired complex I function through expression of the STAT3‐independent NADH dehydrogenase NDI1 from yeast. All of these approaches to restoring a WT NAD+/NADH ratio in STAT3‐null cells led to normalized antioxidant gene expression.
NAD+ has been shown previously to modulate nuclear gene expression through a number of mechanisms. The sirtuin (SIRT) family of histone deacetylases require NAD+ as a cofactor for enzymatic activity, thereby linking epigenetic regulation to the abundance of NAD+ [13, 70]. Similarly, C‐terminal binding protein (CtBP) is a NAD+‐regulated transcriptional corepressor [71] that links transcriptional activity to the cellular NAD+/NADH ratio. NAD has also been shown to regulate DNA methylation, with lower levels of NAD correlating with increased methylation and therefore reduced gene transcription [72]. SIRT can also regulate gene expression by deacetylating transcription factors rather than histones, such as FoxO3 and peroxisome proliferator‐activated receptor‐gamma co‐activator‐1alpha (PGC1α) [73]. Sirt3 has been shown to reduce oxidative damage through IDH2 [21]. Sirt3 increases IDH2 activity through deacetylation, thereby increasing mitochondrial NADPH levels and GSSG/GSH ratio. However, the mitochondrial STAT3‐dependent expression of GCLC was minimally affected by inhibition of SIRT1 or SIRT3 (Fig. S4A), although inhibition of these enzymes decreased the expression of a typical SIRT target gene, PGC1α (Fig. S4B) [74, 75, 76].
Our data suggest that the NAD+/NADH ratio modulated by mitochondrial STAT3 through changes in complex I activity regulates antioxidant gene expression. Indeed, NAD‐dependent regulation of antioxidant gene expression has been previously reported [13, 77, 78], and was ascribed to changes in the abundance of the NRF2 transcription factor. However, we did not observe an impairment of NRF2 levels nor changes in NRF2 transcriptional activity in STAT3 KO cells (Fig. 3B,C), suggesting that STAT3 acts on other components of this pathway and not by directly modulating NRF2.
Mitochondrial STAT3 regulates the electron transport chain, including complex I activity [31, 32], but the mechanistic basis for this regulation remains unclear. Our results show that complex I activity and antioxidant gene expression depend on mitochondrial STAT3 Ser727 phosphorylation, previously shown to be critical for supporting malignant transformation [31, 79]. Moreover, our proximity‐labeling studies revealed that mitochondrial STAT3 associates with three subunits of the NADH dehydrogenase arm of complex I, and this interaction was confirmed by examining endogenous proteins. Importantly, we did not observe any changes in the abundance of complex I subunits between STAT3 WT and KO cells, suggesting that STAT3 influences complex I function and not assembly in these cells. Given the low abundance of STAT3 relative to complex I, this interaction is not stoichiometrically equivalent [80], and the majority of complex I subunit proteins were not associated with STAT3. It is possible that STAT3 associates with a subset of complex I that is critical for dehydrogenase activity. For instance, it has been suggested that STAT3 associates with respiratory supercomplexes that are important for reducing electron leak and thereby limiting ROS generation [81].
TNBC is the most aggressive form of breast cancer, lacking the expression of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2. STAT3 has been shown to play an important pro‐oncogenic role in TNBC [82] and to be a potential therapeutic target [83], with studies indicating the presence of hyperactivated STAT3, driven by the phosphorylation of tyrosine 705. In addition, S727‐phosphorylated mitochondrial STAT3 has also been implicated in breast cancer [84].
We observed a significant requirement for mitochondrial STAT3 in the maintenance of clonogenecity and wound healing in MDA‐MB‐231, confirming that the mitochondrial fraction of STAT3 plays an important role in driving tumorigenecity in TNBC. Importantly, the requirement for mitochondrial STAT3 in these processes could be circumvented by modulation of NAD+ levels through supplementation with NAM or restoration of dehydrogenase activity. These results could have therapeutic implications, as it has been shown that targeting NAD+ metabolism can sensitize tumor cells to drug and radiation therapy [85, 86], adding another potential rationale for targeting mitochondrial STAT3 during cancer therapy [40].
5. Conclusions
Overall, our study presents a novel role for mitochondrial STAT3 in regulation of cellular redox balance through modulation of antioxidant gene expression as a consequence of complex I activity. Reduced complex I activity in the absence of mitochondrial STAT3 impaired regeneration of NAD+. Perturbed NAD+/NADH ratios failed to maintain the basal expression of antioxidant genes, leading to a diminished cellular reducing capacity, a more oxidized cellular environment, increased vulnerability to oxidative stress, and reduced cellular attributes of malignancy. These results implicate NAD+ as a retrograde signal relaying the status of mitochondrial metabolism to the nucleus, and they highlight the potential of mitochondrial STAT3 as a target for cancer chemotherapy.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
TL and DEL conceived and designed the project. TL, JA, and BU acquired the data. TL, JA, BU, MA, LB, and DEL analyzed and interpreted the data; and TL, LB, and DEL wrote the paper.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/1878‐0261.12928.
Supporting information
Fig. S1. Characterization of mutant and reconstituted cell lines. (A) Western blot to confirm MDA‐MB‐231 STAT3 KO and reconstitution of KO cells with WT, MTS (WT), or MTS (S727A) STAT3 (left panel: cytosolic fraction; right panel: mitochondrial fraction). Mitochondrial STAT3 levels are normalized to SDHA. (B) Complex II activity for STAT3 WT, KO, KO + MTS (WT), and KO + MTS (S727A) cells. (C) Western blot to confirm expression levels of complex I subunits in MDA‐MB‐231 STAT3 WT and KO cells. Student’s t‐test: ****= P < 0.0001, ***= P < 0.001, **= P < 0.01, *= P < 0.05, ns = not significant, n = 3. MW are expressed in KDa. Error bars represent ± SD.
Fig. S2. (A) Glutathione levels in WT and mutant cell lines. GSH levels measured in STAT3 WT, KO and KO + MTS cells. (B) Growth of cells is unaffected by STAT3. Growth curve for MDA‐MB‐231 STAT3 WT and KO cells over 4 d measured using crystal violet. (C) Induction of SOCS3 gene expression depends on nuclear STAT3. mRNA expression of nuclear STAT3 target gene SOCS3 in MDA‐MB‐231 STAT3 WT, KO, KO + WT, and KO + MTS cells. Student’s t‐test: ****= P < 0.0001, ***= P < 0.001, **= P < 0.01, *= P < 0.05, ns = not significant, n = 3. Error bars represent ± SD.
Fig. S3. Antioxidant gene expression does not require SIRT1 or SIRT3 activity. (A) mRNA expression of GCLC in MDA‐MB‐231 STAT3 WT or KO cells treated with 200 μm EX‐527 (SIRT1 inhibitor) or 200 μm 3‐TYP (SIRT3 inhibitor) for 24 h. (B) mRNA expression of PGC1α in MDA‐MB‐231 STAT3 WT cells treated with 200 μm EX‐527 or 200 μm 3‐TYP for 24 h. Student’s t‐test: ****= P < 0.0001, ***= P < 0.001, **= P < 0.01, *= P < 0.05, ns = not significant, n = 3. Error bars represent ± SD.
Fig. S4. Specificity control for STAT3‐BioID pull‐down. Streptavidin sepharose was used to pull‐down biotinylated proteins from mitochondrial lysates of MDA‐MB‐231 STAT3 WT cells expressing MTS‐BioID treated with 50 μm biotin for 18 h, and complex I components (NDUFV2, NDUFS2, NDUFAF2) were identified by immunoblotting (left). Pull‐down with streptavidin sepharose from mitochondrial lysates expressing MTS‐BioID and treated with 50 μm biotin for 18 h showing near quantitative recovery of biotinylated proteins (right).
Table S1. Primer sequences for RNA quantification by qRT‐PCR.
Data S1. MS/MS data for peptides copurifying with STAT3‐BioID.
Acknowledgements
We would like to thank Drs. Navdeep S. Chandel (Northwestern University), Gregory David, Thales Papagiannakopoulos, Sarah LeBoeuf, Richard Possemato, Isabelle Marié, Debapriya Basu, and Harold K. Elias (NYU School of Medicine) for reagents, helpful discussions, and valuable scientific input. This work was supported by the National Institutes of Health (R01 AI28900 to DEL). Work in the Proteomics Laboratory is subsidized in part by NYU Langone Health and by the Laura and Isaac Perlmutter Comprehensive Cancer Center support grant P30CA016087 from the National Cancer Institute.
References
- 1. Liou GY & Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44, 479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weinberg F & Chandel NS (2009) Reactive oxygen species‐dependent signaling regulates cancer. Cell Mol Life Sci 66, 3663–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES et al. (2011) Oncogene‐induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bansal A & Simon MC (2018) Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol 217, 2291–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jozkowicz A, Was H & Dulak J (2007) Heme oxygenase‐1 in tumors: is it a false friend? Antioxid Redox Signal 9, 2099–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ross D & Siegel D (2017) Functions of NQO1 in cellular protection and CoQ10 metabolism and its potential role as a redox sensitive molecular switch. Front Physiol 8, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sabharwal SS & Schumacker PT (2014) Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat Rev Cancer 14, 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Santidrian AF, Matsuno‐Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, Yagi T & Felding‐Habermann B (2013) Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest 123, 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kudin AP, Bimpong‐Buta NY, Vielhaber S, Elger CE & Kunz WS (2004) Characterization of superoxide‐producing sites in isolated brain mitochondria. J Biol Chem 279, 4127–4135. [DOI] [PubMed] [Google Scholar]
- 10. Kushnareva Y, Murphy AN & Andreyev A (2002) Complex I‐mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation‐reduction state. Biochem J 368, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kussmaul L & Hirst J (2006) The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci USA 103, 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Houtkooper RH, Canto C, Wanders RJ & Auwerx J (2010) The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 31, 194–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang T & Kraus WL (2010) SIRT1‐dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim Biophys Acta 1804, 1666–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J, Hong Y, Cao W, Yin S, Sh H & Ying W (2019) SIRT2, ERK and Nrf2 Mediate NAD+ treatment‐induced increase in the antioxidant capacity of PC12 cells under basal conditions. Front Mol Neurosci 12, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R et al. (2016) NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. [DOI] [PubMed] [Google Scholar]
- 16. Schwer B, North BJ, Frye RA, Ott M & Verdin E (2002) The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide‐dependent deacetylase. J Cell Biol 158, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX & Finkel T (2008) A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA 105, 14447–14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cimen H, Han MJ, Yang Y, Tong Q, Koc H & Koc EC (2010) Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 49, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Finley LW, Haas W, Desquiret‐Dumas V, Wallace DC, Procaccio V, Gygi SP & Haigis MC (2011) Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS One 6, e23295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shimazu T, Hirschey MD, Hua L, Dittenhafer‐Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt FW, Denu JM et al. (2010) SIRT3 deacetylates mitochondrial 3‐hydroxy‐3‐methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab 12, 654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM & Prolla TA (2010) Sirt3 mediates reduction of oxidative damage and prevention of age‐related hearing loss under caloric restriction. Cell 143, 802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W et al. (2010) Sirt3‐mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 40, 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang Y, Cimen H, Han MJ, Shi T, Deng JH, Koc H, Palacios OM, Montier L, Bai Y, Tong Q et al. (2010) NAD+‐dependent deacetylase SIRT3 regulates mitochondrial protein synthesis by deacetylation of the ribosomal protein MRPL10. J Biol Chem 285, 7417–7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qiu X, Brown K, Hirschey MD, Verdin E & Chen D (2010) Calorie restriction reduces oxidative stress by SIRT3‐mediated SOD2 activation. Cell Metab 12, 662–667. [DOI] [PubMed] [Google Scholar]
- 25. Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu Y, Guo X, Ji L, Huang Q, Zhang H et al. (2017) MCU‐dependent mitochondrial Ca(2+) inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 36, 5897–5909. [DOI] [PubMed] [Google Scholar]
- 26. Levy DE & Lee CK (2002) What does Stat3 do? J Clin Invest 109, 1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C & Darnell JE Jr (1999) Stat3 as an oncogene. Cell 98, 295–303. [DOI] [PubMed] [Google Scholar]
- 28. Yang J, Chatterjee‐Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE & Stark GR (2005) Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res 65, 939–947. [PubMed] [Google Scholar]
- 29. Liu C‐Y, Tseng L‐M, Su J‐C, Chang K‐C, Chu P‐Y, Tai W‐T, Shiau C‐W & Chen K‐F (2013) Novel sorafenib analogues induce apoptosis through SHP‐1 dependent STAT3 inactivation in human breast cancer cells. Breast Cancer Res 15, R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain‐Qimron N, Kim JJ, Choudhury SA, Maruyama R et al. (2011) The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(‐) stem cell‐like breast cancer cells in human tumors. J Clin Invest 121, 2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC & Levy DE (2009) Mitochondrial STAT3 supports Ras‐dependent oncogenic transformation. Science 324, 1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A et al. (2009) Function of mitochondrial Stat3 in cellular respiration. Science 323, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carbognin E, Betto RM, Soriano ME, Smith AG & Martello G (2016) Stat3 promotes mitochondrial transcription and oxidative respiration during maintenance and induction of naive pluripotency. EMBO J 35, 618–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guanizo AC, Fernando CD, Garama DJ & Gough DJ (2018) STAT3: a multifaceted oncoprotein. Growth Factors 36, 1–14. [DOI] [PubMed] [Google Scholar]
- 35. Macias E, Rao D, Carbajal S, Kiguchi K & DiGiovanni J (2014) Stat3 binds to mtDNA and regulates mitochondrial gene expression in keratinocytes. J Invest Dermatol 134, 1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meier JA & Larner AC (2014) Toward a new STATe: the role of STATs in mitochondrial function. Semin Immunol 26, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garama DJ, Harris TJ, White CL, Rosello FJ, Abdul‐Hay M, Gough DJ & Levy DE (2015) A synthetic lethal interaction between glutathione synthesis and mitochondrial reactive oxygen species provides a tumor specific vulnerability dependent on STAT3. Mol Cell Biol 35, 3646–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carballo M, Conde M, El Bekay R, Martin‐Nieto J, Camacho MJ, Monteseirin J, Conde J, Bedoya FJ & Sobrino F (1999) Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J Biol Chem 274, 17580–17586. [DOI] [PubMed] [Google Scholar]
- 39. Liu Z & Butow RA (2006) Mitochondrial retrograde signaling. Annu Rev Genet 40, 159–185. [DOI] [PubMed] [Google Scholar]
- 40. Brambilla L, Lahiri T, Cammer M & Levy DE (2020) STAT3 inhibitor OPB‐51602 is cytotoxic to tumor cells through inhibition of complex I and ROS induction. iScience 23, 101822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim DI & Roux KJ (2016) Filling the void: proximity‐based labeling of proteins in living cells. Trends Cell Biol 26, 804–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mita P, Wudzinska A, Sun X, Andrade J, Nayak S, Kahler DJ, Badri S, LaCava J, Ueberheide B, Yun CY et al. (2018) LINE‐1 protein localization and functional dynamics during the cell cycle. Elife 7, e30058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peled M, Tocheva AS, Sandigursky S, Nayak S, Philips EA, Nichols KE, Strazza M, Azoulay‐Alfaguter I, Askenazi M, Neel BG et al. (2018) Affinity purification mass spectrometry analysis of PD‐1 uncovers SAP as a new checkpoint inhibitor. Proc Natl Acad Sci USA 115, E468–E477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St‐Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY et al. (2013) The CRAPome: a contaminant repository for affinity purification‐mass spectrometry data. Nat Methods 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK et al. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A et al. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP & Tamayo P (2015) The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Du Y, Villeneuve NF, Wang XJ, Sun Z, Chen W, Li J, Lou H, Wong PK & Zhang DD (2008) Oridonin confers protection against arsenic‐induced toxicity through activation of the Nrf2‐mediated defensive response. Environ Health Perspect 116, 1154–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Indo HP, Davidson M, Yen H‐C, Suenaga S, Tomita K, Nishii T, Higuchi M, Koga Y, Ozawa T & Majima HJ (2007) Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 7, 106–118. [DOI] [PubMed] [Google Scholar]
- 50. Hirst J, Kin MS & Pryde KR (2008) The production of reactive oxygen species by complex I. Biochem Soc Trans 36, 976–980. [DOI] [PubMed] [Google Scholar]
- 51. Karamanlidis G, Lee CF, Garcia‐Menendez L, Kolwicz SC, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W & Tian R (2013) Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab 18, 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gyulkhandanyan AV, Feeneyà CJ & Pennefather PS (2003) Modulation of mitochondrial membrane potential and reactive oxygen species production by copper in astrocytes. J Neurochem 87, 448–460. [DOI] [PubMed] [Google Scholar]
- 53. Vomund S, Schafer A, Parnham MJ, Brune B & von Knethen A (2017) Nrf2, the master regulator of anti‐oxidative responses. Int J Mol Sci 18, 2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, Cichy J, Kukreja RC, Dulak J, Lesnefsky EJ et al. (2011) Mitochondrial‐targeted Signal Transducer and Activator of Transcription 3 (STAT3) Protects against ischemia‐induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem 286, 29610–29620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cho J, Hur JH, Graniel J, Benzer S & Walker DW (2012) Expression of yeast NDI1 rescues a drosophila complex I assembly defect. PLoS One 7, e50644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cronin‐Furman EN, Barber‐Singh J, Bergquist KE, Yagi T & Trimmer PA (2019) Differential effects of yeast NADH Dehydrogenase (Ndi1) expression on mitochondrial function and inclusion formation in a cell culture model of sporadic Parkinson’s disease. Biomolecules 9, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schöndorf DC, Ivanyuk D, Baden P, Sanchez‐Martinez A, Cicco SD, Yu C, Giunta I, Schwarz LK, Napoli GD, Panagiotakopoulou V et al. (2018) The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson’s disease. Cell Rep 23, 2976–2988. [DOI] [PubMed] [Google Scholar]
- 58. Taguchi K & Yamamoto M (2017) The KEAP1‐NRF2 system in cancer. Front Oncol 7, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS, Alvarez SW, Wu WL, Karakousi TR, Zavitsanou AM et al. (2017) Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 6, e28083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Snyder M, Huang XY & Zhang JJ (2011) Signal transducers and activators of transcription 3 (STAT3) directly regulates cytokine‐induced fascin expression and is required for breast cancer cell migration. J Biol Chem 286, 38886–38893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR & Ma’ayan A (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wirth C, Brandt U, Hunte C & Zickermann V (2016) Structure and function of mitochondrial complex I. Biochim Biophys Acta 1857, 902–914. [DOI] [PubMed] [Google Scholar]
- 63. Reczek CR & Chandel NS (2017) The two faces of reactive oxygen species in cancer. Annu Rev Cancer Biol 1, 79–98. [Google Scholar]
- 64. Meier JA, Hyun M, Cantwell M, Raza A, Mertens C, Raje V, Sisler J, Tracy E, Torres‐Odio S, Gispert S et al. (2017) Stress‐induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci Signal 10, eaag2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cheng X, Peuckert C & Wolfl S (2017) Essential role of mitochondrial Stat3 in p38(MAPK) mediated apoptosis under oxidative stress. Sci Rep 7, 15388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sarafian TA, Montes C, Imura T, Qi J, Coppola G, Geschwind DH & Sofroniew MV (2010) Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro . PLoS One 5, e9532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Qureshi MA, Haynes CM & Pellegrino MW (2017) The mitochondrial unfolded protein response: signaling from the powerhouse. J Biol Chem 292, 13500–13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bohovych I & Khalimonchuk O (2016) Sending out an SOS: mitochondria as a signaling hub. Front Cell Dev Biol 4, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. van der Knaap JA & Verrijzer CP (2016) Undercover: gene control by metabolites and metabolic enzymes. Genes Dev 30, 2345–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Houtkooper RH, Pirinen E & Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang Q, Piston DW & Goodman RH (2002) Regulation of corepressor function by nuclear NADH. Science 295, 1895–1897. [DOI] [PubMed] [Google Scholar]
- 72. Chang J, Zhang B, Heath H, Galjart N, Wang X & Milbrandt J (2010) Nicotinamide adenine dinucleotide (NAD)‐regulated DNA methylation alters CCCTC‐binding factor (CTCF)/cohesin binding and transcription at the BDNF locus. Proc Natl Acad Sci USA 107, 21836–21841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Daitoku H, Sakamaki J & Fukamizu A (2011) Regulation of FoxO transcription factors by acetylation and protein‐protein interactions. Biochim Biophys Acta 1813, 1954–1960. [DOI] [PubMed] [Google Scholar]
- 74. Amat R, Planavila A, Chen SL, Iglesias R, Giralt M & Villarroya F (2009) SIRT1 controls the transcription of the peroxisome proliferator‐activated receptor‐gamma Co‐activator‐1alpha (PGC‐1alpha) gene in skeletal muscle through the PGC‐1alpha autoregulatory loop and interaction with MyoD. J Biol Chem 284, 21872–21880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, Fang F & Chang Y (2010) Sirtuin 3, a new target of PGC‐1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 5, e11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shi T, Wang F, Stieren E & Tong Q (2005) SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem 280, 13560–13567. [DOI] [PubMed] [Google Scholar]
- 77. Aquilano K, Baldelli S, Pagliei B, Cannata SM, Rotilio G & Ciriolo MR (2013) p53 orchestrates the PGC‐1alpha‐mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid Redox Signal 18, 386–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Olmos Y, Sanchez‐Gomez FJ, Wild B, Garcia‐Quintans N, Cabezudo S, Lamas S & Monsalve M (2013) SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC‐1alpha complex. Antioxid Redox Signal 19, 1507–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gough DJ, Koetz L & Levy DE (2013) The MEK‐ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras‐mediated transformation. PLoS One 8, e83395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Phillips D, Reilley MJ, Aponte AM, Wang G, Boja E, Gucek M & Balaban RS (2010) Stoichiometry of STAT3 and mitochondrial proteins: Implications for the regulation of oxidative phosphorylation by protein‐protein interactions. J Biol Chem 285, 23532–23536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rincon M & Pereira FV (2018) A new perspective: mitochondrial Stat3 as a regulator for lymphocyte function. Int J Mol Sci 19, 1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Qin J‐J, Yan L, Zhang J & Zhang W‐D (2019) STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review. J Exp Clin Cancer Res 38, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lou C, Chen Y, Zhang J, Yang B & Zhao H (2019) Eupalinolide J suppresses the growth of triple‐negative breast cancer cells via targeting STAT3 signaling pathway. Front Pharmacol 10, 1071. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 84. Zhang Q, Raje V, Yakovlev VA, Yacoub A, Szczepanek K, Meier J, Derecka M, Chen Q, Hu Y, Sisler J et al. (2013) Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J Biol Chem 288, 31280–31288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gujar AD, Le S, Mao DD, Dadey DY, Turski A, Sasaki Y, Aum D, Luo J, Dahiya S, Yuan L et al. (2016) An NAD+‐dependent transcriptional program governs self‐renewal and radiation resistance in glioblastoma. Proc Natl Acad Sci USA 113, E8247–E8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lewis JE, Singh N, Holmila RJ, Sumer BD, Williams NS, Furdui CM, Kemp ML & Boothman DA (2019) Targeting NAD(+) metabolism to enhance radiation therapy responses. Semin Radiat Oncol 29, 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Characterization of mutant and reconstituted cell lines. (A) Western blot to confirm MDA‐MB‐231 STAT3 KO and reconstitution of KO cells with WT, MTS (WT), or MTS (S727A) STAT3 (left panel: cytosolic fraction; right panel: mitochondrial fraction). Mitochondrial STAT3 levels are normalized to SDHA. (B) Complex II activity for STAT3 WT, KO, KO + MTS (WT), and KO + MTS (S727A) cells. (C) Western blot to confirm expression levels of complex I subunits in MDA‐MB‐231 STAT3 WT and KO cells. Student’s t‐test: ****= P < 0.0001, ***= P < 0.001, **= P < 0.01, *= P < 0.05, ns = not significant, n = 3. MW are expressed in KDa. Error bars represent ± SD.
Fig. S2. (A) Glutathione levels in WT and mutant cell lines. GSH levels measured in STAT3 WT, KO and KO + MTS cells. (B) Growth of cells is unaffected by STAT3. Growth curve for MDA‐MB‐231 STAT3 WT and KO cells over 4 d measured using crystal violet. (C) Induction of SOCS3 gene expression depends on nuclear STAT3. mRNA expression of nuclear STAT3 target gene SOCS3 in MDA‐MB‐231 STAT3 WT, KO, KO + WT, and KO + MTS cells. Student’s t‐test: ****= P < 0.0001, ***= P < 0.001, **= P < 0.01, *= P < 0.05, ns = not significant, n = 3. Error bars represent ± SD.
Fig. S3. Antioxidant gene expression does not require SIRT1 or SIRT3 activity. (A) mRNA expression of GCLC in MDA‐MB‐231 STAT3 WT or KO cells treated with 200 μm EX‐527 (SIRT1 inhibitor) or 200 μm 3‐TYP (SIRT3 inhibitor) for 24 h. (B) mRNA expression of PGC1α in MDA‐MB‐231 STAT3 WT cells treated with 200 μm EX‐527 or 200 μm 3‐TYP for 24 h. Student’s t‐test: ****= P < 0.0001, ***= P < 0.001, **= P < 0.01, *= P < 0.05, ns = not significant, n = 3. Error bars represent ± SD.
Fig. S4. Specificity control for STAT3‐BioID pull‐down. Streptavidin sepharose was used to pull‐down biotinylated proteins from mitochondrial lysates of MDA‐MB‐231 STAT3 WT cells expressing MTS‐BioID treated with 50 μm biotin for 18 h, and complex I components (NDUFV2, NDUFS2, NDUFAF2) were identified by immunoblotting (left). Pull‐down with streptavidin sepharose from mitochondrial lysates expressing MTS‐BioID and treated with 50 μm biotin for 18 h showing near quantitative recovery of biotinylated proteins (right).
Table S1. Primer sequences for RNA quantification by qRT‐PCR.
Data S1. MS/MS data for peptides copurifying with STAT3‐BioID.