

ABSTRACT
The kidneys’ integrative responses to heat stress aid thermoregulation, cardiovascular control, and water and electrolyte regulation. Recent evidence suggests the kidneys are at increased risk of pathological events during heat stress, namely acute kidney injury (AKI), and that this risk is compounded by dehydration and exercise. This heat stress related AKI is believed to contribute to the epidemic of chronic kidney disease (CKD) occurring in occupational settings. It is estimated that AKI and CKD affect upwards of 45 million individuals in the global workforce. Water and electrolyte disturbances and AKI, both of which are representative of kidney-related pathology, are the two leading causes of hospitalizations during heat waves in older adults. Structural and physiological alterations in aging kidneys likely contribute to this increased risk. With this background, this comprehensive narrative review will provide the first aggregation of research into the integrative physiological response of the kidneys to heat stress. While the focus of this review is on the human kidneys, we will utilize both human and animal data to describe these responses to passive and exercise heat stress, and how they are altered with heat acclimation. Additionally, we will discuss recent studies that indicate an increased risk of AKI due to exercise in the heat. Lastly, we will introduce the emerging public health crisis of older adults during extreme heat events and how the aging kidneys may be more susceptible to injury during heat stress.
Keywords: Renal physiology, hyperthermia, physical work, hypohydration, kidney function, AKI, aged, older adults
Introduction
The kidneys are highly vascularized organs that receive approximately 20% of cardiac output at rest and are central to many homeostatic functions including the regulation of blood pressure, water and electrolytes, and acid/base balance. As such, physiological challenges to these regulatory processes often elicit compensatory responses from the kidneys. For instance, heat stress causes a multitude of physiological actions in the kidneys to maintain blood pressure, conserve water and electrolytes, and redirect blood flow away from the kidneys to the skin to offload heat and promote heat loss. Moreover, there is an elevated demand on these systems when heat stress is combined with dehydration and/or exercise, and the consequence of these combined demands can be physiological or pathophysiological in nature.
Our understanding of the kidneys has evolved over thousands of years, but the effects of heat stress on the kidneys have only been well described within the last century. Terminology for urine and the kidneys first appeared in Sumerian writings around 3,000 BC [1]. Nearly 3,000 years later, the Greek physician Galen of Pergamum famously described the filtering capacity of the kidneys and pondered the function of the kidneys to produce urine [2-4], which provided foundational knowledge that was further clarified in the 1500 and 1600s with the improved depictions of human anatomy [1] and advances in microscopy [5]. During this time, the link between anatomy and physiological function of the kidneys was starting to be described [2,6,7], which is probably best highlighted in the observations of the anatomist Lorenzo Bellini who described that, when the renal tubules were compressed, if “you are not afraid to present this to your tongue you will discover a certain saltiness and, in some, the taste of urine (pg. 20)” [3,8]. Technological advances over the next three centuries led to discoveries of the nephron [9], while understanding of the biophysical and biochemical principles underlying glomerular filtration and body fluid balance were advanced by the preeminent physiologists Carl Ludwig [10] and Ernest Starling [11]. To our knowledge, it wasn’t until 1898 that the first published reports of kidney function and environmental temperature appeared in which it was reported that urinary protein content varied inversely cool ambient temperatures [12].
The first description of the effects of heat stress on kidney function can be found in the 1904 text Physiology and Pathology of the Urine, in which it was described that “ in hot weather … the proportion in urine to liquids swallowed is much less (pg. 1)” [13]. It wasn’t until 1916 that the first direct experimental evidence of the effect of heat stress on kidney function was published, where it was reported that urine output decreases during heat stress [14]. These early findings were furthered by observations that sodium reabsorption by the kidneys varies with urine output and sweat losses [15], data that ultimately provide the foundation for our understanding of the effects of heat stress on kidney function.
The public health challenges of heat stress
The increased frequency, severity, and duration of heat waves caused by climate change [16] and the urban heat island phenomenon [17] will likely increase the physiological demands placed upon the kidneys [18]. Heat stress, which invokes increases in core body temperature, particularly when coupled with dehydration (i.e., a hypertonic, hypovolemic state), amplifies processes that may result in kidney-related pathology, the most notable of which is acute kidney injury (AKI), which is generally defined as an acute reduction in kidney function (AKI) [19-21].
The interaction between heat stress and AKI was first identified in adults aged <45 years undergoing military physical training (i.e., exercise) in a hot environment [20-23]. These relatively isolated occurrences, however, proved to be illustrative of a larger problem taking place in occupational settings in which workers are regularly exposed to heat stress [19,24,25]. This is highlighted by observations that large populations of agricultural workers in Central America, India and Sri Lanka, amongst others, experience recurrent episodes of AKI that are strongly associated with heat stress, which has been hypothesized to be a leading etiological cause of the epidemic of chronic kidney disease (CKD, a progressive loss of kidney function caused by irreversible kidney damage) of unknown origin occurring in these regions [26]. Data in a recent meta-analysis indicate that 15% of people who typically work in the heat experience CKD or AKI [25]. The prevalence of this heat stress related-kidney pathology is alarming due to the large number of people that are or will be exposed to occupational heat stress. For example, according to the International Labour Organization [27], an estimated 280 million people are employed as agricultural workers in Central America, India and Sri Lanka, while the U.S. Bureau of Labor Statistics data [28] indicate that ~15 million Americans work outdoors in occupations that involve regular exposure to heat (e.g., agriculture, forestry, construction). Therefore, an estimated 42 million people in Central America, India and Sri Lanka and ~2.3 million workers in the United States are currently at risk for CKD or AKI due to their occupation.
In addition to occupational settings, which typically involve younger adults 18-45 years old, older adults aged 65 years or more are also likely at risk of AKI associated with heat stress. Furthermore, compared to younger adults, older adults have disproportional increases in morbidity and mortality during heat waves [29-41], which are defined as periods of unusually hot weather lasting two or more days [42]. Emerging epidemiological evidence consistently demonstrates the kidneys’ susceptibility to heat stress in older adults. This is highlighted by observations that the top two causes of hospitalizations in older adults during heat waves are fluid/electrolyte disturbances [32,35,41], a risk factor for AKI [43], and AKI [32-36,41,44]. Notably, this public health challenge is likely to be magnified in the coming decades given the simultaneous rise in the frequency, intensity, and duration of heat waves [45] and growth in the number of older adults [46].
The combination of heat stress and AKI is an emerging public health problem affecting both younger and older populations. There is a need to understand the mechanisms underlying epidemiological observations to identify and examine strategies to mitigate the risk of AKI during heat stress. At present, this understanding is in its infancy. For instance, the mechanisms by which heat stress can elicit AKI, without consideration for biological age, have just started to be elucidated over the past ~5 years in both rodent [47-61] and human [18,19,24,26,54,62-67] models. Moreover, to our knowledge, heat stress provoked AKI has never been experimentally studied in the context of aging.
Objective
The objective of this comprehensive narrative review is to present the physiological and potential pathophysiological effects of heat stress on the kidneys in both younger and older adults. The kidneys are involved in many integrative physiological responses to heat stress, which are further modulated by the mode of heat stress, the magnitude of dehydration, heat acclimation, age, and many other factors. Therefore, we will first introduce the physiology of heat stress and dehydration. Next, we will discuss kidney function and AKI, with some attention paid to the experimental methodologies employed to study the physiological and pathophysiological processes. This background information will provide context to accomplish our objective, which is to comprehensively review the physiological effects of heat stress from the perspective of kidney health in both younger and older adults. A narrative review was carried out given the purposefully broad scope of this review, which makes conducting a systematic review inappropriate [68].
Defining heat stress
In healthy humans, resting core temperature fluctuates over a relatively narrow range of 36.5-37.5°C [69]. Heat stress is defined as net heat load to which a person is exposed and is a function of environmental factors, physical exertion, and clothing. During heat stress, the net heat load is positive causing a rise in core body temperature (i.e., hyperthermia). Heat stress can occur passively, where heat gain is increased despite no change in metabolism, or with exercise, where the rate of metabolic heat production is increased [70]. Thermoregulation aims to balance this heat gain with heat loss [71]. Heat loss responses include cutaneous vasodilation and sweating, with the evaporation of sweat being the most powerful mode of heat loss [70,72]. The ability of heat loss responses to offload heat dictates the extent of hyperthermia and is a function of both the magnitude of heat stress and thermoregulatory processes. By extension, heat stress can be classified into two categories – compensable vs. uncompensable. Compensable heat stress occurs when the maximal evaporative capacity permitted by the environment exceeds the evaporative heat loss requirements necessary to promote heat balance. In this state, core temperature may rise, but will ultimately plateau once heat loss matches heat gained [73]. In contrast, uncompensable heat stress occurs when the maximal evaporative capacity permitted by the environment is lower than the evaporative heat loss required to maintain heat balance. This situation results in a state of continual body heat storage and core temperature will rise indefinitely if the uncompensable conditions persist [74].
There are a variety of methods in which heat stress can be experimentally induced in laboratory settings. Thus, careful consideration is necessary when designing, interpreting, and comparing across experiments examining the physiological effects of heat stress. Decisions associated with selecting a heat stress model should consider whether the focus of the study requires high internal validity or high external validity (Figure 1). For instance, studies requiring a high internal validity would be focused on examining causal relations between increases in skin and/or core body temperature and physiological outcomes. As a result, in such studies there is a need to experimentally control the magnitude of increases in body temperature. In contrast, if high external validity is necessary, then it is important to simulate the conditions encountered in real life scenarios. The compromise, however, is that there is less experimental control of body temperature due to interindividual variation.
Figure 1.
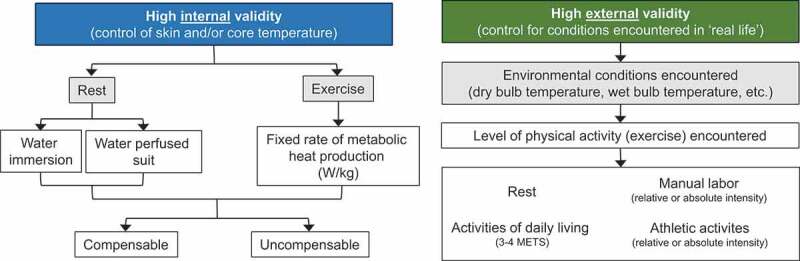
Examples of common methods of studying heat stress in a laboratory setting delineated by internal (left) or external (right) validity. Abbreviations – METS: metabolic equivalents
For studies seeking high internal validity, tight control of skin and/or core temperature can be achieved in several ways. In such instances, selecting the mode of heat stress and the magnitude of heat stress (i.e., compensable vs. uncompensable) will depend on the nature of the research question. For example, studies interested in examining responses to passive heat stress will typically use warm/hot water immersion or water-perfused suits. Warm/hot water immersion (typically ~39-42°C) invokes immediate rises in skin temperature and often provokes a rapid rise in core temperature owing to the high thermal conductivity of water. In this instance, the extent of hyperthermia will depend on the temperature of the water and the desire for compensable or uncompensable conditions. With water immersion, the hydrostatic pressure of the water stimulates increases in central blood volume and readily alters central and peripheral hemodynamic responses, particularly in the kidneys [75]. This likely confounds the utility of employing water immersion as a modality to study the independent effects of heat stress on the kidneys. The water-perfused suit method utilizes a tube-lined suit that is perfused with hot water (typically ~40-50°C) to produce relatively quick and reproducible increases in skin and/or core temperature, the magnitude of which can be relatively easily manipulated depending on whether compensable or uncompensable conditions are desired. Given these advantages, the water-perfused suit is likely the more preferred form of passive heat stress [70]. For studies focused on heat stress during exercise, reproducible increases in core temperature both within and between groups (in healthy people) can be sought by clamping the rate of metabolic heat production normalized to body mass [76]. The environmental conditions in which this exercise is conducted can then be predicated on the desire for compensable or uncompensable heat stress.
For studies seeking high external validity, the goal is to simulate the conditions encountered in real world situations. For example, studies focused on understanding and mitigating the risk of AKI in agricultural workers may want to replicate the conditions to which these people are regularly exposed. Lucas et al. [77] reported that in a single work-shift agricultural workers in Central America spend ~6 hours exposed to a wet bulb globe temperature between 26 and 32°C while working at ~54% of their estimated heart rate maximum. These conditions can be readily simulated in an environmental chamber, which often also permits the simulation of various types of exercise (e.g., upper vs. lower body), work-to-rest ratios, and other variables that are encountered in occupational settings. It is also possible to simulate conditions of a heat wave, which may have more relevance to the general public. In practice, the U.S. National Weather Service issues an Excessive Heat Warning when the heat index, a function of dry and wet bulb temperatures [78], exceeds either 41°C for more than 3 hours during the day or 46°C over any period during the day [79]. These environmental conditions, particularly when studied over consecutive days and coupled with low levels of exercise to simulate activities of daily living (usually 3-4 metabolic equivalents), may provide a unique laboratory model to study the physiological responses to heat waves and/or the evaluation of health and safety countermeasures [80,81]. This may be particularly relevant for older adults given that morbidity in older adults is worsened with increased heat wave duration [32,35]. Moreover, recent findings demonstrate that the magnitude of increase in core temperature is greater on the second of consecutive days of exercise in a hot environment [82,83] and that this effect may be exacerbated in older adults [84].
Physiology and assessment of body water regulation
In humans, water accounts for ~73% of fat free body mass [85]. Under normal conditions, elimination of water from the body is primarily routed through the kidneys with a lesser amount of water loss occurring through sweat loss, evaporation from the skin, respiration, and the gastrointestinal tract (e.g., in feces). The kidneys have a direct role in maintaining euhydration, which is defined as a state of optimal body water content [86]. This is highlighted by the fact that euhydration is maintained with total water intakes ranging from 1.3 to 7.9 L/24 h to maintain plasma osmolality within a normal range (285-295 mOsm/kg) among individuals, in varying environments and circumstances [87]. The process of losing body water is termed dehydration, whereas the state of deficient body water caused by acute or chronic dehydration is known as hypohydration [86].
Humans have a tremendous capacity to produce sweat, with maximal sweat rate often approaching or even exceeding 2 L/h [88]. Thus, during heat stress, dehydration is relatively common given that the large volume of water lost as sweat is often not sufficiently replaced with drinking even when fluids are readily available [89]. Because sweat is hypoosmotic compared to plasma, the resulting body water losses cause intracellular dehydration and result in water loss from both the intra and extracellular fluid spaces, which is ultimately characterized by a state of hyperosmolality and hypovolemia [90]. Objective determination of dehydration (and/or hypohydration) can be ambiguous given that there is no universal gold standard assessment for dehydration, particularly given the high monetary cost associated with direct measurements of total body water [91]. That said, a dehydrated state can be reliably identified by monitoring changes in two or more markers over time [92]. For example, conservative estimates indicate that intracellular dehydration (i.e., hyperosmotic, hypovolemia) can be identified with 95% confidence using a combination of specific anthropometric, blood and urine-based measures [93-95] (Table 1).
Table 1.
Examples of methods for objective determination of hydration status
The state of hyperosmotic, hypovolemia brought about by intracellular dehydration causes a cascade of hormonal responses to promote water conservation including the activation of the renin-angiotensin-aldosterone system and the release of vasopressin [96] (Table 2). Decreases in blood volume, such as with dehydration, are detected by baroreceptors in the systemic vasculature resulting in increased renal sympathetic nerve activity (RSNA) and renin secretion. One of the important consequences of increased plasma renin is the production of angiotensin II, which binds to receptors located in many organs [96]. Activating the angiotensin II type 1 receptor (AT1R) results in many physiological actions that are important during dehydration. In the kidneys, activation of the AT1R located on the basolateral surface of proximal tubules increases sodium reabsorption, and thus water reabsorption, by stimulating Na+/K+ ATPase and the Na+/H+ exchanger NHE3 [97,98]. Additionally, AT1R activation causes vasoconstriction in vascular tissues, including both the renal afferent and efferent arterioles [99,100]. Angiotensin II also contributes to the sensation of thirst through activation of the AT1R in the subfornical organ of the brain [101,102]. The final major role of angiotensin II in the classical renin-angiotensin-aldosterone system is to stimulate aldosterone secretion by the glomerulosa cells of the adrenal cortex through AT1R activation [103]. Aldosterone acts on mineralocorticoid receptors in the distal convoluted tubule and collecting ducts to increase NaCl reabsorption and K+ excretion [104] (Table 2).
Table 2.
Summary of potential neuro-hormonal responses to heat stress (with or without dehydration) and the primary physiological actions, anatomical locations, and physiological outcomes
| Neuro-hormonal effector | Primary physiological actions | Primary anatomical locations | Primary physiological outcomes |
|---|---|---|---|
| Renal sympathetic nerve activation | Vasoconstriction (at higher levels of activation only) Na+ reabsorption Renin release |
Afferent and efferent arterioles Proximal and distal tubules Afferent and efferent arterioles (juxtaglomerular cells) |
|
| Vasopressin | Translocation of Aquaporin-2 Na+ reabsorption Vasoconstriction |
Collecting ducts Distal tubules (distal straight tubule) Efferent arterioles |
|
| Aldosterone | Na+ reabsorption | Distal convoluted tubules, collecting ducts |
|
| Angiotensin II | Vasoconstriction Na+ reabsorption Aldosterone release |
Afferent and efferent arterioles Proximal tubules Adrenal cortex |
|
Dehydration also increases circulating vasopressin (also known as antidiuretic hormone or ADH). Vasopressin acts on type 2 vasopressin receptors (V2) to stimulate NaCl reabsorption by the thick ascending limbs of Henle and promote the localization of the water channel, aquaporin-2, on the apical membrane of the collecting duct to promote water reabsorption [105]. These processes are initiated when the increased plasma osmolality [106] and/or sodium concentration [107] is detected in the brain, and is also stimulated by reductions in blood volume [108]. Ultimately, these processes result in decreased urine output and increased urine concentration [109]. Vasopressin also exhibits vasoconstricting actions in the kidneys by activating V1 receptors in vascular smooth muscle, primarily in efferent arterioles, to reduce renal blood flow [110], which together with angiotensin II is important in maintaining blood pressure during dehydration [111-117] (Table 2).
It is also worth noting that recent work provides support for a localized autocrine/paracrine renin-angiotensin-aldosterone system that operates quasi-independently from the classical renin-angiotensin-aldosterone response [118]. For instance, Wang et al. [119] demonstrated that the intrarenal renin-angiotensin-aldosterone system contributes to increased fluid reabsorption in the distal tubule during fluid deprivation by activating prostaglandin E2 receptor 4, which independently stimulates the (pro)renin receptor and renin, a pathway that has also been shown to complement the actions of in upregulating aquaporin-2 expression [120].
Physiology and assessment of kidney function
A large portion of cardiac output is delivered to the kidneys via the renal artery, which branches into a series of smaller arteries and arterioles and is ultimately filtered by the functional unit of the kidneys, the nephron (Figure 2). The control of renal blood flow (discussed below in Renal blood flow) and glomerular filtration rate (GFR, discussed in Glomerular filtration rate) are important functions of the kidneys to regulate homeostatic processes that are stressed during heat stress, such as blood pressure and water and electrolyte regulation. A multitude of techniques can be used to quantify kidney function, including measures of clearance, renal blood flow, and urine production and concentration. The strengths and limitations of these techniques must be understood when developing and interpreting experiments examining the physiological responses of the kidneys [121]. This section will introduce measures of kidney function that are relevant to physiological or pathophysiological responses to heat stress.
Figure 2.
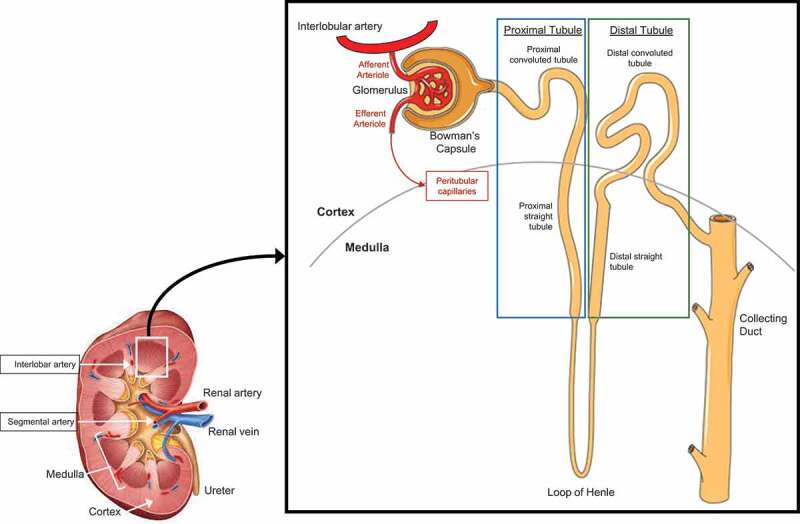
Simplified anatomy of the vascular system of the human kidney and the human nephron
Autonomic control of kidney function
The renal sympathetic nerves have potent actions on kidney function. Innervation sites for sympathetic nerves are located along the major resistance vessels of the kidney (i.e., interlobar, arcuate, and interlobular arteries), the afferent and efferent arterioles, at the juxtaglomerular apparatus, and heterogeneously distributed across the proximal and distal tubules, the thick ascending limb of the loop of Henle, and the collecting duct [122-125] (Figure 2). Thus, the renal sympathetic nerves have important roles in modifying renal blood flow, glomerular filtration rate (GFR), tubular sodium and water reabsorption, and mediating the release of vasoconstricting hormones that increase blood pressure in response to various stimuli.
Measuring RSNA is not possible in humans without highly invasive surgical procedures. Therefore, understanding of the responses of human RSNA is based on direct measures from animal studies (e.g., rodents, rabbits), renal denervation in humans (e.g., kidney transplant, renal sympathetic denervation in patients with resistant hypertension), or approximations based on changes in muscle sympathetic nerve activity and/or renal norepinephrine spillover. Norepinephrine is the primary neurotransmitter released by the renal sympathetic nerves, which also release co-transmitters such as neuropeptide Y and ATP to mediate changes in kidney function [122]. The degree to which muscle sympathetic nerve activity accurately reflects that of RSNA is not known given that there is not a consensus method on how to measure RSNA in humans [126]. However, tests evoking sympathetic activation (e.g., cold pressor test, hand-grip exercise, tilting) have been used to probe changes in renal vascular control [127-132].
Renal denervation causes increases in sodium and water excretion in rats [133,134] and in some patient populations, including those with resistant hypertension [135]. Renal sympathetic nerve stimulation activates adrenoreceptors located in the renal tubular epithelial cells and granular cells of the juxtaglomerular apparatus to cause these cells to reabsorb sodium and release renin, respectively [122]. α1-adrenoreceptor activation in the proximal tubule epithelial cells causes increased activity in Na+/H+ exchanger and NHE3, promoting fluid absorption [136].
In general, renal blood flow (described below in Renal blood flow) is reduced with increased RSNA, including that resulting from general reflex-mediated activation of the sympathetic nervous system (e.g., chemoreceptor or baroreceptor activation) [137-140]. Increased RSNA decreases renal blood flow due to vasoconstriction of the afferent and efferent arterioles and to a lesser degree the interlobular arteries [122]. That said, DiBona and Kopp [141] have also demonstrated a graded response of the kidneys to RSNA with lower frequencies of RSNA increasing renin secretion without changes in sodium handling or renal hemodynamics. At relatively moderate increases in RSNA, that remain subthreshold to alter renal hemodynamics, sodium reabsorption occurs alongside renin secretion. Relatively higher increases in RSNA reduce renal blood flow and glomerular filtration rate, which together with increases in renin secretion, provide a potent stimulus for conserving water (i.e., antidiuresis) and sodium (i.e., antinatriuresis).
The effects of RSNA are likely modulated, at least in part, by angiotensin II. The modulatory role of angiotensin II is supported by evidence demonstrating that the antidiuretic and antinatriuretic effects with lower levels of RSNA were blunted when inhibiting angiotensin-converting enzyme [142] and restored with angiotensin II infusion [143]. The modulatory role of nitric oxide, which is generated by nitric oxide synthase (NOS) enzymes, on RSNA is less clear based on the differing effects reported in the literature. Three isoforms of NOS are located throughout various regions of the kidney, including endothelial NOS in the endothelial cells of the renal vasculature and glomerular capillaries [144,145], neuronal NOS within the renal sympathetic nerves [146], renal tubules and macula densa [147], and inducible NOS in the renal medulla [148]. It has been speculated that nitric oxide may have differing roles at various anatomical sites within the kidneys, such that nitric oxide at the postjunctional membrane, or at vascular and tubular epithelial cells, may depress neurally mediated changes in kidney function, whereas nitric oxide at the prejunctional membrane facilitates the release of norepinephrine [122].
Renal blood flow
Renal blood flow broadly describes blood flow in the renal artery to the kidneys and regional blood flow within the kidneys (e.g., cortical, medullary). Classically, renal blood flow is measured using clearance techniques that measure renal plasma flow, such as para-aminohippurate (PAH) clearance (Table 3). These techniques are founded on the basis that kidney function is the net action of input (i.e., renal artery blood flow) and output, which is comprised of renal venous blood flow and urinary excretion [121]. In this sense, clearance is defined as the “volume of plasma per unit time from which all of a specific substance is removed (pg. 36)” and is defined as Ux·V/Px, where Ux and Px are the concentration of substance ‘x’ in the urine and plasma, respectively, and V is urine flow rate [149]. Renal blood flow is related to renal plasma flow by the hematocrit, such that renal blood flow is equal to renal plasma flow divided by (1-hematocrit) [RBF=RPF/(1-Hct)]. PAH clearance has traditionally been used as the gold standard measure for renal artery plasma flow. Approximately 90% of PAH, which is filtered at the glomeruli and secreted by the tubules, is extracted from the blood by the kidneys [150], with several factors precluding the complete extraction of PAH from the blood, including periglomerular shunts and limitations in secretion of PAH in cortical areas and in regions of reduced perfusion such as the proximal tubules in the medulla [151]. For these reasons, PAH clearance is often referred to in the literature as ‘effective’ renal plasma flow because it provides an approximation of renal plasma flow without requiring a renal venous blood sample [152]. Determination of PAH clearance is an invasive technique that requires a priming dose and sustained infusion to maintain constant PAH concentration in the plasma [152-154]. Classic methods of measuring PAH clearance use the bladder clearance technique, which requires the collection of urine either through spontaneous voiding or catheterization in order to determine the concentration of PAH in the urine [154]. The alternative constant-infusion technique eliminates the need to collect urine, which was often a source of error (e.g., contaminated urine during spontaneous voiding) [155], and assumes that, for substances such as PAH that are neither metabolized nor stored by the body, when plasma PAH is constant, the rate of excretion of Ux·V must be the same as (and therefore can be substituted by) the rate of infusion. However, this requires equilibrium between rate of infusion and rate of excretion of PAH that may not be possible in shorter duration studies [156]. To overcome this limitation, investigators typically correct for changes in plasma concentration of PAH by accounting for plasma concentrations at the beginning and end of the clearance period [157-160].
Table 3.
Common methods for measuring renal blood flow and glomerular filtration rate in humans
| Measure | Method | Advantages | Disadvantages |
|---|---|---|---|
| Renal blood flow | Para-aminohippurate clearance | Gold standard measure of renal plasma flow | Not a direct assessment of arterial blood flow, expensive, invasive, unable to measure dynamic changes, may require timed urine samples. |
| Doppler ultrasound | Reliable, affordable, non-invasive, index of arterial blood flow, can measure dynamic changes | Can only measure blood velocity (not blood flow*) in renal, segmental, and/or interlobular arteries, has not been validated against other measures | |
| Magnetic resonance imaging | Direct assessment of renal blood flow and/or oxygenation, non-invasive | Not feasible in some experimental settings, expensive | |
| Glomerular filtration rate | Inulin clearance** | Gold standard | Expensive, requires timed urine samples |
| Creatinine clearance | Uses an endogenous substance, affordable | Requires timed urine samples, may be inaccurate during intense exercise and/or dehydration | |
| Estimated glomerular filtration rate | Simple, affordable, spot check of an endogenous substance without need to collect urine | May be inaccurate when glomerular filtration rate is rapidly changing |
Despite improvements in technology, several technical challenges hinder the direct quantification of renal artery blood flow and/or regional renal blood flow (i.e., not the net renal blood flow) in experimental settings. In humans, various magnetic resonance imaging (MRI) based methods (e.g., dynamic contrast-enhanced, blood oxygen level-dependent, arterial spin labeling) are promising technologies, but may be cumbersome or infeasible during experimental heat stress. Another approach is Doppler ultrasound, which is often used in clinical and research settings to assess kidney hemodynamics. Doppler ultrasound is appealing because of its noninvasive nature and the ability to quickly render and capture real-time images [121,127,161-168], with acute vascular responses measured within seconds in similar regions within and between subjects [121]. Data obtained using the Doppler ultrasound technique have been found to be reliable both within and between days [166]. However, the diameter of the vessels of the renal vasculature cannot be accurately measured with Doppler ultrasound. Thus, in its current form, this technology can only measure blood velocity, assuming that blood velocity is proportionate to blood flow. This assumption is only valid provided that the diameter of renal and segmental arteries does not change between assessment periods and is reinforced by the fact that the renal and segmental arteries are conduit, not resistance, vessels [128]. In support of this, changes in renal artery blood flow have been found to be dependent on changes in blood velocity and not changes in vessel diameter during infusion of a vasoconstrictor (adenosine) or vasodilators (isosorbide dinitrate, papaverine, dopamine, and fenoldopam) [169,170]. To the contrary, however, there is some evidence that during exercise the diameter of the renal artery decreases [171,172], but these measurements were made using ultrasound techniques that have not yet been replicated by other laboratories.
There is great interest in accurately quantifying changes in renal blood flow because it is a highly controlled variable that has implications for the regulation of blood pressure and water and electrolytes. Thus, it is also important to note that the kidneys have an intrinsic ability to maintain blood flow at varying arterial pressures (i.e., autoregulate). Renal blood flow autoregulation is mediated by actions of the afferent arterioles and interlobular arteries and their myogenic response to constrict or relax in response to changes in perfusion pressure [173-175]. Approximately, 50% of the total autoregulatory response [176,177] rapidly occurs within 3-10 seconds [178,179], which is contributed to by unloading of the renal baroreceptors and tubuloglomerular feedback provided by the juxtaglomerular apparatus [180,181]. Tubuloglomerular feedback also results in renin release by the afferent arterioles in response to sensation of decreased NaCl delivery to the macula densa in the distal tubule [182], which ultimately ensures a relatively stable renal blood flow and glomerular filtration rate (see Glomerular filtration rate). These neural (discussed previously in Autonomic control of kidney function), hormonal (discussed previously in PHYSIOLOGY AND ASSESSMENT OF BODY WATER REGULATION), and autoregulatory mechanisms offer a complex and highly redundant control of renal blood flow to maintain homeostasis utilizing many systems.
Glomerular filtration rate
In clinical settings and most research studies, kidney function is quantified as a function of GFR, which provides a measure of the ability of the kidneys to filter blood. The kidneys typically function at ~75% of their maximum filtration capacity in healthy individuals, which allows the kidneys to preserve GFR in conditions that otherwise reduce GFR, such as a loss of nephron mass or partial loss of individual nephron filtering ability (e.g., as occurs with aging) [183]. Thus, the maintenance of kidney function through hyperfiltration reflects a reserve capacity of the kidneys to increase GFR and highlights that there is no linear relation between nephron number and GFR. This reserve capacity can be assessed as the difference between peak (or ‘stressed’) GFR and baseline GFR, and is often referred in the literature as renal functional reserve [184-186]. The usefulness of GFR is highlighted by when renal mass is decreased, such as in kidney damage, hyperfiltration increases as a compensatory mechanism to maintain GFR within normal ranges, even with losses up to 50% of functional renal tissue [187].
An ideal measure of GFR requires the calculation of clearance of a substance that is freely filtered by the kidneys and is completely excreted in the urine without undergoing metabolism, tubular secretion or reabsorption [188] (Table 3). Inulin clearance is considered the gold standard measurement for GFR [150] and involves continuous inulin infusion and urine collection, but is cumbersome and expensive given that the cost of inulin has increased [189]. There is also evidence supporting the utility of measuring GFR with renal clearance of chromium 51-labeled ethylenediaminetetraacetic acid (51Cr-EDTA) and renal clearance of iohexol [189]. Additionally, mannitol, a 6-carbon alcohol, is an osmotic diuretic that is freely filtered across the glomerulus [190,191] and, with a particular preparation, has been reported to reflect a similar GFR as inulin clearance in healthy adults [192].
Creatinine clearance is probably the most common means of estimating GFR. Creatinine is released into the circulation at a relatively steady rate from the nonenzymatic dehydration of muscle creatine [193]. Approximately 98% of creatine in the body, which is primarily synthesized in the liver, is stored in muscle with ~1.6% of total body creatine being converted to creatinine daily [194,195]. Using creatinine as a marker of GFR has advantages because it is endogenously produced and can be assessed with routine clinical techniques in the blood and urine. However, creatinine clearance is not considered to be a perfect marker of GFR. For instance, creatinine clearance assumes there are no changes in muscle mass or muscle damage during the protocol. Moreover, creatinine clearance requires the use of timed urine samples, which may not always be feasible. Finally, tubular secretion of creatinine can result in overestimations of GFR by ≥20% compared to inulin clearance [196,197]. To our knowledge, the accuracy of creatinine clearance derived measures of GFR compared to that measured with inulin clearance during exercise or heat stress has never been explored. That said, Sjöström et al. [198] have reported that creatinine clearance overestimates GFR by ~5% compared to inulin clearance following dehydration induced by furosemide, which increases water loss through increased isosmotic urine production [199]. However, following rehydration, creatinine clearance overestimates GFR by nearly 50% [198]. Thus, experiments that involve a rehydration protocol, such as during or following exercise in the heat, may result in an overestimation of GFR as tubular secretion of creatinine is increased.
In most clinical settings, GFR is estimated from serum concentrations of creatinine or cystatin C, another endogenous filtration marker, using specialized equations that often adjust for variables such as age, race, sex, body size, blood urea nitrogen, and albumin [121]. These equations utilize the assumption that creatinine or cystatin C are produced endogenously at a constant rate and thus, an increase in these markers is reflective of a decrease in GFR [121,200]. While this approach is convenient, the accuracy of serum creatinine or cystatin C derived estimates of GFR is limited when there are rapid changes in GFR such as during exercise [201,202]. As such, estimated GFR calculated by both creatinine and cystatin C during exercise underestimates decreases in GFR compared to that calculated from creatinine clearance [202].
Water and electrolyte regulation
The kidneys are vital in regulating body fluid volume and composition and there are many systemic and intrarenal mechanisms underlying water and electrolyte regulation. The ability of the kidneys to concentrate urine arises from interactions among the active transport of NaCl from the thick ascending limbs (i.e., sodium reabsorption), water permeability of the collecting ducts, delivery rate of NaCl and water to the loop of Henle, and the volume of tubular fluid delivered to the medullary collecting ducts, which increases osmotic water transport across the epithelium of the collecting duct [203]. Most of the techniques used to quantify changes in water and electrolyte regulation require collection of timed urine samples and, depending on the measurements, blood draws (e.g., fractional excretion calculations, hormones). Thus, the measurements presented in this section typically reflect a systemic response to changes in water and electrolyte regulation.
Urine production can be quantified by volume or flow rate. The extent of urine concentration can be assessed via osmolality, specific gravity, and urinary creatinine concentration. Decreases in urine production and/or increases in the extent of urine concentration are typical physiological responses of the kidneys to mitigate water and electrolyte losses, such as during heat stress. In healthy humans, the excretion of urine by the kidneys has large variability. ‘Normal’ 24 hour urine volumes can range from ~0.5 L to ~25.0 L with osmolality ranging from as high as 1400 mOsm/kg (with low urine volumes) to as low as 40 mOsm/kg (with high urine volumes). This modulation of urine volume and concentration aids in the maintenance of extracellular volume within narrow limits across a wide range of daily water and sodium intakes [204]. Calculations can be made to quantify kidney function via clearance, where the handling of a substance by the kidneys is calculated as the amount of substance excreted in the urine (a product of urine concentration of the substance and urine flow rate) subtracted from the filtered load (a product of GFR and plasma concentration of the substance). The result of this calculation will quantify whether there is net secretion (a positive value), net reabsorption (negative), or neither secretion nor reabsorption (zero) [121]. The concept of clearance is also used to calculate free water clearance as CH20=V∙(1-[Uosm/Posm]), where V is equal to urine flow rate and Uosm and Posm are the osmolality of the urine and plasma, respectively. Free water clearance quantifies how dilute the urine is relative to the plasma. For example, a positive free water clearance in a dehydrated individual would be a sign of pathology where the ability to conserve water is impaired. Another useful calculation includes the fractional excretion of electrolytes, which is calculated using urinary and plasma concentrations of creatinine as FEx=100∙([Ux/Px]/[Ucreatinine/Pcreatinine]), where Ux and Px represent the concentration of substance x in the urine and plasma, respectively. Notably, a low fractional excretion of sodium (i.e., <1%) is typical for euhydrated subjects with normal kidney function and moderate salt intake [205].
Additional markers of kidney function
Various other blood and urine markers can be used to inform interpretation of kidney function. Proteinuria, which can portend a reduction in GFR, can be assessed by measuring total protein or albumin in the urine [206]. Proteinuria may be indicative of increased glomerular permeability, a reduced capacity for reabsorption due to damage or dysfunction in the renal proximal tubules, or saturation of the renal tubules capacity for reabsorption due to overflow of normal or abnormal proteins that are produced in increased amounts [206]. Urine microscopy can be used to determine content of the urinary sediment, such as presence of erythrocytes (i.e., hematuria), casts, or crystals [206]. Blood urea nitrogen (BUN) is a metabolic waste product from dietary protein catabolism and turnover of tissue proteins, which increases in concentration with excessive tissue catabolism, such as fever or severe burns [206]. Measuring BUN may provide insight into potential dysfunction or pathology within the glomeruli. However, interpretation of increased BUN is limited during conditions eliciting increases in the tubular reabsorption of urea (e.g., decreased renal perfusion from heat stress). Kidney biopsies, although highly invasive and impractical for most experimental studies, can provide morphological assessment of the kidney, and have been previously used in suspected heat stress-related nephropathy in agricultural workers [207].
Physiology and assessment of acute kidney injury
Definition of acute kidney injury
Acute kidney injury (AKI) is defined as a rapid (hours to days) decrease in kidney function (e.g., GFR), with increasing concentrations of products of nitrogen metabolism in the plasma (e.g., creatinine, urea) that may also manifest with decreased urine output [208]. Okusa and Portilla [209] describe AKI as a “summation of temporally activated systems that together result in inflammation, activation of cell death pathways, tubular obstruction, back leak, altered glomerular hemodynamics and a loss of the GFR (pg. 912).” Thus, AKI represents a wide range of pathophysiological responses of varying severity and causes. The consensus term AKI replaced the older terminology of acute renal failure (ARF), because ARF suggested a dichotomy between normal kidney function and organ damage/failure, whereas AKI better reflects that even small and transient decrements in kidney function can result in deleterious outcomes [210]. Indeed, even rapid recovery (≤2 days) from one episode of the mildest form of clinically diagnosed AKI (i.e., Stage 1 AKI) is associated with a 43% increased risk of Stage 3 or higher CKD within one year [211]. This risk for the development of CKD is exacerbated with increased severity and/or length of recovery from AKI [211].
Typically, the cause of AKI is divided into three pathophysiologic categories: prerenal, intrinsic, and postrenal (obstructive). AKI of prerenal origins is the most common indication of AKI and results from hypoperfusion of the kidneys occurring secondary to reductions in true or effective arterial blood volume [209]. This hypoperfusion increases RSNA, activates the renin-angiotensin-aldosterone system, and stimulates vasopressin release. Ultimately, renal blood flow and GFR are reduced and an ischemic environment is created in the renal vasculature. Intrinsic AKI is most commonly caused by ischemia or sepsis, with primary epithelial cell injury most commonly occurring in the proximal tubule [209]. The most distal S3 segment of the proximal tubule is particularly susceptible to ischemic injury [212] due to marked hypoperfusion in the medullary region and the limited ability to undergo anaerobic glycolysis [209]. Postrenal AKI is the least common type of AKI and occurs due to obstruction of the ureters, bladder outlet, or urethra [209].
In clinical settings, AKI is evaluated through a review of medical history, physical examination, review of laboratory tests, imaging of the kidneys, and kidney biopsy if deemed appropriate [213]. Changes in kidney function are quantified via measures of GFR (usually estimated via spot assessment of serum creatinine or serum cystatin C) and/or urine output to identify the severity of AKI (i.e., staging) according to criteria established by the Acute Kidney Injury Network (AKIN) [214], Kidney Disease: Improving Global Outcomes (KDIGO) Classification [215], or the Acute Dialysis Quality Initiative (ADQI): Risk, Injury, Failure, Loss of Kidney Function and End-Stage Kidney Disease (RIFLE) classification [216]. However, spurious causes of increased serum creatinine or decreased urine output, such as that occurring from exercise in the heat, must be excluded before a diagnosis of AKI is made. Thus, these classifications are limited in their ability to classify AKI during heat stress, especially when combined with dehydration and/or exercise, because of the normal physiological response of the kidneys to reduce renal blood flow, GFR, and urine output. Therefore, it is likely inappropriate to classify subjects as experiencing AKI from singular measurements of serum creatinine in the post-exercise period in studies of heat stress, because it is not possible to distinguish between a clinical AKI-event (a serious pathological diagnosis) and normal, physiological increases in serum creatinine. Importantly, in human subjects research, it is unethical to experimentally induce AKI when investigating the pathophysiology of the kidneys following exercise in the heat. Thus, an alternative approach is required to understand the pathophysiology underlying the potential for AKI following exercise in the heat. Due to the limitations associated with serum creatinine and urine output-based assessments of AKI, there is great interest in utilizing biomarkers that are more specific to the kidneys [19]. Before detailing how the aggregate findings of these biomarkers can be interpreted, a brief introduction to a few relevant AKI biomarkers is warranted.
Biomarkers of acute kidney injury
Insulin-like growth factor binding protein 7 and tissue inhibitor of metalloproteinase 2
In renal epithelial cells, insulin-like growth factor binding protein 7 (IGFBP7) and tissue inhibitor of metalloproteinase 2 (TIMP-2) induce cell cycle arrest during AKI through autocrine and paracrine mechanisms [217-219]. IGFBP7 and TIMP-2 can be measured in the urine and both have been shown to outperform >330 biomarkers in predicting AKI within 6-12 hours, including neutrophil gelatinase-associated lipocalin (NGAL), kidney injury molecule-1 (KIM-1), interleukin-18 (IL-18) and liver type fatty acid binding protein (L-FABP) in predicting the incidence of AKI in clinical settings [220]. Moreover, the mathematic product of IGFBP7 and TIMP-2 ([IGFBP7•TIMP-2]) has an FDA-approved indication as a tool for assessing the risk for AKI (better known as NEPHROCHECK®) [221], which is the only AKI biomarker to have received this designation [221]. Thus, [IGFBP7•TIMP-2] is likely the best available marker of the risk of AKI (Figure 3). Moreover, independently examining changes in IGFBP7 and TIMP-2 provides insights into the potential location of tubular injury given that IGFBP7 is preferentially secreted by proximal tubule epithelia and TIMP-2 is secreted to a larger degree by distal tubule epithelia [222] (Figure 3).
Figure 3.
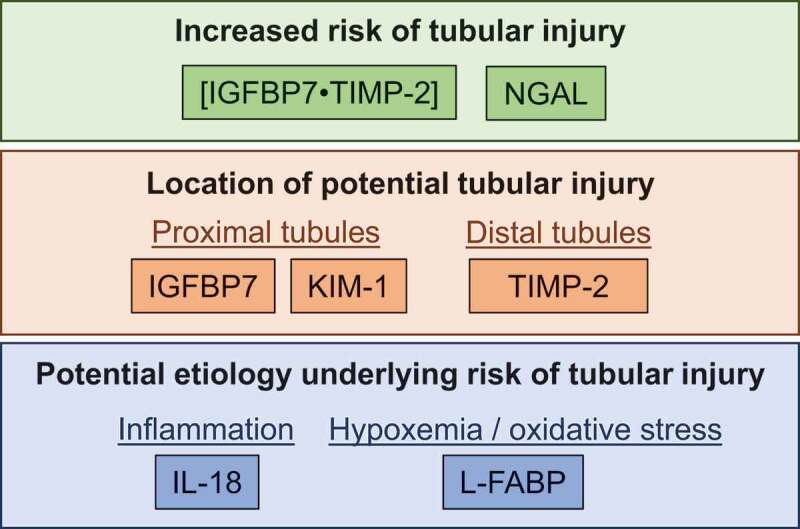
Example of how increases in urinary biomarkers of acute kidney injury can be used to identify the relative risk of tubular injury, the location of this potential injury, and the etiology that may be underlying the risk of tubular injury. Abbreviations – [IGFBP7•TIMP-2]: the product of insulin-like growth factor binding protein 7 (IGFBP7) and tissue inhibitor metalloproteinase-2 (TIMP-2), KIM-1: kidney injury molecule-1, IL-18: interleukin-18, L-FABP: liver-type fatty acid binding protein
Neutrophil gelatinase-associated lipocalin
NGAL is the most widely studied biomarker and is measured in the plasma as a general indicator of renal ischemia or in the urine as a marker of general tubular injury [223-226]. NGAL functions in the immune response as a bacteriostatic agent to form a complex with iron-binding siderophores to enhance the delivery of iron [227], blunt iron-catalyzed damage [228] and ultimately convert renal progenitor cells into tubule epithelial [229]. NGAL expression in renal tubular epithelial cells is upregulated following ischemia-induced AKI [230]. Increases in plasma NGAL are often interpreted as an indication of renal ischemia [223] given that it is mainly expressed in the kidneys, but this conclusion may be clouded by the fact that NGAL is also expressed in hepatic and cardiac tissues, albeit to a lesser degree than in the kidneys [231]. NGAL in the circulation is filtered by the glomeruli and reabsorbed by the proximal tubules [232]. Increases in NGAL in the urine are likely more indicative of NGAL production in the kidneys, coupled with reductions in tubular NGAL reabsorption, to provide utility as a marker of general tubular injury [223,233,234] (Figure 3).
Kidney injury molecule-1
KIM-1 is a transmembrane glycoprotein that is expressed at low levels in the healthy kidneys but becomes highly expressed with intrinsic AKI [235,236]. During AKI, KIM-1 is mainly upregulated in proximal tubule cells, which is reflected by increases in urinary KIM-1 [237,238]. Thus, increases in urinary KIM-1 likely indicate proximal tubule injury, although the etiology is nonspecific (Figure 3).
Interleukin-18
Urinary IL-18, a proinflammatory cytokine, increases with ischemic or nephrotoxic AKI that occur within 6 hours of tubular injury [225,239]. IL-18 is produced in the intercalated cells of the collecting ducts of healthy kidneys [240], but is more broadly expressed in tubular epithelial cells as part of the inflammatory cascade induced by AKI [241]. Thus, urinary IL-18 is likely a marker of general tubular injury and subsequent inflammatory pathway activation (Figure 3).
Liver type fatty acid-binding protein
L-FABP is expressed in the proximal tubule and can be detected in the urine within 6 hours of ischemic kidney injury [242-244]. L-FABP is prophylactically expressed to protect against oxidative stress induced by peroxisomal metabolism [245], particularly in the presence of hypoxia given that the human L-FABP gene contains a hypoxia-inducible factor 1⍺ response element [246]. Therefore, L-FABP may provide an indication that the mechanism of AKI is related to proximal tubule hypoxia and the development of oxidative stress (Figure 3).
Interpretation of acute kidney injury biomarkers
The measurement of AKI biomarkers was intended to identify AKI in clinical situations, in which large and sustained elevations would be interpreted as kidney damage [247,248]. However, increases in these AKI biomarkers are also unexpectedly observed in various settings (e.g., endurance exercise [249-254],). In these cases, the elevations in AKI biomarkers are transient and of a lower magnitude than those observed in clinical situations [220,224,230,246,255,256]. These small and transient increases in AKI biomarkers are often interpreted as meaningful but are not likely indicative of a clinical AKI event. Rather, increases in AKI biomarkers in these non-clinical situations likely reflects an increased potential to develop AKI because some underlying pathophysiological processes taking place [19]. This interpretation is consistent with the idea that small and transient increases in AKI biomarkers are indicative of acute kidney stress [257]. Accordingly, elevations in AKI biomarkers represents a state in which there is an increased risk of developing AKI and that the magnitude of this risk is proportional to the magnitude of elevations in AKI biomarkers. Notably, this approach allows for the assessment of AKI risk between imposed experimental conditions. However, the extent to which the relative risk of AKI reflects absolute risk AKI is unknown.
Currently, a consensus AKI biomarker does not exist. However, as previously discussed, only urinary [IGFBP7•TIMP-2] has an FDA approved indication for the spot assessment of the risk of AKI [258], suggesting that this might be the best single indicator of examining the AKI risk. There is likely utility in employing a battery of AKI biomarkers to quantify the risk of AKI because AKI biomarkers can also be employed to report the anatomical location of this risk and to understand the etiology underlying this potential pathology. For instance, differential peak increases in urinary NGAL and/or urinary [IGFBP7•TIMP-2] may be used to identify the presence of the potential for tubular injury, isolated assessment of IGFBP7, KIM-1 and TIMP-2 may provide insights regarding whether the potential tubular injury is isolated to the proximal tubules (IGFBP7, KIM-1) and/or distal tubules (TIMP-2), and assessment of urinary L-FABP and IL-18 could provide an indication that AKI risk is related to tubular hypoxia and the development of oxidative stress (L-FABP) and/or inflammatory activation (IL-18) (Figure 3). It is also likely important to consider the response of AKI biomarkers after correcting for urinary concentrating ability, e.g., using measures of urine flow rate [65] or osmolality [252,254]. For example, urinary [IGFBP7•TIMP-2] appears to be elevated in healthy individuals with normal kidney function at higher urine osmolality (>600 mOsm/kg), which suggests that correcting to the concentration of the urine may improve the specificity of the biomarker [259,260]. To correct to osmolality or urine flow rate, it is critical to obtain precisely timed urine samples to accurately assess the excretion rates of AKI biomarkers. Thus, correcting for urine production or concentration may not be practical outside of laboratory-controlled conditions. Finally, the timing of when biological samples are collected during the post-exercise periods is important due to the differences in the kinetic response of biomarkers in the blood and urine [63,261]. Therefore, future studies should include sample collections beyond the initial post-exercise period to ensure that a delayed latency in urine AKI biomarkers is not missed due to study design [261,262]. Notably, there is not yet a consensus for best practices for collecting AKI biomarkers beyond the post-exercise period.
Physiological response of the kidneys to heat stress
Passive heat stress
Renal blood flow
In 1943, Byfield et al. [263] reported that a four hour exposure to a 37.5°C, 19% relative humidity environment decreased renal plasma flow by ~30% in three patients with either essential hypertension (n=2) or glomerulonephritis (n=1). These findings were confirmed six years later in healthy adults whereby renal plasma flow decreased by ~38% following seated exposure in a 50°C environment [264]. Since this time, these passive heat stress induced reductions in renal plasma flow have been replicated in many studies [159,160,265-271]. Collectively, depending on the intensity or duration of the heat stress, increases in skin temperature and/or increases in core temperature ranging from ~0.5-2.0°C decreased renal plasma flow by 15-30% [272]. As demonstrated in a classic study by Rowell [271], the redistribution of blood flow away from the kidneys with passive heat stress is thought to be critical to maintain blood pressure at a time when skin blood flow tremendously increases [273].
Activation of the sympathetic nervous system appears to be the primary mechanism by which heat stress causes vasoconstriction in the kidneys [272,274]. In support of this, heat stress-induced increases in RSNA have been reported in rats [275-280] and cats [281] across a wide range of core temperatures. Furthermore, increases in RSNA also occur with partial body heating, such as with the rat’s tail [282-285]. Importantly, there are additional redundant pathways (e.g., renin-angiotensin-aldosterone system, vasopressin, arterial baroreflex) that can induce renal vasoconstriction, but there is currently no direct evidence investigating the interdependent effects of all these systems on renal vasoconstriction during heat stress. That said, Eisman and Rowell [286] reported that renal vasoconstriction during whole body heating in baboons was largely attenuated with infusion of propranolol, which blocks the β-adrenergic release of renin, or with saralasin, which competes with angiotensin II for AT1R receptor-binding. This evidence suggests that angiotensin II also plays an important role in the renal vasoconstrictor response to heat stress. To our knowledge, despite evidence that vasopressin has vasoconstrictor actions in the kidneys [287], there has not been a study to further elucidate the mechanism by which vasopressin may induce renal vasoconstriction during heat stress. Indirect evidence suggests that this renal vasoconstriction to heat stress is not caused by the arterial baroreflex, due to evidence suggesting that vasomotor tone in other visceral vascular beds (e.g., the splanchnic circulation) are not modified by the arterial baroreflex in humans [288].
In addition to reductions in renal blood flow, there may also be a heterogenous redistribution of intrarenal blood flow in response to heat stress. In support of this, Miyamoto [289] reported hyperthermia-induced reductions in cortical renal blood flow and the maintenance of medullary renal blood flow in dogs. Notably, this experimental approach used extreme heat stress with increases in core temperature exceeding 4°C (end core temperature of 42-43°C). Whether heat stress elicits a heterogenous redistribution of intrarenal blood flow in humans when increases in core temperature are in the physiological range remains unknown.
Glomerular filtration rate
Studies investigating the effect of passive heat stress on GFR in healthy adults have shown variable results (Table 4). The collective interpretation of these findings is difficult due to the use of various heat stress models (e.g., dry vs. humid conditions, environmental chamber vs. water-perfused suit), differences in clearance techniques used to measure GFR (e.g., inulin, mannitol, creatinine), the position of the subject during the exposure (i.e., seated vs. supine), lack of core temperature data reported in some studies, and that some studies have subjects drinking water throughout the protocol, which is likely necessary to maintain urine flow rate, but has the consequence of preventing dehydration. For instance, while Byfield et al. [263] reported no changes in GFR (inulin clearance) following a two hour passive exposure in a 37.5°C, 19% relative humidity environment, Radigan and Robinson [32] reported a 21% decrease in GFR (mannitol clearance) in healthy adults following a 60 min resting exposure in a reclined position in a hot and dry environment (50°C, ~17% relative humidity). Notably, subjects drank 1.2-1.5 L of water during the protocol in the latter study. In a follow-up study using the same experimental model, Smith et al. [290] reported contrasting results, where GFR was only decreased during heat stress when the subjects became markedly dehydrated. However, it is not clear if the ~12% decrease in GFR in the dehydrated group was strictly due to the dehydration or the markedly higher core temperatures reached with dehydration (38.5°C vs. 37.3°C). In hot and humid conditions, Kenney [269] reported that GFR was reduced to 108 ± 5 mL/min in a 43°C, 60% relative humidity environment compared to a GFR of 131 ± 15 mL/min elicited by exposure to a 28°C, 80% relative humidity environment. Using a Finnish sauna bath exposure (~67°C, 15% relative humidity environment), Haapanen [266] reported GFR (inulin clearance) decreased by ~20% following ~50-60 min supine rest that resulted in an end rectal temperature of 38.3°C. Finally, passive heat stress invoked by an environmental chamber [291] and a water-perfused suit [292] have demonstrated no changes in GFR when assessed using creatinine clearance.
Table 4.
Summary of studies investigating the effects of passive heat stress on glomerular filtration rate
| Reference | Number of subjects (demographics) | Environmental conditions | Duration of exposure | Water during protocol (volume) |
Change in body weight | Peak core temperature | Method of assessment | Change in GFR from baseline | Magnitude of change in GFR from baseline (% and absolute) |
|---|---|---|---|---|---|---|---|---|---|
| Byfield et al. 1943 [263] | 3 (men, healthy, aged 14-37 years) |
37.5°C,19% or 55% RH | 2-4 hours | Not reported | Not reported | Not reported | Inulin clearance | ↔ A | |
| Radigan and Robinson 1949 [264] | 5 (men, healthy, age not reported) |
50°C, 17% RH | 1 hour | Yes (1.2-1.5 L) |
Not reported | Not reported | Mannitol clearance | ↓ | −21% (−22 ml/min) |
| Smith et al. 1951 [290] | 6 (men, healthy, age not reported) |
50°C, 14% RH | 1 hour | Yes (1.2 L) No |
Not reported -4.6% |
37.3°C 38.5°C |
Mannitol clearance | ↔ B,C ↓ B,C |
−12% (−11 ml/min) |
| Kenney 1952 [269] | Not reported (men) |
38.3°C, 71% RH 43.3°C, 71% RH |
Not reported | Not reported | Not reported | Not reported | Not reported | ↓ B,D ↓ B,D |
−16% (−21 ml/min) -18% (−23 ml/min) |
| Haapanen 1958 [266] | 6 14 (men, healthy, aged 20-48 years) |
68°C, 12% RH 67°C, 15% RH |
48 minutes 58 minutes |
Water only (Not reported) Water + Saline (Not reported) |
Not reported | 38.2°C 38.3°C |
Inulin clearance (single injection) Inulin clearance (continuous infusion) |
↓ ↓ |
−21% (−24 ml/min) -19% (−23 ml/min) |
| Melin et al. 2001 [291] | 8 (men, healthy, aged 27 ± 4 years) |
Environmental chamberE | ~2 hours | No | −2.8% | 38.0°C | Creatinine clearance | ↔ | |
| Ravanelli et al. 2020 [292] | 12 (7 men, 5 women, healthy, aged 26 ± 5 years) |
Water-perfused suit (50°C) | 59 minutes | No | −1.1% | 38.3°C | Creatinine clearance | ↔ |
Abbreviations – RH: relative humidity
AAccording to text by authors. Variable responses reported within small sample size. Subjects were not exposed to the same duration or environmental conditions.
BComparisons were made in different conditions on separate days (i.e., did not compare pre- versus post-condition).
CSubjects drank water in one trial and were dehydrated in the other trial.
DCalculated as a change from 27.8°C, 80.7% RH condition.
EEnvironmental conditions were adjusted from 45°C, 70% RH to 50°C, 30% RH to clamp auditory canal temperature at 38.0°C until 2.8% body mass loss was achieved.
It is reasonable to speculate that the large variability between studies examining the effects of passive heat stress on GFR exists due to the previously described differences in study design. However, there also exists two potential physiological reasons, albeit speculative, that may explain this variance. First, as previously described (see Autonomic control of kidney function), there is a graded response of the kidneys to RSNA where decreases in GFR are only observed at relatively high frequencies of RSNA [141]. In support of this hypothesis, Low et al. [293] have previously shown that with increasing hyperthermia there is a graded response of muscle sympathetic nerve activity, a technique typically interpreted to reflect general sympathetic nervous system outflow. Thus, it may be that the magnitude and/or duration of heat stress in these previous studies elicited increases in RSNA that were subthreshold of the frequency required to alter GFR. A second potential mechanism that may explain some of the variability between studies is renal functional reserve (see Glomerular filtration rate), which provides a stopgap for the kidneys to maintain GFR, in the presence of a reduced renal perfusion, through hyperfiltration. However, to our knowledge, the interaction between renal function reserve in maintaining GFR during heat stress has never been studied. Collectively, given the extremely variable findings and models used between studies, further exploration into the response of GFR during passive heat stress is warranted.
Water and electrolyte regulation
It has been well documented that heat stress and dehydration elicit numerous physiological responses to conserve water and maintain electrolyte balance. In 1921, Adolph [15] observed reductions in urine output associated with higher volumes of sweat during heat stress. These heat stress-induced reductions in urine output with heat stress are associated with decreases in urinary sodium and chloride excretion, which is indicative of an increased renal reabsorption of electrolytes [294]. These changes happen rapidly, such that sodium excretion rates have been observed to fall below 20 µequiv./min and urine flow rates below 0.3 mL/min within 30 minutes of the onset of sweating during exposure to a 41°C environment [295]. This increased electrolyte reabsorption and decreases in urine production are likely mediated by the actions of the renin-angiotensin-aldosterone system [296-300] and increases in circulating vasopressin [301].
Autonomic control
Tests evoking non-exercise sympathoexcitatory stimulation (e.g., cold pressor test, head-up tilting, mental stress) can be used to probe alterations in renal vascular control under various environmental and physiological stressors. For example, during normothermia mental stress results in a 14% decrease in renal plasma flow and 48% increase in renal vascular resistance [302]. Similar findings have been reported using the cold pressor test [127] and head-up tilting [160]. Heat stress likely modulates renal vascular control to sympathetic stimulation in a manner dependent on the magnitude and/or duration of heat stress and the technique used to provoke sympathetic stimulation. For instance, there are no changes in renal vascular conductance elicited by forearm heating that increased muscle temperature by ~5°C and skin temperature by ~9°C, but did not increase core temperature [130]. By contrast, renal vascular control to sympathetic stimulation is likely altered at various levels of whole body heat stress. For example, twenty minutes of head-up tilting during heat stress that increased core temperature by ~0.4°C caused a heightened increase in renal vascular resistance compared to when head-up tilt was completed during normothermia [160]. By contrast, our laboratory has identified that increases in vascular resistance in both the renal and segmental arteries (Doppler ultrasound) during the cold pressor test were attenuated during passive heat stress that increased core temperature by ~1.2°C [303]. The increase in muscle sympathetic nerve activity during a sympathetic stimulus is not affected by passive heat stress [304]. Thus, it is likely that the attenuated increase in renal vascular resistance during sympathetic activation with passive heat stress is due to reduced vasoconstrictor responsiveness to a given level of sympathetic activation, such as occurs in nonrenal vascular beds where increases in local vasodilators, such as nitric oxide and ATP, evoke a sympatholytic effect [305]. The reason for the heightened increase in renal vascular resistance with head-up tilt [160] and attenuated increase in renal vascular resistance with the cold pressor test [303] remains unknown, but may be attributed to differential increases in core temperature (0.4°C vs. 1.2°C) and/or the sympathoexcitatory stimulus (head-up tilt vs. cold pressor test).
Exercise in the heat
Independent effect of exercise on kidney function
Exercise is an independent modulator of kidney function. Therefore, it is important to introduce the effect of exercise on kidney function prior to describing the interactions of exercise and heat stress. As such, it is well established that renal blood flow decreases during exercise [306] and that decreases in renal blood flow are linearly and inversely related to relative exercise intensity, which ultimately aid blood pressure regulation [307]. Renal venous overflow of norepinephrine, a surrogate measure of RSNA, increases at higher exercise intensities, suggesting that renal vasoconstriction during exercise is at least partially sympathetically mediated, with the activation of the renin-angiotensin system likely also playing a role [308]. GFR is either maintained or increases with light exercise (~25% maximal oxygen uptake (O2max)), but begins to decrease from moderate (~40% O2max) to heavy exercise (>80% O2max) [309]. Moreover, to offset plasma volume losses due to hydrostatic pressure, osmotic pressure and/or sweat losses with exercise, these reductions in GFR are coupled with increased sodium reabsorption, which reduces sodium excretion as exercise intensity increases [309]. In addition to renal vasoconstriction, the kidneys release several compounds into the systemic circulation during exercise, including renin and norepinephrine [307]. Interestingly, changes in kidney function during exercise are mostly proportional to the relative workload. Thus, exercise training does not appear to affect the kidney responses to exercise, with the exception being when comparisons are made between individuals of differing levels of aerobic fitness during exercise at the same absolute workload [307]. In these instances, changes in renal blood flow of individuals with higher fitness will be less for a given absolute workload, because the relative intensity is lower. Thus, trained individuals exhibit less renal vasoconstriction in response to the same absolute exercise intensity [310]. This is perhaps best highlighted by data from Ho et al. [157], who observed reduced renal vasoconstriction for a given absolute work intensity in older adults (~65 years) following 4 weeks of intense aerobic exercise training.
Renal blood flow
Radigan and Robinson [264] were the first to show a decrease in renal plasma flow with 60 minutes of exercise in the heat (50°C) compared to exercise in a 21°C environment, with the majority of the augmented reductions in renal plasma flow occurring within the first 20 minutes of exercise. Strikingly, in the over 70 years since these observations were published, the precise latency and magnitude of the exaggerated reductions in renal blood flow during exercise in the heat remain ill-defined due to lack of available techniques that can measure both rapid and dynamic changes in renal blood flow and are practically feasible to utilize during exercise in the heat. Nevertheless, in a follow-up study, Smith et al. [290] identified that renal plasma flow was further decreased during exercise in the heat in dehydrated men compared to when they were well hydrated. Since these early studies, reductions in renal plasma flow as a physiological response to exercise have been consistently reported during exposure to warm (30°C, 60% humidity) [158] or hot (36°C) [157,311] environments.
Glomerular filtration rate
Reductions in GFR are exacerbated when exercise is carried out in the heat [264,290]. Moreover, exercise induced reductions in GFR are ~50% and ~44% lower when dehydrated compared to when euhydrated in a moderate and hot environment, respectively [290]. Interestingly, even incomplete fluid replacement may abolish these effects of dehydration on GFR. For instance, Otani et al. [312] identified that voluntary fluid ingestion (replacing ~32% estimated sweat loss), partial fluid ingestion (replacing ~50%) and full fluid replacement (100%) all attenuated reductions in creatinine clearance, but there were no differences between fluid replacement strategies. Similarly, our laboratory recently found that replacing 100% of body weight loss by drinking water throughout two hours of exercise in the heat attenuates reductions in creatinine clearance by ~16% compared to a trial without fluid replacement [63]. We also identified that attenuating the rise in core temperature via upper body skin cooling better maintained creatinine clearance during exercise in the heat compared to drinking water, which suggests that cooling during exercise in the heat may better support GFR compared to drinking [63]. However, more evidence is needed with gold standard measures of GFR (e.g., inulin clearance). The mechanisms by which reductions in GFR are exaggerated during exercise in the heat and further accentuated by dehydration remain largely unexplored. However, the GFR response to exercise in the heat is unlikely related to the actions of angiotensin II. This is supported by evidence that angiotensin converting enzyme inhibitors (ACEi), which block the conversion of angiotensin I to angiotensin II, do not affect creatinine clearance compared to placebo during exercise in the heat [313]. Thus, the simplest explanation may be that these greater reductions in GFR are caused by decreases in renal blood flow such that the glomeruli can only filter the blood that they receive.
Water and electrolyte regulation
Increases in circulating vasopressin are nearly two-fold higher following exercise when compared to passive heat stress sufficient to elicit equivalent increases in core temperature [301]. This suggests that exercise and heat stress have synergistic effects on vasopressin release. In general, circulating aldosterone and vasopressin increase in a graded manner with hypohydration and exercise intensity during exercise in the heat [314]. The same is likely also true for renin release and the actions of angiotensin II such that ACEi attenuates the rise in plasma aldosterone during exercise in the heat [313]. However, ACEi does not differentially affect urine flow rate or urinary sodium excretion [313]. Nevertheless, the hormonal milieu associated with exercise in the heat clearly supports conditions of water and electrolyte conservation. To the contrary, however, exercise in the heat is a condition in which the capacity of the kidneys to conserve water and electrolytes is relatively impaired. When exercise intensity exceeds 60% O2max, there is a decline in urine concentrating ability that is reflected in elevations in free water clearance despite reductions in urine flow rate [309,315-317]. Melin et al. [318] have reported that hypohydration prior to engaging in moderate exercise in the heat decreased urine concentrating ability despite augmented increases in circulating vasopressin. Otani et al. [312] extended these findings by demonstrating that only full fluid replacement was sufficient to mitigate decrements in urine concentrating ability during prolonged exercise in a hot environment, where reductions in urine concentrating ability persisted when replacing ~50% of sweat loss with drinking water. This finding is paradoxical because a relatively more dilute urine is being produced (i.e., increased free water clearance) despite the overall reduction in urine output during exercise in the heat. It has been speculated that exercise and/or heat stress induced reductions in the medullary osmotic gradient, likely occurring secondary to sympathetic activation, may compromise the ability of the kidneys to concentrate the urine [291,312,318]. Importantly, such a mechanism indicates that urine specific gravity may not be an accurate measurement of hydration status during or following intense exercise. Notably, urea is also an important factor in the urine concentrating mechanism [319]. However, to our knowledge, no studies have investigated how urea may alter the ability of the kidneys to concentrate the urine during exercise in the heat.
Autonomic control
To our knowledge, the autonomic control of renal blood flow and/or kidney function during exercise in the heat has never been explored. A reexamination of data from Smith et al. [290] indicates that after core temperature is increased by ~0.8°C acute increases in renal vascular resistance (PAH clearance) are attenuated during treadmill walking, which activates the sympathetic nervous system. In line with these findings, it is possible that the activation of the sympathetic nervous during exercise in the heat results in a blunted increase in renal vascular resistance, as is observed during passive heat stress [303]. However, this has never been reported in the literature.
Pathophysiological responses of the kidneys to heat stress and exercise in the heat
Proteinuria
Post-exercise proteinuria is a well-documented phenomenon that appears to be more dependent on exercise intensity compared to exercise duration [320,321]. Poortmans & Vanderstraeten [322] suggested that urine albumin excretion is caused by increased glomerular permeability and/or saturation of the renal tubular capacity. In most cases, this post-exercise proteinuria appears to be transient in nature and typically resolves within 24-48 hours [323]. However, excessive proteinuria, which is typically unlikely during normal exercise, can result in progressive glomerular injury, tubulointerstitial damage, and renal interstitial inflammation [324,325]. Although proteinuria can occur with fever [326], it is unlikely that proteinuria is elicited by moderate passive heat stress [292]. However, proteinuria is likely more prevalent with exercise in the heat. For instance, the incidence of proteinuria in agricultural workers in hot climates ranges between 5-30% depending on the region [327-330]. Despite that this proteinuria is typically transient, proteinuria may still be a risk in some individuals during occupational (e.g., agricultural work, construction, firefighting) [331-333] and exercise heat stress [334]. Our laboratory has reported that when exercise intensity and duration are fixed, greater magnitudes of hyperthermia and dehydration during exercise in the heat exacerbate post-exercise proteinuria [63].
Acute kidney injury
Emerging evidence suggests that the kidneys may be at risk of AKI during exercise in the heat [18,26,52,55,64-66,335-337]. This point is highlighted by the findings from Schrier et al. [20] that previously healthy military recruits were clinically diagnosed with AKI that resulted in hospitalization following exercise in the heat. The investigators subsequently estimated that ~10% of all AKI cases under treatment at the Walter Reed General (Military) Hospital from 1960-1966 were caused by exercise in the heat [21]. In a more recent retrospective analysis, Donham et al. [22] reported that ~40% of the active-duty United States military service members hospitalized with exertional heat stroke between 2007-2014 were diagnosed with AKI.
Findings from a rodent model indicated that limiting dehydration by drinking water during recurring bouts of heat stress limits the extent of kidney damage [52]. Likewise, using a similar rodent model, Sato et al. [60] have provided evidence indicating that the extent of hyperthermia during repeated bouts of heat stress may modulate the extent of kidney damage. These rodent models employed passive heat stress. To our knowledge, no studies have examined the effect of passive heat stress on AKI risk in humans. Nevertheless, the rodent data from these studies and others are advantageous for examining the mechanisms associated with kidney damage from heat stress.
Exercise places additional stress on the kidneys beyond that of heat stress and dehydration. Thus, the compounding interactions of exercise in the heat with dehydration places a large stress on the kidneys, which increases the risk of AKI as reflected by elevations in AKI biomarkers. For example, Junglee et al. [65] reported that a 1.3°C increase in core temperature and ~1% body weight loss resulted in elevated serum creatinine, reduced urine flow rate, and increased plasma NGAL. Our laboratory extended these findings and reported that longer exercise durations in the heat, which produce greater rises in core temperature and magnitudes of dehydration, resulted in larger increases in serum creatinine and plasma NGAL [64]. McDermott et al. [66] reported similar findings with elevations in serum creatinine and serum NGAL following 6 hours of exercise in the heat that resulted in a 1.6% reduction in body weight.
Based on this background, in a recent study we sought to determine the relative importance of attenuating the development of hyperthermia and/or dehydration during exercise in the heat on the magnitude of AKI biomarker responses [63]. In four quasi-randomized exercise trials in a 40°C, 30% relative humidity environment, subjects received either water to match losses in body weight to remain euhydrated (Drinking), continuous cooling via a suit top perfusing 2°C water (Cooling), both interventions (Cool+Drink), or no intervention (Control). (Figure 4). We found the greatest reductions in kidney function and largest increases in AKI biomarkers in the Control trial, which elicited the greatest magnitudes of hyperthermia and dehydration, as evidenced by greater increases in serum creatinine, urine albumin, plasma NGAL, urine NGAL, and urine IGFBP7, and greater reductions in urine flow rate. Further examination of these data indicates that the greatest increase in core temperature and magnitude of dehydration (Control trial) also elicited the highest peak [IGFBP7•TIMP-2] levels, independent of time, and that alleviating hyperthermia (Cooling trial) or dehydration (Drinking trial) resulted in peak [IGFBP7•TIMP-2] levels that were not different from when both hyperthermia and dehydration were at their lowest (Cool+Drink trial) (Figure 4). Collectively, these findings support that peak [IGFBP7•TIMP-2] is sensitive to hyperthermia and dehydration evoked by exercise in the heat, that the risk of AKI is greatest when the largest magnitudes of hyperthermia and dehydration are combined, and that this risk is alleviated by mitigating hyperthermia and/or dehydration. Interestingly, there were no differences in urine TIMP-2 between trials, either peak (Figure 4) or over time. Based on the preferential secretion of IGFBP7 in the proximal tubule and TIMP-2 in the distal tubule [222], we interpreted these dichotomous responses of urine IGFBP7 and TIMP-2 as indicating that the proximal tubules are the likely site of renal pathophysiology provoked by greater magnitudes of hyperthermia and dehydration during exercise in the heat [63].
Figure 4.
Continuous upper body cooling and drinking to replace sweat losses to minimize dehydration (Cool+Drink), which attenuated the rise in core temperature and reductions in body weight during 2 hours of exercise in the heat (A), attenuates peak urinary [IGFBP7•TIMP-2] compared to cooling alone (Cool), drinking alone (Drinking) or a condition where drinking or cooling were not permitted (Control) (B). This observation is mostly explained by higher peak urinary IGFBP7 concentrations in the Control trial compared to all other conditions (C) and not differential increases in peak urinary TIMP-2 (D). Data are presented as mean (SD). *indicates different from Control (P<0.05), + indicates different from Cooling (P≤0.03). B-D: Data were reanalyzed from Chapman et al. [63]. Data are presented as box and whisker plots. Data were analyzed using a one-way repeated measures (RM) analysis of variance (ANOVA) with actual p-values reported accordingly. Pairwise comparisons were made using two tailed least significant difference post hoc tests. P-values are shown for differences from Control. Abbreviations – Δ: change, [IGFBP7•TIMP-2]: the product of insulin-like growth factor binding protein 7 (IGFBP7) and tissue inhibitor metalloproteinase-2 (TIMP-2)
This potential tubular injury is likely caused by numerous mechanisms primarily stemming from reductions in renal blood flow. The importance of renal blood flow in the etiology underlying AKI risk from exercise in the heat has never been formally established. However, a retrospective analysis of data collected over three studies published from our laboratory that measured both core temperature and plasma NGAL [62-64], a marker of the potential for renal ischemia [223], suggests that renal blood flow likely plays an important role. For instance, the change in core temperature was found to explain a small (~13%) but significant portion of the variance associated with changes in plasma NGAL during exercise in the heat [338] (Figure 5). This analysis also indicated that the relation between increases in core temperature and changes in plasma NGAL is stronger when subjects are dehydrated compared to when euhydrated. This observation provides preliminary evidence of the potential for a magnifying effect of dehydration on AKI risk during heat stress, a tentative conclusion that is consistent with the dose-dependent nature of the relation between hydration status and renal blood flow during exercise in the heat [290] and that the risk of AKI is greatest when dehydration occurs alongside increases in core temperature [63] (Figure 4).
Figure 5.
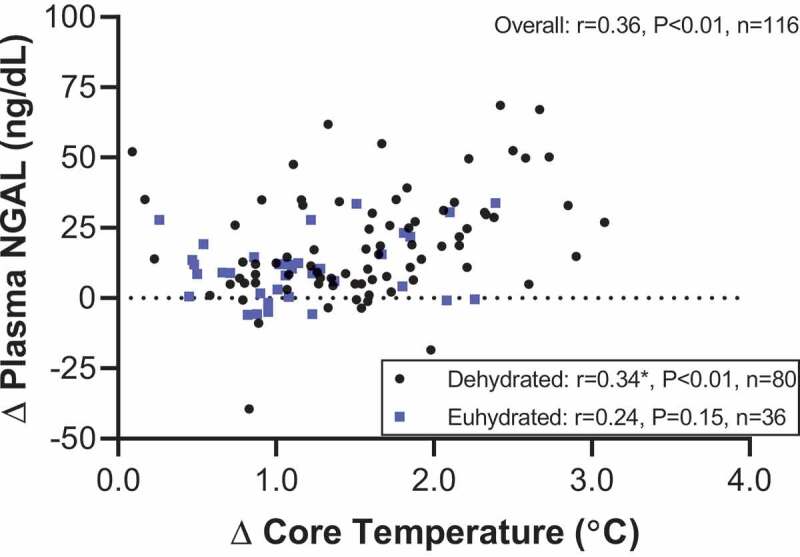
Pearson correlation between the change (Δ) in core temperature evoked by exercise in the heat and the change in plasma neutrophil gelatinase-associated lipocalin (NGAL), a marker of the potential for renal ischemia. Raw data were obtained across three published studies from our laboratory [62-64]. Inset box: Pearson correlations were also conducted separately for trials in which subjects were dehydrated and euhydrated. The resulting correlation coefficients were compared via the methods of Meng et al. [336]. *indicates that the change in core temperature explained more variance in the change in plasma NGAL when subjects were dehydrated compared to when euhydrated (P=0.03)
The mechanisms by which reductions in renal blood flow during exercise in the heat increases the risk of AKI are likely multifactorial (Figure 6). Data from animal models demonstrate that reductions in renal blood flow during heat stress initiates a selective redistribution of blood flow away from the renal cortex [289,339], which house the majority of the proximal tubules [340]. This decreased cortical blood flow can impair oxygen delivery to this anatomical location [341], which occurs despite an increased demand for oxygen caused by the activation of the Na+/K+ pump that is necessary for fluid conservation [291,312]. As a result, tissue oxygenation can become compromised, which can limit ATP production that is necessary for normal cell functioning [60]. Consistent with the hypothesis that dehydration magnifies the risk of AKI during heat stress is that with a background of dehydration, proximal tubule ATP demand is further increased due to an increased drive for water and electrolyte conservation [291,312]. It is also important to note that increases in circulating uric acid, which is generated as an end product of purine metabolism and the breakdown of ATP [342], is common during heat stress and dehydration [63,64], which may also contribute to the increased AKI risk. For instance, in intracellular environments, uric acid is a pro-oxidant that increases the risk of AKI if it accumulates in the circulation (i.e., hyperuricemia) [343]. Moreover, uric acid further increases the demand for intracellular ATP in renal proximal tubular cells [53,344] and can independently decrease renal blood flow [345]. The importance of uric acid as a key modulator of AKI risk with heat stress and dehydration is highlighted by data demonstrating that treatment of mice with allopurinol, a xanthine oxidase inhibitor that reduces serum urate concentrations, decreases the extent of kidney damage caused by recurrent heat stress and dehydration exposures [58]. Finally, it has recently been proposed that the activation of these potentially pathophysiological pathways that ultimately result in ATP depletion is magnified in the presence of a background of inflammation [67]. Indeed, exercise in the heat is a pro-inflammatory state, with the magnitude of inflammation increasing as a function of elevations in core temperature [346], which may provide a background environment conducive to augmenting AKI risk (Figure 6).
Figure 6.
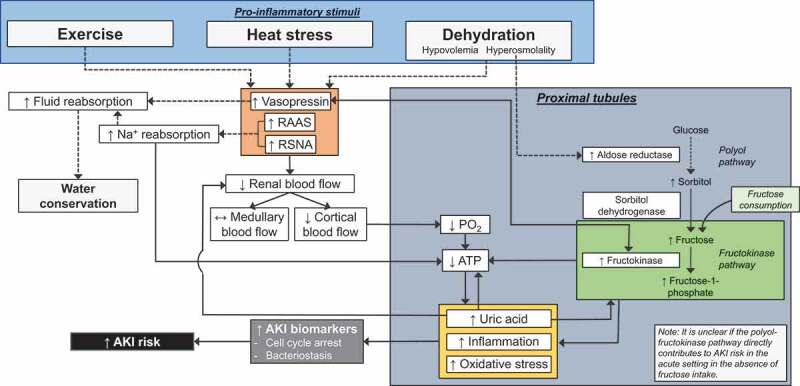
Potential mechanisms by which exercise, increases in core temperature and/or dehydration may increase the risk of acute kidney injury (AKI) while also promoting fluid conservation. Dashed lines indicate known beneficial physiological responses. Solid lines indicate potential pathogenic processes. Abbreviations – RAAS: renin-angiotensin-aldosterone system, RSNA: renal sympathetic nerve activity, PO2: partial pressure of oxygen, ATP: adenosine triphosphate
In recurrent heat stress animal models, increased fructose metabolism in the proximal tubules has also been shown to elicit kidney damage caused by fructokinase activity occurring secondary to endogenous fructose production via the polyol-fructokinase pathway or when consuming fructose sweetened drinks [47-52,57,58,60,61]. The polyol-fructokinase pathway is activated by increased plasma osmolality (Figure 6), which stimulates the conversion of glucose into sorbitol via the enzyme aldose reductase to protect the renal medullary cells against the hypertonic environment [347]. However, this initial protective response has been shown to stimulate fructose metabolism in the proximal tubules with recurrent heat stress and dehydration because of the conversion of sorbitol to fructose [348]. In recurrently heat stressed animals, kidney damage occurs as a secondary effect of fructokinase activity, which in the proximal tubules reduces ATP availability and promotes oxidative stress, inflammation and the production of uric acid [47-52,57,58,60,61], the latter of which can also stimulate the polyol-fructokinase pathway [58,349]. Additionally, the polyol-fructokinase pathway is both partially mediated by, and also mediates, vasopressin release [49,50,57,61]. Strikingly, the importance of the polyol-fructokinase pathway in the potential for kidney injury is demonstrated by data showing that fructokinase knock out mice do not demonstrate kidney damage caused by recurrent heat stress and dehydration [52].
Given the protective nature of sorbitol generation by the enzyme aldose reductase in the initial steps of the polyol-fructokinase pathway [347], it is unclear whether acute activation of this pathway and the potential pathophysiology associated with increased activity of renal fructokinase, contributes to the increased AKI risk during a single bout of heat stress and dehydration, as has been observed in humans [63-65]. Supplemental data published by Garcia-Arroyo et al. [49] indicate that measurable increases in cortical fructokinase are not observed until the third day of repeated heat exposure, which suggests that this pathway is not likely activated during a single bout of passive heat stress and dehydration, but the influence of exercise remains unknown. Unfortunately, however, to our knowledge it is not possible to quantify the activation of the polyol-fructokinase pathway in humans. That said, evidence does support that the polyol-fructokinase pathway can be pathogenic in humans with consumption of a fructose containing soft drink, which exogenously increases substrate for the polyol-fructokinase pathway (Figure 6). Indeed, data from our laboratory indicate that, compared to drinking the same volume of water, consuming a soft drink sweetened with high fructose corn syrup during and following four hours of exercise in the heat exacerbates increases in serum creatinine, reductions in urine flow rate, and elevations in urinary NGAL [62]. Additionally, we reported greater increases in copeptin (a surrogate measure of vasopressin) and serum uric acid, which supports the previously described animal data suggesting the polyol-fructokinase pathway as a mechanism for increased AKI risk with heat stress. In a follow-up study we identified that vascular resistance in the kidneys (Doppler ultrasound) was increased during rest and with sympathetic activation only with soft drinks sweetened with high fructose corn syrup and not those artificially or sucrose-sweetened [167]. This increased renal vasoconstriction appears to be mediated by simultaneous increases in both uric acid and copeptin (i.e., vasopressin). Thus, in total, recent evidence from our laboratory supports data from animal models and indicates that high fructose corn syrup sweetened soft drinks have direct, acute effects on the kidneys [167] which may present a greater risk of AKI during heat stress [62]. Notably, this may have important ramifications regarding fluid prescription in the workplace or during heat waves because of the prevalence of consumption of drinks sweetened with fructose (e.g., soft drinks, fruit juices). That said, some of these drinks contain compounds that may be protective against AKI, such as antioxidants (e.g., vitamin C in fruit juices). The protective effect of antioxidants has been demonstrated by data obtained in rats where antioxidant supplementation protected against kidney damage caused by recurrent heat stress when consuming a fructose sweetened drink [48]. To our knowledge, the potentially protective effects of antioxidant supplements or naturally occurring antioxidants has not been studied in humans at risk of AKI due to exercise in the heat.
Optimal hydration beverages for alleviating the risk of acute kidney injury
Given the etiology underlying the increased risk of AKI during exercise in the heat (Figure 6), there is likely a need to identify optimal hydration strategies. We previously described evidence collectively indicating that hydration practices likely need to occur during the exercise period to minimize the extent of dehydration (and hyperthermia) to reduce the risk of AKI and that, to date, rehydration after heat exposure has not proven effective (see Acute Kidney Injury). In addition to hydration timing, the type of hydration beverage used in occupational settings is of great concern for regulatory agencies whose purpose serves to protect the health and safety of workers. The importance of varying the contents of hydration beverages is highlighted by published recommendations to prevent or alleviate the development of dehydration under conditions of occupational heat stress [350-352]. For instance, the U.S. National Institute for Occupational Safety and Health (NIOSH) recommends drinking 237 mL of cool (<15°C) fluid every 15-20 min to ensure that decreases in body weight are <1.5% during physical work in the heat [352]. Water is recommended if work duration is less than 2 hours, but if work duration is 2 hours or more, NIOSH recommends consuming a beverage containing carbohydrates and electrolytes to the replace electrolytes lost from sweating [352]. However, an important knowledge gap exists because it is not known if these occupational recommendations alleviate the risk of AKI. That said, data obtained from mice exposed to recurrent heat stress [52] and data from our laboratory during a single bout of exercise in the heat [62,63] demonstrate that drinking a volume of water sufficient to prevent dehydration during the heat exposure is protective against AKI. Therefore, if the occupational hydration recommendations stave off dehydration, they should be effective in, at least partially, alleviating AKI risk during exercise in the heat. Moreover, experimental evidence supports that water may be an adequate hydration beverage for preventing AKI provided that water is consumed in a sufficient quantity to prevent dehydration. However, there may be a ceiling effect to the benefits of consuming solely water such that overconsumption of water can lead to hyponatremia [92], which may be associated with increased AKI risk during outdoor work in the heat [353]. However, to our knowledge no studies have directly examined optimal hydration beverages with regards to alleviating AKI risk during exercise in the heat. Notably, identification of optimal fluid replacement beverages in the context of AKI risk is likely multifaceted and depends on many factors including beverage osmolality and composition, and both the duration and magnitude of heat exposure [88].
Based on our understanding of the potential mechanisms by which AKI risk is elevated during exercise in the heat (Figure 6), optimal hydration beverages should likely aim to limit the extent of dehydration and minimize the length of time in a dehydrated state. Thus, the optimal hydration beverage is likely a hypotonic solution containing both carbohydrates and electrolytes (i.e., sodium) [354]. For instance, hypotonic solutions are absorbed in the intestines at a faster rate than hypertonic solutions [355]. In fact, intake of hypertonic solutions can decrease plasma volume owing to an increased net secretion of water into the intestinal lumen during digestion [356]. Moreover, hypotonic solutions containing carbohydrates provide the fastest rate of intestinal absorption due to the active co-transport of solute from the intestinal lumen into the mucosa [355,357]. Notably, hypotonic beverages containing sodium or the addition of sodium to a carbohydrate containing beverages does not modify intestinal fluid absorption [358]. That said, there may be a benefit to ingesting electrolytes during exercise in the heat to alleviate the risk of AKI. For example, workplace data indicate that the consumption of an electrolyte solution during agriculture work in the heat alleviates the risk of AKI [333]. These observations may be explained by decreasing the demand for sodium reabsorption in the kidneys, which would theoretically alleviate ATP demand in the proximal tubules, and sodium-mediated improvements in renal fluid retention in the post-work period [359]. However, this remains speculative. Clearly, therefore, observations from controlled experimental studies are required to identify hydration beverages optimized to avert the risk of AKI during exercise in the heat.
Rhabdomyolysis and acute kidney injury
In both healthy individuals and patients, exercise-induced rhabdomyolysis is a potentially life-threatening condition caused by numerous factors, including but not limited to direct muscle injury, unaccustomed exercise, ischemia, and electrolyte or endocrinological abnormalities [360]. During exertional rhabdomyolysis, myoglobin is one of several muscle proteins released directly into the blood stream following extensive damage to the muscle. Importantly, because myoglobin is cleared by the kidneys, if rhabdomyolysis is left untreated, myoglobin can cause AKI by either precipitating in the kidney or by its direct nephrotoxic actions that stimulate oxidative stress [360]. Muscle damaging exercise increases creatinine content in the blood, which cautions the use of serum creatinine-based estimates of GFR during exercise inducing injury to the muscle [65]. Additionally, dehydration and nonsteroidal anti-inflammatory drugs (NSAID) use during heat stress are associated with increased risk of exertional rhabdomyolysis [361-363]. The incidence of secondary complications of AKI to exertional rhabdomyolysis is not well described in the literature but appears to be a rare occurrence [361,364]. However, this risk appears to be elevated in individuals during heat stress and those with a prior heat injury [365-367].
The risk of AKI during exercise in the heat may be increased in the presence of muscle damage [65] such as occurs in many occupational settings that are often exposed to heat stress, e.g., burnt sugarcane harvesting [368]. For example, a study in Brazilian sugarcane workers observed that serum creatinine based-measures of AKI were associated with increases in serum creatine kinase, a marker of muscle damage, and reductions in serum sodium [353]. These workers consumed large volumes of water (~5-10 L) during the work shift and, with reductions in serum sodium of ~6 mmol/L, it is unclear if the risk of AKI was increased due to muscle damage, hyponatremia, or other environmental factors [353]. That said, further observations demonstrate that declines in kidney function from pre- to post-work shift are not associated with rhabdomyolysis in Guatemalan sugarcane workers [369], a population at risk for AKI and CKD [24]. Additionally, adaptations in skeletal muscle mitigate further damage following the initial bout of exercise-induced muscle damage [370]. Thus, it is likely that the contribution of rhabdomyolysis to AKI from occupational heat stress may be minimal, given the resiliency of skeletal muscle to adapt to repeated exercise (e.g., as occurs in manual labor situations). However, despite the limitations associated with serum creatinine based measures of AKI during exercise in the heat, that markers of muscle damage have been associated with indices of AKI- in a subset of agricultural workers [353] suggests that further investigation is warranted into kidney-related risks associated with muscle damage in occupational settings. As previously mentioned, NSAID use is associated with an increased risk of rhabdomyolysis during exertional heat stress. Ibuprofen exacerbates reductions in GFR during exercise in the heat by reducing prostaglandins [311]. However, they do not appear to differentially elevate plasma NGAL [66]. Whether these observations remain consistent when AKI biomarkers are measured in the urine, which is more specific to the kidneys, remains unknown. Thus, further investigation into the potential nephrotoxic effects of NSAID use during heat stress is warranted.
Thermal burns and acute kidney injury
One might consider a thermal burn as the most severe form of heat stress. A burn is an injury, typically to the skin, that can be caused by extreme heat or other factors such as radiation, electricity, or chemicals. The morbidity associated with burn injuries is high, as evidenced by ~40% of the ~410,000 burn injuries occurring in the United States requiring hospitalization in 2008, and that ~11 million people worldwide suffered burn injuries requiring medical attention in 2004 [371]. In a recent meta-analysis, Folkestad et al. [372] reported that the incidence of AKI is ~38% in burn patients admitted to the hospital. The prognosis is bleak as mortality rates approach upwards of 80% in burn patients with AKI [373]. Proteinuria is a common finding in burn patients [374,375]. These findings are likely due to systemic pathophysiological changes in the hypermetabolic phase [376], which is a profound catabolic response in patients with burns covering ≥20% of total body surface area that can persist anywhere from six months postburn or beyond two years with more severe burn injuries [377,378]. The initial 48 hours of the postburn period is characterized by reductions in metabolic rate and GFR [379,380]. Additionally, renal blood flow is reduced during this period in burn patients and, with the hypovolemia associated with the loss of fluid from the burn wound and intercompartmental fluid shifts into the interstitium, oliguria is observed following a burn [381]. This severe volume depletion in the vasculature in burn patients is associated with elevated concentrations of vasopressin that are 50-fold greater than normal upon hospital admission [382] and requires large resuscitation volumes of fluid to stabilize the patient [378]. It has also been shown that thermal burns may elicit a heterogenous redistribution of intrarenal blood flow, where both moderate (70°C hot water) and severe (90°C hot water) scalding on 30% of the body surface area of dogs for four minutes elicits profound reductions in outer and inner cortical blood flow with no changes in medullary blood flow [383].
In contrast to the initial postburn period, the hypermetabolic phase is characterized by an increase in GFR [380,384] that is likely due to an increased glomerular permeability from the release of inflammatory factors [385-387]. Additionally, renal blood flow is elevated in burn patients during the hypermetabolic phase [388]. Moreover, autopsies by Goodwin et al. [388] revealed a nearly 60% higher kidney weight in patients who died 60 days or more after burn injury compared to individuals who died within 48 hours of their burn. It was subsequently concluded that this increase in kidney weight was primarily due to cellular hypertrophy and hyperplasia [388], which is consistent with the increased work demands imposed on the kidneys during the postburn hypermetabolic phase. Crum et al. [382] reported profound decreases in circulating vasopressin following 48 hours of treatment in burn patients, but plasma atrial natriuretic peptide increased from days 3 to 5 during the hypermetabolic phase. With this background, there remains a paucity of research regarding changes in renal vascular control during the various phases of burn recovery, and if the kidneys are particularly susceptible to additional exposures of heat stress or subsequent burns.
Heat adaptation and the kidneys
Background
Repeated exposures to heat stress that sufficiently elevate core temperature elicit phenotypic alterations (i.e., heat adaptation) that reduce the physiological challenges of subsequent heat exposures. These adaptations, some of which can be observed following as little as three consecutive days of heat exposure, can be induced from heat stress in natural (i.e., heat acclimatization) or artificial (i.e., heat acclimation) environments, and have been demonstrated to improve thermoregulation [389], exercise performance [72,390,391], and markers of cardiometabolic health [392].
There are several methodological approaches to induce heat adaptation, including (but not limited to) fixed work-rate exercise in the heat, where exercise intensity is clamped throughout, self-regulated exercise in the heat, where individuals control work rate based on their own perception of discomfort, and controlled hyperthermia, where core temperature is clamped during the acclimation protocol and can be accomplished in both resting and exercising situations. The fixed work-rate, and to a lesser degree the self-regulated, approaches are commonly used because of their ease of use in group settings (e.g., military personnel). However, both approaches are limited in that the thermal load will differ amongst individuals and, as heat adaptations occur throughout the acclimation period, the magnitude of physiological strain imposed by the heat stressor is attenuated with each heat stress exposure [390]. Notably, the controlled-hyperthermia approach may better optimize heat adaptations because the magnitude of the thermal impulse is maintained [389,390].
As described by Sawka et al. [72], the thermoregulatory adaptations to repeated heat stress are numerous and include lower basal and exercise core temperatures [393,394], lower exercise skin temperatures [395], and improvements in sweating [396] and skin blood flow [397]. These improvements in thermoregulation coincide with alterations in the processes regulating water and electrolyte homeostasis, many of which are influenced by the kidneys, although our collective understanding remains incomplete.
Renal blood flow
To our knowledge only one study has examined the potential adaptive effects of heat acclimation on renal blood flow. In this study, Zappe et al. [398] found that four consecutive days of exercise in the heat did not affect renal plasma flow during rest, exercise in the heat or during recovery in young healthy adults. The four day heat acclimation protocol was sufficient to stimulate plasma volume expansion, suggesting that there was evidence of heat adaptation. That said, the limited duration largely leaves the question regarding whether renal blood flow is modified with heat acclimation in humans unanswered. Nevertheless, data in animals indicate that heat acclimation may actually reduce basal renal plasma flow. Jones et al. [399] reported that after correcting for reductions in renal mass, renal plasma flow is decreased by 15% in the heat acclimated hamster. Additionally, the investigators found that heat acclimation decreased the fractional distribution of cardiac output to the kidneys by 27% and increased renal norepinephrine concentration by 41%, indicating a greater sympathetic outflow to the kidneys following heat acclimation [399]. These findings were further supported by data in rats where three weeks of heat acclimation at ~35°C reduced renal plasma flow by 56% [400].
Glomerular filtration rate
The effect of heat acclimation on GFR has not been fully elucidated, which is likely due to inconsistent findings from the few studies that exist. For instance, there are only two studies that have investigated the effects of passive heat acclimation on GFR. Data in humans demonstrate that seven days of passive, controlled hyperthermia did not alter GFR (creatinine clearance) during normothermia or heat stress [292]. By contrast, Chayoth et al. [400] found that three weeks of passive-heat acclimation in rats lowered GFR (inulin clearance) by ~61%. To further complicate the interpretation, however, the three week heat acclimation protocol caused a ~15% loss in both body and kidney weight. Thus, it is not clear if the results of this study are due to morphological or physiological reasons. Therefore, the effects of passive heat acclimation on GFR remain largely unknown.
Exercise heat acclimation protocols have also revealed variable effects on GFR. Interpreting these studies is difficult due to the differences in the duration and stimulus of heat acclimation across these studies. Furthermore, a gold standard technique (e.g., inulin clearance) has not been used to assess GFR following exercise heat acclimation. In this regard, the current literature has assessed GFR as estimated by serum creatinine (i.e., estimated GFR) or with creatinine clearance. For instance, it has been recently reported that a 23 day heat acclimatization protocol in military personnel attenuated reductions in estimated GFR during exercise in the heat [401]. These findings may be partially supported by data from Schrier et al. [21], who found in four military recruits during summer training in San Antonio, Texas, USA (i.e., heat acclimatization) that creatinine clearance following moderate intensity exercise in the heat was greater towards the end of the acclimatization protocol compared to the earlier phases. However, the authors also note that the increase in serum creatinine (indicative of a reduction in GFR) following a bout of high intensity exercise was not different at 10 days compared to 6 weeks of training. Notably, data before this 10 day period were not reported and, therefore, subjects may have already presented with a fully heat acclimated phenotype by this point in their training [390], which clouds the interpretation of these findings. Nevertheless, recent evidence indicates that reductions in estimated GFR are not altered with six days of heat acclimation [402]. Therefore, it may be that heat acclimation needs to be of a sufficient duration or magnitude before reductions in GFR to exercise heat stress are attenuated. Further research is warranted.
Water and electrolyte regulation
Heat adaptation causes several phenotypic alterations that are beneficial towards maintaining water and electrolyte regulation, despite augmenting sweat losses. These adaptations include an increased total body water, plasma volume expansion, reduced electrolyte losses, and improved thirst response during exercise in the heat [72,389-391,403] These adaptations are at least partially mediated by adaptations in the kidneys. For instance, plasma volume expansion with heat adaptation is elicited via two mechanisms [404] including the conservation of electrolytes by the kidneys and sweat glands, and increased colloidal pressure through increases in plasma protein content [405].
Some of the best early evidence of improvements in renal electrolyte conservation with heat acclimation were identified by Smiles and Robinson [406] who found that individuals on a low sodium diet (i.e., a sodium deficit) experienced marked reductions in urinary sodium excretions with heat acclimation. By contrast, however, individuals on a higher sodium diet during the exercise heat acclimation protocol did not change urinary sodium excretion rate from pre-acclimation values. These findings were later confirmed by Allsopp et al. [407]. Importantly, a study by Francesoni et al. [408] provided evidence that the type of heat acclimation exposure modifies the electrolyte conserving effects. In this study, exercise heat acclimation reduced 24-hour urinary excretions of sodium and potassium. However, passive heat acclimation in the same environmental conditions slightly decreased 24-hour urinary excretion of potassium but did not alter sodium excretion [408]. This unchanged sodium excretion following a short-term passive heat acclimation has also been demonstrated following seven days of hot water immersion using the controlled-hyperthermia approach [292].
The idea that heat adaptation promotes the conservation of electrolytes has prompted studies investigating the effects of heat acclimation on fluid regulatory hormones. For example, heat acclimation does not alter basal plasma concentrations of renin [409-413] nor vasopressin [409,410,414-417]. In response to exercise in the heat, increases in plasma concentrations of renin have been reported to be attenuated [409,412], unchanged [411,413], or potentially increased [410]. It is worth noting that with the exception of one study [413] that used a controlled-hyperthermia approach, these studies controlled for work intensity, which may have limited the magnitude of the heat adaptation stimulus and may account for some of the variability between studies. There are also conflicted findings regarding vasopressin release during exercise in the heat following heat acclimation with reports that vasopressin is decreased [414], unchanged [409,415], or slightly increased [410]. Highlighting this variability, Mudambo et al. [416] reported that heat acclimation attenuates increases in vasopressin during exercise when fluid is not provided, but when allowed to drink water the post-acclimation response of vasopressin was not different from pre-acclimation. This suggests that body fluid status modulates the effect of exercise on vasopressin release following heat acclimation. Overall, while the effect of heat acclimation on resting circulating concentrations of renin and vasopressin are consistent, the response of these hormones to exercise following acclimation requires further study.
To our knowledge, no studies have examined the effect of heat acclimation on resting angiotensin II concentrations in humans. By contrast, over a dozen studies have investigated the effects of heat acclimation on plasma aldosterone, which is likely attributed to the effects of aldosterone at improving electrolyte reabsorption in the sweat gland [72,389]. Despite the plethora of studies, the literature is conflicted with studies reporting heat acclimation increases [409,410,413,418,419] or does not change [411,412,414,415,417,420-422] basal plasma aldosterone concentrations. The cause of this discrepancy between studies is not inherently clear but may be contributed to by differences in the experimental protocols, such as the method of heat acclimation, mode of exercise, training status of subjects, and differences in sodium balance and hydration status. That said, a subset of these studies were included in a meta-analysis from Tyler et al. [390] whose analysis indicated that, despite the large variability between studies, exercise heat acclimation may induce a small positive effect (+25 ± 35%) on resting plasma aldosterone concentrations.
While it appears that heat acclimation increases circulating aldosterone, the literature better supports that sensitivity to aldosterone is increased with heat acclimation. This is supported by studies reporting elevated concentrations of plasma sodium and/or plasma osmolality to exercise in the heat following heat acclimation that is accompanied by attenuated [401,410] or similar [409] post-acclimation increases in plasma aldosterone during exercise in the heat compared to pre-acclimation. Additionally, an increased sensitivity to aldosterone is supported by heat acclimation studies demonstrating attenuated increases (by ~20-30%) in plasma aldosterone to exercise in the heat [412,414,422] accompanied with unaltered plasma sodium responses compared to pre-acclimation [414,422]. In contrast, others have reported that end exercise plasma aldosterone is increased with heat acclimation, but this is likely related to a relative sodium deficit experienced by highly trained individuals during the experimental intervention [418] or as a result of a lower sodium diet [413]. This is supported by the findings of Allsopp et al. [407] who found that five days of heat acclimation increases plasma aldosterone to exercise in the heat when subjects are on a low sodium diet (1,500 mg/day) and that these increases were abolished with a high sodium diet (8,000 mg/day). The modulatory role of sodium intake is further supported by the study of Davies et al. [413] who identified that consuming a saline drink attenuated increases in plasma aldosterone during exercise in the heat following 11 days of heat acclimation. In addition to dietary sodium, the effect of heat acclimation on the fluid regulatory hormonal responses is also modulated by hydration status. Francesconi et al. [419] reported that heat acclimation over 10 days attenuated increases in plasma renin activity and aldosterone during exercise in the heat while dehydrated, further supporting the role of increased aldosterone sensitivity with heat stress. By contrast, it has also been reported that hypohydration during five days of heat acclimation exacerbates increases in plasma aldosterone and vasopressin during exercise heat stress [415]. The reason for these different hormonal profiles is not clear. It may be that these differences can be explained by achieving a more fully heat acclimated phenotype with 10 days of heat acclimation [419]. However, other differences between the studies such as the use of the controlled-hyperthermia approach (compared to fixed-work intensity) or differences in cardiorespiratory fitness levels may also contribute [412,414,418,422]. Nevertheless, despite a minority of studies showing divergent results, the literature supports an increase in aldosterone sensitivity with heat acclimation [389].
The enhanced sensitivity to plasma aldosterone and conservation of electrolytes with heat acclimation stimulates an interesting question regarding whether the urine concentrating ability of the kidneys is augmented with heat adaptation. To our knowledge, only one study has investigated the effects of heat acclimation on urine concentrating ability. This study found that increases in free water clearance to passive heat stress were attenuated after seven days of passive heat acclimation, a finding representative of a relative improvement in urine concentrating ability [292]. The mechanism for these findings remains elusive, but could be related to the enhanced plasma aldosterone sensitivity or improved structural integrity within the nephron.
Pathophysiology and acute kidney injury
Despite the protective effects of heat adaptation, few studies have directly investigated the potential pathophysiological benefits of heat acclimation protocols on the kidneys, and the findings are often conflicting. For example, Ravanelli et al. [292] demonstrated that the incidence of albuminuria during passive heat stress in otherwise healthy individuals was reduced following passive heat acclimation. By contrast, Schrier et al. [21] found that the prevalence of proteinuria was higher at the end of 42 days of summer training in military recruits compared to after 10 days of training, which may indicate that exercise training is a more powerful stimulus driving proteinuria compared to heat stress.
To our knowledge, only three studies have attempted to investigate the efficacy of heat acclimation on reducing the risk of AKI. These investigations employed changes in serum creatinine as their primary endpoint, which is likely a flawed approach (see section PHYSIOLOGY AND ASSESSMENT OF ACUTE KIDNEY INJURY). Despite this limitation, there is still value in such studies as they provide the groundwork from which future studies can be designed employing AKI biomarkers.
In the first of these studies, American Football players were observed over 10 days of preseason training (i.e., heat acclimatization) and serum creatinine was compared at days zero, 5, and 10, from which estimated GFR was calculated [423]. Using this approach, it was found that over 43% of the athletes were classified as at risk of AKI on day 10. However, despite the intent of the study, whether heat acclimatization altered this response remained unknown given that changes in estimated GFR were not calculated within a given day. Rather all comparisons were made with data obtained on day zero. Moreover, it is likely that exercise intensity and duration varied on these measurement days, which further confounds the results. Omassoli et al. [401] later showed stronger evidence that increases in serum creatinine with exercise in the heat were attenuated following 23 days of heat acclimatization and that the extent of attenuations was sufficient to reduce the clinical stratification of AKI according to the traditional clinical classifications. Recently, these findings have been found to be supported by additional evidence that six days of heat acclimation may reduce the rise in serum creatinine caused by high intensity exercise in the heat to such an extent so as to decrease the incidence of AKI defined by traditional clinical criteria [402].
Despite the many beneficial adaptations brought about by heat acclimation, there are, in theory, several mechanisms by which the risk of AKI could be modified with heat acclimation. When examined in isolation, some of the kidney related adaptations with heat acclimation will likely be beneficial and others deleterious. Ultimately, the balance between the positive and negative renal adaptations will determine the effects of heat acclimation on AKI risk during heat stress (Figure 7). To date, the effects of heat acclimation on AKI risk during heat stress remains largely unexplored. For instance, there is a complete lack of information regarding how heat acclimation induced changes in renal blood flow, GFR, and/or renal ATP demand interact to modify AKI risk during heat stress. This is a very important gap in the literature that requires investigation.
Figure 7.
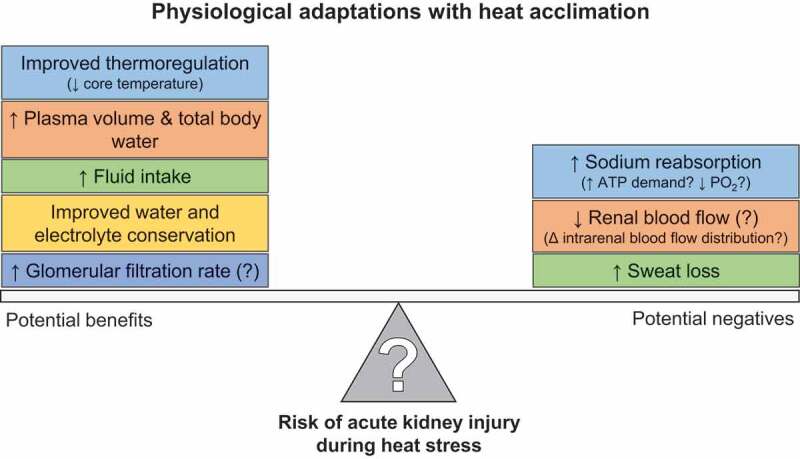
Potential beneficial (on left) and deleterious (on right) adaptations with heat acclimation that may modify the risk of acute kidney injury during heat stress
The kidneys and aging
Background
Kidney function gradually decreases with advancing age [424]. Thus, there is an increased prevalence of CKD in older adults [425], with approximately 45% of adults aged ≥70 years in the United States having CKD [426]. Additionally, polypharmacy is a risk factor for kidney damage [427], which further complicates matters with ~70% of individuals aged 75-84 years receiving ≥5 medications per day [428]. Furthermore, the incidence of AKI in adults ≥65 years is 10 times greater compared to younger adults aged <40 years [429]. Strikingly, approximately one-third of older adults ≥65 years who survive AKI do not fully recover kidney function [430]. Moreover, aging is associated with impairments in thermoregulation and fluid regulation during heat stress that also likely contributes not only to an increased incidence of heat related illnesses and mortality during heat waves, but also AKI [30,32-34,36,40,431,432].
Physiology of aging kidneys
The decline in kidney function associated with healthy aging is caused by many factors, including changes in anatomical structure and renal blood flow regulation. Davies et al. [433] were one of the first to identify that older adults (>60 years) have ~20% lower basal GFR (inulin clearance) and ~30% lower basal renal plasma flow compared to younger adults (<40 years). Larger disparities were observed between age groups for each decade beyond 60 years. This was subsequently followed up by investigations identifying that renal mass decreases with age such that after the age of 50 years kidney parenchymal volume declines by ~10% each decade, with larger decreases in men compared to women [434-436]. In addition to these morphological changes, the reduced kidney function with aging is also contributed to by a progressive loss of functional nephron mass [437]. For instance, Denic et al. [438] reported a 48% decrease in functional glomeruli (nonsclerotic) and a 15% increase in the number of nonfunctional glomeruli (globally sclerotic) with decreases in cortical volume of 16% in healthy adults >70 years compared to those aged 18-29 years. Biopsies from healthy living kidney donors older than 60 years revealed that 36% had >10% glomerulosclerosis with 63% of those individuals having tubular atrophy [439]. Thus, nephrosclerosis, which describes an aggregate of global glomerulosclerosis, arteriosclerosis, interstitial fibrosis and tubular atrophy, increases even with healthy aging [440].
Kenney and colleagues [157-160,311,398,441] have consistently demonstrated that basal renal blood flow is lower in healthy older adults compared to their younger counterparts using PAH clearance techniques. Furthermore, hormone replacement therapy in postmenopausal women does not appear to change basal renal blood flow compared to untreated control subjects [442] despite altering vascular control in other vascular beds [443,444]. Additionally, Doppler ultrasound derived measures of renal artery vascular resistance support that aging is associated with enhanced vasoconstrictor responses to sympathetic stimulation [165,445], which are sustained longer after a given stimulus compared to younger adults [158,446]. Finally, rodent data indicate that an augmented basal activation of the polyol-fructokinase pathway, which we previously described as a pathophysiological pathway during heat stress (see Acute kidney injury), may also contribute to reductions in kidney function observed with aging [56].
Given these impairments in kidney function, it is perhaps not surprising that water and electrolyte regulation is altered in older adults. For instance, there is an increased risk of dehydration and hyper- or hyponatremia in older adults [447]. According to Beck [448], a 70 kg male aged 75 years would have 7-8 liters less total body water than a younger person due to changes in body composition with aging (e.g., increased fat mass). Furthermore, plasma volume is reduced up to 21% in older adults compared to younger adults [449]. This decreased blood volume is further exacerbated by a ~50% reduction in urine concentrating ability in older adults [450], despite increased vasopressin secretion [451] and higher osmoreceptor sensitivity [452]. That this deficit in urine concentrating ability in older adults occurs despite higher vasopressin concentrations suggests that the urine concentrating impairment is predominantly due to factors intrinsic to the kidneys [453]. Indeed, data from a rat model indicate that aging impairs the upregulation of the water channel aquaporin-2, which reduces the ability to reabsorb water in the renal tubules [454]. Further evidence indicates a reduced medullary concentrating gradient [455] and a decreased ability to conserve sodium [456], which also contributes to the deficit in urine concentrating ability with older age. For example, Macias Nunez et al. [457] have reported that in adults older than 60 years there is a 30% reduction in sodium reabsorption in the distal tubule compared to younger adults, but no differences between ages in proximal tubule reabsorption. With this background, it is perhaps not surprising that basal plasma aldosterone is reduced in older adults [458]. Notably, it is believed that elevations in mineralocorticoid receptor sensitivity, which have been observed in vascular smooth muscle cells in older rats [459], does not fully compensate for reductions in circulating aldosterone [460].
Fluid regulation is a function of both fluid output (e.g., urine production) and fluid intake (i.e., drinking). Thus, it is also notable that older adults consistently display impairments in thirst and consume less water for a given level of dehydration compared to younger adults [453,461-463]. Ultimately, these fluid regulation impairments are attributed to a decreased ability to physiologically detect reductions in circulating blood volume and increases in plasma osmolality [460,461], which are caused by reductions in baroreceptor sensitivity and/or altered sensitivity to the fluid regulatory hormones [460,461].
Physiological responses of aging kidneys to heat stress
Renal blood flow
The systemic cardiovascular response to passive or exercise heat stress is attenuated in older adults. For instance, it has been regularly observed that older adults have an impaired ability to increase skin blood flow during heat stress [464-467], which (in theory) contributes to the augmented rate of heat gain in older adults during heat exposure [30]. Minson et al. [159] identified that renal plasma flow was lower during both normothermia (1137 vs. 873 mL/min) and passive heat stress (1.5-2°C increase in core temperature; 847 vs. 674 mL/min) in older adults compared to younger adults. Interestingly, however, the magnitude of renal vasoconstriction was attenuated in older adults. That is, in addition to having lower basal renal blood flow, older adults have an attenuated ability to decrease renal blood flow during heat stress despite lower absolute flow rates in this vascular bed compared to younger adults [159]. Hormone replacement therapy in postmenopausal women does not appear to change renal vasoconstrictor response to passive heat stress compared to women not taking hormone replacement therapy [442].
Despite attenuated increases in cardiac output and less redistribution of blood flow from the renal and splanchnic vascular beds, blood pressure is relatively well-maintained in heat stressed older adults during sympathetic activation (i.e., head-up tilt) due to augmented increases in splanchnic vascular resistance, but not renal vascular resistance [160], the latter of which further supports an altered control of renal blood flow during heat stress in older adults. This differential control of renal blood flow in older adults is unlikely to be due to differences in activation of the sympathetic nervous system given that aging does not differentially modify muscle sympathetic nerve activity or plasma norepinephrine concentrations during passive heat stress [468]. That said, the possibility remains that for a given magnitude of sympathetic activation the renal vascular response in older adults is blunted owing to decreased adrenergic receptor sensitivity, which is known to be diminished with aging [469]. Moreover, it is also possible that regional differences in sympathetic nerve activity, especially within the renal nerves, contribute to the differential renal vascular control in older versus younger adults. In support of this latter point, in healthy aged rats increases in RSNA [470] and sympathetic discharge pattern [277] during heat stress are drastically attenuated compared to their young counterparts. Further studies demonstrated that the altered RSNA during heat stress in older rats is mediated by age-related changes in brain stem neural circuits [278] and an attenuated ability to withdraw rostral ventral lateral medulla GABA tone to activate renal sympathetic nerve discharge [471].
The altered RSNA with aging and heat stress also translates to differential renal blood flow responses during exercise in the heat. Kenney and Zappe [158] were the first to report that compared to younger men, older men have an attenuated ability to redistribute blood flow from the renal vasculature (40% vs. 12% reductions in renal blood flow) during 90 min of cycling exercise at 50% O2max in a 30°C, 60% relative humidity environment, observations that were independent of aerobic fitness or physical activity levels. In an extension of this study, Ho et al. [157] found that in young adults aerobic fitness augments increases in skin blood flow during exercise in the heat as a function of a higher cardiac output and a greater redistribution of blood flow away from the renal and splanchnic vascular beds. However, in older adults, aerobic fitness does not have these same modulatory effects as attenuated reductions in renal and splanchnic blood flow to exercise in the heat persist even in aerobically fit older adults [157]. The ability to restore renal blood flow to basal levels following cessation of exercise in the heat is also impaired in older adults compared to younger adults [158,311]. For example, Farquhar and Kenney [311] observed that renal plasma flow was ~25% lower than baseline values in older adults even after a 45 minute recovery period that followed 60 minutes of exercise at 60% O2max in a 36°C, 36% relative humidity environment and caused rectal temperature to increase by 1.5°C. In the same study [311], the younger control group exhibited complete recovery of renal plasma flow following 45 minutes of rest. Notably, the length of time required for renal blood flow to return to pre-exercise levels following exercise in the heat remains unknown in older adults.
Glomerular filtration rate
Few studies have examined the effect of aging on GFR during heat stress. For instance, we were unable to find a study that examined how older age may alter the GFR response to passive heat stress. On one hand, this is not completely surprising, given the large variability between the relatively few studies that have been performed in younger adults (Table 4). On the other hand, this is an important gap in the literature because GFR is a foundational element of kidney function. Moreover, to our knowledge, only one study has examined the effects of aging on GFR during exercise in the heat. In this study, Farquhar and Kenney (1999) found that older adults demonstrated attenuated reductions in GFR normalized to body surface area compared to younger adults (~30% vs. ~40%). Similar to the response of renal blood flow, the absolute values of GFR were lower following exercise in the heat in older adults compared to younger controls (~60 ml/min/1.73m2 vs. ~78 ml/min/1.73m2) despite attenuated reductions in GFR during exercise, which was likely explained by a lower pre-exercise GFR in older adults (~85 ml/min/1.73m2 vs. ~132 ml/min/1.73m2) [311]. In total, the effects of heat stress on GFR in older adults remains largely unexplored.
Water and electrolyte regulation
Older adults are at a greater risk for hyperthermia and dehydration during passive [472] and exercise heat stress [463], which is contributed to by decrements in thirst perception [473]. This is highlighted by data from Miescher and Fortney [472], who compared fluid regulation in older and younger men during heat stress. It was subsequently identified that older men had higher basal plasma osmolality, but they reported to be less thirsty during passive heat stress despite having larger increases in plasma osmolality (+5 mOsmol/kg vs. +1 mOsmol/kg), greater reductions in plasma volume (−11% vs. −5%), and larger increases in rectal temperature (+1.3°C vs. +0.6°C) compared to the younger men. This reduced thirst in older adults has been found to translate to fluid intake during heat stress [472] and during dehydration in the absence of heat stress [453,463] that is not sufficient to restore body water to baseline levels. Notably, this inadequate fluid intake in older adults is accompanied by an attenuated sodium appetite with aging following fluid deprivation [474,475]. The attenuated sodium intake during dehydration may increase the need of the tubules to reabsorb sodium, a process that is impaired with aging [457]. As noted above (see Physiology of aging kidneys), the reduced fluid intake with heat stress in older adults may be worsened by an attenuated capacity to concentrate urine during heat stress [463], a process that is contributed to by reduced renal responsiveness to vasopressin, decreased aldosterone production, and altered sodium handling with aging [473]. Interestingly, however, for a given volume of fluid ingested, older adults better retain fluid compared to their younger counterparts, which is at least partially explained by altered renal sodium handling and reductions in GFR [476]. Whether this conclusion remains valid during or following heat stress is unknown, but this may have important ramifications for hydration regimens targeted at older adults during heat stress.
Heat adaptation and older adults
To our knowledge two studies have attempted to investigate if heat acclimation mitigates the risk of dehydration in older adults, which would potentially occur via improvements in thirst sensation, plasma volume expansion, and water and electrolyte handling. Takamata et al. [477] found that a six day heat acclimation protocol (fixed work intensity) improves thirst perception during exercise in the heat in older adults. However, these higher ratings of thirst did not translate to greater fluid intakes in older adults [477]. Furthermore, unlike the younger control group, older adults did not present with an expansion of blood or plasma volume following the six day heat acclimation [477], findings that were consistent with a previous observation following a four day heat acclimation protocol in older adults [398]. Interestingly, vasopressin secretion for a given increase in plasma osmolality during exercise in the heat was increased with six days of heat acclimation in older adults, but there were no changes in aldosterone [477], although this is not always consistently observed [398]. Moreover, a four day heat acclimation protocol did not affect GFR or renal plasma flow during or following exercise in the heat in older adults and had little impact on urine concentrating ability or renal sodium handling [398]. Collectively, there is agreement between the two studies conducted to date that there is little evidence that heat acclimation improves renal water and electrolyte regulation. This conclusion is likely a function of the heat acclimation protocols employed. Thus, future studies should employ longer heat acclimation protocols and/or controlled-hyperthermia approach to elicit a more fully heat acclimated phenotype to determine the potential utility of heat acclimation at improving water and electrolyte regulation in older adults.
Acute kidney injury
Older adults are highly susceptible to extreme heat events and have disproportional increases in morbidity and mortality during heat waves [29-41]. The top two causes of hospitalizations during heat waves in older adults are related to the kidneys – fluid and electrolyte disturbances [32,35,41] and AKI [32-36,41,44]. Notably, fluid and electrolyte disturbances is a risk factor for AKI [43] and likely reflects the myriad of deficiencies faced by the aging kidneys to defend against dehydration (see Physiology of aging kidneys). Our understanding of the risk of AKI independent of aging is rapidly developing [19]. By comparison, the pathophysiological response of the kidneys to heat stress in older adults has never been directly investigated. Importantly, understanding the mechanisms by which heat stress may increase the risk of dehydration and/or AKI in older adults would provide critical insight into the interventions that may help alleviate these risks during heat waves.
A recent observational study found evidence that processes underlying the increased risk of AKI during heat stress in younger adults, may also be at play in older adults such that increases in plasma NGAL, which reflects the risk of renal ischemia [223], are independently associated with elevations in ambient temperature in free living older adults [478]. Given this limited evidence of an analogous pathophysiology, there are likely three mechanisms by which aging increases the risk of AKI during heat stress (Figure 8). First, as described above (see Physiology of aging kidneys) kidney function is reduced with aging, which is contributed to by morphological changes [85,434,436-440], reductions in renal blood flow [157-160,311,398,441] and augmented basal activation of the polyol-fructokinase pathway [56]. These deleterious processes may be magnified by heat stress, such that absolute renal blood flow is even lower in older adults during heat stress compared to younger adults [159]. Ultimately, this manifests as an increased severity of AKI and poor recovery following AKI [430,479]. Second, as thoroughly discussed above, water and electrolyte regulation is altered with aging, which collectively increases in the risk of developing dehydration during heat stress [461,473,480]. Notably, dehydration increases the risk of developing AKI during heat stress [63,64]. Third, temperature regulation is impaired in older adults [481]. This is manifested as decreases heat loss stemming from impairments in sweating and skin blood flow [481] but is also contributed to by delays in voluntary decisions to seek cooling [482-484]. In total, this age dependent deterioration of temperature regulation increases the likelihood of the development of hyperthermia [63,64], which increases the risk of developing AKI during heat stress [466,485,486]. Clearly, aggregation of these factors presents a compelling case for older adults being at an increased risk of AKI during heat stress. Perhaps more importantly, however, understanding interactions between temperature regulation, water and electrolyte regulation, and kidney function in the context of aging provides a framework by which interventions can be systematically identified and examined to safeguard the kidney health of older adults during a heat wave.
Figure 8.
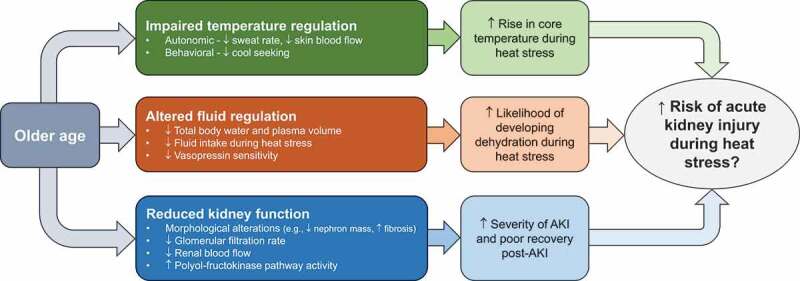
Factors that may exacerbate the risk of acute kidney injury during heat stress in older adults
Summary
The kidneys play an integral role in maintaining homeostasis. Heat stress challenges many physiological systems that are impacted by the kidneys. We have just begun to understand the dynamic, rapid adjustments of the kidneys to heat stress, including reductions in renal blood flow. Interestingly, despite that glomerular filtration rate is typically regarded as the primary measure of kidney function, a consensus does not exist for how glomerular filtration rate may be altered by heat stress. In this narrative review, we have described that the kidneys are susceptible to heat stress, and that an emerging body of evidence links heat stress to kidney-related pathology, such as acute kidney injury. Moreover, this kidney-related pathology is not unique to occupational settings, as older adults are at risk for hospitalization during heat stress for complications related to the kidneys, including water and electrolyte disturbances and AKI. This review has highlighted that our understanding of the kidneys’ physiological response to heat stress in older adults is magnitudes smaller compared to the body of literature in younger adults, and that the pathophysiological response of the kidneys to heat stress in older adults is essentially non-existent. In light of the public health challenge that heat stress imposes on the kidneys, we have highlighted many important gaps in the literature that represent exciting opportunities for future exploration to expand our understanding of kidney physiology and more importantly, protect kidney health for the global population.
Funding Statement
Preparation of this manuscript and some of the original research efforts discussed herein were supported by awards from the Carl V. Gisolfi Memorial Fund from the American College of Sports Medicine Foundation (#17-00580), the Mark Diamond Research Fund from the University at Buffalo (no. SP-19-04), and the U.S. Centers for Disease Control and Prevention (R01OH011528).
Abbreviations
- ACEi
Angiotensin converting enzyme inhibitor
- AKI
Acute kidney injury
- ARF
Acute renal failure
- AT1R
Angiotensin II type 1 receptor
- CKD
Chronic kidney disease
- GFR
Glomerular filtration rate
- IGFBP7
Insulin-like growth factor binding protein 7
- IL-18
Interleukin-18
- KIM-1
Kidney injury molecule-1
- L-FABP
Liver type fatty acid-binding protein
- NGAL
Neutrophil gelatinase-associated lipocalin
- NHE3
Sodium-hydrogen antiporter 3
- NIOSH
U.S. National Institute for Occupational Safety and Health
- NOS
Nitric oxide synthase
- NSAID
Nonsteroidal anti-inflammatory drug
- PAH
Para-aminohippurate
- RSNA
Renal sympathetic nerve activity
- TIMP-2
Tissue inhibitor of metalloproteinase-2
- O2max
Maximal oxygen uptake
- V1
Vasopressin type 1 receptors
- V2
Vasopressin type 2 receptors
Disclosure statement
No conflicts of interest, financial or otherwise, are declared by the authors.
References
- 1.Mujais SK, McConathy DA.. The kidney in the graphic arts. Am J Nephrol. 1985;5(5):375–377. [DOI] [PubMed] [Google Scholar]
- 2.Fine LG . A 2000-year history of nephrology: 10 enduring scientific landmarks. J Nephrol. 2013;26(Suppl. 22): 6–17. [DOI] [PubMed] [Google Scholar]
- 3.Gottschalk CW, Berliner RW, Giebisch GH. Renal physiology: people and ideas. New York, NY: Springer; 2013. [Google Scholar]
- 4.Greydanus D, Kadochi M. Reflections on the Medical History of the Kidney: From Alcmaeon of Croton to Richard Bright - Standing on the Shoulders of Giants. J Integr Nephrol Androl. 2016;3(4):101–108. [Google Scholar]
- 5.Fine LG . Eustachio’s discovery of the renal tubule. Am J Nephrol. 1986;6(1): 47–50. [DOI] [PubMed] [Google Scholar]
- 6.Malpighi M De Viscera Structura Exercitatio Anatomica. 1666.
- 7.Mezzogiorno A, Mezzogiorno V. Marcello Malpighi (1628-1694). Am J Nephrol. 1997;17(3–4):269–273. [DOI] [PubMed] [Google Scholar]
- 8.Bellini L, Blasius GL. Exercitatio anatomica de structura et usu renum. Sumptibus Andreae Frisii; 1662.
- 9.Bowman W. On the structure and use of the Malpighian bodies of the kidney, with observations on the circulation through that gland. Proc Roy Soc Lond. 1843;4:374–377. [Google Scholar]
- 10.Lombard WP. The life and work of Carl Ludwig. Science. 1916;44(1133):363–375. [DOI] [PubMed] [Google Scholar]
- 11.Starling EH. On the Absorption of Fluids from the Connective Tissue Spaces. J Physiol. 1896;19(4):312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sollmann T, McComb E. Preliminary Observations on a Case of Physiological Albuminuria. J Exp Med. 1898;3(1):137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mann JD. Physiology and Pathology of the Urine: With Methods for Its Examination. Glasgow, Scotland: Griffin; 1904. [Google Scholar]
- 14.Haldane JS, Priestley JG. The regulation of excretion of water by the kidneys: I. J Physiol. 1916;50(5):296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adolph EF. The regulation of the water content of the human organism. J Physiol. 1921;55(1–2):114–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stocker TF, Qin D, Plattner G-K, et al. Climate change 2013: The physical science basis. Contribution of working group I to the fifth assessment report of the intergovernmental panel on climate change. 2013;1535.
- 17.Heaviside C, Macintyre H, Vardoulakis S. The Urban Heat Island: Implications for Health in a Changing Environment. Curr Environ Health Rep. 2017;4(3):296–305. [DOI] [PubMed] [Google Scholar]
- 18.Johnson RJ, Sanchez-Lozada LG, Newman LS, et al. Climate Change and the Kidney. Ann Nutr Metab. 2019;74(Suppl 3):38–44. [DOI] [PubMed] [Google Scholar]
- 19.Schlader ZJ, Hostler D, Parker MD, et al. The Potential for Renal Injury Elicited by Physical Work in the Heat. Nutrients. 2019;11:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schrier RW, Henderson HS, Tisher CC, et al. Nephropathy associated with heat stress and exercise. Ann Intern Med. 1967;67(2):356–376. [DOI] [PubMed] [Google Scholar]
- 21.Schrier RW, Hano J, Keller HI, et al. Renal, metabolic, and circulatory responses to heat and exercise. Studies in military recruits during summer training, with implications for acute renal failure. Ann Intern Med. 1970;73(2):213–223. [DOI] [PubMed] [Google Scholar]
- 22.Donham BP, Frankfurt SB, Cartier RA, et al. Low Incidence of Death and Renal Failure in United States Military Service Members Hospitalized with Exertional Heat Stroke: A Retrospective Cohort Study. Mil Med. 2020;185(Supplement_1):362–367. [DOI] [PubMed] [Google Scholar]
- 23.Carter R, Cheuvront SN, Williams JO, et al. Epidemiology of hospitalizations and deaths from heat illness in soldiers. Med Sci Sports Exerc. 2005;37(8):1338–1344. [DOI] [PubMed] [Google Scholar]
- 24.Wesseling C, Glaser J, Rodriguez-Guzman J, et al. Chronic kidney disease of non-traditional origin in Mesoamerica: a disease primarily driven by occupational heat stress. Rev Panam Salud Publica. 2020;44:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flouris AD, Dinas PC, Ioannou LG, et al. Workers’ health and productivity under occupational heat strain: a systematic review and meta-analysis. Lancet Planet Health. 2018;2(12):e521–e531. [DOI] [PubMed] [Google Scholar]
- 26.Johnson RJ, Wesseling C, Newman LS. Chronic kidney disease of unknown cause in agricultural communities. N Eng J Med. 2019;380(19):1843–1852. [DOI] [PubMed] [Google Scholar]
- 27.Free and open access to labour statistics [Internet] . International Labour Organization. 2019. [accessed 2020 April 7]. Available from: https://ilostat.ilo.org/data/.
- 28.Statistics USBoL . Employed persons by detailed industry and age, 2013 annual averages. 2016.
- 29.Anderson BG, Bell ML. Weather-related mortality: how heat, cold, and heat waves affect mortality in the United States. Epidemiol. 2009;20(2):205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kenney WL, Craighead DH, Alexander LM. Heat waves, aging, and human cardiovascular health. Med Sci Sports Exerc. 2014. October;46(10):1891–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oudin Åström D, Bertil F, Joacim R. Heat wave impact on morbidity and mortality in the elderly population: a review of recent studies. Maturitas. 2011;69(2):99–105. [DOI] [PubMed] [Google Scholar]
- 32.Bobb JF, Obermeyer Z, Wang Y, et al. Cause-specific risk of hospital admission related to extreme heat in older adults. JAMA. 2014;312(24):2659–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim Y-H, So R, Lee C, et al. Ambient temperature and hospital admissions for acute kidney injury: A time-series analysis. Sci Total Environ. 2018;616:1134–1138. [DOI] [PubMed] [Google Scholar]
- 34.McTavish RK, Richard L, McArthur E, et al. Association between high environmental heat and risk of acute kidney injury among older adults in a northern climate: a matched case-control study. Am J Kidney Dis. 2018;71(2):200–208. [DOI] [PubMed] [Google Scholar]
- 35.Hopp S, Dominici F, Bobb JF. Medical diagnoses of heat wave-related hospital admissions in older adults. Prev Med. 2018May;110:81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim E, Kim H, Kim YC, et al. Association between extreme temperature and kidney disease in South Korea, 2003–2013: Stratified by sex and age groups. Sci Total Environ. 2018;642:800–808. [DOI] [PubMed] [Google Scholar]
- 37.Semenza JC, McCullough JE, Flanders WD, et al. Excess hospital admissions during the July 1995 heat wave in Chicago. Am J Prev Med. 1999;16(4):269–277. [DOI] [PubMed] [Google Scholar]
- 38.Rey G, Jougla E, Fouillet A, et al. The impact of major heat waves on all-cause and cause-specific mortality in France from 1971 to 2003. Int Arch Occup Environ Health. 2007. July;80(7):615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michelozzi P, Accetta G, De Sario M, et al. High temperature and hospitalizations for cardiovascular and respiratory causes in 12 European cities. Am J Respir Crit Care Med. 2009;179(5):383–389. [DOI] [PubMed] [Google Scholar]
- 40.Kenny GP, Yardley J, Brown C, et al. Heat stress in older individuals and patients with common chronic diseases. Cmaj. 2010;182(10):1053–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basu R, Pearson D, Malig B, et al. The effect of high ambient temperature on emergency room visits. Epidemiology. 2012;813–820. [DOI] [PubMed] [Google Scholar]
- 42.NOAA . National Weather Service: Heat Wave. 2009. [cited 2019; accessed 2020 August 5]. Available from: https://w1.weather.gov/glossary/index.php?letter=h
- 43.Brennan M, O’Keeffe ST, Mulkerrin EC. Dehydration and renal failure in older persons during heatwaves-predictable, hard to identify but preventable?. Age Ageing. 2019;48(5):615–618. [DOI] [PubMed] [Google Scholar]
- 44.Xu Z, Hu X, Tong S, et al. Heat and risk of acute kidney injury: An hourly-level case-crossover study in queensland, Australia. Environ Res. 2020March;182:109058. [DOI] [PubMed] [Google Scholar]
- 45.Diffenbaugh NS, Field CB. Changes in ecologically critical terrestrial climate conditions. Science. 2013;341(6145):486–492. [DOI] [PubMed] [Google Scholar]
- 46.Vincent GK, Velkoff VA. The next four decades: The older population in the United States: 2010 to 2050. US Department of Commerce, Economics and Statistics Administration, US …; 2010. (1138).
- 47.Garcia-Arroyo FE, Cristobal M, Arellano-Buendia AS, et al. Rehydration with soft drink-like beverages exacerbates dehydration and worsens dehydration-associated renal injury. Am J Physiol Regul Integr Comp Physiol. 2016. July 1;311(1):R57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia-Arroyo FE, Gonzaga G, Munoz-Jimenez I, et al. Antioxidant supplements as a novel mean for blocking recurrent heat stress-induced kidney damage following rehydration with fructose-containing beverages. Free Radic Biol Med. 2019September;141:182–191. [DOI] [PubMed] [Google Scholar]
- 49.Garcia-Arroyo FE, Munoz-Jimenez I, Gonzaga G, et al. A Role for Both V1a and V2 Receptors in Renal Heat Stress Injury Amplified by Rehydration with Fructose. Int J Mol Sci. 2019;20:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia-Arroyo FE, Tapia E, Blas-Marron MG, et al. Vasopressin Mediates the Renal Damage Induced by Limited Fructose Rehydration in Recurrently Dehydrated Rats. Int J Biol Sci. 2017;13(8):961–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Milagres T, Garcia-Arroyo FE, Lanaspa MA, et al. Rehydration with fructose worsens dehydration-induced renal damage. BMC Nephrol. 2018. July 13;19(1):180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roncal Jimenez CA, Ishimoto T, Lanaspa MA, et al. Fructokinase activity mediates dehydration-induced renal injury. Kidney Int. 2014. August;86(2):294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roncal-Jimenez C, Garcia-Trabanino R, Barregard L, et al. Heat Stress Nephropathy From Exercise-Induced Uric Acid Crystalluria: A Perspective on Mesoamerican Nephropathy. Am J Kidney Dis. 2016;67(1):20–30. [DOI] [PubMed] [Google Scholar]
- 54.Roncal-Jimenez C, Lanaspa MA, Jensen T, et al. Mechanisms by Which Dehydration May Lead to Chronic Kidney Disease. Ann Nutr Metab. 2015;66(Suppl 3):10–13. [DOI] [PubMed] [Google Scholar]
- 55.Roncal-Jimenez CA, Garcia-Trabanino R, Wesseling C, et al. Mesoamerican Nephropathy or Global Warming Nephropathy?. Blood Purif. 2016;41(1–3):135–138. [DOI] [PubMed] [Google Scholar]
- 56.Roncal-Jimenez CA, Ishimoto T, Lanaspa MA, et al. Aging-associated renal disease in mice is fructokinase dependent. Am J Physiol Renal Physiol. 2016. October 1;311(4):F722–f730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roncal-Jimenez CA, Milagres T, Andres-Hernando A, et al. Effects of exogenous desmopressin on a model of heat stress nephropathy in mice. Am J Physiol Renal Physiol. 2017. March 1;312(3):F418–F426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roncal-Jimenez CA, Sato Y, Milagres T, et al. Experimental heat stress nephropathy and liver injury are improved by allopurinol. Am J Physiol Renal Physiol. 2018. September 1;315(3):F726–F733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanchez-Lozada LG, Andres-Hernando A, Garcia-Arroyo FE, et al. Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J Biol Chem. 2019. March 15;294(11):4272–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sato Y, Roncal-Jimenez CA, Andres-Hernando A, et al. The increase of core temperature affected the progression of kidney injury by repeated heat stress exposure. Am J Physiol Renal Physiol. 2019;317(5):F111–F1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Song Z, Roncal-Jimenez CA, Lanaspa-Garcia MA, et al. Role of fructose and fructokinase in acute dehydration-induced vasopressin gene expression and secretion in mice. J Neurophysiol. 2017;117(2):646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chapman CL, Johnson BD, Sackett JR, et al. Soft drink consumption during and following exercise in the heat elevates biomarkers of acute kidney injury. Am J Physiol Regul Integr Comp Physiol. 2019. March 1;316(3):R189–R198. [DOI] [PubMed] [Google Scholar]
- 63.Chapman CL, Johnson BD, Vargas NT, et al. Both hyperthermia and dehydration during physical work in the heat contribute to the risk of acute kidney injury. J Appl Physiol. 2020;128(4):715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schlader ZJ, Chapman CL, Sarker S, et al. Firefighter Work Duration Influences the Extent of Acute Kidney Injury. Med Sci Sports Exerc. 2017;49(8):1745–1753. [DOI] [PubMed] [Google Scholar]
- 65.Junglee NA, Di Felice U, Dolci A, et al. Exercising in a hot environment with muscle damage: effects on acute kidney injury biomarkers and kidney function. Am J Physiol Renal Physiol. 2013;305(6):F813–20. [DOI] [PubMed] [Google Scholar]
- 66.McDermott BP, Smith CR, Butts CL, et al. Renal stress and kidney injury biomarkers in response to endurance cycling in the heat with and without ibuprofen. J Sci Med Sport. 2018;21(12):1180–1184. [DOI] [PubMed] [Google Scholar]
- 67.Hansson E, Glaser J, Jakobsson K, et al. Pathophysiological Mechanisms by which Heat Stress Potentially Induces Kidney Inflammation and Chronic Kidney Disease in Sugarcane Workers. Nutrients. 2020;12(6):1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Misra DP, Agarwal V. Systematic reviews: challenges for their justification, related comprehensive searches, and implications. J Korean Med Sci. 2018;33:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Webb P. The physiology of heat regulation. Am J Physiol. 1995;268(4 Pt 2):R838–50. [DOI] [PubMed] [Google Scholar]
- 70.Crandall CG, Wilson TE. Human cardiovascular responses to passive heat stress. Compr Physiol. 2011;5(1):17–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schlader ZJ, Vargas NT. Regulation of body temperature by autonomic and behavioral thermoeffectors. Exerc Sport Sci Rev. 2019;47(2):116–126. [DOI] [PubMed] [Google Scholar]
- 72.Sawka MN, Leon LR, Montain SJ, et al. Integrated physiological mechanisms of exercise performance, adaptation, and maladaptation to heat stress. Compr Physiol. 2011. October;1(4):1883–1928. [DOI] [PubMed] [Google Scholar]
- 73.Kenny GP, Jay O. Thermometry, calorimetry, and mean body temperature during heat stress. Compr Physiol. 2013. October;3(4):1689–1719. [DOI] [PubMed] [Google Scholar]
- 74.Cheung SS, McLellan TM, Tenaglia S. The thermophysiology of uncompensable heat stress. Sports Med. 2000;29(5):329–359. [DOI] [PubMed] [Google Scholar]
- 75.Pendergast DR, Moon RE, Krasney JJ, et al. Human physiology in an aquatic environment. Compr Physiol. 2011;5(4):1705–1750. [DOI] [PubMed] [Google Scholar]
- 76.Cramer MN, Jay O. Selecting the correct exercise intensity for unbiased comparisons of thermoregulatory responses between groups of different mass and surface area. J Appl Physiol. 2014. May 1;116(9):1123–1132. [DOI] [PubMed] [Google Scholar]
- 77.Lucas RA, Bodin T, García-Trabanino R, et al., editors. Heat stress and workload associated with sugarcane cutting-an excessively strenuous occupation!. 2015. Extreme Physiol Med: BioMed Central; p.1–2. [Google Scholar]
- 78.Rothfusz L The heat index question. National Weather Service Technical Attachment. 1990.
- 79.Hawkins MD, Brown V, Ferrell J. Assessment of NOAA National Weather Service Methods to Warn for Extreme Heat Events. Weather Clim Soc. 2016;9(1):5–13. [Google Scholar]
- 80.Cramer MN, Huang M, Moralez G, et al. Keeping older individuals cool in hot and moderately humid conditions: wetted clothing with and without an electric fan. J Appl Physiol. 2020;128(3):604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Morris NB, English T, Hospers L, et al. The Effects of Electric Fan Use Under Differing Resting Heat Index Conditions: A Clinical Trial. Ann Intern Med. 2019. November 5;171(9):675–677. [DOI] [PubMed] [Google Scholar]
- 82.Schlader ZJ, Hostler DC, Hostler D. Heat strain is exacerbated by consecutive days of fire supression. Med Sci Sport Exer. 2017;49:999–1005. [DOI] [PubMed] [Google Scholar]
- 83.Pryor RR, Pryor JL, Vandermark LW, et al. Exacerbated heat strain during consecutive days of repeated exercise sessions in heat. J Sci Med Sport. 2019;22(10):1084–1089. [DOI] [PubMed] [Google Scholar]
- 84.Notley SR, Meade RD, D’Souza AW, et al. Heat Loss Is Impaired in Older Men on the Day after Prolonged Work in the Heat. Med Sci Sports Exer. 2018;50(9):1859–1867. [DOI] [PubMed] [Google Scholar]
- 85.Wang Z, Deurenberg P, Wang W, et al. Hydration of fat-free body mass: review and critique of a classic body-composition constant. Am J Clin Nutr. 1999. May;69(5):833–841. [DOI] [PubMed] [Google Scholar]
- 86.McDermott BP, Anderson SA, Armstrong LE, et al. National Athletic Trainers’ Association Position Statement: Fluid Replacement for the Physically Active. J Athl Train. 2017. September;52(9):877–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Armstrong LE, Johnson EC. Water intake, water balance, and the elusive daily water requirement. Nutrients. 2018;10(12):1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Baker LB. Physiology of sweat gland function: The roles of sweating and sweat composition in human health. Temperature. 2019;6(3):211–259. doi: 10.1080/23328940.2019.1632145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Greenleaf JE, Sargent F. Voluntary dehydration in man. J Appl Physiol. 1965;20(4):719–724. [DOI] [PubMed] [Google Scholar]
- 90.Sawka MN, Montain SJ, Latzka WA. Hydration effects on thermoregulation and performance in the heat. Comp Biochem Physiol A Mol Integr Physiol. 2001. April;128(4):679–690. [DOI] [PubMed] [Google Scholar]
- 91.Cheuvront SN, Kenefick RW, Charkoudian N, et al. Physiologic basis for understanding quantitative dehydration assessment. Am J Clin Nutr. 2013. March;97(3):455–462. [DOI] [PubMed] [Google Scholar]
- 92.Sawka MN, Burke LM, Eichner ER, et al. American College of Sports Medicine position stand. Exercise and fluid replacement. Med Sci Sports Exerc. 2007;39(2):377–390. [DOI] [PubMed] [Google Scholar]
- 93.Cheuvront SN, Kenefick RW. Dehydration: physiology, assessment, and performance effects. Compr Physiol. 2014. January;4(1):257–285. [DOI] [PubMed] [Google Scholar]
- 94.Fischbach FT, Dunning MB. A manual of laboratory and diagnostic tests. Philadelphia, PA: Lippincott Williams & Wilkins; 2009. [Google Scholar]
- 95.Cheuvront SN, Ely BR, Kenefick RW, et al. Biological variation and diagnostic accuracy of dehydration assessment markers. Am J Clin Nutr. 2010;92(3):565–573. [DOI] [PubMed] [Google Scholar]
- 96.Sparks MA, Crowley SD, Gurley SB, et al. Classical Renin-Angiotensin system in kidney physiology. Compr Physiol. 2014. July;4(3):1201–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cogan MG. Angiotensin II: a powerful controller of sodium transport in the early proximal tubule. Hypertension. 1990;15(5):451–458. [DOI] [PubMed] [Google Scholar]
- 98.Geibel J, Giebisch G, Boron WF. Angiotensin II stimulates both Na(+)-H+ exchange and Na+/HCO3- cotransport in the rabbit proximal tubule. Proc Natl Acad Sci USA. 1990. October;87(20):7917–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ito M, Oliverio MI, Mannon PJ, et al. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc Natl Acad Sci USA. 1995;92(8):3521–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Casellas D, Carmines PK, Dupont M, et al. Arteriolar renin and vascular effects of angiotensin II in juxtamedullary nephrons. Kidney Int Suppl. 1990;30:S60–4. [PubMed] [Google Scholar]
- 101.Davisson RL, Oliverio MI, Coffman TM, et al. Divergent functions of angiotensin II receptor isoforms in the brain. J Clin Invest. 2000. July;106(1):103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Matsuda T, Hiyama TY, Niimura F, et al. Distinct neural mechanisms for the control of thirst and salt appetite in the subfornical organ. Nat Neurosci. 2017;20(2):230–241. [DOI] [PubMed] [Google Scholar]
- 103.SPÄT A, HUNYADY L. Control of Aldosterone Secretion: A Model for Convergence in Cellular Signaling Pathways. Physiol Rev. 2004;84(2):489–539. [DOI] [PubMed] [Google Scholar]
- 104.Pearce D, Bhalla V. Aldosterone and mineralcorticoid receptors: renal and extrarenal roles. Brenner and Rector’s The Kidney. Philadelphia, PA: Elsevier; 2019. p. 335–356. [Google Scholar]
- 105.Danziger J, Zeidel ML. Osmotic homeostasis. Clin J Am Soc Nephrol. 2015. May 7;10(5):852–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Watso JC, Farquhar WB. Hydration Status and Cardiovascular Function. Nutrients. 2019;11(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kinsman BJ, Browning KN, Stocker SD. NaCl and osmolarity produce different responses in organum vasculosum of the lamina terminalis neurons, sympathetic nerve activity and blood pressure. J Physiol. 2017;595(18):6187–6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bankir L, Bichet DG, Morgenthaler NG. Vasopressin: physiology, assessment and osmosensation. J Intern Med. 2017;282(4):284–297. [DOI] [PubMed] [Google Scholar]
- 109.Bouby N, Fernandes S. Mild dehydration, vasopressin and the kidney: animal and human studies. Eur J Clin Nutr. 2003;57(Suppl 2):S39–46. [DOI] [PubMed] [Google Scholar]
- 110.Skorecki KL, Brown D, Ercolani L, et al. Molecular mechanisms of vasopressin action in the kidney. Compr Physiol. 2010;Suppl 25:1185–1218. [Google Scholar]
- 111.Aisenbrey GA, Handelman WA, Arnold P, et al. Vascular effects of arginine vasopressin during fluid deprivation in the rat. J Clin Invest. 1981;67(4):961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Andrews CE, Brenner BM. Relative contributions of arginine vasopressin and angiotensin II to maintenance of systemic arterial pressure in the anesthetized water-deprived rat. Circ Res. 1981;48(2):254–258. [DOI] [PubMed] [Google Scholar]
- 113.Burnier M, Biollaz J, Brunner DB, et al. Blood pressure maintenance in awake dehydrated rats: renin, vasopressin, and sympathetic activity. Am J Physiol. 1983. August;245(2):H203–9. [DOI] [PubMed] [Google Scholar]
- 114.Rockhold RW, Share L, Crofton JT, et al. Cardiovascular response to vasopressin vasopressor antagonist administration during water deprivation in the rat. Neuroendocrinology. 1984;38(2):139–144. [DOI] [PubMed] [Google Scholar]
- 115.Schwartz J, Reid IA. Role of vasopressin in blood pressure regulation in conscious water-deprived dogs. Am J Physiol. 1983;244(1):R74–7. [DOI] [PubMed] [Google Scholar]
- 116.Trippodo NC, Coleman TG, Cowley AW Jr., et al. Angiotensin II antagonists in dehydrated rabbits without baroreceptor reflexes. Am J Physiol. 1977. February;232(2):H110–3. [DOI] [PubMed] [Google Scholar]
- 117.Woods RL, Johnston CI. Contribution of vasopressin to the maintenance of blood pressure during dehydration. Am J Physiol. 1983;245(5 Pt 1):F615–21. [DOI] [PubMed] [Google Scholar]
- 118.Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006. July;86(3):747–803. [DOI] [PubMed] [Google Scholar]
- 119.Wang F, Lu X, Peng K, et al. Antidiuretic Action of Collecting Duct (Pro)Renin Receptor Downstream of Vasopressin and PGE2 Receptor EP4. J Am Soc Nephrol. 2016;27(10):3022–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang T, Physiology XC. Pathophysiology of the Intrarenal Renin-Angiotensin System: An Update. J Am Soc Nephrol. 2017. April;28(4):1040–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Beierwaltes WH, Harrison-Bernard LM, Sullivan JC, et al. Assessment of renal function; clearance, the renal microcirculation, renal blood flow, and metabolic balance. Compr Physiol. 2013;3(1):165–200. [DOI] [PubMed] [Google Scholar]
- 122.Johns EJ, Kopp UC, DiBona GF. Neural control of renal function. Compr Physiol. 2011;1(2):731–767. [DOI] [PubMed] [Google Scholar]
- 123.Barajas L, Liu L, Powers K. Anatomy of the renal innervation: intrarenal aspects and ganglia of origin. Can J Physiol Pharmacol. 1992;70(5):735–749. [DOI] [PubMed] [Google Scholar]
- 124.Ljungqvist A, Wagermark J. The adrenergic innervation of intrarenal glomerular and extra-glomerular circulatory routes. Nephron. 1970;7(3):218–229. [DOI] [PubMed] [Google Scholar]
- 125.Fourman J. The adrenergic innervation of the efferent arterioles and the vasa recta in the mammalian kidney. Experientia. 1970;26(3):293–294. [DOI] [PubMed] [Google Scholar]
- 126.Nair GKK, Masse S, Asta J, et al. The need for and the challenges of measuring renal sympathetic nerve activity. Heart Rhythm. 2016;13(5):1166–1171. [DOI] [PubMed] [Google Scholar]
- 127.Schlader ZJ, Chapman CL, Benati JM, et al. Renal Hemodynamics During Sympathetic Activation Following Aerobic and Anaerobic Exercise. Front Physiol. 2018;9:1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Conboy EE, Fogelman AE, Sauder CL, et al. Endurance training reduces renal vasoconstriction to orthostatic stress. Am J Physiol Renal Physiol. 2010. February;298(2):F279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kuipers NT, Sauder CL, Kearney ML, et al. Interactive effect of aging and local muscle heating on renal vasoconstriction during isometric handgrip. Am J Physiol Renal Physiol. 2009;297(2):F327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kuipers NT, Sauder CL, Kearney ML, et al. Changes in forearm muscle temperature alter renal vascular responses to isometric handgrip. Am J Physiol Heart Circ Physiol. 2007;293(6):H3432–H3439. [DOI] [PubMed] [Google Scholar]
- 131.Momen A, Leuenberger UA, Ray CA, et al. Renal vascular responses to static handgrip: role of muscle mechanoreflex. Am J Physiol Heart Circ Physiol. 2003;285(3):H1247–53. [DOI] [PubMed] [Google Scholar]
- 132.Ray CA, Carter JR. Effects of aerobic exercise training on sympathetic and renal responses to mental stress in humans. Am J Physiol Heart Circ Physiol. 2010;298(1):H229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.DiBona GF, Sawin LL. Effect of renal nerve stimulation on NaCl and H2O transport in Henle’s loop of the rat. Am J Physiol. 1982;243(6):F576–80. [DOI] [PubMed] [Google Scholar]
- 134.Rogenes PR, Gottschalk CW. Renal function in conscious rats with chronic unilateral renal denervation. Am J Physiol. 1982. February;242(2):F140–8. [DOI] [PubMed] [Google Scholar]
- 135.Poss J, Ewen S, Schmieder RE, et al. Effects of renal sympathetic denervation on urinary sodium excretion in patients with resistant hypertension. Clin Res Cardiol. 2015;104(8):672–678. [DOI] [PubMed] [Google Scholar]
- 136.Liu F, Gesek FA. alpha(1)-Adrenergic receptors activate NHE1 and NHE3 through distinct signaling pathways in epithelial cells. Am J Physiol Renal Physiol. 2001;280(3):F415–25. [DOI] [PubMed] [Google Scholar]
- 137.DiBona GF, Johns EJ. A study of the role of renal nerves in the renal responses to 60 degree head-up tilt in the anaesthetized dog. J Physiol. 1980;299:117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.DiBona GF, Zambraski EJ, Aguilera AJ, et al. Neurogenic control of renal tubular sodium reabsorption in the dog: a brief review and preliminary report concerning possible humoral mediation. Circ Res. 1977. May;40(5 Suppl 1):I127–30. [PubMed] [Google Scholar]
- 139.Zambraski EJ, Dibona GF, Kaloyanides GJ. Effect of sympathetic blocking agents on the antinatriuresis of reflex renal nerve stimulation. J Pharmacol Exp Ther. 1976. August;198(2):464–472. [PubMed] [Google Scholar]
- 140.Huang C, Johns EJ. Role of ANG II in mediating somatosensory-induced renal nerve-dependent antinatriuresis in the rat. Am J Physiol. 1998;275(1):R194–202. [DOI] [PubMed] [Google Scholar]
- 141.DiBona GF, Kopp UC. Neural control of renal function. Physiol Rev. 1997;77(1):75–197. [DOI] [PubMed] [Google Scholar]
- 142.Handa RK, Johns EJ. Interaction of the renin-angiotensin system and the renal nerves in the regulation of rat kidney function. J Physiol. 1985;369:311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Johns EJ. The role of angiotensin II in the antidiuresis and antinatriuresis induced by stimulation of the sympathetic nerves to the rat kidney. J Auton Pharmacol. 1987;7(3):205–214. [DOI] [PubMed] [Google Scholar]
- 144.Bachmann S, Bosse HM, Mundel P. Topography of nitric oxide synthesis by localizing constitutive NO synthases in mammalian kidney. Am J Physiol. 1995;268(5 Pt 2):F885–98. [DOI] [PubMed] [Google Scholar]
- 145.Mattson DL, Wu F. Nitric oxide synthase activity and isoforms in rat renal vasculature. Hypertension. 2000;35(1 Pt 2):337–341. [DOI] [PubMed] [Google Scholar]
- 146.Liu GL, Liu L, Barajas L. Development of NOS-containing neuronal somata in the rat kidney. J Auton Nerv Syst. 1996;58(1–2):81–88. [DOI] [PubMed] [Google Scholar]
- 147.Wu F, Park F, Cowley AW Jr., et al. Quantification of nitric oxide synthase activity in microdissected segments of the rat kidney. Am J Physiol Renal Physiol. 1999;276(6):F874–81. [DOI] [PubMed] [Google Scholar]
- 148.Kone BC, Baylis C. Biosynthesis and homeostatic roles of nitric oxide in the normal kidney. Am J Physiol Renal Physiol. 1997;272(5 Pt 2):F561–78. [DOI] [PubMed] [Google Scholar]
- 149.Eaton D, Pooler J. Vander’s Renal Physiology. 9th ed. New York, NY: McGraw-Hill Education; 2018. [Google Scholar]
- 150.Smith HW. The kidney: structure and function in health and disease. Vol. 1. USA: Oxford University Press; 1951. [Google Scholar]
- 151.Navar LG, Maddox D, Munger KA. The renal circulations and glomerular ultrafiltration. Brenner & rector’s the kidney. 2019:80–114.
- 152.Smith HW, Goldring W, Chasis H. The measurement of the tubular excretory mass, effective blood flow, and filtration rate in the normal human kidney. J Clin Invest. 1938;17(3):263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Goldring W, Chasis H, Ranges HA, et al. Relations of effective renal blood flow and glomerular filtration to tubular excretory mass in normal man. J Clin Invest. 1940;19(5):739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chasis H, Redish J, Goldring W, et al. The use of sodium p-aminohippurate for the functional evaluation of the human kidney. J Clin Invest. 1945;24(4):583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cole BR, Giangiacomo J, Ingelfinger JR, et al.. Measurement of renal function without urine collection. A critical evaluation of the constant-infusion technic for determination of inulin and para-aminohippurate. N Engl J Med. 1972. November 30;287(22):1109–1114. [DOI] [PubMed] [Google Scholar]
- 156.Van Acker BA, Koomen GC, Arisz L. Drawbacks of the constant-infusion technique for measurement of renal function. Am J Physiol Renal Physiol. 1995;268(4):F543–52. [DOI] [PubMed] [Google Scholar]
- 157.Ho CW, Beard JL, Farrell PA, et al. Age, fitness, and regional blood flow during exercise in the heat. J Appl Physiol. 1997;82(4):1126–1135. [DOI] [PubMed] [Google Scholar]
- 158.Kenney WL, Zappe DH. Effect of age on renal blood flow during exercise. Aging (Albany NY). 1994;6(4):293–302. [DOI] [PubMed] [Google Scholar]
- 159.Minson CT, Wladkowski SL, Cardell AF, et al. Age alters the cardiovascular response to direct passive heating. J Appl Physiol. 1998;84(4):1323–1332. [DOI] [PubMed] [Google Scholar]
- 160.Minson CT, Wladkowski SL, Pawelczyk JA, et al. Age, splanchnic vasoconstriction, and heat stress during tilting. Am J Physiol Regul Integr Comp Physiol. 1999;276(1):R203–R212. [DOI] [PubMed] [Google Scholar]
- 161.Boddi M, Sacchi S, Lammel RM, et al. Age-related and vasomotor stimuli-induced changes in renal vascular resistance detected by Doppler ultrasound. Am J Hypertens. 1996;9(5):461–466. [DOI] [PubMed] [Google Scholar]
- 162.Bossard G, Bourgoin P, Corbeau J, et al. Early detection of postoperative acute kidney injury by Doppler renal resistive index in cardiac surgery with cardiopulmonary bypass. Br J Anaesth. 2011;107(6):891–898. [DOI] [PubMed] [Google Scholar]
- 163.Dewitte A, Coquin J, Meyssignac B, et al. Doppler resistive index to reflect regulation of renal vascular tone during sepsis and acute kidney injury. Crit Care. 2012;16(5):R165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Drew RC, Blaha CA, Herr MD, et al. Muscle mechanoreflex activation via passive calf stretch causes renal vasoconstriction in healthy humans. Am J Physiol Regul Integr Comp Physiol. 2017;312(6):R956–R964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Momen A, Leuenberger UA, Handly B, et al. Effect of aging on renal blood flow velocity during static exercise. Am J Physiol Heart Circ Physiol. 2004;287(2):H735–H740. [DOI] [PubMed] [Google Scholar]
- 166.Chapman CL, Johnson BD, Hostler D, et al. Reliability and agreement of human renal and segmental artery hemodynamics measured using Doppler ultrasound. J Appl Physiol. 2020;128(3):627–636. [DOI] [PubMed] [Google Scholar]
- 167.Chapman CL, Grigoryan T, Vargas NT, et al. High-fructose corn syrup-sweetened soft drink consumption increases vascular resistance in the kidneys at rest and during sympathetic activation. Am J Physiol Renal Physiol. 2020;318(4):F1053–F1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chapman CL, Schlader ZJ, Reed EL, et al. Renal and segmental artery hemodynamic response to acute, mild hypercapnia. Am J Physiol Regul Integr Comp Physiol. 2020;318(4):R822–R827. [DOI] [PubMed] [Google Scholar]
- 169.Marraccini P, Fedele S, Marzilli M, et al. Adenosine-induced renal vasoconstriction in man. Cardiovasc Res. 1996;32(5):949–953. [PubMed] [Google Scholar]
- 170.Manoharan G, Pijls NH, Lameire N, et al. Assessment of renal flow and flow reserve in humans. J Am Coll Cardiol. 2006;47(3):620–625. [DOI] [PubMed] [Google Scholar]
- 171.Kawakami S, Yasuno T, Matsuda T, et al. Association between exercise intensity and renal blood flow evaluated using ultrasound echo. Clin Exp Nephrol. 2018;22(5):1061–1068. [DOI] [PubMed] [Google Scholar]
- 172.Kotoku K, Yasuno T, Kawakami S, et al. Effect of exercise intensity on renal blood flow in patients with chronic kidney disease stage 2. Clin Exp Nephrol. 2019;23(5):621–628. [DOI] [PubMed] [Google Scholar]
- 173.Takenaka T, Suzuki H, Okada H, et al. Mechanosensitive cation channels mediate afferent arteriolar myogenic constriction in the isolated rat kidney. J Physiol. 1998;511:245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. J Physiol. 1902;28(3):220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Holstein-Rathlou NH, Marsh DJ. A dynamic model of renal blood flow autoregulation. Bull Math Biol. 1994;56(3):411–429. [DOI] [PubMed] [Google Scholar]
- 176.Takenaka T, Harrison-Bernard LM, Inscho EW, et al. Autoregulation of afferent arteriolar blood flow in juxtamedullary nephrons. Am J Physiol Renal Physiol. 1994. November;267(5 Pt 2):F879–87. [DOI] [PubMed] [Google Scholar]
- 177.Just A. Mechanisms of renal blood flow autoregulation: dynamics and contributions. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R1–17. [DOI] [PubMed] [Google Scholar]
- 178.Just A, Ehmke H, Toktomambetova L, et al. Dynamic characteristics and underlying mechanisms of renal blood flow autoregulation in the conscious dog. Am J Physiol Renal Physiol. 2001;280(6):F1062–71. [DOI] [PubMed] [Google Scholar]
- 179.Walker M 3rd, Harrison-Bernard LM, Cook AK, et al. Dynamic interaction between myogenic and TGF mechanisms in afferent arteriolar blood flow autoregulation. Am J Physiol Renal Physiol. 2000;279(5):F858–65. [DOI] [PubMed] [Google Scholar]
- 180.Navar LG. Integrating multiple paracrine regulators of renal microvascular dynamics. Am J Physiol Renal Physiol. 1998;274(3):F433–44. [DOI] [PubMed] [Google Scholar]
- 181.Schnermann J, Homer W. Smith Award lecture. The juxtaglomerular apparatus: from anatomical peculiarity to physiological relevance. J Am Soc Nephrol. 2003;14(6):1681–1694. [DOI] [PubMed] [Google Scholar]
- 182.Slotki I, Skorecki K. Disorders of sodium balance. Brenner and Rector’s The Kidney. Philadelphia, PA: Elsevier; 2019. p. 390–442. [Google Scholar]
- 183.De Moor B, Vanwalleghem JF, Swennen Q, et al. Haemodynamic or metabolic stimulation tests to reveal the renal functional response: requiem or revival?. Clin Kidney J. 2018;11(5):623–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bosch JP, Saccaggi A, Lauer A, et al. Renal functional reserve in humans: effect of protein intake on glomerular filtration rate. Am J Med. 1983;75(6):943–950. [DOI] [PubMed] [Google Scholar]
- 185.Fliser D, Zeier M, Nowack R, et al. Renal functional reserve in healthy elderly subjects. J Am Soc Nephrol. 1993;3(7):1371–1377. [DOI] [PubMed] [Google Scholar]
- 186.Ronco C, Chawla LS. Glomerular and Tubular Kidney Stress Test: New Tools for a Deeper Evaluation of Kidney Function. Nephron. 2016;134(3):191–194. [DOI] [PubMed] [Google Scholar]
- 187.Sharma A, Mucino MJ, Ronco C. Renal functional reserve and renal recovery after acute kidney injury. Nephron Clin Practice. 2014;127(1–4):94–100. [DOI] [PubMed] [Google Scholar]
- 188.Gaspari F, Perico N, Remuzzi G. Application of newer clearance techniques for the determination of glomerular filtration rate. Curr Opin Nephrol Hypertens. 1998;7(6):675–680. [DOI] [PubMed] [Google Scholar]
- 189.Soveri I, Berg UB, Björk J, et al. Measuring GFR: A Systematic Review. Am J Kidney Dis. 2014. September 01;64(3):411–424. [DOI] [PubMed] [Google Scholar]
- 190.Nissenson AR, Weston RE, Kleeman CR. Mannitol. West J Med. 1979. October;131(4):277–284. [PMC free article] [PubMed] [Google Scholar]
- 191.Barry KG, Berman AR. Mannitol Infusion. N Eng J Med. 1961;264(21):1085–1088. [DOI] [PubMed] [Google Scholar]
- 192.Smith WW, Finkelstein N, Smith HW. Renal excretion of hexitols (sorbitol, mannitol, and dulcitol) and their derivatives (sorbitan, isomannide, and sorbide) and of endogenous creatinine-like chromogen in dog and man. J Biol Chem. 1940;135(1):231–250. [Google Scholar]
- 193.Borsook H, Dubnoff JW. The hydrolysis of phosphocreatine and the origin of urinary creatinine. J Biol Chem. 1947. May;168(2):493–510. [PubMed] [Google Scholar]
- 194.Levey A, Perrone R, Madias N. Serum Creatinine and Renal Function. Annu Rev Med. 1988;39(1):465–490. [DOI] [PubMed] [Google Scholar]
- 195.Heymsfield SB, Arteaga C, McManus C, et al. Measurement of muscle mass in humans: validity of the 24-hour urinary creatinine method. Am J Clin Nutr. 1983. March;37(3):478–494. [DOI] [PubMed] [Google Scholar]
- 196.Sterner G, Frennby B, Mansson S, et al. Determining ‘true’ glomerular filtration rate in healthy adults using infusion of inulin and comparing it with values obtained using other clearance techniques or prediction equations. Scand J Urol Nephrol. 2008;42(3):278–285. [DOI] [PubMed] [Google Scholar]
- 197.Shemesh O, Golbetz H, Kriss JP, et al. Limitations of creatinine as a filtration marker in glomerulopathic patients. Kidney Int. 1985;28(5):830–838. [DOI] [PubMed] [Google Scholar]
- 198.Sjostrom PA, Odlind BG, Wolgast M. Extensive tubular secretion and reabsorption of creatinine in humans. Scand J Urol Nephrol. 1988;22(2):129–131. [DOI] [PubMed] [Google Scholar]
- 199.Romano G, Bortolotti N, Falleti E, et al. The influence of furosemide on free water clearance. Panminerva Med. 1999. June;41(2):103–108. [PubMed] [Google Scholar]
- 200.Levey AS, Stevens LA. Estimating GFR using the CKD epidemiology collaboration (CKD-EPI) creatinine equation: more accurate GFR estimates, lower CKD prevalence estimates, and better risk predictions. Am J Kidney Dis. 2010;55(4):622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hostetter TH, Levey AS, Stevens LA. Clinical impact of reporting estimated glomerular filtration rates. Clin Chem. 2010;56(9):1381–1383. [DOI] [PubMed] [Google Scholar]
- 202.Poortmans JR, Gulbis B, De Bruyn E, et al. Limitations of serum values to estimate glomerular filtration rate during exercise. Br J Sports Med. 2013;47(18):1166–1170. [DOI] [PubMed] [Google Scholar]
- 203.Sands J, Layton H, Fenton R. Urine concentration and dilution. Brenner and Rector’s The Kidney. 2011;1:326–352. [Google Scholar]
- 204.Stanhewicz AE, Kenney WL. Determinants of water and sodium intake and output. Nutr Rev. 2015;73:73–82. [DOI] [PubMed] [Google Scholar]
- 205.Steiner RW. Interpreting the fractional excretion of sodium. Am J Med. 1984;77(4):699–702. [DOI] [PubMed] [Google Scholar]
- 206.Alan S, Chertow GM, Luyckx V, et al. Brenner and Rector’s The Kidney E-Book. Philadelphia, PA: Elsevier Health Sciences; 2019. [Google Scholar]
- 207.Wijkström J, Leiva R, Elinder C-G, et al. Clinical and pathological characterization of Mesoamerican nephropathy: a new kidney disease in Central America. Am J Kidney Dis. 2013;62(5):908–918. [DOI] [PubMed] [Google Scholar]
- 208.Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012;380(9843):756–766. [DOI] [PubMed] [Google Scholar]
- 209.Okusa MD, Portilla D. Pathophysiology of acute kidney injury. Brenner and Rector’s The Kidney. Philadelphia, PA: Elsevier; 2019. p. 906–939. [Google Scholar]
- 210.Weisbord SD, Palevsky PM. Prevention and management of acute kidney injury. Brenner and Rector’s The Kidney. Philadelphia, PA: Elsevier; 2019. p. 940–977. [Google Scholar]
- 211.Heung M, Steffick DE, Zivin K, et al. Acute Kidney Injury Recovery Pattern and Subsequent Risk of CKD: An Analysis of Veterans Health Administration Data. Am J Kidney Dis. 2016;67(5):742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Venkatachalam MA, Bernard DB, Donohoe JF, et al. Ischemic damage and repair in the rat proximal tubule: differences among the S1, S2, and S3 segments. Kidney Int. 1978;14(1):31–49. [DOI] [PubMed] [Google Scholar]
- 213.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334(22):1448–1460. [DOI] [PubMed] [Google Scholar]
- 214.Mehta RL, Kellum JA, Shah SV, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11(2):R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kellum JA, Lameire N, Aspelin P, et al. Kidney disease: improving global outcomes (KDIGO) acute kidney injury work group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl. 2012;2(1):1–138. [Google Scholar]
- 216.Bellomo R, Ronco C, Kellum JA, et al. Acute renal failure–definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8(4):R204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Seo DW, Li H, Qu CK, et al. Shp-1 mediates the antiproliferative activity of tissue inhibitor of metalloproteinase-2 in human microvascular endothelial cells. J Biol Chem. 2006;281(6):3711–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wajapeyee N, Serra RW, Zhu X, et al. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132(3):363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Yang QH, Liu DW, Long Y, et al. Acute renal failure during sepsis: potential role of cell cycle regulation. J Infect. 2009. June;58(6):459–464. [DOI] [PubMed] [Google Scholar]
- 220.Kashani K, Al-Khafaji A, Ardiles T, et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care. 2013;17(1):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Endre ZH, Pickering JW. Acute kidney injury: cell cycle arrest biomarkers win race for AKI diagnosis. Nat Rev Nephrol. 2014. December;10(12):683–685. [DOI] [PubMed] [Google Scholar]
- 222.Emlet DR, Pastor-Soler N, Marciszyn A, et al. Insulin-like growth factor binding protein 7 and tissue inhibitor of metalloproteinases-2: differential expression and secretion in human kidney tubule cells. Am J Physiol Renal Physiol. 2017;312(2):F284–F296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Koyner JL, Parikh CR. Clinical utility of biomarkers of AKI in cardiac surgery and critical illness. Clin J Am Soc Nephrol. 2013. June;8(6):1034–1042. [DOI] [PubMed] [Google Scholar]
- 224.Mishra J, Dent C, Tarabishi R, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet. 2005. April 2- 8;365(9466):1231–1238. [DOI] [PubMed] [Google Scholar]
- 225.Parikh CR, Coca SG, Thiessen-Philbrook H, et al. Postoperative biomarkers predict acute kidney injury and poor outcomes after adult cardiac surgery. J Am Soc Nephrol. 2011. September;22(9):1748–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Nickolas TL, O’Rourke MJ, Yang J, et al. Sensitivity and specificity of a single emergency department measurement of urinary neutrophil gelatinase-associated lipocalin for diagnosing acute kidney injury. Ann Intern Med. 2008;148(11):810–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Goetz DH, Holmes MA, Borregaard N, et al. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10(5):1033–1043. [DOI] [PubMed] [Google Scholar]
- 228.Horwitz LD, Sherman NA, Kong Y, et al. Lipophilic siderophores of mycobacterium tuberculosis prevent cardiac reperfusion injury. Proc Natl Acad Sci USA. 1998. April 28;95(9):5263–5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Mori K, Lee HT, Rapoport D, et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J Clin Invest. 2005;115(3):610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Mishra J, Ma Q, Prada A, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003;14(10):2534–2543. [DOI] [PubMed] [Google Scholar]
- 231.Chakraborty S, Kaur S, Guha S, et al. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim Biophys Acta. 2012;1826(1):129–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Hvidberg V, Jacobsen C, Strong RK, et al. The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase-associated lipocalin with high affinity and mediates its cellular uptake. FEBS Lett. 2005;579(3):773–777. [DOI] [PubMed] [Google Scholar]
- 233.Schrezenmeier E, Barasch J, Budde K, et al. Biomarkers in acute kidney injury–pathophysiological basis and clinical performance. Acta Physiol. 2017;219(3):556–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Schmidt-Ott KM, Mori K, Li JY, et al. Dual action of neutrophil gelatinase-associated lipocalin. J Am Soc Nephrol. 2007;18(2):407–413. [DOI] [PubMed] [Google Scholar]
- 235.Ichimura T, Bonventre JV, Bailly V, et al. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem. 1998;273(7):4135–4142. [DOI] [PubMed] [Google Scholar]
- 236.Prozialeck W, Vaidya V, Liu J, et al. Kidney injury molecule-1 is an early biomarker of cadmium nephrotoxicity. Kidney Int. 2007;72(8):985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ichimura T, Hung CC, Yang SA, et al. Kidney injury molecule-1: a tissue and urinary biomarker for nephrotoxicant-induced renal injury. Am J Physiol Renal Physiol. 2004;286(3):F552–63. [DOI] [PubMed] [Google Scholar]
- 238.Han WK, Bailly V, Abichandani R, et al. Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int. 2002;62(1):237–244. [DOI] [PubMed] [Google Scholar]
- 239.Wu H, Craft ML, Wang P, et al. IL-18 contributes to renal damage after ischemia-reperfusion. J Am Soc Nephrol. 2008;19(12):2331–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Gauer S, Sichler O, Obermüller N, et al. IL-18 is expressed in the intercalated cell of human kidney. Kidney Int. 2007;72(9):1081–1087. [DOI] [PubMed] [Google Scholar]
- 241.Franke EI, Vanderbrink BA, Hile KL, et al. Renal IL-18 production is macrophage independent during obstructive injury. PLoS One. 2012;7(10):e47417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kashani K, Cheungpasitporn W, Ronco C. Biomarkers of acute kidney injury: the pathway from discovery to clinical adoption. Clin Chem Lab Med. 2017;55(8):1074–1089. [DOI] [PubMed] [Google Scholar]
- 243.Parikh CR, Thiessen-Philbrook H, Garg AX, et al. Performance of kidney injury molecule-1 and liver fatty acid-binding protein and combined biomarkers of AKI after cardiac surgery. Clin J Am Soc Nephrol. 2013;8(7):1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Maatman RG, Van Kuppevelt TH, Veerkamp JH. Two types of fatty acid-binding protein in human kidney. Isolation, characterization and localization. Biochem J. 1991;273:759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wang G, Gong Y, Anderson J, et al. Antioxidative function of L‐FABP in L‐FABP stably transfected Chang liver cells. Hepatology. 2005;42(4):871–879. [DOI] [PubMed] [Google Scholar]
- 246.Yamamoto T, Noiri E, Ono Y, et al. Renal L-type fatty acid–binding protein in acute ischemic injury. J Am Soc Nephrol. 2007;18(11):2894–2902. [DOI] [PubMed] [Google Scholar]
- 247.Kashani K, Kellum JA. Novel biomarkers indicating repair or progression after acute kidney injury. Curr Opin Nephrol Hypertens. 2015;24(1):21–27. [DOI] [PubMed] [Google Scholar]
- 248.Murray PT, Mehta RL, Shaw A, et al. Potential use of biomarkers in acute kidney injury: report and summary of recommendations from the 10th Acute Dialysis Quality Initiative consensus conference. Kidney Int. 2014;85(3):513–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.McCullough PA, Chinnaiyan KM, Gallagher MJ, et al. Changes in renal markers and acute kidney injury after marathon running. Nephrology. 2011;16(2):194–199. [DOI] [PubMed] [Google Scholar]
- 250.Hoffman MD, Stuempfle KJ, Fogard K, et al. Urine dipstick analysis for identification of runners susceptible to acute kidney injury following an ultramarathon. J Sports Sci. 2013;31(1):20–31. [DOI] [PubMed] [Google Scholar]
- 251.Hou SK, Chiu YH, Tsai YF, et al. Clinical Impact of Speed Variability to Identify Ultramarathon Runners at Risk for Acute Kidney Injury. PLoS One. 2015;10(7):e0133146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Bongers C, Alsady M, Nijenhuis T, et al. Impact of acute versus repetitive moderate intensity endurance exercise on kidney injury markers. Physiol Rep. 2017;5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Mansour SG, Verma G, Pata RW, et al. Kidney Injury and Repair Biomarkers in Marathon Runners. Am J Kidney Dis. 2017;70(2):252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Bongers C, Alsady M, Nijenhuis T, et al. Impact of acute versus prolonged exercise and dehydration on kidney function and injury. Physiol Rep. 2018;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Supavekin S, Zhang W, Kucherlapati R, et al. Differential gene expression following early renal ischemia/reperfusion. Kidney Int. 2003;63(5):1714–1724. [DOI] [PubMed] [Google Scholar]
- 256.Lin X, Yuan J, Zhao Y, et al. Urine interleukin-18 in prediction of acute kidney injury: a systemic review and meta-analysis. J Nephrol. 2015;28(1):7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Katz N, Ronco C. Acute kidney stress–a useful term based on evolution in the understanding of acute kidney injury. Crit Care. 2016;20:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Vijayan A, Faubel S, Askenazi DJ, et al. Clinical use of the urine biomarker [TIMP-2]×[IGFBP7] for acute kidney injury risk assessment. Am J Kidney Dis. 2016;68(1):19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Noto A, Cortegiani A, David A. NephroCheck: should we consider urine osmolality?. Crit Care. 2019;23(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hahn RG, Zdolsek J. Nephrocheck(®) results should be corrected for dilution. Acta Anaesthesiol Scand. 2017;61(2):261–262. [DOI] [PubMed] [Google Scholar]
- 261.Chapman CL, Schlader ZJ. Assessing the risk of acute kidney injury following exercise in the heat: Timing is important. Temperature. 2020;1–3. doi: 10.1080/23328940.2020.1741333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Wołyniec W, Ratkowski W, Renke J, et al. Changes in Novel AKI Biomarkers after Exercise. A Systematic Review. Int J Mol Sci. 2020;21:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Byfield G, Telser S, Keeton R. Renal blood flow and glomerular filtration: as influenced by environmental temperature changes. JAMA. 1943;121(2):118–123. [Google Scholar]
- 264.Radigan LR, Robinson S. Effects of environmental heat stress and exercise on renal blood flow and filtration rate. J Appl Physiol. 1949;2(4):185–191. [DOI] [PubMed] [Google Scholar]
- 265.Abramson N. Effect of heat stress upon human renal function. Aerospace Med. 1967;38:234–238. [Google Scholar]
- 266.Haapanen E, editor. Effects of the Finnish sauna bath on the electrolyte excretions and the renal clearances. Annales medicinae experimentalis et biologiae Fenniae. 1958;42:39–46. [PubMed] [Google Scholar]
- 267.Kaufmann W, Nieth H, Schlitter J. Renal hemodynamics in exogenous heat load. Klinische Wochenschrift. 1964;42:39–46. [DOI] [PubMed] [Google Scholar]
- 268.Nieth H, Kaufmann W. Renal hemodynamics and excretory function of the kidney in essential and renal hypertension in warm environmental conditions. Verhandlungen der Deutschen Gesellschaft fur Kreislaufforschung. 1963;28:251. [PubMed] [Google Scholar]
- 269.Kenney R. The effect of hot, humid environments on the renal function of West Africans. J Physiol. 1952;118(2):25P. [PubMed] [Google Scholar]
- 270.Redisch W, Wertheimer L, Delisle C, et al. Comparison of various vascular beds in man; their responses to a simple vasodilator stimulus. Circulation. 1954. January;9(1):63–67. [DOI] [PubMed] [Google Scholar]
- 271.Rowell LB, Brengelmann GL, Blackmon JR, et al. Redistribution of blood flow during sustained high skin temperature in resting man. J Appl Physiol. 1970;28(4):415–420. [DOI] [PubMed] [Google Scholar]
- 272.Johnson JM, Proppe DW. Cardiovascular adjustments to heat stress. Compr Physiol. 2010;Suppl 14:215–243. [Google Scholar]
- 273.Johnson JM, Minson CT, Kellogg DL Jr.. Cutaneous vasodilator and vasoconstrictor mechanisms in temperature regulation. Compr Physiol. 2014. January;4(1):33–89. [DOI] [PubMed] [Google Scholar]
- 274.Rowell LB. Cardiovascular adjustments to thermal stress. Compr Physiol. 2011;Suppl 8:967–1023. [Google Scholar]
- 275.Kenney MJ, Barney CC, Hirai T, et al. Sympathetic nerve responses to hyperthermia in the anesthetized rat. J Appl Physiol. 1995;78(3):881–889. [DOI] [PubMed] [Google Scholar]
- 276.Kenney MJ, Claassen DE, Bishop MR, et al. Regulation of the sympathetic nerve discharge bursting pattern during heat stress. Am J Physiol Regul Integr Comp Physiol. 1998;275(6):R1992–2001. [DOI] [PubMed] [Google Scholar]
- 277.Kenney MJ, Fels RJ. Sympathetic nerve regulation to heating is altered in senescent rats. Am J Physiol Regul Integr Comp Physiol. 2002;283(2):R513–20. [DOI] [PubMed] [Google Scholar]
- 278.Kenney MJ, Fels RJ. Forebrain and brain stem neural circuits contribute to altered sympathetic responses to heating in senescent rats. J Appl Physiol. 2003;95(5):1986–1993. [DOI] [PubMed] [Google Scholar]
- 279.Kenney MJ, Musch TI, Weiss ML. Renal sympathetic nerve regulation to heating is altered in rats with heart failure. Am J Physiol Heart Circ Physiol. 2001;280(6):H2868–75. [DOI] [PubMed] [Google Scholar]
- 280.Kenney MJ, Pickar JG, Weiss ML, et al. Effects of midbrain and spinal cord transections on sympathetic nerve responses to heating. Am J Physiol Regul Integr Comp Physiol. 2000;278(5):R1329–38. [DOI] [PubMed] [Google Scholar]
- 281.Ninomiya I, Fujita S. Reflex effects of thermal stimulation on sympathetic nerve activity to skin and kidney. Am J Physiol. 1976;230(2):271–278. [DOI] [PubMed] [Google Scholar]
- 282.DiBona GF, Jones SY. Reflex effects on components of synchronized renal sympathetic nerve activity. Am J Physiol Renal Physiol. 1998;275(3):F441–6. [DOI] [PubMed] [Google Scholar]
- 283.DiBona GF, Sawin LL. Renal hemodynamic effects of activation of specific renal sympathetic nerve fiber groups. Am J Physiol Regul Integr Comp Physiol. 1999;276(2):R539–49. [DOI] [PubMed] [Google Scholar]
- 284.Kopp UC, Smith LA, DiBona GF. Facilitatory role of efferent renal nerve activity on renal sensory receptors. Am J Physiol Renal Physiol. 1987;253(4 Pt 2):F767–77. [DOI] [PubMed] [Google Scholar]
- 285.DiBona GF, Sawin LL, Jones SY. Differentiated sympathetic neural control of the kidney. Am J Physiol Regul Integr Comp Physiol. 1996;271(1 Pt 2):R84–90. [DOI] [PubMed] [Google Scholar]
- 286.Eisman MM, Rowell LB. Renal vascular response to heat stress in baboons–role of renin-angiotensin. J Appl Physiol Respir Environ Exerc Physiol. 1977. October;43(4):739–746. [DOI] [PubMed] [Google Scholar]
- 287.Azzawi SA, Shirley DG. The effect of vasopressin on renal blood flow and its distribution in the rat. J Physiol. 1983;341:233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Rowell LB, Detry JR, Profant GR, et al. Splanchnic vasoconstriction in hyperthermic man–role of falling blood pressure. J Appl Physiol. 1971;31(6):864–869. [DOI] [PubMed] [Google Scholar]
- 289.Miyamoto M. Renal cortical and medullary tissue blood flow during experimental hyperthermia in dogs. Thermal Med. 1994;10(1):78–89. [Google Scholar]
- 290.Smith JH, Robinson S, Pearcy M. Renal responses to exercise, heat and dehydration. J Appl Physiol. 1952;4(8):659–665. [DOI] [PubMed] [Google Scholar]
- 291.Melin B, Koulmann N, Jimenez C, et al. Comparison of passive heat or exercise-induced dehydration on renal water and electrolyte excretion: the hormonal involvement. Eur J Appl Physiol. 2001;85(3–4):250–258. [DOI] [PubMed] [Google Scholar]
- 292.Ravanelli N, Barry H, Schlader ZJ, et al. Impact of passive heat acclimation on markers of kidney function during heat stress. Exp Physiol. 2020. [DOI] [PubMed] [Google Scholar]
- 293.Low DA, Keller DM, Wingo JE, et al. Sympathetic nerve activity and whole body heat stress in humans. J Appl Physiol. 2011;111(5):1329–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Kanter GS. Heat and excretion in man. J Appl Physiol. 1955;7(5):533–536. [DOI] [PubMed] [Google Scholar]
- 295.Macfarlane WV, Robinson K, Howard B, et al. Heat, salt and hormones in panting and sweating animals. Nature. 1958;182(4636):672–673. [DOI] [PubMed] [Google Scholar]
- 296.Bailey RE, Bartos D, Barton F, et al. Activation of aldosterone and renin secretion by thermal stress. Experientia. 1972;28(2):159–160. [DOI] [PubMed] [Google Scholar]
- 297.Kosunen KJ, Pakarinen AJ, Kuoppasalmi K, et al. Plasma renin activity, angiotensin II, and aldosterone during intense heat stress. J Appl Physiol. 1976;41(3):323–327. [DOI] [PubMed] [Google Scholar]
- 298.Follenius M, Brandenberger G, Reinhardt B, et al. Plasma aldosterone, renin activity, and cortisol responses to heat exposure in sodium depleted and repeleted subjects. Eur J Appl Physiol Occup Physiol. 1979;41(1):41–50. [DOI] [PubMed] [Google Scholar]
- 299.Finberg JP, Katz M, Gazit H, et al. Plasma renin activity after acute heat exposure in nonacclimatized and naturally acclimatized man. J Appl Physiol. 1974. May;36(5):519–523. [DOI] [PubMed] [Google Scholar]
- 300.Collins KJ, Weiner JS. Endocrinological aspects of exposure to high environmental temperatures. Physiol Rev. 1968;48(4):785–839. [DOI] [PubMed] [Google Scholar]
- 301.Convertino VA, Greenleaf JE, Bernauer EM. Role of thermal and exercise factors in the mechanism of hypervolemia. J Appl Physiol. 1980;48(4):657–664. [DOI] [PubMed] [Google Scholar]
- 302.Tidgren B, Hjemdahl P. Renal responses to mental stress and epinephrine in humans. Am J Physiol Renal Physiol. 1989;257(4):F682–F689. [DOI] [PubMed] [Google Scholar]
- 303.Chapman CL, Benati JM, Johnson BD, et al. Renal and segmental artery hemodynamics during whole body passive heating and cooling recovery. J Appl Physiol. 2019;127(4):974–983. [DOI] [PubMed] [Google Scholar]
- 304.Cui J, Shibasaki M, Low DA, et al. Heat stress attenuates the increase in arterial blood pressure during the cold pressor test. J Appl Physiol. 2010;109(5):1354–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Schlader ZJ, Wilson TE, Crandall CG. Mechanisms of orthostatic intolerance during heat stress. Auton Neurosci. 2016;196:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Barclay JA, Cooke WT, Kenney RA, et al. The effects of water diuresis and exercise on the volume and composition of the urine. Am J Physiol. 1947;148(2):327–337. [DOI] [PubMed] [Google Scholar]
- 307.Zambraski E. The Renal System. In: Farrell PA, Joyner MJ, Caiozzo V, editors. ACSM’s advanced exercise physiology. Philadelphia, PA: Wolters Kluwer Health Adis (ESP). 2011. p. 551–563. [Google Scholar]
- 308.Tidgren B, Hjemdahl P, Theodorsson E, et al. Renal neurohormonal and vascular responses to dynamic exercise in humans. J Appl Physiol. 1991;70(5):2279–2286. [DOI] [PubMed] [Google Scholar]
- 309.Freund BJ, Shizuru EM, Hashiro GM, et al. Hormonal, electrolyte, and renal responses to exercise are intensity dependent. J Appl Physiol. 1991;70(2):900–906. [DOI] [PubMed] [Google Scholar]
- 310.McAllister RM. Adaptations in control of blood flow with training: splanchnic and renal blood flows. Med Sci Sports Exerc. 1998;30(3):375–381. [DOI] [PubMed] [Google Scholar]
- 311.Farquhar WB, Kenney WL. Age and renal prostaglandin inhibition during exercise and heat stress. J Appl Physiol. 1999;86(6):1936–1943. [DOI] [PubMed] [Google Scholar]
- 312.Otani H, Kaya M, Tsujita J. Effect of the volume of fluid ingested on urine concentrating ability during prolonged heavy exercise in a hot environment. J Sports Sci Med. 2013;12(1):197–204. [PMC free article] [PubMed] [Google Scholar]
- 313.Mittleman KD. Influence of angiotensin II blockade during exercise in the heat. Eur J Appl Physiol Occup Physiol. 1996;72(5–6):542–547. [DOI] [PubMed] [Google Scholar]
- 314.Montain SJ, Laird JE, Latzka WA, et al. Aldosterone and vasopressin responses in the heat: hydration level and exercise intensity effects. Med Sci Sports Exerc. 1997. May;29(5):661–668. [DOI] [PubMed] [Google Scholar]
- 315.Kachadorian WA, Johnson RE. Renal responses to various rates of exercise. J Appl Physiol. 1970;28(6):748–752. [DOI] [PubMed] [Google Scholar]
- 316.Refsum HE, Stromme SB. Relationship between urine flow, glomerular filtration, and urine solute concentrations during prolonged heavy exercise. Scand J Clin Lab Invest. 1975;35(8):775–780. [DOI] [PubMed] [Google Scholar]
- 317.Wade CE, Claybaugh JR. Plasma renin activity, vasopressin concentration, and urinary excretory responses to exercise in men. J Appl Physiol Respir Environ Exerc Physiol. 1980;49(6):930–936. [DOI] [PubMed] [Google Scholar]
- 318.Melin B, Jimenez C, Savourey G, et al. Effects of hydration state on hormonal and renal responses during moderate exercise in the heat. Eur J Appl Physiol Occup Physiol. 1997;76(4):320–327. [DOI] [PubMed] [Google Scholar]
- 319.Yang B, Bankir L. Urea and urine concentrating ability: new insights from studies in mice. Am J Physiol Renal Physiol. 2005;288(5):F881–96. [DOI] [PubMed] [Google Scholar]
- 320.Poortmans JR, Labilloy D. The influence of work intensity on postexercise proteinuria. Eur J Appl Physiol Occup Physiol. 1988;57(2):260–263. [DOI] [PubMed] [Google Scholar]
- 321.Poortmans JR, Mathieu N, De Plaen P. Influence of running different distances on renal glomerular and tubular impairment in humans. Eur J Appl Physiol Occup Physiol. 1996;72(5–6):522–527. [DOI] [PubMed] [Google Scholar]
- 322.Poortmans JR, Vanderstraeten J. Kidney function during exercise in healthy and diseased humans. An update. Sports Med. 1994;18(6):419–437. [DOI] [PubMed] [Google Scholar]
- 323.Poortmans JR. Exercise and renal function. Sports Med. 1984;1(2):125–153. [DOI] [PubMed] [Google Scholar]
- 324.Abbate M, Zoja C, Remuzzi G. How does proteinuria cause progressive renal damage?. J Am Soc Nephrol. 2006;17(11):2974–2984. [DOI] [PubMed] [Google Scholar]
- 325.Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med. 1998;339(20):1448–1456. [DOI] [PubMed] [Google Scholar]
- 326.Reuben DB, Wachtel TJ, Brown PC, et al. Transient proteinuria in emergency medical admissions. N Engl J Med. 1982;306(17):1031–1033. [DOI] [PubMed] [Google Scholar]
- 327.Raines N, Gonzalez M, Wyatt C, et al. Risk factors for reduced glomerular filtration rate in a Nicaraguan community affected by Mesoamerican nephropathy. MEDICC Rev. 2014;16(2):16–22. [DOI] [PubMed] [Google Scholar]
- 328.O’Donnell JK, Tobey M, Weiner DE, et al. Prevalence of and risk factors for chronic kidney disease in rural Nicaragua. Nephrol Dial Transplant. 2011;26(9):2798–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Peraza S, Wesseling C, Aragon A, et al. Decreased kidney function among agricultural workers in El Salvador. Am J Kidney Dis. 2012;59(4):531–540. [DOI] [PubMed] [Google Scholar]
- 330.Laws RL, Brooks DR, Amador JJ, et al. Changes in kidney function among Nicaraguan sugarcane workers. Int J Occup Environ Health. 2015;21(3):241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Wesseling C, Aragon A, Gonzalez M, et al. Heat stress, hydration and uric acid: a cross-sectional study in workers of three occupations in a hotspot of Mesoamerican nephropathy in Nicaragua. BMJ Open. 2016;6(12):e011034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Yasuda N, Ruby BC. Assessment of urinary protein composition in response to consecutive days of wildland firefighting. Int J Occup Saf Ergon. 2019;25(1):27–34. [DOI] [PubMed] [Google Scholar]
- 333.Laws RL, Brooks DR, Amador JJ, et al. Biomarkers of Kidney Injury Among Nicaraguan Sugarcane Workers. Am J Kidney Dis. 2016;67(2):209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Shephard RJ. Exercise proteinuria and hematuria: current knowledge and future directions. J Sports Med Phys Fitness. 2016;56(9):1060–1076. [PubMed] [Google Scholar]
- 335.Correa-Rotter R, Wesseling C, Johnson RJ. CKD of unknown origin in Central America: the case for a Mesoamerican nephropathy. Am J Kidney Dis. 2014. March;63(3):506–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Garcia-Trabanino R, Jarquin E, Wesseling C, et al. Heat stress, dehydration, and kidney function in sugarcane cutters in El Salvador–A cross-shift study of workers at risk of Mesoamerican nephropathy. Environ Res. 2015;142:746–755. [DOI] [PubMed] [Google Scholar]
- 337.Wesseling C, Aragon A, Gonzalez M, et al. Kidney function in sugarcane cutters in Nicaragua–A longitudinal study of workers at risk of Mesoamerican nephropathy. Environ Res. 2016;147:125–132. [DOI] [PubMed] [Google Scholar]
- 338.Meng X-L, Rosenthal R, Rubin DB. Comparing correlated correlation coefficients. Psychol Bull. 1992;111(1):172. [Google Scholar]
- 339.Walker LA, Gerber JG, Frolich JC, et al. Redistribution of intrarenal blood flow following ADH administration: lack of inhibition by blockade of prostaglandin in cyclooxygenase. Prostaglandins Med. 1978;1(4):295–303. [DOI] [PubMed] [Google Scholar]
- 340.Zhuo JL, Li XC. Proximal nephron. Compr Physiol. 2013;3(3):1079–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Hope A, Tyssebotn I. The effect of water deprivation on local renal blood flow and filtration in the laboratory rat. Circ Shock. 1983;11(2):175–186. [PubMed] [Google Scholar]
- 342.Sanchez-Lozada LG, Rodriguez-Iturbe B, Kelley EE, et al. Uric acid and Hypertension: An Update with Recommendations. Am J Hypertens. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Ejaz AA, Johnson RJ, Shimada M, et al. The Role of Uric Acid in Acute Kidney Injury. Nephron. 2019;142(4):275–283. [DOI] [PubMed] [Google Scholar]
- 344.Xiao J, Zhang X, Fu C, et al. Impaired Na(+)-K(+)-ATPase signaling in renal proximal tubule contributes to hyperuricemia-induced renal tubular injury. Exp Mol Med. 2018;50(3):e452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Sanchez-Lozada LG, Tapia E, Santamaria J, et al. Mild hyperuricemia induces vasoconstriction and maintains glomerular hypertension in normal and remnant kidney rats. Kidney Int. 2005;67(1):237–247. [DOI] [PubMed] [Google Scholar]
- 346.Selkirk GA, McLellan TM, Wright HE, et al. Mild endotoxemia, NF-kappaB translocation, and cytokine increase during exertional heat stress in trained and untrained individuals. Am J Physiol Regul Integr Comp Physiol. 2008;295(2):R611–23. [DOI] [PubMed] [Google Scholar]
- 347.Madero M, Garcia-Arroyo FE, Sanchez-Lozada LG. Pathophysiologic insight into MesoAmerican nephropathy. Curr Opin Nephrol Hypertens. 2017;26(4):296–302. [DOI] [PubMed] [Google Scholar]
- 348.Johnson RJ, Rodriguez-Iturbe B, Roncal-Jimenez C, et al. Hyperosmolarity drives hypertension and CKD–water and salt revisited. Nat Rev Nephrol. 2014;10(7):415–420. [DOI] [PubMed] [Google Scholar]
- 349.Huang Z, Hong Q, Zhang X, et al. Aldose reductase mediates endothelial cell dysfunction induced by high uric acid concentrations. Cell Commun Signal. 2017;15(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Safety O, Administration H. Protecting workers from heat illness. OSHA-NIOSH Infosheet. 2011. Available from: http://www.cdc.gov/niosh/docs/2011-174/pdfs/2011-174.pdf
- 351.Centers for Disease Control Prevention . Preventing heat-related illness or death of outdoor workers. Workplace Solutions: 2013. 2014.
- 352.Jacklitsch B, Williams W, Musolin K, et al. NIOSH criteria for a recommended standard: occupational exposure to heat and hot environments. US Department of Health and Human Services, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication. 2016; 106.
- 353.Paula Santos U, Zanetta DM, Terra-Filho M, et al. Burnt sugarcane harvesting is associated with acute renal dysfunction. Kidney Int. 2015. April;87(4):792–799. [DOI] [PubMed] [Google Scholar]
- 354.Baker LB, Jeukendrup AE. Optimal composition of fluid‐replacement beverages. Compr Physiol. 2011;4(2):575–620. [DOI] [PubMed] [Google Scholar]
- 355.Gisolfi C, Summers R, Schedl H, et al. Intestinal water absorption from select carbohydrate solutions in humans. J Appl Physiol. 1992;73(5):2142–2150. [DOI] [PubMed] [Google Scholar]
- 356.Evans GH, Shirreffs SM, Maughan RJ. Acute effects of ingesting glucose solutions on blood and plasma volume. Br J Nutr. 2009;101(10):1503–1508. [DOI] [PubMed] [Google Scholar]
- 357.Gisolfi CV, Summers R, Schedl H, et al. Effect of sodium concentration in a carbohydrate-electrolyte solution on intestinal absorption. Med Sci Sports Exerc. 1995;27(10):1414–1420. [PubMed] [Google Scholar]
- 358.Gisolfi CV, Lambert GP, Summers RW. Intestinal fluid absorption during exercise: role of sport drink osmolality and [Na+]. Med Sci Sports Exerc. 2001;33(6):907–915. [DOI] [PubMed] [Google Scholar]
- 359.Shirreffs SM, Taylor AJ, Leiper JB, et al. Post-exercise rehydration in man: effects of volume consumed and drink sodium content. Med Sci Sports Exerc. 1996;28(10):1260. [DOI] [PubMed] [Google Scholar]
- 360.Rawson ES, Clarkson PM, Tarnopolsky MA. Perspectives on Exertional Rhabdomyolysis. Sports Med. 2017;47(Suppl 1):33–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Clarkson PM. Exertional rhabdomyolysis and acute renal failure in marathon runners. Sports Med. 2007;37(4–5):361–363. [DOI] [PubMed] [Google Scholar]
- 362.Gardner JW, Kark JA. Fatal rhabdomyolysis presenting as mild heat illness in military training. Mil Med. 1994;159(2):160–163. [PubMed] [Google Scholar]
- 363.Landau ME, Kenney K, Deuster P, et al. Exertional rhabdomyolysis: a clinical review with a focus on genetic influences. J Clin Neuromuscul Dis. 2012;13(3):122–136. [DOI] [PubMed] [Google Scholar]
- 364.Huynh A, Leong K, Jones N, et al. Outcomes of exertional rhabdomyolysis following high-intensity resistance training. Intern Med J. 2016;46(5):602–608. [DOI] [PubMed] [Google Scholar]
- 365.Knapik JJ, O’Connor FG. Exertional Rhabdomyolysis: Epidemiology, Diagnosis, Treatment, and Prevention. J Spec Oper Med. 2016;16(3):65–71. [DOI] [PubMed] [Google Scholar]
- 366.Oh RC, Arter JL, Tiglao SM, et al. Exertional rhabdomyolysis: a case series of 30 hospitalized patients. Mil Med. 2015;180(2):201–207. [DOI] [PubMed] [Google Scholar]
- 367.Tan W, Herzlich BC, Funaro R, et al. Rhabdomyolysis and myoglobinuric acute renal failure associated with classic heat stroke. South Med J. 1995;88(10):1065–1068. [DOI] [PubMed] [Google Scholar]
- 368.Barbosa CMG, Terra-Filho M, de Albuquerque ALP, et al. Burnt Sugarcane Harvesting – Cardiovascular Effects on a Group of Healthy Workers, Brazil. PLOS ONE. 2012;7(9):e46142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Sorensen CJ, Butler-Dawson J, Dally M, et al. Risk Factors and Mechanisms Underlying Cross-Shift Decline in Kidney Function in Guatemalan Sugarcane Workers. J Occup Environ Med. 2019;61(3):239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Brown SJ, Child RB, Day SH, et al. Exercise-induced skeletal muscle damage and adaptation following repeated bouts of eccentric muscle contractions. J Sports Sci. 1997;15(2):215–222. [DOI] [PubMed] [Google Scholar]
- 371.Burns .: World Health Organization; 2018. [accessed 2020 April 21]. Available from: https://www.who.int/news-room/fact-sheets/detail/burns
- 372.Folkestad T, Brurberg KG, Nordhuus KM, et al. Acute kidney injury in burn patients admitted to the intensive care unit: a systematic review and meta-analysis. Crit Care. 2020;24(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Clark A, Neyra JA, Madni T, et al. Acute kidney injury after burn. Burns. 2017;43(5):898–908. [DOI] [PubMed] [Google Scholar]
- 374.Yu H, Cooper EH, Settle JA, et al. Urinary protein profiles after burn injury. Burns Incl Therm Inj. 1983;9(5):339–349. [DOI] [PubMed] [Google Scholar]
- 375.Mariano F, Camussi G. Unravelling the enigma of proteinuria in burn patients. Crit Care. 2012;16(6):184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Jaehde U, Sorgel F. Clinical pharmacokinetics in patients with burns. Clin Pharmacokinet. 1995;29(1):15–28. [DOI] [PubMed] [Google Scholar]
- 377.Porter C, Tompkins RG, Finnerty CC, et al. The metabolic stress response to burn trauma: current understanding and therapies. Lancet. 2016;388(10052):1417–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Greenhalgh DG. Management of Burns. N Engl J Med. 2019;380(24):2349–2359. [DOI] [PubMed] [Google Scholar]
- 379.Wolfe RR. Review: acute versus chronic response to burn injury. Circ Shock. 1981;8(1):105–115. [PubMed] [Google Scholar]
- 380.Loirat P, Rohan J, Baillet A, et al. Increased glomerular filtration rate in patients with major burns and its effect on the pharmacokinetics of tobramycin. N Engl J Med. 1978;299(17):915–919. [DOI] [PubMed] [Google Scholar]
- 381.Aikawa N, Wakabayashi G, Ueda M, et al. Regulation of renal function in thermal injury. J Trauma. 1990;30(12 Suppl):S174–8. [DOI] [PubMed] [Google Scholar]
- 382.Crum RL, Dominic W, Hansbrough JF, et al. Cardiovascular and neurohumoral responses following burn injury. Arch Surg. 1990;125(8):1065–1069. [DOI] [PubMed] [Google Scholar]
- 383.Kirkebo A, Haugan A, Tyssebotn I. Blood flow heterogeneity in the renal cortex during burn shock in dogs. Acta Physiol Scand. 1985;123(2):205–213. [DOI] [PubMed] [Google Scholar]
- 384.Zdolsek HJ, Kagedal B, Lisander B, et al. Glomerular filtration rate is increased in burn patients. Burns. 2010;36(8):1271–1276. [DOI] [PubMed] [Google Scholar]
- 385.Mariano F, Cantaluppi V, Stella M, et al. Circulating plasma factors induce tubular and glomerular alterations in septic burns patients. Crit Care. 2008;12(2):R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Yamada Y, Endo S, Nakae H, et al. Examination of soluble Fas (sFas) and soluble Fas ligand (sFasL) in patients with burns. Burns. 2003;29(8):799–802. [DOI] [PubMed] [Google Scholar]
- 387.Arturson G, Mellander S. Acute Changes in Capillary Filtration and Diffusion in Experimental Burn Injury. Acta physiol scand. 1964;62(4):457–463. [DOI] [PubMed] [Google Scholar]
- 388.Goodwin CW, Aulick LH, Becker RA, et al. Increased Renal Perfusion and Kidney Size in Convalescent Burn Patients. JAMA. 1980;244(14):1588–1590. [PubMed] [Google Scholar]
- 389.Taylor NA. Human heat adaptation. Compr Physiol. 2011;4(1):325–365. [DOI] [PubMed] [Google Scholar]
- 390.Tyler CJ, Reeve T, Hodges GJ, et al. The Effects of Heat Adaptation on Physiology, Perception and Exercise Performance in the Heat: A Meta-Analysis. Sports Med. 2016;46(11):1699–1724. [DOI] [PubMed] [Google Scholar]
- 391.Périard JD, Racinais S, Sawka MN. Adaptations and mechanisms of human heat acclimation: Applications for competitive athletes and sports. Scand J Med Sci Sports. 2015;25(Suppl 1):20–38. [DOI] [PubMed] [Google Scholar]
- 392.Brunt VE, Howard MJ, Francisco MA, et al. Passive heat therapy improves endothelial function, arterial stiffness and blood pressure in sedentary humans. J Physiol. 2016;594(18):5329–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Buono MJ, Heaney JH, Canine KM. Acclimation to humid heat lowers resting core temperature. Am J Physiol Regul Integr Comp Physiol. 1998;274(5):R1295–R1299. [DOI] [PubMed] [Google Scholar]
- 394.Eichna LW, Park CR, Nelson N, et al. Thermal regulation during acclimatization in a hot, dry (desert type) environment. Am J Physiol. 1950;163(3):585–597. [DOI] [PubMed] [Google Scholar]
- 395.Lorenzo S, Halliwill JR, Sawka MN, et al. Heat acclimation improves exercise performance. J Appl Physiol. 2010;109(4):1140–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Nadel ER, Pandolf KB, Roberts MF, et al. Mechanisms of thermal acclimation to exercise and heat. J Appl Physiol. 1974;37(4):515–520. [DOI] [PubMed] [Google Scholar]
- 397.Roberts MF, Wenger CB, Stolwijk JA, et al. Skin blood flow and sweating changes following exercise training and heat acclimation. J Appl Physiol. 1977;43(1):133–137. [DOI] [PubMed] [Google Scholar]
- 398.Zappe D, Bell G, Swartzentruber H, et al. Age and regulation of fluid and electrolyte balance during repeated exercise sessions. Am J Physiol Regul Integr Comp Physiol. 1996;270(1):R71–R79. [DOI] [PubMed] [Google Scholar]
- 399.Jones SB, Musacchia XJ, Tempel GE. Mechanisms of temperature regulation in heat-acclimated hamsters. Am J Physiol. 1976;231(3):707–712. [DOI] [PubMed] [Google Scholar]
- 400.Chayoth R, Kleinman D, Kaplanski J, et al. Renal clearance of urea, inulin, and p-aminohippurate in heat-acclimated rats. J Appl Physiol Respir Environ Exerc Physiol. 1984;57(3):731–732. [DOI] [PubMed] [Google Scholar]
- 401.Omassoli J, Hill NE, Woods DR, et al. Variation in renal responses to exercise in the heat with progressive acclimatisation. J Sci Med Sport. 2019;22(9):1004–1009. [DOI] [PubMed] [Google Scholar]
- 402.Pryor RR, Pryor JL, Vandermark LW, et al. Acute Kidney Injury Biomarker Responses to Short-Term Heat Acclimation. Int J Environ Res Public Health. 2020;17:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Périard JD, Travers GJS, Racinais S, et al. Cardiovascular adaptations supporting human exercise-heat acclimation. Auton Neurosci. 2016;196:52–62. [DOI] [PubMed] [Google Scholar]
- 404.Patterson MJ, Stocks JM, Taylor NA. Sustained and generalized extracellular fluid expansion following heat acclimation. J Physiol. 2004;559(Pt 1):327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Harrison MH. Effects on thermal stress and exercise on blood volume in humans. Physiol Rev. 1985;65(1):149–209. [DOI] [PubMed] [Google Scholar]
- 406.Smiles KA, Robinson S. Sodium ion conservation during acclimatization of men to work in the heat. J Appl Physiol. 1971;31(1):63–69. [DOI] [PubMed] [Google Scholar]
- 407.Allsopp AJ, Sutherland R, Wood P, et al. The effect of sodium balance on sweat sodium secretion and plasma aldosterone concentration. Eur J Appl Physiol Occup Physiol. 1998;78(6):516–521. [DOI] [PubMed] [Google Scholar]
- 408.Francesconi R, Maher J, Bynum G, et al. Recurrent heat exposure: effects on levels of plasma and urinary sodium and potassium in resting and exercising men. Aviat Space Environ Med. 1977;48(5):399–404. [PubMed] [Google Scholar]
- 409.Nielsen B, Hales JR, Strange S, et al. Human circulatory and thermoregulatory adaptations with heat acclimation and exercise in a hot, dry environment. J Physiol. 1993;460:467–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Nielsen B, Strange S, Christensen NJ, et al. Acute and adaptive responses in humans to exercise in a warm, humid environment. Pflugers Arch. 1997;434(1):49–56. [DOI] [PubMed] [Google Scholar]
- 411.Armstrong LE, Francesconi RP, Kraemer WJ, et al. Plasma cortisol, renin, and aldosterone during an intense heat acclimation program. Int J Sports Med. 1989;10(1):38–42. [DOI] [PubMed] [Google Scholar]
- 412.Finberg JP, Berlyne GM. Modification of renin and aldosterone response to heat by acclimatization in man. J Appl Physiol Respir Environ Exerc Physiol. 1977. April;42(4):554–558. [DOI] [PubMed] [Google Scholar]
- 413.Davies JA, Harrison MH, Cochrane LA, et al. Effect of saline loading during heat acclimatization on adrenocortical hormone levels. J Appl Physiol Respir Environ Exerc Physiol. 1981;50(3):605–612. [DOI] [PubMed] [Google Scholar]
- 414.Garrett AT, Goosens NG, Rehrer NJ, et al. Induction and decay of short-term heat acclimation. Eur J Appl Physiol. 2009;107(6):659–670. [DOI] [PubMed] [Google Scholar]
- 415.Garrett AT, Goosens NG, Rehrer NJ, et al. Short-term heat acclimation is effective and may be enhanced rather than impaired by dehydration. Am J Hum Biol. 2014;26(3):311–320. [DOI] [PubMed] [Google Scholar]
- 416.Mudambo KS, Coutie W, Rennie MJ. Plasma arginine vasopressin, atrial natriuretic peptide and brain natriuretic peptide responses to long-term field training in the heat: effects of fluid ingestion and acclimatization. Eur J Appl Physiol Occup Physiol. 1997;75(3):219–225. [DOI] [PubMed] [Google Scholar]
- 417.Shido O, Sugimoto N, Tanabe M, et al. Core temperature and sweating onset in humans acclimated to heat given at a fixed daily time. Am J Physiol Regul Integr Comp Physiol. 1999;276(4):R1095–101. [DOI] [PubMed] [Google Scholar]
- 418.Garrett AT, Creasy R, Rehrer NJ, et al. Effectiveness of short-term heat acclimation for highly trained athletes. Eur J Appl Physiol. 2012;112(5):1827–1837. [DOI] [PubMed] [Google Scholar]
- 419.Francesconi RP, Sawka MN, Pandolf KB. Hypohydration and heat acclimation: plasma renin and aldosterone during exercise. J Appl Physiol Respir Environ Exerc Physiol. 1983;55(6):1790–1794. [DOI] [PubMed] [Google Scholar]
- 420.Sunderland C, Morris JG, Nevill ME. A heat acclimation protocol for team sports. Br J Sports Med. 2008;42(5):327–333. [DOI] [PubMed] [Google Scholar]
- 421.Houmard JA, Costill DL, Davis JA, et al. The influence of exercise intensity on heat acclimation in trained subjects. Med Sci Sports Exerc. 1990;22(5):615–620. [DOI] [PubMed] [Google Scholar]
- 422.Kirby CR, Convertino VA. Plasma aldosterone and sweat sodium concentrations after exercise and heat acclimation. J Appl Physiol. 1986;61(3):967–970. [DOI] [PubMed] [Google Scholar]
- 423.Divine JG, Clark JF, Colosimo AJ, et al. American Football Players in Preseason Training at Risk of Acute Kidney Injury Without Signs of Rhabdomyolysis. Clin J Sport Med. 2018. [DOI] [PubMed] [Google Scholar]
- 424.O’Sullivan ED, Hughes J, Ferenbach DA. Renal Aging: Causes and Consequences. J Am Soc Nephrol. 2017;28(2):407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Stevens PE, O’Donoghue DJ, de Lusignan S, et al. Chronic kidney disease management in the United Kingdom: NEOERICA project results. Kidney Int. 2007;72(1):92–99. [DOI] [PubMed] [Google Scholar]
- 426.Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298(17):2038–2047. [DOI] [PubMed] [Google Scholar]
- 427.Formica M, Politano P, Marazzi F, et al. Acute Kidney Injury and Chronic Kidney Disease in the Elderly and Polypharmacy. Blood Purif. 2018;46(4):332–336. [DOI] [PubMed] [Google Scholar]
- 428.Onder G, Bonassi S, Abbatecola AM, et al. High Prevalence of Poor Quality Drug Prescribing in Older Individuals: A Nationwide Report From the Italian Medicines Agency (AIFA). J Gerontol A Biol Sci Med Sci. 2013;69(4):430–437. [DOI] [PubMed] [Google Scholar]
- 429.Baraldi A, Ballestri M, Rapana R, et al. Acute renal failure of medical type in an elderly population. Nephrol Dial Transplant. 1998;13(Suppl 7):25–29. [DOI] [PubMed] [Google Scholar]
- 430.Schmitt R, Coca S, Kanbay M, et al. Recovery of Kidney Function After Acute Kidney Injury in the Elderly: A Systematic Review and Meta-analysis. Am J Kidney Dis. 2008;52(2):262–271. [DOI] [PubMed] [Google Scholar]
- 431.Bunker A, Wildenhain J, Vandenbergh A, et al. Effects of Air Temperature on Climate-Sensitive Mortality and Morbidity Outcomes in the Elderly; a Systematic Review and Meta-analysis of Epidemiological Evidence. EBioMedicine. 2016;6:258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Kenney WL. Thermoregulation at rest and during exercise in healthy older adults. Exerc Sport Sci Rev. 1997;25:41–76. [PubMed] [Google Scholar]
- 433.Davies DF, Shock NW. Age changes in glomerular filtration rate, effective renal plasma flow, and tubular excretory capacity in adult males. J Clin Invest. 1950;29(5):496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Glodny B, Unterholzner V, Taferner B, et al. Normal kidney size and its influencing factors - a 64-slice MDCT study of 1.040 asymptomatic patients. BMC Urol. 2009;9:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Wang X, Vrtiska TJ, Avula RT, et al. Age, kidney function, and risk factors associate differently with cortical and medullary volumes of the kidney. Kidney Int. 2014;85(3):677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Roseman DA, Hwang SJ, Oyama-Manabe N, et al. Clinical associations of total kidney volume: the Framingham Heart Study. Nephrol Dial Transplant. 2017;32(8):1344–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Hollenberg NK, Adams DF, Solomon HS, et al. Senescence and the renal vasculature in normal man. Circ Res. 1974;34(3):309–316. [DOI] [PubMed] [Google Scholar]
- 438.Denic A, Lieske JC, Chakkera HA, et al. The Substantial Loss of Nephrons in Healthy Human Kidneys with Aging. J Am Soc Nephrol. 2017;28(1):313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Rule AD, Semret MH, Amer H, et al., editors. Association of kidney function and metabolic risk factors with density of glomeruli on renal biopsy samples from living donors. Mayo Clin Proc; 2011;86(4):282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Rule AD, Amer H, Cornell LD, et al. The association between age and nephrosclerosis on renal biopsy among healthy adults. Ann Intern Med. 2010;152(9):561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Kenney WL, Ho CW. Age alters regional distribution of blood flow during moderate-intensity exercise. J Appl Physiol. 1995;79(4):1112–1119. [DOI] [PubMed] [Google Scholar]
- 442.Dunbar SL, Kenney WL. Effects of hormone replacement therapy on hemodynamic responses of postmenopausal women to passive heating. J Appl Physiol. 2000;89(1):97–103. [DOI] [PubMed] [Google Scholar]
- 443.Volterrani M, Rosano G, Coats A, et al. Estrogen acutely increases peripheral blood flow in postmenopausal women. Am J Med. 1995;2(99):119–122. [DOI] [PubMed] [Google Scholar]
- 444.Crook D, Meire H, Gangar K, et al. Pulsatility index in internal carotid artery in relation to transdermal oestradiol and time since menopause. Lancet. 1991;338(8771):839–842. [DOI] [PubMed] [Google Scholar]
- 445.Patel HM, Mast JL, Sinoway LI, et al. Effect of healthy aging on renal vascular responses to local cooling and apnea. J Appl Physiol. 2013;115(1):90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Castellani S, Ungar A, Cantini C, et al. Excessive vasoconstriction after stress by the aging kidney: inadequate prostaglandin modulation of increased endothelin activity. J Lab Clin Med. 1998;132(3):186–194. [DOI] [PubMed] [Google Scholar]
- 447.Morley JE. Dehydration, Hypernatremia, and Hyponatremia. Clin Geriatr Med. 2015;31(3):389–399. [DOI] [PubMed] [Google Scholar]
- 448.Beck LH, Lavizzo-Mourey R. Geriatric hypernatremia. Ann Intern Med. 1987;107(5):768–769. [DOI] [PubMed] [Google Scholar]
- 449.Davy KP, Seals DR. Total blood volume in healthy young and older men. J Appl Physiol. 1994;76(5):2059–2062. [DOI] [PubMed] [Google Scholar]
- 450.Sands JM. Urine concentrating and diluting ability during aging. J Gerontol A Biol Sci Med Sci. 2012;67(12):1352–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Cowen LE, Hodak SP, Verbalis JG. Age-associated abnormalities of water homeostasis. Endocrinol Metab Clin North Am. 2013;42(2):349–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Abramow M, Beauwens R, Cogan E. Cellular events in vasopressin action. Kidney Int Suppl. 1987;21:S56–66. [PubMed] [Google Scholar]
- 453.Phillips PA, Rolls BJ, Ledingham JG, et al. Reduced thirst after water deprivation in healthy elderly men. N Engl J Med. 1984;311(12):753–759. [DOI] [PubMed] [Google Scholar]
- 454.Catudioc-Vallero J, Sands JM, Klein JD, et al. Effect of age and testosterone on the vasopressin and aquaporin responses to dehydration in Fischer 344/Brown-Norway F1 rats. J Gerontol A Biol Sci Med Sci. 2000;55(1):B26–34. [DOI] [PubMed] [Google Scholar]
- 455.Combet S, Geffroy N, Berthonaud V, et al. Correction of age-related polyuria by dDAVP: molecular analysis of aquaporins and urea transporters. Am J Physiol Renal Physiol. 2003;284(1):F199–208. [DOI] [PubMed] [Google Scholar]
- 456.Epstein M, Hollenberg NK. Age as a determinant of renal sodium conservation in normal man. J Lab Clin Med. 1976;87(3):411–417. [PubMed] [Google Scholar]
- 457.Macias Nunez JF, Garcia Iglesias C, Bondia Roman A, et al. Renal handling of sodium in old people: a functional study. Age Ageing. 1978;7(3):178–181. [DOI] [PubMed] [Google Scholar]
- 458.Weidmann P, De Myttenaere-Bursztein S, Maxwell MH, et al. Effect on aging on plasma renin and aldosterone in normal man. Kidney Int. 1975;8(5):325–333. [DOI] [PubMed] [Google Scholar]
- 459.Krug AW, Allenhöfer L, Monticone R, et al. Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension. 2010;55(6):1476–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Begg DP. Disturbances of thirst and fluid balance associated with aging. Physiol Behav. 2017;178:28–34. [DOI] [PubMed] [Google Scholar]
- 461.Kenney WL, Chiu P. Influence of age on thirst and fluid intake. Med Sci Sports Exerc. 2001. September;33(9):1524–1532. [DOI] [PubMed] [Google Scholar]
- 462.Stachenfeld NS, Mack GW, Takamata A, et al. Thirst and fluid regulatory responses to hypertonicity in older adults. Am J Physiol Regul Integr Comp Physiol. 1996;271(3 Pt 2):R757–65. [DOI] [PubMed] [Google Scholar]
- 463.Mack GW, Weseman CA, Langhans GW, et al. Body fluid balance in dehydrated healthy older men: thirst and renal osmoregulation. J Appl Physiol. 1994;76(4):1615–1623. [DOI] [PubMed] [Google Scholar]
- 464.Kenney WL. Control of heat-induced cutaneous vasodilatation in relation to age. Eur J Appl Physiol Occup Physiol. 1988;57(1):120–125. [DOI] [PubMed] [Google Scholar]
- 465.Kenney WL, Morgan AL, Farquhar WB, et al. Decreased active vasodilator sensitivity in aged skin. Am J Physiol Heart Circ Physiol. 1997;272(4):H1609–14. [DOI] [PubMed] [Google Scholar]
- 466.Sagawa S, Shiraki K, Yousef MK, et al. Sweating and cardiovascular responses of aged men to heat exposure. J Gerontol. 1988;43(1):M1–8. [DOI] [PubMed] [Google Scholar]
- 467.Kenney WL, Tankersley CG, Newswanger DL, et al. Age and hypohydration independently influence the peripheral vascular response to heat stress. J Appl Physiol. 1990;68(5):1902–1908. [DOI] [PubMed] [Google Scholar]
- 468.Gagnon D, Schlader ZJ, Crandall CG. Sympathetic activity during passive heat stress in healthy aged humans. J Physiol. 2015;593(9):2225–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Seals DR, Dinenno FA. Collateral damage: cardiovascular consequences of chronic sympathetic activation with human aging. Am J Physiol Heart Circ Physiol. 2004;287(5):H1895–905. [DOI] [PubMed] [Google Scholar]
- 470.Stauss HM, Morgan DA, Anderson KE, et al. Modulation of baroreflex sensitivity and spectral power of blood pressure by heat stress and aging. Am J Physiol Heart Circ Physiol. 1997;272(2 Pt 2):H776–84. [DOI] [PubMed] [Google Scholar]
- 471.Kenney MJ. Medullary regulation of visceral sympathetic nerve discharge at peak hyperthermia in aged F344 rats. Auton Neurosci. 2014;186:32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Miescher E, Fortney SM. Responses to dehydration and rehydration during heat exposure in young and older men. Am J Physiol. 1989. November;257(5 Pt 2):R1050–6. [DOI] [PubMed] [Google Scholar]
- 473.Begg DP. Disturbances of thirst and fluid balance associated with aging. Physiol Behav. 2017;178:28–34. [DOI] [PubMed] [Google Scholar]
- 474.Quirós Cognuck S, Reis WL, Silva MS, et al. Sex-and age-dependent differences in the hormone and drinking responses to water deprivation. Am J Physiol Regul Integr Comp Physiol. 2020;318(3):R567–R578. [DOI] [PubMed] [Google Scholar]
- 475.Begg DP, Sinclair AJ, Weisinger RS. Reductions in water and sodium intake by aged male and female rats. Nutr res. 2012;32(11):865–872. [DOI] [PubMed] [Google Scholar]
- 476.Wolf ST, Stanhewicz AE, Clarke MM, et al. Age-related differences in water and sodium handling after commercial hydration beverage ingestion. J Appl Physiol. 2019;126(4):1042–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Takamata A, Ito T, Yaegashi K, et al. Effect of an exercise-heat acclimation program on body fluid regulatory responses to dehydration in older men. Am J Physiol Regul Integr Comp Physiol. 1999;277(4):R1041–50. [DOI] [PubMed] [Google Scholar]
- 478.Honda T, Manjourides J, Suh H. Daily ambient temperature is associated with biomarkers of kidney injury in older Americans. Environ Res. 2019;179:108790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Anderson S, Eldadah B, Halter JB, et al. Acute kidney injury in older adults. J Am Soc Nephrol. 2011;22(1):28–38. [DOI] [PubMed] [Google Scholar]
- 480.Koch CA, Fulop T. Clinical aspects of changes in water and sodium homeostasis in the elderly. Rev Endocr Metab Disord. 2017;18(1):49–66. [DOI] [PubMed] [Google Scholar]
- 481.Kenney WL, Munce TA. Invited review: aging and human temperature regulation. J Appl Physiol. 2003;95(6):2598–2603. [DOI] [PubMed] [Google Scholar]
- 482.Schlader ZJ, Coleman GL, Sackett JR, et al. Behavioral thermoregulation in older adults with cardiovascular co-morbidities. Temperature. 2018;5(1):70–85. doi: 10.1080/23328940.2017.1379585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Taylor NA, Allsopp NK, Parkes DG. Preferred room temperature of young vs aged males: the influence of thermal sensation, thermal comfort, and affect. J Gerontol A Biol Sci Med Sci. 1995;50(4):M216–21. [DOI] [PubMed] [Google Scholar]
- 484.Crowe JP, Moore RE. Proceedings: Physiological and behavioural responses of aged men to passive heating. J Physiol. 1974;236(1):43p–45p. [PubMed] [Google Scholar]
- 485.Stapleton JM, Larose J, Simpson C, et al. Do older adults experience greater thermal strain during heat waves?. Appl Physiol Nutr Metab. 2014. March;39(3):292–8. [DOI] [PubMed] [Google Scholar]
- 486.Kenny GP, Poirier MP, Metsios GS, et al. Hyperthermia and cardiovascular strain during an extreme heat exposure in young versus older adults. Temperature. 2017;4(1):79–88. doi: 10.1080/23328940.2016.1230171 [DOI] [PMC free article] [PubMed] [Google Scholar]