

Abstract
DNA replication is laden with obstacles that slow, stall, collapse, and break DNA replication forks. At each obstacle, there is a decision to be made whether to bypass the lesion, repair or restart the damaged fork, or to protect stalled forks from further demise. Each “decision” draws upon multitude of proteins participating in various mechanisms that allow repair and restart of replication forks. Specific functions for many of these proteins have been described and an understanding of how they come together in supporting replication forks is starting to emerge. Many questions, however, remain regarding selection of the mechanisms that enable faithful genome duplication and how “normal” intermediates in these mechanisms are sometimes funneled into “rogue” processes that destabilize the genome and lead to cancer, cell death, and emergence of chemotherapeutic resistance. In this review we will discuss molecular mechanisms of DNA damage bypass and replication fork protection and repair. We will specifically focus on the key players that define which mechanism is employed including: PCNA and its control by posttranslational modifications, translesion synthesis DNA polymerases, molecular motors that catalyze reversal of stalled replication forks, proteins that antagonize fork reversal and protect reversed forks from nucleolytic degradation, and the machinery of homologous recombination that helps to reestablish broken forks. We will also discuss risks to genome integrity inherent in each of these mechanisms.
Keywords: DNA replication, genome stability, replication fork protection, replication fork reversal, template switching, translesion synthesis, PCNA, translesion synthesis DNA polymerases, HLTF, SHPRH, SMARCAL1, ZRANB3, RAD51, RAD52, BRCA2, RPA
1. Introduction
The process of duplicating an organism’s genome is an essential and highly regulated series of events involving numerous proteins and multi-protein complexes. In eukaryotes, DNA replication is initiated at origins, and two replication forks proceed in opposite directions from each origin until running into forks originating from the adjacent origin (1). Both incomplete replication and over-replication (i.e. replicating a part of genome more than once) can have genome destabilizing consequences (2–7). DNA replication forks are constantly encountering obstacles to normal replication which cause replication fork slowing and/or stalling (8). Prolonged stalling of replication in cells leads to fork collapse, which involves disassembly of the replisome, fork reversal and/or breakage generating DNA double strand breaks (DSB), and can lead to gross chromosomal rearrangements, and genomic instability. To preserve genome stability, cells employ multiple mechanisms, which respond to replication stress by allowing the cell to stabilize or reverse forks, fix or bypass DNA damage, and restart replication. In addition, replication fork recovery can allow more time for DNA repair mechanisms such as base excision repair or nucleotide excision repair to excise the damage prior to fork restart.
DNA replication forks are blocked by many obstacles including DNA lesions, inter-strand crosslinks, protein-DNA crosslinks, single strand breaks (SSB), DSB, ongoing RNA transcription, depletion of nucleotides, and overexpression of oncogenes (3,4,7) (Figure 1). All of these DNA barriers cause the replication fork to stall upon encountering the blockage. It is at this point that the cell initiates fork recovery mechanisms. The nature of the roadblock can, at least in part, dictate the fate of the fork: will it be stabilized, collapsed, or broken, and what mechanism the cell will employ to repair the fork or restart replication. For example, as will be discussed later, runoff replication through an SSB or a topoisomerase I cleavage complexes can result in one-ended DSBs that are repaired by homologous recombination (9,10), while SSBs bound by PARP1 cause replication fork reversal (11). Various alternative mechanisms to overcome blocked replication can be utilized with the primary signal appearing to be the presence of ssDNA gaps at stalled forks due to helicase-polymerase uncoupling (12,13). The recovery mechanisms utilized by the cell come in several general categories: damage bypass including translesion synthesis (TLS) and template switching (TS), fork reversal involving fork remodeling proteins, and fork breakage resulting in generation of one-ended DSB breaks. The proteins involved in these different mechanisms have specific activities and interactions that lead cells to choose one mechanism over another. While several authoritative reviews have described some aspects of the lesion bypass and fork reversal machinery (2,7,8,14–23), the field is moving forward in a rapid pace with more information becoming available, especially regarding the mechanisms and proteins that protect replication forks. This review will focus on what proteins determine and control the mechanism or “choice” of replication fork recovery and will highlight outstanding questions regarding these mechanisms.
Figure 1. Replication forks encounter numerous impediments to their progression.
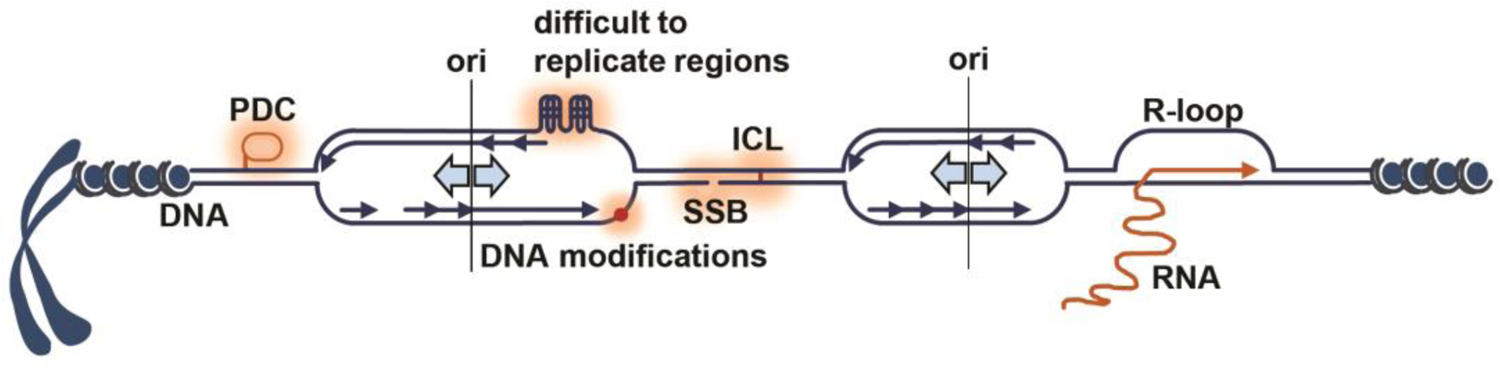
On linear eukaryotic chromosomes, DNA replication forks move bidirectionally from replication origins (ori) and can stall or brake due to encounter with protein DNA complexes (PDC), modified nucleobases, difficult to replicate regions, single-strand breaks (SSBs), interstrand DNA crosslinks (ICLs) and ongoing RNA transcription.
2. Damage bypass mechanisms
Damage bypass (Figure 2a&b) is one of several recovery mechanisms cells have to deal with stalled replication forks at sites of DNA lesions. Cells have developed two methods to bypass various forms of DNA damage; one method using “specialized” DNA polymerases and another that utilizes the newly synthesized sister DNA strand.
Figure 2. Cells employ multiple mechanisms to process, repair and restart stalled and damaged DNA replication forks.
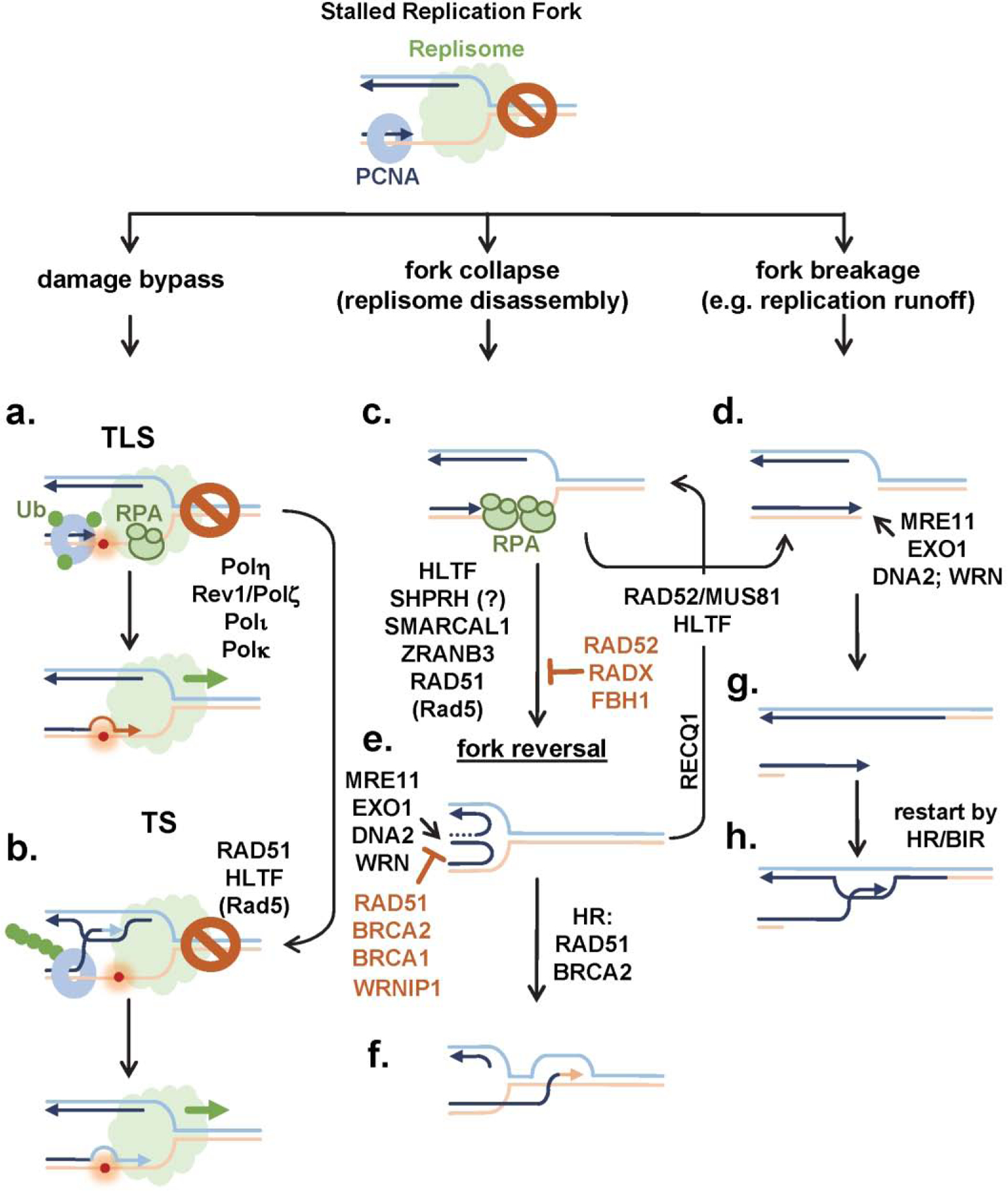
Nature of the replication stalling event in part defines the mechanism of fork protection and restart. a.) Translesion DNA synthesis (TLS) is triggered by ubiquitylation of PCNA and is carried out by specialized DNA polymerases. b). Poly-ubiquitylation of PCNA promotes template switching (TS). Stalled forks can also recruit fork reversal motors (c.) or enzymes that cleave replication forks (d.). e.) Fork reversal generates so-called “chicken foot” structure whose double strand end requires protection from uncontrolled resection by DSB resection machinery, but which can also serve to repair the fork by homologous recombination (HR; f.). g.) One ended DSB generated by fork cleavage or running into SSB is also processed by the DSB resection machinery allowing the fork to be repaired by HR (h.).
One of the major post-translational modifications that occurs at stalled replication forks is the mono-ubiquitylation of PCNA by the RAD6-RAD18 complex: specifically, the addition of ubiquitin to the K164 residue on PCNA. Ub-PCNA is seen in cells experiencing replication stress after hydroxyurea treatment, which depletes dNTPs (24,25). Destabilization of normal replication due to mutations in DNA polymerases ε and δ as well as other replication-associated proteins also elicits PCNA ubiquitylation (22,26). Furthermore, PCNA is mono-ubiquitylated when there is an overexpression of oncogenes that lead to dysfunctional DNA replication (22). Another major source of replication stress is the presence of DNA lesions in the template strand, which causes fork stalling and Ub-PCNA modification. DNA damage induced by UV irradiation, methyl methanesulfonate (MMS) treatment, H2O2 oxidative stress, 4-nitroquinoine 1-oxide generated bulky adducts, crosslinking agents such as mitomycin C, and numerous chemotherapeutic agents, all promote Ub-PCNA modification (22). However, agents that induce DSBs, such as ionizing radiation, bleomycin, or endonuclease cleavage, do not result in ubiquitylated PCNA (22). This post-translational modification of PCNA appears to be a major signal to initiate a recovery response to replication stress induced by the accumulation of ssDNA at stalled replication forks.
One of the recovery mechanisms that cells utilize after Ub-PCNA modification is damage bypass or post replication repair (PRR). PRR seems to be controlled by two non-mutually exclusive aspects: the proteins present at the stalled replication fork and the type of DNA lesion present in the template strand. Here, is where this recovery mechanism branches into either translesion synthesis (TLS) or template switching (TS).
2.1. Translesion synthesis (TLS)
TLS is the most versatile mechanism the cell has to handle the stalling of replication forks at sites of DNA damage. This mechanism, however, can be somewhat mutagenic (27,28). The mutagenic potential of TLS is reduced by the diversity and specificity of the specialized TLS DNA polymerases. Whether or not the cell utilizes TLS prior to other recovery mechanisms at stalled replication forks is not clear and still remains to be determined.
In TLS, the replicative DNA polymerase is replaced by a specialized TLS DNA polymerase that can bypass the DNA lesion. Each of the TLS polymerases incorporates nucleotides opposite damaged DNA templates thereby bypassing a group of specific DNA lesions (see (29) for a comprehensive review). For example, Pol η can incorporate two A’s opposite T-T dimers and 6–4 photoproducts induced by UV irradiation. In humans, loss of Pol η leads to a genetic condition called xeroderma pigmentosum variant form, which makes these individuals more susceptible to UV induced skin cancers and other forms of cancer (30,31). There are five DNA polymerases that carry out most TLS in mammals (32–36). Polymerase eta (pol η), polymerase iota (pol ι), polymerase kappa (pol κ), and REV1 are members of the Y-family of polymerases. Polymerase zeta (pol ζ) is a member of the B-family of polymerases. Each of these enzymes has one or more cognate lesions (32,36), the types of DNA damage that the given polymerase has evolved to bypass efficiently. Moreover, each of these enzymes has unique mechanistic and structural features that allow them to accommodate their cognate lesions.
Pol η is found in all eukaryotes. The absence of pol η leads to increased sensitivity to UV radiation and increased rates of UV-induced mutagenesis (37). These effects arise because the cognate lesions of pol η include thymine dimers (38,39). Steady state and pre-steady state kinetics studies have shown that the bypass of thymine dimers by pol η is generally accurate and efficient (38,39). X-ray crystal structures of the catalytic domain of pol η show that this enzyme has a more spacious active site than replicative polymerases and can readily accommodate a bulky thymine dimer (40). These structures also show that the incoming nucleotide forms Watson-Crick base pairs with the template residue during nucleotide incorporation (Figure 3a).
Figure 3. Structures of damaged DNA templates and incoming dNTPs in the active sites of translesion synthesis polymerases.
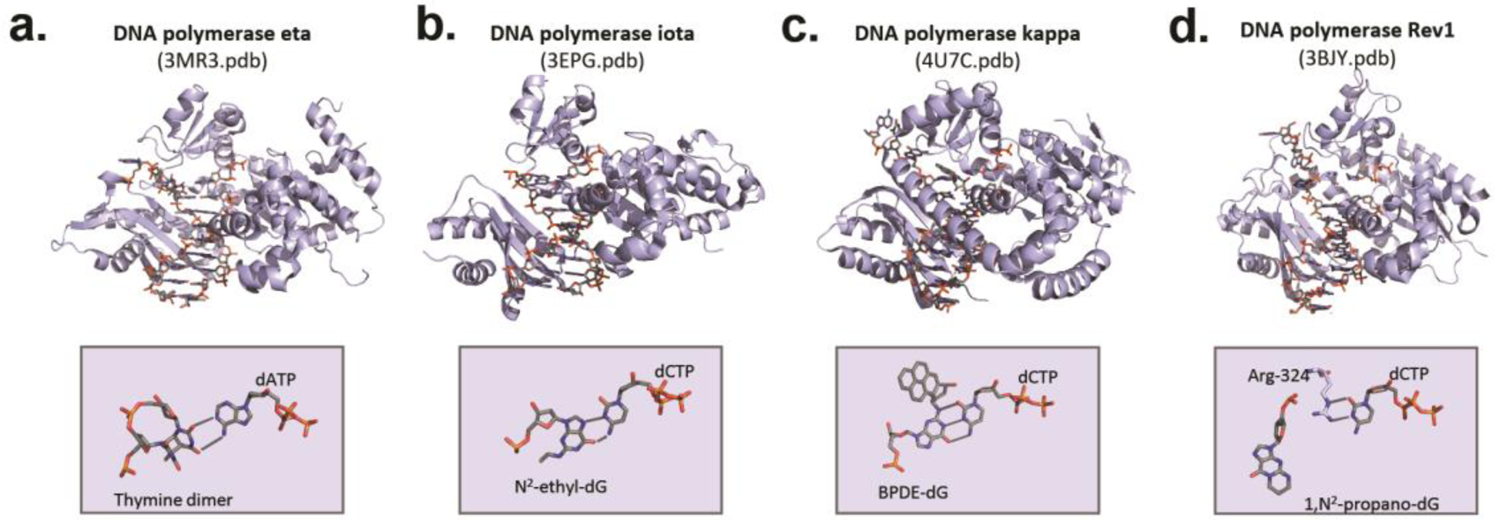
a.) DNA polymerase η incorporating dATP opposite the 5’ T of a thymine dimer (173). b.) DNA polymerase iota incorporating dCTP opposite an N2-ethyl-guanine (44). c.) DNA kappa iota incorporating dCTP opposite a benzo(a)pyrene diol epoxide (BPDE)-adducted guanine (174). d.) Rev1 incorporating dCTP opposite a 1,N2-propano-guanine (65).
Pol ι is found in insects and higher eukaryotes including mammals. The absence of pol ι in mice leads to increased frequencies of urethane-induced lung cancers (41,42), and the loss of pol ι in human cells leads to increased sensitivity to oxidative stress (43). These effects are likely because the cognate lesions of pol ι include minor-groove purine adducts and exocyclic guanine adducts that are not capable of forming Watson-Crick base pairs (44,45). Kinetics studies with pol ι have shown that the mechanism of nucleotide incorporation is different on template purines than it is on template pyrimidines (46–48). Incorporation is generally accurate and efficient on template purines, but is highly inaccurate and inefficient on template pyrimidines. Structures of the catalytic domain of pol ι show that when pol ι binds to DNA containing a purine template, the template forms a Hoogsteen base pair with the incoming nucleotide (49,50). When pol ι binds to DNA containing a pyrimidine template, steric clashes in the active site reduce the efficiency of nucleotide incorporation (Figure 3b).
Pol κ is found in higher eukaryotes including mammals. The loss of pol κ in mice leads to increased spontaneous mutation rates and increased sensitivity to benzo(a)pyrene (BP) (51). Pol κ appears to play two distinct roles in TLS depending on the type of DNA lesion. First, it efficiently incorporates nucleotides opposite minor-groove and exocyclic purine adducts (52). Second, it efficiently extends from nucleotides inserted opposite a wide range of DNA lesions by other TLS polymerases (53,54). Kinetics studies show that pol κ incorporates nucleotides with moderate fidelity and that its cognate lesions do not present any barriers to nucleotide incorporation (55,56). Structures of pol κ show that, unlike pol ι, this enzyme forms Watson-Crick base pairs between the incoming nucleotide and the template residue (57,58) (Figure 3c).
REV1 is found in all eukaryotes. In mice, depletion of REV1 leads to a reduction in the frequency of BP-induced lung cancers (59). This suggests that REV1 plays an important role in the error-prone bypass of BP-induced DNA lesions. Furthermore, depletion of REV1 leads to an increased sensitivity of tumors to cisplatin and cyclophosphamide (60). This suggests that REV1 plays an important role in the bypass of DNA lesions induced by these common anti-cancer drugs. The cognate lesions of REV1 include abasic sites and many minor-groove and exocyclic guanine adducts (61–63). Kinetics studies showed that REV1 greatly prefers to incorporate C opposite all four non-damaged templates as well as a wide range of damaged templates (61–63). Structures of REV1 showed the basis for the extraordinarily unusual substrate specificity of this enzyme. The template base is flipped out of the DNA helix into a pocket on the protein. A conserved arginine side chain forms hydrogen bonds with the incoming dCTP (64–66). As a result, the REV1 protein itself is the template for nucleotide incorporation (Figure 3d).
Pol ζ is found in all eukaryotes. In yeast, this enzyme is composed of the Rev3 catalytic subunit as well as two Rev7, one Pol31, and one Pol32 accessory subunits (67)(68). In addition, pol ζ is frequently associated with Rev1 in yeast. Loss of pol ζ leads to a defect in DNA-damage-induced mutagenesis (27). Kinetics studies have shown that this enzyme is a promiscuous extender from primer-terminal mispairs and aberrant primer-termini containing template lesions (69). The structure of Pol ζ revealed the basis for this promiscuous extension ability. Unlike other polymerases, Pol ζ does not contact the primer-terminal base pair, thereby allowing extension (68,70).This has led to the notion that the role of pol ζ in TLS is to extend from nucleotides inserted opposite a wide range of DNA lesions by other TLS polymerases.
One of the major, unanswered questions regarding TLS is how the most appropriate TLS polymerase is selected to carry out bypass of different types of DNA lesions. A simple kinetic selection model for choosing the best TLS polymerase has recently been developed (15). According to this model, multiple TLS polymerases compete with one another for bypassing the DNA lesion. Polymerase selection is achieved because of the relative rates by which each TLS polymerase incorporates nucleotides opposite different types of DNA damage and the relative rates by which each TLS polymerase dissociates from the DNA substrate prior to incorporating nucleotides. When a given polymerase binds the DNA substrate, it will preferentially incorporate nucleotides only if the DNA damage is a cognate lesion for that polymerase. If it is not a cognate lesion, the polymerase will preferentially dissociate from the DNA without incorporating nucleotides allowing other TLS polymerases to attempt to bypass the lesion. Continuing in such a manner, the most appropriate TLS polymerase will be selected to carryout lesion bypass the vast majority of the time.
2.2. Template switching (TS)
The term template switching (TS) describes a set of molecular events where synthesis of a nascent DNA strand first uses one DNA strand as a template and then jumps to copying another strand. Within the context of PRR, TS can be quite accurate (Figure 2b). Under other circumstances, and especially during break induced DNA replication (BIR), TS displays high potential to cause extensive mutagenesis and genome rearrangements (16). Figure 4 depicts two examples of how TS can result in increased copy number of chromosome segments (Figure 4a) (71) and deletions when an active fork converges on a collapsed fork resulting in an inter-fork annealing (Figure 4b) (72). TS mechanisms in each of these cases, (PRR, BIR, and inter-fork annealing) differ in the proteins involved and thereby in their mechanism.
Figure 4. Possible mutagenic consequences of template switching.
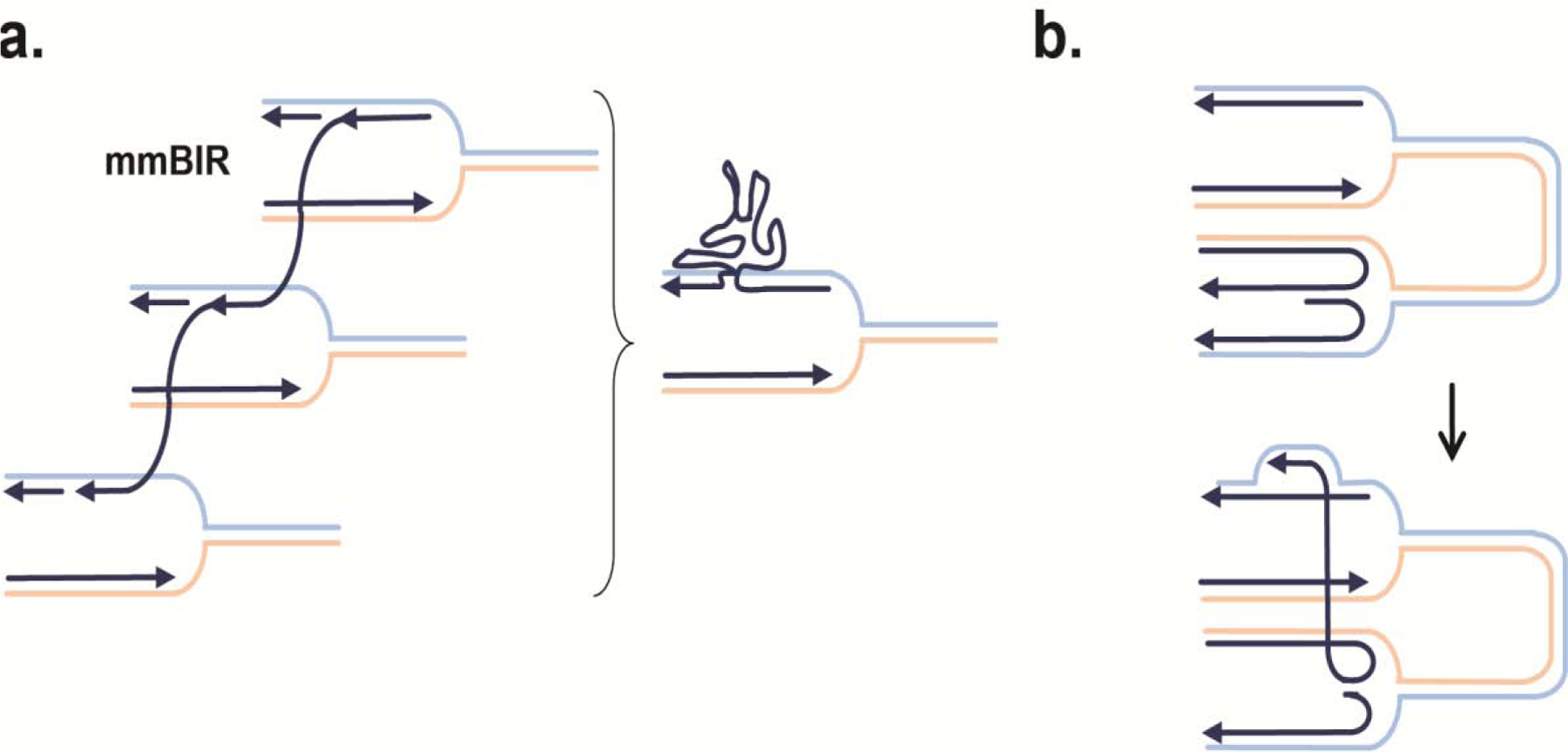
a.) BIR is prone to template switching, which can yield large insertions and rearrangements when multiple forks are involved. b.) TS between merging forks can result in deletions.
In PRR, TS is generally signaled by the polyubiquitination of PCNA (poly-Ub-PCNA) on K164. In budding yeast, poly-Ub-PCNA is carried out by an E2-E3 ubiquitin conjugating-ligase complex composed of Rad5 (E3 ligase) and Ubc13-Mms2 (E2 conjugating enzyme). Rad5 generates K63 poly-ubiquitin linkages onto the mono-ubiquitinated form of PCNA present at the stalled replication forks. This post-translational modification of PCNA signals the cell to switch from the TLS to TS. Rad5 has also been shown to bind to Rad18, the E2 conjugating enzyme involved in mono-ubiquitination of PCNA, and TLS polymerases (73). It has been suggested that it is Rad5 that recruits TLS polymerases to stalled replication forks (74). In addition to Rad5 having ubiquitin ligase activity, it is also an ATP-dependent DNA motor belonging to the SWI/SNF helicase family (75) that can catalyze fork reversal and strand invasion (76–79). Fork reversal entails the conversion of a stalled 3-way replication fork junction into a 4-way junction by annealing the newly synthesized DNA strands forming a so-called chicken-foot structure (Figure 2c&e).
The Rad5 protein contains several conserved structural domains: a RING domain, SWI/SNF helicase domain, and a N-terminal HIRAN domain. The HIRAN domain contains an OB- fold (oligonucleotide/oligosaccharide binding fold) characteristic of many eukaryotic ssDNA binding proteins, but with the ability to accommodate 3’-ssDNA ends. It is the HIRAN domain that provides substrate specificity of Rad5 and its human homolog, HLTF, by interacting with the free 3’-OH of ssDNA overhangs from duplex DNA at stalled replication forks (22,80–85). Rad5 can catalyze fork reversal with the help of both the HIRAN and helicases domains (76,86). Furthermore, the HIRAN domain is required for interaction of Rad5 with PCNA (87).
In humans, there are two Rad5 homologs, HLTF and SHPRH, which are both able to convert Ub-PCNA to poly-Ub-PCNA (88–91). Both HLTF and SHPRH form complexes with RAD18, under non-replication stress conditions. Both of these proteins are also SWI/SNF helicase-like DNA motors that have dsDNA dependent ATPase activity at stalled replication forks (85).
HLTF is more closely related to Rad5 containing all of the same structural domains; whereas, SHPRH has two domains not present in Rad5 and HLTF. These are a H15 linker histone domain and a PHD-finger domain, which are both associated with binding to chromatin. Like Rad5, HLTF can generate a chicken-foot structure allowing for bypass of DNA lesions. HLTF can reverse stalled replication forks by capturing the free 3’-OH of the nascent strand (Figure 5a) (80). This enables HLTF to carry out fork reversal by binding the parental duplex ahead of the fork and the leading strand behind the fork (84). Several in vitro and cell-based studies have examined the ability of HLTF to reverse stalled replication forks. In vitro experiments using a circular plasmid containing a fork structure established that fork reversal by HLTF could proceed for hundreds of bases (85). Alterations to the HIRAN domain, by either complete deletion or point mutations inhibited HLTF’s ability to reverse forked substrates; however, the ATPase and ubiquitin ligase activities were not affected (85). Furthermore, replacing the 3’-OH of the nascent leading strand with a PO4 also blocked fork reversal by HLTF (84).
Figure 5. Replication fork protection.
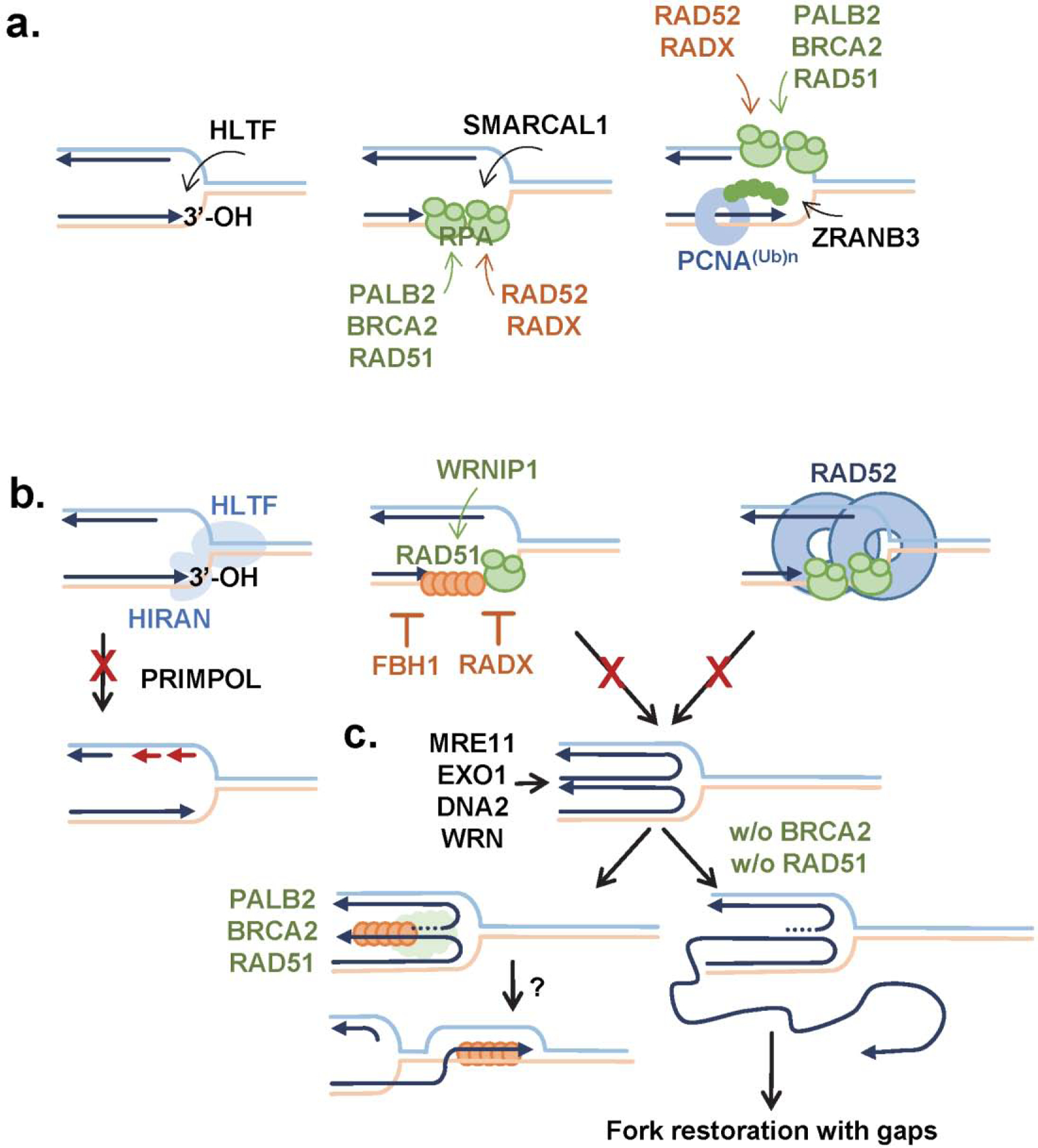
a.) Fork reversal motors and fork protection proteins are recruited to demised forks by different structural features and proteins already present at the fork. b.) HIRAN domain of HLTF interacts with 3’-OH group of the nascent strand; in the absence of HLTF, forks progression depends on repriming by PRIMPOL. FBH1 and RADX prevent RAD51 binding at replication forks and subsequent fork reversal, while RAD52 gatekeeper function restricts access by the fork reversal motors. c.) Recombination machinery, and especially BRCA2 and RAD51 protect reversed forks from excessive degradation, which can have genome destabilizing consequences.
The cellular consequences of deletion of HLTF and mutation of its HIRAN domain, however, are quite different. A recent cell-based study demonstrated that cells lacking HLTF were unable to undergo fork reversal and failed to slow DNA replication under conditions of cellular stress (92). Not surprisingly, these HLTF-deficient cells exhibit a reduction in DSBs and increased survival upon replication stress (92). Replication fork and S-phase progression in the absence of HLTF is achieved primarily by activity of PRIMPOL primase-polymerase (92), which mitigates excessive fork degradation (Figure 5b) (93). However, REV1 enables continued replication in cells expressing HLTF HIRAN mutants (92). It is clear from multiple studies that the HIRAN domain in both Rad5 and HLTF plays a major role in reversing and recognizing stalled replication forks.
The helicase domains of HLTF and Rad5 play a central role in fork reversal by facilitating dsDNA translocase activity, which drives the simultaneous unwinding and annealing of the nascent and parental strands. In addition, the helicase domain of HLTF can promote strand invasion and catalyze the formation of D-loop structures that are RAD51 independent in vitro (94). This is not the case for the yeast Rad5 protein where D-loop formation requires Rad51. D-loop formation by HLTF or Rad5/Rad51 may provide a means for the cell to move past the DNA damage (Figures 2b and 2f). D-loop formation can allow to bypass DNA damage at stalled fork (Figure 2b). It can also happen after the arm formed by annealing of the nascent strands in the chicken-foot is resected generating an ssDNA overhang. This overhang can invade duplexed DNA ahead of the fork, or a homologous/homeologous sequence nearby, creating a D-loop in essence similar to a BIR event (Figure 2f). Several questions remain including: what DNA fork structures does HLTF remodel or generate? Does HLTF create a D-loop by using the newly synthesized strand as a template or by using the reversed fork? What governs the choice between the chicken-foot versus a D-loop resolution? The mechanism of how HLTF regresses replication forks is starting to present a clearer, yet somewhat complex, picture of what is happening at stalled forks that enable the TS branch of PRR.
Less is known about the function of the other human Rad5 homolog SHPRH, which is also present at stalled replication forks. Loss of SHPRH has been observed in several cancers including melanoma, ovarian, cervical, breast, and pancreatic. In addition, several tumor cell lines have a truncated version or mutations of SHPRH effecting normal protein function (95). SHPRH lacks a HIRAN domain but does have ubiquitin ligase activity and can convert Ub-PCNA to poly-UB-PCNA in association with Ubc13-Mms2 (91). As stated earlier, SHPRH can bind chromatin via its PHD domain, which appears to interact with histones that are trimethylated on lysine residues. SHPRH has been shown to bind both dsDNA and nucleosomes with a preference for binding to nucleosomes with extra-nucleosomal DNA (96). Furthermore, SHPRH can interact with various E2 enzymes to ubiquitinate histones and itself, thus suggesting that SHPRH may remodel nucleosomes adjacent to stalled replication forks and self-modulate its own activity (96). Although SHPRH is a SWI/SNF helicase, it has not yet been biochemically shown to reverse stalled replication forks. Whether SHPRH has chromatin remodeling activities and how this activity is connected to fork recovery at stalled replication forks remains to be determined.
The loss of either HLTF or SHPRH has been shown to increase chromosomal abnormalities and genetic instability (89,90). Loss of HLTF from cells via siRNA knockdowns results in enhanced sensitivity to UV-induced mutagenesis; whereas, loss of SHPRH results in increased sensitivity to MMS-induced mutagenesis, with the reverse not being the case for either protein (97). This study by Lin and colleagues showed a dynamic set of protein interactions occurring at the replication fork in treated cells depending on the type DNA damage. As previously stated both HLTF and SHPRH can interact with RAD18 and it appears that which RAD18 complex is formed may determine which TLS polymerase is recruited (97). The interaction between SHPRH and RAD18 was enhanced after MMS treatment but reduced after UV treatment due to a shift to HLTF-RAD18 complex formation. The preference for the SHPRH-RAD18 complex seems to promote the interaction of SHPRH with Pol κ and Pol η to a lesser extent. These researchers went on further to show that HLTF is degraded in an ubiquitin dependent manner upon MMS treatment and that SHPRH-RAD18 complex formation coincided with HLTF degradation. Furthermore, after UV treatment there appeared to be a moderate increase in the association of HLTF and SHPRH and this association suppresses the SHPRH-Rad18 interaction. Lastly, HLTF promotes Ub-PCNA modification and the recruitment of Pol η to stalled replication forks after UV-irradiation. This last observation implies the involvement of HLTF in both TLS and TS branches of the damage bypass recovery mechanism as well as the possibility that Pol η is used for synthesis from a D-loop after TS (98). This study along with others suggests a dynamic interplay of proteins at the stalled replication fork that is affected by the type of DNA lesions, proteins present, and post-translational modifications of the various proteins. Furthermore, there seems to be a branching of the TS mechanism with UV-induced lesions eliciting a recovery response mediated by HLTF and MMS-induced lesions eliciting a recovery response mediated by SHPRH. The TLS polymerases that are recruited to specific types of DNA damage appear to also have preferences for interaction partners and their post-translational modifications. For example, SHPRH prefers Pol κ and its association with Rad18 allows it to poly-ubiquitylate PCNA; furthermore, Pol κ has multiple ubiquitin binding sites that could interact with poly-Ub-PCNA providing additional substrate specificity for this polymerase to bypass the lesion.
3. Fork reversal
One useful outcome of replication fork reversal is that it enables the cell to bypass DNA damage by incorporating the correct nucleotides using the newly synthesized sister strand as the template instead of the lesion containing strand (99). The ability of cells to reverse stalled replication forks requires the disassembly of the replisome; however, it also provides a protection mechanism against fork breakage by, for example, preventing a run off replication through single strand breaks (SSBs). Poly [ADP-ribose] polymerase 1 (PARP1) bound to SSBs triggers replication fork reversal, which in turn prevents DSB formation (11), a feature that reflects the original basis for targeting PARP1 in BRCA-deficient cancers (100). Fork reversal also protects from fork cleavage by MUS81 structure selective nuclease, which has higher enzymatic activity on 3-way replication fork structures than on 4-way junctions (101). Although fork reversal can allow the cell time to repair or bypass DNA damage, it can also be deleterious in its own right if not protected. Considering that the central arm of the reversed fork is produced by annealing of the newly synthesized DNA strands that can terminate in a double-stranded end resembling a DSB, this structure can than attract nucleases and helicases involved in processing DSBs, such as MRE11, EXO1, WRN and DNA2 (8,102–104) (Figure 2e). If not controlled, this nucleolytic processing of the nascent DNA in the reversed fork results in generation of massive amounts of ssDNA, which can result in restoration of forks with gaps and genetic instability (105,106).
Although Rad5, HLTF, and SHPRH can reverse stalled replication forks that encounter DNA damage, a group of proteins that handle fork reversal is larger and also includes fork remodeling enzymes SMARCAL1 and ZRANB3, and proteins that protect the reversed fork from nucleolytic degradation (Figure 2c and Figure 5a). SMARCAL1 and ZRANB3 are also members of the SWI/SNF helicase family with ATPase-dependent fork remodeling. Both of these proteins contain helicase domains that can anneal RPA coated ssDNA bubbles and reverse chicken-foot DNA structures (107–111). Like other SWI/SNF family members, SMARCAL1 and ZRANB3 have substrate recognition domains (SRD). SMARCAL1 contains two HARP SRDs and a RPA binding domain; whereas, ZRANB3 has a HARP-like SRD, HNH nuclease domain, and two PCNA interacting motifs, PIP and APIM. SMARCAL1 and ZRANB3 are non-redundant proteins having different substrate specificities, which is in part governed by the fork junction and the additional proteins associated at the stalled fork.
In addition to reversing stalled replication forks, SMARCAL1 can migrate chicken-foot structures, similar to HLTF, and can bind to dsDNA at the stalled fork (23,107). SMARCAL1 prefers to bind ssDNA gaps present at stalled forks. SMARCAL1 can also restore 3-way replication forks from chicken-foot structures that have been remodeled by ZRANB3, HLTF, and RAD51 (23,108,112). Its interaction with RPA, specifically the RPA32 subunit, at the stalled replication forks is what recruits SMARCAL1 to sites of replication stress (23). RPA’s presence at the stalled replication forks is required for and directs SMARCAL1’s fork remodeling activities, either fork reversal or fork restoration. First, let’s consider what happens when RPA is bound to a ssDNA gap on the leading strand. In this situation, SMARCAL1 is stimulated to reverse the stalled replication fork generating a chicken-foot structure. At the same time, the opposite reaction or fork restoration activity of SMARCAL1 is inhibited. When RPA is bound to the ssDNA gap on lagging DNA strand, SMARCAL1 is blocked from reversing the stalled replication fork and fork restoration or generation of the normal 3-way DNA junction is favored (23). Another interesting observation from comparison for substrate specificity of SMARCAL1 and HLTF suggested that while SMARCAL1 tends prefer forks with the leading strand gaps, HLTF processes forks with lagging strand gaps (84). Lastly, SMARCAL1 can dissolve D-loop structures, but not prevent their formation by RAD51 (109). Interestingly as stated earlier, HLTF can catalyze D-loop formation, which could be dissolved by SMARCAL1 or ZRANB3. These observations seem to suggest a potential way for the cell to shuttle certain stalled replication forks between the different recovery mechanisms in order to efficiently resolve the blockage and restore replication.
ZRANB3 is another helicase protein involved in the fork reversal mechanism. Cells lacking ZRANB3 showed increased levels of spontaneous sister chromatid exchange indicating an involvement of this protein at replication forks and maintenance of genomic stability (109). Like SMARCAL1, ZRANB3 can remodel stalled replication forks by both fork reversal and fork restoration although it displays different substrate specificities (Figure 5a) (108,109). Loss of the helicase activity of ZRANB3 completely impairs the ability of ZRANB3 to slow and reverse replication forks in cells treated with DNA damaging agents (113). Thus, providing further evidence that ZRANB3 is an essential player at stalled replication forks that aids in the prevention of genomic instability in response to DNA damage.
ZRANB3 contains PCNA binding motifs, PIP and APIM domains, that allow it to interact with PCNA and a NZF domain that allows it to interact with poly-Ub-PCNA (109,114,115). It’s through these interactions with PCNA that ZRANB3 is recruited, recognizes, and promotes reversal of damaged replication forks (116,117) (Figure 5a). Mutations within the PIP or APIM domains of ZRANB3 that inhibit its interaction with PCNA block replication fork remodeling by slowing fork progression in response to DNA damage (113). Binding of ZRANB3 to poly-Ub-PCNA is not required for recruitment to stalled replication forks but instead promotes stabilization of the fork (109). This finding suggests that ZRANB3 may mediate fork restoration or restart in addition to slowing fork progression and fork reversal.
RPA also appears to regulate ZRANB3’s ability to remodel stalled replication forks although it lacks a RPA binding domain. ZRANB3 is able to reverse forks containing gaps on the lagging strand regardless if RPA is coating the ssDNA or not. However, lagging strand gaps coated with RPA inhibits ZRANB3’s ability to facilitate restoration of stalled forks or the regeneration of a normal 3-way DNA junction. The situation is different with regards to the leading strand. RPA coated leading strand gaps inhibit ZRANB3 fork reversal activity; whereas, the lack of RPA coating this strand does not affect fork reversal by ZRANB3 (108). Exactly how RPA regulates ZRANB3 fork-remodeling activities remains unknown and requires additional research. In addition to PCNA interaction motifs, ZRANB3 also has a HNH domain that provides the protein with an ATP-dependent endonuclease activity on branched DNA substrates (115). Specifically, this endonuclease activity is able to cut the DNA 5’ of a lesion on the leading strand leaving a free 3’-OH that can act as a primer end for strand invasion and displacement of the DNA lesion (115). This free 3’-OH on the leading strand could also be a substrate for fork reversal activity by HLTF. So, through a coordinated mechanism that involves ZRANB3 endonuclease activity together with HLTF, both enzymes could promote fork reversal, damage bypass, and replication fork restoration. Interestingly, ZRANB3, like SMARCAL1, can resolve pre-formed D-loop structures; however, ZRANB3 has an addition function over SMARCAL1 in that it can prevent D-loop formation by RAD51 (109). As previously stated, HLTF can catalyze the formation of D-loops without RAD51 coating the ssDNA. All of these observations are pointing to other possible mechanisms of how these proteins maybe coordinated to enable fork reversal and restoration. Additional research is necessary to parse out the exact mechanisms and under what conditions one mechanism is favored over the other.
3.1. Replication fork restoration
After replication forks are reversed into a 4-way structure and DNA damage is bypassed or repaired, cells need a way to restart DNA replication and restore the replication fork. In addition to using HR (Figure 2f), fork restoration can also be accomplished by migrating the reversed fork back into the 3-way junction (Figure 2e→c). Several fork reversal motors discussed above have been shown to catalyze migration of the 4-way junction in both directions. Additionally, RECQ1 DNA helicase specifically acts on the reversed fork and promotes fork restoration. RECQ1’s ability to restore an active replication fork is dependent on its ATPase activity and can be inhibited by PARP1 until PARP1 self-parylation releases RECQ1 from the RECQ1-PARP1 complex (118). Thus, PARP1 not only promotes fork reversal, but also acts to protect reversed replication forks allowing for chicken-foot structures to accumulate and not be restored prior to repair or bypass of the DNA damage.
3.2. Replication fork protection
As we mentioned earlier, reversed replication forks can have positive and negative effects on genome stability. The main process that, when misregulated, can have genome destabilizing consequences is MRE11-dependent resection of the double strand ends at the reversed fork by end resection machinery, which includes EXO1, WRN and DNA2 enzymes (8,102–104). If not resected, this DNA end can also serve as a potent inducer of chromosome rearrangements similar to DSBs produced by replication fork breakage (119,120). One can imagine two general mechanisms of restricting DNA degradation at stalled replication forks: protecting the double strand end generated by fork reversal and preventing fork reversal altogether (Figure 5 b&c).
Tumor suppressor and recombination mediator BRCA2 plays an important role in protecting reversed replication forks from MRE11-dependent degradation (121). BRCA2’s function at replication forks, however, is somewhat different from its function in HR and involves different set of interactions with RAD51 recombinase (122). RAD51 is a central player in HR. Both in HR and at replication forks, its active form is a nucleoprotein filament assembled on ssDNA. Assembly of the RAD51 filament is aided by the recombination mediator BRCA2 and RAD51 paralogs (123,124), antagonized by antirecombinases (125) and the heteroduplex rejection machinery (126), and regulated by post translational modifications including phosphorylation by c-ABL (127–129). RAD51 function at replication forks is complex. RAD51 is recruited to uncoupled replication forks with extended regions of ssDNA and promotes fork reversal in response to genotoxic stress (12). Upon fork reversal, it is involved in protection of nascent ssDNA from degradation (121). When too much fork reversal depletes factors that protect nascent DNA from MRE11-dependent degradation, fork degradation can be overcome by RAD51 overexpression (106). Furthermore, it is the DNA binding ability of RAD51, and not its ability to mediate DNA strand exchange, that is required for both fork protection and stimulating fork reversal (130). The ability of RAD51 to be loaded onto ssDNA at reversed replication forks is facilitated by BRCA2, which helps RAD51 to compete with RPA for ssDNA binding. There are two regions of BRCA2 that directly interact with RAD51: 8 BRC motifs and a region of the C-terminus (122,131). Interaction of BRCA2 with RAD51 via the BRC motifs is necessary for RAD51 function in HR. In contrast, fork-related function of BRCA2 involves its C-terminus, which binds the interface of adjacent RAD51 proteins assembled in the nucleoprotein filament, thereby stabilizing the filament, and promoting protection of reversed replication forks (122). Fork protection can be modulated by the phosphorylation of the C-terminus of BRCA2 thus disrupting its ability to stabilize RAD51 nucleoprotein filament (121,132,133). In addition, a point mutation in RAD51, T131P, destabilizes the RAD51 filament and cells expressing this RAD51 mutant are unable to protect reversed replication forks from degradation (134). Another protein, BOD1L also appears to stabilize the RAD51 filament, possibly through a direct interaction, and promotes fork protection by suppressing of FBH1 and BLM helicase activities (135). Finally, Werner helicase interacting protein 1, WRNP1, prevents uncontrolled MRE11-dependent degradation of nascent DNA presumably by stabilizing the RAD51 nucleoprotein filament (136).
Among the negative regulators of RAD51 nucleoprotein filament at replication forks is FBH1, an F-box helicase 1, a bifunctional enzyme with DNA helicase and E3 ubiquitin ligase activities (137). Human FBH1 displays both pro and anti-recombinogenic activities (138–141) controlled through self-ubiquitylation as a part of SCF (SKP1, CUL1 and F-box) ubiquitin ligase complex (142). It also ubiquitylates RAD51 in vitro and this posttranslational modification regulates RAD51’s subcellular localization with accumulation of non-Ub forms in the nucleus after hydroxyurea treatment (143). FBH1 is one of many proteins that is recruited to sites of DNA damage by PCNA and is involved in the fork breakage mechanism in cooperation with MUS81 (144,145). In human cells, MUS81 exists in two subcomplexes, MUS81/EME1 and MUS81/EME2; the latter complex is responsible for the fork cleavage in S-phase (146). DSBs and apoptosis promoted by FBH1 in cooperation with MUS81 nuclease in response to replication stress protect melanocytes from oncogenic transformation (147,148). Downregulation or deletion of FBH1 can compensate for loss of factors that stabilize the RAD51 nucleoprotein filament, such as WRNIP1 in human cells (136) and Rad52 in fission yeast (140,141). Unloading of PCNA also limits FBH1 antirecombinogenic activity and thereby allows loading of recombination proteins at damaged forks (149).
Another recently identified protein; RADX can also prevent MUS81 cleavage of stalled replication forks, antagonize the formation of RAD51 filaments, and inhibit fork reversal (Figure 2c & 5b) (150). RADX is an ssDNA binding protein whose DNA binding ability is required for protecting/stabilizing the fork. RADX can compete with RAD51 for ssDNA (151,152) by condensing RPA-coated ssDNA, thus interfering with RAD51 loading (150,152). Loss or inactivation of RADX in cells facilitates the accumulation of RAD51 at the forks, which is important for fork reversal and preventing excessive MRE11-dependent degradation of reversed forks (150,151).
Similar to RADX, DNA repair protein RAD52 is involved in fork protection upstream of the reversal. RAD52 is recruited to parental ssDNA at stalled forks in cells (106). In vitro, it interacts with both ssDNA and dsDNA by wrapping the DNA around the oligomeric protein ring (153,154). Through these interactions, RAD52 acts as a gatekeeper of stalled replication forks by configuring them into structures refractory to reversal by RAD51, SMARCAL1 and ZRANB3 (105,106). What appears to be at play here is a complex dance between RAD52 and fork-remodeling proteins, such as SMARCAL1, where RAD52 acts as a timer to facilitate fork protection from reversal, but eventually leading to fork breakage and repair. Notably, the gatekeeper function of RAD52 is important not only under pathological condition of replication stress, but also during normal conditions, as excessive fork reversal followed by MRE11-dependent degradation has been observed upon depletion or pharmacological inhibition of RAD52 even in the absence of replication stress (106).
While both acting upstream of replication fork reversal, the function of RAD52 and RADX is not fully equivalent. Depletion or inhibition of RAD52 results in excessive loading of RAD51, SMARCAL1 and ZRANB3 at stalled replication forks, ensuing fork reversal and MRE11-dependent degradation leading to fork restoration with gaps and genome instability (106). RAD51 loading at forks in the absence of RADX seems to interfere with reversal and eventually leads to MUS81 -dependent DSBs (Figure 2d) (150), thus shuttling the cell towards HR or fork breakage recovery mechanism.
4. Fork breakage
The final fork recovery mechanism involves fork breakage whereby one of the fork arms is detached leaving a one-ended DSB (155–157) (Figure 2d). Such forks can be then recovered with the help of HR or HR-related BIR/MiDAS, both in S-phase and during mitosis (16,158,159) (Figure 2g–h).
In addition to replication fork breakage due to encountering SSBs, detachment of the fork arm can be catalyzed enzymatically by structure selective nucleases, such as MUS81 (146). Multiple proteins control the activity of MUS81 nuclease at stalled forks (146,160–164). As discussed earlier, FBH1 promotes MUS81-dependent DSBs and apoptosis. In addition to its gatekeeper function at stalled forks (106) and its role in recruiting MRE11 to unprotected reversed forks (165), RAD52 in human and fission yeast stimulates DSB formation and these DSBs are important for cell survival in the absence of replication checkpoints (166,167). Depletion of BRCA2 and replication stress elicited by hydroxyurea treatment also results in RAD52/MUS81 dependent DSBs (168). Notably, studies in different organisms have identified the role for RAD52 not only in replication fork breakage, but also in its subsequent repair by BIR and MiDAS mechanisms (158,159,169). These observations suggest a very complex set of cellular functions for a protein with a small set of biochemical activities. Similar to other proteins involved in selection of damage tolerance and replication restoration mechanisms, the cellular functions of RAD52 may be controlled by the nature of the DNA substrate, interacting partners, and posttranslational modifications. For example, phosphorylation of human RAD52 at Y104 by cABL kinase modifies its interactions with DNA and enhances its DNA strand annealing activity in vitro (170). While a small acidic protein DSS1, known as an interacting partner of BRCA2, affects the RAD52 function in BIR (171).
5. Conclusions
What has become clear from the extensive research into replication fork stalling is that cells have numerous ways to deal with replication barriers whether they are initiating a DNA damage response or reversal of the replication fork to bypass the barrier. The choice of the recovery mechanism appears to be a complex interplay within the cell consisting of three non-exclusive ways that these choices are made at stalled replication forks. The first is by the composition of the proteins present at the stalled fork and whether or not these proteins are post-translationally modified in response to stalled replication. The second way is by the structure of the stalled replication fork itself, such as the presence and the size of the leading or lagging strand gap, as well as the nature and location of the lesion, for which proteins will recognize or have substrate specificity. The third is the time during the cell cycle when a stalled fork occurs and persists. Notably, many of the lesions can be bypassed over multiple cell division cycles, with every cycle using a different mechanism and possibly producing multiple alternative alleles and different mutation signatures (172).
Acknowledgements:
We gratefully acknowledge the support by the NIH/NIGMS R35GM131704, NIH/NCI R01 CA232425, DOD/CDMRP BC180227P1 and 1836351 EAGER to M.S., and NIH/NIGMS GM081433 to M.T.W. We thank members of the Spies’ and Washington’s labs for many fruitful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Okazaki R, Okazaki T, Sakabe K, Sugimoto K, and Sugino A (1968) Mechanism of DNA chain growth. I. Possible discontinuity and unusual secondary structure of newly synthesized chains. Proceedings of the National Academy of Sciences of the United States of America 59, 598–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berti M, Cortez D, and Lopes M (2020) The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nature reviews. Molecular cell biology; [DOI] [PubMed] [Google Scholar]
- 3.Berti M, and Vindigni A (2016) Replication stress: getting back on track. Nature structural & molecular biology 23, 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franchitto A, and Pichierri P (2014) Replication fork recovery and regulation of common fragile sites stability. Cellular and molecular life sciences : CMLS 71, 4507–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuzminov A (2016) Chromosomal Replication Complexity: A Novel DNA Metrics and Genome Instability Factor. PLoS genetics 12, e1006229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Macheret M, and Halazonetis TD (2015) DNA replication stress as a hallmark of cancer. Annual review of pathology 10, 425–448 [DOI] [PubMed] [Google Scholar]
- 7.Rickman K, and Smogorzewska A (2019) Advances in understanding DNA processing and protection at stalled replication forks. The Journal of cell biology 218, 1096–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neelsen KJ, and Lopes M (2015) Replication fork reversal in eukaryotes: from dead end to dynamic response. Nature reviews. Molecular cell biology 16, 207–220 [DOI] [PubMed] [Google Scholar]
- 9.Cortés-Ledesma F, and Aguilera A (2006) Double-strand breaks arising by replication through a nick are repaired by cohesin-dependent sister-chromatid exchange. EMBO Rep 7, 919–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pardo B, Moriel-Carretero M, Vicat T, Aguilera A, and Pasero P (2020) Homologous recombination and Mus81 promote replication completion in response to replication fork blockage. EMBO Rep 21, e49367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, and Lopes M (2012) Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nature structural & molecular biology 19, 417–423 [DOI] [PubMed] [Google Scholar]
- 12.Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M (2015) Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. The Journal of cell biology 208, 563–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byun TS, Pacek M, Yee MC, Walter JC, and Cimprich KA (2005) Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes & development 19, 1040–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marians KJ (2018) Lesion Bypass and the Reactivation of Stalled Replication Forks. Annual review of biochemistry 87, 217–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Powers KT, and Washington MT (2018) Eukaryotic translesion synthesis: Choosing the right tool for the job. DNA repair 71, 127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakofsky CJ, and Malkova A (2017) Break induced replication in eukaryotes: mechanisms, functions, and consequences. Critical reviews in biochemistry and molecular biology 52, 395–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ripley BM, Gildenberg MS, and Washington MT (2020) Control of DNA Damage Bypass by Ubiquitylation of PCNA. Genes 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Croteau DL, Popuri V, Opresko PL, and Bohr VA (2014) Human RecQ helicases in DNA repair, recombination, and replication. Annual review of biochemistry 83, 519–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng X, and Zhao X (2017) Replication fork regression and its regulation. FEMS yeast research 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quinet A, Lemacon D, and Vindigni A (2017) Replication Fork Reversal: Players and Guardians. Molecular cell 68, 830–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elserafy M, Abugable AA, Atteya R, and El-Khamisy SF (2018) Rad5, HLTF, and SHPRH: A Fresh View of an Old Story. Trends in genetics : TIG 34, 574–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gallo D, and Brown GW (2019) Post-replication repair: Rad5/HLTF regulation, activity on undamaged templates, and relationship to cancer. Critical reviews in biochemistry and molecular biology 54, 301–332 [DOI] [PubMed] [Google Scholar]
- 23.Poole LA, and Cortez D (2017) Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Critical reviews in biochemistry and molecular biology 52, 696–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies AA, Huttner D, Daigaku Y, Chen S, and Ulrich HD (2008) Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein a. Molecular cell 29, 625–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, and Ulrich HD (2005) Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Molecular cell 19, 123–133 [DOI] [PubMed] [Google Scholar]
- 26.Northam MR, Garg P, Baitin DM, Burgers PM, and Shcherbakova PV (2006) A novel function of DNA polymerase zeta regulated by PCNA. The EMBO journal 25, 4316–4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence CW (2004) Cellular functions of DNA polymerase zeta and Rev1 protein. Advances in protein chemistry 69, 167–203 [DOI] [PubMed] [Google Scholar]
- 28.Sale JE (2013) Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb Perspect Biol 5, a012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McVey M, Khodaverdian VY, Meyer D, Cerqueira PG, and Heyer WD (2016) Eukaryotic DNA Polymerases in Homologous Recombination. Annual review of genetics 50, 393–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson RE, Kondratick CM, Prakash S, and Prakash L (1999) hRAD30 mutations in the variant form of xeroderma pigmentosum. Science 285, 263–265 [DOI] [PubMed] [Google Scholar]
- 31.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, and Hanaoka F (1999) The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 399, 700–704 [DOI] [PubMed] [Google Scholar]
- 32.Lehmann AR, Niimi A, Ogi T, Brown S, Sabbioneda S, Wing JF, Kannouche PL, and Green CM (2007) Translesion synthesis: Y-family polymerases and the polymerase switch. DNA repair 6, 891–899 [DOI] [PubMed] [Google Scholar]
- 33.Prakash S, Johnson RE, and Prakash L (2005) Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annual review of biochemistry 74, 317–353 [DOI] [PubMed] [Google Scholar]
- 34.Sale JE, Lehmann AR, and Woodgate R (2012) Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nature reviews. Molecular cell biology 13, 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Washington MT, Carlson KD, Freudenthal BD, and Pryor JM (2010) Variations on a theme: eukaryotic Y-family DNA polymerases. Biochimica et biophysica acta 1804, 1113–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, and Walker GC (2009) Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiology and molecular biology reviews : MMBR 73, 134–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson RE, Prakash S, and Prakash L (1999) Requirement of DNA polymerase activity of yeast Rad30 protein for its biological function. The Journal of biological chemistry 274, 15975–15977 [DOI] [PubMed] [Google Scholar]
- 38.Washington MT, Johnson RE, Prakash S, and Prakash L (2000) Accuracy of thymine-thymine dimer bypass by Saccharomyces cerevisiae DNA polymerase eta. Proceedings of the National Academy of Sciences of the United States of America 97, 3094–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Washington MT, Prakash L, and Prakash S (2003) Mechanism of nucleotide incorporation opposite a thymine-thymine dimer by yeast DNA polymerase eta. Proceedings of the National Academy of Sciences of the United States of America 100, 12093–12098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silverstein TD, Johnson RE, Jain R, Prakash L, Prakash S, and Aggarwal AK (2010) Structural basis for the suppression of skin cancers by DNA polymerase eta. Nature 465, 1039–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iguchi M, Osanai M, Hayashi Y, Koentgen F, and Lee GH (2014) The error-prone DNA polymerase iota provides quantitative resistance to lung tumorigenesis and mutagenesis in mice. Oncogene 33, 3612–3617 [DOI] [PubMed] [Google Scholar]
- 42.Lee GH, and Matsushita H (2005) Genetic linkage between Pol iota deficiency and increased susceptibility to lung tumors in mice. Cancer science 96, 256–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petta TB, Nakajima S, Zlatanou A, Despras E, Couve-Privat S, Ishchenko A, Sarasin A, Yasui A, and Kannouche P (2008) Human DNA polymerase iota protects cells against oxidative stress. The EMBO journal 27, 2883–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pence MG, Blans P, Zink CN, Hollis T, Fishbein JC, and Perrino FW (2009) Lesion bypass of N2-ethylguanine by human DNA polymerase iota. The Journal of biological chemistry 284, 1732–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Washington MT, Minko IG, Johnson RE, Wolfle WT, Harris TM, Lloyd RS, Prakash S, and Prakash L (2004) Efficient and error-free replication past a minor-groove DNA adduct by the sequential action of human DNA polymerases iota and kappa. Molecular and cellular biology 24, 5687–5693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tissier A, Frank EG, McDonald JP, Iwai S, Hanaoka F, and Woodgate R (2000) Misinsertion and bypass of thymine-thymine dimers by human DNA polymerase iota. The EMBO journal 19, 5259–5266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Washington MT, Johnson RE, Prakash L, and Prakash S (2004) Human DNA polymerase iota utilizes different nucleotide incorporation mechanisms dependent upon the template base. Molecular and cellular biology 24, 936–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Yuan F, Wu X, and Wang Z (2000) Preferential incorporation of G opposite template T by the low-fidelity human DNA polymerase iota. Molecular and cellular biology 20, 7099–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nair DT, Johnson RE, Prakash L, Prakash S, and Aggarwal AK (2005) Human DNA polymerase iota incorporates dCTP opposite template G via a G.C + Hoogsteen base pair. Structure 13, 1569–1577 [DOI] [PubMed] [Google Scholar]
- 50.Nair DT, Johnson RE, Prakash L, Prakash S, and Aggarwal AK (2006) An incoming nucleotide imposes an anti to syn conformational change on the templating purine in the human DNA polymerase-iota active site. Structure 14, 749–755 [DOI] [PubMed] [Google Scholar]
- 51.Ogi T, Shinkai Y, Tanaka K, and Ohmori H (2002) Polkappa protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. Proceedings of the National Academy of Sciences of the United States of America 99, 15548–15553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi JY, Angel KC, and Guengerich FP (2006) Translesion synthesis across bulky N2-alkyl guanine DNA adducts by human DNA polymerase kappa. The Journal of biological chemistry 281, 21062–21072 [DOI] [PubMed] [Google Scholar]
- 53.Haracska L, Prakash L, and Prakash S (2002) Role of human DNA polymerase kappa as an extender in translesion synthesis. Proceedings of the National Academy of Sciences of the United States of America 99, 16000–16005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Washington MT, Johnson RE, Prakash L, and Prakash S (2002) Human DINB1-encoded DNA polymerase kappa is a promiscuous extender of mispaired primer termini. Proceedings of the National Academy of Sciences of the United States of America 99, 1910–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson RE, Prakash S, and Prakash L (2000) The human DINB1 gene encodes the DNA polymerase Poltheta. Proceedings of the National Academy of Sciences of the United States of America 97, 3838–3843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohashi E, Bebenek K, Matsuda T, Feaver WJ, Gerlach VL, Friedberg EC, Ohmori H, and Kunkel TA (2000) Fidelity and processivity of DNA synthesis by DNA polymerase kappa, the product of the human DINB1 gene. The Journal of biological chemistry 275, 39678–39684 [DOI] [PubMed] [Google Scholar]
- 57.Jha V, and Ling H (2017) Structural basis of accurate replication beyond a bulky major benzo[a]pyrene adduct by human DNA polymerase kappa. DNA repair 49, 43–50 [DOI] [PubMed] [Google Scholar]
- 58.Lone S, Townson SA, Uljon SN, Johnson RE, Brahma A, Nair DT, Prakash S, Prakash L, and Aggarwal AK (2007) Human DNA polymerase kappa encircles DNA: implications for mismatch extension and lesion bypass. Molecular cell 25, 601–614 [DOI] [PubMed] [Google Scholar]
- 59.Dumstorf CA, Mukhopadhyay S, Krishnan E, Haribabu B, and McGregor WG (2009) REV1 is implicated in the development of carcinogen-induced lung cancer. Molecular cancer research : MCR 7, 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xie K, Doles J, Hemann MT, and Walker GC (2010) Error-prone translesion synthesis mediates acquired chemoresistance. Proceedings of the National Academy of Sciences of the United States of America 107, 20792–20797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choi JY, and Guengerich FP (2008) Kinetic analysis of translesion synthesis opposite bulky N2- and O6-alkylguanine DNA adducts by human DNA polymerase REV1. The Journal of biological chemistry 283, 23645–23655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pryor JM, and Washington MT (2011) Pre-steady state kinetic studies show that an abasic site is a cognate lesion for the yeast Rev1 protein. DNA repair 10, 1138–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Washington MT, Minko IG, Johnson RE, Haracska L, Harris TM, Lloyd RS, Prakash S, and Prakash L (2004) Efficient and error-free replication past a minor-groove N2-guanine adduct by the sequential action of yeast Rev1 and DNA polymerase zeta. Molecular and cellular biology 24, 6900–6906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nair DT, Johnson RE, Prakash L, Prakash S, and Aggarwal AK (2005) Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science 309, 2219–2222 [DOI] [PubMed] [Google Scholar]
- 65.Nair DT, Johnson RE, Prakash L, Prakash S, and Aggarwal AK (2008) Protein-template-directed synthesis across an acrolein-derived DNA adduct by yeast Rev1 DNA polymerase. Structure 16, 239–245 [DOI] [PubMed] [Google Scholar]
- 66.Rechkoblit O, Kolbanovskiy A, Landes H, Geacintov NE, and Aggarwal AK (2017) Mechanism of error-free replication across benzo[a]pyrene stereoisomers by Rev1 DNA polymerase. Nature communications 8, 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Makarova AV, Stodola JL, and Burgers PM (2012) A four-subunit DNA polymerase zeta complex containing Pol delta accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic acids research 40, 11618–11626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Malik R, Kopylov M, Gomez-Llorente Y, Jain R, Johnson RE, Prakash L, Prakash S, Ubarretxena-Belandia I, and Aggarwal AK (2020) Structure and mechanism of B-family DNA polymerase ζ specialized for translesion DNA synthesis. Nature structural & molecular biology [DOI] [PMC free article] [PubMed]
- 69.Johnson RE, Washington MT, Haracska L, Prakash S, and Prakash L (2000) Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature 406, 1015–1019 [DOI] [PubMed] [Google Scholar]
- 70.Washington MT, and Gildenberg MS (2020) Structure of DNA polymerase ζ: capturing the getaway driver. Nature structural & molecular biology [DOI] [PMC free article] [PubMed]
- 71.Hastings PJ, Lupski JR, Rosenberg SM, and Ira G (2009) Mechanisms of change in gene copy number. Nature reviews. Genetics 10, 551–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morrow CA, Nguyen MO, Fower A, Wong IN, Osman F, Bryer C, and Whitby MC (2017) Inter-Fork Strand Annealing causes genomic deletions during the termination of DNA replication. eLife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ulrich HD, and Jentsch S (2000) Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. The EMBO journal 19, 3388–3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gallo D, Kim T, Szakal B, Saayman X, Narula A, Park Y, Branzei D, Zhang Z, and Brown GW (2019) Rad5 Recruits Error-Prone DNA Polymerases for Mutagenic Repair of ssDNA Gaps on Undamaged Templates. Molecular cell 73, 900–914 e909 [DOI] [PubMed] [Google Scholar]
- 75.Beyer DC, Ghoneim MK, and Spies M (2013) Structure and Mechanisms of SF2 DNA Helicases. Advances in experimental medicine and biology 767, 47–73 [DOI] [PubMed] [Google Scholar]
- 76.Blastyak A, Pinter L, Unk I, Prakash L, Prakash S, and Haracska L (2007) Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Molecular cell 28, 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gangavarapu V, Haracska L, Unk I, Johnson RE, Prakash S, and Prakash L (2006) Mms2-Ubc13-dependent and -independent roles of Rad5 ubiquitin ligase in postreplication repair and translesion DNA synthesis in Saccharomyces cerevisiae. Molecular and cellular biology 26, 7783–7790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Unk I, Hajdu I, Blastyak A, and Haracska L (2010) Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA repair 9, 257–267 [DOI] [PubMed] [Google Scholar]
- 79.Blastyak A, Hajdu I, Unk I, and Haracska L (2010) Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Molecular and cellular biology 30, 684–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kile AC, Chavez DA, Bacal J, Eldirany S, Korzhnev DM, Bezsonova I, Eichman BF, and Cimprich KA (2015) HLTF’s Ancient HIRAN Domain Binds 3’ DNA Ends to Drive Replication Fork Reversal. Molecular cell 58, 1090–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Korzhnev DM, Neculai D, Dhe-Paganon S, Arrowsmith CH, and Bezsonova I (2016) Solution NMR structure of the HLTF HIRAN domain: a conserved module in SWI2/SNF2 DNA damage tolerance proteins. Journal of biomolecular NMR 66, 209–219 [DOI] [PubMed] [Google Scholar]
- 82.Hishiki A, Hara K, Ikegaya Y, Yokoyama H, Shimizu T, Sato M, and Hashimoto H (2015) Structure of a Novel DNA-binding Domain of Helicase-like Transcription Factor (HLTF) and Its Functional Implication in DNA Damage Tolerance. The Journal of biological chemistry 290, 13215–13223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gildenberg MS, and Washington MT (2019) Conformational flexibility of fork-remodeling helicase Rad5 shown by full-ensemble hybrid methods. PloS one 14, e0223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chavez DA, Greer BH, and Eichman BF (2018) The HIRAN domain of helicase-like transcription factor positions the DNA translocase motor to drive efficient DNA fork regression. The Journal of biological chemistry 293, 8484–8494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Achar YJ, Balogh D, Neculai D, Juhasz S, Morocz M, Gali H, Dhe-Paganon S, Venclovas C, and Haracska L (2015) Human HLTF mediates postreplication repair by its HIRAN domain-dependent replication fork remodelling. Nucleic acids research 43, 10277–10291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shin S, Hyun K, Kim J, and Hohng S (2018) ATP Binding to Rad5 Initiates Replication Fork Reversal by Inducing the Unwinding of the Leading Arm and the Formation of the Holliday Junction. Cell reports 23, 1831–1839 [DOI] [PubMed] [Google Scholar]
- 87.Fan Q, Xu X, Zhao X, Wang Q, Xiao W, Guo Y, and Fu YV (2018) Rad5 coordinates translesion DNA synthesis pathway by recognizing specific DNA structures in saccharomyces cerevisiae. Current genetics 64, 889–899 [DOI] [PubMed] [Google Scholar]
- 88.Unk I, Hajdu I, Fatyol K, Hurwitz J, Yoon JH, Prakash L, Prakash S, and Haracska L (2008) Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proceedings of the National Academy of Sciences of the United States of America 105, 3768–3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Motegi A, Liaw HJ, Lee KY, Roest HP, Maas A, Wu X, Moinova H, Markowitz SD, Ding H, Hoeijmakers JH, and Myung K (2008) Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proceedings of the National Academy of Sciences of the United States of America 105, 12411–12416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Motegi A, Sood R, Moinova H, Markowitz SD, Liu PP, and Myung K (2006) Human SHPRH suppresses genomic instability through proliferating cell nuclear antigen polyubiquitination. The Journal of cell biology 175, 703–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Unk I, Hajdu I, Fatyol K, Szakal B, Blastyak A, Bermudez V, Hurwitz J, Prakash L, Prakash S, and Haracska L (2006) Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proceedings of the National Academy of Sciences of the United States of America 103, 18107–18112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bai G, Kermi C, Stoy H, Schiltz CJ, Bacal J, Zaino AM, Hadden MK, Eichman BF, Lopes M, and Cimprich KA (2020) HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Molecular cell [DOI] [PMC free article] [PubMed]
- 93.Quinet A, Tirman S, Jackson J, Šviković S, Lemaçon D, Carvajal-Maldonado D, González-Acosta D, Vessoni AT, Cybulla E, Wood M, Tavis S, Batista LFZ, Méndez J, Sale JE, and Vindigni A (2020) PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Molecular cell 77, 461–474.e469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Burkovics P, Sebesta M, Balogh D, Haracska L, and Krejci L (2014) Strand invasion by HLTF as a mechanism for template switch in fork rescue. Nucleic acids research 42, 1711–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sood R, Makalowska I, Galdzicki M, Hu P, Eddings E, Robbins CM, Moses T, Namkoong J, Chen S, and Trent JM (2003) Cloning and characterization of a novel gene, SHPRH, encoding a conserved putative protein with SNF2/helicase and PHD-finger domains from the 6q24 region. Genomics 82, 153–161 [DOI] [PubMed] [Google Scholar]
- 96.Bruhl J, Trautwein J, Schafer A, Linne U, and Bouazoune K (2019) The DNA repair protein SHPRH is a nucleosome-stimulated ATPase and a nucleosome-E3 ubiquitin ligase. Epigenetics & chromatin 12, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lin JR, Zeman MK, Chen JY, Yee MC, and Cimprich KA (2011) SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Molecular cell 42, 237–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, and West SC (2005) Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Molecular cell 20, 783–792 [DOI] [PubMed] [Google Scholar]
- 99.Atkinson J, and McGlynn P (2009) Replication fork reversal and the maintenance of genome stability. Nucleic acids research 37, 3475–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 [DOI] [PubMed] [Google Scholar]
- 101.Mukherjee S, Wright WD, Ehmsen KT, and Heyer WD (2014) The Mus81-Mms4 structure-selective endonuclease requires nicked DNA junctions to undergo conformational changes and bend its DNA substrates for cleavage. Nucleic acids research 42, 6511–6522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, Cejka P, Stewart S, Lopes M, and Vindigni A (2015) DNA2 drives processing and restart of reversed replication forks in human cells. The Journal of cell biology 208, 545–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, and Vindigni A (2017) MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nature communications 8, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Técher H, Baldi G, Shen R, Ciccia A, Pellegrini L, Krejci L, and Costanzo V (2017) Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Molecular cell 67, 867–881.e867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Malacaria E, Honda M, Franchitto A, Spies M, and Pichierri P (2020) Physiological and Pathological Roles of RAD52 at DNA Replication Forks. Cancers 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Malacaria E, Pugliese GM, Honda M, Marabitti V, Aiello FA, Spies M, Franchitto A, and Pichierri P (2019) Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nature communications 10, 1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, Eichman BF, and Cortez D (2012) SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes & development 26, 151–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Betous R, Couch FB, Mason AC, Eichman BF, Manosas M, and Cortez D (2013) Substrate-selective repair and restart of replication forks by DNA translocases. Cell reports 3, 1958–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ, Izhar L, Petit SA, Adamson B, Yoon JC, Kowalczykowski SC, Livingston DM, Haracska L, and Elledge SJ (2012) Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Molecular cell 47, 396–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yusufzai T, and Kadonaga JT (2008) HARP is an ATP-driven annealing helicase. Science 322, 748–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yusufzai T, and Kadonaga JT (2010) Annealing helicase 2 (AH2), a DNA-rewinding motor with an HNH motif. Proceedings of the National Academy of Sciences of the United States of America 107, 20970–20973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bhat KP, Betous R, and Cortez D (2015) High-affinity DNA-binding domains of replication protein A (RPA) direct SMARCAL1-dependent replication fork remodeling. The Journal of biological chemistry 290, 4110–4117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang JW, Holland CL, Zwicky K, Herrador R, Jacobs H, Cortez D, Ciccia A, Penengo L, and Lopes M (2017) Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Molecular cell 67, 882–890 e885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yuan J, Ghosal G, and Chen J (2012) The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Molecular cell 47, 410–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Weston R, Peeters H, and Ahel D (2012) ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes & development 26, 1558–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sotiriou SK, and Halazonetis TD (2019) Remodeling Collapsed DNA Replication Forks for Cancer Development. Cancer research 79, 1297–1298 [DOI] [PubMed] [Google Scholar]
- 117.Puccetti MV, Adams CM, Kushinsky S, and Eischen CM (2019) Smarcal1 and Zranb3 Protect Replication Forks from Myc-Induced DNA Replication Stress. Cancer research 79, 1612–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, Marino F, Lucic B, Biasin V, Gstaiger M, Aebersold R, Sidorova JM, Monnat RJ Jr., Lopes M, and Vindigni A (2013) Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nature structural & molecular biology 20, 347–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Elliott B, and Jasin M (2002) Double-strand breaks and translocations in cancer. Cellular and molecular life sciences : CMLS 59, 373–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Halazonetis TD, Gorgoulis VG, and Bartek J (2008) An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355 [DOI] [PubMed] [Google Scholar]
- 121.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Esashi F, Galkin VE, Yu X, Egelman EH, and West SC (2007) Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nature structural & molecular biology 14, 468–474 [DOI] [PubMed] [Google Scholar]
- 123.Zelensky A, Kanaar R, and Wyman C (2014) Mediators of Homologous DNA Pairing. Cold Spring Harbor Perspectives in Biology 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Prakash R, Zhang Y, Feng W, and Jasin M (2015) Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harbor Perspectives in Biology 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Daley JM, Gaines WA, Kwon Y, and Sung P (2014) Regulation of DNA Pairing in Homologous Recombination. Cold Spring Harbor Perspectives in Biology [DOI] [PMC free article] [PubMed]
- 126.Spies M, and Fishel R (2015) Mismatch Repair during Homologous and Homeologous Recombination. Cold Spring Harbor Perspectives in Biology 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Slupianek A, Schmutte C, Tombline G, Nieborowska-Skorska M, Hoser G, Nowicki MO, Pierce AJ, Fishel R, and Skorski T (2001) BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Molecular cell 8, 795–806 [DOI] [PubMed] [Google Scholar]
- 128.Yuan Z-M, Huang Y, Ishiko T, Nakada S, Utsugisawa T, Kharbanda S, Wang R, Sung P, Shinohara A, Weichselbaum R, and Kufe D (1998) Regulation of Rad51 Function by c-Abl in Response to DNA Damage. Journal of Biological Chemistry 273, 3799–3802 [DOI] [PubMed] [Google Scholar]
- 129.Subramanyam S, Ismail M, Bhattacharya I, and Spies M (2016) Tyrosine phosphorylation stimulates activity of human RAD51 recombinase through altered nucleoprotein filament dynamics. Proceedings of the National Academy of Sciences of the United States of America 113, E6045–e6054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mason JM, Chan YL, Weichselbaum RW, and Bishop DK (2019) Non-enzymatic roles of human RAD51 at stalled replication forks. Nature communications 10, 4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Galkin VE, Esashi F, Yu X, Yang S, West SC, and Egelman EH (2005) BRCA2 BRC motifs bind RAD51-DNA filaments. Proceedings of the National Academy of Sciences of the United States of America 102, 8537–8542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Davies OR, and Pellegrini L (2007) Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nature structural & molecular biology 14, 475–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, and West SC (2005) CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 434, 598–604 [DOI] [PubMed] [Google Scholar]
- 134.Zadorozhny K, Sannino V, Belan O, Mlcouskova J, Spirek M, Costanzo V, and Krejci L (2017) Fanconi-Anemia-Associated Mutations Destabilize RAD51 Filaments and Impair Replication Fork Protection. Cell reports 21, 333–340 [DOI] [PubMed] [Google Scholar]
- 135.Higgs MR, Reynolds JJ, Winczura A, Blackford AN, Borel V, Miller ES, Zlatanou A, Nieminuszczy J, Ryan EL, Davies NJ, Stankovic T, Boulton SJ, Niedzwiedz W, and Stewart GS (2015) BOD1L Is Required to Suppress Deleterious Resection of Stressed Replication Forks. Molecular cell 59, 462–477 [DOI] [PubMed] [Google Scholar]
- 136.Leuzzi G, Marabitti V, Pichierri P, and Franchitto A (2016) WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. The EMBO journal 35, 1437–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Simandlova J, Zagelbaum J, Payne MJ, Chu WK, Shevelev I, Hanada K, Chatterjee S, Reid DA, Liu Y, Janscak P, Rothenberg E, and Hickson ID (2013) FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. The Journal of biological chemistry 288, 34168–34180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fugger K, Mistrik M, Danielsen JR, Dinant C, Falck J, Bartek J, Lukas J, and Mailand N (2009) Human Fbh1 helicase contributes to genome maintenance via pro- and anti-recombinase activities. The Journal of cell biology 186, 655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chiolo I, Saponaro M, Baryshnikova A, Kim JH, Seo YS, and Liberi G (2007) The human F-Box DNA helicase FBH1 faces Saccharomyces cerevisiae Srs2 and postreplication repair pathway roles. Molecular and cellular biology 27, 7439–7450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Morishita T, Furukawa F, Sakaguchi C, Toda T, Carr AM, Iwasaki H, and Shinagawa H (2005) Role of the Schizosaccharomyces pombe F-Box DNA helicase in processing recombination intermediates. Molecular and cellular biology 25, 8074–8083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Osman F, Dixon J, Barr AR, and Whitby MC (2005) The F-Box DNA helicase Fbh1 prevents Rhp51-dependent recombination without mediator proteins. Molecular and cellular biology 25, 8084–8096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Masuda-Ozawa T, Hoang T, Seo YS, Chen LF, and Spies M (2013) Single-molecule sorting reveals how ubiquitylation affects substrate recognition and activities of FBH1 helicase. Nucleic acids research 41, 3576–3587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chu WK, Payne MJ, Beli P, Hanada K, Choudhary C, and Hickson ID (2015) FBH1 influences DNA replication fork stability and homologous recombination through ubiquitylation of RAD51. Nature communications 6, 5931. [DOI] [PubMed] [Google Scholar]
- 144.Bacquin A, Pouvelle C, Siaud N, Perderiset M, Salome-Desnoulez S, Tellier-Lebegue C, Lopez B, Charbonnier JB, and Kannouche PL (2013) The helicase FBH1 is tightly regulated by PCNA via CRL4(Cdt2)-mediated proteolysis in human cells. Nucleic acids research 41, 6501–6513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fugger K, Chu WK, Haahr P, Kousholt AN, Beck H, Payne MJ, Hanada K, Hickson ID, and Sorensen CS (2013) FBH1 co-operates with MUS81 in inducing DNA double-strand breaks and cell death following replication stress. Nature communications 4, 1423. [DOI] [PubMed] [Google Scholar]
- 146.Pepe A, and West SC (2014) MUS81-EME2 promotes replication fork restart. Cell reports 7, 1048–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jeong YT, Rossi M, Cermak L, Saraf A, Florens L, Washburn MP, Sung P, Schildkraut CL, and Pagano M (2013) FBH1 promotes DNA double-strand breakage and apoptosis in response to DNA replication stress. The Journal of cell biology 200, 141–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jeong YT, Cermak L, Guijarro MV, Hernando E, and Pagano M (2013) FBH1 protects melanocytes from transformation and is deregulated in melanomas. Cell cycle (Georgetown, Tex.) 12, 1128–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tamang S, Kishkevich A, Morrow CA, Osman F, Jalan M, and Whitby MC (2019) The PCNA unloader Elg1 promotes recombination at collapsed replication forks in fission yeast. eLife 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dungrawala H, Bhat KP, Le Meur R, Chazin WJ, Ding X, Sharan SK, Wessel SR, Sathe AA, Zhao R, and Cortez D (2017) RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Molecular cell 67, 374–386 e375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bhat KP, Krishnamoorthy A, Dungrawala H, Garcin EB, Modesti M, and Cortez D (2018) RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell reports 24, 538–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang H, Schaub JM, and Finkelstein IJ (2020) RADX condenses single-stranded DNA to antagonize RAD51 loading. bioRxiv, 2020.2005.2015.098996 [DOI] [PMC free article] [PubMed]
- 153.Grimme JM, Honda M, Wright R, Okuno Y, Rothenberg E, Mazin AV, Ha T, and Spies M (2010) Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic acids research 38, 2917–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Saotome M, Saito K, Yasuda T, Ohtomo H, Sugiyama S, Nishimura Y, Kurumizaka H, and Kagawa W (2018) Structural Basis of Homology-Directed DNA Repair Mediated by RAD52. iScience 3, 50–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, Maas A, Essers J, Hickson ID, and Kanaar R (2007) The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nature structural & molecular biology 14, 1096–1104 [DOI] [PubMed] [Google Scholar]
- 156.Franchitto A, Pirzio LM, Prosperi E, Sapora O, Bignami M, and Pichierri P (2008) Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. The Journal of cell biology 183, 241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Petermann E, Orta ML, Issaeva N, Schultz N, and Helleday T (2010) Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Molecular cell 37, 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sotiriou SK, Kamileri I, Lugli N, Evangelou K, Da-Ré C, Huber F, Padayachy L, Tardy S, Nicati NL, Barriot S, Ochs F, Lukas C, Lukas J, Gorgoulis VG, Scapozza L, and Halazonetis TD (2016) Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Molecular cell 64, 1127–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bhowmick R, Minocherhomji S, and Hickson ID (2016) RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Molecular cell 64, 1117–1126 [DOI] [PubMed] [Google Scholar]
- 160.Benitez A, Yuan F, Nakajima S, Wei L, Qian L, Myers R, Hu JJ, Lan L, and Zhang Y (2014) Damage-dependent regulation of MUS81-EME1 by Fanconi anemia complementation group A protein. Nucleic acids research 42, 1671–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kai M, Boddy MN, Russell P, and Wang TS (2005) Replication checkpoint kinase Cds1 regulates Mus81 to preserve genome integrity during replication stress. Genes & development 19, 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kim SM, and Forsburg SL (2020) Active Replication Checkpoint Drives Genome Instability in Fission Yeast mcm4 Mutant. Molecular and cellular biology 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Osman F, and Whitby MC (2007) Exploring the roles of Mus81-Eme1/Mms4 at perturbed replication forks. DNA repair 6, 1004–1017 [DOI] [PubMed] [Google Scholar]
- 164.Rondinelli B, Gogola E, Yücel H, Duarte AA, van de Ven M, van der Sluijs R, Konstantinopoulos PA, Jonkers J, Ceccaldi R, Rottenberg S, and D’Andrea AD (2017) EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nature cell biology 19, 1371–1378 [DOI] [PubMed] [Google Scholar]
- 165.Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, and Lopes M (2017) Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nature communications 8, 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Murfuni I, Basile G, Subramanyam S, Malacaria E, Bignami M, Spies M, Franchitto A, and Pichierri P (2013) Survival of the replication checkpoint deficient cells requires MUS81-RAD52 function. PLoS genetics 9, e1003910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Doe CL, Osman F, Dixon J, and Whitby MC (2004) DNA repair by a Rad22-Mus81-dependent pathway that is independent of Rhp51. Nucleic acids research 32, 5570–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hengel SR, Malacaria E, Folly da Silva Constantino L, Bain FE, Diaz A, Koch BG, Yu L, Wu M, Pichierri P, Spies MA, and Spies M (2016) Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. eLife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, and Halazonetis TD (2014) Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343, 88–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Honda M, Okuno Y, Yoo J, Ha T, and Spies M (2011) Tyrosine phosphorylation enhances RAD52-mediated annealing by modulating its DNA binding. The EMBO journal 30, 3368–3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Stefanovie B, Hengel SR, Mlcouskova J, Prochazkova J, Spirek M, Nikulenkov F, Nemecek D, Koch BG, Bain FE, Yu L, Spies M, and Krejci L (2020) DSS1 interacts with and stimulates RAD52 to promote the repair of DSBs. Nucleic acids research 48, 694–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Aitken SJ, Anderson CJ, Connor F, Pich O, Sundaram V, Feig C, Rayner TF, Lukk M, Aitken S, Luft J, Kentepozidou E, Arnedo-Pac C, Beentjes SV, Davies SE, Drews RM, Ewing A, Kaiser VB, Khamseh A, López-Arribillaga E, Redmond AM, Santoyo-Lopez J, Sentís I, Talmane L, Yates AD, Semple CA, López-Bigas N, Flicek P, Odom DT, and Taylor MS (2020) Pervasive lesion segregation shapes cancer genome evolution. Nature [DOI] [PMC free article] [PubMed]
- 173.Biertümpfel C, Zhao Y, Kondo Y, Ramón-Maiques S, Gregory M, Lee JY, Masutani C, Lehmann AR, Hanaoka F, and Yang W (2010) Structure and mechanism of human DNA polymerase eta. Nature 465, 1044–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jha V, Bian C, Xing G, and Ling H (2016) Structure and mechanism of error-free replication past the major benzo[a]pyrene adduct by human DNA polymerase κ. Nucleic acids research 44, 4957–4967 [DOI] [PMC free article] [PubMed] [Google Scholar]