

Summary
Glioblastoma multiforme (GBM) is an aggressive brain tumor for which current immunotherapy approaches have been unsuccessful. Here, we explore the mechanisms underlying immune evasion in GBM. By serially transplanting GBM stem cells (GSCs) into immunocompetent hosts, we uncover an acquired capability of GSCs to escape immune clearance by establishing an enhanced immunosuppressive tumor microenvironment. Mechanistically, this is not elicited via genetic selection of tumor subclones, but through an epigenetic immunoediting process wherein stable transcriptional and epigenetic changes in GSCs are enforced following immune attack. These changes launch a myeloid-affiliated transcriptional program, which leads to increased recruitment of tumor-associated macrophages. Furthermore, we identify similar epigenetic and transcriptional signatures in human mesenchymal subtype GSCs. We conclude that epigenetic immunoediting may drive an acquired immune evasion program in the most aggressive mesenchymal GBM subtype by reshaping the tumor immune microenvironment.
Keywords: neural stem cell, glioblastoma, immune evasion, macrophage, DNA methylation, interferon signaling, epigenetics, immunoediting, chemokine, syngeneic
Graphical abstract
Highlights
-
•
Disease-relevant TME is recapitulated in immunocompetent GBM mouse models
-
•
Stable reconfiguration of the transcriptome occurs in GSCs following immune attack
-
•
Immune evasive GSCs deploy “myeloid mimicry” to establish a myeloid-enriched TME
-
•
Acquired transcriptional changes consistent with a process of epigenetic immunoediting
Glioblastoma stem cells deploy a myeloid mimicry program through epigenetic immunoediting, rather than subclonal selection, to drive a myeloid-enriched tumor microenvironment, thereby enabling immune evasion and tumor progression.
Introduction
Glioblastoma multiforme (GBM) is an incurable form of adult brain cancer with a dismal patient prognosis. Limited therapeutic options are available for GBMs as they are often detected late, they are highly infiltrative, and they display significant inter- and intra-tumoral genetic heterogeneity (Aldape et al., 2019). Many GBM driver mutations have been identified following genome-wide sequencing. The PI3K- and mitogen-activated protein kinase (MAPK)-associated signaling pathways are frequently co-activated in GBM through activation or amplification of EGFR or PDGFRA and are often accompanied by mutation of the tumor suppressors NF1, TP53, and/or PTEN (Brennan et al., 2013).
GBMs also display heterogeneity in their transcriptional circuits and epigenetic landscapes. Three major transcriptional signatures of GBM have been reported: proneural (PN), classical (CL), and mesenchymal (MES) (Verhaak et al., 2010; Wang et al., 2017). However, individual GBM tumors contain mixtures of cells with each of these subtype signatures, and the proportion of cells with each subtype can shift upon disease relapse following therapy (Brennan et al., 2013; Neftel et al., 2019; Patel et al., 2014; Wang et al., 2017). Single-cell RNA sequencing (RNA-seq) and lineage tracing studies have indicated that GBM cells can display plasticity and transit between cell states, suggesting that the tumor microenvironment (TME), as well as genetic and neurodevelopmental programs, may influence these transcriptional signatures (Neftel et al., 2019). GBM subtypes should not, therefore, be viewed as stable, distinct disease entities; rather, they reflect different proportions of various cell states that can shift throughout tumor development.
Neftel et al. (2019) have proposed four distinct tumor cell states in GBM: three of these—neural stem cell-like, oligodendrocyte progenitor cell-like, and astrocyte-like— mirror cell types within a neurodevelopmental differentiation hierarchy. However, a fourth cell state, termed “mesenchymal-like,” does not seem to correspond to a specific cell type. The mesenchymal GBM subtype has the poorest survival rates and displays increased immune infiltration within the TME, relative to other subtypes (Wang et al., 2017). This increased immune infiltrate correlates with loss of NF1; however, this is neither necessary nor sufficient to explain the emergence of a mesenchymal signature (Verhaak et al., 2010), suggesting additional processes must contribute to the development of an immunosuppressive TME. Understanding the etiology of the GBM mesenchymal-like cell state and the origins of its associated transcriptional subtype will be important for elucidating mechanisms of immune evasion (Lim et al., 2018).
Mechanisms underpinning the establishment of a pro-tumorigenic immune microenvironment in GBM remain unclear. To date, a lack of tractable and immunocompetent models of GBM have hampered efforts to dissect the relationship between GBM and the TME and the relative contribution of genetic, epigenetic, and environmental perturbations in directing tumor cell states. Here, we address the origins and mechanisms of immune evasion phenotypes in mesenchymal GBM by engineering a set of isogenic mouse GBM-initiating cells. Serial transplantation of transformed cells through syngeneic, immunocompetent hosts allowed us to monitor and dissect the progressive transcriptional and epigenetic changes that occur as GBM cells acquire immune evasion capabilities.
Our findings reveal a mechanism involving epigenetic immunoediting, in which exposure to immune attack leads to stable changes in transcriptional circuits, including critical myeloid-affiliated transcription factors and other immune-related pathways, in GBM cells. This occurs in the absence of clonal genetic selection and is likely enforced by sustained signaling from recruited tumor associated macrophages (TAMs). These acquired transcriptional circuits are stabilized by epigenetic changes and, therefore, persist in tumor cell descendants. We demonstrate that this leads to remodeling of the TME and the establishment of a protumorigenic microenvironment.
Results
Engineered GSCs acquire immune evasion capabilities upon serial transplantation through immunocompetent hosts
GBM is driven by cells with neural stem cell (NSC) characteristics (Lathia et al., 2015). Genetically engineered mouse models and genome analysis of patient samples indicates that endogenous NSCs can be a cell-of-origin for GBM (Alcantara Llaguno et al., 2016; Lee et al., 2018). We, therefore, engineered a set of isogenic GBM stem cells (GSCs) from NSCs isolated from C57BL/6 J (BL6) mice, by introducing one of five well established genetic GBM driver mutations: EGFRvIII or PDGFRA overexpression or CRISPR/Cas-mediated ablation of Nf1, Pten, or Trp53 (Figures 1A and S1A–S1G; Table S1; Bressan et al., 2017; Conti et al., 2005; Robertson et al., 2019). These “single hit” knockout GSCs expressed typical NSC markers (NESTIN and SOX2) and maintained the ability to differentiate into neurons and astrocytes (Figure S1H).
Figure 1.

Engineered GSCs acquire immune evasion capabilities upon serial transplantation through immunocompetent hosts
(A) NSC isolation from BL6 mice and engineering of GBM driver mutations.
(B) Immunoblots showing NF1 and PTEN expression in NP cells versus wild-type BL6 NSCs.
(C) Immunoblot confirming overexpression of EGFRvIII in NPE cells.
(D) Representative stereomicroscope images of GFP+ NPE tumors in NSG mice (whole brain live imaging; top, GFP; bottom, GFP/bright field [BF] overlay, n = 15).
(E) H&E staining of NPE tumors in NSG (upper panel), scale bar, 50 μm; immunofluorescence of common NSC (GFAP/Nestin) and proliferation (Ki67) markers in NPE tumors (lower panel), scale bar, 20 μm.
(F) Reverse-phase protein array (RPPA) analysis of common cancer driver pathways in wild-type BL6 NSCs versus mutant cell lines.
(G) Representative bioluminescent imaging of NPE tumor progression in vivo in BL6 recipients. Number of days post-surgery noted above images.
(H) Representative stereomicroscope images of GFP+ NPE tumors in BL6 hosts (whole brain live imaging; top, GFP; bottom, GFP/bright field [BF] overlay).
(I) Experimental design for tumor cell derivation and serial transplantation of NPE cells in NSG and BL6 mice.
(J) Survival curves following orthotopic transplantation of: wild-type NSCs into BL6 mice (n = 4, turquoise curve); NPE (n = 15, orange curve), or NPE-IE (n = 12, green curve) into NSG mice; NPE (n = 19, yellow curve), NPE-BL6-TD (n = 27, light purple curve), or NPE-IE (n = 33, dark purple curve) into BL6 mice.
See also Figure S1.
Figure S1.

Engineering GBM driver mutations in adult mouse neural stem cells to create GBM initiating cell lines, related to Figure 1 and Table S1
(A) Immunoblots of EGFRvIII (left panel) and PDGFRa (right panel) overexpression in NSCs following transfection with the PB-Transposon plasmids.
(B) PCR genotyping of clonal Pten Knock-Out (KO) lines to confirm successful gene targeting.
(C) Immunoblot of Pten KO NSC lines confirms loss of PTEN protein expression.
(D) PCR genotyping of clonal Nf1 KO lines to confirm successful gene targeting.
(E) Immunoblot of Nf1 KO NSC lines confirms loss of NF1 protein expression.
(F) PCR genotyping of clonal Trp53 KO lines to confirm successful gene targeting.
(G) ICC analysis of Trp53 KO clones confirms loss of TRP53 expression, scale bar = 20μm.
(H) CRISPR KO of Pten, Nf1 and Trp53 in NSCs does not affect the expression of the common NSC markers Nestin or SOX2, or the ability of NSCs to respond to differentiation cues (growth factor withdrawal and/or BMP addition), scale bar = 20μm.
(I) Orthotopic transplantation of NP cells into NOD-scid-gamma (NSG) mice provides a premalignant model (whole brain live imaging shown; top, GFP; bottom, overlay of GFP and brightfield (BF), representative images of n = 5 mice).
(J) Immunoblots of the GBM driver mutations (NF1, PTEN and EGFRvIII) in engineered NPE-Mx (multiplex) NSCs.
(K) Top: Orthotopic transplantation of NPE-Mx cells leads to tumour formation in NSG mice (whole brain live imaging shown; top, GFP; bottom, overlay of GFP and brightfield (BF)). Bottom: Survival curve of NSG mice orthotopically transplanted with NPE-Mx cells (n = 3 mice).
(L) Immunoblots of NF1, PTEN, and EGFR expression in NPE-Mx-TD (tumour-derived) polyclonal and clonal lines versus parental NSCs.
(M) Immunoblot of PDGFRa and PTEN in PPP NSCs.
(N) ICC confirming reduction of TRP53 expression in PPP cells versus parental NSCs (left panel) and quantification of Trp53-expressing cells by ICC (right panel; student’s t-test ∗∗∗ p≤0.001, error bars represent SEM), scale bar = 20μm.
(O) ICC of PPP mutant NSC lines for NSC markers (Sox2, Nestin) in self-renewing, EGF/FGF containing media and differentiation markers (GFAP, Tuj1) in differentiation conditions (BMP or -EGF/-FGF), scale bar = 20μm.
(P) Bioluminescent IVIS imaging of PPP tumour progression in vivo in NSG and BL6 recipients. Number of days post-surgery is noted above each image.
(Q) Survival curve of NSG (n = 4) and BL6 (n = 15) mice transplanted with PPP cells
(R) Flow cytometric analysis of the NPE-BL6-TD cells confirms that no CD45+ cells are detectable in vitro as compared to unstained NPE-BL6-TD and bone marrow-derived cells.
(S) Quantification of tumour incidence in NSG and BL6 mice transplanted with NPE, NPE-BL6-TD and NPE-IE cell lines. Numbers above bars denote actual tumour occurrence in all transplants.
(T) Confluence analysis of NPE and NPE-IE cells indicates no significant difference in proliferation rates (p = 0.2888).
(U) Karyotyping of parental subsequently engineered NSCs (>10 cell spreads counted per cell line, bars represent the mean value, error bars represent SEM, dots represent individual counts).
Next, we generated combinations of mutations to model the mesenchymal subtype. First, we created double mutant NSCs, via co-deletion of Nf1 and Pten (“NP” cells) (Figures 1A and 1B). These mutations occur together frequently in human mesenchymal GBM. Orthotopic transplantation of GFP+ NP cells into immunocompromised (NOD-scid-gamma [NSG]) mice led to the formation of small, benign growths (Figure S1I). Only when further engineered with EGFRvIII overexpression—forming a triple mutant cell line termed “NPE” (Figures 1A and 1C)—did we observe extremely aggressive tumor growth and infiltration in vivo (Figure 1D) with the expected hallmarks of GBM (Figure 1E). Concomitant mutation of Nf1 and Pten alongside EGFRvIII overexpression (termed NPE-Multiplex [NPE-Mx]) in an independent BL6 NSC line confirmed the requirement of this triple combination for tumor formation (Figures S1J–S1L). We also generated a tumor-initiating cell line corresponding to the proneural subtype, termed “PPP,” with PDGFRA overexpression alongside Trp53 and Pten mutations (Figures S1M–S1O). PPP tumor models were much less aggressive than their NPE counterparts (Figures S1P and S1Q).
Reverse-phase protein array (RPPA) analysis confirmed the expected activation of both RTK and PI3K signaling pathways in mutant NSCs, including elevated ERK, Ras, Raf, mTor, Akt, and Src expression (Figure 1F). We noted differences between NPE and PPP cells, with EGFRvIII overexpression stimulating increased pERK and STAT3 expression in the NPE mesenchymal model, consistent with previous reports in human tumors (Carro et al., 2010). The proneural subtype of GBM is known to have a reduced immune infiltrate and a less immunosuppressive TME (Wang et al., 2017). Given this, and the much more aggressive nature of NPE cells and their correspondence to the most immune evasive mesenchymal GBM subtype, we decided to focus on the NPE model to explore immune evasion mechanisms.
We confirmed that NPE cells are tumorigenic in immunocompetent, syngeneic BL6 hosts (Figures 1G and 1H), allowing us to model interactions between GBM cells and the immune TME. Tumors formed in parallel in immunocompromised NSG mice served as an important reference control (Figure 1I). Indeed, in contrast to the ∼3-week survival observed in all NSG hosts, NPE tumors arose in BL6 mice at a slower rate than in NSG mice (25–50 days versus 15–20 days, respectively) (Figure 1J, yellow curve versus orange curve), and the majority of BL6 hosts survived long-term, with no detectable tumor (Figures 1G). These data are consistent with the BL6 host immune system responding to NPE cells and constraining their tumor formation.
We reasoned that cells with acquired immune evasion capabilities may be enriched by deriving primary GSC cultures from tumors that evade immune surveillance in BL6 mice following serial transplantation of these through fresh hosts. We therefore established fresh cultures from the NPE tumors of BL6 mice (termed NPE-BL6-TD [tumor-derived]) (Figure 1I). NPE-BL6-TD cells in vitro retained expression of NSC markers and contained no contaminating CD45+ immune cell populations (Figure S1R). NPE-BL6-TD cells were transplanted into fresh, secondary BL6 recipients (Figure 1I) and formed tumors with increased frequency (Figures 1J, light purple curve, and S1S). GSC cultures re-derived from these secondary tumors (herein termed NPE-IE [immune evasive]) were then transplanted into fresh BL6 hosts (tertiary recipients) (Figure 1I). BL6 mice transplanted with NPE-IE cells displayed increased tumor formation with significantly worse survival than previously transplanted lines (Figures 1J, dark purple curve, and S1S). However, NPE and NPE-IE cells showed similar tumorigenicity when transplanted in control NSG mice, and we detected no proliferative advantage for NPE-IE cells in vitro or evident genomic instability (Figures 1J, green curve versus orange curve, S1T, and S1U). Together, these results indicate that NPE-IE cells have acquired immune evasion capabilities, which accounts for their increased tumor formation in BL6 recipients. This is consistent with a form of immunoediting (Schreiber et al., 2011), whereby sustained immune attack leads to the emergence of cells with increased ability to evade immune surveillance.
Immune evasive NPE-IE tumors possess a highly immunosuppressive microenvironment
To confirm if the NPE-IE tumors were more aggressive due to an ability to escape from T cell clearance, we depleted CD8 T cell populations in BL6 mice with NPE, NPE-BL6-TD, and NPE-IE tumors (Figure S2A). In all cases, we observed accelerated tumor growth and a progressive increase in tumor penetrance. This confirms that tumors generated from the NPE line and its derivatives do not undergo antigen loss, and they remain under considerable pressure from CD8 T cell-mediated clearance, from which they are actively escaping.
Figure S2.

Immune profiling of NPE and NPE-IE tumors reveals recapitulation of human disease and dynamic immune populations, related to Figure 2
(A) Survival analysis of BL6 mice orthotopically injected with NPE (left), NPE-BL6-TD (center) or NPE-IE (right) cells and subjected to IP injection of anti-CD8 or isotype matched control (IgG2b). NPE: n = 6 mice + ɑCD8, n = 5 mice + ɑIgG2b; p = 0.1002. NPE-BL6-TD: n = 6 mice + ɑCD8, n = 4 mice + ɑIgG2b; p = 0.1705. NPE-IE: n = 12 + ɑCD8, n = 10 + ɑIgG2b; p = 0.0171).
(B) Example gating strategy to determine immune cell populations in normal and tumor-burdened whole brains.
(C) Quantification of the proportions of major immune cell populations in normal whole brains and tumor-burdened brains.
(D) Quantification of PD1+ TIM3+ CD4 and CD8 T cells in whole brains of non-transplanted BL6 mice versus those with tumors from transplanted NPE or NPE-IE cells; n = 4 brains analyzed for each condition.
(E) Representative images of cell classification training used for macrophage quantification in NPE/NPE-IE fluorescent IHC images.
(F) Quantification of Iba1+ F4/80+ macrophage populations as fraction of the total cell population (right). p values calculated with one-way ANOVA, n = 3 – 5 brains from each condition analyzed.
Next, using flow cytometry, we characterized and compared immune cell types present in non-tumor bearing brains versus those harboring NPE or NPE-IE tumors. In tumor-bearing brains, we observed major changes in the total brain immune cell repertoire (Figures 2A–2D, S2B, and S2C), including a significant increase in macrophages and CD8/CD4 T cells, markers of both M1- and M2-like macrophages, and an apparent decrease in microglia. Further, we observed lymphoid populations in tumor-bearing brains display markers of dysfunction, such as PD-1 and TIM3 expression (Figure S2D). Importantly, fluorescent immunohistochemistry of tumor and adjacent tissue suggested that the dominating macrophage populations (F4/80+Iba1+) are localized to the tumor mass and are not generally increased throughout the brain (Figures 2E, 2F, S2E, and S2F).
Figure 2.

Whole brain immune population profiling reveals recapitulation of human GBM
(A) Multi-parametric flow cytometry uniform manifold approximation and projection (UMAP) of immune cell populations (CD45+) in whole brains of non-transplanted BL6 mice versus those with NPE or NPE-IE tumors; n = 4 brains per condition.
(B) Quantification of macrophage (left) and microglia (right) populations in (A).
(C) Quantification of CD8 T cell (left), CD4 Treg (center), and FOXP3− CD4 T cell populations in (A).
(D) Multi-parametric flow cytometry UMAP of myeloid populations (CD45+/CD11b+) in whole brains of non-transplanted BL6 mice versus those with NPE or NPE-IE tumors; n = 4 per condition.
(E) Representative fluorescent IHC of macrophage populations (F4/80+, Iba1+) in NPE or NPE-IE tumors/adjacent tissue; (n = 3–5 per condition), scale bar, 200 μm.
(F) Quantification of Iba1+ F4/80+ populations in (E) shown as frequency per mm2. One-way ANOVA, n = 3–5 brains from each condition analyzed.
See also Figure S2.
We directly isolated the tumor mass to further characterize the tumor immune microenvironment. Multiparametric flow cytometry data revealed increased immune cell infiltration in NPE-IE tumors relative to NPE tumors (Figures 3A, 3B, S3A, and S3B). Importantly, there was a significant increase in monocytic-myeloid derived suppressor cells (M-MDSCs) and macrophages in NPE-IE tumors (Figures 3C and 3D), with tumor-infiltrating macrophages expressing significantly higher levels of PD-L1 than microglia (Figures 3E and S3C). We did not observe evidence of a phenotypic macrophage switch in NPE-IE tumors when compared with NPE tumors; rather, macrophages in each case displayed classical phagocytic and antigen presenting phenotypes (i.e., CD86+ and CD11c+, Figure S3D), alongside the immunosuppressive marker PD-L1 (Figures 3E and S3E). There were no major changes noted in natural killer (NK) or dendritic cell populations (Figures S3F–S3H).
Figure 3.

Immune evasive NPE-IE tumors possess a highly immunosuppressive microenvironment
(A) Fast interpolation-based t-distributed stochastic neighborhood embedding (FIt-SNE) maps of cell populations in NPE and NPE-IE tumors.
(B) Quantification of cell population frequencies in (A) as proportion of total live cell population (n = 4 per condition).
(C) Flt-SNE maps of myeloid (CD11b+) cells in NPE and NPE-IE tumors.
(D) Quantification of macrophage, M-MDSCs, and microglia frequency in (C) as proportion of total live cell population (n = 4 per condition).
(E) PD-L1 median fluorescent intensity (MFI) quantification on microglia and macrophages in NPE and NPE-IE tumors (n = 4 per condition).
(F) Bioluminescent imaging of NPE-IE tumor progression in BL6 in vivo following intraperitoneal (i.p.) injection of aCSF-1R or PBS.
(G) Survival analysis of BL6 mice orthotopically injected with NPE-IE cells and subjected to i.p. injection of αCSF-1R (n = 17) or PBS (n = 11).
(H) Quantification of CD8+/CD4+ T cell population frequencies in NPE and NPE-IE tumors as a proportion of total live cell population in (A) (n = 6 per condition).
(I) Phenotypic marker expression of CD8+/CD4+ T cell subsets from (A) (n = 6 per condition).
For Flt-SNE plots, data were generated from 150,000 live cells randomly sampled from 3 tumors per condition (50,000 live events shown per tumor).
See also Figure S3.
Figure S3.

Immune evasive NPE-IE tumors possess a highly immunosuppressive TME, related to Figure 3
(A) & (B) Gating strategy to define myeloid (A) and lymphoid (B) populations in NPE and NPE-IE tumors.
(C) Representative histograms of PD-L1 expression on microglia and macrophages in NPE-IE tumors (linked to Figure 3E). Control represents fully stained sample minus anti-PD-L1 antibody.
(D) Representative histograms of CD206, CD86 and CD11c expression on macrophages from NPE and NPE-IE tumors. Data derived from 3 tumors randomly down sampled for 50,000 live cells each. Control represents fully stained sample minus either anti-CD206, anti-CD86 or anti-CD11c antibodies.
(E) Flow cytometry quantification of the frequency of macrophages, M-MDSC and microglia positive for expression of PD-L1
(F) Flow cytometry quantification of the frequency of NK cells as a percentage of live cells.
(G) Flow cytometry quantification of the frequency of CD11b+ DCs as a percentage of live cells.
(H) Flow cytometry quantification of CD11b+ DCs positive for expression of PD-L1.
(I) Survival analysis of NSG mice orthotopically injected with NPE-IE cells subjected to IP injection of aCSF-1R or PBS (n = 5 mice + ɑCSF-1R; n = 3 mice +PBS; p = 0.2367).
Blockade of CSF-1R signaling diminishes the capacity for immune evasion in NPE-IE cells
Previous reports have identified the macrophage colony stimulating factor 1 receptor (CSF-1R) as a potentially promising target for the treatment of gliomas (Pyonteck et al., 2013). To determine whether the increased macrophages recruited to NPE-IE tumors support immune evasion, we next depleted these cells in BL6 mice using a blocking antibody targeting CSF-1R. This resulted in increased survival and even clearance of large, well-established NPE-IE tumors (Figures 3F and 3G). There was no significant impact of this treatment in immunocompromised NSG transplanted controls (Figure S3I). Thus, tumor-associated macrophages play a key role in sustaining the growth and immune evasive qualities of NPE-IE tumors in BL6 mice.
Although there was no significant difference in the frequency of various T cell populations within NPE-IE tumors (Figure 3H), we did identify an increase in both CD8 and CD4 T cell populations positive for the markers PD-1 and LAG3 (Figure 3I). This indicates that the NPE-IE TME exhibits an elevated state of T cell dysfunction and enhanced immunosuppressive functions (Burugu et al., 2018). As mentioned, macrophages in both tumor types were highly positive for expression of the PD-1 ligand, PD-L1 (Figure 3E), and we found microglia were positive for expression of the LAG3 ligand, MHC-II (Figure 2D), suggesting the presence of additional immunosuppressive pathways beyond TAM recruitment. Our analyses highlight the utility of these GBM models, which reflect the features and immune repertoire of human mesenchymal GBM. We find that NPE-derived tumors can progressively escape CD8 T cell clearance and establish an increasingly myeloid-enriched and pro-tumorigenic TME, with accompanying T cell exhaustion.
Immune evasive cells undergo significant transcriptional reconfiguration following immune attack
To uncover the mechanism of acquired immune evasion observed in NPE-IE cells, we characterized their genome, transcriptome, and epigenome. Importantly, both karyotyping (Figure S1U) and whole genome sequencing (WGS) of the NPE and NPE-IE cells revealed no significant genetic disruptions—either ploidy, structural, or point mutations—when compared to parental, wild-type NSCs (Figure S4A). This suggests that it is not clonal evolution or classic genetic immunoediting processes that explain the acquired immune evasion properties observed in NPE-IE tumors. We reasoned that NPE-IE cells may have acquired other cell-intrinsic and stable changes in gene expression that support immune evasion.
Figure S4.

Transcriptional and epigenetic reconfiguration occurs across mouse samples, with DNA hypomethylation occurring at key immune-associated genes, related to Figures 4 and 5
(A) WGS copy number heatmap of log2 ratios of coverage for NPE and NPE-IE lines demonstrates genetic stability of mouse lines (gain of chrX evident as CNVs were called against male reference).
(B) Principal Component Analysis (PCA) of RNA-seq data (top 500 most variable genes) for all cell line samples.
(C) Heatmap of Z-scaled normalized counts for genes specific to all cell lines engineered with Nf1 loss ordered by log2 fold change (NF1 KO samples Vs. all others) (top). Z-scaled normalized counts of specific genes shown in heatmap across all mutants (Red trend line indicates mean Z-scaled expression) (bottom).
(D) Pairwise comparisons of CpG methylation changes between lines (excluding NP double mutant) as density scatterplots, and DMR bar plots – highlighting the predominant hypomethylation within tumor derived samples.
(E) Rank plots of promoter DMRs (+/− 2kb TSS) displaying hypomethylation in NPE-NSG-TD lines versus NPE samples (DMRs with > 50% methylation loss, overlapping genes within immune and interferon GO terms are highlighted in blue).
(F) Promoter DMR methylation (%) across lines for Irf8, Nt5e and Cd274, genes (top), and correlation with RNA-seq normalized counts (bottom) (SCC, and p value reported).
(G) RNA-seq normalized counts of Nt5e across analyzed lines.
We performed mRNA-seq analysis on our panel of cells, including single mutant lines and those derived from NSG tumors (a reference control for in vivo microenvironment exposure, without immune attack). Strikingly, of the single mutant lines, Nf1 loss stood out as having a transcriptome closest to the fully transformed NPE mutants (Figure S4B). Nf1 loss alone revealed enrichment of several gene signatures relevant to GBM, including notable activation of angiogenic and cell migration pathways (Figure S4C; Table S2). This is consistent with increased angiogenic signatures observed in the mesenchymal subtype (Verhaak et al., 2010; Wang et al., 2017) and suggests that loss of Nf1 may “prime” cells for malignant transformation.
Importantly, however, the immune evasive lines (NPE-BL6-TD and NPE-IE) acquired significantly different transcriptional patterns that were not explained by Nf1 loss alone; these included many immune-associated genes and Gene Ontology (GO) terms (Figures 4A–4C). We noted upregulation of several chemokines in NPE-IE cells (Figures 4D–4G; Table S3), particularly Ccl9, which has been previously linked to the establishment of pro-tumorigenic microenvironments (Kortlever et al., 2017) and could explain the increased myeloid cell content of NPE-IE tumors. Another interesting candidate upregulated in cells derived from immunocompetent hosts was Irf8 (interferon regulatory factor 8) (Figure 4B). Activation of Irf8 expression was surprising, because this is a myeloid-specific master transcription factor that is typically exclusively expressed in hematopoietic cells (Driggers et al., 1990) and has known roles in myeloid lineage specification and macrophage differentiation (Holtschke et al., 1996; Tamura et al., 2000). Irf8 is normally silent in NSCs, thus, following immune attack, NPE cells can inappropriately “hijack” expression of a myeloid master regulatory transcription factor. Together, these findings demonstrate that the in vivo immune attack triggers significant transcriptional changes in NPE cells.
Figure 4.

Immune evasive cells undergo significant transcriptional reconfiguration following immune attack
(A) PCA (principal component analysis) of mRNA-seq data (top 500 most variable genes) from NPE cells and derivative lines
(B) Heatmap of Z scaled normalized counts for genes specific to NPE-BL6-TD and NPE-IE lines ordered by log2 fold change (NPE-BL6-TD and NPE-IE versus all others) (top). Z scaled normalized counts of specific genes shown in (B) across all mutants (red trend line indicates mean Z scaled expression) (bottom).
(C) Heatmap of ssGSEA enrichment for select immune associated GO signatures across cell lines (top panel) and ssGSEA enrichment for Verhaak subtypes (proneural [PN], classical [CL], mesenchymal [MES]) (bottom) (-log10 p values (red/blue), simplicity scores (white/gray), and ssGSEA enrichment (red/yellow/blue) reported).
(D) Gene set enrichment analysis (GSEA) plot of chemokine-mediated signaling pathway for genes differentially expressed between NPE-IE and NPE-BL6-TD samples (enrichment score [ES] and false discovery rate [FDR] reported).
(E) Volcano plot of differentially expressed genes between NPE-BL6-TD and NPE-IE. Ccl9 highlighted as gene with highest log2 fold change.
(F) Normalized read counts of selected chemokines (Ccl9, Ccl6, and Ccl2).
(G) Forward phase protein array analysis of selected chemokines (CCL9, CCL6, and CCL2).
Immune evasion in NPE-IE lines is underpinned by epigenetic immunoediting
Given that the transcriptional changes induced following in vivo immune attack are stably retained following ex vivo expansion, we reasoned that epigenetic changes may have occurred. We profiled genome-wide DNA methylation patterns using reduced representation bisulfite sequencing (RRBS). Principal-component analysis (PCA) analysis revealed three distinct groups (Figure 5A), and we identified striking hypomethylation in cells transplanted in vivo (Figures 5B and S4D). The NPE-BL6-TD and NPE-IE lines clustered particularly closely by PCA (Figure 5A) and demonstrated widespread hypomethylation in comparison to NPE cells (Figure 5C). Consistent with mRNA-seq data, we found many differentially demethylated genes in immune evasive cells were related to immune processes (Figures 5D and S4E).
Figure 5.

Immune evasion in NPE-IE lines is underpinned by epigenetic immunoediting
(A) PCA of RRBS CpG methylation (top 25% most variable CpG sites).
(B) Density heatmap of CpG methylation (%) across lines.
(C) Density scatterplots of CpG methylation (%) (left) and differentially methylated region (DMR) bar plots (right) (red, hypermethylated DMRs; blue, hypomethylated DMRs) in NPE-BL6-TD and NPE-IE lines versus NPE (% DMR methylation change reported).
(D) Rank plots of promoter DMRs (±2 kb TSS) displaying hypomethylation in NPE-BL6-TD (top) and NPE-IE (bottom) versus NPE samples (DMRs with >50% methylation loss, overlapping genes within immune and interferon GO terms are highlighted in blue).
(E) Mean CpG methylation (%) tracks for profiled samples around Irf8 transcriptional start site.
See also Figure S4.
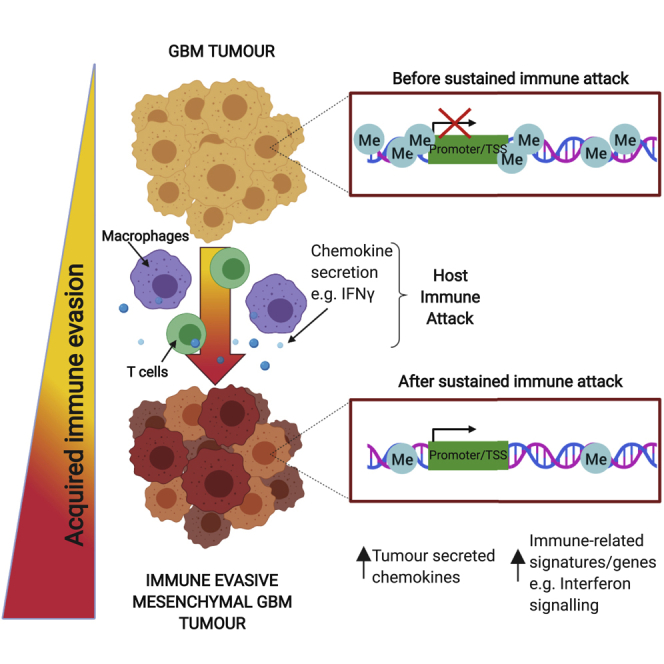
One of the most significant differentially methylated regions occurred within the Irf8 promoter region and gene body, with methylation progressively erased in the NPE-BL6-TD and NPE-IE lines, consistent with increased transcriptional activation (Figures 5D, 5E, and S4F). These changes in DNA methylation patterns suggest a process of “epigenetic immunoediting,” in which transcriptional changes imposed by immune attack are stabilized and selected for in those cells with increased immune evasive qualities. This would lead to highly immune evasive and transcriptionally altered descendants. Interestingly, we identified methylation loss at other well-known immune evasion regulators, including Nt5e (CD73), and Cd274 (PDL1) (Figure S4F), in line with these genes being primed for reactivation in vivo. Indeed, we see transcriptional activation of Nt5e in the NPE-IE tumor cells (Figure S4G), which has been recently identified as a critical GBM immunotherapy target that is elevated in TAMs (Goswami et al., 2020).
Irf8 is responsive to interferon gamma and TAMs in NPE cells in vitro and is important for immune evasion
Irf8 is known to be induced in macrophages via interferon gamma (IFNγ)-induced STAT1 signaling (Contursi et al., 2000). “Response to IFNγ” was an enriched GO term in the NPE-IE lines (Figure 6A), so we postulated that chronic IFNγ signals from the immune TME (Mojic et al., 2017) may stimulate activation of Irf8 expression in NPE cells. We tested this directly in vitro by exposing immune-naive NPE cells to IFNγ. Irf8 transcription was indeed activated in these cells, and we found that Irf8 can be stimulated more quickly and to higher levels in NPE-BL6-TD or NPE-IE cells following IFNγ treatment (Figures 6B, 6C, and S5A). This is consistent with priming for rapid transcriptional activation following DNA methylation loss at the Irf8 locus. Type I IFN signaling (induced by IFNα/β treatment) was not capable of inducing expression of Irf8 to the same extent as IFNγ in immune-naive NPE cells in vitro (Figure S5B). Furthermore, the JAK/STAT inhibitor, tofacitinib, reversed expression of IRF8 in NPE cells but not in NPE-IE cells, suggesting a JAK/STAT-independent mechanism is operating in NPE-IE cells to sustain high Irf8 expression (Figure 6D). Using an mCherry knockin reporter line for Irf8 expression, we confirmed that Irf8 transcription can be induced broadly in NPE cells following IFNγ treatment (Figures S5C and S5D). Moreover, profiling of directly isolated GFP+ NPE-BL6-TD and NPE-NSG-TD tumor cells confirmed activation of key genes occurs prior to in vitro expansion (e.g., Irf8, H2-Ab1) (Figures 6E, S5E, and S5F). Exposure of NPE cells to an immunocompetent in vivo environment is, therefore, sufficient to induce these observed transcriptional changes.
Figure 6.

Irf8 is responsive to IFNγ and TAMs in NPE cells in vitro and is important for immune evasion
(A) GSEA of IFNγ response signature in NPE-IE cells (enrichment score [ES] and FDR reported).
(B) RT-qPCR analysis of Irf8 expression in NPE, NPE-BL6-TD and NPE-IE cells in vitro ± IFNγ treatment.
(C) Representative immunoblot of IRF8 expression in NPE cell panel ± IFNγ time series treatment in vitro (n = 3).
(D) Representative immunoblot of IRF8 and pSTAT1 expression in NPE cell panel with IFNγ/Tofacitinib treatment in vitro (n = 3).
(E) qRT-PCR analysis of Irf8 expression in NPE cells in vitro versus GFP+ cells derived directly from NPE tumors in NSG/BL6 hosts. Points represent technical duplicates of cells isolated from individual animals.
(F) Schematic of NPE cells co-culture with immune populations derived from NPE tumors.
(G) RT-qPCR analysis of Irf8 expression in NPE cells co-cultured as in (F) (paired t test).
(H) Representative bioluminescent imaging of NPE-IE/NPE-IE-Irf8KO tumor progression in BL6 in vivo (Irf8KO clone G4 shown).
(I) Survival of BL6 mice orthotopically transplanted with NPE-IE cells versus clonally derived NPE-IE-Irf8KO lines (NPE-IE, n = 12; NPE-IE Irf8KO G4, n = 15; NPE-IE Irf8KO K6, n = 10; NPE-IE Irf8KO E6, n = 6). p values for survival of each Irf8KO line versus parental NPE-IE: ∗∗parental versus Irf8KO G4, p = 0.0047; ∗parental versus Irf8KO K6, p = 0.0323; parental versus Irf8KO E6, p = 0.0857.
RT-qPCR data representative of at least 3 biological replicates performed in technical duplicates; relative quantification (RQ) to NPE untreated sample.
See also Figure S5.
Figure S5.

Irf8 is responsive to IFNγ and TAMs in NPE and PPP cells in vitro and is important for immune evasion, related to Figure 6
(A) Immunoblot analysis of Irf8 expression in NPE cells and subsequently tumor-derived lines with and without in vitro IFNγ treatment (representative of n = 3 experiments).
(B) RT-qPCR analysis of Irf8 (left) and Ifih1 (right, confirms stimulation of IFN signaling) expression in untreated (UT) NPE, NPE-BL6-TD and NPE-IE cells versus in vitro treatment with IFNα/β (a, b, respectively). Error bars represent SEM, RQ (relative quantification) relative to NPE untreated conditions.
(C) Schematic of Irf8-mCherry reporter design.
(D) Flow cytometric analysis of mCherry expression in NPE-Irf8-mCherry reporter lines ± IFNγ treatment versus Parental NPE lines with adjunct histograms showing population distributions.
(E) Gating strategy employed to isolate GFP+ tumor cells, CD45+CD3+ T cells and CD45hi/lo F4/80+ myeloid cells from NPE tumors in NSG or BL6 host mice. Related to Figure 6E.
(F) RT-qPCR analysis of selected target gene expression (H2-Ab1, H2-Q7 and Ifi47) in ‘immune naive’ NPE cells in vitro compared with cells derived directly from NPE tumors in NSG and BL6 hosts. Each point represents technical duplicates of cells isolated from individual animals. Related to Figure 6E
(G) Heatmap illustrating expression levels of selected target gene expression (Irf8, H2-Q10, H2-Ab1, H2-Q7 and Ifi47) in untreated NPE cells versus those co-cultured with NPE tumor-derived immune populations in vitro. Gene expression was determined by RT-qPCR and is displayed as normalized expression values from technical duplicates of n = 3 biological replicates, related to Figure 6G.
(H) Heatmap illustrating expression levels of selected target gene expression (Irf8, H2-Q10 and H2-Ab1) in untreated PPP cells versus those co-cultured with NPE tumor-derived immune populations in vitro. Gene expression was determined by RT-qPCR and is displayed as normalized expression values from technical duplicates of n = 3 biological replicates.
(I) PCR genotyping of Irf8 locus in NPE-IE lines versus clonally derived NPE-IE Irf8 KO lines confirms successful gene targeting.
(J) Immunoblot of IRF8 expression in NPE-IE lines in the presence or absence of IFNγ treatment in vitro.
To determine the immune cell types within the TME of NPE-IE tumors that might be driving this transcriptional reconfiguration, we isolated immune cells from NPE tumors and co-cultured these with parental NPE cells in vitro (Figures 6F and S5E). Strikingly, our data revealed that infiltrating macrophage populations (F4/80+, CD45hi) can stimulate similar transcriptional changes in immune naive NPE cells to those observed in NPE-BL6-TD and NPE-IE cells (Figures 6G and S5G). This suggests that infiltrating macrophages could be the source of signals in vivo, such as IFNγ, driving this response. Further, although the proneural PPP cells did not generate tumors efficiently in vivo or display immune evasion capabilities, we reasoned that PPP cells may still respond to the NPE tumor-derived immune cell co-culture, as in patient tumors the proneural subtype can shift to mesenchymal. In vitro co-culture of PPP cells with immune cells isolated from NPE tumors suggested that similar transcriptional programs can be activated in proneural models (Figure S5H), consistent with plasticity of subtype signatures reported by Neftel et al., (2019).
We next generated NPE-IE Irf8 knockout cell lines. These showed a less aggressive phenotype, and tumors emerged with similar kinetics to mice transplanted with immune-naive NPE cells (Figures 6H, 6I, S5I, and S5J). Taken together, our data suggest that activation of Irf8 in our model is an important contributor to immune evasion in NPE-IE cells and may occur via IFNγ-mediated activation in vivo.
Human GSCs display two predominant transcriptional subtypes and mesenchymal GSCs are defined by IFN signaling
To assess whether our findings have relevance to human disease, we performed RNA-seq and molecular subtyping of 36 low passage, patient-derived GSC cultures, using non-negative matrix factorization (NNMF) to identify key transcriptional meta-gene modules that distinguish them (Gaujoux and Seoighe, 2010; Figures S6A and S6B). By profiling GSCs expanded in vitro, we can define potential immune-induced gene expression changes without the confounding contamination of immune cells within fresh tumor samples. Two distinct subtypes were identified: S1, which we termed the non-mesenchymal immune signature (Non-MESImm), and S2, which we termed the mesenchymal-immune signature (MESImm) (Figure 7A). Enrichment of these meta-gene modules were associated with published signatures by Wang et al. (2017), based on whole tumor transcriptomes (Figure 7A, top panel). A distinct transcriptional profile was associated with the MESImm subtype that included GO terms associated with IFN signaling as a dominant feature (Figures 7B and 7C; Table S4).
Figure S6.

Non-negative matrix factorization (NMF) of bulk RNA-seq in patient-derived GSCs identifies two distinct subtypes, related to Figure 7
(A) NMF of bulk RNA-seq highlights two predominant subgroups within patient-derived GBM GSCs. Consensus matrices for each NMF model across different ranks, or number of considered meta-gene modules (Metagenes, Consensus cluster assignment, Silhouette score to assess cluster quality, -log10 p values for Verhaak subtypes, and average connectivity across runs, are reported).
(B) Consensus matrices for different algorithms of rank 2 highlight stability across approaches (left). Cophenetic score computed for each number of considered meta-genes (dotted line represents random data), highlight two subgroups as optimal (right).
(C) Heatmap of ssGSEA enrichment for MESImm signature across engineered mouse cell lines (-log10 p values (Red/Blue), and ssGSEA enrichment (red/yellow/blue) are reported).
(D) PCA of publicly available scRNA-seq data of adult GBM cells (Neftel et al., 2019). Separation along PC2 is associated with IFN signaling genes (highlighted in black), and mesenchymal subtype genes (CD44, CHI3L1, and VIM highlighted in red).
(E) scRNA-seq ssGSEA enrichment for select immune GO terms across adult GBM cells ordered by MESImm subtype p value.
(F) Heatmap of Z-score TPM scRNA-seq of adult GBM cells for IFN genes highlighted in Figure 7B (top), and IRF family members (bottom), ordered by MESImm subtype p value. Differential expression between MESImm and Non-MESImm samples log2 fold change, percentile of effect size, and adjusted p values are reported.
(G) Heatmap of Spearman’s rank correlation coefficient for gene sets from (S6F) with previously published subtypes (Non-MESImm, MESImm highlighted in red).
Figure 7.

Human GSCs display two predominant major transcriptional subtypes, one of which is defined by IFN signaling and hypomethylation and is similar to the mesenchymal subtype from Verhaak et al., 2010
(A) Metagene (S1 Non-MESImm and S2 MESImm) enrichment and Verhaak subtype classification (PN/CL/MES, ssGSEA -log10 p values reported) across human GSCs.
(B) Heatmap of Z-scaled normalized counts for genes specific to metagene modules (S1 Non-MESImm/S2 MESImm). IFN associated genes specific to S2 MESImm module highlighted.
(C) Pathways enriched within S1 Non-MESImm/S2 MESImm signatures (∗terms of interest).
(D) Expression of candidate genes of interest (CCL2, IRF1, IRF7, and IRF8) in human Non-MESImm versus MESImm GSCs.
(E) Density heatmap of CpG methylation levels (beta values) in human GSCs.
(F) Empirical cumulative distribution function (ECDF) for DNA methylation levels (beta values) across GSCs (red, MESImm; blue, Non- MESImm).
(G) Bar plot of DMRs between MESImm and Non-MESImm GSCs (red, hypermethylated DMRs; blue, hypomethylated DMRs, DMR methylation % change reported).
(H) Density scatterplot of CpG methylation (beta values) between MESImm and Non-MESImm GSCs.
See also Figures S6 and S7 and Table S4.
Notably, we saw upregulation of several interferon regulatory factor family members in MESImm-enriched GSCs, including IRF8 and IRF7; however, a more striking association was seen for IRF1 (Figure 7D). Human IRF1 is also an IFNγ-responsive transcription factor that has been reported to function alongside hIRF8 in the development of myeloid cells (Langlais et al., 2016). Furthermore, we also identify increased expression of CCL2 in the MESImm signature, which may account for increased immune populations observed in mesenchymal GBM (Figures 7B and 7D; Qian et al., 2011). Finally, we confirmed enrichment of this MESImm signature in our engineered NPE cells, which increased as the cell lines acquired immune evasion capabilities (i.e., NPE-IE cells) (Figure S6C). Thus, related signatures to those uncovered in our mouse models are also observed in MESImm human GBMs. Importantly, these transcriptional signatures are stable and heritable in long term culture, in the absence of IFN signals; this is consistent with epigenetic events underlying a stable change to the transcriptome.
We validated our MESImm and Non-MESImm subtypes by comparing our findings with an analysis of independent, publicly available single-cell RNA-seq (scRNA-seq) data (Neftel et al., 2019). Similarly to human patient-derived GSC cultures, these GBM cells could be defined along a single axis of variation, separating MESImm and Non-MESImm cells. This axis was marked by expression of well-known mesenchymal genes such as CD44, CHI3L1, and VIM, as well as IFN-associated genes defined in the MESImm signature (Figure S6D). MESImm cells were enriched for IFN GO terms, IFN associated genes, and IRF family members, and the MESImm signature was consistent with previously defined mesenchymal transcriptional subtypes (Figures S6E–S6G and S7A; Wang et al., 2017, 2019; Neftel et al., 2019). This confirms that interferon-driven signatures are present within mesenchymal patient tumors.
Figure S7.

Comparison of the MESImm subtype with published subtypes across single cell data—with hypomethylation evident in MESImm samples, related to Figure 7
(A) Heatmap of -log10 ssGSEA p values for subtypes across single cell RNA-seq cohort (Neftel et al., 2019).
(B) Scatterplot of ssGSEA enrichment scores and mean methylation (beta-values) for each sample (red: MESImm, blue: Non- MESImm), with correlation coefficients reported.
(C) Genome tracks of key DMRs for IRF1, CCL2, and IRF8 (red: MESImm, blue: Non- MESImm).
(D) Methylation (beta-value) at each CpG site within each corresponding DMR (trend lines denote LOESS curves).
Human MESImm GBMs display similar DNA methylation changes to NPE-IE cells
To assess if human GBMs enriched for the MESImm module may also have DNA methylation changes reflective of the mouse models, we profiled DNA methylation patterns in patient-derived GSCs and observed a decreased level of DNA methylation in the MESImm subtype (Figures 7E–7H). Furthermore, single-sample gene set enrichment analysis (ssGSEA) of our patient-derived GSCs demonstrated that low levels of DNA methylation observed in the MESImm subtype correlated with previously published mesenchymal signatures, as well as the signatures reported here, and could be associated with activation of relevant immune-related pathways such as IFNγ response (Figure S7B). Notably, in the MESImm samples, we identified a loss of methylation specifically at CpG islands associated with those genes and pathways identified in our mouse models, including IRF1, CCL2, and IRF8 (Figures S7C and S7D). These findings are consistent with human GBM cells having undergone a similar process of transcriptional reconfiguration and epigenetic changes to those observed in the mouse models. Human GBMs may, therefore, also undergo epigenetic immunoediting to stabilize this immune evasive state, using similar mechanisms to those identified in the NPE-IE mouse cells.
Discussion
The specific combination of genetic, epigenetic, and microenvironmental cues that confers the varied transcriptional states observed in GBM cells remains poorly understood. Here, we have shown that a key component of the previously reported “mesenchymal” signature is a transcriptional module acquired in GBM cells following immune attack that is functionally important in facilitating immune evasion. Reconfigured transcription factor expression profiles and altered transcriptional circuits lead to the creation of an enhanced myeloid-enriched, immunosuppressive microenvironment, which is a feature of the most aggressive mesenchymal GBM subtype. This is accompanied by DNA methylation changes, and our results suggest a form of epigenetic immunoediting—rather than sub-clonal genetic diversification and selection—can support a striking gain in immune evasion capabilities. Similar transcriptional and DNA methylation changes are observed in human GBM cells of the mesenchymal subtype, suggesting this process of epigenetic immunoediting may occur in patients.
To date, there has been a lack of preclinical mouse models to explore mechanisms of immune evasion in GBM (Robertson et al., 2019). The set of isogenic cells we report here enables rapid production of tumors that recapitulate hallmarks of human GBM, most notably the immune landscape, in syngeneic, immunocompetent mouse models. A range of immune evasion pathways that we identified in the mouse NPE models are relevant to human disease, primarily the recruitment of pro-tumorigenic myeloid cells, including tumor cell activation of CD73 (Goswami et al., 2020) and markers of T cell exhaustion such as PD-1 and LAG3 (Mohme and Neidert, 2020). Using these models, we have been able to dissect the transcriptional and epigenetic changes that result from immune attack and identify mechanisms that then underpin acquired immune evasion. This has not been previously possible using human xenografts, primary tumor profiling, or existing syngeneic mouse models. NPE-IE cells are a renewable and easily shared primary cell line. The GFP-luciferase reporter enables tracking of tumor cells in live animals and facile recovery of tumor cells. This should be a useful model for the research community to deeply dissect GBM immune biology and support translational studies of immunotherapies.
We observed increased infiltration of macrophages with various phenotypes, which is consistent with recent reports that macrophages mount a multifaceted response in human GBM (Klemm et al., 2020). Enrichment of M-MDSCs was evident in our NPE-IE model, and this myeloid progenitor population has been implicated in glioma progression and is known to be highly immunosuppressive (Mi et al., 2020). Furthermore, we observed a phenotypic shift in the microglia populations present in tumor burdened brains, with MHC-II expression indicative of microglial activation. GBM is regarded as a relatively immunologically “cold” tumor, with its low mutational burden resulting in limited availability of neoantigens to support T cell recognition and effective anti-tumor immunity. Consistent with human disease, the presence of NPE and NPE-IE tumors in mice leads to increased infiltration of both CD4+ immunosuppressive Treg cells and CD8+ T cells into the brain, providing a unique opportunity to explore these populations in GBM. We find that T cell populations display elevated co-expression of markers associated with exhaustion (PD-1, LAG3) and regulatory function (FOXP3, LAG3) in NPE-IE tumors, which is complimented by increased expression of PD-L1 and MHC-II (PD-1 and LAG3 ligands) on macrophages and microglia, respectively. These inhibitory pathways likely contribute to immune escape of NPE-IE tumors and may represent potential therapeutic targets either alone or in combination.
Our engineered GSCs were found to upregulate Irf8. Because Irf8 is normally restricted to myeloid cells, our findings demonstrate the ability of GSCs to hijack myeloid-related transcriptional modules for immune evasion and chemokine expression. The acquired ability of GSCs to secrete chemokines following extensive immune attack may explain the increased presence of immunosuppressive cells in the NPE-IE models as well as the MESImm GBM subtype, with both CCL2 and CCL9 previously implicated in the recruitment of inflammatory monocytes (Qian et al., 2011; Kortlever et al., 2017).
Because the engineered NPE lines remained genetically stable (we deliberately avoided mutation of Trp53), it is unlikely that genetic events, clonal selection, or classic genetic immunoediting can explain our observations. Instead, our data indicate the existence of a process of epigenetic immunoediting, whereby following exposure to an in vivo environment and subsequently, immune attack, GSCs undergo site-specific DNA methylation changes alongside concomitant transcriptional changes that lead to activation of several immune-related signatures. The extent of these changes may vary across the tumor population, with selective pressures then enabling those with increased chemokine secretion to survive immune clearance following macrophage recruitment. The resulting reconfigured transcriptional circuits and modules are stabilized by DNA methylation changes, enhancing immune evasion.
Wang et al. (2017) previously demonstrated the mesenchymal signatures of bulk tumors are tumor cell intrinsic and not simply a feature of contaminating immune cells or associated endothelial cells. However, it remained unclear how this signature is acquired, because, unlike other GBM subtypes, mesenchymal GBM does not reflect a normal neurodevelopmental state and is not precisely defined by specific genetic events—although it clearly correlates with NF1 loss. Our findings do not diminish the importance of NF1 loss or related pathways in contributing to the mesenchymal subtype signature. Indeed, our Nf1−/− NSCs clearly show profound transcriptional changes and upregulation of angiogenesis related pathways. Such GBM driver mutations are a prerequisite for tumorigenesis and help establish the TME. However, we clearly demonstrate using unbiased functional and mechanistic studies that immune attack itself is driving a major component of the changes required for effective immune evasion, and contributing key modules to the mesenchymal signature. Interestingly, we uncover a potential self-reinforcing feedback loop, wherein immune attack drives tumor cells to recruit myeloid cells, which in turn expose cells to even further increased IFNγ. This ultimately results in stable epigenetic changes that are propagated in the expanding tumor population. These transcriptional changes are a functionally important component of immune evasion signatures in mesenchymal GBM cells. Further mechanistic dissection of this relationship, in particular the signals provided by the macrophages that drive this phenotype, may provide a framework to support development of TAM-targeting GBM therapies.
Therefore, we propose that Nf1 loss is a key mutation that primes NPE cells and facilitates the acquired transcriptional and epigenetic changes that occur in response to immune attack. This is supported by recent findings that Nf1−/− neurons stimulate downstream T cell and microglial activation that is essential for low-grade glioma growth (Guo et al., 2020). The specific downstream events of Nf1 loss, and whether these effects are due to excessive MAPK signaling or other effector pathways, remain unknown. Our initial observations for wild-type NSCs suggests these cannot tolerate chronic IFNγ and will fail to expand in culture, whereas mutation of Nf1 can overcome this, suggesting it provides a key step in tolerating the subsequent transcriptional changes imposed by immune attack and associated epigenetic reconfigurations.
Based on our data, the specific signals that trigger the observed transcriptional signatures are likely to include IFN. IFN signaling has a well-recognized role in orchestrating anti-tumor responses (Dunn et al., 2006). However, our data support a model in which prolonged exposure to IFNγ may be subverted to the benefit of the tumor cells by facilitating the acquisition of a pro-tumorigenic TME. We show that IFNγ can drive similar transcriptional changes in NPE cells in vitro, and many of the genes enriched in the NPE-IE cells are downstream effectors of this signaling pathway. However, we cannot rule out that there are contributions from other cytokines in vivo, such as TNFα (Bhat et al., 2013).
Our data suggest that recruited TAMs may be a source of a sustained IFNγ signal; however, other immune cells are known to produce IFNγ and may also contribute. Interestingly, two recent studies have shown that IFNγ can diffuse over long-ranges, indicating that tumor cells may be exposed to IFNγ throughout the TME (Hoekstra et al., 2020; Thibaut et al., 2020). Furthermore, previous studies have demonstrated the ability of in vitro IFNγ exposure to lead to epigenome changes at the chromatin level (Benci et al., 2016). It is likely that IFNγ is, therefore, a major inducer of the acquired immune evasive properties of NPE-IE cells. However, further studies will be needed to fully define the source, timing, and action of signals responsible for transcriptional changes imposed in tumor cells with the MESImm signature.
Overall, our findings suggest the observed gradient, or continuum, of subtype identities in GBM tumors can be explained by the extent to which the tumor immune microenvironment has eroded their epigenetic landscape and altered transcription factor regulatory networks. This may explain the detection of “hybrid” states by Neftel et al. (2019) that might represent partially edited states. It is also noteworthy that ECM and wound healing-associated gene sets correlate with the interferon signaling signatures in our MESImm subtype (data not shown), which is consistent with recent reports (Richards et al., 2021). Activation of wound-healing signatures may relate to physiologically relevant transcriptional changes that occur in regeneration and repair, such as is seen in reactive astrocytosis. Taken together, this implies that the activation of wound healing or injury response programs may accompany the activation of immune/interferon-related pathways; however, whether these programs are related or co-dependent in the establishment of a mesenchymal phenotype remains unclear.
We conclude that transcriptional and epigenetic reconfiguration and the co-opting of myeloid lineage-specific transcription factors and transcriptional modules is a tumor cell-intrinsic response that occurs following immune attack in the TME. We propose this is exploited in GBM to avoid immune detection and clearance. It will be of great interest to determine if similar epigenetic immunoediting processes that we have identified here in GBM also operate in other brain tumors or carcinomas.
Limitations of study
We acknowledge that WGS of more NPE-IE lines would allow us to more definitively rule out the effects of point mutations in driving the immune evasion response reported here. It will also be interesting to further dissect the contributions of different components of the TME on the plasticity of GBM cells using in vivo and in vitro assays. In particular, exploring the relative contribution of macrophage recruitment, retention, and polarization, as well as the roles of other immune cell populations, requires extensive further studies. Although our analysis of the human signatures is consistent with our conclusions, further direct testing of the influence of the immune cells or cytokines on their transcriptional state is necessary. Deeper characterization and testing of our proneural (PPP) model will be needed to determine how specific the pathways we identify here are to the mesenchymal subtype. Finally, our data strongly indicate a role for IFNγ in mediating the emergence of the mesenchymal-like signatures reported throughout our study, however, it will be necessary to confirm the effects of this cytokine and potentially others in vivo and in vitro.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FC: 41BBL-BB700 (Clone TKS-1) | BD | Cat#: 550532 |
| FC: CD103-UV395 (Clone M290) | BD | Cat#: 741083 |
| FC: CD11b-BUV661 (Clone M1/70) | BD | Cat#: 565080 |
| FC: CD11b-BV510 (Clone M1/70) | Biolegend | Cat#: 101263 |
| FC: CD11C-BV786 (Clone N418) | Biolegend | Cat#: 117336 |
| FC: CD11c-PE/Dazzle (Clone N418) | Biolegend | Cat#: 117348 |
| FC: CD140a-BV650 (Clone APA5) | Biolegend | Cat#: 740531 |
| FC: CD16/32 Blocking Antibodies (Clone 2.4G2) | BioXCell | Cat#: BE0307 |
| FC: CD19-BUV737 (Clone 1D3) | BD | Cat#: 564296 |
| FC: CD206-BV650 (Clone C068C2) | Biolegend | Cat#: 141723 |
| FC: CD24-BUV496 (Clone M1/69) | BD | Cat#: 564664 |
| FC: CD62L-BUV395 (Clone MEL-14) | Biolegend | Cat#: 740218 |
| FC: CD3-BUV737 (Clone 7A2) | BD | Cat#: 564380 |
| FC: CD3-BV711 (Clone 17A2) | Biolegend | Cat#: 100241 |
| FC: CD31-BV605 (Clone 390) | Biolegend | Cat#: 102427 |
| FC: CD4-BV510 (Clone GK1.5) | Biolegend | Cat#: 100449 |
| FC: CD44-BV650 (Clone 1MF) | Biolegend | Cat#: 103049 |
| FC: CD45-AF700 (Clone 30-F11) | Biolegend | Cat#: 103128 |
| FC: CD45-BUV805 (Clone HI30) | BD | Cat#: 560106 |
| FC: CD68-BV421 (Clone FA-11) | Biolegend | Cat#: 137017 |
| FC: CD8-BUV737 (Clone 53-67) | Biolegend | Cat#: 100241 |
| FC: CD86-FITC (Clone GL1) | Biolegend | Cat#: 11-0862-82 |
| FC: CD86-PE/Cy7 (Clone GL-1) | Biolegend | Cat#: 105014 |
| FC: F4/80-AF700 (Clone BM8) | Biolegend | Cat#: 123130 |
| FC: F4/80-BV421 (Clone BM8) | Biolegend | Cat#: 123130 |
| FC: FOXP3-eFlour 450 (CloneFJK.16 s) | eBiosciences | Cat#: 48-5773-82 |
| FC: FOXP3-BV421 (clone FJK-16S) | eBiosciences | Cat#: 20210613 |
| FC: GITRL-BV510 (Clone MIH44) | BD | Cat#: 563367 |
| FC: ICOSL-PE (HK5.3) | eBiosciences | Cat#: 12-5985-82 |
| FC: LAG3-PE-Cyanine7 (Clone eBioC9B7W) | eBiosciences | Cat#: 25-2231-82 |
| FC: Ly6C (Clone AL-21) | BD | Cat#: 563011 |
| FC: Ly6C-PerCP/Cy5.5 (Clone HK1.4) | Biolegend | Cat#: 128012 |
| FC: Ly6G-BUV395 (Clone 1A8) | Biolegend | Cat#: 583978 |
| FC: Ly6G-BUV563 (Clone IA8) | BD | Cat#: 565707 |
| FC: MHCII-BV711 (Clone M5-114-15-2) | eBiosciences | Cat#: 107643 |
| FC: MMR-PE (Clone C068C2) | Biolegend | Cat#: 128012 |
| FC: NK1.1-PE (Clone PK136) | Biolegend | Cat#: 18970 |
| FC: OX40L-PeCy7 (Clone RM134L) | Biolegend | Cat#: 108813 |
| FC: PD1-BV605 (Clone 29F.1A12) | Biolegend | Cat#: 135220 |
| FC: PDL1-BV711 (Clone 10F.9G2) | Biolegend | Cat#: 124319 |
| FC: PDL1-PECF590 (Clone 10F.9G2) | Biolegend | Cat#: 124324 |
| FC: PDL2-APC (Clone TY25) | Biolegend | Cat#: 107210 |
| FC: SIRPα-APC (Clone P84) | BD | Cat#: 560106 |
| FC: ST2-APC (Clone RMST2-2) | eBiosciences | Cat#: 17-9335-82 |
| FC: TIM3-PE/Dazzle 594 (Clone B8.2C12) | Biolegend | Cat#: 134014 |
| FC: TIM3-BV605 (RMT3-23) | Biolegend | Cat#: 119721 |
| FC: γδTCR-PerCP/Cy5.5 (CloneGL3) | Biolegend | Cat#: 118118 |
| IB: Actin | Santa Cruz | Cat#: sc-1616 |
| IB: GAPDH | Ambion | Cat#: AM4300 |
| IB: IRF8 | eBiosciences | Cat#: 14-7888-82 |
| IB: NF1 | Santa Cruz | Cat#: sc-67 |
| IB: PDGF Receptor Alpha | Cell Signaling Technology | Cat#: 3174 |
| IB: Phospho-EGF Receptor Tyr1068 | Cell Signaling Technology | Cat#: 3777 |
| IB: Phospho-STAT1 | Cell Signaling Technology | Cat#: 9167 |
| IB: Rabbit PTEN | Cell Signaling Technology | Cat#: 9556 |
| IF: Rat CD45 | Novus | Cat#: NB100-77417SS |
| IF: F4/80 | BioRad | Cat#: MCA497GA |
| IF: GFP | Abcam | Cat#: ab13970 |
| IF: Iba1 | Abcam | Cat#: ab178846 |
| IF: GFAP | Sigma Aldrich | Cat#: G3893 |
| IF: Nestin | DSHB | Cat#: Rat-401 |
| IF: p53 | Cell Signaling Technology | Cat#: 2524 |
| IF: Ki-67 | ThermoFisher | Cat#: MA5-14520 |
| IF: Sox2 | Abcam | Cat#: ab2492 |
| IF: Tuj1 | BioLegend | Cat#: 801202 |
| IN VIVO: CD8 (Clone YTS 169.4) | 2BScientific/BioXCell | Cat#: BE0117 |
| IN VIVO: CSF1R | BioXCell | Cat#: BP0213 |
| IN VIVO: IgG2b | 2BScientific/BioXCell | Cat#: BE0090 |
| Biological samples | ||
| Glioma Tissue and Derived Cells | Glioma Cellular Genetics Resource, CRUK, UK | http://gcgr.org.uk |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM/HAMS-F12 | Sigma | Cat#: D8437 |
| Pen/Strep | GIBCO | Cat#:15140-122 |
| Glucose | Sigma Aldrich | Cat#: G8644 |
| MEM-NEAA (100X) | GIBCO | Cat#: 11140-035 |
| BSA Solution | GIBCO | Cat#:15260-037 |
| Beta Mercaptoethanol | GIBCO | Cat#: 31350-010 |
| B27 Supplement (50X) | LifeTech/GIBCO | Cat#: 17504-044 |
| N2 Supplement (100X) | LifeTech/GIBCO | Cat#: 17502-048 |
| Recombinant Mouse EGF | Peprotech | Cat#: 315-09 |
| Recombinant Human FGF | Peprotech | Cat#: 100-18b |
| Laminin | Cultrex | Cat#: 3446-005-01 |
| Accutase | Sigma Aldrich | Cat#: A6964 |
| Glutamine | GIBCO | Cat#: 25030-021 |
| Mouse Recombinant IFNα | R&D Systems | Cat#: 10149-IF |
| Mouse Recombinant IFNβ | R&D Systems | Cat#: 8234-MB |
| Mouse Recombinant IFNγ | Peprotech | Cat#: 315-05 |
| Mouse Recombinant BMP4 | Peprotech | Cat#: 5020-BP |
| Colcemid | GIBCO | Cat#: 15210-040 |
| Potassium Chloride | Sigma Aldrich | Cat#: P3911 |
| Methanol | Fisher Scientific | Cat#: 13298233 |
| DAPI | Thistle Scientific | Cat#: 30-45-01 |
| SG Cell Line Transfection Kit | Lonza | Cat#: V4XC-3032 |
| Blasticidin | Invivogen | Cat#: ANT-BL-1 |
| Hygromycin B | Life Technologies | Cat#: 10687010 |
| DMSO | Sigma Aldrich | Cat#: 276855 |
| dNTPs | Thermo Scientific | Cat#: R0191 |
| LongAMP Taq Polymerase | NEB | Cat#: M0323 |
| Paraformaldehyde Powder 95% | Sigma | Cat#: 158127 |
| Triton X-100 | Merck Life Sciences | Cat#: X-100 |
| Goat Serum | Sigma Aldrich | Cat#: G6767 |
| Milk Powder | Marvel | N/A |
| Tween 20 | Cambridge Bioscience | Cat#: TW0020 |
| SuperScript III | Invitrogen | Cat#: 18080093 |
| Sodium Azide | Fisher Scientific | Cat#: 12615117 |
| PBS Tablets | Sigma Aldrich | Cat#: P4417 |
| Ethanol | VWR | Cat#: 20821-330 |
| FluoroSave Reagent | Calbiochem | Cat#: 345789 |
| Liberase | Roche | Cat#: 05401119001 |
| DNase | Sigma Aldrich | Cat#: 101041590001 |
| Percoll | GE Healthcare | Cat#: 17-0891-01 |
| RPMI | GIBCO | Cat#: 11875 |
| Collagenase D | Roche | Cat#: COLLD-RO |
| Red Blood Cell Lysis Buffer | Abcam | Cat#: ab204733 |
| HBSS | GIBCO | Cat#: 14170120 |
| FBS | GIBCO | Cat#: 10270-106 |
| Zombie NIR | BioLegend | Cat#: 423105 |
| TruStain Fc Block | BioLegend | Cat#: 101319 |
| SuperG Blocking Buffer | Grace Bio Labs | Cat#: 105100 |
| IRDye 800CW Streptavidin | LI-COR Biosciences | Cat#: 926-32230 |
| Taqman Universal PCR Master Mix | Applied Biosystems | Cat#: 4305719 |
| D-Luciferin potassium salt | Cambridge Bioscience | Cat#: CAY14681 |
| Critical commercial assays | ||
| DNeasy Blood and Tissue Kit | QIAGEN | Cat#: 69506 |
| RNeasy Mini Kit | QIAGEN | Cat#: 74104 |
| CytoFix/Cytoperm kit | BD | Cat#: 554714 |
| Deposited data | ||
| Raw and analyzed RNA-seq data (mouse) | This manuscript | GEO: GSE165386 |
| Raw and analyzed RNA-seq data (human) | Glioma Cellular Genetics Resource, CRUK, UK | https://gcgr.org.uk |
| Raw and analyzed RRBS data (mouse) | This manuscript | GEO: GSE165389 |
| Raw and analyzed EPIC array data (human) | Glioma Cellular Genetics Resource, CRUK, UK | https://gcgr.org.uk |
| Raw WGS data (mouse) | This manuscript | GEO: GSE165390 |
| Raw and analyzed scRNA-seq (human) | Neftel et al., 2019 | GEO: GSM3828672 |
| Experimental models: cell lines | ||
| C57BL/6 J Neural Stem Cells | Steven Pollard Lab, Edinburgh, UK | N/A |
| NPE and other BL6 NSC-derived lines | This manuscript | N/A |
| GCGR Human Glioma Stem Cells | This paper, Glioma Cellular Genetics Resource, CRUK, UK | N/A |
| Experimental models: organisms/strains | ||
| Mouse: BL6 (C57BL/6 J) | Charles River (original source, colony bred in house) | Cat#: 632C57BL/6J |
| Mouse: NSG (NOD-scid-gamma) | Charles River (original source, colony bred in house) | Cat#: 614NSG |
| Oligonucleotides | ||
| sgRNA pairs (see STAR Methods, Table S1) | This manuscript, Integrated DNA Technologies | https://eu.idtdna.com/pages |
| crRNA: Irf8-p2A-mCherry Reporter (see STAR Methods, Table S1) | This manuscript, Twist Biociences | N/A |
| Primer pairs (see STAR Methods, Tables S5 and S6) | This manuscript, Sigma | N/A |
| TaqMan Gene expression assays (see STAR Methods, Table S7) | ThermoFisher | N/A |
| Recombinant DNA | ||
| pET28a/Cas9-Cys | Addgene | Cat#: 53261 |
| PB-CAG-GFP-LUC-Ires-Bsd | This manuscript | N/A |
| pU6-(BbsI)_CBh-Cas9-T2A-mCherry | Addgene | Cat#: 64324 |
| PB-PyCAG-EGFRvIII-Hygro | This manuscript | N/A |
| PyCAG-PDGFRalpha-Ires-Bsd | This manuscript | N/A |
| pDONR221 | Invitrogen | Cat#: 12536017 |
| Software and algorithms | ||
| GraphPad Prism 9.0 | GraphPad Software, Inc | https://www.graphpad.com/ |
| FlowJo | FlowJo 10 | https://www.flowjo.com/ |
| Fiji/ImageJ | Open Source | https://imagej.net/Fiji |
| BioRender | BioRender | https://biorender.com/ |
| TrimGalore (version 0.5.0) | Martin, 2011 | https://github.com/FelixKrueger/TrimGalore |
| kallisto (version 0.44.0) | Bray et al., 2016 | https://pachterlab.github.io/kallisto/ |
| R Package: tximport (version 1.8.0) | Soneson et al., 2015 | https://bioconductor.org/packages/release/bioc/html/tximport.html |
| R Package: DESeq2 (version 1.27.32) | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R Package: clusterProfiler (version 3.15.4) | Yu et al., 2012 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| R Package: ChIPpeakAnno (version 3.22.0) | Zhu et al., 2010 | https://www.bioconductor.org/packages/release/bioc/html/ChIPpeakAnno.html |
| R Package: NMF (version 0.22.0) | Gaujoux and Seoighe, 2010 | https://cran.r-project.org/web/packages/NMF/index.html |
| R Package: ReactomePA (version 1.31.0) | Yu and He, 2016 | https://www.bioconductor.org/packages/release/bioc/html/ReactomePA.html |
| GSEA (version 3.0) | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| ssGSEA (version 1.0) | Wang et al., 2017 | https://secure.jbs.elsevierhealth.com/action/getSharedSiteSession?redirect=https%3A%2F%2Fwww.cell.com%2Fcancer-cell%2Ffulltext%2FS1535-6108%2817%2930253-2&rc=0 |
| trimRRBSdiversityAdaptCustomers.py (version 1.11) | NuGEN Technologies | https://github.com/nugentechnologies/NuMetRRBS |
| nudup.py (version 2.3) | NuGEN Technologies | https://github.com/tecangenomics/nudup |
| Bismark (version 0.16.3) | Krueger and Andrews, 2011 | https://www.bioinformatics.babraham.ac.uk/projects/bismark/ |
| R Package: DSS (version 2.36.0) | Park and Wu, 2016 | https://bioconductor.org/packages/release/bioc/html/DSS.html |
| R Package: minfi (version 1.34.0) | Fortin et al., 2014 | https://bioconductor.org/packages/release/bioc/html/minfi.html |
| R Package: DMRCate (version 2.2.3) | Peters et al., 2015 | https://bioconductor.org/packages/release/bioc/html/DMRcate.html |
| R Package: scran (version 1.16.0) | Lun, McCarthy and Marioni, 2016 | https://bioconductor.org/packages/release/bioc/html/scran.html |
| bcbio-nextgen (version 1.2.3) | bcbio | https://github.com/bcbio/bcbio-nextgen |
| Atropos (version 1.1.25) | Didion, Martin and Collins, 2017 | https://github.com/jdidion/atropos |
| Bowtie2 (version 2.3.5.1) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| samblaster (version 0.1.25) | Faust and Hall, 2014 | https://github.com/GregoryFaust/samblaster |
| VarDict (version 1.6) | Lai et al., 2016b | https://github.com/AstraZeneca-NGS/VarDict |
| Strelka2 (version 2.9.10) | Kim et al., 2018 | https://github.com/Illumina/strelka |
| Mutect2 (gatk version 3.8) | Cibulskis et al., 2013 | https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2 |
| freebayes (version 1.1.0.46) | Garrison and Marth, 2012 | https://github.com/freebayes/freebayes |
| HaplotypeCaller (gatk version 3.8) | DePristo et al., 2011 | https://gatk.broadinstitute.org/hc/en-us/articles/360037225632-HaplotypeCaller |
| Manta (version 1.6.0) | Chen et al., 2016 | https://github.com/Illumina/manta |
| CNVkit (version 0.9.6) | Talevich et al., 2016 | https://cnvkit.readthedocs.io/en/stable/ |
| R Package: VariantAnnotation (version 1.34.0) | Obenchain et al., 2014 | https://bioconductor.org/packages/release/bioc/html/VariantAnnotation.html |
| R Package: TxDb.Mmusculus.UCSC.mm10.knownGene (version 3.4.4) | Team BC, 2016 | https://www.bioconductor.org/packages/release/data/annotation/html/TxDb.Mmusculus.UCSC.mm10.knownGene.html |
| R Package: TxDb.Hsapiens.UCSC.hg38.knownGene (version 3.4.0) | Team BC, 2016 | https://bioconductor.org/packages/release/data/annotation/html/TxDb.Hsapiens.UCSC.hg38.knownGene.html |
| R Package: BSgenome.Mmusculus.UCSC.mm10.masked (version 1.3.99) | Team BC, 2016 | http://bioconductor.org/packages/release/data/annotation/html/BSgenome.Mmusculus.UCSC.mm10.masked.html |
| IGV (version 2.8.2) | Thorvaldsdóttir et al., 2013 | http://software.broadinstitute.org/software/igv/ |
| R v4.0.0 | The R Project for Statistical Computing | https://www.r-project.org/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Steven Pollard (steven.pollard@ed.ac.uk)
Materials availability
All reagents generated in this study (including cell lines and plasmids) are available on request from S.M.P.
Data and code availability
The accession numbers for the mouse mRNA-seq, RRBS, and WGS datasets reported in this paper are available to download from the Gene Expression Omnibus (GEO) with the following accession codes: GSE165386, GSE165389 and GSE165390. Human RNA-seq, and EPIC array datasets will be made available for download at http://gcgr.org.uk. Additional code is available upon request.
Experimental model and subject details
Mice and in vivo procedures
All treatments and procedures on mice were performed in accordance with protocols approved by Home Office UK guidelines in a designated facility under a project license to S.M.P., (PC0395462) at the University of Edinburgh. Mice were maintained on a regular diet in a pathogen-free facility on a 12-hr light/dark cycle with unlimited access to food and water. Intracranial transplantation of GSCs was performed using a stereotaxic frame to inject 200,000 cells resuspended in 2 μl of NSC media into 6- to 8-week-old male NOD/SCID/GAMMA (NSG) or C57BL/6 SCRM (BL6) mouse striatum, following administration of isoflourane general anesthesia and Rimadyl analgesic. Coordinates were 0.6mm anterior and 1.5mm lateral to the Bregma and 2.4 mm deep. Cell preparation and procedures were done as previously described in Pollard et al. (2009). Bioluminescent imaging was performed 20 minutes after D-Luciferin (50 mg/kg) subcutaneous injection, using the IVIS Lumina LT Series III instrument (Perkin Elmer). For all antibody blocking experiments, animals were randomly assigned to test and control groups. Blocking antibodies for CD8, CSF1R and IgG2b were administered by intraperitoneal (IP) injection at doses and frequencies described in the method details.
Cell culture
General cell culture
Mouse and human cells were cultured in serum-free basal medium supplemented with N2 and B27 (Life Technologies), laminin (Cultrex, 2 mg/ml) and growth factors EGF and FGF2 (Peprotech, 10 ng/ml). Cells were maintained as previously described in Pollard et al. (2009).
NSC differentiation to astrocytes
Cells were seeded 4 × 103 cells/ cm2 in NS culture media without EGF or FGF2 and with BMP4 (10 ng/ml). For neuronal differentiation we seeded cells at 8 × 103 cells/cm2 or 4 × 103 cells/cm2 for the BL6-NP and BL6-NPE in NS basal media without growth factors. Media was replenished every 3‐4 days. Immunostaining for differentiation markers was carried out on day 3 for astrocytes, and day 10 for neurons, post-differentiation initiation.
Co-culture of NPE cells with tumor derived immune cells
3x104 NPE cells were seeded in NSC culture media in a 12-well plate at least 6 hours prior to introduction of immune cells to allow attachment. F4/80 + and CD3+ immune cells were isolated from tumors and collected by flow (detailed below). F4/80 + cells were resuspended in ‘minimal macrophage media’ (DMEM high glucose (GIBCO), 10% FBS (GIBCO), 1% pen/strep (GIBCO)) and CD3 cells were resuspended in ‘minimal T cell media’ (complete RPMI 1640 medium (GIBCO) containing 1% pen/strep (GIBCO), 50 μM β-mercaptoethanol (GIBCO), and 1% L-glutamine (GIBCO)) before being seeded in a 3.0 μm pore polycarbonate membrane transwell insert (SLS, #3402) containing the appropriate medium. Co-culture experiments were carried out overnight (16 – 20 hours).
Cell growth analysis
NPE and NPE-IE cells were seeded in NSC culture media in a 24-well plate and growth curves were determined using Incucyte (Essen Bioscience) live cell imaging system.
Method details
Design and construction of CRISPR sgRNAs and PiggyBac plasmids
For gene knockouts, two gRNAs were designed to remove or generate indels, these were introduced to the most disease relevant gene regions in the case of GBM drivers. CRISPR sgRNAs were designed using the Optimized CRISPR Design tool (https://zlab.bio/guide-design-resources). Sequences are provided in Table S1. For construction of sgRNA-encoding plasmids, single-stranded oligonucleodies (IDT) containing the guide sequence of the sgRNAs were annealed, phosphorylated and ligated into BsaI site of U6-BsaI-sgRNA backbone (kindly provided by S. Gerety, Sanger Institute) or U6-BbsI-CBh-Cas9-T2A-mCherry backbone (this was a gift from Ralf Kuehn, Addgene plasmid # 64324) (Chu et al., 2015).
PiggyBac expression vectors were constructed via Gateway cloning apart from the EGFRvIII construct. The LUC-2A-GFP was PCR amplified from existing plasmids (gift from M. Pule, UCL). Human PDGFRα was amplified from cDNA of the G144 cell line (Pollard et al., 2009), PCR products were cloned into Gateway pDONR™221 using BP cloning. The cassettes were then delivered into the intermediate targeting vectors via Gateway LR cloning. Plasmids were sequence verified. EGFRvIII was amplified from existing plasmids (gift from A. Medvinsky lab) incorporating BrsGI restriction sites to clone into a PB-CAG-Ires-Hygro (gift from Keisuke Kaji). Primers used for construction are provided in Table S5.
Recombinant Cas9 plasmid and production
pET28a/Cas9-Cys was a gift from Hyongbum Kim (Ramakrishna et al., 2014). BL21(DE3) cells (New England Biolabs, C2527) were transformed with the plasmid pET28a/Cas9-Cys (Addgene, Cambridge, USA, plasmid #53261) using standard protocols. Cas9 protein expression was induced with 0.5 mM IPTG (Isopropyl β-D-1-thiogalactopyranoside) (Fisher, 10715114) and bacterial cells were incubated overnight at 20°C. Bacterial pellets were resuspended in 20 mL of lysis buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 1 mM TCEP, 5 mM imidazole pH 8.0), sonicated and loaded on a HisTrap HP 5 mL column (GE, 17-5248-01). Cas9 protein was collected in elution buffer (20 mM Tris-HCl pH 8.0, 250 mM NaCl, 10% glycerol, 1 mM TCEP, 250 mM imidazole pH 8.0). Fractions containing Cas9 protein were pooled and loaded into a HiPrep 26/10 Desalting Column (GE, 28-4026-52) to equilibrate in Cas9 buffer (20 mM HEPES-KOH pH 7.5, 150 mM KCl, 1 mM TCEP). Purified Cas9 protein was further concentrated using Vivaspin columns (Vivaspin20, VS2021) as per the user-guide instructions.
Cell line derivation
Mouse NS cell lines were derived from adult C57BL/6 SCRM subventricular zone as previously described (Conti et al., 2005; Sun et al., 2008). To obtain tumor cell lines, tumors were dissected from mouse brains and a piece of tumor was transferred into a 35 cm2 well plate with 2 mL of accutase. Tissue was minced into small pieces with scalpels and incubated 30 min at 37°C. The accutase with the tissue was transferred into a falcon before adding 6 mL of serum-free basal medium. Tissue was dissociated with a glass pipette very gently several times. After centrifugation for 5 min at 500 rcf., the cell pellet was resuspended in complete media and plated in a T25 flask. Cell debris was removed the following day and fresh media was added. Human brain tumor samples were dissociated for cell culture as described by Pollard et al. (2009). Briefly, tumor samples were dissociated into single cells using physical dissociation and accutase incubation for 15 – 20 minutes at 37°C, followed by the addition of serum-free basal medium and centrifugation for 5 min at 500 rcf. The cell pellet was resuspended in complete media and plated in a laminin coated flask. Cell debris was removed the following day and fresh media was added.
Cell transfection
NS cells were transfected using the DN 100 program of a 4D nucleofection system (Lonza). 1.5 × 106 cells were resuspended in 100 μl of SG transfection solution (Lonza) containing a maximum of 3 μg DNA. For CRISPR targeting, ratios of gRNAS and Cas9 were 1:1:2. For transgene overexpression, piggyBac transposase pBase and PB vectors were used in a 1:1 ratio. For multiplex experiments, ratios were equal and the amount of DNA used was 5 μg. Media was changed 6 hours after transfection. Cells were incubated for 48 hours post-transfection prior to FACS sorting.
Drug selection and clonal expansion
Cells with stable integration of the PiggyBac plasmids were selected for 5-7 days using 5 μg/ml blasticidin or 100 μg/ml hygromycin. For individual TSG knockout experiments, GFP positive cells were enriched by FACS single cell sorting into 96-well plates and collected in tubes (bulk population). The Irf8 clonal knockout lines were established using the iotaSciences isoCell system. Clones were expanded after 10-15 days post single cell disposition and transferred into 2 × 96-well replica plates and expanded for cell banking and gDNA extraction. For NPE-Multiplex experiments, GFP and mCherry double positive cells were sorted and collected in tubes. These cells were expanded and maintained for 5 days with hygromycin and 6 days with blasticidin and transplanted in mice without picking colonies. After tumor formation, a cell line was derived, NPE-Mx-TD, and individual colonies were picked into 2 × 24-well replica plates and expanded for cell banking and gDNA extraction.
Genotyping of knockout clones
Genomic DNA from individual clones was isolated from 24-well plates using DNeasy Blood & Tissue Kit (QIAGEN). 100-150 ng DNA was used in a 25 μL PCR reaction. PCR reactions comprised 0.3 μL DMSO (100% v/v, Sigma), 1 μL dNTPs (10 mM, Thermo Fisher Scientific), 5 μL LongAMP buffer (5x NEB), 1 μL LongAMP Taq (NEB), and 1 μL of each primer (at 10 μM concentration). Primer sequences are provided in Table S6. Genotyping primers flanking the targeting site of Cas9 were used to identify potential indels and directly assessed by Sanger sequencing traces from the resulting PCR products.
Immunocytochemistry
Cells were fixed in 4% PFA for 10 min and then blocked in 0.2% Triton X-100 and 3% Goat Serum. Cells were incubated in primary antibodies overnight at 4°C followed by incubation with appropriate secondary antibodies at room temperature for 1 hour. DNA was stained with a final incubation of cells in 4’,6-diamidino-2-phenylindole (DAPI). The specific dilution of primary antibodies used are as follows: mouse Nestin (1:10; DSHB, Rat-401), rabbit Sox2 (1:200; abcam ab2494), mouse GFAP (1:200; Sigma G3893), rabbit Ki-67 (1:200; Thermo Fisher MA5-14520), Tuj1 (1:1000; Biolegend, 801202), mouse p53 (1:500; Cell Signaling Technology 2524).
Immunoblotting
Immunoblotting was performed using standard protocols. Antibodies were diluted in blocking solution (5% milk powder in TBS-T or 3% BSA/1% PVP in TBS-T). Protein detection was performed using peroxidase-coupled secondary antibodies (Invitrogen) and application of homemade ECL before protein detection by exposure to X-ray films or imaging using the ChemiDock system (BioRad). The following primary antibodies were used: rabbit NF1 (1:500; Santa Cruz sc-67), rabbit PTEN (1:1000; CST 9556), rabbit phospho-EGF Receptor Tyr1068 (1:1000; CST 3777), rabbit PDGFRa (1:1000; CST 3174), mouse GAPDH (1:50000; Ambion, AM4300), mouse IRF8 (1:500; eBiosciences 14-7888-82), ACTIN (1:1000; Santa Cruz sc-1616), rabbit pSTAT1 (1:1000, CST 9167).
RT-qPCR
RT-qPCR was performed using standard protocols. RNA was isolated and gDNA eliminated using the RNeasy Mini Kit (QIAGEN, 74104). cDNA was synthesized using SuperScript III (Invitrogen) and RT-qPCR was performed using the QuantStudio 7 Flex (Applied Biosystems) instrument. Each sample/gene combination was run in duplicate or triplicate. TaqMan assays used are provided in Table S7.
RPPA analysis
Cell lysates were prepared and analyzed as previously described (Teo et al., 2018). Briefly, medium was removed from the plates and cells were rinsed twice with PBS. Cells were lysed on ice for 20 minutes with occasional shaking every 5 minutes. Cell lysates were collected with a scraper and clarified by centrifugation at 18,000 rcf. for 10 min at 4°C. Clarified supernatants in biological triplicate were adjusted to 2 mg/mL concentration and printed onto nitrocellulose-coated slides (Grace Bio-Labs) in a dilution series (four serial 2-fold dilutions) in technical triplicate using an Aushon 2470 arrayer (Aushon Biosystems). Slides were blocked, probed with validated primary antibodies and detected with DyLight 800-conjugated secondary antibodies (New England BioLabs). Slides were read using an InnoScan 710-IR scanner (Innopsys) and quantified using Mapix (Innopsys). Relative fluorescence intensities were normalized to respective FastGreen-stained spots (total protein), and data were computationally analyzed as previously described (Byron, 2017; Teo et al., 2018).
In vivo antibody administration
Animals were randomly assigned to test and control groups for all antibody blocking experiments. For CD8 T Cell Blockade, monoclonal anti-CD8a (Clone YTS 169.4, Cat # BE0117) and anti-IgG2b (Clone LTF-2 Cat # BE0090) antibodies were purchased from 2BScientific (BioXCell) and 20 μg of ɑCD8/IgG2b resuspended in 200 μl of PBS was administered via IP injection on alternate days, three times per week (Monday, Wednesday, Friday) for three weeks, beginning 3 days post orthotopic transplantation of tumor cells, to ensure continuous CD8+ T cell depletion. Anti-mouse CSF1R (CD115) was a kind gift from Sergio Quezada (BioXCell Cat # BP0213). Mice received three doses of 200 μg of anti-CSF1R resuspended in 200 μl of PBS on alternate days, beginning 5 days post-orthotopic transplantation of tumor cells.
Mouse Brain Fixation, Histopathology and Immunohistochemistry
Brains were fixed in 4% PFA overnight at 4°C, then rinsed several times with PBS and stored in PBS +0.05% Sodium Azide. For Histopathology procedures, brains were transferred into 70% Ethanol media and then embedded in paraffin for processing. 10 μm coronal slices were prepared forhematoxylin and eosin (H&E) staining. For immunohistochemistry of fixed brain tissue, 20 μm vibratome slices were transferred into a 24-well plate. Slices were incubated at room temperature for 30 min in blocking solution (0.2% Triton X-100 and 3% Goat Serum). Primary antibodies were incubated overnight at 4°C as follows: nestin (1:10; DSHB, Rat-401), chicken GFAP (1:100, BioLegend 829401), rabbit Ki-67 (1:100 Thermo Fisher MA5-14520), GFP (1:300 Abcam 13970). After three washes with PBS, slices were incubated with appropriate Alexa Fluor secondary antibodies (1:1000, Life technologies) and DAPI (1:2000, Sigma D9542) for 2 hours. Slices were washed three times and were mounted on a slide with FluoroSave™ Reagent (345789, Calbiochem). Slices were examined with a confocal microscope (Leica TCS SP8).
Multiplex Immunofluorescence
Staining of deparaffinized 5 μm FFPE sections was performed on automated tissue staining system (Bond-Rx, Leica Microsystems, CA) in the SURF facility at the University of Edinburgh. Primary antibodies; GFP (ab13970, Abcam Cambridge, MA, dilution 1:200); F4/80 (MCA497GA, BioRad, Hercules, CA, dilution 1:100); CD45 (NB100-77417SS, Novus, Manchester, UK, dilution, 1:100); Iba1 (ab178846, Abcam, dilution 1:250) were used to detect tumor cells and macrophages. Cells were labeled by using a tyramide signal amplification kit (Opal 7-color automation kit, NEL797001KT, Akoya BioSci. Menlo Park, CA). Opal 520 (1:150 dilution), Opal 570 (1:300, dilution), Opal 620 (1:150, dilution), Opal 650 (1:300 dilution), reactive fluorophores were used. Slides were counter stained with DAPI (Sigma-Aldrich, St. Louis, Missouri) and mounted by Prolong Diamond Antifade Mountant (P36965, Thermofisher Sci., Waltham, MA). Whole slide brain sections were imaged using the Vectra 3.0 multispectal imaging system (Perkin Elmer) at 20X magnification.
Image quantification
Images are analyzed in Inform Image analysis software (Version 2.9.4). To spectrally unmix emission spectra of each fluorophore, a spectral library and spectral unmixing algorithm was created using by using autofluorescent and single Opal dye-stained images. Background area were segmented from the tissue and removed from the analysis. The tumor regions was identified by using H&E staining and by staining GFP staining and at least 5 randomly selected ROI regions inside the tumor area was analyzed per tissue (20x object lens). Images were processed for nuclear detection (DAPI+ nuclear objects) and cytoplasm detection (2-pixel wide from nuclear detection). Using built-in machine learning algorithm, cell phenotype in the tumor region was trained by using Iba-1, F4/80 and CD45 markers. At the end of batch processing, the quality of phenotype identification was checked by using processed and component RGB images. Incorrectly classified images and damaged tissues were removed from the analysis. The total number of Iba+ and Iba-1 F4/80+ positive cells per mm2 of tissue and and total cells was calculated from the sum of the total images used per tissue.
Preparation of brain cell suspension for flow cytometry
For whole brain analysis, brains were dissected from mice and mechanically disaggregated using scissors, followed by enzymatic digestion using liberase (Roche Cat# 05401119001) and DNase (Sigma, Cat: 101041590001). Following enzymatic digestion cells suspension underwent Percoll (GE Healthcare, ref 17-0891-01) gradient separation to obtain cell suspensions enriched on hematopoietic cells. Cell suspensions were incubated 10 minutes with a blocking mixture of mouse, rat and calf serum containing anti-CD16/32 blocking antibodies (Clone 2.4G2, Bioxcell, Cat: BE0307). Cells were washed with PBS and incubated with a mixture of flow cytometry monoclonal antibodies per 20 minutes. In order to detect intracellular epitopes cells were fixed and permeabilized using the BD CytoFix/Cytoperm kit (BD, Cat: 554714) following the manufacturer protocol. Cells were resuspended in PBS and acquired using the FACSymphony. Flow cytometry data was compensated and down sampled to 5000 events using FlowJo. Data from down-sampled FCS files was then clustered using Cytofkit and FlowSom restricted to 20 clusters. Clusters were then visualized using Uniform Manifold Approximation and Projection (UMAP). All data was analyzed using R version 3.6.2. Antibodies for flow cytometry were purchased from Biolegend, BD and ThermoFisher Scientific (eBioscience): CD103-UV395 (Clone M290), CD24-BUV496 (Clone M1/69), Ly6G-BUV563 (Clone IA8), CD11b-BUV661 (Clone M1/70), CD19-BUV737 (Clone 1D3), CD3-BUV737 (Clone 7A2), CD45-BUV805 (Clone HI30), CD68-BV421 (Clone FA-11), GITRL-BV510 (Clone MIH44), Ly6C (Clone AL-21), CD206-BV650 (Clone C068C2), MHCII-BV711 (Clone M5-114-15-2), CD11C-BV786 (Clone N418), CD86-FITC (Clone GL1), 41BBL-BB700 (Clone TKS-1), ICOSL-PE (HK5.3), PDL1-PECF590 (Clone 10F.9G2), OX40L-PeCy7 (Clone RM134L), SIRPα-APC (Clone P84), F4/80-AF700 (Clone BM8).
For analysis of intra-tumoral immune cell populations, GFP + tumors were dissected from brains and placed in 1 mL RPMI in a 60 mm cell culture dish and mechanically dissociated using a sterile scalpel. 2 mL of DMEM supplemented with 2 mg/ml collagenase D and 10 μg/mL DNase 1 was added to the tumor sample which was further dissociated by pipetting before transfer to a 50 mL Falcon tube and incubation at 37°C, with shaking at 200 rpm for 30 min. Following enzymatic digestion, 10 mL of PBS was added to the tumor cell suspension before centrifugation. All centrifugation steps were carried out at 500 rcf. for 5 min at 4°C unless otherwise stated. The supernatant was discarded, and pelleted tumor cells were resuspended in 10 mL of cold PBS and passed through a cell strainer into a fresh 50 mL Falcon tube, with a further 5 mL cold PBS used to encourage as many remaining cells as possible to pass through the strainer. Cells were then centrifuged and resuspended in 5 mL of 1X Red Blood Cell Lysis Buffer, incubated at room temperature for 5 min and pelleted again. The pellet was then resuspended in 10 mL MACs Buffer (1X HBSS + 2% FBS) and repelleted before decanting MACs Buffer.
To remove myelin, cells were resuspended in remaining residual MACs Buffer and 20 μl of myelin magnetic beads were added to the cell suspension before a 15 min incubation at 4°C. A further 5 mL of MACs Buffer was added to the cells before centrifugation and resuspension in 1 mL of MACs Buffer before transfer to a 15 mL Falcon tube and myelin removal using a MACs machine. Following this, cells were washed with 5 mL PBS, centrifuged and resuspended in a final volume of 200 μl before transfer of each sample to an individual well of a 96-well plate. For controls (unstained/viability only), two 20 μl aliquots were removed from each tumor cell sample and placed in independent wells. The plates were sealed (to prevent cross-contamination) and centrifuged. With exception of the sample to be used as an unstained control, cell pellets were resuspended in 100 μl of a 1:1000 dilution of Zombie NIR in PBS as a viability stain (unstained sample resuspended in PBS only) and incubated for 20 min at 4°C. Plates were again sealed and centrifuged followed by two washes of samples in FACS buffer. Pelleted samples were then blocked (50 μl of 1:200 CD16/32 in FACS buffer) at 4°C for 15 min in the dark. Cells were then stained by adding 50 μl of primary antibody mix to the pre-existing Fc block solution and incubated for 30 min at 4°C in the dark before pelleting and washing in twice in FACS buffer. Samples being stained for myeloid cell populations were then resuspended in FACS buffer and transferred to FACS tubes for analysis. Samples being stained for T cell population markers were permeabilized overnight in fixation/permeabilization buffer at 4°C and washed twice the following morning in 1X permeabilization buffer before incubation in 100 μl of anti-FOXP3 for 30 min at 4°C in the dark. Finally, these samples were pelleted, washed twice in FACS buffer and resuspended in FACS buffer and transferred to FACS tubes for analysis. To generate tSNE maps, Flow cytometry data was randomly down sampled to 50,000 events per tumor for three independent tumors and the resulting data concatenated into a single dataset using FlowJo. Data from the resulting fcs file was then clustered using the tSNE plugin in FlowJo with the following settings: Auto(opt-SNE), Iterations: 1000, KNN algorithm: Exact (vantage point tree), gradient algorithm: Flt-SNE. To identify clusters on the tSNE map, concatenated data was subject to conventional gating as detailed in Figure S3 and applied to the tSNE map using FlowJo layout editor. All data was analyzed using FlowJo version 10 and R version 4.0.2.
Antibodies for flow cytometry were purchased from Biolegend, BD and ThermoFisher Scientific (eBioscience). CD45-AF700 (Clone 30-F11). For the T cell Panel: CD3-BV711 (Clone 17A2), CD8-BUV737 (Clone 53-67), CD4-BV510 (Clone GK1.5), TIM3-PE/Dazzle 594 (Clone B8.2C12), CD62L-BUV395 (Clone MEL-14), CD44-BV650 (Clone 1MF), ST2-APC (Clone RMST2-2), γδTCR-PerCP/Cy5.5 (CloneGL3), FOXP3-eFlour 450 (CloneFJK.16 s), LAG3-PE-Cyanine7 (Clone eBioC9B7W), PD1-BV605 (Clone 29F.1A12), NK1.1-PE (Clone PK136). For the myeloid cell panel: F4/80-BV421 (Clone BM8), CD11b-BV510 (Clone M1/70), Ly6G-BUV395 (Clone 1A8), CD31-BV605 (Clone 390), PDL1-BV711 (Clone 10F.9G2), Ly6C-PerCP/Cy5.5 (Clone HK1.4), MMR-PE (Clone C068C2), CD11c-PE/Dazzele (Clone N418), CD86-PE/Cy7 (Clone GL-1), PDL2-APC (Clone TY25), CD140a-BV650 (Clone APA5).
To isolate immune cells from tumors for co-culture experiments, GFP + tumors were dissected from brains, placed in bijou tube with 1 mL accutase and mechanically dissociated with a sterile blade, pipetting and disruption with the IKA Ultra TurraxTM Tube Drive followed by incubation at 37°C for 30min. Next, 6ml of wash media was added to the tumor cell suspension before centrifugation (all centrifugation steps were carried out at 500 rcf. for 5 min at 4°C unless otherwise stated), the supernatant was discarded, and cells were incubated for 10 minutes at room temperature in RBC lysis buffer (abcam). After centrifugation, the tumor cell suspension was washed once with ice cold PBS + 0.1% BSA and then filtered through a 70 μm cell strainer (Falcon). Tumor suspension was resuspended in cold PBS + 0.1% BSA and Fc block was added at a concentration of 1:100 for 5 minutes at room temperature. The suspension was washed once, before resuspension in 100 μL PBS + 0.1% BSA and suitable antibodies for staining and incubated for 30 minutes, at 4°C in the dark. Tumor suspension was then washed twice and resuspended in FACS buffer for sorting. Antibodies used were: CD45 (1:200, Biolegend), F4/80 (1:100, eBioscience) and CD3 (1:200, Biolegend).
Forward-Phase Protein Arrays (FPPA)
Media was conditioned by cells for 48 hours. Antibody microarrays were printed in-house using an Aushon BioSystems’ 2470 array printing platform. Microarrays were blocked for 1 hour with SuperG™ Blocking Buffer (Grace Bio Labs) at room temperature on a rocker. Media from samples were centrifuged at 1000 rcf. for 5 minutes at 4°C. Supernatants were added to microarrays for 12 hours at 4°C. Microarrays were washed three times for 5 minutes in TBS-T and blocked for 10 minutes with SuperG™ Blocking Buffer at room temperature on an orbital shaker, then washed again three times for 5 minutes in TBS-T. Detection antibodies (1:500 antibody diluted in 5% BSA/PBST, 1% SuperG™ Blocking Buffer) mixtures were made in plates and 2 μL of each antibody was applied to each well of the microarrays. Microarrays were clamped and 50 μL of each antibody was added to corresponding microarray wells. Microarrays were incubated for 1 hour on a flat surface. Clamps were removed and microarrays were washed three times for 5 minutes in TBS-T. Microarrays were then blocked for 10 minutes with SuperG™ Blocking Buffer at room temperature on a rocker and again washed three times for 5 minutes in PBS-T. 3 mL of IRDye® 800CW Streptavidin (LI-COR Biosciences) diluted 1 in 5000 in PBST supplemented with 5% BSA, 1% SuperG™ Blocking Buffer. Microarrays were covered and incubated on a rocker at room temperature for 45 minutes then washed for 5 minutes, three times in PBS-T followed by three 5 min PBS washes and then washed with distilled water. Microarrays were dried then scanned on the InnoScan 710 high resolution Microarray scanner (Innopsys Life Sciences). Data was normalized for protein concentration and background fluorescence in Microsoft Excel. Graphs were calculated using Prism (Graphpad).
RNA-seq sample preparation
All samples were run in triplicate. RNA was extracted from cells using the QIAGEN RNeasy kit.
For mouse samples, sample concentration and quality was assessed using the Aligent Tapestation and Nanodrop, only samples with a RIN value of 9.6 or above were used. Library prep and mRNA sequencing of mouse samples was performed by University of Edinburgh Clinical Research Facility Genetics Core. Further quality control was performed by the facility using the Qubit 2.0 Fluorometer and the Qubit RNA BR assay. Library prep was performed with 500ng of starting material using the Illumina TruSeq Stranded mRNA Sample Preparation kit. Briefly, poly-A containing mRNA molecules were purified using poly-T oligo attached magnetic beads. These were fragmented, reverse transcribed and further purified to separate blunted dscDNA for adaptor ligation. Library amplification of 12 cycles was used before purification with AMPure XP beads and quantification with the Qubit dsDNA HS assay and QC using the Aligent Bioanalyser. Sequencing was performed using the Illumina NextSeq 500/550 High-Output v2 kit on the NextSeq 550 platform. 48 libraries were pooledin equimolar amounts and run across 3 High-Output Flow Cells.
For human samples, RNA samples were run on Bioanalyzer RNA 6000 nano chips to determine RIN values and sample concentrations were measured using Qubit RNA BR assay and 200 ng of starting material was used. RNaseq libraries were prepared using the KAPA mRNA Hyper prep kit with KAPA SeqCap Adapters. Fragmentation was done at 94C for 6 minutes and library amplification of 12 cycles. Each library was quantified using Qubit dsDNA HS assay and average fragment size was determined using Bioanalyzer DNA 1000 or DNA HS. Molarity of each library was calculated using the Qubit and BioA results and then normalized to ∼10 nM and pooled, 24 libraries per pool. Each pool was then quantified using Qubit dsDNA HS assay and average fragment size determined using Bioanalyzer DNA HS. Pooled library molarity was determined using Qubit and BioA. The libraries were sequenced on Illumina HiSeq 2500 instrument. Dilute and denature was done according to manufacturer’s instructions for “Dilute and Denature Libraries for cBot Clustering,” Standard normalization method. Each 24-library pool was run on two lanes of a HiSeq High Output flow cell. All human GSCs were processed at passage 3 from derivation.
Reduced Representation Bisulphite Sequencing (RRBS) Sample preparation
All samples were run in triplicate. DNA was extracted from cells using the QIAGEN DNeasy Blood and Tissue kit and sample concentration and quality was assessed using the Nanodrop. Samples were confirmed to have appropriate quality by ensuring 260/280 values were 1.84-1.96 and 260/230 values of 1.93-2.21. RRBS was performed by the University of Edinburgh Clinical Research Facility Genetics Core. 200ng of each sample was processed with the Ovation RRBS Methyl-Seq System 1- 16 (Tecan Genomics Inc, #0353). 0.5ng of unmethylated λDNA (New England Biolabs, #N3013S) was spiked into each DNA sample to allow assessment of the bisulfite-conversion reaction efficiency. DNA was digested using the methylation-insensitive restriction enzyme, MspI, and fragments were directly ligated to cohesive-ended adapters without the requirement for blunting or A-tailing. Bisulfite-conversion was performed with the Epitect Fast Bisulfite Conversion Kit. Bisulfite- treated adaptor-ligated fragments were then amplified by PCR (9 cycles) before purification using Agencourt RNAClean® XP beads. RRBS libraries were quantified using the Qubit 2.0 fluorometer and the Qubit dsDNA HS assay and were assessed for size and quality using the Agilent Bioanalyser with the DNA HS Kit. Sequencing was performed using the NextSeq 500/550 High-Output v2.5 Kit on the NextSeq 550 platform. Libraries were combined in three equimolar pools of five and run on three high output flow cells, with ∼1% PhiX Control library
Whole genome sequencing (WGS) sample preparation
Genomic DNA (gDNA) was extracted from cells using the QIAGEN DNeasy Blood and Tissue kit and submitted to Edinburgh Genomics for processing. Genomic DNA was quantified using Quant-iT Picogreen reagent, Lambda Standard DNA and a Molecular Devices, Spectramax XPS Gemini plate reader. Quality of gDNA was evaluated using an AATI Fragment Analyzer and the Standard Sensitivity Genomic DNA Analysis Kit. Genomic DNA samples > 1000ng and a quality score > 5 pass sample QC. Genomic DNA samples were then pre-normalized to fall within 20-100ng/μL concentration range required for Illumina SeqLab TruSeq PCR Free library preparation method using the Hamilton MicroLab STAR. Libraries were prepared using Illumina SeqLab specific TruSeq PCR-Free High Throughput library preparation kits in conjunction with the Hamilton MicroLab STAR and Clarity LIMS X Edition according to standard protocols. Genomic DNA was sheared to a 450bp mean insert size using a Covaris LE220 focused-ultrasonicator. Inserts were blunt ended, A-tailed, size selected and had TruSeq adapters ligated onto the ends. Insert size for each library was evaluated using the Caliper GX Touch with a HT DNA 1k/12K/HI SENS LabChip and HT DNA HI SENS Reagent Kit to ensure that mean fragment sizes fell between 300 bp and 800 bp. Library concentration was normalized to 1.5nM before being denatured for clustering and sequencing at 300pM using a Hamilton MicroLab STAR with Genologics Clarity LIMS X (4.2) Edition. Libraries were clustered onto a HiSeqX Flow cell v2.5 on cBot2s and the clustered flow cell was transferred to a HiSeqX for sequencing using a HiSeqX Ten Reagent kit v2.5.
Bioinformatics Analysis
RNA-seq data processing and differential expression
RNA sequencing reads were trimmed with TrimGalore (version 0.5.0) and aligned to a reference genome using the pseudo aligner Kallisto with default parameters (version 0.44.0) (Martin, 2011; Bray et al., 2016). For mouse and human samples mm10, and hg38 were used respectively. Read counts were summarized via the R package Tximport (version 1.8.0) (Soneson et al., 2015) and subsequent normalization was completed via DESeq2 (version 1.27.32) (Love et al., 2014). Reported normalized read counts, unless otherwise stated, are as regularised-logarithm transformation (rlog) (Love et al., 2014). Differential expression was completed using DESeq2 with log2 fold change shrinkage (Love et al., 2014). Genes with less than 1 read count across samples were pre-filtered. Multiple testing was accounted for using the Benjamini-Hochberg procedure to calculate false discovery rates (FDRs) (Benjamini and Hochberg, 1995). Principal component analysis (PCA) was completed on the top 500 genes with greatest variance across samples. Where heatmaps of normalized expression are clustered this was completed using Ward’s method and Euclidean distance (Murtagh and Legendre, 2014).
RNA-seq - Identifying genes specific to tumor derived mutants
Genes specific to NPE tumor derived (NPE-BL6-TD and NPE-IE), and NF1 mutant samples (NF1, NP, NPE, NPE-BL6-TD, NPE-NSG-TD, NPE-IE) were identified via the Distal binarization approach as previously described (Corces et al., 2018). In brief an intra-group median and inter-quartile range (IQR) were calculated for each gene. For each gene, groups were then ranked by median expression across replicates with BL6 WT samples as a baseline. For any gene where the median expression of a group was higher than 1.5 times the IQR than the group preceding it in rank, this gene was labeled specific to this sample group. All samples ranked higher than this group based on gene expression were also labeled as gene specific. Genes were further filtered such that only specific genes that were also differentially expressed (FDR < 0.05, LFC > 1) between relevant comparisons (NPE tumor derived samples Vs. All others, and NF1 mutant samples Vs. All others) were retained (Love et al., 2014).
RNA-seq - Defining human subtypes
Genes with low expression (< 10 counts across all groups, with < 1 count per sample) and low variability of counts per million reads (CPM) across GSC samples, as calculated by DESeq2, were filtered (SD < 5 across groups, and coefficient of variation < 0.25). A final 9291 genes were subsequently used for downstream analysis. The NMF package in R (version 0.22.0) was used to perform the brunet implementation of non-negative matrix factorisation using default parameters (Brunet et al., 2004; Gaujoux and Seoighe, 2010). The highest cophenetic score was used as a criterion to obtain the optimal number of latent factors (Subtypes: S1 Non-MESImm, S2 MESImm). Two distinct subtypes were consistently obtained when running NMF with different algorithms and returned the most stable consensus clustering when compared with other rank numbers (Lee and Seung, 2001; Brunet et al., 2004; Pascual-Montano et al., 2006). To extract gene features for each subtype the most specific genes were selected (S1 Non- MESImm: 589, S2 MESImm: 235 genes) with the Kim and Park method as implemented in the NMF package (Kim and Park, 2007). Subtype enrichment scores were derived as the column normalized S1/S2 score (%) for each sample from the NMF coefficient matrix. For downstream analysis subtype labels (S1 Non-MESImm, or S2 MESImm) were applied to each sample as the subtype with the highest enrichment score.
RNA-seq - Overrepresentation and gene set enrichment analysis
R packages clusterprofiler, (version 3.15.4) and ReactomePA (version 1.31.0) were used to find enriched gene sets for genes specific to tumor derived mouse samples, and human subtypes S1 Non-MESImm, or S2 MESImm (Yu et al., 2012; Yu and He, 2016). Where gene ratio is reported this is the number of genes in each pathway or GO term enriched in a gene list of interest (e.g., S1 or S2) divided by the total number of genes in the gene list of interest. Gene set enrichment analysis (GSEA) was completed for differentially expressed gene sets via the Broad institute’s GSEA Preranked tool from the GSEA software package 3.0 (Subramanian et al., 2005). For human samples the Gene ontology (GO) database for Biological process, Cellular component and Molecular process, was utilized as a gene set database (Harris et al., 2004). For mouse samples an equivalent GO database was derived (Lai et al., 2016a). Single sample gene set enrichment analysis (ssGSEA), with simplicity scores, was completed using a recent implementation of the ssGSEA algorithm by Wang et al., with selected immune GO terms, and glioma subtype gene sets (Mesenchymal, Classical, and Proneural) (Barbie et al., 2009; Wang et al., 2017).
Reduced representation bisulfite sequencing
Reads were trimmed with TrimGalore (version 0.5.0). NuGen diversity adaptors were removed with a Python script supplied by NuGen “trimRRBSdiversityAdaptCustomers.py” (version 1.11 https://github.com/nugentechnologies/NuMetRRBS) (Martin, 2011). Reads were then aligned to the mm10 in silico bisulfite transformed genome with the bisulphite sequencing aligner Bismark (version 0.16.3 with Bowtie version 2.2.6 using parameters: -N 0 -L 20) (Krueger and Andrews, 2011). PCR duplicates were then removed using a Python script from NuGen “nudup.py” (version 2.3 https://github.com/tecangenomics/nudup). Methylation estimation was completed using the Bismark methylation extractor ignoring the first two bases of read 2 to minimize common methylation call bias at these sites. Initial visualization with PCA was completed on the top 25% of CpGs with the highest variance of percentage methylation. No batch effect was observed following PCA. Identification of differentially methylated loci (DMLs) was completed with the R package DSS (version 2.36.0) in multifactor mode to account for RRBS batch (Park and Wu, 2016). FDR was used to account for multiple testing (Benjamini and Hochberg, 1995). Differentially methylated regions (DMRs) were called by merging adjacent statistically significant DMLs within 50bp of one another, and filtering for DMRs with a minimum length of 50bp and with greater than 3 significant CpG sites. CpG island tracks were retrieved from the UCSC table browser service as a BED file (Karolchik et al., 2004). Percentage methylation, DMRs, DMLs, and CpG island tracks were visualized using the IGV genome browser (Thorvaldsdóttir et al., 2013). The annotation of DMRs was completed using the R package ChIPpeakAnno (version 3.22.0) – where DMRs were attributed to nearest gene promoter/transcription start site (TSS) (Zhu et al., 2010). TSS coordinates were defined as ± 2kb of sequence surrounding the start of the first exon. Promoter rank plots were filtered to include DMRs overlapping promoter regions and associated hypomethylated immune genes were highlighted in blue (> 50% methylation loss, overlapping immune and interferon GO terms: GO:0006955, GO:0034341, GO:0034340, GO:0035456, GO:0035455, and GO:0019882). Density heatmaps were produced using the ‘densityHeatmap’ function from the R package ComplexHeatmap (version 2.4.2), and clustering was completed using Euclidean distance and Ward’s method (Murtagh and Legendre, 2014; Gu, Eils and Schlesner, 2016).
Infinium® MethylationEPIC BeadChip
Pre-processing and normalization from idat files to beta values was completed via the R package minfi (version 1.34.0) with combined functional and Noob normalization (Triche et al., 2013; Fortin et al., 2014, 2017). Poor quality probes, and those that could not be mapped to the hg38 genome, were filtered as previously described (Maksimovic et al., 2016; Zhou et al., 2017). Classification of human EPIC array samples were completed via the online portal https://www.molecularneuropathology.org/mnp. IDH mutants were removed prior to downstream methylation analysis (Capper et al., 2018). DMLs and DMRs were identified via the R Package DMRCate (version 2.2.3) using limma (version 3.43.11) (Peters et al., 2015; Ritchie et al., 2015). Annotation of DMRs was completed via the R package ChIPpeakAnno by assigning DMRs to nearest TSS (+/− 2kb) (Zhu et al., 2010). Density heatmaps were produced using the ‘densityHeatmap’ function from the R package ‘ComplexHeatmap’, and clustering was completed using Euclidean distance with Ward’s method (Murtagh and Legendre, 2014; Gu et al., 2016).
Single-cell RNA-seq
Smart-seq2 single cell RNA-seq normalized counts (Transcripts per million, TPM) were downloaded from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) (GEO accession: GSM3828672) for 7930 glioma cells, from 28 separate tumors (Neftel et al., 2019). Contaminating cells (Macrophages, T Cells, and Oligodendrocytes) were filtered as previously described (Neftel et al., 2019). Paediatric samples were then filtered prior to downstream analysis. PCA was completed on the top 2000 most variable genes. As in the bulk RNA-Seq analysis ssGSEA was completed on each cell for glioma transcriptional subtypes, and immune GO terms. Where subtype labels are applied to individual cells this was completed using ssGSEA - where an S2 MESImm subtype was applied to cells with a MESImm p value < 0.05, otherwise a S1 Non-MESImm label was applied. Differential expression between MESImm and Non- MESImm labeled cells was completed using the ‘findMarkers’ function from the R package scran (version 1.16.0) using default parameters (Lun et al., 2016).
Whole genome sequencing
Whole genome sequencing (WGS) of BL6 WT, NPE and NPE-IE lines was completed using the best practises WGS paired cancer normal pipeline bcbio (bcbio-nextgen version 1.2.3) (https://github.com/bcbio/bcbio-nextgen). Adaptors were trimmed with the read trimmer Atropos (version 1.1.25) (Didion et al., 2017), a variant of Cutadapt, and aligned to the mm10 genome using the aligner Bowtie2 (version 2.3.5.1) (Langmead and Salzberg, 2012). An additional human EGFRvIII contig was included to capture human reads from the EGFRvIII transgene present in NPE and NPE-IE lines. PCR duplicates were marked with samblaster (version 0.1.25) (Faust and Hall, 2014), and GATK base quality score recalibration was completed on the aligned BAM files (gatk version 3.8) (DePristo et al., 2011). Single nucleotide variant (SNV) calling was completed using an ensemble-based approach using VarDict (version 1.6), Strelka2 (version 2.9.10), and Mutect2 (gatk version 3.8) for somatic calling, and freebayes (version 1.1.0.46), GATK HaplotypeCaller (gatk version 3.8), and Strelka2 (version 2.9.10) for germline calling (DePristo et al., 2011; Garrison and Marth, 2012; Cibulskis et al., 2013; Lai et al., 2016b; Kim et al., 2018). Germline calls were used to filter final somatic calls. Variants were combined and included in the final call if present in 2 out of 3 variant callers (bcbio min_allele_fraction: 2). Variant calls were targeted at exonic regions as defined by the R package TxDb.Mmusculus.UCSC.mm10.knownGene (version 3.4.4). Structural variants (SVs) were called with Manta (version 1.6.0) (Chen et al., 2016). Structural variants that did not pass default Manta filters were removed. Break-end (BND) calls for NPE and NPE-IE samples were either imprecise and without split read evidence or had less than 5 split reads for the alternative allele, and hence were not considered for further analysis. Copy number variants (CNVs) were assessed with CNVkit (version 0.9.6) (Talevich et al., 2016). To accommodate parameter flexibility, CNVKit was run outside of bcbio using the “wgs” option, targeted at transcript regions as defined by the R package TxDb.Mmusculus.UCSC.mm10.knownGene. Absolute copy number calls were completed with the “clonal” option to account for known high purity of cell line samples and copy number regions were filtered using confidence interval filtering. SNV and SVs were annotated using the R packages VariantAnnotation (version 1.34.0), TxDb.Mmusculus.UCSC.mm10.knownGene, and BSgenome.Mmusculus.UCSC.mm10.masked (version 1.3.99) (Obenchain et al., 2014). Following annotation SNV calls were filtered again to remove those within noncoding regions, those that were synonymous, as well as those with an allele frequency (AF) < 0.25. Final, filtered, SNV, SV, and CNV calls were overlapped with known cancer driver genes as defined by the COSMIC gene census (https://cancer.sanger.ac.uk/census), converted to mouse homologs via MGI batch query (http://www.informatics.jax.org/batch) (Sondka et al., 2018; Bult et al., 2019).
Quantification and statistical analysis
Statistical analysis was performed on GraphPad or using R (version 4.0.0). Numerical data presented in charts is displayed as mean ± SEM and analyzed using unpaired t tests unless otherwise stated in corresponding legends of the article. Statistical analysis of survival data was performed on GraphPad using the Log Rank (Mantel-Cox) test. Significance is noted in figures or figure legends; p values are denoted as follows: ∗ < 0.05, ∗∗ < 0.01, ∗∗∗ < 0.001, ∗∗∗∗ < 0.0001, n.s. > 0.05.
Acknowledgments
The authors gratefully acknowledge the expertise and assistance of several core facilities at the University of Edinburgh, including the BVS animal facility (in particular, Jacek Mendrychowski and James Todd), the flow and genomic cytometry facility (particularly Fiona Rossi), the Tissue Culture facility at the Centre for Regenerative Medicine, the Clinical Research Facility Genetics core, the Host and Tumour Profiling Unit for RPPA, and UCL Cancer Institute genomics core (particularly Pawan Dhami and Heli Vaikkinen). We thank our colleague Pooran Dewari for his input on CRISPR engineering design strategies and the Elaine Emmerson laboratory at the Centre for Regenerative Medicine for providing reagents. Schematics presented in this manuscript were created with BioRender. Patient-derived models and associated data were generated by the Cancer Research UK Centre Accelerator Award (A21922). S.M.P. is a Cancer Research UK Senior Research Fellow (A17368). E. Gangoso was supported by a Postdoctoral Fellowship from Fundación Ramón Areces (Spain). L.B. and F.G.C. were supported by the Cancer Research UK Brain Tumour Award (A28592). S.R. was supported by Erasmus programme KA103 and a Student mobility HE-SMP-P traineeship grant. G.M. was supported by the Cancer Research UK Centre Accelerator Award (A21922). M.A.M.-T. was supported by a Postdoctoral EMBO Long-Term Fellowship (EMBO ALTF 439-2014). C.A.K. was supported by a Medical Research Council cross-disciplinary fellowship (MC_UU_00009/2). A.S. is supported by Cancer Research UK (C39669/A25919). S. Brandner is supported by the Department of Health’s NIHR Biomedical Research Centre’s funding scheme.
Author contributions
Conceptualization, E. Gangoso, L.B., B.S., A.S., and S.M.P.; Methodology, L.B., E. Gangoso, B.S., N.A., and S.M.P.; Formal Analysis, B.S., C.-A.K., L.C., and J.H.; Investigation, L.B., E. Gangoso, S.R., F.G.C., N.M., A.B., E. Güç, K.M.F., N.A., V.G., C.B., M.A.M.-T., and I.F.S.; Resources, G.M., P.M.B., P.B., S. Beck, S. Brandner., S.P., S.A.Q., and J.W.P.; Data Curation, B.S. and C.-A.K.; Writing – Original Draft, L.B., E. Gangoso, and S.M.P.; Writing – Review and Editing, L.B., B.S., and S.M.P.; Visualization, L.B., B.S., and S.M.P.; Supervision, J.W.P., S.A.Q., D.S., M.F., A.S., and S.M.P.; Project Administration, S.M.P.; Funding Acquisition, J.W.P., S.A.Q., D.S., M.F., A.S., and S.M.P.
Declaration of interests
S.M.P is a co-founder and shareholder of Cellinta., a biotechnology start-up that is developing cancer therapeutics, including for glioblastoma, and acts as an advisor to the company. J.W.P is a co-founder and shareholder of Macomics. S.A.Q. is co-founder and shareholder and Chief Scientific Officer for Achilles Therapeutics. The other authors declare no competing interests.
Published: April 14, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.03.023.
Supplemental information
References
- Alcantara Llaguno S.R., Xie X., Parada L.F. Cell of Origin and Cancer Stem Cells in Tumor Suppressor Mouse Models of Glioblastoma. Cold Spring Harb. Symp. Quant. Biol. 2016;81:31–36. doi: 10.1101/sqb.2016.81.030973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldape K., Brindle K.M., Chesler L., Chopra R., Gajjar A., Gilbert M.R., Gottardo N., Gutmann D.H., Hargrave D., Holland E.C. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019;16:509–520. doi: 10.1038/s41571-019-0177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie D.A., Tamayo P., Boehm J.S., Kim S.Y., Moody S.E., Dunn I.F., Schinzel A.C., Sandy P., Meylan E., Scholl C. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benci J.L., Xu B., Qiu Y., Wu T.J., Dada H., Twyman-Saint Victor C., Cucolo L., Lee D.S.M., Pauken K.E., Huang A.C. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell. 2016;167:1540–1554.e12. doi: 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. A Stat. Soc. 1995;57:289–300. [Google Scholar]
- Bhat K.P.L., Balasubramaniyan V., Vaillant B., Ezhilarasan R., Hummelink K., Hollingsworth F., Wani K., Heathcock L., James J.D., Goodman L.D. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24:331–346. doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Brennan C.W., Verhaak R.G.W., McKenna A., Campos B., Noushmehr H., Salama S.R., Zheng S., Chakravarty D., Sanborn J.Z., Berman S.H., TCGA Research Network The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressan R.B., Dewari P.S., Kalantzaki M., Gangoso E., Matjusaitis M., Garcia-Diaz C., Blin C., Grant V., Bulstrode H., Gogolok S. Efficient CRISPR/Cas9-assisted gene targeting enables rapid and precise genetic manipulation of mammalian neural stem cells. Development. 2017;144:635–648. doi: 10.1242/dev.140855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet J.-P., Tamayo P., Golub T.R., Mesirov J.P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. USA. 2004;101:4164–4169. doi: 10.1073/pnas.0308531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult C.J., Blake J.A., Smith C.L., Kadin J.A., Richardson J.E., Mouse Genome Database Group Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019;47(D1):D801–D806. doi: 10.1093/nar/gky1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burugu S., Dancsok A.R., Nielsen T.O. Emerging targets in cancer immunotherapy. Semin. Cancer Biol. 2018;52:39–52. doi: 10.1016/j.semcancer.2017.10.001. [DOI] [PubMed] [Google Scholar]
- Byron A. Clustering and Network Analysis of Reverse Phase Protein Array Data. Methods Mol. Biol. 2017;1606:171–191. doi: 10.1007/978-1-4939-6990-6_12. [DOI] [PubMed] [Google Scholar]
- Capper D., Jones D.T.W., Sill M., Hovestadt V., Schrimpf D., Sturm D., Koelsche C., Sahm F., Chavez L., Reuss D.E. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555:469–474. doi: 10.1038/nature26000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro M.S., Lim W.K., Alvarez M.J., Bollo R.J., Zhao X., Snyder E.Y., Sulman E.P., Anne S.L., Doetsch F., Colman H. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Schulz-Trieglaff O., Shaw R., Barnes B., Schlesinger F., Källberg M., Cox A.J., Kruglyak S., Saunders C.T. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–1222. doi: 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- Chu V.T., Weber T., Wefers B., Wurst W., Sander S., Rajewsky K., Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti L., Pollard S.M., Gorba T., Reitano E., Toselli M., Biella G., Sun Y., Sanzone S., Ying Q.L., Cattaneo E., Smith A. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 2005;3:e283. doi: 10.1371/journal.pbio.0030283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contursi C., Wang I.M., Gabriele L., Gadina M., O’Shea J., Morse H.C., 3rd, Ozato K. IFN consensus sequence binding protein potentiates STAT1-dependent activation of IFNgamma-responsive promoters in macrophages. Proc. Natl. Acad. Sci. USA. 2000;97:91–96. doi: 10.1073/pnas.97.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces M.R., Granja J.M., Shams S., Louie B.H., Seoane J.A., Zhou W., Silva T.C., Groeneveld C., Wong C.K., Cho S.W. The chromatin accessibility landscape of primary human cancers. Science. 2018;362:eaav1898. doi: 10.1126/science.aav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didion J.P., Martin M., Collins F.S. Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ. 2017;5:e3720. doi: 10.7717/peerj.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driggers P.H., Ennist D.L., Gleason S.L., Mak W.H., Marks M.S., Levi B.Z., Flanagan J.R., Appella E., Ozato K. An interferon gamma-regulated protein that binds the interferon-inducible enhancer element of major histocompatibility complex class I genes. Proc. Natl. Acad. Sci. USA. 1990;87:3743–3747. doi: 10.1073/pnas.87.10.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn G.P., Koebel C.M., Schreiber R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006;6:836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- Faust G.G., Hall I.M. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics. 2014;30:2503–2505. doi: 10.1093/bioinformatics/btu314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin J.-P., Labbe A., Lemire M., Zanke B.W., Hudson T.J., Fertig E.J., Greenwood C.M., Hansen K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15:503. doi: 10.1186/s13059-014-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin J.-P., Triche T.J., Jr., Hansen K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017;33:558–560. doi: 10.1093/bioinformatics/btw691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E., Marth G. Haplotype-based variant detection from short-read sequencing. arXiv. 2012 arXiv:1207.3907. [Google Scholar]
- Gaujoux R., Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics. 2010;11:367. doi: 10.1186/1471-2105-11-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami S., Walle T., Cornish A.E., Basu S., Anandhan S., Fernandez I., Vence L., Blando J., Zhao H., Yadav S.S. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020;26:39–46. doi: 10.1038/s41591-019-0694-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Guo X., Pan Y., Xiong M., Sanapala S., Anastasaki C., Cobb O., Dahiya S., Gutmann D.H. Midkine activation of CD8 + T cells establishes a neuron-immune-cancer axis responsible for low-grade glioma growth. Nature Communications. 2020;11 doi: 10.1038/s41467-020-15770-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M.A., Clark J., Ireland A., Lomax J., Ashburner M., Foulger R., Eilbeck K., Lewis S., Marshall B., Mungall C., Gene Ontology Consortium The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra M.E., Bornes L., Dijkgraaf F.E., Philips D., Pardieck I.N., Toebes M., Thommen D.S., van Rheenen J., Schumacher T.N.M. Long-distance modulation of bystander tumor cells by CD8+ T cell-secreted IFNγ. Nat. Can. 2020;1:291–301. doi: 10.1038/s43018-020-0036-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtschke T., Löhler J., Kanno Y., Fehr T., Giese N., Rosenbauer F., Lou J., Knobeloch K.P., Gabriele L., Waring J.F. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–317. doi: 10.1016/s0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Park H. Sparse non-negative matrix factorizations via alternating non-negativity-constrained least squares for microarray data analysis. Bioinformatics. 2007;23:1495–1502. doi: 10.1093/bioinformatics/btm134. [DOI] [PubMed] [Google Scholar]
- Kim S., Scheffler K., Halpern A.L., Bekritsky M.A., Noh E., Källberg M., Chen X., Kim Y., Beyter D., Krusche P., Saunders C.T. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods. 2018;15:591–594. doi: 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- Klemm F., Maas R.R., Bowman R.L., Kornete M., Soukup K., Nassiri S., Brouland J.-P., Iacobuzio-Donahue C.A., Brennan C., Tabar V. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell. 2020;181:1643–1660.e17. doi: 10.1016/j.cell.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortlever R.M., Sodir N.M., Wilson C.H., Burkhart D.L., Pellegrinet L., Brown Swigart L., Littlewood T.D., Evan G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell. 2017;171:1301–1315.e14. doi: 10.1016/j.cell.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F., Andrews S.R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L., Hennessey J., Bares V., Son E.W., Ban Y., Wang W. GSKB: A gene set database for pathway analysis in mouse. bioRxiv. 2016 doi: 10.1101/082511. [DOI] [Google Scholar]
- Lai Z., Markovets A., Ahdesmaki M., Chapman B., Hofmann O., McEwen R., Johnson J., Dougherty B., Barrett J.C., Dry J.R. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016;44:e108. doi: 10.1093/nar/gkw227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais D., Barreiro L.B., Gros P. The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J. Exp. Med. 2016;213:585–603. doi: 10.1084/jem.20151764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia J.D., Mack S.C., Mulkearns-Hubert E.E., Valentim C.L.L., Rich J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.D., Seung H.S. Proceedings of the 13th International Conference on Neural Information Processing Systems (NIPS’00) MIT Press; 2001. Algorithms for non-negative matrix factorization; pp. 535–541. [Google Scholar]
- Lee J.H., Lee J.E., Kahng J.Y., Kim S.H., Park J.S., Yoon S.J., Um J.-Y., Kim W.K., Lee J.-K., Park J. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature. 2018;560:243–247. doi: 10.1038/s41586-018-0389-3. [DOI] [PubMed] [Google Scholar]
- Lim M., Xia Y., Bettegowda C., Weller M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018;15:422–442. doi: 10.1038/s41571-018-0003-5. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun A.T.L., McCarthy D.J., Marioni J.C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res. 2016;5:2122. doi: 10.12688/f1000research.9501.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksimovic J., Phipson B., Oshlack A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Res. 2016;5:1281. doi: 10.12688/f1000research.8839.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10. [Google Scholar]
- Mi Y., Guo N., Luan J., Cheng J., Hu Z., Jiang P., Jin W., Gao X. The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front. Immunol. 2020;11:737. doi: 10.3389/fimmu.2020.00737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohme M., Neidert M.C. Tumor-Specific T Cell Activation in Malignant Brain Tumors. Front. Immunol. 2020;11:205. doi: 10.3389/fimmu.2020.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojic M., Takeda K., Hayakawa Y. The Dark Side of IFN-γ: Its Role in Promoting Cancer Immunoevasion. Int. J. Mol. Sci. 2017;19:89. doi: 10.3390/ijms19010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtagh F., Legendre P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? J. Classif. 2014;31:274–295. [Google Scholar]
- Neftel C., Laffy J., Filbin M.G., Hara T., Shore M.E., Rahme G.J., Richman A.R., Silverbush D., Shaw M.L., Hebert C.M. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178:835–849.e21. doi: 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenchain V., Lawrence M., Carey V., Gogarten S., Shannon P., Morgan M. VariantAnnotation: a Bioconductor package for exploration and annotation of genetic variants. Bioinformatics. 2014;30:2076–2078. doi: 10.1093/bioinformatics/btu168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y., Wu H. Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics. 2016;32:1446–1453. doi: 10.1093/bioinformatics/btw026. [DOI] [PubMed] [Google Scholar]
- Pascual-Montano A., Carazo J.M., Kochi K., Lehmann D., Pascual-Marqui R.D. Nonsmooth nonnegative matrix factorization (nsNMF) IEEE Trans. Pattern Anal. Mach. Intell. 2006;28:403–415. doi: 10.1109/TPAMI.2006.60. [DOI] [PubMed] [Google Scholar]
- Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters T.J., Buckley M.J., Statham A.L., Pidsley R., Samaras K., V Lord R., Clark S.J., Molloy P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6. doi: 10.1186/1756-8935-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard S.M., Yoshikawa K., Clarke I.D., Danovi D., Stricker S., Russell R., Bayani J., Head R., Lee M., Bernstein M. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Pyonteck S.M., Akkari L., Schuhmacher A.J., Bowman R.L., Sevenich L., Quail D.F., Olson O.C., Quick M.L., Huse J.T., Teijeiro V. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B.-Z., Li J., Zhang H., Kitamura T., Zhang J., Campion L.R., Kaiser E.A., Snyder L.A., Pollard J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishna S., Kwaku Dad A.B., Beloor J., Gopalappa R., Lee S.K., Kim H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014;24:1020–1027. doi: 10.1101/gr.171264.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards L.M., Whitley O.K.N., MacLeod G., Cavalli F.M.G., Coutinho F.J. Gradient of Developmental and Injury Response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat. Cancer. 2021;2:157–173. doi: 10.1038/s43018-020-00154-9. [DOI] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson F.L., Marqués-Torrejón M.-A., Morrison G.M., Pollard S.M. Experimental models and tools to tackle glioblastoma. Dis. Model. Mech. 2019;12:dmm040386. doi: 10.1242/dmm.040386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R.D., Old L.J., Smyth M.J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Sondka Z., Bamford S., Cole C.G., Ward S.A., Dunham I., Forbes S.A. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer. 2018;18:696–705. doi: 10.1038/s41568-018-0060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C., Love M.I., Robinson M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Pollard S., Conti L., Toselli M., Biella G., Parkin G., Willatt L., Falk A., Cattaneo E., Smith A. Long-term tripotent differentiation capacity of human neural stem (NS) cells in adherent culture. Mol. Cell. Neurosci. 2008;38:245–258. doi: 10.1016/j.mcn.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Talevich E., Shain A.H., Botton T., Bastian B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016;12:e1004873. doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T., Nagamura-Inoue T., Shmeltzer Z., Kuwata T., Ozato K. ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity. 2000;13:155–165. doi: 10.1016/s1074-7613(00)00016-9. [DOI] [PubMed] [Google Scholar]
- Teo K., Gómez-Cuadrado L., Tenhagen M., Byron A., Rätze M., van Amersfoort M., Renes J., Strengman E., Mandoli A., Singh A.A. E-cadherin loss induces targetable autocrine activation of growth factor signalling in lobular breast cancer. Sci. Rep. 2018;8:15454. doi: 10.1038/s41598-018-33525-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibaut R., Bost P., Milo I., Cazaux M., Lemaître F., Garcia Z., Amit I., Breart B., Cornuot C., Schwikowski B., Bousso P. Bystander IFN-γ activity promotes widespread and sustained cytokine signaling altering the tumor microenvironment. Nat. Can. 2020;1:302–314. doi: 10.1038/s43018-020-0038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triche T.J., Jr., Weisenberger D.J., Van Den Berg D., Laird P.W., Siegmund K.D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013;41:e90. doi: 10.1093/nar/gkt090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak R.G.W., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P., Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Babikir H., Müller S., Yagnik G., Shamardani K., Catalan F., Kohanbash G., Alvarado B., Di Lullo E., Kriegstein A. The Phenotypes of Proliferating Glioblastoma Cells Reside on a Single Axis of Variation. Cancer Discov. 2019;9:1708–1719. doi: 10.1158/2159-8290.CD-19-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Hu B., Hu X., Kim H., Squatrito M., Scarpace L., deCarvalho A.C., Lyu S., Li P., Li Y. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell. 2017;32:42–56.e6. doi: 10.1016/j.ccell.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., He Q.-Y. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016;12:477–479. doi: 10.1039/c5mb00663e. [DOI] [PubMed] [Google Scholar]
- Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Laird P.W., Shen H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017;45:e22. doi: 10.1093/nar/gkw967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.J., Gazin C., Lawson N.D., Pagès H., Lin S.M., Lapointe D.S., Green M.R. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics. 2010;11:237. doi: 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the mouse mRNA-seq, RRBS, and WGS datasets reported in this paper are available to download from the Gene Expression Omnibus (GEO) with the following accession codes: GSE165386, GSE165389 and GSE165390. Human RNA-seq, and EPIC array datasets will be made available for download at http://gcgr.org.uk. Additional code is available upon request.