

Abstract
Hereditary tyrosinemia type I (HT1) results from the loss of fumarylacetoacetate hydrolase (FAH) activity and can lead to lethal liver injury (LLI). Therapeutic options for HT1 remain limited. The FAH−/− pig, a well-characterized animal model of HT1, represents a promising candidate for testing novel therapeutic approaches to treat this condition. Here, we report an improved single-step method to establish a biallelic (FAH−/−) mutant porcine model using CRISPR-Cas9 and cytoplasmic microinjection. We also tested the feasibility of rescuing HT1 pigs through inactivating the 4-hydroxyphenylpyruvic acid dioxygenase (HPD) gene, which functions upstream of the pathogenic pathway, rather than by directly correcting the disease-causing gene as occurs with traditional gene therapy. Direct intracytoplasmic delivery of CRISPR-Cas9 targeting HPD before intrauterine death reprogrammed the tyrosine metabolism pathway and protected pigs against FAH deficiency-induced LLI. Characterization of the F1 generation revealed consistent liver-protective features that were germline transmissible. Furthermore, HPD ablation ameliorated oxidative stress and inflammatory responses and restored the gene profile relating to liver metabolism homeostasis. Collectively, this study not only provided a novel large animal model for exploring the pathogenesis of HT1, but also demonstrated that CRISPR-Cas9-mediated HPD ablation alleviated LLI in HT1 pigs and represents a potential therapeutic option for the treatment of HT1.
Keywords: HPD ablation, tyrosinemia type I, lethal liver injury, pig, metabolic reprogramming, oxidative stress, liver hemeostasis, CRISPR/Cas9
Graphical abstract
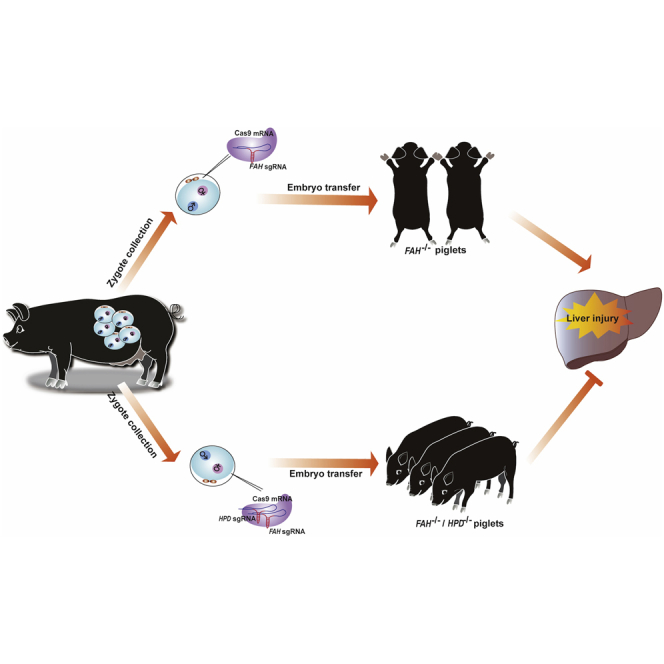
By CRISPR-mediated editing in pig embryos, Gu and colleagues provide a possibility for long-term protection against liver injury of hereditary tyrosinemia type I (HT1) by genetic blocking of 4-hydroxyphenylpyruvate acid dioxygenase HPD, a gene upstream of the pathogenic pathway, rather than by directly correcting disease-causing gene as traditional gene therapy.
Introduction
Hereditary tyrosinemia type I (HT1), an autosomal recessive hereditary disease, is caused by a deficiency of fumarylacetoacetate hydrolase (FAH), the last enzyme in the tyrosine degradation pathway.1,2 To date, approximately 100 HT1 causative mutations have been identified in the FAH gene. The morbidity of HT1 is around 1/100,000 live births worldwide, but cases appear to cluster in certain regions, such as Saguenay-Lac-St-Jean in Canada.3 During HT1 pathogenesis, tyrosine and its electrophilic byproducts, such as fumarylacetoacetate (FAA) and succinylacetone (SA), accumulate to toxic levels, which severely damages, and even kills, cells in the liver, kidneys, and other organs.4,5 The acute form of HT1 generally presents with fatal liver failure in early infancy (before 6 months of life), while cirrhosis and hepatocellular carcinoma (HCC) in early childhood are characteristic clinical presentations in the chronic phase.6, 7, 8 Accordingly, therapeutic efforts have focused on treating liver injury as it is the leading cause of death in HT1 patients.
2-(2-Nitro-4-trifluoro-methylben-zyol)-1,3 cyclohexanedione (NTBC or Nitisinone), an inhibitor of 4-hydroxyphenylpyruvate acid dioxygenase (HPD), the second enzyme in the tyrosine catabolism pathway, combined with a low tyrosine/phenylalanine diet, has been applied to rescue the HT1-associated lethal liver injury (LLI) by preventing the accumulation of toxic metabolites.9,10 However, this therapeutic strategy requires the initiation of medication at a very young age and must be continued indefinitely, which is inconvenient, costly, and not curative. In addition, some studies have already reported that NTBC cannot fully normalize the gene-expression profile of the liver of HT1 patients and the incidence of HCC in this population is still significantly greater than that of normal people.11,12 Following the advent of CRISPR technology, efforts have been directed toward genetically modifying liver cells to treat HT1. In vivo liver-targeted gene therapy based on adeno-associated vectors (AAVs) and homology-directed repair (HDR) has been successfully used to treat HT1 in rodent models.13, 14, 15 However, this procedure only repaired a fraction of the affected hepatocytes, which is inadequate for addressing the concerns regarding HCC. Moreover, high-dose intravenous vector administration has the potential for inducing severe toxicity in clinical trials, a complication that has already been observed in nonhuman primates and pigs.16
Alternatively, the phenotype of Fah-deficient mice has been successfully rescued by crossing Fah−/− and HT3 (Hpd−/−) mice, with the resulting double mutants (Fah−/−/Hpd−/−) showing normal liver histology.17 Similarly, using the CRISPR-Cas9 system, Pankowicz et al.18 knocked out the Hpd gene in adult HT1 mice, which converted lethal HT1 into a mild form of the disease (HT3) through metabolic reprogramming. Although Hpd deletion showed some therapeutic benefits in ameliorating liver injury symptoms in HT1 mouse models, whether this is accompanied by the normalization of gene expression in the liver remains unclear. In addition, the limitations of using rodents to model human diseases, including differences in physiological characteristics, metabolic function, immune responses, and organ size, have become increasingly apparent.19, 20, 21 These disadvantages make it difficult to evaluate the safety and efficacy of in-vivo gene therapy for long-term preclinical evaluation. The pig (Sus scrofa domestica) represents a more clinically-relevant model of human disease and has the potential for accelerating the pace of transforming gene editing into a therapeutic strategy to treat various human disorders.22, 23, 24 Heterozygous FAH mutant pigs have been generated through rAAV-mediated gene knockout, following which homozygote FAH−/− pigs were obtained by outbreeding of the FAH+/− pigs.25,26 Although feasible, this method is also time-consuming and not cost-effective for generating large animal models of human diseases, as pigs take at least 6 months to reach sexual maturity. Moreover, FAH−/− homozygous pigs require NTBC therapy to prevent potentially fatal liver disease in the fetus during pregnancy, and continuous medication is needed to maintain the therapeutic effect after birth.
The aim of this study was to generate FAH biallelic mutant pigs by a more efficient single-step method and investigate whether the genetic blocking of the HPD gene in a pig model can ameliorate the acute LLI phenotype associated with HT1.
Results
The single-step generation of FAH−/− pigs via embryo injection
The FAH gene encodes the enzyme FAH and is exclusively expressed in the liver and kidney (Figure 1A).27 Two single guide RNAs (sgRNAs) targeting FAH (sgRNA-Fah1 targeting exon 2 and sgRNA-Fah2 targeting exon 5) were cloned into the pGL3-U6-sgRNA-PGK-Puro vector (Figure 1B). Pig zygotes were injected with a mixture of 50 ng/μL sgRNA (25 ng/μL of each sgRNA) and 100 ng/μL Cas9 mRNA, and 180 injected embryos were transplanted into ten surrogate sows (Table 1). Three surrogate sows were confirmed to be pregnant on day 20 and kept on 2 mg/kg NTBC per day until delivery, yielding a total of four piglets (Figures 1C and 1D). Genotyping by PCR and Sanger sequencing showed that all four founder piglets harbored gene modifications in the FAH gene locus (Figure 1E). Of the four founder piglets, three (75%; founders #1, #3, and #4) displayed a complete loss of the FAH gene and one (founder #2) showed multiple allelic mutations, indicating that the piglet was mosaic (Table 1). To mimic the acute LLI phenotype associated with the HT1 disorder, we administered no NTBC to either nursing sows or piglets after birth. Surprisingly, the FAH−/− piglets experienced severe failure to thrive and died of acute liver injury soon after birth, while the FAH+/− piglet displayed a normal phenotype (Figure 1F). Liver tissue was collected from the deceased mutant piglets and the FAH protein level was detected by western blotting and immunohistochemistry. As expected, no FAH protein was detected in the liver tissue of the biallelic mutant piglets, in contrast to the abundant expression observed in wild-type (WT) piglets (Figures 1G and 1H); this indicated that the FAH gene had been inactivated due to the frameshift (short indels of various lengths) mutation resulting from the nonhomologous end-joining (NHEJ) repair system. In addition, no off-target mutations were observed at potential off-target sites in the FAH−/− pigs (Figure S1).
Figure 1.

The generation of FAH−/− pigs via direct zygote injection
(A) FAH mRNA levels were determined in the liver, heart, spleen, lung, kidney, brain, and intestine of healthy pigs (n = 6). (B) Schematic diagram of the target sequence at the FAH locus. The sgRNA sequences are underlined and highlight in red and the protospacer adjacent motif (PAM) sequences are highlighted in blue. (C) Schematic overview of the single-step generation of FAH mutant pigs. (D) Top panel: representative photograph of microinjection into the cytoplasm of one-cell-stage zygotes; bottom panel, representative photograph of newborn FAH−/− piglets. (E) Sanger sequencing of mutant sequences from four newborn piglets. The targeted sequences are underlined and shown at the top. The mutations are shown in red. Deletions and insertions are denoted as “Δ” and “+” plus the number of base pairs, respectively. (F) Kaplan-Meier survival curve for FAH−/− and FAH+/− pigs. (G) Western blotting and quantification of FAH and GAPDH protein levels in liver tissue of FAH−/− founders (#1, #3, #4). Data are presented as means ± SD. ∗∗∗p < 0.001. (H) Immunohistochemical staining for FAH in liver sections derived from FAH−/− pigs showing the absence of FAH expression. Scale bar, 200 μm (left) and 50 μm (right).
Table 1.
Summary of gene-edited pigs
| Target gene | Number of zygotes | Number of 2-cell (%)a | Transferred embryos | Number of recipients | Number of pregnancies (%)b | Number of born | Number of FAH mutant (%)c | Number of HPD mutant (%)c |
|---|---|---|---|---|---|---|---|---|
| FAH | 199 | 182 (91.5) | 180 | 10 | 3(30) | 4 | 3 (75) | – |
| FAH and HPD | 68 | 63 (92.6) | 63 | 4 | 2(50) | 6 | 5 (83.3) | 6 (100) |
| Total | 14 | 5 (35.7) | 9 | – | – | |||
Calculated from the number of zygotes.
Calculated from the number of recipients.
Calculated from the number of born.
Generation and characterization of double-mutant (FAH−/−/HPD−/−) pigs
The HPD enzyme has a role in tyrosine catabolism in most organisms and is mainly present in the liver in mammals (Figure S2A).28 Multiple sgRNAs specifically targeting HPD and FAH were designed to generate double-knockout pigs (Figures 1B and 2A). To investigate the toxicity and specificity of the sgRNAs, we first validated the sgRNAs in vitro. A mixture containing 100 ng/μL of Cas9 mRNA and 50 ng/μL of total sgRNA was injected into the cytoplasm of pig zygotes and the embryos were then cultured in vitro to the blastocyst stage. As shown in Table S1, the in vitro blastocyst formation rate of zygotes injected with multiple sgRNAs (86.67%,13/15; 80%, 12/15) and zygotes injected with water (86.67%, 13/15) did not differ, suggesting that the co-delivery of multiple sgRNAs elicited low or no toxicity during early pig embryo development. The FAH and HPD mutation rates were 60% and 80%, respectively, when each gene was individually targeted, whereas all five analyzed embryos carried double mutations at the target sites when 2 sgRNAs for each gene were injected. Next, 63 embryos microinjected using this system were transferred to four estrus-synchronized recipients, two of which became pregnant. One recipient was administered 2 mg/kg NTBC throughout the pregnancy while the other was not (Figure S2B). A total of six healthy piglets were obtained (Figure 2B; Table 1). Genotyping of the piglets revealed that all six harbored site-specific insertions/deletions (indels) in the FAH and HPD target sites (Figure 2C). Five of the six piglets showed biallelic modification of both genes (83.3%), and one (#3) showed a biallelic modification of the HPD gene and a monoallelic mutation in the FAH gene. Notably, the absence of the protein expression of FAH (Figures 2D and 2E) and HPD (Figures 2D and 2F) in the liver tissue of the double mutants was confirmed by immunoblotting and immunohistochemistry analysis. Moreover, no off-target mutations were detected at potential off-target loci in the double mutants (Figure S3).
Figure 2.

The generation of FAH−/−/HPD−/− double-knockout pigs
(A) Schematic diagram of the target sequence at the HPD locus. The sgRNA sequences are underlined and highlighted in red and the PAM sequences are highlighted in blue. (B) Representative photographs of FAH−/−/HPD−/− founder piglets (2 months old). (C) Sanger sequencing results of the target regions at the FAH and HPD loci in FAH−/−/HPD−/− founders. The targeted sequences (FAH and HPD) are underlined and shown at the top. The mutations are shown in red. Deletions and insertions are denoted as “Δ” and “+” plus the number of base pairs, respectively. (D–F) FAH and HPD protein expression as detected by western blotting (D), immunohistochemistry (scale bar, 200 μm [left panel] and 50 μm [right panel]; E) and immunofluorescence (scale bar, 50 μm; F). The results show that the FAH−/−/HPD−/− founders were negative for FAH and HPD protein expression in liver tissue, whereas FAH−/− pigs expressed a reduced amount of HPD compared with that in wild-type (WT) pigs. Data are presented as means ± SD. ∗∗∗p < 0.001.
FAH−/−/HPD−/− piglets were healthy and did not display the LLI phenotype
FAH−/− piglets died shortly (4–7 days) after birth with the withdrawal of NTBC administration (Figure 3A). However, the double mutants survived healthily, except for one piglet that died 1 day after birth likely due to visceral hemorrhage caused by sow crushing (Figure 3A). Long-term monitoring (24 months) revealed no obvious symptoms of hepatocellular injury or body weight loss in the FAH−/−/HPD−/− pigs (Figure 3B). Several blood biochemical parameters relating to liver function were also measured in both FAH−/− and FAH−/−/HPD−/− piglets. Compared with the WT controls, the levels of alanine transaminase (ALT; p < 0.05), aspartate aminotransferase (AST; p < 0.001), and alkaline phosphatase (ALP; p < 0.05) were significantly increased in FAH−/− pigs, whereas no significant differences were seen in FAH−/−/HPD−/− pigs (Figures 3C–3E), suggesting that the double-mutant pigs did not manifest severely impaired liver function. Examination of the bodies of the dead FAH−/− piglets further revealed that their livers were enlarged and experienced bleeding compared with age-matched WT piglets, while the double mutants appeared to be normal (Figure 3F). Moreover, hematoxylin and eosin (H&E) staining revealed obvious histopathological changes in FAH−/− pigs, including extensive hepatocyte necrosis, inflammatory cell infiltration, and cytoplasmic vacuolization, indicative of severe hepatic lobular injury. In contrast, in FAH−/−/HPD−/− pigs, most hepatocytes were morphologically normal, there was less inflammatory cell infiltration, and polyhedral hepatocytes were distributed radially from the central vein (Figure 3G). Altogether, these results demonstrated that the double mutants were protected against LLI.
Figure 3.

HPD ablation ameliorated liver injury and rescued the lethality associated with HT1
(A) Kaplan-Meier survival curve for FAH−/− pigs (n = 3), FAH−/−/HPD−/− pigs (n = 5), and WT pigs (n = 5). (B) The body weight of FAH−/−/HPD−/− pigs and WT pigs (n = 4 per group). (C–E) The serum levels of ALT (C), AST (D), and ALP (E) (n = 3 per group). (F and G) Representative images of livers (F) and hematoxylin and eosin (H&E) staining in liver samples (G) of WT (left), FAH−/− (middle), and FAH−/−/HPD−/− pigs (right). Scale bar, 100 μm (top) and 50 μm (bottom). (H) Quantitative PCR analysis of hepatic TNF-α, IL1β, and IL-6 gene expression. (I–K) Representative western blot results for TNF-α, IL-6, and the NF-κB p-p65 subunit in liver tissue lysates and the semi-quantification results. Beta-actin served as loading control. Data are reported as means ± SD; n.s., not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The double mutants showed a reduced inflammatory response
The release of multiple inflammatory cytokines is an important marker of the progression of liver injury.29,30 As expected, FAH−/− pigs exhibited significantly higher mRNA levels of tumor necrosis factor alpha (TNF-α), interleukin 1 beta (IL-1β), and IL-6 compared with those in both WT and FAH−/−/HPD−/− pigs (Figure 3H). Similarly, the increased protein expression of TNF-α and IL-6 in FAH−/− pigs relative to WT pigs was suggestive of an extensive inflammatory response; in contrast, the expression of these proteins was significantly reduced in the double mutants (Figures 3I and 3J). Phosphorylated p65 (p-p65), a major component of nuclear factor κB (NF-κB), is responsible for NF-κB transactivation, which is associated with inflammatory cytokine regulation.31 Here, we assessed the levels of p-p65 using western blotting. The relative protein expression of NF-κB p-p65 was significantly decreased in double-mutant pigs (FAH−/−/HPD−/−) when compared with that in FAH−/− pigs (Figure 3K). These results showed that HPD ablation protected against FAH deficiency-induced LLI and that this protective effect was associated with the NF-κB-mediated inflammatory pathway.
Metabolic correction of the tyrosine degradation pathway in the double mutants
Quantitative analysis of blood amino acids was carried out in FAH−/−, double mutant (FAH−/−/HPD−/−), and WT control pigs. Urine tyrosine catabolites were assessed only in double-mutant and WT pigs as insufficient urine was collected due to the quick death of the FAH−/− piglets. The mean plasma concentrations of tyrosine and phenylalanine were significantly greater in FAH−/− pigs than in WT pigs (Figures 4A and 4B); however, in the double mutants, the tyrosine concentration was decreased compared with that in FAH−/− pigs, although the level remained higher than that seen in the WT controls (Figure 4A). Interestingly, the mean serum phenylalanine levels in the double-mutant pigs were similar to those of WT pigs (Figure 4B). This was unexpected because phenylalanine synthesis occurs upstream of the metabolic block induced by HPD ablation. Meanwhile, the Phe/Tyr ratio was similar between FAH−/− and FAH−/−/HPD−/− pigs but was significantly lower than that of WT controls (Figure 4C). Analysis of the urine tyrosine catabolite profile of the double mutant pigs revealed an accumulation of 4-hydroxyphenylacetic acid and 4-hydroxyphenyllactic acid, but not 4-hydroxyphenylpyruvic acid, compared with WT pigs (Figure S4). SA was not detected. FAA accumulation has been reported to be an upstream regulator of enzymes in the tyrosine degradation pathway via negative feedback loops.32 We, therefore, ascertained whether the impaired expression of genes and enzymes involved in tyrosine metabolism was corrected following the loss of HPD. As expected, the protein expression of FAH and HPD was undetectable in the double mutants (Figure 4D). Interestingly, HPD protein expression was also significantly reduced in the FAH−/− pigs (Figures 2F and 4D). Moreover, FAH−/− pigs showed a significant decrease in the mRNA expression levels of tyrosine aminotransferase (TAT), homogentisate 1,2-dioxygenase (HGD), and glutathione S-transferase zeta 1 (GSTZ1) compared with those of the controls. Meanwhile, the expression of these genes increased significantly, although only half of the WT controls, in double mutants compared with FAH−/− pigs (Figure 4E). Western blot results indicated that the expression levels of the above proteins showed a consistent increase, although they did not return to WT levels in the livers of FAH−/−/HPD−/− pigs (Figures 4F–4H). This suggested that the loss of HPD led to the reprogramming of the tyrosine degradation pathway and the partial restoration of the expression of tyrosine metabolism-related enzymes in the double mutants.
Figure 4.

HPD deletion corrected the tyrosine metabolism profile of FAH−/− pigs
(A–C) Comparison of serum tyrosine levels (A), phenylalanine levels (B), and the Phe/Tyr ratio (C) between FAH−/−/HPD−/− (n = 5) and FAH−/− pigs (n = 3). WT pigs (n = 5) served as control. (D) Western blotting assay to assess the expression levels of FAH and HPD protein in in the liver tissue of each group. (E) The mRNA levels of the tyrosine metabolism-related genes TAT, HGD, and GSTZ1 in liver tissues. (F–H) Representative western blotting results for TAT (F), HGD (G), and GSTZ1 (H); semi-quantification results are also shown. Data are reported as means ± SD; n.s., not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Oxidative damage was reduced in the livers of FAH−/−/HPD−/− pigs
In HT1 patients, the accumulation of electrophilic metabolites causes oxidative damage to hepatocytes,33 giving rise to the abnormal liver function manifestations associated with this disorder. In this study, to evaluate the effect of knocking out HPD on the oxidative stress response, we assessed the levels of the oxidative stress marker 4-hydroxynonenal (4HNE) by western blot and immunohistochemistry. As expected, compared with WT pigs, the protein expression of 4HNE was significantly increased in FAH−/− pigs, whereas the opposite was seen in the double mutants (Figures 5A and 5B). Subsequently, we searched the STRING database and identified a potential oxidative stress-related protein-protein interaction (PPI) network involving the Kelch-like ECH-associated protein 1/ nuclear erythroid 2-like 2 transcription factor (KEAP1/NRF2) signaling pathway (Figure 5C). In line with the PPI data, the mRNA expression levels of NRF2 and NRF2 target genes (except glutamate cysteine ligase modifier, GCLM) were significantly increased in FAH−/− pigs, whereas that of KEAP1 was significantly decreased when compared with WT controls (Figure 5D). In the double mutants, however, the mRNA levels of NRF2, NAD(P)H:quinone oxidoreductase (NQO1), HO-1, glutamate–cysteine ligase catalytic (GCLC), and glutathione peroxidase (GPX1) were significantly decreased compared with those in FAH−/− pigs and were similar to those of the controls (Figure 5D). A western blot assay confirmed these observations (Figure 5E). Considering that the nuclear translocation of NRF2 is a key indicator of the oxidative stress response, we explored the effect of HPD ablation on KEAP1 expression and NRF2 activation. As expected, total NRF2 and nuclear NRF2 levels were markedly higher in FAH−/− pigs than in WT pigs, whereas the levels of KEAP1 and cytoplasmic NRF2 were reduced (Figure 5F). In contrast, no significant difference was detected in the expression of KEAP1, cytoplasmic/nuclear NRF2, or total NRF2 between FAH−/−/HPD−/− pigs and WT controls (Figure 5F). Moreover, TUNEL staining showed that the number of apoptotic hepatocytes was significantly greater in FAH−/− pigs than in the double mutants or WT controls (Figures 5G and 5H). Western blotting and immunohistochemistry analysis also showed that the expression of AFP, a genetic marker of hepatocyte precursors and/or genetic abnormalities in HCC, was significantly increased in FAH−/− pigs when compared with that in FAH−/−/HPD−/− or WT pigs (Figures 5I and 5J). Taken together, these findings suggested that HPD ablation can correct the expression of oxidative stress-related genes, as well as mitigate the apoptosis of hepatocytes and the induction of AFP in FAH−/−/HPD−/− pigs.
Figure 5.

HPD ablation reduced oxidative stress and apoptosis in the liver of HT1 pigs
(A and B) The lipid peroxidation byproduct 4HNE was detected by western blot (A) and immunohistochemistry (B) to assess the oxidative stress level in each group. (C) Protein-protein interaction (PPI) network diagram of proteins related to NRF2. (D) The relative mRNA expression of KEAP1, NRF2, and NRF2 target genes (such as NQO1, HO-1, GCLM, and GCLC) in the livers of FAH−/− and FAH−/−/HPD−/− pigs. WT pigs served as control. (E) Western blotting assay to assess the expression levels of NRF2-target proteins in total cell extracts; beta-actin served as the reference. (F) Western blotting analysis of the expression levels of KEAP1, total NRF2, nuclear (N)-NRF2, and cytoplasmic (C)-NRF2 in the liver tissue of each group. (G and H) TUNEL staining (G) and TUNEL-positive cell analysis (H) to assess the effect of HPD ablation on FAH deficiency-induced hepatocyte apoptosis. (I and J) Representative immunoblot (I) and immunohistochemical analysis (J) of the AFP protein expression level in individual pigs from each group and the quantification results. Scale bar, 50 μm. Data are reported as means ± SD; n.s., not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The profile of liver development-related genes was restored in FAH−/−/HPD−/− pigs
Studies have shown that the expression of hepatocyte-specific enzymes and liver-enriched transcription factors (LETFs) is greatly decreased in the livers of FAH-deficient humans and mice.12,34,35 Accordingly, we addressed whether genetically blocking HPD can rescue the expression of LETFs in FAH−/−/HPD−/− pigs. To this end, we measured the mRNA levels of three LETFs—hepatocyte nuclear factor 1 (HNF1), HNF4, and CCAAT/enhancer-binding protein (C/EBPA)—in FAH−/−, FAH−/−/HPD−/−, and age-matched WT pigs. As expected, the qPCR results indicated that the expression of the LETFs was significantly suppressed in the livers of FAH-deficient pigs (Figures 6A–6C). In contrast, the mRNA expression of HNF1, HNF4, and C/EBPA was restored in the double mutants, albeit only partially when compared with that of WT pigs (Figures 6A–6C). Importantly, the protein expression level of HNF1β was also significantly higher in FAH−/−/HPD−/− pigs than in FAH−/− pigs (Figure 6D). Next, peripheral blood was collected for blood glucose analysis. We found that the blood glucose level of FAH−/− pigs was lower than 1.0 mmol/L, whereas the double mutants displayed levels that ranged from 2.8 to 6.2 mmol/L (Figure 6E). This indicated that the ablation of HPD ameliorated the severe postpartum hypoglycemia. We also examined the mRNA and protein expression of multiple enzymes related to hepatic glucose metabolism and amino acid turnover, such as phosphoenolpyruvate carboxykinase 1 (PCK1), glucose-6-phosphatase (G6PC), succinate dehydrogenase (SDH), and methionine adenosyltransferase (MAT1A), in the liver tissues of the pigs. Compared with WT controls, the expression of these important metabolic enzymes was significantly downregulated in FAH−/− pigs; however, the expression levels of these enzymes were restored to varying levels in the double mutants (Figures 6F–6J).
Figure 6.

Liver homeostasis was improved in FAH−/−/HPD−/− pigs compared with that in FAH−/− pigs
(A–C) mRNA levels of the liver-enriched transcriptional factors HNF1 (A), HNF4 (B), and C/EBPα (C). (D) Representative western blot results for HNF1β and the semi-quantification results. (E) Blood glucose concentrations of newborn piglets from each group. (F and G) The mRNA levels of the glucose regulation-related genes PCK1 (F) and G6PC (G) in the liver. (H and I) The mRNA levels of the amino acid turnover regulation-related genes SDHA (H) and MAT1A (I) in the liver. (J) Representative image and semiquantitative analysis of the expression of the liver homeostasis-associated proteins PCK1, G6PC, and SDHA in WT, FAH−/−, and FAH−/−/HPD−/− pigs. Data are reported as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The liver-protective features of the double mutants were transmissible to the F1 generation
Given that the CRISPR-Cas9-induced FAH and HPD gene mutations were transmissible via the germline, we wondered whether the liver-protective effects could be transmitted to the next generation. To this end, FAH−/−/HPD−/− F0 pigs were inbred, yielding nine F1 generation pigs (Figure 7A). Ear tissues and semen samples were isolated from F1 generation pigs for Sanger sequencing. Genotype analysis showed that all nine F1 generation pigs carried FAH and HPD allelic mutations transmitted from the F0 generation (Figure S5). Notably, for F1 #2 and F1 #7, mutations in the FAH and HPD loci were also detected in their sperm DNA (Figure 7B). In addition, western blotting results confirmed the absence of FAH and HPD expression in the liver tissue of the F1 pigs, similar to that observed in the F0 double mutants (Figure 7C). Furthermore, the levels of enzymes in the tyrosine degradation pathway, such as TAT, HGD, and GSTZ1, were also significantly higher in the F1 double-mutant generation than in the FAH knockout pigs. This suggested that the tyrosine metabolic profile had also been reprogrammed in the F1 generation (Figure 7D). Next, we collected peripheral blood from F1 double mutants to measure liver function-related indicators, and found no differences in the levels of ALT, AST, ALP, TP, and ALB between the F1 double mutants and WT pigs (Figures 7E–7G; Figure S6). Picrosirius red and H&E staining of liver tissue further confirmed the biochemical test results (Figure 7H), indicating that double-mutant F1 generation pigs shared liver-protective phenotypes similar to those of F0 pigs.
Figure 7.

Characterization of the F1 double-mutant generation
(A) Pedigree showing the intercrossing of the F0 FAH−/−/HPD−/− pigs to produce the F1 generation. Filled squares indicate FAH−/−/HPD−/− males, filled circles indicate FAH−/−/HPD−/− females. (B) Sanger sequencing chromatograms for sperm DNA from WT, F1- #1, and F1- #7 pigs. The blue arrow indicates the overlapping peaks in target sites. (C) Western blotting analysis of the FAH and HPD protein expression in liver tissues of WT, FAH−/−, and FAH−/−/HPD−/− F1 pigs. (D) Representative images and semiquantitative analysis of the protein expression levels of TAT, HGD, and GSTZ1 in the liver tissues of each group as detected by western blotting. (E–G) Serum ALT, AST, and ALP levels were determined in WT, FAH−/−/HPD−/− founders (F0), and FAH−/−/HPD−/− F1 generation pigs. (H) H&E and picrosirius red staining showing normal liver tissue architecture, without obvious hepatic fibrosis, of the FAH−/−/HPD−/− founders (F0) and F1 generations. Scale bar, 100 μm. (I) A schematic diagram depicting the mechanism involved in genetically blocking HPD to protect against liver injury in HT1. Data are reported as means ± SD; n.s., not significant; ∗∗∗p < 0.001.
Discussion
In the present study, we report an improved method for the single-step generation of FAH biallelic mutant pigs and demonstrate for the first time that the silencing of HPD can prevent the LLI phenotype associated with FAH deficiency in large mammals. Moreover, we also provide evidence for the reprogramming of the tyrosine catabolism pathway and restoration of the liver gene-expression pattern in FAH−/−/HPD−/− pigs.
Species differences between rodents and humans, as emphasized by gene therapy approaches to treat Duchenne muscular dystrophy (DMD) and cystic fibrosis (CF),36,37 highlight that the safety and efficacy of CRISPR-Cas9-mediated in vivo gene therapy should be evaluated in a large animal model before clinical application. Therefore, in our present study, a novel FAH biallelic mutant pig model mimicking human HT1 was generated by intracytoplasmic microinjection combined with CRISPR-Cas9 technology. We devised a procedure that was optimized for oestrus synchronization and vaginal smear staining, thereby ensuring that one-cell-stage embryos could be employed to generate FAH biallelic mutant pigs. Additionally, NTBC was administrated to surrogate sows immediately after the confirmation of pregnancy to avoid homozygous intrauterine death. Our data showed that FAH−/− pigs can be generated in a single step without the need for outbreeding between FAH heterozygotes, which greatly shortens the breeding cycle and reduces costs. Notably, FAH deficiency in pigs results in lethality in utero, rendering NTBC administration a requirement for sustained gestation. In humans, however, the loss of FAH activity is not believed to cause in utero lethality, whereas liver injury is a common manifestation.38 Without NTBC administration, our FAH-deficient pigs died with extensive liver damage and exhibited many of the clinical and pathological features of the human acute HT1 disease, such as failure to thrive, hypertyrosinemia, hepatomegaly, and hypoglycemia.39 FAH-deficient pigs may serve as an ideal animal model to study novel treatment strategies for HT1.
Although NTBC treatment reduces the incidence of LLI and prolongs the survival of HT1 patients, it does not address the significantly greater incidence of HT1-associated HCC in this population.40, 41, 42 As in humans, NTBC administration can ameliorate the tyrosinemia phenotype of Fah−/− mice; however, it does not normalize liver gene expression.12,43 Orthotopic liver transplantation remains the only curative therapy for patients with acute or advanced HT1. However, liver transplantation is largely limited by the difficulty of surgery, a shortage of liver donors, and the need for life-long immunosuppressive treatment.44, 45, 46 Consequently, there is an urgent need for other effective and safe therapeutic strategies for the treatment of patients with HT1 and similar metabolic liver diseases. Although therapeutic gene deletion or gene correction represents a possible alternative, this approach requires evaluation and validation through a large number of preclinical trials, especially in large animals. The fact that NTBC functions partially by inhibiting the activity of the HPD enzyme, and that the ablation of the Hpd gene rescued the lethality associated with Fah−/− mice, prompted us to investigate whether genetically blocking the HPD gene could serve as an effective strategy in preventing LLI and promoting the survival of HT1 pigs. We previously described a genetic pig model with a mutation in the HPD locus that showed elevated blood tyrosine levels and a large accumulation of tyrosine derivatives in the urine.47 Importantly, the absence of HPD in pigs did not lead to liver damage and was not lethal, which is consistent with the phenotype of Hpd−/− mice.48 To evaluate the feasibility of using HPD ablation to treat HT1 in a pig model, we generated FAH−/−/HPD−/− pigs by direct intracytoplasmic delivery of CRISPR-Cas9 and sgRNAs targeting the FAH and HPD genes. As predicted, metabolic reprogramming by the knocking out of the HPD gene converted HT1 to HT3, a milder form of the disorder. The phenotypes of the FAH−/−/HPD−/− pigs were similar to those of HPD−/− pigs, namely, tyrosine levels remain high but liver morphology and histology are normal. Although high levels of tyrosine in vivo can damage tissues such as the eyes and skin, we did not observe this in our double-mutant pigs, but longer-term evaluation may be needed to identify potential additional complications. Off-target modifications at undesired genomic sites also remain a critical concern with the use of the CRISPR-Cas9 system.49 In the present study, no off-target events were detected at the tested potential off-targets (top 10) in the genome of FAH−/−/HPD−/− pigs, as verified by Sanger sequencing. Nevertheless, whole-genome sequencing using accurate and sensitive off-target profiling techniques such as genome-wide unbiased indentification of DSBs evaluated by sequencing (GUIDE-seq) and circularization for in vitro reporting of cleavage effects by sequencing (CIRCLE-seq) would be required to completely exclude the presence of unexpected mutations.50,51 Combined, our data support that completely blocking the tyrosine catabolism pathway by HPD deletion can protect against liver injury and prevent lethality in HT1 pigs.
Large alterations in gene-expression patterns in the liver represent an important index of liver damage in both HT1 patients and HT1 animal models.6,52,53 The failure of NTBC to normalize the HT1-induced alterations in hepatic gene expression may underlie the devastating progression of HT1.12,43,54 There was a prominent recovery of the expression of tyrosine metabolism-related enzymes in FAH−/−/HPD−/− pigs. These results implied that dysregulated tyrosine metabolism may be specific to FAH−/− pigs and patients with more severe forms of HT1. In addition, various transcripts encoding proteins necessary for systematic homeostasis, including glucose production, amino acid turnover, and LETFs.35,55,56 were not induced in FAH−/− pigs, and may have contributed to their neonatal death. Notably, although both the phenotype and the suppression of the expression of genes involved in liver homeostasis were alleviated in FAH−/−/HPD−/− pigs, the expression of several genes, including TAT, HNF, and G6PC, was not fully restored to WT levels with HPD ablation. The promoter region of the HPD gene reportedly contains binding sites for several liver-specific transcription factors, including HNF1, HNF4, and C/EBP.57 The reduced expression of these factors in double mutants may be due to the absence of HPD gene. Moreover, liver-specific enzymes such as G6P, TAT, and PCK are normally inducible by glucocorticoids and/or cyclic AMP (cAMP) around the time of parturition.58 Therefore, we suspect that the perinatal activation of the genes that code for these enzymes is subject to tissue-specific and hormonal regulation and is likely to be a complex process sensitive to interference at many levels. Further elucidation of the mechanism underlying the incomplete restoration of the expression of the above-mentioned genes, such as genome-wide analysis, may provide additional information on the effects of HPD deletion on newborn HT1 piglets.
Inflammation is an essential factor in the pathogenesis of liver injury.59 NF-κB, a redox-sensitive transcriptional regulator, is associated with the transcription of genes related to the inflammation response and inflammatory cytokine production.60,61 The accumulation of toxic metabolites in hepatocytes stimulates inflammatory cell infiltration and the release of proinflammatory cytokines, ultimately resulting in liver damage. In FAH−/− pigs, the activation of NF-κB-mediated transcription elicited a significantly stronger inflammatory response following NTBC withdrawal. Meanwhile, there was a notable reduction in the expression of genes coding for inflammatory cytokines in FAH−/−/HPD−/− pigs, indicating that genetically blocking HPD inhibited the activation of the NF-κB signal. Additionally, it is known that the accumulation of electrophilic intermediates, such as FAA, can activate oxidative stress responses and trigger apoptosis.33,38 NRF2, a key regulator of numerous genes encoding antioxidative response enzymes, can be induced under oxidative stress.62,63 Under physiological conditions, NRF2 and KEAP1 are complexed and sequestered in the cytoplasm. Following oxidative or electrophilic stress, NRF2/KEAP1 separate, allowing NRF2 to translocate to the nucleus, where it binds to antioxidant response elements (AREs) and mediates responses to oxidative stress.64 The data presented here revealed that FAH−/− pigs exhibit a striking oxidative stress response, characterized by the activation of the 4HNE and NRF2 pathways. These changes were accompanied by extensive hepatocyte apoptosis and robust AFP induction. In the double mutants, however, the expression of a relatively large number of genes involved in oxidative stress responses was significantly ameliorated. Our data preliminarily showed that genetically blocking HPD can largely eliminate the effects of HT1-associated inflammation and oxidative stress, thereby reducing hepatocyte apoptosis.
Although our findings demonstrated that HPD ablation elicited encouraging therapeutic effects in FAH−/− pigs, this study was preliminary and had several limitations. First, the sample size was relatively small. Second, potential off-target effects call for further concern. Third, the global gene-expression pattern of FAH−/−/HPD−/− pigs should be further assessed. Finally, the co-disruption of genes in the embryos simultaneously created the disease and the correction event and did not directly treat preexisting HT1. A recent study reported the in utero, CRISPR-mediated therapeutic editing of metabolic genes. A nonsense mutation was introduced into the HPD gene in utero to permanently disrupt gene function, resulting in the rescue of LLI in HT1 mice.65 This report provides further evidence that disrupting the HPD gene may be a safe and reliable alternative therapy for the treatment of HT1 and encourages the development of optimized methods for gene editing in large animal models. In summary, our data provided proof-of-principle that HPD gene silencing represents a potential alternative therapeutic strategy for the treatment of HT1, i.e., a one-time treatment that provides long-term protection against liver injury. Our results further suggest that the reprogramming of the tyrosine metabolism pathway in vivo by genetically blocking HPD may protect against FAH deficiency-induced LLI by ameliorating oxidative stress and NF-κB-dependent inflammatory responses (Figure 7I).
Materials and methods
Animals and animal care
All pigs used in this research were raised in the Laboratory Animal Center, Southern Medical University (Guangzhou, China). The animal use protocol listed below followed the Guidelines on Animal Care and Use established by the Southern Medical University Animal Care and Use Committee (Animal Welfare Assurance L2019199). All surgery was performed under anesthesia and all efforts were made to minimize animal suffering. The number of animals used was kept to a minimum by the experimental design.
Vector construction and in vitro transcription
gRNAs targeting the WT sequences of the porcine FAH and HPD genes were designed using the online CRISPR design tool (http://crispor.tefor.net/). Oligos for the generation of sgRNA expression plasmids were annealed and cloned into the BsaI sites of pGL3-U6-sgRNA-PGK-Puro that contains a T7 promoter using primers listed in Table S2. The T7-sgRNA PCR products were purified and transcribed in vitro using the MEGA Shortscript T7 Kit (AM1354, Life Technologies, USA). The pST1374-NLS-flag-linker-Cas9 plasmid was linearized with AgeI, and Cas9 mRNA was synthesized using the mMESSAGE mMACHINE T7 Ultra Kit (AM1345, Life Technologies). The sgRNAs and the Cas9 mRNA were purified by phenol:chloroform extraction and ethanol precipitation using standard methods and eluted in 40∼80 μL of RNase-free water.
Estrus synchronization and the collection of porcine zygotes
The protocol for estrus synchronization and the collection of porcine zygotes was described in detail in our previous study.66 Briefly, pubertal Tibet minipigs (approximately 7–9 months of age, 30–35 kg body weight) with normal estrus cycles served as donors for embryo collection. Estrus was synchronized by feeding 20 mg/day/pig Altrenogest (Beijing Keyifeng Biotech Develop, China) for 18 days. Crystal violet staining of vaginal smears was performed to assess the estrus state and natural mating was implemented 5–7 days after drug withdrawal. Before the surgical procedure, the animals were fasted for 12 h. Anesthesia (18–24 h after the last mating) was induced by intramuscular administration of 30–45 mg/kg pentobarbital sodium. The ovary and uterus were exposed by a lower abdominal incision and single-cell-stage embryos were flushed with Dulbecco’s phosphate-buffered saline solution (DPBS; 21-031-CVR, Corning, USA) containing 1% bovine serum albumin (BSA).
Pig embryo injection and transfer
A mixture of Cas9 mRNA (100 ng/μL) and sgRNA (50 ng/μL) was microinjected into the cytoplasm of porcine zygotes on a heated platform using a FemtoJet microinjector (Eppendorf, Hamburg, Germany). Injected zygotes were returned to the PZM3 medium for culture and then immediately surgically transferred into synchronized foster mother sows using an embryo transfer catheter, as previously described.67 Pregnancies were confirmed using Pregnancy Test Kits (Q/CPWHS 022-2000, Beijing Wanhua Bioengineering, China) and by ultrasound at 19–21 days after embryo transfer. To prevent the intrauterine death of FAH homozygotes, we administered pregnant sows 2 mg/kg NTBC per day until delivery.
The detection of mutant genes
Genomic DNA was extracted from the ear tissues of newborn piglets and semen samples of double-mutant boars using the TIANamp Genomic DNA Kit (DP304, TIANGEN, China) following the manufacturer’s instructions and PCR amplified using PrimerSTAR HS DNA polymerase (R010A, Takara Bio, Japan). The PCR products were gel purified and cloned using a pMD-19 cloning kit (3271-C1, Takara Bio). Ten colonies were picked from each transformation and Sanger sequenced to detect mutations. The primers used for amplifying the targeted fragments are listed in Table S3.
Assay to detect off-target effects
Potential off-target loci were predicted using an online CRISPR design tool (http://crispor.tefor.net/) and Cas-OFFinder (http://www.rgenome.net/cas-offinder/). Ten sites with <5 total mismatches were chosen as potential off-target sites for testing. The selected off-target sites were PCR amplified and Sanger sequenced. The primers used for amplifying the off-target sites are listed in Tables S4–S7.
Blood biochemical analysis
Venous Blood was obtained from the ear vein of the pigs and collected in normal tubes. Serum was separated using standard protocols and analyzed for ALT, AST ALP, ALB, and TP concentrations by Automatic Chemistry Analyzer (7600-020, HITACHI, Japan) using commercial kits and following the manufacturer’s instructions.
H&E staining, immunohistochemistry, and immunofluorescence
Liver samples from WT and mutant pigs were dehydrated via a graded ethanol series and fixed in 4% paraformaldehyde overnight. After paraffin embedding, the samples were sectioned into 5-μm thick slices for H&E staining, immunostaining, and immunofluorescence analyses. H&E staining was performed using standard protocols. For immunohistochemistry, paraffin-embedded sections were treated with 1 M citrate buffer (pH 6.0) for 20 min followed by the blocking of endogenous peroxidase activity using 5% hydrogen peroxide. Sections were incubated at 4°C overnight with a rabbit anti-FAH primary antibody (1:200, PA5-42049, Thermo). Immunofluorescence staining was performed on liver sections as previously described68 using an anti-HPD primary antibody (ab232906, Abcam).
TUNEL staining
TUNEL staining was performed on paraformaldehyde-fixed and paraffin-embedded liver sections according to the manufacturer’s instructions (11684795910, Roche, Switzerland) to detect hepatic cell apoptosis. The sections were observed under a fluorescence microscope (Nikon Eclipse Ti-SR and Nikon DS-U3) and the fluorescence intensity was quantified using ImageJ software.
Western blotting
For western blot analysis, liver tissue samples were homogenized in 1 mL of lysis buffer (KeyGEN BioTECH, China) and protein concentrations were quantified using a bicinchoninic acid assay kit (KeyGEN BioTECH). Equal amounts of total protein (40 μg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Temecula, CA, USA). The membranes were blocked with 5% BSA for 1 h and then incubated with primary antibodies at 4°C overnight. Anti-GAPDH and anti-beta-actin antibodies were used as internal controls. The antibodies used in this study are listed in Table S8.
RNA extraction and quantitative real-time PCR
Total RNA was extracted from liver tissues using TRIzol reagent (Invitrogen, Life Technologies, USA). cDNA was synthesized using the PrimeScript RT Reagent Kit (Takara). Quantitative real-time PCR was performed in a CFX96 Real-Time System (Bio-Rad) with SYBR Premix Ex Taq (Takara) according to the manufacturer’s instructions. The gene-specific primers used are shown in Table S9. Relative gene-expression levels were normalized to that of GAPDH and quantified using the 2−ΔΔCq method.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis
Amino acid analysis was performed on dried blood spots using an ABI 3200 Q-TRAP LC-MS/MS system (Applied Biosystems) as previously described.69 Briefly, 25 μL of blood was applied to an S&S Grade 903 filter, dried for 3 h, and then sealed in a plastic bag at −20°C until testing. A 3-mm diameter disc was obtained from each dried blood card and placed in a 96-well polyprolene plate. Methanol (100 μL) containing isotopic internal standards was added to each well followed by incubation at 45°C for 45 min with gentle agitation. After 20 min at room temperature, 75 μL aliquots were transferred to a new plate and covered with an aluminum membrane before use.
Gas chromatography-mass spectrometry (GC/MS) analysis
Urine organic metabolites were analyzed using standard methodologies.70 Briefly, urine was collected using metabolic cages and urine specimens containing 0.1 mg of creatinine and 5 mg of tropic acid (TA; internal standard) were incubated with 2 units of urease at 37°C for 30 min to degrade the urea. Oximation was performed with hydroxylamine hydrochloride at pH > 12, before adjusting the pH to 1. The supernatant was transferred to a 1.8-mL vial and gently dried under a nitrogen stream at 60°C. The dry residue was derivatized to trimethylsilyl (TMS) ethers by treatment with 100 μL of BSTFA-TMCS (10:1, v/v) for 30 min at 80°C, following which the mixture was loaded onto a GC-MS QP-2010 Ultrasystem for analysis.
Statistical analysis
Statistical analysis was performed using GraphPad PRISM 8.0 (GraphPad Software, San Diego, CA, USA) or SPSS version 22 (SPSS, Chicago, IL, USA). Quantitative data were expressed as means ± SD. The Student’s t test or the Mann-Whitney U-test was performed for comparisons between two groups. One-way ANOVA with either the Bonferroni procedure (parametric) or the Kruskal-Wallis test (non-parametric) was used for comparisons among multiple groups. A p value ≤0.05 was considered statistically significant.
Data and materials availability
Data are available on reasonable request from the corresponding author.
Acknowledgments
We are grateful to all participants of the group at Songshan Lake Pearl Laboratory Animal Sci. & Tech. Co., Ltd. We also thank Xuran Yang for support of metabolic studies. This work was supported by the International Science and Technology Cooperation Project (grant number 2011DFA33290 to W.-W.G.), Jiangmen Introduced Innovative Scientific Research Team Program (grant number 2017TD02 to W.-W.G.), and Guangdong Basic and Applied Basic Research Fund Project (grant number 2019A1515011503 to Y.G.T.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
Conceived and designed the experiments, W.G. and P.G.; performed the experiments, P.G., Q.Y., B.C., Y.-n.B., T.X., C.L., X.Y., Y.L., and X.T.; analyzed the data: P.G. and Q.Y.; contributed reagents/materials/analysis tools, W.L., Y.T., and H.L.; wrote the paper, P.G. and Q.Y.; edited and revised manuscript, P.G., W.G. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.04.002.
Supplemental information
References
- 1.Lindblad B., Lindstedt S., Steen G. On the enzymic defects in hereditary tyrosinemia. Proc. Natl. Acad. Sci. USA. 1977;74:4641–4645. doi: 10.1073/pnas.74.10.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gagné R. [Genetic counseling: experience of 4 years] Union Med. Can. 1978;107:391–393. [PubMed] [Google Scholar]
- 3.Angileri F., Bergeron A., Morrow G., Lettre F., Gray G., Hutchin T., Ball S., Tanguay R.M. Geographical and ethnic distribution of mutations of the fumarylacetoacetate hydrolase gene in hereditary tyrosinemia type 1. JIMD Rep. 2015;19:43–58. doi: 10.1007/8904_2014_363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jorquera R., Tanguay R.M. Fumarylacetoacetate, the metabolite accumulating in hereditary tyrosinemia, activates the ERK pathway and induces mitotic abnormalities and genomic instability. Hum. Mol. Genet. 2001;10:1741–1752. doi: 10.1093/hmg/10.17.1741. [DOI] [PubMed] [Google Scholar]
- 5.Bliksrud Y.T., Ellingsen A., Bjørås M. Fumarylacetoacetate inhibits the initial step of the base excision repair pathway: implication for the pathogenesis of tyrosinemia type I. J. Inherit. Metab. Dis. 2013;36:773–778. doi: 10.1007/s10545-012-9556-0. [DOI] [PubMed] [Google Scholar]
- 6.Tanguay R.M., Valet J.P., Lescault A., Duband J.L., Laberge C., Lettre F., Plante M. Different molecular basis for fumarylacetoacetate hydrolase deficiency in the two clinical forms of hereditary tyrosinemia (type I) Am. J. Hum. Genet. 1990;47:308–316. [PMC free article] [PubMed] [Google Scholar]
- 7.Morrow G., Tanguay R.M. Biochemical and clinical aspects of hereditary tyrosinemia type 1. Adv. Exp. Med. Biol. 2017;959:9–21. doi: 10.1007/978-3-319-55780-9_2. [DOI] [PubMed] [Google Scholar]
- 8.Fernández-Lainez C., Ibarra-González I., Belmont-Martínez L., Monroy-Santoyo S., Guillén-López S., Vela-Amieva M. Tyrosinemia type I: clinical and biochemical analysis of patients in Mexico. Ann. Hepatol. 2014;13:265–272. [PubMed] [Google Scholar]
- 9.Lindstedt S., Holme E., Lock E.A., Hjalmarson O., Strandvik B. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet. 1992;340:813–817. doi: 10.1016/0140-6736(92)92685-9. [DOI] [PubMed] [Google Scholar]
- 10.Bartlett D.C., Lloyd C., McKiernan P.J., Newsome P.N. Early nitisinone treatment reduces the need for liver transplantation in children with tyrosinaemia type 1 and improves post-transplant renal function. J. Inherit. Metab. Dis. 2014;37:745–752. doi: 10.1007/s10545-014-9683-x. [DOI] [PubMed] [Google Scholar]
- 11.Al-Dhalimy M., Overturf K., Finegold M., Grompe M. Long-term therapy with NTBC and tyrosine-restricted diet in a murine model of hereditary tyrosinemia type I. Mol. Genet. Metab. 2002;75:38–45. doi: 10.1006/mgme.2001.3266. [DOI] [PubMed] [Google Scholar]
- 12.Luijerink M.C., Jacobs S.M.M., van Beurden E.A.C.M., Koornneef L.P., Klomp L.W.J., Berger R., van den Berg I.E. Extensive changes in liver gene expression induced by hereditary tyrosinemia type I are not normalized by treatment with 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC) J. Hepatol. 2003;39:901–909. doi: 10.1016/s0168-8278(03)00433-1. [DOI] [PubMed] [Google Scholar]
- 13.Yin H., Xue W., Chen S., Bogorad R.L., Benedetti E., Grompe M., Koteliansky V., Sharp P.A., Jacks T., Anderson D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014;32:551–553. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang L., Wang L., Huo Y., Chen X., Yin S., Hu Y., Zhang X., Zheng R., Geng H., Han H. Amelioration of an inherited metabolic liver disease through creation of a De Novo start codon by cytidine base editing. Mol. Ther. 2020;28:1673–1683. doi: 10.1016/j.ymthe.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li N., Gou S., Wang J., Zhang Q., Huang X., Xie J., Li L., Jin Q., Ouyang Z., Chen F. CRISPR/Cas9-mediated gene correction in newborn rabbits with hereditary tyrosinemia type I. Mol. Ther. 2021;29:1001–1015. doi: 10.1016/j.ymthe.2020.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinderer C., Katz N., Buza E.L., Dyer C., Goode T., Bell P., Richman L.K., Wilson J.M. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther. 2018;29:285–298. doi: 10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Endo F., Kubo S., Awata H., Kiwaki K., Katoh H., Kanegae Y., Saito I., Miyazaki J., Yamamoto T., Jakobs C. Complete rescue of lethal albino c14CoS mice by null mutation of 4-hydroxyphenylpyruvate dioxygenase and induction of apoptosis of hepatocytes in these mice by in vivo retrieval of the tyrosine catabolic pathway. J. Biol. Chem. 1997;272:24426–24432. doi: 10.1074/jbc.272.39.24426. [DOI] [PubMed] [Google Scholar]
- 18.Pankowicz F.P., Barzi M., Legras X., Hubert L., Mi T., Tomolonis J.A., Ravishankar M., Sun Q., Yang D., Borowiak M. Reprogramming metabolic pathways in vivo with CRISPR/Cas9 genome editing to treat hereditary tyrosinaemia. Nat. Commun. 2016;7:12642. doi: 10.1038/ncomms12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogers C.S., Stoltz D.A., Meyerholz D.K., Ostedgaard L.S., Rokhlina T., Taft P.J., Rogan M.P., Pezzulo A.A., Karp P.H., Itani O.A. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321:1837–1841. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan S., Tu Z., Liu Z., Fan N., Yang H., Yang S., Yang W., Zhao Y., Ouyang Z., Lai C. A huntingtin knockin pig model recapitulates features of selective neurodegeneration in Huntington’s disease. Cell. 2018;173:989–1002.e13. doi: 10.1016/j.cell.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang J.T., Cho B., Ryu J., Ray C., Lee E.J., Yun Y.J., Ahn S., Lee J., Ji D.Y., Jue N. Biallelic modification of IL2RG leads to severe combined immunodeficiency in pigs. Reprod. Biol. Endocrinol. 2016;14:74. doi: 10.1186/s12958-016-0206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lunney J.K. Advances in swine biomedical model genomics. Int. J. Biol. Sci. 2007;3:179–184. doi: 10.7150/ijbs.3.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roura E., Koopmans S.J., Lallès J.P., Le Huerou-Luron I., de Jager N., Schuurman T., Val-Laillet D. Critical review evaluating the pig as a model for human nutritional physiology. Nutr. Res. Rev. 2016;29:60–90. doi: 10.1017/S0954422416000020. [DOI] [PubMed] [Google Scholar]
- 24.Evers M.M., Miniarikova J., Juhas S., Vallès A., Bohuslavova B., Juhasova J., Skalnikova H.K., Vodicka P., Valekova I., Brouwers C. AAV5-miHTT gene therapy demonstrates broad distribution and strong human mutant huntingtin lowering in a Huntington’s disease minipig model. Mol. Ther. 2018;26:2163–2177. doi: 10.1016/j.ymthe.2018.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hickey R.D., Lillegard J.B., Fisher J.E., McKenzie T.J., Hofherr S.E., Finegold M.J., Nyberg S.L., Grompe M. Efficient production of Fah-null heterozygote pigs by chimeric adeno-associated virus-mediated gene knockout and somatic cell nuclear transfer. Hepatology. 2011;54:1351–1359. doi: 10.1002/hep.24490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hickey R.D., Mao S.A., Glorioso J., Lillegard J.B., Fisher J.E., Amiot B., Rinaldo P., Harding C.O., Marler R., Finegold M.J. Fumarylacetoacetate hydrolase deficient pigs are a novel large animal model of metabolic liver disease. Stem Cell Res. (Amst.) 2014;13:144–153. doi: 10.1016/j.scr.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.St-Louis M., Tanguay R.M. Mutations in the fumarylacetoacetate hydrolase gene causing hereditary tyrosinemia type I: overview. Hum. Mutat. 1997;9:291–299. doi: 10.1002/(SICI)1098-1004(1997)9:4<291::AID-HUMU1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 28.Rüetschi U., Cerone R., Pérez-Cerda C., Schiaffino M.C., Standing S., Ugarte M., Holme E. Mutations in the 4-hydroxyphenylpyruvate dioxygenase gene (HPD) in patients with tyrosinemia type III. Hum. Genet. 2000;106:654–662. doi: 10.1007/s004390000307. [DOI] [PubMed] [Google Scholar]
- 29.Gong S., Lan T., Zeng L., Luo H., Yang X., Li N., Chen X., Liu Z., Li R., Win S. Gut microbiota mediates diurnal variation of acetaminophen induced acute liver injury in mice. J. Hepatol. 2018;69:51–59. doi: 10.1016/j.jhep.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yagi H., Soto-Gutierrez A., Navarro-Alvarez N., Nahmias Y., Goldwasser Y., Kitagawa Y., Tilles A.W., Tompkins R.G., Parekkadan B., Yarmush M.L. Reactive bone marrow stromal cells attenuate systemic inflammation via sTNFR1. Mol. Ther. 2010;18:1857–1864. doi: 10.1038/mt.2010.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pereira S.G., Oakley F. Nuclear factor-kappaB1: regulation and function. Int. J. Biochem. Cell Biol. 2008;40:1425–1430. doi: 10.1016/j.biocel.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 32.Langlois C., Jorquera R., Finegold M., Shroads A.L., Stacpoole P.W., Tanguay R.M. Evaluation of dichloroacetate treatment in a murine model of hereditary tyrosinemia type 1. Biochem. Pharmacol. 2006;71:1648–1661. doi: 10.1016/j.bcp.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 33.Dieter M.Z., Freshwater S.L., Miller M.L., Shertzer H.G., Dalton T.P., Nebert D.W. Pharmacological rescue of the 14CoS/14CoS mouse: hepatocyte apoptosis is likely caused by endogenous oxidative stress. Free Radic. Biol. Med. 2003;35:351–367. doi: 10.1016/s0891-5849(03)00273-9. [DOI] [PubMed] [Google Scholar]
- 34.Scott C.R. The genetic tyrosinemias. Am. J. Med. Genet. C. Semin. Med. Genet. 2006;142C:121–126. doi: 10.1002/ajmg.c.30092. [DOI] [PubMed] [Google Scholar]
- 35.Kelsey G., Ruppert S., Beermann F., Grund C., Tanguay R.M., Schütz G. Rescue of mice homozygous for lethal albino deletions: implications for an animal model for the human liver disease tyrosinemia type 1. Genes Dev. 1993;7:2285–2297. doi: 10.1101/gad.7.12a.2285. [DOI] [PubMed] [Google Scholar]
- 36.Zhou Z.P., Yang L.L., Cao H., Chen Z.R., Zhang Y., Wen X.Y., Hu J. In vitro validation of a CRISPR-mediated CFTR correction strategy for preclinical translation in pigs. Hum. Gene Ther. 2019;30:1101–1116. doi: 10.1089/hum.2019.074. [DOI] [PubMed] [Google Scholar]
- 37.Moretti A., Fonteyne L., Giesert F., Hoppmann P., Meier A.B., Bozoglu T., Baehr A., Schneider C.M., Sinnecker D., Klett K. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020;26:207–214. doi: 10.1038/s41591-019-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grompe M. The pathophysiology and treatment of hereditary tyrosinemia type 1. Semin. Liver Dis. 2001;21:563–571. doi: 10.1055/s-2001-19035. [DOI] [PubMed] [Google Scholar]
- 39.Chinsky J.M., Singh R., Ficicioglu C., van Karnebeek C.D.M., Grompe M., Mitchell G., Waisbren S.E., Gucsavas-Calikoglu M., Wasserstein M.P., Coakley K., Scott C.R. Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet. Med. 2017;19:1380. doi: 10.1038/gim.2017.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Spronsen F.J., Bijleveld C.M.A., van Maldegem B.T., Wijburg F.A. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifluoro- methylbenzoyl)-1, 3-cyclohexanedione treatment. J. Pediatr. Gastroenterol. Nutr. 2005;40:90–93. doi: 10.1097/00005176-200501000-00017. [DOI] [PubMed] [Google Scholar]
- 41.Santra S., Baumann U. Experience of nitisinone for the pharmacological treatment of hereditary tyrosinaemia type 1. Expert Opin. Pharmacother. 2008;9:1229–1236. doi: 10.1517/14656566.9.7.1229. [DOI] [PubMed] [Google Scholar]
- 42.Mayorandan S., Meyer U., Gokcay G., Segarra N.G., de Baulny H.O., van Spronsen F., Zeman J., de Laet C., Spiekerkoetter U., Thimm E. Cross-sectional study of 168 patients with hepatorenal tyrosinaemia and implications for clinical practice. Orphanet J. Rare Dis. 2014;9:107. doi: 10.1186/s13023-014-0107-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka Y., Nakamura K., Matsumoto S., Kimoto Y., Tanoue A., Tsujimoto G., Endo F. Gene expression profiles of homogentisate-treated Fah-/- Hpd-/-mice using DNA microarrays. Mol. Genet. Metab. 2006;89:203–209. doi: 10.1016/j.ymgme.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 44.Fagiuoli S., Daina E., D’Antiga L., Colledan M., Remuzzi G. Monogenic diseases that can be cured by liver transplantation. J. Hepatol. 2013;59:595–612. doi: 10.1016/j.jhep.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 45.Wertheim J.A., Petrowsky H., Saab S., Kupiec-Weglinski J.W., Busuttil R.W. Major challenges limiting liver transplantation in the United States. Am. J. Transplant. 2011;11:1773–1784. doi: 10.1111/j.1600-6143.2011.03587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Curnock R., Heaton N.D., Vilca-Melendez H., Dhawan A., Hadzic N., Vara R. Liver transplantation in children with propionic acidemia: medium-term outcomes. Liver Transpl. 2020;26:419–430. doi: 10.1002/lt.25679. [DOI] [PubMed] [Google Scholar]
- 47.Gu P., Chen B., Xu T., Liu W., Deng J., Xu M., Yuan J., Chen E., Gu W. Generation of a Bama minipig model of hereditary tyrosinemia type III by modifying the Hpd gene. Chin. J. Comp. Med. 2019;29:11–16. [Google Scholar]
- 48.Endo F., Awata H., Katoh H., Matsuda I. A nonsense mutation in the 4-hydroxyphenylpyruvic acid dioxygenase gene (Hpd) causes skipping of the constitutive exon and hypertyrosinemia in mouse strain III. Genomics. 1995;25:164–169. doi: 10.1016/0888-7543(95)80122-3. [DOI] [PubMed] [Google Scholar]
- 49.Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai S.Q., Nguyen N.T., Malagon-Lopez J., Topkar V.V., Aryee M.J., Joung J.K. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruppert S., Kelsey G., Schedl A., Schmid E., Thies E., Schütz G. Deficiency of an enzyme of tyrosine metabolism underlies altered gene expression in newborn liver of lethal albino mice. Genes Dev. 1992;6:1430–1443. doi: 10.1101/gad.6.8.1430. [DOI] [PubMed] [Google Scholar]
- 53.Angileri F., Roy V., Morrow G., Scoazec J.Y., Gadot N., Orejuela D., Tanguay R.M. Molecular changes associated with chronic liver damage and neoplastic lesions in a murine model of hereditary tyrosinemia type 1. Biochim. Biophys. Acta. 2015;1852:2603–2617. doi: 10.1016/j.bbadis.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 54.Molchanov S., Gryff-Keller A. Inhibition of 4-hydroxyphenylpyruvate dioxygenase by 2-[2-nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione. Acta Biochim. Pol. 2009;56:447–454. [PubMed] [Google Scholar]
- 55.Gluecksohn-Waelsch S. Genetic control of morphogenetic and biochemical differentiation: lethal albino deletions in the mouse. Cell. 1979;16:225–237. doi: 10.1016/0092-8674(79)90001-1. [DOI] [PubMed] [Google Scholar]
- 56.Loose D.S., Shaw P.A., Krauter K.S., Robinson C., Englard S., Hanson R.W., Gluecksohn-Waelsch S. Trans regulation of the phosphoenolpyruvate carboxykinase (GTP) gene, identified by deletions in chromosome 7 of the mouse. Proc. Natl. Acad. Sci. USA. 1986;83:5184–5188. doi: 10.1073/pnas.83.14.5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Awata H., Endo F., Matsuda I. Structure of the human 4-hydroxyphenylpyruvic acid dioxygenase gene (HPD) Genomics. 1994;23:534–539. doi: 10.1006/geno.1994.1540. [DOI] [PubMed] [Google Scholar]
- 58.Montoliu L., Blendy J.A., Cole T.J., Schütz G. Analysis of the cAMP response on liver-specific gene expression in transgenic mice. Fundam. Clin. Pharmacol. 1994;8:138–146. doi: 10.1111/j.1472-8206.1994.tb00790.x. [DOI] [PubMed] [Google Scholar]
- 59.Kumar V., Kumar V., Luo J., Mahato R.I. Therapeutic potential of OMe-PS-miR-29b1 for treating liver fibrosis. Mol. Ther. 2018;26:2798–2811. doi: 10.1016/j.ymthe.2018.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Espín-Palazón R., Traver D. The NF-κB family: Key players during embryonic development and HSC emergence. Exp. Hematol. 2016;44:519–527. doi: 10.1016/j.exphem.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y.F., Xu X., Fan X., Zhang C., Wei Q., Wang X., Guo W., Xing W., Yu J., Yan J.L., Liang H.P. A cell-penetrating peptide suppresses inflammation by inhibiting NF-κB signaling. Mol. Ther. 2011;19:1849–1857. doi: 10.1038/mt.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tu W., Wang H., Li S., Liu Q., Sha H. The anti-inflammatory and anti-oxidant mechanisms of the Keap1/Nrf2 are signaling pathway in chronic diseases. Aging Dis. 2019;10:637–651. doi: 10.14336/AD.2018.0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liang K.J., Woodard K.T., Weaver M.A., Gaylor J.P., Weiss E.R., Samulski R.J. AAV-Nrf2 promotes protection and recovery in animal models of oxidative stress. Mol. Ther. 2017;25:765–779. doi: 10.1016/j.ymthe.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marhenke S., Lamlé J., Buitrago-Molina L.E., Cañón J.M.F., Geffers R., Finegold M., Sporn M., Yamamoto M., Manns M.P., Grompe M., Vogel A. Activation of nuclear factor E2-related factor 2 in hereditary tyrosinemia type 1 and its role in survival and tumor development. Hepatology. 2008;48:487–496. doi: 10.1002/hep.22391. [DOI] [PubMed] [Google Scholar]
- 65.Rossidis A.C., Stratigis J.D., Chadwick A.C., Hartman H.A., Ahn N.J., Li H., Singh K., Coons B.E., Li L., Lv W. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat. Med. 2018;24:1513–1518. doi: 10.1038/s41591-018-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen B., Gu P., Jia J., Liu W., Liu Y., Liu W., Xu T., Lin X., Lin T., Liu Y. Optimization strategy for generating gene-edited tibet minipigs by synchronized oestrus and cytoplasmic microinjection. Int. J. Biol. Sci. 2019;15:2719–2732. doi: 10.7150/ijbs.35930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Petersen B., Lucas-Hahn A., Oropeza M., Hornen N., Lemme E., Hassel P., Queisser A.L., Niemann H. Development and validation of a highly efficient protocol of porcine somatic cloning using preovulatory embryo transfer in peripubertal gilts. Cloning Stem Cells. 2008;10:355–362. doi: 10.1089/clo.2008.0026. [DOI] [PubMed] [Google Scholar]
- 68.Xie Y., Lv X., Ni D., Liu J., Hu Y., Liu Y., Liu Y., Liu R., Zhao H., Lu Z., Zhou Q. HPD degradation regulated by the TTC36-STK33-PELI1 signaling axis induces tyrosinemia and neurological damage. Nat. Commun. 2019;10:4266–4277. doi: 10.1038/s41467-019-12011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turgeon C., Magera M.J., Allard P., Tortorelli S., Gavrilov D., Oglesbee D., Raymond K., Rinaldo P., Matern D. Combined newborn screening for succinylacetone, amino acids, and acylcarnitines in dried blood spots. Clin. Chem. 2008;54:657–664. doi: 10.1373/clinchem.2007.101949. [DOI] [PubMed] [Google Scholar]
- 70.Purevsuren J., Hasegawa Y., Kobayashi H., Endo M., Yamaguchi S. Urinary organic metabolite screening of children with influenza-associated encephalopathy for inborn errors of metabolism using GC/MS. Brain Dev. 2008;30:520–526. doi: 10.1016/j.braindev.2008.01.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.