

Abstract
The locus coeruleus (LC) is one of the essential chemoregulatory and sleep–wake (S–W) modulating centers in the brain. LC neurons remain highly active during wakefulness, and some implicitly become silent during rapid eye movement (REM) sleep. LC neurons are also involved in CO2-dependent modulation of the respiratory drive. Acid-sensing ion channels (ASICs) are highly expressed in some brainstem chemosensory breathing regulatory areas, but their localization and functions in the LC remain unknown. Mild hypercapnia increases the amount of non-REM (NREM) sleep and the number of REM sleep episodes, but whether ASICs in the LC modulate S–W is unclear. Here, we investigated the presence of ASICs in the LC and their role in S–W modulation and the state transition from NREM to REM sleep. Male Wistar rats were surgically prepared for chronic polysomnographic recordings and drug microinjections into the LC. The presence of ASIC-2 and ASIC-3 in the LC was immunohistochemically characterized. Microinjections of amiloride (an ASIC blocker) and APETx2 (a blocker of ASIC-2 and -3) into the LC significantly decreased wakefulness and REM sleep, but significantly increased NREM sleep. Mild hypercapnia increased the amount of NREM and the number of REM episodes. However, APETx2 microinjection inhibited this increase in REM frequency. These results suggest that the ASICs of LC neurons modulate S–W, indicating that ASICs could play an important role in vigilance-state transition. A mild increase in CO2 level during NREM sleep sensed by ASICs could be one of the determinants of state transition from NREM to REM sleep.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12264-020-00625-0.
Keywords: Acid-sensing ion channels, Carbon dioxide, Hypercapnia, NREM sleep, REM sleep
Introduction
The locus coeruleus (LC) plays an essential role in the modulation of physiological processes, including the CO2-dependent modulation of respiratory drive and sleep–wakefulness [1–7]. The LC potentiates ventilation in response to an increase in the systemic CO2 level [8]. In vitro studies in neonates have demonstrated that the majority of LC neurons (> 80%) show increased activity in response to a reduced pH [8–11].The intrinsic property of LC neurons to sense changes in CO2/H+ levels remains intact in cultured LC neurons [12]. In adult and vagotomized rats, the firing pattern of most LC neurons shows central respiratory modulation: some neurons fire during the inspiratory phase and others fire during the post-inspiratory phase [13]. In addition, these neurons can be activated by stimulating peripheral chemoreceptors [13]. Moreover, lessoning of brainstem norepinephrinergic (NE-ergic) neurons, including the LC, largely attenuates both normal breathing and the breathing induced by hypercapnia [14–16]. The acidosis induced by microinjecting acetazolamide into the LC enhances phrenic nerve activity and the breathing rate [8, 17]. The LC modulates hypercapnia-induced ventilator responses via both electrical (gap junctions) and chemical (such as glutamatergic, serotonergic and orexinergic) synapses [18, 19]. Studies have shown that microinjection of the gap junction blocker carbenoxolone, an ionotropic glutamate receptor antagonist kynurenic acid, or the 5-HT1A receptor antagonist WAY-100635 into the LC alters hypercapnia-induced pulmonary ventilation [18–20]. Interestingly, inhibition of orexin-1 receptors in the LC during the active circadian phase attenuates the hypercapnia-induced chemoreflex during wakefulness but not during sleep [21]. Therefore, these results suggest that the LC is one of the CO2-dependent respiratory modulatory centers, and plays an essential role in the vigilant state-mediated changes in the respiratory pattern.
LC neurons also exhibit vigilant state-dependent changes in their firing properties [1, 4, 22]: they are highly active during wakefulness, much less active during non-rapid eye movement (NREM) sleep, and virtually silent during REM sleep [22, 23]. Studies have suggested that activated LC neurons are involved in the maintenance of wakefulness, while deactivated LC neurons facilitate the generation of NREM and REM sleep [4, 24–27]. Most LC neurons are norepinephrinergic (NE-ergic) and a deficiency of dopamine beta-hydroxylase (an enzyme that converts dopamine to NE) augments NREM sleep, while significantly decreasing wakefulness and REM sleep [28, 29]. Similarly, brainstem neuronal lesions with conjugated saporin anti-dopamine β-hydroxylase alter ventilation and CO2-mediated chemosensitivity [14]. Therefore, the LC is also one of the centers that function in CO2-mediated changes in S–W states.
Changes in systemic CO2 levels or extracellular pH in the brainstem area can alter sleep–wakefulness [4, 30]. For example, mild hypercapnia induced by 2%–4% CO2 in inspired air increases the amount of NREM sleep and the frequency of REM sleep episodes [4, 31], whereas severe hypercapnia induced by > 5% CO2 in inspired air significantly induces wakefulness and decreases NREM and REM sleep [4, 31]. On the other hand, the systemic CO2 levels significantly increase during NREM sleep, possibly because of the reduced breathing rate in this state [32–34]. Infusion of acidic solution through microdialysis in the dorsal raphe nucleus (DRN) induces arousals from sleep [35]. Interestingly, the loss of 5-hydroxytryptamine (5-HT) receptors in the DRN aborts CO2-mediated arousal from sleep [35, 36]. CO2-induced arousal also activates glutamatergic neurons in the lateral parabrachial nucleus, and deletion of the vesicular glutamate transporter-2 gene aborts CO2-induced arousal [37]. Therefore, these results indicate that altered pH in the chemosensory areas of the brain predisposes an individual to state transitions between wake and sleep. However, the underlying mechanism is not known.
Studies have revealed that several receptors, transporters, and ion channels, including acid-sensing ion channels (ASICs) in brainstem chemosensory neurons, are involved in the detection of extracellular pH fluctuations [5, 38–40]. The ASIC is one of the most sensitive ion channels in detecting changes in pH [41], primarily because of its low pH-threshold, which makes it possible to detect pH changes in a very narrow physiological range [41]. In addition to pH sensing, ASICs are involved in the modulation of other physiological and behavioral processes such as learning and memory [42], pain perception [43], and neurodegenerative disorders [44, 45]. ASICs are expressed in several brain areas such as solitary nucleus, ventral medulla, amygdala, hippocampus, and hypothalamus [39, 46, 47]. The LC plays an essential role in sleep–wakefulness and CO2-mediated respiratory drive across the vigilant state. However, it remains unknown whether ASICs in the LC function in sleep–wake modulation and the hypercapnia-mediated influence on sleep–wakefulness. We have proposed earlier that mild CO2 accumulation during prolonged NREM sleep could be one of the factors that initiate the state transition from NREM to REM sleep [4]. Moreover, ASICs can detect slight changes in pH, and the LC isa part of the central CO2-chemoregulatory system. Based on these findings, we tested whether ASICs are present on LC neurons and if they are also involved in S–W regulation, and finally whether they play a role in the mild hypercapnia-induced state transition from NREM to REM sleep.
Materials and Methods
In this study, we used male Wistar rats (n = 22) weighing 250 g–300 g. Rats were obtained from the Central Laboratory Animal Resources facility and kept for a week in the school’s animal room for habituation. The rats were maintained under a 12-hlight-dark cycle with lights on at 07:00 and lights off at 19:00. Room temperature was maintained at 23 °C ± 1 °C. Food and water were given ad libitum. All procedures and protocols were approved by the Institutional Animal Ethics Committee (IAEC Protocol #14/2015) of Jawaharlal Nehru University, New Delhi, India.
To address our hypotheses, we conducted three experiments: Experiment-I to characterize the presence of ASICs on LC neurons; Experiment-II to investigate their role in sleep–wake modulation; and Experiment III to investigate their role in the mild hypercapnia-induced state transition from NREM to REM sleep.
Experiment-I
Immunohistochemical Localization of ASIC-2 and ASIC-3 on NE-Ergic Neurons in the LC
For immunohistochemical localization of ASIC-2 and ASIC-3 on LC NE-ergic neurons, each rat (n = 7) was deeply anesthetized and transcardially perfused with ice-cold 0.1 M phosphate-buffered saline (PBS) followed by 4% paraformaldehyde in 0.1 mol/L PBS. The brain was extracted and kept overnight in 4% paraformaldehyde, followed by 30% sucrose for 2–3 days. Coronal brain sections were cut at 40 μm on a cryostat (Thermo Fisher Scientific). The sections were kept in 0.1 mol/L PBS overnight to wash off the formaldehyde traces and incubated in a blocking solution (4% goat serum in 0.1 mol/L PBS with 0.3% Triton-X) for 2 h at room temperature. After washing with 0.1 mol/L PBS (3 times, 5 min each), the sections were incubated with primary antibodies against ASIC-2 (1:500, ACCN2-OSR00098W, Thermo Fisher Scientific) or ASIC-3 (1:1000, Anti Sodium Channel ASIC-3-S5070, Sigma Aldrich) and anti-tyrosine hydroxylase (1:1000, AB1542, Millipore) for 72 h at 4 °C. After primary incubation, the sections were washed (3 times, 10 min each) with 0.1 mol/L PBS and then incubated in anti- rabbit Alexa-480 secondary antibodies (1:1200, Thermo Fisher Scientific) for ASICs and anti-sheep Alexa-555 for tyrosine hydroxylase (1:1200, Thermo Fisher Scientific) for 24 h in the dark at 4 °C. After washing with 0.1 mol/L PBS (3times, 10 min each), the sections were mounted on glass slides with Fluoroshield mounting medium (Sigma Aldrich) under coverslips. Slides were observed under fluorescence microscope (Olympus-B53 Model, Japan) at 10X magnifications for localization of ASICs in the LC.
Experiment-II
Role of ASICs in the LC in S–W Modulation
Surgical Procedures for Polysomnographic Recording and Cannula Implantation
Rats were surgically prepared for chronic S–W recording and cannula implantation for drug infusions using a previously reported procedure [48–51]. Briefly, rats were anesthetized with 4% isoflurane (Baxter Healthcare, India) using a facemask. The head was shaved and fixed in stereotaxic apparatus. A midline incision was made with a sterile surgical blade and the skull exposed for electrode implantation. Two pairs of small, stainless-steel screw electrodes were affixed to the skull above the frontal and parietal cortices to record the electroencephalogram (EEG). Three electrodes (flexible wires, insulated except at the tip) were implanted in the dorsal neck muscles to record the bipolar electromyogram (EMG) (the third electrode was implanted as a safeguard). One screw electrode was fixed lateral to the midline in the nasal bone as a reference. For microinjection of drugs into the LC, 24-gauge stainless steel guide cannulas were implanted bilaterally at the co-ordinates AP: − 9.8 mm, ML: 1.3 mm, and DV: 6 mm with reference to bregma [52]. The guide cannulae were placed 1 mm above the LC to prevent any mechanical damage to LC neurons and fixed to the skull with acrylic dental cement. Styles were inserted into the guide cannulas to prevent occlusion. The free ends of the EEG, EMG, and reference electrodes were soldered to a 9-pin miniature connector and cemented to the skull with dental acrylic. Finally, the neck skin was sutured, and the rat was removed from the stereotaxic apparatus. Each rat was treated with dexamethasone (1.5 mg/kg) for 3–4 days post-operation to reduce brain inflammation and Nebasulf powder (antibiotic) was used to control potential infection. Each rat was allowed one week to recover from surgery.
Experimental Procedure
We studied the effects of amiloride (a universal blocker of ASICs) and APETx2 (a specific blocker of ASIC-2 and ASIC-3) in the LC on sleep–wake architecture. After recovery from surgery, rats were randomly divided into two groups: (1) the amiloride microinjected group (n = 7) in which a low and a high dose of Amiloride were microinjected into the LC after a gap of 48 h in random order; and (2) the APETx2 microinjected group (n = 8) in which a low and a high dose of APETx2 were microinjected into the LC after a gap of 48 h in random order. A gap of 48 h was chosen because the half-life of both drugs is ~ 3–9 h. Hence, during the 48-h period, the residual drugs were completely washed out from the site of injection [53, 54].
Polysomnographic Recordings
Sleep–wakefulness was recorded in a sleep recording cage (38 cm × 28 cm × 20 cm, length, width, and height) placed in a well-ventilated and sound dampened (dark-colored) Plexiglas chamber (122 cm × 60 cm × 60 cm) illuminated at 20 Lux to minimize external disturbances. The rats were provided with food and water in the sleep recording chamber. For the first two days (days 1 and 2), rats were habituated to the chamber for 6 h (11:00–17:00), during which they were tethered to the recording cable through a commutator. The EEG and EMG signals were examined in a computer using Somnologica Science software and Embla A10 (MedcareFlaga, Iceland). After habituation, sleep–wakefulness was recorded for two days (days 3 and 4) as baseline. On days 5, 6 and 7, sleep–wake was recorded after microinjections of either vehicle or low/high doses of drugs. Microinjections were always performed after a gap of one day between two injections in a randomized fashion. EEG signals were processed with high-pass 0.1 Hz and low-pass 40 Hz filters, while EMG signals were processed with high-pass 10 Hz and low-pass 90 Hz filters digitized at 100 Hz sampling rate. The recordings were saved in a computer for offline analysis.
Microinjections of Amiloride and APETx2 into the LC
We microinjected the ASIC blockers, amiloride and APETx2 into the LC (pH 7.4; 200 nL). Two different doses of amiloride (2 mmol/L and 20 mmol/L) were prepared by dissolution in DMSO and then dilution with distilled water (pH 7.4) as in previous reports [39, 55, 56]. Similarly, 0.5 µmol/L and 1 µmol/L of APETx2 (Tocris Bioscience, UK) were prepared in 0.9% sterile saline solution and the pH was adjusted to 7.4. The doses of APETx2 were based on its inhibitory potency on ASICs of the neurons, which ranges from 63 nmol/L to 2 µmol/L [57]. In the control groups, 200 nL of either distilled water with DMSO or 0.9% sterile saline solution was microinjected as vehicle in the respective groups. The drugs or vehicles were injected bilaterally into the LC with a microinfusion pump (PHD Ultra Syringe Pumps, Harvard Apparatus) and an injector cannula (30-gauge; 18 mm long). The injector cannulas were connected to Hamilton syringes (100 µL) through PE tubing, and inserted into the guide cannulas in semi-restrained animals. The injection lasted > 2 min, at a flow rate of 100 nL/min. The injector cannula was left in the guide cannula for 2 min before withdrawal, and the stylet was returned to the guide cannula. Sleep–wake recording was started immediately.
Experiment-III
Role of LC ASICs in the Mild Hypercapnia-Induced State Transition from NREM to REM Sleep
Polysomnographic Recording in the CO2 Chamber
In another group, the rats were prepared for chronic sleep–wake recording using a similar protocol to Experiment II. The rats were divided into four groups (n = 5 rats per group): vehicle alone, CO2 + vehicle, CO2 + APETx2, and APETx2 only. Sleep–wake was recorded in the CO2 chamber in the presence of either room air or 4% CO2 at constant inflow and outflow. Each rat was first habituated to the air-tight chamber infused with normal room air for two days. On day 3, baseline sleep was recorded for 4 h (11:00–15:00) after vehicle microinjection into the LC. On days 4 and 5, sleep–wake was recorded in the presence of 4% CO2 for an initial 1 h after microinjection of either vehicle or APETx2 into the LC. The doses and volume of APETx2 used were also the same as those in Experiment II. Thereafter, normal room air was infused into the chamber, and the recording was continued for another 3 h. Thus, sleep–wake was recorded for a total of 4 h (1 h during 4% CO2 infusion and 3 h without CO2). CO2 was not infused during the initial 1 h in the Vehicle only and APETx2 only groups. Recordings were saved in the computer for offline analysis.
Histology
At the end of Experiments II and III, each rat was sacrificed with an overdose of cocktail anesthesia (80 mg/kg Ketamine and 40 mg/kg Xylazine). Each rat was transcardially perfused with 0.9% saline and 10% formalin for 15 min each. The brain was removed and stored in 10% formalin for further histological analysis. Prior to histology, the brain was immersed in sucrose solution for a day or two at room temperature. Coronal sections were cut at 40 µm on a cryostat (Thermo Fisher Scientific, USA) and mounted on subbed glass slides. The sections were then stained with 0.1% cresyl violet, and the cannula injection sites were identified under microscope (Fig. 1). The injection sites in 3 rats in Experiment II were found to be outside the LC. The data of these three animals were used as an additional control (Fig. S1).
Fig. 1.
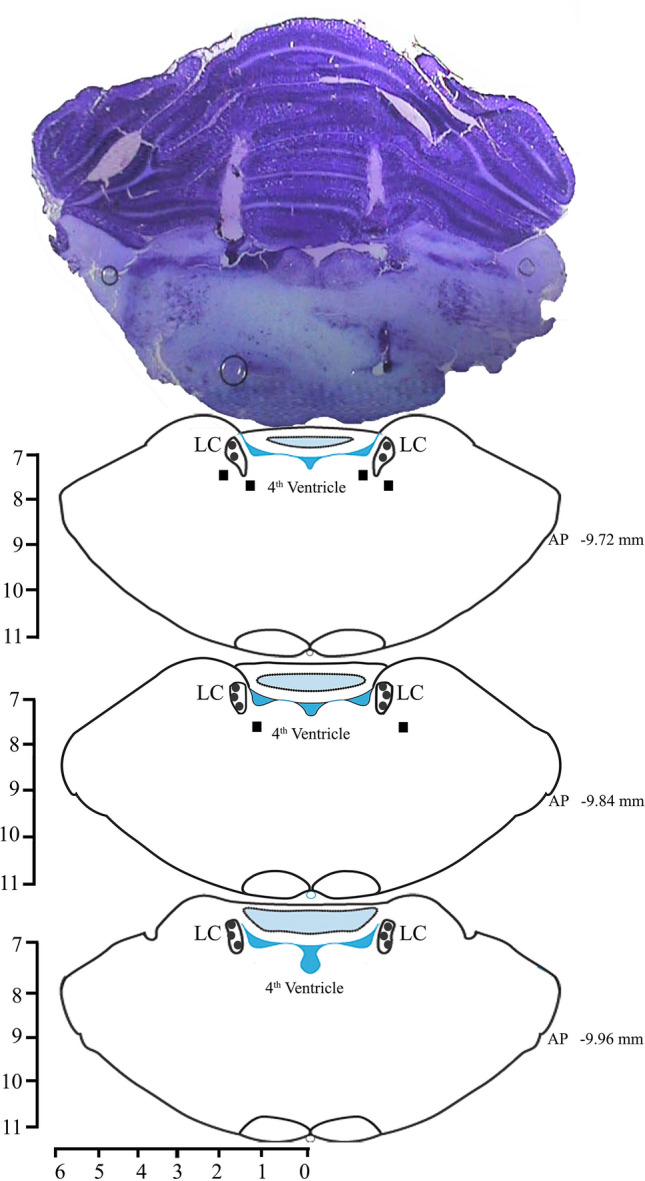
Photomicrograph of a 40 µm section stained with cresyl violet and reconstructions of the sites of injection within (filled circles) and outside (filled boxes) the LC.
Data Analysis
The polysomnographic recordings in experiments-II and III were scored offline using Somnologica Science Software (MedcareFlaga, Iceland). Sleep recordings were manually scored in 4-s epochs, using the standard criteria for the rat [48]. Low-voltage and high-frequency EEG waves associated with increased motor activity were marked as awake. Epochs of high-voltage, low-frequency EEG waves with prominent delta waves (0.5–4 Hz), and decreased motor activity we remarked as NREM sleep, while epochs with low-voltage, high-frequency EEG waves with a prominent theta peak (5–9 Hz) and neck muscle atonia were marked as REM sleep. The total times spent awake, and in NREM and REM sleep were calculated and expressed as total mean percentages and hourly mean percentages of the total recording time. In all our experiments, the same animal served as a self control. The changes in the amount of different vigilance states between treatments (low and high doses) and drug types (amiloride and APETx2) were compared statistically using two-way ANOVA followed by the Bonferroni post hoc test. The number and average duration of NREM as well as REM sleep episodes in treatments and drug types were also calculated and statistically compared using two-way ANOVA followed by the Bonferroni post hoc test. The hypercapnia-mediated changes in S–W and its parameters were compared with normocapnia using one-way RM ANOVA.
Results
Immunohistochemical Localization of ASIC-2 and ASIC-3 on NE-Ergic and non-NE-Ergic Neurons in the LC
Our immunohistochemical results showed that ASIC-2 and ASIC-3 were present on both NE-ergic and non-NE-ergic neurons in the LC. We characterized ASIC-2 and ASIC-3 receptors on the NE-ergic LC neurons using double-labeling immunohistochemical methods. We used specific primary antibodies against ASIC-2 and ASIC-3 as well as an antibody against the tyrosine hydroxylase (TH) enzyme to label LC NE-ergic neurons. We observed ASIC-2 and TH co-labeled neurons as well as ASIC-3 and TH co-labeled neurons in the LC (Fig. 2). However, some TH-negative neurons were also labeled with the ASIC antibodies. Although most ASIC-2+ neurons were TH+ but some ASIC-3+ neurons were also TH-negative (Fig. 2) (co-ordinates: – 9.7 mm to – 10.0 mm from bregma).The expression levels of ASIC-2+ and ASIC-3+ neurons from the rostro-caudal plane in the LC were similar. For a negative control, we incubated the histological sections with secondary antibodies only (the sections were not incubated with the ASIC primary antibodies) to show that the binding of secondary antibodies was specific to primary antibodies. We found no expression of ASIC-2 and ASIC-3 in the LC region in the absence of primary antibodies (Fig. 2). This demonstrated that the expression was specific to the reactivity of the primary antibodies with ASIC-2 and ASIC-3. These antibodies have been used to show the presence of ASICs in other brain areas [54, 55]. Thus, ASIC-2 and ASIC-3 are present on LC neurons.
Fig. 2.

Localization of ASIC-2 and ASIC-3 on LC neurons. Representative fluorescent images showing immunoreactive ASIC-2 (upper panels) and ASIC-3 (lower panels) cells in the LC region (rostro-caudal co-ordinates: –9.7 to –10.0 mm from bregma). Negative control, fluorescence images without primary antibodies; green, ASIC-2- (upper panels) and ASIC-3- (lower panels) positive neurons; red, TH-positive neurons in the LC; yellow, ASIC-2 (upper merged panel) and ASIC-3 (lower merged panel) neurons co-labeled with TH; inset, ASIC and TH co-labeled neurons in a magnified view.
Role of ASICs in the LC in S–W Modulation
Microinjections of the ASIC blockers amiloride and APETx2 into the LC increased NREM sleep and decreased wakefulness and REM sleep.
The total amounts of wakefulness, NREM, and REM sleep (out of the total recording time) in the amiloride and APETx2 microinjected groups are shown in Fig. 3. Two-way ANOVA revealed that these microinjections significantly decreased wakefulness and REM sleep. However, the amount of NREM sleep increased significantly compared with the vehicle groups. For the changes in the amount of wakefulness and NREM sleep in the amiloride and APETx2 microinjected groups, two-way ANOVA showed significant main effects of drugs (low and high doses) only, but no significant interaction between types of drugs (Amiloride vs. APETx2).
Fig. 3.
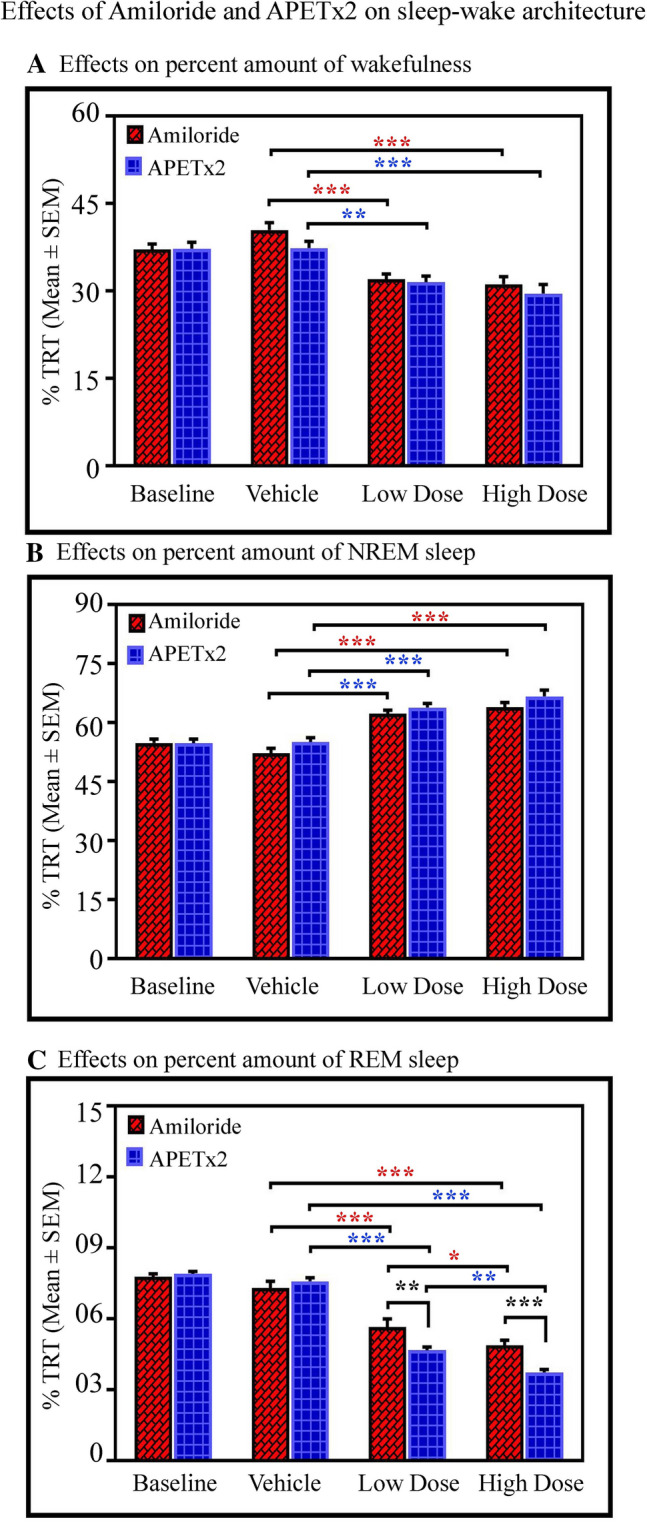
Effects of amiloride and APETx2 microinjections into the LC on sleep–wake architecture as shown in the total recording time (TRT) bar graph. Microinjections of amiloride (n = 7) and APETx2 (n = 8) into the LC (A) decreases wakefulness (P < 0.001, F(3,59) = 23.63, two-way ANOVA), (B) increases NREM sleep (P < 0.001, F(3,59) = 49.89, two-way ANOVA), and (C) decreases REM sleep (P < 0.001, F(3,59) = 163.03 two-way ANOVA).
The post hoc comparison demonstrated that the low (2 mmol/L) and high (20 mmol/L) doses of amiloride decreased wakefulness by 20% (Bonferroni test P < 0.001; Cohen’s d = 2.72; power = 0.99 at alpha level 0.05) and by 22%, (Bonferroni test P < 0.001; Cohen’s d = 8.01; power = 1 at alpha level 0.05) respectively, compared with the vehicle group. Similarly, in the APETx2 group, post hoc comparison showed that the amount of wakefulness decreased by 16% (Bonferroni test, P < 0.01; Cohen’s d = 1.82; power = 0.93 at alpha level 0.05) and by 21% (Bonferroni test, P < 0.001; Cohen’s d = 1.99; power = 0.94 at alpha level 0.05) in the low (0.5 µmol/L) and high (1 µmol/L) dose groups respectively compared with the vehicle group (Fig. 3A).
The amount of NREM sleep significantly increased after microinjections of amiloride and APETx2 into the LC. The post hoc comparison demonstrated that the low dose of amiloride increased NREM by 19.3% (Bonferroni test, P < 0.001; Cohen’s d = 3.47; power = 1 at alpha level 0.05) and the high dose increased it by 22% (Bonferroni test, P < 0.001; Cohen’s d = 3.66; power = 1 at alpha level 0.05) compared with the vehicle group (Fig. 3B). Similarly, in the APETx2 microinjected groups, the post hoc comparison showed that NREM increased by 16% (Bonferroni test, P < 0.001; Cohen’s d = 2.83; power = 1 at alpha level 0.05) in the low dose group, and by 21% (Bonferroni test, P < 0.001; Cohen’s d = 2.45; power = 0.99 at alpha level 0.05) in the high dose group compared with the vehicle group. The two-way ANOVA did not show any interaction between high and low doses of drugs on NREM (Fig. 3B).
Microinjections of amiloride and APETx2 into the LC significantly decreased REM sleep. Two-way ANOVA revealed significant effects of drug treatments (P < 0.01; F(1,59) = 9.84) and drug types (P < 0.001; F(3,59) = 163.03). Two-way ANOVA also revealed a significant interaction between treatment versus types (high and low doses vs. amiloride and APETx2) (P < 0.001; F(3,59) = 7.42). The post hoc comparison demonstrated that the low dose of amiloride decreased REM by 23% compared with vehicle (Bonferroni test, P < 0.001; Cohen’s d = 2.01; power = 0.94 at alpha level 0.05), while the high dose decreased REM by 33% compared with vehicle (Bonferroni test, P < 0.001; Cohen’s d = 3.64; power = 1 at alpha level 0.05) (Fig. 3C). Similarly, post hoc comparison showed that the low dose of APETx2 decreased REM by 39% compared with vehicle (Bonferroni test, P < 0.001; Cohen’s d = 7.50; power = 1 at alpha level 0.05), while the high dose decreased REM by 51% compared to vehicle (Bonferroni test, P < 0.001; Cohen’s d = 10.43; power = 1 at alpha level 0.05). Two-way ANOVA also demonstrated significant effects of drug types, and the post hoc comparison revealed that REM decreased in the APETx2 low-dose group compared to the amiloride low-dose group (Bonferroni test, P < 0.001; Cohen’s d = 1.75; power = 0.78 at alpha level 0.05). Similarly, REM sleep was decreased in the APETx2 high-dose group compared to the amiloride high-dose group (Bonferroni test, P < 0.001; Cohen’s d = 2.46; power = 0.99 at alpha level 0.05) (Fig. 3C). In three rats, the sites of injection were not in the LC but in the vicinity (Fig. 1). In these rats, we did not find changes in the percentages of wakefulness, NREM and REM in the groups microinjected with low and high doses of amiloride and APETx2 compared to their baseline and vehicle groups (Fig. S1). These data suggested that only the ASICs in the LC influence S–W architecture but not those located in the vicinity. Although we injected a minimum of 200 nL of drugs, it was still not possible to rule out micro-spill over into the ventricle because the injection sites were near the ventricles.
Hourly expression of the amounts of wakefulness, NREM, and REM sleep in the amiloride and APETx2 microinjected groups are shown in Tables 1, 2 and 3. Hourly expression of wakefulness in the amiloride groups showed a decreasing trend with significant changes at 2 h and 3 h in the low-dose group and 5 h and 6 h in the high-dose group compared to the vehicle group (Table 1). Similarly, in the APETx2 groups, hourly expression of wakefulness decreased significantly at 3 h and 6 h in the low-dose group and at 3 h in the high-dose group compared to the vehicle group (Table 1). In the amiloride groups, there was an increasing trend in NREM sleep at every hour with a significant increase at 2 h and 3 h in the low-dose group, and at 2 h, 3 h and 6 h in the high-dose group compared to the vehicle group (Table 2). Similarly, in the APETx2 groups, we also found an increasing trend in NREM sleep at every hour with a significant increase at 3 h and 6 h in the low-dose group and at 2 h, 3 h, 4 h, and 6 h in the high-dose group compared to the vehicle group (Table 2). Hourly expression of REM sleep in the Amiloride groups showed a decline with a significant change at 3 h, 4 h, and 5 h only in the high-dose group. A similar decreasing trend was also found in the APETx2 groups, with significant change at 2 h, 4 h, and 5 h in the low-dose group, and at 2 h, 3 h, 5 h, and 6 h in the high-dose groups compared with the vehicle group (Table 3).
Table 1.
Hourly expression of wakefulness (% Total Recording Time).
| Hour 1 | Hour 2 | Hour 3 | Hour 4 | Hour 5 | Hour 6 | |
|---|---|---|---|---|---|---|
| Amiloride microinjected animals | ||||||
| Baseline | 65.48 ± 7.82 | 31.68 ± 5.25 | 35.76 ± 4.21 | 35.76 ± 5.13 | 33.82 ± 4.75 | 38.50 ± 4.02 |
| Vehicle | 67.35 ± 6.05 | 41.64 ± 6.87 | 33.25 ± 4.15 | 35.57 ± 4.15 | 37.00 ± 8.17 | 36.00 ± 6.05 |
| Low dose | 50.19 ± 8.42 | 28.26 ± 4.71** | 22.00 ± 2.82* | 28.27 ± 7.85 | 29.59 ± 5.11 | 33.95 ± 4.62 |
| High dose | 50.32 ± 9.21 | 18.58 ± 6.26 | 26.72 ± 4.10 | 33.49 ± 6.12 | 34.01 ± 5.18* | 23.94 ± 2.74** |
| APETx2 microinjected animals | ||||||
| Baseline | 60.36 ± 7.019 | 19.67 ± 3.75 | 28.80 ± 5.34 | 36.79 ± 6.21 | 31.48 ± 4.91 | 34.03 ± 6.34 |
| Vehicle | 61.04 ± 8.05 | 22.22 ± 4.10 | 31.17 ± 4.15 | 36.16 ± 4.84 | 37.40 ± 7.74 | 41.76 ± 6.33 |
| Low dose | 50.70 ± 6.26 | 22.63 ± 5.87 | 19.34 ± 3.13* | 31.28 ± 5.88 | 36.58 ± 3.24 | 27.45 ± 4.45** |
| High dose | 40.04 ± 5.69 | 15.87 ± 2.11 | 17.17 ± 4.81* | 28.27 ± 4.90 | 37.07 ± 6.61 | 37.36 ± 3.83 |
(*P < 0.05, **P < 0.01, one- way RM ANOVA followed by Tukey’s post hoc test).
Table 2.
Hourly expression of NREM sleep (% TRT) in the.
| Hour 1 | Hour 2 | Hour 3 | Hour 4 | Hour 5 | Hour 6 | |
|---|---|---|---|---|---|---|
| Amiloride microinjected animals | ||||||
| Baseline | 32.72 ± 6.99 | 60.05 ± 6.93 | 55.03 ± 3.86 | 52.11 ± 3.83 | 65.99 ± 3.62 | 53.80 ± 4.06 |
| Vehicle | 31.09 ± 5.42 | 54.41 ± 7.05 | 57.29 ± 4.53 | 56.55 ± 3.65 | 59.21 ± 4.71 | 59.95 ± 4.90 |
| Low dose | 48.57 ± 8.21 | 69.58 ± 4.75** | 71.27 ± 3.38* | 65.54 ± 7.28 | 60.91 ± 4.86 | 57.05 ± 3.68 |
| High dose | 46.98 ± 8.86 | 76.75 ± 5.54 | 68.96 ± 3.54* | 62.28 ± 5.81 | 59.83 ± 4.74 | 68.78 ± 2.33** |
| APETx2 microinjected animals | ||||||
| Baseline | 38.49 ± 6.92 | 70.47 ± 3.56 | 61.59 ± 4.29 | 52.92 ± 4.95 | 57.71 ± 4.41 | 47.02 ± 5.13 |
| Vehicle | 37.70 ± 7.95 | 68.15 ± 2.65 | 56.93 ± 3.21 | 53.68 ± 5.05 | 55.76 ± 6.66 | 58.19 ± 5.46 |
| Low dose | 48.33 ± 5.92 | 73.42 ± 6.36 | 73.22 ± 3.17** | 62.71 ± 5.92 | 58.45 ± 2.98 | 66.79 ± 3.67** |
| High dose | 55.38 ± 5.32 | 79.72 ± 2.16* | 76.09 ± 4.26* | 66.47 ± 3.92* | 59.01 ± 6.55 | 59.43 ± 3.26* |
*P < 0.05, **P < 0.01, one-way RM-ANOVA followed by Tukey’s post hoc test.
Table 3.
Hourly expression of REM sleep (% TRT).
| Amiloride | Hour 1 | Hour 2 | Hour 3 | Hour 4 | Hour 5 | Hour 6 |
|---|---|---|---|---|---|---|
| Amiloride microinjected animals | ||||||
| Baseline | 1.80 ± 1.16 | 8.27 ± 3.28 | 9.20 ± 1.57 | 8.51 ± 1.58 | 8.60 ± 1.64 | 7.55 ± 1.90 |
| Vehicle | 1.56 ± 0.88 | 6.57 ± 1.24 | 9.46 ± 1.01 | 7.89 ± 1.73 | 8.99 ± 2.85 | 9.87 ± 2.34 |
| Low dose | 1.25 ± 0.71 | 2.16 ± 0.79 | 6.73 ± 1.55 | 6.19 ± 1.99 | 9.50 ± 2.81 | 8.13 ± 1.74 |
| High dose | 2.69 ± 1.36 | 4.66 ± 1.98 | 4.31 ± 1.51* | 4.22 ± 1.01* | 6.15 ± 1.19* | 7.14 ± 1.64 |
| APETx2 microinjected animals | ||||||
| Baseline | 1.14 ± 0.57 | 9.85 ± 1.93 | 9.59 ± 1.98 | 10.28 ± 3.20 | 10.80 ± 1.38 | 5.93 ± 1.32 |
| Vehicle | 0.83 ± 0.43 | 8.59 ± 1.72 | 10.90 ± 2.08 | 10.15 ± 2.00 | 6.83 ± 2.05 | 6.78 ± 1.94 |
| Low dose | 0.57 ± 0.15 | 4.77 ± 1.37* | 7.42 ± 2.00 | 6.00 ± 0.99* | 4.97 ± 1.14** | 4.59 ± 1.05 |
| High dose | 1.52 ± 0.66 | 4.19 ± 1.22* | 4.65 ± 1.29* | 5.25 ± 1.62 | 3.92 ± 1.12** | 2.86 ± 1.07* |
*P < 0.05, **P < 0.01, one-way RM-ANOVA followed by Tukey’s post hoc test.
The changes in the sleep–wake parameters are shown in Fig. 4. The average length and number of wakefulness episodes did not change (Figs. 4A, B). Two-way ANOVA, however, showed main effects of drugs on the length of NREM episodes only (P < 0.05; F(3,59) = 5.23) (Fig. 4C), and the numbers of NREM sleep episodes did not change (Fig. 4D).The post hoc comparison showed that the length of NREM sleep episodes increased in the APETx2 high-dose group compared to the vehicle group (Bonferroni test, P < 0.05; Cohen’s d = 1.90; power = 0.85 at alpha level 0.05)(Fig. 4C). The length of REM sleep episodes did not change significantly with the drug types (Fig. 4E). Moreover, two-way ANOVA revealed main effects of the drug (P < 0.001; F(3,59) = 9.65) on the number of REM sleep episodes(Fig. 4F). The post hoc comparison showed that the number of REM sleep episodes was less in theAPETx2 high-dose group than in the vehicle group (Bonferroni test, P < 0.01; Cohen’s d = 2.25; power = 0.99 at alpha level 0.05) (Fig. 4F).
Fig. 4.

Effects of amiloride and APETx2 microinjections into the LC on sleep–wake parameters. Two-way ANOVA reveals no significant effect of the drugs on wake episode length (A) and episode number (B). Blocking ASICs with a high dose of APETx2 (C) increases the length of NREM sleep episodes compared to the vehicle group (P < 0.05), however, the number of NREM sleep episodes does not change (D). On the other hand, APETx2 does not change the length of REM sleep episodes (E), whereas a high dose of APETx2 decreases the number of REM sleep episodes (F) compared to the vehicle group (P < 0.01) (*P < 0.05, **P < 0.01, Bonferroni post hoc test).
Role of LC ASICs in the Mild Hypercapnia-Induced State Transition from NREM to REM Sleep
Next, we investigated the effects of hypercapnia on S–W architecture and the effects of APETx2 microinjection into the LC on the CO2-induced changes in S–W. The rats were divided into four groups (n = 5 rats per group): (a) vehicle only, (b) CO2 (4%) + vehicle, (c) CO2 + APETx2, and (d) APETx2 alone [comparable animals were used in all groups; data from the vehicle-only group are shown as normocapnia and from the CO2 + vehicle group as mild hypercapnia (4% CO2) in Fig. 5].
Effects of hypercapnia on S–W architecture
Fig. 5.
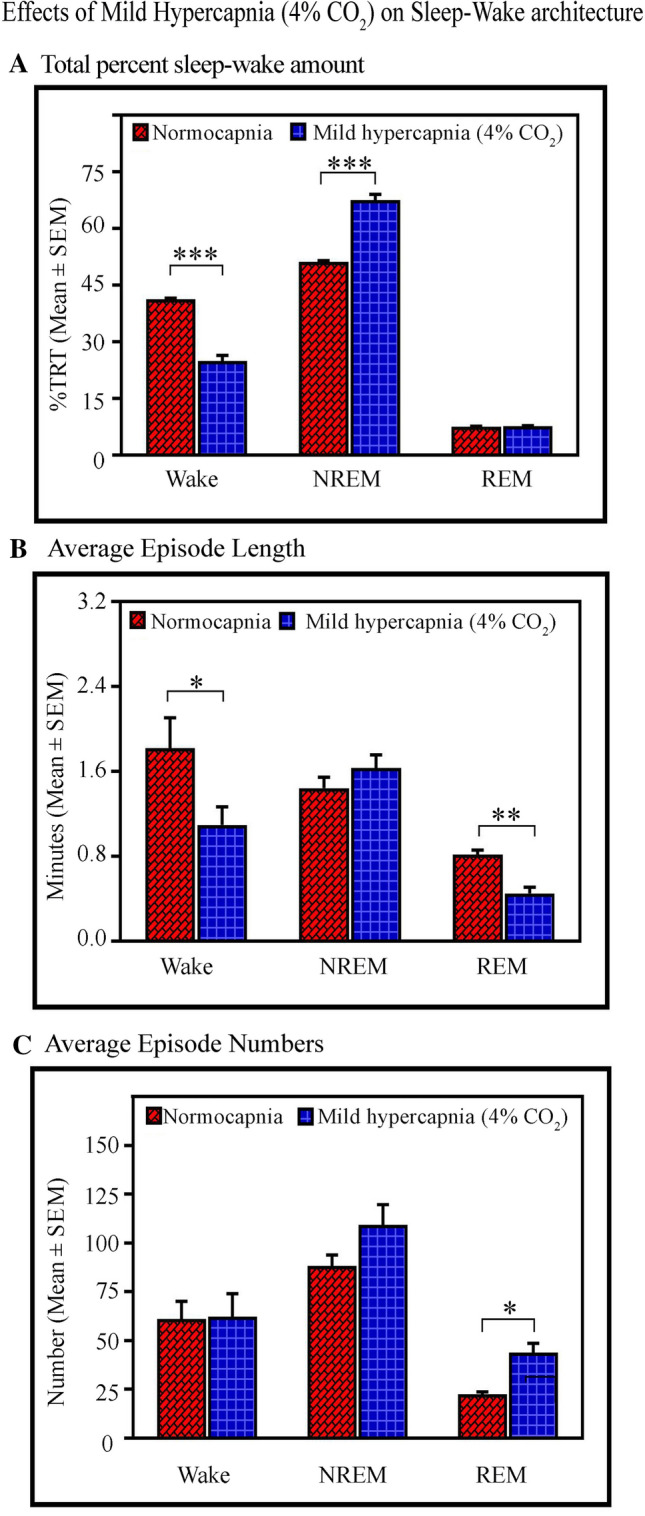
Effects of mild hypercapnia on sleep–wake architecture. A One-hour exposure to 4% CO2 (mild hypercapnia) decreases waking (P < 0.001), and increases NREM sleep (P < 0.001) compared to normocapnia. Nevertheless, the amount of REM does not change. B The mild hypercapnia decreases the length of wakefulness episodes (P < 0.05) but does not change the length of NREM episodes. The length of REM episodes also decreases (P < 0.01). C The mild hypercapnia does not influence the numbers of wake and NREM episodes, but the number of REM sleep episodes significantly increases [(P < 0.05). (*P < 0.05, **P < 0.01 (Bonferroni post hoc test)].
First, we evaluated the effects of hypercapnia on the amount and parameters of S–W (Fig. 5). One hour of exposure to 4% CO2 significantly influenced the S–W architecture. The amount of wakefulness decreased by 40% (Bonferroni test, P < 0.001; F(1,9) = 144.64; Cohen’s d = 8.35; power = 1 at alpha 0.05), while the amount of NREM sleep increased by 32% (Bonferroni test, P < 0.001; F(1,9) = 136.80; Cohen’s d = 8.14; power = 1 at alpha 0.05) in mild hypercapnia compared to normocapnia. Nevertheless, the amount of REM sleep did not change (Fig. 5A). One-way RM ANOVA demonstrated a significant effect on the length of wakefulness episodes (Bonferroni test, P < 0.05; F(1,9) = 7.94; Cohen’s d = 1.5; power = 0.54 at alpha 0.05) and REM sleep only (Bonferroni test, P < 0.01; F(1,9) = 25.98; Cohen’s d = 3.32; power = 0.98 at alpha 0.05), but not on the length of NREM sleep episodes (Fig. 5B). Mild hypercapnia did not influence the numbers of waking and NREM sleep episodes (Fig. 5C), but the number of REM sleep episodes increased (Bonferroni test, P < 0.05; F(1,9) = 17.87; Cohen’s d = 3.22; power = 0.98 at alpha 0.05) (Fig. 5C).
-
2.
Effects of APETx2 on the CO2-induced changes in S–W architecture
Two-way ANOVA revealed a significant interaction between treatment and types [treatment: CO2 and non-CO2; types: vehicle and APETx2) on the amount of wakefulness (P < 0.001; F(1,19) = 20.16), NREM sleep (P < 0.001; F(1,19) = 38.16), and REM sleep (P < 0.001; F(1,19) = 161.91). Further, the post hoc comparison demonstrated that the amount of wakefulness decreased in the CO2 + vehicle (Bonferroni test, P < 0.001; Cohen’s d = 8.35; power = 1 at alpha level 0.05), CO2 + APETx2 (Bonferroni test, P < 0.001; Cohen’s d = 11.72; power = 1 at alpha level 0.05), and APETx2-only (Bonferroni test, P < 0.001; Cohen’s d = 5.26; power = 1 at alpha level 0.05) groups compared to the vehicle-only group (Fig. 6A). On the other hand, the post hoc comparison showed that NREM sleep increased in the CO2 + vehicle (Bonferroni test, P < 0.001; Cohen’s d = 8.14; power = 1 at alpha level 0.05), CO2 +APETx2 (Bonferroni test, P < 0.001; Cohen’s d = 13.11; power = 1 at alpha level 0.05), and APETx2-only (Bonferroni test, P < 0.001; Cohen’s d = 6.92; power = 1 at alpha level 0.05) groups compared with the vehicle-only group (Fig. 6A). Interestingly, the post hoc comparison demonstrated that REM sleep did not decrease in the CO2 + vehicle and CO2 + APETx2 groups compared with the vehicle-only group. However, it decreased in the APETx2-only group compared with the vehicle-only group (Bonferroni test, P < 0.001; Cohen’s d = 14.00; power = 1 at alpha level 0.05), CO2 + vehicle (Bonferroni test, P < 0.001; Cohen’s d = 17.22; power = 1 at alpha level 0.05), and CO2 + APETx2 (Bonferroni test, P < 0.001; Cohen’s d = 9.53; power = 1 at alpha level 0.05) groups (Fig. 6A).
Fig. 6.
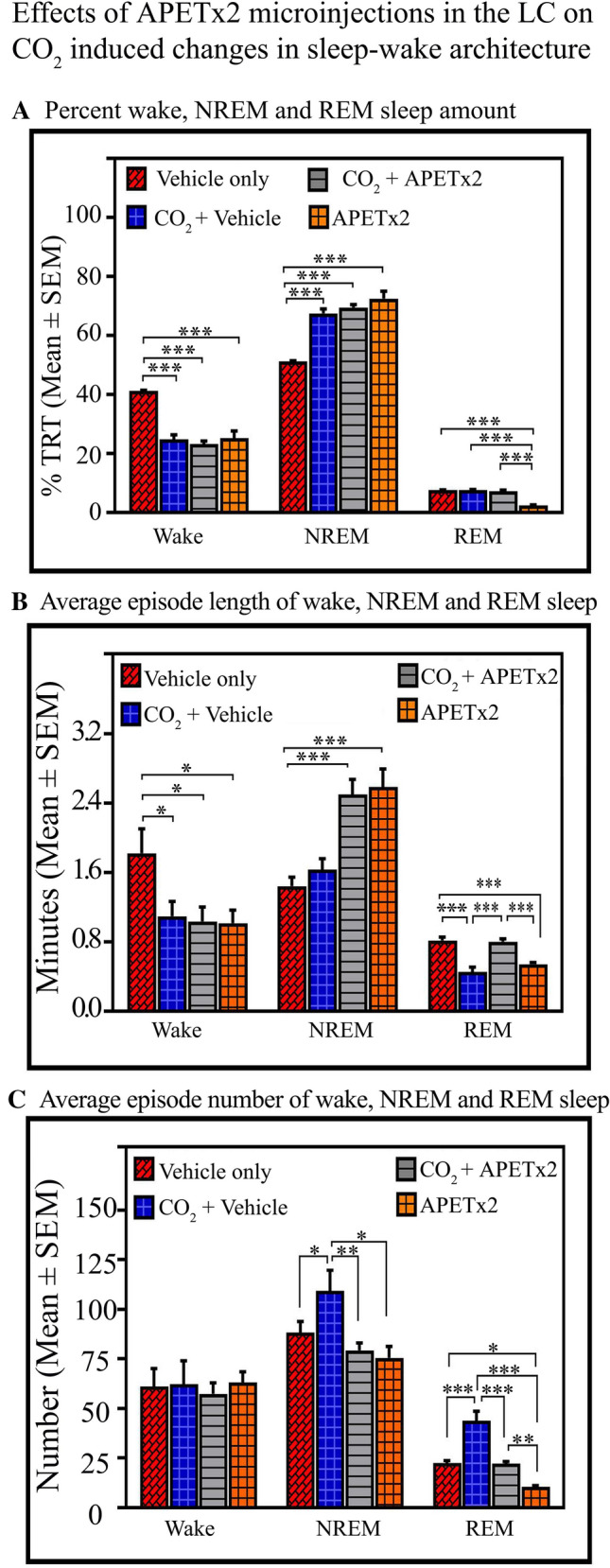
Effects of APETx2 microinjection into the LC on 4% CO2-induced changes in sleep–wake architecture. A Wakefulness amount decreased in the CO2 + vehicle (P < 0.001), CO2 + APETx2 (P < 0.001), and APETx2-only (P < 0.001) groups compared to the vehicle-only group. NREM sleep increases in the CO2 + vehicle (P < 0.001), CO2 + APETx2 (P < 0.001), and APETx2-only (P < 0.001) groups compared with the vehicle-only group. However, REM sleep does not change in the CO2 + vehicle and CO2 + APETx2 groups compared with the vehicle-only group. It significantly decreases in the APETx2-only group compared with the vehicle-only (P < 0.001), CO2 + vehicle (P < 0.001), and CO2 + APETx2 (P < 0.001) groups. B The average length of wakefulness episodes decreases in the CO2 + vehicle (P < 0.05), CO2 + APETx2 (P < 0.05), and APETx2-only (P < 0.05) groups compared to the vehicle-only group. The average length of NREM sleep episodes increases in the CO2 + APETx2 (P < 0.001) and APETx2-only (P < 0.001) groups compared with the vehicle-only group. It, however, does not change in the CO2 + vehicle group compared with the vehicle only group. The average length of REM sleep episodes does not change in the CO2 + APETx2 group compared with the vehicle-only group. However, it decreases in the CO2 + vehicle (P < 0.001) and in the APETx2-only group (P < 0.001) compared with the vehicle-only group. The average length of REM sleep episodes is also higher in the CO2 + APETx2 group than in the CO2 + vehicle (P < 0.001) and APETx2-only groups (P < 0.001). C The average number of awake episodes does not change; however, the number of NREM sleep episodes increases in the CO2 + vehicle (P < 0.05) compared to the vehicle-only group. It does not change in the CO2 + APETx2 and APETx2-only groups compared to the vehicle-only group. The average number of NREM sleep episodes is also higher in the CO2 + vehicle group compared with the CO2 + APETx2 (P < 0.01) and APETx2-only groups (P < 0.05). The number of REM sleep episodes increases in the CO2 + vehicle (P < 0.001) and decreases in the APETx2-only (P < 0.05) groups compared with the vehicle-only group. In the CO2 + APETx2 group, the number of REM sleep episodes is comparable to the vehicle-only group. However, it is less than in the CO2 + vehicle (P < 0.001) and more than in the APETx2-only (P < 0.01) group (*P < 0.05, **P < 0.01, ***P < 0.001, Bonferroni post hoc test.
Similarly, two-way ANOVA revealed a significant interaction between treatment and types on the average length of wakefulness episodes (P < 0.05; F(1,19) = 4.51), NREM sleep (P < 0.001; F(1,19) = 41.73), and REM sleep (P < 0.001; F(1,19) = 59.68) (Fig. 6B). The post hoc comparison demonstrated that the average length of wakefulness episodes decreased in the CO2 + vehicle (Bonferroni test, P < 0.05; Cohen’s d = 1.50; power = 0.54 at alpha level 0.05), CO2 + APETx2 (Bonferroni test, P < 0.05; Cohen’s d = 1.58; power = 0.59 at alpha level 0.05), and APETx2-only (Bonferroni test, P < 0.05; Cohen’s d = 1.67; power = 0.63 at alpha level 0.05) groups compared to the vehicle-only group (Fig. 6B). On the other hand, the post hoc comparison showed that the average length of NREM sleep episodes increased in the CO2 + APETx2 (Bonferroni test, P < 0.001; Cohen’s d = 3.53; power = 0.99 at alpha level 0.05), and APETx2-only (Bonferroni test, P < 0.001; Cohen’s d = 3.38; power = 0.98 at alpha level 0.05) groups compared with the vehicle-only group. It, however, did not increase in the CO2 + vehicle group compared with the vehicle-only group (Fig. 6B). Interestingly, the post hoc comparison demonstrated that the average length of REM sleep episodes did not change in the CO2 + APETx2 group compared with the vehicle-only group. However, it decreased in the CO2 + vehicle (Bonferroni test, P < 0.001; Cohen’s d = 3.23; power = 0.98 at alpha level 0.05) and in the APETx2-only groups (Bonferroni test, P < 0.001; Cohen’s d = 3.94; power = 1 at alpha level 0.05) compared with the vehicle-only group. The average length of REM sleep episodes was also greater in the CO2 + APETx2 group (Bonferroni test, P < 0.001; Cohen’s d = 3.50; power = 0.99 at alpha level 0.05) than in the CO2 + vehicle and APETx2-only groups (Bonferroni test, P < 0.001; Cohen’s d = 4.33; power = 1 at alpha level 0.05) (Fig. 6B). These results showed that ASIC blockade abolishes the CO2-mediated effect on the length of REM episodes. Similar effects did not occur on the length of awake and NREM sleep episodes.
Two-way ANOVA showed no significant interaction between treatment and types on the average number of wakefulness episodes. Nevertheless, for the numbers of NREM sleep episodes, the two-way ANOVA only revealed a significant interaction between treatments (P < 0.001; F(1,19) = 9.68) and no interaction between types. The post hoc comparison demonstrated that the number of NREM sleep episodes increased in the CO2 + vehicle (Bonferroni test, P < 0.05; Cohen’s d = 1.51; power = 0.43 at alpha level 0.05) compared to the vehicle-only group (Fig. 6C). However, it did not change in the CO2 + APETx2 and APETx2-only groups compared with the vehicle-only group. The average number of NREM sleep episodes was higher in the CO2 + vehicle group than in the CO2 + APETx2 (Bonferroni test, P < 0.01; Cohen’s d = 1.88; power = 0.74 at alpha level 0.05) and APETx2-only groups (Bonferroni test, P < 0.05; Cohen’s d = 1.86; power = 0.73 at alpha level 0.05) (Fig. 6C).
For the number of REM sleep episodes, two-way ANOVA revealed main effects on treatments (P < 0.001; F(1,19) = 43.42)and types (P < 0.001; F(1,19) = 41.38). The post hoc comparison demonstrated that the number of episodes increased in the CO2 + vehicle (Bonferroni test, P < 0.001; Cohen’s d = 3.22; power = 0.99 at alpha level 0.05) and decreased in the APETx2-only (Bonferroni test, P < 0.05; Cohen’s d = 6.97; power = 1 at alpha level 0.05) groups compared with the vehicle-only group. In the CO2 +APETx2 group, the number of REM sleep episodes was comparable to the vehicle-only group. However, it was less than in the CO2 + vehicle (Bonferroni test, P < 0.001; Cohen’s d = 3.22; power = 0.99 at alpha level 0.05) group and more than in the APETx2-only (Bonferroni test, P < 0.01; Cohen’s d = 6.59; power = 1 at alpha level 0.05) group (Fig. 6C). We again found that CO2 was unable to induce the effect on the number of REM sleep episodes if ASICs were blocked. These results demonstrate that ASIC blockade with APETx2 nullifies the CO2-induced changes in the length and number of REM sleep episodes. Such an interaction did not occur in wake and NREM sleep parameters. Overall, these results suggest that CO2 modulates REM sleep generation through ASICs, and the effects on wake and NREM sleep are likely independent of their interaction.
Discussion
Our immunohistochemical results demonstrated the presence of two types of acid-sensing channels, ASIC-2 and ASIC-3, mostly on NE-ergic LC neurons. We further found that ASICs on the NE-ergic LC neurons modulated sleep–wakefulness as well as hypercapnia-induced NREM-REM sleep state transition. Inhibition of ASICs in the LC with two different inhibitors amiloride (a universal ASIC inhibitor) and APETx2 (a specific inhibitor of ASIC-2 and ASIC-3) significantly decreased wakefulness and REM sleep. NREM sleep, however, was significantly increased at the same time. The blockade of ASICs in the LC failed to influence the length and number of wake episodes, the number of NREM sleep episodes and the length of REM sleep episodes. Nevertheless, the length of NREM sleep episode was increased and the number of REM sleep episodes was decreased significantly. These results suggest that ASICs in the LC are involved in the maintenance, but not the generation, of NREM sleep. Moreover, they may also be involved in the generation of REM sleep rather than its maintenance. In addition, mild hypercapnia (4% CO2) significantly decreased waking and increased NREM sleep, but did not influence the amount of REM (Fig. 5). Infusion of 4% CO2 significantly increased the number of REM sleep episodes, yet significantly decreased the length of awake and REM sleep episodes. Interestingly, ASIC blockade with APETx2 in the LC during mild hypercapnia abolished the effects of 4% CO2 on the length and number of REM sleep episodes, but the impact on waking and NREM sleep and its parameters were an independent effect of CO2 and APETx2 (Fig. 6). These results suggest that ASICs in the LC are involved in the CO2-mediated effects on REM sleep only.
Several studies have reported that ASICs are widely present in the central nervous system. In the brain, several sub-classes of ASICs such as ASIC1a, ASIC2a, ASIC2b, and ASIC3 have been reported [39, 42, 55, 58]. In the chemosensory areas of the brainstem, ASIC-1, ASIC-2, and ASIC-3 have been found in the nucleus tractus solitarius (NTS), ventro-lateral medulla (VLM), and trapezoid nucleus, where they are involved in chemoregulatory processes [46, 59–61]. Inhibition of ASICs in the NTS with amiloride significantly decreases the breathing rate as well as impairing the hypercapnia-mediated augmentation of ventilation rate [39, 46]. Similarly, inhibition of ASICs by amiloride and psalmotoxin-1 in the VLM impairs the CO2-mediated increase in breathing rate [55, 62]. The expression levels of ASIC-1, ASIC-2 and ASIC-3 vary considerably under hypoxic conditions in the trapezoid body as well as in the lateral paragigantocellular nucleus [59]. Besides the brainstem chemosensory areas, ASICs in the hypothalamus also play an essential role in the acidosis-mediated augmentation of breathing rate [55]. ASIC-1, ASIC-2, and ASIC-3 expression have also been reported in the hypothalamus, and their inhibition with Amiloride impairs the hypercapnia-induced ventilatory rate [55, 63]. These studies demonstrate that ASICs are involved in the modulation of central chemoregulatory processes.
In our study, we found that wakefulness and REM sleep significantly decreased, while NREM sleep significantly increased, after amiloride and APETx2 microinjection into the LC. The majority of LC neurons are NE-ergic, and they are involved in the induction of wakefulness [1, 64]. The optogenetic stimulation of NE-ergic neurons induces wakefulness [25, 65]. Similarly, blocking NE reuptake in the LC by inhibiting NE transporters with phentermine and reboxetine also induces wakefulness [66, 67]. On the other hand, the inactivation of LC neurons by photo-inhibition or microinjection of the α-2 NE receptor agonist clonidine (an anti-hypertensive drug) augments NREM sleep and slow-wave activity during NREM sleep [25, 68]. Further, LC lesions [14] and the microinjection of interleukin-1β and tumor necrosis factor into the LC (causing peptide-mediated inhibition of NE-ergic neurons) increase NREM sleep [69]. Therefore, these findings suggest that selective inhibition of NE-ergic neurons either optogenetically, peptide-mediated, or through its own receptors augments NREM sleep [25]. In addition, it has been reported that LC lesions significantly decrease the numbers of both NREM and REM sleep episodes [70, 71]. These studies thus suggest that NE-ergic neurons in the LC play an important role in sleep consolidation.
Previous studies have shown that brain acidosis affects the binding affinity of NE [72, 73].The binding affinity of NE-ergic receptors with their ligands decreases during respiratory arrhythmia or acid–base abnormalities [74]. A low pH induces receptor protonation, which causes the formation of a low-affinity state of NE receptors [73]. All these studies suggest that a higher dose of NE should be administered at the time of cardio-pulmonary resuscitation [74]. In addition, the number of NE-ergic receptors declines at synapses under low pH or hypoxia, suggesting the pH-dependent down-regulation of receptor density [75, 76]. Prolonged sleep deprivation also alters the binding affinity of β-NE-ergic receptors in the brain [77], and the global blockade of such binding inhibits NE release as well as inducing profound sleepiness [78]. These studies clearly suggest that an acidic milieu, as well as an abnormal sleep load, profoundly alter the NE-receptor binding affinity, which in turn, alters the manifestations of sleep.
During the initial phase of sleep, the cycle length remains short, with only a few episodes of REM sleep. However, as sleeping time progresses, the length of the cycle increases along with a notable increase in REM sleep frequency [4, 79]. The frequency of REM sleep increases as the length of NREM sleep episodes become longer [4, 79]. Given that the reduced ventilatory rate during a prolonged NREM sleep episode may cause mild hypercapnia [4], the appearance of REM sleep would necessarily be required to maintain normocapnia, and ASICs may also be required in the state transition from NREM to REM sleep. Mild hypercapnia increases the amount of NREM sleep as well as REM sleep frequency [31], but the underlying mechanism is not clear. The LC provides NE to almost every area in the brain [80] and modulates neuronal functions primarily through α1-, α2-, and β-NE receptors [78]. Studies have shown that α1- and β-receptors are localized post-synaptically, while α2-receptors are present both pre- and post-synaptically [78]. Activation of presynaptic α2- receptors in the LC inhibits NE release and induces sleep [78, 81, 82], while the simultaneous blockade of α1- and β-receptors elicits profound sedation [78]. Therefore, a plausible explanation for the increased NREM sleep induced by mild hypercapnia is the decrease in receptor affinity and/or down-regulation of NE receptor density in the LC caused by mild hypercapnia [74–76], which in turn may inhibit the neuronal activity and NE release and subsequently induce NREM sleep. In addition, mild hypercapnia increases the REM sleep frequency, possibly through the modulation of GABA-ergic transmission. LC neurons are reportedly inhibited by GABA neurons during REM sleep, primarily through pontine REM-active glutamatergic/cholinergic neurons [83], and the functions of GABA receptors can be altered by mild hypercapnia [72]. Studies have reported that ASICs play an essential role in maintaining GABA-ergic synaptic strength by increasing the GABA-ergic inward current [84]. Hence, ASICs in the LC are likely to be involved in the de-protonation of GABA receptors under a hypercapnic state. Therefore, the modulation of GABA-ergic neurons by ASICs in the LC may be involved in the transition from NREM to REM sleep (Fig. 7). Coinciding with such a proposition, our results also showed increased numbers of NREM and decreased mild-hypercapnia-mediated REM sleep episodes after blocking ASICs in the LC [4], which has physiological implications as well. A persistent NREM sleep–mediated hypercapnic state exists in the nervous system, and this needs to be quickly overcome by increasing the breathing rate during either wakefulness or REM sleep [4]. Frequent arousal from NREM sleep is not a normal physiological state, and leads to medical conditions such as sleep apnea [4]. Hence, mild hypercapnia would induce more REM sleep, and excess CO2can then be eliminated by increasing the breathing rate without causing wakefulness. Therefore, one function of REM sleep is to maintain a normal brain CO2 level for sustained and unperturbed sleep [4].
Fig. 7.

A model illustrating the possible mechanism of state transition from NREM to REM sleep. The LC modulates NREM sleep primarily through α1-, α2-, and β-NE-ergic receptors. The deactivation of post-synaptic α1- and β-receptors or the activation of pre-synaptic α2 autoreceptors within or outside the LC induces NREM sleep. Prolonged NREM sleep causes mild hypercapnia, which, in turn, augments NREM sleep, possibly by protonating NE-ergic receptors (protonation causes receptor deactivation). Although the generation of NREM sleep and mild-hypercapnia seem to be complementary to each other, the nervous system does not remain in a persistent NREM sleep-like hypercapnic state. Hence, the system may undergo mandatory state transition, preferably to REM sleep, to restore normocapnic conditions (increased breathing during REM sleep restores normocapnia). Mild hypercapnia does not influence the amount of REM sleep, but increases its frequency. Recurrent REM sleep occurs through the activation of pontine GABA-ergic transmission. The pontine REM-active glutamatergic/cholinergic neurons initiate REM sleep by inhibiting LC REM-OFF neurons through GABAergic inter-neurons. ASICs play an essential role in maintaining the GABA-ergic synaptic strength by increasing the GABA-ergic inward current and de-protonating the GABA receptors during hypercapnia. Hence, the activation of GABA-ergic receptors in the LC may generate a state transition from NREM to REM. Thus, the increased breathing rate during REM sleep restores normocapnic conditions.
It has been shown that in some brain regions such as the NTS, VLM, hypothalamus, and amygdala, ASICs under low pH cause neuronal depolarization, but do not induce depolarization under neutral pH (7.4) [39, 46, 55, 85]. Therefore, it is likely that blocking ASICs in the LC under reduced pH (during hypercapnia) would cause hyperpolarization in LC NE-ergic neurons. In our study, the latency of NREM sleep did not change, but its episode length increased significantly after blocking ASICs with APETx2. These results suggest that ASICs per se may not be involved in the generation of NREM sleep, but may contribute to its maintenance as well as an efficient state transition from NREM to REM sleep. Although the role of ASICs in the modulation of LC neurons is not known, patch-clamp recordings in the future would provide in-depth information about alterations in the ASIC-mediated currents of LC neurons.
In summary, our results show that the chemosensory machinery of the LC may use ASICs to initiate pH-dependent signaling cascades in neurons to modulate sleep–wake architecture. We report that ASIC-2 and ASIC-3 are present in the LC, and they are involved in sleep–wake modulation. These findings support the notion that sleep and ionic homeostasis are interlinked [86]. The study provides insights into the mechanisms of state transition from NREM to REM sleep. Our results suggest that one of the functions of REM sleep is to maintain normocapnia and avoid frequent arousals from sleep. Our study may further provide potential therapeutic treatment strategies for various sleep-related disorders, including sleep apnea, congenital central hypoventilation syndrome, and sudden infant death syndrome where chemosensory and sleep–wake neural circuitries jointly play an active role [87, 88].
Supplementary Information
Acknowledgements
This work was supported by the Department of Science and Technology-Cognitive Science Initiative project funded to Sushil K Jha. We also acknowledge support from Department of Biotechnology (DBT), Department of Science and Technology-(PURSE), Universities for Potential of Excellence (UPOE II) and University Grants Commission-Special Assistance Programme), and JNU funds to Sushil K Jha.
References
- 1.Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep–waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aston-Jones G, Gonzalez M, Doran S. Role of the locus coeruleus-norepinephrine system in arousal and circadian regulation of the sleep–wake cycle. In: Schwartz MA, Frazer A, editors. Brain norepinephrine: Neurobiology and therapeutics (Ordway GA. New York, USA: Cambridge University Press; 2007. pp. 157–195. [Google Scholar]
- 3.Dean JB, Kinkade EA, Putnam RW. Cell-cell coupling in CO(2)/H(+)-excited neurons in brainstem slices. Respir Physiol. 2001;129:83–100. doi: 10.1016/S0034-5687(01)00284-5. [DOI] [PubMed] [Google Scholar]
- 4.Madan V, Jha SK. A moderate increase of physiological CO2 in a critical range during stable NREM sleep episode: a potential gateway to REM sleep. Front Neurol. 2012;3:19. doi: 10.3389/fneur.2012.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Putnam RW. CO2 chemoreception in cardiorespiratory control. J Appl Physiol. 2010;108:1796–1802. doi: 10.1152/japplphysiol.01169.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu N, Fu C, Yu H, Wang Y, Shi L, Hao Y, et al. Respiratory control by phox2b-expressing neurons in a locus coeruleus-prebötzinger complex circuit. Neurosci Bull. 2020 doi: 10.1007/s12264-020-00519-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu Y, Shi P, Gao Z. Norepinephrine from the locus coeruleus regulates microglia dynamics during wakefulness. Neurosci Bull. 2020;36:554–556. doi: 10.1007/s12264-019-00457-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gargaglioni LH, Hartzler LK, Putnam RW. The locus coeruleus and central chemosensitivity. Respiratory Physiology & Neurobiology. 2010;173:264–273. doi: 10.1016/j.resp.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pineda J, Aghajanian GK. Carbon dioxide regulates the tonic activity of locus coeruleus neurons by modulating a proton- and polyamine-sensitive inward rectifier potassium current. Neuroscience. 1997;77:723–743. doi: 10.1016/S0306-4522(96)00485-X. [DOI] [PubMed] [Google Scholar]
- 10.Filosa JA, Dean JB, Putnam RW. Role of intracellular and extracellular pH in the chemosensitive response of rat locus coeruleus neurones. J Physiol. 2002;541:493–509. doi: 10.1113/jphysiol.2001.014142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oyamada Y, Ballantyne D, Mückenhoff K, Scheid P. Respiration-modulated membrane potential and chemosensitivity of locus coeruleus neurones in the in vitro brainstem-spinal cord of the neonatal rat. J Physiol. 1998;513(Pt 2):381–398. doi: 10.1111/j.1469-7793.1998.381bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson SM, Haxhiu MA, Richerson GB. GFP-expressing locus ceruleus neurons from Prp57 transgenic mice exhibit CO2/H + responses in primary cell culture. J Appl Physiol. 1985;2008(105):1301–1311. doi: 10.1152/japplphysiol.90414.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guyenet PG, Koshiya N, Huangfu D, Verberne AJ, Riley TA. Central respiratory control of A5 and A6 pontine noradrenergic neurons. Am J Physiol. 1993;264:R1035–1044. doi: 10.1152/ajpregu.1993.264.6.R1035. [DOI] [PubMed] [Google Scholar]
- 14.Li A, Nattie E. Catecholamine neurones in rats modulate sleep, breathing, central chemoreception and breathing variability. J Physiol. 2006;570:385–396. doi: 10.1113/jphysiol.2005.099325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Carvalho D, Patrone LGA, Marques DA, Vicente MC, Szawka RE, Anselmo-Franci JA, et al. Participation of locus coeruleus in breathing control in female rats. Respir Physiol Neurobiol. 2017;245:29–36. doi: 10.1016/j.resp.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Oliveira LM, Tuppy M, Moreira TS, Takakura AC. Role of the locus coeruleus catecholaminergic neurons in the chemosensory control of breathing in a Parkinson’s disease model. Exp Neurol. 2017;293:172–180. doi: 10.1016/j.expneurol.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 17.Coates EL, Li A, Nattie EE. Widespread sites of brain stem ventilatory chemoreceptors. J Appl Physiol. 1985;1993(75):5–14. doi: 10.1152/jappl.1993.75.1.5. [DOI] [PubMed] [Google Scholar]
- 18.de Carvalho D, Patrone LGA, Taxini CL, Biancardi V, Vicente MC, Gargaglioni LH. Neurochemical and electrical modulation of the locus coeruleus: contribution to CO2 drive to breathe. Front Physiol 2014, 5. [DOI] [PMC free article] [PubMed]
- 19.Quintero MC, Putnam RW, Cordovez JM. Theoretical perspectives on central chemosensitivity: CO2/H + -sensitive neurons in the locus coeruleus. PLoS Comput Biol. 2017;13:e1005853. doi: 10.1371/journal.pcbi.1005853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patrone LG, Bicego KC, Hartzler LK, Putnam RW, Gargaglioni LH. Cardiorespiratory effects of gap junction blockade in the locus coeruleus in unanesthetized adult rats. Respir Physiol Neurobiol. 2014;190:86–95. doi: 10.1016/j.resp.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Vicente MC, Dias MB, Fonseca EM, Bícego KC, Gargaglioni LH. Orexinergic system in the locus coeruleus modulates the CO2 ventilatory response. Pflugers Arch. 2016;468:763–774. doi: 10.1007/s00424-016-1793-x. [DOI] [PubMed] [Google Scholar]
- 22.Jha SK, Mallick BN. Modulation of REM sleep by non-REM sleep and waking areas in the brain. In: Mallick BN, McCarley RW, Pandi-Perumal SR, editors. Rapid Eye Movement Sleep: Regulation and Function (Morrison AR. Cambridge: Cambridge University Press; 2011. pp. 173–182. [Google Scholar]
- 23.Aston-Jones G, Shipley MT, Chouvet G, Ennis M, van Bockstaele E, Pieribone V, et al. Afferent regulation of locus coeruleus neurons: anatomy, physiology and pharmacology. Prog Brain Res. 1991;88:47–75. doi: 10.1016/S0079-6123(08)63799-1. [DOI] [PubMed] [Google Scholar]
- 24.Braun CMJ, Pivik RT. Effects of locus coeruleus lesions upon sleeping and waking in the rabbit. Brain Research. 1981;230:133–151. doi: 10.1016/0006-8993(81)90397-8. [DOI] [PubMed] [Google Scholar]
- 25.Carter ME, Yizhar O, Chikahisa S, Nguyen H, Adamantidis A, Nishino S, et al. Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nature Neuroscience. 2010;13:1526–1533. doi: 10.1038/nn.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaitin KI, Bliwise DL, Gleason C, Nino-Murcia G, Dement WC, Libet B. Sleep disturbance produced by electrical stimulation of the locus coeruleus in a human subject. Biol Psychiatry. 1986;21:710–716. doi: 10.1016/0006-3223(86)90235-0. [DOI] [PubMed] [Google Scholar]
- 27.Pirhajati Mahabadi V, movahedin m, Mazaheri Z, Semnanian S, Mirnajafi_zadeh J, Faizi M. Effects of bilateral lesion of the locus coeruleus on the sleep–wake cycle in the rat. Physiol Pharmacol 2015, 19: 22–30.
- 28.Ouyang M, Hellman K, Abel T, Thomas SA. Adrenergic signaling plays a critical role in the maintenance of waking and in the regulation of REM sleep. J Neurophysiol. 2004;92:2071–2082. doi: 10.1152/jn.00226.2004. [DOI] [PubMed] [Google Scholar]
- 29.Yamaguchi H, Hopf FW, Li SB, de Lecea L. In vivo cell type-specific CRISPR knockdown of dopamine beta hydroxylase reduces locus coeruleus evoked wakefulness. Nat Commun. 2018;9:5211. doi: 10.1038/s41467-018-07566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ayas NT, Brown R, Shea SA. Hypercapnia can induce arousal from sleep in the absence of altered respiratory mechanoreception. Am J Respir Crit Care Med. 2000;162:1004–1008. doi: 10.1164/ajrccm.162.3.9908040. [DOI] [PubMed] [Google Scholar]
- 31.Fraigne JJ, Dunin-Barkowski WL, Orem JM. Effect of hypercapnia on sleep and breathing in unanesthetized cats. Sleep. 2008;31:1025–1033. [PMC free article] [PubMed] [Google Scholar]
- 32.Robin ED, Whaley RD, Crump CH, Travis DM. Alveolar gas tensions, pulmonary ventilation and blood pH during physiologic sleep in normal subjects. J Clin Invest. 1958;37:981–989. doi: 10.1172/JCI103694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gothe B, Altose MD, Goldman MD, Cherniack NS. Effect of quiet sleep on resting and CO2-stimulated breathing in humans. J Appl Physiol Respir Environ Exerc Physiol. 1981;50:724–730. doi: 10.1152/jappl.1981.50.4.724. [DOI] [PubMed] [Google Scholar]
- 34.Horner RL, Rivera MP, Kozar LF, Phillipson EA. The ventilatory response to arousal from sleep is not fully explained by differences in CO(2) levels between sleep and wakefulness. J Physiol. 2001;534:881–890. doi: 10.1111/j.1469-7793.2001.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith HR, Leibold NK, Rappoport DA, Ginapp CM, Purnell BS, Bode NM, et al. Dorsal raphe serotonin neurons mediate CO2-induced arousal from sleep. J Neurosci. 2018;38:1915–1925. doi: 10.1523/JNEUROSCI.2182-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buchanan GF, Smith HR, MacAskill A, Richerson GB. 5-HT2A receptor activation is necessary for CO2-induced arousal. J Neurophysiol. 2015;114:233–243. doi: 10.1152/jn.00213.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaur S, Saper CB. Neural circuitry underlying waking up to hypercapnia. Front Neurosci. 2019;13:401. doi: 10.3389/fnins.2019.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui N, Zhang X, Tadepalli JS, Yu L, Gai H, Petit J, et al. Involvement of TRP channels in the CO2 chemosensitivity of locus coeruleus neurons. J Neurophysiol. 2011;105:2791–2801. doi: 10.1152/jn.00759.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song N, Guan R, Jiang Q, Hassanzadeh CJ, Chu Y, Zhao X, et al. Acid-sensing ion channels are expressed in the ventrolateral medulla and contribute to central chemoreception. Sci Rep. 2016;6:38777. doi: 10.1038/srep38777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Peng H, Veasey SC, Ma J, Wang GF, Wang KW. Blockade of Na +/H + exchanger type 3 causes intracellular acidification and hyperexcitability via inhibition of pH-sensitive K + channels in chemosensitive respiratory neurons of the dorsal vagal nucleus in rats. Neurosci Bull. 2014;30:43–52. doi: 10.1007/s12264-013-1373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kersh AE, Hartzler LK, Havlin K, Hubbell BB, Nanagas V, Kalra A, et al. pH regulating transporters in neurons from various chemosensitive brainstem regions in neonatal rats. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1409–1420. doi: 10.1152/ajpregu.91038.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, et al. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002;34:463–477. doi: 10.1016/S0896-6273(02)00661-X. [DOI] [PubMed] [Google Scholar]
- 43.Deval E, Gasull X, Noel J, Salinas M, Baron A, Diochot S, et al. Acid-sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacol Ther. 2010;128:549–558. doi: 10.1016/j.pharmthera.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 44.Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, et al. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13:1483–1489. doi: 10.1038/nm1668. [DOI] [PubMed] [Google Scholar]
- 45.Wong HK, Bauer PO, Kurosawa M, Goswami A, Washizu C, Machida Y, et al. Blocking acid-sensing ion channel 1 alleviates Huntington’s disease pathology via an ubiquitin-proteasome system-dependent mechanism. Hum Mol Genet. 2008;17:3223–3235. doi: 10.1093/hmg/ddn218. [DOI] [PubMed] [Google Scholar]
- 46.Huda R, Pollema-Mays SL, Chang Z, Alheid GF, McCrimmon DR, Martina M. Acid-sensing ion channels contribute to chemosensitivity of breathing-related neurons of the nucleus of the solitary tract. J Physiol. 2012;590:4761–4775. doi: 10.1113/jphysiol.2012.232470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ziemann AE, Allen JE, Dahdaleh NS, Drebot II, Coryell MW, Wunsch AM, et al. The amygdala is a chemosensor that detects carbon dioxide and acidosis to elicit fear behavior. Cell. 2009;139:1012–1021. doi: 10.1016/j.cell.2009.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jha SK, Mallick BN. Presence of α-1 norepinephrinergic and GABA-A receptors on medial preoptic hypothalamus thermosensitive neurons and their role in integrating brainstem ascending reticular activating system inputs in thermoregulation in rats. Neuroscience. 2009;158:833–844. doi: 10.1016/j.neuroscience.2008.10.038. [DOI] [PubMed] [Google Scholar]
- 49.Kumar T, Jha SK. Sleep deprivation impairs consolidation of cued fear memory in rats. PLoS one. 2012;7:e47042. doi: 10.1371/journal.pone.0047042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uebele VN, Nuss CE, Santarelli VP, Garson SL, Barrow JC, Stauffer SR, et al. T-type calcium channels regulate cortical plasticity in vivo. Neuroreport. 2009;20:257. doi: 10.1097/WNR.0b013e3283200111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tripathi S, Jha SK. Short-term total sleep deprivation alters delay-conditioned memory in the rat. Behav Neurosci. 2016;130:325. doi: 10.1037/bne0000136. [DOI] [PubMed] [Google Scholar]
- 52.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates: Hard Cover Edition. Elsevier Science, 2007.
- 53.Durham-Lee JC, Mokkapati VUL, Johnson KM, Nesic O. Amiloride improves locomotor recovery after spinal cord injury. J Neurotrauma. 2011;28:1319–1326. doi: 10.1089/neu.2011.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gilbert HTJ, Hodson N, Baird P, Richardson SM, Hoyland JA. Acidic pH promotes intervertebral disc degeneration: acid-sensing ion channel -3 as a potential therapeutic target. Sci Rep. 2016;6:37360. doi: 10.1038/srep37360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song N, Zhang G, Geng W, Liu Z, Jin W, Li L, et al. Acid sensing ion channel 1 in lateral hypothalamus contributes to breathing control. PloS one. 2012;7:e39982–e39982. doi: 10.1371/journal.pone.0039982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qureshi MF, Jha SK. Proton pump inhibition increases rapid eye movement sleep in the rat. Biomed Res Int. 2014;2014:162314. doi: 10.1155/2014/162314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Diochot S, Baron A, Rash LD, Deval E, Escoubas P, Scarzello S, et al. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J. 2004;23:1516–1525. doi: 10.1038/sj.emboj.7600177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wemmie JA, Price MP, Welsh MJ. Acid-sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci. 2006;29:578–586. doi: 10.1016/j.tins.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 59.Cao XL, Chen Q, Zhou H, Tang YH, Xu JG, Zheng Y. Expression of acid-sensing ion channels in neurons of trapezoid body and lateral paragigantocellular nuclei in rat brain, and effects of intermittent hypoxia on their expression. Sichuan Da Xue Xue Bao Yi Xue Ban. 2009;40:662–666. [PubMed] [Google Scholar]
- 60.Detweiler ND, Vigil KG, Resta TC, Walker BR, Jernigan NL. Role of acid-sensing ion channels in hypoxia- and hypercapnia-induced ventilatory responses. PLOS one. 2018;13:e0192724. doi: 10.1371/journal.pone.0192724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin LH, Jones S, Talman WT. Cellular Localization of Acid-Sensing Ion Channel 1 in Rat Nucleus Tractus Solitarii. Cell Mol Neurobiol. 2018;38:219–232. doi: 10.1007/s10571-017-0534-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krishtal O. The ASICs: signaling molecules? Modulators? Trends Neurosci. 2003;26:477–483. doi: 10.1016/S0166-2236(03)00210-8. [DOI] [PubMed] [Google Scholar]
- 63.Meng QY, Wang W, Chen XN, Xu TL, Zhou JN. Distribution of acid-sensing ion channel 3 in the rat hypothalamus. Neuroscience. 2009;159:1126–1134. doi: 10.1016/j.neuroscience.2009.01.069. [DOI] [PubMed] [Google Scholar]
- 64.Aston-Jones G, Chiang C, Alexinsky T. Discharge of noradrenergic locus coeruleus neurons in behaving rats and monkeys suggests a role in vigilance. Prog Brain Res. 1991;88:501–520. doi: 10.1016/S0079-6123(08)63830-3. [DOI] [PubMed] [Google Scholar]
- 65.Breton-Provencher V, Sur M. Active control of arousal by a locus coeruleus GABAergic circuit. Nat Neurosci. 2019;22:218–228. doi: 10.1038/s41593-018-0305-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kocsis B, Li S, Hajos M. Behavior-dependent modulation of hippocampal EEG activity by the selective norepinephrine reuptake inhibitor reboxetine in rats. Hippocampus. 2007;17:627–633. doi: 10.1002/hipo.20299. [DOI] [PubMed] [Google Scholar]
- 67.Roth JD, Rowland NE. Efficacy of administration of dexfenfluramine and phentermine, alone and in combination, on ingestive behavior and body weight in rats. Psychopharmacology (Berl) 1998;137:99–106. doi: 10.1007/s002130050598. [DOI] [PubMed] [Google Scholar]
- 68.Berridge CW, Page ME, Valentino RJ, Foote SL. Effects of locus coeruleus inactivation on electroencephalographic activity in neocortex and hippocampus. Neuroscience. 1993;55:381–393. doi: 10.1016/0306-4522(93)90507-C. [DOI] [PubMed] [Google Scholar]
- 69.De Sarro G, Gareri P, Sinopoli VA, David E, Rotiroti D. Comparative, behavioural and electrocortical effects of tumor necrosis factor-α and interleukin-1 microinjected into the locus coeruleus of rat. Life Sci. 1997;60:555–564. doi: 10.1016/S0024-3205(96)00692-3. [DOI] [PubMed] [Google Scholar]
- 70.Schwartz MD, Nguyen AT, Warrier DR, Palmerston JB, Thomas AM, Morairty SR, et al. Locus coeruleus and tuberomammillary nuclei ablations attenuate hypocretin/orexin antagonist-mediated REM sleep. eNeuro 2016, 3. [DOI] [PMC free article] [PubMed]
- 71.Pirhajati V, Movahedin M, Mazaheri Z, Semnanian S, Mirnajafi-Zadeh J, Faizi M. Effects of bilateral lesion of the locus coeruleus on the sleep–wake cycle in the rat. Physiol Pharmacol. 2015;19:22–30. [Google Scholar]
- 72.Hugel S, Kadiri N, Rodeau JL, Gaillard S, Schlichter R. pH-dependent inhibition of native GABA(A) receptors by HEPES. Br J Pharmacol. 2012;166:2402–2416. doi: 10.1111/j.1476-5381.2012.01956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gende OA, Camilion de Hurtado MC, Cingolani HE. [Effect of pH changes on the binding of agonists and antagonists to the adrenergic beta receptor]. Acta Physiol Pharmacol Latinoam 1985, 35: 205–216. [PubMed]
- 74.Modest VE, Butterworth JFt. Effect of pH and lidocaine on beta-adrenergic receptor binding. Interaction during resuscitation? Chest 1995, 108: 1373–1379. [DOI] [PubMed]
- 75.Santala M, Saarikoski S, Castren O, Parviainen M. Lymphocyte beta-2-adrenoceptors and plasma catecholamines in fetal hypoxia. Gynecol Obstet Invest. 1990;30:150–154. doi: 10.1159/000293246. [DOI] [PubMed] [Google Scholar]
- 76.Marsh JD, Margolis TI, Kim D. Mechanism of diminished contractile response to catecholamines during acidosis. Am J Physiol. 1988;254:H20–27. doi: 10.1152/ajpcell.1988.254.1.C20. [DOI] [PubMed] [Google Scholar]
- 77.Hipolide DC, Tufik S, Raymond R, Nobrega JN. Heterogeneous effects of rapid eye movement sleep deprivation on binding to alpha- and beta-adrenergic receptor subtypes in rat brain. Neuroscience. 1998;86:977–987. doi: 10.1016/S0306-4522(98)00067-0. [DOI] [PubMed] [Google Scholar]
- 78.Berridge CW, Schmeichel BE, España RA. Noradrenergic modulation of wakefulness/arousal. Sleep Med Rev. 2012;16:187–197. doi: 10.1016/j.smrv.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Madan V, Jha SK. Sleep alterations in mammals: Did aquatic conditions inhibit rapid eye movement sleep? Neurosci Bull. 2012;28:746–758. doi: 10.1007/s12264-012-1285-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Foote SL, Bloom FE, Aston-Jones G. Nucleus locus ceruleus: new evidence of anatomical and physiological specificity. Physiol Rev. 1983;63:844–914. doi: 10.1152/physrev.1983.63.3.844. [DOI] [PubMed] [Google Scholar]
- 81.Miyazaki S, Uchida S, Mukai J, Nishihara K. Clonidine effects on all-night human sleep: opposite action of low- and medium-dose clonidine on human NREM-REM sleep proportion. Psychiatry Clin Neurosci. 2004;58:138–144. doi: 10.1111/j.1440-1819.2003.01207.x. [DOI] [PubMed] [Google Scholar]
- 82.De Sarro GB, Ascioti C, Froio F, Libri V, Nisticò G. Evidence that locus coeruleus is the site where clonidine and drugs acting at alpha 1- and alpha 2-adrenoceptors affect sleep and arousal mechanisms. Br J Pharmacol. 1987;90:675–685. doi: 10.1111/j.1476-5381.1987.tb11220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu X, Franks NP, Wisden W. Sleep and sedative states induced by targeting the histamine and noradrenergic systems. Front Neural Circuits 2018, 12. [DOI] [PMC free article] [PubMed]
- 84.Storozhuk M, Kondratskaya E, Nikolaenko L, Krishtal O. A modulatory role of ASICs on GABAergic synapses in rat hippocampal cell cultures. Mol Brain. 2016;9:90. doi: 10.1186/s13041-016-0269-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.González-Garrido A, Vega R, Mercado F, López IA, Soto E. Acid-sensing ion channels expression, identity and role in the excitability of the cochlear afferent neurons. Front Cell Neurosci. 2015;9:483. doi: 10.3389/fncel.2015.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mir FA, Jha SK, Jha VM. The Role of Sleep in Homeostatic Regulation of Ionic Balances and Its Implication in Cognitive Functions. In: Jha VM, editor. Sleep, Memory and Synaptic Plasticity (Jha SK. Singapore: Springer Singapore; 2019. pp. 77–106. [Google Scholar]
- 87.Soto E, Ortega-Ramírez A, Vega R. Protons as messengers of intercellular communication in the nervous system. Front Cell Neurosci. 2018;12:342. doi: 10.3389/fncel.2018.00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wiemann M, Frede S, Tschentscher F, Kiwull-Schone H, Kiwull P, Bingmann D, et al. NHE3 in the human brainstem: implication for the pathogenesis of the sudden infant death syndrome (SIDS)? Adv Exp Med Biol. 2008;605:508–513. doi: 10.1007/978-0-387-73693-8_89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.