

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive form of cancer with high mortality. The cellular origins of PDAC are largely unknown; however, ductal cells, especially centroacinar cells (CACs), have several characteristics in common with PDAC, such as expression of SOX9 and components of the Notch-signaling pathway. Mutations in KRAS and alterations to Notch signaling are common in PDAC, and both these pathways regulate the transcription factor SOX9. To identify genes regulated by SOX9, we performed siRNA knockdown of SOX9 followed by RNA-seq in PANC-1s, a human PDAC cell line. We report 93 differentially expressed (DE) genes, with convergence on alterations to Notch-signaling pathways and ciliogenesis. These results point to SOX9 and Notch activity being in a positive feedback loop and SOX9 regulating cilia production in PDAC. We additionally performed ChIP-seq in PANC-1s to identify direct targets of SOX9 binding and integrated these results with our DE gene list. Nine of the top 10 downregulated genes have evidence of direct SOX9 binding at their promoter regions. One of these targets was the cancer stem cell marker EpCAM. Using whole-mount in situ hybridization to detect epcam transcript in zebrafish larvae, we demonstrated that epcam is a CAC marker and that Sox9 regulation of epcam expression is conserved in zebrafish. Additionally, we generated an epcam null mutant and observed pronounced defects in ciliogenesis during development. Our results provide a link between SOX9, EpCAM and ciliary repression that can be exploited in improving our understanding of the cellular origins and mechanisms of PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common and aggressive form of pancreatic cancer. PDAC is the ninth most commonly diagnosed cancer in women and 10th in men; however, it has the fourth highest mortality rate (1). There are several reasons for the low survival rate in PDAC patients including: late detection of the tumor (2), a predicted early spread of cancer cells during tumor progression (3) and PDAC’s inherent chemoresistance (reviewed in (4)).
PDAC was originally named after the duct-like morphology of the cancer cells; however, the actual cellular origin is still unknown. Indeed, PDAC in mouse models can be induced by oncogene overexpression in a number of different pancreatic cell types (5–7). Broadly speaking, the cells of the pancreas are classified by their function, being either endocrine or exocrine. The endocrine pancreas is comprised of cell clusters, called islets, which secrete hormones into the blood that aid metabolic regulation. The functional unit of the exocrine pancreas is called the acinus (pl. acini); it consists of spherically organized acinar cells that secrete zymogen into small intercalated ducts. These intercalated ducts are lined by squamous epithelial cells. The most terminal of these intercalated ductal cells is actually positioned within the acini, and hence called centroacinar cells (CACs) (8). CACs are found in all vertebrates and have a very distinct morphology; they constitutively express genes associated with stem-cell maintenance, such as the components of Notch signaling and homologs of SOX9 (9–11). In zebrafish, CACs act as facultative progenitors, differentiating to replace lost pancreatic endocrine cells as required (12,13). Although there is limited evidence that CACs act as progenitors in vivo in mammals (9,14), murine terminal ductal cells do have considerable potency in vitro (15).
Although CACs and other ductal cells seem like good candidates for the cellular origins of PDAC, these cells are vastly outnumbered in the pancreas by acinar cells. The early stages of tumor initiation in the pancreas involve the loss of cells with acinar characteristics and their replacement by cells with both ductal morphology and ductal marker gene expression (16). This early step is called acinar-to-ductal metaplasia (ADM). Following oncogenic activation, preneoplasmic lesions called pancreatic intraepithelial neoplasias (PanINs) are thought to accumulate mutations over time before progressing into PDAC (17). Together these observations suggest that the origins of PDAC could be acinar cells, which first have to transition through a duct-like state during the early stages of transformation.
Most PDAC is caused by sporadic somatic mutations where the biggest known risk factor in accumulating these mutations is pancreatitis (inflammation and destruction of the pancreas) (18). About 5–10% of PDAC cases have a hereditary component (19), with a significant proportion of these familial cases caused by a genetic susceptibility to developing pancreatitis. Sequencing of PDAC samples has demonstrated that the majority (92%) of these tumors contain activating mutations in the GTPase, KRAS (20–22). Other commonly affected molecular pathways found in PDAC samples include G1/S checkpoint machinery (78%), TGF-β signaling (47%) and histone modification (24%) (22).
Constitutive KRAS activity leads to aberrant cell proliferation and differentiation (reviewed in (23)) as well as activation of SOX9 (24). SOX9 encodes a transcription factor and gets its name from being a member of the SRY-related box (SOX) gene family. All Sox genes contain a high mobility group (HMG) DNA-binding domain (25). SOX9 also has several other conserved domains including a homodimerization domain and a C-terminal activation domain (26). Besides being an activator of transcription, SOX9 can also repress transcription (27,28). This repressive activity is likely to involve the recruitment of repressor cofactors (29). SOX9 is expressed in nearly all the organs in a human and haploinsufficiency causes campomelic dysplasia—a syndrome that affects multiple structures leading to heart complications, skeletal abnormalities (30), sex reversal (31,32) and, importantly, malformed pancreatic islets (33).
Evidence from animal models has shown that SOX9 homologs play an important role during pancreas development in maintaining pancreas progenitor potency and regulating endocrine differentiation (23,24). Dysregulation of SOX9 in mature pancreas, however, is implicated in accelerating PDAC formation and progression (16). Another hallmark of PDAC is the activation of the Notch-signaling pathway (34), which also activates SOX9 expression (11). SOX9 is thought to play multiple and critical roles in PDAC initiation and progression. Using transgenic mouse models, Kopp et al. demonstrated that overexpression of Sox9 in acinar cells destabilized their cellular state, as evidenced by loss of acinar markers and conversely, gain of ductal markers (16). These changes in gene expression are consistent with the initiation of ADM. Furthermore, these authors showed that there was a significant synergy in PDAC formation when both KRAS and Sox9 were manipulated in pancreatic epithelium. Finally, Kopp et al. showed that deleting Sox9 blocked production of PanINs and PDAC in mice even when oncogenic KRAS was overexpressed in pancreatic epithelium (16).
As mentioned, whether SOX9-expressing CACs or other ductal cells normally contribute to PDAC in patients is still unknown. Results from a mouse model utilizing solely overexpression of oncogenic KRAS suggested that ductal cells are actually refractory to becoming PDAC (16). In contrast, more complex models using oncogenic KRAS activity in concert with loss of function of tumor suppressor genes suggest that ductal cells (including CACs) are actually ‘primed’ for PDAC transformation (5,7). Whatever the cellular origin, CACs naturally express genes and utilize molecular pathways that are instrumental in PDAC initiation and progression (16,35–37).
To further elucidate the role of SOX9 in pancreas and PDAC biology, we utilized two very different model systems: 1) A human cell line named PANC-1 (38) and 2) the developing zebrafish pancreas. As PANC-1 s are derived from a resected PDAC, they express high levels of SOX9 and are a useful source of both RNA and chromatin to facilitate genomic studies into PDAC biology. To identify effectors of SOX9 transcriptional activity in PANC-1 s, we integrated RNA-seq and ChIP-seq to identify direct transcriptional targets of SOX9. Zebrafish have two homologs of SOX9, namely: sox9a and sox9b. However, sox9b is the only homolog expressed in the pancreas, allowing phenotypic consequences of both haploinsufficiency and loss of function to be studied in the pancreas without side effects from gross abnormalities elsewhere in the organism (10).
In this paper, we report on direct transcriptional targets of SOX9 function in PANC-1 s and start to investigate the in vivo implication of some the downstream SOX9-dependent biological pathways during development.
Results
SOX9 modulates the transcription of proliferation and cilia genes in PANC-1s
To identify transcripts dependent on SOX9 activity, we performed RNA-seq on PANC-1s that had been transfected with either a SOX9 siRNA or a control siRNA. SOX9 siRNA-dependent loss of SOX9 protein was verified using both western blotting of cell lysate (Fig. 1A) and immunofluorescence detection in fixed cells (Fig. 1B). Total RNA was extracted from SOX9 siRNA and control siRNA transfected PANC-1s and sequenced. Upon SOX9 knockdown, we identified 93 differentially expressed (DE) genes with 60 genes being upregulated and 33 downregulated (Fig. 1C,Supplementary Material, Table S1). We confirmed the predicted top five upregulated and top five downregulated DE genes using qRT-PCR (Fig. 1D). Nine out of 10 of these predicted DE genes were validated in this assay; the one exception was SKIV2L. Although SKIV2L had the largest fold change of downregulation in our RNA-seq experiment, this gene was the closest to the cut-off P-value set for acceptability—and likely represents a false positive.
Figure 1.
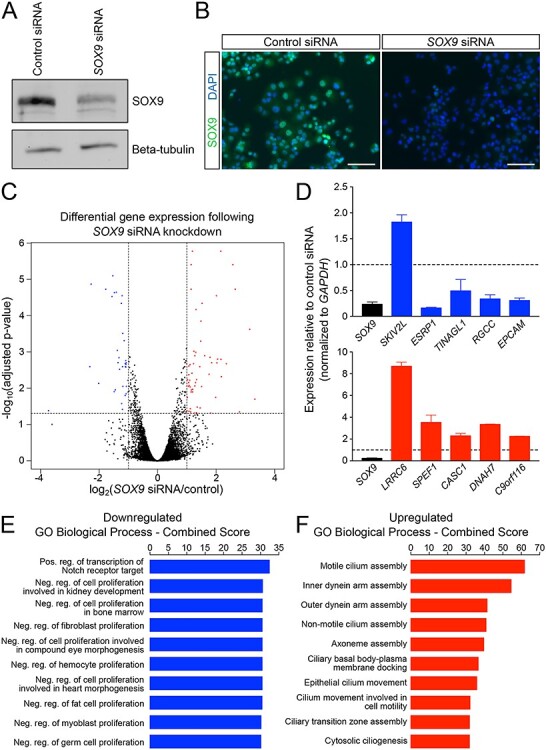
Knockdown of SOX9 results in an increase of ciliary gene expression and a decrease in expression of genes negatively regulating proliferation. (A) Western blot and (B) immunofluorescence confirms knockdown of SOX9 protein following SOX9 siRNA treatment. (C) A volcano plot of adjusted P-value versus fold change upon SOX9 knockdown indicates that 93 genes exhibit significantly altered expression (33 decreased and 60 increased). (D) Quantitative PCR confirms all but one (SKIV2L) of the top five upregulated and top five downregulated genes observed with RNA-seq. Error bars are standard deviation from three biological replicates. (E) GO analysis of downregulated genes reveals a role for SOX9 in Notch signaling as well as in the regulation of proliferation. (F) GO analysis of upregulated genes is enriched for ciliary development and function. Neg, Negative; Pos, Positive; reg, regulation. Scale bar size is 100 μM (B).
To assess the biological consequences of our DE genes, we explored their individual functions and gene ontology (GO) terms. Genes that are downregulated following SOX9 knockdown include genes with established roles in cancer motility (ESRP1, (39)), cell–cell adhesion (TINAGL1, (40,41), obesity and insulin resistance (RGCC, (42)), and cancer stem cell (CSC) maintenance (EPCAM, (43)). Downregulated genes were collectively enriched for biological processes associated with Notch signaling and the negative regulation of proliferation (Fig. 1E). As a whole, upregulated genes were enriched for processes associated with cilia development, assembly and movement (Fig. 1F), suggesting that SOX9 typically suppresses these processes in PANC-1 s. Indeed the most highly upregulated gene following SOX9 knockdown was LRRC6, which is required for the normal axoneme (core of cilia) formation (44,45).
For additional validation of our results, we analyzed publicly available RNA-seq data from 178 pancreatic adenocarcinoma samples in The Cancer Genome Atlas (TCGA). In all these samples, SOX9 is highly expressed and as such, we predicted to observe directions of effect opposite to those observed in our SOX9 siRNA-induced knockdown experiments. We clustered our differentially expressed genes based on their expression patterns in the tumor samples and observe that in general upregulated genes clustered together and were lowly expressed in the tumor samples, and downregulated genes clustered together and with SOX9 and are highly expressed (Fig. 2A). Furthermore, at an individual gene level, we correlated SOX9 expression with each of the top 10 DE genes to examine how closely the genes are co-regulated in these tumor samples and observed there to be a high degree of correlation between the differentially expressed genes and SOX9 expression (Table 1). For example, CCDC13 is a gene that encodes a centriolar satellite protein that is essential for ciliogenesis (46). This gene that is upregulated with SOX9 knockdown has the strongest degree of negative correlation with SOX9 expression (r = −0.50), consistent with SOX9 negatively regulating CCDC13 expression not only in PANC-1 s but also across PDAC samples (Fig. 2B). Conversely, expression of the downregulated gene Epithelial splicing regulatory protein 1 (ESRP1), is strongly positively correlated with SOX9 expression (r = 0.67), further suggesting that SOX9 positively regulates this gene’s expression (Fig. 2C). Overall, these data serve to validate observations from our genome wide RNA-seq analyses and provide additional support for our observed transcriptional targets of SOX9. Importantly, these results support the biological relevance of our in vitro model and suggest that the genes we identify as targets of SOX9 are relevant to the human disease state.
Figure 2.
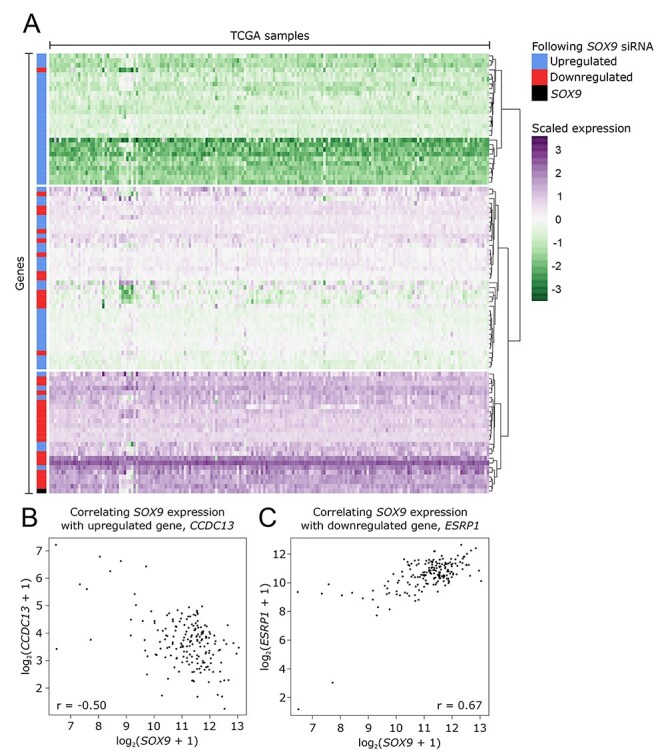
Expression data from PDAC samples corroborate the expression patterns seen in PANC-1s. (A) Expression analysis of the TCGA pancreatic adenocarcinoma sample data set indicate that genes upregulated following siRNA knockdown of SOX9 in PANC-1s tend to cluster together and are lowly expressed in these ex vivo samples. Furthermore, downregulated genes with SOX9 knockdown in PANC-1s tend to cluster together and with SOX9 and are highly expressed. (B) At an individual gene level, CCDC13, a gene that is upregulated following SOX9 knockdown, is negatively correlated with SOX9 expression, suggesting that SOX9 negatively regulates this gene. (C) Conversely, ESRP1, a gene that is downregulated following SOX9 knockdown, is positively correlated with SOX9 expression, suggesting that SOX9 positively regulates this gene.
Table 1.
Comparison of SOX9-dependent, SOX9-bound and duct-expressed genes.
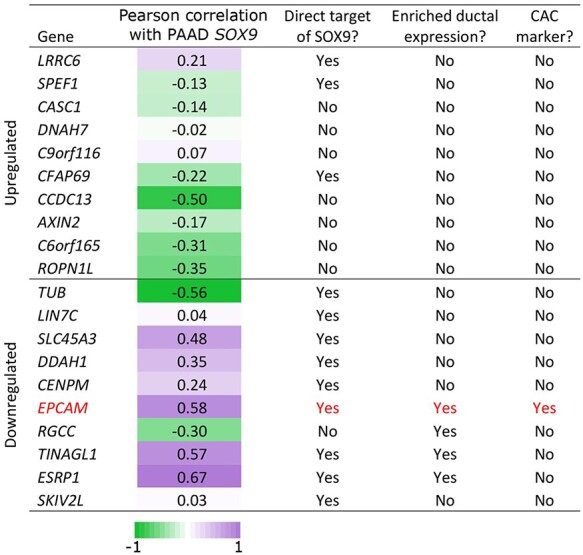
|
SOX9 binding occurs primarily at transcription start sites and regulates pancreatic functions
To identify SOX9-responsive regulatory regions, we undertook anti-SOX9 ChIP-seq using PANC-1 chromatin. Following pull-down, sequencing and alignment, our analysis identified 47 858 SOX9 binding sites in PANC-1s. Next, we analyzed the sequence underlying the top 1000 most significant SOX9 binding events to identify transcription factor motifs present at these sites. The top enriched motif was a ‘head-to-head’ palindrome with high similarity to the SOX9 consensus motif (Fig. 3A). This result supports previous observations in chondrocytes that SOX9 can function as a homodimer (47). The second highest enriched motif matched the binding sequence for FOS::JUN (Fig. 3B), which has been previously reported to bind in conjunction with SOX9 in chondrocytes (48). Similar to findings from other groups (49–51), we observe an enrichment of SOX9 binding events at gene promoters (8.8% of SOX9 peaks, n = 4219 versus the 1.1% of peaks expected; Fig. 3C), with diminishing proportions of SOX9 binding events occurring as the distance from the transcriptional start site increases (Fig. 3D).
Figure 3.
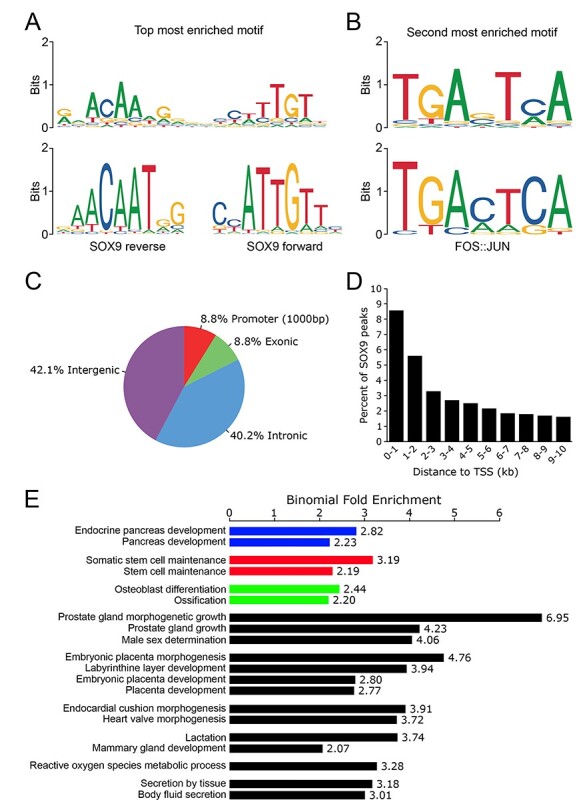
Characteristics of SOX9 gene regulation include promoter proximal binding and regulation of genes important in pancreatic biology. (A) The most common motif seen at SOX9 binding sites is head-to-head SOX9 binding sequences indicating binding as a homodimer. (B) The second most common motif at SOX9 binding sites is recognized by FOS::JUN. (C) SOX9 binding is enriched at promoters (≤1000 base-pairs upstream of a transcription start site). (D) As distance from the transcriptional start site increases, the proportion of SOX9 binding events decreases. (E) The nearest genes to SOX9 binding sites are enriched for GO terms related to known SOX9 biology, including endocrine pancreas functions.
To interrogate the potential outcome of SOX9 binding in PANC-1s, we performed functional annotation of the genes proximal to SOX9 binding sites. We found many GO biological processes that matched SOX9’s known roles (Fig. 3E). These include 1) ‘endocrine pancreas development’, consistent with the known function of SOX9 activity in the pancreas during embryogenesis (9,49,52) and 2) ‘stem-cell maintenance’, reflecting the participation of SOX9 in maintaining pancreatic progenitor identity (37,53,54). And 3) ‘ossification and osteoblast differentiation’ reflecting known functions for SOX9 that are normally restricted to bone development (55,56) but may become dysregulated in cancer (57).
Direct targets of SOX9 and overlap with known pancreatic ductal genes
We compared our datasets of genes affected by SOX9 knockdown (RNA-seq) to genes associated with SOX9 binding (ChIP-seq data). Of the genes upregulated after SOX9 knockdown, a minority (43%) was shown to be bound by SOX9. In the top 10 upregulated genes, there is only evidence for three genes to be directly bound by SOX9. This is in contrast to the genes downregulated by SOX9 knockdown where a majority are bound by SOX9 (64%) and nine of the top 10 downregulated genes are putative direct targets of SOX9 (Table 1).
As PANC-1s are originally derived from a PDAC (38), we were interested to determine if PANC-1s possess similarities to normal ductal cells—a possible cellular origin for this tumor (58,59). To do this, we compared our datasets to two previously published bulk RNA-seq experiments that were performed on two populations of cells taken from adult zebrafish pancreas. Ghaye et al. used an nkx6.1 fluorescent reporter to isolate by FACS a ‘ductal’ cell population (including CACs) from adult transgenic zebrafish (13,60); whereas, Delaspre et al. used a Notch-responsive fluorescent reporter to specifically isolate CACs (12). Sox9b was shown to be enriched in both the resulting ‘ductal’ and ‘CAC’ transcriptomes, reflecting the known expression of this gene in adult pancreas (10). Table 1 summarizes the comparison of the PANC-1 data generated here with the previous zebrafish-transcriptome work. Following siRNA-induced SOX9 knockdown in PANC-1 s, none of the top 10 upregulated genes have expression in either zebrafish pancreatic ducts or CACs. In contrast 4/10 of the ‘downregulated genes’ have enriched expression in pancreatic ductal cells (Table 1). Together, these results are consistent with sox9b-expressing ducts being enriched for homologues of SOX9 direct targets in PANC-1s. One of these homologues, epcam, was also shown to be enriched in the zebrafish CAC transcriptome (Table 1). These results suggest that our in vitro model derived from a PDAC reflects the normal biology of sox9b-expressing CACs.
SOX9 directly regulates expression of EPCAM
There are several biological connections between EpCAM and SOX9. The expression of both these genes in healthy pancreas is restricted to the ducts (61), whereas, expression of both genes in tumors is enriched in cancer stem cells CSCs (57,62–65). Accordingly, we examined the relationship between SOX9 and EpCAM in greater detail. Examining the ChIP-seq (red) and RNA-seq (blue) reads from our PANC-1 experiments at the EPCAM locus (black), we interrogated how SOX9 might be regulating EPCAM. First, through ChIP-seq, we identified a SOX9 binding event overlapping the EPCAM transcriptional start site and promoter (Fig. 4A). Consistent with SOX9 regulation of EPCAM, following siRNA knockdown of SOX9, EPCAM transcription is greatly diminished (Fig. 4A). To verify this observation, we took the additional step of performing immunofluorescence to detect EpCAM and SOX9 in PANC-1s transfected with either control or SOX9 siRNA. In control-transfected cells, SOX9 was expressed in all cell nuclei and EpCAM was expressed at the plasma membrane in all cells, albeit with varying intensity (Fig. 4B). Following SOX9 knockdown, SOX9 expression was reduced or absent in nuclei, and EpCAM expression too was reduced or absent (Fig. 4C). Altogether, these results support the conclusion that SOX9 directly binds the EPCAM locus, enhancing EPCAM production in PANC-1s. Next, we examined the co-regulation of SOX9 and EPCAM in the TCGA pancreatic adenocarcinoma samples, as previously described. In doing so, we observe a strong positive correlation of SOX9 and EPCAM expression (r = 0.58; Fig. 4D), consistent with SOX9 being a positive regulator of EPCAM transcription in multiple PDACs and not just an artifact of PANC-1s derivation or in vitro maintenance.
Figure 4.
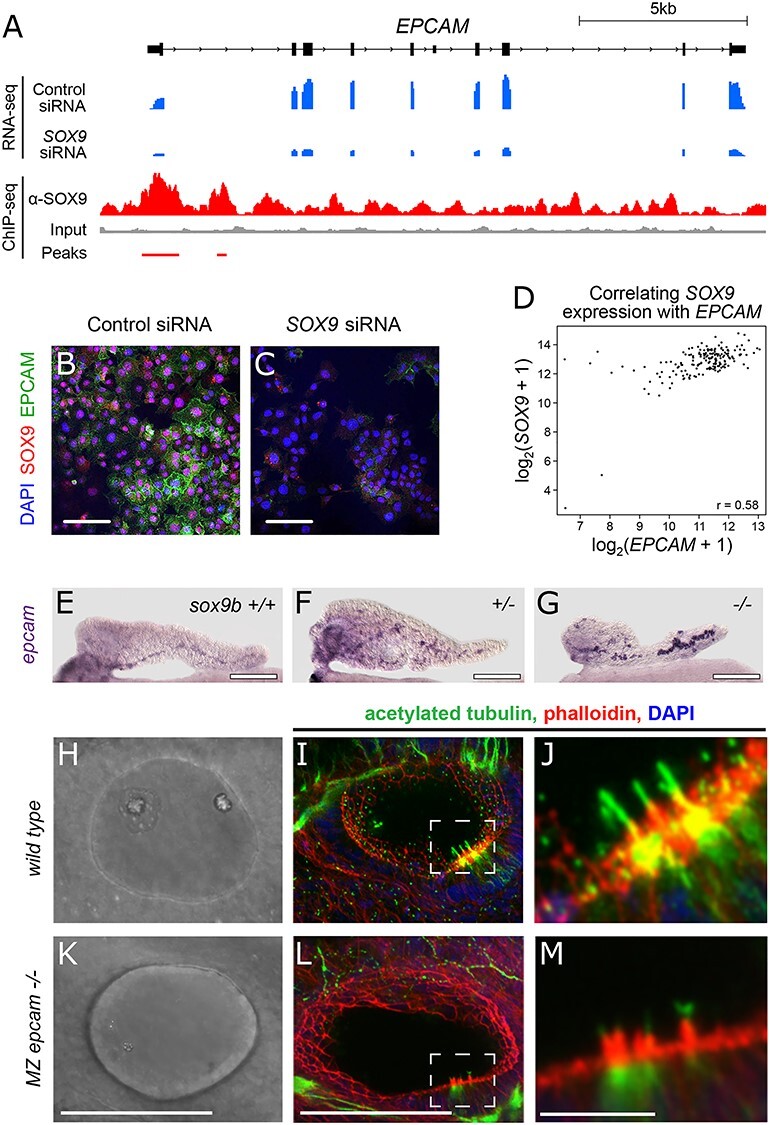
EPCAM is direct target of SOX9. (A) RNA-seq (blue) and ChIP-seq (red) reads over the EPCAM locus confirm siRNA-mediated EPCAM knockdown and SOX9 binding at the transcription start site. (B, C) Immunofluorescence detection of SOX9 and EPCAM in PANC-1 s treated with control siRNA (B) and SOX9 siRNA (C). (D) Positive correlation of EPCAM and SOX9 expression in TCGA pancreatic adenocarcinoma samples. (E-G) Whole mount in situ hybridization to detect epcam expression in the pancreas of 5 dpf zebrafish with the following sox9b genotypes: (E) +/+, (F) +/− and (G) −/− (H–M) cilia phenotype in MZ epcamjh79 mutants: Bright-field images of otic vesicles in wild-type (H) and MZ epcam mutants (K). Immunofluorescent detection of acetylated tubulin (green), phalloidin (red) and DAPI (blue), using confocal microscopy, in wild-type (I) and MZepcam mutants (L). Dashed insets indicate areas enlarged in (J) and (M) to visualize cilia. Scale bar 100 μm (B, C, E, F, G, H, I, K and L) and 5 μm (J, M).
SOX9 has many distinct roles during pancreas organogenesis, including maintaining a bipotent endocrine/ductal progenitor population (reviewed (66)). To test whether SOX9 regulates EPCAM expression during development, we performed in situ hybridizations on pancreata dissected from larval zebrafish at 5 days postfertilization (dpf).
At this stage of development, acinar cells surround a large principal islet and extend posteriorly to form a pancreatic tail. The ductal cells are organized equatorial to the islet and run down the center of the pancreatic tail (8,67,68). At these larval stages, all the ductal cells are Notch responsive, express sox9b, and are akin to early CACs (8).
To detect if levels of Sox9b activity affect epcam expression, we utilized a zebrafish line containing the mutant null allele sox9bfh313 (10,69). Heterozygous sox9bfh313 fish (hereafter sox9b+/−) were incrossed and whole mount in situ hybridization (wmish) was performed to detect epcam transcript in the progeny at 5 dpf. Epcam expression is detected in the ductal region of pancreata in all three genotypes (Fig. 4E–G). Epcam expression is harder to discern in wild-type pancreata (n = 33, representative image in Fig. 4E); the signal is noticeably stronger in heterozygous pancreata (Fig. 4F, n = 55) and strongest in the CACs of mutant homozygotes (Fig. 4G, n = 23). Previous studies showed that Sox9b−/− homozygotes have fewer CACs than either heterozygotes (53) or wild types (10,69). As the strongest epcam staining occurs in the pancreata with the least CACs, we conclude that loss of Sox9b causes enhanced epcam expression. As in the case of PDAC, there is a clear correlation in expression between a SOX9 homologue and an EPCAM homologue suggesting transcriptional regulation—although SOX9 activity is consistent with a role as a repressor in the developing pancreas and an activator in PDAC.
To learn more about Epcam function, we generated a null allele (epcam jh79) using CRISPR/Cas9 technology in zebrafish (Supplementary Material, Fig. S1). Null homozygous epcam mutants are viable and have morphologically normal pancreata (data not shown). Indeed, there are only a few gross morphological abnormalities. Using alternative mutant alleles of epcam, other groups have previously reported that one clear abnormality is in the developing the ear (70). By 1 dpf, wild-type embryos have very distinctive otic vesicles, each containing two large otoliths (Fig. 4H); whereas, otoliths in mutants are either absent or extremely small (Fig. 4K). This defect was most pronounced in the otic vesicles of maternal zygotic (MZ) epcamjh79 homozygotes (hereafter MZ mutants). This ear defect is only transient and by 2.5 dpf, mutants display otoliths within the size range seen in wild-type siblings (data not shown). Otolith formation is linked to cilia function in the otic epithelium (71). Accordingly, we performed immunofluorescent staining against acetylated tubulin to detect cilia in the developing ear of 1 dpf wild-type (Fig. 4I and J) and MZ mutant embryos (Fig. 4L and M). On close quantification by confocal microscopy, all MZ mutants (n = 20) had aberrant cilia. At 1 dpf, a wild-type otic vesicle (n = 18) has on average 6.17 ± 0.44 cilia, whereas MZ mutants have significantly fewer 2.55 ± 0.3, P < 0.01, two-tailed t test (Supplementary Material, Fig. S2A). When present in the MZ mutants, cilia were significantly shorter (P < 0.01, two-tailed t test) at an average length of 4.01 ± 0.2 microns—compared to wild-type cilia at 5.44 ± 0.09 microns (Supplementary Material, Fig. S2B). To see if the aberrant cilia phenotype was conserved in other epithelial, we looked at two other structures previously described as highly ciliated, namely the developing olfactory pits (72) and pronephric duct (73). We saw a lack of cilia in the developing olfactory pit and the distal pronephric epithelium (Supplementary Material, Fig. S2C and D). Together these results indicate that Sox9b activity negatively regulates epcam expression in the pancreas and that EpCAM is required for normal ciliogenesis in multiple epithelia.
Discussion
To better understand the role of SOX9 in PDAC, we set out to obtain a list of genes regulated by this transcription factor in PANC-1 s. We performed two experiments: 1) a knock down with RNAi to get a list of differentially expressed genes that depend on SOX9 levels and 2) ChIP-seq using a SOX9 antibody to find the regions of the genome bound by SOX9. In our first experiment, we achieve a reduction in SOX9 expression to approximately 25% normal levels and consequently, we identified 93 DE genes (Supplementary Material, Table S1). While we achieve a robust decrease in SOX9 levels, perhaps a larger number of differentially regulated genes could be identified with knock-out studies, as suggested in Stöckl et al. (74). These genes fit into two groups—those downregulated upon SOX9 knock down and those upregulated upon SOX9 knock down. It is assumed that in PANC-1 s, the former group of DE genes is normally positively regulated by SOX9 activity and those in the latter group are normally repressed by SOX9 activity.
Gene ontology pointed to biological processes positively regulated by SOX9 that included Notch-signaling target activation and negative regulation of cell proliferation. In murine pancreatic progenitors during development, it has been shown that Notch signaling regulates Sox9 expression (11,75). Our results show that SOX9 also positively regulates Notch-signal transduction suggesting a positive feedback loop between SOX9 and Notch. Notch signaling is thought to have multiple roles in the induction and progression of pancreatic tumors (76). Cells in human PDACs respond to Notch signaling (34), and several experiments in mice suggest an oncogenic-like function for Notch (77,78). However, our results do not point to a simple model where Notch signaling and SOX9 act in concert to drive PDAC formation. Most GO biological terms associated with the downregulated DE genes point to SOX9 positively regulating repressors of cell proliferation—a characteristic predicted for a tumor suppressor. Indeed, a connection between SOX9 activity and low levels of proliferation has been made in several different systems (79–81) and further work is needed to tease out the complicated relationship between SOX9, Notch and tumorigenesis.
From our GO results, the top biological processes repressed by SOX9 activity were all involved in some aspect of cilia biology. Primary cilia are organelles instrumental in sensing signals from the extracellular environment. Biliary epithelial cells (BECs) of the liver share many similarities with intercalated duct cells and CACs of the pancreas. When expression of both Sox9 and Sox4 were removed from mouse BECs, these cells failed to form primary cilia (82). Pancreatic duct cells in Sox9-null homozygous mice also failed to develop primary cilia (11). Our results now provide a more detailed picture of the cilia-related genes altered when SOX9 is lost. What is intriguing, however, is our finding that SOX9 acts to repress rather than enhance ciliogenesis in PANC-1 s. Clearly, there is an inverted relationship between SOX9 function and cilia in cancer versus development.
We compared our results from PANC-1 s to PDAC samples listed in TCGA. In doing so, we showed that the relationship found in PANC-1 s between SOX9 expression and many of the DE genes was also present in multiple PDAC samples. This correlation lends support to our conclusion that the DE genes are dependent on SOX9 function and also suggests that the SOX9 regulation we have uncovered is not unique to just PANC-1 s. To distinguish direct from downstream targets of SOX9 activity, we performed ChIP-seq in PANC-1s. In contrast to previous work in other cell systems (50) wherein identifying a robust consensus SOX9 motif was hindered by an apparently low-sequence specificity of SOX9 binding, we detected the enrichment of a canonical SOX9 binding consensus sequence (head-to-head SOX9 motif) under peaks. The presence of enriched SOX9 sites is taken as strong validation of the results. We observe an enrichment of SOX9 binding peaks at gene promoters and investigated whether there is evidence of direct SOX9 binding at the promoters of the genes identified as differentially regulated following SOX9 knockdown (83).
Of the top 10 downregulated genes following SOX9 knockdown, nine were also bound at their promoters by SOX9 in our ChIP-seq data. These genes would be predicted to be positively regulated by SOX9, and indeed, we know several of these transcripts are expressed in sox9b-expressing pancreatic ducts in the zebrafish. One of these genes, EpCAM, is expressed in CACs, a ductal cell subtype with the characteristics of facultative progenitors (8). As with the cilia genes mentioned above, the relationship between SOX9 function and EpCAM expression seems to be inverted when comparing the situation in PANC-1 s to an in vivo zebrafish model system. There are several explanations that could account for this switch from activator to repressor. SOX9 (and homologs) are known to regulate transcription by binding cis-regulatory elements in the genome, but it is cell-specific cofactors that determine whether Sox9 will act as a transactivator or repressor (28,29,84). Our results are consistent with different species-specific cofactors being used, or different cofactors being used in healthy versus cancerous cells. These observations require further investigation as such changes in cofactor use could provide a potential therapeutic route.
The EpCAM gene itself encodes a 40 kDa transmembrane glycoprotein that is often used as a biomarker for carcinoma (85), and in common with SOX9, it is enriched in CSCs (86). In most carcinomas (including pancreatic), high EpCAM expression correlates with low survival rate (reviewed in (87) and (88). Although a wealth of data is available for EpCAM function in vitro, exactly what if any role EpCAM plays in carcinogenesis is still under debate (43). As its full name (epithelial cell adhesion molecule) implies, this protein was originally believed to be a homophilic cell adhesion molecule; however, recent biochemical work has refuted this role (89). Furthermore, EpCAM is cleaved multiple times and on either side of the membrane and the intra-cellular portion can modulate the function of PKC (90) and even enter the nucleus, where it can interact with transcription factors, such as LEF1 (91). We provide the first evidence that loss of function in EpCAM leads to a cilia defect. In MZ epcam mutants, every otic vesicle displays a significant delay in otolith production concurrent with a cilia defect—both in number and length at 1 dpf. Future studies must address if regulating cilia is a general role for EpCAM or specific to the otic vesicle. It will also be interesting to determine whether this role in ciliogenesis is related to EpCAM’s association with decreased survival rate in cancer or whether EpCAM expression is solely a marker of carcinogenesis.
Loss of cilia can impact cell biology profoundly as it disrupts normal paracrine signaling—a classic hallmark of carcinogenesis (92). PanIN cells induced by the overexpression of KRAS in transgenic mice were shown to be abnormally devoid of primary cilia (93). In another mouse model, Kif3a (essential cilia gene) was conditionally deleted in the pancreas leading to ADM and pancreatitis (94). Pancreatitis (inflammation and destruction of the pancreas) is the leading known risk factor for developing PDAC (18). Considering all these data together, we hypothesize a direct link between KRAS activity, SOX9 activation and cilia-gene repression—a potential cancer-driving pathway that warrants closer investigation.
Materials and Methods
RNA-seq library preparation and sequencing
PANC-1s were transfected in 24 well plates with either 25 nM of control siRNA (Dharmacon catalog # D-001210-03-05) or 25 nM of SOX9 siRNA (Dharmacon catalog # M-021507-00-0005) using Lipofectamine 3000 (Thermo). After 48 h, transfected cells were pooled (four wells per replicate) and harvested, two replicates from each siRNA condition, and total RNA was isolated using a Qiagen RNeasy kit. RNA-seq libraries were created using the Illumina TruSeq Stranded Total RNA Sample Prep Kit. RNA-seq libraries were pooled and sequenced on the Illumina HiSeq 2000 to a minimum depth of 60 million 2 × 100 bp reads per library.
RNA-seq alignment, quantification and analysis
Reads were aligned to hg19 genome with HISAT2 (v2.0.5; (95)) and visualized with the Integrative Genomics Viewer (96,97). Statistical analyses were performed using both R (98). Gene expression was quantified using the ‘featureCount’ function of the Rsubread package (v1.28.1; (99)) to count read overlap with RefSeq genes. Genes with greater than one read across all four samples were submitted to DESeq2 (v1.18.1; (100)) to identify genes differentially expressed across conditions (absolute(log2(fold change)) > 1, adjusted P-value <0.05). To generate the volcano plot, each gene’s log2(fold change) was plotted against the –log10(adjusted P-value), with genes meeting our criteria for significantly differentially expressed being plotted in red (upregulated) or blue (downregulated). Genes significantly up- and downregulated were submitted to Enrichr (101,102). The GO Biological Process (2017b) was ranked on the basis of the combined score. Differentially expressed genes were annotated as being directly bound by SOX9 if there was a SOX9 binding event within 1 kb (upstream or downstream) of the transcriptional start site (RefSeq, hg19).
ChIP-seq library preparation and sequencing
PANC-1s were grown in DMEM supplemented with 10% FBS at 37°C with 5.0% CO2 and passaged at 70–80% confluency. Two biological replicates of ChIP-seq were performed. Briefly; approximately 2.0 × 108 cells were crosslinked in 11% formaldehyde and stopped with 2.5 M glycine before being washed in 1 × PBS, lysed and sonicated for 35 min in a Bioruptor at 4°C to achieve a fragment size of approximately 200 bp. An input fraction was set aside, and the rest of the lysate was then incubated with 10 μg anti-SOX9 (AB5535, Millipore) overnight at 4°C. Antibody-bound chromatin was then purified using Protein G Dynabeads (Thermo), and crosslinking was reversed overnight at 65°C. ChIP-seq libraries were created using the Illumina TruSeq DNA Sample Prep Kit and quantified using Quant-iT PicoGreen dsDNA assay (Invitrogen). ChIP-seq libraries were pooled and sequenced on the Illumina HiSeq to a minimum depth of 59 million, 1 × 50 bp reads per library.
ChIP-seq alignment, peak calling and analysis
Reads were aligned to hg19 with Bowtie2 (v2.2.5; (103)) in—local mode following TruSeq adapter removal and quality filtering with fastx toolkit (v0.0.14). Following alignment, reads with mapping score < MAPQ30, reads aligning to the mitochondria and duplicate reads were removed with SAMtools (v1.3.1; (104)). ChIP-seq replicates were combined, and peaks were called on this joint file with MACS2 (v2.1.1.20160309; (105)) using ‘callpeak’. Peaks with q-value >10−3 and those overlapping ENCODE blacklists were removed (106). These peaks were annotated for their genomic location using CEAS (v1.0.0; (107) of the Cistrome analysis pipeline (108) and the distance of each peak to the nearest gene’s transcriptional start site was quantified. The top 1000 most significant SOX9 peaks by q-value were submitted to SeqPos (v1.0.0) under default parameters. The top resulting position weight matrices were matched to motifs in the JASPAR database (109). These same 1000 peaks were submitted to GREAT (v3.0.0; (110)) under default settings except that the association rule was expanded such that ‘proximal’ was defined as 5 kb both upstream and downstream. The top 20 GO Biological Process terms by binomial rank were chosen for display and manually grouped by function.
Western blot confirmation of SOX9 knockdown
PANC-1s were cultured in 6-well plates and transfected with 100 nM control siRNA (catalogue number above) or 100 nM SOX9 siRNA (catalogue number above). After 48 h, cells were washed with PBS, isolated in RIPA buffer with complete, EDTA-free protease inhibitor (Roche) and vortexed. Supernatant was collected after centrifugation. Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Scientific), and 10 μg of protein was run on an any kD Mini-PROTEAN TGX Precast Protein Gel (Bio-Rad). Transfer from gel to membrane was carried out at 45 V for 90 min, and then the membrane was blocked for 1 h before overnight incubation in rabbit anti-Sox9 (Santa Cruz sc-20 095 1:500). Membrane was then washed three times and incubated in anti-rabbit HRP (Cell Signaling 7074S 1:2500) for 1 h. Signal was developed using SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) and exposure on a ChemiDoc-It2 for 5 min. Membrane was stripped using Restore Western Blot Stripping Buffer (Thermo Scientific), and repeated staining plus development was performed as described above using rabbit anti-beta-tubulin (Cell Signaling 2128 1:1000) primary, exposure for 2 min 30 s.
Antibody staining
PANC-1s were grown on gelatin-coated coverslips for 48 h after siRNA transfection and fixed in 4% paraformaldehyde buffered in 1X PBS. Following 4 × 5 min washes in 1X PBS, coverslips were blocked in PBST +10% FBS for 1 h at room temperature and permeablized in 0.5% Triton in PBS for 20 min, incubated with primary antibodies (rabbit anti-SOX9 Santa Cruz sc-20 095 1:250, mouse anti-EPCAM Santa Cruz sc-66 020 1:100) at 4°C overnight. Coverslips were washed 4 × 5 min in blocking, then incubated in secondary antibody (Alexa Fluor 488 donkey anti-rabbit, Alexa Fluor 488 donkey anti-mouse, Cy3 donkey anti-rabbit, all 1:500, Jackson ImmunoResearch 711-546-152, 715-456-150, 711-166-152, respectively) at 4°C overnight before 4 × 5 min final PBST washes and a brief DAPI (1:2500 in PBS) stain. Images were collected using a Nikon A1-si Laser Scanning Confocal microscope.
Zebrafish EpCAM mutant and wild-type embryos at 1 dpf were fixed in 4% paraformaldehyde overnight at 4°C. Afterwards, the embryos were washed 3 × 10 min in 0.1% TritonX-100 PBS (PBST). The embryos were then blocked in PBST +10% FBS for 1 h and incubated overnight at 4°C with mouse anti-acetylated alpha tubulin (Sigma 1:500) and Alexa-Fluor 568 phalloidin (1:40). After, another 3 × 10 min washes with PBST, the embryos were incubated with Alexa Fluor 488 donkey anti-mouse (1:500) for 3 h at room temperature. After another 3 × 10 min washes with PBST, the embryos were covered with hard set Vectashield mounting medium containing DAPI. The embryos were then mounted and imaged on a Leica SP8 confocal microscope.
Live imaging of embryos
Embryos at 1 dpf were anesthetized and mounted in low melt agarose. Their otoliths were then imaged on a Leica SP8 confocal in brightfield.
Quantitative PCR confirmation of differentially expressed genes
PANC-1s were cultured in 12-well plates and transfected with 100 nM control siRNA (catalogue number above) or 100 nM SOX9 siRNA (catalogue number above). After 48 h, RNA was isolated using Qiagen RNeasy Kit (with DNase digestion step) and cDNA was synthesized using Superscript III (Thermo) with random hexamer primers. Three biological replicates of the quantitative PCR reactions were run in technical triplicate following the default SYBR green cycling conditions on an Applied Biosystems Viia 7 using 2× Power SYBR Green Master Mix (Applied Biosystems). Expression was calculated using the ΔΔCT method normalized to GAPDH expression and control siRNA transfected cells. Primer sequences can be found in Supplementary Material, Table S2.
Correlation with TCGA pancreatic adenocarcinoma expression patterns
mRNA sequencing from 178 pancreatic adenocarcinoma samples from the TCGA was accessed and downloaded on March 2, 2018. Read counts were log2 normalized after addition of a pseudocount, and a heatmap (111) was generated for all significantly upregulated and downregulated genes and SOX9’s expression patterns in these samples, using hierarchical clustering to group these selected genes into three clusters, and scaling the expression values by column. Individual gene correlation with SOX9 expression was calculated using the Pearson correlation method, and for visualization, normalized expression values were plotted.
in situ hybridization
sox9bfh313 heterozygotes were in-crossed, and subsequent embryos were maintained in E3 media. NOTE: the sequence of epcam is highly polymorphic, and all fish used to generate larvae were first screened for homozygosity across the locus. At 5 dpf, zebrafish larvae were euthanized on ice. Cadavers were cut (transverse) into two at the level of the cloaca. The tail of each larvae was placed in a well of a 96 well plate and digested for genomic DNA prep (112). The remaining fish was placed into 4% PFA in a separate 96-well plate (in the same well position as tail) overnight at 4°C. Tail DNA was genotyped (10). As required, pancreata of known genotypes were dissected and placed into separate tubes ready for whole mount in situ (WMISH) protocol (68). The original WMISH protocol was modified for dissected larval zebrafish pancreata (See Supplemental Materials and Methods for modifications).
The epcam riboprobe (1.2 kb) was synthesized from larval cDNA (derived from sibling fish) using the following primers: forward—GGCCAGAGAGGGGATATCTT, reverse—GTTAATCCAATTGAAGAGAAGC. To make a stock riboprobe of epcam, the transcription reaction was carried out for 2 h using 1 μg of DNA in a total volume of 20 μL. The probe was transcribed using Dig rNTP labeling mix (Roche), and the transcribed riboprobe was cleaned using the RNAeasy elution kit following the manufacturers protocol (Qiagen). To establish a working epcam probe for WMISH, the probe was diluted 1:100 in hybridization buffer. Images were collected on a Zeiss Axioplan2.
Data Availability
ChIP-sequencing and RNA-sequencing data will be available at the Gene Expression Omnibus (GEO) under the accession number GSE167590.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors would like to thank Wei Huang for help and advice with methodology.
Conflict of Interest statement.
The authors declare no conflict of interest.
Contributor Information
Hannah E Edelman, McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, 470 Miller Research Building, Baltimore, MD 21205, USA.
Sarah A McClymont, McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, 470 Miller Research Building, Baltimore, MD 21205, USA.
Tori R Tucker, Department of Developmental and Cell Biology, University of California, Irvine, Natural Sciences II, CA 92697, USA.
Santiago Pineda, Department of Developmental and Cell Biology, University of California, Irvine, Natural Sciences II, CA 92697, USA.
Rebecca L Beer, McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, 470 Miller Research Building, Baltimore, MD 21205, USA.
Andrew S McCallion, McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, 470 Miller Research Building, Baltimore, MD 21205, USA.
Michael J Parsons, McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, 470 Miller Research Building, Baltimore, MD 21205, USA; Department of Developmental and Cell Biology, University of California, Irvine, Natural Sciences II, CA 92697, USA.
Funding
The NIH (R01DK080730 to M.J.P., R01MH106522 to A.S.M.). The results presented here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga
References
- 1. American Cancer Society . (2020). Cancer Facts & Figures 2020. Atlanta: American Cancer Society. [Google Scholar]
- 2. Niederhuber, J.E., Brennan, M.F. and Menck, H.R. (1995) The National Cancer Data Base report on pancreatic cancer. Cancer, 76, 1671–1677. [DOI] [PubMed] [Google Scholar]
- 3. Rhim, A.D., Mirek, E.T., Aiello, N.M., Maitra, A., Bailey, J.M., McAllister, F., Reichert, M., Beatty, G.L., Rustgi, A.K., Vonderheide, R.H.et al. (2012) EMT and dissemination precede pancreatic tumor formation. Cell, 148, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adamska, A., Elaskalani, O., Emmanouilidi, A., Kim, M., Abdol Razak, N.B., Metharom, P. and Falasca, M. (2018) Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. Adv Biol Regul, 68, 77–87. [DOI] [PubMed] [Google Scholar]
- 5. Bailey, J.M., Hendley, A.M., Lafaro, K.J., Pruski, M.A., Jones, N.C., Alsina, J., Younes, M., Maitra, A., McAllister, F., Iacobuzio-Donahue, C.A. and Leach, S.D. (2016) p53 mutations cooperate with oncogenic Kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene, 35, 4282–4288. [DOI] [PubMed] [Google Scholar]
- 6. Aguirre, A.J., Bardeesy, N., Sinha, M., Lopez, L., Tuveson, D.A., Horner, J., Redston, M.S. and DePinho, R.A. (2003) Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev., 17, 3112–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee, A.Y.L., Dubois, C.L., Sarai, K., Zarei, S., Schaeffer, D.F., Sander, M. and Kopp, J.L. (2019) Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut, 68, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beer, R.L., Parsons, M.J. and Rovira, M. (2016) Centroacinar cells: at the center of pancreas regeneration. Dev. Biol., 413, 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kopp, J.L., Dubois, C.L., Schaffer, A.E., Hao, E., Shih, H.P., Seymour, P.A., Ma, J. and Sander, M. (2011) Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development, 138, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Manfroid, I., Ghaye, A., Naye, F., Detry, N., Palm, S., Pan, L., Ma, T.P., Huang, W., Rovira, M., Martial, J.A.et al. (2012) Zebrafish sox9b is crucial for hepatopancreatic duct development and pancreatic endocrine cell regeneration. Dev. Biol., 366, 268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shih, H.P., Kopp, J.L., Sandhu, M., Dubois, C.L., Seymour, P.A., Grapin-Botton, A. and Sander, M. (2012) A notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development, 139, 2488–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Delaspre, F., Beer, R.L., Rovira, M., Huang, W., Wang, G., Gee, S., Vitery, M.D., Wheelan, S.J. and Parsons, M.J. (2015) Centroacinar cells are progenitors that contribute to endocrine pancreas regeneration. Diabetes, 64, 3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghaye, A.P., Bergemann, D., Tarifeno-Saldivia, E., Flasse, L.C., Von Berg, V., Peers, B., Voz, M.L. and Manfroid, I. (2015) Progenitor potential of nkx6.1-expressing cells throughout zebrafish life and during beta cell regeneration. BMC Biol., 13, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Solar, M., Cardalda, C., Houbracken, I., Martin, M., Maestro, M.A., De Medts, N., Xu, X., Grau, V., Heimberg, H., Bouwens, L. and Ferrer, J. (2009) Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev. Cell, 17, 849–860. [DOI] [PubMed] [Google Scholar]
- 15. Rovira, M., Scott, S.G., Liss, A.S., Jensen, J., Thayer, S.P. and Leach, S.D. (2010) Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc. Natl. Acad. Sci. U. S. A., 107, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kopp, J.L., vonFigura, G., Mayes, E., Liu, F.F., Dubois, C.L., Morris, J.P.t., Pan, F.C., Akiyama, H., Wright, C.V., Jensen, K.et al. (2012) Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell, 22, 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hruban, R.H., Goggins, M., Parsons, J. and Kern, S.E. (2000) Progression model for pancreatic cancer. Clin. Cancer Res., 6, 2969–2972. [PubMed] [Google Scholar]
- 18. Lowenfels, A.B., Maisonneuve, P., Cavallini, G., Ammann, R.W., Lankisch, P.G., Andersen, J.R., Dimagno, E.P., Andren-Sandberg, A. and Domellof, L. (1993) Pancreatitis and the risk of pancreatic cancer. International pancreatitis study group. N. Engl. J. Med., 328, 1433–1437. [DOI] [PubMed] [Google Scholar]
- 19. Permuth-Wey, J. and Egan, K.M. (2009) Family history is a significant risk factor for pancreatic cancer: results from a systematic review and meta-analysis. Fam. Cancer, 8, 109–117. [DOI] [PubMed] [Google Scholar]
- 20. Almoguera, C., Shibata, D., Forrester, K., Martin, J., Arnheim, N. and Perucho, M. (1988) Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell, 53, 549–554. [DOI] [PubMed] [Google Scholar]
- 21. Jones, S., Zhang, X., Parsons, D.W., Lin, J.C., Leary, R.J., Angenendt, P., Mankoo, P., Carter, H., Kamiyama, H., Jimeno, A.et al. (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science, 321, 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bailey, P., Chang, D.K., Nones, K., Johns, A.L., Patch, A.M., Gingras, M.C., Miller, D.K., Christ, A.N., Bruxner, T.J., Quinn, M.C.et al. (2016) Genomic analyses identify molecular subtypes of pancreatic cancer. Nature, 531, 47–52. [DOI] [PubMed] [Google Scholar]
- 23. di Magliano, M.P. and Logsdon, C.D. (2013) Roles for KRAS in pancreatic tumor development and progression. Gastroenterology, 144, 1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou, H., Qin, Y., Ji, S., Ling, J., Fu, J., Zhuang, Z., Fan, X., Song, L., Yu, X. and Chiao, P.J. (2018) SOX9 activity is induced by oncogenic Kras to affect MDC1 and MCMs expression in pancreatic cancer. Oncogene, 37, 912–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stevanovic, M., Lovell-Badge, R., Collignon, J. and Goodfellow, P.N. (1993) SOX3 is an X-linked gene related to SRY. Hum. Mol. Genet., 2, 2013–2018. [DOI] [PubMed] [Google Scholar]
- 26. Lefebvre, V. and Dvir-Ginzberg, M. (2017) SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect. Tissue Res., 58, 2–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garside, V.C., Cullum, R., Alder, O., Lu, D.Y., Vander Werff, R., Bilenky, M., Zhao, Y., Jones, S.J., Marra, M.A., Underhill, T.M. and Hoodless, P.A. (2015) SOX9 modulates the expression of key transcription factors required for heart valve development. Development, 142, 4340–4350. [DOI] [PubMed] [Google Scholar]
- 28. Leung, V.Y., Gao, B., Leung, K.K., Melhado, I.G., Wynn, S.L., Au, T.Y., Dung, N.W., Lau, J.Y., Mak, A.C., Chan, D. and Cheah, K.S.E. (2011) SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet., 7, e1002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kamachi, Y. and Kondoh, H. (2013) Sox proteins: regulators of cell fate specification and differentiation. Development, 140, 4129–4144. [DOI] [PubMed] [Google Scholar]
- 30. OMIM . (# 114290, 2017). Johns Hopkins University, https://omim.org/.
- 31. Wagner, T., Wirth, J., Meyer, J., Zabel, B., Held, M., Zimmer, J., Pasantes, J., Bricarelli, F.D., Keutel, J., Hustert, E.et al. (1994) Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell, 79, 1111–1120. [DOI] [PubMed] [Google Scholar]
- 32. Foster, J.W., Dominguez-Steglich, M.A., Guioli, S., Kwok, C., Weller, P.A., Stevanovic, M., Weissenbach, J., Mansour, S., Young, I.D., Goodfellow, P.N.et al. (1994) Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature, 372, 525–530. [DOI] [PubMed] [Google Scholar]
- 33. Piper, K., Ball, S.G., Keeling, J.W., Mansoor, S., Wilson, D.I. and Hanley, N.A. (2002) Novel SOX9 expression during human pancreas development correlates to abnormalities in Campomelic dysplasia. Mech. Dev., 116, 223–226. [DOI] [PubMed] [Google Scholar]
- 34. Miyamoto, Y., Maitra, A., Ghosh, B., Zechner, U., Argani, P., Iacobuzio-Donahue, C.A., Sriuranpong, V., Iso, T., Meszoely, I.M., Wolfe, M.S.et al. (2003) Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell, 3, 565–576. [DOI] [PubMed] [Google Scholar]
- 35. Kopinke, D., Brailsford, M., Shea, J.E., Leavitt, R., Scaife, C.L. and Murtaugh, L.C. (2011) Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development, 138, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nishikawa, Y., Kodama, Y., Shiokawa, M., Matsumori, T., Marui, S., Kuriyama, K., Kuwada, T., Sogabe, Y., Kakiuchi, N., Tomono, T.et al. (2019) Hes1 plays an essential role in Kras-driven pancreatic tumorigenesis. Oncogene, 38, 4283–4296. [DOI] [PubMed] [Google Scholar]
- 37. Seymour, P.A., Freude, K.K., Tran, M.N., Mayes, E.E., Jensen, J., Kist, R., Scherer, G. and Sander, M. (2007) SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc. Natl. Acad. Sci. U. S. A., 104, 1865–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lieber, M., Mazzetta, J., Nelson-Rees, W., Kaplan, M. and Todaro, G. (1975) Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer, 15, 741–747. [DOI] [PubMed] [Google Scholar]
- 39. Ishii, H., Saitoh, M., Sakamoto, K., Kondo, T., Katoh, R., Tanaka, S., Motizuki, M., Masuyama, K. and Miyazawa, K. (2014) Epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) suppress cancer cell motility via different mechanisms. J. Biol. Chem., 289, 27386–27399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tajiri, Y., Igarashi, T., Li, D., Mukai, K., Suematsu, M., Fukui, E., Yoshizawa, M. and Matsumoto, H. (2010) Tubulointerstitial nephritis antigen-like 1 is expressed in the uterus and binds with integrins in decidualized endometrium during postimplantation in mice. Biol. Reprod., 82, 263–270. [DOI] [PubMed] [Google Scholar]
- 41. Li, D., Mukai, K., Suzuki, T., Suzuki, R., Yamashita, S., Mitani, F. and Suematsu, M. (2007) Adrenocortical zonation factor 1 is a novel matricellular protein promoting integrin-mediated adhesion of adrenocortical and vascular smooth muscle cells. FEBS J., 274, 2506–2522. [DOI] [PubMed] [Google Scholar]
- 42. Cui, X.B., Luan, J.N., Ye, J. and Chen, S.Y. (2015) RGC32 deficiency protects against high-fat diet-induced obesity and insulin resistance in mice. J. Endocrinol., 224, 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schnell, U., Cirulli, V. and Giepmans, B.N. (2013) EpCAM: structure and function in health and disease. Biochim. Biophys. Acta, 1828, 1989–2001. [DOI] [PubMed] [Google Scholar]
- 44. Horani, A., Ferkol, T.W., Shoseyov, D., Wasserman, M.G., Oren, Y.S., Kerem, B., Amirav, I., Cohen-Cymberknoh, M., Dutcher, S.K., Brody, S.L.et al. (2013) LRRC6 mutation causes primary ciliary dyskinesia with dynein arm defects. PLoS One, 8, e59436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kott, E., Duquesnoy, P., Copin, B., Legendre, M., Dastot-Le Moal, F., Montantin, G., Jeanson, L., Tamalet, A., Papon, J.F., Siffroi, J.P.et al. (2012) Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet., 91, 958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Staples, C.J., Myers, K.N., Beveridge, R.D., Patil, A.A., Howard, A.E., Barone, G., Lee, A.J., Swanton, C., Howell, M., Maslen, S.et al. (2014) Ccdc13 is a novel human centriolar satellite protein required for ciliogenesis and genome stability. J. Cell Sci., 127, 2910–2919. [DOI] [PubMed] [Google Scholar]
- 47. Bernard, P., Tang, P., Liu, S., Dewing, P., Harley, V.R. and Vilain, E. (2003) Dimerization of SOX9 is required for chondrogenesis, but not for sex determination. Hum. Mol. Genet., 12, 1755–1765. [DOI] [PubMed] [Google Scholar]
- 48. He, X., Ohba, S., Hojo, H. and McMahon, A.P. (2016) AP-1 family members act with Sox9 to promote chondrocyte hypertrophy. Development, 143, 3012–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shih, H.P., Seymour, P.A., Patel, N.A., Xie, R., Wang, A., Liu, P.P., Yeo, G.W., Magnuson, M.A. and Sander, M. (2015) A gene regulatory network cooperatively controlled by Pdx1 and Sox9 governs lineage allocation of foregut progenitor cells. Cell Rep., 13, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kadaja, M., Keyes, B.E., Lin, M., Pasolli, H.A., Genander, M., Polak, L., Stokes, N., Zheng, D. and Fuchs, E. (2014) SOX9: a stem cell transcriptional regulator of secreted niche signaling factors. Genes Dev., 28, 328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shi, Z., Chiang, C.I., Labhart, P., Zhao, Y., Yang, J., Mistretta, T.A., Henning, S.J., Maity, S.N. and Mori-Akiyama, Y. (2015) Context-specific role of SOX9 in NF-Y mediated gene regulation in colorectal cancer cells. Nucleic Acids Res., 43, 6257–6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Seymour, P.A., Freude, K.K., Dubois, C.L., Shih, H.P., Patel, N.A. and Sander, M. (2008) A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev. Biol., 323, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huang, W., Beer, R.L., Delaspre, F., Wang, G., Edelman, H.E., Park, H., Azuma, M. and Parsons, M.J. (2016) Sox9b is a mediator of retinoic acid signaling restricting endocrine progenitor differentiation. Dev. Biol., 418, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scott, C.E., Wynn, S.L., Sesay, A., Cruz, C., Cheung, M., Gomez Gaviro, M.V., Booth, S., Gao, B., Cheah, K.S., Lovell-Badge, R. and Briscoe, J. (2010) SOX9 induces and maintains neural stem cells. Nat. Neurosci., 13, 1181–1189. [DOI] [PubMed] [Google Scholar]
- 55. Dy, P., Wang, W., Bhattaram, P., Wang, Q., Wang, L., Ballock, R.T. and Lefebvre, V. (2012) Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev. Cell, 22, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bi, W., Deng, J.M., Zhang, Z., Behringer, R.R. and deCrombrugghe, B. (1999) Sox9 is required for cartilage formation. Nat. Genet., 22, 85–89. [DOI] [PubMed] [Google Scholar]
- 57. Kawai, T., Yasuchika, K., Ishii, T., Miyauchi, Y., Kojima, H., Yamaoka, R., Katayama, H., Yoshitoshi, E.Y., Ogiso, S., Kita, S.et al. (2016) SOX9 is a novel cancer stem cell marker surrogated by osteopontin in human hepatocellular carcinoma. Sci. Rep., 6, 30489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee, A.Y.L., Dubois, C.L., Sarai, K., Zarei, S., Schaeffer, D.F., Sander, M. and Kopp, J.L. (2019) Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut, 68, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shi, C., Pan, F.C., Kim, J.N., Washington, M.K., Padmanabhan, C., Meyer, C.T., Kopp, J.L., Sander, M., Gannon, M., Beauchamp, R.D.et al. (2019) Differential cell susceptibilities to Kras(G12D) in the setting of obstructive chronic pancreatitis. Cell. Mol. Gastroenterol. Hepatol., 8, 579–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tarifeno-Saldivia, E., Lavergne, A., Bernard, A., Padamata, K., Bergemann, D., Voz, M.L., Manfroid, I. and Peers, B. (2017) Transcriptome analysis of pancreatic cells across distant species highlights novel important regulator genes. BMC Biol., 15, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cirulli, V., Crisa, L., Beattie, G.M., Mally, M.I., Lopez, A.D., Fannon, A., Ptasznik, A., Inverardi, L., Ricordi, C., Deerinck, T.et al. (1998) KSA antigen ep-CAM mediates cell-cell adhesion of pancreatic epithelial cells: morphoregulatory roles in pancreatic islet development. J. Cell Biol., 140, 1519–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bao, B., Azmi, A.S., Aboukameel, A., Ahmad, A., Bolling-Fischer, A., Sethi, S., Ali, S., Li, Y., Kong, D., Banerjee, S.et al. (2014) Pancreatic cancer stem-like cells display aggressive behavior mediated via activation of FoxQ1. J. Biol. Chem., 289, 14520–14533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Imrich, S., Hachmeister, M. and Gires, O. (2012) EpCAM and its potential role in tumor-initiating cells. Cell Adh. Migr., 6, 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li, C., Heidt, D.G., Dalerba, P., Burant, C.F., Zhang, L., Adsay, V., Wicha, M., Clarke, M.F. and Simeone, D.M. (2007) Identification of pancreatic cancer stem cells. Cancer Res., 67, 1030–1037. [DOI] [PubMed] [Google Scholar]
- 65. Ma, X.L., Hu, B., Tang, W.G., Xie, S.H., Ren, N., Guo, L. and Lu, R.Q. (2020) CD73 sustained cancer-stem-cell traits by promoting SOX9 expression and stability in hepatocellular carcinoma. J. Hematol. Oncol., 13, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Seymour, P.A. (2014) Sox9: a master regulator of the pancreatic program. Rev. Diabet. Stud., 11, 51–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yee, N.S., Lorent, K. and Pack, M. (2005) Exocrine pancreas development in zebrafish. Dev. Biol., 284, 84–101. [DOI] [PubMed] [Google Scholar]
- 68. Parsons, M.J., Pisharath, H., Yusuff, S., Moore, J.C., Siekmann, A.F., Lawson, N. and Leach, S.D. (2009) Notch-responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech. Dev., 126, 898–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Delous, M., Yin, C., Shin, D., Ninov, N., Debrito Carten, J., Pan, L., Ma, T.P., Farber, S.A., Moens, C.B. and Stainier, D.Y. (2012) Sox9b is a key regulator of pancreaticobiliary ductal system development. PLoS Genet., 8, e1002754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Slanchev, K., Carney, T.J., Stemmler, M.P., Koschorz, B., Amsterdam, A., Schwarz, H. and Hammerschmidt, M. (2009) The epithelial cell adhesion molecule EpCAM is required for epithelial morphogenesis and integrity during zebrafish epiboly and skin development. PLoS Genet., 5, e1000563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stooke-Vaughan, G.A., Huang, P., Hammond, K.L., Schier, A.F. and Whitfield, T.T. (2012) The role of hair cells, cilia and ciliary motility in otolith formation in the zebrafish otic vesicle. Development, 139, 1777–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Reiten, I., Uslu, F.E., Fore, S., Pelgrims, R., Ringers, C., Diaz Verdugo, C., Hoffman, M., Lal, P., Kawakami, K., Pekkan, K.et al. (2017) Motile-cilia-mediated flow improves sensitivity and temporal resolution of olfactory computations. Curr. Biol., 27, 166–174. [DOI] [PubMed] [Google Scholar]
- 73. Kramer-Zucker, A.G., Wiessner, S., Jensen, A.M. and Drummond, I.A. (2005) Organization of the pronephric filtration apparatus in zebrafish requires Nephrin, Podocin and the FERM domain protein mosaic eyes. Dev. Biol., 285, 316–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Stockl, S., Lindner, G., Li, S., Schuster, P., Haferkamp, S., Wagner, F., Prodinger, P.M., Multhoff, G., Boxberg, M., Hillmann, A.et al. (2020) SOX9 knockout induces polyploidy and changes sensitivity to tumor treatment strategies in a chondrosarcoma cell line. Int. J. Mol. Sci., 21, 7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hosokawa, S., Furuyama, K., Horiguchi, M., Aoyama, Y., Tsuboi, K., Sakikubo, M., Goto, T., Hirata, K., Tanabe, W., Nakano, Y.et al. (2015) Impact of Sox9 dosage and Hes1-mediated notch signaling in controlling the plasticity of adult pancreatic duct cells in mice. Sci. Rep., 5, 8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Avila, J.L. and Kissil, J.L. (2013) Notch signaling in pancreatic cancer: oncogene or tumor suppressor? Trends Mol. Med., 19, 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. De La, O.J., Emerson, L.L., Goodman, J.L., Froebe, S.C., Illum, B.E., Curtis, A.B. and Murtaugh, L.C. (2008) Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. U. S. A., 105, 18907–18912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Plentz, R., Park, J.S., Rhim, A.D., Abravanel, D., Hezel, A.F., Sharma, S.V., Gurumurthy, S., Deshpande, V., Kenific, C., Settleman, J.et al. (2009) Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology, 136, 1741–1749 e1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Formeister, E.J., Sionas, A.L., Lorance, D.K., Barkley, C.L., Lee, G.H. and Magness, S.T. (2009) Distinct SOX9 levels differentially mark stem/progenitor populations and enteroendocrine cells of the small intestine epithelium. Am. J. Physiol. Gastrointest. Liver Physiol., 296, G1108–G1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Drivdahl, R., Haugk, K.H., Sprenger, C.C., Nelson, P.S., Tennant, M.K. and Plymate, S.R. (2004) Suppression of growth and tumorigenicity in the prostate tumor cell line M12 by overexpression of the transcription factor SOX9. Oncogene, 23, 4584–4593. [DOI] [PubMed] [Google Scholar]
- 81. Panda, D.K., Miao, D., Lefebvre, V., Hendy, G.N. and Goltzman, D. (2001) The transcription factor SOX9 regulates cell cycle and differentiation genes in chondrocytic CFK2 cells. J. Biol. Chem., 276, 41229–41236. [DOI] [PubMed] [Google Scholar]
- 82. Poncy, A., Antoniou, A., Cordi, S., Pierreux, C.E., Jacquemin, P. and Lemaigre, F.P. (2015) Transcription factors SOX4 and SOX9 cooperatively control development of bile ducts. Dev. Biol., 404, 136–148. [DOI] [PubMed] [Google Scholar]
- 83. Ohba, S., He, X., Hojo, H. and McMahon, A.P. (2015) Distinct transcriptional programs underlie Sox9 regulation of the mammalian chondrocyte. Cell Rep., 12, 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Geraldo, M.T., Valente, G.T., Nakajima, R.T. and Martins, C. (2016) Dimerization and transactivation domains as candidates for functional modulation and diversity of Sox9. PLoS One, 11, e0156199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Went, P., Vasei, M., Bubendorf, L., Terracciano, L., Tornillo, L., Riede, U., Kononen, J., Simon, R., Sauter, G. and Baeuerle, P.A. (2006) Frequent high-level expression of the immunotherapeutic target Ep-CAM in colon, stomach, prostate and lung cancers. Br. J. Cancer, 94, 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Al-Hajj, M., Wicha, M.S., Benito-Hernandez, A., Morrison, S.J. and Clarke, M.F. (2003) Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U. S. A., 100, 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. van der Gun, B.T., Melchers, L.J., Ruiters, M.H., deLeij, L.F., McLaughlin, P.M. and Rots, M.G. (2010) EpCAM in carcinogenesis: the good, the bad or the ugly. Carcinogenesis, 31, 1913–1921. [DOI] [PubMed] [Google Scholar]
- 88. Patriarca, C., Macchi, R.M., Marschner, A.K. and Mellstedt, H. (2012) Epithelial cell adhesion molecule expression (CD326) in cancer: a short review. Cancer Treat. Rev., 38, 68–75. [DOI] [PubMed] [Google Scholar]
- 89. Gaber, A., Kim, S.J., Kaake, R.M., Bencina, M., Krogan, N., Sali, A., Pavsic, M. and Lenarcic, B. (2018) EpCAM homo-oligomerization is not the basis for its role in cell-cell adhesion. Sci. Rep., 8, 13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Maghzal, N., Vogt, E., Reintsch, W., Fraser, J.S. and Fagotto, F. (2010) The tumor-associated EpCAM regulates morphogenetic movements through intracellular signaling. J. Cell Biol., 191, 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Maetzel, D., Denzel, S., Mack, B., Canis, M., Went, P., Benk, M., Kieu, C., Papior, P., Baeuerle, P.A., Munz, M. and Gires, O. (2009) Nuclear signalling by tumour-associated antigen EpCAM. Nat. Cell Biol., 11, 162–171. [DOI] [PubMed] [Google Scholar]
- 92. Liu, H., Kiseleva, A.A. and Golemis, E.A. (2018) Ciliary signalling in cancer. Nat. Rev. Cancer, 18, 511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Seeley, E.S., Carriere, C., Goetze, T., Longnecker, D.S. and Korc, M. (2009) Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res., 69, 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cano, D.A., Sekine, S. and Hebrok, M. (2006) Primary cilia deletion in pancreatic epithelial cells results in cyst formation and pancreatitis. Gastroenterology, 131, 1856–1869. [DOI] [PubMed] [Google Scholar]
- 95. Kim, D., Langmead, B. and Salzberg, S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nat. Methods, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Robinson, J.T., Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E.S., Getz, G. and Mesirov, J.P. (2011) Integrative genomics viewer. Nat. Biotechnol., 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Thorvaldsdottir, H., Robinson, J.T. and Mesirov, J.P. (2013) Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform., 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. R_Core_Team . (2017) The R Project for Statistical Computing. www.r-project.org/.
- 99. Liao, Y., Smyth, G.K. and Shi, W. (2013) The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res., 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kuleshov, M.V., Jones, M.R., Rouillard, A.D., Fernandez, N.F., Duan, Q., Wang, Z., Koplev, S., Jenkins, S.L., Jagodnik, K.M., Lachmann, A.et al. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res., 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chen, E.Y., Tan, C.M., Kou, Y., Duan, Q., Wang, Z., Meirelles, G.V., Clark, N.R. and Ma'ayan, A. (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics, 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Langmead, B. and Salzberg, S.L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhang, Y., Liu, T., Meyer, C.A., Eeckhoute, J., Johnson, D.S., Bernstein, B.E., Nusbaum, C., Myers, R.M., Brown, M., Li, W. and Liu, X.S. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Consortium, E.P. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Shin, H., Liu, T., Manrai, A.K. and Liu, X.S. (2009) CEAS: cis-regulatory element annotation system. Bioinformatics, 25, 2605–2606. [DOI] [PubMed] [Google Scholar]
- 108. Liu, T., Ortiz, J.A., Taing, L., Meyer, C.A., Lee, B., Zhang, Y., Shin, H., Wong, S.S., Ma, J., Lei, Y.et al. (2011) Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol., 12, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Khan, A., Fornes, O., Stigliani, A., Gheorghe, M., Castro-Mondragon, J.A., van derLee, R., Bessy, A., Cheneby, J., Kulkarni, S.R., Tan, G.et al. (2018) JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res., 46, D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. McLean, C.Y., Bristor, D., Hiller, M., Clarke, S.L., Schaar, B.T., Lowe, C.B., Wenger, A.M. and Bejerano, G. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol., 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kolde, R. (2016) Pretty Heatmaps. R Project. https://CRAN.R-project.org/package=pheatmap.
- 112. Meeker, N.D., Hutchinson, S.A., Ho, L. and Trede, N.S. (2007) Method for isolation of PCR-ready genomic DNA from zebrafish tissues. Biotechniques, 43, 610–612, 614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-sequencing and RNA-sequencing data will be available at the Gene Expression Omnibus (GEO) under the accession number GSE167590.