

Abstract
The common kidney disease diabetic nephropathy (DN) accounts for significant morbidity and mortality in patients with diabetes, and its effective diagnosis in incipient stages is still lacking. Renal fibrosis is the main pathological feature of DN. Cell division autoantigen 1 (CDA1), a phosphorylated protein encoded by TSPYL2 on the X chromosome, plays a fibrogenic role by modulating the transforming growth factor-β (TGF-β) signaling, but the exact mechanism remains unclear. TGF-β signaling has been recognized as the key factor in promoting the development and progression of DN. At present, strict control of blood sugar and blood pressure can significantly lower the development and progression of DN in the early stages, and many studies have shown that blocking TGF-β signaling can delay the progress of DN. However, TGF-β is a multifunctional cytokine. Its direct intervention may result in increased side effects. Therefore, the targeted intervention of CDA1 not only can block the TGF-β signaling pathway but also can reduce these side effects. In this article, we review the main physiological roles of CDA1, with particular attention to its effect and potential mechanism in the renal fibrosis of DN.
1. Introduction
Diabetes is one of the most important noncommunicable diseases that seriously threaten human health at present. Its prevalence rate is increasing year by year, and it has reached epidemic proportions worldwide. The rate of kidney disease diabetic nephropathy (DN), one microvascular complication of diabetes, continues to increase with the growing incidence of diabetes [1–3]. DN has become one of the main causes of end-stage renal disease (ESRD) and the death of diabetic patients [4]. It also lacks effective biological diagnostic biomarkers in the early stage. Microalbuminuria is the earliest recognized and noninvasive diagnostic indicator of DN. However, it is unable to be used as a marker for DN diagnosis in young patients and nonalbuminuric DN or to predict the risk from advanced stages in the progression of chronic kidney disease (CKD) [5]. DN is usually diagnosed too late and irreversible, meaning it cannot be effectively treated. Currently, the main treatment in the early stage of DN is too tightly control blood sugar, blood lipids, and blood pressure, as well as practicing lifestyle changes [6, 7]. Although some reports suggest that renin-angiotensin-aldosterone system inhibitors (RAASi) [8, 9], sodium-glucose cotransporter 2 inhibitors (SGLT-2i) [10–12], glucagon-like peptide-1 receptor agonists (GLP-1 RA) [10, 13, 14], and endothelin receptor antagonists (ERAs) [15–17] have renoprotective properties in delaying the progression of DN, they cannot stop DN progression to end-stage renal failure. This is because of the persistent secondary pathological processes [18]. Therefore, it is necessary to develop new treatments to delay the occurrence and development of DN.
The pathogenesis of DN is complex and multifactorial. Renal fibrosis is the final pathological change in DN [19], characterized by the accumulation of extracellular matrix (ECM) [20]. TGF-β is considered to be a core pathway leading to renal fibrosis [21]. In various disease models, inhibition of TGF-β1 or its downstream signaling pathway significantly restricted renal fibrosis, whereas overexpression of TGF-β1 induces renal fibrosis [22]. Many studies have shown that blocking the TGF-β signaling pathway can delay the progression of DN. But TGF-β1, a member of the TGF-β superfamily, is a pleiotropic cytokine with a wide variety of physiological roles that include not only regulating tissue fibrosis but also regulating many biological responses, including wound healing [23], cell proliferation, cell differentiation, ECM production and remodeling, chemotaxis, growth factor and hormone production [24], angiogenesis and hematopoiesis [25], immune regulation [26], and cell apoptosis and autophagy [27]. Direct blocking of TGF-β signaling pathway may result in many other adverse events, such as prenatal lethality [28], excessive inflammatory response [29] and immune dysregulation [30], defective hematopoiesis and vasculogenesis [28], and delayed wound healing [31]. Hence, partial blockading of TGF-β may block only its antifibrotic effect and reduce the occurrence of other side effects. Studies have suggested that CDA1 (also known as TSPYL2, TSPX, Se20-4, NP79, CINAP, and DENTT) has a synergistic effect with TGF-β, which activates the TGF-β signaling pathway and promotes the occurrence and development of renal fibrosis in DN [32, 33]. However, the mechanism by which CDA1 enhances the TGF-β signaling pathway is not yet clear. CDA1 is not an essential component of the TGF-β signaling pathway, and TGF-β signaling cannot be completely blocked by the deletion of functional CDA1. Healthy CDA1 KO mice show normal growth and reproduction without abnormal phenotype [33], but blockade of CDA1 does not affect any essential processes mediated by TGF-β signaling, such as wound healing is unknown. Therefore, targeting CDA1 is a potential new strategy to delay the development of DN. It is worth mentioning that a prototype peptide inhibitor of CDA1 can safely and efficiently delay key renal parameters associated with diabetic-induced renal fibrosis in experimental animal models. However, it remains unclear whether this kind of research strategy can be reproduced in humans to delay the progression of DN.
2. Overview on CDA1
Chai et al. [34, 35] identified a novel protein in the serum of a patient with discoid lupus erythematosus. They named it CDA1 (cell division autoantigen 1). CDA1 is encoded by TSPYL2 (testis-specific Y-encoded-like protein 2) on the X chromosome. Its cDNA is composed of 2,808 base pairs, of which 2,079 base pairs of open reading frame encode 693 amino acids (aa) with a predicted molecular polypeptide of 79.43 KD and a pI of 4.26. The structure of CDA1 is shown in Figure 1. The antigen information of CDA1 shows that its antigen position is between 390 and 527 aa, with a length of 138 aa (Figure 2). Also, the amino acid sequences of CDA1 in humans and other species have been compared to each other, indicating that CDA1 is highly conserved among different species (Figure 3). Research from the Chai group suggests that CDA1 localizes in the nucleus of HeLa cells. However, since CDA1 has been detected in the cytoplasm, nucleus, or certain tissues of both adult mice and monkeys, it may shuttle between the cytoplasm and the nucleus [36, 37]. Biochemical studies show that CDA1 is a phosphorylated protein with multiple phosphorylation sites [34].
Figure 1.
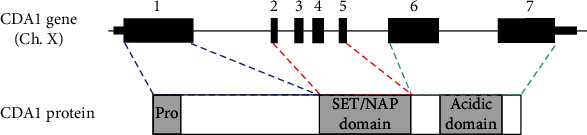
The gene and protein structure of CDA1. The CDA1 gene contains 7 exons, exon 1 encodes the N-terminal proline-rich domain, exons 2-5 encode the SET/NAP domain, and exons 6 and 7 encode the C-terminal acid domain.
Figure 2.
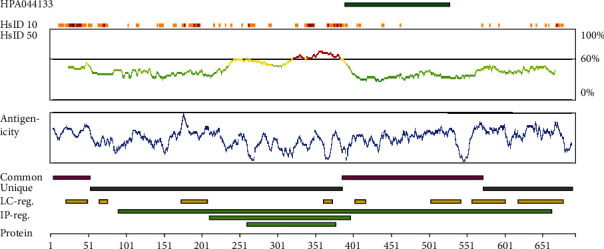
Antigenicity of CDA1. At the top of the diagram, the green bar shows the position of the antigen, with a length of 138 aa, located between 390 and 527 aa (This picture comes from The Human Protein Atlas (https://www.proteinatlas.org/)).
Figure 3.
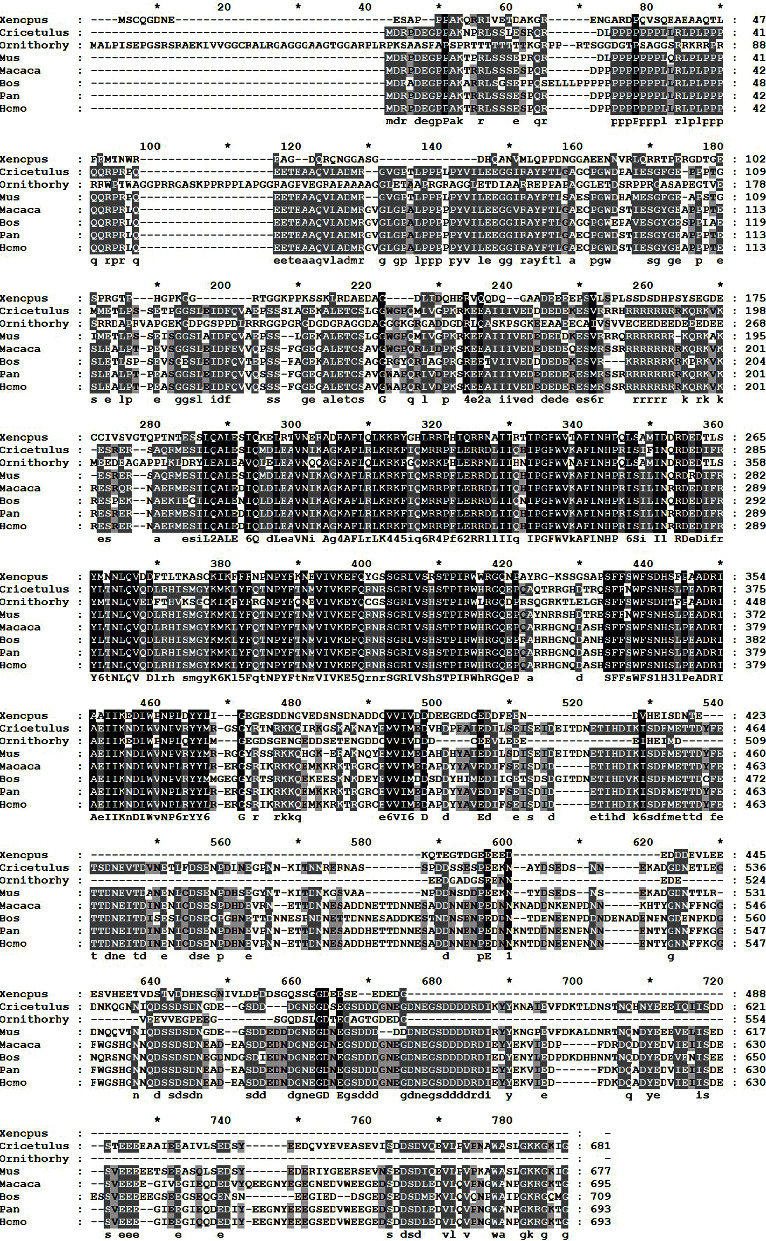
Multiple alignments of CDA1 between different species. Comparison of the amino acid sequence of CDA1 in humans and other species such as Xenopus, Cricetulus, Ornithorhy, Mus, Macaca, Bos, and Pan. In this alignment, the amino acids of other species are all identical to humans represented by black shadowing, and those of other species are, respectively, identical to humans represented by gray shadowing.
Ozbun et al. [36, 37] found that CDA1 expresses in several tissues of the adult monkey and mouse (Figure 4). To further clarify the distribution and localization of CDA1 in humans, they observed its expression in adult monkey tissues by RT-PCR, Western blot, Northern blot analysis, and immunohistochemical staining and found that its highest expression in the brain and testis is followed by the adrenal gland, ovary, prostate, lung, and mammary gland. In addition, the level of CDA1 expression in the colon, kidney, lung, stomach, pancreas, and intestine is lower than that in the adrenal gland, cerebral cortex, and ovary. In adult monkey and developing mouse kidney, CDA1 expresses in the renal cortex and medullary tubules, but not in the glomeruli [36, 37]. Conversely, it is worth noting that the other two references reported that CDA1 not only expressed in glomerular podocytes in spontaneously hypertensive rats (SHRs) kidney and healthy human kidney but also suggested that CDA1 expression was significantly increased in human renal biopsy samples from both individuals with diabetic kidney disease and nondiabetic sclerotic renal disease [32, 33]. However, we still do not know why the CDA1 expression is different in the glomeruli with normal circumstances and diabetic condition. Moreover, CDA1 is also detectable in epithelial cells, fibroblasts, endothelial cells, smooth muscle cells, and chondrocytes in the monkey, and its expression is observed in many neurons including the peripheral and parasympathetic nervous systems of the central nervous system [36].
Figure 4.

The CDA1 gene expression in different tissue and cells. CDA1 expression is highest in the brain and testis, followed by the ovary, adrenal gland, prostate, lungs, kidneys, and breasts, and is very low in the liver and pancreas (This picture is from Human Proteome Map (http://www.humanproteomemap.org/)).
3. Function of CDA1
CDA1 exerts a variety of physiological functions, such as inhibiting tumorigenesis, inhibiting cell growth and proliferation, functioning as a cutaneous T-cell lymphoma specific antigen, maintaining normal function of the human heart, and promoting brain development (Figure 5).
Figure 5.

Summary of CDA1 function. CDA1, also known as TSPYL2, TSPX, DENTT, CINAP, Se20-4, and NP79, plays a positive or negative regulatory role in a variety of diseases such as various tumors, diabetes, DN, and atherosclerosis.
3.1. Role in Tumorigenesis
CDA1 is widely expressed in normal tissues, but is downregulated in various types of cancer, including lung cancer [38], glioma [39], liver cancer [40], and prostate cancer [41]. Delbridge et al. [42] searched for a source of the TSPY gene associated with the gonadoblastoma formation (GBY) factor, a transcribed gene homologous to TSPY on the X chromosome of human and mouse. This source was identified and named as TSPX. The Y-localized TSPY and its X-homologue TSPX derived from the same ancestral genes but act as a protooncogene and a tumor suppressor gene, respectively. The C-terminal acidic domain (CAD) of TSPX is vital for the tumor suppressor function but is not contained in TSPY [43]. Tao et al. [44] have shown that TSPX serves a vital role in the maintenance of G1 checkpoint upon DNA damage. TSPX is also required for the induction of p21 transcription while the loss of TSPX results in cell cycle defect in the case of DNA damage. In addition, the CAD of TSPX also inhibits cyclin B1/CDK1 phosphorylation activity and performs a normative function in regulating cell-cycle progression at the G2/M stage [45]. Recent studies have also shown that TSPX is an important X-linked tumor suppressor gene in prostate cancer. Overexpression of TSPX could significantly inhibit cell proliferation and induce cell death in a prostate cancer cell line LNCaP. TSPX overexpression downregulated multiple oncogenes in a CAD-dependent manner and upregulated many tumor suppressors in a CAD-independent manner [41]. Besides, TSPX could interact with androgen receptors (AR) and inhibit the AR transactivation of target genes in a CAD-dependent manner [46]. Because androgen and AR play a fundamental role in the development of prostate cancer, TSPX might act as a modular for androgen and AR activities in the prostate. These results suggested that TSPX plays an important role in the initiation and progression of prostate cancer, and its CAD is essential for tumor suppressor functions.
Ozbun [38] has reported the discovery of a new gene in the TGF-β1 responsive epithelial non-small-cell lung cancer (NSCLC) cell line NCI-H727 and is named differentially expressed nucleolar TGF-β1 target gene (DENTT). These findings suggest that this new gene may be a target gene for TGF-β 1-mediated response, which is closely related to tumorigenesis, especially lung cancer. A subsequent study reported that messenger RNA (mRNA) and protein levels of DENTT were significantly downregulated in human lung tumors, human lung cancer cell lines, and mouse lung tumor models, and overexpression of DENTT significantly suppressed cell growth and clonogenic potential in lung and breast cell lines. In addition, DENTT mRNA and protein levels were elevated after ectopic expression of DENTT or treatment with TGF-β1 in lung cancer cells. In summary, their results suggested that DENTT played a suppressor role in tumor development [47]. Magni [48] has reported that CDA1 is a tumor suppressor and plays a pivotal role in regulating cell growth and DNA damage response. It is well known that P53 is an important tumor suppressor gene in humans, and its deletion or mutation increases the susceptibility of tumors. Upon DNA damage, P53 promotes transcription of specific genes, leading to cell cycle arrest, apoptosis, or senescence. P53 is a target protein of lysine acetyltransferase P300, and nicotinamide adenine dinucleotide- (NAD-) dependent deacetylase SIRT1 negatively regulates the activity of p300 activity toward p53, making P53 deacetylated and thus preventing P53-induced apoptosis. When DNA damage occurs, CDA1 inhibits SIRT1 to promote p53 acetylation and initiate p53-dependent cell death, thereby inhibiting tumorigenesis.
Chai et al. [34, 35] reported that overexpression of CDA1 in HeLa cells arrests cell growth and inhibits DNA synthesis. They found that the mutation of two CDK consensus phosphorylation sites abolished the function of CDA1 inhibited cell growth. This finding indicates that CDA1 acts a negative regulator of cell growth. Further research suggested that CDA1 plays an antiproliferation role by activating p53 and MEK/ERK1/2 MAPK pathway to upregulate p21Waf1/CIP1 transcription [49].
SPYL2/REST complex has been found to have an important inhibitory effect on epithelial cancers, and its antiproliferative effect is achieved by activating and enhancing TGF-β signaling. Epping et al. [50] reported that CDA1 is a hugely important part of the REST/NRSF transcriptional complex and plays a role in tumor inhibition by interacting with REST to enhance TGF- β signaling.
3.2. As a Cutaneous T-Cell Lymphoma-Specific Antigen
Eichmuller et al. [51] found a new gene, named as se20-4 (accession numbers: AF273046), in the serum and tissues of patients with cutaneous T-cell lymphoma (CTCL) when using SEREX (serological identification of recombinantly expressed genes) method to identify cutaneous T lymphoma-specific antigens. This identified antigen se20-4 showed high homology to the already known gene CDA1; they were the same gene (GenBank database accession numbers: AB015345). Further studies have confirmed that se20-4 is also present in tumor tissues and cell lines such as melanoma, leukemia [52], and lymphoma [53] and is a nuclear targeting protein.
3.3. As Differentially Expression Genes in Human Heart Tissues
Sun et al. [54] identified three novel genes in the genes differentially expressed between normal human and congenital heart defect (CHD) heart tissues by RNA arbitrarily primed-PCR (RAP-PCR) and RT-PCR, including NP79 (coding for a nuclear protein of 79KD), which is likely to be related to the normal function of the human heart. NP79 (accession numbers: AF273046) is identical to H. sapiens CDA1 mRNA and is also a clone for cutaneous T-cell lymphoma-associated antigen isolated from a normal testis cDNA library. Since NP79 is highly expressed in the adult heart, it is speculated that it plays a role in maintaining the normal function of the human heart.
3.4. Role in Brain Development
Wang et al. [55] identified a new gene named CINAP (CASK-Interacting Nucleosome Assembly Protein), from an adult rat brain library in a yeast two-hybrid screen using the guanylate kinase (GK) domain of CASK as baits. CINAP contains the NAP domain of the NAP/SET/TSPY family common characteristic structure. Thus, CINAP belongs to this family and is a homologous mouse gene of CDA1. CDA1 was found to be a nuclear protein mainly present in the cerebral cortex. CDA1, Tbr-1 (an essential transcription factor in cerebral cortex development), and CASK (acts as a coactivator for Tbr-1) form a complex in the nucleus of neurons to regulate in response to neuronal synaptic activity and chromatin assembly [55, 56]. Several pieces of research have shown that mutations or deletion of CDA1 are associated with varying degrees of intellectual disability [57–59], suggesting that CDA1 may play an important role in learning and behavior. According to a study, CDA1 KO mice showed higher locomotor and exploratory activities compared to their wild-type littermates, but the molecular mechanism is unclear [60]. Glutamate pathology is involved in neurodevelopmental conditions, and CDA1 regulates the expression of genes encoding glutamate receptors. CDA1 KO mice may have neurodevelopmental and behavioral abnormalities due to the disruption of glutamate signaling, mainly manifested as sensorimotor gating impairment, mild hyperactivity, and hypersensitivity to the dopamine agonist amphetamine [61]. Subsequently, the study found that compared with wild type littermates, the expression of H3K27me3 (a key histone modification important for brain development and neuronal function) in the hippocampus of CDA1 KO mice was upregulated. In addition, the expression of H3K27 methyltransferase enhancer of zeste 2 (EZH2) target genes important for neuron function was downregulated. Chromatin immunoprecipitation (ChIP) reveals that hemagglutinin-tagged CDA1 coexist in EZH2 in the target promoter of human neuroblastoma cells. These findings indicated that CDA1 interacted with EZH2 and enhanced the expression of H3K27me3-labeled neuronal genes to achieve normal neuronal maturation and function [62]. All the above results indicate that CDA1 is a critical gene for neurodevelopment. In addition, crosstalk between CDA1 and TGF-β signaling has been described [35], signaling by the TGF-β family playing a central role in many aspects of nervous system development and function [63], suggested that TGF-β-CDA1 signaling pathway could be essential in regulating neurogenesis and neurological function.
4. CDA1 and Renal Fibrosis in DN
It has been found that CDA1 plays a positive or negative regulatory role in a large number of different diseases (Table 1), and it can play a nonnegligible role in DN renal fibrosis by enhancing the TGF-β signaling pathway, but its specific mechanism is not yet clear.
Table 1.
Studies of CDA1 in different diseases.
| Disease type | Strategy | Regulation | Research object | Pathway/process | Function | References |
|---|---|---|---|---|---|---|
| Lung cancer | Transfection | CDA1↓ | (1) In vivo: mice; (2) in vitro: A549 cells; (3) human lung tumor | Cyclin B-CDK1↓; TGF-β1↓ | ↓Cell growth; ↓cell migration | [47] |
| Breast cancer | Transfection | CDA1↓ | (1) In vivo: mice; (2) in vitro: MCF-7 cells | Cyclin B-CDK1↓ | ↓Cell growth; ↓cell migration | [47] |
| Prostate cancer | Lentivirus | CDA1↓ | (1) In vitro: LNCaP cells, PC3 cells, DU145 cells; (2) human prostate | Cyclin B-CDK1↓ | ↓Androgen receptor; ↓oncogenes; ↑tumor suppressors | [41] |
| Malignant glioma | Inhibitors | CDA1↓ | (1) In vitro: T98G cells; (2) human glioblastoma, human multiforme, human glioma, human astrocytoma, human oligodendroglioma | Hypermethylation | ↓Cell growth; ↓cell migration | [64] |
| Hepatocellular carcinoma | Transfection | CDA1↓ | In vitro: 293T cells, HuH7 cells | Ubiquitin-proteasome ↑; hepatitis B viral protein (HBx)↓ | ↓Cell growth; ↓cell migration | [65] |
| Aortic aneurysms | Gene knockout | CDA1↓ | (1) In vivo: mice; (2) human abdominal aortic aneurysms | TGF-β1↓ | ↓Aneurysm formation in human; ↓aneurysm severity in mice | [66] |
| Diabetic nephropathy | Gene knockout; adenoviral | CDA1↓ | (1) In vivo: mice; (2) in vitro: HK-2 cells; (3) human renal biopsy | TGF-β ↓ | ↓Renal fibration | [32, 33, 67] |
| Diabetic atherosclerosis | Gene knockdown adenoviral | CDA1↓ | (1) In vivo: mice; (2) in vitro: vascular smooth muscle cells | CDA1↓ | ↓Atherosclerosis | [68] |
| Attention deficit hyperactivity disorder | Gene knockdown | TSPYL2↓ | In vivo: mice | Glutamate receptors↓ | ↑Activity; ↓prepulse inhibition | [61] |
| Neurodevelopmental psychiatric disorders | Gene knockdown | TSPYL2↓ | (1) In vivo: mice; (2) in vitro: HEK293 cells | Glutamate↓; GluN2A↓, GluN2B↓; BDNF↓, Egr3↓, Grin2c↓ | ↓Learning and behavior | [59, 62] |
| Intellectual disability | Genetic studies | Mutations in SPYL2 | Mild nonsyndromic ID | Synaptic signaling↓ | ↓Memory skills and language development | [57, 58] |
↓: decrease; ↑: increase.
4.1. CDA1 Promotes the Occurrence and Development of Renal Fibrosis in DN
Chai's team [32] has found that CDA1 expression level in the kidneys of diabetic animal models was elevated. In addition, compared with healthy kidneys, the expression of CDA1 was significantly increased in the kidneys of patients with DN and patients with nondiabetic sclerotic renal disease [33]. In vivo assays, they used spontaneously hypertensive rats (SHRs) and the ApoE−/− mouse as a model of DN. The results showed that the mRNA levels of TGF-β, TGF-β type I receptor (TβRI), TGF-β type II receptor (TβRII), connective tissue growth factor (CTGF), collagen I (COL1), III (COL3), IV (COL4), and fibronectin (FN) were significantly increased compared to their nondiabetic controls. In vitro assays forced expression of CDA1 in HK-2 cells resulted in TGF-β signaling pathway activation by upregulation of these genes, while knockdown of CDA1 in HK-2 cells significantly reduced TGF-β signaling and downregulated the expression of these genes. Moreover, recombined TGF-β treatment of CDA1-overexpressing HK-2 cells resulted in further activation of TGF-β signaling, and its downstream of ECM genes such as COL1 and COL3, but CDA1 knockdown effectively blocked TGF-β-stimulated expression of collagen genes. These results suggested that the molecular synergy between CDA1 and TGF-β underpin TGF-β signaling and its target gene expression [32]. Furthermore, compared with CDA1 wild-type (WT) mice, the expression of TGF-β, TβRI, TβRII, and key target genes of TGF-β associated with diabetes including CTGF, α smooth muscle actin (α-SMA), COL1, COL3, and COL4, as well as monocyte chemoattractant protein-1(MCP-1) and vascular cell adhesion molecule 1(VCAM-1) and FN were significantly attenuated in diabetic CDA1 KO mice. CDA1 deletion reduced the glomerular and tubulointerstitial injury indexes in CDA1/ApoE gene dKO mice. Deletion of CDA1 disrupted TGF-β signaling, failing TGF-β to stimulate the expression of its target genes such as COL1 and COL3 in primary kidney cells isolated from CDA1 WT and KO mice. CDA1 deficiency in diabetic mice attenuated TGF-β signaling, leading to a reduction in ECM accumulation in the kidney [33]. These results suggest that CDA1 plays a vital role in the development of DN. Thus CDA1 may increase the production of ECM protein in the kidney of diabetic patients by enhancing TGF-β signaling, thereby promoting the development of renal fibrosis of DN. Studies have also shown that CDA1 appears to be a significant molecule not only in DN but also in other diabetic vascular complications such as atherosclerosis [68]. Li et al. [66] indicated that CDA1 plays a key role in the protective effect of diabetes on aneurysms. However, the specific mechanism by which CDA1 enhances the TGF-β signaling pathway and promotes renal fibrosis in DN deserves further investigation.
4.2. The Possible Mechanism of CDA1 Promoting Renal Fibrosis in DN through the TGF-β Signaling Pathway
Downstream of the TGF-β signaling pathway includes canonical (Smad-based) and noncanonical (non-Smad-based) signaling pathways. The canonical pathway is mainly through Smads signaling pathway. Non-Smad-based pathways include MAPKs (including ERK, p38, and JNK), NF-κB, PI3K-AKT, and Rho-like GTPase [69]. CDA1 may be involved in these pathways of TGF-β to promote renal fibrosis in DN (Figure 6).
Figure 6.
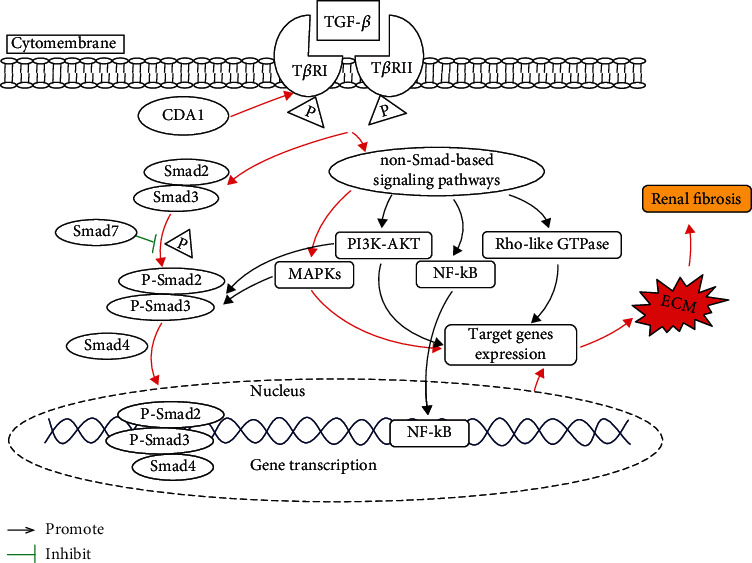
The possible mechanism of crosstalk between CDA1 and TGF-β promoting renal fibrosis in DN. The canonical TGF-β signaling pathway and MAPK signaling pathway marked as red arrow have been demonstrated to be associated with CDA1 expression in diabetic nephropathy, while pathways marked as black related with CDA1 expression remain unknow.
4.2.1. Through the TGF-β/Smad Signaling Pathway
TGF-β is a bioactive polypeptide and a member of the transforming growth factor superfamily. Its three isoforms including TGF-β1, TGF-β2, and TGF-β3 have been successively cloned in mammals [70], mainly through the mechanism of autocrine or paracrine to reach the corresponding target organs to perform their multiple functions [71]. TGF-β binds to and gathers together with serine/threonine kinase receptors (mainly including two subtypes TβRI and TβRII with similar structures) on the cell membrane to initiate signaling [72]. The subtype of TGF-β in the kidneys is mainly TGF-β1, with studies suggesting that TGF-β1 or its signaling downstream signaling pathway is the major pathogenic factor leading to onset or progression of forms of renal fibrosis [22]. The main mechanisms of TGF-β1 inducing renal fibrosis include (1) directly inducing the synthesis of ECM, including COL1 and FN, from the transcription level [73]; (2) inducing the imbalance of matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinase (TIMPs) to inhibit the degradation of ECM [74]; (3) directly affecting renal intrinsic cells: it can induce mesangial cell proliferation and collagen secretion, promote epithelial and podocyte damage, and aggravate inflammation and secondary fibrosis, etc. [75]; (4) promoting the transdifferentiation and proliferation of myofibroblasts from various sources such as pericyte, fibroblast, epithelial cells, and macrophages, as well as mediating fibrosis responses [76]. Usually, the downstream effector transcription factor of TGF-β signaling pathway is Smad. They exist in the cytoplasm, with their main function to transduce TGF-β signals from the cell membrane into the nucleus. The sequential phosphorylation reaction process sees TGF-β activating Smad. TβRII of TGF-β binds to the ligand and phosphorylates TβRI, which in turn phosphorylates Smad2/3 (membrane-receptor-activated Smad, R-Smad), then undergoes homotrimerization and formation of heteromeric complexes with the Smad4 (comediator Smad, co-Smad). This complex moves into the nucleus and interacts with various transcription factors to regulate the transcription of target genes [77]. Activated TβRI phosphorylates R-Smad, initiating the TGF-β signaling pathway as well as activating Smad7 (inhibitory Smad, I-Smad). Smad2 and Smad3 are two main downstream regulators of TGF-β 1-mediated tissue fibrosis, and Smad7 acts as an inhibitory regulator of the TGF-β1/Smad pathway to prevent TGF-β 1-mediated fibrosis [78]. Several previous studies have proven that renal fibrosis can be attenuated by regulating the TGFβ/Smad3 signaling pathway [79–82]. Chai et al. found that CDA1 enhances TGF-β signaling by regulating TβRI and increases phosphorylation of Smad3 in HK-2 cells, primary kidney cells from CDA1 WT mice, tubular cells, and glomerular podocytes in ApoE KO and CDA1/ApoE dKO mice, which result in accumulation of ECM in the kidney, resulting in the development of DN [32, 33]. Therefore, CDA1 may promote the development of diabetic renal fibrosis through the TGF-β/Smads signaling pathway.
4.2.2. Through TGF-β/MAPK Signaling Pathway
Mitogen-activated protein kinases (MAPKs) are important non-Smad-dependent downstream signaling pathways of TGF-β. Studies have found that MPPK's family members including extracellular signal-regulated kinase (ERK), mitogen-activated protein kinase (p38), and C-Jun N-terminal kinases (JNK) were significantly involved in renal fibrosis. Studies have demonstrated that TGF-β1 could activate MAPKs, then induced apoptosis in renal podocytes thereby accelerating DN progression [83–85]. Lakshmanan et al. [86] found that p38 MAPK, ERK, and JNK were activated in streptozotocin- (STZ-) induced diabetic kidney disease in mice, and these protein expressions were upregulated. Furthermore, crosstalk between ERK, JNK, and p38 MAPKs and Smad signaling pathways could synergistically enhance the expression of TGF-β-induced profibrotic genes in various types of renal cells to promote renal fibrosis [87–90]. It has been found that some drugs can reverse renal fibrosis by inhibiting the MAPK pathway [91, 92]. Studies have suggested that CDA1 could increase the phosphorylation of ERK/MAPK in animal models of DN and increase the accumulation of ECM in the kidney, leading to the development of DN. Conversely, CDA1 deletion attenuated TGF-β signaling pathway by attenuating phosphorylation of Smad3 and ERK/MAPK in primary kidney cells from CDA1 WT mice, thereby reducing renal ECM accumulation [32, 33]. As a result, CDA1 may promote the development of diabetic renal fibrosis through the TGF-β/MAPKs signaling pathway.
4.2.3. Through TGF-β/NF-κB Signaling Pathway
Nuclear factor kB (NF KB) is a vital transcription factor, which not only mediates various immune and inflammatory reactions but also participates in a wide range of biological processes, including cell proliferation, differentiation, autophagy, and senescence [93]. TGF-β can activate NF-κB and mediate transcriptional activation of TGF-β target genes in a variety of cell types [93]. It has been found that NF-κB signaling can participate in the occurrence of renal fibrosis. In the priming phase of renal fibrosis, direct renal tubular epithelial cell injury or cellular stimuli triggers the production of various proinflammatory molecules driven by activation of NF-κB signaling, leading to the recruitment of inflammatory cells, thus mediates inflammatory response and promotes the occurrence of renal fibrosis [94, 95]. NF-KB signaling pathway could mediate hypoxia-induced inflammatory responses, ECN accumulation, and oxidative stress leading to renal fibrosis [96]. These results suggest that NF-κB signaling pathway plays a crucial role in the development of renal fibrosis. CDA1 can promote renal fibrosis by regulating TGF-β signaling, and NF-κB can serve as the downstream signaling pathway of TGF-β. Therefore, it is speculated that CDA1 can promote the progression of diabetic nephropathy through the TGF-β/NF-κB signaling pathway. However, there is no direct evidence that CDA1 is involved in renal fibrosis of DN through the TGF-β/NF-κB signaling pathway.
4.2.4. Through TGF-β/PI3K-AKT Signaling Pathway
TGF-β and phosphoinositide 3-kinase/protein kinase B (PI3K-Akt) signaling pathway controls a variety of cellular responses, including glucose homeostasis, cell proliferation, apoptosis, migration, and survival [93, 97]. Studies have reported that extensive crosstalk between TGF-β and the PI3K/AKT signaling pathway plays a key role in tumor progression. In the early stages of cancer, the activated PI3K/AKT pathway antagonizes TGF-β/Smad induced the cytostatic or apoptosis response, whereas, in advanced cancers, both two pathways synergistically cooperate in promoting the invasiveness of cancer cells [97]. The PI3K/AKT signaling pathway not only regulates the occurrence of tumors but is also involved in the regulation of renal fibrosis. Runyan et al. [98] found that the crosstalk between Smad and PI3K/Akt pathway may contribute to TGF-β-induced glomerular matrix accumulation. PI3K-AKT signaling pathway could mediate hypoxia-induced fibroblast activation, collagen synthesis, and EMT regulation leading to renal fibrosis [91]. Several studies have confirmed that inhibition of the PI3K/AKT signaling pathway can alleviate renal interstitial fibrosis [99–101]. These results suggest that the PI3K/AKT signaling pathway crosstalk with the TGF-β signaling pathway plays a pivotal role in the progression of tumors and renal interstitial fibrosis. CDA1 can regulate renal fibrosis and inhibit tumorigenesis through the TGF-β signaling pathway. Therefore, it is speculated that CDA1 can promote the progression of DN through the TGF-β/PI3K-AKT signaling pathway. Direct evidence of CDA1 participating in the development of renal fibrosis via the TGF-β/PI3K-AKT signaling pathway is still lacking. This question needs to be further clarified in subsequent studies.
4.2.5. Through TGF-β/Rho-Like GTPases Signaling Pathway
Rho GTPases are members of the Ras superfamily, while the three best characterized mammalian members are Rho, Rac, and Cdc42 [102]. ROCK, also known as Rho-associated kinase, belongs to the serine/threonine-protein kinase. ROCK is the most important and characteristic Rho downstream target effector molecule and is widely expressed in human body. It has many functions such as regulating cell contraction, motility, cell division, adhesion, and proliferation, and it participates in the occurrence and development of a variety of diseases [103]. Studies have demonstrated that DN is associated with the activation of the Rho/ROCK signaling pathway [104]. The application of ROCK inhibitors can delay the development of renal interstitial fibrosis [105–108]. Furthermore, TGF-β has been discovered to induce activation of Rho, Rac, and Cdc42 in different cell systems. However, most studies have concentrated on the role of RhoA and its effector kinase ROCK in TGF-β-induced epithelial-to-mesenchymal transdifferentiation (EMT) [109]. It was found that TGF-β rapidly activates RhoA-dependent signaling pathways in epithelial cells and promotes the occurrence of EMT by regulating cytoskeletal remodeling and activating SMA promoter [110, 111]. CDA1 regulates the TGF-β signaling pathway, while Rho-like GTPase is a Smad-independent downstream signaling pathway of TGF-β. Thus, CDA1 may promote the development of diabetic renal fibrosis through TGF-β/Rho-like GTPase signaling pathway. However, there is currently no evidence that CDA1 promotes the development of renal fibrosis of DN through the TGF-β/Rho-like signaling pathway.
4.3. Current Method for Targeting CDA1 to Treat Renal Fibrosis of DN
Targeting CDA1 has been demonstrated to attenuate renal fibrosis-related genes expression by regulating TGF-β signaling in several animal models and in in vitro studies. Chai's team [33] generated CDA1 KO mice by genetic deletion of Tspyl2. They found that CDA1 KO mice had no significant effect on animal health, while no obvious abnormal phenotypes were found during reproduction. Genetic deletion of CDA1 was found to significantly reduce the expression of diabetes-associated renal matrix accumulation and profibrosis genes both in vivo and in vitro [32, 33], confirming that CDA1 is an important and effective potential target in DN.
Recently, Chai et al. [67] isolated and identified a novel protein interacting with CDA1 from a human testis testicular cDNA library using yeast two-hybridization screening, which was called CDA1 binding protein 1 (CDA1BP1). They found that CDA1 and CDA1BP1 were expressed in human and mouse kidneys, as well as human renal tubular HK-2 cells. CDA1BP1 binds to CDA1 and enhances TGF-β signaling to exert its profibrotic effect. They utilized the model of diabetic by using STZ-induced male ApoE KO, WT, and CDA1BP1 KO mice, all with the background of C57BL6. The expression level of COL1, COL3, FN, and tumor necrosis factor-α (TNF-α) were significantly reduced in diabetic CDA1BP1 KO mice renal. Also, CDA1BP1 siRNA knockdown reduced the level of COL1 and COL3 in HK-2 cells with or without TGF-β treatment. Furthermore, Chai's team generated a hybrid peptide inhibitor CHA-061, which contains a short CDA1BP1 sequence with an ability to bind to CDA1, and a cell penetrating peptide [112, 113]. CHA-061 treatment significantly attenuated or blocked the expression of diabetes-associated profibrotic genes TGF-β1, TGF-β2, TβRI, TβRII, and CTGF, sclerotic genes such as COL1, COL3, COL4, FN, and MMP2, proinflammatory genes such as TNF-α, C-reactive protein (CRP), MCP-1, intercellular adhesion molecules-1 (ICAM1), and vascular cell adhesion molecule (VCAM1) in the kidneys. It also reduced renal ECM accumulation and glomerular injury index. No significant side effects were observed during the study. These studies suggested that CDA1/CDA1BP1 axis targeting by genetic and pharmacological approaches is a safe and effective method to attenuate pathological hallmarks and to inhibit renal fibrosis of experimental DN.
5. Conclusion
CDA1 is distributed in various tissues of the human body exerting various biological functions, such as inhibiting cell growth and inhibiting tumor proliferation. It also plays a crucial role in renal fibrosis of DN. Quite a few studies have shown that CDA1 can enhance renal and vascular TGF-β signaling and promote the development of renal fibrosis in DN. However, the specific mechanism remains unclear. CDA1 may act on TβRI, induce Smad phosphorylation, and activate the ERK MAPK pathway to promote renal fibrosis and atheroma formation in DN. No abnormal phenotype was observed in CDA1 KO DN mice, which were able to develop and grow normally, with only mild kidney damage. Therefore, CDA1 may be a safe and effective therapeutic target for delaying DN and other mediated renal fibrosis induced by TGF-β. Recently, prototype peptide inhibitors of CDA1 have been developed with their safety and efficacy having been verified genetically and pharmacologically. In the future, more drugs need to be developed to target CDA1 in the treatment of DN. In addition, due to the complexity of the TGF-β signaling pathway, whether CDA1 enhances TGF-β signaling through other mechanisms requires further validation in future studies.
Acknowledgments
We thank Dr. James Flowers for the helpful suggestions. We are thankful for the financial support of the Joint Fund of Hubei Provincial Health Commission (WJ2019H561), National Nature Science Foundation of China (nos. 32071445 and 81501330), and the Science Foundation of CTGU (KJ2014B066).
Contributor Information
Bin Hu, Email: hubinhxy@163.com.
Changbai Liu, Email: cbliu@ctgu.edu.cn.
Hu Wang, Email: biomed_wang@yahoo.com.
Conflicts of Interest
The authors indicate no potential conflicts of interest.
Authors' Contributions
LLC, JW, CBL, BH, and HW conducted the literature review and initial draft. LLC, JW, and HW conceived the figures. All authors contributed to outline conceiving, reviewing, and figures and manuscript editing. Manuscript submission was approved by HW, CBL, and BH. All authors read and approved the final manuscript version. LinLin Chen and Jiao Wu contributed equally to this work.
References
- 1.Wild S., Roglic G., Green A., Sicree R., King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Guariguata L., Whiting D. R., Hambleton I., Beagley J., Linnenkamp U., Shaw J. E. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Research and Clinical Practice. 2014;103(2):137–149. doi: 10.1016/j.diabres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Brenneman J., Hill J., Pullen S. Emerging therapeutics for the treatment of diabetic nephropathy. Bioorganic & Medicinal Chemistry Letters. 2016;26(18):4394–4402. doi: 10.1016/j.bmcl.2016.07.079. [DOI] [PubMed] [Google Scholar]
- 4.Chan G. C., Tang S. C. Diabetic nephropathy: landmark clinical trials and tribulations. Nephrology, Dialysis, Transplantation. 2016;31(3):359–368. doi: 10.1093/ndt/gfu411. [DOI] [PubMed] [Google Scholar]
- 5.Papadopoulou-Marketou N., Chrousos G. P., Kanaka-Gantenbein C. Diabetic nephropathy in type 1 diabetes: a review of early natural history, pathogenesis, and diagnosis. Diabetes/metabolism research and reviews. 2017;33(2) doi: 10.1002/dmrr.2841. [DOI] [PubMed] [Google Scholar]
- 6.Gnudi L., Coward R. J. M., Long D. A. Diabetic nephropathy: perspective on novel molecular mechanisms. Trends in Endocrinology and Metabolism. 2016;27(11):820–830. doi: 10.1016/j.tem.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Lim A. Diabetic nephropathy – complications and treatment. International journal of nephrology and renovascular disease. 2014;7:361–381. doi: 10.2147/IJNRD.S40172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koszegi S., Molnar A., Lenart L., et al. RAAS inhibitors directly reduce diabetes-induced renal fibrosis via growth factor inhibition. The Journal of Physiology. 2019;597(1):193–209. doi: 10.1113/JP277002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roscioni S. S., Heerspink H. J., de Zeeuw D. The effect of RAAS blockade on the progression of diabetic nephropathy. Nature Reviews. Nephrology. 2014;10(2):77–87. doi: 10.1038/nrneph.2013.251. [DOI] [PubMed] [Google Scholar]
- 10.Warren A. M., Knudsen S. T., Cooper M. E. Diabetic nephropathy: an insight into molecular mechanisms and emerging therapies. Expert Opinion on Therapeutic Targets. 2019;23(7):579–591. doi: 10.1080/14728222.2019.1624721. [DOI] [PubMed] [Google Scholar]
- 11.Zou H., Zhou B., Xu G. SGLT2 inhibitors: a novel choice for the combination therapy in diabetic kidney disease. Cardiovascular Diabetology. 2017;16(1):p. 65. doi: 10.1186/s12933-017-0547-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alicic R. Z., Johnson E. J., Tuttle K. R. SGLT2 inhibition for the prevention and treatment of diabetic kidney disease: a review. American Journal of Kidney Diseases. 2018;72(2):267–277. doi: 10.1053/j.ajkd.2018.03.022. [DOI] [PubMed] [Google Scholar]
- 13.Vitale M., Haxhi J., Cirrito T., Pugliese G. Renal protection with glucagon-like peptide-1 receptor agonists. Medicina (Kaunas) 2020;54:91–101. doi: 10.1016/j.coph.2020.08.018. [DOI] [PubMed] [Google Scholar]
- 14.Thomas M. C. The potential and pitfalls of GLP-1 receptor agonists for renal protection in type 2 diabetes. Diabetes & metabolism. 2017;43(Supplement 1):2S20–22S27. doi: 10.1016/S1262-3636(17)30069-1. [DOI] [PubMed] [Google Scholar]
- 15.Kohan D. E., Pollock D. M. Endothelin antagonists for diabetic and non-diabetic chronic kidney disease. British Journal of Clinical Pharmacology. 2013;76(4):573–579. doi: 10.1111/bcp.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kohan D. E., Barton M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney International. 2014;86(5):896–904. doi: 10.1038/ki.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Georgianos P. I., Agarwal R. Endothelin A receptor antagonists in diabetic kidney disease. Current Opinion in Nephrology and Hypertension. 2017;26(5):338–344. doi: 10.1097/MNH.0000000000000342. [DOI] [PubMed] [Google Scholar]
- 18.Huynh P., Chai Z. Transforming growth factor β (TGFβ) and related molecules in chronic kidney disease (CKD) Clinical Science (London, England) 2019;133(2):287–313. doi: 10.1042/CS20180438. [DOI] [PubMed] [Google Scholar]
- 19.Zeng L. F., Xiao Y., Sun L. A glimpse of the mechanisms related to renal fibrosis in diabetic nephropathy. Advances in Experimental Medicine and Biology. 2019;1165:49–79. doi: 10.1007/978-981-13-8871-2_4. [DOI] [PubMed] [Google Scholar]
- 20.Ma T. T., Meng X. M. TGF-β/Smad and renal fibrosis. Advances in Experimental Medicine and Biology. 2019;1165:347–364. doi: 10.1007/978-981-13-8871-2_16. [DOI] [PubMed] [Google Scholar]
- 21.Meng X. M., Tang P. M., Li J., Lan H. Y. TGF-β/Smad signaling in renal fibrosis. Frontiers in Physiology. 2015;6:p. 82. doi: 10.3389/fphys.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng X. M., Nikolic-Paterson D. J., Lan H. Y. TGF-β: the master regulator of fibrosis. Nature Reviews. Nephrology. 2016;12(6):325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 23.Shah M., Revis D., Herrick S., et al. Role of elevated plasma transforming growth factor-β1 levels in wound healing. The American Journal of Pathology. 1999;154(4):1115–1124. doi: 10.1016/S0002-9440(10)65364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moses H. L., Roberts A. B., Derynck R. The discovery and early days of TGF-β: a historical perspective. Cold Spring Harbor perspectives in biolog. 2016;8(7, article a021865) doi: 10.1101/cshperspect.a021865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goumans M. J., Liu Z., ten Dijke P. TGF-β signaling in vascular biology and dysfunction. Cell Research. 2009;19(1):116–127. doi: 10.1038/cr.2008.326. [DOI] [PubMed] [Google Scholar]
- 26.Morikawa M., Derynck R., Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harbor Perspectives in Biology. 2016;8(5, article a021873) doi: 10.1101/cshperspect.a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie F., Ling L., van Dam H., Zhou F., Zhang L. TGF-β signaling in cancer metastasis. Acta biochimica et biophysica Sinica. 2018;50(1):121–132. doi: 10.1093/abbs/gmx123. [DOI] [PubMed] [Google Scholar]
- 28.Dickson M. C., Martin J. S., Cousins F. M., Kulkarni A. B., Karlsson S., Akhurst R. J. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development. 1995;121(6):1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 29.Kulkarni A. B., Huh C. G., Becker D., et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(2):770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christ M., McCartney-Francis N. L., Kulkarni A. B., et al. Immune dysregulation in TGF-beta 1-deficient mice. Journal of Immunology. 1994;153(5):1936–1946. [PubMed] [Google Scholar]
- 31.Crowe M. J., Doetschman T., Greenhalgh D. G. Delayed wound healing in immunodeficient TGF-β1 knockout mice. The Journal of Investigative Dermatology. 2000;115(1):3–11. doi: 10.1046/j.1523-1747.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- 32.Tu Y., Wu T., Dai A., et al. Cell division autoantigen 1 enhances signaling and the profibrotic effects of transforming growth factor-β in diabetic nephropathy. Kidney International. 2011;79(2):199–209. doi: 10.1038/ki.2010.374. [DOI] [PubMed] [Google Scholar]
- 33.Chai Z., Dai A., Tu Y., et al. Genetic deletion of cell division autoantigen 1 retards diabetes-associated renal injury. Journal of the American Society of Nephrology. 2013;24(11):1782–1792. doi: 10.1681/ASN.2013010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chai Z., Sarcevic B., Mawson A., Toh B. H. SET-related cell division autoantigen-1 (CDA1) arrests cell growth. The Journal of Biological Chemistry. 2001;276(36):33665–33674. doi: 10.1074/jbc.M007681200. [DOI] [PubMed] [Google Scholar]
- 35.Toh B. H., Tu Y., Cao Z., Cooper M. E., Chai Z. Role of cell division autoantigen 1 (CDA1) in cell proliferation and fibrosis. Genes (Basel) 2010;1(3):335–348. doi: 10.3390/genes1030335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ozbun L. L., Martínez A., Jakowlew S. B. Differentially expressed nucleolar TGF-β1 target (DENTT) shows tissue-specific nuclear and cytoplasmic localization and increases TGF-β1-responsive transcription in primates. Biochimica et Biophysica Acta (BBA)-Gene Structure and Expression. 2005;1728(3):163–180. doi: 10.1016/j.bbaexp.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 37.Ozbun L. L., Martinez A., Angdisen J., et al. Differentially expressed nucleolar TGF-beta1 target (DENTT) in mouse development. Developmental Dynamics. 2003;226(3):491–511. doi: 10.1002/dvdy.10257. [DOI] [PubMed] [Google Scholar]
- 38.Ozbun L. L., You L., Kiang S., Angdisen J., Martinez A., Jakowlew S. B. Identification of differentially expressed nucleolar TGF-β1 target (DENTT) in human lung cancer cells that is a new member of the TSPY/SET/NAP-1 superfamily. Genomics. 2001;73(2):179–193. doi: 10.1006/geno.2001.6505. [DOI] [PubMed] [Google Scholar]
- 39.Eyler C. E., Wu Q., Yan K., et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell. 2011;146(1):53–66. doi: 10.1016/j.cell.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kido T., Lo R. C., Li Y., et al. The potential contributions of a Y-located protooncogene and its X homologue in sexual dimorphisms in hepatocellular carcinoma. Human Pathology. 2014;45(9):1847–1858. doi: 10.1016/j.humpath.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 41.Kido T., Li Y., Tanaka Y., Dahiya R., Chris Lau Y. F. The X-linked tumor suppressor TSPX downregulates cancer-drivers/oncogenes in prostate cancer in a C-terminal acidic domain dependent manner. Oncotarget. 2019;10(15):1491–1506. doi: 10.18632/oncotarget.26673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Delbridge M. L., Longepied G., Depetris D., et al. TSPY, the candidate gonadoblastoma gene on the human Y chromosome, has a widely expressed homologue on the X-implications for Y chromosome evolution. Chromosome Research. 2004;12(4):345–356. doi: 10.1023/B:CHRO.0000034134.91243.1c. [DOI] [PubMed] [Google Scholar]
- 43.Lau Y.-F. C., Li Y., Kido T. Battle of the sexes: contrasting roles of testis-specific protein Y-encoded (TSPY) and TSPX in human oncogenesis. Asian Journal of Andrology. 2019;21(3):260–269. doi: 10.4103/aja.aja_43_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tao K. P., Fong S. W., Lu Z., Ching Y. P., Chan K. W., Chan S. Y. TSPYL2 is important for G1 checkpoint maintenance upon DNA damage. PLoS One. 2011;6(6, article e21602) doi: 10.1371/journal.pone.0021602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y., Lau Y. F. TSPY and its X-encoded homologue interact with cyclin B but exert contrasting functions on cyclin-dependent kinase 1 activities. Oncogene. 2008;27(47):6141–6150. doi: 10.1038/onc.2008.206. [DOI] [PubMed] [Google Scholar]
- 46.Clift D., McEwan W. A., Labzin L. I., et al. A method for the acute and rapid degradation of endogenous proteins. Cell. 2017;171(7):1692–1706.e18. doi: 10.1016/j.cell.2017.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kandalaft L. E., Zudaire E., Portal-Nunez S., Cuttitta F., Jakowlew S. B. Differentially expressed nucleolar transforming growth factor-beta1 target (DENTT) exhibits an inhibitory role on tumorigenesis. Carcinogenesis. 2008;29(6):1282–1289. doi: 10.1093/carcin/bgn087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Magni M., Buscemi G., Maita L., et al. TSPYL2 is a novel regulator of SIRT1 and p300 activity in response to DNA damage. Cell Death & Differentiation. 2019;26(5):918–931. doi: 10.1038/s41418-018-0168-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tu Y., Wu W., Wu T., et al. Antiproliferative autoantigen CDA1 transcriptionally up-regulates p21Waf1/Cip1 by activating p53 and MEK/ERK1/2 MAPK pathways. The Journal of Biological Chemistry. 2007;282(16):11722–11731. doi: 10.1074/jbc.M609623200. [DOI] [PubMed] [Google Scholar]
- 50.Epping M. T., Lunardi A., Nachmani D., et al. TSPYL2 is an essential component of the REST/NRSF transcriptional complex for TGF _β_ signaling activation. Cell Death and Differentiation. 2015;22(8):1353–1362. doi: 10.1038/cdd.2014.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eichmuller S., Usener D., Dummer R., Stein A., Thiel D., Schadendorf D. Serological detection of cutaneous T-cell lymphoma-associated antigens. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(2):629–634. doi: 10.1073/pnas.98.2.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guinn B. A., Gilkes A. F., Woodward E., et al. Microarray analysis of tumour antigen expression in presentation acute myeloid leukaemia. Biochemical and Biophysical Research Communications. 2005;333(3):703–713. doi: 10.1016/j.bbrc.2005.05.161. [DOI] [PubMed] [Google Scholar]
- 53.Huang S., Preuss K.-D., Xie X., Regitz E., Pfreundschuh M. Analysis of the antibody repertoire of lymphoma patients. Cancer Immunology, Immunotherapy. 2002;51(11-12):655–662. doi: 10.1007/s00262-002-0320-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun G., Yuen Chan S., Yuan Y., et al. Isolation of differentially expressed genes in human heart tissues. Biochimica et Biophysica Acta. 2002;1588(3):241–246. doi: 10.1016/S0925-4439(02)00171-0. [DOI] [PubMed] [Google Scholar]
- 55.Wang G. S., Hong C. J., Yen T. Y., et al. Transcriptional modification by a CASK-interacting nucleosome assembly protein. Neuron. 2004;42(1):113–128. doi: 10.1016/S0896-6273(04)00139-4. [DOI] [PubMed] [Google Scholar]
- 56.Wang T. F., Ding C. N., Wang G. S., et al. Identification of Tbr-1/CASK complex target genes in neurons. Journal of Neurochemistry. 2004;91(6):1483–1492. doi: 10.1111/j.1471-4159.2004.02845.x. [DOI] [PubMed] [Google Scholar]
- 57.Vasli N., Ahmed I., Mittal K., et al. Identification of a homozygous missense mutation in LRP2 and a hemizygous missense mutation in TSPYL2 in a family with mild intellectual disability. Psychiatric Genetics. 2016;26(2):66–73. doi: 10.1097/YPG.0000000000000114. [DOI] [PubMed] [Google Scholar]
- 58.Moey C., Hinze S. J., Brueton L., et al. Xp11.2 microduplications including IQSEC2, TSPYL2 and KDM5C genes in patients with neurodevelopmental disorders. European Journal of Human Genetics. 2016;24(3):373–380. doi: 10.1038/ejhg.2015.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsang K. H., Lai S. K., Li Q., et al. The nucleosome assembly protein TSPYL2 regulates the expression of NMDA receptor subunits GluN2A and GluN2B. Scientific Reports. 2015;4(1):p. 3654. doi: 10.1038/srep03654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chung W. C., Huang T. N., Hsueh Y. P. Targeted deletion of CASK-interacting nucleosome assembly protein causes higher locomotor and exploratory activities. Neurosignals. 2011;19(3):128–141. doi: 10.1159/000327819. [DOI] [PubMed] [Google Scholar]
- 61.Li Q., Chan S. Y., Wong K. K., et al. Tspyl2 loss-of-function causes neurodevelopmental brain and behavior abnormalities in mice. Behavior Genetics. 2016;46(4):529–537. doi: 10.1007/s10519-015-9777-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu H., Peng L., So J., et al. TSPYL2 regulates the expression of EZH2 target genes in neurons. Molecular Neurobiology. 2019;56(4):2640–2652. doi: 10.1007/s12035-018-1238-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meyers E. A., Kessler J. A. TGF-β family signaling in neural and neuronal differentiation, development, and function. Cold Spring Harbor perspectives in biology. 2017;9(8) doi: 10.1101/cshperspect.a022244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim T. Y., Zhong S., Fields C. R., Kim J. H., Robertson K. D. Epigenomic profiling reveals novel and frequent targets of aberrant DNA methylation-mediated silencing in malignant glioma. Cancer Research. 2006;66(15):7490–7501. doi: 10.1158/0008-5472.CAN-05-4552. [DOI] [PubMed] [Google Scholar]
- 65.Kido T., Ou J. H., Lau Y. F. The X-linked tumor suppressor TSPX interacts and promotes degradation of the hepatitis B viral protein HBx via the proteasome pathway. PLoS One. 2011;6(7, article e22979) doi: 10.1371/journal.pone.0022979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li J., Huynh P., Dai A., et al. Diabetes reduces severity of aortic aneurysms depending on the presence of cell division autoantigen 1 (CDA1) Diabetes. 2018;67(4):755–768. doi: 10.2337/db17-0134. [DOI] [PubMed] [Google Scholar]
- 67.Chai Z., Wu T., Dai A., et al. Targeting the CDA1/CDA1BP1 Axis retards renal fibrosis in experimental diabetic nephropathy. Diabetes. 2019;68(2):395–408. doi: 10.2337/db18-0712. [DOI] [PubMed] [Google Scholar]
- 68.Pham Y., Tu Y., Wu T., et al. Cell division autoantigen 1 plays a profibrotic role by modulating downstream signalling of TGF-beta in a murine diabetic model of atherosclerosis. Diabetologia. 2010;53(1):170–179. doi: 10.1007/s00125-009-1555-9. [DOI] [PubMed] [Google Scholar]
- 69.Heldin C. H., Moustakas A. Signaling receptors for TGF-β family members. Cold Spring Harbor Perspectives in Biology. 2016;8(8) doi: 10.1101/cshperspect.a022053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Govinden R., Bhoola K. D. Genealogy, expression, and cellular function of transforming growth factor-β. Pharmacology & Therapeutics. 2003;98(2):257–265. doi: 10.1016/S0163-7258(03)00035-4. [DOI] [PubMed] [Google Scholar]
- 71.Hong S., Lee H. J., Kim S. J., Hahm K. B. Connection between inflammation and carcinogenesis in gastrointestinal tract: focus on TGF-beta signaling. World Journal of Gastroenterology. 2010;16(17):2080–2093. doi: 10.3748/wjg.v16.i17.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shi Y., Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/S0092-8674(03)00432-X. [DOI] [PubMed] [Google Scholar]
- 73.Samarakoon R., Overstreet J. M., Higgins S. P., Higgins P. J. TGF-β1 → SMAD/p53/USF2 → PAI-1 transcriptional axis in ureteral obstruction-induced renal fibrosis. Cell and Tissue Research. 2012;347(1):117–128. doi: 10.1007/s00441-011-1181-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meng X.-M., Tang P. M.-K., Li J., Lan H. Y. TGF-β/Smad signaling in renal fibrosis. Frontiers in Physiology. 2015;6:p. 82. doi: 10.3389/fphys.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lopez-Hernandez F. J., Lopez-Novoa J. M. Role of TGF-β in chronic kidney disease: an integration of tubular, glomerular and vascular effects. Cell and Tissue Research. 2012;347(1):141–154. doi: 10.1007/s00441-011-1275-6. [DOI] [PubMed] [Google Scholar]
- 76.Meng X. M., Wang S., Huang X. R., et al. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death & Disease. 2016;7(12, article e2495) doi: 10.1038/cddis.2016.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu P., Liu J., Derynck R. Post-translational regulation of TGF-β receptor and Smad signaling. FEBS Letters. 2012;586(14):1871–1884. doi: 10.1016/j.febslet.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hu H. H., Chen D. Q., Wang Y. N., et al. New insights into TGF-β/Smad signaling in tissue fibrosis. Chemico-Biological Interactions. 2018;292:76–83. doi: 10.1016/j.cbi.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 79.Zeisberg M., Hanai J., Sugimoto H., et al. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nature Medicine. 2003;9(7):964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 80.Ji X., Wang H., Wu Z., et al. Specific inhibitor of Smad3 (SIS3) attenuates fibrosis, apoptosis, and inflammation in unilateral ureteral obstruction kidneys by inhibition of transforming growth factor β (TGF-β)/Smad3 signaling. Medical science monitor: international medical journal of experimental and clinical research. 2018;24:1633–1641. doi: 10.12659/MSM.909236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.You Y. K., Luo Q., Wu W. F., et al. Petchiether A attenuates obstructive nephropathy by suppressing TGF-β/Smad3 and NF-κB signalling. Journal of Cellular and Molecular Medicine. 2019;23(8):5576–5587. doi: 10.1111/jcmm.14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang Y., Meng X. M., Huang X. R., Lan H. Y. The preventive and therapeutic implication for renal fibrosis by targetting TGF-β/Smad3 signaling. Clinical Science (London, England) 2018;132(13):1403–1415. doi: 10.1042/CS20180243. [DOI] [PubMed] [Google Scholar]
- 83.Schiffer M., Bitzer M., Roberts I. S., et al. Apoptosis in podocytes induced by TGF-β and Smad7. The Journal of Clinical Investigation. 2001;108(6):807–816. doi: 10.1172/JCI200112367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Loeffler I., Wolf G. Transforming growth factor-beta and the progression of renal disease. Nephrology, Dialysis, Transplantation. 2014;29(Supplement 1):i37–i45. doi: 10.1093/ndt/gft267. [DOI] [PubMed] [Google Scholar]
- 85.Gruden G., Perin P. C., Camussi G. Insight on the pathogenesis of diabetic nephropathy from the study of podocyte and mesangial cell biology. Current Diabetes Reviews. 2005;1(1):27–40. doi: 10.2174/1573399052952622. [DOI] [PubMed] [Google Scholar]
- 86.Lakshmanan A. P., Thandavarayan R. A., Watanabe K., et al. Modulation of AT-1R/MAPK cascade by an olmesartan treatment attenuates diabetic nephropathy in streptozotocin-induced diabetic mice. Molecular and Cellular Endocrinology. 2012;348(1):104–111. doi: 10.1016/j.mce.2011.07.041. [DOI] [PubMed] [Google Scholar]
- 87.Dennler S., Prunier C., Ferrand N., Gauthier J. M., Atfi A. c-Jun inhibits transforming growth factor β-mediated transcription by repressing Smad3 transcriptional activity. The Journal of Biological Chemistry. 2000;275(37):28858–28865. doi: 10.1074/jbc.M910358199. [DOI] [PubMed] [Google Scholar]
- 88.Hayashida T., Decaestecker M., Schnaper H. W. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-beta-dependent responses in human mesangial cells. The FASEB Journal. 2003;17(11):1576–1578. doi: 10.1096/fj.03-0037fje. [DOI] [PubMed] [Google Scholar]
- 89.Leivonen S. K., Chantry A., Hakkinen L., Han J., Kahari V. M. Smad3 mediates transforming growth factor-β-induced collagenase-3 (matrix metalloproteinase-13) expression in human gingival fibroblasts. The Journal of Biological Chemistry. 2002;277(48):46338–46346. doi: 10.1074/jbc.M206535200. [DOI] [PubMed] [Google Scholar]
- 90.Leivonen S. K., Hakkinen L., Liu D., Kahari V. M. Smad3 and extracellular signal-regulated kinase 1/2 coordinately mediate transforming growth factor-β-induced expression of connective tissue growth factor in human fibroblasts. The Journal of Investigative Dermatology. 2005;124(6):1162–1169. doi: 10.1111/j.0022-202X.2005.23750.x. [DOI] [PubMed] [Google Scholar]
- 91.Li Z., Liu X., Wang B., et al. Pirfenidone suppresses MAPK signalling pathway to reverse epithelial-mesenchymal transition and renal fibrosis. Nephrology (Carlton) 2017;22(8):589–597. doi: 10.1111/nep.12831. [DOI] [PubMed] [Google Scholar]
- 92.Zhou X., Bai C., Sun X., et al. Puerarin attenuates renal fibrosis by reducing oxidative stress induced-epithelial cell apoptosis via MAPK signal pathways in vivo and in vitro. Renal Failure. 2017;39(1):423–431. doi: 10.1080/0886022X.2017.1305409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Luo K. Signaling cross talk between TGF-β/Smad and other signaling pathways. Cold Spring Harbor Perspectives in Biology. 2017;9(1, article a022137) doi: 10.1101/cshperspect.a022137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chuang P. Y., Menon M. C., He J. C. Molecular targets for treatment of kidney fibrosis. Journal of Molecular Medicine (Berlin, Germany) 2013;91(5):549–559. doi: 10.1007/s00109-012-0983-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nature Reviews. Nephrology. 2011;7(12):684–696. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu M., Ning X., Li R., et al. Signalling pathways involved in hypoxia-induced renal fibrosis. Journal of Cellular and Molecular Medicine. 2017;21(7):1248–1259. doi: 10.1111/jcmm.13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang L., Zhou F., ten Dijke P. Signaling interplay between transforming growth factor-β receptor and PI3K/AKT pathways in cancer. Trends in Biochemical Sciences. 2013;38(12):612–620. doi: 10.1016/j.tibs.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 98.Runyan C. E., Schnaper H. W., Poncelet A. C. The phosphatidylinositol 3-kinase/Akt pathway enhances Smad3-stimulated mesangial cell collagen I expression in response to transforming growth factor-β1. The Journal of Biological Chemistry. 2004;279(4):2632–2639. doi: 10.1074/jbc.M310412200. [DOI] [PubMed] [Google Scholar]
- 99.Wang J. L., Chen C. W., Tsai M. R., et al. Antifibrotic role of PGC-1α-siRNA against TGF-β1-induced renal interstitial fibrosis. Experimental Cell Research. 2018;370(1):160–167. doi: 10.1016/j.yexcr.2018.06.016. [DOI] [PubMed] [Google Scholar]
- 100.Yang C., Chen Z., Yu H., Liu X. Inhibition of disruptor of telomeric silencing 1-like alleviated renal ischemia and reperfusion injury-induced fibrosis by blocking PI3K/AKT-mediated oxidative stress. Drug Design, Development and Therapy. 2019;13:4375–4387. doi: 10.2147/DDDT.S224909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang X., Lu H., Xie S., et al. Resveratrol suppresses the myofibroblastic phenotype and fibrosis formation in kidneys via proliferation-related signalling pathways. British Journal of Pharmacology. 2019;176(24):4745–4759. doi: 10.1111/bph.14842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bishop A. L., Hall A. Rho GTPases and their effector proteins. Biochemical Journal. 2000;348(2):241–255. doi: 10.1042/bj3480241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Amano M., Nakayama M., Kaibuchi K. Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 2010;67(9):545–554. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Peng F., Wu D., Gao B., et al. RhoA/Rho-kinase contribute to the pathogenesis of diabetic renal disease. Diabetes. 2008;57(6):1683–1692. doi: 10.2337/db07-1149. [DOI] [PubMed] [Google Scholar]
- 105.Komers R. Rho kinase inhibition in diabetic kidney disease. British Journal of Clinical Pharmacology. 2013;76(4):551–559. doi: 10.1111/bcp.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nagatoya K., Moriyama T., Kawada N., et al. Y-27632 prevents tubulointerstitial fibrosis in mouse kidneys with unilateral ureteral obstruction. Kidney International. 2002;61(5):1684–1695. doi: 10.1046/j.1523-1755.2002.00328.x. [DOI] [PubMed] [Google Scholar]
- 107.Kolavennu V., Zeng L., Peng H., Wang Y., Danesh F. R. Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes. 2008;57(3):714–723. doi: 10.2337/db07-1241. [DOI] [PubMed] [Google Scholar]
- 108.Baba I., Egi Y., Utsumi H., Kakimoto T., Suzuki K. Inhibitory effects of fasudil on renal interstitial fibrosis induced by unilateral ureteral obstruction. Molecular Medicine Reports. 2015;12(6):8010–8020. doi: 10.3892/mmr.2015.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xu J., Lamouille S., Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Research. 2009;19(2):156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bhowmick N. A., Ghiassi M., Bakin A., et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Molecular Biology of the Cell. 2001;12(1):27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Masszi A., di Ciano C., Sirokmány G., et al. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. American Journal of Physiology. Renal Physiology. 2003;284(5):F911–F924. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- 112.Liu H., Zeng F., Zhang M., et al. Emerging landscape of cell penetrating peptide in reprogramming and gene editing. Journal of Controlled Release. 2016;226:124–137. doi: 10.1016/j.jconrel.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 113.Wu J., Li J., Wang H., Liu C. B. Mitochondrial-targeted penetrating peptide delivery for cancer therapy. Expert Opinion on Drug Delivery. 2018;15(10):951–964. doi: 10.1080/17425247.2018.1517750. [DOI] [PubMed] [Google Scholar]