

Abstract
A key hallmark of cancer, altered metabolism, is central to cancer pathogenesis and therapy resistance. Robust glutamine metabolism is among cellular processes regulating tumor progression and responsiveness to therapy in a number of cancers, including melanoma and breast cancer. Among mechanisms underlying the increase in glutamine metabolism in tumors is enhanced glutamine uptake mediated by the glutamine transporters, with SLC1A5 (also known as ASCT2) shown to play a predominant role. Correspondingly, increased SLC1A5 expression coincides with poorer survival in breast cancer and melanoma patients. Therefore, we performed an image-based screen to identify small-molecules that are able to prevent the localization of SLC1A5 to the plasma membrane without impacting cell shape. From 7,000 small molecules, 9 were selected as hits, of which one (IMD-0354) qualified for further detailed functional assessment. IMD-0354 was confirmed as a potent inhibitor of glutamine uptake that attained sustained low intracellular glutamine levels. Concomitant with its inhibition of glutamine uptake, IMD-0354 attenuated mTOR signaling, suppressed 2D and 3D growth of melanoma cells, and induced cell cycle arrest, autophagy and apoptosis. Pronounced effect of IMD-0354 was observed in different tumor derived cell lines, compared with non-transformed cells. RNAseq analysis identified the unfolded protein response, cell cycle and DNA damage pathways to be affected by IMD-0354. Combination of IMD-0354 with GLS1 or LDHA inhibitors enhanced melanoma cell death. In vivo, IMD-0354 suppressed melanoma growth in a xenograft model. As a modulator of glutamine metabolism, IMD-0354 may serve as an important therapeutic and experimental tool that deserves further examination.
Keywords: SLC1A5, glutamine metabolism, mTOR, melanoma, UPR
Introduction
Glutamine is a semi-essential and the most abundant amino acid in the vasculature (1). Along with its requirement for protein synthesis, glutamine is essential for the biosynthesis of nucleotides, glutathione, fatty acids and non-essential amino acids glutamate, aspartate, and asparagine (2). In addition to its biosynthetic activity, glutamine has been shown to facilitate the uptake of essential amino acids, maintenance of mTORC1 activity and c-MYC translation (3–5). Through its anaplerotic assimilation into the TCA cycle, glutamine also provides the reducing power to fuel mitochondrial respiration and energy production (2,6).
Following the identification of c-MYC as a regulator of the expression of mitochondrial glutaminase (GLS1) (7) and glutamine transporter SLC1A5 (8), proteins frequently overexpressed in tumors, there has been a considerable interest in targeting glutamine metabolism for therapeutic benefit (9–11). Increase in glutamine dependency was also noted in melanoma tumors that established resistance to BRAF inhibitor therapy (12). Likewise, a shift in glutamine metabolism from oxidation to reductive carboxylation, a change that is catalyzed by HIF-1-SIAH2-mediated targeted degradation of the TCA cycle enzyme α-ketoglutarate dehydrogenase to promote lipid synthesis, is an important component of reprogrammed metabolism exhibited by tumor cells (13). Our earlier studies have identified increase in glutamine uptake as a mechanism of resistance to taxane therapy in triple negative breast cancer, highlighting the importance of glutamine carrier proteins in such tumors (14). In all, these studies provided the foundation for exploring the possible use of glutamine uptake inhibitors, as means to attenuate tumor growth and tumor support by its microenvironment.
Small molecule GLS1 inhibitors, including CB-839, which suppress glutamine catalysis to glutamate, have shown some success in the pre-clinical models of different tumor types (15–17), including possible synthetic lethality with Myc-driven chemoresistant cancers (18). CB-839 has also been evaluated in multiple clinical trials (19). However, given glutaminolysis-independent activities of glutamine, including uptake of essential amino acids for the maintenance of mTORC1 activity (3), biosynthesis of asparagine and nucleotides, and protein synthesis (20), it is expected that attenuation of glutamine uptake would be required to achieve a complete cessation of glutamine-mediated pro-tumorigenic activities.
Among the various glutamine carrier proteins which facilitate glutamine uptake, SLC1A5 has been identified as the most effective glutamine transporter in a number of tumor models (14,21,22), prompting the development of small-molecule inhibitors to block SLC1A5-mediated glutamine uptake (23–25). However, demonstrable concerns surrounding the specificity of these inhibitors for SLC1A5, with inhibition of other SLC family of transporters, and potential negative impact on the proliferation of normal tissues, remain to be addressed (26).
In light of the limitations of the current class of SLC1A5 inhibitors (26), we utilized high-content screening to identify IMD-0354, a small molecule that impairs plasma membrane localization of SLC1A5, a unique approach to selectively suppress cellular glutamine uptake. IMD-0354 enhanced glucose metabolism, culminating in increased lactate generation, as a compensatory metabolic route in glutamine-restricted SLC1A5 inhibited cells. IMD-0354 effectively inhibited growth of tumor cells in culture as in preclinical models.
Materials and Methods
Cell culture
The following cell lines were used in this study: Human epidermoid carcinoma cell line A431, lung adenocarcinoma cell line A549, and renal carcinoma cell line A498 were obtained from ATCC and cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM, HyClone) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Human fibroblast cell line GM00083 was obtained from Coriell Institute for Medical Research, Inc. (New Jersey, US) and cultured in Minimum Essential Medium Eagle (Corning) supplemented with 15% fetal bovine serum and 1% penicillin-streptomycin. Human melanoma cell lines A375, Mel-501, Lu1205, UACC903, WM1366, WM3629, MeWo, Mel-5, Mel-28, mouse melanoma cell line SW1 (obtained from ATCC, the Wistar Institute, TGen, NCI), and human melanocytes H3A (obtained from the Welcome Trust Functional Genomic Cell Bank; EV Sviderskaya, DC Bennett) were cultured in DMEM described above. Human prostate cancer cell line LNCap, 22RV1, PC3, human colon cancer cell line HCT116, human pancreatic cancer cell line MiaPaCa were grown in RPMI 1640 medium (Corning) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Cell cultures were maintained in cell culture incubator at 37°C in 5% CO2. Mycoplasma testing was performed regularly using MycoAlert Mycoplasma detection kit as per manufacturer’s instruction (Lonza, Switzerland). For cell line authentication, short tandem repeat (STR) analysis was performed with the Genomic Shared Resources at SBP. Allele profiles for each line were matched those maintained in the Expasy Cellosaurus STR database (https://web.expasy.org/cellosaurus/) using CLASTR v1.4.4.
Reagents
IMD-0354, TPCA-1, Baricitinib were purchased from Toronto Research Chemicals (Ontario, Canada). Torin 1, rapamycin, Ruxolitinib, Y-27632 were obtained from Selleckchem (Houston, TX), SU11274 and (S)-H-1152 were obtained from Cayman Chemical (Ann Arbor, Michigan). Human TNF-α was purchased from Cell Signaling Technology. pLX304, pLX304-V5-SLC1A5 and pLX304-V5-SLC38A2 were purchased from DNASU Plasmid Repository (Phoenix, CA). All siRNAs were purchased from Sigma-Aldrich. For SLC1A5, siRNA IDs are SASI_Hs01_00162267 and SASI_Hs01_00162267. For IKK1, siRNA IDs are SASI_Hs01_00206921 and SASI_Hs01_00206922. For IKK2, siRNA IDs are SASI_Hs01_00156170 and SASI_Hs01_00156172.
Compound treatment, Western blot analysis and antibodies
To determine the effect of selected compounds, cells were plated at 50% confluency and grown overnight in 6-well plates. Test compounds at different concentrations were added to the plates and incubated for different time (depends on experiments). For phospho-IKK/phospho-IκB experiments, cells were plated as described above and grown overnight. Compounds were added to cells for 1h prior to stimulation with 10 ng/ml TNFα. Cells were then rinsed with PBS and lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 10 μg/ml aprotinin, 1 μg/ml pepstatin A, 10 μg/ml leupeptin, 2 mM phenylmethylsulphonyl fluoride, 2.5 mM sodium orthovanadate). Protein concentration was determined using Coomassie Plus Protein Assay Reagent (Thermo Scientific). Equal amounts of cell lysate proteins (50 μg) were separated on SDS-PAGE and transferred to PVDF membrane. Membranes were blocked with 3% BSA/TBST, incubated with primary antibodies and secondary antibodies. Detection and quantifications were made using Odyssey Infrared Imaging System (LiCor Biosciences). Antibodies against phospho- IKKα/β (Ser176/180), IKK, phospho-IκB (Ser32), IκB, phospho-p70S6K (Thr389), p70S6K, CHOP, BIP, IRE1, PARP, LC3, p21, p27, SLC1A5, SLC7A5 and E-Cadherin were purchased from Cell Signaling Technology. Antibodies α-tubulin, GAPDH were obtained from Santa Cruz Biotechnology. Secondary antibodies were goat anti-rabbit Alexa-680 F(ab’)2 (Molecular Probes) and goat anti-mouse IRDye 800 F(ab’)2 (Rockland Immunochemicals). All antibodies were used according to the suppliers’ recommendations.
Screen of SLC1A5 inhibitors by High Content Screen (HCS)
For identification of SLC1A5 inhibitors, the benchtop microscope slide based immunofluorescence SLC1A5 assay was adapted for high-throughput high content screening in 384-well plate format. Briefly, A431 cells were seeded in full culture medium at 3,000 cells per well in PerkinElmer (Waltham, MA) CellCarrier-384 Ultra microplates and allowed to attach overnight at 37°C in 5% CO2. Control and screening library compounds were added using a Labcyte (San Jose, CA) Echo acoustic liquid handler and the plates were returned to the incubator for ~18 hours. Cells were fixed in 4% paraformaldehyde for 30 minutes followed by a combined blocking and permeabilization step using PBS with 0.1% Triton-X and 3% BSA for 1 hour. Cells were stained using anti-SLC1A5 (Sigma-Aldrich) primary antibody at 1:1,000 in PBST for 2 hours, and fluorescently labeled with Alexa Fluor® 488 (AF488) secondary antibody at 1:400 and HCS CellMask™ Orange at 1:10,000 in PBST for one hour. Finally, the nuclear counterstain DAPI was added at 100ng/ml and plates were sealed. Plates were imaged on a PerkinElmer (Waltham, MA) Opera Phenix™ High Content Screening System using a 20× 0.4NA air objective and 4 fields per well. Images were analyzed using a custom-built PerkinElmer Harmony® software analysis script (details shown in Fig 2b), and the resulting data was normalized, quality-controlled, and further analyzed using Genedata (Basel, Switzerland) Screener software with HCS extension. Data was deposited in and linked to chemical compound library information using CBIS (ChemInnovation, San Diego, CA) chemical-biology database software.
Figure 2. Screen for SLC1A5 Inhibitors by High Content Screening and Validation of Top Hits.

(a) A431 cells were treated with vehicle or ER stress inducer thapsigargin (2 μM) for 24h followed by IF staining. Scale bar, 20 μm. (b) Image analysis scheme for HCS assay read-outs with “Fraction of Int. in PM” used as primary HCS parameter. Metrics are calculated per cell and averaged for each well to create well-level assay read-outs. (PM = Plasma Membrane, Int. = Intensity). (c) Summary of primary HCS. Results of primary screen with hit selection criteria indicated by dotted lines and hits selected for follow-up indicated by labels, screen results histogram shown in the inset, demonstrating good assay performance as indicated by average plate Z’ value > 0.5. (d) Chemical structure of IMD-0354. (e) A431 cells were treated with IMD-0354 (10 μM) for 24h and stained for SLC1A5. (f) A431 cells were treated with vehicle, 1 μM or 5 μM of IMD-0354 for 24h. Cells lysates were fractionated and analyzed by western blotting for indicated proteins.
Compound libraries selected for screening included the 1,200 approved drugs Prestwick Library (Prestwick Chemical) at 5 μM, SBPs ~600 kinase inhibitors collection (Selleck Chemicals, Cayman Chemical, EMD Biosciences) at 0.5 μM, and the 5,000 synthetic natural product inspired compounds NatX library (Analyticon Discovery) at 10 μM screening concentrations. All plates contained a set of positive and negative control wells with 0.05% DMSO utilized as negative (vehicle) control and 150nM Thapsigargin in 0.05% DMSO as positive control. 6,929 compounds were tested in the screening campaign in singlets as recommended for assay Z’-values > 0.5 (27) and the screen showed good assay performance as indicated by a Z’-value of 0.57 (Fig 2c). 120 compound wells with cell counts <20% as compared to vehicle controls (0.05% DMSO) were considered overtly toxic and removed from further consideration. Using a hit criterion for the assay read-out “Fraction Intensity on Plasma Membrane” of % activity < −40% (normalized to 0%=DMSO, −100%=TG controls), 122 initial hits were identified. Further review of cell shape parameters and visual evaluation of the hit images to remove hits significantly affecting cell morphology resulted in a reduced hit set of 32 compounds. Considering the compounds known mechanisms-of-action or pathways and number of hits per mechanism/pathway, a subset of 9 compounds was selected for further evaluation in functional assays.
siRNA transfection
1×105 cells were seeded overnight per well in 6-well plates. Negative control or si-RNA targeting the transcript of interest was transfected utilizing jetPRIME® transfection reagent, as per manufacturer’s instructions (Polyplus, NY, USA).
Immunofluorescence microscopy
Cells were grown in 96-well black plate and treated as indicated in the text. Cells were then fixed with paraformaldehyde in PBS for 10 min at room temperature. After fixation, samples were permeabilized with 0.5% Triton X-100 in PBS for 15 min and thereafter blocked with 10% fetal calf serum in PBS containing 0.1% Triton X-100. Cells were incubated for 1h each at 25°C with primary and secondary antibodies and 15 min with DAPI (4′,6-diamidino-2-phenylindole). Cells were then visualized with an inverted fluorescence microscope (Olympus). Anti-SLC1A5 antibody for HCS and IF was obtained from Sigma-Aldrich. Anti-LC3 antibody was obtained from Cell Signaling Technology. Secondary antibodies used were Alexa 594- or 488-labeled anti-rabbit or anti-mouse IgG antibodies (Invitrogen).
3H-Glutamine uptake assay
Cells were treated with different concentrations of compounds for 15 min before the radiotracer L-[3,4,3H] glutamine (Perkin Elmer) (5 μCi/ml) was added. Transport measurements were terminated after 15 min by three rapid washes with ice-cold PBS. Intracellular glutamine was extracted with PBS containing 0.2 % SDS and 0.2N NaOH. After 1h incubation at room temperature, lysates were neutralized with 2N HCl and quantified using liquid scintillation counter (Beckman LS6500).
Proliferation assay
All cell lines were seeded (4,000 cells in 100 μL per well) in 96-well plates. Cells were allowed to attach overnight. Tested compounds were serially diluted using media from stock solutions (10 mM) and were added to cells. Tests were performed in triplicate, and each microplate included media and DMSO control wells. Cell viability was assessed using ATPlite according to manufacturer’s protocol. Cell growth inhibition was calculated as a percentage of DMSO-treated controls and plotted against the compound concentration.
Colony formation assay
For colony formation assays, cells were plated in triplicate (500 cells/well) in 6-well plates or 100 cells/well in 24-well plates and grown overnight before compounds were added. After 1–2 weeks, depending on cell line, colonies were stained with Accustain Crystal Violet solution (Sigma-Aldrich) for 30 min. Plates were rinsed with water and images were acquired by scanning. Colony formation efficiency was calculated relative to the number of colonies in control (DMSO)-treated wells.
Hanging drop spheroid culture and treatment
Briefly, cells were resuspended in fresh medium and counted. Cell suspensions of 20 μL (200 cells) were pipetted into each well of the hanging drop plate. Plate was covered with lid and put upside down in a container with wet filter paper to keep the cells hydrated. The plate was maintained at 37°C in humidified incubator with 5% CO2 to allow the spheroids to form. Cells were imaged after three days to check for aggregation and cell proliferation. After spheroids were formed, they were transferred to 24-well plate, which was plated with 0.5 ml of medium on top of 0.7% agar layer in each well. Different concentrations of IMD-0354 was added to wells and the growth of spheroids was monitored every two days under microscope.
Reverse transcription and real-time qPCR.
Total RNA was extracted using a total RNA miniprep kit (Sigma) with the On-column DNase I digestion step included. cDNA was synthesized using cDNA kit from Applied Biosystems according the manufacturer’s protocol. Real-time PCR was performed on a Bio-Rad CFX Connect Real-Time System using FastStart Universal SYBR Green Master from Bio-Rad. H3A was used as an internal control. Quantitative PCR reactions were performed in triplicates. PCR primers were designed using PrimerBank (http://pga.mgh.harvard.edu/primerbank).
13C metabolite labeling and amino acid quantification
Cell extraction and GC-MS analysis for metabolite quantification were performed as described (28). For GC-MS analysis, intracellular metabolite amounts are expressed as nmol per cell sample (cells from one well of six-well plates; approximately 0.5 × 106 to 1.0 × 106 cells) and (in Fig 3e) normalized to controls. Glucose (Glc), lactate, and glutamine (Gln) in culture medium samples were measured using YSI 2950 analyzer (YSI OH, USA). For Glc and Gln estimation in the media, the starting concentrations of Glc and Gln were 24.75 and 4.0 mM, respectively, which is their standard concentration in high-glucose Dulbecco’s modified Eagle’s medium (DMEM).
Figure 3. IMD-0354 Treatment Inhibits Glutamine Uptake and Suppresses Associated Amino Acid Biosynthesis.

(a) A431 cells were treated with vehicle, IMD-0354 (5 μM or 10 μM) for 1h or 24h followed by 3H-glutamine uptake assay. (b) A431 and A375 cells were treated with increasing concentrations of IMD-0354 for 1h followed by 3H-glutamine uptake assay. (c) A431 cells were treated with 2 μM of IMD-0354 for 72h, intracellular glutamine and glutamate levels were determined by GC-MS. (d) A431 cells were treated with 2 μM of IMD-0354 for 72h, glutamine level in media was determined by GC-MS. (e, f) A431 cells were treated with the indicated concentrations of IMD-0354 for 6h followed by GC-MS-based analysis. Statistical analysis was performed by one-way ANOVA for the comparison of more than two groups. Unpaired t-test was used for the comparison of two groups. Data are shown as the mean ± SD, n = 3. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Cell cycle and cell death by Flow Cytometry
A375 cells were treated as indicated in the text. Cells were trypsinized and fixed in 70% ice-cold ethanol for 60 min, washed in phosphate-buffered saline, and incubated for 30 min at 25°C in propidium iodide (PI) buffer (10 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 50 μg of PI per ml, and 10 μg of RNase A/ml). The stained cells were acquired by the FACSort flow cytometer (BD Biosciences), and the DNA content was analyzed using FlowJo software.
Cell fractionation
A375 cells were treated as indicated in Results. Cell fractionation was performed using Cell Fractionation Kit (#9038, Cell Signaling) according to manufacturer’s protocol. Cytoplasmic and membrane proteins were then subjected to western blotting analysis.
RNAseq
A375 cells were treated as indicated in the text. Total RNA was extracted using a total RNA miniprep kit (Sigma) with the On-column DNase I digestion step included. Libraries were prepared from isolated total RNA using the QuantSeq 3’ mRNA-Seq Library Prep Kit FWD for Illumina from Lexogen, (Vienna, Austria). Barcoded libraries were pooled and single end sequenced (1X75) on the Illumina NextSeq 500 using the High output V2.5 kit (Illumina Inc., San Diego, CA). Read data was processed and multiplexed with the BlueBee Genomics Platform (BlueBee, San Mateo, CA).
RNA-Seq data processing
Lexogen 3’ mRNA-Seq sequencing reads were processed using cutadapt v1.18 (29) to remove adapter remnants. Sequencing reads were then aligned using STAR aligner version 2.7 (30) to human genome version 38 and Ensemble gene annotation version 84. RSEM v1.3.1 (31) was used to estimate gene expression. Estimated read counts from different groups were compared using the R Bioconductor package DESeq2 following generalized linear model based on negative binomial distribution (32). Genes with Benjamini-Hochberg–corrected p value< 0.05 and fold change >=2 or <=0.5 were selected as significantly differentially expressed genes.
Differentially expressed genes were uploaded to Ingenuity Pathway Analysis (IPA, November 2019) (Qiagen, Redwood City, USA) for further analyses. Top canonical pathways and upstream regulators were exported to illustrate the biological mechanism. Enrichment results of different comparisons were compared to the published data sets using IPA Analysis Match function. RNAseq data has been uploaded to the GEO public database (GSE165899).
Animal studies
All animal studies were conducted in the SBP Medical Discovery Institute Animal Facility in accordance with the Institutional Animal Care and Use Committee guidelines (IACUC # 18–079). Mice were maintained in a pathogen-free environment with free access to food. Six-week-old males C3H mice were purchased from Charles River Laboratories and allowed to acclimatize for 1 week. SW1 cells (0.5×106, suspended in 150 μl sterile PBS) were injected into the subcutaneous tissue of the flank. When tumors reached ~150 mm3, mice were randomized and sorted into treatment groups (n=8 animal/group). Vehicle or IMD-0354 (20 mg/kg) was administered daily for 3 weeks intraperitoneally. Tumor volume was measured twice a week with linear calipers and calculated using the formula: ([length in millimeters × (width in millimeters)2]/2). After the mice were sacrificed, tumors were frozen or fixed in 10% formalin and embedded in paraffin for immunohistochemistry. Snap frozen tumors were utilized for protein and RNA extraction for further analysis.
Statistics and reproducibility
Statistical significance between two groups was assessed by the unpaired Student’s t-test. Ordinary one-way ANOVA was used to analyze more than two groups. Two-way ANOVA was utilized to analyze cell proliferation at multiple timepoints. GraphPad Prism 7 and 8 software (Graphpad, La Jolla, CA) was used for to perform all statistical calculations. All cell culture experiments were performed three times. Data is presented as mean ± SD (unless noted otherwise in the figure legends) and a P value less than 0.05 was considered statistically significant.
Results
SLC1A5 Inhibition Attenuates Glutamine Uptake and Growth of Melanoma and Skin Epidermoid Carcinoma Cells
To assess the importance of SLC1A5, a key glutamine carrier protein in melanoma cells, we inhibited its expression in melanoma cell line A375, using two specific siRNA. Efficiency of SLC1A5 inhibition was monitored at the level of RNA and protein expression (Fig S1a and S1b), and glutamine uptake by 3H-labelled glutamine. 3H-glutamine uptake was effectively inhibited in cells transfected with siSLC1A5, compared to control cells (Fig 1a). At later time points, cells harboring siSLC1A5 exhibited attenuated cell proliferation and colony formation (Fig 1b and 1c). Since we were interested in identifying small molecule inhibitors of glutamine uptake that work through perturbation of SLC1A5 membrane localization, we searched for cell lines with high SLC1A5 expression and single-layer clustered or cuboidal growth characteristics. Such cell lines would enable microscopic visualization of the plasma membrane, which is paramount for an image-based high content screen (HCS) quantifying subcellular localization of the membrane protein SLC1A5. Immunofluorescence (IF) analysis of different cell lines identified the skin epidermoid carcinoma cell line A431 as the best candidate for a high throughput screen, displaying pronounced SLC1A5 membrane localization in microscope images (Fig S1c). Consistent with the requirement of SLC1A5 in A375 cells, its silencing in A431 cells (Fig S1d) resulted in reduced expression of the protein (Fig 1d) and inhibited 3H-glutamine uptake (Fig 1e), with concomitant decrease in cell growth (Fig 1f) and colony formation (Fig 1g). These results demonstrate the important role SLC1A5 plays in cellular glutamine uptake and key cellular functions, reflected in 2D and 3D growth, both in melanoma and skin epidermoid carcinoma cells.
Figure 1. Inhibition of SLC1A5 Expression Suppresses Glutamine Uptake, Cell Growth and Colony Formation in Melanoma A375 Cells and Skin Epidermoid Carcinoma A431 Cells.
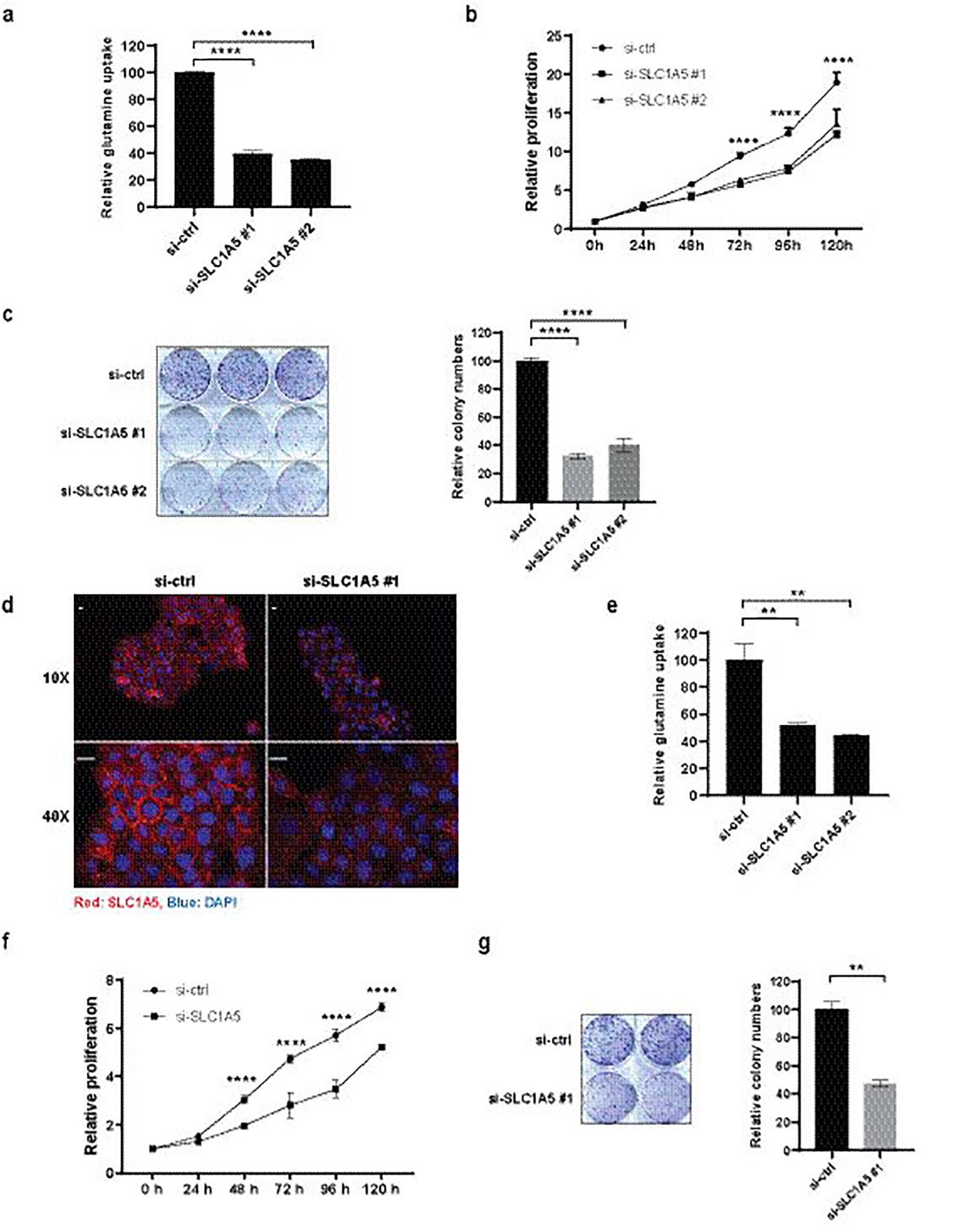
(a) A375 cells were transfected with si-ctrl or si-SLC1A5 for 48h followed by 3H-glutamine uptake assay. (b) A375 cells were transfected with si-ctrl or si-SLC1A5 and cell proliferation was measured for the indicated duration by ATPlite. (c) A375 cells were transfected with si-ctrl or si-SLC1A5 and colony formation assay was performed (left) and quantified (right). (d) A431 cells were transfected with si-ctrl or si-SLC1A5 for 48h followed by immunofluorescence staining with anti-SLC1A5 (red) and DAPI (blue). Scale bar, 20 μm. (e) A431 cells were transfected with si-ctrl or si-SLC1A5 for 48h followed by 3H-glutamine uptake assay. (f) A431 cells were transfected with si-ctrl or si-SLC1A5 and cell proliferation was measured for the indicated duration by ATPlite.
(g) A431 cells were transfected with si-ctrl or si-SLC1A5 and colony formation assay was performed (left) and quantified (right). Statistical analysis was performed by two-way ANOVA for time-dependent proliferation changes and by one-way ANOVA for the comparison of more than two groups. Data are shown as the mean ± SD, n = 3. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. In (b) and (f), statistical comparison is only shown for the treatments that showed a significant change.
High Content Screen for SLC1A5 Inhibitors
The data obtained with A431 cells prompted the design of a microscopy-based high throughput screen for small molecule inhibitors that would reduce the localization of SLC1A5 at the plasma membrane without significantly altering cell morphology. Given our findings that thapsigargin (TG) triggers degradation of SLC1A5 (14), we validated its possible use as a positive control in A431 cells. Indeed, TG led to marked inhibition of SLC1A5 expression (Fig 2a). To enable a microscopy-based screen, an Opera Phenix HCS system in combination with the Harmony HCS software package was used to determine the cellular fraction of SLC1A5 intensity on the membrane (Fig 2b). A dose-dependent effect of TG on SLC1A5 membrane localization was established as part of the HCS assay implementation (Fig S2a).
Using ~7000 small molecules from kinase inhibitor and approved drug libraries as well as a natural product inspired library, a high content microscopy-based screen was performed to identify those affecting SLC1A5 localization at the plasma membrane in A431 cells (Fig 2c). Overtly toxic compounds were excluded (Fig S2b) and top hits (SLC1A5 PM Intensity < −40% Activity) that impacted SLC1A5 membrane distribution, but not cell morphology, were selected (Fig 2c; - Table S1). Of those, 9 compounds (marked in red in Table S1, structure and reference information of selected compounds in Table S2) were subjected to independent validation using IF and 3H-glutamine uptake. Of these, IMD-0354 (Fig 2d) appeared to exhibit the most striking effect, impairing plasma membrane localization of SLC1A5 (Fig 2e), without impacting the membrane distribution of plasma membrane marker phalloidin (Fig S2c) and ATP1A1 (Fig S2d). IMD-0354 did not affect plasma membrane localization of other key members of the SLC family of carrier proteins SLC38A2 and SLC7A5 (Fig 2f), ruling out a general attenuation in membrane localization of SLC carrier proteins. Notably, IMD-0354 was the only compound that inhibited 3H-glutamine uptake in both A375 and A431 cells (Fig S2e). Exposure of A431 cells to IMD0354 showed a dose dependent dislocation of SLC1A5 from the plasma membrane (Fig S2f), suggesting a direct effect of the drug on its membrane localization. These findings identify IMD-0354 as a small molecule inhibiting SLC1A5 localization at the plasma membrane in skin epidermoid and melanoma cells.
IMD-0354 Inhibits Glutamine Uptake and Suppresses Glutamine-Dependent Amino Acid Pathways
To verify the effect of IMD-0354 on glutamine uptake, A431 cells were treated with increasing concentrations of IMD-0354 and 3H-glutamine uptake was measured after 1h and 24h. Inhibition of glutamine uptake was evidenced 1h following IMD-0354 treatment and was sustained after 24h (Fig 3a). Notably, this inhibition could also be seen as early as 15 min exposure to IMD-0354 (Fig S3). Dose dependent inhibition of glutamine uptake was also observed in A375 cells (Fig 3b), early as 1 and 3 min following IMD-0354 treatment of the melanoma cultures UACC903 and A375 (Fig S3b). These observations substantiated the inhibition of glutamine uptake by IMD-0354 in cancer cells.
To determine whether IMD-0354 affects other glutamine carrier proteins, we surveyed the expression of 10 such carrier proteins in A375 melanoma cells. Among those SLC1A5, SLC7A5, and SLC38A2 were the primary expressed carrier proteins (Fig S3c). We next monitored the expression of these proteins in melanoma cells that were subjected to IMD-0354 treatment, finding that the expression of SLC38A2 was unaffected compared with minor changes in the expression of SLC1A5 and SLC7A5 (Fig S3d). To further assess possible changes in the expression of SLC38A2, which is the main glutamine carrier protein expressed in addition to SLC1A5 in these melanoma cells, we monitored possible changes in its expression following KD of SLC1A5. Notably, inhibition of SLC1A5 expression using the corresponding siRNA, did not affect the expression of SLC38A2 (Fig S3e). Inhibition of SLC1A5 expression increased the transcription of SLC7A5, an expected outcome given its ability to compensate for reduced glutamine uptake. These findings substantiate a primary role of SLC1A5 in glutamine uptake in melanoma cells, consistent with its reported role in other cancer cell types (14,21,22).
One of the routes intracellular glutamine adopts is its conversion to glutamate, a reaction catalyzed by glutaminase. Expectedly, Gas Chromatography-Mass Spectrometry (GC-MS)-based analysis of A431 cells treated with IMD-0354 showed decrease in intracellular levels of glutamine and glutamate (Fig 3c). Correspondingly, consistent with its suppressed intracellular levels, glutamine levels increased in the media (Fig 3d). IMD-0354 did not induce any significant shift in the intracellular levels of essential amino acids, crucially ruling out any unintended suppression in the activity of other related amino acid transporters (Fig 3e). Aspartate biosynthesis, catalyzed by glutamic-oxaloacetic transaminase 2 (GOT2), requires oxaloacetate and glutamate as substrates, and aspartate and glutamine are precursors for asparagine biosynthesis. Therefore, suppressed glutamine and glutamate levels in IMD-0354-treated A431 cells (Fig 3c) decreased intracellular aspartate and asparagine levels (Fig 3f).
IMD-0354 Attenuates mTOR, but not IKK-NF-κB Signaling
SLC1A5-mediated glutamine uptake is important for the maintenance of mTORC1 activity, an effect that is uncoupled from glutamine metabolism (33). To determine whether IMD-0354 affects mTORC1 function, A375 cells were treated with IMD-0354 (1h, 3h and 24h) and the phosphorylation of p70S6K, a direct substrate of mTORC1, was examined. Following IMD-0354 treatment, phosphorylation of p70S6K was attenuated (Fig 4a). Among the initial hits in our screen was a Rho-associated kinase inhibitor ((S)-H-1152), which perturbed plasma membrane localization of SLC1A5, albeit to a lesser extent as seen for IMD-0354 (Fig S2e). Consistently, this inhibitor exhibited less pronounced suppression of mTORC1 activity (Fig 4a). These results support the effect of IMD-0354 on the canonical glutamine uptake signaling pathway which engages mTORC1 signaling.
Figure 4. IMD-0354 Suppresses mTOR Signaling without Impacting IκK Pathway.
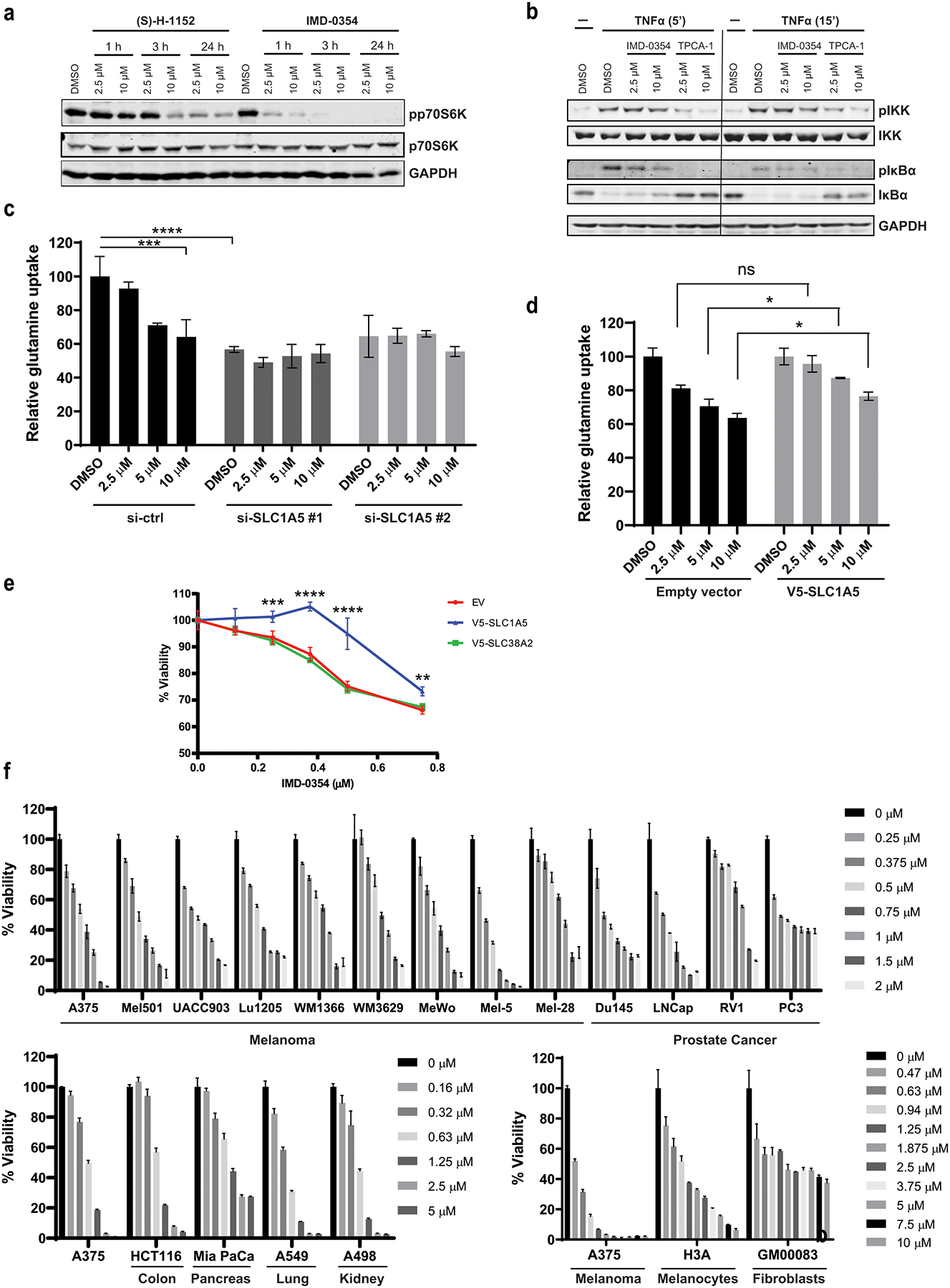
(a) A375 cells were treated with IMD-0354 or (S)-H-1152 as indicated followed by western blotting for the indicated proteins. (b) A375 cells were pre-treated with the indicated concentrations of IMD-0354 or IKK-2 inhibitor TPCA-1 for 1h followed by treatment with 10 ng/ml TNFα for 5 min or 15 min. Cells were harvested and tested for the indicated proteins by western blotting. (c) A375 cells were transfected with si-ctrl or si-SLC1A5 for 48h followed by 3H-glutamine uptake assay in the presence of indicated concentrations of IMD-0354. (d) A375 cells were transfected with empty vector or V5-SLC1A5 for 48h followed by 3H-glutamine uptake assay in the presence of indicated concentrations of IMD-0354. (e) A375 cells were transfected with EV, V5-SLC1A5 or V5-SLC38A2 for 24h followed by treatment with indicated concentrations of IMD-0354. Cell viability was measured by ATPlite after 48h. (f) Melanoma, prostate, pancreatic, colon and lung cancer cells lines, immortalized (H3A) melanocytes and non-transformed fibroblasts (GM00038) were treated with indicated concentrations of IMD-0354. Cell viability was measured by ATPlite after 72h. Statistical analysis was performed by two-way ANOVA for viability assay and one-way ANOVA for the comparison of more than two groups. Data are shown as the mean ± SD, n = 3. ns, not significant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
IMD-0354 was reported to inhibit IKKβ in human mastocytoma cells (HMC-1) (34). To test the possible effect of IMD-0354 on the NF-κB pathway, the ability of IMD-0354 to suppress NF-κB signaling was compared with TPCA-1, an independent IKK inhibitor V (35), also identified in our screen. TNFα stimulation, expectedly, increased IκB phosphorylation, an effect attenuated by TPCA-1 but not by IMD-0354 (Fig 4b). Correspondingly, TPCA-1 but not IMD-0354 upregulated levels of IκB, a negative regulator of NF-κB signaling (Fig 4b). Similarly, IMD-0354 failed to suppress NF-κB signaling in A431 cells (Fig S4a). To evaluate a possible link between NF-κB signaling and glutamine uptake, we genetically silenced IKK-1 and IKK-2 using corresponding siRNA in A375 and A431 cells, which were then subjected to 3H-glutamine uptake assay. Knockdown of IKK did not affect glutamine uptake either in A431 or A375 cells (Fig S4b). Moreover, genetic inhibition of NF-κB pathway components failed to enhance attenuation of glutamine uptake elicited by IMD-0354 (Fig S4c). Taken together, these results suggest that inhibition of glutamine uptake by IMD-0354 is uncoupled from the NF-κB pathway components.
At higher concentrations, IMD was reported to impact additional protein kinases, including MEK and p38 MAPK (34). Notably, neither of these kinases was affected by IMD-0354 (Fig S4d). Further, neither of these kinase inhibitors, nor inhibitors to other cellular signaling pathways attenuated glutamine uptake, compared with the effect of IMD-0354 (Fig S4e). These findings substantiate the effect of IMD-0354 on glutamine metabolism, independent of previously reported signaling associated with this small molecule.
To further establish the mechanism of the effect of IMD-0354 on glutamine uptake, we monitored changes in glutamine uptake upon inhibition or ectopic expression of SLC1A5. Expectedly, siRNA-mediated SLC1A5 knockdown effectively attenuated 3H-glutamine uptake on its own, however, it did not significantly accentuate the inhibition of glutamine uptake elicited by IMD-0354 (Fig 4c). Moreover, overexpression of SLC1A5 partially restored glutamine uptake inhibited by IMD-0354 (Fig 4d). Further, ectopic expression of SLC1A5, but not SLC38A2, attenuated the effect of IMD-0354 on melanoma cell viability (Fig 4e). Taken together, these results suggest that inhibition of glutamine uptake by IMD-0354 is largely mediated by SLC1A5.
To determine whether the effect of IMD-0354 is limited to the melanoma cells studied here, we set to monitor the viability of other cancer and non-transformed cell lines following treatment with this inhibitor. Extending the analyses to additional melanoma, as well as prostate, pancreatic, colon, lung and kidney tumor-derived cell lines revealed that IMD-0354 effectively inhibited the viability of diverse tumor cell lines, in a dose dependent manner (Fig 4f). Studying the effect of IMD-0354 on non-transformed immortalized melanocytes and a fibroblast cell line revealed effective dose dependent inhibition, albeit, to lesser degree. While a dose of 1.25 μM IMD-0354 caused over 90% inhibition of A375 cell viability, it was limited to ~40% for the non-transformed cells (Fig 4f). Further, while higher concentrations of IMD-0354 effectively suppressed melanoma cell viability, the degree of inhibition in fibroblast cell line GM00038 was limited to 40% (Fig 4f).
IMD-0354 Suppresses Key Characteristics of Tumor Cells
Recognizing the effect of the inhibition of glutamine uptake on melanoma growth (Fig 1b, 1c; (28,36)), we further assessed the effect of IMD-0354 on tumor cell growth in 2D and 3D cultures. Along with its effect on multiple tumor cell lines (Fig 4f), IMD-0354 treatment led to a dose-dependent inhibition of cell proliferation and colony formation in both A375 and A431 tumor cell lines (Fig 5a, 5b) as well as melanoma spheroid formation (Fig 5c), representative of tumor architecture in vivo.
Figure 5. IMD-0354 Treatment Inhibits Melanoma Cell Growth, Induces Cell Cycle Arrest, Apoptosis and Autophagy.

(a) A431 and A375 cells were treated with the indicated concentrations of IMD-0354. Cell viability was measured by ATPlite after 72h. (b) A431 and A375 cells were plated at low density (500 cells/well in 6-well plates) and grown in medium containing indicated concentrations of IMD-0354. The number of colonies formed after 10 days in culture was determined by crystal violet staining (left) and quantified (right). (c) A375 cell spheroids were treated with indicated concentrations of IMD-0354. Images were taken under bright field microscope after three days (left) and quantified (right). (d) A375 cells were treated with indicated concentrations of IMD-0354 for 24h. Cells were stained with propidium iodide (PI) and cell cycle was analyzed by FACS.
(e) A375 cells were treated with IMD-0354 (2 μM) for 24h. Cells were stained with anti-LC3 and DAPI. Scale bar, 20 μm. (f) A375 cells were seeded in transwell and treated with IMD-0354 (0.25 μM or 1 μM) for 16h. Migrated cells were visualized by crystal violet staining. Statistical analysis was performed by one-way ANOVA for the comparison of more than two groups. Unpaired t-test was used for the comparison of two groups. Data are shown as the mean ± SD, n = 3. **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Among mechanisms reported to underlie the effect of glutamine uptake inhibition by attenuated SLC1A5 expression on tumor cell growth is cell cycle suppression, often reflected in G1 arrest (36–38). Indeed, treatment of melanoma cells with IMD-0354 resulted in increased fraction of cells in the G1 phase (from 40% to over 50%, within the range of 0.32 – 1.25 μM), with a corresponding decrease in S and G2 phases of the cell cycle (Fig 5d). Consistent with our earlier work showing prolonged attenuation of glutamine uptake by inhibition of SLC1A5 inducing autophagy (14), treatment with IMD-0354 increased staining of LC3 punctae, a key feature of autophagic cells (Fig 5e). Prolonged deprivation of glutamine or higher concentration of IMD-0354 induced apoptosis, as determined by increased fraction of cells in the sub-G1 phase (Fig 5d, at concentration of 2.5 – 5 μM). Corresponding to these changes was increase in the expression of cell cycle inhibitory proteins p21 and p27; the expression of the processed form of LC3; and the expression of apoptotic markers, including PARP and CHOP (Fig S4f). Notably, the effect of IMD-0354 was not limited to attenuated growth, as Boyden chamber transwell assay revealed a notable inhibition of A375 cell migration (Fig 5f), an effect also seen in response to SLC1A5 suppression (39). Collectively, these observations support the inhibitory effect of IMD-0354 on key phenotypic characteristics of tumor cells.
RNAseq Maps Molecular Changes Underlying IMD-0354 effect in Melanoma
To identify unbiased changes in cellular signaling that are caused by IMD-0354, RNAseq analysis was carried out on A375 cells that were subjected to three different concentrations (0.32 μM, 0.63 μM and 1.25 μM) of IMD-0354 over 24h and 48h period. This analysis identified 480 genes that were upregulated and 516 genes that were downregulated (FC>2; BHP<0.05) (Fig 6a, 6b). Since the analysis was performed on cells subjected to distinct conditions (different concentrations of IMD-0354 for different durations), we were able to select for the common changes, narrowing the number of upregulated (n=59) and downregulated (n=40) genes (Fig 6b). Top genes regulated by IMD-0354 treatment are implicated in cell cycle and DNA damage response pathways, as well as UPR signaling (Fig 6c), changes that were previously linked to curtailed glutamine metabolism (24,36,40). Correspondingly, upstream components predicted to impose the changes identified following IMD-0354 treatment are regulatory components of cell cycle (Fig 6d) as well as central metabolic regulatory proteins, including c-Myc and the UPR signaling component CHOP (DDIT3) (Fig 6e). A vast majority of the final set of IMD-0354-responsive genes were implicated in key cell cycle and ROS/UPR signaling (Fig 6f). Notably, integration of the major changes identified in this analysis revealed glutamine metabolism as a major downregulated (P=9.2e-4) and AKT signaling as a key upregulated signaling pathway (P=2.5e-4), providing independent support for the impact of IMD-0354 on glutamine metabolism (Fig 6e).
Figure 6. Transcriptomic Profiling of IMD-0354-Treated A375 Cells.
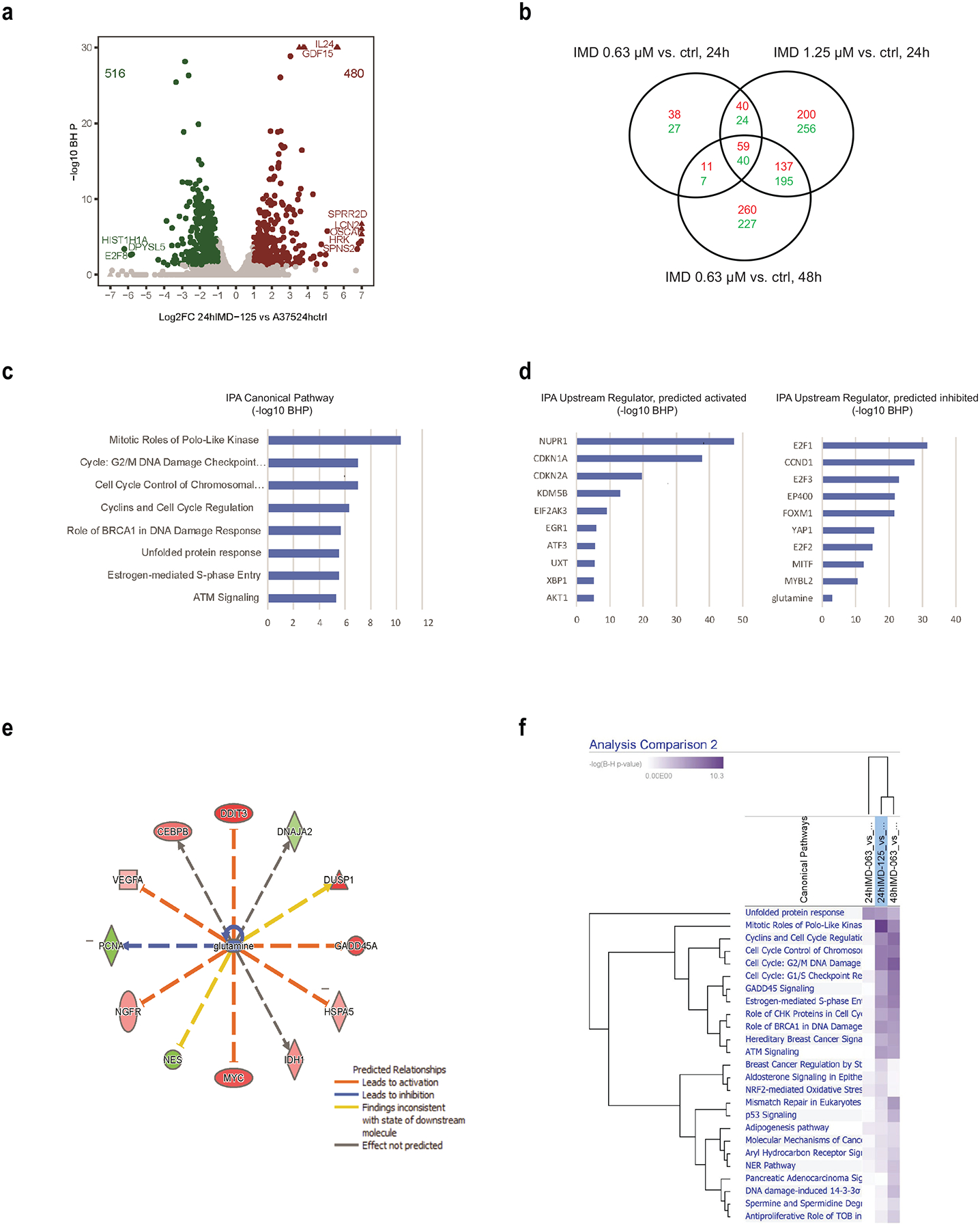
A375 cells were treated with IMD-0354 (0.32 μM, 0.64 μM or 1.25 μM) for 24h and 0.64 μM for 48h. RNA was extracted and subjected to RNAseq analysis. (a) Volcano plot for RNA-Seq gene differential expression analysis for A375 cells treated with 1.25 μM of IMD-0354 for 24h versus DMSO control. Gene differential expression was decided based on Fold Change (FC) >=2 or <=0.5, and Benjamini & Hochberg (BH) corrected P <0.05. Red circles are significantly up-regulated genes, and green circles are significantly down-regulated genes. Triangles are genes out of the boundaries of the graph. (b) Venn diagram for intersection of differentially expressed genes from different doses and treatment times of IMD-0354 on A375 vs. DMSO controls. Red numbers are numbers of up-regulated genes. Green numbers are numbers of down-regulated genes. (c, d) Top canonical pathways and upstream regulators from Ingenuity Pathway Analysis for A375 cells treated with IMD-0354 (1.25 μM) for 24h vs. DMSO control. (e) Network of glutamine regulated genes in A375 cells treated with IMD-0354 (1.25 μM) for 24h vs. DMSO control. (f) Top canonical pathways from Ingenuity Pathway Analysis for 59 upregulated and 40 downregulated genes identified in (b).
To further understand IMD-0354 mechanism-of-action, the molecular profiles from RNA-Seq were compared to public data sets. The signaling pathways and regulators predicted to be associated with response to IMD-0345 were first compared to the small-molecule inhibitor treatment signatures from the Library of Integrated Network-Based Cellular Signatures (LINCS). Small molecules targeting CHK1, PI3K, GSK-3β showed a gene expression signature (suppressed E2F and MYC signaling, and activated TP53, TNF, IL1B and IL6 pathways) that was similar to the one obtained with IMD-0354 treatment (Fig S5a). Next, IMD-0345 profiles were compared to different skin melanoma subtypes characterized by somatic mutations (Fig S5b). Interestingly, these melanoma subtypes showed opposite z-scores for similar set of genes (listed above), suggesting that IMD-0345 may reverse the signaling pathways important for these tumor subtypes. Interestingly, the small molecule targets and somatic mutations in the melanoma subtypes converged on common canonical pathways, including PTEN, STAT3 and NF-κB signaling pathways (Fig S5c), indicating an essential role of IMD-0345 in interrupting these key pathways for tumor development and progression. qRT-PCR analysis confirmed the changes identified by RNAseq in key regulatory nodes (UPR, cell cycle/DNA damage and ROS) (Fig 7a–c). The transcriptional signature observed following IMD-0354 treatment was also seen upon genetic inhibition of SLC1A5 in A375 cells (Fig S6a) or upon deprivation of glutamine from media of cultured melanoma cells (Fig S6b), further substantiating that the effect of IMD-0354 is consistent with the inhibition of glutamine uptake.
Figure 7. Validation of Genes Identified by RNAseq After IMD-0354 Treatment.
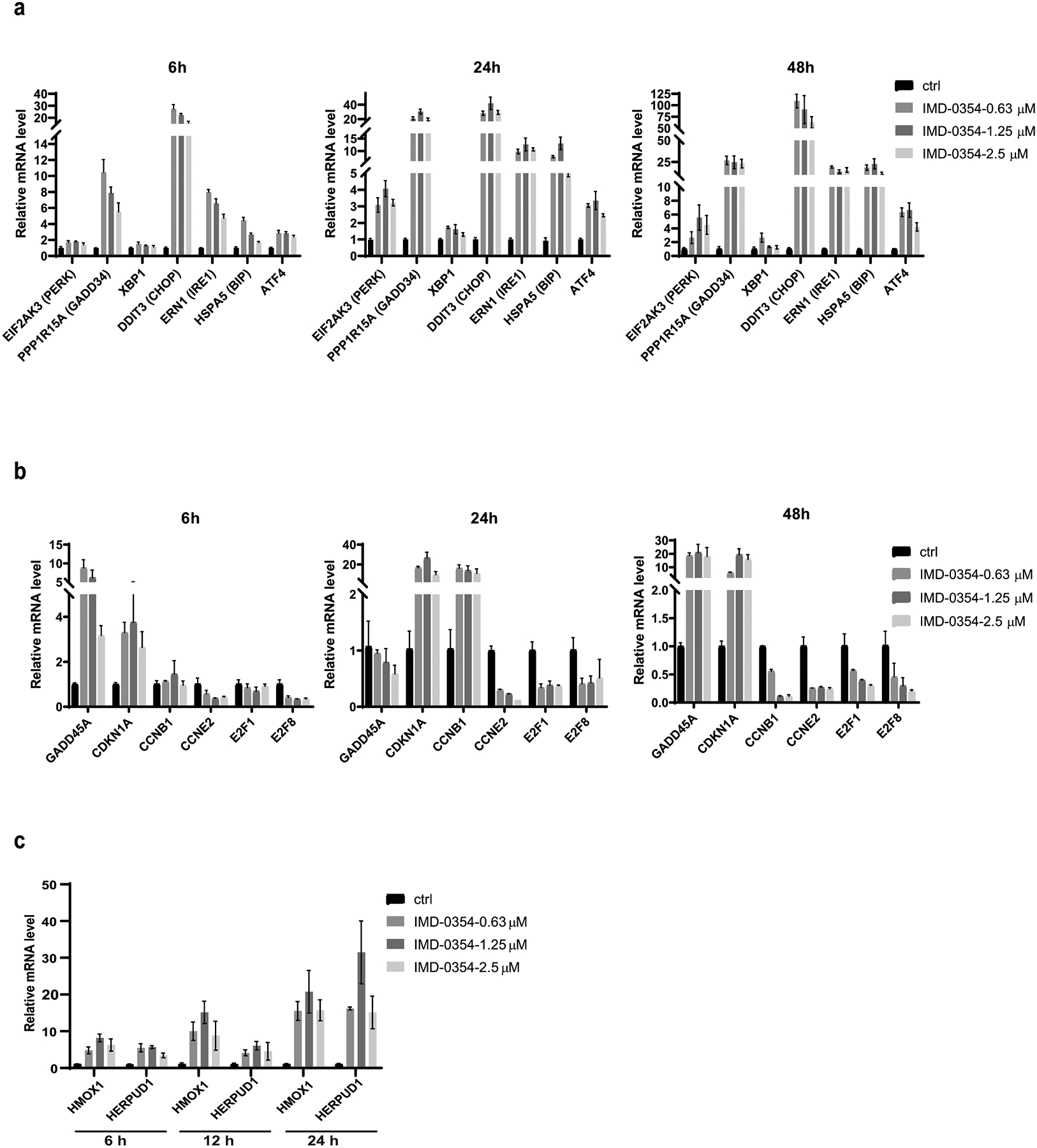
(a-c) A375 cells were treated as indicated with IMD-0354. RNA was extracted and subjected to qPCR analysis against (a) UPR-related genes (b) cell cycle/DNA damage-related genes and (c) ROS-related genes.
The identification of UPR as key signaling pathway and its key component ATF4, led us to explore the importance of ATF4 for IMD-0354 response. Indeed, knockdown of ATF4 effectively attenuated the cytotoxic effect of IMD-0354 (Fig 8a). Further, activation of UPR components seen following IMD-0354 treatment, was effectively blocked in cells that were subjected to siATF4 (Fig 8b). These finding substantiate the importance of UPR with ATF4 as central player, in glutamine metabolism and the effect of IMD-0354 on this pathway.
Figure 8. Characterization of IMD-0354 Effect on Melanoma in Culture and In Vivo.
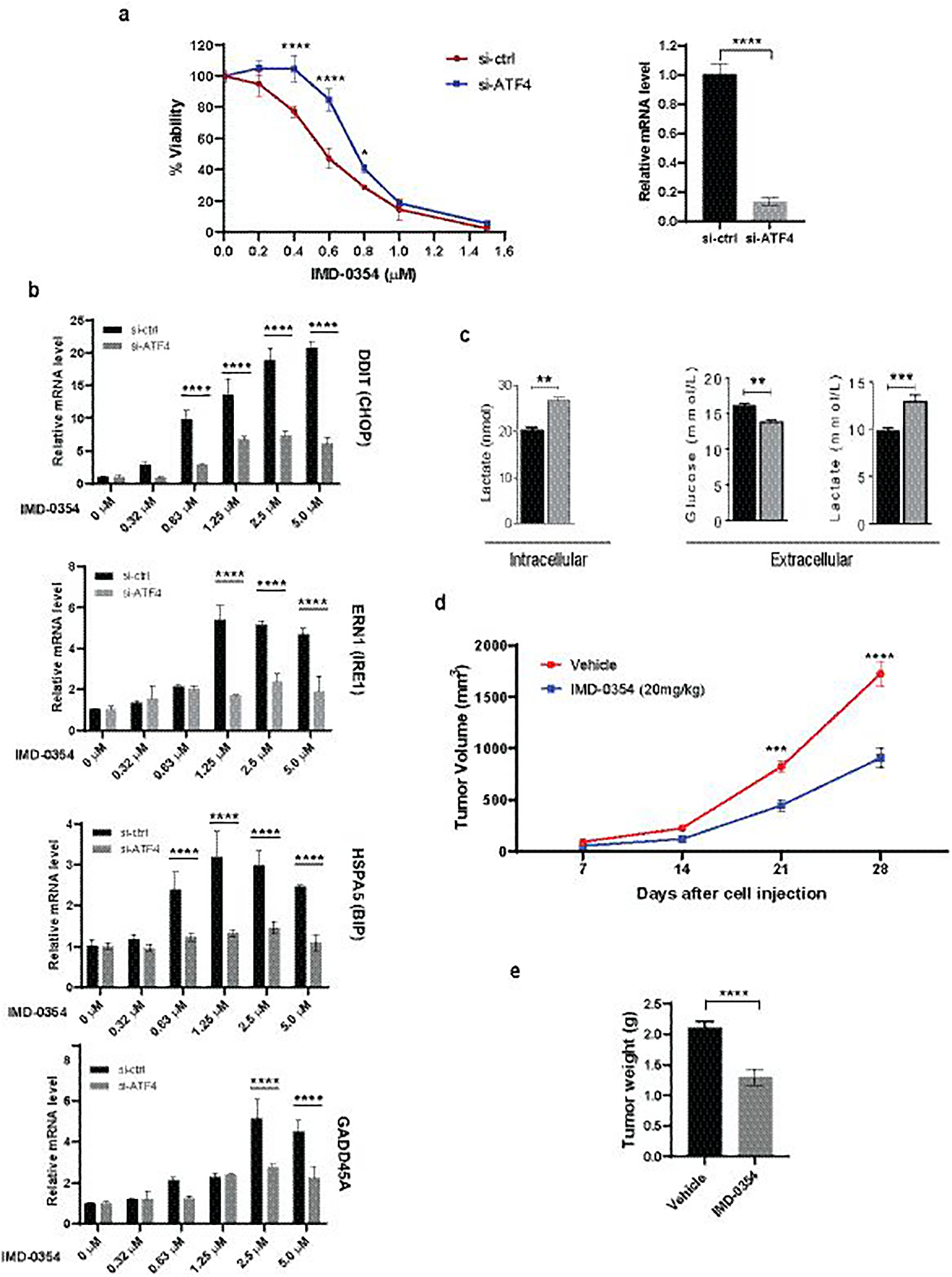
(a) A375 cells were transfected with si-ctrl or si-ATF4 and treated with increasing concentrations of IMD-0354. Cell proliferation was measured after 72h by ATPlite (left). ATF4 knockdown efficiency was confirmed by qPCR (right). (b) A375 cells were transfected with si-ctrl or si-ATF4 for 48h followed by treatment with IMD-0354 for 6h at indicated concentrations. RNA was extracted and subjected to qPCR analysis for UPR-related genes. (c) IMD-0354 treatment enhanced lactate production and suppressed glucose uptake. A431 cells were treated with IMD-0354 (2 μM) for 72h. Extracellular glucose and both extracellular and intracellular lactate levels were determined by GC-MS. (d, e) SW1 cells were injected into the right flank of male C3H/HeJ mice. After tumor establishment (~150mm3), mice were treated with 20 mg/kg of IMD-0354 daily intraperitoneally. Tumor volume was monitored for three weeks (d). Tumors were weighed at the end of the experiment (e). Statistical analysis was performed by two-way ANOVA for viability assay and tumor growth. Unpaired t-test was used for the comparison of two groups. Data are shown as the mean ± SD, n = 3. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. For animal experiment, statistical analysis was performed by two-way ANOVA for time-dependent tumor volume changes (d) and unpaired t-test was used for the comparison of end point tumor weight (e). Data are shown as the mean ± SEM, n = 8. ***P ≤ 0.001, ****P ≤ 0.0001.
Inhibition of Melanoma Growth by Combined Treatment with IMD-0354 and LDHAi or GLS1i
Our previous work has shown that inhibition of Lactate Dehydrogenase Isoform A (LDHA), the enzyme responsible for aerobic glycolysis –through catalysis of pyruvate oxidation to lactate– results in an expansive pro-survival response (5). This includes upregulation of SLC1A5 and glutamine uptake in melanoma cells, as part of metabolic reprogramming in response to LDHA inhibition (5). To examine whether suppression of glutamine uptake by IMD-0354 may conversely affect aerobic glycolysis, first, IMD-0354 -treated A431 cells were tested for intracellular lactate levels by GC-MS-based analysis. Also, the media in which these cells were cultured and treated with IMD-0354 was analyzed for glucose and lactate levels. IMD-0354 enhanced both intracellular and the media levels of lactate, while curtailing levels of glucose in the media (Fig 8c). Together, these data suggested glycolytic compensation (glucose utilization) for suppressed glutamine uptake caused by IMD-0354. This observation prompted us to examine whether co-inhibition of SLC1A5 and LDHA may have greater impact on tumor cell proliferation. Indeed, genetic inhibition of both SLC1A5 and LDHA, using corresponding siRNAs, led to a more effective suppression of melanoma cell proliferation and colony formation (Fig S7a and S7b). Given these observations, we assessed the effect of combined IMD-0354 and GNE-140 (a specific LDHA inhibitor (41)) treatment on the viability of melanoma cells. Treatment of BRAF mutant human as well as N-Ras mutant mouse melanoma cells with the combination of IMD-0354 and GNE-140 led to a more pronounced suppression of their viability as compared to the effect of either treatment alone (Fig S7c).
Given that cellular glutamine metabolism is dependent on its uptake (primarily facilitated by SLC1A5) as well as its assimilation (mediated by GLS1), we asked whether a combination of the glutamine uptake and assimilation inhibitors would achieve more effective inhibition of melanoma cell proliferation. Inhibition of glutamine uptake was performed by either genetic inhibition of SLC1A5 or by using the pharmacological inhibitor identified and characterized in this study, IMD-0354. Combination of either IMD-0354 or siSLC1A5 with CB-839 led to an additive (~20%) inhibition of melanoma cell growth (Fig S7d). These finding substantiate the importance of IMD-0354 in inhibition of glutamine uptake and its implication for glutamine metabolism requirement for melanoma proliferation.
Evaluation of IMD-0354 in mice
We next assessed the effect of IMD-0354 on melanoma tumor growth in vivo. SW1 melanoma cells harboring activating NRAS mutation were injected into syngeneic C3H mice, which were treated one week later with IMD-0354 (20 mg/kg) daily for the duration of the study. Treatment of mice with IMD-0354 inhibited SW1 melanoma xenograft growth as compared to the vehicle group (Fig 8d, 8e). Immunohistochemistry performed on the tumor sections confirmed that the tumors from IMD-0354-treated animals exhibited inhibition of proliferation (Ki67 staining) and mTOR activity (pS6 expression as a marker), concomitant with increased apoptosis (caspase 3 cleavage staining) compared to the vehicle-treated tumor group (Fig S8). These observations substantiate the findings and the characterization of IMD-0354 in cultured melanoma cell lines.
Discussion
Cancer cells exhibit high biosynthetic and energetic demands, requiring ample nutrient availability (42). While glucose is the chief source of carbons needed for macromolecular biosynthesis and ATP generation, glutamine has emerged as another key nutrient for cancer cells (2). Studies in a number of cancer types have demonstrated how glutamine uptake and metabolism is upregulated to selectively promote tumor growth and resistance to therapy (12,43). Our own work has demonstrated the importance of glutamine metabolism for breast cancer responsiveness to taxane therapy, whereby increased glutamine uptake was shown to confer resistance to paclitaxel (14). These observations supported the rationale for identifying novel inhibitors to suppress glutamine metabolism. Although glutaminase inhibitor (CB-839) that blocks anaplerotic entry of glutamine carbons into the TCA cycle is currently being evaluated in a number of clinical trials, attaining durable clinical response is likely to require also overcoming extramitochondrial functions of glutamine. Therefore, we set our focus on possible inhibition of glutamine uptake, searching for small molecules that would inhibit one of the key glutamine carrier proteins, SLC1A5. Here we report a microscopy-based screen of cancer cells for small molecules that can interfere with the membrane localization of SLC1A5 and hence compromise its function in glutamine uptake. IMD-0354 was identified as a top hit, which was confirmed to attenuate the uptake of glutamine in a number of different cancer cell lines. We report the implications of IMD-0354-mediated impairment of SLC1A5 function on (i) melanoma growth in culture and in vivo, using xenografts; (ii) define cellular pathways that are perturbed and causally involved in growth suppression exerted by IMD-0354; and (iii) unveil underlying mechanistic and functional parallels between IMD-0354-mediated and other glutamine metabolism-targeting strategies. While demonstrating the therapeutic benefit of limiting glutamine access of melanoma cells, our studies identify enhanced aerobic glycolysis as the mechanism of cellular metabolic rewiring in IMD-0354-treated cells. Importantly, our investigations go on to show how co-inhibiting aerobic glycolysis via LDHA inhibition can overcome metabolic escape of IMD-0354-treated melanoma cells and achieve effective suppression of cell proliferation.
IMD-0354, previously shown to suppress IKKβ activity, blocks nuclear translocation of NF-κB (34). Our screen identified two IKK inhibitors, which initially suggested a putative role of NF-κB signaling in glutamine uptake. Yet, our detailed investigations, utilizing an additional chemical inhibitor of IKK and the latter’s genetic inhibition ruled out a putative role of IKK-NF-κB pathway in glutamine uptake. These data suggest that in melanoma alternative pathways are regulated by IMD-0354. Notably, in response to glutamine deprivation, IKKβ was shown to interact with and phosphorylate 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase isoform 3 (PFKFB3), a major driver of aerobic glycolysis. This phosphorylation resulted in inhibition of PFKFB3 activity, and aerobic glycolysis, with co-inhibition of IKKβ and glutamine metabolism resulting in synergistic killing of cancer cells (44). It is therefore conceivable that a concomitant suppression of glutamine uptake and alleviation of IKKβ-mediated suppression of glycolysis by IMD-0354, could render glycolysis as a synthetic vulnerability of glutamine-deprived melanoma cells, sensitizing them to the inhibition of aerobic glycolysis. This idea is supported by our studies, showing stronger anti-proliferative effect of combined IMD-0354 and LDHA inhibitor. Although glycolytic enhancement through modulation of PFKFB3 activity is plausible, the precise mechanism underlying the ability of IMD-0354 to enhance glycolytic pathway and thus mark it as a potential synthetic lethal partner for concurrent suppression of glutamine uptake remains to be established. That a combination of IMD-0354 and CB-839, curtailing both uptake and assimilation of glutamine, led to an effective inhibition of melanoma cell proliferation further supports the importance of glutamine metabolism block in melanoma growth and the complementary effect of blocking glutamine uptake on its de novo synthesis.
Our RNAseq studies followed by the validation of gene expression changes implicated cellular processes/pathways previously linked to altered glutamine metabolism, including DNA damage response signaling, cell cycle, autophagy, programmed cell death, and aspartate biosynthesis (45–47). Computational analyses pointed to the importance of UPR and in particular ATF4, with experiments establishing the functional requirement of ATF4 in IMD-0354-mediated growth suppression. As IMD-0354 impairs plasma membrane distribution of SLC1A5, components of the post-translational machinery, plasma membrane localization, and anchoring stand as likely candidates for IMD-0354 effect.
How specific is IMD-0354 for SLC1A5 and glutamine uptake? Through a number of independent studies, we have demonstrated that IMD-0354 inhibits glutamine uptake, which is primarily mediated by SLC1A5. We have excluded the effect of IMD-0354 on SLC38A2, another key glutamine carrier protein. Within the range of concentrations tested, our studies also exclude the effect of IMD-0354 on the NF-κB and p38/MAPK pathways, that were previously suggested to serve as IMD-0354 targets. Mapping gene expression profiles, coupled with changes in cell death and autophagy programs, seen following IMD-0354 treatment, are consistent with independent studies which reported similar changes following inhibition of glutamine uptake. With these, one cannot exclude the possibility that IMD-0354 may also affect additional cellular targets, which may complement those associated with glutamine inhibition. Targeting of any given metabolic pathway is limited given the dynamic interplay among different metabolic signaling. Thus, targeting glutamine uptake is expected to impact other metabolic pathway, a point we experimentally addressed by combination of glutamine uptake and LDH inhibition. We have also demonstrated a partial rescue of IMD-0354-mediated suppression of melanoma cell proliferation by overexpression of SLC1A5. Notably, such rescue was limited to a narrow range of IMD-0354 concentrations, likely due to non-specific effects of high IMD-0354 concentrations, an aspect deserving further studies.
Rampant metabolic re-wiring in cancer cells poses a major challenge to nutrient restriction approaches. While clearly an impediment to achieving durable therapeutic responses, greater understanding of cellular adaptive mechanisms to amino acid restriction approaches have started to reveal actionable vulnerabilities that are guiding development of effective combination treatments (41,48,49). Our studies with IMD-0354 have highlighted the compensatory role of enhanced glucose metabolism (aerobic glycolysis). These findings complement previous reports that show suppression of aerobic glycolysis –by LDHA inhibition– increasing dependence on enhanced glutamine uptake and increased TCA cycle activity, forming rationale for combining LDHA inhibitors with the Complex I inhibitors of the mitochondrial electron transport chain (41,50). While our findings of the interplay between glutamine and glucose metabolism and the effectiveness of combining IMD-0354 with LDHA inhibition, form basis for further pre-clinical testing of this combination, currently there are no effective LDHA inhibitors for in vivo use. As MAPK pathway is constitutively active in a large majority of melanomas and is a well-established regulator of glucose metabolism (51), in part through LDHA regulation (5), combining IMD-0354 with FDA-approved MAPK signaling inhibitors presents a potentially viable therapeutic option that should be further investigated in pre-clinical studies.
Supplementary Material
Acknowledgments
We thank members of the Bioinformatic, Metabolomic, and the animal shared facilities as well as the members of the CPCCG for their help in carrying out different aspects of these studies. Support by NCI grant R35CA197465, DOD grant CA1810216, MRA grant 509524 to Z. Ronai are gratefully acknowledged. Support through grant P30 CA030199 to Shared Resource Facilities at the Sanford Burnham Prebys NCI designated Cancer Center is gratefully acknowledged.
Footnotes
Financial Information
No conflict of interest declared for this study. ZAR is a co-founder and serves as a scientific advisor to Pangea Therapeutics. All other authors declare no competing interests. A patent application detailing the findings disclosed in this manuscript has been filed by SBP Discovery.
REFERENCES
- 1.Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018;10(11) doi 10.3390/nu10111564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 2016;16(10):619–34 doi 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009;136(3):521–34 doi 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yue M, Jiang J, Gao P, Liu H, Qing G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep 2017;21(13):3819–32 doi 10.1016/j.celrep.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Pathria G, Scott DA, Feng Y, Sang Lee J, Fujita Y, Zhang G, et al. Targeting the Warburg effect via LDHA inhibition engages ATF4 signaling for cancer cell survival. EMBO J 2018;37(20) doi 10.15252/embj.201899735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010;29(3):313–24 doi 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009;458(7239):762–5 doi 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao X, Petrashen AP, Sanders JA, Peterson AL, Sedivy JM. SLC1A5 glutamine transporter is a target of MYC and mediates reduced mTORC1 signaling and increased fatty acid oxidation in long-lived Myc hypomorphic mice. Aging Cell 2019;18(3):e12947 doi 10.1111/acel.12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi YK, Park KG. Targeting Glutamine Metabolism for Cancer Treatment. Biomol Ther (Seoul) 2018;26(1):19–28 doi 10.4062/biomolther.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019;366(6468):1013–21 doi 10.1126/science.aav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016;35(24):3201–8 doi 10.1038/onc.2015.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baenke F, Chaneton B, Smith M, Van Den Broek N, Hogan K, Tang H, et al. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol Oncol 2016;10(1):73–84 doi 10.1016/j.molonc.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun RC, Denko NC. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab 2014;19(2):285–92 doi 10.1016/j.cmet.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeon YJ, Khelifa S, Ratnikov B, Scott DA, Feng Y, Parisi F, et al. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell 2015;27(3):354–69 doi 10.1016/j.ccell.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee P, Malik D, Perkons N, Huangyang P, Khare S, Rhoades S, et al. Targeting glutamine metabolism slows soft tissue sarcoma growth. Nat Commun 2020;11(1):498 doi 10.1038/s41467-020-14374-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katt WP, Lukey MJ, Cerione RA. Starving the Devourer: Cutting Cancer Off from Its Favorite Foods. Cell Chem Biol 2019;26(9):1197–9 doi 10.1016/j.chembiol.2019.09.005. [DOI] [PubMed] [Google Scholar]
- 17.Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Magyar C, Braas D, et al. Targeted Inhibition of EGFR and Glutaminase Induces Metabolic Crisis in EGFR Mutant Lung Cancer. Cell Rep 2017;18(3):601–10 doi 10.1016/j.celrep.2016.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen YA, Hong J, Asaka R, Asaka S, Hsu FC, Suryo Rahmanto Y, et al. Inhibition of the MYC-regulated glutaminase metabolic axis is an effective synthetic lethal approach for treating chemoresistant cancers. Cancer Res 2020. doi 10.1158/0008-5472.CAN-19-3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song M, Kim SH, Im CY, Hwang HJ. Recent Development of Small Molecule Glutaminase Inhibitors. Curr Top Med Chem 2018;18(6):432–43 doi 10.2174/1568026618666180525100830. [DOI] [PubMed] [Google Scholar]
- 20.Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017;3(3):169–80 doi 10.1016/j.trecan.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Zhao T, Li Z, Wang L, Yuan S, Sun L. The role of ASCT2 in cancer: A review. Eur J Pharmacol 2018;837:81–7 doi 10.1016/j.ejphar.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Wang C, Wu J, Wang Z, Yang Z, Li Z, Deng H, et al. Glutamine addiction activates polyglutamine-based nanocarriers delivering therapeutic siRNAs to orthotopic lung tumor mediated by glutamine transporter SLC1A5. Biomaterials 2018;183:77–92 doi 10.1016/j.biomaterials.2018.08.035. [DOI] [PubMed] [Google Scholar]
- 23.Schulte ML, Khodadadi AB, Cuthbertson ML, Smith JA, Manning HC. 2-Amino-4-bis(aryloxybenzyl)aminobutanoic acids: A novel scaffold for inhibition of ASCT2-mediated glutamine transport. Bioorg Med Chem Lett 2016;26(3):1044–7 doi 10.1016/j.bmcl.2015.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med 2018;24(2):194–202 doi 10.1038/nm.4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Geldermalsen M, Quek LE, Turner N, Freidman N, Pang A, Guan YF, et al. Benzylserine inhibits breast cancer cell growth by disrupting intracellular amino acid homeostasis and triggering amino acid response pathways. BMC Cancer 2018;18(1):689 doi 10.1186/s12885-018-4599-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Broer A, Fairweather S, Broer S. Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Front Pharmacol 2018;9:785 doi 10.3389/fphar.2018.00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 1999;4(2):67–73 doi 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 28.Ratnikov B, Aza-Blanc P, Ronai ZA, Smith JW, Osterman AL, Scott DA. Glutamate and asparagine cataplerosis underlie glutamine addiction in melanoma. Oncotarget 2015;6(10):7379–89 doi 10.18632/oncotarget.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin YC. Why you should read Dr. Cramer’s perspective. J Comput Aided Mol Des 2011;25(3):195–6 doi 10.1007/s10822-011-9414-4. [DOI] [PubMed] [Google Scholar]
- 30.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323 doi 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cormerais Y, Massard PA, Vucetic M, Giuliano S, Tambutte E, Durivault J, et al. The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J Biol Chem 2018;293(8):2877–87 doi 10.1074/jbc.RA117.001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka A, Konno M, Muto S, Kambe N, Morii E, Nakahata T, et al. A novel NF-kappaB inhibitor, IMD-0354, suppresses neoplastic proliferation of human mast cells with constitutively activated c-kit receptors. Blood 2005;105(6):2324–31 doi 10.1182/blood-2004-08-3247. [DOI] [PubMed] [Google Scholar]
- 35.Podolin PL, Callahan JF, Bolognese BJ, Li YH, Carlson K, Davis TG, et al. Attenuation of murine collagen-induced arthritis by a novel, potent, selective small molecule inhibitor of IkappaB Kinase 2, TPCA-1 (2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide), occurs via reduction of proinflammatory cytokines and antigen-induced T cell Proliferation. J Pharmacol Exp Ther 2005;312(1):373–81 doi 10.1124/jpet.104.074484. [DOI] [PubMed] [Google Scholar]
- 36.Wang Q, Beaumont KA, Otte NJ, Font J, Bailey CG, van Geldermalsen M, et al. Targeting glutamine transport to suppress melanoma cell growth. Int J Cancer 2014;135(5):1060–71 doi 10.1002/ijc.28749. [DOI] [PubMed] [Google Scholar]
- 37.Hassanein M, Hoeksema MD, Shiota M, Qian J, Harris BK, Chen H, et al. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res 2013;19(3):560–70 doi 10.1158/1078-0432.CCR-12-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Q, Hardie RA, Hoy AJ, van Geldermalsen M, Gao D, Fazli L, et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol 2015;236(3):278–89 doi 10.1002/path.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu J, Chen M, Tao Z, Gao S, Li Y, Cao Y, et al. Effects of targeting SLC1A5 on inhibiting gastric cancer growth and tumor development in vitro and in vivo. Oncotarget 2017;8(44):76458–67 doi 10.18632/oncotarget.19479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L, Liu Y, Zhao TL, Li ZZ, He JY, Zhang BJ, et al. Topotecan induces apoptosis via ASCT2 mediated oxidative stress in gastric cancer. Phytomedicine 2019;57:117–28 doi 10.1016/j.phymed.2018.12.011. [DOI] [PubMed] [Google Scholar]
- 41.Boudreau A, Purkey HE, Hitz A, Robarge K, Peterson D, Labadie S, et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat Chem Biol 2016;12(10):779–86 doi 10.1038/nchembio.2143. [DOI] [PubMed] [Google Scholar]
- 42.Keibler MA, Wasylenko TM, Kelleher JK, Iliopoulos O, Vander Heiden MG, Stephanopoulos G. Metabolic requirements for cancer cell proliferation. Cancer Metab 2016;4:16 doi 10.1186/s40170-016-0156-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hernandez-Davies JE, Tran TQ, Reid MA, Rosales KR, Lowman XH, Pan M, et al. Vemurafenib resistance reprograms melanoma cells towards glutamine dependence. J Transl Med 2015;13:210 doi 10.1186/s12967-015-0581-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reid MA, Lowman XH, Pan M, Tran TQ, Warmoes MO, Ishak Gabra MB, et al. IKKbeta promotes metabolic adaptation to glutamine deprivation via phosphorylation and inhibition of PFKFB3. Genes Dev 2016;30(16):1837–51 doi 10.1101/gad.287235.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan HWS, Sim AYL, Long YC. Glutamine metabolism regulates autophagy-dependent mTORC1 reactivation during amino acid starvation. Nat Commun 2017;8(1):338 doi 10.1038/s41467-017-00369-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tran TQ, Ishak Gabra MB, Lowman XH, Yang Y, Reid MA, Pan M, et al. Glutamine deficiency induces DNA alkylation damage and sensitizes cancer cells to alkylating agents through inhibition of ALKBH enzymes. PLoS Biol 2017;15(11):e2002810 doi 10.1371/journal.pbio.2002810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alkan HF, Walter KE, Luengo A, Madreiter-Sokolowski CT, Stryeck S, Lau AN, et al. Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab 2018;28(5):706–20 e6 doi 10.1016/j.cmet.2018.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gwinn DM, Lee AG, Briones-Martin-Del-Campo M, Conn CS, Simpson DR, Scott AI, et al. Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 2018;33(1):91–107 e6 doi 10.1016/j.ccell.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pathria G, Lee JS, Hasnis E, Tandoc K, Scott DA, Verma S, et al. Translational reprogramming marks adaptation to asparagine restriction in cancer. Nat Cell Biol 2019;21(12):1590–603 doi 10.1038/s41556-019-0415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chaube B, Malvi P, Singh SV, Mohammad N, Meena AS, Bhat MK. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget 2015;6(35):37281–99 doi 10.18632/oncotarget.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov 2014;4(4):423–33 doi 10.1158/2159-8290.CD-13-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.