

Abstract
This paper describes a new method for the transannular functionalization of the γ-C-H bonds in alicyclic amines to install C(sp3)–halogen, oxygen, nitrogen, boron, and sulfur bonds. The key challenge for this transformation is controlling the relative rate of Cγ–H versus Cα–H functionalization. We demonstrate that this selectivity can be achieved by pre-complexation of the substrate with Pd prior to the addition of oxidant. This approach enables the use of diverse oxidants that ultimately install various heteroatom functional groups at the γ–position with high site- and diastereoselectivity.
Keywords: C–H activation, Palladium, Alicyclic amines, Relative rates
Entry for the Table of Contents
An approach to selectively transform Cγ–H bonds of alicyclic amines to Cγ(sp3)–halogen, oxygen, nitrogen, boron, and sulfur bonds is described. This method is enabled via the pre-complexation of the amine substrate with palladium followed by the addition of oxidant to yield bioactive motifs functionalized at the γ–position.
Alicyclic amines bearing various substitution patterns are common structural motifs in bioactive molecules.[1] Conventional synthetic routes to these structures require multi-step sequences to assemble the appropriately functionalized alicyclic amine cores.[2] Approaches involving the late-stage C–H functionalization of pre-assembled alicyclic amines would complement existing synthetic routes and thus streamline the diversification of these motifs. Over the past several decades, numerous methods have been developed for functionalization at the activated Cα–H position of alicyclic amines (Scheme 1a, kα).[3] These studies have shown that the proximity of the Cα–H bond to nitrogen greatly enhances its reactivity towards oxidative functionalization.[4] For example, C(sp3)–H bonds α to nitrogen have relatively low bond dissociation energies (~90 kcal/mol).[5] Furthermore, oxidation of nitrogen to a radical cation renders the Cα–H site highly acidic (pKa ~ 16) relative to unactivated C(sp3)–H bonds.[6] In contrast, the C(sp3)–H bonds that are remote from nitrogen (for example, Cγ–H) are typically much less reactive than Cα–H, making it significantly more challenging to selectively target these sites.
Scheme 1.
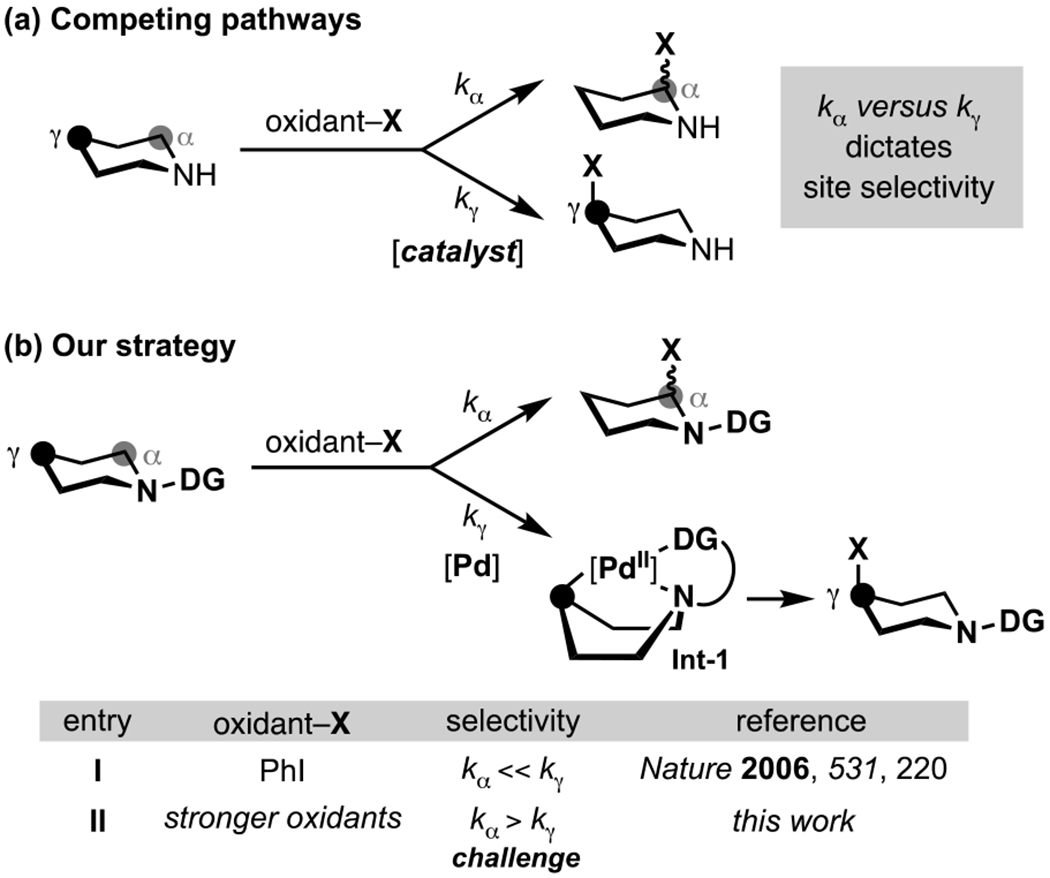
(a) Competing Cα–H versus Cγ–H (b) Our strategy.
Conceptually, the selective γ-functionalization of alicyclic amines requires controlling the relative reactivity of the Cα–H (Scheme 1a, kα) versus Cγ–H sites (Scheme 1a, kγ). To date, most successful efforts have achieved selectivity through modification of the substrate. Common strategies involve (a) blocking the Cα–H sites with other substituents (thus decreasing kα),[7] (b) protonating the amine nitrogen to electronically deactivate Cα–H (thus decreasing kα),[8] or (c) employing a directing group to accelerate Cγ–H functionalization (increasing kγ).[9] In an example of the latter, our group recently demonstrated that installing a directing group on the amine nitrogen can enable transannular Cγ–H activation via a boat-like intermediate (Int-1, Scheme 1b).[10] When the Pd catalyst for this transformation is paired with a mild aryl iodide (ArI) oxidant, kγ is significantly greater than kα. As such, directed transannular C–H arylation outcompetes background α-functionalization (Scheme 1b, entry I).
An important goal for enhancing the utility of this transformation is to broaden the scope of functional groups that can be introduced at Cγ. In principle, this can be achieved by replacing the aryl iodide with an alternative oxidant (oxidant–X) that is designed to transfer the functional group of interest (X). However, in practice, changing to alternative, more kinetically reactive oxidants (for example, N-halosuccinimides, hypervalent iodine reagents, electrophilic fluorinating reagents) results in a dramatic increase in kα, such that the background α-functionalization pathway predominates (Scheme 1b, entry II; vide infra for examples). In this report, we present a strategy to address this challenge that leverages the in situ formation of Pd(II) amine complexes to enable selective transannular Cγ–H functionalization with a wide range of oxidants.
Initial studies targeted the Pd-catalyzed transannular Cγ–H bromination of 1-A with N-bromosuccinimide (NBS). Notably, NBS has been successfully employed in related Pd-catalyzed ligand-directed C(sp3)–H bromination reactions (of non-amine containing substrates),[11] while 1-A was shown to be an effective substrate for transannular Cγ–H arylation with PhI. At 100 °C in tert-amyl alcohol, 1-A reacts with PhI to afford the Cγ–H phenylation product in 30% yield, with no detectable background α-functionalization products (kα << kγ). However, when NBS was used in place of PhI under otherwise analogous catalytic conditions, none of the Cγ–H bromination product γ-Br was detected (Scheme 2). Instead, α-oxidation products α-N and α-O were formed in 30% and 30% yield, respectively (Scheme 2).[12] When this reaction was conducted in the absence of Pd catalyst, α-N and α-O were obtained in nearly identical yields of 25% and 31%. These results demonstrate that with NBS, the rate of background α-oxidation (kα) is significantly greater than that of Pd-catalyzed γ-oxidation (kγ).
Scheme 2.
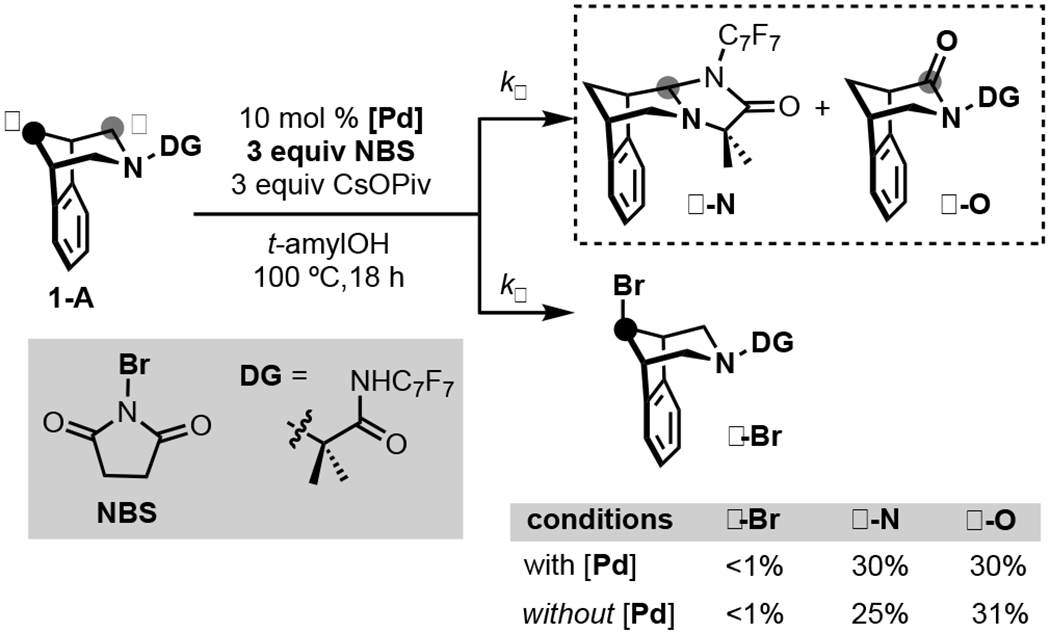
Pd-catalyzed C–H bromination with NBS.
We hypothesized that these relative rates might be reversed by pre-assembling a complex between substrate 1-A and Pd (Scheme 3a).[13] This proposal was predicated on our previous report showing that γ-H/D exchange is fast at the isolable Pd-complex 2-A (occurring at temperatures as low as 40 ºC).[13] This suggests that pre-complexation to Pd could enhance kγ versus kα in the NBS reactions. Indeed, the treatment of 1 equiv of complex 2-A with 1 equiv of NBS in t-amylOH at 100 ºC for 18 h led to the selective formation of γ-Br in 50% yield (Scheme 3a). Only traces (<1%) of α-N/α-O were detected in this reaction. γ-Br was formed as a single regio- and stereoisomer, as determined by NMR spectroscopy. As discussed below, this stereochemistry suggests that Cγ–Br bond formation occurs via an inner sphere process with retention of configuration. Changing the solvent to MeCN led to a higher (70%) yield of γ-Br, again with <1% of α-N/α-O.
Scheme 3.
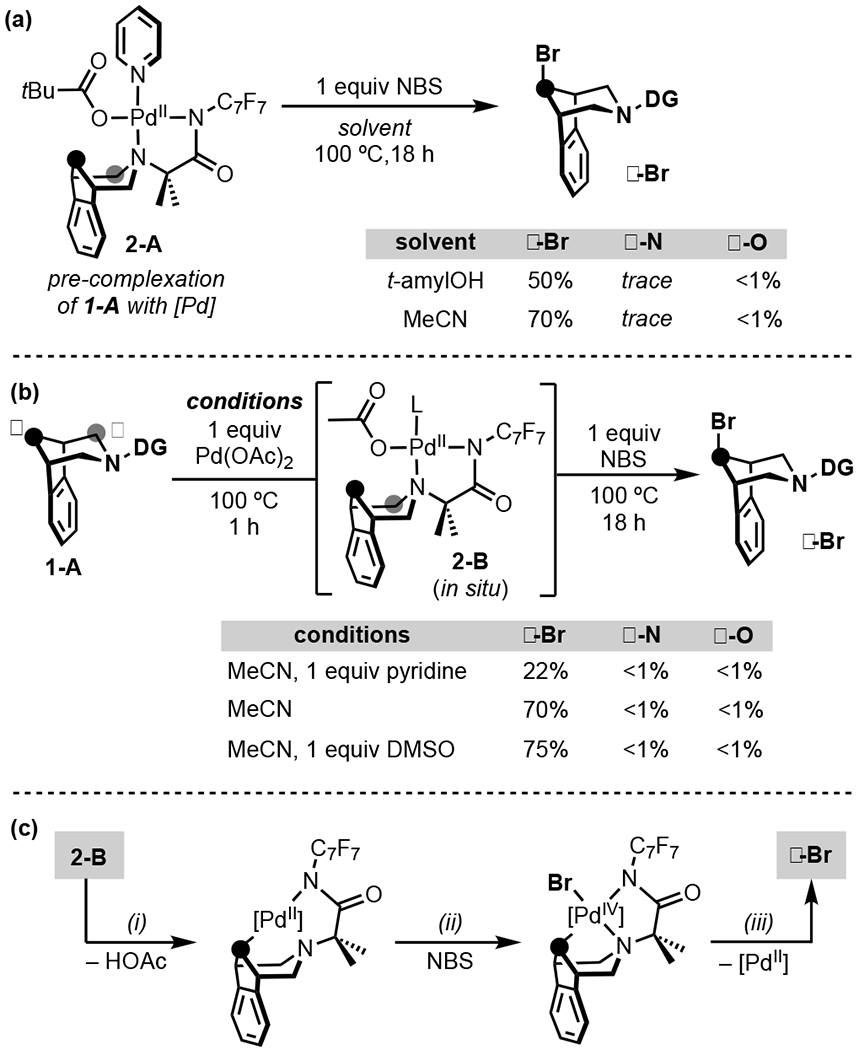
(a) γ-Br with complex 2-A (b) In situ method for γ-Br. (c) Proposed pathway.
To render this approach more practical, we next pursued a 2-step 1-pot approach to the in situ assembly/γ-functionalization of a 1-A/Pd complex. First, 1 equiv of 1-A, 1 equiv of Pd(OAc)2, and 1 equiv of pyridine were stirred at 100 ºC for 1 h in MeCN. NBS (1 equiv) was then added, and the mixture was heated at 100 ºC for an additional 18 h. This afforded a modest 22% yield of γ-Br with <1% of α-N/α-O (Scheme 3b). Conducting the analogous reaction in the absence of pyridine gave 70% yield of γ-Br, and the addition of 1 equiv of DMSO further improved the yield to 75% while maintaining high selectivity (<1% of α-N/α-O).[14]
A proposed pathway for this sequence based on literature precedent for the individual steps is shown in Scheme 3c. Initial coordination of 1-A to Pd(OAc)2 affords 2-B, where L is likely MeCN or DMSO.[13],[15] Acetate-assisted transannular Cγ–H activation[10c], [16] (Scheme 3c, i) is followed by oxidation of this alkyl PdII intermediate to PdIV with NBS (Scheme 3c, ii).[17] C(sp3)–Br bond-forming reductive elimination from this highly reactive PdIV center[18] then proceeds via an inner sphere mechanism with retention of configuration at carbon[19] to afford the product γ-Br (Scheme 3c, iii).
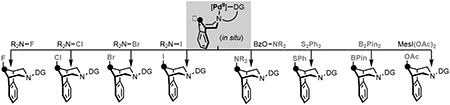
We next explored the use of a series of different oxidants in this 2-step, 1-pot protocol in order to install diverse functional groups at the γ-position. As shown in Scheme 4, this approach enabled the formation of C–O, C–S, C–N, C–F, C–Cl, C–I, and C–B bonds in high γ-selectivity and modest to good isolated yields. The site- and stereoselectivity of each functionalization was established via 1H NMR spectroscopy (all products) as well as X-ray crystallography (for γ-I, γ-F, γ-OAc). In all cases, the major product derived from Cγ–H functionalization with retention of configuration during the C–X bond-forming step.[20]
Scheme 4.
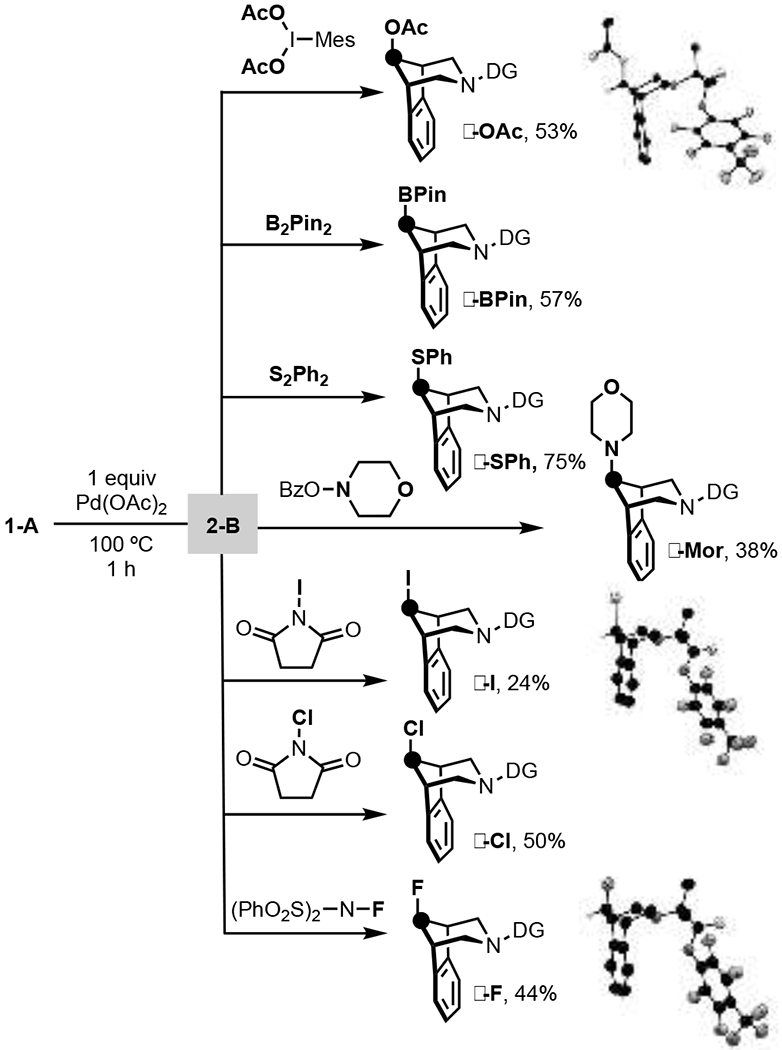
γ-functionalizations with in situ method.
Finally, we evaluated the scope of Cγ–H functionalization with respect to alicyclic amine substrates. The borylation reaction with B2Pin2 was selected for this study based on the versatility of the boronate ester products (which can be readily transformed into amines, alcohols, or C–C bonds).[21] As shown in Scheme 5, nitro, amino, cyano, chloro, bromo, boronate ester, and amide substituents were all well tolerated. Other bicyclic amines, including those derived from the bioactive molecules varenicline (10-BPin) and cytisine (12-BPin) also reacted to afford Cγ–H borylated products with high selectivity.[22]
Scheme 5.
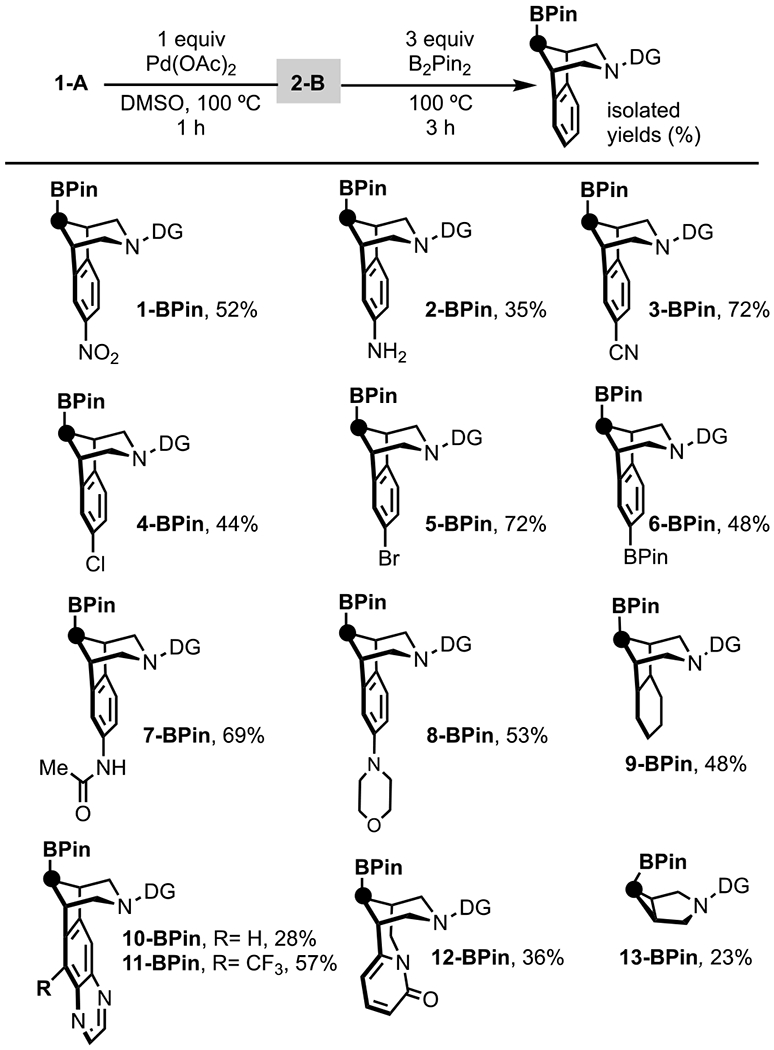
Scope of Cγ–BPin functionalization.
In summary, this report describes a strategy for the selective Cγ–H oxidation of alicyclic amines via pre-formation of amine-Pd complexes. This pre-complexation increases the relative rate of the desired Cγ–H activation versus competing background Cα–H oxidation. This work adds to a growing suite of methods in which the use of stoichiometric Pd enables selective late-stage diversification of complex organic molecules.[23] While catalytic processes are often favored by the organic chemistry community, this stoichiometric approach provides rapid and selective access to numerous challenging-to-synthesize alicyclic amine derivatives. In the context of, for example, medicinal chemistry, the speed, selectivity, and diversity of products generated via this approach counterbalance the cost of the Pd. Ultimately, we anticipate that pre-complexation could prove valuable for tuning selectivity in other reactions of alicyclic amines as well as in metal-mediated C–H functionalizations of more diverse substrates.
Supplementary Material
Acknowledgements
We thank Dr. Jeff. W. Kampf for carrying out X-ray crystallographic analyses as well as James Windak for high-resolution mass spectrometry analyses. We thank Dr. Mark A. Mantell for performing Parr Reactor experiments, and Dr. Scott M. Thullen for helpful discussions. EA acknowledges the US National Science Foundation for a graduate fellowship. This work was supported by the NIH (R35 GM136332).
References
- [1].a) Vitaku E, Smith DT, Njardarson JT, J. Med. Chem . 2014, 57, 10257–10274; [DOI] [PubMed] [Google Scholar]; b) Taylor RD, MacCoss M, Lawson ADG, J. Med. Chem . 2014, 57, 5845–5859. [DOI] [PubMed] [Google Scholar]
- [2].Källström S, Leino R, Bioorg. Med. Chem. 2008, 16, 601–635. [DOI] [PubMed] [Google Scholar]
- [3].For examples of Cα—H of alicyclic amines, see:a) Pastine SJ, Gribkov D, Sames D, J. Am. Chem. Soc. 2006, 128, 14220–14221; [DOI] [PubMed] [Google Scholar]; b) Campos KR, Chem. Soc. Rev. 2007, 36, 1069–1084; [DOI] [PubMed] [Google Scholar]; c) Mitchell EA, Peschiulli A, Lefevre N, Meerpoel L, Maes BUW, Chem. - Eur. J . 2012, 18, 10092–10142; [DOI] [PubMed] [Google Scholar]; d) Shi L, Xia W, Chem. Soc. Rev. 2012, 41, 7687–7697; [DOI] [PubMed] [Google Scholar]; e) He J, Hamann LG, Davies HML, Beckwith REJ, Nat. Commun. 2015, 6, 5943; [DOI] [PubMed] [Google Scholar]; f) Spangler JE, Kobayashi Y, Verma P, Wang D-H, Yu J-Q, J. Am. Chem. Soc. 2015, 137, 11876–11879; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Chen W, Ma L, Paul A, Seidel D, Nat. Chem . 2017, 10, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].For examples of amines undergoing side reactions in presence of metal catalysts and oxidants, see:a) Murahashi S, Naota T, Yonemura K, J. Am. Chem. Soc . 1988, 110, 8256–8258; [Google Scholar]; b) Venkataramanan NS, Kuppuraj G, Rajagopal S, Coord. Chem. Rev. 2005, 249, 1249–1268; [Google Scholar]; c) Park J, Morimoto Y, Lee Y-M, You Y, Nam W, Fukuzumi S, Inorg. Chem. 2011, 50, 11612–11622; [DOI] [PubMed] [Google Scholar]; d) Liu P, Liu Y, Wong EL-M, Xiang S, Che C-M, Chem. Sci. 2011, 2, 2187–2195; [Google Scholar]; e) Cai XW, Sha M, Guo CP, Pan RM, Asian J Chem . 2012, 24, 3781–3784; [Google Scholar]; f) Genovino J, Lütz S, Sames D, Touré BB, J. Am. Chem. Soc . 2013, 135, 12346–12352; [DOI] [PubMed] [Google Scholar]; g) Ling Z, Yun L, Liu L, Fu X, Chem. Commun . 2013, 49, 4214–4216; [DOI] [PubMed] [Google Scholar]; h) Malik HA, Taylor BLH, Kerrigan JR, Grob JE, Houk KN, Du Bois J, Hamann LG, Patterson AW, Chem. Sci . 2014, 5, 2352–2361; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Kim S, Ginsbach JW, Lee JY, Peterson RL, Liu JJ, Siegler MA, Sarjeant AA, Solomon EI, Karlin KD, J. Am. Chem. Soc. 2015, 137, 2867–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wayner DDM, Clark KB, Rauk A, Yu D, Armstrong DA, J. Am. Chem. Soc . 1997, 119, 8925–8932. [Google Scholar]
- [6].Chu JCK, Rovis T, Angew. Chem. Int. Ed. 2018, 57, 62–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].For remote C–H functionalization blocking Cα–H sites on alicylic amines, see:a) McNally A, Haffemayer B, Collins BS, Gaunt MJ, Nature 2014, 510, 129–133; [DOI] [PubMed] [Google Scholar]; b) Calleja J, Pla D, Gorman TW, Domingo V, Haffemayer B, Gaunt MJ, Nature Chemistry 2015, 7, 1009–1016. [DOI] [PubMed] [Google Scholar]
- [8].For remote C−H functionalization protonating nitrogen on alicylic amines, see:a) Annese C, D’Accolti L, De Zotti M, Fusco C, Toniolo C, Williard PG, Curcu R, J. Org. Chem. 2010, 75, 4812–4816; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mbofana CT, Chong E, Lawniczak J, Sanford MS, Org. Lett. 2016, 18, 4258–4261; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schultz DM, Lévesque F, DiRocco DA, Reibarkh M, Ji Y, Joyce LA, Dropinski JF, Sheng H, Sherry BD, Davies IW, Angew. Chem. Int. Ed . 2017, 56, 15274–15278; [DOI] [PubMed] [Google Scholar]; d) Lee M, Sanford MS, Org. Lett . 2017, 19, 572–575; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) White CM, Zhao J, J. Am. Chem. Soc. 2018, 140, 13988–14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].For remote C−H functionalization using directing groups, see:a) Antermite D, Bull JA, Synthesis 2019, 51, 3171–3204; [Google Scholar]; b) He C, Whitehurst WG, Gaunt MJ, Chem 2019, 5, 1031–1058; [Google Scholar]; c) Li Z, Dechantsreiter M, Dandapani S, J. Org. Chem. 2020, 85, 6747–6760. [DOI] [PubMed] [Google Scholar]
- [10].a) Topczewski JJ, Cabrera PJ, Saper NI, Sanford MS, Nature 2016, 531, 220–224; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cabrera PJ, Lee M, Sanford MS, J. Am. Chem. Soc. 2018, 140, 5599–5606; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dewyer AL, Zimmerman PM, ACS Catal. 2017, 7, 5466–5477. [Google Scholar]
- [11].Zhu R-Y, Saint-Denis TG, Shao Y, He J, Sieber JD, Senanayake CH, Yu J-Q, J. Am. Chem. Soc . 2017, 16, 5724–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Even at 50 °C (under otherwise analogous conditions to Scheme 2), the yields of γ-Br, α-N, and α-O were 0, 12, and 15%, respectively. [Google Scholar]
- [13].Aguilera EY, Sanford MS, Organometallics 2019, 38, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Other L-type ligands were also evaluated (e.g. bipyridine, PPh3) and all gave poor yields of γ-Br. [Google Scholar]
- [15].In our hands, complex 2-B with L = DMSO proved challenging to isolate, providing further impetus for generating this putative intermediate in situ. [Google Scholar]
- [16].a) Lapointe D, Fagnou K, Chem. Lett . 2010, 39, 1118–1126; [Google Scholar]; b) Gary JB, Sanford MS, Organometallics 2011, 30, 6143–6149; [Google Scholar]; c) Ackermann L, Chem. Rev . 2011, 111, 1315–1345; [DOI] [PubMed] [Google Scholar]; d) Gorelsky SL, Lapointe D, Fagnou K, J. Org. Chem. 2012, 77, 658–668; [DOI] [PubMed] [Google Scholar]; e) Gorelsky SI, Coord. Chem. Rev. 2013, 257, 153–164. [Google Scholar]
- [17].Whitfield SR, Sanford MS, J. Am. Chem. Soc. 2007, 129, 15142–15143. [DOI] [PubMed] [Google Scholar]
- [18].Racowski J, Sanford MS in Topics in Organometallic Chemistry, Vol. 53, (Eds.: Canty A), Springer, 2011, pp. 61–84. [Google Scholar]
- [19].a) Racowski JM, Gary BJ, Sanford MS, Angew. Chem. Int. Ed . 2012, 51, 3414–3417; [DOI] [PubMed] [Google Scholar]; b) Chen Y-Q, Singh S, Wu Y, Wang Z, Hao W, Verma P, Qiao JX, Sunoj RB, Yu J-Q, J. Am. Chem. Soc . 2020, 142, 9966–9974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Compound α-N was formed as a minor product (15% yield) with N-iodosuccinimide. With N-chlorosuccinimide, N-fluorobenzenesulfonimide, iodomesitylene diacetate, morpholino benzoate, B2Pin2, and S2Ph2, compound α-N was also detected but in <10% yield. [Google Scholar]
- [21] a).Larsen MA, Hartwig JF, J. Am. Chem. Soc. 2014, 136, 4287–4299; [DOI] [PubMed] [Google Scholar]; b) Fyfe JWB, Watson AJB, Chem 2017, 3, 31–55; [Google Scholar]; c) Xu L, Wang G, Zhang S, Wang H, Wang L, Liu L, Li P, Tetrahedron 2017, 73, 7123–7157. [Google Scholar]
- [22].With piperidines as the substrate, only starting material and α-functionalization was observed (see Table 3 in SI for examples). [Google Scholar]
- [23].Uehling MR, King RP, Krska SW, Cernak T, Buchwald SL, Science 2019, 363, 405–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.