

Summary
Immunogenic tumor cell death enhances anti-tumor immunity. However, the mechanisms underlying this effect are incompletely understood. We established a system to induce tumor cell death in situ and investigated its effect on dendritic cell (DC) migration and T cell responses using intravital photolabeling in mice expressing KikGR photoconvertible protein. We demonstrate that tumor cell death induces phagocytosis of tumor cells by tumor-infiltrating (Ti)-DCs, and HMGB1-TLR4 and ATP-P2X7 receptor signaling-dependent Ti-DC emigration to draining lymph nodes (dLNs). This led to an increase in anti-tumor CD8+ T cells of memory precursor effector phenotype and secondary tumor growth inhibition in a CD103+ DC-dependent manner. However, combining tumor cell death induction with lipopolysaccharide treatment stimulated Ti-DC maturation and emigration to dLNs but did not improve tumor immunity. Thus, immunogenic tumor cell death enhances tumor immunity by increasing Ti-DC migration to dLNs where they promote anti-tumor T cell responses and tumor growth inhibition.
Subject areas: Immunology, Cell Biology, Cancer
Graphical abstract
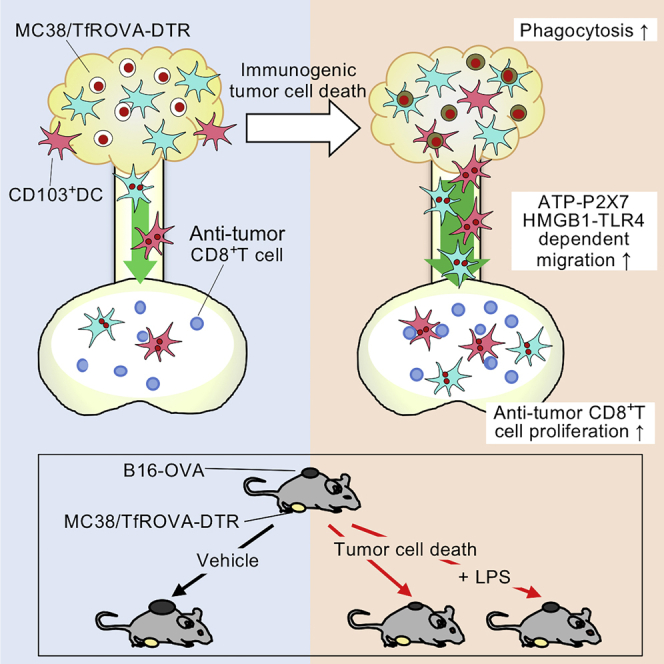
Highlights
-
•
Immunogenic cell death (ICD) promotes egress of tumor-infiltrating (Ti)-DCs to dLNs
-
•
ICD induced Ti-DC migration to dLNs utilizes P2X7R and HMGB1 signaling pathways
-
•
LPS treatment attenuates the anti-tumor effects of ICD
-
•
CD103+ DCs are required at the time of ICD for suppression of secondary tumor growth
Immunology ; Cell Biology ; Cancer
Introduction
Chemotherapy and radiation therapy induce tumor cell death (Galluzzi et al., 2017; Montico et al., 2018; Zitvogel et al., 2008) and stimulate anti-tumor immunity through immunogenic cell death (ICD) and increased supply of tumor antigens. In ICD, damage-associated molecular patterns (DAMPs) molecules, calreticulin (CRT), heat shock protein (HSP)-70, and HSP-90 are expressed on the plasma membrane of dying tumor cells, and adenosine triphosphate (ATP) and high-mobility group box (HMGB) 1 are released from dying tumor cells. ATP released from oxaliplatin-killed tumor cells stimulates dendritic cells (DCs) in tumors via P2X7 receptor (P2X7R) and enhances systemic anti-tumor immunity induced in draining lymph nodes (dLNs) (Ghiringhelli et al., 2009). Similarly, HMGB1 released from anthracycline-treated dying tumor cells has robust adjuvant-like effects achieved by stimulating toll-like receptor (TLR) 4 and inducing maturation and antigen processing by DCs (Fucikova et al., 2011; Hato et al., 2014). Furthermore, injection of doxorubicin-killed tumor cells or intra-tumoral injection of doxorubicin suppressed tumor growth in vivo (Casares et al., 2005). In these cases, induction of anti-tumor CD8+ T cells by DCs that migrated from tumor sites to dLNs is crucial for anti-tumor immunity (Durgeau et al., 2018). Both direct and indirect presentation pathways have been reported to play a substantial role in CD8+ T cell priming in dLNs. Direct priming involves phagocytosis of spontaneously generated dying tumor cells by tumor-infiltrating (Ti)-DCs, followed by migration to dLNs and subsequent presentation of tumor antigens to CD8+ T cells (Abbas et al., 2015; Sadozai et al., 2017). Migratory CD103+ DCs have been reported to play a critical role in inducing anti-tumor CD8+ T cell immune responses (Roberts et al., 2016; Salmon et al., 2016) by shuttling tumor antigens to dLNs and directly presenting antigen-derived peptides with major histocompatibility complex (MHC) class I molecules to stimulate CD8+ T cells. On the other hand, indirect priming involves CD8α+ lymph node-resident DCs (LNDCs), which receive tumor antigens from migratory CD103+ and CD103– CD11b+ DCs (cDC2) (Roberts et al., 2016) and then present tumor antigens to CD8+ T cells. Thus, recruitment to tumors and subsequent emigration of immune cells, especially CD103+ and CD103– Ti-DCs, from tumors to dLNs are crucial steps for effective anti-tumor immunity. CD103– DCs (cDC2) can also promote tumor growth regression by inducing Th17 responses (Laoui et al., 2016). Therefore, both CD103+ Ti-DCs and CD103– Ti-DCs have important roles in anti-tumor immunity and possess different functions and need to be analyzed separately.
Notably, other DC subsets such as CD11c+CD11b+Ly6Chi monocyte-derived inflammatory dendritic cells (MoDCs) act in situ (within tumors) to mediate ICD-induced inhibition of primary tumor growth (Ma et al., 2013). ATP released from killed tumor cells induces intratumoral recruitment and differentiation of MoDCs. They then phagocytose dying tumor cells and promote antigen presentation within tumors.
Adjuvants, such as TLR ligands, CpG oligodeoxynucleotide (Pashenkov et al., 2006), and lipopolysaccharide (LPS) (Shetab Boushehri et al., 2018) have been utilized to enhance anti-tumor immunity. However, given that TLR ligand stimulation induces DC maturation, it is unclear what effect TLR ligands in combination with ICD have on maturation and migration of Ti-DCs’ subsequent stimulation of anti-tumor T cell responses.
Tumor-infiltrating effector CD8+ T cells can be subdivided into 3 populations based on expression of cell surface molecules KLRG1 and CD127. KLRG1– CD127+ memory precursor effector cells (MPECs) possess similar effector functions to KLRG1+ CD127– short-lived effector cells (SLECs), but persist for longer than SLECs. Thus, MPECs would provide a more sustained anti-tumor response (Joshi et al., 2007). In addition, KLRG1+ CD127+ double-positive effector cells (DPECs) have been reported to provide strong anti-tumor immunity and promote tumor growth control in B16 melanoma (Kobayashi et al., 2015). Consistent with an important role for DPECs, treatment with agonistic antibody against 4-1BB, a co-stimulatory molecule expressed on activated T cells, led to increased DPECs and enhanced anti-tumor immunity (Kobayashi et al., 2015). Thus, elucidating the effect of ICD on generation of effector and memory CD8+ T cells is crucial to understanding how ICD affects anti-tumor immunity.
Despite the importance of Ti-DC migration for the anti-tumor response, it is not clear how this migration is affected by ICD. Specifically, we currently lack quantitative and qualitative information about influx and retention of CD103+ and CD103– Ti-DCs within tumors and their emigration to dLN both in the steady state and following ICD in situ. Furthermore, it is unknown how molecules secreted by immunogenic dying tumor cells regulate migration and function of CD103+ and CD103– Ti-DCs. Understanding how ICD regulates endogenous CD103+ and CD103– Ti-DC and CD8+ T cell migration on its own and in combination with adjuvants will provide insights into the mechanism of ICD-elicited anti-tumor immunity.
One of the confounding factors in uncovering the effect of ICD on the immune response is that chemotherapeutic drugs are immunosuppressive making it difficult to delineate the effects of ICD and chemotherapy on hematopoietic cells. To circumvent this limitation, we established a system where adenocarcinoma cells were engineered to express human diphtheria toxin (DT) receptor (DTR) (Saito et al., 2001) making it possible to induce ICD and generate tumor cell antigens in situ while avoiding immunosuppressive effects of chemotherapy. To monitor immune cell emigration from tumors quantitatively, we utilized mice expressing a photoconvertible protein KikGR to label and track tumor-infiltrating cells (Tomura et al., 2014; Torcellan et al., 2017). Here we implanted modified adenocarcinoma cells in mice expressing a photoconvertible protein and elucidated the effect of ICD on endogenous Ti-DC dynamics in tumor and migration and function, as well as tumor antigen-specific T cell response and anti-tumor immunity.
Results
Immunogenic tumor cell death induction in situ enhances Ti-DC phagocytosis of dying tumor cells
Although immunization with killed tumor cells can enhance host tumor immunity (Apetoh et al., 2007; Asano et al., 2011), how this process affects endogenous Ti-DC dynamics and subsequent anti-tumor immune response is not known due to the difficulty in analyzing these cells in situ in intact tumors. To address this issue, we established a model for inducing and visualizing tumor cell death. We generated colon adenocarcinoma MC38 cell line expressing (1) membrane-bound ovalbumin by fusing it to transferrin receptor (TfROVA) (Teasdale et al., 1992), (2) DTR for tumor cell death induction (Saito et al., 2001), and (3) nuclei-localized red fluorescent protein tandem Keima (Kogure et al., 2006) for visualizing tumor cell nuclei (MC38/TfROVA-DTR). First, we examined whether DT-induced cell death demonstrated typical signs of ICD. Cultured MC38/TfROVA-DTR cells were treated with DT to induce cell death. We then analyzed the supernatants and detected increased CRT expression as well as higher concentration of HMGB1 and ATP (Figures 1A and S1A). Similarly, DT treatment increased ATP concentration in cell culture supernatants from DTR-expressing NIH/3T3 and 3LL cell lines (Figure S1B). These results indicate that DT-induced cell death is ICD.
Figure 1.

Induction of immunogenic tumor cell death enhances Ti-DC phagocytosis
(A) MC38/TfROVA-DTR tumors were treated with DT or vehicle. Proportions of CRT+PI– cells (left) and concentration of HMGB-1 (center) and ATP (right) in culture supplement are shown. Bar graphs show means ± SEM of pooled data from two independent experiments (CRT: n = 7, HMGB-1: n = 4, ATP: n = 6). ∗p < 0.05, ∗∗p < 0.01 (Mann-Whitney U test).
(B and C) MC38/TfROVA-DTR tumor-bearing CD11c-YFP mice 24 h after DT or vehicle treatment. Two-photon microscopic images of tumors (B); the number of Keima+CD45– cells in tumors was calculated using data obtained from flow cytometric analysis (C). Scale bars, 50 μm. Bar graph shows means ± SEM of pooled data from two independent experiments (n = 8). ∗∗∗p < 0.001 (Mann-Whitney U test).
(D and E) MC38/TfROVA-DTR tumor-bearing XCR1-Venus mice 24 h after DT or vehicle treatment. Fluorescence microscopic images (D), the number of Venus+ MHC II+ CD103+ cells in tumors (left), and Keima+ Venus+ MHC II+ CD103+ cells in tumors (right) (E). Scale bars, 50 μm, Bar graphs show means ± SEM of pooled data from two independent experiments (n = 6−7). ∗p < 0.05, ∗∗∗p < 0.001 (Mann-Whitney U test).
Next, we determined whether DT injection induces death of MC38/TfROVA-DTR cells and phagocytosis of dying tumor cells by tumor-infiltrating immune cells in vivo. MC38/TfROVA-DTR cells were inoculated in CD11c-YFP reporter mice in which CD11c+ cells express YFP (Lindquist et al., 2004). Five days following tumor cell inoculation, YFP+ cells were interspersed among Keima+ tumor cells, but only a few YFP+ cells phagocytosed tumor cells (Figure 1B). However, 24 h after induction of ICD by intraperitoneal injection of DT, the number of live tumor cells in treated tumors decreased to approximately 25% of the starting number (Figure 1C). Furthermore, we observed fragmented Keima+ nuclei, some of which were contained inside of YFP+ cells suggesting that tumor-infiltrating immune cells phagocytosed tumor cell debris (Figure 1B). As CD11c can be expressed by several subsets of myeloid cells in tumors (Ma et al., 2013), we identified YFP+ cell subsets in tumors in CD11c-YFP mice. YFP+ cells in tumors consisted of approximately 50% MoDCs, 25% CD11c+ macrophages, less than 10% CD103– Ti-DCs, and less than 1% CD103+ Ti-DCs (Figure S2A).
It is thought that CD103+ Ti-DCs phagocytose dying tumor cells and mature and migrate to dLNs (Böttcher and Reis e Sousa, 2018). Previous studies reported that CD103+ DCs are localized at edge of the tumor mass (Broz et al., 2014); however it is unclear where CD103+ DCs acquire tumor antigen. To shed light on this and demonstrate that ICD increases phagocytosis by CD103+ DCs in tumors, we analyzed the effect of ICD on CD103+ DC localization and phagocytosis using XCR1-Venus reporter mice that express Venus fluorescent protein selectively in CD103+ DCs and CD8α+ LNDCs (Yamazaki et al., 2013). Consistent with previous reports (Broz et al., 2014), 5 days after MC38/TfROVA-DTR inoculation, we observed Venus+ cells only at the edge of the tumor (Figure 1D). However, 24 h after ICD induction, we observed Venus+ cells containing fragmented Keima+ nuclei that have infiltrated the tumor mass (Figure 1D). Furthermore, Venus+ cells were decreased, whereas Keima+ Venus+ cells tended to increase (Figures 1E and S2C). Thus, these results suggest that ICD induces CD103+ Ti-DC penetration of tumors, which may promote DC phagocytosis of dying tumor cells.
Immunogenic tumor cell death induction decreases the number of tumor-remaining migratory Ti-DCs
To analyze Ti-DC kinetics in tumors, we utilized photoconvertible KikGR mice. KikGR protein is initially green (KikGR-Green), but can be irreversibly photoconverted to red (KikGR-Red) by exposure to violet light (Tomura et al., 2014). Thus, after photolabeling cells within tumors and analyzing cellular content at specific time points by flow cytometry, we can distinguish cells that have been retained in tumors following photoconversion from cells that recently entered tumors and track cell migration of photoconverted cells to other anatomical sites. Tumor-infiltrating cells in KikGR mice inoculated with MC38/TfROVA-DTR cells 5 days earlier were photoconverted and the number of photoconverted, and non-photoconverted cells per gram of tumor was assessed 24 h later (Figures 2A and 2B).
Figure 2.

Immunogenic tumor cell death decreases the number of CD103+ and CD103– Ti-DCs retained in tumors
(A) Experimental setup for the analysis of tumor DC turnover and migration to dLNs. Tumor-infiltrating KikGR+ cells were photoconverted by exposure to violet light, and at the same time, tumor cell death was induced by DT administration. Cells in tumors and dLNs were analyzed 24 h later.
(B) KikGR cells were detected in photoconverted and non-photocoverted tumors and inguinal dLNs in KikGR mice immediately after photoconversion. Data are representative of at least three independent experiments.
(C) Number of total and KikGR-Red MoDCs, F4/80+CD11c+ macrophage, neutrophil, CD103− Ti-DCs, CD103+ Ti-DCs, and CD103– DCs in MC38/TfROVA-DTR tumors in KikGR mice 24 h following photoconversion and vehicle or DT treatment. Bar graph shows means ± SEM of pooled data from two independent experiments (n = 8). Numbers in bar graphs indicate frequencies of KikGR-Red population (%) in each subset. ∗p < 0.05 (Mann-Whitney U test).
In the absence of ICD, 24 h after photoconversion, the proportion of KikGR-Red myeloid cells retained in tumors varied by subsets: MoDCs (14%), F4/80+CD11c+ macrophage (30%), neutrophil (35%), CD103−Ti-DCs (29%), and CD103+ Ti-DCs (39%) (Figure 2C and S3). This indicates that a large proportion of cells in each subset entered the tumors during the previous 24 h. When ICD was induced and tumors were photoconverted, the number of total and KikGR-Red tumors retained MoDCs, F4/80+CD11c+ macrophages, and neutrophils was not altered, suggesting that ICD had limited effect on the kinetics of these subsets (Figure 2C). On the other hand, although ICD did not decrease the total number of CD103− DCs, it did lead to a reduction in the number of KikGR-Red CD103− DCs. Furthermore, ICD decreased both the number of total and KikGR-Red CD103+ Ti-DCs (Figure 2C). These results suggest that ICD induction has a greater impact on the kinetics of Ti-DCs than other immune subsets.
Immunogenic tumor cell death stimulates Ti-DC migration to dLN
We next examined whether ICD can enhance Ti-DC migration to dLN. As shown in Figure 2B, no KikGR-Red cells were detected in inguinal dLN immediately after photoconversion, indicating that photoconversion was restricted to the tumor mass. Twenty-four hours following photoconversion, we detected KikGR-Red cells among migratory DCs (CD11cint MHC class IIhigh), but not among LNDCs (CD11chigh MHC class IIint) (Figures 3A and S4A).
Figure 3.

Tumor cell death enhances Ti-DC migration to dLNs
(A) Representative flow cytometry data of KikGR signals in CD11chighMHC class IIint LNDCs and CD11c+MHC class IIhigh migratory DCs in the dLN of KikGR mice 24 h after photoconversion (left). Proportions of KikGR-Red cells in each DC subset are shown (right). Bar graph shows means ± SEM of pooled data from four independent experiments (n = 8). ∗∗p < 0.01 (Mann-Whitney U test).
(B) Representative fluorescence intensity graph of CellTrace Violet in OT-I T cells co-cultured for 72 h with sorted DC subsets from dLNs of tumor-bearing KikGR mice 24 h after photoconversion. Numbers in histogram plots indicate proportion of proliferating cells identified by CellTrace Violet dilution (left). Bar graph shows summarized data of proportion of proliferating OT-I T cells (right). Bar graph shows means ± SEM of pooled data from two independent experiments (n = 3). ∗∗p < 0.01, ∗∗∗p < 0.001 (one-way ANOVA with Holm-Sidak's multiple comparisons test).
(C) Number of KikGR-Red cells in MHC class IIhigh total migratory DCs and DC subsets in dLNs from vehicle or DT-treated tumor-bearing KikGR mice at indicated time points following photoconversion. Bar graph shows means ± SEM of pooled data from three (24, 48 h) or one (72 h) independent experiment (n = 2−8). ∗∗p < 0.01 (Mann-Whitney U test).
To confirm that Ti-DCs that migrated to dLNs have the capacity to induce tumor antigen-specific T cell responses, KikGR-Red and KikGR-Green migratory DCs and LNDCs were purified from dLNs and mixed with OVA-specific OT-I T cells that were labeled with CellTrace Violet to track cell division. We found that KikGR-Red migratory DCs strongly induced proliferation of tumor-specific T cells when compared with KikGR-Green migratory DCs. LNDCs did not induce T cell proliferation (Figure 3B). These results suggest that migratory Ti-DCs are the main tumor-antigen-presenting DC subset in this model.
As migratory DCs in LNs consist of CD103+, CD103–, and CD326+ (classified as Langerhans cells [Henri et al., 2010]) subsets (Tomura et al., 2014), we analyzed the expression of these markers by DCs 24 h after photoconversion to identify which subsets emigrated from photoconverted tumors. Our analysis detected KikGR-Red cells in all migratory DC subsets, and CD103– DCs were the major subset of migratory Ti-DCs (Figure S4B). The highest number of tumor-emigrating DCs in all subsets was detected in dLNs at 48 h following tumor photoconversion (Figure 3C). Notably, induction of ICD substantially increased the number of KikGR-Red Ti-DCs of each phenotype found in dLNs after 24 h. To confirm that this effect is not restricted to a specific cancer model, we demonstrated that DT-induced cell death increased CD103+ and CD103– Ti-DC migration to dLNs from 3LL-DTR tumor in KikGR mice 24 h following photoconversion and DT treatment (Figure S5). These results suggest that ICD stimulates Ti-DC egress from tumors leading to rapid relocation of a large number of Ti-DCs to dLNs. Furthermore, this provides direct evidence of CD103+ Ti-DC migration from tumors to dLNs. This is particularly significant because this subset has been shown to play a dominant role in antigen trafficking to dLNs and subsequent presentation of tumor antigens to CD8+ T cells (Roberts et al., 2016).
Mechanism of immunogenic tumor cell death-induced DC migration
In ICD, ATP, HMGB1, and other DAMPs molecules released from dying cells stimulate the immune response (Krysko et al., 2012). ATP released from dying tumor cells interacts with Ti-DCs via one of the ATP receptors, P2X7R, and enhances anti-tumor immunity (Ghiringhelli et al., 2009). HMGB1 activates immune cells by binding to TLR4 (Apetoh et al., 2007). We hypothesized that DAMPs may promote tumor immunity by enhancing Ti-DC migration to dLNs. Therefore, we used MC38/TfROVA-DTR to test this hypothesis by examining whether Ti-DC migration was modulated via HMGB1-TLR4 and/or ATP-P2X7R pathways. When tumors were photoconverted and ICD was induced, mice were simultaneously injected peritoneally with P2X7R antagonists, oxidized ATP (oATP) (Mutini et al., 1999; Zhao et al., 2016) or A-740003 (De Marchi et al., 2019). KikGR-Red migratory DCs were analyzed 24 h later (Figure S6). We found that injection of both antagonists counteracted ICD-induced increase in KikGR-Red total migratory, CD103+ DCs, and CD103– DCs in dLNs, reducing migration to levels similar to steady-state migration. Glycyrrhizin (Gly), blocker of HMGB1 binding to TLR4 (Mollica et al., 2007; Yan et al., 2017), inhibited migration of KikGR-Red total migratory and CD103+ Ti-DCs and tended to inhibit that of CD103 DCs. In addition, anti-HMGB1 antibody significantly inhibited migration of KikGR-Red total migratory, CD103+, and CD103– Ti-DCs (Figure 4). These results suggested that enhanced CD103+ and CD103– Ti-DC migration to dLNs elicited by ICD utilizes P2X7R and HMGB1 signaling pathways.
Figure 4.

Tumor cell death enhanced CD103+ Ti-DC migration to dLNs is inhibited by ATP-P2X7R, HMGB1-TLR4, Gαi-protein, and S1PR1 pathway blockade
Inhibitors were administrated together with DT at the time of tumor photoconversion and analyzed 24 h later. A number of KikGR-Red cells in total migratory DCs and DC subsets in dLNs. Bar graph shows means ± SEM of pooled data from at least two independent experiments (n = 8-16). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (one-way ANOVA with Holm-Sidak's multiple comparisons test).
Steady-state Ti-DC migration is dependent on Gαi-protein-coupled chemokine receptor, CCR7, and thus can be blocked by an inhibitor of Gαi signaling, pertussis toxin (PTX) (Roberts et al., 2016). We examined whether enhanced Ti-DC migration after ICD induction is similarly suppressed by PTX treatment. PTX injection before tumor photoconversion and ICD induction significantly reduced KikGR-Red total migratory and CD103+ and CD103– Ti-DC egress to dLNs, to a lower level than migration in the non-DT-treated group (Figure 4). This is probably due to the inhibitory effect of PTX on DC migration in steady state as well as on ICD-induced Ti-DC migration.
T cell migration from normal and inflamed peripheral tissues utilizes S1P-S1PR1 pathway (Ledgerwood et al., 2008; Nakanishi et al., 2017; Pappu et al., 2007; Tomura et al., 2010). However, the contribution of this pathway to DC egress from tissues is controversial (Lan et al., 2005). Furthermore, it is not known whether S1P-S1PR1 pathway contributes to Ti-DC egress after ICD induction. To test this, we administered FTY-720, which blocks the S1P-S1PR1 pathway. This treatment inhibited migration of total migratory DCs, CD103+, and CD103– Ti-DCs to dLNs (Figure 4). These results suggested that enhanced migration of Ti-DCs after ICD induction is chemokine signaling dependent, and CD103+ DCs use S1P1 pathway to egress tumors. Notably, CD103+ DCs were more sensitive to FTY-720 inhibition than CD103– DCs, suggesting that CD103– DCs use additional signaling pathways to egress tumors.
Partial tumor cell death accelerates Ti-DC migration from tumors to dLNs
Our results indicate that inducing tumor cell death in a large proportion of tumor cells accelerates Ti-DC turnover within tumors and migration to dLNs. However, anti-tumor treatments, such as chemotherapy, do not typically kill all tumor cells. Thus, we next elucidated the effect of ICD on anti-tumor responses in a situation where most of the tumor cells survive and tumor mass remains. To do this we examined whether a lower proportion of killed tumor cells could also enhance myeloid cell phagocytosis and Ti-DC migration to dLNs. A mixture of MC38/TfROVA-DTR and MC38 cells (mixed in the 3:7 ratio) was inoculated in CD11c-YFP mice as in Figure 1B. Similarly to our earlier findings, YFP+ cells infiltrated tumors, but almost no YFP+ cells phagocytosed Keima-labeled tumor cells. However, after partial ICD was induced, YFP+ cells phagocytosed dying tumor cells (Figure 5A). DT administration in this system led to death of more than 85% of Keima+ MC38/TfROVA-DTR tumor cells (Figure 5B).
Figure 5.

Partial tumor cell death induction accelerates Ti-DC migration
Tumors consisted of MC38/TfROVA-DTR and MC38 cells (mixed in the 3:7 ratio).
(A and B) Organs from CD11c-YFP mice bearing mixed tumors were analyzed 24 h after DT or vehicle treatment. Fluorescent microscopic images of tumors (A), number of Keima+CD45– cells (B). While arrows indicate YFP+ cells that phagocytosed dying tumor cells. Scale bar, 50 μm. Bar graph shows means ± SEM of pooled data from two independent experiments (n = 6−7). ∗∗∗p < 0.001 (Mann-Whitney U test).
(C) Number of KikGR-Red cells in MHC class IIhigh total migratory DCs and DC subsets in dLNs from vehicle or DT-treated KikGR mice bearing mixed tumors at indicated time points following photoconversion. Bar graphs show means ± SEM of pooled data from two independent experiments (n = 7−10). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (Mann-Whitney U test).
To analyze Ti-DC dynamics in tumors, KikGR mice were inoculated with a mixture of MC38/TfROVA-DTR and MC38 cells (3:7 ratio), and tumors were photoconverted as in Figure 2A. Time course study revealed that ICD induction modestly, but significantly, enhanced migration of total migratory DC and all Ti-DC subsets to the dLN (Figure 5C).
Immunogenic tumor cell death enhances tumor antigen-specific T cell proliferation
TLR ligands have been utilized as adjuvants to enhance immune responses. TLR4 ligand, LPS, induces DC maturation (Castiello et al., 2011). Furthermore, LPS analogs have been developed for use as adjuvants to enhance anti-tumor immune responses (Shetab Boushehri et al., 2018). As shown earlier, endogenous TLR4 ligand, HMGB-1 released from dying tumor cells induced Ti-DC migration. Although, LPS is not released by dying tumor cells, it is important to study how this adjuvant affects DC migration and function in relation to ICD, because multiple studies have reported conflicting roles of LPS in modulating DC function (Klaska et al., 2017; Shen et al., 2008). To address these issues, we first examined the effect on Ti-DC migration in our system. Tumors grown from MC38/TfROVA-DTR and MC38 cells (mixed in the 3:7 ratio) in KikGR mice were photoconverted and LPS was injected directly into these tumors. Twenty-four hours later, we observed that the number of KikGR-Red total migratory Ti-DCs and CD103– Ti-DCs in dLNs was increased to a much greater extent by LPS treatment than by ICD induction. Furthermore, DC migration was further enhanced by combining LPS treatment with ICD induction (Figure 6A).
Figure 6.

Ti-DC migration and antigen-specific CD8+ T cell proliferation after tumor cell death induction and LPS treatment
DT (intraperitoneal) and/or LPS (intratumoral) were administered to KikGR mice bearing mixed MC38/TfROVA-DTR and MC38 tumors (3:7 ratio) at time of photoconversion.
(A) Number of KikGR-Red cells in DCs in the dLNs from vehicle, DT-, and/or LPS-treated KikGR mice 24 h after tumor photoconversion. Bar graphs show means ± SEM of pooled data from at least nine independent experiments (n = 18−20). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (Kruskal-Wallis test with Dunn's comparisons test).
(B) Representative fluorescent intensity graph of CellTrace Violet of OT-I T cells, the proportion of proliferating OT-I T cells among CD8+ T cells, and the cell number of proliferating OT-I T cells in the dLNs 45 h after transfer to tumor bearing mice 24 h after vehicle, DT, and/or LPS treatment. Bar graphs show means ± SEM of pooled data from at least four independent experiments (n = 7−11). ∗p < 0.05. (Kruskal-Wallis test with Dunn's comparisons test).
We also observed that expression of costimulatory molecules CD80 and CD86 was the lowest in CD8α+ LNDCs and highest in KikGR-Red migratory CD103+ and CD103– Ti-DCs regardless of treatment. In addition, CD86 expression in KikGR-Red cells was further upregulated after LPS injection compared with ICD induction alone. In contrast, whereas MHC class I expression was lower in CD8α+ LNDCs and higher in CD103+ and CD103– DCs, its levels were not altered after LPS injection (Figure S7).
We next examined whether enhanced Ti-DC migration together with increased MHC class I and co-stimulatory molecule expression on Ti-DCs led to augmented proliferation of tumor-specific T cells in vivo. CellTrace Violet-labeled OVA-specific OT-I T cells were adoptively transferred into MC38/TfROVA-DTRs and MC38 tumor-bearing mice and analyzed two days later. We observed that ICD induction, but not LPS, increased CD8+ T cell proliferation (Figure 6B). Furthermore, combining LPS with ICD tended to reduce the number of proliferating tumor-specific CD8+ T cells compared with ICD induction alone. These results suggest that ICD can enhance anti-tumor CD8+ T cell proliferation in vivo. However, LPS-induced maturation and migration of Ti-DCs did not boost this further but may have inhibited anti-tumor T cell responses induced by ICD.
Immunogenic tumor cell death promotes antigen-specific CD8+ effector T cell responses in dLNs and at distal tumor sites
Increased tumor-antigen-specific T cell proliferation after ICD induction in dLNs prompted us to investigate the effect of ICD on distal tumors and systemic immunity. At first, we analyzed endogenous tumor-antigen-specific T cells following ICD. Wild-type mice were inoculated with two tumors: MC38/TfROVA-DTR and MC38 cells (injected in the 3:7 ratio), and a distal secondary tumor, B16 melanoma cells expressing the same antigen (ovalbumin, B16-OVA). Five days later ICD was induced in the MC38 tumor (Figure 7A) and OVA-specific endogenous T cells were detected with H-2Kb OVA tetramer-SIINFEKL. ICD induction led to a greater number of OVA-specific T cells in B16-OVA tumors and draining axillary LNs 2 and 10 days after ICD induction compared with untreated animals (Figure 7B). Next, we elucidated the effect of ICD on CD8+ T cell effector/memory subsets 2 days after ICD induction (Figures 7C and S8). We observed that more MPECs were present in B16-OVA tumors after ICD induction, whereas DPECs and MPECs were significantly elevated in B16-OVA dLNs. These results indicate that inducing ICD can promote anti-tumor T cell responses.
Figure 7.

Partial tumor cell death induction increases tumor antigen-specific effector T cells
(A) Experimental setup for the analysis of endogenous tumor-antigen specific T cells after partial tumor cell death induction. Mixture of MC38/TfROVA-DTR and MC38 cells (3:7 ratio) was intradermally inoculated into abdominal skin, and B16-OVA tumor cells were subcutaneously inoculated into flank skin of C57BL/6 mice 5 days before tumor cell death induction. Tumor cell death was induced at day 0. Two, five, and ten days later, endogenous tumor-antigen-specific T cells were analyzed.
(B) Number of OVA tetramer+ CD8+ T cells at indicated time points following DT or vehicle treatment. Line graphs show means ± SEM of pooled data from at least two independent experiments (n = 8−12). ∗p < 0.05 (Mann-Whitney U test).
(C) Number of DPEC, MPEC, and SLEC in OVA tetramer+ CD8+ T cells 2 days following vehicle or DT treatment. Bar graphs show means ± SEM of pooled data from two independent experiments (n = 8). As values of two samples of DEPCs in axillary LN from Vehicle group and 1 sample of MEPCs in Tumor from Vehicle and DT groups were zero, these data were not shown in these figures. ∗p < 0.05, ∗∗p < 0.01 (Mann-Whitney U test).
Inducing immunogenic tumor cell death in primary tumors leads to the suppression of secondary tumor growth in a CD103+ DC-dependent manner
We hypothesized that the increase in tumor antigen-specific T cells in secondary tumors following primary tumor cell death induction may affect secondary tumor growth. Thus, we evaluated anti-tumor immunity after ICD induction, LPS injection, and combination of both treatments by quantitating growth of secondary B16-OVA tumors. Mice were injected with a mixture of MC38/TfROVA-DTR and MC38 cells (3:7 ratio) and B16-OVA cells (as in Figure 7A) and 5 days later treated with DT, LPS, or DT and LPS as previously. We observed that partial ICD significantly suppressed B16-OVA growth, whereas treatment with LPS alone or in combination with ICD had only a slight effect on tumor growth (Figure 8A). The observed effect was not due to the direct suppression of B16-OVA growth by DT because B16-OVA growth was not altered in mice without MC38/TfROVA-DTRs regardless of DT treatment (Figure 8B). It was also not due to an abscopal effect because growth of B16 tumors not expressing OVA was not altered with or without partial cell death of MC38/TfROVA-DTRs (Figure 8C). These results suggested that consistent with increased tumor antigen-specific T cell proliferation (Figure 6B), inducing ICD leads to enhanced anti-tumor immunity. In contrast to ICD induction alone, LPS-induced Ti-DC maturation was less effective in controlling tumor growth. Furthermore, addition of LPS reduced the effect of ICD.
Figure 8.

Tumor cell death induction suppresses secondary tumor growth in CD103+ DCs and cell migration-dependent manner
(A, D, and E) A mixture of MC38/TfROVA-DTR and MC38 cells (3:7 ratio) and B16-OVA tumor cells were intradermally and subcutaneously inoculated into abdominal and flank skin of C57BL/6 mice (A and D), or XCR1-DTR+/– or XCR1-DTR–/– wild type littermate mice (E), respectively.
(B) MC38 and B16-OVA tumor cells were intradermally and subcutaneously inoculated into abdominal and flank skin of C57BL/6 mice, respectively.
(C) A mixture of MC38/TfROVA-DTR and MC38 cells (3:7 ratio) and B16 tumor cells were intradermally and subcutaneously inoculated into abdominal and flank skin of C57BL/6 mice, respectively. Five days following tumor inoculation, tumor-bearing mice were treated with vehicle, LPS, DT, and LPS + DT (A), vehicle and DT (B, C, and E), PTX, DT, and PTX + DT (D). Line graphs show means ± SEM of pooled data of B16-OVA (A, B, D, E) or B16 (C) volumes obtained in a representative experiment (A: n = 6−8, B: n = 5 or 6, C: n = 12, D: n = 8−10, E: n = 6−12). All experiments were performed at least two times. v.s. Vehicle (A, E) or PTX (D) ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; v.s. DT + LPS (A), DT + PTX (D), or DT-treated XCR1-DTR+/– (E); †p < 0.05, ††p < 0.01, †††p < 0.001 (two-way ANOVA with Sidak's multiple comparisons test).
As Ti-DC emigration to dLNs was Gαi dependent (Figure 4), we examined whether inhibiting this migration will abrogate increased anti-tumor responses following ICD induction. To test this, we injected PTX at the same time as ICD was induced with DT (DT + PTX). Tumors grew significantly faster in mice treated with PTX even when ICD was induced with DT compared with mice that were treated with DT alone (Figure 8D). These results suggested that Ti-DC migration to dLNs is important for enhancing tumor immunity following ICD.
However, as PTX blocks migration of other immune cells as well as DCs, we looked for a specific contribution of CD103+ DCs at the time of ICD to improve anti-tumor immune response following tumor cell death induction. We generated XCR1-DTR+/– mice, where XCR1+ cells express DTR, and thus a single DT injection transiently depletes CD103+ migratory DCs and CD8α+ LNDCs (Yamazaki et al., 2013). By concurrent ICD induction and transient removal of CD103+ DCs, we investigated the importance of CD103+ DCs during ICD induction. XCR1-DTR+/– or XCR1-DTR–/– wild type mice were inoculated with a mixture of MC38/TfROVA-DTRs and MC38 cells and B16-OVA as previously. Five days later tumor-bearing mice were treated with DT or vehicle. Compared with XCR1-DTR+/− control mice that received vehicle treatment (ICD(−),CD103+ DC non-depleted), growth of B16-OVA tumors was strongly inhibited in XCR1-DTR–/– mice treated with DT (ICD(+), CD103+DC non-depleted). However, transient depletion of CD103+ DCs in XCR1-DTR+/− mice following DT administration (ICD(+), CD103+ DC depleted) attenuated the tumor growth inhibitory effect of ICD to a level close to that of XCR1-DTR+/- vehicle treatment control mice (Figure 8E). These results suggested that CD103+ DCs are necessary at the time of ICD induction to improve systemic anti-tumor immune responses following ICD.
Discussion
In this study, by combining in vivo ICD induction via DT treatment with intravital photolabeling in KikGR mice, we have established a system that allowed us to exclude the confounding effects of chemo- and radiotherapy and quantify cell migration during ICD induction. Taking advantage of this system, we examined kinetics of Ti-DC and other myeloid cell turnover in tumors, characterized retention and egress of different subsets in response to ICD, and showed that DCs were more sensitive to ICD enhancement compared with other myeloid cells. Furthermore, we show that inducing ICD in situ accelerated CD103+ Ti-DC phagocytosis, turnover within tumors, and emigration to dLNs. This led to increased tumor antigen-specific CD8+ T cells and suppression of secondary tumor growth. DAMPs molecules released by dying tumor cells, including ATP and HMGB1, accelerated CD103+ and CD103– Ti-DC migration from tumors to dLNs via P2X7R and TLR4 signaling pathways. However, combining tumor cell death induction with LPS treatment stimulated Ti-DC maturation and emigration to dLNs but reduced the effect of tumor cell death-induced tumor growth suppression. Finally, we demonstrate that immune cell migration at the time of ICD induction is crucial for tumor cell death-induced suppression of tumor growth. Furthermore, our results add to a substantial body of literature on ICD in tumors and suggest that our findings about the effect of ICD on myeloid cell turnover, migration, and function are useful in understanding tumor immunity where ICD is induced by chemo- or radiotherapy.
Using tumor-bearing KikGR mice, we showed that only a small proportion of MoDCs remains in the tumors after 24 h, regardless of ICD induction. This indicated that the majority of MoDCs in tumors turnover within 24 h and the effect of ICD on MoDC dynamics is limited. Future studies will identify the exact fate of Ti-MoDCs: whether they die in situ and/or migrate to dLNs. In contrast to MoDCs, ICD reduced the number of KikGR-Red CD103+ and CD103− Ti-DCs remaining in tumors after 24 h. Concomitant with this, the number of KikGR-Red DCs in dLNs peaked early and started to decline by 48 h following tumor photoconversion and ICD induction. Notably, KikGR-Red Ti-DCs did not accumulate in dLNs. We previously reported that the lifetime of skin migratory DC subsets in dLNs is less than 1 day in the tape-stripping model of inflammation and skin migratory DCs do not egress the dLN (Tomura et al., 2014). It is plausible that the lifetime of Ti-DCs is similarly short in the inflammatory milieu of the tumor microenvironment and Ti-DCs that migrated to the dLN would then die in situ without egressing the dLN.
In addition, we analyzed phenotypes of Ti-DCs in dLN by comparing KikGR-Red migratory DCs that emigrated from tumors to KikGR-Green migratory DCs that either migrated from tumors before photoconversion or came to dLNs from other sites. Tumor-derived migratory DCs in dLNs expressed higher level of co-stimulatory molecules and had a higher capacity to induce proliferation of naive antigen-specific CD8+ T cells compared with that of KikGR-Green migratory DCs. These results suggest that the tumor microenvironment, which includes inflammatory cytokines in the tumor parenchyma, may stimulate DC function.
We observed that a small number of CD103+ DCs were localized at the edge of solid tumors and almost no CD103+ DCs were observed within the tumor mass. This result is in line with an earlier report that CD103+ DCs are sparse in proximity to tumor margins but plentiful in distal regions in PYMT mice (Broz et al., 2014). However, after inducing ICD, CD103+ DCs accumulated inside the tumor and phagocytosed dying tumor cells. ATP accelerates DC migration (Sáez et al., 2017), whereas DAMPs signals promote opsonization or phagocytosis of dying cancer cells (Gardai et al., 2005; Garg et al., 2012a, 2012b). Thus, it is plausible that DAMPs molecules released from dying tumor cells induce DC accumulation within tumor and promote phagocytosis by CD103+ DCs.
PTX strongly inhibited CD103+ DC migration to dLNs. In contrast, blocking ATP-P2X7R or HMGB1-TLR4 pathways abrogated ICD induced increase in CD103+ DC migration but only to basal level (without induced tumor cell death). These results suggest that the molecular mechanisms of spontaneous Ti-DC migration are regulated through pathways additional to ATP-P2X7R or HMGB1-TLR4.
TLR4 ligand, LPS, stimulated DC maturation and migration as well as CD86 upregulation, but did not promote generation of antigen-specific CD8+ T cells in dLNs. It is possible that in the case of simultaneous ICD induction and LPS injection, Ti-DCs lose phagocytic activity due to maturation (Wilson et al., 2006) even in the presence of dying tumor cells. In addition to DC maturation, LPS induces DC apoptosis by NAFT activation through CD14 (Zanoni et al., 2009), suggesting that the strong stimulation of TLR4 by LPS accelerated Ti-DC death and shortened the antigen presentation period of DCs. These could lead to a lower efficiency of antigen presentation and antigen-specific T cell stimulation in dLNs and attenuation of ICD-induced tumor regression. Alternatively, LPS may counteract the anti-tumor effect of ICD by inducing IL-10 and DC tolerance (endotoxin-tolerized DCs) (Klaska et al., 2017). LPS can induce IL-10-secreting DCs that promote generation of CD62L-expressing Tregs and suppress experimental autoimmune uveoretinitis (Lau et al., 2008).
Inducing ICD in situ led to an increase in antigen-specific CD8+ T cells in secondary tumors. Specifically, we observed increased numbers of MPECs and DEPCs. MPECs contribute to long-term anti-tumor responses (Kobayashi et al., 2015). They phenotypically resemble SLECs and produce comparable level of IFN-γ (Kobayashi et al., 2015). In addition, it has been proposed that DPECs may represent a long-lived effector cell population with capacity for proliferation and potential to become memory cells (Steffensen et al., 2013). In fact, DEPCs were the major population in B16-OVA tumors in our model, and it is possible that the anti-tumor effect of ICD is mediated through enhanced MPEC and DPEC response. Increased number of these cells may lead to effective long-term anti-tumor immunity.
Indirect presentation pathways, where CD103– DCs carry the antigen to dLNs, play a role in priming CD8+ T cells. It has been reported that CD103– DCs (cDC2) have the highest Th17-inducing ability (Laoui et al., 2016) among DC subsets, and thus it is possible that CD103– DCs enhance anti-tumor immune response via Th17 generation. Th17 cells have also been reported to acquire MHC class I+ tumor antigen-derived peptides by trogocytosis and stimulating antigen-specific CD8+ T cells (Ankathatti Munegowda et al., 2011). Although the role of MoDCs in the ICD-induced suppression of distal tumor growth is unclear, an earlier study suggested that MoDCs rather than CD103+ DCs are responsible for the ICD-induced anti-cancer effects in the primary tumor (Ma et al., 2013). Thus, it is possible that enhanced CD103– DC migration to dLNs induced by ICD and MoDCs in primary tumors together contribute to increased systemic anti-tumor immunity. However, we think that, at least in our system, the contribution of non-CD103+ DC subsets, such as CD103– DCs and MoDCs, to systemic anti-tumor immune responses following ICD induction is limited. On the other hand, the requirement for CD103+ DCs at the time of ICD induction, coupled with a report that tumor growth suppression elicited by killed tumor cell immunization is completely abrogated in BATF3−/− mice, which lack CD103+ DCs (Jaime-Sanchez et al., 2020), suggests that CD103+ DCs are the main subset responsible for inhibition of secondary tumor growth. Although a specific tumor system was used in our study to demonstrate the effect of ICD on turnover and migration of antigen-presenting cells in cancer, our findings that CD103+ DCs migrating from the tumor to the dLN are important for enhancing the systemic anti-tumor immune response induced by ICD are applicable to other tumor settings where ICD is induced in situ by chemotherapy or radiotherapy.
We have demonstrated the effect of ICD on anti-tumor immune responses by eliminating the immunosuppressive effects of chemotherapy and irradiation. In clinic, attempts have been made to use dying immunogenic cells induced by chemotherapy or irradiation therapy to enhance anti-tumor immunity (Wang et al., 2018). However, rapid Ti-DC turnover in our tumor model suggests that continuous supply of DC precursors from the bone marrow is crucial for induction and maintenance of Ti-DCs involved in stimulating anti-tumor antigen-specific CD8+ T cell response. The supply of Ti-DC precursor cells may be affected by bone marrow suppression. Thus, anti-tumor therapy should aim to minimize bone marrow suppression to achieve enhancement of the anti-tumor response by ICD.
Limitations of the study
A potential caveat of this work is that the effects of ICD on anti-tumor responses with or without LPS treatment have been examined in vivo in one tumor model only.
Resource availability
Lead contact
Requests for further information and and reagents should be directed to and will be fulfilled by the lead contact, Michio Tomura (michio.tomura@gmail.com).
Materials availability
Materials generated in this study are available upon request.
Data and code availability
This study did not generate/analyze datasets or code.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
This work was supported in part by JSPS Grants-in-Aid for Scientific Research in Innovative Areas ‘‘Analysis and Synthesis of Multidimensional Immune Organ Network’’ (#24111007); JSPS Grants-in-Aid for Scientific Research (B) (#16H05087); Special Coordination Funds for Promoting Science and Technology of the Japanese Government; National Health and Medical Research Council Australia project grants GNT1106043 (T.C.), National Breast Cancer Foundation grant IIRS-19-027 (T.C.), Pankind Foundation grant (T.C.), Perpetual Impact grant (T.C.), and Astellas Pharma Inc. through the Formation of Innovation Centers for the Fusion of Advanced Technologies Program.
Author contributions
T.M., S.U., R.I, I.Y., Y.N., K.M., Y.K, T.C., and M.T. designed experiments. T.M., K.Kitagawa, Y.H., and M.U. performed experiments. H.H. and T.K. prepared XCR1-Venus mice and XCR1-DTR mice. T.H. and K.Kabashima prepared CD11c-YFP mice. T.M. and M.T. analyzed data. T.M., T.C., and M.T. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102424.
Supplemental information
References
- Abbas A., Lichtman A., Pillai S. International Edition, 8th. Elsevier Saunders; Philadelphia: 2015. Cellular and Molecular Immunology. [Google Scholar]
- Ankathatti Munegowda M., Deng Y., Mulligan S.J., Xiang J. Th17 and Th17-stimulated CD8+ T cells play a distinct role in Th17-induced preventive and therapeutic antitumor immunity. Cancer Immunol. Immunother. 2011;60:1473–1484. doi: 10.1007/s00262-011-1054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apetoh L., Ghiringhelli F., Tesniere A., Obeid M., Ortiz C., Criollo A., Mignot G., Maiuri M.C., Ullrich E., Saulnier P. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- Asano K., Nabeyama A., Miyake Y., Qiu C.-H., Kurita A., Tomura M., Kanagawa O., Fujii S., Tanaka M. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity. 2011;34:85–95. doi: 10.1016/j.immuni.2010.12.011. [DOI] [PubMed] [Google Scholar]
- Böttcher J.P., Reis e Sousa C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer. 2018 doi: 10.1016/j.trecan.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz M.L., Binnewies M., Boldajipour B., Nelson A.E., Pollack J.L., Erle D.J., Barczak A., Rosenblum M.D., Daud A., Barber D.L. Article dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26:638–652. doi: 10.1016/j.ccell.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casares N., Pequignot M.O., Tesniere A., Ghiringhelli F., Roux S., Chaput N., Schmitt E., Hamai A., Hervas-Stubbs S., Obeid M. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005;202:1691–1701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castiello L., Sabatino M., Jin P., Clayberger C., Marincola F.M., Krensky A.M., Stroncek D.F. Monocyte-derived DC maturation strategies and related pathways: a transcriptional view. Cancer Immunol. Immunother. 2011;60:457–466. doi: 10.1007/s00262-010-0954-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marchi E., Orioli E., Pegoraro A., Sangaletti S., Portararo P., Curti A., Colombo M.P., Di Virgilio F., Adinolfi E. The P2X7 receptor modulates immune cells infiltration, ectonucleotidases expression and extracellular ATP levels in the tumor microenvironment. Oncogene. 2019;38:3636–3650. doi: 10.1038/s41388-019-0684-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durgeau A., Virk Y., Corgnac S., Mami-Chouaib F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front. Immunol. 2018;9:14. doi: 10.3389/fimmu.2018.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fucikova J., Kralikova P., Fialova A., Brtnicky T., Rob L., Bartunkova J., Špíšek R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011;71:4821–4833. doi: 10.1158/0008-5472.CAN-11-0950. [DOI] [PubMed] [Google Scholar]
- Galluzzi L., Buqué A., Kepp O., Zitvogel L., Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017;17:97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- Gardai S.J., McPhillips K.A., Frasch S.C., Janssen W.J., Starefeldt A., Murphy-Ullrich J.E., Bratton D.L., Oldenborg P.A., Michalak M., Henson P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005 doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Garg A.D., Krysko D.V., Vandenabeele P., Agostinis P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol. Immunother. 2012;61:215–221. doi: 10.1007/s00262-011-1184-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A.D., Krysko D.V., Verfaillie T., Kaczmarek A., Ferreira G.B., Marysael T., Rubio N., Firczuk M., Mathieu C., Roebroek A.J.M. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012;31:1062–1079. doi: 10.1038/emboj.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiringhelli F., Apetoh L., Tesniere A., Aymeric L., Ma Y., Ortiz C., Vermaelen K., Panaretakis T., Mignot G., Ullrich E. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1Β-dependent adaptive immunity against tumors. Nat. Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- Hato S.V., Khong A., De Vries I.J.M., Lesterhuis W.J. Molecular Pathways : the immunogenic effects of platinum- based chemotherapeutics. Clin. Cancer Res. 2014 doi: 10.1158/1078-0432.CCR-13-3141. [DOI] [PubMed] [Google Scholar]
- Henri S., Poulin L.F., Tamoutounour S., Ardouin L., Guilliams M., De Bovis B., Devilard E., Viret C., Azukizawa H., Kissenpfennig A., Malissen B. CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J. Exp. Med. 2010;207:189–206. doi: 10.1084/jem.20091964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaime-Sanchez P., Uranga-Murillo I., Aguilo N., Khouili S.C., Arias M.A., Sancho D., Pardo J. Cell death induced by cytotoxic CD8 + T cells is immunogenic and primes caspase-3-dependent spread immunity against endogenous tumor antigens. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2020-000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N.S., Cui W., Chandele A., Lee H.K., Urso D.R., Hagman J., Gapin L., Kaech S.M. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaska I.P., Muckersie E., Martin-Granados C., Christofi M., Forrester J.V. Lipopolysaccharide-primed heterotolerant dendritic cells suppress experimental autoimmune uveoretinitis by multiple mechanisms. Immunology. 2017;150:364–377. doi: 10.1111/imm.12691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Doff B.L., Rearden R.C., Leggatt G.R., Mattarollo S.R. NKT cell-targeted vaccination plus anti-4-1BB antibody generates persistent CD8 T cell immunity against B cell lymphoma. Oncoimmunology. 2015;4:e990793. doi: 10.4161/2162402X.2014.990793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogure T., Karasawa S., Araki T., Saito K., Kinjo M., Miyawaki A. A fluorescent variant of a protein from the stony coral Montipora facilitates dual-color single-laser fluorescence cross-correlation spectroscopy. Nat. Biotechnol. 2006;24:577–581. doi: 10.1038/nbt1207. [DOI] [PubMed] [Google Scholar]
- Krysko D.V., Garg A.D., Kaczmarek A., Krysko O., Agostinis P., Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- Lan Y.Y., De Creus A., Colvin B.L., Abe M., Brinkmann V., Coates P.T.H., Thomson A.W. The sphingosine-1-phosphate receptor agonist FTY720 modulates dendritic cell trafficking in vivo. Am. J. Transpl. 2005;5:2649–2659. doi: 10.1111/j.1600-6143.2005.01085.x. [DOI] [PubMed] [Google Scholar]
- Laoui D., Keirsse J., Morias Y., Van Overmeire E., Geeraerts X., Elkrim Y., Kiss M., Bolli E., Lahmar Q., Sichien D. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat. Commun. 2016;7:1–17. doi: 10.1038/ncomms13720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A.W.T., Biester S., Cornall R.J., Forrester J.V. Lipopolysaccharide-activated IL-10-secreting dendritic cells suppress experimental autoimmune uveoretinitis by MHCII-dependent activation of CD62L-expressing regulatory T cells. J. Immunol. 2008;180:3889–3899. doi: 10.4049/JIMMUNOL.180.6.3889. [DOI] [PubMed] [Google Scholar]
- Ledgerwood L.G., Lal G., Zhang N., Garin A., Esses S.J., Ginhoux F., Merad M., Peche H., Lira S.A., Ding Y. The sphingosine 1-phosphate receptor 1 causes tissue retention by inhibiting the entry of peripheral tissue T lymphocytes into afferent lymphatics. Nat. Immunol. 2008;9:42–53. doi: 10.1038/ni1534. [DOI] [PubMed] [Google Scholar]
- Lindquist R.L., Shakhar G., Dudziak D., Wardemann H., Eisenreich T., Dustin M.L., Nussenzweig M.C. Visualizing dendritic cell networks in vivo. Nat. Immunol. 2004;5:1243–1250. doi: 10.1038/ni1139. [DOI] [PubMed] [Google Scholar]
- Ma Y., Adjemian S., Mattarollo S.R., Yamazaki T., Aymeric L., Yang H., Portela Catani J.P., Hannani D., Duret H., Steegh K. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38:729–741. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Mollica L., De Marchis F., Spitaleri A., Dallacosta C., Pennacchini D., Zamai M., Agresti A., Trisciuoglio L., Musco G., Bianchi M.E. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem. Biol. 2007;14:431–441. doi: 10.1016/j.chembiol.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Montico B., Nigro A., Casolaro V., Dal Col J. Immunogenic apoptosis as a novel tool for anticancer vaccine development. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19020594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutini C., Falzoni S., Ferrari D., Chiozzi P., Morelli A., Baricordi O.R., Collo G., Ricciardi-Castagnoli P., Di Virgilio F. Mouse dendritic cells express the P2X7 purinergic receptor: characterization and possible participation in antigen presentation. J. Immunol. 1999;163:1958–1965. [PubMed] [Google Scholar]
- Nakanishi Y., Ikebuchi R., Chtanova T., Kusumoto Y., Okuyama H., Moriya T., Honda T., Kabashima K., Watanabe T., Sakai Y., Tomura M. Regulatory T cells with superior immunosuppressive capacity emigrate from the inflamed colon to draining lymph nodes. Mucosal Immunol. 2017;11:437–448. doi: 10.1038/mi.2017.64. [DOI] [PubMed] [Google Scholar]
- Pappu R., Schwab S.R., Cornelissen I., Pereira J.P., Regard J.B., Xu Y., Camerer E., Zheng Y.W., Huang Y., Cyster J.G., Coughlin S.R. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316:295–298. doi: 10.1126/science.1139221. [DOI] [PubMed] [Google Scholar]
- Pashenkov M., Goëss G., Wagner C., Hörmann M., Jandl T., Moser A., Britten C.M., Smolle J., Koller S., Mauch C. Phase II trial of a toll-like receptor 9-activating oligonucleotide in patients with metastatic melanoma. J. Clin. Oncol. 2006;24:5716–5724. doi: 10.1200/JCO.2006.07.9129. [DOI] [PubMed] [Google Scholar]
- Roberts E.W., Broz M.L., Binnewies M., Headley M.B., Nelson A.E., Wolf D.M., Kaisho T., Bogunovic D., Bhardwaj N., Krummel M.F. Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. 2016;30:324–336. doi: 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadozai H., Gruber T., Hunger R.E., Schenk M. Recent successes and future directions in immunotherapy of cutaneous melanoma. Front. Immunol. 2017;8:1617. doi: 10.3389/fimmu.2017.01617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáez P.J., Vargas P., Shoji K.F., Harcha P.A., Lennon-Duménil A.-M., Sáez J.C. ATP promotes the fast migration of dendritic cells through the activity of pannexin 1 channels and P2X 7 receptors. Sci. Signal. 2017;10:eaah7107. doi: 10.1126/scisignal.aah7107. [DOI] [PubMed] [Google Scholar]
- Saito M., Iwawaki T., Taya C., Yonekawa H., Noda M., Inui Y., Mekada E., Kimata Y., Tsuru A., Kohno K. Diphtheria toxin receptor-mediated conditional and targeted cell ablation in transgenic mice. Nat. Biotechnol. 2001;19:746–750. doi: 10.1038/90795. [DOI] [PubMed] [Google Scholar]
- Salmon H., Idoyaga J., Rahman A., Leboeuf M., Remark R., Jordan S., Casanova-Acebes M., Khudoynazarova M., Agudo J., Tung N. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. 2016;44:924–938. doi: 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H., Tesar B.M., Walker W.E., Goldstein D.R. Dual signaling of MyD88 and TRIF is critical for maximal TLR4-induced dendritic cell maturation. J. Immunol. 2008;181:1849–1858. doi: 10.4049/jimmunol.181.3.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetab Boushehri M.A., Abdel-Mottaleb M.M.A., Béduneau A., Pellequer Y., Lamprecht A. A nanoparticle-based approach to improve the outcome of cancer active immunotherapy with lipopolysaccharides. Drug Deliv. 2018;25:1414–1425. doi: 10.1080/10717544.2018.1469684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen M.A., Holst P.J., Steengaard S.S., Jensen B.A.H., Bartholdy C., Stryhn A., Christensen J.P., Thomsen A.R. Qualitative and quantitative analysis of adenovirus type 5 vector-induced memory CD8 T cells: not as bad as their reputation. J. Virol. 2013;87:6283–6295. doi: 10.1128/jvi.00465-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teasdale R.D., D’Agostaro G., Gleeson P.A. The signal for golgi retention of bovine β1,4-galactosyltransferase is in the transmembrane domain. J. Biol. Chem. 1992;267:4084–4096. [PubMed] [Google Scholar]
- Tomura M., Hata A., Matsuoka S., Shand F.H.W., Nakanishi Y., Ikebuchi R., Ueha S., Tsutsui H., Inaba K., Matsushima K. Tracking and quantification of dendritic cell migration and antigen trafficking between the skin and lymph nodes. Sci. Rep. 2014;4:1–11. doi: 10.1038/srep06030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomura M., Itoh K., Kanagawa O., Alerts E. Naive CD4+ T lymphocytes circulate through lymphoid organs to interact with endogenous antigens and upregulate their function. J. Immunol. 2010;184:4646–4653. doi: 10.4049/jimmunol.0903946. [DOI] [PubMed] [Google Scholar]
- Torcellan T., Hampton H.R., Bailey J., Tomura M., Brink R., Chtanova T. In vivo photolabeling of tumor-infiltrating cells reveals highly regulated egress of T-cell subsets from tumors. Proc. Natl. Acad. Sci. U. S. A. 2017 doi: 10.1073/pnas.1618446114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Ju X., Wang J., Fan Y., Ren M., Zhang H. Immunogenic cell death in anticancer chemotherapy and its impact on clinical studies. Cancer Lett. 2018 doi: 10.1016/j.canlet.2018.08.028. [DOI] [PubMed] [Google Scholar]
- Wilson N.S., Behrens G.M.N., Lundie R.J., Smith C.M., Waithman J., Young L., Forehan S.P., Mount A., Steptoe R.J., Shortman K.D. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat. Immunol. 2006;7:165–172. doi: 10.1038/ni1300. [DOI] [PubMed] [Google Scholar]
- Yamazaki C., Sugiyama M., Ohta T., Hemmi H., Hamada E., Sasaki I., Fukuda Y., Yano T., Nobuoka M., Hirashima T. Critical roles of a dendritic cell subset expressing a chemokine receptor, XCR1. J. Immunol. 2013;190:6071–6082. doi: 10.4049/jimmunol.1202798. [DOI] [PubMed] [Google Scholar]
- Yan B., Chen F., Xu L., Xing J., Wang X. HMGB1-TLR4-IL23-IL17A axis promotes paraquat-induced acute lung injury by mediating neutrophil infiltration in mice. Sci. Rep. 2017;7:597. doi: 10.1038/s41598-017-00721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I., Ostuni R., Capuano G., Collini M., Caccia M., Ronchi A.E., Rocchetti M., Mingozzi F., Foti M., Chirico G. CD14 regulates the dendritic cell life cycle after LPS exposure through NFAT activation. Nature. 2009;460:264–268. doi: 10.1038/nature08118. [DOI] [PubMed] [Google Scholar]
- Zhao R., Liang D., Sun D. Blockade of extracellular ATP effect by oxidized ATP effectively mitigated induced mouse experimental autoimmune uveitis (EAU) PLoS One. 2016;11:e0155953. doi: 10.1371/journal.pone.0155953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L., Apetoh L., Ghiringhelli F., Kroemer G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets or code.