

Abstract
Purpose of review
The aim of this review is to summarize recent advances on development of in vivo preclinical models of adrenocortical carcinoma (ACC).
Recent findings
Significant progress has been achieved in the underlying molecular mechanisms of adrenocortical tumorigenesis over the last decade, and recent comprehensive profiling analysis of ACC tumors identified several genetic and molecular drivers of this disease. Therapeutic breakthroughs, however, have been limited because of the lack of preclinical models recapitulating the molecular features and heterogeneity of the tumors. Recent publications on genetically engineered mouse models and development of patient-derived ACC xenografts in both nude mice and humanized mice now provide researchers with novel tools to explore therapeutic targets in the context of heterogeneity and tumor microenvironment in human ACC.
Summary
We review current in-vivo models of ACC and discuss potential therapeutic opportunities that have emerged from these studies.
Keywords: adrenocortical carcinoma, genetically engineered mouse models, in-vivo research models, patient-derived xenograft mouse models
INTRODUCTION
Adrenocortical tumors present with the spectrum of cell growth, transformation, and tumorigenesis ranging from benign disease including adrenocortical hyperplasia and adenoma (ACA) to malignant tumors such as adrenocortical carcinoma (ACC) [1,2]. Although ACAs are common (up to 4% of the population with age) [3], ACCs are rare and aggressive tumors with an estimated incidence of 0.5–2 cases/million per year [4]. Surgery is the first-line therapy for patients with ACC; however, relapse or persistent disease are frequent as the majority of patients present with locally invasive or metastatic disease. Patients with advanced disease are treated with cytotoxic etoposide, doxorubicin, and cisplatin (EDP) chemotherapy in combination with mitotane, an adrenolytic, with limited responses [5]. There are no targeted therapeutic alternatives for patients with ACC and given the dismal 5 years survival rate of ~35%, management of patients with ACC remains a significant challenge [5,6▪▪].
In other types of human malignancies, preclinical research models, including in vitro cell lines and in vivo mouse models, have allowed researchers to examine the molecular and cellular underpinnings of a disease. Although appropriate to dissect the signaling pathways and molecular mechanisms of particular driver mutations or gene targets, in vitro cell line models often fail to fully recapitulate patient tumor characteristics and tumor microenvironment, which can lead to discordance between laboratory results and clinical outcomes. Similar to in vitro models, cell line-derived xenograft mouse models (Fig. 1A), which have been the workhorse of tumor biology research, are characterized by a lack of tumor heterogeneity and patient-specific genetic alternations to fully recapitulate the primary tumor. In recent years, patient-derived xenograft (PDX; Fig. 1B) and genetically engineered mouse models (GEMM; Fig. 2) have gained popularity because of their retention of tumor heterogeneity, microenvironment and many genetic, molecular, and histopathological traits. To date, several mouse models of benign adrenocortical tumors (hyperplasias or adenomas) have been generated, which partly recapitulate characteristics of human adrenocortical tumors [1,2]. Development of mouse models of ACC have been even more challenging given the rarity of the disease and heterogeneity of the tumors.
FIGURE 1.
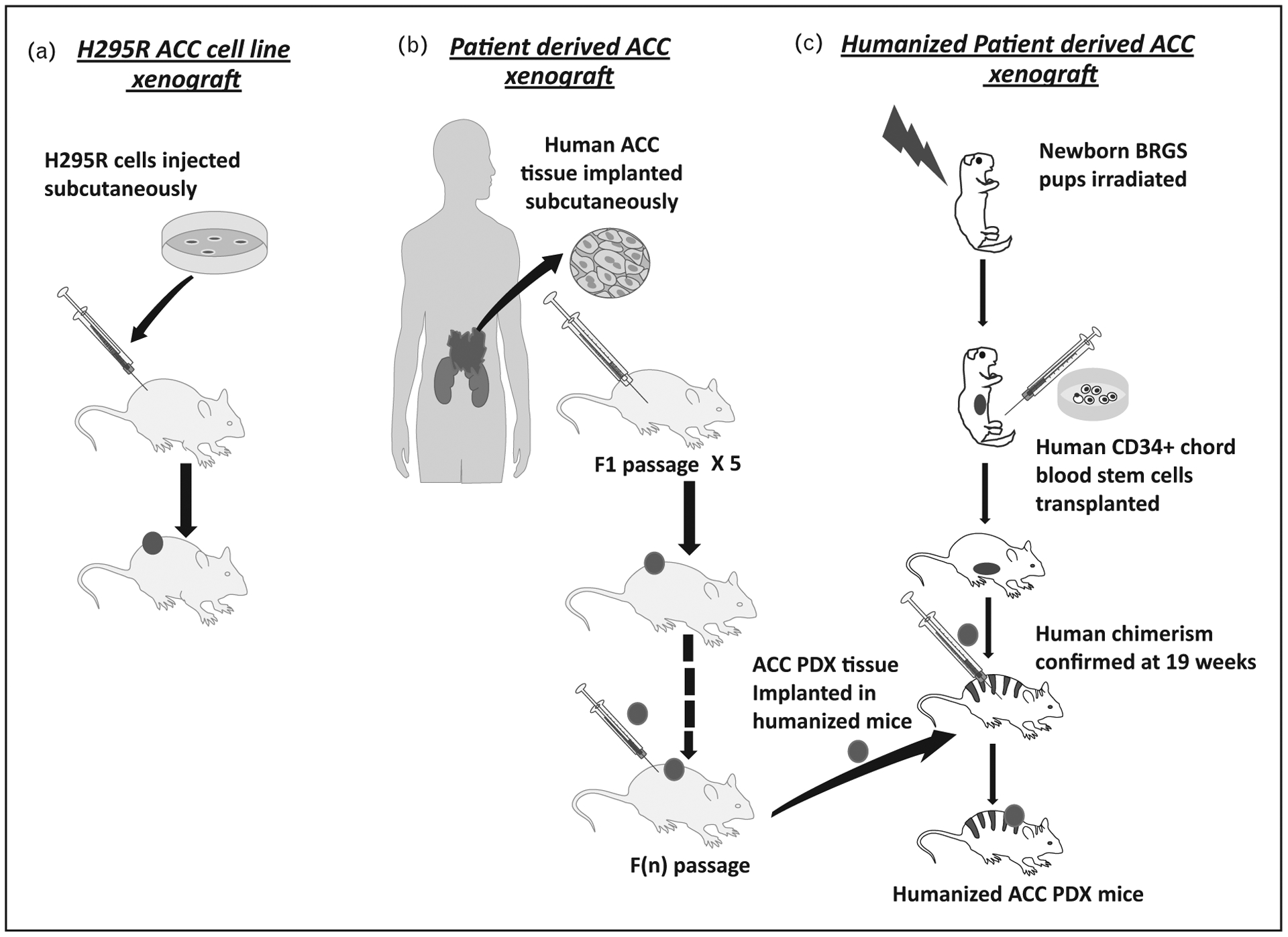
Development of xenograft models of adrenocortical carcinoma (ACC). (A) Generation of H295R cell-derived xenograft. (B) Development of ACC patient-derived xenograft (PDX) mouse model in nu/nu athymic mice. (C) Generation of humanized mice and development of humanized mouse ACC PDX models.
FIGURE 2.
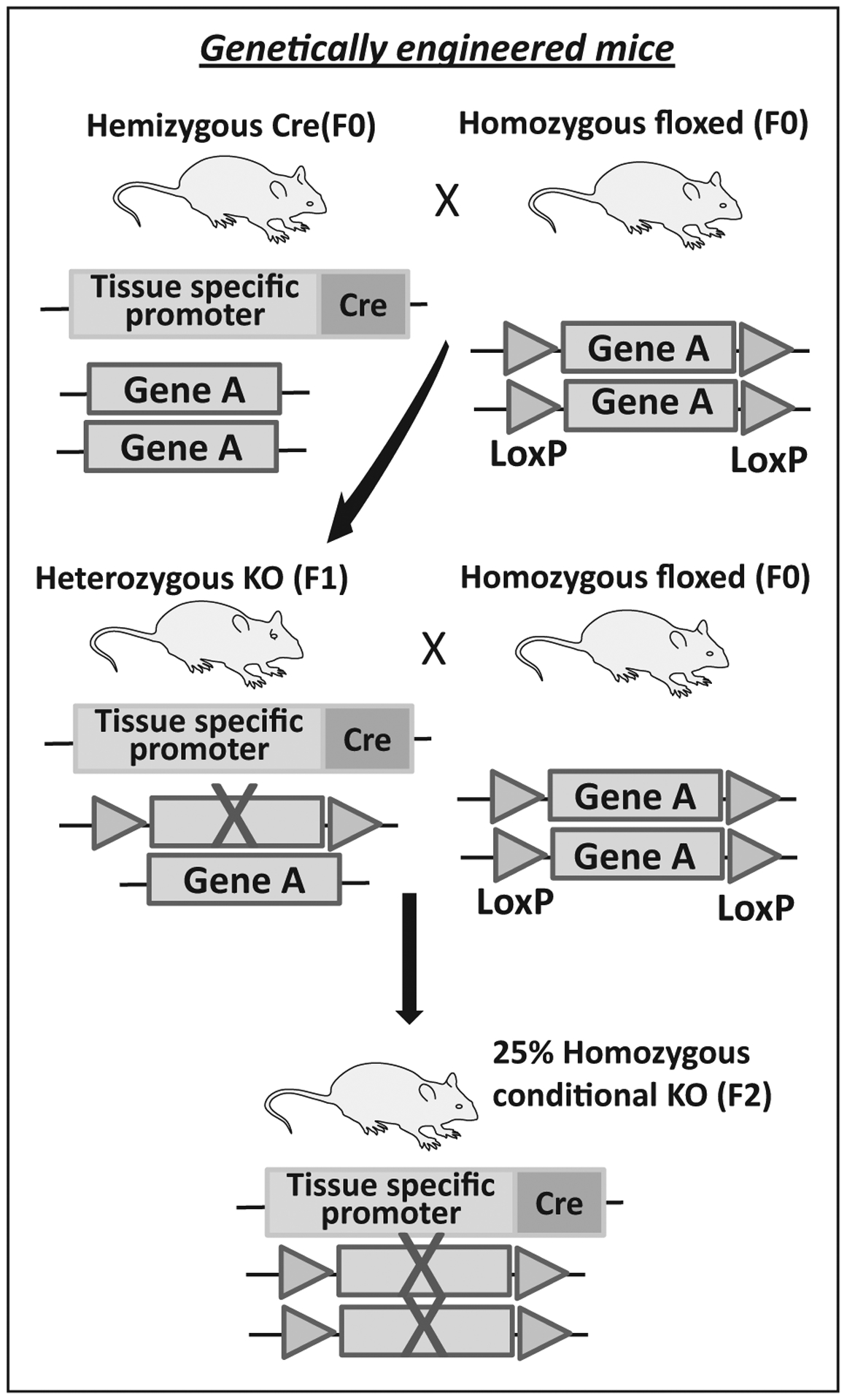
Model of development of genetically engineered mouse model (GEMM).
Pan-genomic The Cancer Genome Atlas (TCGA) characterization of ACCs reported three ACC tumor subtypes with distinct transcriptomic profiles, somatic alterations, whole genome doubling (WGD) and CpG island methylator signatures (CIMP), corresponding to different clinical phenotypes [7,8]. The most common somatic variants reported in ACCs involved gene alterations in CTNNB1, TP53, and CDKN2A, followed by RB1, MEN1, ZNRF3, and TERT. At the transcript level, nearly all ACC tumors have overexpression of IGF2 [6▪▪,8,9]. Considering the genetic variability, no single model can replicate the molecular heterogeneity of ACC; thus, multiple models will need to be developed and applied to gain comprehensive understanding of the disease.
Over the last decade several genetically engineered mouse models of p53 pathway inactivation and Wnt/β catenin signaling activation have been generated but few simulate human ACC tumorigenesis [10]. Alternatively, xenograft models using H295R cells have been used to test novel therapeutics with modest success.
In this review, we will briefly discuss the previously genetically engineered mouse models and xenograft models of ACC and provide an update on the most recent model developments in the ACC field.
PREVIOUS GENETICALLY ENGINEERED MOUSE MODELS AND XENOGRAFTS USED IN MODELING ADRENOCORTICAL CARCINOMA
Because overexpression of IGF2 and activation of Wnt/β catenin signaling pathway are the most frequent alternations in ACC tumors [9,11], many of the early genetically engineered mouse models (GEMM) were focused on these changes [7–9,12]. Despite creation of models with high basal levels of IGF2 and mildly increased proliferation, neither mice models with IGF2 overexpression under the phosphoenol pyruvate carboxykinase (PEPCK) promoter or adrenal cortex-specific (Adigf2) mice developed adrenocortical carcinomas [13,14]. These data demonstrated that IGF2 is less likely an oncogenic driver of ACC tumors and may instead be required for tumor maintenance and/or progression. To model initiation events of ACC, mouse models were generated with either alteration in the β-catenin gene itself or one of its negative regulators, such as APC [15,16]. The ΔCat mice were generated by floxing out the third exon of the β-catenin gene via steroidogenic cell-specific expression of Cre recombinase in the adrenal cortex [15–17]. Excising the third exon prevented β-catenin degradation and led to constitutive activation of β-catenin target genes mimicking Wnt signaling. Most of ΔCat mice developed adrenal hyperplasia and dysplasia with older mice developing benign aldosterone secreting tumors with certain malignant characteristics such as neovascularization and regional invasion [15]. Another approach to target Wnt activation utilized APC knockout mouse models, where exon 14 of APC was deleted and targeted with the steroidogenic specific Sf-1 Cre recombinase resulting in β-catenin stabilization in the adrenal cortex [12]. These mice, similar to the ΔCat model, developed hyperplasia and microscopic adenomas, but no malignant transformation. Additional GEMM models where ΔCat was crossed with IGF2 overexpressing mice [14] or where APC knockout mice were crossed with animals bearing a loss of imprinting at the Igf2/H19 locus to cause IGF2 overexpression [12] developed severe adrenocortical hyperplasia or adenomas, with the latter producing one ACC.
Several ACC cell line xenografts and PDX have also been developed to study ACC [18,19]. Historically, most utilized the ACC cell line NCI-H295R (Fig. 1A) [19]. Such xenografts, displayed clone-dependent heterogeneity [18]. In contrast to cell line-derived xenografts, most PDX in other malignancies retain patient tumor characteristics and would have advantages for our goal to understand ACC pathophysiology. The first PDX of pediatric ACC (SJACC3) was developed by implantation of an adrenal mass collected from a 11-year-old patient bearing a germline TP53 mutation (G245C) [20]. The first adult ACC PDX model MUC-1 was established, along with a corresponding cell line, derived from a metastatic ACC neck lesion [18]. In the initial reports, these models were used to evaluate the effectiveness of several cytotoxic chemotherapies as proof-of concept studies [18,20,21].
RECENT UPDATES IN GENETICALLY ENGINEERED MICE MODELS OF ADRENOCORTICAL CARCINOMA
In addition to mutations in the Wnt signaling pathway, ACCs are characterized by frequent alterations in the TP53 gene/pathway [7,8,22]. About 25–30% of sporadic ACCs in adults carry somatic mutation or loss of heterozygosity at the TP53 locus [23,24]. To date, no adrenal-specific models of TP53 loss has been published and no ACC tumor formation has been reported in any other mouse models of TP53 dysfunction. The relevance of loss of TP53-dependent checkpoint control in ACC development was first studied in adrenal insufficient Acd (mutation in adrenal dysplasia gene) mice model with a TP53 null background [25,26]. In a more relevant transgenic mouse model, p53 ablation was modeled by adrenal targeting of the Simian virus 40 (SV40) large T antigen (TAg) expressed under the control of the adrenal cortex-specific Akr1b7 promoter [27]. In this model, 100% of founder males developed bilateral adrenal tumors at the age of 8 months that displayed stage-specific malignant characteristics from 2 to 8 months. High-grade malignancy was observed after 6 months of age and was characterized by a high Ki-67 index, overexpression of cyclin E, and histone methyl transferase EZH2 with loss of paternally imprinted H19 and evidence of distant metastasis to lung and liver. These tumors also demonstrated evidence for spontaneous Wnt/β catenin pathway activation [27]. Activation of the mTOR pathway was confirmed as an early step in the tumorigenic process, a pathway often found activated in a cohort of patients with ACC. At 8 months of age, all tumors were functional and secreted excess corticosterone [27]. Recently, a mouse model targeting ZNRF3, a negative regulator of the Wnt signaling, has been reported [28▪▪]. Large-scale ACC genomic studies have identified as much as 20% genetic alterations in ZNRF3 [7,22]. ZNRF3 has been identified as a transmembrane E3 ubiquitin ligase responsible for degradation of the frizzled receptor (FZD) and therefore subsequent brake in Wnt signaling [29,30]. With an aim to elucidate the effect of loss of ZNRF3 on adrenal cortex homoeostasis, Basham et al. [28▪▪] developed ZNRF3 knockout mice by crossing a SF1-Cre mice with Znrf3-floxed mice, resulting in a mice lacking functional ZNRF3 protein in the adrenal cortex. These mice showed marked adrenal hyperplasia at 6 weeks of age because of proliferative expansion of the zona fasciculata and a loss of the normal Wnt/β-catenin gradient. Although these mice did not demonstrate progression from hyperplasia to adrenocortical carcinoma, this is clinically relevant model recapitulating loss of ZNNRF3 as one of the most common genetic alterations in ACC. Future characterization of this model is likely to give us a better understanding of the disease pathophysiology.
UPDATE ON PATIENT-DERIVED XENOGRAFT MODEL OF ADRENOCORTICAL CARCINOMA
Since the initial studies to establish PDX models of human tumorigenesis [31], the development and application of PDX models in cancer research continue to grow [32–36]. Given the sporadic nature of ACC, PDX mice provide a unique opportunity to understand molecular heterogeneity underlying the disease. We have recently characterized two new PDX models, CU-ACC1 and CU-ACC2, via subcutaneous implantation of patient tumor tissues in athymic nu/nu mice models (Fig. 1B) [37▪]. CU-ACC1 was derived from a sporadic ACC metastasis to the perinephric region and CU-ACC2 was derived from an ACC liver metastasis in a patient with Lynch syndrome characterized by the germline deletion of exons 1–6 in the mismatch repair gene MSH2. The PDXs were extensively characterized and adrenocortical origin was confirmed with expression adrenocortical markers including inhibin alpha and steroidogenic factor 1 (SF-1). Immunohistological analysis of the PDXs tumor tissue was similar to the matching patients’ tumor characteristics. Immunohistochemistry revealed loss of MSH2 in the CU-ACC2 PDX tissues consistent with patient tumor characteristic. Whole exomic sequencing of the patient tumors and the PDX models identified a known ACC-associated mutation in CTNNB1 (p.G34R) in CU-ACC1 and a TP53 (G245S) mutation in CU-ACC2. In-silico predictions for both mutations were damaging and have been reported in other human cancers [38,39]. Global transcriptome profiling was performed in both PDXs and matching human tumors and revealed clustering of matching tissues and transcriptome expression characteristic for ACC. Corresponding cell lines from the PDX models were developed, and have been utilized by ourselves and others to explore novel therapeutic targeted therapy in ACC [37▪,40▪,41▪].
DEVELOPMENT OF HUMANIZED MICE MODEL OF ADRENOCORTICAL CARCINOMA
With the current advances in immunotherapeutic modalities and their effectiveness in several malignancies, studying unique human responses to anti-bodies targeted against tumor-associated proteins or immune checkpoint inhibitors have become critical [42]. Humanized mice models with immune-deficient mice engrafted with human cells or tissues have served as a preclinical conduit for several of these research areas. Although most ACCs are sporadic, a subset of ACC tumors harbor either germline or somatic mutations in DNA mismatch repair genes, or mismatch repair components [43]. Recent approval of the anti-PD1 inhibitor, pembrolizumab, for mismatch repair deficient or high microsatellite instability solid tumors [44], has advanced therapeutic possibilities for subset of patients with ACC. We recently reported the development of the first humanized ACC PDX mouse model, and analyzed the effects of pembrolizumab on tumor growth and changes in infiltrating lymphocytes and immune cells in the peripheral lymph organs in comparison to changes in immune markers of the matching patient with advanced ACC [45▪].
The humanized mouse model was created by intravenous or intrahepatic injection of CD34+ human umbilical chord blood cells into sublethally irradiated newborn BRGs (BALB/c-Rag2null Il2rγnull SirpaNOD) pups [45▪]. The human chimerism was confirmed at week 19 and a previously established PDX from a liver metastasis of a Lynch patient (CU-ACC2-M2B PDX) was implanted in the humanized mice to develop the humanized CU-ACC2-M2B PDX (Fig. 1C). Although this model is specific to the T-cell population with an overall low abundancy of human myeloid, monocytes, or dendritic cells, it provided an comprehensive analyses of the human immune system activating and inhibitory proteins, and expression of inflammatory factors with establishing a first ACC humanized model. These type of models can now be used for evaluating response to immunotherapy alone or in combination with mitotane or novel therapeutics in different types of ACC tumors [45▪].
USING IN-VIVO RESPONSES TO MODEL THERAPEUTIC ALTERNATIVES IN ADRENOCORTICAL CARCINOMA
There are limited therapeutic options for patients with progressive ACC, with mitotane as the only FDA approved modality. The recent development of new in-vivo ACC models present opportunities for testing of potential therapeutic alternatives. Using the AdTAg model of spontaneous ACC, the efficacy of rapamycin dependent mTORC1 inhibition was tested in a three week short-term or a 3 months long-term treatment. In line with effects on tumor response both with short-term and long-term treatment, significantly induced apoptosis in tumor cells, reduced their proliferation and normalized cortisol level [27]. The AdTAg model belongs to the more aggressive subclass of ACC tumors identified through transcriptional signatures. Incidentally this group is also marked by higher expression of mitotic cell-cycle genes and aneuploidy.
Given that gene expression analysis in recently developed CU-ACC1 and CU-ACC2 PDX models show similar upregulation of genes involved in cell-cycle pathway, several upregulated mitotic kinases were targeted using available small molecule inhibitors in this recently developed preclinical models of sporadic ACC [40▪,41▪]. Targeting the mitotic kinase PBK in in vitro and in vivo models of ACC inhibited many of the malignant properties, induced apoptosis and colony formation, and significantly reduced ACC tumorigenic growth [41▪]. Similarly, targeting MELK, another mitotic kinase, caused apoptosis and inhibited proliferation and clonogenicity in in vitro assays using multiple ACC cell lines, and in newly established PDX models [40▪,46▪]. More recently, testing the effects of pembrolizumab in humanized mice models have led to a prototype that can be effectively used to further future studies matching PDX response to that of patients with ACC with a similar molecular signature. Clinical observations suggest that mitotane treatment has variable outcomes in patients with ACC and possibly affects the tumor microenvironment. In our recently published clinical study, limited patient data suggest a benefit of pembrolizumab in combination with mitotane in patients with ACC irrespective of the microsatellite stability or mismatch repair status [47▪]. Further progress with humanized ACC PDX models will allow preclinical evaluation of such combinatorial therapies and enhance understanding of ACC pathophysiology and microenvironment.
CONCLUSION
In the field of preclinical and co-clinical studies, in vivo genetically engineered models and PDX models are commonly considered superior to cell line-derived xenograft, which lacks original tumor heterogeneity because of selective proliferation over numerous passages. Genetically engineered mouse models have been invaluable for the process of understanding tumor initiation and relapse but can be less predictable for studying drug efficacies. Cytogenetic profile of cancers derived from mouse cells cannot completely mimic human cancer genome instability, alterations in activating pathways, or the tumor microenvironment [48]. In comparison, PDX tumor models maintain patient tumor molecular signatures and tumor heterogeneity with minimal drift. It has been shown that PDX tumors show comparable treatment response to those performed clinically [48]. Given the rarity of ACC, patient recruitment is difficult and most clinical trials of ACC span several years limiting therapeutic advances for patients with ACC. Future developments of ACC PDX models and additional humanized mice PDX models with varied molecular signatures could provide a path towards more ‘avatar’ PDX models for patients with ACC [49–52]. In cases of recurrence and metastasis, these ‘avatar’ PDX models can be used to investigate sensitivity of all chemotherapy and possible targeted drugs alone or in combination and also can be used for drug biomarker screenings.
KEY POINTS.
Genetically engineered mice models of ACC are able to recapitulate cohorts of human ACC tumors and help researchers to understand the pathophysiology of ACC development, maintenance, and metastasis.
Patient-derived xenograft models of ACC represents diverse molecular signatures of ACC and can be used for therapeutic screening for a more concordant clinical outcomes.
Humanized mice models of ACC PDXs provide new opportunities to test the effects of immunotherapy in conjunction with other established or novel treatment regimens.
Financial support and sponsorship
This work was supported by NIH K08CA222620 (to K.K.V.), Cancer League of Colorado Award (to K.K.V., M.E.W., and A.K.), Golfers Against Cancer (to K.K.V.), Veterans Affairs Merit Review Award 001 (to M.E.W.).
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
- 1.Leccia F, Batisse-Lignier M, Sahut-Barnola I, et al. Mouse models recapitulating human adrenocortical tumors: what is lacking? Front Endocrinol 2016; 7:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basham KJ, Hung HA, Lerario AM, Hammer GD. Mouse models of adrenocortical tumors. Mol Cell Endocrinol 2016; 421:82–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bovio S, Cataldi A, Reimondo G, et al. Prevalence of adrenal incidentaloma in a contemporary computerized tomography series. J Endocrinol Invest 2006; 29:298–302. [DOI] [PubMed] [Google Scholar]
- 4.Else T, Kim AC, Sabolch A, et al. Adrenocortical carcinoma. Endocr Rev 2014; 35:282–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varghese J, Habra MA. Update on adrenocortical carcinoma management and future directions. Curr Opin Endocrinol Diabetes Obes 2017; 24:208–214. [DOI] [PubMed] [Google Scholar]
- 6. ▪▪.Mohan DR, Lerario AM, Hammer GD. Therapeutic targets for adrenocortical carcinoma in the genomics era. J Endocr Soc 2018; 2:1259–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a review providing an up-to-date summary of genetic and epigenetic factors implicated in ACC and covers therapeutic strategies in light of the genomic landscape of ACC.
- 7.Assie G, Letouze E, Fassnacht M, et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 2014; 46:607–612. [DOI] [PubMed] [Google Scholar]
- 8.Zheng S, Cherniack AD, Dewal N, et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 2016; 29:723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giordano TJ, Kuick R, Else T, et al. Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin Cancer Res 2009; 15:668–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gahete MD, Jimenez-Vacas JM, Alors-Perez E, et al. Mouse models in endocrine tumors. J Endocrinol 2018; 240:R73–R96. [DOI] [PubMed] [Google Scholar]
- 11.Ribeiro TC, Latronico AC. Insulin-like growth factor system on adrenocortical tumorigenesis. Mol Cell Endocrinol 2012; 351:96–100. [DOI] [PubMed] [Google Scholar]
- 12.Heaton JH, Wood MA, Kim AC, et al. Progression to adrenocortical tumorigenesis in mice and humans through insulin-like growth factor 2 and beta-catenin. Am J Pathol 2012; 181:1017–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weber MM, Fottner C, Schmidt P, et al. Postnatal overexpression of insulin-like growth factor II in transgenic mice is associated with adrenocortical hyperplasia and enhanced steroidogenesis. Endocrinology 1999; 140:1537–1543. [DOI] [PubMed] [Google Scholar]
- 14.Drelon C, Berthon A, Ragazzon B, et al. Analysis of the role of Igf2 in adrenal tumour development in transgenic mouse models. PLoS One 2012; 7:e44171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berthon A, Sahut-Barnola I, Lambert-Langlais S, et al. Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum Mol Genet 2010; 19:1561–1576. [DOI] [PubMed] [Google Scholar]
- 16.Lambert-Langlais S, Val P, Guyot S, et al. A transgenic mouse line with specific Cre recombinase expression in the adrenal cortex. Mol Cell Endocrinol 2009; 300:197–204. [DOI] [PubMed] [Google Scholar]
- 17.Harada N, Tamai Y, Ishikawa T, et al. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J 1999; 18:5931–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hantel C, Shapiro I, Poli G, et al. Targeting heterogeneity of adrenocortical carcinoma: evaluation and extension of preclinical tumor models to improve clinical translation. Oncotarget 2016; 7:79292–79304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rainey WE, Bird IM, Mason JI. The NCI-H295 cell line: a pluripotent model for human adrenocortical studies. Mol Cell Endocrinol 1994; 100(1–2):45–50. [DOI] [PubMed] [Google Scholar]
- 20.Pinto EM, Morton C, Rodriguez-Galindo C, et al. Establishment and characterization of the first pediatric adrenocortical carcinoma xenograft model identifies topotecan as a potential chemotherapeutic agent. Clin Cancer Res 2013; 19:1740–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satoh K, Zhang L, Zhang Y, et al. Identification of niclosamide as a novel anticancer agent for adrenocortical carcinoma. Clin Cancer Res 2016; 22:3458–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Reynies A, Assie G, Rickman DS, et al. Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J Clin Oncol 2009; 27:1108–1115. [DOI] [PubMed] [Google Scholar]
- 23.Libe R, Groussin L, Tissier F, et al. Somatic TP53 mutations are relatively rare among adrenocortical cancers with the frequent 17p13 loss of heterozygosity. Clin Cancer Res 2007; 13:844–850. [DOI] [PubMed] [Google Scholar]
- 24.Reincke M, Karl M, Travis WH, et al. p53 mutations in human adrenocortical neoplasms: immunohistochemical and molecular studies. J Clin Endocrinol Metab 1994; 78:790–794. [DOI] [PubMed] [Google Scholar]
- 25.Beamer W, Sweet H, Bronson R, et al. Adrenocortical dysplasia: a mouse model system for adrenocortical insufficiency. J Endocrinol 1994; 141:33–43. [DOI] [PubMed] [Google Scholar]
- 26.Else T, Trovato A, Kim A, et al. Genetic p53 deficiency partially rescues the adrenocortical dysplasia phenotype at the expense of increased tumorigenesis. Cancer Cell 2009; 15:465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Batisse-Lignier M, Sahut-Barnola I, Tissier F, et al. P53/Rb inhibition induces metastatic adrenocortical carcinomas in a preclinical transgenic model. Oncogene 2017; 36:4445–4456. [DOI] [PubMed] [Google Scholar]
- 28. ▪▪.Basham KJ, Rodriguez S, Turcu AF, et al. A ZNRF3-dependent Wnt/beta-catenin signaling gradient is required for adrenal homeostasis. Genes Dev 2019; 33(3–4):209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study focuses on the effects of ZNRF3 knockout on adrenal gland homoeostasis. The model provides important preclinical information on how the Wnt signaling is controlled during adrenal gland development by the negative regulator ZNRF3, which is one of the most altered gene in ACC.
- 29.Hao HX, Xie Y, Zhang Y, et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012; 485:195–200. [DOI] [PubMed] [Google Scholar]
- 30.Nusse R, Clevers H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017; 169:985–999. [DOI] [PubMed] [Google Scholar]
- 31.Kim MP, Evans DB, Wang H, et al. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc 2009; 4:1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hidalgo M, Amant F, Biankin AV, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov 2014; 4:998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobrolecki LE, Airhart SD, Alferez DG, et al. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev 2016; 35:547–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jung J, Seol HS, Chang S. The generation and application of patient-derived xenograft model for cancer research. Cancer Res Treat 2018; 50:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moiola CP, Lopez-Gil C, Cabrera S, et al. Patient-derived xenograft models for endometrial cancer research. Int J Mol Sci 2018; 19:pii: E2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu C, Li X, Liu P, et al. Patient-derived xenograft mouse models: a high fidelity tool for individualized medicine. Oncol Lett 2019; 17:3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. ▪.Kiseljak-Vassiliades K, Zhang Y, Bagby SM, et al. Development of new preclinical models to advance adrenocortical carcinoma research. Endocr Relat Cancer 2018; 25:437–451. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports development of two novel PDXs and cell lines of ACC. Two models are distinct in their genetic profile and add to the repertoire of preclinical models for ACC.
- 38.Gao C, Wang Y, Broaddus R, et al. Exon 3 mutations of CTNNB1 drive tumorigenesis: a review. Oncotarget 2018; 9:5492–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor Perspect Biol 2010; 2:a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. ▪.Kiseljak-Vassiliades K, Zhang Y, Kar A, et al. Elucidating the role of the maternal embryonic leucine zipper kinase (MELK) in adrenocortical carcinoma. Endocrinology 2018; 159:2532–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper explores the role the MELK kinase and its inhibitor OTSSP167 in three ACC cell lines and shows that targeting ACC cell lines with OTSSP167 inhibits proliferation and other malignant growth properties.
- 41. ▪.Kar A, Zhang Y, Yacob B, et al. Targeting PDZ binding kinase is antitumorigenic in novel preclinical models of ACC. Endocr Relat Cancer 2019; 26:765–778. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a first ever study of targeted therapeutics in a PDX of ACC. The study explores the role of the mitotic kinase PBK in ACC tumorigenesis and shows that targeting PBK is antitumorigenic in a patient-derived model of ACC.
- 42.O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol 2019; 16:151–167. [DOI] [PubMed] [Google Scholar]
- 43.Raymond VM, Everett JN, Furtado LV, et al. Adrenocortical carcinoma is a lynch syndrome-associated cancer. J Clin Oncol 2013; 31:3012–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (New York, NY) 2017; 357:409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. ▪.Lang J, Capasso A, Jordan KR, et al. Development of an adrenocortical cancer humanized mouse model to characterize anti-PD1 effects on tumor microenvironment. J Clin Endocrinol Metab 2020; 105:. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the development of the first ACC humanized mice model of the CU-ACC2 and defines biomarkers of response to pembrolizumab and compares it to the CU-ACC2 patient on pembrolizumab therapy. The model serves as a prototype for future ACC studies exploring tumor responses to immunotherapy as a single treatment modality or in combination with other therapeutics in context of an immune mileiu.
- 46. ▪.Kar A, Zhang Y, Yacob B, et al. SUN-337 antitumorigenic effects of the maternal leucine zipper kinase (MELK) inhibitor, OTSSP167, in preclinical in vivo models of adrenocortical carcinomas (ACC). J Endocr Soc 2019; 3(Supplement_1.). [Google Scholar]; This study reports development of another ACC PDX model CU-ACC9 and also establishes the antitumorigenic effects of the MELK inhibitor OTSSP167 on in vivo studies of CU-ACC1 and CU-ACC9 PDXs.
- 47. ▪.Head L, Kiseljak-Vassiliades K, Clark TJ, et al. Response to immunotherapy in combination with mitotane in patients with metastatic adrenocortical cancer. J Endocr Soc 2019; 3:2295–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]; This clinical study reports six patients treated with pembrolizumab in combination with mitotane, and suggests potential synergistic effect of the combination therapy.
- 48.Herter-Sprie GS, Kung AL, Wong KK. New cast for a new era: preclinical cancer drug development revisited. J Clin Invest 2013; 123:3639–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weroha SJ, Becker MA, Enderica-Gonzalez S, et al. Tumorgrafts as in vivo surrogates for women with ovarian cancer. Clin Cancer Res 2014; 20:1288–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bousquet G, Feugeas JP, Ferreira I, et al. Individual xenograft as a personalized therapeutic resort for women with metastatic triple-negative breast carcinoma. Breast Cancer Res 2014; 16:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Z, Chen Z, Wang J, et al. Mouse avatar models of esophageal squamous cell carcinoma proved the potential for EGFR-TKI afatinib and uncovered Src family kinases involved in acquired resistance. J Hematol Oncol 2018; 11:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garralda E, Paz K, Lopez-Casas PP, et al. Integrated next-generation sequencing and avatar mouse models for personalized cancer treatment. Clin Cancer Res 2014; 20:2476–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]