

Summary
Poor maternal diet increases the risk of obesity and type 2 diabetes in offspring, adding to the ever-increasing prevalence of these diseases. In contrast, we find that maternal exercise improves the metabolic health of offspring, and here we demonstrate that this occurs through a vitamin D receptor-mediated increase in placental superoxide dismutase 3 (SOD3) expression and secretion. SOD3 activates an AMPK/TET signaling axis in fetal offspring liver, resulting in DNA demethylation at the promoters of glucose metabolic genes, enhancing liver function, and improving glucose tolerance. In humans, SOD3 is upregulated in serum and placenta from physically active pregnant women. The discovery of maternal exercise-induced cross talk between placenta-derived SOD3 and offspring liver provides a central mechanism for improved offspring metabolic health. These findings may lead to novel therapeutic approaches to limit the transmission of metabolic disease to the next generation.
Graphical Abstract

Introduction
370 million people worldwide have type 2 diabetes (T2D) in 2015 and this number is projected to rise to 630 million by 2045 (Zheng et al., 2018). Risk patterns for both obesity and T2D can originate as a consequence of alteration in metabolism during critical windows of prenatal development (Duque-Guimaraes and Ozanne, 2013; Glastras et al., 2018; Reynolds et al., 2015). Importantly, the occurrence of obese mothers of reproductive age serve as a prominent risk factor for the transmission of obesity to subsequent generations (Agarwal et al., 2018; Elshenawy and Simmons, 2016; van Dijk et al., 2015; Vickers, 2014). Determining an effective means to reduce a vicious cycle of metabolic dysfunction from mother to offspring has an important impact on population health.
Physical activity has major benefits for people with T2D, and there is strong epidemiological evidence that regular exercise can prevent the onset of this disease (Knowler et al., 2002; Tuomilehto et al., 2001). Our group and others have indicated that maternal exercise improves markers of metabolic health in adult offspring including improved glucose tolerance and insulin sensitivity (Carter et al., 2012; Carter et al., 2013; Kusuyama et al., 2020; Laker et al., 2014; Ribeiro et al., 2017; Sheldon et al., 2016; Stanford et al., 2015; Stanford et al., 2017; Wasinski et al., 2015). We also established that the liver is the major organ responsible for governing the improved metabolic function in offspring from exercise-trained mothers (Stanford et al., 2017). Since maternal exercise changed the profile of hepatic genes in offspring, we hypothesized that an epigenetics is responsible for the translational effects of maternal exercise that lead to improved hepatic function in offspring. Previous studies suggest that metabolic programming may be partially reversed through interventions during phases of developmental plasticity, as long as offspring maintain their capacity for biological reprogramming (Zheng et al., 2014), although both the mechanism of induction and its reversal remain incompletely understood. During the most plastic phase of fetal development where offspring are exposed to the intrauterine environment, the placenta interfaces between the mother and fetus by not only supplying nutrients and oxygen, but also transmitting signals from mother to fetus, that are crucial determinants of fetal reprogramming (Sferruzzi-Perri and Camm, 2016). We postulated that maternal diet and exercise can alter secretion of placental endocrine factors that can increase or decrease the risk of altered glucose metabolism in later life of offspring.
Here we show that maternal exercise increases placental expression and release of superoxide dismutase 3 (SOD3) in both rodents and humans. SOD3 then acts as a regulator which activates 5′ AMP-activated protein kinase (AMPK)-isocitrate dehydrogenase (IDH)-alpha-ketoglutarate (αKG)-ten-eleven translocation (TET) methylcytosine demethylase in offspring liver. This causes demethylation of DNA in the promoter of genes in fetal liver, resulting in epigenetic reprograming and improving metabolism in offspring. This cross talk between the placenta and liver by exercise provides a new pathway for regulating offspring metabolic health and a means to counteract the risk of metabolic disease being propagated across generations.
Results
Maternal exercise induces DNA demethylation in the promoters of glucose metabolic genes in fetal liver of offspring.
We previously showed that maternal exercise by voluntary wheel running during gestation improved metabolic gene expression in offspring liver at 52 weeks (Stanford et al., 2017). To determine if this adaptation is an early life event, we analyzed the expression of metabolic genes in the livers of offspring at 52 weeks, isolated hepatocytes from 4 weeks, and newborns (day 0), and hepatoblasts at embryonic day 13.5 (E13.5). Maternal exercise increased the gene expression related to pyruvate metabolism (Pklr, Pcx), Krebs cycle (Pdha1, Pdk4, Cs, Idh3a, Mdh2), and fatty acid metabolism (Cd36, Fatp4, Acox1, Cpt1a) in the offspring liver at 52 weeks, an effect that was present in offspring hepatocytes at 4 weeks, day 0, and E13.5 hepatoblasts (Sup. Fig. 1A–D). The expression of gluconeogenesis genes (G6pc, Fbp1, Pgc1a, Pck1) was suppressed by maternal exercise in the offspring liver at younger ages, but not in older offspring. Corresponding to changed gene profile, RNA-seq and KEGG pathway analysis showed that maternal exercise upregulated several metabolic pathways including fatty acid metabolism, glycerolipid metabolism, insulin signaling, pyruvate metabolism, and sucrose metabolism (Sup. Fig. 1E). Interestingly, maturity onset diabetes of the young pathway was downregulated by maternal exercise. In contrast to offspring liver, maternal exercise had no effect on glucose metabolic genes (Foxo1, Glut4, and Pdk4) in offspring subcutaneous and visceral white adipose tissue (WAT), gastrocnemius, soleus, or triceps skeletal muscles (Sup. Fig. 2A). Collectively, these changes in the gene expression profile of offspring liver during early development suggest that an epigenetic mechanism may be responsible for the beneficial effects of maternal exercise on offspring.
DNA methylation is an epigenetic change that results in suppressed gene expression with a CpG-dense promoter (Luo et al., 2018). Methylated cytosine (5-methylcytosine: 5-mC) is not stable and DNA demethylation occurs by the conversion of 5-mC to 5-hydroxymethylcytosine (5-hmC) (Wu and Zhang, 2017). To test the hypothesis that the effects of maternal exercise on hepatic gene expression are induced by DNA demethylation, we measured the total 5-mC and 5-hmC in genomic DNA in the hepatocytes of 4-week-old offspring from dams that were either sedentary or trained and fed a chow or high fat diet (HFD) (Fig. 1A & B). Maternal exercise decreased 5-mC and increased 5-hmC in the hepatocytes of offspring from chow-fed dams. Offspring hepatocytes from sedentary dams fed a HFD had higher levels of 5-mC and a lower 5-hmC. Importantly, the effects of maternal HFD on offspring hepatocyte methylation status were eliminated if the dams had been trained. To verily the DNA methylation levels of specific sequences, we performed methylation-specific PCR targeting liver metabolic genes which have CpG island in the -1000 upstream region (Fig. 1C). Methylation levels of the top 2 apparent CpG islands at the promoter of Pcx, Cs, Idh3a, Mdh2, Ogdh, Fatp4 and Cpt1a were significantly higher in the offspring hepatocytes from HFD-fed dams compared with the offspring of chow diet-fed dams, whereas offspring hepatocytes from trained dams had reduced DNA demethylation of these sites. In accordance with decreased 5-mC levels, 5-hmC DNA immunoprecipitation qPCR showed that 5-hmC levels at the same promoter regions of Pcx, Cs, Idh3a, Mdh2, Ogdh, Fatp4, and Cpt1a were increased by maternal exercise in offspring hepatocytes from chow-fed dams (Fig. 1D). HFD feeding decreased the generation of 5-hmC, and maternal exercise reversed this effect. We also analyzed gene expression and levels of promoter methylation in CpG islands of the top-ranked upregulated genes in the list for differential expression genes of KEGG pathway analysis. Maternal exercise increased the mRNA expression of Fads2, a gene involved in the biosynthesis of unsaturated fatty acids, and Aldh3a2, a gene involved in limonene/pinene degradation (Sup. Fig. 1F) and decreased 5-mC levels at the promoter of these genes (Sup. Fig. 1G). Conversely, maternal exercise did not affect mRNA expression and DNA methylation in housekeeping genes including Gapdh and Tbp (Sup. Fig. 1H & I). These data show that maternal exercise specifically induced the conversion of 5-mC to 5-hmC at the promoter of several types of hepatic genes in offspring liver, suggesting that DNA demethylation increases liver metabolic gene expression in spite of maternal HFD.
Figure 1: Maternal Exercise Induces DNA Demethylation in the Promoter of Hepatic Genes in Offspring.
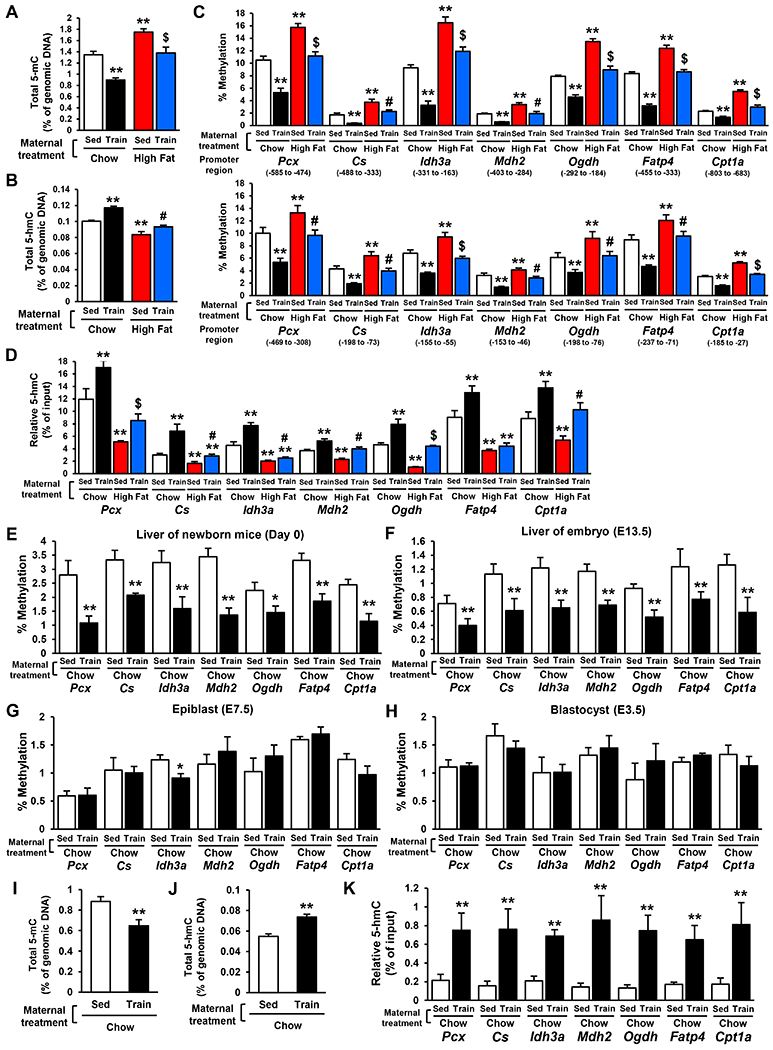
A, B, I, J) Total 5-mC (A, I) and 5-hmC (B, J) in genomic DNA from 4-week-old (A, B) and E13.5 (I, J) offspring livers. C-H, K) The relative 5-mC (C, E-H) and 5-hmC levels (D, K) at the promoter of specific liver metabolic genes in 4-week-old offspring hepatocytes (C, D), Day 0 (E) and E13.5 (F, K) offspring livers, epiblasts (G), and blastocysts (H) from dams that were either sedentary or trained and fed a chow HFD. (n=6, **P < 0.01 vs Sed-Chow, #P < 0.05 vs Sed-High Fat, $P < 0.01 vs Sed-High Fat)
To examine whether DNA methylation changes are caused by cell type remodeling, we analyzed histology in 4-week-old offspring liver from dams that were either sedentary or trained and fed a chow or HFD (Sup. Fig. 2B). Maternal diet and exercise did not change cell composition, fibrosis, or lipid deposition in offspring liver. The Markers of hepatic cells including hepatocytes, macrophages, fibroblasts, and hepatic biliary cells was not affected by both maternal diet and exercise in offspring liver (Sup. Fig. 2C). These results indicate that maternal exercise-induced changes in 5-mC/5-hmC were not caused by cell type remodeling of offspring liver.
Next, we explored the developmental time course by which maternal exercise induces the conversion of 5-mC to 5-hmC in the offspring hepatic genes. Maternal exercise decreased the 5-mC levels of promoters from hepatic genes in the offspring liver from trained mother at Day 0 (Fig. 1E) and E 13.5 (Fig. 1F), but not in offspring at the epiblast (E 7.5) (Fig. 1G) and the blastocyst (E 3.5) stages of development (Fig. 1H). Fetal liver from E13.5 offspring of trained dams exhibited decreased total 5-mC (Fig. 1I), increased total 5-hmC (Fig. 1J), and increased 5-hmC generation at promoters of hepatic genes (Fig. 1K). These results show that the effect of maternal exercise on DNA demethylation is first observed 13.5 days post conception and is maintained through the rest of gestation.
Maternal exercise activates the TET signaling in primary hepatoblasts.
DNA demethylation is mediated through TET enzymes (Wu and Zhang, 2017). We found that mRNA (Fig. 2A) and protein (Fig. 2B) expression of Tet1 and Tet2 were increased in E13.5 offspring liver from trained dams. In contrast, the expression of Tet3 and DNA methyltransferase (Dnmt1, Dnmt3a, Dnmt3b), were not affected by maternal exercise. The binding rate of TET1 and TET2 to the promoter region of Pcx, Mdh2, Ogdh and Fatp4 was also increased by maternal exercise in E13.5 offspring liver (Fig. 2C). Additionally, the amount of αKG, a positive regulator for TET activity (Wu and Zhang, 2017), was increased in E13.5 offspring liver from trained dams (Fig. 2D), as were IDH1 and IDH2, enzymes involved in αKG production (Fig. 2E). Given the roles of AMPK in many of the exercise benefits (Richter and Ruderman, 2009), we hypothesized that AMPK regulates TET signaling in the offspring livers, and found that AMPKα1/2 and (Fig. 2F) and acetyl-CoA carboxylase (ACC) phosphorylation (Fig. 2G) was significantly increased in fetal liver from trained dams. We next isolated hepatoblasts from embryos of sedentary control dams, and treated them with the AMPK activators, AICAR and metformin. The amount of αKG (Fig. 2H) and the mRNA expression of Tet, Idh (Fig. 2I), and liver metabolic genes were increased by both AICAR and metformin treatment (Fig. 2J). Next, we transiently transfected hepatoblasts with Tet1 or Tet2-specific siRNA and stimulated cells with A769662, a specific AMPK activator.
Figure 2: Maternal Exercise Activates the TET Signaling in Liver and Hepatoblasts of Offspring.
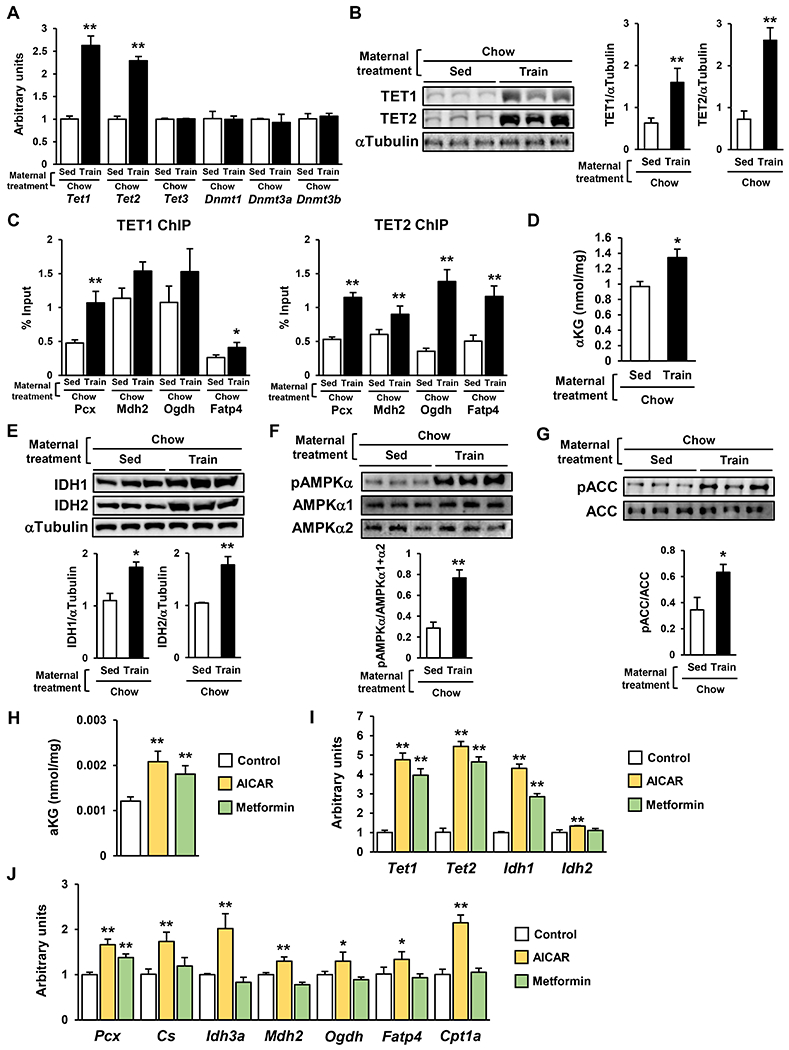
A-B) mRNA expression of Tet and Dnmt (A) and protein expression of TET1 and TET2 (B) in E 13.5 offspring livers from sedentary or trained dams. (n=6, *P < 0.05, **P < 0.01) C) The binding of TET1 and TET2 to the promoter region of liver metabolic genes in E 13.5 offspring liver from sedentary or trained dams. D) The amount of αKG in E13.5 offspring liver from sedentary or trained dams. E-G) The relative protein expression of IDH1 and IDH2 (E) and the phosphorylation of AMPK-α (F) and ACC (G) in the liver of E13.5 offspring from sedentary or trained dams. H-J) The amount of αKG (H) and mRNA expression of Tet, Idh (I), and glucose metabolic genes (J) in 100 μM AICAR or 100 μM metformin-treated hepatoblasts. (n=5, *P < 0.05, **P < 0.01)
Tet1 and Tet2 knockdown inhibited A769662-stimulated hepatic gene expression and DNA demethylation at the promoter of liver metabolic genes (Sup. Fig. 3A & B). Taken together, these data suggest that maternal exercise activates AMPK-IDH-αKG-TET signaling in offspring liver, in order to confer its beneficial metabolic effects.
TET signal upregulates glucose and fatty acid metabolism in primary hepatocytes.
We induced transient overexpression of TET1 and TET2 in primary hepatocytes (Sup. Fig. 4A & B), and analyzed mRNA expression of the hepatic genes that had shown increased-promoter 5-hmC levels in offspring liver as a result of maternal exercise training (Sup. Fig. 4C). TET1 overexpression increased mRNA expression of Ogdh and Fatp4. On the other hand, TET2 overexpression had broad effects on mRNA increases in Pcx, Cs, Idh3a, Ogdh, and Fatp4. The expression of Ogdh and Fatp4 expression was enhanced by combination of TET1 and TET2. Basal and insulin-stimulated rates of glucose production were decreased by the overexpression of TET2 but not TET1, whereas glucagon-mediated hepatocyte glucose production was suppressed by both TET1 and TET2 overexpression (Sup. Fig. 4D). Combined overexpression of TET1 and TET2 resulted in suppressed glucose production under all three conditions. Both TET1 and TET2 overexpression decreased oleic acid-induced cellular content of triglycerides (Sup. Fig. 4E), whereas only TET2 overexpression decreased oleic acid-induced cholesterol accumulation (Sup. Fig. 4F). To validate the basal TET activity in hepatic function, we treated Tet1 or Tet2-specific siRNA transfected hepatocytes (Sup. Fig. 4G) with dimethyl-α-KG (DM-αKG), a cell-permeable type of αKG (Sup. Fig. 4H). DM-αKG-induced increases of glucose metabolic gene expression were suppressed by Tet1 or Tet2 siRNA. Moreover, the decreased glucose production by DM-αKG under basal, insulin, and glucagon-treated conditions was also blocked by Tet1 or Tet2 knockdown (Sup. Fig. 4I). Similarly, the inhibition of Tet1 and Tet2 impaired triglyceride (Sup. Fig. 4J) and cholesterol (Sup. Fig. 4K) metabolism in both basal and DM-αKG-treated hepatocytes. Collectively, these data indicate that high expression and activity of TET enhance hepatic function as characterized by improvements in glucose and fatty acid metabolism.
Trained dams produce a placenta-derived protein, Sod3, that induces activation of AMPK-Tet signaling.
Because maternal exercise induced DNA demethylation in offspring liver at embryological stage E13.5, we reasoned that the serum from trained dams may include circulating factors that induce the observed improvement of hepatic function in hepatoblasts. To test this hypothesis, we isolated hepatoblasts from E13.5 livers born to sedentary control dams. The cells were then stimulated with serum collected from sedentary or trained pregnant females at day 13.5 of pregnancy, or trained non-pregnant females. Serum from sedentary, pregnant dams did not affect the mRNA expression of Tet, Idh, and glucose metabolic genes in hepatoblasts (Fig. 3A). Similarly, serum from trained non-pregnant females had no effect on the expression of these genes. However, treatment with serum from trained pregnant dams increased expression of Tet, Idh and glucose metabolic genes. Likewise, only pregnant, exercise-trained, dam-derived serum significantly increased the phosphorylation level of AMPK in hepatoblasts (Fig. 3B). 80 °C heat-treated serum failed to activate the AMPK-TET axis (Fig. 3C), suggesting that the candidate factor is a protein. These results indicate that the stimulation of hepatoblasts with serum from trained dams mimics the effects of maternal exercise on offspring hepatic gene expression and AMPK-TET signaling.
Figure 3: Serum Derived from Trained Dams Activates AMPK-TET Signaling in Primary Hepatoblasts via SOD3.
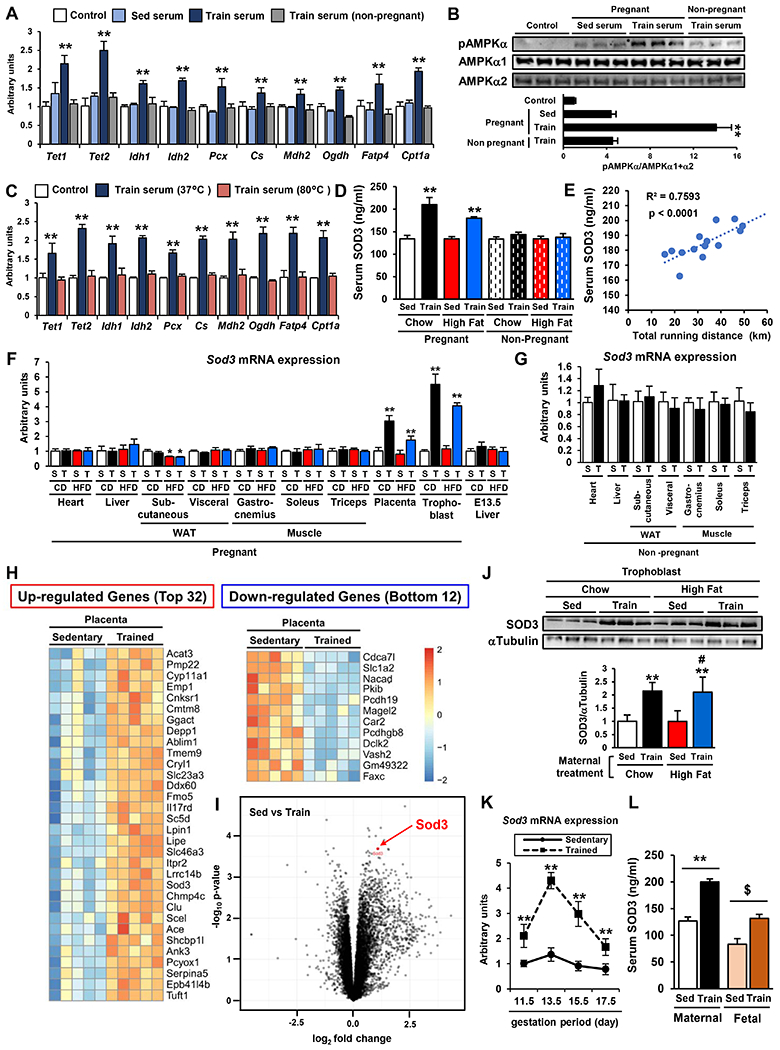
A-B) The mRNA expression of Tet, Idh, and liver metabolic genes (A) and the phosphorylation level of AMPKα (B) in hepatoblasts treated with 10% serum from sedentary or trained pregnant females at 13.5 d.p.c or trained non-pregnant females for 24h. (n=6, **P < 0.01 vs Sed serum) C) The expression of Tet, Idh, and liver metabolic genes in hepatoblasts treated with serum from trained dams after heat treatment at 37 or 80 °C. (n=3, **P < 0.01 vs 80 °C) D) The serum SOD3 in sedentary or trained, chow or HFD fed, pregnant or non-pregnant female at 13.5 d.p.c. (n=6, **P < 0.01 vs Sed-Chow-Pregnant) E) The correlation between serum SOD3 and total running distance in trained dams. (n=14) F-G) Sod3 mRNA expression in various types of tissues and E13.5 offspring livers from sedentary or trained pregnant (F) or non-pregnant (G) females. (CD; chow diet, HFD; high fat diet, S; sedentary, T; Train, n=6, *P < 0.05 vs S-CD, **P < 0.01 vs S-CD) H-I) RNA-seq from the placenta of sedentary or trained dam. (n=5) J) The protein levels of SOD3 in trophoblasts from chow or HFD fed and sedentary or trained dams (n=3, **P < 0.01 vs Sed-Chow, #P <0.01 vs Sed-High Fat) K) The mRNA expression of Sod3 in the placenta from sedentary or trained dams plotted throughout gestation (n=6, **P < 0.01 vs sed) L) Maternal and fetal serum levels of SOD3 in sedentary or trained dams at 13.5 dp.c. (n=7, **P < 0.01 vs Maternal Sed, $P < 0.01 vs Fetal Sed)
To identify candidates that transmit the beneficial effects of maternal exercise on fetal hepatoblasts, serum from sedentary or trained pregnant dams was analyzed by LC-MS/MS. We found a total of 577 proteins in these serum samples with 78 of these proteins being increased 1.5-fold or more in serum from trained dams. Of these 78 proteins, 19 were identified as secretory proteins using the GO database. Because most maternal proteins do not traverse the placental barrier (Sibley et al., 2010; Syme et al., 2004), we hypothesized that the placenta is the organ that can directly transmit the circulating factor to fetal blood, and therefore is likely the source of the factor influencing fetal hepatic function. 4 proteins were highly expressed in the placenta, and only 1 of these proteins, SOD3 has been reported to increase AMPK phosphorylation (Hong et al., 2018). We therefore selected SOD3 as the best candidate to mediate the transmission of the beneficial effects of maternal exercise on offspring.
We analyzed SOD3 protein in the serum from dams and non-pregnant females that were sedentary or trained, chow or HFD-fed at day 13.5 of pregnancy (Fig. 3D). Exercise training increased the serum level of SOD3 in both chow and HFD-fed pregnant females. On the other hand, neither diet nor exercise affected SOD3 levels in non-pregnant females. Serum SOD3 was positively correlated with total daily running distance from day 0 to 13.5 of pregnancy (Fig. 3E). We analyzed Sod3 mRNA expression in multiple tissues from dams including heart, liver, subcutaneous and visceral WAT, gastrocnemius, soleus, triceps skeletal muscle, placenta, and trophoblasts which are the main component of the placenta (Fig. 3F). Exercise during pregnancy increased Sod3 gene expression only in the placenta and trophoblasts. Sod3 expression was not changed in the liver of E13.5 embryos, which were collected at the same time that the pregnant dams were euthanized. Sod3 expression was not changed in these organs from trained non-pregnant females (Fig. 3G). RNA-seq of the placentas from sedentary or trained dams showed that the exercise-induced increases in Sod3 expression were highly significant and Sod3 ranked in the top 22 exercise-responding genes in the placenta (Fig. 3H & I). Protein expression of SOD3 was significantly increased in trophoblasts from trained dams fed either a chow or HFD (Fig. 3J). We also found that maternal exercise-induced Sod3 gene expression in placenta peaked at day 13.5 in the time line of gestation (Fig. 3K). In accordance with the increases of SOD3 in the serum from trained dams, maternal exercise increased the levels of SOD3 in fetal serum (Fig. 3L). These findings demonstrate that maternal exercise markedly increases placental SOD3 secretion into both maternal and fetal blood, making this protein a candidate for mediating the beneficial effects of exercise on offspring liver.
To examine whether SOD3 mimics the maternal exercise-induced AMPK-TET axis and DNA demethylation, hepatoblasts from chow or HFD sedentary dams were treated with recombinant SOD3 protein. The mRNA expression Tet, Idh, and glucose metabolic genes was significantly increased by SOD3 treatment of hepatoblasts from chow-fed dams (Fig. 4A). SOD3 treatment almost fully reversed the suppressive effects of the HFD on hepatic gene expression. Human hepatoblasts treated with recombinant SOD3 protein also showed increased expression of TET, IDH, and glucose metabolic genes (Fig. 4B). TET1 enzymatic activity was upregulated by SOD3 treatment in mouse hepatoblasts (Fig. 4C). We then analyzed the effects of SOD3 on DNA demethylation at the promoter of glucose metabolic genes in hepatoblasts from sedentary chow-fed dams, and found that SOD3 treatment decreased 5-mC levels (Fig. 4D) and increased 5-hmC levels (Fig. 4E) at the CpG island of these genes. Tet1 and Tet2 knockdown significantly attenuated SOD3-induced liver metabolic gene expression in hepatoblasts (Fig. 4F). SOD3 treatment increased AMPKα phosphorylation in both mouse and human hepatoblasts (Fig. 4G), although the level of AMPKα phosphorylation was lower in SOD3-stimulated hepatoblasts compared with 100 μM AICAR or 100 μM metformin-treated hepatoblasts. Transient transfection of hepatoblasts with AMPKα1 or AMPKα2-specific siRNA suppressed basal and SOD3-induced mRNA expression of Tet and Idh (Fig. 4H). Likewise, treatment of the cells with compound C, an inhibitor of AMPK, blocked the effects of SOD3. Taken together, these results suggest that SOD3 promotes liver metabolic gene expression through DNA demethylation by way of an AMPK-TET axis.
Figure 4: SOD3 Treatment Mimics the Effects of Maternal Exercise in Primary Hepatoblasts.
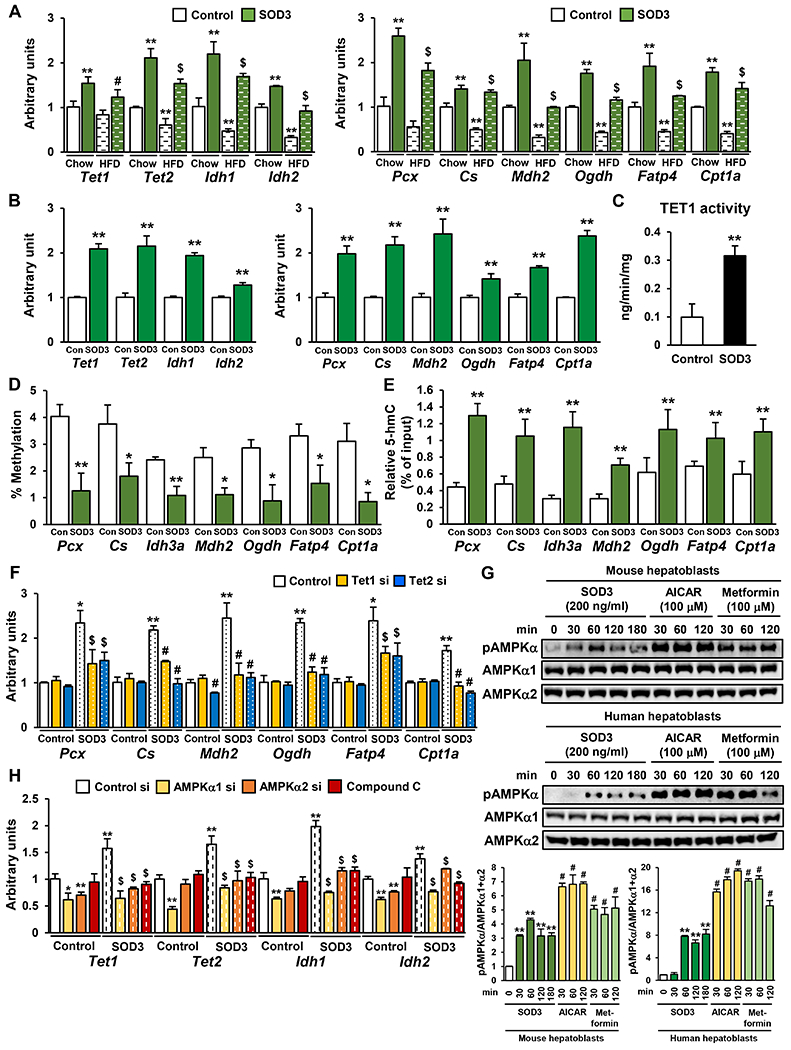
A-B) The expression of Tet, Idh, and liver metabolic genes in mouse hepatoblasts from chow and HFD fed dams (A), and human hepatoblasts (B) treated with 200 ng/ml SOD3 for 24h. (n=3, **P < 0.01 vs Control-Chow, #P < 0.05 vs Control-HFD, $P < 0.01 vs Control-HFD) C-E) TET1 activity (C), 5-mC (D) and 5-hmC levels (E) at the promoter of liver metabolic genes in SOD3-treated mouse hepatoblasts. (n=3, *P < 0.05, **P < 0.01) F) The effects of Tet1 and Tet2 knockdown on SOD3-induced glucose metabolic gene expression in mouse hepatoblasts. (n=3, *P < 0.05 vs Control si-Control, **P < 0.01 vs Control si-Control, $P <0.05 vs Control si-SOD3) G) The phosphorylation of AMPKα in SOD3, AICAR, or metformin-treated mouse and human hepatoblasts. (n=3, **P < 0.01 vs 0 min, #P < 0.01 vs Sod3 30 min) H) The effects of Tet1 and Tet2 knockdown or treament with Compound C on SOD3-induced Tet and Idh mRNA expression in mouse hepatoblasts. (n=3, *P < 0.05 vs Control si-Control, **P < 0.01 vs Control si-Control, $P <0.05 vs Control si-Sod3)
Placenta-specific Sod3 deletion abolishes the beneficial effects of maternal exercise on offspring.
To determine the role of placental SOD3 in the beneficial effects of maternal exercise on offspring metabolic health, we used a trophoblast specific protein α (Tpbpa)/adenosine deaminase (Ada) Cre/loxP system (Zhou et al., 2012) to generate trophoblast-specific Sod3 knockout (Sod3−/−). Average and cumulative running distance during both preconception and gestation were not different between C57BL/6 and flox control (Sod3f/f) (Sup. Fig. 5A & B). SOD3 protein expression was increased by maternal exercise in the placenta from Sod3f/f mice; however, Sod3−/− exhibited a dramatic decrease of SOD3 in the placenta of both sedentary and trained dams (Fig. 5A). Next, we mated sedentary or trained Sod3f/f female mice with Tpbpa/Ada Cre+/−; Sod3f/f male mice to generate both Sod3−/− or Sod3f/f placenta in the same dams (Fig. 5B). The percentage of pups for each genotype within each litter was no differences in the number of offspring between genotypes (Sod3−/−; 46±11%; Sod3f/f; 54±11%). Similarly, there was no difference in the female/male ratio between genotypes (Sod3−/−; 51±17% female; Sod3f/f; 53±11% female). Tet1, Tet2, Cs, Mdh2, Fatp4, and Cpt1a mRNA expression was attenuated in Sod3−/− offspring liver in E13.5 from untrained dams (Fig. 5C). The maternal exercise-induced increase in Tet, Idh and liver metabolic gene expression was fully blocked in Sod3−/− offspring liver. Training of dams did not decrease 5-mC levels in E13.5 livers of Sod3−/− offspring (Sup. Fig. 5C) and 5-hmC was lower in Sod3−/− offspring liver from sedentary and trained dams (Sup. Fig. 5D). Likewise, 5-mC levels in the promoter regions of the hepatic gene was increased in Sod3−/− offspring liver from sedentary dams relative to Sod3f/f controls (Fig. 5D), and was not decreased by maternal exercise. Correspondingly, 5-hmC levels in these promoters was suppressed in the Sod3−/− liver from sedentary dams and was not increased by maternal exercise (Fig. 5E). AMPKα phosphorylation in the offspring liver from trained dams was also inhibited in Sod3−/− (Fig. 5F).
Figure 5: The Effects of Maternal Exercise on the TET-AMPK Signaling Axis and Glucose Metabolism in Offspring Liver are Significantly Impaired by Trophoblast-Specific Sod3 Knockout.
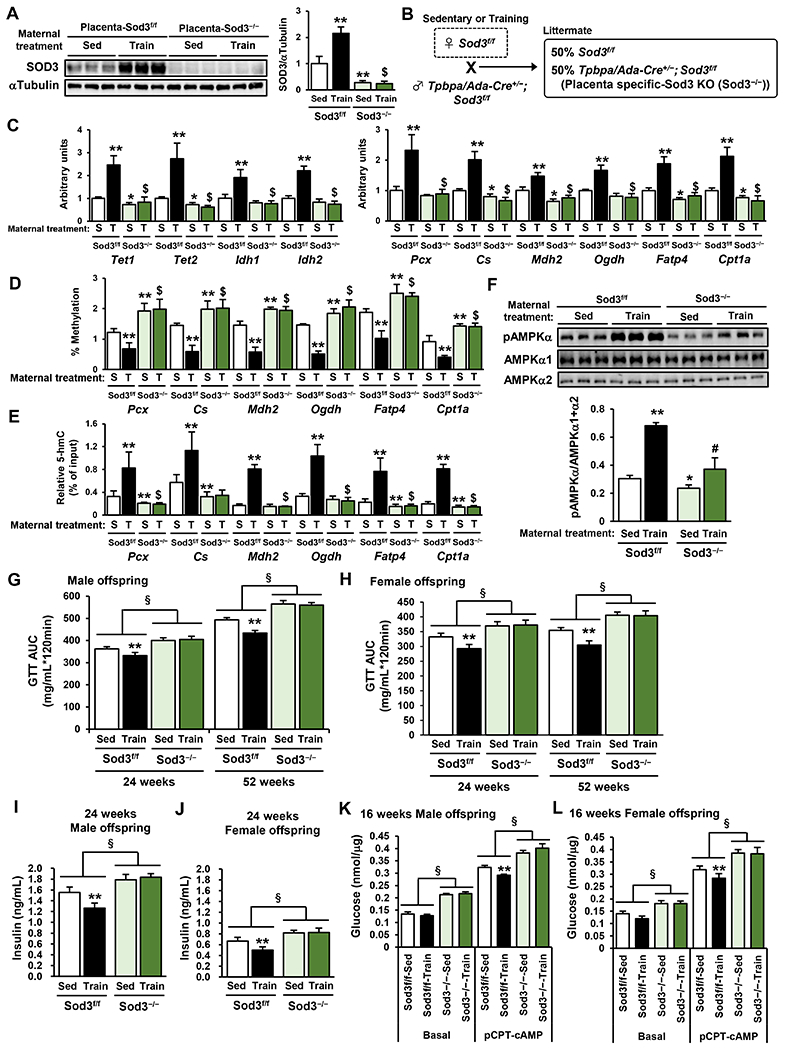
A) SOD3 protein expression in Sod3−/− or Sod3f/f placenta from sedentary or trained dams. (n=6, **P < 0.01 vs Sed- Sod3f/f $P < 0.01 vs Train- Sod3f/f) B) Breeding method to generate both Sod3−/− and Sod3f/f placentae in the same dams. C) The effects of placenta-specific Sod3−/− on the expression of Tet, Idh, and liver metabolic genes (C), the levels of 5-mC (D) and 5-hmC (E) at the promoter of liver metabolic genes, and the phosphorylation levels of AMPKα (F) in E13.5 offspring liver from sedentary or trained dams. (S; Sed, T; Train, n=6, **P < 0.01 vs Sed- Sod3f/f, #P < 0.05 vs Sed-Sod3−/−, $P < 0.01 vs Sed-Sod3−/−) G-H) Glucose tolerance was measured at 24 and 52 weeks of age in Sod3f/f or Sod3−/− offspring from sedentary or trained dams. Glucose area under the curve (AUC) of male (G) and female (H) offspring are shown, respectively. I-J) Fasting serum insulin concentrations of male (I) and female (J) Sod3f/f or Sod3−/− offspring at 24 weeks. K-L) Glucose production in hepatocytes from 16-week-old male (K) and female (L) Sod3f/f or Sod3−/− offspring of sedentary or trained dams. Data are expressed as means ± SEM. (n=3, **P<0.01, §P<0.01 effect of genotype)
We next analyzed metabolic function of adult Sod3−/− offspring. Consistent with our previous data showing that the effects of maternal exercise manifest at later offspring ages (Stanford et al., 2015; Stanford et al., 2017), at 16 weeks of age, male and female Sod3f/f mice from trained dams showed a tendency for improved glucose tolerance as indicated by the glucose excursion curve (Sup. Fig. 5E & F) and area under the curve (Sup. Fig. 5G & H). However, at this age, and at older offspring ages (Fig. 5G & H & Sup. Fig. 5I–L), the Sod3−/− mice had significantly impaired glucose tolerance compared to Sod3f/f mice. Fasting basal glucose levels were also worsened at 52 weeks of age in male and female Sod3f/f mice (Sup. Fig. 5M & N). Maternal exercise improved glucose tolerance in male and female Sod3f/f offspring at 24 and 52 weeks of age, but there was no effect of maternal exercise in male and female Sod3−/− offspring (Fig. 5G & H). Fasting plasma insulin concentrations were higher in male and female Sod3−/− offspring at 24 weeks of age (Fig. 5I & J). Maternal exercise reduced insulin concentrations in Sod3f/f offspring, but this effect was negated in male and female Sod3−/− offspring (Fig. 5I & J). To determine the involvement of placental Sod3 in hepatic function, we measured glucose production in hepatocytes from Sod3f/f or Sod3−/− male and female 16-week-old offspring of sedentary or trained dams (Fig. 5K & L). Basal and pCPT-cAMP-mediated glucose production was increased in hepatocytes from Sod3−/− offspring. Moreover, the maternal exercise-induced decrease in glucose production under pCPT-cAMP conditions was blocked in Sod3−/− hepatocytes. Furthermore, hepatocytes from 16-week-old Sod3−/− offspring showed decreased expression of Pklr, Pcx, Pfkl, Pdha1, Pdk4, Cs, Idh3a, Mdh2, Ogdh, Cd36, Fatp4, Acox1, Cpt1a, Lcad, Mcad and increased expression of G6pc (Sup. Fig. 5O & P). The positive effect of maternal exercise on the expression of these key hepatic genes was also diminished in Sod3−/− offspring livers.
To further examine the importance of SOD3 secretion from placenta for the benefits of maternal exercise on offspring liver, we blocked SOD3 activity using diethyldithiocarbamate trihydrate (DETCA), a SOD3 specific inhibitor (Rahden-Staroń et al., 2012), during hepatic development of offspring of sedentary or trained dams. First, we used in utero development system (Yamada et al., 2008) to injected DETCA or saline into offspring liver at day E13.5. Dams continued to be sedentary or trained for 2 days after DETCA injection, and fetal livers were collected at E15.5 (Sup. Fig. 6A). DETCA treatment significantly inhibited maternal exercise-induced mRNA expression of Tet, Idh, and glucose metabolic genes in hepatoblasts (Sup. Fig. 6B). Next, we treated offspring liver at E13.5 with DETCA or saline using an in exo utero developmental system (Yamada et al., 2008). Dams were sedentary or trained for 5 days after DETCA injection. The caesarean sectioned-newborn mice at day 18.5 were parented by sedentary foster mothers (Sup. Fig. 6C) and glucose production was measured in hepatocytes of male and female 4-week-old offspring. The maternal exercise-induced decrease in glucose production under pCPT-cAMP conditions was blocked in hepatocytes from DETCA-treated dams (Sup. Fig. 6D & E). Taken together, these results show that SOD3 is an exercise-induced, placenta-derived protein that improves glucose homeostasis via epigenetic changes in offspring liver.
Vitamin D receptor (VDR) signaling is necessary to induce Sod3 expression in the placenta.
RNA-seq revealed that 26 transcription factors were differently expressed the placenta from trained dams (Sup. Fig. 7A & B). Among these proteins, 12 transcription factors (Elf3, Foxa2, Foxp1, Gata4, Irf6, Klf5, Klf15, Ppara, Stat5b, Vdr, Zic1) have the potential binding sites to activate the Sod3 promoter, as determined using the JASPAR. We established HEK293 stably transfected with reporter plasmids containing various lengths of the 5’ upstream region of the mouse Sod3 gene and these cells were transfected with overexpression plasmids of 12 candidate transcription factors. Reporter activity was increased by VDR overexpression in HEK293 mSod3 containing −999 to −0 and −1999 to −1000 upstream regions (Sup. Fig. 7C). Forkhead box P1 (FoxP1) overexpression also increased reporter activity in in HEK293 mSod3 containing -1999 to -1000, although to a lower level. Based on these data, we determined the effects of maternal exercise on protein levels of VDR, retinoid X receptor α (RXRα), a coactivator of VDR, and FoxP1 in trophoblasts, and found that only VDR protein was increased by training (Fig. 6A). We overexpressed FLAG-tagged mouse VDR and Myc-tagged mouse RXRα in HEK293 mSod3 containing −999 to −0 and −1999 to −1000 upstream regions (Sup. Fig. 7D) and then treated the cells with vitamin D (Sup. Fig. 7E). Vitamin D treatment resulted in a dose-dependent increase in Sod3 promoter activity in the reporter cells. Combining vitamin D treatment with RXRα overexpression further upregulated vitamin D-induced Sod3 promoter activation. Likewise, the combination of VDR overexpression and vitamin D treatment increased the protein expression of SOD3 in mouse primary trophoblasts (Fig. 6B) and three types of human trophoblast lines, HTR8, JAR, and JEG3 (Fig. 6C).
Figure 6: Normal Vitamin D Levels are Necessary for the Beneficial Effects of Maternal Exercise on Offspring Liver.

A) Protein expression of VDR, RXRα and FoxP1 in the placenta of sedentary or trained dams at 13.5 day (n=3, **P < 0.01 vs Sed-Chow). B-C) SOD3 protein expression in mouse primary trophoblasts (B), HTR8, JAR, and JEG3 (C) with transient overexpression of FLAG-tagged VDR 10 nM vitamin D for 24h. (n=3, **P < 0.01 vs DMSO-Control). D-G) Serum levels of 25(OH) vitamin D (D), placental Vdr and Sod3 mRNA expression (E), serum levels of SOD3 (F), and Tet, Idh, and glucose metabolic gene expression of E13.5 offspring liver (G) from normal (1000 U), low (100 U), or high (10000 U) levels of vitamin D-diet-fed, sedentary or trained dams at 0 or 13.5 day of pregnancy. (n=6, *P < 0.05 vs Normal VD Sed, **P < 0.01 vs Normal VD Sed, $P < 0.01 vs Normal VD Train).
To test the involvement of vitamin D in exercise-regulated placental Sod3 expression in vivo, maternal exercise was combined with dietary manipulation containing normal vitamin D (1000 U), low vitamin D (100 U), or high vitamin D (10000 U). At day 0 and day 13.5 of gestation, serum vitamin D levels were 66% lower in low vitamin D-fed dams and 350% higher in high vitamin D-fed dams compared to normal vitamin D-fed dams (Fig. 6D). Maternal exercise did not affect serum vitamin D levels in any of the three dietary conditions. We found that high vitamin D feeding enhanced the effects of maternal exercise on placental Sod3 expression (Fig. 6E), serum SOD3 concentrations (Fig. 6F), and mRNA expression of Tet, Idh, and glucose metabolic genes (Fig. 6G) in E13.5 offspring liver. Conversely, a low vitamin D impaired the otherwise up-regulated effects of maternal exercise on the placenta and offspring liver (Fig. 6E–G). These results indicate that maternal exercise-induced expression of VDR and vitamin D signaling mediates placental Sod3 expression, and normal vitamin D levels are necessary for the beneficial effects of maternal exercise on offspring liver gene expression.
SOD3 is upregulated in the placenta from active pregnant women.
Next, we sought to translate the involvement of the placental SOD3 secretion to human physiology. First, we collected the serum of pregnant women followed at the Aarhus University Hospital during the 1st (1-13 weeks), 2nd (14-27 weeks), and 3rd (28-40 weeks) trimesters. Interestingly, serum SOD3 level was significantly higher in the 3rd trimester compared to both the 1st and 2nd trimesters (Fig. 7A), and positively correlated with placental weight (Fig. 7B). However, because of high baseline physical activity in this cohort, it was difficult to meaningfully classify individual pregnant women as active or non-active, resulting in no discernible relationship between daily exercise intensity and serum SOD3 level (Fig. 7C). To examine the effects of daily physical activity on SOD3 expression, we sampled from a cohort of pregnant Canadian women who wore accelerometers during pregnancy to track daily physical activity. In this observational study, women were classified as either active or non-active using the Adult Canadian Physical Activity Guidelines (Mottola et al., 2018), which recommend 150 min of moderate-to-vigorous intensity physical activity (MVPA) per week accumulated in 10 min bouts. The serum concentration of SOD3 was significantly increased in active pregnant women when measured at the 2nd trimester (Fig. 7D). The mRNA expression of SOD3 was also significantly upregulated in the placenta of active women collected after parturition (Fig. 7E). Placental SOD3 expression was positively correlated with daily MVPA in pregnant women (Fig. 7F). Collectively, these results show that placental maturation and daily physical activity during pregnancy are positively associated in placental SOD3 in women.
Figure 7: The Effects of Pregnancy Stage and Physical Activity on the Level of SOD3 in Pregnant Women.

A) Serum SOD3 in 1st (1-13 weeks, n=395), 2nd (14-27 weeks, n=366), and 3rd trimesters (28-40 weeks, n=342) of pregnancy. (**P < 0.01) B) Correlation between the serum SOD3 in 3rd trimester and the placental weight at parturition. C) Relationship between the serum SOD3 at 1st, 2nd, and 3rd trimester and daily MVPA. D) Serum SOD3 in physically inactive or active pregnant women at 2nd trimester. E) Sod3 mRNA expression in placenta from physically inactive or active pregnant women at parturition. (n=9, *P < 0.05) F) Correlation between placental Sod3 expression and daily MVPA.
Discussion
Through numerous animal-based studies, maternal exercise has emerged as a potentially promising approach to protect offspring from metabolic disorders (Carter et al., 2012; Carter et al., 2013; Laker et al., 2014; Ribeiro et al., 2017; Sheldon et al., 2016; Stanford et al., 2015; Stanford et al., 2017; Wasinski et al., 2015). While these studies have advanced our knowledge of the physiological response of offspring to maternal exercise, elucidating the molecular mechanisms that mediate the beneficial effects of maternal exercise on offspring glucose homeostasis are essential for developing strategies to prevent the transmission of metabolic disease. Here, we establish that the mechanism for these metabolic effects involves adaptations to the placenta, leading to the secretion of SOD3. We find that SOD3 mediates the transmission of the effects of maternal exercise to offspring through activation of AMPK-TET axis that controls DNA demethylation and gene expression in offspring liver, resulting in improved glucose homeostasis. Placenta-specific Sod3−/− mice are equally glucose intolerant if sedentary or trained, meaning that basal exposure of placental SOD3 to fetal liver is essential for normal glucose homeostasis in offspring.
Exercise in pregnancy has multiple effects on the placenta including vascularization (Son et al., 2019), distribution of nutrient transporters (Mangwiro et al., 2019), and autocrine signaling (Mangwiro et al., 2018). Here, we find that numerous known secretory proteins, including SOD3, are increased in placenta of trained mouse and human mothers, suggesting that the placenta functions as a secretory organ in response to exercise during pregnancy. This is interesting because we know that in the absence of pregnancy, exercise can result in the secretion of cytokines from various organs such as skeletal muscle (Eckel, 2019; Whitham and Febbraio, 2016) and WAT (Mika et al., 2019; Takahashi et al., 2019), and that these factors play important roles in whole body metabolism. This concept is now expanded to include exercise regulation of placental secretory factors. With respect to fetal development, endocrine action by fetal-facing trophoblasts in the placenta is reported to have significant effects on fetal development and subsequent disease risk (Bronson and Bale, 2016; Burton et al., 2016). Our finding, that exercise-induces SOD3 secretion during gestation, makes SOD3 an extremely unique signal because of its ability to be transmitted to embryos, enabling direct exposure of fetal liver to this exercise-induced protein during a critical window in fetal development and programming.
SOD3 has been recognized as a secretory enzyme that dismutates O2•− into H2O2 and oxygen (Wang et al., 2018). Studies of non-pregnant mice have reported that exercise of varying intensities and durations increase Sod3 levels in aorta (Fukai et al., 2000; Hitomi et al., 2008) and skeletal muscle (Hitomi et al., 2008). The roles of exercise-induced SOD3 have been hypothesized to counteract the formation of reactive oxygen species generated as a by-product of oxygen metabolism. We report a new role of SOD3, as an exercise-induced, pregnancy-specific protein and a signal transducer that leads to DNA demethylation. Given that recent studies show that SOD3 functions in kinase activation (Hong et al., 2018; Kemp et al., 2010; Laukkanen, 2016) and gene regulation (Call et al., 2017; Mira et al., 2018; Sah et al., 2016), an emerging concept in biology is that SOD3 plays a fundamental role in numerous cellular functions.
We found that the effects of SOD3 on DNA demethylation were mediated through an AMPK-IDH-αKG-TET axis in hepatoblasts. One previous study has shown that increased αKG levels are correlated with an increase in 5-hmC levels (Yang et al., 2014). AMPK-mediated αKG production is also required for the TET-induced brown adipocyte differentiation (Yang et al., 2016). Interestingly, pharmacological or physiological activation of AMPK can phosphorylate and stabilize TET2, preventing reduction of 5-hmC (Wu et al., 2018). These findings suggest that AMPK-TET axis regulates the epigenome, which ultimately controls cellular physiology and its response to offspring environmental alterations. Given the role of exercise as a significant metabolic inducer of AMPK activation (Richter and Ruderman, 2009), AMPK-mediated regulation of DNA methylation in response to exercise provides an example of how physical activity can affect gene expression. This mechanism explains how exercise can impart a lasting impact, or “exercise memory” on an organism and its tissues. We have not yet identified the specific receptor of SOD3 and the upstream pathway for AMPK. Further investigation is needed to elucidate the complete molecular mechanism surrounding exercise-mediated SOD3 expression and to explore the biological roles of SOD3.
We showed that both TET1 and TET2 were involved in SOD3-induced DNA demethylation in hepatoblasts. TET1 activity is common at promoter sites enriched with CpG islands marked by histone H3 lysine 4 trimethylation (H3K4me3) and H3K27me3 (Wu et al., 2011; Xu et al., 2011). Conversely, TET2 recruitment is primarily mediated with other coupling factors, which are mainly tissue-specific transcription factors (Greenberg and Bourc'his, 2019; Wu and Zhang, 2017). We showed that maternal exercise does not affect the expression of housekeeping genes. These results suggest that the specificity of DNA demethylation at the promoter sites of hepatic genes is regulated by the coupling of TET2 and hepatoblast-specific transcriptional regulator(s) in liver. Additionally, recent studies report that the majority of tissue-specific DNA demethylation occurs at enhancer regions through the combined activity of TET2 and transcription factors (Rasmussen et al., 2019; Sardina et al., 2018). Collectively, DNA demethylation of offspring liver resulting from maternal exercise via SOD3 seems to be due to cooperation between both TET1 and TET2, not only in promoter regions but also in some enhancer regions. Further study is needed to determine the mechanism behind TET enzyme recruitment to liver chromatin.
We found that offspring DNA methylation/demethylation occurred later in pregnancy (day 13.5), but was not affected by maternal exercise in the earlier blastocyst or epiblast stages, which are important stage for embryonic development (Greenberg and Bourc’his, 2019). In accordance, our previous study reported no differences in the rate of conception, sex distribution, or litter size between sedentary dams and trained dams (Stanford et al., 2015; Stanford et al., 2017). In the current study, we found that maternal exercise-induced Sod3 expression in the placenta peaks around 13.5 d.p.c. At this point in the gestational time line, the development of the placental vasculature and the differentiation of trophoblasts are already complete (Rossant and Cross, 2001). This point in gestation also corresponds with the developmental stage of the offspring liver that includes the proliferation and differentiation of hepatoblasts (Gordillo et al., 2015). If we apply our mouse data to human physiology (Carter et al., 2012; Carter et al., 2013; Laker et al., 2014; Ribeiro et al., 2017; Sheldon et al., 2016; Stanford et al., 2015; Stanford et al., 2017; Wasinski et al., 2015), the critical window for maternal exercise to exert its beneficial effects seems to be between the 2nd and 3rd trimester in pregnancy (Otis and Brent, 1954). Accordingly, we found that the serum levels of SOD3 was significantly increased in active pregnant women when measured in the 2nd trimester compared to sedentary control. Of note, our previous study showed that the maximum effects of maternal exercise to improve glucose homeostasis in offspring occurred when maternal exercise was performed both before and during gestation (Stanford et al., 2015). This suggests that exercise before and during the early stage of pregnancy may affect whole body metabolism that can enhance the secretion of SOD3 in pregnancy. The positive effects of daily physical activity on placental SOD3 induction during pregnancy may be clinically translated into an exercise program to benefit pregnant women and their children.
Together, these findings suggest that SOD3 may provide new therapeutic approaches to prevent the transmission of increased risk of metabolic disease from mother to offspring. We found that human hepatoblasts treated with SOD3 protein showed increased hepatic gene expression correlating with DNA demethylation at the promoter of these genes, however in a clinical application, SOD3 would need to be present in umbilical cord blood in order to overcome the placental barrier. In this regard, if SOD3 is to be considered as a therapeutic, maternal exercise is the most efficient way to treat the fetus with placenta-derived SOD3. Our data suggest that serum concentrations of SOD3 could be a valuable biomarker for assessing the benefits of exercise training during gestation and may guide optimization of exercise programs for pregnant women.
Another finding from this work that has important clinical significance is that vitamin D-VDR signaling is essential for maternal exercise to induce placental Sod3 expression. Previous studies using rodent models reported that offspring of mothers with low serum vitamin D during pregnancy have impaired metabolic phenotypes later in life, such as insulin resistance (Ideraabdullah et al., 2019). Maternal vitamin D deficiency has also been reported to significantly increase global DNA methylation in adult offspring liver leading to hepatic inflammation (Zhang et al., 2014). Importantly, we found that in order for maternal exercise to result in epigenetic changes in the offspring liver, the maternal diet must contain normal or high levels of vitamin D. Moreover, neither the intake of high vitamin D in the absence of exercise nor exercise without sufficient dietary vitamin D increased placental SOD3. Future work will be important to determine the exercise-induced stimulators of VDR-Sod3 expression in trophoblasts in order to maximize the benefits of exercise to pregnant women and their offspring as well as enhance the monitoring of maternal and fetal health during gestation.
In conclusion, SOD3 is an exercise-induced, vitamin D-regulated, placenta-derived protein that improves hepatic gene expression and glucose metabolism in offspring liver and regulates in vivo glucose homeostasis in offspring. SOD3 activates an AMPK-IDH-αKG-TET signaling pathway, which is the mechanism controlling DNA demethylation in offspring liver. This study has uncovered the molecular and epigenetic mechanisms that underlie the profound effects of maternal exercise on offspring metabolic health, and provides insight onto the critical roles of the placenta in exercise physiology and its ability to confer significant metabolic benefits that can be propagated across generations.
Limitations of Study
All mouse experiments were performed using only the C57BL/6 strain and therefore results cannot be generalized to other strains. For the human studies, the pregnant women were healthy and recruited solely from the regions of Aarhus or Ottawa, making generalization to all women not possible. We measured placental SOD3 at the time of birth and this may not have been the time of peak of placental SOD3 in the women since the highest level of placental SOD3 in the mice occurred during the second trimester. Maternal exercise-induced SOD3 activated AMPK-TET axis in offspring liver, however the specific mechanisms underlying AMPK activation by SOD3 have not been determined. Although we observed an HFD-mediated effect on DNA methylation, Sod3 expression was not affected, and instead implicates other unidentified factors in diet-induced DNA methylation. Due to the importance of the finding that serum from trained pregnant dams and umbilical cord blood can mimic the effects of maternal exercise on offspring, in vivo SILAC would be another means to find additional critical circulating factors mediating the transmission of exercise benefits. Comprehensive omics analysis including transcriptome and 5-mC/5-hmC-DIP seq should be done to examine the global epigenetic changes in tissues from offspring. Finally, it is potentially important to determine whether knocking out AMPK in fetal liver would affect glucose metabolism in adulthood.
STAR Methods
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joji Kusuyama (joji.kusuyama.c1@tohoku.ac.jp)
Material availability
Mouse lines generated in this study are available from the lead contact upon request.
Data and code availability
There are neither new datasets/code generated nor restrictions for use of the materials in this paper.
Experimental model and subject details
Mouse models
We used the Tpbpa/Ada Cre/loxP system to generate trophoblast-specific Sod3 knockout (Sod3−/−) and flox control (Sod3f/f) mice. Sod3f/f mice were obtained from the University of Colorado (Lob et al., 2011; Nozik-Grayck et al., 2014), and Tpbpa/Ada Cre mice were generated in the University of Texas at Houston (Zhou et al., 2012). Eight to twelve-week old male and female Sod3−/− or Tpbpa/Ada Cre were used for experiments. Tails were genotyped with the following primer sequences. Sod3 flox: forward GAT GAC CTG GGA ACA TGA TGG AGA GG; and reverse GCC TGT TCT GCT AAG CTC TCA CAA AC. Tpbpa/Ada Cre: forward CGG TCT CTG AGA GCC ATC; and reverse CCC TGA ACA TGT CCA TCA. Cre for qPCR: forward TTT CCA TAT TGC AGA ACG AAA ACG; and reverse CAG GCT AAG TGC CTT CTC TAC A. Internal control for qPCR: forward TTT AAG TGG CTT GCC ATT TCT GG; and reverse TCA TCC TAC AGT GGA AGG ATT CAC.
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the Joslin Diabetes Center and were in accordance with National Institutes of Health (NIH) guidelines. 8-week-old C57BL/6 virgin female mice and 7 to 12-week-old Sod3f/f mice were fed a chow (21% kcal from fat; F5020, Lab Diet), high-fat (60% kcal from fat; D12492, Research Diets Inc.), or vitamin D-supplemented diet (AIN-93G, containing normal (1000U), low (100U), or high (10000U) vitamin D, Envigo) for 2 weeks preconception, during gestation, and until pup weaning. Mice were additionally divided into two subgroups: trained (mice housed with running wheels preconception and during gestation) or sedentary (housed in static cages). Male breeders were 10-week-old C57BL/6 mice or 10 to 12-week-old Sod3f/f Tpbpa/Ada Cre+/− mice maintained on a chow diet and were sedentary. To control for potential differences in sires, breeding was done as harems. Litters were culled to six mice, and offspring were chow-fed and housed in static cages (sedentary) from birth onwards.
Primary cell cultures
For primary embryonic mouse hepatoblast and human hepatoblast culture, fetal liver was gently pulled away from the E13.5 embryo and cleaned with forceps to remove associated fibrotic tissues. Collected livers were mechanically disrupted by pipetting in 10 ml of 0.5 mg/ml collagenase type 2 in HEPES buffer (150 mM NaCl, 10 mM HEPES, 5 mM KCl, 1.5 mM Na2HPO4, and pH 7.65), and incubated at 37°C with shaking for 20 min. The enzymatic digestion was stopped by the addition of 15 ml of D-MEM. Cell suspension was filtered through a 100 μm cell strainer and centrifuged at 150 x g for 5 min. Cell pellet was incubated in 10 ml of hemolysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.5 M EDTA, and pH 7.4) for 5 min at room temperature. After centrifugation at 150 x g for 5 min, the cell pellet was resuspended in 250 to 500 <l of Calcium-HEPES buffer (5 mM CaCl2 and 2% FBS in HEPES buffer) to adjust the cell number to 5 to 25 x 107/ml. Hepatoblasts were immunomagnetically sorted using Biotin-conjugated CD324 (E-cadherin) antibody (13-3249-82, eBioscience) and EasySep Release Mouse Biotin Positive Selection Kit (17655, STEMCELL Technologies) according to the manufacturer’s instruction. CD324-positive selected cells were resuspended with Kubota’s Hepatoblast Growth medium (11002, Phoenix Songs). 2 x 105 cells were cultured in collagen-coated plates. Human hepatoblasts were obtained from Phoenix Songs (13002-010, cell line ID: FL-1010) cultured with Kubota’s Hepatoblast Growth medium in Liver Biomatrix Suspension-coated plates (12005-012, Phoenix Songs). Cells were treated with 100 μM AICAR (A-8129, Sigma), 100 μM metformin (D150959, Sigma), A769662 (11900, Cayman Chemical) or 200 ng/ml recombinant SOD3 protein (provided by Dr. Eva Nozik-Grayck from the University of Colorado) with or without 10 μM compound C (11967, Cayman Chemical) for the indicated times. Transfection of 3 unique siRNA duplexes specific to Tet1 (SR423353, Origene), Tet2 (SR423294, Origene), AMPKα1 (Lian et al., 2015), AMPKα2 si (Lian et al., 2015), and control (SR30004, Origene) was performed using Lipofectamine RNAiMAX (13778-030, Invitrogen) for 24 h.
For primary hepatocyte culture, each mouse was anesthetized by pentobarbital. A 30G needle was inserted into the exposed inferior vena cava, and the portal vein was cut for drainage. The first perfusion was performed with 25 ml of EGTA solution (150 mM NaCl, 25 mM Tricine, 5 mM KCl, 0.5 mM EGTA, 0.4 mM KH2PO4, 0.3 mM Na2HPO4, pH 7.4) at 2.2 ml/min flow rate. Subsequently, the second perfusion was continued with 25 ml of Digestion buffer (10 mg of collagenase type 2 and 2% penicillin/streptomycin in Earle’s Balanced Salt Solution (EBSS)) at 2.2 ml/min flow rate. Digested liver was minced with sterilized scissors in 10 ml of D-MEM. The cell suspension was filtered through a 100 μm cell strainer and centrifuged at 150 x g for 3 min. The pellet was resuspended with 15 ml of D-MEM and mixed with 10 ml of 90% Percoll (17-0891-02, GE Healthcare) in HBS (pH 7.05). After centrifugation at 250 x g for 10 min, the cell pellet was washed with D-MEM and resuspended with 10 ml of D-MEM. 1 x 106 cells were cultured in collagen-coated plates. The purity of hepatocytes was evaluated by the mRNA expression of cell-type specific markers (Sup. Fig. 4A). Transfection of pcDNA3-Flag-Tet1 (70129, Addgene) and pcDNA3-Flag-Tet2 (60939, Addgene) was performed using jetPEI-Hepatocyte (102-05N, Polyplus-transfection) according to the manufacturer’s instruction. Transfection of siRNA duplexes was performed with or without 5 mM DM-αKG (349631, Sigma-Aldrich).
For collection of blastocysts, pregnant mice at 3.5 day post coitum (d.p.c.) were euthanized by cervical dislocation. The reproductive organs including ovary, oviduct and two uterine horns were collected as one intact piece and were placed into a 35-mm petri dish filled with prewarmed Embryo MAX M2 medium (MR-015-D, EMD Millipore). The fat pads were removed, and the oviduct and the ovary were cut off from uterine horn. A 27 G needle attached to a 1 ml syringe was filled with prewarmed Embryo MAX M2 medium inserted into one uterine horn, and blastocysts were flushed out with medium twice. This same step was repeated with the other horn. The blastocysts were transferred into 10 μl of prewarmed Embryo MAX M2 medium using a micropipette and washed with several additional drops to clear away any residual uterine debris. DNA methylation was determined in the collected blastocysts with an EZ DNA Methylation-Direct Kit (D5020, Zymo Research) according to the manufacturer’s instructions.
For collection of epiblasts, pregnant mice at 7.5 d.p.c were euthanized by cervical dislocation. The uterine tube was collected from the reproductive organs and placed into a 60-mm petri dish filled with prewarmed Embryo MAX M2 medium (MR-015-D, EMD Millipore). After removing the gonadal fat pads from the uterine tube, each deciduum was dissected from the uterine muscle layers to reveal each embryo using micro forceps under a stereomicroscope. After transferring each embryo to a new 35-mm petri dish filled with Embryo MAX M2 medium, the embryo was cut at the embryonic/extraembryonic boundary. The embryonic fragment which includes epiblast with overlying visceral endoderm was transferred to 10 μl of cold Embryo MAX M2 medium. The visceral ectoderm was peeled away from the epiblasts by micropipette. DNA methylation was determined in the collected epiblasts using an EZ DNA Methylation-Direct Kit (D5020, Zymo Research) according to the manufacturer’s instructions.
Cell lines
HTR8 and JAR, human trophoblast cell lines, were obtained from ATCC (Manassas, VA) and maintained in RPMI-1640 medium (ATCC) containing 10% fetal bovine serum, 50 units/ml penicillin, and 50 mg/ml streptomycin. JEG3, a human trophoblast cell line, was also obtained from ATCC and maintained in EMEM with similar condition.
Human studies
In the Danish cohort, the study was approved by the Regional Committee on Health Research Ethics (j.nr. M-20100048). Written informed consent was obtained from all participants before inclusion. Participants did not receive any financial reimbursement for participating in the study. A total of 400 women were recruited at routine antenatal care visits between gestational week 10 and 13 at Aarhus University Hospital. Women were eligible if they were ≥18 years of age with a singleton pregnancy, had no movement-impairing disabilities and understood Danish. Exclusion criteria were age<18 years, multifetal pregnancy, any physical disability affecting their movement and inadequate knowledge of the Danish language. The final study population was comparable with normal pregnant women regarding age, parity and BMI. Three times during pregnancy, at gestational weeks 10-15, 18-24 and 32-38, participants attended study-related examinations. During each visit they were weighed by a trained nurse and had blood samples drawn. Additionally, the participants had and their daily physical activity measured for the seven days following each visit. Physical activity was measured using Sense Wear Armband Pro3 (SWA) (Bodymedia, Inc., Pittsburgh, PA, USA). SWA was worn over the triceps on the right arm for seven consecutive days, except during bathing and water activities. Sense Wear Professional software (version 6.1) was used to compute energy expenditure, steps per day and metabolic equivalent of task (MET). Days with less than 12 hours of wear time were excluded.
In the Canadian cohort, participants gave informed written consent to participate in either the PhysicaL Activity and diEtary implicatioNs Throughout pregnAncy (PLACENTA) study (file number: H11-15-29) or the Acute Exercise study (file number: H-06-18-634), at the University of Ottawa. Both studies were approved by the Research Ethics Board of the University of Ottawa and conformed with all aspects of the Declaration of Helsinki. Pregnant and non-pregnant women were recruited from the Ottawa region for participation in the Chronic Exercise study. Women aged 18-40 years, with a pre-pregnancy body mass index considered normal or overweight (18.5 – 29.9 kg/m2), and with no contraindication to exercise were included. Those with hypertension, diabetes, or untreated thyroid disease were excluded. Only women in the second trimester (between 13-28 weeks gestation) of pregnancy were included. Free-living physical activity measurements were obtained prospectively from participants for seven consecutive days, during the second trimester (between 24-28 weeks gestation) using an omni-directional Actical® accelerometer (Phillips Respironics, Montreal, QC) worn on the right hip during daytime, except during bathing and aquatic activities. The data reduction and analysis were harmonized with the Canadian Health Measures Survey approach (Colley et al., 2010). The 9 most physical active and the 9 least physically active participants (as determined by their second trimester Actical metrics) were included in our analyses. Classification of the women as “active” or “inactive” was performed using the Adult Canadian physical activity guidelines which recommends 150 min of MVPA per week accumulated in 10-min bouts (Tremblay et al., 2011). Serum samples were collected during the 2nd trimester. The placenta was collected within 30 min of delivery. Biopsied samples of placental villous tissue of approximately 0.5 × 0.5 cm thickness were rinsed in PBS and placed immediately in RNAlater (QIAGEN). After refrigeration for 24 h, the RNAlater was removed and the samples were stored at −80 °C for RNA analysis.
Method details
Trophoblast isolation by cell sorting
Placentas were mechanically minced with scissors, and digested with 10 ml of PluriSTEM Dispase-II (SCM133, EMD Millipore) at 37 °C, shaking for 60 min, followed by the addition of 10 ml of HBSS containing 10% FBS and 25 μg/ml DNase I and incubated at 37 °C for 30 min. Cell suspensions were filtered through 50 μm cell strainers and overlaid on 6 ml of Ficoll-Paque PREMIUM 1.084 (17-5446-02, GE Healthcare). After centrifugation at 700 x g for 20 min, the cell layer was washed with DMEM and resuspended with staining buffer (0.5% BSA, 2mM EDTA, and 0.05% NaN3 in PBS). In order to prevent nonspecific binding of antibodies, single cell suspensions were treated with anti-mouse CD16/32 (clone:93, 101302, BioLegend) and True-Stain Monocyte Blocker (426102, BioLegend) at room temperature for 15 min. The cells were then stained with monoclonal antibodies including FITC-conjugated anti-mouse CD45 (clone:30F-11, 103107, BioLegend), PE/Cy7-conjugated anti-mouse H-2Kb/H-2Db (clone:28-8-6, 114615, BioLegend), PE-conjugated anti-mouse CD31 (clone:MEC13.3, 102507, BioLegend), and APC/Fire750-conjugated anti-mouse CD54 (clone: YN1/1.7.4, 116125, BioLegend) at 4 °C for 20 min. Cells were washed and resuspended with staining buffer with DAPI (1 μg/ml). Placental trophoblast cells (CD45−, H-2Kb/H-2Db−, CD3U−, CD54−) were sorted by flow cytometry (Moflo, Beckman Coulter) at the Joslin Diabetes Center Flowcytometry Core Lab. The gate parameters were determined according to staining with isotype controls.
Dam-derived serum stimulation
Blood was collected via cardiac pucture from both sedentary and trained dams at day 13.5 of pregnancy and 13.5-day non-pregnant trained females, respectively under isoflurane anesthesia. Serum was collected, ah quoted and stored at −80 °C for future use. Primary hepatoblasts isolated from E13.5 embryos from sedentary-chow-fed dams were treated with 10% serum for the indicated times. For heat inactivation studies, serum was incubated at 37°C or 80°C for 10 min .
In vitro glucose production assay
Primary hepatocytes were serum-starved for 12 h and incubated in Krebs-Henseleit-HEPES (KHH) buffer or Mediuml99 (Life Technologies) containing 0.6% BSA, 20 mM sodium lactate, and 5 mM sodium pyruvate with or without 100 μM glucagon (G2044, Sigma-Aldrich), 0.1 mM pCPT-cAMP (C3912, Sigma-Aldrich), and 10 μM insulin (I0516 Sigma-Aldrich) for 4 h. Glucose in culture medium was determined using Glucose Hexokinase Reagent (2821, Eagle Diagnostics) and normalized to total protein levels. Normalized values were used as an index to estimate glucose production.
In vitro fatty acid metabolism assay
Primary hepatocytes were treated with 500 μM oleic acid /3% fatty acid-free BSA or 30% fatty acid-free BSA alone for 24 h. Total cellular triglyceride (TR0100, Sigma Aldrich) and cholesterol (K623-100, BioVision) were analyzed according to manufacturer’s instruction.
Methylation-specific PCR (MSP)
Genomic DNA was isolated from cells and tissues by Quick-gDNA MiniPrep (D3024, Zymo Research). Complete non-methylated DNA was prepared by genomic DNA amplification using REPLI-g Mini Kits (150025, Qiagen). Complete methylated DNA was prepared from amplified DNA using Sss I methyltransferase reaction (M0226, New England BioLabs). Bisulfite conversion of DNA was performed using EZ DNA Methylation Kit (D5001, Zymo Research) according to the manufacturer’s instructions. Quantitative MSP was performed using the pair of methylation primers (M-primers) and unmethylation primers (U-primer) targeting each promoter regions. M-primers and U-primers were designed using Methyl Primer Express Software (Applied Biosystems). The primer sequences used in PCR are shown in Supplemental table 1. To determine the absolute number of copies of methylated or non-methylated DNA, PCR product dilutions ranging from 101 to 108 copies were used to generate standard curves.
5-hmC DNA immunoprecipitation qPCR (5-hmC DIP-qPCR)
10 μg genomic DNA in 100 μl TE buffer was sheared by sonication 6 times for a duration of 1 min each at 25% amplitude power with 30 sec cooling on ice between each pulse. After adjusting the total volume to 480 μl with TE buffer, DNA was heated at 95 °C for 10 min and immediately incubated on ice for 5 min. 80 μl of DNA sample was stored for use as input, and then 100 μl of 5X IP buffer (700 mM NaCl, 50 mM Na3PO4 (pH 7.0), and 0.25% Triton X-100) was added to 400 μl of DNA sample. 250 μl of each DNA sample was incubated with 2.5 μl of 5-hmC antibody (39769, Active Motif) or control normal rabbit IgG (2729, Cell Signaling Technology) overnight at 4 °C. DNA-antibody mixtures were captured by Dynabeads Protein G (10004D, Thermo Fisher Scientific) according to the manufacturer’s instructions. Captured DNA was washed 3 times with 1X IP buffer, and extracted with 250 μl of digestion buffer (50 mM Tris-HCl (pH 8.0), 10 mM EDTA, and 0.5% SDS) add 100 μg of proteinase K at 55 °C for 4 h. DNA was collected by phenol/chloroform/isoamyl alcohol extraction, and precipitated in the presence of 20 μg of glycerol. Finally, DNA was dissolved in 50 μl of TE buffer and subjected to qPCR. Primer sequences used in qPCR for 5-hmC DIP are Pcx: forward TAT AGA TTT TAG GTG GTA TTG GT and reverse TAA AAC AAC ACT ACC AAA CAC AC, Cs: TTT GGT ATT ATA GTG GGT TTT TTG T and reverse AAA ATA AAA CTC CAA CAC ACA AC, Idh3a: forward TGG GAT TGT AAA GTT TTA AGG GGT and reverse CTT TTA ATA ACA CAC AAA ACA A, Mdh2: forward AAT TTA GAG TTT GTT TTT TGG GT and reverse CAC TTC CAA AAC CTA ACA CTA AA, Ogdh: forward GTG GGT TTT AAA GTT GTA GT and reverse ACA CTT CCA AAA CTT ATT AA, Fatp4: forward GGT ATA GAA GGT GGT GTT GTT and reverse CCA AAC CTC AAT ACA TCT AAC, Cptla: forward GTT TTT TGT GTG TTG TTA GT and reverse AAA AAC AAC AAA ATC AAA A..
Chromatin immunoprecipitation (ChIP)
1 x 108 primary hepatoblasts isolated from E13.5 embryonic livers from sedentary or trained dams were cross-linked with 1 ml of 1% formalin in PBS with 10% FBS for 10 min at 25 °C. The fixing reaction was stopped by adding 100 μl of 2.5 M Glycine. After discarding the supernatant, the cellular pellet was washed twice with 500 μl of NP-40 lysis buffer (10 mM NaCl, 10 mM Tris-HCl (pH 7.5), 0.5% NP-40, and protease inhibitor cocktail), and lysed with 100 μl of SDS lysis buffer (50 mM Tris-HCl (pH 8.0), 10 mM EDTA (pH 8.0), 1% SDS, and protease inhibitor cocktail) on ice for 10 min. After incubation, cell lysates were diluted with 400 μl of 1% Triton buffer (150 mM NaCl, 15 mM Tris-HCl (pH 8.0), 1 mM EDTA (pH 8.0), 1% Triton X-100, and protease inhibitor cocktail). The chromatin was sheared by sonication 6 times for 1 min at 25% amplitude power with 30 sec cooling on ice between each pulse. After adding 600 μl of 1% Triton buffer, 100 μl of sample lysates were used as input. Primary antibodies against TET1 (ab191698, abeam) and TET2 (92529, Cell Signaling Technology) or control normal rabbit IgG (2729, Cell Signaling Technology) were captured on Dynabeads M-280 Sheep Anti-Rabbit IgG (11203D, Thermo Fisher Scientific) according to the manufacturer’s instructions. Chromatin fractions were immunoprecipitated with either Dynabeads-anti-TET1 or Dynabeads-anti-TET2, or Dynabeads-IgG overnight at 4 °C. Immune complexes were washed with buffers in the order of low salt buffer (150 mM NaCl, 20 mM Tris-HCl (pH 8.0), 2 mM EDTA, 0.1% SDS, and 1% Triton X-100), high salt buffer (500 mM NaCl, 20 mM Tris-HCl (pH 8.0), 2 mM EDTA, 0.1% SDS, and 1% Triton X-100), LiCl buffer (25 mM LiCl, 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1% NP-40, and 1% Sodium Deoxycholate) and TE buffer, and extracted with 200 μl of salting buffer (400 mM NaCl, 20 mM Tris-HCl (pH 8.0), 10 mM EDTA, and 1% SDS). Cross-linking was reversed with 0.05% SDS and 50 μg RNase A at 65 °C for overnight and digested with 100 μg of proteinase K at 55 °C for 4 h. DNA was collected by phenol/chloroform/isoamyl alcohol extraction, and precipitation in the presence of 20 μg of glycerol. Finally, DNA was dissolved in 50 μl of TE buffer and subjected to qPCR. For each reaction, 1% of cross-link released chromatin was saved and reversed at 65 °C for 6 h followed by proteinase K digestion and DNA extraction, and recovered DNA was used as input control. Primer sequences used in qPCR for ChIP are shown in Supplemental table 2.
Quantitative reverse transcription PCR (RT-qPCR)
Total RNA was isolated with TRI reagent (TR118, Molecular Research Center), and reverse transcribed with High Capacity Reverse Transcription Kits (4368814, Applied Biosystems). Complementary DNA was amplified with Power SYBR Green PCR Master Mix (4367659, Applied Biosystems) using an ABI 7900HT real-time PCR system. For each gene, mRNA expression was calculated relative to that of Rpl13a. Primer sequences used in RT-qPCR for mice and human samples are presented in Supplemental table 3.
ELISA and biochemical assays
Total level of 5-mC and 5-hmC in genomic DNA was determined using MethylFlash Global DNA methylation (5-mC) ELISA Easy Kit (P-1030, EpiGentek) and Global 5-hmC Quantification kit (55018, Active Motif), respectively. Cellular content of αKG was determined by alpha KG Assay Kit (ab83431, abeam). Serum levels of SOD3 was determined using an ELISA, for mouse serum (OKCD01107, Aviva System Biology), for human serum (MBS2701262, MyBioSource). TET1 activity was analyzed by Epigenase 5mC-Hydroxylase TET Activity/Inhibition Assay Kit (P-3086, EpiGentek). Plasma insulin were measured using Ultra Sensitive Mouse Insulin ELISA kits (90080, Crystal Chem Inc.). Plasma 25(OH) vitamin D were measured using total 25-OH Vitamin D IVD ELISA kit (RDKAP1971, R&D systems). All assays were performed according to the manufacturer’s protocols.
Western blotting
Cells and tissues were lysed in PLC lysis buffer (50 mM HEPES (pH 7.0), 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10 mM Na4P2O7, 10% Glycerol, 1% Triton X-100, 0.1% Na3VO4, and protease inhibitor mixture) or RIPA lysis buffer (150 mM NaCl, 1.0% Nonidet P-40, 0.5% deoxycholic acid, 0.1% SDS, 50 mM Tris (pH 8.0), 0.1% Na3VO4, and protease inhibitor mixture), and protein concentrations were determined by Bradford assay (Bio-Rad). Protein lysates were separated by 7.5 to 12.5 % SDS-PAGE gel and transferred to nitrocellulose membranes. The membranes were blocked for 1 h in 5% fat free milk in TBST (20 mM Tris-HCl (pH 7.6), 0.15 M sodium chloride, and 0.1% Tween 20), washed three times with TBST, and incubated for overnight with primary antibodies that probe for TET1 (abl91698, abeam), TET2 (ab124297, abeam), αTubulin (2125, Cell Signaling Technology), IDH1 (8137, Cell Signaling Technology), IDH2 (56439, Cell Signaling Technology), pAMPKα (2531, Cell Signaling Technology), AMPKα1 (07-350, EMD Millipore), AMPKα2 (07-363, EMD Millipore), ACC (3676, Cell Signaling Technology), pACC (11818, Cell Signaling Technology), VDR (12550, Cell Signaling Technology), RXRα (3085, Cell Signaling Technology), FoxP1 (4402, Cell Signaling Technology), FLAG (F1804 Sigma Aldrich), and Myc (2272, Cell Signaling Technology) in TBST. After three washes in TBST, the membranes were incubated for 1 h with horseradish peroxidase-conjugated anti-rabbit immunoglobulin (Millipore) diluted 1:5000 in 5% fat free milk in TBST, and washed three times in TBST. The blots were developed with the enhanced chemiluminescence substrate (0RT2755, PerkinElmer Life Sciences) according to the manufacturer’s instructions, and imaged with a ChemiDoc Touch System (Bio-Rad).
Protein sequence analysis by LC-MS/MS
Serum from sedentary or trained dams at day 13.5 pregnancy was separated by 10 % SDS-PAGE gel and stained with Coomassie Blue. Excised gel bands were cut into approximately 1 mm3 pieces. Gel pieces were then subjected to a modified in-gel trypsin digestion procedure (Shevchenko et al., 1996). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide as described previously (Peng and Gygi, 2001). Peptide sequences were determined by matching protein databases with the acquired fragmentation pattern by the software program, Sequest (Thermo Fisher Scientific) (Eng et al., 1994). All databases include a reversed version of all the sequences and the data was filtered to between a one and two percent peptide false discovery rate.
RNA sequencing
The samples were quantified using an Agilent 4200 Tapestation instrument, with a corresponding Agilent High Sensitivity RNA assay. The resulting RIN (RNA Integrity Number) scores and concentrations were taken into account for qualifying samples to proceed. The samples were then normalized to 2 ng/μL, before using KAPA’s mRNA HyperPrep library preparation kit. A final cleanup was performed using Aline PCRCLean DX beads in a 0.63x SPRI-based cleanup. The resulting purified libraries were run on an Agilent 4200 Tapestation instrument, with a corresponding Agilent High Sensitivity D1000 ScreenTape assay to visualize the libraries and to check the size and concentration of the library. Values obtained from this assay were used to normalize all samples in equimolar ratio. The pool was denatured and loaded onto an Illumina NextSeq instrument, with an Illumina NextSeq High Output 150-cycle kit to obtain Paired-End 75bp reads. The pool was loaded at 1.9 pM, with 5% PhiX spiked in to serve as a sequencing control. The basecall files were demultiplexed through the BioPolymer Facility’s pipeline, and the resulting FASTQ files were used in subsequent analysis.
Intraperitoneal glucose tolerance test (ipGTT)
Mice were fasted for 11 h (21:00 to 08:00) with free access to drinking water. A baseline blood sample was collected from the tails of fully conscious mice, followed by intraperitoneal injection of glucose (2 g glucose/kg body weight), and blood was taken from the tails for glucose measurements at 15, 30, 60, and 120 min using an Infinity Glucose meter.
Histology
Mouse livers were stored in 10% phosphate-buffered formalin (pH 7.4) and processed for paraffin embedding after demineralization in Plank-Rychlo solution. Serial sections were cut at 5–7 μm thickness and stained with haematoxylin and eosin for histometry.
Generation of Sod3 promoter constructs and promoter assay
Mouse and human Sod3 genomic DNA containing various lengths of the 5’ upstream region (−999 to −0, −1999 to −1000, −2999 to −2000, −3999 to −3000) of the putative transcriptional initiation site were cloned by PCR from C57BL/6 and human genomic DNA. PCR products were ligated into a pCR-Blunt II-TOPO vector (Life technologies, Carlsbad, CA), and then the inserts were subcloned into a luciferase reporter vector, pNL2.1 (Promega, San Luis Obispo, CA). The resultant plasmids were stably co-transfected with G418-resistant plasmid into HEK293 cells by Lipofectamine 3000 (L3000015, Invitrogen) according to the manufacturer’s instructions, followed by selection with 1.0 mg/ml G418. As a negative control cell line, pNL2.1 vector was stably transfected into HEK293 cells. After G418 selection, three individual cell lines were isolated for each plasmid. Each HEK293 pNL2.1 Sod3 line was transiently transfected with pcDNA 3.1 overexpression vectors of Elf3, Foxa2, Foxp1, Gata4, Irf6, Klf5, Klf15, Ppara, Stat5b, Vdr, Zic1, or RXRα with or without 2 or 10 nm of vitamin D for 24h. Relative luciferase activities were measured by the Nano-Glo Luciferase Assay System (Promega) according to the manufacturer’s instructions.
In utero and exo utero developmental system
Manipulation and operation on live embryo of mice at E13.5 was performed as described previously (Yamada et al., 2008). Briefly, 8-week-old C57BL/6 virgin female mice were fed a chow (21% kcal from fat; F5020, Lab Diet) for 2 weeks preconception and during gestation. Mice were additionally divided into two subgroups: trained (mice housed with running wheels preconception and during gestation) or sedentary (housed in static cages). Male breeders were 10-week-old C57BL/6 mice maintained on a chow diet and were sedentary. The pregnant mice at E13.5 were anesthetized and the abdominal wall was incised at the midline. In each dam, three embryos on one side of the uterine horn were designated as the experimental group (DETCA group; see below). The three embryos on the other side of the uterine horn were designated as controls (saline group). Diethyldithiocarbamate trihydrate (DETCA) was dissolved in ultrapure water (100 μM), and 1 μl of solution was intraperitoneally injected near the liver of each embryo through the uterine wall (in utero) in the DETCA group. Similarly, the same amount of saline was injected as the vehicle in the controls. In the case of exo utero developmental system, a longitudinal incision was made along the uterine wall to expose the fetuses covered by the embryonic membrane, and DETCA or saline were directly injected through the fetal membrane. After the recovery from anesthesia, the dams went back to their respective cages (sedentary or trained). For the in utero experiment fetal livers were collected at E15.5 (Sup. Fig. 6A). For the exo utero experiment, the embryos at E18.5 were removed from the abdomen, and resuscitated newborns were parented by C57BL/8 foster mothers (Sup. Fig. 6C). Litters were culled to three mice per treatment (DETCA or saline), and offspring were chow-fed and housed in static cages (sedentary) from birth onwards.
Bioinformatic analysis
Reads were aligned to the mouse transcriptome from Ensembl (version. 94) by using kallisto (Bray et al., 2016) and the transcript counts were converted to gene counts by using tximport (Soneson et al., 2015). Gene counts were transformed to log2-counts per million (LogCPM), their mean-variance relationship was estimated, their weights were computed with voom (Law et al., 2014), and their differential expression was assessed using linear modeling with the R package limma (Ritchie et al., 2015). Volcano plots were prepared with R package ggplot2. Gene sets based on Kyoto Encyclopedia of Genes and Genomes (KEGG) were downloaded from the Molecular Signatures Database and the gene set enrichment was tested by using the limma Roast method. This method computed the average t-statistic from testing differential expression over all genes in a pathway, and then it compared this average to averages from many “rotations” of the data set to calculate pathway p-values. P-values were corrected using the Benjamini-Hochberg false discovery rate (FDR), and FDR <0.25 was considered statistically significant.
Quantification and statistical analysis
Statistical analysis
The data are means ± SEM. Statistical significance was defined as P < 0.05 or 0.01 and determined by one- or two-way ANOVA, with Tukey and Bonferroni post hoc analysis. For experiments that were carried out at various ages, statistical analyses were determined based on the control group at that specific time point, and comparisons among ages were not analyzed.
Supplementary Material
Supplemental Table 1: List of primer sequences used for MSP, related to Figure 1, 4, 5. M primers are used for detecting methylated DNA sequences. U primers are used for detecting unmethylated DNA sequences.
Supplemental Table 2: List of primer sequences used for 5hmC-DIP, related to Figure 1, 2, 4, 5.
Supplemental Table 3: List of primer sequences used for RT-qPCR in mouse and human samples, related to Figure 2, 3, 4, 5, 6, 7.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD324 (E-Cadherin) Monoclonal Antibody (DECMA-1), Biotin | eBioscience | Cat# 13-3249-82; RRID: AB_1659688 |
| CD16/32 (clone:93) | BioLegend | Cat# 101302; RRID: AB_312801 |
| FITC-conjugated anti-mouse CD45 (clone:30F-11) | BioLegend | Cat# 103107; RRID: AB_312972 |
| PE/Cy7-conjugated anti-mouse H-2Kb/H-2Db (clone:28-8-6) | BioLegend | Cat# 114615 RRID: AB_2750195 |
| PE-conjugated anti-mouse CD31 (clone:MEC13.3) | BioLegend | Cat# 102507 RRID: AB_312914 |
| APC/Fire750-conjugated anti-mouse CD54 (clone:YN1/1.7.4) | BioLegend | Cat# 116125 RRID: AB_2716073 |
| 5-hmC antibody | Active Motif | Cat# 39769; RRID: AB_10013602 |
| Normal Rabbit IgG | Cell Signaling Technology | Cat# 2729; RRID: AB_1031062 |
| TET1 | abcam | Cat# ab191698; RRID:AB_2858250 |
| TET2 | Cell Signaling Technology | Cat# 92529 RRID:AB_2800188 |
| TET2 | abcam | Cat# ab124297 RRID:AB_2722695 |
| αTubulin | Cell Signaling Technology | Cat# 2125 RRID:AB_2619646 |
| IDH1 | Cell Signaling Technology | Cat# 8137 RRID:AB_10950504 |
| IDH2 | Cell Signaling Technology | Cat# 56439 RRID:AB_2799511 |
| PAMPKα | Cell Signaling Technology | Cat# 2531 RRID:AB_330330 |
| AMPKα1 | EMD Millipore | Cat# 07-350 RRID:AB_310542 |
| AMPKα2 | EMD Millipore | Cat# 07-363 RRID:AB_310553 |
| ACC | Cell Signaling Technology | Cat# 3676 RRID:AB_2219397 |
| Pacc | Cell Signaling Technology | Cat# 11818 RRID:AB_330337 |
| VDR | Cell Signaling Technology | Cat# 12550 RRID:AB_2637002 |
| RXRα | Cell Signaling Technology | Cat# 3085 RRID:AB_11140620 |
| FoxP1 | Cell Signaling Technology | Cat# 4402 RRID:AB_10545755 |
| FLAG M2 | Sigma Aldrich | Cat# F1804 RRID:AB_262044 |
| Myc | Cell Signaling Technology | 2272 RRID: AB_10692100 |
| Biological Samples | ||
| Human Tissue Samples (serum and placenta) | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Kubota’s Hepatoblast Growth medium | Phoenix Songs | Cat# 11002 |
| Liver Biomatrix Suspension-coated plates | Phoenix Songs | Cat# 12005-012 |
| AICAR | Sigma-Aldrich | Cat# D150959 |
| Metformin | Sigma-Aldrich | Cat# A-8129 |
| A769662 | Cayman Chemical | Cat# 11900 |
| recombinant SOD3 protein | Nozik-Grayck et al., 2014 | N/A |
| Compound C | Cayman Chemical | Cat# 11967 |
| Lipofectamine RNAiMAX | Invitrogen | Cat# 13778-030 |
| Percoll | GE Healthcare | Cat# 17-0891-02 |
| jetPEI-Hepatocyte | Polyplus-transfection | Cat# 102-05N |
| DM-αKG | Sigma-Aldrich | Cat# 349631 |
| Embryo MAX M2 medium | EMD Millipore | Cat# MR-015-D |
| PluriSTEM Dispase-II | EMD Millipore | Cat# SCM133 |
| Ficoll-Paque PREMIUM 1.084 | GE Healthcare | Cat# 17-5446-02 |
| True-Stain Monocyte Blocker | BioLegend | Cat# 426102 |
| Glucagon | Sigma-Aldrich | Cat# G2044 |
| pCPT-cAMP | Sigma-Aldrich | Cat# C3912 |
| Insulin | Sigma-Aldrich | Cat# I0516 |
| Lipofectamine 3000 | Invitrogen | Cat# L3000015 |
| Critical Commercial Assays | ||
| EasySep Release Mouse Biotin Positive Selection Kit | STEMCELL Technologies | Cat# 17655 |
| EZ DNA Methylation-Direct Kit | Zymo Research | Cat# D5020 |
| Glucose Hexokinase Reagent | Eagle Diagnostics | Cat# 2821 |
| Triglyceride Determination Kit | Sigma-Aldrich | Cat# TR0100 |
| Total Cholesterol and Cholesteryl Ester Colorimetric Assay Kit II | BioVision | Cat# K623-100 |
| Quick-gDNA MiniPrep | Zymo Research | Cat# D3024 |
| REPLI-g Mini Kits | Qiagen | Cat# 150025 |
| Sss I methyltransferase | New England BioLabs | Cat# M0226 |
| EZ DNA Methylation Kit | Zymo Research | Cat# D5001 |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat# 10004D |
| Dynabeads M-280 Sheep Anti-Rabbit IgG | Thermo Fisher Scientific | Cat# 11203D |
| TRI reagent | Molecular Research Center | Cat# TR118 |
| High Capacity Reverse Transcription Kits | Applied Biosystems | Cat# 4368814 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4367659 |
| MethylFlash Global DNA methylation (5-mC) ELISA Easy Kit | EpiGentek | Cat# P1030 |
| Global 5-hmC Quantification kit | Active Motif | Cat# 55108 |
| alpha KG Assay Kit | abcam | Cat# ab83431 |
| SOD3 ELISA Kit (Mouse) | Aviva System Biology | Cat# OKCD01107 |
| Human Superoxide Dismutase 3, Extracellular (SOD3) ELISA Kit | MyBioSource | Cat# MBS2701262 |
| Epigenase 5mC-Hydroxylase TET Activity/Inhibition Assay Kit | EpiGentek | Cat# P-3086 |
| Ultra Sensitive Mouse Insulin ELISA Kit | Crystal Chem Inc. | Cat# 90080 |
| Total 25-OH Vitamin D IVD ELISA kit | R&D systems | Cat# RDKAP1971 |
| Enhanced chemiluminescence substrate | PerkinElmer Life Sciences | Cat# 0RT2755 |
| Nano-Glo Luciferase Assay System | Promega | Cat# PRN1110 |
| Experimental Models: Cell Lines | ||
| Human hepatoblasts (cell line ID: FL-1010) | Phoenix Songs | Cat# 13002-010 |
| HEK293 | ATCC | N/A |
| HTR8 | ATCC | N/A |
| JAR | ATCC | N/A |
| JEG3 | ATCC | N/A |
| Experimental Models: Organisms/Strains | ||
| Sod3f/f mice | Lob et al., 2011 | N/A |
| Tpbpa/Ada Cre mice | Zhou et al., 2012 | N/A |
| Tpbpa/Ada Cre; Sod3f/f mice | This paper | N/A |
| C57BL/6 | Charles Liver | N/A |
| Oligonucleotides | ||
| Tet1 siRNA | Origene | Cat# SR423353 |
| Tet2 siRNA | Origene | Cat# SR423294 |
| AMPKα1 siRNA | Lian et al., 2015 | N/A |
| AMPKα2 siRNA | Lian et al., 2015 | N/A |
| Control siRNA | Origene | Cat# SR30004 |
| A full list of primers is in Table S3-S6 | This Paper | N/A |
| Recombinant DNA | ||
| pcDNA3-Flag-Tet1 | Wang et al Cell Rep. 2014, Addgene | Cat# 70129 |
| pcDNA3-Flag-Tet2 | Wang et al Cell Rep. 2014, Addgene | Cat# 60939 |
| pCR-Blunt II-TOPO | Life technologies | N/A |
| pNL2.1 | Promega | N/A |
| Software and Algorithms | ||
| Methyl Primer Express Software | Applied Biosystems | N/A |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Prism 8 | GraphPad | N/A |
| Other | ||
| Chow diet | Lab Diet | Cat# F5020 |
| High-fat diet | Research Diets Inc. | Cat# D12492 |
| AIN-93G, containing normal (1000U) vitamin D | Envigo | Custom |
| AIN-93G, containing low (100U) vitamin D | Envigo | Custom |
| AIN-93G, containing high (1000U) vitamin D | Envigo | Custom |
| Wheel running cage | Nalgene | N/A |
| Moflo flow cytometry | Beckman Coulter | N/A |
| SenseWear Armband Pro3 (SWA) | Bodymedia, Inc | N/A |
| omni-directional Actical® accelerometer | Phillips Respironics | N/A |
SOD3 is increased in serum and placenta from physically active pregnant mice and women
Maternal exercise-activated VDR signaling is necessary for placental Sod3 expression
SOD3/AMPK/TET signals demethylation of glucose metabolic gene promoters in fetal liver
Placental SOD3 is required for the benefits of maternal exercise on offspring health
Kusuyama et al. define a novel mechanism for placental transmission of improved metabolic health to offspring. SOD3 is identified as an exercise-induced and vitamin D-regulated placenta-derived protein that improves glucose homeostasis in offspring. SOD3 activates an AMPK/TET signaling axis resulting in epigenetic changes to metabolic genes in offspring liver.
Acknowledgement
This work was supported by NIH grant Awards R01DK101043 (to L.J.G.), P30DK036836 (DRC to Joslin Diabetes Center), 1R35HL139726 (to E.N.G.), K23DK114550 (to R.J.W.M.), R01DK106193 (to P.E.M.), by American Diabetes Association (training grant number 1-17-PMF-009 (to A.B. A-W)), by Canadian Institutes of Health Research MOP142298 (to K.B.A.), by CIHR#142298 (to N.H.V.), by NSERC RGPIN-2017-05457 (to K.B.A.), by Société Francophone du Diabété (to C.A. and L.G.), by Novo Nordisk Foundation (to P.G.O.), by P. Carl Petersen Foundation (to M.M.), and by Sapere Aude Research Leader Grant from the Danish Council for Independent Research # 4183-00384 (to N.J.). J.K. was supported by research fellowships from Sunstar Foundation, JSPS Overseas Research Fellowships, Kanae foundation for the promotion of medical science, and Meiji Yasuda life foundation of health and welfares. We thank Dr. Ross Tomaino from Taplin Mass Spectrometry Facility, Dr. Ashley Ciulla from Biopolymers Facility, Drs. Hui Pan and Jonathan Dreyfuss from the Joslin Bioinformatics and Biostatistics Core, Angela Wood from Joslin Flowcytometry Core, and Laura Hemandez-Lagunas from University of Colorado. We thank Drs. Pasquale Nigro, Maria Vamvini, Susana Rovira Llopis, David Presby, C. Ronald Kahn, Sarah Lessard, Tara MacDonald, Christiano Alves, Jeffrey Richard and Leslie Rowland for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing financial interests
The authors have declared no conflict of interests.
References
- Agarwal P, Morriseau TS, Kereliuk SM, Doucette CA, Wicklow BA, and Dolinsky VW (2018). Maternal obesity, diabetes during pregnancy and epigenetic mechanisms that influence the developmental origins of cardiometabolic disease in the offspring. Critical reviews in clinical laboratory sciences 55, 71–101. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nature biotechnology 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Bronson SL, and Bale TL (2016). The Placenta as a Mediator of Stress Effects on Neurodevelopmental Reprogramming. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 41, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton GJ, Fowden AL, and Thornburg KL (2016). Placental Origins of Chronic Disease. Physiological reviews 96, 1509–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call JA, Donet J, Martin KS, Sharma AK, Chen X, Zhang J, Cai J, Galarreta CA, Okutsu M, Du Z, et al. (2017). Muscle-derived extracellular superoxide dismutase inhibits endothelial activation and protects against multiple organ dysfunction syndrome in mice. Free radical biology & medicine 113, 212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter LG, Lewis KN, Wilkerson DC, Tobia CM, Ngo Tenlep SY, Shridas P, Garcia-Cazarin ML, Wolff G, Andrade FH, Charnigo RJ, et al. (2012). Perinatal exercise improves glucose homeostasis in adult offspring. American journal of physiology. Endocrinology and metabolism 303, E1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter LG, Qi NR, De Cabo R, and Pearson KJ (2013). Maternal exercise improves insulin sensitivity in mature rat offspring. Medicine and science in sports and exercise 45, 832–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley R, Connor Gorber S, and Tremblay MS (2010). Quality control and data reduction procedures for accelerometry-derived measures of physical activity. Health reports 21, 63–69. [PubMed] [Google Scholar]
- Duque-Guimaraes DE, and Ozanne SE (2013). Nutritional programming of insulin resistance: causes and consequences. Trends in endocrinology and metabolism: TEM 24, 525–535. [DOI] [PubMed] [Google Scholar]
- Eckel J (2019). Myokines in metabolic homeostasis and diabetes. Diabetologia 62, 1523–1528. [DOI] [PubMed] [Google Scholar]
- Elshenawy S, and Simmons R (2016). Maternal obesity and prenatal programming. Molecular and cellular endocrinology 435, 2–6. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Fukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G, and Harrison DG (2000). Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. The Journal of clinical investigation 105, 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glastras SJ, Chen H, Pollock CA, and Saad S (2018). Maternal obesity increases the risk of metabolic disease and impacts renal health in offspring. Bioscience reports 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordillo M, Evans T, and Gouon-Evans V (2015). Orchestrating liver development. Development (Cambridge, England) 142, 2094–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg MVC, and Bourc’his D (2019). The diverse roles of DNA methylation in mammalian development and disease. Nature reviews. Molecular cell biology. [DOI] [PubMed] [Google Scholar]
- Hitomi Y, Watanabe S, Kizaki T, Sakurai T, Takemasa T, Haga S, Ookawara T, Suzuki K, and Ohno H (2008). Acute exercise increases expression of extracellular superoxide dismutase in skeletal muscle and the aorta. Redox report : communications in free radical research 13, 213–216. [DOI] [PubMed] [Google Scholar]
- Hong YA, Lim JH, Kim MY, Kim Y, Park HS, Kim HW, Choi BS, Chang YS, Kim TY, and Park CW (2018). Extracellular Superoxide Dismutase Attenuates Renal Oxidative Stress Through the Activation of Adenosine Monophosphate-Activated Protein Kinase in Diabetic Nephropathy. Antioxidants & redox signaling 28, 1543–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideraabdullah FY, Belenchia AM, Rosenfeld CS, Kullman SW, Knuth M, Mahapatra D, Bereman M, Levin ED, and Peterson CA (2019). Maternal vitamin D deficiency and developmental origins of health and disease (DOHaD). The Journal of endocrinology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp K, Hares K, Mallam E, Heesom KJ, Scolding N, and Wilkins A (2010). Mesenchymal stem cell-secreted superoxide dismutase promotes cerebellar neuronal survival. Journal of neurochemistry 114, 1569–1580. [DOI] [PubMed] [Google Scholar]
- Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, and Nathan DM (2002). Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. The New England journal of medicine 346, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusuyama J, Alves-Wagner AB, Makarewicz NS, and Goodyear LJ (2020). Effects of maternal and paternal exercise on offspring metabolism. Nature metabolism 2, 858–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laker RC, Lillard TS, Okutsu M, Zhang M, Hoehn KL, Connelly JJ, and Yan Z (2014). Exercise prevents maternal high-fat diet-induced hypermethylation of the Pgc-1alpha gene and age-dependent metabolic dysfunction in the offspring. Diabetes 63, 1605–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukkanen MO (2016). Extracellular Superoxide Dismutase: Growth Promoter or Tumor Suppressor? Oxidative medicine and cellular longevity 2016, 3612589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome biology 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian K, Du C, Liu Y, Zhu D, Yan W, Zhang H, Hong Z, Liu P, Zhang L, Pei H, et al. (2015). Impaired adiponectin signaling contributes to disturbed catabolism of branched-chain amino acids in diabetic mice. Diabetes 64, 49–59. [DOI] [PubMed] [Google Scholar]
- Lob HE, Vinh A, Li L, Blinder Y, Offermanns S, and Harrison DG (2011). Role of vascular extracellular superoxide dismutase in hypertension. Hypertension (Dallas, Tex. : 1979) 58, 232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Hajkova P, and Ecker JR (2018). Dynamic DNA methylation: In the right place at the right time. Science (New York, N.Y.) 361, 1336–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangwiro YTM, Cuffe JSM, Briffa JF, Mahizir D, Anevska K, Jefferies AJ, Hosseini S, Romano T, Moritz KM, and Wlodek ME (2018). Maternal exercise in rats upregulates the placental insulin-like growth factor system with diet- and sex-specific responses: minimal effects in mothers bom growth restricted. The Journal of physiology 596, 5947–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangwiro YTM, Cuffe JSM, Mahizir D, Anevska K, Gravina S, Romano T, Moritz KM, Briffa JF, and Wlodek ME (2019). Exercise initiated during pregnancy in rats bom growth restricted alters placental mTOR and nutrient transporter expression. The Journal of physiology 597, 1905–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika A, Macaluso F, Barone R, Di Felice V, and Sledzinski T (2019). Effect of Exercise on Fatty Acid Metabolism and Adipokine Secretion in Adipose Tissue. Frontiers in physiology 10, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira E, Carmona-Rodriguez L, Perez-Villamil B, Casas J, Fernandez-Acenero MJ, Martinez-Rey D, Martin-Gonzalez P, Heras-Murillo I, Paz-Cabezas M, Tardaguila M, et al. (2018). SOD3 improves the tumor response to chemotherapy by stabilizing endothelial HIF-2alpha. Nature communications 9, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottola MF, Davenport MH, Ruchat SM, Davies GA, Poitras VJ, Gray CE, Jaramillo Garcia A, Barrowman N, Adamo KB, Duggan M, et al. (2018). 2019 Canadian guideline for physical activity throughout pregnancy. British journal of sports medicine 52, 1339–1346. [DOI] [PubMed] [Google Scholar]
- Nozik-Grayck E, Woods C, Taylor JM, Benninger RK, Johnson RD, Villegas LR, Stenmark KR, Harrison DG, Majka SM, Irwin D, et al. (2014). Selective depletion of vascular EC-SOD augments chronic hypoxic pulmonary hypertension. American journal of physiology. Lung cellular and molecular physiology 307, L868–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis EM, and Brent R (1954). Equivalent ages in mouse and human embryos. The Anatomical record 120, 33–63. [DOI] [PubMed] [Google Scholar]
- Peng J, and Gygi SP (2001). Proteomics: the move to mixtures. Journal of mass spectrometry : JMS 36, 1083–1091. [DOI] [PubMed] [Google Scholar]
- Rahden-Staroń I, Grosicka-Maciqg E, Kurpios-Piec D, Czeczot H, Grzela T, and Szumilo M (2012). The effects of sodium diethyldithiocarbamate in fibroblasts V79 cells in relation to cytotoxicity, antioxidative enzymes, glutathione, and apoptosis. Archives of toxicology 86, 1841–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen KD, Berest I, Kebetaler S, Nishimura K, Simon-Carrasco L, Vassiliou GS, Pedersen MT, Christensen J, Zaugg JB, and Helin K (2019). TET2 binding to enhancers facilitates transcription factor recruitment in hematopoietic cells. Genome research 29, 564–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds CM, Gray C, Li M, Segovia SA, and Vickers MH (2015). Early Life Nutrition and Energy Balance Disorders in Offspring in Later Life. Nutrients 7, 8090–8111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro TA, Tofolo LP, Martins IP, Pavanello A, de Oliveira JC, Prates KV, Miranda RA, da Silva Franco CC, Gomes RM, Francisco FA, et al. (2017). Maternal low intensity physical exercise prevents obesity in offspring rats exposed to early ovemutrition. Scientific reports 7, 7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter EA, and Ruderman NB (2009). AMPK and the biochemistry of exercise: implications for human health and disease. The Biochemical journal 418, 261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossant J, and Cross JC (2001). Placental development: lessons from mouse mutants. Nature reviews. Genetics 2, 538–548. [DOI] [PubMed] [Google Scholar]
- Sah SK, Park KH, Yun CO, Kang KS, and Kim TY (2016). Effects of Human Mesenchymal Stem Cells Transduced with Superoxide Dismutase on Imiquimod-Induced Psoriasis-Like Skin Inflammation in Mice. Antioxidants & redox signaling 24, 233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardina JL, Collombet S, Tian TV, Gomez A, Di Stefano B, Berenguer C, Brumbaugh J, Stadhouders R, Segura-Morales C, Gut M, et al. (2018). Transcription Factors Drive Tet2-Mediated Enhancer Demethylation to Reprogram Cell Fate. Cell stem cell 23, 727–741 e729. [DOI] [PubMed] [Google Scholar]
- Sferruzzi-Perri AN, and Camm EJ (2016). The Programming Power of the Placenta. Frontiers in physiology 7, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon RD, Nicole Blaize A, Fletcher JA, Pearson KJ, Donkin SS, Newcomer SC, and Rector RS (2016). Gestational exercise protects adult male offspring from high-fat diet-induced hepatic steatosis. Journal of hepatology 64, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, and Mann M (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Analytical chemistry 68, 850–858. [DOI] [PubMed] [Google Scholar]
- Sibley CP, Brownbill P, Dilworth M, and Glazier JD (2010). Review: Adaptation in placental nutrient supply to meet fetal growth demand: implications for programming. Placenta 31 Suppl, S70–74. [DOI] [PubMed] [Google Scholar]
- Son JS, Liu X, Tian Q, Zhao L, Chen Y, Hu Y, Chae SA, de Avila JM, Zhu MJ, and Du M (2019). Exercise prevents the adverse effects of maternal obesity on placental vascularization and fetal growth. The Journal of physiology 597, 3333–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford KI, Lee MY, Getchell KM, So K, Hirshman MF, and Goodyear LJ (2015). Exercise before and during pregnancy prevents the deleterious effects of maternal high-fat feeding on metabolic health of male offspring. Diabetes 64, 427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford KI, Takahashi H, So K, Alves-Wagner AB, Prince NB, Lehnig AC, Getchell KM, Lee MY, Hirshman MF, and Goodyear LJ (2017). Maternal Exercise Improves Glucose Tolerance in Female Offspring. Diabetes 66, 2124–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syme MR, Paxton JW, and Keelan JA (2004). Drug transfer and metabolism by the human placenta. Clinical pharmacokinetics 43, 487–514. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Alves CRR, Stanford KI, Middelbeek RJW, Pasquale N, Ryan RE, Xue R, Sakaguchi M, Lynes MD, So K, et al. (2019). TGF-beta2 is an exercise-induced adipokine that regulates glucose and fatty acid metabolism. Nature metabolism 1, 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay MS, Warburton DE, Janssen T, Paterson DH, Latimer AE, Rhodes RE, Kho ME, Hicks A, Leblanc AG, Zehr L, et al. (2011). New Canadian physical activity guidelines. Applied physiology, nutrition, and metabolism = Physiologic appliquee, nutrition et metabolisme 36, 36–46; 47–58. [DOI] [PubMed] [Google Scholar]
- Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H., Ilanne-Parikka P, Keinanen-Kiukaanniemi S, Laakso M, Louheranta A, Rastas M, et al. (2001). Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. The New England journal of medicine 344, 1343–1350. [DOI] [PubMed] [Google Scholar]
- van Dijk SJ, Tellam RL, Morrison JL, Muhlhausler BS, and Molloy PL. (2015). Recent developments on the role of epigenetics in obesity and metabolic disease. Clinical epigenetics 7, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers MH (2014). Early life nutrition, epigenetics and programming of later life disease. Nutrients 6, 2165–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Branicky R, Noe A, and Hekimi S (2018). Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. The Journal of cell biology 217, 1915–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasinski F, Bacurau RF, Estrela GR, Klempin F, Arakaki AM, Batista RO, Mafra FF, do Nascimento LF, Hiyane MI, Velloso LA, et al. (2015). Exercise during pregnancy protects adult mouse offspring from diet-induced obesity. Nutrition & metabolism 12, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitham M, and Febbraio MA (2016). The ever-expanding myokinome: discovery challenges and therapeutic implications. Nature reviews. Drug discovery 15, 719–729. [DOI] [PubMed] [Google Scholar]
- Wu D, Hu D, Chen H, Shi G, Fetahu IS, Wu F, Rabidou K, Fang R, Tan L, Xu S, et al. (2018). Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 559, 637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, Zhao K, Sun YE, and Zhang Y (2011). Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 473, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, and Zhang Y (2017). TET-mediated active DNA demethylation: mechanism, function and beyond. Nature reviews. Genetics 18, 517–534. [DOI] [PubMed] [Google Scholar]
- Xu Y, Wu F, Tan L, Kong L, Xiong L, Deng J, Barbera AJ, Zheng L, Zhang H, Huang S, et al. (2011). Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Molecular cell 42, 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Hatta T, and Otani H (2008). Mouse exo utero development system: protocol and troubleshooting. Congenital anomalies 48, 183–187. [DOI] [PubMed] [Google Scholar]
- Yang H, Lin H, Xu H, Zhang L, Cheng L, Wen B, Shou J, Guan K, Xiong Y, and Ye D (2014). TET-catalyzed 5-methylcytosine hydroxylation is dynamically regulated by metabolites. Cell research 24, 1017–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Liang X, Sun X, Zhang L, Fu X, Rogers CJ, Berim A, Zhang S, Wang S, Wang B, et al. (2016). AMPK/alpha-Ketoglutarate Axis Dynamically Mediates DNA Demethylation in the Prdm16 Promoter and Brown Adipogenesis. Cell metabolism 24, 542–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Chu X, Huang Y, Li G, Wang Y, Li Y, and Sun C (2014). Maternal vitamin D deficiency during pregnancy results in insulin resistance in rat offspring, which is associated with inflammation and Ikappabalpha methylation. Diabetologia 57, 2165–2172. [DOI] [PubMed] [Google Scholar]
- Zheng J, Xiao X, Zhang Q, and Yu M (2014). DNA methylation: the pivotal interaction between early-life nutrition and glucose metabolism in later life. The British journal of nutrition 112, 1850–1857. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Ley SH, and Liu FB (2018). Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nature reviews. Endocrinology 14, 88–98. [DOI] [PubMed] [Google Scholar]
- Zhou CC, Chang J, Mi T, Abbasi S, Gu D, Huang L, Zhang W, Kellems RE, Schwartz RJ, and Xia Y (2012). Targeted expression of Cre recombinase provokes placental-specific DNA recombination in transgenic mice. PloS one 7, e29236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: List of primer sequences used for MSP, related to Figure 1, 4, 5. M primers are used for detecting methylated DNA sequences. U primers are used for detecting unmethylated DNA sequences.
Supplemental Table 2: List of primer sequences used for 5hmC-DIP, related to Figure 1, 2, 4, 5.
Supplemental Table 3: List of primer sequences used for RT-qPCR in mouse and human samples, related to Figure 2, 3, 4, 5, 6, 7.
Data Availability Statement
There are neither new datasets/code generated nor restrictions for use of the materials in this paper.