

SERINC5 (S5) is a host cell restriction factor (RF) that impairs the infectivity of HIV-1 particles in target cell lines. To assess the potential physiological relevance of this restriction, we assessed the effects of S5 on HIV-1 infection of relevant primary human target cells.
KEYWORDS: HIV-1 Nef, SERINC5, primary cells, proinflammatory cytokines, HIV-1 Nef
ABSTRACT
HIV-1 has to overcome physical barriers posed by host cell restriction factors (RFs) for efficient replication. Some RFs, including Trim5α and tetherin, trigger antiviral signaling in addition to directly impairing HIV replication. SERINC5 (S5) is an RF that is incorporated into HIV-1 particles to potently impair their infectivity and is efficiently antagonized by the viral pathogenesis factor Nef. Since effects of S5 on HIV-1 infectivity were mostly studied in reporter cell lines, we analyzed the effects of S5 during infection of primary HIV-1 target cells. In activated CD4+ T lymphocytes, virion incorporation of S5 only moderately impaired virion infectivity and was not associated with altered innate immune recognition. In contrast, in monocyte-derived macrophages, S5 virion incorporation potentiated the production of proinflammatory cytokines with very potent but donor-dependent effects on virion infectivity. Nef counteracted effects of S5 on both cytokine production and virion infectivity. Similar S5-induced cytokine production was observed in immature monocyte-derived dendritic cells. Notably, S5-mediated enhancement of cytokine production was not linked to the efficacy of productive infection and could be overcome by using vesicular stomatitis virus glycoprotein (VSV-G) but not infectivity restriction-insensitive HIV-1 Env for cell entry. Moreover, inhibiting entry of S5-negative HIV-1 ΔNef particles increased proinflammatory cytokine production comparably to virion incorporation of S5. Together, these results describe the sensitization of noninfectious HIV-1 particles to proinflammatory cytokine production by myeloid target cells as an additional and Nef-sensitive activity of S5. Moreover, the study reveals important cell-type and donor-dependent differences in the sensitivity of HIV target cells for antiviral effects of S5.
IMPORTANCE SERINC5 (S5) is a host cell restriction factor (RF) that impairs the infectivity of HIV-1 particles in target cell lines. To assess the potential physiological relevance of this restriction, we assessed the effects of S5 on HIV-1 infection of relevant primary human target cells. We found that effects of S5 on infection of CD4+ T lymphocytes were negligible. In myeloid target cells, however, virion incorporation of S5 potently suppressed infectivity and promoted innate immune recognition of HIV-1 particles characterized by proinflammatory cytokine production. Both effects were not observed in cells of all donors analyzed, were exerted independently of one another, and were counteracted by the HIV-1 pathogenesis factor Nef. These results identify the sensitization of HIV-1 particles for innate immune recognition by myeloid target cells as a novel activity of S5 and emphasize the need to study RF function in the context of primary target cells and taking donor variabilities into account.
INTRODUCTION
Upon transmission to a new host, human immunodeficiency virus (HIV) faces numerous defense mechanisms that need to be bypassed to ensure efficient virus replication and spread. This includes adaptive responses, such as lysis of infected cells by cytotoxic T or natural killer cells, as well as humoral responses with the potential of generating neutralizing antibodies (1, 2). However, in the initial phase of infection, innate immune mechanisms are of critical importance and aim at fending off virus infection as well as at priming subsequent adaptive responses (3, 4). Key effectors of innate immunity to HIV infection include the cell-intrinsic ability to elicit signaling triggered by the sensing of viral structures (pathogen-associated molecular patterns [PAMPs]). Such signaling induces antiviral states in infected and bystander cells and drives expression of physical barriers to virus replication (so-called “restriction factors” [RFs]), whose expression is often induced by antiviral signaling. Intense research in the past decade identified that human cells dispose of an impressive array of RFs that collectively target virtually any step of HIV replication, from virus entry and intracellular replication steps to the release of new viral progeny (5, 6). For the prominent RFs Trim5α and tetherin, it was further identified that their respective roles as physical barriers to incoming HIV capsids or virions budding from assembly sites are coupled to the ability to elicit antiviral signaling (7–9). Some RFs hence are capable of directly restricting HIV replication as well as facilitating the establishment of an antiviral state. Because of the collective antiviral potency of host cell RFs, productive infection of such target cells by HIV requires specific countermeasures, which typically are provided by the expression of a viral antagonist that inactivates the RF or the ability of the virus to tolerate modifications in the structure targeted by the RF.
With Vpu, Vpr, Vif, and Nef, HIV-1 encodes four accessory proteins that are dispensable for virus growth in cell culture but are important pathogenic determinants in vivo. While the role of Vpu, Vpr, or Vif as RF antagonists is well established, it was only a few years ago that proteins of the serine incorporator (SERINC) family were identified as host cell RFs that are antagonized by the HIV-1 pathogenesis factor Nef (10, 11). In particular, SERINC5 (S5) potently reduces the infectivity of HIV-1 particles when expressed during virus production. Independently of the reported ability of SERINC proteins to alter membrane lipid composition (12, 13), S5 seems to act by impairing fusion of HIV particles with target cells (10, 11, 14). Recent data indicate that this activity results from the incorporation of S5 into virions, where it interferes with the fusogenic potential of the viral glycoprotein Env (14–16). The antagonism of this activity by Nef reflects its ability to reduce virion incorporation of the RF but also a yet to be identified mechanism to inactivate virion-associated S5 (10, 11, 17). Whether S5 impacts HIV replication by effects beyond its ability to reduce virion infectivity has not yet been explored.
Suppression of HIV-1 virion infectivity by S5 that was ectopically expressed during virus production has unambiguously been reported by a number of studies that produced HIV from 293T cells and quantified the infectivity of produced virions on single-round reporter cell lines, such as TZM-bl cells (10, 11, 14, 15, 17–21). The ability of S5 to affect HIV-1 replication in primary target cells, however, has not been studied in much detail. One study observed a restriction to virion infectivity by S5 in primary human CD4+ T cells, but the magnitude of this effect was moderate and did not amplify over several rounds of infection (10). This suggests that S5 may specifically act on infection with cell-free virus, while cell-cell transmission, which constitutes the more efficient mode of transmission in CD4+ T cell cultures (22–24), may be relatively insensitive to S5. However, S5 was recently demonstrated to potently impair the spread of murine leukemia virus in mice (25), which depends on efficient cell-cell transmission (26–28). Collectively, these results indicated that S5 may exert additional antiviral functions to impairing the infectivity of cell-free virions and warranted investigating the effects of S5 during infection of primary target cells. We therefore set out in this study to address whether the S5 restriction to cell-free infection is relevant in primary human target cells and to test if S5 has an impact on innate immune reactions to HIV-1 particles of these cells.
RESULTS
SERINC5 affects infectivity but not innate recognition of HIV particles in activated CD4+ T cells.
The aim of this study was to assess the impact of S5 incorporation into HIV-1 particles for infection and innate immune recognition in primary target cells. In a first series of experiments, we focused on CD4+ T cells as targets. To this end, wild-type (WT) and nef-negative (ΔNef) variants of CXCR4 (X4)-tropic HIV-1 NL4-3SF2nef were produced in 293T cells cotransfected with an expression plasmid for S5 or a control vector. As expected (10, 11, 17), testing of the relative infectivity of these virus stocks in single rounds of infection on TZM-bl reporter cells revealed that the presence of S5 during virus production did not significantly impair the infectivity of the WT but decreased the infectivity of ΔNef particles more than 10-fold (Fig. 1A, upper panel). Consistent with an S5-mediated restriction of virion infectivity and antagonism thereof by Nef, the viral protein markedly reduced the amounts of the RF that were incorporated into virus particles (Fig. 1A, lower panel). These viruses, normalized for the amount of virus particles as determined by reverse transcriptase (RT) activity quantification, were then used to infect primary human CD4+ T cells that had been activated with phytohemagglutinin (PHA) and interleukin-2 (IL-2) for 3 days, and infection rates were determined by fluorescence-activated cell sorter (FACS) analysis for intracellular p24 3 days postinfection (p.i.) (Fig. 1B). S5 did not have significant effects on WT infection efficiencies in cells from nine donors (Fig. 1C). Infection rates of ΔNef virus were slightly lower in cells from some donors but overall similar to that of WT. Of note, expression of S5 during virus production only moderately reduced infection of cells from some donors with ΔNef virus (4.2× reduction [e.g., donor 6]), while in cells from other donors, the presence of S5 did not have any effect on infection efficiencies (1.2× reduction [e.g., donor 5]). Overall, S5 reduced infection rates of ΔNef virus in a statistically significant manner, but the effects were less pronounced than in TZM-bl cells and not observed in cells from all donors.
FIG 1.

SERINC5 affects infectivity but not innate recognition of HIV-1 particles in activated CD4+ T cells. (A, upper panel) Relative infectivity of X4-tropic HIV-1 particles produced in 293T cells transfected with a SERINC5 HA-tagged expression plasmid or a control vector. Particle infectivity was measured by infecting TZM-bl reporter cells and normalizing to the amount of RT activity of the respective virus stock as determined by SG-PERT assay (mean ± SD from three independent virus stocks). (Lower panel) Representative Western blot analysis of these virions for the presence of SERINC5.HA, Nef, and HIV-1 p24. (B) Schematic of experimental flow. CD4+ T cells isolated from buffy coats were activated for 3 days with IL-2/PHA and then infected with the virus characterized in panel A. Three days p.i., the cells were analyzed by flow cytometry (p24 in panel C and MxA in panel E) and RT-qPCR (TNF-α mRNA in panel F). Presented are data points for cells from individual donors, with the mean value for each experimental group indicated by a black line. (C) Intracellular p24 levels relative to WT-infected cultures (set to 100%, with the percentage of p24+ cells ranging from 23% to 80% between donors). Shown are mean values from duplicate infections. The fold changes between the ΔNef ± S5 conditions are indicated for donors 5 and 6 as representatives of donors that display no or moderate sensitivity to S5-mediated restriction of HIV-1 infectivity. NI, noninfected. (D) Cytokine expression in MDMs from donors 5 and 6 as a heat map indicating changes of expression over NI (set to 1). (E) Relative levels of intracellular MxA shown as fold change over noninfected cells (NI) (set to 1). (F) Relative TNF-α mRNA levels represented as fold change over NI (set to 1). (G and H) Pearson’s correlation between S5-mediated restriction of infectivity and induction of intracellular MxA (E) or TNF-α mRNA (F) levels upon infection with ΔNef+S5 particles (compared to ΔNef). Statistics (Student's t test): n.s., nonsignificant; *, P < 0.05; **, P < 0.01.
The same cells were also tested for an innate immune response to exposure with the four different HIV-1 variants. To identify functionally relevant differences in innate immune responses, we submitted the cell culture supernatants of these infection experiments to a comprehensive analysis of cytokine production (Fig. 1D). The results revealed subtle effects of S5 on the production of GRO alpha and IL-6 in the context of WT infection. More importantly, cytokine production of cells infected with ΔNef particles, which are more sensitive to S5 than WT particles, was unaffected by the presence of S5 during virus production. Since the analysis of cytokine production may have missed more subtle differences on the transcriptional level, we assessed two effector functions of innate immune recognition of HIV-1 in CD4+ T cells: intracellular MxA was analyzed by fluorescence-activated cell sorter (FACS) to score for induction of interferon-stimulated gene expression (29) (Fig. 1E), and tumor necrosis factor alpha (TNF-α) mRNA levels were quantified by quantitative reverse transcriptase PCR (RT-qPCR) as a measure of induced cytokine expression (Fig. 1F). As expected, infection with WT resulted in an increase of intracellular MxA levels, and the magnitude of this induction varied between cells from different donors. However, the presence of S5 or Nef did not affect MxA expression levels (Fig. 1E). Similarly, donor variability was observed for the extent of TNF-α induction following HIV-1 infection (Fig. 1F). In cells from two of the four donors tested (donors 6 and 7), the lack of Nef appeared to reduce TNF-α expression, but importantly, S5 did not have any effect on TNF-α mRNA levels. Consistently, the reduction of infectivity of ΔNef particles by S5 (calculated as ratio between the infectivity of ΔNef particles and S5-containing ΔNef particles [ΔNef+S5]) was not at all or weakly correlated with intracellular levels of MxA (Fig. 1G) or TNF-α (Fig. 1H), respectively. Since the trend toward a correlation between TNF-α expression and virion infectivity was based on a very moderate increase of TNF-α, we consider it unlikely to be a meaningful mechanistic link. In activated primary CD4+ T cells, S5 thus exerts a moderate effect on virion infectivity in cells from some but not all donors, and this effect can be antagonized by Nef but is not associated with altered innate immune recognition.
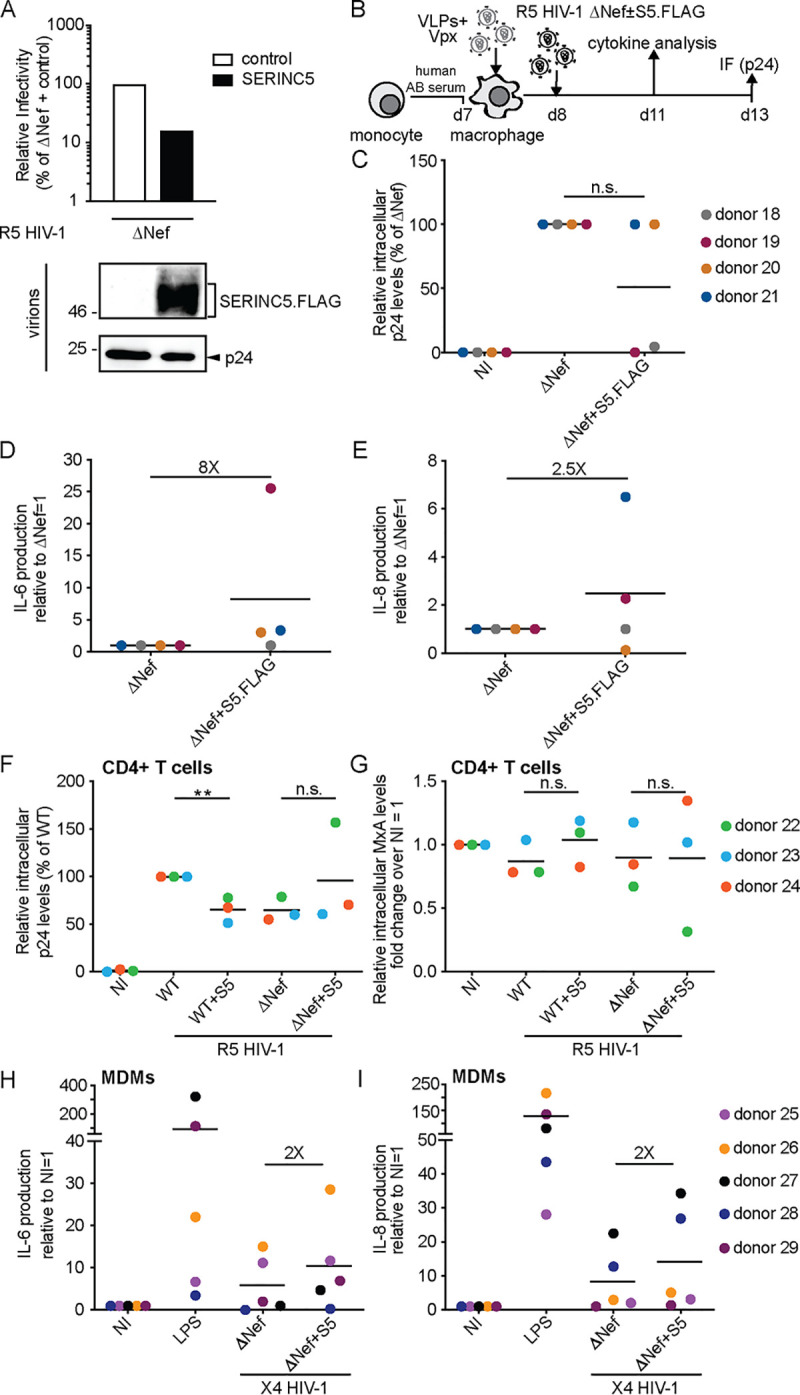
SERINC5 enhances proinflammatory cytokine production by monocyte-derived macrophages in response to infection with HIV-1 ΔNef particles.
We next sought to test effects of S5 on HIV-1 particle infectivity and innate recognition using monocyte-derived macrophages (MDMs) as target cells. To infect these cells, we generated in 293T cells stocks of CCR5 (R5)-tropic HIV-1 WT and ΔNef variants in the presence or absence of S5. Controlling for effects on particle infectivity on TZM-bl cells (Fig. 2A, upper panel) and virion incorporation (Fig. 2A, lower panel) confirmed efficient infectivity restriction by S5 and its antagonism by Nef. These HIV-1 particles were then used to infect MDMs that had been differentiated from primary monocytes with human AB serum. Prior to infection, MDMs were transduced with lentiviral vectors carrying Vpxmac239 (virus-like particles [VLPs] containing Vpx) for 18 h to degrade the restriction factor SAMHD1 and promote productive infection of HIV-1 particles (30) (Fig. 2B). Productively infected cells were quantified 5 days p.i. by immunostaining of intracellular p24CA. In initial experiments, ΔNef particles produced in the absence or presence of S5 were used and revealed again two different donor-dependent effects (Fig. 2C): while in cells from one subgroup of donors, S5 did not have any negative effect on infection rates (e.g., donor 12), the presence of S5 during virus production reduced infection up to 100-fold in cells from other donors (e.g., donor 14). For six of these experiments, we were able to conduct a broad characterization of cytokine production associated with infection at day 3 p.i. (Fig. 2D). This analysis revealed that the incorporation of S5 in ΔNef particles was associated with profound changes in cytokine production characterized by increased production of proinflammatory cytokines such as granulocyte-macrophage colony-stimulating factor (GM-CSF), GRO alpha, IL-6, IL-8, and TNF-α (see Fig. 2E to J, left panels, for plots of individual cytokines). Possibly due to the low sensitivity of detection for this cytokine, production of alpha interferon (IFN-α) was not observed. This induction varied in magnitude for each cytokine between cells from different donors, and cells of one of the six donors tested did not respond with increased cytokine production (donor 16). Overall, however, the presence of S5 in HIV-1 particles was associated with the induction of a complex mixture of proinflammatory cytokines, while production of many other cytokines remained unaffected or was even downregulated (e.g., IP-10) by the presence of S5. Notably, effects of S5 on infection rates and cytokine production in response to infection were not correlated (see Fig. 2E to J, right panels, for plots of individual cytokines).
FIG 2.

SERINC5 sensitizes HIV-1 ΔNef particles for increased production of proinflammatory cytokines by monocyte-derived macrophages (MDMs). (A, upper panel) Relative infectivity of R5-tropic HIV-1 particles produced in 293T cells analyzed as described in the legend to Fig. 1A (mean ± SD from three independent virus stocks). (Lower panel) Representative Western blot analysis of these virions for incorporation of SERINC5.HA, Nef, and HIV-1 p24. (B) Schematic of experimental flow. Macrophages were differentiated from monocytes with human AB serum for 7 days. The cells were then transduced with VLPs+Vpx, infected the next day, and analyzed for productive infection (C) or cytokine production from the cell culture supernatant (D) 5 or 3 days p.i., respectively. (C) Relative intracellular levels of p24 as quantified by immunofluorescence (with ΔNef set as 100%). Shown are values of individual infections, with the percentage of p24+ cells ranging from 0.6% to 30% between donors. Data points are from cells of individual donors, with the mean indicated by a black line. The fold changes between the ΔNef ± S5 conditions are indicated for donors 12 and 14 as representatives of donors that display no or pronounced sensitivity to S5-mediated restriction of HIV-1 infectivity. NI, noninfected. (D) Cytokine production of MDMs infected with HIV-1 ΔNef+S5 particles normalized to ΔNef. Data are displayed as log2 fold change, with red, black, and blue, respectively, indicating upregulated, unaltered, or reduced cytokine production. (E to J) Effect of virion incorporation of S5 on the production of individual cytokines (left panels, presented as fold change over ΔNef, set to 1). Shown are data points from cells of individual donors, with the mean value of all donors indicated by a black line. Also shown is Pearson’s correlation between the infectivity restriction by S5 and the S5-mediated induction of each cytokine (right panels). Statistics (Student's t test): n.s., nonsignificant; *, P < 0.05.
The S5-HA (hemagglutinin) construct may have induced antigen-specific memory responses in cells from donors that underwent infection with or vaccination against influenza A virus. We therefore repeated these experiments with an S5 construct tagged with a synthetic FLAG tag that is not expected to trigger immune responses. The results matched those obtained with S5-HA: the presence of the RF during virus production impaired the infectivity of ΔNef particles in TZM-bl cells (Fig. 3A), while MDMs from 2 out of 4 donors analyzed were sensitive to the S5-mediated reduction in infection rates (Fig. 3B and C). Similarly, proinflammatory cytokine production was induced in MDMs from some but not all donors by ΔNef particles produced in the presence of S5-FLAG (Fig. 3D (IL-6) and E (IL-8). Moreover, the differential response of CD4+ T cells and MDMs to S5-containing HIV-1 described above could in principle reflect differences not only between these cell types but also between the CXCR4- and CCR5-tropic viruses used for infection. We therefore repeated these experiments infecting CD4+ T cells with CCR5-tropic HIV-1 and MDMs with CXCR4-tropic HIV-1 (Fig. 3F to I). Although this resulted in significantly lower rates of infection (see absolute values in the figure legend), CD4+ T cells were again insensitive to infection suppression (Fig. 3F) and enhancement of innate immune recognition (Fig. 3G) by S5, while production of IL-6 and IL-8 by MDMs was induced by S5-containing HIV-1 ΔNef (Fig. 3H and I). Analysis of effects of S5 on the infectivity of X4 HIV-1 in MDMs was precluded by very low infection rates (below 0.5% infected cells). Altogether, these results revealed that in MDM target cells, production of HIV-1 particles in the presence of S5 can potently restrict their infectivity in a donor-dependent manner but can also facilitate innate immune recognition of these particles resulting in enhanced proinflammatory cytokine production.
FIG 3.
The SERINC5-mediated induction of proinflammatory cytokines in MDMs is not mediated by the HA tag and independent of viral tropism. (A, upper panel) Relative infectivity of R5-tropic HIV-1 particles produced in 293T cells transfected with a SERINC5 FLAG-tagged expression plasmid or a control vector and analyzed as described in the legend to Fig. 1A. (Lower panel) Representative Western blot analysis of these virions for incorporation of SERINC5.FLAG and HIV-1 p24. (B) Schematic of experimental flow as in Fig. 2B. (C) Relative intracellular levels of p24 as quantified by immunofluorescence (with ΔNef set as 100% and 0.2 to 4% p24+ cells). Shown are data points from cells of individual donors, with the mean indicated by a black line. (D and E) Effect of virion incorporation of S5.FLAG on the production of individual cytokines (IL-6 in panel D and IL-8 in panel E) presented as fold change over ΔNef (set to 1). Shown are data points from cells of individual donors, with the mean indicated by a black line. (F) Activated CD4+ T cells were infected with the R5 HIV-1 variants from Fig. 2A. Intracellular p24 levels are relative to WT-infected cultures (set to 100%, with the percentage of p24+ cells ranging from 16% to 27% between donors). Shown are data points from cells of individual donors, with the mean value of all donors indicated by a black line. (G) Relative levels of intracellular MxA shown as fold change over noninfected (NI; set to 1). (H) MDMs were infected with X4 HIV-1 variants from Fig. 1A. Quantification of IL-6 in the supernatant of MDMs is displayed as relative to ΔNef (set to 1). Shown are data points from cells of individual donors, with the mean of all donors indicated by a black line. (I) Quantification of IL-8 concentration. The fold change between the ΔNef ± S5 conditions is indicated. Statistics (Student's t test): n.s., nonsignificant; **, P < 0.01.
SERINC5 also promotes proinflammatory cytokine production in response to HIV-1 ΔNef particles by monocyte-derived immature dendritic cells.
To investigate whether the S5-mediated priming of MDMs for increased production of proinflammatory cytokines in response to challenge with ΔNef particles is a common characteristic of myeloid target cells, we also challenged monocyte-derived immature dendritic cells (MDDCs) with our R5 HIV-1 variants. Immature MDDCs were differentiated from peripheral monocytes by GM-CSF and IL-4, incubated with lentiviral vectors carrying Vpxmac239 to degrade SAMHD1, and analyzed by flow cytometry for intracellular p24 at day 4 p.i. (Fig. 4A). In the cells from four donors analyzed, we did not observe adverse effects of S5 on productive infection and even observed a slight promotion of infection by S5 in cells from donor 33 (Fig. 4B). In contrast, the presence of S5 in ΔNef particles induced the production of proinflammatory cytokines 2 days p.i. in a similar manner to MDMs (Fig. 4C, D, and F). Again, the magnitude of production varied between cells from different donors, and cells from one of the four donors failed to respond to S5-containing ΔNef particles. Consistent with the results in MDMs, cytokine production and infection rates were not correlated (Fig. 4E and G). These results suggest that MDDCs are insensitive to the S5-mediated restriction to HIV-1 particle infectivity. However, the induction of proinflammatory cytokines in response to infection with S5-containing ΔNef particles seems to be a shared property of MDMs and MDDCs.
FIG 4.

SERINC5 also sensitizes HIV-1 ΔNef particles for increased production of proinflammatory cytokines by immature monocyte-derived dendritic cells (MDDCs). (A) Schematic of experimental flow. Immature dendritic cells were differentiated from monocytes by GM-CSF and IL-4 for 6 days. The cells were then transduced with VLPs+Vpx, infected 2 days later (duplicate infections per donor), and analyzed for productive infection (B) or cytokine production from the cell culture supernatant (D) 4 or 2 days p.i., respectively. Shown are data points from cells of individual donors (mean of duplicate infections), with the mean of all donors indicated by a black line. (B) Relative intracellular p24 levels as analyzed by flow cytometry (with ΔNef set as 100% and the percentage of p24+ cells ranging from 2% to 9% between donors). NI, noninfected. (C) Cytokine production of MDDCs infected with HIV-1 ΔNef+S5 particles normalized to ΔNef. Data are displayed as log2 fold change, with red, black, and blue, respectively, indicating upregulated, unaltered, or reduced cytokine production. (D to G) Quantification of cytokine production (IL-6 in panel D and TNF-α in panel F) relative to HIV-1 ΔNef-infected cultures (set to 1). Shown are data points from cells of individual donors, with the mean indicated by a black line. Also shown is Pearson’s correlation between the ratio of infectivity of ΔNef to ΔNef+S5 and the induction of cytokine production by HIV-1 ΔNef+S5 particles (E and G). Statistics (Student's t test): n.s., nonsignificant; *, P < 0.05.
Nef antagonizes the SERINC5-mediated induction of proinflammatory cytokines in MDMs.
We next sought to investigate if the increase in proinflammatory cytokine production in response to ΔNef particles by S5 can be antagonized by Nef and compared the cytokine profiles of MDMs infected with WT or ΔNef particles produced in the absence or presence of S5 (Fig. 5A). Only samples from cells of three donors that displayed enhanced proinflammatory cytokine production upon virion incorporation of S5 in the absence of Nef were included in this analysis (Fig. 5B). In contrast, S5 had no appreciable effect on the cytokine production of MDMs infected with WT particles (see Fig. 5B for comparison to uninfected cultures and Fig. 5C for a comparison of supernatants from MDMs infected with ΔNef+S5 or WT+S5 virions, respectively). Effects of S5 on infection rates were again subject to marked donor variability (Fig. 5D), and as shown for IL-6 as an example, induced cytokine production was not correlated with the efficacy of infection (Fig. 5E and F). We conclude that Nef is able to antagonize the ability of S5 to render HIV-1 particles more susceptible for proinflammatory cytokine production by MDM target cells.
FIG 5.

Nef antagonizes SERINC5-mediated induction of proinflammatory cytokine production in MDMs. (A) Schematic of experimental flow (analogous to Fig. 2B, but with the addition of HIV-1 WT ± S5). (B and C) Cytokine expression by MDMs from donors 35, 36, and 37 as a heat map. (B) The color gradient covers up to >10-fold change of expression over noninfected (NI; set to 1). (C) Data are displayed as log2 fold change for MDMs infected with HIV-1 ΔNef+S5 particles relative to the ΔNef control, with red, black, and blue indicating, respectively, upregulated, unaltered, or reduced cytokine production. (D) Quantification of intracellular p24 levels in infected MDM cultures relative to WT-infected cultures (set to 100%, with the percentage of p24+ cells ranging from 4% to 49% between donors). (E) IL-6 production by MDMs infected with ΔNef+S5 relative to that by MDMs infected with WT+S5 (set to 1) (D to F) Shown are data points from cells of individual donors, with the mean of all donors indicated by a black line. (F) Pearson’s correlation between the WT+S5/ΔNef+S5 infectivity ratio and the induction of IL-6 upon infection with ΔNef+S5 particles. Statistics (Student's t test): n.s., nonsignificant; **, P < 0.01.
Cell entry of HIV-1 ΔNef particles via VSV-G but not HIV-1 Env resistant to the infectivity restriction by SERINC5 circumvents SERINC5-mediated enhancement of proinflammatory cytokine production by MDMs.
The sensitivity of virus particles to S5-mediated restriction of infectivity is determined by the envelope (Env) glycoprotein (10, 11, 15, 31), and pseudotyping of HIV particles with the VSV glycoprotein (VSV-G) alleviates the suppression of particle infectivity imposed by the incorporation of S5 into virions (10, 11, 14). We therefore tested if the use of VSV-G for cell entry would also affect the enhancement of proinflammatory cytokine production of S5-containing HIV particles by MDMs. ΔNef particles were produced from 293T cells in the presence or absence of S5 and with the addition of VSV-G. As expected, the presence of S5 during virus production did not impair the infectivity of VSV-G-pseudotyped ΔNef particles, despite robust virion incorporation of the RF (Fig. 6A, upper and lower panels). VSV-G-pseudotyped and native ΔNef particles were then used to infect MDMs, and infection rates and cytokine production were quantified (Fig. 6B). Infection rates of particles carrying VSV-G were not reduced by the presence of S5 and in one case (donor 15) even markedly increased (Fig. 6C). Similarly, the boost in production of proinflammatory cytokines induced by S5 for native HIV-1 particles was largely abrogated by VSV-G pseudotyping (see Fig. 6D for the overall cytokine panel and Fig. 6E for IL-6), and infection rates were not correlated with cytokine production (Fig. 6F and G).
FIG 6.

Pseudotyping of HIV-1 ΔNef particles with VSV-G prevents SERINC5-mediated proinflammatory cytokine production by MDMs. (A, upper panel) Relative infectivity of HIV-1 ΔNef particles pseudotyped with VSV-G produced in 293T cells analyzed as described in the legend to Fig. 1A. (Lower panel) Representative Western blot analysis of these virions for incorporation of SERINC5.HA, Nef, and HIV-1 p24. (B) Schematic of experimental flow (as in Fig. 2B, but including additional infections of MDMs with VSV-G HIV-1). (C) Quantification of intracellular p24 levels in MDMs relative to cultures infected with ΔNef particles (set to 100%, with the percentage of p24+ cells ranging from 1% to 6% between donors). Shown are data points from cells of individual donors, with the mean of all donors indicated by a black line. (D) Cytokine production of MDMs infected with HIV-1 Env or VSV-G containing ΔNef+S5 particles relative to the corresponding ΔNef control. Data are displayed as log2 fold change, with red, black, and blue, respectively, indicating upregulated, unaltered, or reduced cytokine production. (E) Quantification of IL-6 concentration in the supernatant of the MDMs analyzed in panels C and D relative to the corresponding ΔNef (set to 1). Shown are data points from cells of individual donors, with the mean of all donors indicated by a black line. (F and G) Pearson’s correlation between the ΔNef/ΔNef+S5 infectivity ratio and induction of IL-6 production by ΔNef+S5 particles containing HIV-1 Env (F) or VSV-G (G). Statistics (Student's t test): n.s., nonsignificant; **, P < 0.01.
Since these results suggested that VSV-G pseudotyping of ΔNef virions overrides their S5-mediated sensitization for proinflammatory cytokine production by MDMs, we asked which cytokine signature is induced by HIV-1 particles that carry an S5-resistant Env variant. To this end, we employed HIV-1 AD8, whose Env protein has been shown to be resistant to S5-mediated infectivity impairment (15) and generated a Nef-negative version thereof. As expected, HIV-1 AD8 ΔNef particles produced in the presence of S5 incorporated the RF, but their infectivity on TZM-bl target cells was not reduced compared to that of particles produced in the absence of S5 (Fig. 7A) Infection of MDMs (Fig. 7B) revealed that the presence of S5 during virus production had variable and donor-specific effects on rates of productive infection ranging from strong inhibition over no effect to a marked increase (Fig. 7C). As previously observed with HIV-1 carrying an Env that is sensitive to S5-mediated infectivity impairment, the production of cytokines such as IL-6 and IL-8 was increased when AD8 ΔNef particles produced in the presence of S5 were used for infection (Fig. 7D and E). Together, these results show that substituting HIV-1 Env with an unrelated viral glycoprotein for entry protects HIV-1 particles from effects of S5 on virion infectivity, as well as target cell cytokine production. However, enhanced production of proinflammatory cytokines is observed even when S5 effects on virion infectivity are circumvented by the use of an HIV-1 Env variant that is resistant to the S5 infectivity restriction.
FIG 7.

The infectivity of impairment-resistant HIV-1 AD8 Env does not circumvent the SERINC5-induced production of proinflammatory cytokines in MDMs. (A, upper panel) Relative infectivity of HIV-1 AD8 ΔNef particles produced in 293T cells transfected with a SERINC5 expression plasmid or a control vector. Particle infectivity was measured by infecting TZM-bl reporter cells and normalized to the amount of RT activity of the respective virus stock as determined by SG-PERT assay. (Lower panel) Representative Western blot analysis of these virions for presence of SERINC5.HA and HIV-1 p24. (B) Schematic of experimental flow (as in Fig. 2B, but with infection with HIV-1 AD8). (C) Relative intracellular levels of p24 as quantified by immunofluorescence (with ΔNef set as 100% and the percentage of p24+ cells ranging from 0.6% to 7% between donors). Shown are data points from cells of individual donors, with the mean of all donors indicated by a black line. (D and E) Quantification of IL-6 (D) and IL-8 (E) concentrations in the supernatant of the MDMs analyzed in panel C. NI, noninfected. The fold change between the ΔNef ± S5 conditions is indicated. Statistics (Student's t test): n.s., nonsignificant; **, P < 0.01.
Preventing fusion of ΔNef particles promotes proinflammatory cytokine production by MDMs.
Since VSV-G pseudotyping likely drives HIV-1 particles to a different entry pathway than when using HIV-1 Env (32), the fact that VSV-G pseudotyping but not the use of a S5-resistant HIV-1 Env variant abrogated the S5-mediated sensitization for proinflammatory cytokine production by MDMs triggered us to investigate if this immune recognition occurs prior to or post-viral fusion. To this end, we conducted infection experiments to compare untreated MDMs to MDMs that were pretreated and kept in the presence of the fusion inhibitor maraviroc (MVC) (Fig. 8A). Under these conditions, MVC efficiently blocked fusion and productive infection of R5-tropic HIV-1 (data not shown). Cytokine profiles of experiments with cells from 4 donors (see Fig. 8B for the full cytokine panel and Fig. 8C and D for IL-6 and TNF-α) revealed that the addition of MVC to infections with the HIV-1 WT produced in the absence of S5 triggered a moderate increase in proinflammatory cytokine production. This effect was more pronounced for HIV-1 ΔNef particles produced in the absence of S5 (compare WT versus WT+MVC and ΔNef versus ΔNef+MVC). Notably, addition of MVC to infections with HIV-1 ΔNef particles produced in the presence of S5 resulted in an additional marked increase in proinflammatory cytokine production. Again, these effects were subject to donor variability, with the strongest synergistic effects of S5 and MVC in cells from donors that did not display a marked cytokine response to only S5 or MVC (donors 35 and 37). As shown here for IL-6, MVC alone (i.e., in the absence of HIV) did not induce any cytokine production (Fig. 8E). These results suggest that the production of HIV-1 particles in the presence of S5 and blocking HIV-1 Env fusion by MVC lead to comparable and synergistic induction of proinflammatory cytokine production by MDMs and that Nef can antagonize this activity. Finally, we assembled the data obtained for cells from the 12 donors for which full cytokine expression profiles were determined in the course of this study to test if the effects on proinflammatory cytokine production by HIV-1 ΔNef produced in the presence of S5 were statistically significant despite the marked variability between MDMs from different donors (Fig. 8F and G). The results illustrate again that induction of cytokine release is observed in cells of about half of the donors and demonstrate that S5-dependent differences were statistically significant even when considering together results from S5-sensitive as well as S5-insensitive MDMs.
FIG 8.

Preventing fusion of HIV-1 ΔNef particles promotes proinflammatory cytokine production by MDMs. (A) Schematic of experimental flow (as in Fig. 5A, but in the presence or absence of MVC). (B) Cytokine expression in MDMs from donors 35, 36, 37, and 39 as a heat map. The color gradient covers up to a >10-fold change in expression over noninfected (NI; set to 1). Where indicated, cells were treated with MVC, starting from 1 h before infection for the entire experiment. (C and D) Quantification of IL-6 (C) and TNF-α (D) concentrations in the supernatant of infected MDM cultures relative to the NI control (set to 1). Shown are data points from cells of individual donors, with the mean of all donors indicated by a black line. The fold change between the ΔNef ± MVC conditions is indicated. (E) Concentration of IL-6 (relative to NI set to 1) in the supernatant of noninfected MDM cultures treated or not with MVC. Shown are data points from cells of individual donors, with the mean of all donors indicated by a black line. (F and G) Summary of the effects of S5 on proinflammatory cytokine production by HIV-1 ΔNef particles. Shown is the relative production of IL-6 (F) and TNF-α (G) for MDMs infected with HIV-1 ΔNef+S5 particles normalized to ΔNef (set to 1) from all 12 donors for which full cytokine profiles were established in this study. Data points indicate values from cells of individual donors, with the mean value for cells of all donors indicated by a black line. Statistics (Student's t test): n.s., nonsignificant; *, P < 0.05; **, P < 0.01.
DISCUSSION
In this study, we set out to assess the role of S5 in HIV-1 infection in primary target cells. The initial results revealed that the extent of S5-mediated reduction of virion infectivity is highly variable between different target cell types, ranging from no to moderate and high sensitivity for S5 restriction of virion infectivity in MDDCs, CD4+ T cells, and MDMs, respectively. Moreover, a strong donor variability was observed, including MDMs from some donors on which S5 did not impair HIV-1 infection. Since phylogenetic studies document that the overall ability of S5 to affect HIV-1 infection and of lentiviral Nef proteins to antagonize this antiviral activity is highly conserved in evolution (33–35), we considered that S5 may exert antiviral functions in addition to impairing virion infectivity. Other RFs, including Trim5α and tetherin, induce antiviral signaling when encountering their viral target structure (7–9). However, initial experiments, conducted in analogy to tetherin-mediated innate signaling via activation of NF-κB, failed to detect such increased signaling upon expression of S5 during virus production (data not shown). In contrast, we identified that virion incorporation of S5 triggers the production of proinflammatory cytokines in myeloid target cells, albeit again with marked variability between cells from different donors. Expression of Nef or pseudotyping S5-containing virions with VSV-G prevented this sensitization of HIV-1 particles for innate immune recognition by MDMs. Together, these results identify the induction of proinflammatory cytokine production by myeloid target cells in response to challenge with HIV-1 particles as a novel activity of S5.
A key result of our study was the marked difference between primary HIV-1 target cell types in sensitivity to S5-mediated reduction of virion infectivity. In the TZM-bl cells commonly used to measure effects of S5 on HIV-1 particle infectivity, incorporation of S5 reduces virion infectivity by 10- to 1,000-fold (10, 11, 17, 18, 31, 36, 37). In contrast, primary human CD4+ T cells were only moderately sensitive to S5-mediated effects on virion infectivity, with reductions of relative infectivity upon virion incorporation of S5 not exceeding 5-fold. This is in line with the moderate reduction of initial infection rates described by Usami et al. and with the fact that this advantage in replication did not further increase over multiple rounds (10). Moreover, the Göttlinger lab recently reported that in the MOLT-3 T cell line, the positive effect of Nef on HIV-1 replication does not depend on antagonizing S5 or SERINC3 (38). Consistently, CD4+ T cells express only low levels of S5 mRNA, and expression is not regulated by T cell activation or stimulation with alpha interferon or proinflammatory interleukins (39). Collectively, these results suggest that the main effects of S5 with respect to HIV-1 infection are not exerted in CD4+ T cells. In line with the induction of S5 expression upon monocyte differentiation (39), our results rather suggest MDMs as a key cell type in which the presence of S5 can potently (up to over 100-fold) reduce productive HIV-1 infection. MDDCs, in contrast, appear to be insensitive to the virion infectivity restriction by S5. The molecular bases for these differences remain to be determined, but may include differences between these cell types in local densities of binding and entry receptors, the overall route of entry, and/or the innate response to infection (40–43).
The analysis of cytokine production from primary target cells challenged with HIV-1 particles with or without incorporated S5 revealed the promotion of innate immune recognition as a novel consequence of S5 expression during virus production. This effect was observed with MDMs and MDDCs but not CD4+ T cells as target cells, suggesting that the pathways that mediate this enhanced cytokine response are specific to cells of the myeloid lineage. Since the production of the virus stocks compared in individual experiments occurred in parallel using the same set of plasmids and buffers, multiple sets of virus stocks produced at different time points gave similar results, and S5-specific induction of proinflammatory cytokine production was only observed with ΔNef but not WT particles, we conclude that these effects result from specific innate immune recognition and not unspecific activation of MDMs by contaminants (e.g., microvesicles or plasmid DNA). Although the marked variability in the magnitude of proinflammatory cytokine production precluded a more detailed characterization of the underlying molecular mechanisms, this initial description provides a characterization of this novel S5 activity. Of note, productive entry was not required for innate recognition resulting in enhanced cytokine production, which sets this phenomenon apart from previously reported innate sensing events of postentry HIV replication intermediates in MDMs (43–46). It was thus conceivable that the ability of S5 to increase target cell cytokine production could be linked to its impairment of Env fusion. Notably, impairment of fusion of S5-negative HIV-1 particles by MVC had similar effects on cytokine production to virion incorporation of S5. Although by distinct mechanisms involving disruption of Env trimers in the virion (16) and blockade of engagement of the entry coreceptor CCR5 by Env (47), S5 and MVC likely exert similar negative effects on Env-coreceptor interactions that prevent fusion, while maintaining virions bound to the binding receptor CD4. Consistently, the effects of S5 and MVC on cytokine production synergized in a similar way to that previously reported for Env fusion (15). Enhanced cytokine production may thus reflect the routing of HIV-1 particles to a nonproductive uptake pathway that leads to more efficient innate immune recognition of these particles: e.g., sensing of genomic RNA by cellular Toll-like receptors (TLRs). In this scenario, the circumvention of induced cytokine production by VSV-G could reflect that in S5-containing virions, VSV-G interactions with its low-density lipoprotein (LDL) receptor (48–51) dominate over Env-coreceptor interactions and funnel HIV-1 particles to the endocytic uptake pathway used by VSV-G, which apparently does not result in enhanced innate immune recognition of HIV-1 particles.
Since blocking entry of native HIV-1 particles and the production of HIV-1 ΔNef particles in the presence of S5 had synergistic effects on proinflammatory cytokine production, it was very surprising that the effects of S5 on cytokine production were not correlated with an impairment of virion infectivity in cells from most donors. Moreover, the fact that HIV-1 Env that is resistant to the infectivity impairment by S5 did not affect the increase of cytokine production triggered by virus particles produced in the presence of S5 further underscored that effects of S5 on virion infectivity and cytokine production are mechanistically uncoupled. Nevertheless, both effects of S5 appear to be mediated at the entry level. This could, e.g., indicate that conformational changes in Env induced by S5 that do not significantly impair virion infectivity are sufficient to induce more efficient innate recognition of these particles. In this scenario, different thresholds of, e.g., Env-receptor interactions likely govern the efficacy of virus entry and transport to the cellular compartments in which innate immune recognition occurs. Thus, moderate effects on Env-receptor interactions may reroute a small fraction of particles to a sensing-competent uptake pathway to result in significant changes in cytokine production while infection rates remain unaffected. It is also important to consider that the particle population used in this study certainly contains a heterogenous mixture of particles containing divergent S5 levels and displaying variable sensitivity to innate recognition (e.g., containing or lacking viral RNA genomes). Finally, the presented data are consistent with a model in which S5 increases cytokine production in MDM target cells by virtue of its incorporation into virions and is antagonized by Nef by virion exclusion. However, the contribution of alternative mechanisms, such as, e.g., additional S5-dependent modifications of virions, cannot be excluded. The identification of the cellular transport and sensing pathways involved in this process will constitute important aims of future studies.
The exclusive use of primary target cells for infection also allowed us to appreciate that in addition to cell-type-specific differences, important variability exists between the same cell types isolated from different donors in the response to effects of S5 on virion infectivity and cytokine production. This donor variability was particularly pronounced for MDMs, where cells from some donors were entirely resistant to infectivity impairment or enhancement of cytokine production by S5. Of note, HIV-1 infection rates in MDMs are generally known to vary significantly between cells from different donors and even when cells are isolated and differentiated from the same donor on consecutive days (52). The observed donor variability in sensitivity to effects of S5 may thus reflect this plasticity. Although delineating the molecular bases for this differential sensitivity was beyond the scope of this study, the fact that the sensitivity of cells from individual donors to S5 effects on virion infectivity and increased cytokine production were not correlated provided the important insight that both effects can be mechanistically uncoupled. Future studies will investigate whether this donor variability reflects differences in expression of factors involved in the infection and/or innate recognition processes at the genetic or epigenetic level and whether they govern the general susceptibility to S5 or the threshold amounts of S5 to which these cells respond.
In sum, the results of this study identify the sensitization of HIV-1 particles for innate immune recognition resulting in the production of proinflammatory cytokines as a novel activity of S5. The large majority of cell-free retroviral particles are thought to be noninfectious (53–56), and “tagging” them with S5 to induce proinflammatory cytokine production from bystander myeloid targets may be an efficient strategy to activate the innate immune system toward a propagating infection. This property of S5 may thus contribute to the recently described control of retrovirus spread in the infected host (25) and may represent a driving force for the evolutionary conservation of antiviral SERINC proteins and the emergence of viral antagonists.
MATERIALS AND METHODS
Cell culture.
293T and TZM-bl cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 1% penicillin-streptomycin. Primary CD4+ T cells were isolated from buffy coats using the RosetteSep Human CD4+ T cell enrichment kit (StemCell Technologies) according to the manufacturer’s protocol. Buffy coats were obtained from healthy anonymous blood donors at the Heidelberg University Hospital Blood Bank according to the regulations of the local ethics committee. CD4+ T cells were activated using 20 ng/ml IL-2 and 1 μg/ml PHA for 72 h in complete Roswell Park Memorial Institute medium 1640 (RPMI supplemented with 1% penicillin-streptomycin and 10% FCS). To generate monocyte-derived macrophages (MDMs) and monocyte-derived immature dendritic cells (MDDCs), human peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats by Biocoll (Merck Biochrom) density gradient centrifugation using SepMate tubes (StemCell Technologies). CD14+ monocytes were isolated from PBMCs by positive selection using magnetic beads (CD14 MicroBeads; Miltenyi Biotech). The monocytes were seeded in RPMI supplemented with 10% heat-inactivated FCS and antibiotics and subsequently differentiated into macrophages by adding to the medium 5% human AB serum (Sigma-Aldrich) and into immature dendritic cells by adding to the medium 20 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF; PeproTech) and 20 ng/ml interleukin 4 (IL-4; PeproTech).
Plasmids.
The expression plasmid for the hemagglutinin (HA)-tagged version of SERINC5 (pBJ5_SERINC5.intHA) and the empty control vector were received from Heinrich Göttlinger and Massimo Pizzato (10, 11). The pcDNA-based expression plasmid of FLAG-tagged SERINC5 was received from Hendrik Streeck (57). The proviral plasmid pHIV-1NL4-3 Nef stop (ΔNef) was described previously (58), and pHIV-1AD8 Nef stop (ΔNef) was generated by introducing two stop codons in the nef frame after the stop codon of nef into the wild-type provirus HIV-1 AD8 with the help of the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies). The provirus pHIV-1NL4-3 SF2 Nef WT (HIV-1 Nef WT) (59) and the proviral plasmids R5 pHIV-1NL4-3 WT (SF2 Nef WT) and ΔNef encoding an R5-tropic Env glycoprotein that differs from Env NL4-3 by seven point mutations within the V3 loop were described previously (60). The plasmid pcDNA3.1Vpx SIVmac239-Myc and the packaging plasmid containing the Vpx-interacting motif in group-specific antigen protein (pΔR8.9 NSDP) were described elsewhere (61, 62) and provided by Nathaniel Landau. The vector backbone pWPI and the plasmid pMD2.G encoding the vesicular stomatitis virus glycoprotein (VSV-G) were generated by Didier Trono and obtained through Addgene.
Reagents.
The following antibodies and reagents were used: mouse anti-HA (HA.11, clone 16B11; Biolegend), sheep anti-Nef (arp444; NIH AIDS repository), sheep anti-HIV-1 p24 CA antiserum (from Barbara Müller), mouse anti-VSV-G (1667351; Roche Diagnostics), rabbit anti-FLAG (5407; Cell Signaling), rabbit anti-MxA (ab95926; Abcam), KC-57 conjugated with fluorescein isothiocyanate (FITC) (p24; Beckman Coulter), allophycocyanin (APC)-conjugated goat anti-rabbit (Jackson Immuno Research), Alexa Fluor 568-conjugated donkey anti-sheep and Hoechst 33342 (Invitrogen/Thermo Fisher Scientific), and lipopolysaccharide (LPS) from Escherichia coli (L4524; Sigma-Aldrich).
Virus production, infectivity measurements, and infection assays.
Virus production from 293T cells, infectivity measurements using the SG-PERT (SYBR green I-based product-enhanced RT) assay for total viral particles and TZM-bl luciferase reporter assay, and Western blotting for protein expression in virions were performed as described previously (17, 32). Lentiviral vectors carrying Vpxmac239 were produced by cotransfection of pWPI, pcDNA.Vpxmac239, pΔR8.9 NSDP, and VSV-G at a molar ratio of 4:1:3:1. Primary CD4+ T cells were infected (duplicate infections per donor) with 5 × 1010 pU reverse transcriptase (RT) for 4 h. The virus was washed away, and cells were maintained in medium containing IL-2. Primary MDMs were transduced with lentiviral vectors carrying Vpxmac239 corresponding to 1010 pU RT 18 h before infection with 1011 pU RT of HIV-1 R5 (single infections per donor). Infection was stopped at day 5 p.i. by treatment with 3% paraformaldehyde (PFA) for 90 min, and newly synthesized Gag was detected by immunostaining using anti-CA (rabbit) antiserum; nuclei were counterstained with Hoechst. For scoring infectivity, after immunostaining, samples were imaged using a wide-field microscope (Olympus IX81 SIF-3; Xcellence Pro software) with a 10× air immersion objective (NA 0.45). Ten positions were acquired per sample. To quantify infected cells, a previously published script in MatLab was used (30, 63). Primary MDDCs were transduced with lentiviral vectors carrying Vpxmac239 corresponding to 1010 pU RT 18 h before infection with 1011 pU RT of HIV-1 R5 (duplicate infections per donor).
Flow cytometry.
CD4+ T cell pellets were collected 3 days p.i., fixed for 90 min with 3% PFA, and stained with p24 KC-57–FITC antibody and anti-MxA antibody in 0.1% Triton X-100 for 30 min on ice. Cells were then stained with the APC-conjugated goat anti-rabbit secondary antibody for 30 min, protected from light. Samples were measured by flow cytometry in BD FACSVerse with BD FACSuite software. Primary MDDC pellets were collected 4 days p.i., fixed for 90 min with 3% PFA, and stained with p24 KC-57–FITC antibody in 0.1% Triton X-100 for 30 min on ice. Samples were measured by flow cytometry in a BD FACSCelesta with BD FACSDiva software. Gating and mean fluorescent intensity (MFI) analysis were done using FlowJo software 10.4.2, and data were processed with Microsoft Office Excel 2016 and GraphPad Prism 8.0 software.
RT-qPCR.
Total RNA was extracted using the NucleoSpin RNA II kit (Macherey-Nagel) and reverse transcribed using the SuperScript One-Step RT-PCR system (Life technologies) according to the manufacturer’s instructions. cDNA levels were determined by using the SYBR green PCR master mix (Life Technologies), and reactions were performed on an ABI PRISM 7500 sequence detection system (Applied Biosystems) using the following program: 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA was used for normalization of input RNA. Data were analyzed by using the threshold cycle (ΔΔCT) method. The following primers were used: GAPDH Fwd, 5′-GAAGGTGAAGGTCGGAGTC-3′, and Rev, 5′-GAAGATGGTGATGGGATTTC-3′; and TNF-α, Fwd, 5′-CCCAGGGACCTCTCTCTAATC-3′, and Rev, 5′-ATGGGCTACAGGCTTGTCACT-3′.
Cytokine quantification.
The amounts of cytokines and chemokines present in cell culture supernatants were determined by Eve Technologies Corporation using the Discovery Assay: Human Cytokine Array/Chemokine Array 42-Plex. Results are expressed in pg/ml of cytokines/chemokines according to the company protein standard as reported earlier (64). Cell-free supernatants were also analyzed for levels of IL-6 and IL-8 by enzyme-linked immunosorbent assay (ELISA; BD Biosciences) according to the manufacturer's instructions.
Statistical analysis.
Statistical analysis of data sets was carried out using Prism version 8.0 (GraphPad). Statistical significance was calculated using Student's t test and Pearson’s correlation coefficient. The following abbreviations are used in the figure legends: n.s., not significant; *, P < 0.05; **, P < 0.01.
ACKNOWLEDGMENTS
We are grateful to Heinrich Göttlinger, Massimo Pizzato, Bianca Schulte, Hendrik Streeck, and Nathaniel Landau for the gift of reagents, Nadine Tibroni for technical assistance, David Bejarano and Hans-Georg Kräusslich for expertise on and protocols for the production and infection of MDMs, and Kathrin Bajak for help with manuscript preparation and submission.
This project is supported by the Deutsche Forschungsgemeinschaft (DFG [German Research Foundation]; SPP 1923 grants to O.T.F. and F.K. and SFB 1129 [projektnummer 240245660] grant to O.T.F.) and by the German Centre for Infection Research (DZIF [TTU 04.810—An integrated approach for HIV-cure “Shock and Kill”] grant to O.T.F.).
REFERENCES
- 1.Pollara J, Bonsignori M, Moody MA, Pazgier M, Haynes BF, Ferrari G. 2013. Epitope specificity of human immunodeficiency virus-1 antibody dependent cellular cytotoxicity [ADCC] responses. Curr HIV Res 11:378–387. 10.2174/1570162x113116660059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen MS, Shaw GM, McMichael AJ, Haynes BF. 2011. Acute HIV-1 infection. N Engl J Med 364:1943–1954. 10.1056/NEJMra1011874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altfeld M, Gale M, Jr.. 2015. Innate immunity against HIV-1 infection. Nat Immunol 16:554–562. 10.1038/ni.3157. [DOI] [PubMed] [Google Scholar]
- 4.Smed-Sorensen A, Lore K. 2011. Dendritic cells at the interface of innate and adaptive immunity to HIV-1. Curr Opin HIV AIDS 6:405–410. 10.1097/COH.0b013e328349b06b. [DOI] [PubMed] [Google Scholar]
- 5.Colomer-Lluch M, Ruiz A, Moris A, Prado JG. 2018. Restriction factors: from intrinsic viral restriction to shaping cellular immunity against HIV-1. Front Immunol 9:2876. 10.3389/fimmu.2018.02876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strebel K. 2013. HIV accessory proteins versus host restriction factors. Curr Opin Virol 3:692–699. 10.1016/j.coviro.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, Bisiaux A, Albert ML, Strambio-De-Castillia C, Mothes W, Pizzato M, Grutter MG, Luban J. 2011. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472:361–365. 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Portilho DM, Fernandez J, Ringeard M, Machado AK, Boulay A, Mayer M, Muller-Trutwin M, Beignon AS, Kirchhoff F, Nisole S, Arhel NJ. 2016. Endogenous TRIM5alpha function is regulated by SUMOylation and nuclear sequestration for efficient innate sensing in dendritic cells. Cell Rep 14:355–369. 10.1016/j.celrep.2015.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galao RP, Le Tortorec A, Pickering S, Kueck T, Neil SJ. 2012. Innate sensing of HIV-1 assembly by tetherin induces NFkappaB-dependent proinflammatory responses. Cell Host Microbe 12:633–644. 10.1016/j.chom.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Usami Y, Wu Y, Gottlinger HG. 2015. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 526:218–223. 10.1038/nature15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, McCauley SM, Nowosielska A, Antonarakis SE, Luban J, Santoni FA, Pizzato M. 2015. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526:212–217. 10.1038/nature15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inuzuka M, Hayakawa M, Ingi T. 2005. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J Biol Chem 280:35776–35783. 10.1074/jbc.M505712200. [DOI] [PubMed] [Google Scholar]
- 13.Trautz B, Wiedemann H, Luchtenborg C, Pierini V, Kranich J, Glass B, Krausslich HG, Brocker T, Pizzato M, Ruggieri A, Brugger B, Fackler OT. 2017. The host-cell restriction factor SERINC5 restricts HIV-1 infectivity without altering the lipid composition and organization of viral particles. J Biol Chem 292:13702–13713. 10.1074/jbc.M117.797332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sood C, Marin M, Chande A, Pizzato M, Melikyan GB. 2017. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J Biol Chem 292:6014–6026. 10.1074/jbc.M117.777714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beitari S, Ding S, Pan Q, Finzi A, Liang C. 2017. Effect of HIV-1 Env on SERINC5 antagonism. J Virol 91:e02214-16. 10.1128/JVI.02214-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen YC, Sood C, Marin M, Aaron J, Gratton E, Salaita K, Melikyan GB. 2020. Super-resolution fluorescence imaging reveals that serine incorporator protein 5 inhibits human immunodeficiency virus fusion by disrupting envelope glycoprotein clusters. ACS Nano 14:10929–10943. 10.1021/acsnano.0c02699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trautz B, Pierini V, Wombacher R, Stolp B, Chase AJ, Pizzato M, Fackler OT. 2016. The Antagonism of HIV-1 Nef to SERINC5 particle infectivity restriction involves the counteraction of virion-associated pools of the restriction factor. J Virol 90:10915–10927. 10.1128/JVI.01246-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Zhou T, Yang J, Lin Y, Shi J, Zhang X, Frabutt DA, Zeng X, Li S, Venta PJ, Zheng YH. 2017. Identification of SERINC5-001 as the predominant spliced isoform for HIV-1 restriction. J Virol 91:e00137-17. 10.1128/JVI.00137-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulte B, Selyutina A, Opp S, Herschhorn A, Sodroski JG, Pizzato M, Diaz-Griffero F. 2018. Localization to detergent-resistant membranes and HIV-1 core entry inhibition correlate with HIV-1 restriction by SERINC5. Virology 515:52–65. 10.1016/j.virol.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ananth S, Morath K, Trautz B, Tibroni N, Shytaj IL, Obermaier B, Stolp B, Lusic M, Fackler OT. 2019. Multifunctional roles of the N-terminal region of HIV-1SF2Nef are mediated by three independent protein interaction sites. J Virol 94:e01398-19. 10.1128/JVI.01398-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obermaier B, Ananth S, Tibroni N, Pierini V, Shytaj IL, Diaz RS, Lusic M, Fackler OT. 2020. Patient-derived HIV-1 Nef alleles reveal uncoupling of CD4 downregulation and SERINC5 antagonism functions of the viral pathogenesis factor. J Acquir Immune Defic Syndr 85:e23–e26. 10.1097/QAI.0000000000002418. [DOI] [PubMed] [Google Scholar]
- 22.Ahmed SS, Bundgaard N, Graw F, Fackler OT. 2020. Environmental restrictions: a new concept governing HIV-1 spread emerging from integrated experimental-computational analysis of tissue-like 3D cultures. Cells 9:1112. 10.3390/cells9051112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imle A, Kumberger P, Schnellbacher ND, Fehr J, Carrillo-Bustamante P, Ales J, Schmidt P, Ritter C, Godinez WJ, Muller B, Rohr K, Hamprecht FA, Schwarz US, Graw F, Fackler OT. 2019. Experimental and computational analyses reveal that environmental restrictions shape HIV-1 spread in 3D cultures. Nat Commun 10:2144. 10.1038/s41467-019-09879-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agosto LM, Uchil PD, Mothes W. 2015. HIV cell-to-cell transmission: effects on pathogenesis and antiretroviral therapy. Trends Microbiol 23:289–295. 10.1016/j.tim.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Timilsina U, Umthong S, Lynch B, Stablewski A, Stavrou S. 2020. SERINC5 potently restricts retrovirus infection in vivo. mBio 11:e00588-20. 10.1128/mBio.00588-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin J, Li F, Mothes W. 2011. Viral determinants of polarized assembly for the murine leukemia virus. J Virol 85:7672–7682. 10.1128/JVI.00409-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin J, Sherer NM, Heidecker G, Derse D, Mothes W. 2009. Assembly of the murine leukemia virus is directed towards sites of cell-cell contact. PLoS Biol 7:e1000163. 10.1371/journal.pbio.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li F, Jin J, Herrmann C, Mothes W. 2013. Basic residues in the matrix domain and multimerization target murine leukemia virus Gag to the virological synapse. J Virol 87:7113–7126. 10.1128/JVI.03263-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vermeire J, Roesch F, Sauter D, Rua R, Hotter D, Van Nuffel A, Vanderstraeten H, Naessens E, Iannucci V, Landi A, Witkowski W, Baeyens A, Kirchhoff F, Verhasselt B. 2016. HIV triggers a cGAS-dependent, Vpu- and Vpr-regulated type I interferon response in CD4(+) T cells. Cell Rep 17:413–424. 10.1016/j.celrep.2016.09.023. [DOI] [PubMed] [Google Scholar]
- 30.Bejarano DA, Puertas MC, Borner K, Martinez-Picado J, Muller B, Krausslich HG. 2018. Detailed characterization of early HIV-1 replication dynamics in primary human macrophages. Viruses 10:620. 10.3390/v10110620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahi YS, Zhang S, Thappeta Y, Denman A, Feizpour A, Gummuluru S, Reinhard B, Muriaux D, Fivash MJ, Rein A. 2016. Functional interplay between murine leukemia virus Glycogag, Serinc5, and surface glycoprotein governs virus entry, with opposite effects on gammaretroviral and Ebolavirus glycoproteins. mBio 7:e01985-16. 10.1128/mBio.01985-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pizzato M. 2010. MLV glycosylated-Gag is an infectivity factor that rescues Nef-deficient HIV-1. Proc Natl Acad Sci U S A 107:9364–9369. 10.1073/pnas.1001554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heigele A, Kmiec D, Regensburger K, Langer S, Peiffer L, Sturzel CM, Sauter D, Peeters M, Pizzato M, Learn GH, Hahn BH, Kirchhoff F. 2016. The potency of Nef-mediated SERINC5 antagonism correlates with the prevalence of primate lentiviruses in the wild. Cell Host Microbe 20:381–391. 10.1016/j.chom.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Sousa-Pereira P, Abrantes J, Bauernfried S, Pierini V, Esteves PJ, Keppler OT, Pizzato M, Hornung V, Fackler OT, Baldauf HM. 2019. The antiviral activity of rodent and lagomorph SERINC3 and SERINC5 is counteracted by known viral antagonists. J Gen Virol 100:278–288. 10.1099/jgv.0.001201. [DOI] [PubMed] [Google Scholar]
- 35.Kmiec D, Akbil B, Ananth S, Hotter D, Sparrer KMJ, Sturzel CM, Trautz B, Ayouba A, Peeters M, Yao Z, Stagljar I, Passos V, Zillinger T, Goffinet C, Sauter D, Fackler OT, Kirchhoff F. 2018. SIVcol Nef counteracts SERINC5 by promoting its proteasomal degradation but does not efficiently enhance HIV-1 replication in human CD4+ T cells and lymphoid tissue. PLoS Pathog 14:e1007269. 10.1371/journal.ppat.1007269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dai W, Usami Y, Wu Y, Gottlinger H. 2018. A long cytoplasmic loop governs the sensitivity of the anti-viral host protein SERINC5 to HIV-1 Nef. Cell Rep 22:869–875. 10.1016/j.celrep.2017.12.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma S, Lewinski MK, Guatelli J. 2018. An N-glycosylated form of SERINC5 is specifically incorporated into HIV-1 virions. J Virol 92:e00753-18. 10.1128/JVI.00753-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu Y, Olety B, Weiss ER, Popova E, Yamanaka H, Gottlinger H. 2019. Potent enhancement of HIV-1 replication by Nef in the absence of SERINC3 and SERINC5. mBio 10:e01071-19. 10.1128/mBio.01071-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zutz A, Scholz C, Schneider S, Pierini V, Munchhoff M, Sutter K, Wittmann G, Dittmer U, Draenert R, Bogner JR, Fackler OT, Keppler OT. 2020. SERINC5 is an unconventional HIV restriction factor that is upregulated during myeloid cell differentiation. J Innate Immun 12:399–409. 10.1159/000504888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee B, Sharron M, Montaner LJ, Weissman D, Doms RW. 1999. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc Natl Acad Sci U S A 96:5215–5220. 10.1073/pnas.96.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Wilgenburg B, Moore MD, James WS, Cowley SA. 2014. The productive entry pathway of HIV-1 in macrophages is dependent on endocytosis through lipid rafts containing CD4. PLoS One 9:e86071. 10.1371/journal.pone.0086071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nasr N, Maddocks S, Turville SG, Harman AN, Woolger N, Helbig KJ, Wilkinson J, Bye CR, Wright TK, Rambukwelle D, Donaghy H, Beard MR, Cunningham AL. 2012. HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood 120:778–788. 10.1182/blood-2012-01-407395. [DOI] [PubMed] [Google Scholar]
- 43.Silvin A, Manel N. 2015. Innate immune sensing of HIV infection. Curr Opin Immunol 32:54–60. 10.1016/j.coi.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 44.Decalf J, Desdouits M, Rodrigues V, Gobert FX, Gentili M, Marques-Ladeira S, Chamontin C, Mougel M, Cunha de Alencar B, Benaroch P. 2017. Sensing of HIV-1 entry triggers a type I interferon response in human primary macrophages. J Virol 91:e00147-17. 10.1128/JVI.00147-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diget EA, Zuwala K, Berg RK, Laursen RR, Soby S, Ostergaard L, Melchjorsen J, Mogensen TH. 2013. Characterization of HIV-1 infection and innate sensing in different types of primary human monocyte-derived macrophages. Mediators Inflamm 2013:208412. 10.1155/2013/208412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herbein G, Gras G, Khan KA, Abbas W. 2010. Macrophage signaling in HIV-1 infection. Retrovirology 7:34. 10.1186/1742-4690-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu GG, Guo J, Wu Y. 2014. Chemokine receptor CCR5 antagonist maraviroc: medicinal chemistry and clinical applications. Curr Top Med Chem 14:1504–1514. 10.2174/1568026614666140827143745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. 2013. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci U S A 110:7306–7311. 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aiken C. 1997. Pseudotyping human immunodeficiency virus type 1 (HIV-1) by the glycoprotein of vesicular stomatitis virus targets HIV-1 entry to an endocytic pathway and suppresses both the requirement for Nef and the sensitivity to cyclosporin A. J Virol 71:5871–5877. 10.1128/JVI.71.8.5871-5877.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chazal N, Singer G, Aiken C, Hammarskjold ML, Rekosh D. 2001. Human immunodeficiency virus type 1 particles pseudotyped with envelope proteins that fuse at low pH no longer require Nef for optimal infectivity. J Virol 75:4014–4018. 10.1128/JVI.75.8.4014-4018.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Firrito C, Bertelli C, Vanzo T, Chande A, Pizzato M. 2018. SERINC5 as a new restriction factor for human immunodeficiency virus and murine leukemia virus. Annu Rev Virol 5:323–340. 10.1146/annurev-virology-092917-043308. [DOI] [PubMed] [Google Scholar]
- 52.Joseph SB, Arrildt KT, Swanstrom AE, Schnell G, Lee B, Hoxie JA, Swanstrom R. 2014. Quantification of entry phenotypes of macrophage-tropic HIV-1 across a wide range of CD4 densities. J Virol 88:1858–1869. 10.1128/JVI.02477-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDougal JS, Cort SP, Kennedy MS, Cabridilla CD, Feorino PM, Francis DP, Hicks D, Kalyanaraman VS, Martin LS. 1985. Immunoassay for the detection and quantitation of infectious human retrovirus, lymphadenopathy-associated virus (LAV). J Immunol Methods 76:171–183. 10.1016/0022-1759(85)90489-2. [DOI] [PubMed] [Google Scholar]
- 54.Dimitrov DS, Willey RL, Sato H, Chang LJ, Blumenthal R, Martin MA. 1993. Quantitation of human immunodeficiency virus type 1 infection kinetics. J Virol 67:2182–2190. 10.1128/JVI.67.4.2182-2190.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bourinbaiar AS. 1994. The ratio of defective HIV-1 particles to replication-competent infectious virions. Acta Virol 38:59–61. [PubMed] [Google Scholar]
- 56.Rusert P, Fischer M, Joos B, Leemann C, Kuster H, Flepp M, Bonhoeffer S, Gunthard HF, Trkola A. 2004. Quantification of infectious HIV-1 plasma viral load using a boosted in vitro infection protocol. Virology 326:113–129. 10.1016/j.virol.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 57.Molnar S, Wieczorek L, Zemil M, Schulte B, Martinez E, Gift S, Tang L, Streeck H, Gramzinski RA, Michael NL, Joyce G, Polonis VR. 2020. Novel monoclonal antibodies to the SERINC5 HIV-1 restriction factor detect endogenous and virion-associated SERINC5. MAbs 12:1802187. 10.1080/19420862.2020.1802187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schindler M, Wurfl S, Benaroch P, Greenough TC, Daniels R, Easterbrook P, Brenner M, Munch J, Kirchhoff F. 2003. Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J Virol 77:10548–10556. 10.1128/jvi.77.19.10548-10556.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fackler OT, Moris A, Tibroni N, Giese SI, Glass B, Schwartz O, Krausslich HG. 2006. Functional characterization of HIV-1 Nef mutants in the context of viral infection. Virology 351:322–339. 10.1016/j.virol.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 60.Bozek K, Eckhardt M, Sierra S, Anders M, Kaiser R, Krausslich HG, Muller B, Lengauer T. 2012. An expanded model of HIV cell entry phenotype based on multi-parameter single-cell data. Retrovirology 9:60. 10.1186/1742-4690-9-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sunseri N, O'Brien M, Bhardwaj N, Landau NR. 2011. Human immunodeficiency virus type 1 modified to package simian immunodeficiency virus Vpx efficiently infects macrophages and dendritic cells. J Virol 85:6263–6274. 10.1128/JVI.00346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pertel T, Reinhard C, Luban J. 2011. Vpx rescues HIV-1 transduction of dendritic cells from the antiviral state established by type 1 interferon. Retrovirology 8:49. 10.1186/1742-4690-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borner K, Hermle J, Sommer C, Brown NP, Knapp B, Glass B, Kunkel J, Torralba G, Reymann J, Beil N, Beneke J, Pepperkok R, Schneider R, Ludwig T, Hausmann M, Hamprecht F, Erfle H, Kaderali L, Krausslich HG, Lehmann MJ. 2010. From experimental setup to bioinformatics: an RNAi screening platform to identify host factors involved in HIV-1 replication. Biotechnol J 5:39–49. 10.1002/biot.200900226. [DOI] [PubMed] [Google Scholar]
- 64.Kaw S, Ananth S, Tsopoulidis N, Morath K, Coban BM, Hohenberger R, Bulut OC, Klein F, Stolp B, Fackler OT. 2020. HIV-1 infection of CD4 T cells impairs antigen-specific B cell function. EMBO J 39:e105594. 10.15252/embj.2020105594. [DOI] [PMC free article] [PubMed] [Google Scholar]