

Abstract
Objective
To determine whether memory is preserved longitudinally in primary progressive aphasia (PPA) associated with Alzheimer disease (AD) and to identify potential factors that maintain memory despite underlying neurofibrillary degeneration of mediotemporal memory areas.
Methods
Longitudinal memory assessment was done in 17 patients with PPA with autopsy or biomarker evidence of AD (PPA-AD) and 14 patients with amnestic dementia of the Alzheimer type with AD at autopsy (DAT-AD).
Results
In PPA-AD, episodic memory, tested with nonverbal items, was preserved at the initial testing and showed no decline at retesting 2.35 ± 0.78 years later, at which time symptoms had been present for 6.26 ± 2.21 years. In contrast, language functions declined significantly during the same period. In DAT-AD, both verbal memory and language declined with equal severity. Although imaging showed asymmetric left-sided mediotemporal atrophy in PPA-AD, autopsy revealed bilateral hippocampo-entorhinal neurofibrillary degeneration at Braak stages V and VI. Compared to DAT-AD, however, the PPA-AD group had lower incidence of APOE ε4 and of mediotemporal TAR DNA-binding protein 43 (TDP-43) pathology.
Conclusions
Memory preservation in PPA is not just an incidental finding at onset but a core feature that persists for years despite the hippocampo-entorhinal AD neuropathology that is as severe as that of DAT-AD. Asymmetry of mediotemporal atrophy and a lesser impact of APOE ε4 and of TDP-43 on the integrity of memory circuitry may constitute some of the factors underlying this resilience. Our results also suggest that current controversies on memory in PPA-AD reflect inconsistencies in the diagnosis of logopenic PPA, the clinical variant most frequently associated with AD.
ClinicalTrials.gov Identifier
NCT00537004 and NCT03371706.
Primary progressive aphasia (PPA) is diagnosed when language impairment emerges on a background of preserved memory and behavior.1 Approximately 40% of cases represent atypical manifestations of Alzheimer disease (AD) and usually display a logopenic variant of the syndrome. Asymmetric left-sided neurodegeneration accounts for the atypical language impairment.2–9 However, the correlates of memory preservation remain unexplored. This is particularly interesting because of the known vulnerability of mediotemporal memory areas to AD neuropathology. Is the memory preservation in PPA-AD a consistent core feature or a transient finding confined to initial presentation? If the former, what underlies the resilience of memory function to mediotemporal AD neuropathology?
Characterization of memory in PPA-AD is challenging because most tests use word lists and may be failed because of the aphasia. The few investigations using nonverbal memory tasks reported inconsistent conclusions. In 1 study, patients with PPA-AD (6 of 7 logopenic) showed impaired recall of words but not pictures, whereas patients with typical AD dementia (DAT-AD) were impaired in both.10 However, a review of 849 cases concluded that logopenic PPA was associated with verbal and nonverbal memory deficits,11 while another study, also on logopenic aphasics, found both verbal and nonverbal memory deficits as severe as those of typical AD.12
To address these divergent accounts, we assessed the longitudinal course of nonverbal memory in PPA-AD. A second group of patients with DAT-AD allowed us to compare temporal trajectories of memory function and related neuropathologic correlates in these 2 distinct phenotypes of AD neuropathology.
Methods
The PPA-AD Group
The Northwestern PPA Research Program prospectively enrolls participants who volunteer for longitudinal assessments, imaging, and biomarker analyses. Most also agree to brain donation. We searched the database to identify those participants with PPA with autopsy or biomarker evidence of AD who also had at least 2 consecutive visits during which language and memory assessment had been obtained with the same tests. All 17 participants who fit these characteristics were included in this report. They were all right-handed. The diagnosis of PPA had been based on 3 criteria1: (1) adult onset and progressive impairment of language (not just speech), (2) absence of other consequential behavioral or cognitive deficits for approximately the first 2 years, and (3) neurodegenerative disease as the only cause of impairment. The aphasia was then classified according to the 2011 consensus guidelines.13 Confirmation of AD neuropathology was based on autopsy in 8 of the cases, CSF biomarkers in 3, and amyloid PET in 6.
Typical AD Group (DAT)
The control sample included participants enrolled at the Northwestern Alzheimer's Disease Center with AD as the principal neuropathologic diagnosis at postmortem. Selection was based on 3 additional criteria: the clinical diagnosis of typical amnestic probable AD dementia according to the McKhann et al.14 criteria, the presence of at least 2 consecutive visits during which memory and language had been assessed with the same tests, and the absence of disease-causing genetic mutations. Fourteen such participants were identified and made up the DAT-AD group.
Memory Tests
Memory tasks based on words are difficult to interpret in aphasic patients. We therefore chose the Picture Recognition subtest of the Rivermead Behavioral Memory Test (RBMT) to assess nonverbal retentive memory.15 It consists of 10 pictures of common objects presented first in learning trials and then after a 10-minute delay in a yes-no recognition phase in which targets are mixed with 10 foils. A perfect performance would receive a score of 20, based on 10 correct yes and 10 correct no responses. Performance is not influenced by aphasia, and healthy older adults obtain nearly perfect scores.16 Performance is significantly lower in those with mild cognitive impairment and early stages of Alzheimer-type dementias.17
In our research programs, the RBMT is given only to participants with PPA. In the DAT-AD group, memory assessment was based on the Rey Auditory Verbal Learning Test, a test designed to measure episodic memory with verbal items.18,19 Two 15-word lists are used. Recall of the first list is tested after a delay of ≈20 minutes, followed by a yes-no recognition trial containing 15 foils. The maximum score is 15 in the delayed recall condition and 30 in the recognition phase (15 correct yes and 15 correct no responses). Delayed recall of words is likely to be considerably more difficult than recognition of pictures. Analyses were therefore confined to the recognition task, which is structured identically to the RBMT Picture Recognition subtest and is likely to offer a similar level of difficulty.
Language Tests
Global language performance was assessed with the Aphasia Quotient (AQ) of the revised Western Aphasia Battery. It offers an extensively validated measure of aphasia severity.20 The Boston Naming Test (BNT) was used as an additional language test. It assesses word retrieval by requiring the patient to name 60 line drawings of objects of varying familiarity.21 The maximum scores are 100 in the revised Western Aphasia Battery AQ and 60 in the BNT. Participants with both PPA-AD and DAT-AD had BNT scores. The participants with PPA-AD also had AQ scores at all visits.
Clinical Dementia Rating Scale
The Clinical Dementia Rating (CDR)22 global score was used to assess global aspects of functionality in daily activities.
Neuropathology
Postmortem neuropathologic evaluations were available for 8 cases in the PPA-AD group and all cases of the DAT-AD group. Sections from 25 regions from each hemisphere were processed with the Gallyas stain, thioflavin-S, and immunohistochemistry with antibodies to phosphorylated tau, β-amyloid (Aβ), phosphorylated TAR DNA-binding protein 43 (TDP-43), p62, and phosphorylated α-synuclein. Densities of neurofibrillary tangles (NFTs), neuritic Aβ plaques, pathologic TDP-43 aggregates, and tau pathology (Pick bodies, astrocytic plaques, tufted astrocytes, etc) were rated as absent, mild, moderate, or severe according to standardized criteria by experienced neuropathologists (E.H., Q.M., M.F.). NFT distribution was determined by thioflavin-S and the AT8 phosphorylated tau antibody. The areas of interest for this report included the hippocampo-entorhinal area and sections from the middle and inferior frontal gyri, the inferior parietal lobule, and the superior temporal gyrus. The tau and Aβ markers of AD neuropathology were also classified according to the A, B, C guidelines of the National Institute on Aging and the Alzheimer's Association in which the A3B3C3 pattern indicates that AD neuropathologic changes have a high likelihood of being the cause of the dementia.23
Apolipoprotein Genotyping and Cerebrospinal Biomarkers
Apolipoprotein genotyping was done through the National Centralized Repository for Alzheimer's Disease and Related Dementias. Cerebrospinal levels of Aβ42, total tau, and phosphorylated tau were determined commercially through Athena Diagnostics (Marlborough, MA). The results were interpreted as indicative of underlying AD neuropathology if phosphorylated tau levels were >68 pg/mL and the Aβ42 to total tau index was <0.8.24
Amyloid PET
Amyloid PET scans were performed on a Siemens Biograph 40 TruePoint/TrueV PET-CT system (Siemens, Munich, Germany). A CT scan was acquired for attenuation correction followed by a 20-minute dynamic PET acquisition 50 minutes after administration of 370 MBq18 F-florbetapir. A board-certified nuclear medicine physician performed visual interpretations as described in our previous publications.9 This method shows strong test-retest reliability, and in an autopsy study, the visual reads were slightly more reliable than the quantitative algorithms.25 Individuals were considered to have a positive scan, indicative of neuritic amyloid plaques characteristic of AD, if there was increased retention of the tracer in cortical gray matter, defined either as loss of gray-white contrast in at least 2 cortical regions or as intense, focal uptake in at least 1 cortical region.
Structural Imaging
Structural MRI scans were acquired with a 3.0T Siemens scanner and were reconstructed with the FreeSurfer image analysis suite (version 5.1) as previously described.9 Cortical thickness maps of the PPA group were statistically contrasted against those of 35 right-handed age- and education-matched healthy volunteers. Differences in cortical thickness between groups were calculated by conducting a general linear model on every vertex along the cortical surface. A relatively permissive false discovery rate of 0.05 was applied to adjust for multiple comparisons and to detect areas of peak cortical thinning (i.e., atrophy) in PPA compared to controls.26
Statistical Methods
Data were summarized using mean and SD for the memory and language scores. Scores were analyzed by percent change, and the initial visit is considered to be the baseline. Differences in percentage change over time were assessed with a linear mixed-effects model with random intercept. Differences between language and memory scores were assessed with t tests comparing the β estimates from the linear mixed-effects model. All analyses were conducted in R version 3.5.3 (R Foundation for Statistical Computing) and SAS 9.4 software (SAS Institute Inc, Cary, NC).
Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by the Institutional Review Board of Northwestern University, and informed consent was obtained from all participants. The study is listed on ClinicalTrials.Gov with identifiers NCT00537004 and NCT03371706.
Data Availability
Anonymized neuropsychological, imaging, and neuropathologic datasets are available to be shared by request from qualified investigators.
Results
Sample Characteristics of the PPA-AD Group
There were 10 male and 7 female participants(table 1). According to the 2011 classification guidelines, the PPA was logopenic in 11 and agrammatic in 5. One patient (P17) had characteristics that could fit either designation. This distribution is in keeping with the known close correlation of logopenic PPA with AD pathology but also highlights the absence of complete overlap.2 The clinical characteristics of agrammatic PPA associated with AD pathology have been described previously.27 Age at symptom onset ranged from 47 to 74 years with a mean ± SD of 59.41 ± 6.3 years. The interval between the initial and repeat testing varied from 1.5 to 4 years with a mean ± SD of 2.35 ± 0.78 years. Initial testing occurred 3.91 ± 2.04 years after estimated symptom onset and follow-up testing 6.26 ± 2.21 years after symptom onset.
Table 1.
Characteristics of the PPA-AD Group

Sample Characteristics of the DAT-AD Group and Comparison to PPA-AD
There were 8 male and 6 female participants, a distribution very similar to that of the PPA-AD group (table 2). Age at symptom onset (66.28 ± 8.39 years) was greater than in the PPA-AD group (p = 0.014). The interval between the 2 testing sessions (1.7 ± 0.59 years) was shorter than in the PPA-AD group (p = 0.016), but reported symptom duration at first testing (4.28 ± 2.18 years) was not significantly different than in the PPA-AD group. Of particular interest to the interpretation of postmortem data, mean age at death was not significantly different in the 2 groups (72 ± 6.2 vs 77 ± 8.6 years, p = 0.1548). The groups did not differ in years of education. The median CDR score at initial testing was 0.5 for both groups, indicating that the testing in both groups had been done at stages of very mild dementia, at a time when cognitive profiles are more reliably characterized and differentiated.
Table 2.
Characteristics and Comparisons of the PPA-AD and DAT-AD Groups

Comparison of Language to Memory Changes Over Time in PPA-AD
At initial testing, the mean RBMT memory score was 19.71 ± 0.59 of a maximum score of 20 (table 1). This performance confirms the preservation of multimodal episodic memory, a necessary criterion for the PPA diagnosis. Initial language assessment revealed a mean AQ score of 87.76 ± 7.34 of a maximum score of 100 and a mean object naming score of 46.59 ± 9.91 of a maximum score of 60. These values confirm the presence of language impairment because scores in our cognitively normal group for AQ and BNT are 99.7 ± 0.7 and 58.32 ± 1.8, respectively.
In 13 of the participants, the initial memory score was 20 of 20. In 6 of these participants, the score remained 20 of 20 at retest. In 2 participants, initial scores of 19 and 18 improved to 20 of 20 at retest. The 11 logopenic participants showed no specific differences from the 5 with agrammatic PPA. In this subgroup of logopenic participants, 7 of the 11 had a perfect score of 20 of 20 at retest. In the group of 17 as a whole, the mean yearly decreases from baseline were 0.94% for memory, 4.25% for the AQ, and 6.21% for object naming. The yearly decreases were statistically significant for the 2 language tests but not for the memory test. Compared to yearly declines of memory scores, yearly declines were significantly greater for the AQ (p = 0.042) and the BNT (p = 0.026) (table 3).
Table 3.
Longitudinal Change in Memory and Language

Comparison of Language to Memory Changes Over Time in DAT-AD
At initial testing, the Rey Auditory Verbal Learning Test delayed recall memory score was 2.14 ± 3.68 of 15, confirming the severe and salient memory loss despite the relatively mild dementia. Seven of the 14 participants could not remember any of the 15 words after the delay. We therefore used the recognition score for the analyses. The mean recognition score was 20.93 ± 4.45 of 30. The initial BNT score was 45.14 ± 14.53 of 60, confirming the multimodal nature of the dementia in DAT. The mean yearly decline was 2.15% for memory and 4.053% for language, both statistically significant. The 2 progression rates were not different from each other (p = 0.25) (table 3).
Structural Imaging of the Cerebral Cortex in PPA-AD
Systematic structural imaging at each initial visit was available only for the PPA group. Cortical thinning in PPA-AD was more extensive in the language-dominant left hemisphere, where it extended into all components of the language network, including the temporoparietal junction, the inferior frontal gyrus, and the lateral temporal lobe (figure). Of particular importance to this report is that significant mediotemporal thinning in the parahippocampal gyrus appeared to be confined to the left hemisphere.
Figure. Group Atrophy Map of the 17 PPA-AD Cases at the Time of the Initial Visit at False Discovery Rate 0.05.
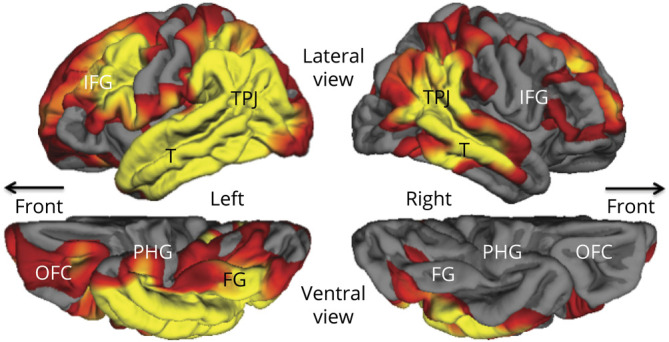
Yellow and red areas show regions of significant cortical thinning (atrophy) with p values ranging from 0.01 (red) to 0.0001 (yellow). FG = fusiform gyrus; IFG = inferior frontal gyrus; OFC = orbitofrontal cortex; PHG = parahippocampal gyrus; PPA-AD = primary progressive aphasia with Alzheimer disease; T = lateral temporal cortex; TPJ = temporoparietal junction.
APOE Genotyping in PPA-AD and DAT-AD
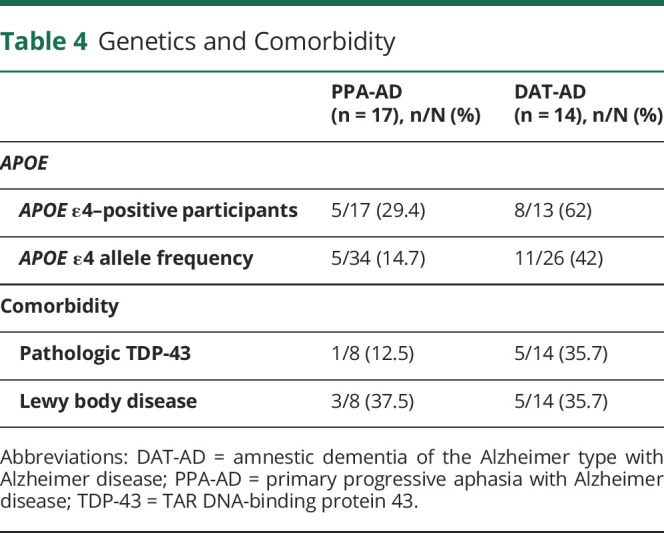
In the PPA-AD group, 5 of 17 (29.4%) of the participants had 1 ε4 allele, and the overall ε4 allele frequency was 14.7% (table 4). All other APOE alleles were of the ε3 type. In the DAT-AD group, 8 of 13 (62%) had an ε4 allele, and the overall ε4 allele frequency was 42% because 3 of the participants had the homozygous 4,4 pattern. The ε4 allele frequency in PPA-AD was at the level reported for control populations, whereas the frequency in DAT-AD was much higher and within the range seen in other autopsy series on typical amnestic AD.28
Table 4.
Genetics and Comorbidity
Neuropathology in the PPA-AD Group
In keeping with AD neuropathology as the principal diagnosis, all 8 cases with PPA-AD had maximum levels of amyloid and tau pathology, namely A3 scores for Aβ plaques, B3 scores for Braak NFT stages, and C3 scores for Consortium to Establish a Registry for Alzheimer's Disease plaque densities (table 5).23 The emphasis in this report is on NFTs rather than amyloid because the NFT density and distribution are more closely correlated with cognitive state. The B3 score (corresponding to Braak stages V–VI) indicates severe NFT pathology in neocortex and in all memory-related parts of the medial temporal lobe, including the hippocampo-entorhinal area. Six of the 8 cases showed pathology that was as severe in the hippocampo-entorhinal areas as in neocortical areas of the frontal, temporal, and parietal lobes. Two of the 8 cases (PPA1 and PPA8) were of the hippocampal-sparing type according to the semiquantitative assessment showing cortical NFT densities generally higher than those in the hippocampo-entorhinal region.29 Nonetheless, even these 2 had extensive neurofibrillary degeneration in the hippocampus and entorhinal cortex, at Braak stages of V to VI.30 Comparisons of the left and right hemispheres showed that the neocortical areas of the language-dominant left hemisphere contained more NFTs in 6 of the 8 cases and more neuritic amyloid plaques in 2 of the 8. No asymmetry of hippocampo-entorhinal pathology was reported. With respect to coexisting pathology, 3 cases had amygdala-predominant Lewy body disease (37.5%), 1 with additional cerebral amyloid angiopathy (table 4). Only 1 case (12.5%) had pathologic TDP-43 aggregates, and they were confined to the mediotemporal lobe. The interval between cognitive retest and autopsy was 3.6 ± 1.6 years.
Table 5.
Alzheimer Neuropathologic Change in the 8 PPA-AD and 14 DAT-AD Cases With Autopsy

Neuropathology in the DAT-AD Group
As in the cases with PPA-AD, all 14 cases with DAT-AD fulfilled the A3B3C3 criteria for the diagnosis of AD, indicative of severe NFT pathology in neocortex and the hippocampo-entorhinal areas (table 5). Bilateral representation of tissue sections was available for the neocortex in 9 of the cases and for the hippocampo-entorhinal region in 7. Asymmetry of cortical NFT was detected in 3 cases, 2 favoring the left and 1 the right. No asymmetry of NFT was detected in the hippocampo-entorhinal region. One of the cases (DAT2) showed a pattern of hippocampal sparing. In 5 of the cases, the hippocampo-entorhinal area had more NFT than cortical areas. In the remaining 8 cases, NFT formation was as severe in the hippocampo-entorhinal area as in neocortical regions. Coexisting pathology was seen in the form of amygdala predominant or limbic (transitional) Lewy bodies in 5 cases (35.7%) and pathologic TDP-43 aggregates confined to mediotemporal cortex in 5 cases (36%), one of which had also hippocampal sclerosis.
Discussion
Longitudinal change of memory and language was assessed in 31 patients with autopsy or biomarker evidence of AD neuropathology, 17 with aphasic dementia (PPA-AD) and 14 with amnestic dementia (DAT-AD). In PPA-AD, the yearly decline was significant for language but not memory. The majority of these patients had the logopenic variant of PPA and displayed the same pattern of memory sparing as shown by the whole group. In DAT-AD, however, memory and language showed equally significant yearly declines, in keeping with the characterization of DAT-AD as an amnestic multidomain dementia. These results raise 2 questions. First, are there factors that underlie the putative resilience of memory in PPA-AD despite the postmortem detection of severe hippocampo-entorhinal degeneration? Second, how can we reconcile our findings with reports of prominent memory impairment in the logopenic variant of progressive aphasia, a syndrome closely associated with PPA-AD11,12?
Stroke and temporal lobectomies show that permanent impairment of episodic memory occurs only when the underlying mediotemporal lesion is bilateral.31 In fact, the episodic memory impairment in DAT-AD is associated with bilateral mediotemporal neurodegeneration.32 Preservation of episodic memory in PPA-AD could therefore be attributed to the unilaterality of mediotemporal degeneration suggested by the figure. However, tau-PET imaging also shows that cortical atrophy in AD is a lagging indicator of underlying neurofibrillary tauopathy33 so that the absence of significant atrophy in the right mediotemporal area of PPA-AD does not necessarily prove the underlying absence of NFTs.
This question could potentially be addressed by postmortem analyses. For example, neocortical NFTs in PPA-AD have been shown to be more numerous in components of the left hemisphere language network than in homologous areas of the right hemisphere, an asymmetry not seen in DAT-AD.4,5 These findings show that the clinically concordant selectivity of NFT distribution is maintained up to the time of death. The picture is less clear when it comes to the status of memory-related areas. Some studies show hippocampo-entorhinal NFTs to be as numerous in PPA-AD as in DAT-AD; others show that NFTs are fewer in the hippocampo-entorhinal areas than in neocortex, in the pattern of hippocampal sparing.4,5,34 Even in cases that fit the hippocampal-sparing pattern,29 the mediotemporal areas generally contain thousands of NFT per cubic millimeter at Braak stages V and VI, and there is no consistent lateral asymmetry suggestive of right-sided mediotemporal sparing.5
In keeping with these findings, our cases with PPA-AD displayed severe and symmetric hippocampo-entorhinal neurofibrillary degeneration all at Braak stages V to VI. With the possible exception of 2 of the cases with hippocampal sparing, the neurodegeneration within the core components of the episodic memory network was as intense in PPA-AD as in DAT-AD, at least as determined by semiquantitative analysis. The relatively short interval of 3.6 ± 1.6 years between the second cognitive testing and death in the PPA-AD group suggests that the NFT pattern at autopsy is unlikely to have been very different from the pattern that existed at the second cognitive testing, during which the mean memory score was 18.18 ± 2.04 of 20.
Although tau-PET in PPA-AD has shown asymmetric left-sided binding in a pattern that mirrors the asymmetric cortical neurodegeneration,6–8,35 it has not revealed patterns of mediotemporal binding that would differentiate PPA-AD from DAT-AD.6 The hypothesis that memory sparing in PPA-AD reflects asymmetric mediotemporal neurofibrillary degeneration, while plausible, remains to be established by longitudinal tau-PET investigations and by neuropathologic evaluations using antibodies differentially sensitive to stages of NFT evolution. Although the figure shows that the structural integrity of mediotemporal cortex, as indexed by atrophy, is asymmetrically compromised, the bilateral neurofibrillary degeneration at autopsy raises the need to look for additional factors that may mediate the preservation of memory in PPA-AD.
The frequency of APOE ε4 in the PPA-AD group was within the range of control populations, whereas that of the DAT-AD group was more than twice as high, in keeping with the fact that APOE ε4 is a major risk factor for AD neuropathology in DAT but not PPA.28 Four of the 13 cases with DAT-AD but none of the cases with PPA-AD also had the homozygous 4,4 pattern of APOE.
APOE has a complex relationship to neuroplasticity and mediotemporal functionality. For example, compensatory synaptogenesis in rodents triggers astrocytic APOE expression36; ApoE-deficient mice display impaired reactive synaptogensis37,38; and the ε4 allele inhibits axonal and dendritic plasticity whereas the ε3 allele has the opposite effect.39 Furthermore, neurologically intact ε4 carriers have smaller hippocampal volumes40 and show a heightened memory-related neural activation that paradoxically predicts greater memory decline over time.41 A study on 229 consecutive patients with a clinical diagnosis of Alzheimer-type dementia also showed that memory was more impaired than naming among ε4 carriers.42
APOE ε4 may therefore magnify the selective vulnerability of the mediotemporal memory network to AD, perhaps by undermining compensatory neuroplasticity and neural efficiency. The low frequency of the ε4 vs the ε3 allele in PPA-AD may therefore provide one of the factors that underlie the heightened resilience of memory to mediotemporal pathology.
Abnormal TDP-43 aggregates characterize forms of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. However, such aggregates also exist as comorbid features in almost 50% of cases with AD, where they are usually confined to mediotemporal areas.43–46 In a separate set of cases, we had reported that mediotemporal TDP-43 was less frequent in PPA-AD (1 of 16) than in DAT-AD (14 of 27).43 In the current group of cases, we also find that fewer cases of PPA-AD (1 of 8) than DAT-AD (5 of 14) had TDP-43 comorbidity. This comparison was not influenced by differing ages of death, which is an important correlate of mediotemporal TDP-43.47 The co-occurrence of TDP-43 is typical in AD and exacerbates memory loss and mediotemporal atrophy.46 These effects are not mediated by hippocampal sclerosis. Mediotemporal TDP-43 is also associated with worse age-related episodic memory changes and accounts for nearly as much of cognitive decline as the NFT.48,49 The lesser frequency of TDP-43 comorbidity may therefore be another factor that makes memory more resilient to mediotemporal neurodegeneration in PPA-AD compared to DAT-AD.
One notable study reported that patients with logopenic progressive aphasia (LPA) had verbal and nonverbal memory deficits that were as severe as those in typical AD and conclude that LPA is an amnestic syndrome.12 The LPA diagnosis in that study was said to have followed the 2011 Gorno-Tempini et al.13 guidelines for logopenic variant of PPA, a syndrome closely associated with AD. One possible explanation for this discrepancy revolves around the application of the 2011 classification guidelines. As indicated in table 1 of that article, memory impairment is an exclusionary criterion for PPA.1,13 Table 4, listing the criteria for logopenic variant PPA, is therefore valid only if the PPA diagnosis had first been established based on table 1. It is quite likely that some investigations have based the diagnosis of LPA exclusively on table 4 of the 2011 Gorno-Tempini et al.13 guidelines in patients who do not necessarily fulfill the PPA criteria. So, the LPA term could be accurate in a descriptive sense but might not correspond to the syndromic designation of logopenic variant PPA. In that generic sense, LPA without PPA would be a most common correlate of typical dementias of AD as shown by the additional naming impairment in our DAT-AD group. This potential terminological conflation may also have influenced meta-analyses that find both verbal and nonverbal memory impairments in logopenic PPA.11 It is therefore essential to ascertain that the specifications of table 1 have been met before applying the criteria listed in table 4 of the 2011 consensus guidelines for the classification of PPA.13
When diagnosed rigorously, the PPA caused by AD constitutes a unique nosologic entity with distinctive anatomic vulnerabilities, genetic risk factors, and neuropathologic comorbid conditions that distinguish it from typical dementias of AD. The relationship of NFTs to cognition is not linear. There is a long presymptomatic phase when NFTs accumulate without overt clinical manifestations. There are also substantial interindividual differences in the resistance to the emergence of NFTs and resilience to their impact on cognition.50 It is quite likely that numerous constitutional and experiential factors compete in tilting the balance toward degeneration vs neuroplasticity during the lengthy cascade of neurodegeneration associated with AD. Some of these factors may lead to asymmetric vulnerability to degeneration; others may underlie reserves that buffer the effects of AD neuropathology; others such as APOE ε4 may decrease the neuroplasticity potential of limbic networks; and still others such as TDP-43 may exacerbate the pathologic effects of the NFT on memory function. The PPA-AD syndrome offers unique opportunities for exploring the biological foundations of these phenomena that interactively modulate the effect of AD neuropathology on cognitive function.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- AQ
Aphasia Quotient
- BNT
Boston Naming Test
- CDR
Clinical Dementia Rating
- DAT-AD
amnestic dementia of the Alzheimer type with AD
- LPA
logopenic progressive aphasia
- NFT
neurofibrillary tangle
- PPA
primary progressive aphasia
- RBMT
Rivermead Behavioral Memory Test
- TDP-43
TAR DNA-binding protein 43
Appendix. Authors
Footnotes
Editorial, page 243
Study Funding
This project was supported in part by R01 DCOO8552 from the National Institute of Deafness and Communication Disorders; P30 AG013854, R01 AG056258, and R01 AG062566 from the National Institute on Aging; and NS075075 from the National Institute of Neurologic Disorders and Stroke; the Davee Foundation; and the Jeanine Jones Fund. 18F-florbetapir doses were provided as nonfinancial support by Avid Radiopharmaceuticals (awarded to E.R.). MRI was performed at the Northwestern University Department of Radiology Center for Translational Imaging.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Mesulam MM. Primary progressive aphasia. Ann Neurol 2001;49:425–432. [PubMed] [Google Scholar]
- 2.Mesulam MM, Weintraub S, Rogalski EJ, Wieneke C, Geula C, Bigio EH. Asymmetry and heterogeneity of Alzheimer and frontotemporal pathology in primary progressive aphasia. Brain 2014;137:1176–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martersteck A, Sridhar J, Rader B, et al. Differential neurocognitive network perturbation in amnestic and aphasic Alzheimer disease. Neurology 2020;94:e699–e704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gefen T, Gasho K, Rademaker A, et al. Clinically concordant variations of Alzheimer pathology in aphasic versus amnestic dementia. Brain 2012;135:1554–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohm DT, Fought AJ, Rademaker A, et al. Neuropathologic basis of in vivo cortical atrophy in the aphasic variant of Alzheimer's disease. Brain Pathol 2020;30:332–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ossenkoppele R, Schonhaut DR, Schöll M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain 2016;139:1551–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Josephs KA, Martin PR, Botha H, et al. [18F]AV-1451 tau-PET and primary progressive aphasia. Ann Neurol 2018;83:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nasrallah IM, Chen YJ, Hsieh MK, et al. 18F-Flortaucipir PET/MRI correlations in nonamnestic and amnestic variants of Alzheimer disease. J Nucl Med 2018;59:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rogalski EJ, Sridhar J, Martersteck A, et al. Clinical and cortical decline in the aphasic variant of Alzheimer's disease. Alzheimers Dement 2019;15:543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kielb S, Cook A, Wieneke C, et al. Neuropathologic correlates of learning and memory in primary progressive aphasia. JAMA Neurol 2016;73:246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eikelboom WS, Janssen N, Jiskoot LC, van den Berg E, Roelofs A, Kessels RPC. Episodic and working memory function in primary progressive aphasia: a meta-analysis. Neurosci Biobehavioral Rev 2018;92:243–254. [DOI] [PubMed] [Google Scholar]
- 12.Ramanan S, Marstaller L, Hodges J, Piguet O, Irish M. Understanding the neural basis of episodic amnesia in logopenic progressive aphasia: a multimodal imaging study. Cortex 2020;125:272–287. [DOI] [PubMed] [Google Scholar]
- 13.Gorno-Tempini ML, Hillis A, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKhann GM, Knopman D, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson B, Cockburn J, Baddeley A, Hiorns R. The development and validation of a test battery for detecting and monitoring everyday memory problems. J Clin Exper Neuropsychol 1989;11:855–870. [DOI] [PubMed] [Google Scholar]
- 16.Cockburn J, Wilson B, Baddeley A, Hiorns R. Assessing everyday memory in patients with dysphasia. Br J Clin Psychol 1990;29:353–360. [DOI] [PubMed] [Google Scholar]
- 17.Kazui H, Matsuda A, Hirono N, et al. Everyday memory impairment of patients with mild cognitive impairment. Demen Geriatr Cogn Disord 2005;19:331–337. [DOI] [PubMed] [Google Scholar]
- 18.Rey A. L'Examen Clinique en Psychologie. Paris: Presses Universitaires se France; 1964. [Google Scholar]
- 19.Lezak MD, Howieson DE, Bigler E, Tranel D. Neuropsychological Assessment. 5th ed. New York: Oxford University Press; 2012. [Google Scholar]
- 20.Kertesz A. Western Aphasia Battery-Revised (WAB-R). Austin: Pearson; 2006. [Google Scholar]
- 21.Kaplan E, Goodglass H, Weintraub S. The Boston Naming Test. Philadelphia: Lea & Febiger; 1983. [Google Scholar]
- 22.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 23.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Alzheimers Demen 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blennow K, Dubois B, Fagan AM, Lewczuk P, De Leon MJ, Hampel H. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer's disease. Alzheimers Demen 2015;11:58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joshi AD, Pontecorvo MJ, Clark CM, et al. Performance characteristics of amyloid PET with florbetapir F 18 in patients with Alzheimer's disease and cognitively normal subjects. J Nucl Med 2012;53:378–384. [DOI] [PubMed] [Google Scholar]
- 26.Genovese CR, Lazar NA, Nichols TE. Thresholding of statistical maps in functional imaging using the false discovery rate. Neuroimage 2002;15:870–878. [DOI] [PubMed] [Google Scholar]
- 27.Rogalski E, Sridhar J, Rader B, et al. Aphasic variant of Alzheimer's disease: clinical, anatomic and genetic features. Neurology 2016;87:1337–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weintraub S, Teylan M, Rader B, et al. APOE is a correlate of phenotypic heterogeneity in Alzheimer disease in a national cohort. Neurology 2020;94:e607–e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murray ME, Graff-Radford NR, Ross OA, petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 2011;10:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 1997;18:351–357. [DOI] [PubMed] [Google Scholar]
- 31.Signoret JL. Memory and amnesias. In: Mesulam MM, ed. Principles of Behavioral Neurology. Philadelphia: FA Davis, 1985: 169–192. [Google Scholar]
- 32.Dickerson BC, Bakkour A, Salat DH, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex 2009;19:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer's disease correlates with the intensity and topography of baseline tau-PET. Sci Transl Med 2020;12:eaau5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petersen C, Nolan AL, de Paula França Resende E, et al. Alzheimer's disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathol 2019;138:597–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia C, Makaretz SJ, Caso C, et al. Association of in vivo [18F]AV-1451 tau PET imaging results with cortical atrophy and symptoms in typical and atypical Alzheimer disease. JAMA Neurol 2017;74:427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poirier J. Apolipoprotein E in animal models of CNS injury and in Alzheimer's disease. Trends Neurosci 1994;17:525–530. [DOI] [PubMed] [Google Scholar]
- 37.Masliah E, Mallory M, Alford M, Veinbergs I, Roses AD. Apolipoprotein E role in maintaining the integrity of the aging central nervous system. In: Roses AD, Weisgraber KH, Christen Y, editors. Apolipoprotein E and Alzheimer's Disease. Berlin: Springer-Verlag; 1996:59–73. [Google Scholar]
- 38.Teter B, Harris-White ME, Frautschy SA, Cole GM. Role of apolipoprotein E and estrogen in mossy fiber sprouting in hippocampal slice cultures. Neuroscience 1999;91:1009–1016. [DOI] [PubMed] [Google Scholar]
- 39.Nathan BP, Jiang Y, Wong GK, Shen F, Brewer GJ, Struble RG. Apolipoprotein E4 inhibits, and apolipoprotein E3 promotes neurite outgrowth in cultured adult mouse cortical neurons through the low-density lipoprotein receptor-related protein. Brain Res 2002;928:96–105. [DOI] [PubMed] [Google Scholar]
- 40.Reiman EM, Uecker A, Caselli RJ, et al. Hippocampal volumes in cognitively normal persons at genetic risk for Alzheimer's disease. Ann Neurol 1998;44:288–291. [DOI] [PubMed] [Google Scholar]
- 41.Bookheimer SY, Strojwas MH, Cohen MS, et al. Patterns of brain activation in people at risk for Alzheimer's disease. N Engl J Med 2000;343:450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Vlies AE, Pijnenburg YAL, Koene T, et al. Cognitive impairment in Alzheimer's is modified by ApoE genotype. Demen Geriatr Cogn Disord 2007;24:98–103. [DOI] [PubMed] [Google Scholar]
- 43.Bigio E, Mishra M, Hatanpaa KJ, et al. TDP-43 pathology in primary progressive aphasia and frontotemporal dementia with pathologic Alzheimer disease. Acta Neuropathol 2010;120:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uchino A, Takao M, Hatsuta H, et al. Incidence and extent of TDP-43 accumulation in aging human brain. Acta Neuropathol Commun 2015;3:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson AC, Dugger BN, Dickson DW, Wang DS. TDP-43 in aging and Alzheimer's disease: a review. Int J Clin Exp Pathol 2011;4:147–155. [PMC free article] [PubMed] [Google Scholar]
- 46.Josephs KA, Whitwell JL, Weigand SD, et al. TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol 2014;127:811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 2019;142:1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nag S, Yu L, Wilson RS, Chen EY, Bennett DA, Schneider JA. TDP-43 pathology and memory impairment in elders without pathologic diagnoses of AD or FTLD. Neurology 2017;88:653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson RS, Yu L, Trojanowski JQ, et al. TDP-43 pathology, cognitive decline and dementia in old age. JAMA Neurol 2013;70:1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rogalski E, Gefen T, Mao Q, et al. Cognitive trajectories and spectrum of neuropathology in superagers: the first 10 cases. Hippocampus 2019;29:458–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized neuropsychological, imaging, and neuropathologic datasets are available to be shared by request from qualified investigators.