

Summary
H3.3G34R-mutant gliomas are lethal tumors of the cerebral hemispheres, with unknown mechanisms of regional specificity and tumorigenicity. We developed a human embryonic stem cell-based model of H3.3G34R-mutant glioma that recapitulates the key features of the tumors, with cell type-specificity to forebrain interneuronal progenitors but not hindbrain precursors. We show that H3.3G34R, ATRX and TP53 mutations cooperatively impact alternative RNA splicing events, particularly suppression of intron retention. This leads to increased expression of components of the Notch pathway, notably NOTCH2NL, a human-specific gene family. We also uncover a parallel mechanism of enhanced NOTCH2NL expression via genomic amplification of its locus in some H3.3G34R-mutant tumors. These findings demonstrate a novel mechanism whereby evolutionary pathways that lead to larger brain size in humans are co-opted to drive tumor growth.
Keywords: High-grade glioma, H3.3G34R, ATRX, TP53, NOTCH2NL, Pluripotent stem cells, cancer models, histone mutant glioma
eTOC Blurb
A human embryonic stem cell model of H3.3G34R mutant gliomas was developed by Tabar and colleagues. The model identifies ventral forebrain interneuron progenitors as putative cells of origin and reveals a role for alternative mRNA splicing and NOTCH2NL in tumorigenesis.
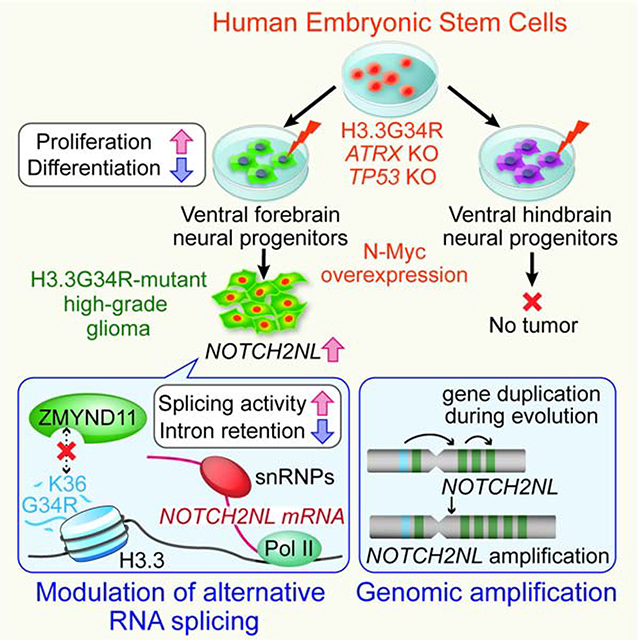
Graphical Abstract
Introduction
Histone H3.3G34R/V are recurrent mutations in the H3F3A gene recently described in a subset of high-grade gliomas (HGG) (Schwartzentruber et al., 2012; Wu et al., 2012). Current data suggest specific spatiotemporal localization to the supratentorial region of the brain in children and young adults. In comparison, histone H3K27M mutations are largely confined to the midline (thalamus, pons and spinal cord), and affect younger children. Multi-dimensional data demonstrate that these histone mutations are associated with specific gene expression signatures, DNA methylation patterns, and mutation profiles (Khuong-Quang et al., 2012; Mackay et al., 2017; Schwartzentruber et al., 2012; Sturm et al., 2012). Contrary to patients’ age distribution (median age of diagnosis at 18), the gene expression profile of H3.3G34R/V-mutant tumors is associated with early stages of brain development, suggesting that these tumors might originate much earlier than the time of diagnosis (Sturm et al., 2012). The vast majority of H3.3G34R/V-mutant tumors harbor loss-of-function mutations in both TP53 and ATRX. These findings suggest that tumorigenicity depends on specific cellular and genetic context; however, neither cell type(s) of origin, nor the molecular mechanism of oncogenic transformation, is clear.
In a previous study, we developed a human embryonic stem cell (hESC)-based model of H3.3K27M-mutant glioma, which demonstrated very potent and cell type-specific effects of the K27M mutation (Funato et al., 2014). Here, we further advance our model system by integrating recent technological advancements in genome editing and region-specific neural spheroid cultures as a means to dissect the molecular mechanism as well as spatial specificity of H3.3G34R-mutant tumors. This newly developed model shows that H3.3G34R, ATRX and TP53 mutations cooperatively block differentiation and maintain proliferation in interneuronal progenitors of the ventral forebrain, but not in hindbrain progenitor cells, reflecting the regional specificity of patient tumors. RNA sequencing shows that the combination of mutations enhances the expression of NOTCH2NL, a human-specific gene family, through suppression of intron retention. NOTCH2NL expands cortical progenitors during human brain development by activating the Notch signaling pathway (Fiddes et al., 2018; Suzuki et al., 2018); however, its oncogenic role has not been investigated. Functional assays show that NOTCH2NL contributes to increased proliferation in vitro and tumorigenicity in vivo. We also found genomic amplification of the NOTCH2NL locus in patient samples, further supporting an oncogenic role in H3.3G34R-mutant tumors.
Results
Development of hESC-based model of H3.3G34R-mutant HGG
To identify the putative cell type of origin, we first analyzed the expression profile of human tumor data in silico. Expression signatures of different cell populations within the developing human brain were extracted from single-cell RNA-seq data (Nowakowski et al., 2017) and enrichment or depletion of these signatures were calculated for each subtype of pediatric glioma (Figure 1A). Gene signatures strongly enriched in H3.3G34R/V-mutant glioma are associated with inhibitory neuronal progenitors mostly originating from the ganglionic eminence (Hu et al., 2017) (Figures 1B and S1A). Genes specifically expressed in H3.3G34R/V-mutant tumors include interneuronal marker genes encoding DLX transcription factors, glutamic acid decarboxylase GAD2, as well as transcription factors highly expressed in the ganglionic eminence of the human fetal brain such as SP8 and ARX (Figure 1C). These data suggest that H3.3G34R/V-mutant HGGs likely originate from interneuronal progenitor cells of the developing ventral forebrain (vFNPCs). During normal brain development, these vFNPCs produce cortical interneurons that migrate from the ventral forebrain to the cortex, where H3.3G34R/V-mutant tumors arise. To efficiently obtain such cells from hESCs, we optimized a previously published differentiation protocol (Figure S1B) (Maroof et al., 2013). HESCs are exposed to a Wnt inhibitor (XAV-939) during neural induction, followed by a sonic hedgehog agonist, to impart ventral forebrain identity, corresponding to the region of the ganglionic eminences. Proper patterning of the cells was confirmed by the expression of relevant genes, including FOXG1, DLX1, SP8, and ARX (Figures 1D and S1C). We also developed a new protocol for the efficient derivation of ventral hindbrain neural progenitor cells (vHNPCs) (Figure S1B), predicated on early exposure to a Wnt agonist (CHIR99021) and retinoic acid to induce ventral caudal patterning. These vHNPCs, unlike their forebrain counterparts, highly express hindbrain markers such as homeobox genes NKX6.1 and HOXB2, but not genes associated with forebrain and/or interneuron specification (Figures 1D and S1C).
Figure 1. Development of hESC-based model of H3.3G34R-mutant HGG.

(A) Schematic illustration of expression profile analysis.
(B) Representative GSEA plots show enrichment of interneuronal expression signature in H3.3G34R mutant tumors compared to H3K27M mutant tumors. MGE-IPC2: Medial Ganglionic Eminence - Intermediate Progenitor Cell - Cluster 2 (see methods).
(C) Table shows a list of genes specifically expressed in H3.3G34R/V-mutant HGG.
(D) Lineage-specific expression of marker genes in ventral forebrain neural progenitor cells (vFNPCs) and in ventral hindbrain neural progenitor cells (vHNPCs) was validated by RT-qPCR. Values are normalized to those of H1 embryonic stem cells (ESCs). Bars indicate mean ± S.E.M. (n = 3–4).
(E) Composite immunohistochemistry images of frozen sections of neural spheroids from each condition show an incremental increase of SOX2-positive NPCs and decrease of ßIII-tubulin (TUJl)-positive neurons by the combination of mutations. Scale bar, 20 μm.
(F-H) Intracellular flow cytometry shows an increase of SOX2-positive NPCs (F), a decrease of TUJl-positive neurons (G), and an increase in the Ki-67 proliferation index (H) by the combination of mutations, in ventral forebrain cultures but not in ventral hindbrain cultures. Bars indicate mean ± S.E.M. (n = 3–6).
(I) Box plot shows the number of rosette structures per spheroid in ventral forebrain cultures. More than 50 spheroids were analyzed for each condition.
(J) Combination of mutations increased the number of rosette structures per spheroid in DKO ventral forebrain (vF) cultures, but not in DKO ventral hindbrain (vH) cultures. (n = 28–32) P values were calculated by two-sided Welch’s t-test (F-H) or Wilcoxon rank-sum test (I and J). *P < 0.05, **P < 0.01, ***P < 0.001, NS, Not Significant.
Over 90% of H3.3G34R/V-mutant HGGs harbor loss-of-function mutations in TP53 and ATRX. To emulate these genetic alterations, TP53 knockout (p53KO) and TP53/ATRX double knockout (DKO) lines were derived from H1 hESCs by CRISPR/Cas9-based genome editing (Shi et al., 2017). Sanger sequencing and targeted exome sequencing confirmed frame-shift mutations in TP53 and ATRX, while other cancer-related genes and putative off-target sites remained intact (Figures S1D and S1E). The knockout cells exhibited significantly reduced mRNA expression of TP53 and ATRX (most likely by nonsense-mediated decay) and loss of protein expression (Figure S1F and S1G). Suppression of the p53-target p21 further confirmed the loss of p53 function. Consistent with previous reports (Garrick et al., 2006), we failed to derive an ATRX single-knockout hESC line due to the essential role of ATRX in hESC propagation (data not shown). Knockout of TP53 and ATRX did not significantly impair ventral patterning and derivation of both vFNPCs and vHNPCs (Figures S1H and S1I). However, constitutive expression of H3.3G34R in DKO hESCs resulted in erratic neuronal differentiation (data not shown). To circumvent this issue, either wildtype H3.3 or H3.3G34R was expressed under the core promoter region of HES5 gene, a well-characterized and widely used approach to achieve NPC-specific expression (Imayoshi et al., 2013; Ziller et al., 2015). The construct allows for monitoring of transgene expression by GFP, which is separated from H3.3 by a T2A self-cleavage peptide (Figure S1J).
HESC-derived lines representing each of the six genotypes (H3.3WT or H3.3G34R in Mock, p53KO or DKO backgrounds) were differentiated into vFNPCs or vHNPCs. The cells were cultured in a medium supplemented with a low dose of epidermal growth factor (EGF) for two weeks, followed by neuronal differentiation for another two weeks. During this maturation period, cells gradually form three-dimensional (3D) spheroids and, at day 46 of differentiation, proliferation and cellular composition were quantified by intracellular flow cytometry and immunohistochemistry. We found that the combination of mutations resulted in an incremental increase in SOX2-positive cells (46.5 ± 4.8% to 71.9 ± 4.4%; p = 0.0036), along with a decrease in ßIII-tubulin (TUJ1)-positive neurons (39.4 ± 5.0% to 5.8 ± 2.1%; p = 0.0059; Figures 1E–G) and a significant increase in proliferation (Ki67) (Figure 1H). These changes were significantly more pronounced in vFNPC compared to vHNPC cultures. Western blotting of the chromatin fraction confirmed the expression and incorporation of the wildtype and mutant histone into chromatin (Figure S1K). Rosette-forming primitive neuroectodermal cells appear transiently during hESC differentiation (Elkabetz et al., 2008), prior to maturation. By day 46 of differentiation, the H3.3G34R mutation leads to a dramatic increase in the number of rosette structures and in the percentage of cells expressing the primitive neuroepithelial marker PLZF in the DKO vFNPC condition (Figures 1I and S1L), while vHNPCs fail to form rosettes under similar conditions (Figure 1J). Taken together, these findings indicate that the H3.3G34R mutation, when combined with ATRX and TP53 mutations, strongly impairs neuronal differentiation of NPCs and enables cells to proliferate despite differentiation cues. Of particular relevance is that these phenotypes are more prominent in the forebrain cells compared to the hindbrain cells, consistent with the anatomical distribution of the H3.3G34R/V-mutant tumors which, to date, have not been reported in the hindbrain.
The H3.3G34R mutation enhances tumorigenicity in a mouse xenograft model
To evaluate the role of the histone mutation and the regional patterning of cells of origin in tumorigenicity, cells were labeled with luciferase and intracranially transplanted into immunodeficient mice (Figure 2A). A challenging issue is the fact that the triple-mutant cells, harboring H3.3G34R, ATRX, and TP53 mutations, do not grow well in vivo (Figure S2A). Consistent with this observation, another group has noted that these three mutations are not sufficient for tumorigenesis in a mouse model of HGG (Pathania et al., 2017). That study and genomics data of H3.3G34R/V-mutant patient tumors strongly suggest the requirement of additional alterations in oncogenes or tumor suppressor genes such as PDGFRA, EGFR, and CDKN2A (Gessi et al., 2013; Pathania et al., 2017; Schwartzentruber et al., 2012; Sturm et al., 2012). In addition, MYCN, which encodes the N-Myc oncogene, is known to be either amplified (Gessi et al., 2013) or upregulated in H3.3G34R/V-mutant tumors (Bjerke et al., 2013; Mackay et al., 2017). We thus introduced N-Myc in vFNPCs and in vHNPCs to facilitate tumor formation in vivo. All animals in the vFNPC-H3.3G34R group reached endpoint clinical criteria by 150 days. Their survival was significantly shorter than animals bearing vFNPCs with the wild type histone and DKO mutations, while vHNPC injections did not impair animal survival (Figures 2B and S2A). Histological assessment identified highly invasive tumors exhibiting high proliferation, extensive nuclear pleomorphism, rosette-like structures and leptomeningeal spread (Figures 2C, 2D, S2B and S2C). The expression of forebrain and interneuron markers was maintained in the tumors (Figure S2D). Immunostaining for N-Myc oncogene showed similar expression levels among histone-wildtype, H3.3G34R-mutant, and patient-derived cell line xenografts (Figure S2E). In addition, there was no apparent change in N-Myc expression by H3.3G34R (Figure S2F). Strikingly, the hindbrain cells did not form tumors even with all four oncogenic perturbations including N-Myc overexpression (Figures 2B, 2C and S2G). Surviving mutant hindbrain cells in the mouse brains showed almost no proliferation (Figures 2C and 2E). These results are consistent with the clinical observation that most H3.3G34R/V-mutant tumors arise in the supratentorial region, but not in the brainstem. In addition to the specific localization, H3.3G34R/V-mutant tumors are known to exhibit CpG hypomethylation (Sturm et al., 2012). We analyzed DNA methylation of the H3.3G34R-mutant xenografts and compared them with sets of patient tumors encompassing histone wildtype, IDH1-mutant, H3K27M-mutant and H3.3G34R/V-mutant subtypes, in addition to normal neural progenitors. The data show low CpG methylation levels in the H3.3G34R-mutant xenografts, which were comparable to those of H3.3G34R-mutant patient tumors (Figure S2H). A key feature of ATRX mutations is their association with alternative lengthening of telomeres (ALT), a feature that we identified by telomere FISH in the xenograft cells as well as sections from a surgical specimen of a human H3.3G34R-mutant HGG (Figure 2F). These results further support the critical role of neurodevelopmental context in the formation of H3.3G34R-mutant tumors.
Figure 2. The H3.3G34R mutation enhances tumorigenicity in a mouse xenograft model.

(A) Schematic illustration of in vivo tumorigenicity assay.
(B) Kaplan-Meier survival curve shows quadruple-mutant vFNPCs (vFNPC-DKO-G34R-MYCN) exhibited worse survival than histone-wildtype cells (vFNPC-DKO-H3.3-MYCN) and quadruple-mutant vHNPCs (n = 6–8).
(C) Immunofluorescence images of representative sections labeled for human-specific nuclear antigen (HNA) and Ki67 (bottom panel), and counter-stained with DAPI. Hindbrain cells formed non-proliferating small cell clusters (right panel, arrowhead).
(D) Immunohistochemistry for human NCAM or human nuclear antigen (HNA) shows infiltration of vFNPC-derived tumor cells into brain parenchyma (top), nuclear pleomorphism and rosette-like structures (bottom).
(E) Quantification of proliferating cells in xenografts. Bars indicate mean ± S.E.M. (n = 3–5).
(F) Fluorescence in situ hybridization (FISH) for telomeres demonstrates alternative lengthening of telomeres (ALT) in the transplanted cells and in patient tumor cells.
Scale bars, 1 mm (C, top and middle); 20 μm (C, bottom); 100 μm (D, top); 20 μm (D, bottom); 10 μm (F). P values were calculated by the log-rank test (B) or two-sided Welch’s t-test (E). *P < 0.05, ***P < 0.001.
H3.3G34R and co-occurring mutations alter gene expression profile and alternative splicing pattern
To reveal the molecular mechanism underlying the observed oncogenic phenotypes, GFP-positive HES5-expressing NPCs were isolated from day 46 spheroids from each genetic background and gene expression profiles were analyzed by RNA sequencing. We then performed a Principal Component Analysis on the ES-derived cells and on two H3.3G34R patient tumor-derived cell lines (Figure 3A). The analysis shows that vFNPCs with all three mutations (i.e. H3.3G34R, ATRX and p53) cluster closely with the patient tumor cell lines (SJHGGx6 and HSJD002) but separately from vFNPCs bearing one, two or no mutations. The vHNPCs cluster together regardless of their mutation status, and separately from all other groups. Patient tumor tissues form a separate cluster regardless of histone mutation status, suggesting a contribution of environmental factors to their gene expression profiles (Figure S3A). Unsupervised hierarchical clustering of gene expression data also demonstrates segregation of the hindbrain cells from the forebrain cells regardless of oncogene expression (Figure S3B). Genes associated with stem cell maintenance are upregulated by the combination of mutations in vFNPCs, including helix-loop-helix genes HES1, HEYL and ID1, while ASCL1, an essential transcription factor for neuronal differentiation, is downregulated (Figures 3B). In the hindbrain cells, these genes are minimally affected by the mutations.
Figure 3. H3.3G34R and co-occurring mutations alter gene expression profile.

(A) Principal component analysis of RNA-seq data shows a distinct expression profile of DKO-G34R cells, which are clustered together with H3.3G34R-mutant patient-derived cell lines (SJHGGx6 and HSJD002) (upper left). Hindbrain cells form a separate cluster regardless of mutation status (upper right).
(B) RT-qPCR confirms the altered expression of the indicated genes by the combination of mutations in vFNPCs, but not in vHNPCs (n = 3–5).
P values were calculated by two-sided Welch’s t test. *P < 0.05, **P < 0.01, ***P < 0.001.
RNA alternative splicing (AS) is a fundamental mechanism of molecular diversity that plays a crucial role in temporo-spatial gene regulation during CNS development (Raj and Blencowe, 2015). The H3.3G34R oncohistone inhibits H3.3K36 trimethylation (me3) in cis, with the total H3K36me3 level unchanged (Lewis et al., 2013). H3.3K36me3 marks are specifically recognized by ZMYND11 (or BS69), which also regulates AS (Guo et al., 2014; Wen et al., 2014). We reasoned that H3.3G34R may in fact impact alternative splicing, leading to dysregulation of progenitor self-renewal and differentiation. We identified AS events that are specifically regulated in the triple-mutant condition, compared to all other genetic conditions (Figure 4A). Upregulated AS events are enriched in exon skipping, especially multiple exon skipping, and depleted of intron retention (IR), while downregulated AS events show the opposite trend (Figures 4A and S4A). This result is consistent with the previous observation that ZMYND11 mainly regulates exon skipping and IR (Guo et al., 2014). Motif analysis around splicing donor and acceptor sites shows consensus splicing sites, indicating that IR is not solely regulated in a sequence-specific manner (Figures S4B). Pathway analysis indicates that most genes regulated by the mutations are known targets of AS (Figure S4C). To further examine the role of AS in the formation of G34R-mutant HGG, we analyzed AS patterns in RNA-Seq data from H3.3G34R-mutant and histone-wildtype tumors that are age- and location-matched (Figure S4D). Genes that are highly expressed (top 10%) in the H3.3G34R-mutant tumors were also differentially spliced compared to histone-wildtype tumors, mostly in the form of intron retention (Figure S4E).
Figure 4. H3.3G34R and co-occurring mutations alter alternative RNA splicing pattern.

(A) Hierarchical clustering of alternative splicing (AS) events upregulated (924 events) or downregulated (853 events) in triple-mutant vFNPCs (DKO-G34R). Color bar at the bottom indicates the types of AS events. The bar chart shows suppression of intron retention (IR) and enrichment in multi-exon skipping (MES) in DKO-G34R cells. AS events upregulated in DKO-G34R cells show opposite trends compared to downregulated AS events.
(B) Intron retention of Notch component genes was impacted in DKO-G34R cells.
(C) RT-PCR shows the changes of intron retention in NOTCH2NL and DLL3.
(D) Quantitative real-time RT-PCR confirms the changes in intron retention. Difference in Percentage Spliced In (ΔPSI) values are shown. PSI for each gene is calculated as the proportion of properly spliced transcripts to all transcripts containing any of the relevant exons. Bars indicate mean ± S.E.M. (n = 4–6).
(E) Box plot shows the expression of the indicated genes in each tumor type, based on published tumor datasets (see methods)
P values were calculated by two-sided paired t-test (D) or two-sided Welch’s t test (E). *P < 0.05, **P < 0.01, NS, Not Significant.
Upregulation of NOTCH2NL genes contributes to tumorigenicity in H3.3G34R-mutant tumors
The Notch pathway is central to major neurodevelopmental events and is known to be involved in stem cell self-renewal and neural cell fate specification; it is also implicated in HGG (Teodorczyk and Schmidt, 2014). Considering that the triple-mutant spheroids showed an increase in SOX2-positive NPCs and a reciprocal decrease in Tuj1-positive neurons (Figures 1E–G), we focused on a potential role for the Notch pathway. In support of this hypothesis, expression profile analysis showed an upregulation of Notch target genes such as HES1 and HEYL (Figure 3B). We also performed AS analysis notably showed that a subset of genes involved in the Notch signaling pathway were subject to suppression of intron retention by the triple mutations (Figure 4B). Suppression of intron retention in these genes was confirmed by both conventional RT-PCR and quantitative real-time RT-PCR (Figures 4C, 4D, S4F and S4G). Among them, NOTCH2NL exhibited the highest change in splicing (Figure 4B). In addition, NOTCH2NL and HES1 had significantly higher expression levels in H3.3G34R/V-mutant patient tumors compared to K27M-mutant and histone-wildtype tumors (Figure 4E).
The NOTCH2NL family consists of four highly homologous genes that have played a key role in the evolution of larger brains and in human-specific cortical development (Fiddes et al., 2018; Suzuki et al., 2018). They are thought to have originated as a non-functional partial genomic duplication of the NOTCH2 gene in the great apes, which was repaired as humans diverged from chimpanzees. Quantitative RT-PCR confirmed that the combination of mutations in our hESC model system led to a decrease in the retention level of the last intron of NOTCH2NL, resulting in an increase in the properly spliced mRNA transcript (Figures 4C, 4D, 5A and 5B). The loss of ATRX and p53 resulted in an increase in appropriate splicing of NOTCH2NL in both vFNPCs and vHNPCs; the addition of H3.3G34R further enhanced it in a statistically significant manner in vFNPCs, but not in vHNPCs (Figure 4D). A similar result was seen in DLL3 gene, suggesting a cell type-specific effect of the mutations (Figure 4D). Consistent with this finding in our model, the level of intron retention of NOTCH2NL is lower in H3.3G34R-mutant tumor cell lines, compared to H3K27M-mutant tumor cell lines (Figures S5A and S5B). ChIP-PCR data showed that the level of H3K36me3 and its specific reader ZMYND11 were both downregulated at the site of intron retention in the triple-mutant condition, while there was an enrichment of H3.3G34R at the same sites in the NOTCH2NL locus (Figure 5C). These data collectively suggest that the suppression of intron retention may be mediated by the loss of H3K36me3 and subsequent reduced binding of ZMYND11. To further examine the role of H3.3-ZMYND11 interaction, the lysine 36 residue in the H3.3 histone tail was substituted with arginine (K36R), which cannot be methylated and therefore does not allow binding of ZMYND11. Wildtype, H3.3G34Ror K36R H3.3 were lentivirally transduced into DKO vFNPCs and expressed under the human PGK1 promoter (Figure S5C). Expression of H3.3K36R in DKO vFNPCs resulted in an increase in proliferation and sphere formation, comparable to the effect of H3.3G34R (Figures S5D and S5E). Both H3.3G34R and H3.3K36R also suppressed the intron retention of NOTCH2NL, and resulted in an increase in its expression and that of Notch target genes HES1 and HEYL (Figures S5F and S5G).
Figure 5. Upregulation of NOTCH2NL genes contributes to tumorigenicity in H3.3G34R-mutant tumors.

(A) RNA-seq read mapping at the last exons of NOTCH2NL gene shows decreased intron retention in triple-mutant vFNPCs (DKO-G34R).
(B) Expression of NOTCH2NL is increased by the mutations in vFNPCs, but not in vHNPCs. Bars indicate mean ± S.E.M. (n = 3–5).
(C) ChIP-PCR shows decreased H3K36me3 and BS69/ZMYND11 binding at the intron retention site of NOTCH2NL (primer pair #2), but not at the first intron (primer pair #1). Occupancy by H3.3G34R is confirmed by a specific antibody. A primer for the first intron of LHX8 was used as a control. Bars indicate mean ± S.E.M. (n = 3–4).
(D) Overexpression of NOTCH2NL (N2NL) enhances proliferation in p53KO vFNPCs, but not in p53KO vHNPCs. Bars indicate mean ± S.E.M. (n = 3–6).
(E) Western blotting confirms specific knockdown of NOTCH2NL by two independent small hairpin RNAs (shRNAs).
(F) Knockdown of NOTCH2NL in H3.3G34R-mutant patient-derived cell line (HSJD002) significantly reduces the total cell number. Bars indicate mean ± S.D. (n = 4).
(G) Limiting dilution assay shows a decrease in sphere-forming capacity by NOTCH2NL knockdown.
(H) Bioluminescence imaging shows suppressed in vivo growth of H3.3G34R-mutant cell line by NOTCH2NL knockdown (n = 4–5). P values were calculated by two-sided Welch’s t test (B, C, D, F, H) or the likelihood-ratio test (G). *P < 0.05, **P < 0.01.
We next carried out gain- and loss-of-function experiments to evaluate the role of NOTCH2NL in gliomagenesis. Overexpression of NOTCH2NL boosted proliferation twofold in the forebrain cells but had the opposite effect in the hindbrain cells (Figures 5D) indicating a cell context-dependent effect. Expression of a NOTCH2NL transgene containing intron 4 did not affect proliferation, sphere formation or the expression of Notch target genes (Figures S5E and S5H–J), further confirming the role of the properly spliced form of NOTCH2NL. We next designed shRNAs targeting the last exon of NOTCH2NL, but not NOTCH2, as confirmed by western blotting (Figure 5E). Knockdown of NOTCH2NL in the H3.3G34R-mutant cell line resulted in a decrease in total cell number as well as in sphere-forming capacity (Figures 5F and 5G). When injected intracranially in immunodeficient mice, the tumor line bearing shNOTCH2NL showed a significantly lower tumor burden, compared to control shRNA (Figure 5H). Taken together, these data suggest a key role for NOTCH2NL in mediating the effects of the combined mutations on proliferation and tumor formation in a regional patterning-dependent manner.
The NOTCH2NL locus located at 1 q21.1 has been subjected to multiple rounds of genome duplication during human evolution. We therefore hypothesized that this locus may be prone to genomic rearrangement during tumor development. Quantitative genomic PCR indicated that H3.3G34R mutant tumor cell lines obtained from surgical samples indeed have a high copy number of NOTCH2NL, whereas H3K27M-mutant tumor or histone-wildtype adult glioblastoma cell lines do not show obvious changes (Figure 6A). Next, we designed fluorescent in situ hybridization (FISH) probes targeting the NOTCH2NL locus and the NOTCH2 locus and confirmed the increased copy number of NOTCH2NL locus in H3.3G34R-mutant tumor tissue as well as in two H3.3G34R-mutant cell lines (Figures 6B and S6A). Array comparative genomic hybridization (aCGH) data further confirms the focal amplifications of the NOTCH2NL locus in 44% of H3.3G34R-mutant tumors and, to a lesser extent, in H3K27M-mutant (22%) and histone-wildtype tumors (6%) (Figures S6B and S6C). Collectively, these data suggest orthogonal mechanisms for amplification of NOTCH2NL activity in these tumors, further supporting its crucial role in driving tumor development.
Figure 6. NOTCH2NL locus is amplified in H3.3G34R-mutant tumors.

(A) Genomic copy number of the NOTCH2NL locus in different cell lines was quantified by genomic real-time PCR. Bars indicate mean ± S.E.M. (n = 3).
(B) FISH assay targeting the NOTCH2NL (green) and the NOTCH2 (red) loci shows an increase in NOTCH2NL copy number in H3.3G34R-mutant patient-derived tumor tissue and in an H3.3G34R-mutant patient-derived cell line. Scale bar, 5 μm.
Discussion
In this study, we developed a model that can recapitulate the specific spatial localization of H3.3G34R/V-mutant HGG to cerebral hemispheres. Our data collectively suggest that ventral forebrain interneuronal progenitors represent a cell type of origin of these tumors; they also support the idea that the initiation of H3.3G34R-mutant tumors starts early, possibly during fetal brain development (Sturm et al., 2012). However, further study is required to demonstrate these findings more definitively. Our model can be a useful platform to further dissect the mechanistic role of H3.3G34R mutation as well as other types of histone mutations. Besides glioma, head and neck squamous cell carcinomas and bone and soft tissue tumors harbor recurrent point mutations in histone H3.3 variant (H3.3K36M in chondroblastoma and H3.3G34W/L in giant cell tumors) (Behjati et al., 2013). Similar to H3.3G34R/V mutations, all of these mutations suppress H3K36me3, but only the H3.3K36M mutation does so in trans (Lewis et al., 2013). One recent study shows that the H3.3G34W mutation alters alternative RNA splicing in giant cell tumors, suggesting a shared role of H3K36me3 in tumorigenesis (Lim et al., 2017). However, these three mutations have not been identified in glioma. Recent studies indicate that H3.3G34R/V mutations not only suppress H3K36me3, but also alter other histone modifications by promoting aberrant PRC2 activity (Jain et al., 2020) and inhibiting the KDM4/JMJD2 family of histone demethylases (Voon et al., 2018). The roles of KDM4/JMJD2 family and their target H3K9me3 in gliomagenesis are not yet elucidated and will require further study. It is also worthwhile to note that, unlike K27M mutations that are also found in canonical histone H3 genes, H3.3G34R/V mutations are only found in the H3F3A gene. Our data suggest that the suppressed binding of a “reader” protein ZMYND11, which can specifically recognize K36me3 on histone H3.3 variants, might be required for the oncogenic phenotype. In support of this idea, expression of H3.3K36R mutant, which cannot be methylated and therefore does not allow binding of ZMYND11, exhibited the same phenotype. Consistent with our findings, ZMYND11 has been shown to affect intron retention and exon skipping in HeLa cells (Guo et al., 2014). However, considering that H3.3K36R mutation has not been identified in glioma, and may lead to additional alterations that are not uncovered here. Further inquiry into the role of H3.3G34R/V is also warranted.
Despite frequent mutations in brain tumors, the role of ATRX in tumorigenesis is still under debate. Knockdown of Atrx in mouse glioma models enhances intracranial tumor growth (Koschmann et al., 2016), while knockdown of ATRX in a human model of IDH1-mutant low-grade glioma reduces proliferation (Modrek et al., 2017). In our model, ATRX mutation results in robust phenotypes in proliferation and differentiation, and these phenotypes are further enhanced by H3.3G34R mutation. On the other hand, the effect of the H3.3G34R mutation alone is limited, indicating a cooperative role of these mutations. ATRX forms a histone chaperone complex with DAXX and deposits histone H3.3 variants to repetitive regions such as endogenous retroviruses (ERVs) and telomeres (Elsässer et al., 2015; Goldberg et al., 2010). The effect of loss of ATRX on the deposition pattern of histone H3.3 variants, especially at the repetitive regions, is still to be elucidated.
Until recently, the NOTCH2NL family was poorly characterized, in part because the genomic locus in which the NOTCH2NL family is located was not properly mapped and annotated to the human genome until the latest genome assembly (hg38). The expression of NOTCH2NL was elevated in patient tumors as well as in our model cells by the combination of mutations, at least in part by suppression of intron retention. Neither splicing regulation nor the oncogenic role of NOTCH2NL has been previously reported. Moreover, we found that the NOTCH2NL locus was amplified in H3.3G34R-mutant cell lines as well as in tumor tissue, suggesting a potential link between human evolution and tumor evolution.
Taken together, this study provides novel insight into the mechanisms of gliomagenesis that are mediated by the H3.3G34R mutation. While the Notch signaling pathway has been implicated in gliomagenesis, NOTCH2NL is a recently described hominid-specific duplicated set of genes with no previously described oncogenic role. We propose that, the H3.3G34R gliomas co-opt this evolutionary pathway, which has been developed to expand the human cortex, by regulating splicing as well as genomic rearrangement of the NOTCH2NL locus and potentially other genes at the 1q.21 locus.
Limitation of study
Mutations found in G34R/V-mutant HGGs, including H3.3G34R, ATRX and TP53 mutations, are all somatic mutations (Schwartzentruber et al., 2012; Wu et al., 2012). Our model system, in which ATRX and TP53 mutations were introduced at ESC stage, is less suitable for the study of timing and order of mutations during gliomagenesis. In addition, the mutant histone is expressed under the HES5 promoter in addition to endogenous histone gene expression, which limits our ability to study the dosage-dependent effect of the oncohistone. Considering the cell type-specific effect of the mutations as well as the diversity of interneurons, further study using more specific promoters/enhancers or targeted knock-in approaches can be considered. While we demonstrate a functional role for NOTCH2NL, the contribution of genomic amplification across H3.3G34R mutant tumors requires further study. The NOTCH2NL family genes are evolutionarily new genes and highly homologous. Because of their high sequence similarity, detection and characterization of each single gene is challenging. Furthermore, the locus in which the NOTCH2NL family is located has a complicated genomic structure due to multiple rounds of genomic duplication during hominid evolution (Lodewijk et al., 2020); therefore, careful analysis and awareness of copy number variations in the general population are required for more extensive analysis.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Viviane Tabar (tabarv@mskcc.org).
Materials Availability
Plasmids and cells generated in this study will be shared upon reasonable request.
Data and Code Availability
The RNA-seq and Methyl Capture Sequencing data generated during this study are available at NCBI Gene Expression Omnibus (GEO) with an accession number GSE147543 and Sequence Read Archive (SRA) with an accession number SRP254041.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human H1 iCas9 ESCs (XY) were a kind gift from Danwei Huangfu Laboratory (MSKCC) (Shi et al., 2017). G34R-mutant patient-derived cell lines SJHGGx6 and HSJD002 were kindly provided by Suzanne J. Baker (St. Jude Children’s Research Hospital) and Ángel Montero Carcaboso (Hospital Sant Joan de Déu Barcelona), respectively. All cells were cultured with respective media in a humidified 37°C, 5% CO2 incubator.
Animal studies
All animal experiments were done in accordance with protocols approved by our Institutional Animal Care and Use Committee and following NIH guidelines for animal welfare. 3-months-old NOD/SCID mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; RRID: IMSR_JAX:005557) were obtained from the Jackson Laboratory and maintained at the designated facility. Male and female animals were evenly assigned to each group.
METHOD DETAILS
Cell culture
H1 hESCs (WA-01) were maintained in Essential 8 FLEX medium (Thermo Fisher) on cell culture dishes coated with recombinant Vitronectin (Thermo Fisher). Human patient-derived cell lines (HSJD002 and SJHGGx6) were maintained in a 1:1 mixture of DMEM/F12 medium and Neurobasal medium (Thermo Fisher) supplemented with B27 without Vitamin A (Thermo Fisher), EGF (20 ng/ml), basic FGF (20 ng/ml, R&D Systems), PDGF-AA and -BB (20 ng/ml, Peprotech) and heparin (10 ng/ml, Stemcell Technologies). HEK293T cells were maintained in DMEM medium (Thermo Fisher) supplemented with 10% fetal bovine serum (FBS).
Embryonic stem cell culture and differentiation
HESCs were differentiated to interneuronal progenitor cells of the ventral forebrain (vFNPCs) by a previously described protocol (Maroof et al., 2013) with minor modifications. 150,000 cells were plated on a 24-well plate coated with Matrigel (1:30 dilution, Corning). On the next day, at day 0, medium was changed to Essential 6 medium (Thermo Fisher) with BMP inhibitor LDN193189 (200 nM, Stemgent), SMAD inhibitor SB431542 (10 μM, Tocris Bioscience), and Wnt inhibitor XAV939 (2 μM, Sigma). From day 6 to 10, cells were cultured in DMEM/F12 medium with N2 supplement (Thermo Fisher Scientific) and NeuroCult SM1 Supplement without Vitamin A (Stemcell Technologies). At day 10, cells were dissociated with Accutase (Innovative Cell Technologies) and replated into culture plate coated with poly-ornithine (PO; 15 μg/ml), laminin (Lam; 1 μg/ml), and fibronectin (FN; 2 μg/ml), and were treated with SAG (200 nM, Cayman Chemical) for 8 days. To differentiate hESCs into ventral hindbrain neural progenitor cells (vHNPCs), cells were treated with retinoic acid (RA, 1 μM) in addition to LDN193189 and SB431542 for 2 days, followed by Wnt activator CHIR99021 (3 μM, Tocris Bioscience) for 2 days. From day 6 to day 10, cells were treated with SAG (200 nM) and FGF8 (100 ng/ml, R&D Systems). At day 10, cells were replated on PO/Lam/FN-coated plate and cultured with DMEM/F12 medium supplemented with N2, NeuroCult, SAG (200 nM) and FGF8 (100 ng/ml) for 8 days. After this patterning period, cells were dissociated with Accutase, re-plated on PO/Lam/FN-coated plate using high-density droplets (~10,000 cells/μl), and cultured with medium supplemented with N2, NeuroCult and epidermal growth factor (EGF; 0.4 ng/ml, R&D Systems) for 14 days, followed by neural differentiation medium supplemented with brain-derived neutrophil factor (BDNF; 20 ng/ml, R&D Systems) and ascorbic acid (AA; 200 μM, Sigma) for another 14 days.
CRISPR/Cas9-based gene editing
H1 iCas9 cells (Shi et al., 2017), which express Cas9 under the control of tetracycline-responsive element (TRE), were used to introduce mutations in ATRX and TP53. Single guide RNAs (sgRNAs) were designed using CRISPRdirect (https://crispr.dbcls.jp)(Naito et al., 2015) to minimize the off-target effect. In addition, Cas-OFFinder (http://www.rgenome.net/cas-offinder/)(Bae et al., 2014) was used to select target sequences that have at least three mismatches to any genomic location except the target sites. Selected target sequences are sgTP53: 5’-GGGAGAGACCGGCGCACAG-3’ and sgATRX: 5’-TGAATTCTATACGATCAAGG-3’. These sgRNAs were cloned into pSico-puro-mCherry plasmid and transferred into hESCs using Nucleofection (Amaxa). After Nutlin-3 treatment to select p53-mutant cells, single-cell clones were isolated by FACS and the genotype of each clone was checked by Sanger sequencing of the genomic DNA. Putative off-target sites that have perfectly matched 12-mer and PAM sequence (2 hits for sgTP53 and no hit for sgATRX) were searched using CRISPRdirect and validated by genomic DNA sequencing. Mutations in TP53 and ATRX as well as the lack of off-target mutations were further confirmed by MSK-IMPACT, a targeted exon sequencing platform that covers 480 cancer-related genes, and calculations of copy number variations and microsatellite instability. Validated hESC lines were lentivirally transduced with histone H3.3-expressing construct and maintained under puromycin selection.
Intracellular Flow Cytometry
Spheroids were dissociated using Papain dissociation kit (Worthington Biochemical) and fixed with Cytofix/Cytoperm solution (BD Biosciences), followed by incubation with Perm/Wash buffer. Cells were stained with fluorescent-labeled antibodies and analyzed on a BD LSRII flow cytometer following the manufacturer’s instructions. Fluorescent-conjugated mouse monoclonal antibodies against SOX2 (O30–678; 1:40), Ki67 (B56; 1:200), and PLZF (R17–809; 1:40) were obtained from BD; TUBB3 (TUJ1) from BioLegend.
In vitro limiting dilution assay
Sphere-forming capacity was assessed by a limiting dilution assay. 10~160 cells were plated into 96-well low-attachment plates. Following 12 days of incubation, spheres with more than 5 cells were counted. P-values were calculated using Extreme Limiting Dilution Analysis (ELDA) software (http://bioinf.wehi.edu.au/software/elda/)(Hu and Smyth, 2009).
Immunohistochemistry
Spheroids and mouse brains were fixed with 4% paraformaldehyde (PFA). After cryopreservation in 30% sucrose in PBS, samples were embedded into a 1:1 solution of OCT compound (Tissue-Tek) and 30% sucrose in PBS and frozen with dry ice. Cryosections of 5 μm thickness were cut from the frozen block using a Cryostat, attached to glass slides and stored at −80°C. For immunofluorescent staining, cryosections were dried, rinsed with PBS and incubated with blocking solution containing 10% FBS and 0.3% Triton X-100 for 1 hour. Antibodies were diluted into the blocking solution and incubated with samples overnight at 4°C. Samples were washed three times with PBS and incubated with secondary antibody diluted in PBS supplemented with 0.1% bovine serum albumin (BSA) followed by nuclear staining with DAPI. After washing with PBS, samples were mounted with Vectashield (Vector) and sealed by cover glass. For 3,3’-Diaminobenzidine (DAB) staining, cryosections were stained by Mouse-On-Mouse IgG HRP polymer Immunodetection Kit and visualized by ImmPACT DAB Peroxidase Substrate (Vector), followed by counterstaining with Hematoxylin. Rat monoclonal antibody against SOX2 (Btjce; 1:200) was obtained from eBioscience. Mouse monoclonal antibody against human NCAM (ERIC-1; 1:200) was obtained from Santa Cruz; human nuclear antigen (HNA; 235–1; 1:200) and Nestin (10C2; 1:200) from Millipore; NKX6–1 (F55A10; 1:100) from DSHB; TUBB3 (TUJ1; 1:400) from BioLegend. Rabbit polyclonal antibodies against Ki67 (ab15580; 1:200) and DLX2 (ab135620; 1:100) were obtained from Abcam; FOXG1 (M227; 1:40) from Takara. Images in Figure 1E are composites of multiple images. Images in the top and middle panels in Figure 2C were captured by Mirax Slide scanner.
In silico analysis of putative cell of origin
For figure 1A–C and S1A, a modified version of Gene Set Enrichment Analysis (GSEA) (Lamb et al., 2006; Subramanian et al., 2005) was performed on the GenePattern server (https://cloud.genepattern.org) to compare the expression profiles in datasets of HGG (GSE34824 and GSE36245) and single-cell RNA-seq dataset obtained from human fetal brains (Nowakowski et al., 2017). For each cell population, the top 100 up and down regulated differentially expressed genes (by one-vs-all strategy) were used as query signatures. Genes on the microarray (Affymetrix Human Genome U133 Plus 2.0) were rank-ordered based on differential expression (signal-to-noise ratio) between G34R/V-mutant tumors and K27M-mutant tumors (G34R/V vs K27M) or histone-wildtype tumors (G34R/V vs WT). The enrichment/depletion score was calculated by subtracting the normalized weighted Kolmogorov-Smirnov statistic of the downregulated query signature from that of the upregulated query signature. P values were calculated by random permutation of the sample labels (n = 1,000) and adjusted for multiple comparisons using Benjamini-Hochberg method.
RNA-seq
Total RNA was extracted with Trizol reagent (Thermo Fisher). Library preparation and next-generation sequencing (NGS; Illumina HiSeq, paired-end) was performed by GENEWIZ. Data processing was carried out within the Galaxy public server (https://usegalaxy.org)(Afgan et al., 2018). After removing low-quality reads and adapter sequences, reads were mapped to the human genome (hg38) using HISAT2 with default parameters. Expression levels of each gene were measured by featureCounts and normalized to TPM (Transcripts Per Kilobase Million). After removing genes with low expression or low variation across samples, expression data were subjected to principal component analysis (PCA) and hierarchical clustering with Ward’s method and Pearson correlation distance using the R software. Detection and quantification of RNA alternative splicing (AS) events was performed using SplAdder with the default parameters (Kahles et al., 2016, 2018). PSI value is calculated as the ratio of the number of reads that represent a given AS event to the number of all reads containing any of its relevant exons. AS events significantly altered (ΔPSI > 10% and p-value < 0.05) in the triple-mutant condition were subjected to further analyses. Hierarchical clustering in Figure 4A and volcano plots in Figure S4A were performed using the R software. Motif analysis in Figure S4B was performed by the WebLogo website (https://weblogo.berkeley.edu/logo.cgi). Pathway analysis in Figure S4C was performed by the DAVID website (https://david.ncifcrf.gov/). The RNA-Seq data were deposited to the NCBI Gene Expression Omnibus (GEO) under the accession code GSE147543.
Analysis of RNA-seq data of patient tumors
RNA-seq dataset of patient tumors was obtained from St. Jude Children Research Hospital (EGAS0000100192) (Wu et al., 2014). H3.3G34R-mutant (n = 4) and age- and location-matched histone-wildtype tumors (n = 8) were selected. All genes were ranked by differential expression (ratio of mean expression values) and top and bottom 10% genes were extracted (up- and downregulated in H3.3G34R-mutant tumors, respectively). AS events were analyzed using SplAdder and alternatively spliced genes (ΔPSI > 10% and p-value < 0.05) were selected and compared to the differentially expressed gene sets.
Analysis of array-based comparative genomic hybridization data
Focal amplification of the NOTCH2NL locus was analyzed in an HGG dataset (Sturm et al., 2012) within the Progenetix public server (https://progenetix.org) (Baudis and Cleary, 2001; Cai et al., 2014). Amplifications that include at least one of the NOTCH2NL family genes and spans less than 5 Mb were selected and mapped to the GRCh38/hg38 assembly using the UCSC genome browser (http://genome.ucsc.edu/).
Analysis of microarray data
Expression profiles of HGG were obtained from the NCBI GEO database (GSE34824 and GSE36245). Expression values of the indicated genes were log-transformed, normalized and shown in a boxplot using R software with default parameters.
DNA methylation analysis
Genomic DNA was extracted from frozen sections of xenografts derived from the quadruple-mutant (i.e., DKO-G34R-MYCN) vFNPCs. hESC-derived vFNPCs were used as a control cell type. To minimize contamination of murine cells, all sections were stained with H&E and tumor parts were micro-dissected. After the quality control process, a sequencing library was created from the genomic DNA using TruSeq Methyl Capture EPIC Library Prep Kit (Illumina) and sequenced by Illumina HiSeq4000. Sequencing data were mapped to the human genome (hg38) using Map with BWA on the galaxy website (https://usegalaxy.org). DNA methylation data of patient tumors were obtained from GSE36278 (Sturm et al., 2012).
Cytogenetic analyses and genomic PCR
Fluorescence in situ hybridization was performed using Green-dUTP-labeled BAC clone RP11–958G12 spanning the NOTCH2NL locus in 1q21.1 and partially matched to the NOTCH2 locus in 1p12. Red-dUTP-labeled BAC clone RP11–79A8 was used to specifically label the NOTCH2 locus. FISH was performed on frozen sections of xenografts and formalin-fixed paraffin-embedded sections of human tumor tissue and patient-derived cell lines with conditions optimized for each sample type. Human B-lymphoma cells served as normal control. To quantify copy number, genomic DNA was extracted from patient-derived cell lines and subjected to quantitative real-time PCR using primer pairs that specifically amplify the NOTCH2NL locus or amplify both NOTCH2 and NOTCH2NL loci. Values are normalized by those of GAPDH located at 12p13.31. Human H1 ESCs and HEK293T cells were used as normal diploid cells.
ChIP-PCR
DKO-vFNPCs constitutively expressing wildtype or G34R-mutant H3.3 were fixed with 1% formaldehyde for 10 min at 25 °C. The reaction was stopped by adding glycine to final concentration of 125 mM. Cells were washed with ice-cold PBS containing protease inhibitors and lysed in lysis buffer (10 mM Tris·HCl, pH 8.0, 150 mM NaCl, 10 mM EDTA, 1% SDS, 0.1% Sodium Deoxycholate) containing proteinase inhibitors. Lysates were sonicated to yield 200–500 bp DNA fragments and diluted with ChIP dilution buffer (10 mM Tris·HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton-X100). 2 μg of anti-H3K36me3 (ab9050; Abcam), anti-BS9 antibody (EP18343; Abcam), or control rabbit IgG was added to the sample and gently mixed for overnight at 4 °C. 50 μL of Protein G Dynabeads (Thermo Fisher) were washed with ChIP dilution buffer and gently mixed with the sample for 1 h. Beads were washed with low-salt wash buffer (10 mM Tris·HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton-X100, 0.1% SDS), high-salt wash buffer (10 mM Tris·HCl, pH 8.0, 500 mM NaCl, 1 mM EDTA, 1% Triton-X100, 0.1% SDS), LiCl wash buffer (10 mM Tris-HCl, pH 8.0, 250 mM LiCl, 1% NP-40, 1% Sodium Deoxycholate, 1 mM EDTA), and TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA). Immunoprecipitated DNA was eluted in elution buffer (1% SDS, 100 mM NaHCO3, 1 mM DTT), treated with proteinase K, and de-cross-linked for 14 h at 65 °C. DNA was purified using the QIAquick PCR Purification Kit (QIAGEN) and subjected to quantitative PCR using SYBR Green Master Mix (Thermo Fisher) on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Values were normalized by those of species-matched IgG control. Sequences of primers are shown in Table S1.
Tumorigenicity assay
All animal experiments were done in accordance with protocols approved by our Institutional Animal Care and Use Committee and following NIH guidelines for animal welfare. Cells were dissociated with Accutase (Innovative Cell Technologies) and the number of live cells was counted by Acridine Orange (AO) and Propidium Iodide (PI) staining (Nexcelom). A papain dissociation kit (Worthington Biochemical) was used for the dissociation of spheroids. Cells were resuspended in PBS and the indicated number of cells was stereotactically transplanted into the brain of NOD/SCID mice. Male and female mice were evenly assigned to each group. Coordinates of the injection site were 2 mm lateral and 2 mm posterior to bregma and 2 mm deep from the head skull. Mice were monitored for up to 6 months. Survival of mice was evaluated by Kaplan-Meier analysis and p-values were calculated using a log-rank test. For in vivo imaging, cells were lentivirally labeled with luciferase and luminescence was measured using IVIS Spectrum In Vivo Imaging System.
Western Blot
Cells were lysed in RIPA Buffer (50 mM Tris-HCl, pH 8.0; 120 mM NaCl; 5 mM EDTA; 0.5% NP-40). Following 30-minute centrifugation at 14000 rpm, the supernatant was collected, and protein concentration was measured by the Bradford Assay (Bio-Rad). Lysates were boiled for 5 minutes in Laemmli sample buffer and separated by electrophoresis on 4–12% Bis-Tris gel in SDS running buffer for 1.5–2 hours and transferred to nitrocellulose membrane. Non-specific protein binding was prevented by blocking the membrane with 5% Blotting-Grade Blocker (BioRad) in TBS-T (0.1% Tween-20 in 1x Tris-buffered saline). The membrane was incubated at 4°C overnight in the blocking buffer with primary antibodies. Mouse monoclonal antibodies against NOTCH2NL (39-Y; 1:2000) and α-tubulin (DM1A; 1:2000) were obtained from Santa Cruz. Rat monoclonal antibody against human influenza hemagglutinin (HA) tag (3F10; 1:2000) was obtained from Roche. Rabbit monoclonal antibodies against NOTCH2 (D76A6; 1:1000) and GAPDH (14C10; 1:1000) were obtained from Cell Signaling. After four washes with TBS-T, the blot was incubated with respective secondary antibodies for Mouse (1:5000) or Rabbit (1:5000) at room temperature for 30 minutes. ECL Prime Western Blotting Detection Kit was used for detection according to the manufacturer’s instruction (Amersham). For re-blotting, antibodies were stripped by Restore Western Blot Stripping Buffer (Thermo Fisher)
Quantitative real-time PCR
Total RNA was extracted using TRIzol (Invitrogen). For each sample, 1 μg of total RNA was reverse-transcribed using the QuantiTect Reverse Transcription Kit (QIAGEN). Amplified material was detected using SYBR Green Master Mix (Thermo Fisher) on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). All results were normalized to a β-Actin control. Sequences of primers are provided upon request.
Vectors and mutagenesis
The human H3F3A ORF was amplified from cDNA of H1 hESCs and fused with human influenza hemagglutinin (HA) tag, T2A self-cleavage sequence and EGFP by PCR extension. The G34R and K36R mutations were introduced using the QuikChange Site-Directed Mutagenesis Kit (Agilent). The HES5 promoter region was amplified from the genomic DNA extracted from H1 hESCs and cloned into pLVX-Puro vector (Clontech) along with H3F3A-T2A-EGFP fusion gene. Wildtype, G34R-mutant, and K36R-mutant H3F3A ORF with C-terminal HA-tag were also cloned into pENTR1A vector (Thermo Fisher). The human NOTCH2NLB ORF was amplified from cDNA of HSJD002 cell line and cloned into pENTR1A vector. The cloned ORFs were transferred to pLenti PGK Puro DEST vector (Addgene #19068)(Campeau et al., 2009) by LR recombination according to the manufacturer’s instruction (Thermo Fisher). For the construction of shRNA-expressing vectors, annealed oligos were cloned into pLKO.1-TRC cloning vector (Addgene #10878)(Moffat et al., 2006). Target sequences are shNOTCH2NL#1: 5’-GAGCTCT GGGAAAGAGACAGG −3’ and shNOTCH2NL#2: 5’-GGTTAATAAAGTGCTTTAAAC −3’. Luciferase-expressing vector (pLenti PGK Blast V5-LUC) was obtained from Addgene (#19166)(Campeau et al., 2009).
Lentivirus production
Lentiviral vectors were transfected in HEK293T cells with packaging vectors (pCMV-dR8.2 and pCMV-VSV-G), in the presence of Polyethylenimine (Polysciences). Viral supernatants were collected 72 h after transfection and viral particles were concentrated by ultracentrifugation at 49,000g for 1.5 h at 4 °C.
QUANTIFICATION AND STATISTICAL ANALYSIS
P values were calculated by Welch’s ANOVA followed by Welch’s t tests for pairwise comparisons, unless indicated otherwise. Numbers of biological replicates for each experiment are shown in figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal NOTCH2NL - WB | Santa Cruz | Cat# sc-100307, RRID:AB_2251318 |
| Rabbit monoclonal NOTCH2 - WB | Cell signaling | Cat# 5732, RRID:AB_10693319 |
| Mouse monoclonal α-tubulin -WB | Santa Cruz | Cat# sc-32293, RRID:AB_628412 |
| Rabbit monoclonal GAPDH -WB | Cell signaling | Cat# 5014, RRID:AB_10693448 |
| Rat monoclonal HA -WB | Roche | Cat# 11867431001, RRID:AB_390919 |
| Rabbit polyclonal histone H3K36me3- WB | Abcam | Cat# ab9050, RRID:AB_306966 |
| Mouse monoclonal histone H3 - WB | Active motif | Cat# 39763, RRID:AB_2650522 |
| Rabbit monoclonal histone H3.3 - WB | Abcam | ab176840, RRID:AB_2715502) |
| PerCP-Cy5.5-conjugated mouse monoclonal SOX2 - Flow cytometry | BD | Cat# 561506, RRID:AB_10646039 |
| Alexa647-conjugated mouse monoclonal TUBB3 - Flow cytometry | BioLegend | Cat# 801210, RRID:AB_2686931 |
| Alexa647-conjugated mouse monoclonal Ki67 - Flow cytometry | BD | Cat# 561126, RRID:AB_10611874 |
| PE-conjugated mouse monoclonal PLZF - Flow cytometry | BD | Cat# 564850, RRID:AB_2738984 |
| Rat monoclonal SOX2- IHC | eBioscience | Cat# 14-9811-95, RRID:AB_2865466 |
| Rabbit polyclonal Ki67 - IHC | Abcam | Cat# ab15580, RRID:AB_443209 |
| Mouse monoclonal TUBB3 - IHC | BioLegend | Cat# 801202, RRID:AB_10063408 |
| Mouse monoclonal Human Nuclear Antigen - IHC | Millipore | Cat# MA5-33098, RRID:AB_2802648 |
| Mouse monoclonal NCAM - IHC | Santa Cruz | Cat# sc-106, RRID:AB_627128 |
| Rabbit polyclonal DLX2 - IHC | Abcam | Cat# ab135620 |
| Goat polyclonal SP8 - IHC | Santa Cruz | Cat# sc-104661 |
| Rabbit polyclonal FOXG1 - IHC | Takara | Cat# M227, RRID:AB_2827749 |
| Mouse monoclonal NKX6-1 - IHC | DSHB | Cat# F55A10, RRID:AB_532378 |
| Mouse monoclonal Nestin - IHC | Millipore | MAB5326, RRID:AB_2251134 |
| Rabbit monoclonal N-Myc - IHC | Cell signaling | Cat# 84406, RRID:AB_2800038 |
| Rabbit polyclonal H3K36me3 - ChIP | Abcam | Cat# ab9050, RRID:AB_306966 |
| Rabbit monoclonal BS69 - ChIP | Abcam | Cat# ab190890 |
| Rabbit monoclonal H3.3G34R - ChIP | Abcam | Cat# ab254402 |
| Rabbit IgG - ChIP | Bethyl Laboratories | P120-101, RRID:AB_479829 |
| HRP-conjugated donkey anti-Rabbit IgG - WB | GE Healthcare | Cat# NA934, RRID:AB_772206 |
| HRP-conjugated sheep anti-Mouse IgG - WB | GE Healthcare | Cat# NA931, RRID:AB_772210 |
| Alexa555-conjugated goat anti-Mouse IgG - IHC | Molecular Probes | Cat# A-21422, RRID:AB_141822 |
| Alexa555-conjugated goat anti-Mouse IgG1 - IHC | Molecular Probes | Cat# A-21127, RRID:AB_141596 |
| Alexa555-conjugated goat anti-Rabbit IgG - IHC | Molecular Probes | Cat# A-21428, RRID:AB_141784 |
| Alexa647-conjugated goat anti-Mouse IgG1 - IHC | Molecular Probes | Cat# A-21240, RRID:AB_141658 |
| Alexa647-conjugated goat anti-Mouse IgG2a - IHC | Molecular Probes | Cat# A-21241, RRID:AB_141698 |
| Alexa647-conjugated goat anti-Rabbit IgG - IHC | Molecular Probes | Cat# A-21245, RRID:AB_141775 |
| Alexa555-conjugated donkey anti-Rabbit IgG - IHC | Molecular Probes | Cat# A-31572, RRID:AB_162543 |
| Alexa647-conjugated donkey anti-Goat IgG - IHC | Molecular Probes | Cat# A-21447, RRID:AB_2535864 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Essential 8 FLEX Medium | Gibco/ThermoFisher | Cat# A2858501 |
| Essential 6 Medium | Gibco/ThermoFisher | Cat# A1516401 |
| DMEM/F12 Medium | Gibco/ThermoFisher | Cat# 11330-032 |
| N2 Supplement | Gibco/ThermoFisher | Cat# 17502-046 |
| B-27 Supplement, minus vitamin A | Gibco/ThermoFisher | Cat# 12587-010 |
| Matrigel Matrix Growth Factor Reduced | Corning | Cat# 354230 |
| LDN-193189 | Stemgent | Cat# 04-0074-10 |
| SB431542 | Tocris | Cat# 1614 |
| XAV-939 | Sigma-Aldrich | Cat# X3004 |
| SAG | Cayman chemical | Cat# 11914 |
| Recombinant Human Epidermal Growth Factor | R&D Systems | Cat# 236-EG-200 |
| Recombinant Human Brain-derived neurotrophic factor | R&D Systems | Cat# 248-BDB-050 |
| Recombinant Human Fibroblast Growth Factor 8-b | R&D Systems | Cat# 423-F8 |
| Accutase | Innovative Cell Technologies | Cat# NC9464543 |
| Neurobasal Medium | Gibco/ThermoFisher | Cat# 21103049 |
| L-Ascorbic Acid | Sigma-Aldrich | Cat# A4403 |
| MEM Non-Essential Amino Acids Solution | Gibco/ThermoFisher | Cat# 11140050 |
| HEPES | Gibco/ThermoFisher | Cat# 15630080 |
| GlutaMAX Supplement | Gibco/ThermoFisher | Cat# 35050061 |
| Y-27632 dihydrochloride | Tocris | Cat# 12-541 |
| Poly-L-Ornithine hydrobromide | Sigma-Aldrich | Cat# P3655 |
| Cultrex mLaminin I | R&D Systems | Cat# 3400-010-02 |
| Fibronectin | ThermoFisher | Cat# 356008 |
| Recombinant Human Vitronectin truncated | ThermoFisher | Cat# A14700 |
| Recombinant Human Platelet-derived Growth Factor AA | Stem Cell Technologies | Cat# 78095 |
| Recombinant Human Platelet-derived Growth Factor BB | Peprotech | Cat# 100-14B |
| 0.2% Heparin Solution | Stem Cell Technologies | Cat #07980 |
| Retinoic Acid | Sigma-Aldrich | Cat# R2625 |
| Critical Commercial Assays | ||
| Papain Dissociation System | Worthington Biochemical Corporation | Cat# LK003150 |
| ImmPACT DAB Peroxidase (HRP) Substrate | Vector Laboratory | Cat# SK-4105 |
| M.O.M. ImmPRESS HRP Polymer Kit | Vector Laboratory | Cat# MP-2400 |
| H&E Staining Kit | Abcam | Cat# ab245880 |
| Deposited Data | ||
| RNA-Seq | this study | GSE147543 |
| EPIC Methyl Capture Seq | this study | PRJNA615218 |
| RNA-seq data of patient tumors | Wu G et al. (2014) | EGAS0000100192 |
| Microarray data of patient tumors | Schwartzentruber J et al, (2012) | GSE34824 |
| Microarray data of patient tumors | Sturm D et al, (2012) | GSE36245 |
| DNA methylation data of patient tumors | Sturm D et al, (2012) | GSE36278 |
| Experimental Models: Cell Lines | ||
| Human: H1 iCas9 Embryonic Stem Cells | Shi ZD et al, (2017) | NA |
| Human: patient-derived glioma cell line SJHGGx6 | St Jude Children’s Hospital | NA |
| Human: patient-derived glioma cell line HSJD002 | Hospital Sant Joan de Déu de Barcelona | NA |
| HEK293T | ATCC | Cat# CRL-3216, RRID:CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NSG (NOD.Cg-Prkdc Il2rg) | The Jackson Laboratory | Cat# JAX:005557, RRID:IMSR_JAX:005557 |
| Oligonucleotides | ||
| sgRNA target sequence: TP53: GGGAGAGACCGGCGCACAG | this study | NA |
| sgRNA target sequence: ATRX: TGAATTCTATACGATCAAGG | this study | NA |
| shRNA target sequence: NOTCH2NL #1: GAGCTCTGGGAAAGAGACAGG | this study | NA |
| shRNA target sequence: NOTCH2NL #2: GGTTAATAAAGTGCTTTAAAC | this study | NA |
| Primers, see Table S1 | this study | NA |
| Recombinant DNA | ||
| pLVX.puro-HES5p-H3.3-T2A-GFP | this study | NA |
| pLVX.puro-HES5p-G34R-T2A-GFP | this study | NA |
| pSico-sgATRX-puro-mCherry | this study | NA |
| pSico-sgTP53-puro-mCherry | this study | NA |
| pLKO.1puro-shNOTCH2NL#1 | this study | NA |
| pLKO.1puro-shNOTCH2NL#2 | this study | NA |
| pLKO.1puro-Non-target shRNA control | Sigma-Aldrich | Cat# SHC016-1EA |
| pLenti.puro-PGK1p-H3.3WT | this study | NA |
| pLenti.puro-PGK1p-H3.3G34R | this study | NA |
| pLenti.puro-PGK1p-H3.3K36R | this study | NA |
| pLenti.puro-PGK1p-NOTCH2NL | this study | NA |
| pLenti.puro-PGK1p-NOTCH2NL-IR | this study | NA |
| pLenti PGK Blast V5-LUC | Addgene | Cat# 19166 |
| Software and Algorithms | ||
| Galaxy | Afgan E et al. 2018 https://usegalaxy.org | |
| FastQC | Version 0.72 | |
| HSAT2 | Version 2.0.5.2 | |
| FeatureCounts | Version 1.6.4 | |
| Map with BWA | Version 0.7.17.4 | |
| SplAdder | Kahles et al, 2016 | Version 2.4.2 |
| R | https://www.r-project.org | Version 3.5.2 |
| RStudio | https://rstudio.com | Version 1.0.143 |
| Image J | https://imagej.nih.gov/ij/ | Version 1.49g |
| FlowJo | Version 9.7.6 | |
| Database for Annotation, Visualization and Integrated Discovery (DAVID) | Huang DW et al. 2009 https://david.ncifcrf.gov | Version 6.8 |
| GenePattern | Reich M et al. 2006 https://cloud.genepattern.org | Version 3.9.11 |
| WebLogo | https://weblogo.berkeley.edu/logo.cgi | Version 2.8.2 |
| Extreme Limiting Dilution Analysis (ELDA) | Hu and Smyth, 2009 http://bioinf.wehi.edu.au/software/elda/ | |
Highlights.
H3.3G34R, ATRX and TP53 mutations transform ventral but not hindbrain precursors
Triple mutant hESC-derived ventral progenitors show alternative mRNA splicing patterns.
Increased expression of human-specific NOTCH2NL in triple mutant ventral progenitors
NOTCH2NL locus is amplified in H3.3G34R-mutant high-grade glioma.
Acknowledgments
We thank Suzanne J. Baker, St. Jude Children’s Research Hospital, and Ángel Montero Carcaboso, Hospital Sant Joan de Déu Barcelona, for the human G34R-mutant cell lines, which are available under material transfer agreements. We also thank Cameron Brennan, Lorenz Studer, Danwei Huangfu, all lab members, and Arclev Academia Strategists Network (AASN) members for discussion and/or providing materials. K.F. was supported by grants from The Functional Genomics Initiative and the Center for Stem Cell Biology at Memorial Sloan Kettering Cancer Center, and by a Druckenmiller Fellowship from the New York Stem Cell Foundation. V.T. is supported by NIH/NCI R01 CA208405. R.C.S. was supported by NIH/NCI F31 CA210408 and by the Louis V. Gerstner, Jr., Graduate School of Biomedical Sciences. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Footnotes
Declaration of interests
The named authors have no relevant conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afgan E, Baker D, Batut B, Van Den Beek M, Bouvier D, Ech M, Chilton J, Clements D, Coraor N, Grüning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Park J, and Kim JS (2014). Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudis M, and Cleary ML (2001). Progenetix.net: an online repository for molecular cytogenetic aberration data. Bioinformatics 17, 1228–1229. [DOI] [PubMed] [Google Scholar]
- Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, et al. (2013). Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet 45, 1479–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, Bax D. a, Carvalho D, Taylor KR, Vinci M, et al. (2013). Histone H3.3 Mutations Drive Pediatric Glioblastoma through Upregulation of MYCN. Cancer Discov 3, 512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Kumar N, Ai N, Gupta S, Rath P, and Baudis M (2014). Progenetix: 12 years of oncogenomic data curation. Nucleic Acids Res 42, D1055–D1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, and Studer L (2008). Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev 22, 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsässer SJ, Noh KM, Diaz N, Allis CD, and Banaszynski LA (2015). Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiddes IT, Lodewijk GA, Mooring M, Bosworth CM, Ewing AD, Mantalas GL, Novak AM, van den Bout A, Bishara A, Rosenkrantz JL, et al. (2018). Human-Specific NOTCH2NL Genes Affect Notch Signaling and Cortical Neurogenesis. Cell 173, 1356–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, and Tabar V (2014). Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science (80-. ). 346, 1529–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrick D, Sharpe JA, Arkell R, Dobbie L, Smith AJH, Wood WG, Higgs DR, and Gibbons RJ (2006). Loss of Atrx affects trophoblast development and the pattern of X-inactivation in extraembryonic tissues. PLoS Genet. 2, e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessi M, Gielen GH, Hammes J, Dörner E, Mühlen A. Zur, Waha A, and Pietsch T (2013). H3.3 G34R mutations in pediatric primitive neuroectodermal tumors of central nervous system (CNS-PNET) and pediatric glioblastomas: Possible diagnostic and therapeutic implications? J. Neurooncol 112, 67–72. [DOI] [PubMed] [Google Scholar]
- Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. (2010). Distinct Factors Control Histone Variant H3.3 Localization at Specific Genomic Regions. Cell 140, 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, Xu W, Mu S, Wen H, Qiu J, et al. (2014). BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol. Cell 56, 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, and Smyth GK (2009). ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 347, 70–78. [DOI] [PubMed] [Google Scholar]
- Hu JS, Vogt D, Sandberg M, and Rubenstein JL (2017). Cortical interneuron development: A tale of time and space. Development 144, 3867–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Shimojo H, Sakamoto M, Ohtsuka T, and Kageyama R (2013). Genetic visualization of notch signaling in mammalian neurogenesis. Cell. Mol. Life Sci 70, 2045–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain SU, Khazaei S, Marchione DM, Lundgren SM, Wang X, Weinberg DN, Deshmukh S, Juretic N, Lu C, Allis CD, et al. (2020). Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc. Natl. Acad. Sci. U. S. A 117, 27354–27364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahles A, Ong CS, Zhong Y, and Rätsch G (2016). SplAdder: Identification, quantification and testing of alternative splicing events from RNA-Seq data. Bioinformatics 32, 1840–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahles A, Lehmann K. Van, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, Caesar-Johnson SJ, et al. (2018). Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 34, 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuong-Quang D-A, Buczkowicz P, Rakopoulos P, Liu X-Y, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, et al. (2012). K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschmann C, Calinescu AA, Nunez FJ, Mackay A, Fazal-Salom J, Thomas D, Mendez F, Kamran N, Dzaman M, Mulpuri L, et al. (2016). ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci. Transl. Med 8, 328ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. (2006). The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science (80-. ). 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski L. a, Garcia B. a, Muir TW, Becher OJ, and Allis CD (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Park JH, Baude A, Yoo Y, Lee YK, Schmidt CR, Park JB, Fellenberg J, Zustin J, Haller F, et al. (2017). The histone variant H3.3 G34W substitution in giant cell tumor of the bone link chromatin and RNA processing. Sci. Rep 7, 13459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodewijk GA, Fernandes DP, Vretzakis I, Savage JE, and Jacobs FMJ (2020). Evolution of human brain-size associated NOTCH2NL genes proceeds towards reduced protein levels. Mol. Biol. Evol msaa104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M, et al. (2017). Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32, 520–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof AM, Keros S, Tyson JA, Ying SW, Ganat YM, Merkle FT, Liu B, Goulburn A, Stanley EG, Elefanty AG, et al. (2013). Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek AS, Golub D, Khan T, Bready D, Prado J, Bowman C, Deng J, Zhang G, Rocha PP, Raviram R, et al. (2017). Low-Grade Astrocytoma Mutations in IDH1, P53, and ATRX Cooperate to Block Differentiation of Human Neural Stem Cells via Repression of SOX2. Cell Rep 21, 1267–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A Lentiviral RNAi Library for Human and Mouse Genes Applied to an Arrayed Viral High-Content Screen. Cell 124, 1283–1298. [DOI] [PubMed] [Google Scholar]
- Naito Y, Hino K, Bono H, and Ui-Tei K (2015). CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 31, 1120–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, et al. (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science (80-. ). 358, 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, Pahlavan P, Henderson S, Mikael LG, Richard-Londt A, Zhang Y, et al. (2017). H3.3K27M Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 32, 684–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B, and Blencowe BJ (2015). Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 87, 14–27. [DOI] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP (2006). GenePattern 2.0. Nat. Genet 38, 500–501. [DOI] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang D-AK, Tonjes M, et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. [DOI] [PubMed] [Google Scholar]
- Shi ZD, Lee K, Yang D, Amin S, Verma N, Li QV, Zhu Z, Soh CL, Kumar R, Evans T, et al. (2017). Genome Editing in hPSCs Reveals GATA6 Haploinsufficiency and a Genetic Interaction with GATA4 in Human Pancreatic Development. Cell Stem Cell 20, 675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones DTW, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, et al. (2012). Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 22, 425–437. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki IK, Gacquer D, Van Heurck R, Kumar D, Wojno M, Bilheu A, Herpoel A, Lambert N, Cheron J, Polleux F, et al. (2018). Human-Specific NOTCH2NL Genes Expand Cortical Neurogenesis through Delta/Notch Regulation. Cell 173, 1370–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teodorczyk M, and Schmidt MHH (2014). Notching on cancer’s door: Notch signaling in brain tumors. Front. Oncol 4, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voon HPJ, Udugama M, Lin W, Hii L, Law RHP, Steer DL, Das PP, Mann JR, and Wong LH (2018). Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nat. Commun 9, 3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, Tanaka K, Ren Y, Xia Z, Wu J, et al. (2014). ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 508, 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet 44, 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, et al. (2014). The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet 46, 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Edri R, Yaffe Y, Gifford C. a, Xing J, Gu H, Kohlbacher O, and Gnirke A (2015). Dissecting neural differentiation regulatory networks through epigenetic footprinting. Nature 518, 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq and Methyl Capture Sequencing data generated during this study are available at NCBI Gene Expression Omnibus (GEO) with an accession number GSE147543 and Sequence Read Archive (SRA) with an accession number SRP254041.