

SUMMARY
Depletion of architectural factors globally alters chromatin structure, but only modestly affects gene expression. We revisit the structure-function relationship using the inactive X chromosome (Xi) as a model. We investigate cohesin imbalances by forcing its depletion or retention using degron-tagged RAD21 (cohesin subunit) or WAPL (cohesin release factor). Cohesin loss disrupts Xi superstructure, unveiling superloops between escapee genes, with minimal effect on gene repression. By contrast, forced cohesin retention markedly affects Xi superstructure, compromises spreading of Xist RNA-Polycomb complexes, and attenuates Xi silencing. Effects are greatest at distal chromosomal ends, where looping contacts with the Xist locus are weakened. Surprisingly, cohesin loss creates an “Xi superloop” and cohesin retention creates “Xi megadomains” on the active X. Across the genome, a proper cohesin balance protects against aberrant inter-chromosomal interactions and tempers Polycomb-mediated repression. We conclude that a balance of cohesin eviction and retention regulates X-inactivation and inter-chromosomal interactions across the genome.
eTOC Blurb
Kriz et al. reveal that cohesin balance tempers Polycomb-mediated inter-chromosomal interactions and gene repression. During X inactivation, cohesin folds the Xi into megadomains, but inhibits superloops. However, too much cohesin compromises Xist-Polycomb spreading and gene silencing. Forced cohesin retention also creates Xi-like megadomains and superloop on the active X.
Graphical Abstract
INTRODUCTION
Classical microscopy studies revealed that mammalian chromosomes fold into large scale structures and occupy discrete nuclear territories in order to fit into this space (Cremer and Cremer, 2001; Fraser et al., 2015; Paulson and Laemmli, 1977). The advent of genome-wide chromosome conformation capture (3C)-based assays revealed how in particular, chromosomes form “loops” at several levels (Paulson and Laemmli, 1977; Rao et al., 2014; Splinter et al., 2006) and partition into “compartments” based on chromatin properties and gene expression states (Rowley and Corces, 2018). Chromosomes have also been proposed to fold into “Topologically associating domains” (TADs) (Dixon et al., 2012; Nora et al., 2012), though single cell Hi-C and microscopy studies reveal that TADs represent population-based averages, rather than stable domains (Beagrie et al., 2017; Bintu et al., 2018; Nagano et al., 2013; Nagano et al., 2017; Stevens et al., 2017). Two factors dynamically regulate chromosome architecture — cohesin and CTCF. Cohesin, a multi-subunit complex, is thought to form a ring-like structure around chromatin and extrudes chromatin loops in an ATP-dependent manner (Davidson et al., 2019; Kim et al., 2019). RAD21 is the essential kleisin subunit of the cohesin complex, which additionally consists of SMC proteins SMC1, SMC3, and either SA1 or SA2 (Nasmyth and Haering, 2009; Peters et al., 2008). CTCF, an 11-zinc finger DNA-binding protein, insulates enhancers from promoters and typically anchors long-range loops (Bell and Felsenfeld, 2000; Rao et al., 2014).
It was hypothesized that 3D genome structures had an important role in gene regulation {Merkenschlager, 2016 #840;Guo, 2015 #841;de Wit, 2015 #842;Lupianez, 2015 #280}. Unexpectedly, a series of loss-of-function studies targeting CTCF, cohesin component RAD21, and cohesin release factor WAPL showed that depletion of each of these factors dramatically alters 3D genome structure, but only modestly impacts gene expression (Haarhuis et al., 2017; Nora et al., 2017; Rao et al., 2017). These results beg the question of what precise role 3D genome organization plays during epigenetic regulation. A caveat is that these studies were conducted in a limited number of cell types and developmental timepoints. Therefore, 3D genome structure could play a more acute and global role during epigenetic regulation than is currently appreciated.
Here we revisit the role of cohesin-mediated architecture using a model epigenetic phenomenon, X chromosome inactivation (XCI) — the process by which female mammals ‘inactivate’ one of their X chromosomes to balance dosage of X-linked genes with males. XCI occurs during differentiation of female mouse embryonic stem cells (mESCs). The process is mediated by Xist, a noncoding RNA which is transcribed from and spreads across the inactive X (Xi), triggering recruitment of repressive factors such as Polycomb Repressive Complexes 1 and 2 (PRC1/2), formation of heterochromatin, and gene silencing (Disteche, 2016; Lee, 2011; Starmer and Magnuson, 2009; Wutz, 2011). XCI provides an extreme example of changes in 3D genome structure. While the active X (Xa) folds similarly to autosomes, the Xi shows greatly weakened TAD structure and appears to be devoid of A/B compartmentalization. The Xi is instead organized into two large (>50Mb) megadomains (Deng et al., 2015; Minajigi et al., 2015; Rao et al., 2014) and the A/B compartments are reorganized into “S1” and “S2” compartments before assuming the super-structure that characterizes the globally silenced chromosome (Wang et al., 2019; Wang et al., 2018). Restructuring from A/B to S1/S2 compartments requires Xist RNA and Polycomb repressive complex 1 (PRC1), and further reorganization of S1/S2 into a single large compartment requires the noncanonical SMC protein, SMCHD1 (Gdula et al., 2019; Wang et al., 2019; Wang et al., 2018), a cohesin and condensin relative (Blewitt et al., 2008).
While SMCHD1 and the S1/S2 structures are required for gene silencing, a remaining pivotal question is whether accompanying structural changes are required for or are merely byproducts of silencing the entire chromosome. Clarifying these questions would impact general understanding of 3D genome regulation, as well as XCI biology. To this aim, here we perturb cohesin levels through targeted degradation of a cohesin component RAD21 and a release factor of cohesin, WAPL, using the dTAG system. WAPL evicts cohesin from DNA in prophase (Gandhi et al., 2006; Kueng et al., 2006) as well as during interphase (Tedeschi et al., 2013), possibly by opening a DNA ‘exit gate’ (Buheitel and Stemmann, 2013; Chan et al., 2012; Huis in ‘t Veld et al., 2014). By degrading either RAD21 or WAPL, we modulate cohesin levels upwards and downwards in a mouse embryonic stem cell (mESCs) model and examine the consequences for loop formation, compartments, and XCI.
RESULTS
Toggling cohesin binding using a RAD21- and WAPL-dTAG degron system
We employed the dTAG degron system (Nabet et al., 2018; Weintraub et al., 2017) to acutely and selectively degrade a cohesin component RAD21 and cohesin release factor, WAPL, in female TST ES cells (Fig. 1A). The female TST ES line carries a Tsix termination cassette on one X chromosome (TsixTST/+) to ensure silencing of the Mus musculus X chromosome in this hybrid ES line (Ogawa et al., 2008) (Fig. 1A). In the dTAG system, the heterobifunctional dTAG molecule (dTAG-13) recruits an endogenous E3 ubiquitin ligase, Cereblon, to degrade FKBP12F36V (herein referred to as FKBPdegron)-tagged proteins. The dTAG degron system thus avoids previously described issues with auxin degrons (Nora et al., 2017), namely difficulties maintaining Tir1 expression and effective degradation during differentiation. The FKBPdegron tag was knocked in at the endogenous STOP codons of the Rad21 and Wapl genes using CRISPR/Cas9 and a homology-directed repair template (Fig. S1A–E). We isolated 2 RAD21-FKBPdegron clones with comparable expression levels to endogenous RAD21 (Fig. 1B, S1B–C). We also isolated 2 WAPL- FKBPdegron clones, although these had different baseline levels of WAPL expression (Fig. 1B, S1D). We later learned that the higher WAPL expression in clone H7 was due to an amplification of the Wapl-FKBPdegron gene. We therefore took extra care to characterize the second clone, H2, which lacked the amplification but had a much lower baseline WAPL level relative to wildtype cells (Fig. S1F). Because clone H7 had a baseline WAPL level closer to wildtype cells, we considered it important to analyze both H7 and H2. Thus, all analyses were carried out with both clones for both RAD21 and WAPL-FKBPdegron, with results agreeing between clones.
Fig.1: Toggling cohesin binding and 3D genome structure during differentiation.

(A) Schematic of dTAG degron system, adapted from (Weintraub et al., 2017) (top) and the TST mouse embryonic stem cell system for X chromosome inactivation (bottom).
(B) Western blots for FKBPdegron-tagged proteins and GADPH loading control in unmodified parental TST (WT) and indicated FKBPdegron lines. Samples treated with DMSO vehicle control (D) or 500nM dTAG-13 (13).
(C) Table of investigated time frames in each degron line. Red, focuses of this study.
(D) Representative tracks of RAD21 binding by calibrated ChIP-seq (cChIP-seq) in RAD21-FKBPdegron lines and WAPL-FKBPdegron lines.
(E) Metaplot and heatmaps of RAD21 binding by cChIP-seq in RAD21-FKBPdegron and WAPL-FKBPdegron lines.
(F) Representative Hi-C interaction maps in RAD21-FKBPdegron lines (10kb resolution, clones merged) at day 7 after 8 hours of dTAG-13 or DMSO control treatment. Cohesin-dependent (black arrows) and cohesin-independent (blue arrows) loops indicated.
(G) Frequency versus distance plots of RAD21-FKBPdegron Hi-C maps (clones merged). Treatment as in (F).
(H) Saddle plot analysis of compartment strength in RAD21-FKBPdegron Hi-C maps (clones merged). Scores represent B-B, B-A, and A-A compartment interaction strength. Treatment as in (F).
(I) Aggregate peak analysis (APA) plots of aggregate Hi-C signal over sets of loops called in dTAG-13 or DMSO RAD21-FKBPdegron Hi-C maps (clones merged). Lower left: average loop enrichment (P2LL ratio). Numbers and percent of dTAG-13 and DMSO loops with both anchors overlapping RAD21, H3K27me3 cChIP peaks or mESC super-enhancers (Whyte et al. 2013) are shown.
(J) Representative Hi-C interaction maps in RAD21-FKBPdegron lines (15kb resolution, clones merged) at day 7 after 3 days of dTAG-13 or DMSO control treatment. Longer cohesin-anchored chromatin loops appearing upon WAPL degradation indicated (black arrows).
(K) Frequency versus distance plots of WAPL-FKBPdegron Hi-C maps (clones merged). Treatment as in (J).
(L) Saddle plot analysis of compartment strength in WAPL-FKBPdegron Hi-C maps (clones merged). Scores as in (H). Treatment as in (J).
(M) APA plots of aggregate Hi-C signal over sets of loops called in dTAG-13 or DMSO WAPL-FKBPdegron Hi-C maps (clones merged). Lower left: average loop enrichment (P2LL ratio). Numbers and percent of dTAG-13 and DMSO loops with both anchors overlapping RAD21, H3K27me3 cChIP peaks in corresponding samples or mESC super-enhancers (Whyte et al. 2013) are shown.
(N) APA plots of aggregate WAPL-FKBPdegron Hi-C signal over sets of cohesin-independent H3K27me3-anchored loops (clones merged). Lower left: average loop enrichment (P2LL ratio).
Western blots and immunofluorescence confirmed that RAD21 and WAPL were rapidly degraded during ES differentiation upon treatment with dTAG-13 but not with the DMSO vehicle (Fig. 1B, S1C–D, G). To study effects on XCI, we performed analyses at days 7 and 10 of differentiation — timepoints corresponding to the mid and late stages of XCI, respectively (Fig. 1C). For RAD21, we chose 8 hours of degradation to compare to previous degron studies (Rao et al., 2017; Rhodes et al., 2020). This degradation timeframe resulted in less impact on the cell cycle (Fig. S2A–B) than experienced in a previous study (Rhodes et al., 2020). For WAPL, we performed both an 8-hour and 3-day degradation, reasoning that it may take time to build up cohesin levels and observe resulting physiological effects. WAPL degradation did not affect cell cycle profiles (Fig. S2C–E), consistent with the idea that separase can cleave cohesin at centromeres in the absence of WAPL, enabling cell cycle progression (Tedeschi et al., 2013).
We then performed calibrated ChIP sequencing (cChIP-seq) to examine effects on cohesin chromatin binding. RAD21 binding was reduced dramatically (Fig. 1D-E). Whereas 29,519 RAD21 significant peaks were observed in DMSO controls, only 351 peaks were present after dTAG-13 treatment in clone C4 (99% decrease). Similarly, whereas 23,659 peaks occurred in DMSO controls, only 780 peaks remained after dTAG-13 treatment in clone G9 (97% decrease), This degree of loss in cohesin binding is comparable to a prior study (Rao et al., 2017). We also performed calibrated ChIP-seq (cChIP-seq) for RAD21 following WAPL degradation. After 8 hours of degradation, we observed increased cohesin binding (Fig. 1E). Specifically we detected 49,130 (clone H2) and 42,086 (clone H7) peaks in DMSO control conditions and 53,480 (clone H2) and 47,866 (clone H7) peaks after dTAG-13 treatment. After 3 days of degradation, 45,415 peaks were called in DMSO control and 66,488 peaks called after dTAG-13 treatment in clone H7 (Fig. 1D-E). In clone H2, similar numbers of peaks were called (73,706 in DMSO and 68,881 in dTAG-13), but became wider on average after dTAG-13 treatment (Fig. S3A). The 3-day increase in cohesin binding was similar to that observed in a previous WAPL knockout study (Haarhuis et al., 2017). Together, these data demonstrate that we can successfully toggle cohesin levels using the RAD21- and WAPL-degron system.
Changes in 3D architecture in response to cohesin eviction and retention
To investigate changes in 3D architecture, we generated in situ Hi-C interaction maps, with a minimum of 360 million read pairs for each day 7 sample (totaling 2.8 billion read pairs and 2.7 billion valid contacts across 14 samples, Table S1) achieving a resolution of 10kb. For RAD21, 8 hours of degradation at day 7 or 10 ablated topological domains evident along diagonals of contact heatmaps (Fig. 1F-G, S3B–D), as expected. By contrast, A/B compartmentalization was not greatly altered (Fig. 1H,S3B), agreeing with previous studies (Rao et al., 2017; Wutz et al., 2017). In contrast, there were intriguing effects on long-range loops, visible in Hi-C matrices as dots of high interaction frequency (Fig. 1F, arrows). HiCCUPs (Durand et al., 2016) and metaloop analysis showed a clear decrease in the number of looping interactions following cohesin depletion (Fig. 1I) — 4,932 (DMSO) versus 983 (dTAG13) at day 7, 2074 versus 289 at day 10.
Previous studies found that ablation of cohesin dysregulates Polycomb target genes in undifferentiated mESCs (Rhodes et al., 2020), with subunits SA1 and SA2 having opposing effects on interactions between Polycomb domains (Cuadrado et al., 2019). We independently observed that a substantial fraction (10–18%) of remaining loops were anchored by H3K27me3 (Fig. 1I). However, most cohesin-independent loops were not anchored by H3K27me3, indicating chromatin features in addition to cohesins and PRC2 are involved in loop formation (Fig. 1I, S3E). Interestingly, metaloop analysis showed that loops persisting after cohesin degradation were strengthened (Fig. 1I). No persistent loops occurred between super-enhancers (SE) (Whyte et al., 2013) (Fig. 1I). SE interactions may occur at lower frequency during cell differentiation than in pluripotent ES cells (Rao et al., 2017; Rhodes et al., 2020). These data indicate that depleting cohesin binding reduces the number of loops across the genome, but persistent loops paradoxically strengthen.
We next examined consequences of increasing cohesin binding by degrading WAPL. When we degraded WAPL for 8 hours (to mirror the timeframe of RAD21 depletion), there was only a modest increase in cohesin binding (Fig. 1E). Thus, we degraded WAPL for 3 days between days 4–7 or days 7–10. These timeframes resulted in an increase in interactions of 1–10Mb (Fig. 1J-K, S3B,F) and a decrease in far-cis compartmental interactions of >10Mb (Fig. 1K-L,S3F). Concurrently, there was a large increase in number of chromatin loops, with HiCCUPs calling 5731 loops in controls versus 8715 loops in WAPL-depleted cells, of which the vast majority were anchored by RAD21 (Fig. 1M). Whereas RAD21-depletion strengthened Polycomb-anchored loops (Fig. 1I), WAPL degradation weakened them (Fig. 1N). The effect appeared to be distance-dependent, as loops spanning >1Mb were weakened more than loops spanning <1Mb (Fig. 1N). Thus, whereas cohesin depletion reduces loop number and strengthens persistent loops, cohesin retention did the opposite.
Polycomb loops protect against effects of cohesin loss
We next investigated effects on gene expression by RNA-seq. After 8-hr RAD21 in day 10 cells, 43 genes were significantly upregulated and 98 were downregulated (adjusted p-value < 0.1), with downregulated genes being enriched for H3K27me3 (Fig. 2A, top, Table S2). These numbers are similar to those reported for other cell types (Rao et al., 2017; Rhodes et al., 2020). We wondered if they may be an underestimate because of the median mRNA half-life of ~10 hr (Greenberg, 1972). We therefore performed a transcriptome-wide analysis of nascent transcription using PRO-seq. However, a similar number of genes were revealed to be differentially expressed after 8 hr of RAD21 depletion on day 7 (Fig. 2A; 37 downregulated, 32 upregulated), reinforcing a modest effect of acute cohesin loss on gene expression. Loss of RAD21-loop anchors did not correlate with gene dysregulation at day 10 (Fig. S4A). At day 7, there was a significant tendency for genes overlapping cohesin-dependent loop anchors to be more upregulated, but again the effect was modest (Fig. S4A).
Fig.2: Polycomb loops protect against gene dysregulation upon cohesin loss, but increase sensitivity to cohesin gain during differentiation.

(A) Genes upregulated or downregulated after indicated time courses of RAD21 degradation (relative to DMSO control) overlapping with H3K27me3 cChIP-seq peaks. All genes shown for reference.
(B) RAD21-FKBPdegron RNA-seq (day 7: PRO-seq) DESeq2 log2-fold (dTAG-13/DMSO) change (left) or log2 FPKM (day 7: FPK, right, averaged between clones) of different categories of expressed (day 7: all) H3K27me3-marked genes. (No) loop: (not) overlapping anchor of H3K27me3-associated, cohesin-independent loop, Hox: Hox gene located in Hox cluster. All genes shown for reference. Significant p-values by Wilcoxon rank test indicated by * (p<7.344e-05), ** (p<0.0006566), *** (p<2.847e-05), **** (p<0.0005113), ***** (p<0.003082), ****** (p<0.0006716), ******* (p<0.001102), ********* (p<0.000146). N.S., not significant.
(C) Average DMSO and dTAG-13 H3K27me3 cChIP-seq signal over upregulated and downregulated genes in day 7 RAD21-FKBPdegron and WAPL-FKBPdegron samples.
(D) Inter-chromosomal Hi-C interaction maps between Hox cluster regions in RAD21-FKBPdegron lines at day 7 and day 10 at indicated resolutions (clones merged). H3K27me3 cChIP-seq signal in day 7 RAD21- FKBPdegron lines shown for reference (scale: 200).
(E) Schematic of inter-chromosomal Hox interactions in mESCs, day 7 and day 10 of differentiation. Bold arrows, interactions detected by Hi-C under control conditions.
(F) Genes upregulated or downregulated upon WAPL degradation from days 7–10 (relative to DMSO control) overlapping with H3K27me3 cChIP-seq peaks. All genes shown for reference.
(G) WAPL-FKBPdegron RNA-seq log2-fold (dTAG-13/DMSO) change of different categories of expressed H3K27me3-marked genes in WAPL-FKBPdegron lines. Categories as in (B). Significant p-values by Wilcoxon rank test indicated by * (p<6.703e-08), ** (p<1.462e-07), *** (p<4.638e-05).
(H) Inter-chromosomal Hi-C interaction maps between Hox cluster regions in WAPL-FKBPdegron lines (clones merged) at indicated resolutions. H3K27me3 cChIP-seq signal in day 7 WAPL- FKBPdegron lines shown for reference (scale: 100).
However, Polycomb targets inside a H3K27me3-marked persistent loop anchor were less downregulated than Polycomb targets not in such loop anchors in day 7 cells (Fig. 2B, bottom). This contrasts with day 0 ES cells, in which Polycomb loops were correlated with increased repression upon cohesin degradation (Rhodes et al., 2020). Further examination revealed that even in the DMSO control, clustered Polycomb targets tended to be more repressed than those not involved in such interactions (Fig. 2B). This suggests that, normally, interactions between Polycomb domains reinforce repression and cohesin depletion makes repression even more robust. Changes in gene expression upon cohesin removal were not correlated with changes in H3K27me3 by cChIP-seq (Fig. 2C, top). Thus, Polycomb target genes are strikingly resistant to cohesin degradation in differentiating ES cells.
Cohesin protects against inter-chromosomal Hox interactions and tempers Polycomb-mediated repression
Among classic Polycomb targets are the 4 Hox clusters, each located on a different chromosome, with each cluster being coordinately regulated in cis to specify anterior-posterior patterning in mammals (Deschamps and Duboule, 2017; Pearson et al., 2005; Wellik, 2007). As Hox regulation is recapitulated during ES differentiation (Lanctot et al., 2007), we studied how architectural organization of Hox genes is affected by cohesin loss. Examination of Hi-C maps revealed an entire network of inter-Hox contacts between chromosomal clusters (Fig. 2D, S3G–I). A small subset of these contacts had been seen previously in ES cells (Lanctot et al., 2007; Schoenfelder et al., 2015). Heretofore unseen, however, are interactions that extend across entire Hox clusters and throughout cell differentiation. Most intriguingly, these trans-interactions strengthened without cohesin (Fig. 2D, S3G–I). A previous RAD21-degron study did not report these contacts (Rhodes et al., 2020). Reanalysis of their Hi-C dataset indicated that they indeed exist in day 0 cells—but only after RAD21 degradation (Fig. S3G). Thus, analysis of our degron-tagged ES cells showed a dynamic and extensive network of Hox-to-Hox contacts (Fig. 2E).
Next we asked how inappropriate cohesin retention affects these Hox dynamics. Because 8-hour WAPL depletion only modestly increased cohesin retention (Fig. S4B–D), we depleted WAPL from days 4–7 of differentiation, where we observed much more cohesin retention (Fig. 1E). RNA-seq revealed differential expression of 2386 genes, of which 981 were downregulated and 1405 were upregulated (adjusted p-value < 0.1, Table S2). WAPL degradation at a later timepoint, days 7–10, resulted in fewer DEGs ( 39 upregulated and 19 downregulated, Fig. S4E). We therefore focused further analysis on the day 4–7 timecourse. As might be expected, genes that are normally anchored by cohesin did not change expression patterns when cohesins were more strongly retained (Fig. S4A). Intriguingly, upregulated DEGs tend to be Polycomb targets, particularly those in H3K27me3-marked loop anchors that persisted after RAD21 degradation (Fig. 2F,G). This was especially evident at the Hox loci, where WAPL depletion and consequent cohesin retention led to a loss of trans-interaction networks and marked increase in gene expression (Fig. 2G-H, S3J). Among the upregulated DEGs were also the Zscan4 and Usp17l families (Figure S4F), markers of the two-cell embryonic development stage (Falco et al., 2007; Macfarlan et al., 2012).Thus, disruption of Polycomb interaction networks, both in cis and in trans, through inappropriate cohesin retention can have catastrophic effects on gene repression.
Taken together, our data indicate that cohesins play an important role in Polycomb regulation. As Polycomb domains have an inherent tendency to cluster (Bonev and Cavalli, 2016; Denholtz et al., 2013; Joshi et al., 2015; Kundu et al., 2017; McLaughlin et al., 2019; Rhodes et al., 2020; Schoenfelder et al., 2015; Vieux-Rochas et al., 2015), we propose that a key role of cohesins is to organize chromatin in two respects: First, to prevent ectopic long-range cis-interactions as well as inappropriate trans-interactions; and second, to temper such interactions when they occur in the normal physiological context. Indeed, when cohesin is ablated, trans-interactions strengthen between chromosomes, as exemplified by inter-Hox interactions. When cohesins are aberrantly retained, such interactions are disrupted, with a dramatic increase in expression of certain Polycomb targets.
Cohesin regulates genes expression associated with super-enhancers in a looping-independent manner
Our RAD21/WAPL degron system enabled us to revisit an important question about dependence of super-enhancers (SEs) on cohesin for which there is disagreement in the field (Rao et al., 2017; Rhodes et al., 2020). To examine proximity of DEGs to SEs, we restricted analysis to expressed genes (FPKM > 0.5), leveraged our PRO-seq data to focus on nascent transcription, and measured distances from SEs to all expressed genes, downregulated DEGs, or upregulated DEGs (Fig. S5A). Significantly, downregulated DEGs in cohesin-depleted cells were closer to super-enhancers (Fig. S5A–B). Approximately 54% of downregulated DEGs were <150kb away, and ~29% were <20kb away (Fig. S5B). Overall, loss of cohesin resulted in ~2-fold downregulation of these targets. To ask whether this could be due to an associated shift in looping interactions, we examined Hi-C maps for interactions between SEs. Meta-loop analysis using APA plots revealed a slight enrichment of aggregate signal over SE anchors, suggesting low-frequency contacts between SEs (Fig. S5C). However, the contacts were not appreciably disrupted by cohesin loss in either day 7 or day 10 cells. These data contrast with those in day 0 ES cells, where SE interactions are cohesin-dependent (Rhodes et al., 2020). We conclude that cohesins play role in regulating SE-associated genes independent of looping in differentiating ES cells.
Xi superloops form between escapees in a cohesin- and Polycomb-independent manner
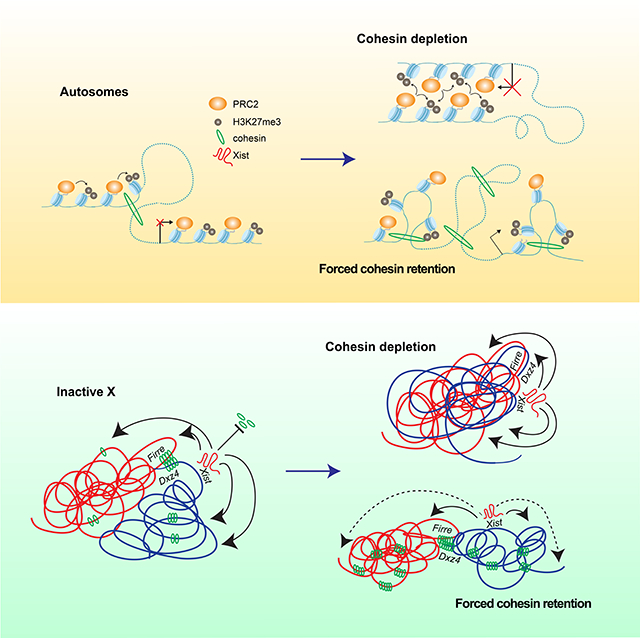
The inactive X chromosome (Xi) is a strong Polycomb target, becoming highly enriched with H3K27me3 in female ES cells undergoing differentiation and XCI (Plath et al., 2003; Silva et al., 2003; Wang et al., 2001; Zhao et al., 2008). XCI brings about major architectural changes to the X chromosome, as Xist RNA evicts up to 90% of cohesins while it spreads along the Xi (Minajigi et al., 2015) (Fig. 3A and 4A, Xi MEF row). Here we followed the Xi from mid (day 7) to late (day 10) XCI and indeed observed 40– 50% loss by days 7 to 10 (Fig. 3B and 4B). Changes in cohesin binding during differentiation are known to correlate with (i) suppression of “TADs” across the Xi (Wang et al., 2018), (ii) appearance of two megadomains with a strong border at the macrosatellite repeat, Dxz4, (Deng et al., 2015; Froberg et al., 2018; Giorgetti et al., 2016; Minajigi et al., 2015; Rao et al., 2014) and (iii) establishment of a ‘superloop’ spanning ~30 Mb between Dxz4 and another repeat element, Firre (Darrow et al., 2016; Hacisuleyman et al., 2014; Horakova et al., 2012; Rao et al., 2014) (Fig. 3A). The superloops are not present on the Xa. Current models postulate that superloops form through loop extrusion of cohesin (Bansal et al., 2019; Bonora et al., 2018), but cohesin’s exact role in XCI is unclear. Does cohesin binding antagonize the establishment of the Xi? Is cohesin eviction required for XCI? Does the remaining cohesin binding have a role in folding the Xi?
Fig.3: Cohesin is required for proper folding of the inactive X chromosome at day 7 of differentiation.

(A) Schematic of expected cohesin levels versus TAD, compartment, and superstructures detected on the X chromosome at different stages of XCI, based on prior studies. Levels corresponding to day 7 DMSO control and dTAG-13 treated cells indicated.
(B) Metaplot and heatmaps of active (Xa) and inactive (Xi) X chromosome RAD21 binding by cChIP-seq in day 7 RAD21-FKBPdegron embryoid bodies treated for 8 hours with 500nM dTAG-13 or DMSO.
(C) TAD scores calculated from day 7 Xa and Xi RAD21-FKBPdegron allelic Hi-C interaction maps binned at 100kb. DMSO and dTAG-13 samples as in (B). Representative Hi-C maps shown for the indicated regions (50kb resolution, clones merged).
(D) Chromosome-wide day 7 Xi RAD21-FKBPdegron allelic Hi-C maps (250kb resolution, clones merged) and log2-ratio map (1Mb resolution). Treatment as in (B). Inset, 50kb resolution.
(E) Day 7 composite X, Xi, and Xa Hi-C maps at Dxz4-Firre superloop (100kb resolution, clones merged). RAD21 and H3K27me3 cChIP-seq at superloop anchors shown for reference. Treatment as in (B).
(F) Day 7 composite X, Xi, and Xa Hi-C maps at Kdm5c-Ogt loop (250kb resolution, clones merged). RAD21 and H3K27me3 cChIP-seq at loop anchors shown for reference. Treatment as in (B).
(G) Day 7 composite X, Xi, and Xa Hi-C maps at Bcor-Bcorl1 loop (100kb resolution, clones merged). RAD21 and H3K27me3 cChIP-seq at loop anchors shown for reference. Treatment as in (B).
(H) First Principal Component (PC1) tracks of day 7 Xa and Xi RAD21-FKBPdegron Hi-C maps. Treatment as in (B).
(I) Saddle plot analysis of compartment strength in RAD21-FKBPdegron Xi and Xa Hi-C maps (clones merged). Scores represent B-B (Xi: S2-S2), B-A (Xi: S2-S1), and A-A (Xi: S1-S1) compartment interaction strength. Treatment as in (B).
(J) Cumulative distribution plots of allelic chromosome X and chromosome 13 expression by PRO-seq in RAD21-FKBPdegron linesTreatment as in (B).
Fig 4. Cohesin is required for proper folding of the inactive X chromosome at day 10 of differentiation.

(A) Schematic of expected cohesin levels versus TAD, compartment, and superstructures detected on the X chromosome at different stages of XCI, based on prior studies. Levels corresponding to DMSO control and dTAG-13 treated cells indicated.
(B) Metaplot and heatmaps of active (Xa) and inactive (Xi) X chromosome RAD21 binding by uncalibrated ChIP-seq in day 10 RAD21-FKBPdegron embryoid bodies treated for 8 hours with 500nM dTAG-13 or DMSO.
(C) TAD scores calculated from day 10 Xa and Xi RAD21-FKBPdegron allelic Hi-C interaction maps binned at 100kb. DMSO and dTAG-13 samples as in (B). Representative Hi-C maps shown for the indicated regions (50kb resolution, clones merged).
(D) Chromosome-wide day 10 Xi RAD21-FKBPdegron Hi-C maps (250kb resolution, clones merged) and log2-ratio map (1Mb resolution). Treatment as in (B).
(E) Chromosome-wide day 10 Xa RAD21-FKBPdegron Hi-C maps (250kb resolution, clones merged) and log2-ratio map (1Mb resolution). Treatment as in (B).
(F) Day 7 composite X, Xi, and Xa Hi-C maps at Dxz4-Firre superloop (100kb resolution, clones merged), Kdm5c-Ogt loop (250kb resolution, clones merged) and Bcor-Bcorl1 loop (100kb resolution, clones merged). RAD21 and H3K27me3 cChIP-seq at superloop anchors shown for reference. Treatment as in (B).
(G) First Principal Component (PC1) tracks of day 7 Xa and Xi RAD21-FKBPdegron Hi-C maps. Treatment as in (B).
(H) Saddle plot analysis of compartment strength in RAD21-FKBPdegron Xi and Xa Hi-C maps (clones merged). Scores represent B-B (Xi: S2-S2), B-A (Xi: S2-S1), and A-A (Xi: S1-S1) compartment interaction strength. Treatment as in (B).
(I) Xi escapee (including Ogt, Bcor1, Bcorl1) gene expression in RAD21-FKBPdegron lines. Significant p-values by Wilcoxon signed rank test indicated by * (p<0.001663), ** (p<0.00232), *** (p<0.0002441). N.S., not significant.
(J) Cumulative distribution plots of allelic chromosome X and chromosome 13 expression by RNA-seq in RAD21-FKBPdegron lines. Treatment as in (B).
Our ability to toggle cohesin levels using RAD21/WAPL degrons provided an opportunity to address these questions (Figs. 3–4). Our in situ Hi-C was performed in a hybrid female ES line (TsixTST/+) carrying a Mus musculus (mus) and a Mus castaneus (cas) X chromosome, with the mus X chromosome bearing a Tsix mutation that ensures inactivation of the X-chromosome in cis (Ogawa et al., 2008). We were therefore able to perform allele-specific analysis to examine the Xa and Xi separately. In day 7 control cells, whereas the Xa showed topological domains, the Xi showed weakened (but not ablated) local domains (Fig. 3C, 4C) and megadomains (Fig. 3D, 4D), as expected. When cohesin was depleted by dTAG-13 treatment, the topological domains were nearly abolished on both Xa and Xi (Fig. 3C, 4C). The loss of interactions observed on the Xa was similar to autosomes (Fig. 4E,1F). On the Xi, the megadomains and Dxz4 border also disappeared in the absence of cohesin (Fig. 3D, 4D). Thus, although cohesin becomes depleted on the Xi during the normal course of XCI, the remaining cohesin is required for folding into megadomains. Notably, Dxz4 contains cohesin-binding sites (Fig. 3E, RAD21 track) that normally persist on the Xi despite Xist-mediated cohesin repulsion (Bonora et al., 2018; Froberg et al., 2018; Giorgetti et al., 2016).
The Firre locus also normally retains cohesin binding on the Xi (Fig. 3E, RAD21 track) (Bonora et al., 2018; Froberg et al., 2018; Giorgetti et al., 2016). It may thereby form a superloop with Dxz4 through cohesin-mediated loop extrusion across a 30Mb (Bansal et al., 2019; Bonora et al., 2018). Intriguingly, our allelic Hi-C map revealed a cohesin-dependent ‘stripe’ extending from Dxz4 (Fig. 3D, inset). This stripe is indicative of a unidirectional loop extrusion (Vian et al., 2018) whereby cohesin remains bound at the Dxz4 anchor while extruding chromatin. However, this stripe extended away from Firre, implying that Dxz4 does indeed anchor loops, but loop extrusion may not be the mechanism by which it engages Firre. Indeed, the Dxz4-Firre superloop was not perturbed by cohesin degradation, but paradoxically became stronger (Fig. 3E, 4F). Not only did the superloop strengthen on the Xi, it became visible on the Xa as well (Fig. 3E, 4F). Thus, surprisingly, the Dxz4-Firre superloop is not cohesin-dependent and is in fact inhibited by cohesin.
Firre and Dxz4 were depleted for H3K27me3 relative to the rest of the Xi (Fig. 3E), arguing against Polycomb-dependent looping for these loci. We noted that Firre belongs to a group of “escapees”, X-linked genes that are not subject to XCI, and surmised that escapee status might render genes resistant to cohesin depletion. Intriguingly, allelic Hi-C analysis revealed a clustering of multiple escapee or Polycomb-depleted genes, including Kdm5c, Ogt, Bcor, and Bcorl1 (Fig. 3F-G and 4F). All such loops were reinforced by RAD21 degradation. While some escapees were previously observed to associate using 4C (Splinter et al., 2011), our high resolution allelic Hi-C maps enabled us to identify these superloops on a chromosome-wide basis and demonstrate that cohesin antagonizes their formation. No replicable changes to escapee expression was detected by PRO-seq at day 7 (8 hours degradation). A trend for decreased escapee expression could be detected by RNA-seq at day 10, but was only significant in one replicate (Fig. 4I). We conclude that escapees form long-range loops through mechanisms other than cohesin-mediated loop extrusion and Polycomb-based co-clustering, and may instead self-associate via active chromatin. We propose that escapee clustering may aid in their persistent expression on the Xi, independently of cohesin and Polycomb.
Transitory S1/S2 compartments on the Xi do not require cohesins
We next investigated effects on Xa and Xi compartments. Prior to XCI, the two X-chromosomes are organized into A/B compartments, much as any autosome. At the onset of XCI, Xist RNA — together with PRC1 that it recruits — fuses the A/B compartments into larger intermediate structures called S1 and S2 compartments (Fig. 3A, middle column) (Wang et al., 2019). Xist then recruits the non-canonical SMC protein, SMCHD1, to merge S1 and S2 compartments into a unique Xi-specific superstructure in which compartments are not visible (Wang et al., 2018). We asked how the transition from A/B to S1/S2 to the Xi superstructure is affected by cohesin perturbations. At days 7 and 10, S1/S2 compartments were visible on the control Xi as “checkerboard” patterns, and also as alternating +/− segments in the 1st Principal Component of the Hi-C map (Fig. 3D,H,4D,G). Classic A/B compartments were evident on the Xa (Fig. 3D,H,4E,G). After 8 hours of cohesin degradation, these patterns were unchanged on both Xa and Xi (Fig. 3I, 4H). There were also no significant effects on Xi silencing at day 7 or 10 (Fig. 3J, 4J). While these findings are in line with the fact that up to 90% of cohesins are normally evicted from the Xi during XCI (Minajigi et al., 2015), they were somewhat unanticipated, given how much residual cohesin binding contributes to the Xi superstructure. Thus, like some Polycomb targets on autosomes, Xi superstructure and transcriptomics appear to be resistant to cohesin loss. We surmise that the Xi’s enrichment for Polycomb marks at least partially underlies the resistance.
Forced cohesin retention disrupts the 3D structure of Xi and creates megadomains on Xa
What would happen to the Xi if we forced cohesin retention? In our WAPL-FKBPdegron lines, 8 hours of WAPL depletion only succeeded in retaining cohesins at the 30Mb centromeric and distal telomeric ends of the Xi (Fig. S4C). We therefore focused analysis on 3 days of WAPL depletion (days 4–7), when cohesins became enriched 2-fold across the Xi (Fig. 5B). A 2-fold enrichment also occurred on the Xa. Interestingly, cohesin levels did not become equalized between the Xi and the Xa (Fig. 5B). This suggests that recruitment of WAPL to the Xi may not be the sole means of cohesin eviction during XCI. It is also possible that degradation from days 4–7 is too late to fully restore cohesin levels. Nevertheless, the increased cohesin levels on the Xi enabled us to examine consequences for Xi architecture and gene expression.
Fig.5: Cohesin retention results in increase of interactions spanning TAD borders and weakening of megadomains and compartments on the Xi.

(A) Schematic of expected cohesin levels versus TAD, compartment, and superstructures detected on the X chromosome at different stages of XCI, based on prior studies. Levels corresponding to day 7 DMSO control and dTAG-13 treated cells indicated.
(B) Metaplot and heatmaps of active (Xa) and inactive (Xi) X chromosome RAD21 binding by cChIP-seq in day 7 WAPL-FKBPdegron embryoid bodies treated for 3 days with 500nM dTAG-13 or DMSO.
(C) TAD scores calculated from day 7 Xa and Xi WAPL-FKBPdegron allelic Hi-C interaction maps binned at 100kb. DMSO and dTAG-13 samples as in (B). Representative Hi-C maps shown for the indicated regions (50kb resolution, clones merged).
(D) Chromosome-wide day 7 Xi WAPLFKBPdegron allelic Hi-C maps (250kb resolution, clones merged) and log2-ratio map (1Mb resolution). Treatment as in (B). Inset, 50kb resolution.
(E) Chromosome-wide day 7 Xa WAPL-FKBPdegron allelic Hi-C maps (250kb resolution, clones merged). Treatment as in (B).
(F) First and third Principal Component (PC1 and PC3) tracks of day 7 Xa and Xi WAPL-FKBPdegron Hi-C maps. Treatment as in (B).
(G) Saddle plot analysis of compartment strength in WAPL-FKBPdegron Xi and Xa Hi-C maps (clones merged). Scores represent B-B (Xi: S2-S2), B-A (Xi: S2-S1), and A-A (Xi: S1-S1) compartment interaction strength. Treatment as in (B).
(H) Zoomed-in day 7 composite X, Xi, and Xa Hi-C maps at Dxz4-Firre superloop (100kb resolution, clones merged). RAD21 and H3K27me3 cChIP-seq at superloop anchors shown for reference. Treatment as in (B).
Intriguingly, forced cohesin retention did not substantially change strength of domains on the Xi (so-called “TADs”; Fig. 5C) — contrasting with the marked attenuation seen when cohesins were depleted (Fig. 3C,4C). Furthermore, the Dxz4 megadomain border remained strong and the architectural stripe anchored at Dxz4 appeared to lengthen (Fig. 5D), consistent with cohesin retention and a unidirectional loop extrusion originating at Dxz4. The preservation of shorter-range looping interactions (Fig. 5C) occurred at the expense of longer-range intra-megadomain interactions (Fig. 5D, right panel – subtractive map). Longer-range interactions were also affected on the Xa (Fig. 5E), in line with increased cohesin retention on the Xa. However, PC1 analysis showed that cohesin retention did not prevent the appearance of transitory S1/S2 compartment structures except for disruptions at the distal centromeric end of the Xi (Fig. 5F, arrow). Saddle plot analysis (Schwarzer et al., 2017) confirmed changes in compartments, with S1-S1 interactions weakening and S1-S2 interactions strengthening (Fig. 5G), which could potentially explain the disruptions to S1/S2 compartments. Finally, no effects on the Dxz4-Firre superloop was observed (Fig. 5H), further enforcing the idea that the superloop is cohesin-independent. Thus, forced cohesin retention resulted in failure to attenuate local looping interactions and disrupted the Xi superstructure without affecting superloops.
Findings for the Xa were equally intriguing. The Xa also enriched for cohesins by 2-fold after WAPL degradation (Fig. 5B). While this retention did not affect topological domains (Fig. 5C), A/B compartments could not be detected in PC1 of the Xa Hi-C map (Fig. 5F). Instead, Xa adopted a megadomain-like pattern, which is typically only seen for the Xi (Fig. 5F, PC3). Saddle plot analysis showed weakened A-A interactions and strengthened A-B interactions (Fig. 5G), which may partially account for the sudden change in the overall Xa structure. Thus, unexpectedly, megadomain organization can emerge independently of XCI.
Failure of cohesin eviction compromises Xist spreading and Xi gene silencing
Previous work demonstrated that merging S1/S2 compartments is critical for Xist to spread and silence genes on the Xi (Wang et al., 2018). Given the changes in compartment structure (Fig. 5), we asked if forced cohesin retention affected Xist spreading and Xi silencing. We performed CHART-seq to map genomic binding sites for Xist RNA in WAPL-depleted cells (dTAG-13 treated between days 4–7). Xist RNA still bound the Xi despite retained cohesins (Fig. 6A and S6A). However, the localization appeared to be compromised, as there was a 2-fold drop in Xist coverage at the centromeric and telomeric ends (Fig. 6A,S6A, Xist Δ track). Notably, these regions are furthest from the Xist locus. In the Hi-C contact map, these were also the regions that tend to lose contact with Xist (Fig. 6A,S6A, top track). Statistical analysis confirmed Xist RNA depletion across the entire chromosome, with greatest effect size at the 30 Mb centromeric and telomeric ends (Fig. 6B, S6B). Concomitantly, H3K27me3 levels decreased up to 4-fold at the Xi ends (Fig. 6A,S6A, H3K27me3 tracks). Statistical analysis confirmed this decrease (Fig. 6C,S6C). These effect sizes are Xi-specific in both biological replicates. While Xist and H3K27me3 coverages decreased significantly in both S1 and S2 compartments, the S1 compartment near the chromosomal ends were most affected (Fig. 6A,S6A, yellow highlights) and the effect size was largest in S1 compartments overall (Fig.S6F–G). These data support the notion that weakened compartmentalization negatively impacts Xist and H3K27me3 spreading.
Fig.6: Retaining cohesin on the Xi early in differentiation leads to XCI defects, especially at the ends of the chromosome.

(A) Tracks of Xi Hi-C contacts to the Xist transcription locus, Xist binding (CHART-seq) and H3K27me3 (cChIP-seq) in WAPL-FKBPdegron lines treated from days 4–7 of differentiation with DMSO (D) or 500nM dTAG-13 (13). Log fold ratio tracks (dTAG-13/DMSO) are also shown (Δ). Principal component corresponding to compartments on the Xi (PC1, positive S1, negative S2) shown for reference.
(B) Boxplot showing Xist coverage over 100kb bins in the centromeric most 30Mb, middle 100Mb, and telomeric most 30Mb of the X chromosome in WAPL-FKBPdegron, clone H7 treated as in (A). Significant p-values by Wilcoxon signed rank test indicated by * (p<2.2e-16).
(C) Boxplots showing allelic H3K27me3 coverage over 100kb bins in the centromeric most 30Mb, middle 100Mb, and telomeric most 30Mb of the X chromosome in WAPL-FKBPdegron, clone H2 treated as in (A). Significant p-values by Wilcoxon signed rank test indicated by * (p<2.2e-16) and ** (p<0.001187). N.S., not significant.
(D) Boxplot showing average Xist density over different classes of genes on the X chromosome in WAPL-FKBPdegron, clone H7 treated as in (A). Significant p-values by Wilcoxon signed rank test indicated by * (p<2.2e-16). N.S., not significant.
(E) Boxplot showing average allelic H3K27me3 density over different classes of genes on the X chromosome in WAPL-FKBPdegron, clone H2 treated as in (A). Significant p-values by Wilcoxon signed rank test indicated by * (p<2.2e-16), ** (p<8.094e-16), *** (p<0.00206), **** (p< 0.0001193). N.S., not significant.
The increased number of longer chromatin loops (Fig. 1K,M) could also play a role in weakened Xist and PRC2 spreading. As looping contacts mainly increased in the 1–10Mb range (Fig. 1K), we examined how spreading was affected within 10Mb around Xist. While Xist coverage decreased within 10Mb, H3K27me3 was not replicably changed (Fig.S6H). Therefore, while additional chromatin loops may inhibit Xist spreading, this does not seem to be sufficient to impede H3K27me3 deposition at short ranges.
Finally, we investigated effects on Xi gene silencing. Xist and H3K27me3 coverages were significantly decreased over genes subject to XCI (Fig. 6D-E,S6D–E). They were also reduced over intergenic regions, but not over escapees which normally lacked Xist and H3K27me3. We performed allele-specific RNA-seq analysis in WAPL-depleted cells (dTAG-13 treated days 4–7) and observed a significant cumulative shift in expression from the Xi (Fig. 7A) — indicating either a failure of inactivation during the establishment phase, or an initial inactivation and then a partial reactivation in later stages. Consistent with compromised spreading of the Xist-PRC2 complex, failure of silencing was greater at the centromeric and telomeric 30Mb ends (Fig. 7B). Examination of specific loci revealed strong (>1.7-fold) upregulation from various genes, including Clcn5, Apex2, Tspan7, Spin2c, and Ap1s2 (Fig. 7C) — all of where were located at the chromosome ends. We conclude that aberrant cohesin retention disrupts the ability of the Xist-PRC2 complex to spread at long range to the ends of the Xi, with consequences for gene silencing.
Fig.7: Selective cohesin eviction and retention are required to maintain Polycomb targets, fold the Xi, and properly silence Xi genes.

(A) Cumulative distribution plots of chromosome-wide Xi expression in 2 WAPL-FKBPdegron clones treated from days 4–7 of differentiation with DMSO or 500nM dTAG-13. P-value calculated by the KS test.
(B) Cumulative distribution plots of Xi expression from the centromeric most 30Mb, middle 100Mb, and telomeric most 30Mb of the chromosome in 2 in 2 WAPL-FKBPdegron clones treated as in (A). P-value calculated by the KS test.
(C) Examples of genes at the far centromeric (Clcn5,Tspan7) and telomeric (Apex2,Ap1s2) ends of the X chromosome. Allelic RNA-seq and H3K27me3 cChIP-seq shown for 2 WAPL-FKBPdegron clones treated as in (A).
(D) Model: cohesin tempering of Polycomb targets on autosomes.
(E) Model: cohesin tempering of Xist and Polycomb spreading on the Xi.
DISCUSSION
Large and finer scale 3D genome structures have long been hypothesized to play a pivotal role in gene regulation (de Wit et al., 2015; Guo et al., 2015; Merkenschlager and Nora, 2016). However, recent degron studies targeting architectural factors and their genomic binding sites have challenged this model. Here, we revisited the genomic structure-function relationship through the lens of XCI and demonstrate that a fine balance between cohesin eviction and retention is necessary to shape the Xi. Additionally, this balance is required to protect against inappropriate inter-chromosomal contacts and to temper Polycomb-mediated gene repression on autosomes.
While it is now established that Xist RNA evicts cohesins and the Xi is generally depleted of architectural factors, it was previously not known whether some cohesins must be retained for proper architectural transformation into a silent chromosome. By toggling cohesin levels at will, we demonstrate that XCI requires that cohesins be evicted from some regions and retained on others. Depleting cohesin ablates the sharp megadomain border, whereas forcing cohesin retention does not compromise the sharp border but reduces intra-megadomain interactions. Likewise, loop domains or “TADs” on the Xi require cohesin. When cohesin is depleted, these structures disappear almost entirely. We also demonstrate that Xi compartments — specifically the S1/S2 compartments — are weakened by stabilization of cohesin. Superloops, such as the those between Dxz4-Firre and between escapees Kdm5c and Ogt, are also antagonized by cohesins and strengthened when cohesins are degraded. Thus, selective regions on the Xi indeed must retain cohesin binding in order to construct the Xi superstructure. An interesting question for future consideration is how Xist evicts one set of cohesins while retaining others on the Xi.
The present work also reveals that Xist and Polycomb spreading and gene silencing all depend on cohesin eviction across the Xi. Forced cohesin retention results in reduced spreading of Xist RNA and H3K27me3 across the whole Xi, especially at the distal ends. Gene silencing was also blunted to varying degrees along the whole chromosome, with the strongest effects again at the Xist-distal ends. Expression of escapee genes, by contrast, is not affected by cohesin retention. However, cohesin depletion led to a surprising reinforcement of clustering of various escapees. The Dxz4-Firre superloop also strengthened upon cohesin depletion. It is possible that the depletion of cohesin that normally occurs on the Xi reinforces clustering of escapees to ensure their continued expression. Intriguingly, a recent study implicated Firre RNA as a regulator of the Xi and autosomes in trans (Fang et al., 2020). Future work will be needed to determine if interactions of the Firre genomic locus could be involved as well.
Our data show that cohesin imbalances also affect the Xa, causing the Xa to adopt some Xi-like structures. Indeed, a megadomain-like pattern appears mysteriously on the Xa when cohesins are aberrantly enriched, suggesting that enhanced local contacts may spontaneously drive partitioning of an X-chromosome into two large domains. Importantly, this means that megadomain organization can emerge independently of Xist and XCI. A Dxz4-Firre interaction also appeared on the Xa after cohesin degradation, also suggesting that the superloop could be a consequence of cohesin depletion rather than XCI.
Finally, our study revealed unexpected effects on autosomes as well. For instance, cohesins antagonize interactions between Polycomb targets and temper Polycomb-mediated repression (Rhodes et al., 2020). Moreover, the effects on Polycomb domains are not limited in cis, as loss of cohesin strengthens clustering of Polycomb targets in trans, as exemplified by trans-interactions among the four Hox gene clusters. Inter-chromosomal bridges between the Hox loci are especially prominent during differentiation, in contrast to mESCs, where such interactions are not detected in control conditions. When cohesins are aberrantly retained, such interactions are disrupted and expression of the Polycomb targets are dramatically upregulated.
Altogether, the relationship between cohesin and gene expression on autosomes is analogous to that observed on the Xi (Fig. 7D-E). On autosomes under wild type conditions, current models postulate that cohesin translocates along chromatin during the process of loop extrusion, passing through Polycomb domains (Fig. 7D, left). Polycomb targets participating in interactions with each other tend to be highly repressed and are therefore indifferent to the presence of cohesin. However, certain Polycomb targets which are not engaged in interactions can become further repressed upon loss of cohesin. As Polycomb-anchored loops appear to be reinforced through cohesin loss, it is possible that this occurs due to increased interactions between previously non-interacting regions (Fig. 7D, middle). As the Xi as a whole is highly repressed and covered with Polycomb that forms self-associating compartments, it is logical that Xi expression would also be indifferent to the loss of cohesin, similar to interacting Polycomb targets on autosomes (Fig. 7E, middle). When WAPL is degraded, cohesin levels increase genome-wide, weakening interactions between Polycomb domains. Such disruptions lead to upregulation of Polycomb targets previously engaged in such interactions (Fig. 7D, right). We also observe upregulation across the Xi. In contrast to autosomes, repression on the Xi is mediated by Xist (Fig. 7E, left). Therefore, we observe the largest impact on gene silencing at the distal ends of the chromosome furthest from Xist, where the most Hi-C contacts are lost as well (Fig. 7E, right). In conclusion, we propose that a key role of cohesins is to organize local chromatin and prevent ectopic long-range cis- and trans-interactions. In doing so, cohesins tempers repression of Polycomb targets as well as X chromosome inactivation.
STAR★Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Jeannie T. Lee (lee@molbio.mgh.harvard.edu).
Materials Availability
Requests for materials generated in this study should be directed to and will be fulfilled by the Lead Contact upon completion of a Material Transfer Agreement.
Data and Code Availability
Original unprocessed blot and microscope images in this manuscript have been deposited at Mendeley Data and are available at: http://dx.doi.org/10.17632/9b83hvkgm5.1
Raw high-throughput sequencing data and processed files for RNA-seq, PRO-seq, cChIP-seq, CHART-seq, and Hi-C reported in this paper have been deposited at GEO under accession number: GSE144834
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
RAD21-FKBPdegron and WAPL-FKBPdegron mESCs were generated by transfecting TsixTST/+ mESCs with (1) a plasmid (pSpCas9-(BB) 2A Puro) expressing Cas9 with an sgRNA targeted to the STOP codon (in the case of Rad21) or to a region in the last codon near the STOP codon (in the case of Wapl) (2) a homology repair template plasmid including 900bp homology arms around the STOP codon, the FKBPdegron tag in a pUC19 backbone and (3) a homology-directed repair plasmid (pMB 1609-pRR-EGFP) with the sgRNA target site cloned in. Specifically, 500,000 TST mESCs were transfected using Lipofectamine 3000 1.5ug of pSpCas9-(BB) 2A Puro, 1.5ug of the non-linearized repair plasmid, and 400ng of pMB 1609-pRR-EGFP. Transfected cells were GFP sorted after 48 hours and the entire GFP positive pool plated. After 5 days, the pool was split and plated at low density, and after 5 more days, clones were picked from the low-density plate in a 96-well plate. Clones were grown to confluency, then triplicate plated in 96-well plates. One 96-well plate was used to PCR screen clones. Clones were then thawed from one of the triplicate plates, expanded, and re-screened by PCR and Sanger sequenced to confirm correct knock-in of the FKBPdegron tag.
METHOD DETAILS
ES cell differentiation and induced degradation
Undifferentiated mESCs were grown on γ-irradiated MEF feeders in medium containing DMEM, high glucose, GlutaMAX supplement, pyruvate, 15% Hyclone FBS (Sigma), 25 mM HEPES pH 7.2–7.5, 1x MEM non-essential amino acids, 1x Pen/Strep, 0.1 mM βME, and 500 U/mL ESGRO recombinant mouse Leukemia Inhibitory Factor (LIF) protein (Sigma, ESG1107) at 37°C with 5% CO2. To induce differentiation in EBs, mESCs were trypsinized and resuspended in the same media without LIF. For the first 3 days of differentiation, EBs were grown in suspension. On day 4 and subsequent days, EBs were grown on gelatinized plates until harvested at day 7 or day 10.
Degradation of RAD21 or WAPL in FKBPdegron lines during differentiation was induced by adding a 1:20,000 dilution of 10mM dTAG-13 in DMSO to cell culture media (500nM final concentration) for the indicated time frames. The small molecule dTAG-13 was synthesized as previously described (Erb et al., 2017; Nabet et al., 2018). For DMSO vehicle controls, the same volume of DMSO alone was added to cell culture media.
Western blot
Cells were counted and ~100,000 cells per sample lysed in 100ul 1X Laemmli buffer. Protein extracts were boiled for 5 min at 95C. Equal cell counts (5–15ul) were loaded and resolved by SDS page. Protein was transferred to an Immobilon-P PVDF membrane (EMD Millipore). After transfer, membrane was blocked with 5% milk in PBST (PBS/0.05% Tween-20) for 30 min at room temperature. Membrane was incubated with primary antibody (1:1000 for RAD21 and WAPL, 1:20,000 for GAPDH) in 5% milk in PBST at 4C overnight, then washed three times with PBST. Membrane was then blocked with secondary antibody-HRP conjugate (1:5000) in 5% milk in PBST for 1 hour at room temperature. Membrane was incubated with Western Lightning Plus-ECL (PerkinElmer) and exposed using the Bio-Rad ChemiDoc MP Imaging System.
Immunofluorescence
Immunofluorescence was performed as described previously with minor modifications (Del Rosario et al., 2019). Briefly, cells on glass slides were first permeabilized 2 min on ice with CSKT (100 mM NaCl, 300 mM Sucrose, 10 mM PIPES pH 6.8, 3 mM MgCl2, 0.5% Triton X-100) and then fixed in PBS 4% paraformaldehyde for 10 min at room temperature followed by a PBS wash step. Fixed cells were blocked (PBS, 0.2% Tween-20, 4% bovine serum albumin (BSA), 2% normal goat serum (NGS) (Sigma)) for 30 min at room temperature and incubated with primary antibody in a humid chamber at 4C overnight. Antibodies were diluted in blocking buffer. Samples were washed with PBS300.2T (PBS supplemented to 300 mM NaCl, 0.2% Tween-20) and incubated with secondary antibody for 1 hour at room temperature. Slides were mounted using Vectashield with DAPI (Vector Labs). Slides were imaged with a Nikon Eclipse 90i microscope with Volocity software (Perkin Elmer).
Antibodies
For ChIP-seq the following antibodies were used: H3K27me3 (GeneTex GTX60892), RAD21 (Abcam ab992). For Western blot: RAD21 (Millipore 05–908), WAPL (Abcam ab70741), GAPDH (CST 5174T). For immunofluorescence: HA (Roche 11867423001).
ChIP-seq and calibrated ChIP-seq
ChIP-seq was performed as described previously with minor modifications (Del Rosario et al., 2019). Briefly, cells were trypsinized, resuspended in 1 million cells/mL of media and crosslinked with 1% formaldehyde for 10 min at room temperature with rotation. Cells were then quenched with 0.2 M glycine for 5 min. Crosslinked cells were washed once with cold PBS, pelleted, and snap frozen in an EtOH/dry ice bath. Cell pellets were stored at −80C until further processing. 5–10 million cross-linked cells per IP were thawed on ice and resuspended in 300ul Lysis Buffer 1 (25mM HEPES pH 7.2–7.5, 25mM EDTA, 0.5% SDS). Nuclei were sonicated (Qsonica Q800 Sonicator) in polystyrene tubes at 40% power setting, 30s ON/30s OFF for a total sonication (ON) time of 6 min. After sonication, 1.2 mL of Lysis Buffer 2 (25mM HEPES pH 7.2–7.5, 187.5mM NaCl, 1.25% Triton X-100, 0.625% Na-deoxycholate, 6.25% glycerol, 1x cOmplete EDTA-free protease inhibitor cocktail) was added. 30ul of Dynabeads Protein G (Thermo Fisher Scientific) per IP were washed 3 times with ChIP Wash Buffer 1 (25mM HEPES pH 7.2–7.5, 150mM NaCl, 5mM EDTA, 1% Triton X-100, 0.5% Na-deoxycholate, 5% glycerol, 1x cOmplete EDTA-free protease inhibitor cocktail) and resuspended in 60ul ChIP Wash Buffer 1. Each IP was blocked with 20ul washed beads for 2 hours at 4C with rotation. The other 40ul of washed beads were incubated with antibody (5ul of RAD21 antibody or 2ul of H3K27me3 antibody/IP) for 2 hours at 4C. 5% input was taken from blocked lysates. Beads conjugated to antibody were added to blocked lysates and IP allowed to proceed overnight at 4C with rotation. IPs were then washed 3 times with cold ChIP Wash Buffer 1, 3 times with cold ChIP Wash Buffer 2 (25mM HEPES pH 7.2–7.5, 300mM NaCl, 5mM EDTA, 1% Triton X-100, 0.5% Na-deoxycholate, 5% glycerol, 1x cOmplete EDTA-free protease inhibitor cocktail). And 1 time with cold TENG (50mM Tris-HCl pH 8, 1mM EDTA, 50mM NaCl, 5% glycerol, 1x cOmplete EDTA-free protease inhibitor cocktail). All washes were performed by inverting tubes and setting aside for ~2 minutes per wash. IPs were then resuspended in 500ul TENG with 100ug RNAse A (Thermo Fisher Scientific) and incubated for 30 min at 37C. IPs were then washed 2 times with cold ChIP Wash Buffer 2 and 2 times with cold TENG without protease inhibitors. IPs were resuspended in 200ul elution buffer (50mM Tris-HCl pH 7.5, 10mM EDTA, 1% SDS) and incubated 15 min at 65C. Eluate was then transferred to a new tube and 198ul TE buffer (Ambion) and 40ug Proteinase K (Sigma) added. Inputs were thawed, TE buffer added to equalize volume to IPs and 200ul elution buffer and 40ug Proteinase K added. Inputs and IPs were then incubated for 2h at 45C and crosslinks reversed overnight at 65C. DNA was purified with phenol-chloroform extraction and resuspended in 50ul TE. Efficiency of ChIP was assayed via qPCR, with samples displaying over 10-fold enrichment of positive regions over negative control prepared for sequencing. Libraries were prepared from ~500ng of input or IP DNA using the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (NEB) according to kit instructions. Experiments were performed in two biological replicates. Input and ChIP-seq libraries were sequenced on Illumina HiSeq, generating 20–50 million paired end 50 or 150 nucleotide reads per sample.
Calibrated ChIP-seq was performed using the same protocol as ChIP-seq except that 50,000 HEK293 cells per 5 million mouse cells were mixed in at the initial lysis step. Cells were counted and equal numbers of cells used per IP.
ChIP-seq and calibrated ChIP-seq analysis
For uncalibrated ChIP-seq, input and ChIP-seq reads were aligned to the 129S1/SvJm (mus) and CAST/Eih (cas) genome as previously described (Kung et al., 2015; Minajigi et al., 2015). PCR duplicates were removed and uniquely mapped reads used for further analysis. Fpm-normalized bigwig tracks were generated for all uniquely mapped (comp), mus-specific (mus), and cas-specific (cas) reads.
Heatmaps and metaplots were generated using deepTools functions computeMatrix and plotHeatmap with the reference point as the summit of the called ChIP peaks.
Calibrated ChIP-seq analysis was performed as previously described (Hu et al., 2015). Briefly, paired-end reads were aligned to a concatenated mouse and human genome (mm9 + hg19) using Bowtie2 with “--no-mixed” and “--no-discordant” options. PCR duplicates were filtered out with SAMBAMBA (Tarasov et al., 2015). Uniquely aligned mouse reads were normalized to human spike-in by subsampling mm9 reads so that total number of hg19 reads was matched between samples and bigWig tracks generated from normalized samples. For calibration of RAD21 cChIP between RAD21-FKBPdegron clones, only reads within hg19 peaks (hg19 tracks combined) were counted for normalization purposes. Subsampling factors were further adjusted based on ratio of hg19 to mm9 reads in matched input samples. After normalization factor was determined, paired end reads were aligned to mus and cas genomes as previously described, except using Bowtie2 with the same parameters as above (with “--no-mixed” and “-no-discordant”) instead of Novoalign. PCR duplicates were filtered out with SAMBAMBA and allelic reads normalized using the same normalization factor as above.
Input subtracted H3K27me3 calibrated ChIP coverage profiles were generated with SPP (Kharchenko et al., 2008) with smoothing using 1kb windows recorded every 500 bp, as previously described (Simon et al., 2013). For input subtraction, inputs from DMSO and dTAG-13 treated samples were combined to ensure equal profiles were subtracted from each calibrated IP.
Heatmaps and metaplots were generated using deepTools functions computeMatrix and plotHeatmap with the reference point as the summit of the called ChIP peaks. Plots of calibrated H3K27me3 signal over upregulated or downregulated genes were calculated using “scale-regions” with the entire gene scaled to 3kb.
To compare the allelic densities of H3K27me3 over different classes of X-linked genes, we computed average densities over gene bodies using multiBigwigSummary from deepTools with default parameters. Non-expressed genes were defined as X-linked genes have 0 RPKM in at least one RNA-seq replicate of DMSO control. Genes subject to XCI were identified as the set of genes have allelic information (see RNA-seq analysis above), with escapees, Xist and Tsix excluded. Escapees were as defined previously (Wang et al., 2018). To compare allelic densities of H3K27me3 over S1 and S2 compartments, we defined S1 compartments as regions having positive value in the first Principal Component calculated in the WAPL-FKBPdegron DMSO control days 4–7 sample and S2 compartments as regions have negative value. S1 and S2 compartments were then binned into 100kb windows using bedtools makewindows, excluding windows that overlapped with unmappable regions as defined previously (Wang et al., 2018). We then we computed average densities over these 100kb bins using multiBigwigSummary from deepTools with default parameters.
RNA-seq
Total RNA was extracted using TRIzol Reagent (Thermo Fisher Scientific) and mRNA isolated using the NEBNext Poly(A) Magnetic Isolation Module (NEB) according to the kit instructions. Two biological replicates of RNA-seq libraries were prepared using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB) according the kit instructions. Libraries were sequenced on the Illumina HiSeq2500 (rapid run), generating 30–50 million 50 nt paired end reads per sample or the Illumina HiSeq4000, generating 16–27 million 150 nt paired end reads per sample.
RNA-seq analysis
RNA-seq reads were aligned allele-specifically to the 129S1/SvJm (mus) and CAST/Eih (cas) genomes using TopHat2 (Kim et al., 2013) as previously described (Kung et al., 2015; Minajigi et al., 2015). PCR duplicates were removed and unique reads mapping to exons of each gene quantified by Homer (Heinz et al., 2010). DESeq 2 was used to call differentially expressed genes with a cut off of adjusted p-value < 0.1. For analyses on all genes, no FPKM threshold was applied, but genes were required to have at least 1 count in one sample. An expression threshold of FKPM > 0.5 was applied for analyses on expressed genes. Expressed genes with a minimum of 13 allelic-specific reads in at least one sample were counted as genes with allelic information (Colognori et al., 2019). For a given gene, fraction mus was calculated as the fraction of allelic reads that were mus-specific (mus / (mus+cas) reads).
Log2FoldChange and log2baseMean from DESeq2 were plotted for indicated classes of genes. H3K27me3 genes were defined as genes overlapping H3K27me3 cChIP-seq peaks in their respective samples (DMSO and dTAG-13 peaks combined). H3K27me3 loop genes were defined as H3K27me3 genes overlapping H3K27me3-bound loop anchors called in RAD21-FKBPdegron dTAG-13 Hi-C maps (i.e., cohesin-independent H3K27me3 anchored loops). For overlap with RAD21 loop anchors, loops were called in respective DMSO Hi-C maps.
PRO-seq library construction
PRO-Seq library construction was performed by the Nascent Transcriptomics Core at Harvard Medical School, Boston, MA. Aliquots of frozen (−80°C) permeabilized cells were thawed on ice and pipetted gently to fully resuspend. An aliquot was removed and diluted 1:10 in 1xPBS for cell counting using an automatic counter (Biorad TC-20) with counts spot checked by manual recount using a hemocytometer as needed. An aliquot of each dilution was also stained with Trypan Blue and counting repeated to determine permeabilization efficiency. For each sample, 1 million permeabilized cells were used for nuclear run-on. For normalization, 50,000 permeabilized Drosophila S2 cells were added to each sample of 1 million cells. Nuclear run on assays and library preparation were then performed essentially as described [Elrod, N.D., et al. (2019) Molecular Cell 76: 738–752.e7] with modifications noted below.
The nuclear run-on buffer was prepared as 4X stock (20mM Tris (pH 8), 20mM MgCl2, 2mM DTT, 600mM KCl, 40uM/ea biotin-11-NTPs (Perkin Elmer), 20U SuperaseIN (Thermo)). The 4X stock was mixed 1:1 with 2% sarkosyl (Sigma) to yield 2X complete run-on mix. The 3’ adapter (RNA oligo: 5’P-GAUCGUCGGACUGUAGAACUCUGAAC-3’InvdT) was pre-adenylated prior to use (5’ DNA adenylation kit, NEB, according to the manufacturer’s instructions). Adenylated oligo was purified by ethanol precipitation, resuspended in water and quality was modification confirmed by electrophoresis on a 15% TBE-Urea gel (Novex) next to appropriate controls. The adapter concentration was adjusted to 10uM and 1uL was used for each ligation which used T4 RNA ligase 2, truncated KQ (NEB) with 15% PEG-8000 in the reaction mix. Reactions were incubated at 16°C overnight. Each ligation reaction was then diluted with 180uL of betaine binding buffer (1.42g of betaine brought to 10mL with binding buffer and sterile filtered) containing 1uL of 100uM blocking oligo (TCCGACGATCCCACGTTCCCGTGG/3InvdT/). The blocking oligo is complementary to and overhangs the 3’ end of the 3’ adapter and reduces 5’/3’ adapter dimer formation during the 5’ adapter ligation step. The blocking oligo has modifications at its 3’ end, to prevent extension by reverse transcriptase. Blocking oligo was also included in final wash solution prior to the 5’ adapter ligation and in the ligation reaction itself, both at 3-fold molar excess over the initial amount of 3’ adapter (i.e. 1uL of 30uM per reaction). The 5’ adapter ligation was performed as in Elrod except PEG-8000 was increased to 15%.
After reverse transcription, cDNA was immediately amplified for 5-cycles (“preCR” step). A PCR cocktail consisting of NEBNext Ultra II Q5 master mix (NEB) and Illumina TruSeq PCR primers (RP-1, common; and RPI-X, indexing) was added to each cDNA and amplification was done according to manufacturer’s suggested 2-step cycling conditions for NGS applications. To determine optimal library amplification, the preCRs were thawed on ice and an aliquot serially diluted. Each dilution was mixed with a PCR cocktail (Q5 DNA polymerase with optional high GC enhancer) containing 10uM/ea of Illumina universal P5 and P7 primers and amplified 15-cycles. The reactions were electrophoresed on a 2.2% agarose gel (SeaKem) cast with SYBR gold (Thermo) and analyzed to determine the optimal number of cycles for final amplification. preCRs were transferred from ice to a pre-heated thermal cycler block and amplified for the appropriate number of additional cycles to reach the total determined in the test amplification.
Pooled libraries were sequenced using Illumina NovaSeq S1 (PE 50) platform.
PRO-seq analysis
Paired-end reads were aligned to a concatenated mouse and fly genome (mm9 + dm6) using Bowtie2 with “--no-mixed” and “--no-discordant” options. DMSO and dTAG-13 samples for each clone were normalized to drosophila spike-in by subsampling mm9 reads so that total number of dm6 reads was matched between samples. For RAD21-FKBPdegron clone G9, downsampling factors corresponded to library size (i.e., were equivalent to downsampling based on total number of reads). For RAD21-FKBPdegron clone C4, downsampling factors were 1 for DMSO and 0.73 for dTAG-13 although library sizes were similar, indicating that there were overall lower levels of nascent transcription in the dTAG-13 sample. Duplicates were not removed to preserve reads corresponding to TSS and promoter proximal PolII positions. Reads mapping to genes (full gene bodies) were quantified by Homer (Heinz et al., 2010), removing miRNA, snoRNA and scaRNAs. DESeq 2 was used to call differentially expressed genes with a cut off of adjusted p-value < 0.1. For analyses on all genes, no FPKM threshold was applied, but genes were required to have at least 1 count in one sample. An expression threshold of FKPM > 0.5 was applied for analyses on expressed genes.
Log2FoldChange and log2baseMean from DESeq2 were plotted for indicated classes of genes. H3K27me3 genes were defined as genes overlapping H3K27me3 cChIP-seq peaks in their respective samples (DMSO and dTAG-13 peaks combined). H3K27me3 loop genes were defined as H3K27me3 genes overlapping H3K27me3-bound loop anchors called in RAD21-FKBPdegron dTAG-13 Hi-C maps (i.e., cohesin-independent H3K27me3 anchored loops). For overlap with RAD21 loop anchors, loops were called in respective DMSO Hi-C maps.
For allelic analysis, PRO-seq reads not mapping to the fly genome (dm6) were aligned to mus and cas genomes using Bowtie2 with the same parameters as above (“--no-mixed” and “--no-discordant”) and downsampled using the same factors as calculated above. Expressed genes with a minimum of 13 allelic-specific reads in at least one sample were counted as genes with allelic information (Colognori et al., 2019). For CDPs, normalized mus and cas counts divided by gene length were plotted.
In situ Hi-C and visualization
In situ Hi-C was done as described previously (Rao et al., 2014) using 2 clones for each degron line and 5 million cells per sample. See Supplemental Table 1 for Hi-C statistics.
Reads were trimmed using cutadapt with the options --adapter=GATCGATC (MboI ligation junction) and --minimum-length=20. Reads of each pair were individually mapped to the mus (Xi) and cas (Xa) reference genomes using novoalign and merged into Hi-C summary files and filtered using HOMER as previously described (Minajigi et al., 2015). Hi-C contact maps were generated from reads in the filtered HOMER mus and cas tag directories using the ‘pre’ command of Juicer tools with the genomeID mm9 (Durand et al., 2016b). The resulting Hi-C contact maps in .hic format were visualized and normalized with the ‘Coverage Square Root’ option in Juicebox version 1.8.8 (Durand et al., 2016a).
In situ Hi-C analysis
Principal component analysis (PCA) was done using the runHiCpca.pl command of HOMER with the filtered HOMER Hi-C tag directory as input and the options -res 100000 -superRes 100000 -genome mm9. Pearson correlation maps were also computed by HOMER using the analyzeHiC command with the filtered HOMER Hi-C tag directory as input and the options -chr chrX -res 200000 –corr.
Loops were called from Hi-C interaction maps using the GPU version of HiCCUPS (Durand et al., 2016) with the following parameters: ‘java -jar juicer_tools_1.13.02.jar hiccups -m 512 -r 25000,50000 -k KR -f .1,.1 -p 1,1 -i 3,3 -t 0.02,1.5,1.75,2 -d 50000,100000’, adding the --ignore_sparsity option when needed. Loops were called from merged Hi-C interaction maps for each condition (DMSO and dTAG-13). Loops were additionally filtered to require at least 27 contacts in the peak center, 2.25 enrichment over the donut neighborhood and an FDR < 0.05 in the donut neighborhood. APA was performed on loops called by HiCCUPS using Juicer (Durand et al., 2016) at 5kb resolution.
For inter-chromosomal Hox interactions, APA was performed at 100kb resolution with the -e option using an input loop list of inter-chromosomal interactions anchored at 500kb intervals centered at Hox clusters.
For super-enhancer interactions, APA was performed at 100kb resolution using an input loop list of interactions anchored at 500kb intervals centered at super-enhancers (Whyte et al., 2013). Super-enhancer intervals were split into 5 quantiles based on distance between anchors.
TAD scores were calculated as described previously. Specifically, 100-kb resolution ‘Balanced’ normalized chromosome Hi-C interaction matrices were extracted from Juicebox using the ‘dump’ function of Juicer tools (juicer dump observed KR name.hic X X BP 100000 name.KR.100kb.dump). Custom shell and R scripts were used to convert this output matrix into the cworld matrix format. Insulation scores were then calculated for the X chromosome using the resulting cworld matrix and the matrix2insulation.pl cworld script with parameters –is 500000 –ids 400000 –im iqrMean–nt 0 –ss 200000 –yb 1.5 –nt 0 –bmoe 0. As described previously, regions of insulation score minima represent regions of locally high insulation and are considered potential TAD boundaries (Crane et al., 2015). TAD scores were then calculated using insulation scores from given samples and TAD borders called previously on the Xa (Colognori et al., 2019) with the cworld script insulation2tads.pl with default parameters.
For saddle plot compartmental analysis, .hic format Hi-C maps were first converted to .mcool format using ‘hic2cool convert’ with default options (Abdennur and Mirny, 2020). Publicly available code from https://cooltools.readthedocs.io/en/latest/notebooks/04_saddle-plots.html was then used to generate saddle plots from .mcool files.
CHART-seq
Xist CHART-seq was performed as previously described (Wang et al., 2018). Briefly, cells were trypsinized, resuspended in 1 million cells/mL of media and crosslinked with 1% formaldehyde for 10 min at room temperature with rotation. Cells were then quenched with 0.2 M glycine for 5 min. Crosslinked cells were washed once with cold PBS, pelleted, and snap frozen in an EtOH/dry ice bath. Cell pellets were stored at −80C until further processing. 25 million cross-linked cells per CHART were thawed on ice and resuspended in 1 mL cold sucrose buffer. (10 mM HEPES pH 7.5, 0.3 M sucrose, 1% Triton X-100, 100 mM potassium acetate, 0.1 mM EGTA) supplemented with 0.5 mM spermidine, 0.15 mM spermine, 1 mM DTT, 1x protease inhibitor cocktail (Sigma, P8340), 10 U/mL RNase inhibitor (Roche). The cell suspension was rotated for 10 min at 4°C, after which it was diluted with additional 2 mL cold sucrose buffer. This was added to a pre-chilled 15-mL glass Wheaton dounce tissue grinder (Thermo Fisher Scientific) that had been pre-rinsed once with RNaseZAP RNase decontamination solution (Thermo Fisher Scientific), three times with DEPC-treated water, and once with 2 mL sucrose buffer. Release of nuclei was accomplished by douncing cells 20 times with a tight pestle, and verified by staining with Trypan Blue Solution (Thermo Fisher Scientific). The nuclear suspension was then layered atop a cushion of 7.5 mL cold glycerol buffer (10 mM HEPES pH 7.5, 25% glycerol, 1 mM EDTA, 0.1 mM EGTA, 100 mM potassium acetate) supplemented with 0.5 mM spermidine, 0.15 mM spermine, 1 mM DTT, 1x cOmplete EDTA-free protease inhibitor cocktail (Roche), and 5 U/mL RNase inhibitor, and centrifuged at 1500 g for 10 min at 4°C. The purified nuclear pellet was resuspended in 6 mL PBS and further cross-linked with 3% formaldehyde for 30 min at room temp with rotation. Afterward, nuclei were pelleted by centrifugation at 1,000 g for 5 min at 4°C and washed three times with cold PBS. Nuclei were resuspended in in 1 mL cold nuclear extraction buffer (50 mM HEPES pH 7.5, 250 mM NaCl, 0.1 mM EGTA, 0.5% sarkosyl, 0.1% sodium deoxycholate, 5 mM DTT, 10 U/mL RNase inhibitor) and incubated for 10 min at 4°C with rotation. Nuclei were then centrifuged at 400 g for 5 min at 4°C and resuspended in 230 μL cold sonication buffer (50 mM HEPES pH 7.5, 75 mM NaCl, 0.1 mM EGTA, 0.5% sarkosyl, 0.1% sodium deoxycholate, 0.1% SDS, 5 mM DTT, 10 U/mL RNase inhibitor) to a final volume of ~270 μL. Nuclei were sonicated for 5 min at 4°C using a Covaris E220e (140W peak incident power, 10% duty factor, 200 cycles/burst). Sonicated chromatin was centrifuged at 16,000 g for 20 min at 4°C to remove debris. The supernatant (~220 μL) was mixed with 110 μL sonication buffer to a final volume of ~330 μL, which was split into two CHART reactions (Xist capture using antisense probes and control capture using sense probes).
For two CHART reactions, 600 μL MyOne Streptavidin C1 beads (Thermo Fisher Scientific) were used, which were pre-washed with 1 mL DEPC-treated water, blocked with 600 μL blocking buffer (33% sonication buffer, 67% 2x hybridization buffer, 500 ng/μL yeast RNA [Thermo Fisher Scientific], 1% BSA) at room temp for 1 hr, washed with 1 mL DEPC-treated water again, and resuspended in 610 μL 1x hybridization buffer (33% sonication buffer, 67% 2x hybridization buffer [see below for composition]). For each CHART, 160 μL chromatin was mixed with 320 μL 2x hybridization buffer (50 mM Tris pH 7.0, 750 mM NaCl, 1% SDS, 1 mM EDTA, 15% formamide, 1 mM DTT, 1 mM PMSF, 1x cOmplete EDTA-free protease inhibitor cocktail, 100 U/mL RNase inhibitor) and pre-cleared with 60 μL pre-blocked MyOne Streptavidin C1 beads at room temp for 1 hr with rotation. Pre-cleared chromatin was separated from beads, with 1% saved as “input” sample. The remaining pre-cleared chromatin (~500 μL) was mixed with 36 pmol of either antisense (Xist-targeting) or sense (control) biotinylated capture probes. We used a pool of 9 capture probes, which were selected from the 11 probes described previously (Simon et al., 2013), excluding probes X.3 and X.8. Hybridization was carried out at 37°C for 4 hr followed by incubation with 240 μL blocked MyOne Streptavidin C1 beads at 37°C for 1 hr. The beads were washed once with 1x hybridization buffer (33% sonication buffer, 67% 2x hybridization buffer) at 37°C for 10 min, five times with wash buffer (10 mM HEPES pH 7.5, 150 mM NaCl, 2% SDS, 2 mM EDTA, 2 mM EGTA, 1 mM DTT) at 37°C for 5 min, and twice with elution buffer (10 mM HEPES pH 7.5, 150 mM NaCl, 0.5% NP-40, 3 mM MgCl2, 10 mM DTT) at 37°C for 5 min. 10% of the final wash was saved as an “RNA capture” sample to estimate the efficiency of Xist RNA capture afterward by qRT-PCR. CHART-enriched DNA was eluted twice in 200 μL elution buffer supplemented with 5 U/μL RNase H (New England BioLabs) at 37°C for 30 min. An “input” sample and CHART DNA were treated with 0.5 mg/mL RNase A (Thermo Fisher Scientific) at 37°C for 1 hr and then with 1% SDS, 10 mM EDTA, and 0.5 mg/mL proteinase K (Sigma) at 55°C for 1 hr. Reversal of crosslinks was performed by addition of 150 mM NaCl (final 300 mM) and incubation at 65°C overnight. DNA was purified by phenol-chloroform extraction and further sheared to < 500-bp fragments using Covaris E220e (140W peak incident power, 10% duty factor, 200 cycles/burst). Sonicated DNA was purified using 1.8x Agencourt AMPure XP beads (Beckman Coulter). Input and CHART-seq libraries were prepared using the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (NEB) according to kit instructions. Libraries were sequenced on Illumina, generating ~50 million 150-nt paired-end reads per sample.
CHART-seq analysis
Input and ChIP-seq reads were aligned to the 129S1/SvJm (mus) and CAST/Eih (cas) genome as previously described (Kung et al., 2015). PCR duplicates were removed and uniquely mapped reads used for further analysis. Fpm-normalized bigwig tracks were generated for all uniquely mapped (comp), mus-specific (mus), and cas-specific (cas) reads. CHART libraries were sampled to equal read depth and input subtracted CHART coverage profiles were generated with SPP (Kharchenko et al., 2008) with smoothing using 1kb windows recorded every 500 bp, as previously described (Simon et al., 2013). After scaling, we computed base 2 logarithm of the ratio of the coverage of DMSO control divided by that of dTAG-13 using bigWigCompare from deepTools with --binSize 1000 and default parameters otherwise. To compare Xist densities over centromeric, middle, and telomeric regions, we split the first 30Mb, middle 100Mb and last 30Mb of the X chromosome into 100kb windows using bedtools makewindows, excluding windows that overlapped with unmappable regions as defined previously (Wang et al., 2018) and computed average Xist densities over these bins using multiBigwigSummary from deepTools with default parameters. To compare the densities of Xist over different classes of X-linked genes, we computed average densities over gene bodies using multiBigwigSummary from deepTools with default parameters. Non-expressed genes were defined as X-linked genes have 0 FPKM in at least one RNA-seq replicate of DMSO control. Genes subject to XCI were identified as the set of genes have allelic information (see RNA-seq analysis above), with escapees, Xist and Tsix excluded. Escapees were as defined previously (Wang et al., 2018). To compare densities of Xist over S1 and S2 compartments, we defined S1 compartments as regions having positive value in the first Principal Component calculated in the WAPL-FKBPdegron DMSO control days 4–7 sample and S2 compartments as regions have negative value. S1 and S2 compartments were then binned into 100kb windows using bedtools makewindows, excluding windows that overlapped with unmappable regions as defined previously (Wang et al., 2018). We then we computed average densities over these 100kb bins using multiBigwigSummary from deepTools with default parameters.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the statistical tests used and the values of n and p are reported in the figures, figure legends, or associated main texts. Statistical significance is determined by the value of p < 0.05 by the indicated tests.
Supplementary Material
Table S1. Statistics for Hi-C datasets, related to Figure 1
Table S2. DESeq2 tables for RNA-seq and PRO-seq datasets, related to Figure 2
HIGHLIGHTS.
Cohesin tempers intra- and inter-chromosomal Polycomb-anchored ‘loops’
Cohesin depletion affects formation of Xi megadomains but not superloops
Forced cohesin retention disrupts Xist-Polycomb spreading and gene silencing
Cohesin loss recapitulates an Xi-like superloop and megadomain on the active X
ACKNOWLEDGEMENTS
We thank J. Froberg and C.-Y. Wang for critical reading of the manuscript, and we thank all lab members for support. We thank N. Gray for support and the generous gift of dTAG-13. The authors would like to thank the Nascent Transcriptomics Core at Harvard Medical School, Boston, MA for performing PRO-seq library construction. Nucleosomes in Fig. 7 were created with BioRender.com. A.J.K. is supported by 1F31HD100109-01 and J.T.L. by grants from the National Institutes of Health (9R01HD097665-09) and the Howard Hughes Medical Institute. B.N. is supported by an American Cancer Society Postdoctoral Fellowship (PF-17-010-01-CDD) and the Katherine L. and Steven C. Pinard Research Fund.
Footnotes
DECLARATION OF INTERESTS
J.T.L. is a cofounder of Translate Bio and Fulcrum Therapeutics, and is also an advisor to Skyhawk Therapeutics. B.N. is an inventor on patent applications related to the dTAG system (WO/2017/024318, WO/2017/024319, WO/2018/148440 and WO/2018/148443).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abdennur N, and Mirny LA (2020). Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal P, Kondaveeti Y, and Pinter SF (2019). Forged by DXZ4, FIRRE, and ICCE: How Tandem Repeats Shape the Active and Inactive X Chromosome. Front Cell Dev Biol 7, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagrie RA, Scialdone A, Schueler M, Kraemer DC, Chotalia M, Xie SQ, Barbieri M, de Santiago I, Lavitas LM, Branco MR, et al. (2017). Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AC, and Felsenfeld G (2000). Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405, 482–485. [DOI] [PubMed] [Google Scholar]
- Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, and Zhuang X (2018). Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blewitt ME, Gendrel AV, Pang Z, Sparrow DB, Whitelaw N, Craig JM, Apedaile A, Hilton DJ, Dunwoodie SL, Brockdorff N, et al. (2008). SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nature genetics 40, 663–669. [DOI] [PubMed] [Google Scholar]
- Bonev B, and Cavalli G (2016). Organization and function of the 3D genome. Nat Rev Genet 17, 661–678. [DOI] [PubMed] [Google Scholar]
- Bonora G, Deng X, Fang H, Ramani V, Qiu R, Berletch JB, Filippova GN, Duan Z, Shendure J, Noble WS, et al. (2018). Orientation-dependent Dxz4 contacts shape the 3D structure of the inactive X chromosome. Nat Commun 9, 1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buheitel J, and Stemmann O (2013). Prophase pathway-dependent removal of cohesin from human chromosomes requires opening of the Smc3-Scc1 gate. EMBO J 32, 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KL, Roig MB, Hu B, Beckouet F, Metson J, and Nasmyth K (2012). Cohesin’s DNA exit gate is distinct from its entrance gate and is regulated by acetylation. Cell 150, 961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colognori D, Sunwoo H, Kriz AJ, Wang CY, and Lee JT (2019). Xist Deletional Analysis Reveals an Interdependency between Xist RNA and Polycomb Complexes for Spreading along the Inactive X. Mol Cell 74, 101–117 e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, and Cremer C (2001). Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet 2, 292–301. [DOI] [PubMed] [Google Scholar]
- Cuadrado A, Gimenez-Llorente D, Kojic A, Rodriguez-Corsino M, Cuartero Y, Martin-Serrano G, Gomez-Lopez G, Marti-Renom MA, and Losada A (2019). Specific Contributions of Cohesin-SA1 and Cohesin-SA2 to TADs and Polycomb Domains in Embryonic Stem Cells. Cell Rep 27, 3500–3510 e3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrow EM, Huntley MH, Dudchenko O, Stamenova EK, Durand NC, Sun Z, Huang SC, Sanborn AL, Machol I, Shamim M, et al. (2016). Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proceedings of the National Academy of Sciences of the United States of America 113, E4504–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson IF, Bauer B, Goetz D, Tang W, Wutz G, and Peters JM (2019). DNA loop extrusion by human cohesin. Science 366, 1338–1345. [DOI] [PubMed] [Google Scholar]
- de Wit E, Vos ES, Holwerda SJ, Valdes-Quezada C, Verstegen MJ, Teunissen H, Splinter E, Wijchers PJ, Krijger PH, and de Laat W (2015). CTCF Binding Polarity Determines Chromatin Looping. Mol Cell 60, 676–684. [DOI] [PubMed] [Google Scholar]
- Del Rosario BC, Kriz AJ, Del Rosario AM, Anselmo A, Fry CJ, White FM, Sadreyev RI, and Lee JT (2019). Exploration of CTCF post-translation modifications uncovers Serine-224 phosphorylation by PLK1 at pericentric regions during the G2/M transition. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Ma W, Ramani V, Hill A, Yang F, Ay F, Berletch JB, Blau CA, Shendure J, Duan Z, et al. (2015). Bipartite structure of the inactive mouse X chromosome. Genome biology 16, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denholtz M, Bonora G, Chronis C, Splinter E, de Laat W, Ernst J, Pellegrini M, and Plath K (2013). Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell 13, 602–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps J, and Duboule D (2017). Embryonic timing, axial stem cells, chromatin dynamics, and the Hox clock. Genes Dev 31, 1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despang A, Schopflin R, Franke M, Ali S, Jerkovic I, Paliou C, Chan WL, Timmermann B, Wittler L, Vingron M, et al. (2019). Functional dissection of the Sox9-Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nat Genet 51, 1263–1271. [DOI] [PubMed] [Google Scholar]
- Disteche CM (2016). Dosage compensation of the sex chromosomes and autosomes. Semin Cell Dev Biol 56, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, and Aiden EL (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, Souza A, Roberts JM, Dastjerdi S, Buckley DL, et al. (2017). Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falco G, Lee SL, Stanghellini I, Bassey UC, Hamatani T, and Ko MS (2007). Zscan4: a novel gene expressed exclusively in late 2-cell embryos and embryonic stem cells. Dev Biol 307, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Bonora G, Lewandowski JP, Thakur J, Filippova GN, Henikoff S, Shendure J, Duan Z, Rinn JL, Deng X, et al. (2020). Trans- and cis-acting effects of Firre on epigenetic features of the inactive X chromosome. Nat Commun 11, 6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemr M, and Buhler M (2015). Single-Step Generation of Conditional Knockout Mouse Embryonic Stem Cells. Cell Rep 12, 709–716. [DOI] [PubMed] [Google Scholar]
- Fraser J, Williamson I, Bickmore WA, and Dostie J (2015). An Overview of Genome Organization and How We Got There: from FISH to Hi-C. Microbiol Mol Biol Rev 79, 347–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froberg JE, Pinter SF, Kriz AJ, Jegu T, and Lee JT (2018). Megadomains and superloops form dynamically but are dispensable for X-chromosome inactivation and gene escape. Nat Commun 9, 5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi R, Gillespie PJ, and Hirano T (2006). Human Wapl is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Curr Biol 16, 2406–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gdula MR, Nesterova TB, Pintacuda G, Godwin J, Zhan Y, Ozadam H, McClellan M, Moralli D, Krueger F, Green CM, et al. (2019). The non-canonical SMC protein SmcHD1 antagonises TAD formation and compartmentalisation on the inactive X chromosome. Nat Commun 10, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgetti L, Lajoie BR, Carter AC, Attia M, Zhan Y, Xu J, Chen CJ, Kaplan N, Chang HY, Heard E, et al. (2016). Structural organization of the inactive X chromosome in the mouse. Nature 535, 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg JR (1972). High stability of messenger RNA in growing cultured cells. Nature 240, 102–104. [DOI] [PubMed] [Google Scholar]
- Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, et al. (2015). CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell 162, 900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarhuis JHI, van der Weide RH, Blomen VA, Yanez-Cuna JO, Amendola M, van Ruiten MS, Krijger PHL, Teunissen H, Medema RH, van Steensel B, et al. (2017). The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 169, 693–707 e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacisuleyman E, Goff LA, Trapnell C, Williams A, Henao-Mejia J, Sun L, McClanahan P, Hendrickson DG, Sauvageau M, Kelley DR, et al. (2014). Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat Struct Mol Biol 21, 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horakova AH, Moseley SC, McLaughlin CR, Tremblay DC, and Chadwick BP (2012). The macrosatellite DXZ4 mediates CTCF-dependent long-range intrachromosomal interactions on the human inactive X chromosome. Hum Mol Genet 21, 4367–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Petela N, Kurze A, Chan KL, Chapard C, and Nasmyth K (2015). Biological chromodynamics: a general method for measuring protein occupancy across the genome by calibrating ChIP-seq. Nucleic Acids Res 43, e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huis in ‘t Veld PJ, Herzog F, Ladurner R, Davidson IF, Piric S, Kreidl E, Bhaskara V, Aebersold R, and Peters JM (2014). Characterization of a DNA exit gate in the human cohesin ring. Science 346, 968–972. [DOI] [PubMed] [Google Scholar]
- Joshi O, Wang SY, Kuznetsova T, Atlasi Y, Peng T, Fabre PJ, Habibi E, Shaik J, Saeed S, Handoko L, et al. (2015). Dynamic Reorganization of Extremely Long-Range Promoter-Promoter Interactions between Two States of Pluripotency. Cell Stem Cell 17, 748–757. [DOI] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, and Park PJ (2008). Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 26, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Shi Z, Zhang H, Finkelstein IJ, and Yu H (2019). Human cohesin compacts DNA by loop extrusion. Science 366, 1345–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziejczyk AA, Kim JK, Tsang JC, Ilicic T, Henriksson J, Natarajan KN, Tuck AC, Gao X, Buhler M, Liu P, et al. (2015). Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell 17, 471–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueng S, Hegemann B, Peters BH, Lipp JJ, Schleiffer A, Mechtler K, and Peters JM (2006). Wapl controls the dynamic association of cohesin with chromatin. Cell 127, 955–967. [DOI] [PubMed] [Google Scholar]
- Kundu S, Ji F, Sunwoo H, Jain G, Lee JT, Sadreyev RI, Dekker J, and Kingston RE (2017). Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation. Mol Cell 65, 432–446 e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung JT, Kesner B, An JY, Ahn JY, Cifuentes-Rojas C, Colognori D, Jeon Y, Szanto A, del Rosario BC, Pinter SF, et al. (2015). Locus-specific targeting to the X chromosome revealed by the RNA interactome of CTCF. Mol Cell 57, 361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanctot C, Kaspar C, and Cremer T (2007). Positioning of the mouse Hox gene clusters in the nuclei of developing embryos and differentiating embryoid bodies. Exp Cell Res 313, 1449–1459. [DOI] [PubMed] [Google Scholar]
- Lee JT (2011). Gracefully ageing at 50, X-chromosome inactivation becomes a paradigm for RNA and chromatin control. Nature reviews Molecular cell biology 12, 815–826. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing S (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luppino JM, Park DS, Nguyen SC, Lan Y, Xu Z, Yunker R, and Joyce EF (2020). Cohesin promotes stochastic domain intermingling to ensure proper regulation of boundary-proximal genes. Nat Genet 52, 840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, Firth A, Singer O, Trono D, and Pfaff SL (2012). Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin K, Flyamer IM, Thomson JP, Mjoseng HK, Shukla R, Williamson I, Grimes GR, Illingworth RS, Adams IR, Pennings S, et al. (2019). DNA Methylation Directs Polycomb-Dependent 3D Genome Re-organization in Naive Pluripotency. Cell Rep 29, 1974–1985 e1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkenschlager M, and Nora EP (2016). CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation. Annu Rev Genomics Hum Genet 17, 17–43. [DOI] [PubMed] [Google Scholar]
- Minajigi A, Froberg JE, Wei C, Sunwoo H, Kesner B, Colognori D, Lessing D, Payer B, Boukhali M, Haas W, et al. (2015). Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, and Fraser P (2013). Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Varnai C, Dudley C, Leung W, Baran Y, Mendelson Cohen N, Wingett S, Fraser P, and Tanay A (2017). Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K, and Haering CH (2009). Cohesin: its roles and mechanisms. Annu Rev Genet 43, 525–558. [DOI] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944 e922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Sun BK, and Lee JT (2008). Intersection of the RNA interference and X-inactivation pathways. Science 320, 1336–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliou C, Guckelberger P, Schopflin R, Heinrich V, Esposito A, Chiariello AM, Bianco S, Annunziatella C, Helmuth J, Haas S, et al. (2019). Preformed chromatin topology assists transcriptional robustness of Shh during limb development. Proc Natl Acad Sci U S A 116, 12390–12399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson JR, and Laemmli UK (1977). The structure of histone-depleted metaphase chromosomes. Cell 12, 817–828. [DOI] [PubMed] [Google Scholar]
- Pearson JC, Lemons D, and McGinnis W (2005). Modulating Hox gene functions during animal body patterning. Nat Rev Genet 6, 893–904. [DOI] [PubMed] [Google Scholar]
- Peters JM, Tedeschi A, and Schmitz J (2008). The cohesin complex and its roles in chromosome biology. Genes Dev 22, 3089–3114. [DOI] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, and Zhang Y (2003). Role of histone H3 lysine 27 methylation in X inactivation. Science 300, 131–135. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320 e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Ventii KH, and Wilkinson KD (2009). Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem 78, 363–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes JDP, Feldmann A, Hernandez-Rodriguez B, Diaz N, Brown JM, Fursova NA, Blackledge NP, Prathapan P, Dobrinic P, Huseyin MK, et al. (2020). Cohesin Disrupts Polycomb-Dependent Chromosome Interactions in Embryonic Stem Cells. Cell Rep 30, 820–835 e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, and Corces VG (2018). Organizational principles of 3D genome architecture. Nat Rev Genet 19, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S, Sugar R, Dimond A, Javierre BM, Armstrong H, Mifsud B, Dimitrova E, Matheson L, Tavares-Cadete F, Furlan-Magaril M, et al. (2015). Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat Genet 47, 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, C HH, Mirny L, et al. (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, Mak W, Zvetkova I, Appanah R, Nesterova TB, Webster Z, Peters AH, Jenuwein T, Otte AP, and Brockdorff N (2003). Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell 4, 481–495. [DOI] [PubMed] [Google Scholar]
- Simon MD, Pinter SF, Fang R, Sarma K, Rutenberg-Schoenberg M, Bowman SK, Kesner BA, Maier VK, Kingston RE, and Lee JT (2013). High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature 504, 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter E, de Wit E, Nora EP, Klous P, van de Werken HJ, Zhu Y, Kaaij LJ, van Ijcken W, Gribnau J, Heard E, et al. (2011). The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes & development 25, 1371–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N, and de Laat W (2006). CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev 20, 2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starmer J, and Magnuson T (2009). A new model for random X chromosome inactivation. Development 136, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens TJ, Lando D, Basu S, Atkinson LP, Cao Y, Lee SF, Leeb M, Wohlfahrt KJ, Boucher W, O’Shaughnessy-Kirwan A, et al. (2017). 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, and Prins P (2015). Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A, Wutz G, Huet S, Jaritz M, Wuensche A, Schirghuber E, Davidson IF, Tang W, Cisneros DA, Bhaskara V, et al. (2013). Wapl is an essential regulator of chromatin structure and chromosome segregation. Nature 501, 564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vian L, Pekowska A, Rao SSP, Kieffer-Kwon KR, Jung S, Baranello L, Huang SC, El Khattabi L, Dose M, Pruett N, et al. (2018). The Energetics and Physiological Impact of Cohesin Extrusion. Cell 175, 292–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieux-Rochas M, Fabre PJ, Leleu M, Duboule D, and Noordermeer D (2015). Clustering of mammalian Hox genes with other H3K27me3 targets within an active nuclear domain. Proc Natl Acad Sci U S A 112, 4672–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Colognori D, Sunwoo H, Wang D, and Lee JT (2019). PRC1 collaborates with SMCHD1 to fold the X-chromosome and spread Xist RNA between chromosome compartments. Nat Commun 10, 2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Jegu T, Chu HP, Oh HJ, and Lee JT (2018). SMCHD1 Merges Chromosome Compartments and Assists Formation of Super-Structures on the Inactive X. Cell 174, 406–421 e425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Mager J, Chen Y, Schneider E, Cross JC, Nagy A, and Magnuson T (2001). Imprinted X inactivation maintained by a mouse Polycomb group gene. Nat Genet 28, 371–375. [DOI] [PubMed] [Google Scholar]
- Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, et al. (2017). YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588 e1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellik DM (2007). Hox patterning of the vertebrate axial skeleton. Dev Dyn 236, 2454–2463. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson I, Kane L, Devenney PS, Flyamer IM, Anderson E, Kilanowski F, Hill RE, Bickmore WA, and Lettice LA (2019). Developmentally regulated Shh expression is robust to TAD perturbations. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz A (2011). Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet 12, 542–553. [DOI] [PubMed] [Google Scholar]
- Wutz G, Varnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, Schoenfelder S, Jessberger G, Muhar M, Hossain MJ, et al. (2017). Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J 36, 3573–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Wu DY, Zheng H, Wang Y, Sun QR, Liu X, Wang LY, Xiong WJ, Wang Q, Rhodes JDP, et al. (2020). Analysis of Genome Architecture during SCNT Reveals a Role of Cohesin in Impeding Minor ZGA. Mol Cell 79, 234–250 e239. [DOI] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, and Lee JT (2008). Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Statistics for Hi-C datasets, related to Figure 1
Table S2. DESeq2 tables for RNA-seq and PRO-seq datasets, related to Figure 2
Data Availability Statement
Original unprocessed blot and microscope images in this manuscript have been deposited at Mendeley Data and are available at: http://dx.doi.org/10.17632/9b83hvkgm5.1
Raw high-throughput sequencing data and processed files for RNA-seq, PRO-seq, cChIP-seq, CHART-seq, and Hi-C reported in this paper have been deposited at GEO under accession number: GSE144834