

Abstract
Despite the boom in biologics over the past decade, the intrinsic instability of these large molecules poses significant challenges to formulation development. Almost half of all pharmaceutical protein products are formulated in the solid form to preserve protein native structure and extend product shelf-life. In this review, both traditional and emerging drying techniques for producing protein solids will be discussed. During the drying process, various stresses can impact the stability of protein solids. However, understanding the impact of stress on protein product quality can be challenging due to the lack of reliable characterization techniques for biological solids. Both conventional and advanced characterization techniques are discussed including differential scanning calorimetry (DSC), solid-state Fourier transform infrared spectrometry (ssFTIR), solid-state fluorescence spectrometry, solid-state hydrogen deuterium exchange (ssHDX), solid-state nuclear magnetic resonance (ssNMR) and solid-state photolytic labeling (ssPL). Advanced characterization tools may offer mechanistic investigations into local structural changes and interactions at higher resolutions. The continuous exploration of new drying techniques, as well as a better understanding of the effects caused by different drying techniques in solid state, would advance the formulation development of biological products with superior quality.
Keywords: protein solids, drying technology, solid-state characterization, process stress, physical stability
Graphical abstract
1. INTRODUCTION
The rapid growth in development of biopharmaceuticals, particularly protein products, in recent decades has been accompanied by challenges in their formulations [1–4]. Most biopharmaceuticals are introduced into the human body through parenteral administration [5]. However, biomolecules are generally less stable in the liquid state than in the solid. The preservation of biopharmaceuticals in solid state has proved to be effective in maintaining molecule stability and extending product shelf-life [6]. The removal of water also facilitates the transportation process. In solid products, the biomolecules are usually dried with disaccharide excipients to protect the structure and ensure stability. The stabilizing mechanisms of excipients are commonly explained by either water replacement theory or vitrification theory [7]. The type of excipient has a great impact on the stability of biomolecules [7–9]. There are multiple drying techniques with assorted drying stresses and efficiency available for solid biopharmaceutical development. Direct solid-state characterization methods for biopharmaceuticals are limited. It can be challenging to fully understand the large biomolecule and the matrix in solids because most characterization techniques are developed for liquids. In general, the development process for the dried biopharmaceuticals is more complicated than for solution products [10]. In this review, we will discuss various aspects regarding manufacturing of biological solids, with a focus on pharmaceutical protein solids.
Lyophilization, or freeze drying, has been used for a long time in the development of biopharmaceuticals in solid state [11]. It is a mature drying method compared to others such as spray drying. However, lyophilization is a time-consuming batch process with extensive energy use. Technologies such as spray drying and spray freeze drying are potential new processing methods to be applied on biologics. It is important to understand their drying efficiency as well as the physico-chemical properties of the dried molecules. In this review, we will discuss the advantages and disadvantages of different types of drying techniques and their suitability for protein products.
One of the key purposes for developing biopharmaceuticals in solid state is to maintain the stability and extend shelf-life. However, the drying process can generate various stresses which promote aggregation and lead to visible and sub-visible particles in the final products [12]. For example, improperly controlled freezing steps in lyophilization could affect drying efficiency and final product quality. High drying temperature in spray drying may induce protein denaturation. The stresses affecting the stability of protein solids are discussed here.
A combination of various solid-state characterization methods is usually necessary to understand the stability of protein solids since different methods provide various aspects or levels of information. Differential scanning calorimetry (DSC) can be used as a routine method for Tg detection of the solid matrix while it does not provide information on protein structure. Solid-state Fourier Transform Infrared Spectrometry (ssFTIR) detects the change of protein secondary structure but its quantitation capability is limited. More detailed information of protein conformation and dynamic could be obtained from advanced approaches such as Solid-state Hydrogen Deuterium Exchange (ssHDX) and Solid-state Nuclear Magnetic Resonance (ssNMR); nevertheless, these methods generally require extra sample preparation or extensive data analysis. The characterization methods for the structure and conformation of protein in the solid state as well as the properties of solid matrix are introduced here.
2. DRYING TECHNOLOGY
2.1. Lyophilization/Freeze Drying (Lyo/FD)
Lyophilization, or freeze drying, is the process of dehydrating under vacuum at low temperatures. The word “lyophilization” is derived from the ancient Greek and means “to make solvent-loving”. The method originated during prehistoric times when it was used by the Eskimo and Aztecs to preserve food [13]. It wasn’t until the 1880s that lyophilization was used in a laboratory setting. The advancement of refrigeration and vacuum technologies and the increased necessity to process heat sensitive antibiotics and blood products in the 1930s enabled the production of the industrial scale freeze dryer to be used in both the food and pharmaceutical industries. Proteins were one of the earliest substances to be lyophilized and the demand for bulk preparations of plasma proteins during World War II paved the path for lyophilization of biomolecules at an industrial scale [13, 14]. Lyophilization has since evolved as the most widely used technique for manufacturing biomolecules with therapeutic and diagnostic applications.
2.1.1. Description of Process
The lyophilization process involves multiple steps. First, sample freezing immobilizes the solution components, reduces the thermal denaturation, and prevents foaming of the product when vacuum is applied. It also creates an ice-crystal matrix in the sample that facilitates the drying of the product. Sample freezing is generally followed by primary drying, where the principle of drying is sublimation of ice. Primary drying is followed by secondary drying, where the moisture adsorbed on the dried sample structure is removed by desorption at the ambient or higher temperature. Sealing the samples in a vacuum or under an inert gas takes place after the secondary drying. The use of vacuum or inert gas excludes reactive and destabilizing atmospheric gases like oxygen or carbon dioxide and prevents the adsorption of moisture [15, 16].
2.1.2. Design of Lyophilization Process
Lyophilization is a batch process that is time-consuming and energy-intensive. A non-optimized freeze drying cycle may lead to even longer drying time and cause damage to the product [17–20]. In order to design an optimized freeze drying cycle, it is important to understand the critical properties of the sample. The critical properties of lyophilized formulations are the stability of the drug and the properties of the excipients used as well as the collapse temperature of the final formulation [21]. The role of the stabilizing excipients will be discussed elsewhere in this theme issue.
The temperature above which the macroscopic structure of the lyophilized product collapses during lyophilization is known as the macroscopic collapse temperature (Tc) [22]. Tc in general is about 2 °C higher than the glass transition temperature (Tg’) of the formulation in the frozen state [23]. If the solute is crystallized in the frozen state then the Tc is about 2 °C higher than the eutectic temperature (Teu). If the frozen solute contains a mixture of amorphous and crystalline matrix then the amorphous matrix will collapse if the product temperature (Tp) is above the Tg’. A macroscopic collapse occurs if both Tg’ and Teu are below the product temperature. If Tg’ is below Tp and Teu is above Tp, then the crystalline matrix provides mechanical support to the amorphous matrix, thereby preventing macroscopic collapse [24]. Tc is close to Teu in a formulation containing amorphous and crystalline matrices while dominated by the crystalline matrix. Therefore, it is important to lyophilize a formulation at a temperature less than Tc [17].
Every step in the lyophilization process takes a different amount of time to finish but all have a significant impact on the final product. For this reason, it is important to understand the freezing, primary drying and secondary drying steps in order to design a process where each of the steps are optimized for a specific formulation.
2.1.2.1. Freezing
Freezing is the first step in the lyophilization process. This step often induces stresses on the protein such as protein-protein interactions which lead to aggregation due to an increase of protein concentration, crystallization of buffer salts leading to pH change, and protein degradation at the ice-water interface. These stresses can be minimized with formulation changes and by designing a proper freeze drying cycle. In the freezing step, numerous freezing methods such as nitrogen freezing, ramped cooling, or loading vials onto a precooled shelf may result in differences in the cooling effect and ice crystal growth size [25]. It has been reported that slower cooling rates (0.5°C/min) led to a larger super-cooling effect and loading vials onto pre-cooled shelves reduces the super-cooling effect but increases the heterogeneity in super-cooling among the vials [25]. Another study indicated that the slower cooling rate increased the potential of phase separation in formulations that were susceptible to phase separation, and led to protein damage and longer freezing times [26]. A moderate cooling rate (1°C/min) that allows moderate levels of supercooling and ice surface area along with fast freezing times to be observed has been suggested [21]. The final freezing temperature of the formulation is dependent on the Tg’ and/or Teu of the formulation and is generally 2°C less than the Tg’ or Teu [21]. Annealing is a step where the product is held at a temperature higher than the final freezing temperature for a certain amount of time [27]. It is usually applied when the freeze drying cycle is designed for formulations with components that could crystallize, such as the bulking agent mannitol [21]. The purpose of annealing is to ensure the crystallizing components can completely crystallize before the primary drying step, as crystallization during the primary drying may lead to vial breakage, and partially crystalized material may fully crystalize during storage [28].
2.1.2.2. Primary Drying
Primary drying is the longest step in the lyophilization process. In this step it is important to optimize the product temperature (Tp) as an increase in the product temperature leads to a decrease in the primary drying cycle time. It is reported that a 1°C increase in Tp can decrease the primary drying time by 13% [17]. Therefore, a balance in Tp should be considered between drying cycle time and product stability. Tp depends on multiple factors such as the formulation, the freeze dryer’s shelf temperature and the chamber pressure. Tp should be below the Tc in order to prevent the product from collapsing. The chamber pressure (Pc) is another crucial parameter in designing the primary drying cycle because it influences the sublimation rate. Generally, a Pc well below the vapor pressure of the ice as the sublimation rate can be represented by the equation
| (1) |
where dm/dt is the rate of ice sublimation (g/hour per vial), Pc is the chamber pressure, Pice is the equilibrium vapor pressure of ice at the sublimation interface temperature (Torr), and Rp and Rs are the resistance to water vapor transport from the sublimation interface (Torr·h/g) due to the dry layer and stopper, respectively [29]. This suggests that the smallest chamber pressure results in the highest sublimation rate. However, it has also been observed that very low chamber pressure may result in greater heterogeneity in the heat transfer among the vials, thereby resulting in larger Tp differences between the vials [30]. An optimum chamber pressure can be calculated using the equation below to obtain a target product temperature (Tp) [21].
| (2) |
where Pc is the chamber pressure (Torr) and Tp is the product temperature (°C).
Developing a shelf temperature - time profile for efficiently achieving the product temperature during primary drying is another important factor in designing an optimized lyophilization cycle. The end point of primary drying can be detected using several methods [30], such as with thermocouples within the vials. However, it has been reported that the vials containing the thermocouples usually have lower levels of super-cooling than the vials without thermocouples, leading to larger ice crystals and faster ice sublimation and therefore, a faster primer drying cycle [31]. Dew point sensors have also been used to detect the vapor composition change and can indicate the end of the primary drying cycle [31].
2.1.2.3. Secondary Drying
The next step in the process is secondary drying, where the adsorbed water is removed through desorption. Depending on the formulation and the primary drying cycle, the primary dried product may contain 5–20% w/w residual water. Secondary drying is performed to reduce the residual water content to the preferred amount, generally less than 1–2 % [21]. To prevent the solid product from collapsing, a slow ramp rate (0.1 – 0.15°C/min) of temperature is preferred. For crystalline products, a slightly higher ramp rate (0.3–0.4°C/min) is suggested. It is reported that the chamber pressure does not affect the desorption rate if the chamber pressure is maintained under 200 mTorr [21]. Secondary drying time is also dependent on the type of solid matrix as it takes more time to desorb water in an amorphous matrix than in a crystalline matrix. It is suggested that the secondary drying should be done at higher temperatures for shorter periods of time rather than lower temperature for longer periods of time [32], as long as higher temperatures do not jeopardize stability.
2.1.2.4. Use of Mathematical Modelling in Lyophilization
In the past, optimization of the lyophilization process was approached empirically through trial and error [21]. Mathematical models based on partial differential equations (PDE) [33–36] and model predictive control (MPC) [37–39] were some of the earlier predictive tools used to optimize the lyophilization cycle. However, determining the values of the many parameters involved was experimentally difficult [40]. Another approach to understanding the lyophilization process and optimizing the cycles is the use of computational fluid dynamics (CFD). Some studies have been recently conducted using CFD to understand the mass and heat transfer within various sections of the lyophilizer such as the drying chamber and the condenser [41, 42]. The parameters of different cycles in lyophilization might differ even for the same product when the operation takes place in a lab-, pilot- or production-scale lyophilizer [40]. This is due to the position of the vials, and change in the mass and heat transfer from the vials in the lyophilizer at different scales. Therefore, an optimized process obtained in the lab- or pilot-scale equipment often may not be transferred directly to production-scale equipment without modifications. Mathematical models based on the dominant parameters in the lyophilizer have been developed to facilitate the scale-up of the process [43–45]. It has been observed that at a higher product temperature there is a reduction in frost layer thickness that offers less resistance to vapor flow within the vial, resulting in greater primary drying efficiency. This observation led to the development of a scale-up theory that focuses on the primary drying [40, 46, 47].
While the use of mathematical models to optimize lyophilization has been helpful, there has also been a development of the in-situ process parameters monitoring technology that can help in modifying and improving these models. The technology is known as process analytical technology (PAT). It can be used to study the temperature and pressure data in the lyophilizer. Some of the PAT tools currently used in lyophilization are listed in Table 1.
Table 1.
PAT for monitoring freeze drying process.
| PAT method | Parameter measured | Integrated into the system | Reference |
|---|---|---|---|
| Thermocouple | Product temperature (Tp) | Yes | [31] |
| Resistance thermal detectors (RTD) | Product temperature (Tp) | Yes | [31] |
| Temperature remote interrogation system | Product temperature (Tp) | Yes | [48] |
| Manometric temperature measurement (MTM) | Sublimation interface temperature (Tice) and Product temperature (Tp) | Yes | [49–52] |
| Valveless monitoring system (VMS) | Product temperature (Tp) | Yes | [53] |
| Temperature measurement by sublimation rate (TMbySR) | Product temperature (Tp) | Yes | [54] |
| Capacitance Manometer | Chamber Pressure (Pc) | Yes | [55, 56] |
| Pirani Gauge | Chamber Pressure (Pc) | Yes | [55] |
| Lyotrack | Vapor saturation in chamber | Yes | [57] |
| Residual gas analyzer | Analysis of gas and vapor in the chamber | Yes | [55, 57–59] |
| Karl Fischer | Moisture content measurement | No | [60] |
| Thermogravimetric analysis (TGA/LOD) | Moisture content measurement | No | [60] |
| Computrac® Vapor Pro® XL | Moisture content measurement | No | [61] |
| Near IR spectroscopy (NIR) | Moisture content measurement | Yes | [62] |
| Raman Spectroscopy | Moisture content measurement | Yes | [62] |
| Tunable diode laser absorption spectroscopy (TDLAS) | Water vapor concentration | Yes | [63] |
2.1.3. Advancements and Future of Lyophilization
2.1.3.1. Microwave-assisted Freeze Drying
Several new technologies are being developed to improve lyophilization. Although currently being tested at a smaller lab-scale level, microwave-assisted freeze drying (MFD) is one such technology. In MFD, freeze drying is assisted with a microwave heat source to increase the drying rate while improving the product quality [64]. In a recent study, the application of MFD in a modified laboratory scale vacuum dryer reduced drying time by over 75%. The study also showed that samples produced by MFD have shown similar stability during accelerated stability studies when compared to conventional freeze drying for monoclonal antibody formulations used in the study [65]. However, more studies are needed before this technology can be used in an industrial scale lyophilizer.
2.1.3.2. Continuous Freeze drying
Continuous manufacturing is gaining popularity in the pharmaceutical industry due to advantages such as operation flexibility, improved manufacturing efficiency, real time quality monitoring and reduction in investment and operating costs [66]. In biopharmaceutical products, around 50% of the FDA and EMA approved drugs are lyophilized solid products despite the high operating costs and long processing time of the lyophilization process [67]. The current lyophilization process is a batch operation with several disadvantages, such as uneven heat transfer causing vial to vial inhomogeneity even within a single batch, and batch to batch product quality variability [68]. While continuous freeze drying in the food industry dates back to the 1960s, the transition of the technology to pharmaceutical products has not prevailed due to strict requirements in regards to the sterility, product quality and accurate dosage at a large-scale industrial production level. The principles behind the operation and current use of these continuous freeze dryers have been discussed in a recent review article by Pisano et al. [69].
Continuous freeze dryers that work on single dosage units appear favorable for biopharmaceutical products. As it is not straightforward for the transition from the existing batch freezer dryers to the continuous freeze dryers, there are only a limited number of concepts that have been developed over the years for continuous freeze drying of unit doses. Shell/spin freezing followed by vacuum drying is one example. During this process, the vial with the solution is rotated while freezing, causing a thin layer to form on the surface and then dry [68]. Becker’s concept [70], Broadwin’s process [71], and Oughton’s freeze dryer [72] all use the shell/spin freezing concept. A recent patent by Corver [73] looks at a new approach to shell/spin freezing followed by continuous drying. Another new continuous freeze drying concept has been proposed in which the vials are suspended over a track that moves through chambers with different temperatures and pressure conditions [74]. This method uses vacuum-induced surface freezing to produce controlled nucleation, thereby reducing vial to vial variability. Overall, continuous freeze drying on single dose units offers a promising way forward to reduce the processing time and the operating cost with potential capability to incorporate process analytic technology (PAT).
2.2. Spray Drying (SD)
Spray drying is a process in which the atomization of a solution, suspension or emulsion into a high-temperature gaseous medium produces dried powders [75]. The dairy [76] and the food [77, 78] industries were among the first to employ this technique at an industrial-scale. In the pharmaceutical industry, producing dried powders through spray drying offers advantages such as consistent powder quality, continuous and controllable processes with high drying efficiency, the availability of a wide range of dryers, and scale-up capability with high output.
2.2.1. Description of the Process
For a laboratory-scale spray dryer, there are four main components: drying chamber, atomizer/nozzle, aspirator, and particle collection cyclone [79]. In a typical spray drying process, a feed stock is pumped through the atomizer/nozzle to produce droplets with a desired size range. The droplet size can be altered with different types of atomizer/nozzles, solution concentration and feed rate of the liquid through the atomizer. The atomized droplets then enter the drying chamber where a high temperature gas stream evaporates the solvent and solid particles are formed. The temperature and the gas for the gas stream should be selected depending on solvent type, material degradation temperature, and glass transition temperature. The dried particles are then collected in a cyclone. The physical properties of the final product, such as particle size, density and morphology, can be altered by changing the process and formulation conditions.
2.2.2. Spray Dryer Setup
Atomization is a critical step in the spray drying process because the particle size, morphology, and residence time of the droplets/particles in the drying chamber are dependent on the atomization of feed solution. While the type of atomizer used has an effect on the atomization process, other factors such as shearing and inertial stresses, viscosity and size distribution of the droplets also influence the angle and velocity of the atomization [80]. There are several atomizer/nozzles available to be used in the spray drying process, including the rotary atomizer, single-fluid nozzle, multi-fluid nozzle, and ultrasonic nozzle. The rotary atomizer comes in two types: atomizer wheel and atomizer disc. The atomization occurs when the feed liquid passes through or across the rotating disc/wheel, where centrifugal forces of the disc break the feed liquid into droplets [81, 82]. The rotary atomizer is favored for viscous solutions because it can produce relatively uniform droplet size for viscous feed liquid. Rotary atomizers also have high feeding capacities, making them suitable for industrial-scale operations [80].
For the single-fluid nozzle, the fluid is pressurized and passed through an orifice, causing the fluid to break into droplets [82]. In a multi-fluid nozzle, the solution feed is atomized by a gas stream (generally air or inert gas) which breaks the feed into droplets at the nozzle tip. These multi-fluid nozzles usually produce fine droplet size (10–100 μm) and are widely used in the pharmaceutical industry [82]. The multi-fluid nozzles include 2-fluid nozzles, where the solution feed is atomized by the gas stream, and 3- or 4- fluid nozzles, [83] where the nozzle can atomize two different feed solutions either for microencapsulation [84] or multifunctional layered microparticles [85]. The ultrasonic nozzle generates droplets by vibrating at high frequency [86], which can produce a relatively larger droplet size range (5–1000 μm) [82]. Another advantage of the ultrasonic nozzle is the minimized risk of nozzle clogging, which can occur with the multi-fluid nozzles [82].
The drying gas flow pattern in the drying chamber of a spray dryer differs depending on design. The flow patterns that are commonly used in a spray dryer system are counter-current, co-current, and mixed flow. In a counter-current setting, the gas enters from the bottom of the chamber and leaves from the top, while the feed solution enters from the top and the dried particles settle at the bottom [87]. In this arrangement, the residence time of the droplets/particles in the drying chamber is more than in the other two arrangements, allowing for higher heat transfer and resulting in better drying. However, this arrangement is not preferred for materials that are heat sensitive. In a co-current setup, both the feed solution and drying gas enter from the top of the drying chamber and the particles are collected at the bottom using a cyclone. This drying gas flow arrangement is common for most pharmaceutical products, especially biopharmaceuticals. Mixed flow is a combination of counter-current and co-current flow patterns. In this setup, the atomization of the feed takes place in the central part of the drying chamber with the atomized feed solution sprayed upwards or downwards based on the thermal sensitivity of the material, while the drying gas enters from the top of the drying chamber [88, 89].
2.2.3. Spray Drying of Biopharmaceutics
Many biopharmaceutical products such as protein- and peptide-based formulations show less stability in the liquid state than in the solid state at the ambient temperature [90]. Lyophilization has been used to produce solid state biopharmaceuticals for a long time. With the development of novel drug delivery systems such as long acting microspheres [91], powders for transdermal delivery [92], and for pulmonary delivery [93], lyophilization can no longer satisfy the product requirements that are necessary for these new delivery methods; thus, the demand for alternative drying techniques increases. Spray drying provides significant control on powder properties and is feasible to scale-up. However, the use of relatively high temperatures for solvent evaporation is a limitation when it comes to drying thermo-sensitive biopharmaceutical products. Mezhericher et al. [94] gave a detailed description of the drying process for a droplet in a spray dryer. While spray drying eliminates some of the stresses in lyophilization, such as ice formation, freeze concentration of solute and pH imbalances, it introduces air-liquid interface and thermal stress. These stresses and their impacts on the stability of biological solids will be discussed in detail in the following section.
Recent work has shown that the influence of drying methods on physical stability (or protein aggregation) of protein solids is highly correlated with protein type and excipient choice [95, 96]. The use of excipients that can preserve hydrogen bonds during the drying process has been extensively studied. Excipients that are expected to satisfy the hydrogen bonding requirements to preserve the secondary structure of the proteins include sugars such as sucrose [97], trehalose [98], and polyols (sorbitol) [99]. Other excipients such as surfactants [100], polymers [101], antioxidants [102], and amino acids [103] also improve the stability of the proteins during spray drying. However, one should be cautious about the potential negative effects of such excipients on the stability of protein solids, especially for those which crystallize during drying. Mannitol is a commonly used excipient for lyophilization formulations due to its ability to form a crystalline matrix that prevents macroscopic collapse of the lyophilized cakes. Use of mannitol by itself as an excipient has been shown to cause phase separation, resulting in poor protein stability [95].
Powder properties are affected by type of excipient, protein/excipient ratio, total solid concentration in the feed solution, inlet/outlet temperature, solution feed rate, atomizer type, and atomizing pressure. Yield, physical and chemical stability, morphology, therapeutic efficiency, and powder flow characteristics are some of the parameters that need to be optimized for spray drying [79]. Often these parameters are inter-related, and optimization of these parameters becomes complicated. Models based on single droplet drying have been used in tandem with experimental setups to understand the heat and mass transfer within a droplet [104, 105]. DoE, CFD, heat and mass transfer modelling have also been used by various research groups to understand the impact of formulation and process in both the lab- and industrial-scale spray dryers [106–108].
In the pharmaceutical industry, spray drying has been used to manufacture biological solids for inhalation and topical products. Insulin, owing to its high thermal stability and fast absorption, is one biopharmaceutical product that has been developed for pulmonary drug delivery. Exubera® spray-dried insulin received FDA approval for pulmonary delivery. The Exubera® formulation consisted of recombinant insulin (60%), mannitol, sodium citrate, glycine and small amounts of sodium hydroxide (for pH adjustment) [109]. While the product was considered to be technically successful in terms of the drug delivery, it was withdrawn from the market within a year due to poor sales [110]. Another pharmaceutical product, Raplixa®, is the first FDA-approved sterile spray drying solid protein product [111].
2.3. Spray Freeze Drying (SFD)
Spray freeze drying is a relatively new drying technique [112] used to manufacture thermo-sensitive food products like whey protein, coffee, maltodextrin, and milk powder [113]. Although spray freeze drying has not been widely adopted to manufacture commercial pharmaceutical products, there is increasing interest in applying spray freeze drying for manufacturing thermo-sensitive biopharmaceutical solids.
2.3.1. Description of Process
In SFD, the feed solution is sprayed into a cold vapor phase or a cryogenic liquid. The atomized droplets freeze either during their movement through the cold vapor phase or upon contact with the cryogenic liquid. The frozen droplets are dried under the same principle as freeze drying. SFD offers the advantage of producing particles for products that are thermo-labile. Various setups have been explored on how to handle the frozen droplets [93, 114–116]. Atmospheric freezing, spray freezing into liquid (SFL) and spray freezing with compressed carbon dioxide are some examples of this process [117–119].
2.3.2. Spray Freeze Drying of Biopharmaceutics
Factors such as the chemical composition, atomization rate, feed rate, and temperature of the cryogenic fluid play crucial roles in ensuring the stability and controlling the density and particle size distribution of the particles [117]. Moreover, the super-cooling phenomena due to the use of a cryogenic liquid may mitigate ice crystallization effects and pH shifts that can be observed in freeze drying. The production of porous spherical particles during SFD increases the surface area and thereby reduces the time for sublimation and secondary drying. The spherical shape of SFD particles results in relatively satisfactory flowability. The porous structure generally leads to low particle density, which contributes to better aerosolization behavior for inhalation products [120].
In a study of the effects of atomizing conditions and variability in formulations containing bovine serum albumin (BSA) on the particle size and stability, particle size was found to be inversely related to the specific surface area and the amounts of BSA aggregates [114, 115]. In a study of spray-dried and spray freeze dried anti-IgE Mab and rhDNase formulations, it was shown that the spray freeze dried powders had better aerosol performance than the spray-dried powders, likely owing to the porous structure of the particles [93]. Other studies on salmon calcitonin [121] and parathyroid hormone [122] also showed that SFD resulted in products with good flow properties. In a recent study conducted by our lab group, we found that the spray freeze dried formulations of lysozyme and myoglobin showed similar stability results to freeze dried formulations in the formulations containing non-crystallizing cryoprotectants (sucrose) as excipient [123]. In another study, insulin with/without tylopaxol and lactose was spray freeze dried [124]. Irrespective of the presence of lyoprotectants, spray freeze dried insulin showed minor to no degradation, suggesting that spray freeze drying is capable of delivering stable solid product [124]. Spray freeze dried influenza vaccines showed better stability compared to the liquid formulations [125]. A study evaluating enzyme activity of trypsinogen after SFD found an [126] approximately 15% loss from initial activity, with an aggregation of 1.4%.
Spray freeze drying is promising in terms of particle morphology control and flow property optimization, making it a favorable drying method for pulmonary delivery. It offers advantages such as rapid freezing and short drying time when compared to freeze drying for biopharmaceutical production. Nevertheless, more research is warranted to fully understand the impact of the process on the physical and chemical stability as well as the therapeutic efficacy of the products.
2.4. Other Emerging Drying Techniques
In addition to the drying techniques mentioned above, other emerging drying techniques for biopharmaceutical solids include super critical fluid drying (SCFD), foam drying and electrostatic spray drying.
In SCFD, materials like carbon dioxide, ethylene or methanol are used above their critical temperature and pressure in order to facilitate dehydration. Compared to ethylene and methanol, carbon dioxide is a non-toxic and non-inflammable material with a critical temperature of approximately 31°C and critical pressure of approximately 73 bar. Two approaches have been proposed to use the SCFD process for biopharmaceutical products [127]. The first is to use a super critical fluid (SCF) as an anti-solvent for water extraction, leading proteins to be concentrated and precipitated, followed by spray freezing and drying to remove the remaining water before obtaining the final product. In the second approach, the SCF dissolved at high pressure is used as a propellant to atomize the feed solution in a two-fluid nozzle. Results have shown that SCFD is capable of generating particles with adjustable sizes and uniform spherical features with acceptable flow properties [128–130]. Additional research is necessary to understand the long-term storage stability and therapeutic efficacy of SCFD-produced products.
Foam drying is another relatively new process for producing biopharmaceutical solids, in which the solution is converted to dried foam [131]. The solution is boiled or foamed under reduced vapor pressure, followed by water evaporation that leads to a solidified foam structure [132]. Foam drying offers the advantage of drying at near-ambient temperatures without ice formation. In a study with Ty21a vaccine, foam drying demonstrated a stability of 42 weeks at 25 °C [133]; while a commercial freeze dried Ty21a vaccine, Vivotif™, showed stability of only 2 weeks at the same temperature [134]. In another study, foam drying of an IgG1 mAb formulation with varying levels of sucrose showed better storage stability than freeze drying and spray drying [135]. The increased storage stability is ascribed to creating products with lower specific surface area and surface accumulation of the protein, [135] as lower surface area is related to lower monomer loss in protein formulations [114]. Although foam drying appears to exhibit better stability, it presents several challenges such as surface tension stresses leading to cavitation of the dried product, reduced water desorption rate resulting in longer drying time, and the risk of freezing due to evaporative cooling. The process usually takes days to complete. The drying kinetics are not fully understood and research of this topic is required to optimize the drying cycle and improve product stability. Scale-up of this process for manufacturing warrants further investigations.
Recently, electrostatic spray drying has been developed as an alternative to traditional spray drying. In this process, an electric charge is applied to the feed solution before spray drying. Within these charged droplets, solvent (usually water) possesses a higher proportion of electrons and attains a greater charge density due to its high-polar nature, while the solute molecules that are generally less polar in nature than the water have fewer electrons. Such an effect leads to a solute-rich core and a solvent-rich shell within the droplet, which improves drying efficiency and facilitates drying at low temperature [136]. Spray dryers with these technologies are commercially available with varied scales from bench-top lab equipment to full-scale for industrial manufacturing [137].
The concept of electrostatic spray drying has been previously evaluated to produce nanoparticles [138], nanosuspensions [137], and microspheres for encapsulation [139]. Commercially available electrostatic spray dryers have been used to study the process of encapsulation for modified food starch, vitamin C, strawberry flavor and sunflower oil [140]. An emulsion containing non-polar active ingredient, polar solvent and carrier material are spray dried using the electrostatic spray dryer. The non-polar active ingredient has fewer electrons and forms the core, while the carrier and solvent pick up more electrons and form the shell through droplet formation, enabling encapsulation of the active ingredient inside the carrier. Since it is relatively new, publications on its use for biologics are rare. More research is warranted to provide a clearer understanding on the mechanisms of how charging affects drying efficiency of pharmaceutical related materials.
Although traditional lyophilization is still the mainstay drying technique for the production of biopharmaceutical solid products for now, alternative technologies have been employed to circumvent key shortcomings of lyophilization. Spray drying appears to be a viable alternative to lyophilization in producing biopharmaceutical solids, particularly for developing inhalation products. FDA-approved sterile spray-dried biologic products are already on the market [109], which eases some concerns on the sterility of spray-dried products. In addition, the emergence of new drying technologies that are specifically designed for biological products will facilitate the development of commercial solid biopharmaceutical products in the future; while there is an urgent demand in research to obtain the fundamental understanding on how the process and formulation affect product quality.
A simplified workflow illustration of lyophilization, spray drying and spray freeze drying is presented in Figure 1.
Figure 1.
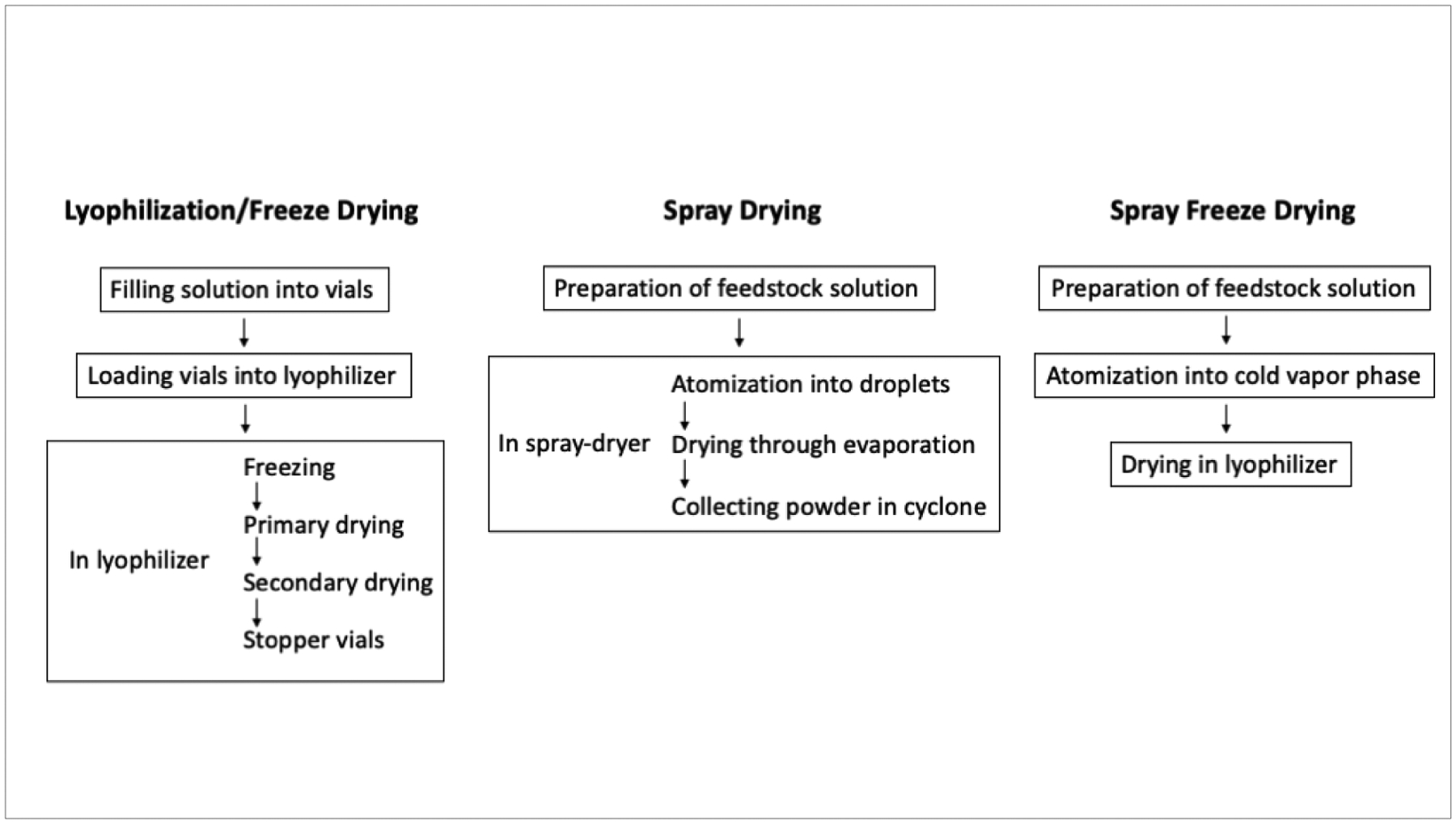
Illustration of lyophilization, spray drying and spray freeze drying processes.
3. STRESSES IN DRYING PROCESS
Proteins undergo a complicated process from the raw material to the end user, including synthesis, purification, formulation, storage and handling [141]. The time needed for the formulation step is largely impacted by the desired dosage form, with different challenges associated with formulations in aqueous solution and solid state [142]. Lyophilization, or freeze drying, is the most common approach to produce dried biologic product [143]. Recently, more drying methods are being explored for biologics, such as spray drying, spray freeze drying, and foam drying [144].
With the rapid growth of protein products in the pharmaceutical industry, understanding how the processing stresses affect protein stability and function is vital for formulation design. The native structure of protein is maintained by different interactions including hydrogen bonds, van der Waals forces, electrostatic interactions, and hydrophobic interactions [145]. These interactions are critical for protein functions such as binding and solubility. It has been a challenge in the development of protein products to protect the native conformation of protein from the processing stresses [142].
The stresses induced by drying processes could alter the native conformation of proteins. The interactions among proteins, or between the protein and its surrounding environment, can be affected by these stresses, resulting in an energetically unfavorable state which leads to further structure disruption [145]. Processing stresses can disrupt the secondary, tertiary, or quaternary protein structure [146], and thus cause protein denaturation.
The manufacturing process exposes proteins to various types of stresses simultaneously that can destabilize the molecules. The final effect to the biologic product is a combination of the exposure time and strength, as well as the selection of the formulation composition [127]. This section presents common stresses from drying methods that induce physical denaturation of the protein structure as well as formulation strategies for preventing protein unfolding and aggregation.
3.1. Temperature
One of the most common causes for protein denaturation is the exposure to extreme temperature changes. The thermodynamics of a protein unfolding process in a two-state model can be described by an equilibrium between a folded native state (N) and an unfolded, denatured state (D) [145]. The equilibrium constant (K) of the equilibrium is represented by the following equilibrium equation:
| (3) |
with K representing the equilibrium constant; [D] and [N] representing the concentrations of denatured and native proteins respectively. Furthermore, the Gibbs free energy between the unfolded and native states, ΔGu can be given by the equation:
| (4) |
where R is the ideal gas constant, T is the temperature, ΔHu is the enthalpy change of unfolding, and ΔSu is the entropy change of unfolding.
The free energy for the globular protein in its folded state is 5–20 kcal/mol higher than its unfolded state [147]. The activation energy needed for unfolding to occur is within the range of 25–150 kcal/mol, which is typically weaker than covalent or ionic bonds [127]. The free energy of protein varies with exposure to temperature changes, the degree of which corresponds with the non-covalent interactions stabilizing the protein structure. The induction of unfolding can be highly temperature-dependent, and is accompanied by a large increase in the heat capacity (given by ΔCp) [148]. Since the changes in enthalpy and entropy of unfolding are both temperature-dependent, these properties can be described as:
| (5) |
| (6) |
where Tm is the midpoint of the thermal unfolding curve; ΔHm and ΔSm are the values of the changes in the enthalpy and entropy of unfolding, respectively, at Tm. These parameters can be inserted into Equation (4) to give:
| (7) |
Furthermore, a modified Gibbs-Helmholtz equation can be used to describe the free energy of unfolding by its dependence on temperature [149]. This theory is derived from the assumption that as the free energy of unfolding approaches zero, the midpoint of the change in entropy approaches ΔHm/Tm, which can be given by the following equation:
| (8) |
Following equation (8), the free energy of unfolding can be plotted as a function of temperature (Figure 2). As shown, the kinetics for protein unfolding is parabolic, with the free energy of unfolding reaching zero at around −30°C and 80°C. When the free energy is negative, protein unfolding is energetically favorable, which can lead to aggregation. The two-state model is not applicable for all the protein folding states. Proteins in intermediate states with partially unfolded structures have been identified [150–152]. These intermediates could act as the initial states for denaturation and aggregation before the environment temperature reaches the calculated unfolding point.
Figure 2.
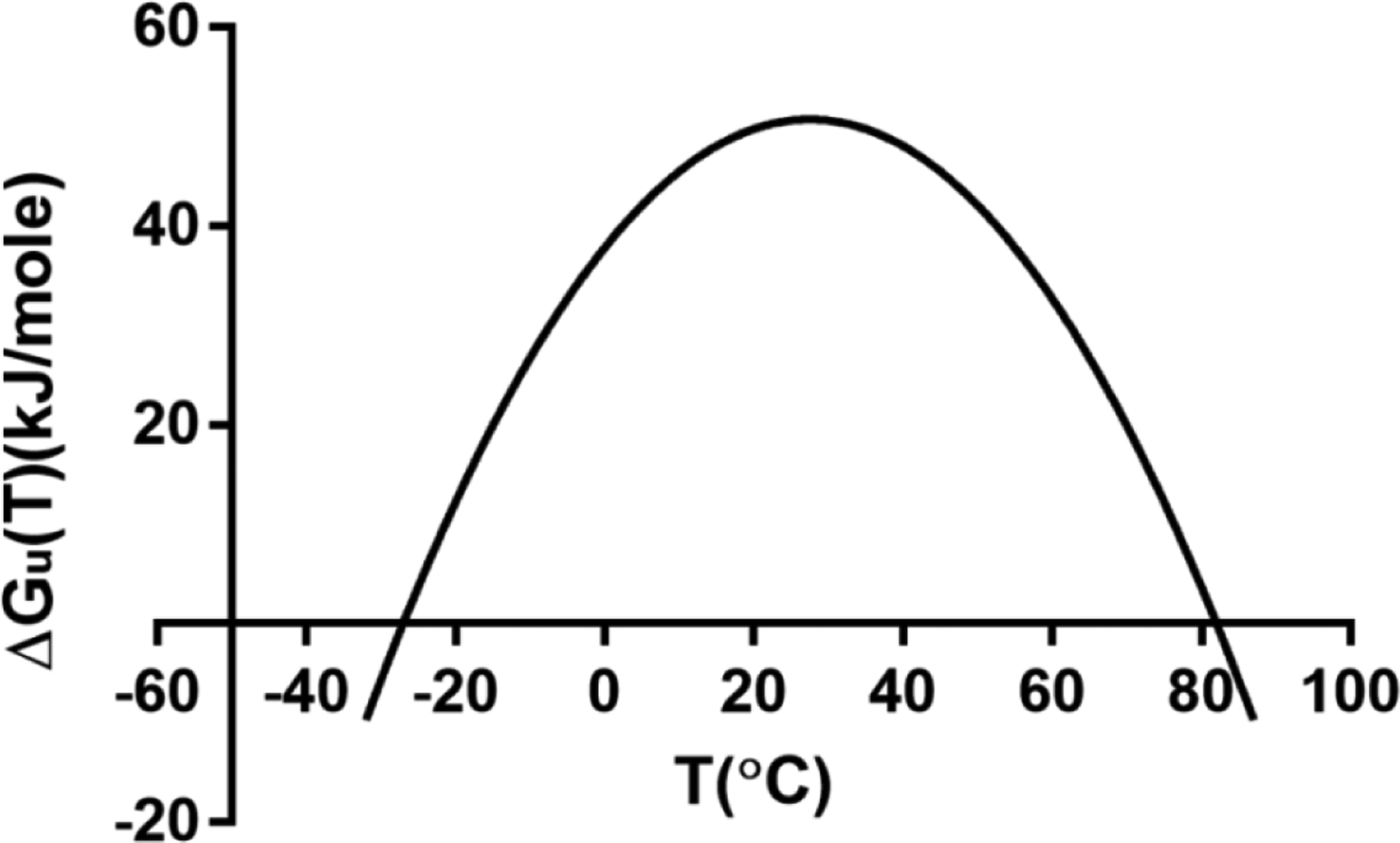
A protein stability curve for the Gibb’s free energy of unfolding as a function of temperature, given in Equation (8). Repurposed from [148].
3.1.1. High Temperature Denaturation
High temperatures during the drying process pose a significant risk to the stability of biologics, including proteins [129]. Drying methods such as spray drying [144] and convective oven drying [153] utilize high temperature. In spray drying, the solution formulation is atomized by air into droplets. After brief exposure to a hot drying gas, water is removed and dry solid particles form. In a convective oven dryer, solutions or dry solids are exposed to elevated temperatures for a set period of time to reach a desired moisture content. The temperature settings used for these drying methods are significantly higher than the denaturation temperature, or melting point, of most proteins, which usually begin to unfold in a range between 40–80°C [154].
As indicated in Figure 1, the unfolding process is energetically favorable at temperatures above 80°C. The kinetic energy of the non-covalent bonds maintaining the protein structure increases with an elevated temperature. The increasing heat from the environment leads to the disruption of intramolecular hydrogen bonding, resulting in protein denaturation [145]. Various intermediates can form through the unfolding process upon drying, which is associated with the drying rate as well as the formulation composition [150]. For example, β-lactoglobulin showed decreased α-helical and β-sheet content and increased disordered structure above 50°C. Aggregation was observed above 60°C with an increase in intermolecular β-sheet content [155]. For thermally-sensitive proteins, irreversible aggregates tend to form quickly upon unfolding due to high temperature.
In spray-dried protein solids, thermal denaturation may not be observed even at the inlet temperature above 100°C because of self-cooling in droplets induced by solvent evaporation [141]. The droplet temperature typically does not exceed the wet bulb temperature, which is the lowest temperature present on the droplet surface through solvent evaporation. The wet bulb temperature increases as the drying process continues, while the temperature inside the droplet maintains at 10–15°C lower [156]. Once the critical concentration on the droplet surface is achieved, secondary drying occurs until it reaches the dry bulb temperature, which is close to the outlet temperature of the spray dryer [141].
The extent of exposure time and high temperature is critical to the level of denaturation observed on the protein molecules. For spray drying, a lower outlet temperature is preferred. It has been well-documented that outlet temperature has a greater impact on protein denaturation and aggregation than inlet temperature [157–159]. The protein primary structure could affect the stability against the effect of high temperature. It has been reported that molecules containing higher content of hydrophobic amino acid residues tend to have an increased stability against thermal stress than those with more hydrophilic residues [141]. Additionally, selecting excipients capable of forming strong hydrogen bonding with the protein also provides protection against denaturation induced by high temperature and improves the overall protein stability [99].
3.1.2. Cold Denaturation
Exposure to low temperatures could lead to protein denaturation with the free energy of unfolding, comparable to that induced by high temperature [160]. Cold denaturation can be induced by the freezing step of lyophilization, or through product storage at temperatures below 0°C. Proteins unfold at low temperatures through different mechanisms than at high temperatures. In an study of β-lactoglobulin, it has been reported that the intermediates forming from thermal stress varied at different temperatures, which indicates that the unfolding mechanism at lower temperature differs from that at a higher temperature [151]. Though not fully understood, it is proposed that unfolding in cold denaturation results from changes in the interactions between water and the nonpolar residues on the protein [161–163]. As the temperature drops below a certain point, it is entropically favorable for water to interact with the hydrophobic amino acids. Consequently, the hydrophobic residues are hydrated, weakening the hydrophobic interactions stabilizing globular protein structure. Such changes in hydrophobicity can lead to the collapse of tertiary structure. In a study of apomyoglobin, protein stability was affected by its hydrophobic core at lower temperatures. Below the denaturation temperature, the helices in the hydrophobic core of apomyoglobin became detached, followed by protein unfolding [152].
The effects of cold denaturation are not fully understood because it is difficult to analyze it at below 0°C in the partially-frozen solution. Even without protein denaturants such as urea, cold denaturation can be found below the freezing point of water [164]. However, the effects of cold denaturation on proteins can be reversible [165]. Structure changes caused by low temperature denaturation can be recovered by thawing the frozen solutions at a higher temperature or reconstituting the lyophilized solids back to aqueous solution. Dried protein solids can be stored at low temperature with the removal of water. With the use of a cryoprotectant such as sucrose or trehalose, protein stability can be further improved to protect against the effect of low temperature in the process [166].
3.2. Surface-induced Denaturation
During processing of protein formulations, it is inevitable that solutions will be exposed to some type of foreign interface, whether by filtration, filling, or drying methods [167]. Exposure to these interfaces can result in interactions of the protein by adsorption, which can lead to shearing conditions and unfolding in relation to its environment. This is due to the amphipathic nature of proteins, which leads to high concentrations orienting at the surface because of their polar and nonpolar side chains [168, 169]. There are two hypotheses regarding the underlying principles of surface-related stresses that lead to unfolding: (1) surface-induced denaturation due to surface adsorption, aggregation of intermediate states, and/or recycling of unfolded proteins into solution; (2) shear-induced denaturation by structural changes of proteins in bulk solution by shear force, aggregation of intermediate states, or solid surface adsorption [127]. In recent studies, it has been found that shear alone is not significant enough to induce protein structural changes, even at conditions higher than those that would be found in traditional manufacturing processes [170]. For unfolding, a force of 20–150 pN is generally required for proteins such as a monoclonal antibody. Shear alone is only capable of generating a force of 0.06 pN, far lower than what is required for protein unfolding [171]. This indicates that another foreign interface in tandem with shear may be critical in generating stress high enough to cause denaturation. Three types of surface-induced stresses found in manufacturing are solid-liquid, air-liquid, and liquid-liquid interfacial stresses.
3.2.1. Solid-liquid Interfacial Stress
During all phases of processing, protein formulations will be exposed to solid surfaces, either in a vial or storage device used for the drug product [172], in membranes during dialysis [173], or the equipment used in the manufacturing process [174]. Whether proteins adsorb to a surface depends on the surface interacting with the protein, and the state of the protein’s bonding interactions, in particular its electrostatic and hydrophobic interactions [170]. If the protein favorably interacts with the solid surface it is being processed in, the protein can adsorb onto the surface. During this adsorption the protein may be folded or partially unfolded, with a higher probability of adsorption for a partially unfolded protein, as the exposure of the hydrophobic residues increases the number of possible interactions with the surface [169, 175]. The initially adsorbed proteins act as nuclei on the interface, whereby additional molecules passing through the solid can attach to these sites, increasing the size of the aggregates.
The stability of the protein structure plays an important role. There are two classes of proteins, based on their stability, that determine which types of surfaces will lead to adsorption: “hard” and “soft” proteins [169]. Hard proteins have a high internal stability, or strong hydrophobic interactions, which help to maintain the protein structure against adsorption. For these types of molecules, adsorption at the solid interface is unlikely on a hydrophilic surface unless there is a strong electrostatic interaction disrupting the protein structure, or unless there is a strong hydrophobic surface. For example, lysozyme has a higher amount of aggregation due to adsorption that occurs on a more hydrophobic surface, polytetrafluoroethylene, than on glass [176]. Soft proteins have a low internal stability, or weaker hydrophobic interactions, which allows for adsorption on any surface with an increase in conformational entropy by unfolding and attaching to a solid interface. For example, a monoclonal antibody has been found to unfold during adsorption to stainless steel [177].
The ionic strength in the solution can affect solid-liquid interfacial stress in the formulation. Increasing the ionic strength can weaken the repulsion interactions found between the protein and solid during processing, leading to aggregation [178]. In addition, it has been found that catalase can bind to hydroxyapatite when protein and surface are both negatively charged due to the high ionic strength of the buffering agent [179].
Interfacial stress caused by proteins or solid breaking off into the solution leads to additional aggregation [168]. Denatured proteins may desorb from the surface and move back into solution, serving as additional nuclei for aggregation or unfolding. The manufacturing equipment can introduce particulates into solution by leaching or shedding of sub-visible particles through regular usage [180]. These particulates can further promote aggregation and denaturation.
Interfacial stress can be limited by choosing materials unfavorable to adsorption for hard proteins, or by controlling the solution pH and ionic strength to prevent electrostatic attractions. For soft proteins, sugars and surfactants can prevent aggregation by reducing the amount of protein interacting with the solid surface, such as preventing interactions with the ice interface during lyophilization [181, 182]. Reducing the flow rate of the material passing through a pump or filtration unit can limit the combined effects of shear with solid-liquid interfacial stress and reduce adsorption[170].
3.2.2. Air-liquid Interfacial Stress
The air-liquid interface is present in nearly all types of processing methods. For example, air atomizes solution into small droplets to control particle size during spray drying and protein solution is exposed to air-liquid interface [183, 184]. Stress at the air-liquid interface can also be induced upon completion of unit operations such as filling, which introduces air headspace [185] and leads to agitation during shipping and handling [186].
When exposed to the air-liquid interface, proteins tend to adsorb at the surface due to their amphiphilic nature. A higher concentration of protein can be found at the interface than in solution [184]. As the protein moves from solution to the surface, unfolding can expose its hydrophobic core to equally align at the air-liquid interface. More denaturation and protein damage can be found at the surface, which increases the tendency for aggregation as protein crowding occurs [79]. Additionally, the increase in protein concentration at the air-liquid interface leads to more interactions among protein molecules with a decrease in free volume, causing additional conformational dissociation and alteration of the tertiary structure. An increase in atomization air flow rate in spray-freeze drying of bovine serum albumin has been found to decrease particle size. However, higher amounts of aggregates were observed with increased protein interactions at the air-liquid interface [114].
Proteins exposed at the particle surface tend to have greater heterogeneity, as suggested by a study of spray-dried bovine serum albumin on its heterogeneity and particle surface composition (see Figure 3) [96]. The effect of the stress from air-liquid interface can be minimized by reducing the air exposure time during the drying process, or by decreasing exposure area by reducing vial headspace [167]. Surfactants can be used to alleviate the stress effect from air-liquid interface in manufacturing [115, 187, 188]. Surfactants are amphipathic, which compete with proteins for adsorption at the surface, decreasing the presence of proteins at the air-liquid interface.
Figure 3.
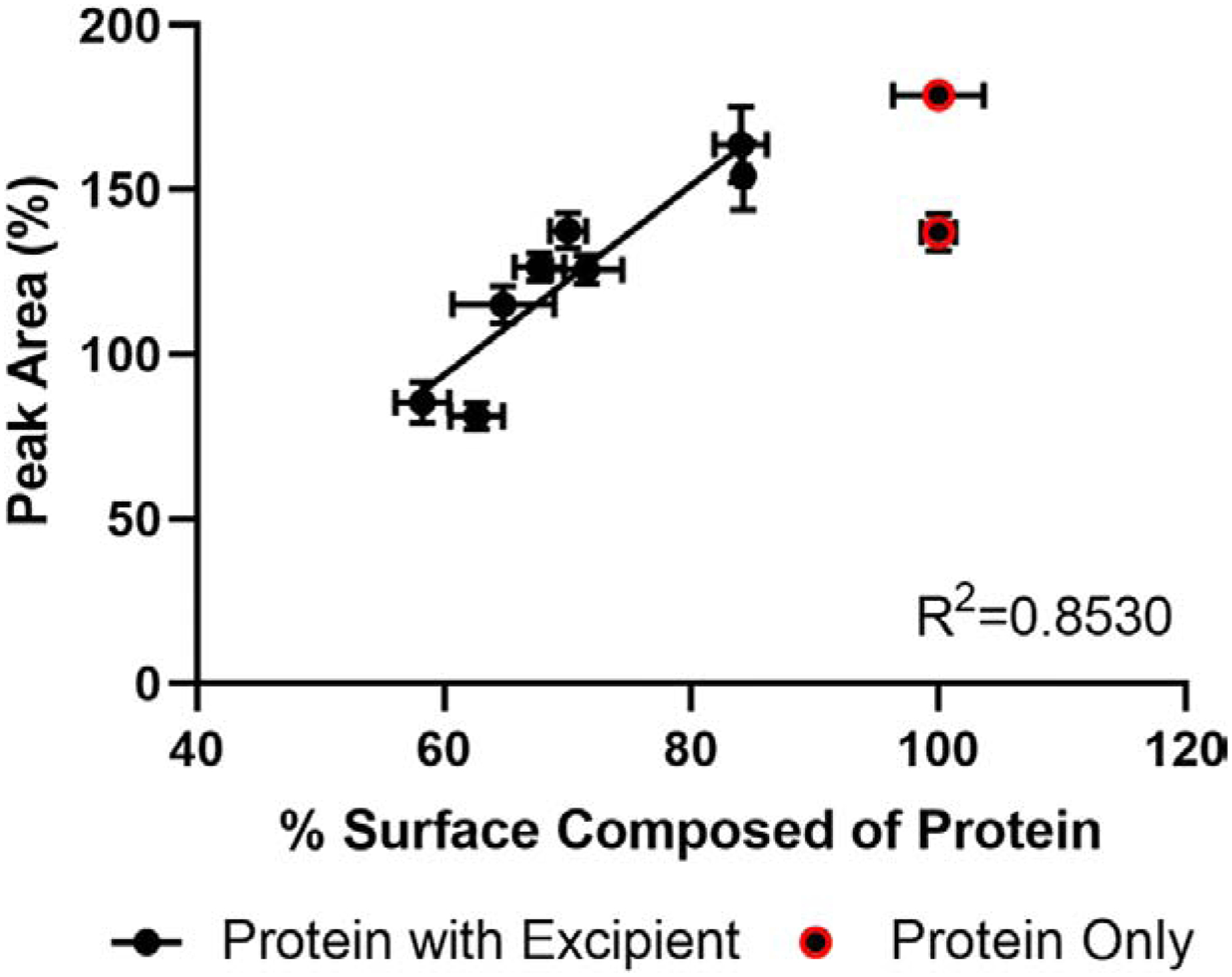
Correlation of percentage surface composition of protein to peak area of the deconvoluted mass envelope relative to an experimentally fully-deuterated sample (Reprinted from [96, 189] with permission from Springer Nature).
3.2.3. Liquid-liquid Interfacial Stress
Proteins can be adsorbed to the interface in a two-fluid system and denatured. Such stress can be found in purification [190], encapsulation with emulsion-based methods [191], or through contamination by foreign liquids [192]. Protein molecules orient themselves at the liquid-liquid interface to reduce surface tension. Proteins will rearrange their native structures and expose hydrophilic and hydrophobic residues in order to reduce free energy at the surface and stabilize the solution [193].
Adsorption at the liquid-liquid interface depends on the properties of the two fluids, particularly when there are significant differences in solubility [194]. In several enzymes, it has been shown that the difference in solvent exposure area and interfacial tension in an aqueous-organic solvent system affects the extent of denaturation [195]. Compared to the solid-liquid and air-liquid interface, higher molecular mobility can be found at the liquid-liquid interface [193]. It promotes aggregation formation with the recycling of denatured proteins from the interface back into the bulk solution.
Excipients can reduce denaturation induced by liquid-liquid interface. Surfactant has been shown to prevent the adsorption of proteins from liquid-liquid interface [196]. Solvents can be carefully selected to prevent unfolding and structure loss. For example, the use of polyethylene glycol-dextran solutions should be avoided in lyophilization because it introduces phase separation in freezing [197]. Moreover, avoiding contamination from silicon oil during manufacturing prevents the formation of liquid-liquid interface and preserves protein structure [192].
3.3. Dehydration/Moisture Content
Proteins in aqueous solutions are hydrated with a fluid hydration shell [198]. The surface area of protein in contact with the hydration shell decreases as water is removed by high temperature drying or sublimation. Drying reduces the number of water molecules interacting with the protein. At the same time, protein molecules tend to be exposed to excipients at a higher extent as the solution becomes more concentrated, and thereby intermolecular interactions between protein and excipient that can stabilize the drug product are increased [145, 199].
Nevertheless, the drying process itself can be detrimental to protein structure. With moisture removal, the free energy of the protein changes, which causes the protein to be less stable. To compensate for the energy difference, protein molecules can rearrange themselves, or aggregate by forming intermolecular interactions with other protein molecules [198]. The increased interactions result in destabilization of protein structure, especially when there is a lack of stabilizing excipients to replace the hydrogen bonding formed by water [200]. Typically, dried protein products exhibit reasonable long-term storage stability when stored appropriately [201]. Increased moisture uptake introduces phase separation of different components in the solid [202]. Extra moisture exposure prior to reconstitution of solids could induce precipitation and aggregation of solid components and decrease protein efficacy [203]. The effect of residue moisture on protein stability needs to be further explored. In a study of a lyophilized monoclonal antibody formulation, the physical stability was compromised in the solid with higher moisture content as Tg decreased and aggregation formation increased. Moderate increase in moisture level of lyophilized solids might have a negligible impact on the chemical stability of proteins based on aspartate isomerization rate [204].
Disaccharide excipients such as sucrose and trehalose are commonly used to stabilize proteins in solid state [205]. The stabilizing effect can be explained by their ability to serve as hydrogen bond replacements for water [205]. In solution, excipients are preferentially excluded from the protein surface. Protein molecules can form strong hydrogen bonds with excipients upon depletion of the hydration shell [206, 207]. The protein structure can be stabilized as unfolding is avoided to reach a thermodynamically favorable state. Ideally, both excipients and protein remain amorphous in a stable solid state [158]. With excess excipient concentration, increased interactions among excipient molecules lead to nucleation or small crystals. These crystals may not have any deleterious effects upon formation in minute amounts [208, 209]. However, they can act as nuclei for other excipient molecules, which can lead to phase separation and could remove available hydrogen bonds for proteins.
3.4. pH
Solution pH is important in determining the interactions of formulation components. pH shifts through manufacturing steps. pH dictates the charge state of exposed amino acids of protein molecules and changes their electrostatic interactions by altering the total charge of the molecule. It affects local and global protein structures as well as solubility [154, 210, 211].
At the isoelectric point (pI), proteins have a net neutral charge and the lowest solubility [210]. Decreasing (more acidic) or increasing (more basic) pH can alter the net charge and increase protein solubility, which can destabilize the protein structure. At higher charged state, there are more electrostatic interactions with nonspecific repulsions among amino acid residues [212]. This causes amino acid sidechains to move apart, which leads to protein unfolding to maintain a state with lower free energy. However, increased electrostatic charges can introduce the formation of salt bridges. Ion pairing between amino acid sidechains with opposite charges forms a strong electrostatic bond stabilizing protein conformation [213].
Highly charged protein molecules develop repulsive interactions among themselves, which stabilize proteins and make aggregation energetically unfavorable in a colloidal solution. However, at pH closer to pI, proteins possess an anisotropic distribution of charges with both negative and positive charges. Dipoles exist and favorably interact with other protein molecules, leading to the formation of aggregates [213].
Solution pH affects protein structures. As mentioned previously, the accuracy of the two-state model of folded and unfolded protein is limited as intermediate states are not included. At extreme pH conditions, proteins can undergo conformational changes where they exist in a partially folded state called the “molten globule” state [214]. The protein molecules maintain their secondary structure similar to their natively folded state, while the overall shape tends to be slightly larger. The tertiary structure cannot be detected. It is assumed that the hydrophobic core of proteins is exposed to the solvent. The molten globule state is useful in understanding the protein folding mechanism. However, it is undesirable in a formulation. Proteins such as staphylococcal nuclease, α-lactalbumin, and β-lactamase have demonstrated pH dependence on the change of secondary and tertiary structure [215–217].
Buffer component and solution pH is critical to ensure reasonable solubility and minimize repulsive interactions. Ideally, solution pH should be far from the pI [210]. At low concentrations of salt, it reduces electrostatic interactions. When there is an excess of salt, it can bind to the protein surface and disrupt protein native conformation [218]. Higher concentration of salt is capable of salting out of the protein from solution. In addition, the processing steps such as freezing in lyophilization affects the pH and ionization strength of the solution [219]. pH shift was observed by freezing phosphate buffered saline (PBS) solution with the crystallization of disodium hydrogen phosphate dodecahydrate (Na2HPO4•12H2O) [220]. Such change has contributed to protein aggregation formation in the formulations of bovine serum albumin and β-galactosiase [221]. In addition, it has been reported recently that protonation and tautomeric states of histidine varied in solids lyophilized from solutions ranging from pH 4.5 to 11.0 as identified by 13C and 15N NMR spectra. The change of histidine was used as a probe to investigate the microenvironmental acidity change in an HPV vaccine [222].
3.5. Freezing
Drying techniques such as lyophilization and spray-freeze drying involve freezing of solutions. Freezing involves a combination of different stresses destabilizing proteins, including cold denaturation, stress at interfaces, phase separation, and changes induced by concentrated components [26, 143, 181, 209]. Freezing a solution follows the steps of super-cooling, ice nucleation, and crystal growth [223]. Super-cooling is a state when the solution maintains liquid below the ice formation temperature. Super-cooled solution solidifies quickly upon the release of latent heat [224]. As super-cooling leads to formation of ice crystals, the proteins and excipients are excluded from the sites of ice nucleation as the solubility decreases, leading to increased interaction among components in the matrix as they are concentrated. The ice nuclei begin to increase in size as the lower temperature is maintained until the water in the system has been crystallized.
Cooling rate dictates the number of ice nuclei and the size of the crystals, which is affected by the formulation composition [225]. A faster cooling rate results in greater nuclei formation and smaller crystals. A fast-freezing step used for lactate dehydrogenase demonstrated larger extent of structural damage and lower activity recovery than for a slow-freezing rate [225]. As smaller ice crystals have a larger surface area for proteins to be adsorbed, formulations with smaller ice crystals tends to lose their secondary and tertiary structures.
Differences in solubility of formulation composition causes phase separation in freezing, and certain components crystallize instead of maintaining in the amorphous matrix [226]. Crystallization of stabilizing excipients reduces stabilizing interactions with proteins, exposing them to cold denaturation stresses and increasing intermolecular interactions. The ionization strength and pH of the solution change in freezing. When the frozen solution loses moisture through drying, hydrophobic interactions decrease and the ionization strength of the solution as well as the charge on the protein increases. Additionally, buffer components can crystallize at different temperatures, leading to significant pH changes. For example, in a phosphate buffer solution, the pH with dibasic sodium salt solution will drop from 7 to 4 between −0.5°C to −9.9°C, whereas only a minor pH shift can be observed in potassium salt solution [219, 227].
Controlling the nuclei size can minimize the negative effects from freezing on protein structure, which can be achieved with controlled ice nucleation in lyophilization [228]. This promotes the formation of larger ice crystals and reduces the overall surface area, resulting in less aggregation formed at ice interfaces. Surfactants such as polysorbate 80 can also be used to reduce the adsorption of proteins at ice interfaces [181].
3.6. Pressure
Proteins are elastic in order to bind to specific sites and adapt to environmental changes [142, 229]. The volume of the protein can be altered with high pressure, resulting in conformational changes. The change in volume as a result of the change in free energy due to pressure can be described in the following equation:
| (9) |
where ΔV is the change in volume, ΔG is the change in Gibbs free energy, and p is the pressure of the system.
The volume change is affected by the space occupied by the atoms of the protein, the volume of the excluded solvent (such as the hydrophobic core), and the volume difference from the interaction between the protein and solvent [230]. High pressures have an effect on all the factors mentioned above and induce protein disassociation or unfolding. The rate of denaturation or disassociation due to pressure can be given by the equation:
| (10) |
where Kd is the equilibrium constant for dissociation or denaturation at pressure p, and Kd0 is the equilibrium constant for structural destabilization at atmospheric pressure [231].
Stabilizing interactions with protein molecules such as ionic and hydrophobic bonding can be disrupted with high pressures. For oligomeric proteins primarily connected by electrostatic and hydrophobic interactions, moderate pressures (0.5–2 kbar) are required for dissociation [232]. Protein unfolding leads to cavitation, exposing the hydrophobic core to the solvent. The formation of these denatured species under high pressures is favorable due to volume reduction, as unfolded protein have higher compressibility than those in the folded state [233].
The pressure required to impact protein stability is typically 2–4 kbar [234], which is not common in standard manufacturing processes. High pressure can be used to concentrate proteins in solution and reduce aggregation through dissociation oligomers. In protein synthesis, increased pressure has been shown to improve the recovery of properly folded proteins from non-native aggregates with and without chaotropic agents [235, 236]. Similarly, proteins have been stabilized at the pressure between 1–3 kbar by dissociating oligomers into monomers [237].
In summary, protein solid products are exposed to various stresses in development and manufacturing, which can cause detrimental effects on protein structure and further promote irreversible damage and aggregation. By understanding the origin and impact of these stresses in manufacturing, strategies in formulation and process design can be utilized to minimize the effects and achieve stable products. In the next section, we will discuss the solid-state characterization techniques that can detect and evaluate stress-induced damage on protein during the drying process.
4. SOLID STATE CHARACTERIZATION TECHNIQUES
Today, monoclonal antibodies (mAb) are popular biotherapeutics [238], and other new biotherapeutics such as mRNA [239] and Cart-T cells [240] are emerging. Due to the unique nature of proteins, their manufacturing process is much more complicated than that of smaller molecules [241]. As we discussed in the previous sections, proteins can be exposed to various stresses throughout the manufacturing process, which can lead to chemical and physical changes [241]. Protein drugs are usually formulated with disaccharide excipients and must be manufactured in solid dosage forms in order to have a reasonable shelf-life [10]. It is critical to ensure that the protein structure remains stable during manufacturing and storage. Understanding the properties of solid matrix containing excipients as well as understanding the interactions between protein and excipients will improve the formulation development process.
Some techniques can be used to detect protein structure changes in the solid samples. For example, solid state Fourier transform infrared spectrometry (ssFTIR) and solid-state fluorescence spectroscopy can be used to measure the secondary and tertiary structure of a protein. There are also methods to characterize the global properties of solids. Differential scanning calorimetry (DSC) measures the glass transition temperature (Tg) of the solid matrix. Tg is routinely determined in formulation development, since protein formulations are expected to remain stable while the storage temperature is below Tg [242]. These techniques can easily be applied on solid samples without extra sample preparation. However, it is difficult to examine solid matrix at the molecular level with these traditional analytical methods. Traditional methods used for small molecules and newly emerging techniques have been applied on biopharmaceutical solids for characterization purposes. Solid-state magnetic resonance (ssNMR) has been used to analyze protein dynamics and phase separation in solid matrix [243, 244]. In addition, solid-state hydrogen deuterium exchange (ssHDX) coupled with mass spectrometry (MS) has been used to study the disruption of protein conformation in solid state, and the results have been correlated with long-term physical stability of proteins in formulations [245]. Unconventional methods such as photolytic cross-linking have also emerged to study the interactions between excipient and protein in the solid state [246]. The choice of characterization technique usually depends on instrument resolution and the information needed. A combination of various analytical tools is commonly used to characterize proteins and the solid matrix [247, 248]. This section focuses on analytical techniques that have been applied on the characterization of protein formulations in the solid state. Their principles as well as advantages and disadvantages are discussed.
4.1. Differential Scanning Calorimetry (DSC)
Pharmaceutical products are often formulated as solids to achieve desired stability [249]. Such protein solids are often amorphous; when they are heated, the system undergoes a transition from amorphous to a rubbery condition [250]. The heat flow between the sample and reference can be used to quantitatively characterize the change in enthalpy and heat capacity. This works well for solids of a pure material when heated at a constant rate, but not for pharmaceutical solids containing several components [251]. There are reversible (e.g. glass transition) and non-reversible (e.g. crystallization) heat events involved when heating amorphous pharmaceutical solids. Thus, modulated DSC (mDSC) has been introduced by adding a temperature modulation to the constant heating rate. The response to multiple heating rates is recorded simultaneously, which helps to separate the thermal events of each component [252]. The state transition from amorphous to rubbery shows as a discontinuity on the diagram of heat-capacity versus temperature, which exhibits as a base line shift. The midpoint of such a shift is defined as the glass transition temperature (Tg) [253].
Most lyophilized products exist in glassy state below the Tg value. A high Tg value is desirable when considering the storage condition of pharmaceutical solids. In the powders of a model protein alkaline phosphatase spray dried with either inulin or trehalose, optimal protein stability was maintained when the storage temperature was 10 °C to 20 °C below Tg [254]. Tg is one of the most important criteria in the development of the lyophilization cycle because it is closely related to determining storage temperature. In a mAb formulation lyophilized with only sucrose, a Tg of ~43 °C was achieved. When different levels of the combination of 2-hydroxypropyl-beta-cyclodextrin (CD), recombinant human albumin, PVP and dextran 40 kDa were added to the formulation, Tg values of 96~200 °C could be obtained. Formulations with sucrose/CD and sucrose/CD/PVP in 9-month storage at 40 °C were better compared to the formulation with only sucrose in characterization tests such as visible/subvisible particles and monomer content [255].
The glass transition temperature of freeze-concentrated solution (Tg’) is another important parameter that can be measured with DSC [256]. Following the freezing step during lyophilization, the components in the formulation remaining uncrystalized were concentrated as the water changed into ice [257]. Crystalized ice serves as a supporting structure in the frozen mixture. Drying above Tg’ leads to loss of microstructure by viscous flow of components with the removal of ice [258]. Therefore, understanding Tg’ is important for the drying cycle development of pharmaceutical solids. The drying temperature for the primary drying step in lyophilization can slightly exceed Tg’ within a certain range. In a study involving four monoclonal antibodies (mAbs), the formulations were dried at temperatures above Tg’ but below the collapse temperature [259]. Product quality and physical or chemical stability were found to be comparable among all formulations in this study. With the elevated drying temperature, pharmaceutically acceptable lyophilized cakes were achieved with significant reduction in drying time [259].
The Tg value determined by DSC is useful when designing the protein formulation for drying cycles. It serves as a routine characterization method for pharmaceutical solids. Excipients that result in a high Tg value are preferable for maintaining stability under unexpected high temperature upon storage. Generally, the drug product should be stored at a temperature below the Tg of the solids. However, in some cases, a high Tg value does not necessarily lead to a more stable protein solid. The aggregation and chemical degradation in lyophilized solids of a mAb in a stability study over 2.5 years were shown to be poorly correlated with Tg [260]. Studies also showed that the correlation between the protein stability and Tg is insignificant when the storage temperature is far below Tg [255, 261]. Additionally, Tg does not provide information on protein structure in the solids. Little can be known about the change in protein structure or kinetics.
4.2. Neutron Scattering
Neutron scattering is one form of vibrational spectroscopy, the calculation of which is based on the measured scattering intensities. It has been used to probe the protein structure and protein-protein interactions in solution [262–264]. Later, the method is applied in pharmaceutical related solids to study the protein dynamics as well as the packing arrangement of dried solids [265, 266]. The coherent and incoherent neutron cross section of each element determine the total scattering intensity. Coherent contribution provides information about the distribution of each atom, which describes molecular shape and morphology. Incoherent contribution results from the neutron spin. Hydrogen (1H) exhibits a significant neutron incoherent cross section and causes major changes in measured scattering when hydrogen is changed to deuterium (2D) [267].
Small-Angle Neutron Scattering (SANS) can be used to acquire protein structural information from the solid samples. Neutrons interact with samples and the intensity of the scattering neutrons as a function of the magnitude of the momentum transfer vector are detected by a 2D detector. Small-angle scattering shares the similar equation as Rayleigh light scattering [267]. SANS was used to investigate the microstructural change of lyophilized and spray-dried solids of a mAb and a Fab [268]. SANS with vapor cell was used on the same formulations to study the microstructural change when the solids were exposed to extra D2O vapor. The use of SANS with a D2O vapor cell enhanced the contrast and allowed for better observation of protein-protein interactions. While exposed to extra moisture, protein clustering was detected in the solids by SANS, which was shown to be reversible aggregate upon reconstitution [268]. In the heat-stressed solid samples, protein clustering was not detected but an increase in aggregate content upon reconstitution was observed. The results suggest that the irreversible protein aggregates formed upon reconstitution in the heat-stressed samples did not result from the reversible aggregates in the solid state [268].
Neutron scattering is also a useful tool to study dynamics of pharmaceutical solids. Changes in atom motions affect velocity of the scattered neutron. The energy distribution of neutrons can be measured to reflect the dynamics. Two types of neutron spectroscopy are available: inelastic neutron scattering and quasielastic neutron scattering (QENS). There is finite energy transfer in inelastic neutron scattering. QENS is a special case of inelastic neutron scattering [267]. In a study involving three IgG1 and two recombinant cytokines, proteins were lyophilized with various amounts of sucrose [269]. Fast local dynamics of solids were characterized by neutron scattering. The change in atom position was characterized by the hydrogen-weighted mean-square atomic displacement. The fast local mobility detected by neutron scattering was shown to be correlated with aggregation formation in solid samples [269].
Detection with neutron scattering does not need special preparation with the solid samples. The measurement can be achieved in-situ. However, this technique is less accessible compared to other characterization methods. The data acquisition process can be time-consuming: potentially more than one day for a single sample [267]. Thus, neutron scattering may not be desirable if quick measurements are needed.
4.3. Solid-state Nuclear Magnetic Resonance Spectroscopy (ssNMR)
ssNMR has been used to study the dynamics of pharmaceutical solids. The detailed principle of function can be found in other reviews of this issue [270–273]. For example, NMR probes atomic motions on MHz and kHz scale, which are laboratory spin-lattice relaxation time (T1) and rotating-frame spin-lattice relaxation time (T1ρ), respectively [274]. The T1 was determined by 13C NMR in a bovine serum γ-globulin (BGG) lyophilized with dextran. The correlation time of each carbon (τc) was calculated accordingly. The spin-spin relaxation time (T2) of BGG and dextran were measured by pulsed 1H NMR, which was used to determine the critical mobility temperature (Tmc). A temperature dependence of τc of dextran methin carbon was found below Tmc. The τc of BGG carbonyl carbon showed similar dependence and decreased when the temperature was above Tmc. The molecular motion of BGG rose above Tmc with the increase of global segmental motion of dextran [275]. In another study, T1ρ of the lysozyme carbonyl carbon was measured in lyophilized solids of lysozyme both with and without sugar. A model with two types of molar motion β-relaxation and γ-relaxation was established to analyze the effect of sugar on molecular mobility. β-relaxation has been found to be more sensitive to changes in sugar content compared to γ-motion in the solids of lysozyme lyophilized with sucrose, trehalose or isomaltose. The β-mobility of lysozyme carbonyl carbon was decreased by sucrose, unaltered by trehalose, and increased by isomaltose [276].
Specific interactions between excipient and protein can be probed with ssNMR by following the changes of specific nuclei. 13C-NMR and 15N-NMR were monitored in the lyophilized solids of anti-CD11a and anti-IgE antibodies with either arginine or glycine as an excipient. The chemical shifts on 13C-NMR and 15N-NMR of arginine and glycine were recorded and compared to the pure lyophilized material. The broadening of the line-widths indicated an amorphous character. The change in chemical shifts suggested intermolecular interactions between the excipient and the protein. 13C-NMR and 15N-NMR spectra showed that there was direct interaction between the guanidine side chain on arginine and the proteins [277].
To stabilize protein in the pharmaceutical solids, one of the prerequisite conditions is that protein and excipient need to be in the same phase. A model IgG was lyophilized with sugars of different molecular weight, ranging from trehalose (342 Da) to dextran (70 kDa). By comparing the 1H T1 and 1H (T1ρ) of protein and sugars, the miscibility of the two components was established at different length scales (20–50 nm for T1 and 2–5 nm for T1ρ). The aggregation tendency was smaller for low weight sugar with better miscibility. A shorter 1H T1 relaxation time was correlated with a higher aggregation rate in the storage samples [244]. Similar results were observed in an IgG1 mAb with different levels of sucrose and trehalose in various pH conditions. Decreased 1H T1 relaxation time was found in formulations that were more stable during storage [278].
ssNMR is a useful tool in providing dynamic information about pharmaceutical solids. Even though it has been used to study protein structure in solution or insoluble amyloid fiber [279–282], it can be complicated to analyze protein structure in pharmaceutical solids due to the amorphous environment. Acquiring data for relaxation time occasionally requires an extended temperature range. The analysis could require extra mathematical calculation and results may be complex to interpret.
4.4. Solid-state Fourier Transform Infrared Spectrometry (ssFTIR)
Stresses created during drying or storage can disrupt the protein native structure, which leads to loss of protein activity [143, 283]. The change in protein secondary structure, such as α-helix and β-sheet, can be detected with solid-state Fourier Transform Infrared Spectrometry (ssFTIR). The infrared spectrometry (IR) detects the vibrational spectroscopy of protein, the bands of which tend to be overlapped. Thus, Fourier self-deconvolution and second derivatization are used to analyze the spectrum signal and resolve the band frequency. The typical vibrational frequency of proteins appears in two regions: amide I region (near 1650 cm−1) and Amide II region (near 1550 cm−1). The amide I region corresponds to the C=O stretching of the peptide bond, which directly indicates protein backbone conformation. The amide II region results from the bending of in-plane N-H, and the stretching vibration of C-N and C-C bonds [284, 285]. Significant band shift and intensity change in the amide I region is constantly used to detect the protein structure disruption of α-helix and β-sheet. The signature band peak is around 1655 cm−1 for α-helix, 1630 and 1690 cm−1 for β-sheet and β-turn [286–288]. Comparison of ssFTIR spectra in different protein formulations could provide an initial insight into protein stability.
ssFTIR spectroscopy has been used to study the stabilizing effect of excipients on protein structure in lyophilized solids. The ssFTIR spectra of lysozyme lyophilized with trehalose was compared with those of lysozyme lyophilized alone and of lysozyme in aqueous solution [289]. Band shift and broadening were observed in the solid sample of lysozyme lyophilized alone. The addition of trehalose as an excipient shifted the amide I band back to the position in the solution sample of lysozyme. The band shapes were more similar in the lysozyme lyophilized with trehalose and the lysozyme in aqueous solution. The amide I band of carboxylate was not detectable with the lyophilized lysozyme alone, but was observed in the aqueous lysozyme and the lyophilized lysozyme with trehalose. Furthermore, different concentrations of trehalose, lactose, or myo-inositol were lyophilized with lysozyme. The band shift in each formulation was used to determine the stabilizing effect of each excipient [289]. In another study, IgG1 lyophilized without any sugar excipient exhibited greater secondary structure disruption in FTIR spectra than in those lyophilized with disaccharide excipients. With an increase in weight ratio of sucrose in the formulation, the peak tended to be more similar to that of the IgG1 in solution, which is considered to be its native structure. Similar results were observed in rHSA lyophilized with different concentrations of sucrose [290].
However, the disruption of secondary structure does not necessarily correlate to the instability of protein formulations. The secondary structure changes of an IgG1 monoclonal antibody in formulations containing sucrose, trehalose, mannitol or histidine were analyzed based on the major peak intensity in the secondary derivative of the spectra [291]. The decrease in intensity was poorly correlated with the aggregation rate, as determined by size exclusion chromatography (SEC) in the long-term storage study [291].
The shift of wavenumber on FTIR spectra is in general difficult to use for quantitation purposes, though curve fitting of the spectra could facilitate the content estimation of certain structures in a protein. The FTIR spectra were collected for the spray-freeze dried IgG solids containing a potential protein stabilizer hydroxypropyl β-cyclodextrin (HPβCD) [290] Protein stability curves. Curve fitting was performed with a mixed Gaussian/Lorentzian function on the second derivatization of the deconvoluted spectra ranging from 1700 to 1600 cm−1. The peak width, height, center and area were used for an estimation of α-helix and β-sheet content in the IgG molecule in the solids. The protein β-sheet content in the spray-dried solids of IgG formulations containing different ratios of trehalose and HPβCD ranged from 65.13% to 77.82%. After storage up to 2 months at 45 °C, the β-sheet content among the formulations ranged from 66.32% to 78.15% [292]. Similar quantitation of secondary structure with FTIR spectra has been used to study the lyophilized recombinant human growth hormone and spray-freeze dried BSA complexed with Zinc [114, 293].
ssFTIR is a relatively simple and quick characterization method for the protein analysis of pharmaceutical solids. The sample amount needed for ssFTIR is in the milligrams. The second derivative spectra easily demonstrate the change in protein secondary structure by overall change of peak shape, such as in wavenumber shift and peak intensity. However, it is difficult to quantitatively describe the correlation between the change in FTIR spectra and the degree of protein structure disruption, especially with only small changes in peak positions. Additionally, ssFTIR analyzes the global change of protein secondary structure in a static motion. It does not indicate minor changes in certain area or provide any dynamic information.
4.5. Solid-state Fluorescence Spectrometry
The aromatic residues on proteins exhibit distinct fluorescence spectrometry, which has long been used in solution to monitor protein activities such as folding and unfolding. The intrinsic fluorescence of tryptophan is especially sensitive to the perturbation of protein structure [294]. Such a method can be applied to solids for evaluating the perturbation of the tertiary structure of proteins. Fully exposed tryptophan residue shows maximum fluorescence at around 334 nm [295]. In order to minimize scattering from the background, a front surface sample holder is typically used. The change in tertiary structure is mainly determined by peak shift, since the concentration dependence behavior of the intensity is unlikely in the solid state [296]. In a study of lyophilized and spray-dried mAb formulations, solid-state fluorescence spectrometry was used to evaluate the effect of drying method on protein structure perturbation [297]. For mannitol formulations, peak shift was detected in the spray-dried solids but not in the lyophilized solids with or without controlled nucleation. Such a peak shift was not detected in the reconstituted solution samples, suggesting that the disruption of tertiary structure in solid state could be reversible [297].
Solid state fluorescence spectroscopy has limited ability to detect protein structure because the detection sensitivity is dependent on the number and location of tryptophan in proteins. In some cases, the minor change in peak shift is not enough to conclude a change in protein structure. Moreover, there is insufficient evidence to show a direct correlation between the disruption of tertiary structure with protein stability in the solid state.
4.6. Solid-state Hydrogen Deuterium Exchange (ssHDX)
Solution hydrogen deuterium exchange (HDX) has been used to study protein structure and its solvent accessibility by evaluating whether a protein maintains its native folded conformation in a solution formulation [298]. When the formulation components are unable to stabilize the protein, its conformation can be disrupted, causing further aggregation or degradation. HDX can be used to detect the conformational change, which indicates the physical stability of the molecule. The hydrogen atoms on the protein are exchangeable with those from H2O in the surrounding environment. When exposed to solution containing deuterium oxide (D2O), hydrogen atoms on the protein can exchange with the deuterium from the environment. Not all the hydrogen atoms on the protein are exchangeable and not all can be detected with mass spectrometry (MS). For example, the exchange reaction kinetic on the side chain is too fast to be measured. Instead, the mass increase detected by MS shows how many amide hydrogen atoms on the backbone exchange with deuterium in the solvent [299]. Amide hydrogens are involved in the intermolecular and intramolecular hydrogen bonding formation, which contributes to overall protein conformation. The deuterium uptake kinetics are associated with protein dynamics in the solution. The difference in the deuterium uptake value indicates changes in hydrogen bonding patterns, which are related to higher-order protein structure, protein-protein interactions and protein-ligand binding. Proteolysis of deuterated protein can provide further information on protein conformation change in local regions [298, 300, 301].
ssHDX can be used as a characterization method for protein structure in the solid formulations. ssHDX is achieved by exposing protein solids to D2O vapor. The reaction is quenched at different time points by flash-freezing the samples in liquid nitrogen. To minimize back exchange with hydrogen, the solid samples are reconstituted with ice cold acidic buffer (pH < 2.5) immediately before MS analysis. It is also recommended to keep the connection tubing and columns prior to MS detector at a cold temperature [302–304]. The completeness of the HDX reaction partly depends on protein size. However, the reaction rate in solid state is significantly slower compared to HDX kinetics in solution [305, 306]. While HDX in solution reaches equilibrium within minutes to hours, the plateau of deuterium uptake may not be observed even after exposing the solid samples to D2O vapor for days. Thus, the data of deuterium uptake at various time points are fitted into mono-exponential or bi-exponential equations to estimate the plateau value. In a study of myoglobin, the deuterium uptake of lyophilized solids containing sucrose or mannitol was fitted into a bi-exponential equation [245]. The myoglobin in sucrose formulation is less deuterated than with the mannitol formulation, suggesting greater structure protection from sucrose. This study has shown that both the extent and kinetic parameters of deuterium incorporation are highly correlated with myoglobin aggregation, which is detected by size exclusion chromatography (SEC) in physical stability studies (see Figure 4). ssHDX data was collected within 10 days while the storage stability study was conducted for up to 1 year. The results indicate that ssHDX could be a promising tool for predicting long-term protein physical stability in a relatively short time [245]. The deconvoluted mass spectra of deuterated protein can provide information on protein heterogeneity in structure. Peak width and area were analyzed in a study of lyophilized and spray-dried IgG1 monoclonal antibody (mAb) formulations [297]. Peak splitting and larger peak areas of deuterated mass envelopes have been observed in sucrose and trehalose formulation processes by spray drying. This suggests the existence of more structural states in spray-dried powder as compared to the lyophilized solids [297]. Such correlation between protein stability and ssHDX result has been found in proteins such as bovine serum albumin, lysozyme, β-lactoglobulin [95, 189].
Figure 4.
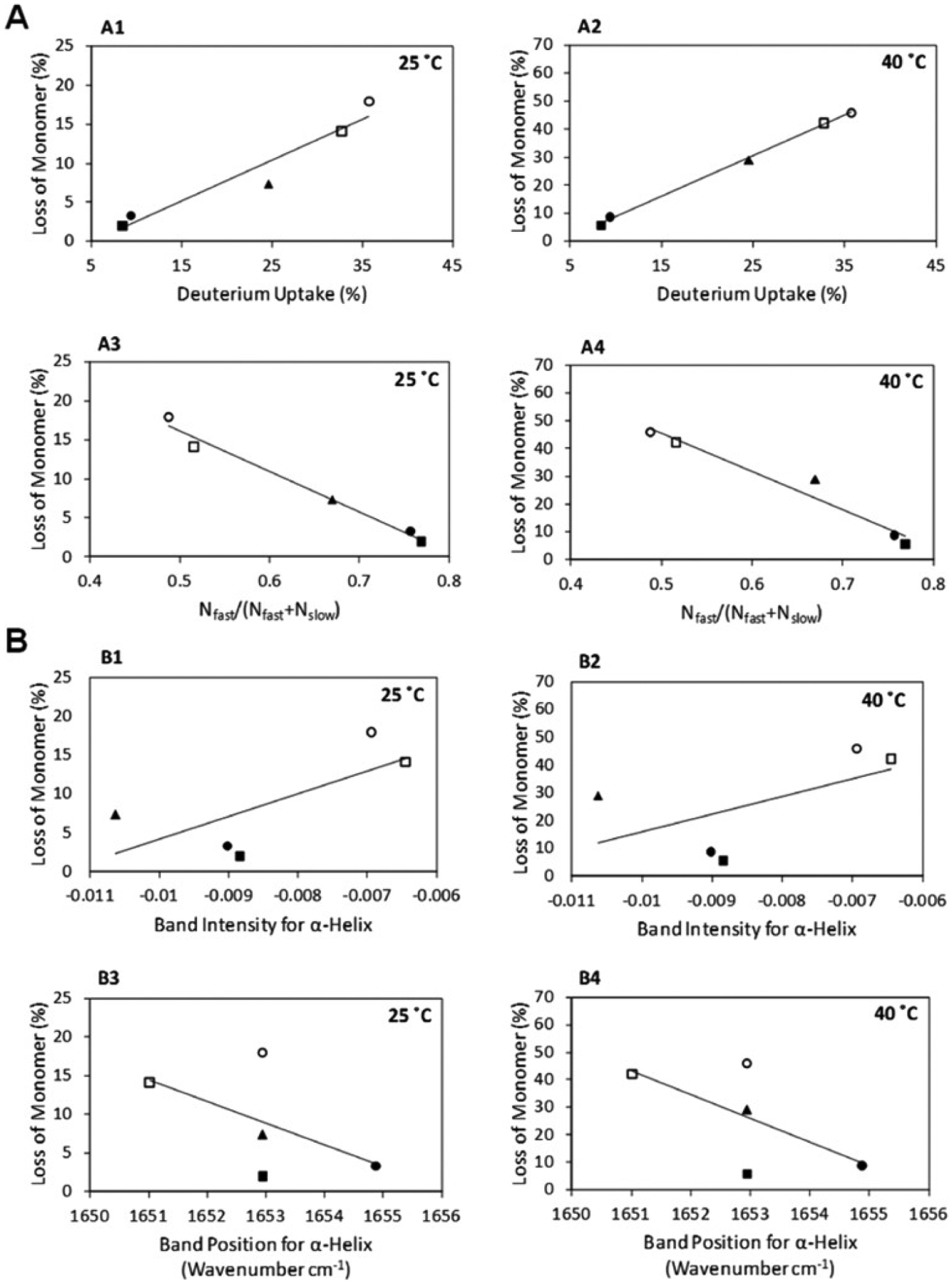
Correlation of Mb aggregation during long-term storage (at t = 360 days) with t = 0 ssHDX-MS (A) and FTIR (B) analysis. The percent loss of Mb monomer after 360 days of storage at 25 and 40 °C as a function of percent deuterium uptake (A1 and A2), Nfast/(Nfast + Nslow) (A3 and A4), band intensity for α-helix (B1 and B2), and band position for α-helix (B3 and B4) in formulations, MbA (closed circle), MbB (closed triangle), MbC (open circle), MbD (open square), and MbE (closed square) (n = 3, ± SE). Reprint from [245] with permission from ACS. Further permissions related to the material excerpted should be directed to the ACS.
ssHDX measures the change in protein backbone, which can be directly altered by the intramolecular and intermolecular hydrogen bonding of protein with the solid matrix. The difference between various excipients may not be detected with ssHDX when the existence of excipients has no impact on protein backbone; or when excipients interact with protein through other types of interactions other than hydrogen bonding. A mAb formulation containing different levels of arginine showed no significant difference in the deuterium uptake of ssHDX results [307]. Nevertheless, the accelerated stability study suggested that the aggregation rate decreased with an increased level of arginine in the formulation [307]. Additionally, the aggregation rate changed when the histidine buffer in the formulation was replaced by the phosphate buffer, the difference of which has not been observed from ssHDX. It was suggested that the charge-based interactions dominated in the lyophilized solid and stabilized mAb. The ionic interactions between amino acid excipient and protein side chain cannot be detected by MS because the back-exchange is too fast [307].
4.7. Solid-state Photolytic Labeling (ssPL)
The protein and excipient involved in hydrogen bonding or charge-based interactions are in close vicinity in the solid state. These interactions are non-covalent and difficult to detect. Solid-state photolytic labeling (ssPL) has been developed as a complementary quantitative analysis method to ssHDX for protein-excipient interactions. An ssPL study generally includes the following steps: 1) incorporating photo-reactive functional groups into either protein or excipient, or using commercialized photo-excipients directly; 2) preparing formulation in solutions or solids; 3) exposing prepared formulations to UV at certain wavelength to initiate photo-reaction; 4) analyzing photo-reaction products to study protein-excipient interactions. These photo-reactive groups remain unreactive through formulation preparation steps and only become reactive when exposed to UV light at certain wavelength. Covalent adducts from protein and excipient in close vicinity in solid state can be analyzed quantitatively with common analytical methods such as MS and HPLC to show the solid matrix environment near the protein.
Proteins can be labeled with photo-reactive functional groups to probe the interactions in the solid state. The interactions between myoglobin and raffinose in the lyophilized solids have been studied with ssPL [308]. Succinimidyl 4,4’-azipentanoate (SDA) was used to transfer the photo-reactive functional group to myoglobin. SDA is a bifunctional probe containing NHS-ester and diazirine group. Diazirine becomes reactive when exposed to UV at 365 nm. NHS ester reacts with primary amine of the lysine side chains on myoglobin and transfers diazirine onto the protein. SDA-labeled myoglobin was prepared with either raffinose or guanidine hydrochloride (Gdn HCl) and lyophilized. The lyophilized formulations were exposed to UV to initiate the reaction and the photo-reaction products were analyzed by MS. Following the analysis of digested myoglobin after photo-reaction, peptide-peptide, peptide-excipient and peptide-water adducts were detected. The raffinose formulation showed more peptide-peptide adducts than the one containing Gdn HCl. In the solids of the Gdn HCl formulation, more protein-protein and protein-water adducts were detected, showing a more disrupted protein structure [308].
One challenge for this method is analyzing the reaction products quantitatively, as it is difficult to establish calibration curves. A model peptide KLQ containing seven amino acids has been used to study the photo-reaction products based on HPLC results [309]. Unexpected products such as peptide-salt and regenerated unlabeled peptide have been detected. Calibration curves have been established and used to analyze reaction products on a concentration basis [309].
Photo-reactive reagents can be incorporated in the solid formulations as excipients in order to study protein-excipient interactions. Photo-leucine (L-2-amino-4,4’-azipentanoic acid) has been used in myoglobin formulations with or without sucrose to study the effect of controlled ice nucleation during the lyophilization process [310]. In this study, the ssHDX kinetics showed similar results in the lyophilized solids with and without controlled ice nucleation. On the other hand, the myoglobin formulation containing sucrose with controlled ice nucleation showed the greatest photo-leucine incorporation as well as more labeled region on the protein. Photo-leucine can be used as a detection reagent to demonstrate formulation differences that cannot be detected by other analytical methods. In order to have a photo-reactive reagent mimicking structures of commonly used sugar excipients, photo-glucosamine has been synthesized and used in lyophilized solids to study peptide-excipient interactions [311]. Minor changes in the formation of peptide-excipient products have been observed in photo-glucosamine formulation when the pH prior to lyophilization was increased. In photo-leucine formulation, the increase in the solution pH led to more peptide-excipient adducts. The results demonstrate that photo-leucine and photo-glucosamine interact differently with the peptide in the solid state [311].
ssPL is relatively new compared to other characterization methods. Photo-reaction products can be complicated for analyzing proteins labeled with photo-reactive functional groups. Compared to photo-reactive proteins, it is more feasible to quantitatively analyze photo-reaction products with photo-reactive excipients incorporated in the formulation to evaluate protein formulations. At the same time, the application of ssPL is limited by the availability of photo-reactive excipient. The commercially available photo-reactive excipients used in the previous studies do not possess structures mimicking those of disaccharides such as sucrose or trehalose. There is a need for photo-reactive excipients with structures that mimic commonly used excipients in protein formulations.
Compared to protein solution formulations, there are relatively fewer characterization methods for pharmaceutical solids. There are solid state characterization methods not detailed that are introduced in this section. Near Infrared Spectroscopy (NIR) can detect the intramolecular hydrogen bonds for α-helix and β-sheet structure of proteins. In the study of a model IgG, the sample chamber of the lyophilizer was equipped with an NIR non-contact probe, which enables an in-line observation of protein unfolding behavior during the lyophilization process [312]. Dielectric relaxation spectroscopy measures the relaxation times of molecules and provides information on molecular mobility of the solid matrix. In a lyophilized formulation with lysozyme, the molecular mobility was reduced by trehalose as detected by dielectric relaxation spectroscopy [313]. At the same time, molecular mobility and miscibility of the solid matrix can be evaluated by other detection methods such as DSC and ssNMR. Characterization methods like ssNMR can provide information about the protein as well as the solid matrix, but data acquisition may take longer than with ssFTIR. ssHDX measures detailed protein structure change and provides kinetic information with extra sample preparation. Sophisticated techniques such as neutron scattering may not be readily accessible while the same information can be obtained from other methods, such as with dielectric relaxation spectrometry. A summary of advantages and disadvantages of the characterization methods mentioned previously has been listed in Table 2. To acquire a thorough understanding of protein behavior and quality, the characterization of pharmaceutical solids usually involves multiple techniques. Which characterization techniques are used depends on the information needed, the accessibility of the instruments, the efficiency of the data acquisition and analysis process, and other practical considerations.
Table 2.
Summary of solid-state characterization methods.
| Characterization Method | What does it measure? | Information Provided | Advantage | Disadvantage |
|---|---|---|---|---|
| DSC | Change in enthalpy and heat capacity when heating the solid sample | Tg; matrix dynamics | Routine measurement; useful for drying cycle development | Global information on matrix; little information on local protein structure |
| Neutron Scattering | Energy and intensity of scattered neutrons | Matrix dynamics; molecule distribution in solid state | In-situ detection without extra sample preparation | Time-consuming data acquisition and analysis; limited accessibility of the technique |
| ssFTIR | Vibrational frequency of chemical bonds shown in fingerprint regions | Protein secondary structure | Fast and easy to access information on protein secondary structure | Global structure information; difficult to quantitatively describe structural disruption |
| Solid-state Fluorescence Spectrometry | Protein intrinsic fluorescence from aromatic residues | Protein tertiary structure | Detect tertiary structure disruption in-situ | Not proved to be correlated with physical stability in protein formulations |
| ssNMR | Chemical shifts of isotopes | Protein conformation; matrix and protein dynamics; protein-excipient interactions | Provide comprehensive information for both protein and matrix in solid state | Data acquisition and interpretation can be time-consuming |
| ssHDX | Change in protein backbone based on the disruption of hydrogen bonding | Disruption of protein conformation; heterogeneity of protein in solid state | Characterize hydrogen bonding patterns between protein and sugar-based excipients; correlated with long-term stability of protein formulations | Poor correlation in formulations with ionic excipients; does not work well to distinguish different drying process |
| ssPL | Distribution of the molecules in the solid matrix near protein | Protein interactions with excipient, salt or residual water | Labeling available for both protein and excipient; does not require specific type of interactions between protein and excipient | Extra sample preparation; complicated quantitation process |
5. CONCLUSION
Solid formulations are valuable alternatives to liquid dosage forms when the stability of the biologic product is a concern. The boom in biologics requires advanced development in drying technologies. Lyophilization, or freeze drying, is still the most commonly used drying technique for biologics despite the fact that it is a time-consuming and energy-inefficient batch process. Depending on the administration route of the final products, extra processing steps may be required after lyophilization. For example, milling is needed for lyophilized solids to generate fine particles for the inhaled products. New drying technologies are needed for higher drying efficiency and specific formulation purposes. Continuous freeze drying, spray drying, spray freeze drying, supercritical fluid drying and electrostatic spray drying have emerged as alternative drying techniques to lyophilization. More studies are needed to investigate the effect of drying methods on scalability for massive manufacturing process, and physico-chemical properties and stability of biologics. There is an urgent need for the pharmaceutical industry to develop, understand and employ novel high-efficiency and continuous techniques for manufacturing biological, especially protein solid products.
Stresses in drying process, including high/low temperature, surface induced denaturation, and sudden pH change are usually process-dependent, which affect molecule stability. Understanding protein stability from formulation development to final product is key for quality assurance. However, characterization methods targeting protein and solid matrix are limited. Solid-state FTIR and solid-state fluorescence spectrometry measure the protein structural change directly; however, they only provide global information without details into local structural change. DSC provides information about Tg, but in many cases it does not indicate protein stability directly. More advanced characterization tools such as ssHDX, ssNMR and ssPL have been employed to understand the molecular interaction and protein structure. Practical considerations such as data acquisition time and instrument accessibility should be considered when choosing characterization techniques. There is a demand to develop characterization platforms that enable process analytic technology and provide collective information to predict protein performance and stability without long-term stability tests. Such platforms are promising to reduce the time and cost for the development of solid protein products while ensuring the quality.
Acknowledgement
This publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institute of Health under Award Number R01AI146160 and R01AI132681. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health.
Abbreviation
- Lyo
Lyophilization
- SD
Spray Drying
- SFD
Spray Freeze Drying
- SCFD
Super Critical Fluid Drying
- Tg
Glass Transition Temperature
- Teu
Eutectic Temperature
- Tc
Collapse Temperature
- CFD
Computational Fluid Dynamics
- PAT
Process Analytical Technology
- MFD
Microwave-assisted Freeze Drying
- FDA
Food and Drug Administration
- EMA
European Medicines Agency
- DoE
Design of Experiments
- DSC
Differential Scanning Calorimetry
- ssNMR
Solid-state Nuclear Magnetic Resonance
- ssHDX
Solid-state Hydrogen Deuterium Exchange
- ssPL
Solid-state Photolytic Labeling
- ssFTIR
Solid-state Fourier Transform Infrared Spectrometry
- mAb
Monoclonal Antibody
- IgG
Immunoglobulin G
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Tiwari G, Tiwari R, Sriwastawa B, Bhati L, Pandey S, Pandey P, Bannerjee SK, Drug delivery systems: An updated review, International journal of pharmaceutical investigation, 2 (2012) 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chen T, Formulation concerns of protein drugs, Drug Development and Industrial Pharmacy, 18 (1992) 1311–1354. [Google Scholar]
- [3].Frokjaer S, Otzen DE, Protein drug stability: a formulation challenge, Nature Reviews Drug Discovery, 4 (2005) 298–306. [DOI] [PubMed] [Google Scholar]
- [4].Shire SJ, Shahrokh Z, Liu J, Challenges in the development of high protein concentration formulations, Journal of Pharmaceutical Sciences, 93 (2004) 1390–1402. [DOI] [PubMed] [Google Scholar]
- [5].Zhou XH, Li Wan Po A, Peptide and protein drugs: I. Therapeutic applications, absorption and parenteral administration, International Journal of Pharmaceutics, 75 (1991) 97–115. [Google Scholar]
- [6].Carpenter JF, Chang BS, Garzon-Rodriguez W, Randolph TW, Rational design of stable lyophilized protein formulations: theory and practice, Rational design of stable protein formulations, Springer; 2002, pp. 109–133. [DOI] [PubMed] [Google Scholar]
- [7].Chang L, Pikal MJ, Mechanisms of protein stabilization in the solid state, Journal of Pharmaceutical Sciences, 98 (2009) 2886–2908. [DOI] [PubMed] [Google Scholar]
- [8].Allison SD, Manning MC, Randolph TW, Middleton K, Davis A, Carpenter JF, Optimization of storage stability of lyophilized actin using combinations of disaccharides and dextran, Journal of pharmaceutical sciences, 89 (2000) 199–214. [DOI] [PubMed] [Google Scholar]
- [9].Sou T, Forbes RT, Gray J, Prankerd RJ, Kaminskas LM, McIntosh MP, Morton DAV, Designing a multi-component spray-dried formulation platform for pulmonary delivery of biopharmaceuticals: the use of polyol, disaccharide, polysaccharide and synthetic polymer to modify solid-state properties for glassy stabilisation, Powder technology, 287 (2016) 248–255. [Google Scholar]
- [10].Wang W, Lyophilization and development of solid protein pharmaceuticals, International journal of pharmaceutics, 203 (2000) 1–60. [DOI] [PubMed] [Google Scholar]
- [11].Carpenter JF, Pikal MJ, Chang BS, Randolph TW, Rational design of stable lyophilized protein formulations: some practical advice, Pharmaceutical research, 14 (1997) 969–975. [DOI] [PubMed] [Google Scholar]
- [12].Philo JS, Arakawa T, Mechanisms of protein aggregation, Current pharmaceutical biotechnology, 10 (2009) 348–351. [DOI] [PubMed] [Google Scholar]
- [13].Adams GD, Lyophilization of vaccines, Vaccine Protocols, Springer; 1996, pp. 167–185. [DOI] [PubMed] [Google Scholar]
- [14].Mackenzie A, A current understanding of the freeze-drying of representative aqueous solutions, Science et Technique du Froid (France), (1985). [Google Scholar]
- [15].Oetjen G-W, Industrial freeze-drying for pharmaceutical applications, Drugs and the pharmaceutical sciences, 96 (1999) 267–335. [Google Scholar]
- [16].Adams G, Freeze-Drying—The Integrated Approach, Pharmaceutical Manufacturing International, (1995) 177–180. [Google Scholar]
- [17].Pikal MJ, Freeze-drying of proteins. Part I: process design, BioPharm, 3 (1990) 18–27. [Google Scholar]
- [18].Beals J, Edwards M, Pikal M, Rinella J Jr, Formulations of obesity protein, USA: Eur Pat Appl (Eli Lilly and co. USA). EP, (1997) 48. [Google Scholar]
- [19].Nail SL, Gatlin LA, Freeze-drying: principles and practice, Pharmaceutical Dosage Forms-Parenteral Medications, CRC Press; 2016, pp. 367–396. [Google Scholar]
- [20].Franks F, Freeze drying: from empiricism to predictability, Cryo-Lett., 11 (1990) 93–110. [PubMed] [Google Scholar]
- [21].Tang XC, Pikal MJ, Design of freeze-drying processes for pharmaceuticals: practical advice, Pharmaceutical research, 21 (2004) 191–200. [DOI] [PubMed] [Google Scholar]
- [22].MacKenzie A, Basic principles of freeze-drying for pharmaceuticals, Bulletin of the parenteral drug association, 20 (1966) 101. [PubMed] [Google Scholar]
- [23].Pikal MJ, Shah S, The collapse temperature in freeze drying: Dependence on measurement methodology and rate of water removal from the glassy phase, International Journal of Pharmaceutics, 62 (1990) 165–186. [Google Scholar]
- [24].Shalaev EY, Franks F, Changes in the physical state of model mixtures during freezing and drying: Impact on product quality, Cryobiology, 33 (1996) 14–26. [Google Scholar]
- [25].Jiang S, Nail SL, Effect of process conditions on recovery of protein activity after freezing and freeze-drying, European Journal of Pharmaceutics and Biopharmaceutics, 45 (1998) 249–257. [DOI] [PubMed] [Google Scholar]
- [26].Heller MC, Carpenter JF, Randolph TW, Protein formulation and lyophilization cycle design: Prevention of damage due to freeze‐concentration induced phase separation, Biotechnology and Bioengineering, 63 (1999) 166–174. [DOI] [PubMed] [Google Scholar]
- [27].Lu X, Pikal MJ, Freeze-drying of mannitol-trehalose-sodium chloride-based formulations: the impact of annealing on dry layer resistance to mass transfer and cake structure, Pharm Dev Technol, 9 (2004) 85–95. [DOI] [PubMed] [Google Scholar]
- [28].Williams NA, Lee Y, Poll GP, Jennings TA, The effects of cooling rate on solid phase transitions and associated vial breakage occurring in frozen mannitol solutions, PDA Journal of Pharmaceutical Science and Technology, 40 (1986) 135–141. [PubMed] [Google Scholar]
- [29].Pikal M, Shah S, Senior D, Lang J, Physical chemistry of freeze-drying: measurement of sublimation rates for frozen aqueous solutions by a microbalance technique, Journal of pharmaceutical sciences, 72 (1983) 635–650. [DOI] [PubMed] [Google Scholar]
- [30].Pikal MJ, Roy M, Shah S, Mass and heat transfer in vial freeze-drying of pharmaceuticals: Role of the vial, Journal of pharmaceutical sciences, 73 (1984) 1224–1237. [DOI] [PubMed] [Google Scholar]
- [31].Roy ML, Pikal MJ, Process control in freeze drying: determination of the end point of sublimation drying by an electronic moisture sensor, PDA Journal of Pharmaceutical Science and Technology, 43 (1989) 60–66. [PubMed] [Google Scholar]
- [32].Pikal M, Shah S, Roy M, Putman R, The secondary drying stage of freeze drying: drying kinetics as a function of temperature and chamber pressure, International journal of pharmaceutics, 60 (1990) 203–207. [Google Scholar]
- [33].Sadikoglu H, Liapis A, Crosser O, Optimal control of the primary and secondary drying stages of bulk solution freeze drying in trays, Drying Technology, 16 (1998) 399–431. [Google Scholar]
- [34].Sadikoglu H, Ozdemir M, Seker M, Optimal control of the primary drying stage of freeze drying of solutions in vials using variational calculus, Drying Technology, 21 (2003) 1307–1331. [Google Scholar]
- [35].Lombraña J, Diaz J, Heat programming to improve efficiency in a batch freeze-drier, The Chemical Engineering Journal, 35 (1987) B23–B30. [Google Scholar]
- [36].Lombraña J, Diaz J, Coupled vacuum and heating power control for freeze-drying time reduction of solutions in phials, Vacuum, 37 (1987) 473–476. [Google Scholar]
- [37].Mahmood M, Mhaskar P, Enhanced stability regions for model predictive control of nonlinear process systems, AIChE journal, 54 (2008) 1487–1498. [Google Scholar]
- [38].Nail SL, Jiang S, Chongprasert S, Knopp SA, Fundamentals of freeze-drying, Development and manufacture of protein pharmaceuticals, Springer; 2002, pp. 281–360. [DOI] [PubMed] [Google Scholar]
- [39].Zavala VM, Biegler LT, Optimization-based strategies for the operation of low-density polyethylene tubular reactors: nonlinear model predictive control, Computers & Chemical Engineering, 33 (2009) 1735–1746. [Google Scholar]
- [40].Fissore D, Barresi AA, Scale-up and process transfer of freeze-drying recipes, Drying Technology, 29 (2011) 1673–1684. [Google Scholar]
- [41].Barresi AA, Rasetto V, Marchisio DL, Use of computational fluid dynamics for improving freeze-dryers design and process understanding. Part 1: Modelling the lyophilisation chamber, European Journal of Pharmaceutics and Biopharmaceutics, 129 (2018) 30–44. [DOI] [PubMed] [Google Scholar]
- [42].Marchisio DL, Galan M, Barresi AA, Use of computational fluid dynamics for improving freeze-dryers design and process understanding. Part 2: Condenser duct and valve modelling, European Journal of Pharmaceutics and Biopharmaceutics, 129 (2018) 45–57. [DOI] [PubMed] [Google Scholar]
- [43].Pikal M, Rambhatla S, Ramot R, The impact of the freezing stage in lyophilization: effects of the ice nucleation temperature on process design and product quality, American Pharmaceutical Review, 5 (2002) 48–53. [Google Scholar]
- [44].Goldman JM, More HT, Yee O, Borgeson E, Remy B, Rowe J, Sadineni V, Optimization of Primary Drying in Lyophilization During Early-Phase Drug Development Using a Definitive Screening Design With Formulation and Process Factors, Journal of Pharmaceutical Sciences, 107 (2018) 2592–2600. [DOI] [PubMed] [Google Scholar]
- [45].Daraoui N, Dufour P, Hammouri H, Hottot A, Model predictive control during the primary drying stage of lyophilisation, Control Engineering Practice, 18 (2010) 483–494. [Google Scholar]
- [46].Pisano R, Fissore D, Barresi AA, Rastelli M, Quality by design: scale-up of freeze-drying cycles in pharmaceutical industry, AAPS PharmSciTech, 14 (2013) 1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kodama T, Sawada H, Hosomi H, Takeuchi M, Wakiyama N, Yonemochi E, Terada K, Optimization of primary drying condition for pharmaceutical lyophilization using a novel simulation program with a predictive model for dry layer resistance, Chemical and Pharmaceutical Bulletin, 62 (2014) 153–159. [DOI] [PubMed] [Google Scholar]
- [48].Schneid S, Gieseler H, Evaluation of a new wireless Temperature Remote Interrogation System (TEMPRIS) to measure product temperature during freeze drying, AAPS PharmSciTech, 9 (2008) 729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tang XC, Nail SL, Pikal MJ, Freeze-drying process design by manometric temperature measurement: design of a smart freeze-dryer, Pharmaceutical research, 22 (2005) 685–700. [DOI] [PubMed] [Google Scholar]
- [50].Kawasaki H, Shimanouchi T, Yamamoto M, Takahashi K, Kimura Y, Scale-up procedure for primary drying process in lyophilizer by using the vial heat transfer and the drying resistance, Chemical and Pharmaceutical Bulletin, 66 (2018) 1048–1056. [DOI] [PubMed] [Google Scholar]
- [51].Velardi SA, Rasetto V, Barresi AA, Dynamic parameters estimation method: advanced manometric temperature measurement approach for freeze-drying monitoring of pharmaceutical solutions, Industrial & Engineering Chemistry Research, 47 (2008) 8445–8457. [Google Scholar]
- [52].Milton N, Pikal MJ, Roy ML, Nail SL, Evaluation of manometric temperature measurement as a method of monitoring product temperature during lyophilization, PDA Journal of Pharmaceutical Science and Technology, 51 (1997) 7–16. [PubMed] [Google Scholar]
- [53].Pisano R, Fissore D, Barresi AA, Noninvasive Monitoring of a Freeze-Drying Process for tert-Butanol/Water Cosolvent-Based Formulations, Industrial & Engineering Chemistry Research, 55 (2016) 5670–5680. [Google Scholar]
- [54].Kawasaki H, Shimanouchi T, Sawada H, Hosomi H, Hamabe Y, Kimura Y, Temperature Measurement by Sublimation Rate as a Process Analytical Technology Tool in Lyophilization, Journal of Pharmaceutical Sciences, 108 (2019) 2305–2314. [DOI] [PubMed] [Google Scholar]
- [55].Nail SL, Johnson W, Methodology for in-process determination of residual water in freeze-dried products, Dev Biol Stand, 74 (1992) 137–150; dicussion 150–131. [PubMed] [Google Scholar]
- [56].Nail SL, Gatlin LA, Advances in control of production freeze dryers, PDA Journal of Pharmaceutical Science and Technology, 39 (1985) 16–27. [PubMed] [Google Scholar]
- [57].Mayeresse Y, Veillon R, Sibille P, Nomine C, Freeze-drying process monitoring using a cold plasma ionization device, PDA-Journal of Pharmaceutical Science and Technology, 61 (2007) 160–174. [PubMed] [Google Scholar]
- [58].Jennings T, Residual gas analysis and vacuum freeze drying, PDA Journal of Pharmaceutical Science and Technology, 34 (1980) 62–69. [PubMed] [Google Scholar]
- [59].Connelly JP, Welch JV, Monitor lyophilization with mass spectrometer gas analysis, PDA Journal of Pharmaceutical Science and Technology, 47 (1993) 70–75. [PubMed] [Google Scholar]
- [60].May JC, Regulatory Control of Freeze-Dried Products: Importance and Evaluation of Residual Moisture, Freeze Drying/Lyophilization of Pharmaceutical and Biological Products, (2016) 288. [Google Scholar]
- [61].Brookfield A, Computrac Vapor Pro XL, 2016. [Google Scholar]
- [62].De Beer T, Vercruysse P, Burggraeve A, Quinten T, Ouyang J, Zhang X, Vervaet C, Remon JP, Baeyens W, In-line and real-time process monitoring of a freeze drying process using Raman and NIR spectroscopy as complementary process analytical technology (PAT) tools, Journal of pharmaceutical sciences, 98 (2009) 3430–3446. [DOI] [PubMed] [Google Scholar]
- [63].Gieseler H, Kessler WJ, Finson M, Davis SJ, Mulhall PA, Bons V, Debo DJ, Pikal MJ, Evaluation of tunable diode laser absorption spectroscopy for in-process water vapor mass flux measurements during freeze drying, Journal of Pharmaceutical Sciences, 96 (2007) 1776–1793. [DOI] [PubMed] [Google Scholar]
- [64].Fan K, Zhang M, Mujumdar AS, Recent developments in high efficient freeze-drying of fruits and vegetables assisted by microwave: A review, Critical reviews in food science and nutrition, 59 (2019) 1357–1366. [DOI] [PubMed] [Google Scholar]
- [65].Gitter JH, Geidobler R, Presser I, Winter G, Significant Drying Time Reduction Using Microwave-Assisted Freeze-Drying for a Monoclonal Antibody, Journal of Pharmaceutical Sciences, 107 (2018) 2538–2543. [DOI] [PubMed] [Google Scholar]
- [66].Kleinebudde P, Khinast J, Rantanen J, Continuous manufacturing of pharmaceuticals, John Wiley & Sons; 2017. [Google Scholar]
- [67].Burns LR, The biopharmaceutical sector’s impact on the US economy: Analysis at the national, state, and local levels, Washington, DC: Archstone Consulting, LLC, (2009). [Google Scholar]
- [68].De Meyer L, Van Bockstal P-J, Corver J, Vervaet C, Remon JP, De Beer T, Evaluation of spin freezing versus conventional freezing as part of a continuous pharmaceutical freeze-drying concept for unit doses, International Journal of Pharmaceutics, 496 (2015) 75–85. [DOI] [PubMed] [Google Scholar]
- [69].Pisano R, Arsiccio A, Capozzi LC, Trout BL, Achieving continuous manufacturing in lyophilization: Technologies and approaches, European Journal of Pharmaceutics and Biopharmaceutics, 142 (2019) 265–279. [DOI] [PubMed] [Google Scholar]
- [70].Becker HI, “Low voltage electrolytic capacitor.” U.S. Patent No. 2,800,616, 1957.
- [71].Broadwin SM, “Centrifugal freeze drying apparatus.” U.S. Patent No. 3,203,108., 1965.
- [72].Oughton DMA, Smith PRJ, MacMichael DBA, Freeze-drying process and apparatus, Google Patents, 1999.
- [73].Corver JAWM, Method and system for freeze-drying injectable compositions, in particular pharmaceutical compositions, August 2012.
- [74].Capozzi LC, Trout BL, Pisano R, From batch to continuous: freeze-drying of suspended vials for pharmaceuticals in unit-doses, Industrial & Engineering Chemistry Research, 58 (2019) 1635–1649. [Google Scholar]
- [75].Peroy SR, “Improvement in drying and concentrating liquid substances by atomizing.” U.S Patent No. 125406A, (1872).
- [76].Schuck P, Spray drying of dairy products: state of the art, Le Lait, 82 (2002) 375–382. [Google Scholar]
- [77].Truong V, Bhandari BR, Howes T, Optimization of cocurrent spray drying process for sugar-rich foods. Part II—Optimization of spray drying process based on glass transition concept, Journal of Food Engineering, 71 (2005) 66–72. [Google Scholar]
- [78].Gharsallaoui A, Roudaut G, Chambin O, Voilley A, Saurel R, Applications of spray-drying in microencapsulation of food ingredients: An overview, Food research international, 40 (2007) 1107–1121. [Google Scholar]
- [79].Ameri M, Maa Y-F, Spray drying of biopharmaceuticals: stability and process considerations, Drying technology, 24 (2006) 763–768. [Google Scholar]
- [80].Ledet GA, Graves RA, Bostanian LA, Mandal TK, Spray-drying of biopharmaceuticals, Lyophilized Biologics and Vaccines, Springer; 2015, pp. 273–297. [Google Scholar]
- [81].Lefebvre AH, McDonell VG, Atomization and sprays, CRC press; 2017. [Google Scholar]
- [82].Williams III RO, Watts AB, Miller DA, Formulating poorly water soluble drugs, Springer; 2016. [Google Scholar]
- [83].Chen R, Takahashi H, Okamoto H, Danjo K, Particle design of three-component system for sustained release using a 4-fluid nozzle spray-drying technique, Chem Pharm Bull (Tokyo), 54 (2006) 1486–1490. [DOI] [PubMed] [Google Scholar]
- [84].Sunderland T, Kelly JG, Ramtoola Z, Application of a novel 3-fluid nozzle spray drying process for the microencapsulation of therapeutic agents using incompatible drug-polymer solutions, Arch Pharm Res, 38 (2015) 566–573. [DOI] [PubMed] [Google Scholar]
- [85].Pabari RM, Sunderland T, Ramtoola Z, Investigation of a novel 3-fluid nozzle spray drying technology for the engineering of multifunctional layered microparticles, Expert Opin Drug Deliv, 9 (2012) 1463–1474. [DOI] [PubMed] [Google Scholar]
- [86].Cal K, Sollohub K, Spray drying technique. I: Hardware and process parameters, Journal of pharmaceutical sciences, 99 (2010) 575–586. [DOI] [PubMed] [Google Scholar]
- [87].Piatkowski M, Zbicinski I, Analysis of the Mechanism of Counter-current Spray Drying, 1970, pp. 89–101. [Google Scholar]
- [88].Masters K, Spray drying handbook, Spray drying handbook., (1985). [Google Scholar]
- [89].Ziaee A, Albadarin AB, Padrela L, Femmer T, O’Reilly E, Walker G, Spray drying of pharmaceuticals and biopharmaceuticals: Critical parameters and experimental process optimization approaches, European Journal of Pharmaceutical Sciences, 127 (2019) 300–318. [DOI] [PubMed] [Google Scholar]
- [90].Maa Y-F, Prestrelski SJ, Biopharmaceutical powders particle formation and formulation considerations, Current pharmaceutical biotechnology, 1 (2000) 283–302. [DOI] [PubMed] [Google Scholar]
- [91].Tracy MA, Development and scale-up of a microsphere protein delivery system, Biotechnology progress, 14 (1998) 108–115. [DOI] [PubMed] [Google Scholar]
- [92].Chen D, Maa Y-F, Haynes JR, Needle-free epidermal powder immunization, Expert Review of Vaccines, 1 (2002) 265–276. [DOI] [PubMed] [Google Scholar]
- [93].Maa Y-F, Nguyen P-A, Sweeney T, Shire SJ, Hsu CC, Protein inhalation powders: spray drying vs spray freeze drying, Pharmaceutical research, 16 (1999) 249–254. [DOI] [PubMed] [Google Scholar]
- [94].Mezhericher M, Levy A, Borde I, Heat and mass transfer of single droplet/wet particle drying, Chemical Engineering Science, 63 (2008) 12–23. [Google Scholar]
- [95].Wilson NE, Topp EM, Zhou QT, Effects of drying method and excipient on structure and stability of protein solids using solid-state hydrogen/deuterium exchange mass spectrometry (ssHDX-MS), International Journal of Pharmaceutics, 567 (2019) 118470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Wilson NE, Mutukuri TT, Zemlyanov DY, Taylor LS, Topp EM, Zhou QT, Surface Composition and Formulation Heterogeneity of Protein Solids Produced by Spray Drying, Pharmaceutical Research, 37 (2020) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Hulse WL, Forbes RT, Bonner MC, Getrost M, Do co-spray dried excipients offer better lysozyme stabilisation than single excipients?, European journal of pharmaceutical sciences, 33 (2008) 294–305. [DOI] [PubMed] [Google Scholar]
- [98].Ógáin ON, Li J, Tajber L, Corrigan OI, Healy AM, Particle engineering of materials for oral inhalation by dry powder inhalers. I—Particles of sugar excipients (trehalose and raffinose) for protein delivery, International journal of pharmaceutics, 405 (2011) 23–35. [DOI] [PubMed] [Google Scholar]
- [99].Maury M, Murphy K, Kumar S, Mauerer A, Lee G, Spray-drying of proteins: effects of sorbitol and trehalose on aggregation and FT-IR amide I spectrum of an immunoglobulin G, European journal of pharmaceutics and biopharmaceutics, 59 (2005) 251–261. [DOI] [PubMed] [Google Scholar]
- [100].Haj-Ahmad RR, Elkordy AA, Chaw CS, Moore A, Compare and contrast the effects of surfactants (Pluronic® F-127 and Cremophor® EL) and sugars (β-cyclodextrin and inulin) on properties of spray dried and crystallised lysozyme, European Journal of Pharmaceutical Sciences, 49 (2013) 519–534. [DOI] [PubMed] [Google Scholar]
- [101].Jacob S, Shirwaikar A, Srinivasan K, Alex J, Prabu S, Mahalaxmi R, Kumar R, Stability of proteins in aqueous solution and solid state, Indian journal of pharmaceutical sciences, 68 (2006). [Google Scholar]
- [102].Akers MJ, DeFelippis MR, Peptides and proteins as parenteral solutions, Pharmaceutical formulation development of peptides and proteins, (2012) 145. [Google Scholar]
- [103].Ajmera A, Scherließ R, Stabilisation of proteins via mixtures of amino acids during spray drying, International Journal of Pharmaceutics, 463 (2014) 98–107. [DOI] [PubMed] [Google Scholar]
- [104].Perdana J, Fox MB, Schutyser MA, Boom RM, Single-droplet experimentation on spray drying: evaporation of a sessile droplet, Chemical Engineering & Technology, 34 (2011) 1151–1158. [Google Scholar]
- [105].Schutyser MA, Perdana J, Boom RM, Single droplet drying for optimal spray drying of enzymes and probiotics, Trends in Food Science & Technology, 27 (2012) 73–82. [Google Scholar]
- [106].Maltesen MJ, Bjerregaard S, Hovgaard L, Havelund S, Van De Weert M, Quality by design–Spray drying of insulin intended for inhalation, European journal of pharmaceutics and biopharmaceutics, 70 (2008) 828–838. [DOI] [PubMed] [Google Scholar]
- [107].Jamaleddine TJ, Ray MB, Application of computational fluid dynamics for simulation of drying processes: A review, Drying technology, 28 (2010) 120–154. [Google Scholar]
- [108].Keshani S, Daud WRW, Nourouzi M, Namvar F, Ghasemi M, Spray drying: An overview on wall deposition, process and modeling, Journal of Food Engineering, 146 (2015) 152–162. [Google Scholar]
- [109].White S, Bennett DB, Cheu S, Conley PW, Guzek DB, Gray S, Howard J, Malcolmson R, Parker JM, Roberts P, EXUBERA®: pharmaceutical development of a novel product for pulmonary delivery of insulin, Diabetes technology & therapeutics, 7 (2005) 896–906. [DOI] [PubMed] [Google Scholar]
- [110].Heinemann L, The failure of exubera: are we beating a dead horse?, Journal of diabetes science and technology, 2 (2008) 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].McKeage K, Raplixa™: a review in improving surgical haemostasis, Clinical drug investigation, 35 (2015) 519–524. [DOI] [PubMed] [Google Scholar]
- [112].Benson SW, Ellis DA, Surface Areas of Proteins. I. Surface Areas and Heats of Absorption1, Journal of the American Chemical Society, 70 (1948) 3563–3569. [DOI] [PubMed] [Google Scholar]
- [113].Ishwarya SP, Anandharamakrishnan C, Stapley AG, Spray-freeze-drying: a novel process for the drying of foods and bioproducts, Trends in Food Science & Technology, 41 (2015) 161–181. [Google Scholar]
- [114].Costantino HR, Firouzabadian L, Hogeland K, Wu C, Beganski C, Carrasquillo KG, Córdova M, Griebenow K, Zale SE, Tracy MA, Protein Spray-Freeze Drying. Effect of Atomization Conditions on Particle Size and Stability, Pharmaceutical Research, 17 (2000) 1374–1382. [DOI] [PubMed] [Google Scholar]
- [115].Costantino HR, Firouzabadian L, Wu C, Carrasquillo KG, Griebenow K, Zale SE, Tracy MA, Protein spray freeze drying. 2. Effect of formulation variables on particle size and stability, Journal of pharmaceutical sciences, 91 (2002) 388–395. [DOI] [PubMed] [Google Scholar]
- [116].Gieseler H, Product morphology and drying behavior delineated by a new freeze-drying microbalance, Verlag nicht ermittelbar, 2004. [Google Scholar]
- [117].Wanning S, Sueverkruep R, Lamprecht A, Pharmaceutical spray freeze drying, International journal of pharmaceutics, 488 (2015) 136–153. [DOI] [PubMed] [Google Scholar]
- [118].Rogers TL, Johnston KP, Williams III RO, Solution-based particle formation of pharmaceutical powders by supercritical or compressed fluid CO2 and cryogenic spray-freezing technologies, Drug development and industrial pharmacy, 27 (2001) 1003–1015. [DOI] [PubMed] [Google Scholar]
- [119].Emami F, Vatanara A, Najafabadi AR, Kim Y, Park EJ, Sardari S, Na DH, Effect of amino acids on the stability of spray freeze-dried immunoglobulin G in sugar-based matrices, European Journal of Pharmaceutical Sciences, 119 (2018) 39–48. [DOI] [PubMed] [Google Scholar]
- [120].Zhu C, Chen J, Yu S, Que C, Taylor LS, Tan W, Wu C, Zhou QT, Inhalable Nanocomposite Microparticles with Enhanced Dissolution and Superior Aerosol Performance, Molecular Pharmaceutics, 17 (2020) 3270–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Poursina N, Vatanara A, Rouini MR, Gilani K, Najafabadi AR, The effect of excipients on the stability and aerosol performance of salmon calcitonin dry powder inhalers prepared via the spray freeze drying process, Acta Pharmaceutica, 66 (2016) 207–218. [DOI] [PubMed] [Google Scholar]
- [122].Poursina N, Vatanara A, Rouini MR, Gilani K, Rouholamini Najafabadi A, Systemic delivery of parathyroid hormone (1–34) using spray freeze-dried inhalable particles, Pharmaceutical Development and Technology, 22 (2017) 733–739. [DOI] [PubMed] [Google Scholar]
- [123].Mutukuri TT, Wilson NE, Taylor LS, Topp EM, Zhou QT, Effects of drying method and excipient on the structure and physical stability of protein solids: Freeze drying vs. spray freeze drying, International Journal of Pharmaceutics, 594 (2021) 120169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Rogers TL, Hu J, Yu Z, Johnston KP, Williams III RO, A novel particle engineering technology: spray-freezing into liquid, International Journal of pharmaceutics, 242 (2002) 93–100. [DOI] [PubMed] [Google Scholar]
- [125].Maa YF, Ameri M, Shu C, Payne LG, Chen D, Influenza vaccine powder formulation development: spray-freeze-drying and stability evaluation, Journal of pharmaceutical sciences, 93 (2004) 1912–1923. [DOI] [PubMed] [Google Scholar]
- [126].Sonner C, Maa YF, Lee G, Spray-freeze-drying for protein powder preparation: Particle characterization and a case study with trypsinogen stability, Journal of pharmaceutical sciences, 91 (2002) 2122–2139. [DOI] [PubMed] [Google Scholar]
- [127].Jovanović N, Bouchard A, Hofland GW, Witkamp G-J, Crommelin DJ, Jiskoot W, Stabilization of proteins in dry powder formulations using supercritical fluid technology, Pharmaceutical research, 21 (2004) 1955–1969. [DOI] [PubMed] [Google Scholar]
- [128].Jovanović N, Bouchard A, Sutter M, Van Speybroeck M, Hofland GW, Witkamp G-J, Crommelin DJ, Jiskoot W, Stable sugar-based protein formulations by supercritical fluid drying, International journal of pharmaceutics, 346 (2008) 102–108. [DOI] [PubMed] [Google Scholar]
- [129].Maltesen MJ, Van De Weert M, Drying methods for protein pharmaceuticals, Drug Discovery Today: Technologies, 5 (2008) e81–e88. [DOI] [PubMed] [Google Scholar]
- [130].Nuchuchua O, Every H, Hofland G, Jiskoot W, Scalable organic solvent free supercritical fluid spray drying process for producing dry protein formulations, European Journal of Pharmaceutics and Biopharmaceutics, 88 (2014) 919–930. [DOI] [PubMed] [Google Scholar]
- [131].Ohtake S, Martin R, Saxena A, Pham B, Chiueh G, Osorio M, Kopecko D, Xu D, Lechuga-Ballesteros D, Truong-Le V, Room temperature stabilization of oral, live attenuated Salmonella enterica serovar Typhi-vectored vaccines, Vaccine, 29 (2011) 2761–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Walters RH, Bhatnagar B, Tchessalov S, Izutsu K-I, Tsumoto K, Ohtake S, Next generation drying technologies for pharmaceutical applications, Journal of pharmaceutical sciences, 103 (2014) 2673–2695. [DOI] [PubMed] [Google Scholar]
- [133].Cryz S Jr, Pasteris O, Varallyay S, Fürer E, Factors influencing the stability of live oral attenuated bacterial vaccines, Developments in biological standardization, 87 (1996) 277–281. [PubMed] [Google Scholar]
- [134].Ohtake S, Martin RA, Saxena A, Lechuga-ballesteros D, Santiago AE, Barry EM, Truong-Le V, Formulation and stabilization of Francisella tularensis live vaccine strain, Journal of pharmaceutical sciences, 100 (2011) 3076–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Cicerone MT, Soles CL, Fast dynamics and stabilization of proteins: binary glasses of trehalose and glycerol, Biophysical journal, 86 (2004) 3836–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].www.chemengonline.com, ELECTROSTATIC SPRAY DRYING PROTECTS HEAT-SENSITIVE PRODUCTS, https://www.chemengonline.com/electrostatic-spray-drying-protects-heat-sensitive-products/, 2017.
- [137].Thakkar S, Misra M, Electrospray drying of docetaxel nanosuspension: A study on particle formation and evaluation of nanocrystals thereof, Journal of Drug Delivery Science and Technology, 60 (2020) 102009. [Google Scholar]
- [138].Gomez A, Bingham D, Juan L.d., Tang K, Production of protein nanoparticles by electrospray drying, Journal of Aerosol Science, 29 (1998) 561–574. [Google Scholar]
- [139].Wang J, Helder L, Shao J, Jansen JA, Yang M, Yang F, Encapsulation and release of doxycycline from electrospray-generated PLGA microspheres: Effect of polymer end groups, International Journal of Pharmaceutics, 564 (2019) 1–9. [DOI] [PubMed] [Google Scholar]
- [140].F. A Joseph P. Szczap and Associates, Aurora, IL, USA, Spray Drying Process Improved Using Low-Temperature Electrostatic Atomization.
- [141].Abdul-Fattah AM, Kalonia DS, Pikal MJ, The challenge of drying method selection for protein pharmaceuticals: product quality implications, Journal of pharmaceutical sciences, 96 (2007) 1886–1916. [DOI] [PubMed] [Google Scholar]
- [142].Ohtake S, Kita Y, Arakawa T, Interactions of formulation excipients with proteins in solution and in the dried state, Advanced Drug Delivery Reviews, 63 (2011) 1053–1073. [DOI] [PubMed] [Google Scholar]
- [143].Bhatnagar BS, Bogner RH, Pikal MJ, Protein Stability During Freezing: Separation of Stresses and Mechanisms of Protein Stabilization, Pharmaceutical Development and Technology, 12 (2007) 505–523. [DOI] [PubMed] [Google Scholar]
- [144].Langford A, Bhatnagar B, Walters R, Tchessalov S, Ohtake S, Drying technologies for biopharmaceutical applications: Recent developments and future direction, Drying Technology, 36 (2018) 677–684. [Google Scholar]
- [145].Hill JJ, Shalaev EY, Zografi G, Thermodynamic and dynamic factors involved in the stability of native protein structure in amorphous solids in relation to levels of hydration, Journal of Pharmaceutical Sciences, 94 (2005) 1636–1667. [DOI] [PubMed] [Google Scholar]
- [146].Casal HL, Köhler U, Mantsch HH, Structural and conformational changes of β-lactoglobulin B: an infrared spectroscopic study of the effect of pH and temperature, Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology, 957 (1988) 11–20. [DOI] [PubMed] [Google Scholar]
- [147].Chi EY, Krishnan S, Randolph TW, Carpenter JF, Physical stability of proteins in aqueous solution: mechanism and driving forces in nonnative protein aggregation, Pharmaceutical research, 20 (2003) 1325–1336. [DOI] [PubMed] [Google Scholar]
- [148].Franks F, Hatley RHM, Friedman HL, The thermodynamics of protein stability: Cold destabilization as a general phenomenon, Biophysical Chemistry, 31 (1988) 307–315. [DOI] [PubMed] [Google Scholar]
- [149].J. BW, A. SJ, Protein stability curves, Biopolymers, 26 (1987) 1859–1877. [DOI] [PubMed] [Google Scholar]
- [150].Moriyama Y, Watanabe E, Kobayashi K, Harano H, Inui E, Takeda K, Secondary Structural Change of Bovine Serum Albumin in Thermal Denaturation up to 130 °C and Protective Effect of Sodium Dodecyl Sulfate on the Change, The Journal of Physical Chemistry B, 112 (2008) 16585–16589. [DOI] [PubMed] [Google Scholar]
- [151].Azuaga AI, Galisteo ML, Mayorga OL, Cortijo M, Mateo PL, Heat and cold denaturation of β-lactoglobulin B, FEBS Letters, 309 (1992) 258–260. [DOI] [PubMed] [Google Scholar]
- [152].Sabelko J, Ervin J, Gruebele M, Cold-Denatured Ensemble of Apomyoglobin: Implications for the Early Steps of Folding, The Journal of Physical Chemistry B, 102 (1998) 1806–1819. [Google Scholar]
- [153].Haque MA, Aldred P, Chen J, Barrow C, Adhikari B, Drying and Denaturation Characteristics of α-Lactalbumin, β-Lactoglobulin, and Bovine Serum Albumin in a Convective Drying Process, Journal of Agricultural and Food Chemistry, 62 (2014) 4695–4706. [DOI] [PubMed] [Google Scholar]
- [154].Wang W, Instability, stabilization, and formulation of liquid protein pharmaceuticals, International Journal of Pharmaceutics, 185 (1999) 129–188. [DOI] [PubMed] [Google Scholar]
- [155].Panick G, Malessa R, Winter R, Differences between the Pressure- and Temperature-Induced Denaturation and Aggregation of β-Lactoglobulin A, B, and AB Monitored by FT-IR Spectroscopy and Small-Angle X-ray Scattering, Biochemistry, 38 (1999) 6512–6519. [DOI] [PubMed] [Google Scholar]
- [156].Masters K, Understanding and applying spray dryers in chemical processing, Powder Bulk Eng, (1990) 36–44. [Google Scholar]
- [157].Mumenthaler M, Hsu CC, Pearlman R, Feasibility Study on Spray-Drying Protein Pharmaceuticals: Recombinant Human Growth Hormone and Tissue-Type Plasminogen Activator, Pharmaceutical Research, 11 (1994) 12–20. [DOI] [PubMed] [Google Scholar]
- [158].Maa Y-F, Costantino HR, Nguyen P-A, Hsu CC, The Effect of Operating and Formulation Variables on the Morphology of Spray-Dried Protein Particles, Pharmaceutical Development and Technology, 2 (1997) 213–223. [DOI] [PubMed] [Google Scholar]
- [159].Haque MA, Adhikari B, Drying and denaturation of proteins in spray drying process, Handbook of industrial drying, (2015) 971–985. [Google Scholar]
- [160].Jaenicke R, Protein structure and function at low temperatures, Philosophical Transactions of the Royal Society of London. B, Biological Sciences, 326 (1990) 535–553. [DOI] [PubMed] [Google Scholar]
- [161].Yoshidome T, Kinoshita M, Hydrophobicity at low temperatures and cold denaturation of a protein, Physical Review E, 79 (2009) 030905. [DOI] [PubMed] [Google Scholar]
- [162].Yoshidome T, Kinoshita M, Physical origin of hydrophobicity studied in terms of cold denaturation of proteins: comparison between water and simple fluids, Physical Chemistry Chemical Physics, 14 (2012) 14554–14566. [DOI] [PubMed] [Google Scholar]
- [163].Dias CL, Ala-Nissila T, Wong-ekkabut J, Vattulainen I, Grant M, Karttunen M, The hydrophobic effect and its role in cold denaturation, Cryobiology, 60 (2010) 91–99. [DOI] [PubMed] [Google Scholar]
- [164].Privalov PL, Cold Denaturation of Protein, Critical Reviews in Biochemistry and Molecular Biology, 25 (1990) 281–306. [DOI] [PubMed] [Google Scholar]
- [165].Lopez CF, Darst RK, Rossky PJ, Mechanistic Elements of Protein Cold Denaturation, The Journal of Physical Chemistry B, 112 (2008) 5961–5967. [DOI] [PubMed] [Google Scholar]
- [166].Tang X, Pikal MJ, The Effect of Stabilizers and Denaturants on the Cold Denaturation Temperatures of Proteins and Implications for Freeze-Drying, Pharmaceutical Research, 22 (2005) 1167–1175. [DOI] [PubMed] [Google Scholar]
- [167].Nitin R, S. RR, Current Perspectives on Stability of Protein Drug Products during Formulation, Fill and Finish Operations, Biotechnology Progress, 24 (2008) 504–514. [DOI] [PubMed] [Google Scholar]
- [168].Cromwell MEM, Hilario E, Jacobson F, Protein aggregation and bioprocessing, The AAPS Journal, 8 (2006) E572–E579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Nakanishi K, Sakiyama T, Imamura K, On the adsorption of proteins on solid surfaces, a common but very complicated phenomenon, Journal of Bioscience and Bioengineering, 91 (2001) 233–244. [DOI] [PubMed] [Google Scholar]
- [170].Thomas CR, Geer D, Effects of shear on proteins in solution, Biotechnology Letters, 33 (2011) 443–456. [DOI] [PubMed] [Google Scholar]
- [171].Bee JS, Stevenson JL, Mehta B, Svitel J, Pollastrini J, Platz R, Freund E, Carpenter JF, Randolph TW, Response of a Concentrated Monoclonal Antibody Formulation to High Shear, Biotechnology and bioengineering, 103 (2009) 936–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Tzannis ST, Hrushesky WJM, Wood PA, Przybycien TM, Adsorption of a Formulated Protein on a Drug Delivery Device Surface, Journal of Colloid and Interface Science, 189 (1997) 216–228. [Google Scholar]
- [173].Truskey GA, Gabler R, DiLEO A, Manter T, The Effect of Membrane Filtration Upon Protein Conformation, PDA Journal of Pharmaceutical Science and Technology, 41 (1987) 180–191. [PubMed] [Google Scholar]
- [174].Tyagi AK, Randolph TW, Dong A, Maloney KM, Hitscherich C, Carpenter JF, IgG particle formation during filling pump operation: A case study of heterogeneous nucleation on stainless steel nanoparticles, Journal of Pharmaceutical Sciences, 98 (2009) 94–104. [DOI] [PubMed] [Google Scholar]
- [175].Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS, Stability of Protein Pharmaceuticals: An Update, Pharmaceutical Research, 27 (2010) 544–575. [DOI] [PubMed] [Google Scholar]
- [176].Colombié S, Gaunand A, Lindet B, Lysozyme inactivation under mechanical stirring: effect of physical and molecular interfaces, Enzyme and Microbial Technology, 28 (2001) 820–826. [DOI] [PubMed] [Google Scholar]
- [177].Bee JS, Davis M, Freund E, Carpenter JF, Randolph TW, Aggregation of a Monoclonal Antibody Induced by Adsorption to Stainless Steel, Biotechnology and bioengineering, 105 (2010) 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Wahlgren M, Arnebrant T, Paulsson MA, The Adsorption from Solutions of β-Lactoglobulin Mixed with Lactoferrin or Lysozyme onto Silica and Methylated Silica Surfaces, 1993.
- [179].Barroug A, Lernoux E, Lemaitre J, Rouxhet PG, Adsorption of Catalase on Hydroxyapatite, Journal of Colloid and Interface Science, 208 (1998) 147–152. [DOI] [PubMed] [Google Scholar]
- [180].Jared B, David C, Suzanne S, L. SJ, Koustuv C, Erwin F, F. CJ, W. RT, Monoclonal antibody interactions with micro‐ and nanoparticles: Adsorption, aggregation, and accelerated stress studies, Journal of Pharmaceutical Sciences, 98 (2009) 3218–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Chang BS, Kendrick BS, Carpenter JF, Surface-induced denaturation of proteins during freezing and its inhibition by surfactants, Journal of Pharmaceutical Sciences, 85 (1996) 1325–1330. [DOI] [PubMed] [Google Scholar]
- [182].Wendorf JR, Radke CJ, Blanch HW, Reduced protein adsorption at solid interfaces by sugar excipients, Biotechnology and Bioengineering, 87 (2004) 565–573. [DOI] [PubMed] [Google Scholar]
- [183].Maa Y-F, Nguyen P-AT, Hsu SW, Spray-Drying of Air–Liquid Interface Sensitive Recombinant Human Growth Hormone, Journal of Pharmaceutical Sciences, 87 (1998) 152–159. [DOI] [PubMed] [Google Scholar]
- [184].Webb SD, Golledge SL, Cleland JL, Carpenter JF, Randolph TW, Surface adsorption of recombinant human interferon‐γ in lyophilized and spray‐lyophilized formulations, Journal of Pharmaceutical Sciences, 91 (2002) 1474–1487. [DOI] [PubMed] [Google Scholar]
- [185].Sharma B, Immunogenicity of therapeutic proteins. Part 2: Impact of container closures, Biotechnology Advances, 25 (2007) 318–324. [DOI] [PubMed] [Google Scholar]
- [186].Kiese S, Papppenberger A, Friess W, Mahler H-C, Shaken, Not Stirred: Mechanical Stress Testing of an IgG1 Antibody, Journal of Pharmaceutical Sciences, 97 (2008) 4347–4366. [DOI] [PubMed] [Google Scholar]
- [187].Wiesbauer J, Prassl R, Nidetzky B, Renewal of the Air–Water Interface as a Critical System Parameter of Protein Stability: Aggregation of the Human Growth Hormone and Its Prevention by Surface-Active Compounds, Langmuir, 29 (2013) 15240–15250. [DOI] [PubMed] [Google Scholar]
- [188].Patapoff TW, Esue O, Polysorbate 20 prevents the precipitation of a monoclonal antibody during shear, Pharmaceutical Development and Technology, 14 (2009) 659–664. [DOI] [PubMed] [Google Scholar]
- [189].Wilson NE, Mutukuri TT, Zemlyanov DY, Taylor LS, Topp EM, Zhou QT, Surface Composition and Formulation Heterogeneity of Protein Solids Produced by Spray Drying, Pharmaceutical Research, 37 (2019) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Mazzola Priscila G, Lopes Andre M, Hasmann Francislene A, Jozala Angela F, Penna Thereza CV, Magalhaes Perola O, Rangel‐Yagui Carlota O, Pessoa A Jr, Liquid–liquid extraction of biomolecules: an overview and update of the main techniques, Journal of Chemical Technology & Biotechnology, 83 (2007) 143–157. [Google Scholar]
- [191].Sah H, Stabilization of proteins against methylene chloride/water interface-induced denaturation and aggregation, Journal of Controlled Release, 58 (1999) 143–151. [DOI] [PubMed] [Google Scholar]
- [192].Jones LS, Kaufmann A, Middaugh CR, Silicone Oil Induced Aggregation of Proteins, Journal of Pharmaceutical Sciences, 94 (2005) 918–927. [DOI] [PubMed] [Google Scholar]
- [193].Brash JL, Horbett TA, Proteins at Interfaces, Proteins at Interfaces II, American Chemical Society; 1995, pp. 1–23. [Google Scholar]
- [194].Owusu RK, Cowan DA, Correlation between microbial protein thermostability and resistance to denaturation in aqueous: organic solvent two-phase systems, Enzyme and Microbial Technology, 11 (1989) 568–574. [Google Scholar]
- [195].Colja L, Sjef B, Kees V, Cees V, Rules for optimization of biocatalysis in organic solvents, Biotechnology and Bioengineering, 30 (1987) 81–87. [DOI] [PubMed] [Google Scholar]
- [196].Miller R, Fainerman VB, Makievski AV, Krägel J, Grigoriev DO, Kazakov VN, Sinyachenko OV, Dynamics of protein and mixed protein/surfactant adsorption layers at the water/fluid interface, Advances in Colloid and Interface Science, 86 (2000) 39–82. [DOI] [PubMed] [Google Scholar]
- [197].Heller MC, Carpenter JF, Randolph TW, Effects of Phase Separating Systems on Lyophilized Hemoglobin, Journal of Pharmaceutical Sciences, 85 (1996) 1358–1362. [DOI] [PubMed] [Google Scholar]
- [198].Timasheff SN, Protein Hydration, Thermodynamic Binding, and Preferential Hydration, Biochemistry, 41 (2002) 13473–13482. [DOI] [PubMed] [Google Scholar]
- [199].Allison SD, Chang B, Randolph TW, Carpenter JF, Hydrogen bonding between sugar and protein is responsible for inhibition of dehydration-induced protein unfolding, Archives of Biochemistry and Biophysics, 365 (1999) 289–298. [DOI] [PubMed] [Google Scholar]
- [200].Oobatake M, Ooi T, Hydration and heat stability effects on protein unfolding A2 - DOYAMA, Masao, in: Kihara J, Tanaka M, Yamamoto R (Eds.) Computer Aided Innovation of New Materials II, Elsevier, Oxford, 1993, pp. 1307–1310. [Google Scholar]
- [201].Zaks A, Protein-water interactions. Role in protein structure and stability, Pharmaceutical Biotechnology, 2 (1992) 249–271. [Google Scholar]
- [202].Costantino HR, Langer R, Klibanov AM, Aggregation of a Lyophilized Pharmaceutical Protein, Recombinant Human Albumin: Effect of Moisture and Stabilization by Excipients, Bio/Technology, 13 (1995) 493. [DOI] [PubMed] [Google Scholar]
- [203].Robert F, Kenneth D, Michael H, Jeremy C, Janet M, Water vapor sorption studies on the physical stability of a series of spray‐dried protein/sugar powders for inhalation, Journal of Pharmaceutical Sciences, 87 (1998) 1316–1321. [DOI] [PubMed] [Google Scholar]
- [204].Breen ED, Curley JG, Overcashier DE, Hsu CC, Shire SJ, Effect of moisture on the stability of a lyophilized humanized monoclonal antibody formulation, Pharmaceutical research, 18 (2001) 1345–1353. [DOI] [PubMed] [Google Scholar]
- [205].Liao Y-H, Brown MB, Quader A, Martin GP, Protective Mechanism of Stabilizing Excipients Against Dehydration in the Freeze-Drying of Proteins, Pharmaceutical Research, 19 (2002) 1854–1861. [DOI] [PubMed] [Google Scholar]
- [206].Carpenter JF, Crowe JH, The mechanism of cryoprotection of proteins by solutes, Cryobiology, 25 (1988) 244–255. [DOI] [PubMed] [Google Scholar]
- [207].Hageman MJ, The Role of Moisture in Protein Stability, Drug Development and Industrial Pharmacy, 14 (1988) 2047–2070. [Google Scholar]
- [208].Andya JD, Maa Y-F, Costantino HR, Nguyen P-A, Dasovich N, Sweeney TD, Hsu CC, Shire SJ, The Effect of Formulation Excipients on Protein Stability and Aerosol Performance of Spray-Dried Powders of a Recombinant Humanized Anti-IgE Monoclonal Antibody1, Pharmaceutical Research, 16 (1999) 350–358. [DOI] [PubMed] [Google Scholar]
- [209].Izutsu K.-i., Kojima S, Excipient crystallinity and its protein-structure-stabilizing effect during freeze-drying, Journal of Pharmacy and Pharmacology, 54 (2002) 1033–1039. [DOI] [PubMed] [Google Scholar]
- [210].Lazaros K, Joe R, Effect of pH and Salts on the Solubility of Egg White Protein, Journal of Food Science, 51 (1986) 1445–1447. [Google Scholar]
- [211].Dill KA, Dominant forces in protein folding, Biochemistry, 29 (1990) 7133–7155. [DOI] [PubMed] [Google Scholar]
- [212].Kristinsson HG, Hultin HO, Changes in Conformation and Subunit Assembly of Cod Myosin at Low and High pH and after Subsequent Refolding, Journal of Agricultural and Food Chemistry, 51 (2003) 7187–7196. [DOI] [PubMed] [Google Scholar]
- [213].Chi EY, Krishnan S, Kendrick BS, Chang BS, Carpenter JF, Randolph TW, Roles of conformational stability and colloidal stability in the aggregation of recombinant human granulocyte colony-stimulating factor, Protein Science : A Publication of the Protein Society, 12 (2003) 903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Kuwajima K, The molten globule state as a clue for understanding the folding and cooperativity of globular‐protein structure, Proteins: Structure, Function, and Bioinformatics, 6 (1989) 87–103. [DOI] [PubMed] [Google Scholar]
- [215].Goto Y, Calciano LJ, Fink AL, Acid-induced folding of proteins, Proceedings of the National Academy of Sciences of the United States of America, 87 (1990) 573–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Fink AL, Calciano LJ, Goto Y, Nishimura M, Swedberg SA, Characterization of the stable, acid‐induced, molten globule-like state of staphylococcal nuclease, Protein Science, 2 (1993) 1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Dolgikh DA, Gilmanshin RI, Brazhnikov EV, Bychkova VE, Semisotnov GV, Venyaminov SY, Ptitsyn OB, α‐Lactalbumin: compact state with fluctuating tertiary structure?, FEBS letters, 136 (1981) 311–315. [DOI] [PubMed] [Google Scholar]
- [218].Arakawa T, Timasheff SN, Mechanism of protein salting in and salting out by divalent cation salts: balance between hydration and salt binding, Biochemistry, 23 (1984) 5912–5923. [DOI] [PubMed] [Google Scholar]
- [219].Van Den Berg L, pH changes in buffers and foods during freezing and subsequent storage, Cryobiology, 3 (1966) 236–242. [Google Scholar]
- [220].Thorat AA, Suryanarayanan R, Characterization of phosphate buffered saline (PBS) in frozen State and after Freeze-drying, Pharmaceutical research, 36 (2019) 98. [DOI] [PubMed] [Google Scholar]
- [221].Thorat AA, Munjal B, Geders TW, Suryanarayanan R, Freezing-induced protein aggregation-Role of pH shift and potential mitigation strategies, Journal of Controlled Release, (2020). [DOI] [PubMed] [Google Scholar]
- [222].Li M, Koranne S, Fang R, Lu X, Williams DM, Munson EJ, Bhambhani A, Su Y, Probing Microenvironmental Acidity in Lyophilized Protein and Vaccine Formulations Using Solid-state NMR Spectroscopy, Journal of Pharmaceutical Sciences, (2020). [DOI] [PubMed] [Google Scholar]
- [223].Bam NB, Randolph TW, Cleland JL, Stability of Protein Formulations: Investigation of Surfactant Effects by a Novel EPR Spectroscopic Technique, Pharmaceutical Research, 12 (1995) 2–11. [DOI] [PubMed] [Google Scholar]
- [224].Shibata H, Vacuum Drying of Porous Solids under Supercooling, Drying Technology, 24 (2006) 541–550. [Google Scholar]
- [225].Enhong C, Yahuei C, Zhanfeng C, R. FP, Effect of freezing and thawing rates on denaturation of proteins in aqueous solutions, Biotechnology and Bioengineering, 82 (2003) 684–690. [DOI] [PubMed] [Google Scholar]
- [226].Izutsu K.-i., Kojima S, Freeze-Concentration Separates Proteins and Polymer Excipients Into Different Amorphous Phases, Pharmaceutical Research, 17 (2000) 1316–1322. [DOI] [PubMed] [Google Scholar]
- [227].Parag K, Elizabeth A, Satish KS, Impact of freezing on pH of buffered solutions and consequences for monoclonal antibody aggregation, Biotechnology Progress, 26 (2010) 727–733. [DOI] [PubMed] [Google Scholar]
- [228].Patel SM, Bhugra C, Pikal MJ, Reduced Pressure Ice Fog Technique for Controlled Ice Nucleation during Freeze-Drying, AAPS PharmSciTech, 10 (2009) 1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Heremans K, Smeller L, Protein structure and dynamics at high pressure1Dedicated to the memory of Gregorio Weber (1916–1997), a pioneer in high pressure biophysics.1, Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology, 1386 (1998) 353–370. [DOI] [PubMed] [Google Scholar]
- [230].Webb JN, Webb SD, Cleland JL, Carpenter JF, Randolph TW, Partial molar volume, surface area, and hydration changes for equilibrium unfolding and formation of aggregation transition state: High-pressure and cosolute studies on recombinant human IFN-γ, Proceedings of the National Academy of Sciences, 98 (2001) 7259–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Silva JL, Foguel D, Royer CA, Pressure provides new insights into protein folding, dynamics and structure, Trends in Biochemical Sciences, 26 (2001) 612–618. [DOI] [PubMed] [Google Scholar]
- [232].Mozhaev VV, Heremans K, Frank J, Masson P, Balny C, High pressure effects on protein structure and function, Proteins: Structure, Function, and Bioinformatics, 24 (1996) 81–91. [DOI] [PubMed] [Google Scholar]
- [233].Savadkoohi S, Kasapis S, High pressure effects on the structural functionality of condensed globular-protein matrices, International Journal of Biological Macromolecules, 88 (2016) 433–442. [DOI] [PubMed] [Google Scholar]
- [234].Royer CA, Revisiting volume changes in pressure-induced protein unfolding, Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology, 1595 (2002) 201–209. [DOI] [PubMed] [Google Scholar]
- [235].Gorovits BM, Horowitz PM, High Hydrostatic Pressure Can Reverse Aggregation of Protein Folding Intermediates and Facilitate Acquisition of Native Structure, Biochemistry, 37 (1998) 6132–6135. [DOI] [PubMed] [Google Scholar]
- [236].Seefeldt MB, Kim Y-S, Tolley KP, Seely J, Carpenter JF, Randolph TW, High-pressure studies of aggregation of recombinant human interleukin-1 receptor antagonist: Thermodynamics, kinetics, and application to accelerated formulation studies, Protein Science : A Publication of the Protein Society, 14 (2005) 2258–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].John R.J. St., Carpenter JF, Randolph TW, High pressure fosters protein refolding from aggregates at high concentrations, Proceedings of the National Academy of Sciences of the United States of America, 96 (1999) 13029–13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Johnson DE, Biotherapeutics: Challenges and Opportunities for Predictive Toxicology of Monoclonal Antibodies, Int J Mol Sci, 19 (2018) 3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Sahin U, Karikó K, Türeci Ö, mRNA-based therapeutics — developing a new class of drugs, Nature Reviews Drug Discovery, 13 (2014) 759–780. [DOI] [PubMed] [Google Scholar]
- [240].June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC, CAR T cell immunotherapy for human cancer, Science, 359 (2018) 1361. [DOI] [PubMed] [Google Scholar]
- [241].Das TK, Narhi LO, Sreedhara A, Menzen T, Grapentin C, Chou DK, Antochshuk V, Filipe V, Stress Factors in mAb Drug Substance Production Processes: Critical Assessment of Impact on Product Quality and Control Strategy, Journal of Pharmaceutical Sciences, 109 (2020) 116–133. [DOI] [PubMed] [Google Scholar]
- [242].Mensink MA, Frijlink HW, van der Voort Maarschalk K, Hinrichs WLJ, How sugars protect proteins in the solid state and during drying (review): Mechanisms of stabilization in relation to stress conditions, European Journal of Pharmaceutics and Biopharmaceutics, 114 (2017) 288–295. [DOI] [PubMed] [Google Scholar]
- [243].Lam Y-H, Bustami R, Phan T, Chan H-K, Separovic F, A Solid-State NMR Study of Protein Mobility in Lyophilized Protein–Sugar Powders, Journal of Pharmaceutical Sciences, 91 (2002) 943–951. [DOI] [PubMed] [Google Scholar]
- [244].Mensink MA, Nethercott MJ, Hinrichs WLJ, van der Voort Maarschalk K, Frijlink HW, Munson EJ, Pikal MJ, Influence of Miscibility of Protein-Sugar Lyophilizates on Their Storage Stability, The AAPS Journal, 18 (2016) 1225–1232. [DOI] [PubMed] [Google Scholar]
- [245].Moorthy BS, Schultz SG, Kim SG, Topp EM, Predicting Protein Aggregation during Storage in Lyophilized Solids Using Solid State Amide Hydrogen/Deuterium Exchange with Mass Spectrometric Analysis (ssHDX-MS), Molecular Pharmaceutics, 11 (2014) 1869–1879. https://pubs.acs.org/doi/1810.1021/mp500005v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Chen Y, Topp EM, Photolytic Labeling and Its Applications in Protein Drug Discovery and Development, Journal of Pharmaceutical Sciences, 108 (2019) 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Wakankar A, Chen Y, Gokarn Y, Jacobson FS, Analytical methods for physicochemical characterization of antibody drug conjugates, mAbs, 3 (2011) 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Guo Y, Shalaev E, Smith S, Physical stability of pharmaceutical formulations: solid-state characterization of amorphous dispersions, TrAC Trends in Analytical Chemistry, 49 (2013) 137–144. [Google Scholar]
- [249].Shire SJ, Formulation and manufacturability of biologics, Current Opinion in Biotechnology, 20 (2009) 708–714. [DOI] [PubMed] [Google Scholar]
- [250].Yoshioka S, Aso Y, Correlations between Molecular Mobility and Chemical Stability During Storage of Amorphous Pharmaceuticals, Journal of Pharmaceutical Sciences, 96 (2007) 960–981. [DOI] [PubMed] [Google Scholar]
- [251].Knopp MM, Löbmann K, Elder DP, Rades T, Holm R, Recent advances and potential applications of modulated differential scanning calorimetry (mDSC) in drug development, European Journal of Pharmaceutical Sciences, 87 (2016) 164–173. [DOI] [PubMed] [Google Scholar]
- [252].Gill P, Sauerbrunn S, Reading M, Modulated differential scanning calorimetry, Journal of Thermal Analysis and Calorimetry, 40 (1993) 931–939. [Google Scholar]
- [253].Chen T, Oakley DM, Thermal analysis of proteins of pharmaceutical interest, Thermochimica Acta, 248 (1995) 229–244. [Google Scholar]
- [254].Grasmeijer N, Stankovic M, de Waard H, Frijlink HW, Hinrichs WLJ, Unraveling protein stabilization mechanisms: Vitrification and water replacement in a glass transition temperature controlled system, Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics, 1834 (2013) 763–769. [DOI] [PubMed] [Google Scholar]
- [255].Haeuser C, Goldbach P, Huwyler J, Friess W, Allmendinger A, Excipients for Room Temperature Stable Freeze-Dried Monoclonal Antibody Formulations, Journal of Pharmaceutical Sciences, 109 (2020) 807–817. [DOI] [PubMed] [Google Scholar]
- [256].Horn J, Friess W, Detection of Collapse and Crystallization of Saccharide, Protein, and Mannitol Formulations by Optical Fibers in Lyophilization, Frontiers in Chemistry, 6 (2018) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Duddu SP, Dal Monte PR, Effect of Glass Transition Temperature on the Stability of Lyophilized Formulations Containing a Chimeric Therapeutic Monoclonal Antibody, Pharmaceutical Research, 14 (1997) 591–595. [DOI] [PubMed] [Google Scholar]
- [258].Her L-M, Nail SL, Measurement of Glass Transition Temperatures of Freeze-Concentrated Solutes by Differential Scanning Calorimetry, Pharmaceutical Research, 11 (1994) 54–59. [DOI] [PubMed] [Google Scholar]
- [259].Depaz RA, Pansare S, Patel SM, Freeze-Drying Above the Glass Transition Temperature in Amorphous Protein Formulations While Maintaining Product Quality and Improving Process Efficiency, Journal of Pharmaceutical Sciences, 105 (2016) 40–49. [DOI] [PubMed] [Google Scholar]
- [260].Moorthy BS, Zarraga IE, Kumar L, Walters BT, Goldbach P, Topp EM, Allmendinger A, Solid-State Hydrogen–Deuterium Exchange Mass Spectrometry: Correlation of Deuterium Uptake and Long-Term Stability of Lyophilized Monoclonal Antibody Formulations, Molecular Pharmaceutics, 15 (2018) 1–11. [DOI] [PubMed] [Google Scholar]
- [261].Pikal MJ, Rigsbee D, Roy ML, Galreath D, Kovach KJ, Wang B, Carpenter JF, Cicerone MT, Solid state chemistry of proteins: II. The correlation of storage stability of freeze-dried human growth hormone (hGH) with structure and dynamics in the glassy solid, Journal of Pharmaceutical Sciences, 97 (2008) 5106–5121. [DOI] [PubMed] [Google Scholar]
- [262].Goupil-Lamy AV, Smith JC, Yunoki J, Parker SF, Kataoka M, High-Resolution Vibrational Inelastic Neutron Scattering: A New Spectroscopic Tool for Globular Proteins, Journal of the American Chemical Society, 119 (1997) 9268–9273. [Google Scholar]
- [263].Gaspar AM, Appavou MS, Busch S, Unruh T, Doster W, Dynamics of well-folded and natively disordered proteins in solution: a time-of-flight neutron scattering study, European Biophysics Journal, 37 (2008) 573–582. [DOI] [PubMed] [Google Scholar]
- [264].Velev OD, Kaler EW, Lenhoff AM, Protein Interactions in Solution Characterized by Light and Neutron Scattering: Comparison of Lysozyme and Chymotrypsinogen, Biophysical Journal, 75 (1998) 2682–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Tsai AM, Neumann DA, Bell LN, Molecular dynamics of solid-state lysozyme as affected by glycerol and water: a neutron scattering study, Biophysical journal, 79 (2000) 2728–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Zanotti J-M, Bellissent-Funel M-C, Parello J, Hydration-Coupled Dynamics in Proteins Studied by Neutron Scattering and NMR: The Case of the Typical EF-Hand Calcium-Binding Parvalbumin, Biophysical Journal, 76 (1999) 2390–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Castellanos MM, McAuley A, Curtis JE, Investigating Structure and Dynamics of Proteins in Amorphous Phases Using Neutron Scattering, Computational and Structural Biotechnology Journal, 15 (2017) 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Koshari SHS, Nayak PK, Burra S, Zarraga IE, Rajagopal K, Liu Y, Wagner NJ, Lenhoff AM, In Situ Characterization of the Microstructural Evolution of Biopharmaceutical Solid-State Formulations with Implications for Protein Stability, Molecular Pharmaceutics, 16 (2019) 173–183. [DOI] [PubMed] [Google Scholar]
- [269].Wang B, Tchessalov S, Cicerone MT, Warne NW, Pikal MJ, Impact of sucrose level on storage stability of proteins in freeze-dried solids: II. Correlation of aggregation rate with protein structure and molecular mobility**This work is a product of the U.S. Government and is not subject to copyright in the United States, Journal of Pharmaceutical Sciences, 98 (2009) 3145–3166. [DOI] [PubMed] [Google Scholar]
- [270].Berendt RT, Sperger DM, Munson EJ, Isbester PK, Solid-state NMR spectroscopy in pharmaceutical research and analysis, TrAC Trends in Analytical Chemistry, 25 (2006) 977–984. [Google Scholar]
- [271].Brown SP, Applications of high-resolution 1H solid-state NMR, Solid State Nuclear Magnetic Resonance, 41 (2012) 1–27. [DOI] [PubMed] [Google Scholar]
- [272].Opella SJ, Stewart PL, Valentine KG, Protein structure by solid-state NMR spectroscopy, Quarterly Reviews of Biophysics, 19 (1987) 7–49. [DOI] [PubMed] [Google Scholar]
- [273].Li M, Xu W, Su Y, Solid-state NMR Spectroscopy in Pharmaceutical Sciences, TrAC Trends in Analytical Chemistry, (2020) 116152. [Google Scholar]
- [274].Aso Y, Yoshioka S, Zhang J, Zografi G, Effect of water on the molecular mobility of sucrose and poly (vinylpyrrolidone) in a colyophilized formulation as measured by 13C-NMR relaxation time, Chemical and pharmaceutical bulletin, 50 (2002) 822–826. [DOI] [PubMed] [Google Scholar]
- [275].Yoshioka S, Aso Y, Kojima S, Sakurai S, Fujiwara T, Akutsu H, Molecular Mobility of Protein in Lyophilized Formulations Linked to the Molecular Mobility of Polymer Excipients, as Determined by High Resolution 13C Solid-State NMR, Pharmaceutical Research, 16 (1999) 1621–1625. [DOI] [PubMed] [Google Scholar]
- [276].Yoshioka S, Forney KM, Aso Y, Pikal MJ, Effect of Sugars on the Molecular Motion of Freeze-Dried Protein Formulations Reflected by NMR Relaxation Times, Pharmaceutical Research, 28 (2011) 3237–3247. [DOI] [PubMed] [Google Scholar]
- [277].Tian F, Middaugh CR, Offerdahl T, Munson E, Sane S, Rytting JH, Spectroscopic evaluation of the stabilization of humanized monoclonal antibodies in amino acid formulations, International Journal of Pharmaceutics, 335 (2007) 20–31. [DOI] [PubMed] [Google Scholar]
- [278].Park J, Nagapudi K, Vergara C, Ramachander R, Laurence JS, Krishnan S, Effect of pH and Excipients on Structure, Dynamics, and Long-Term Stability of a Model IgG1 Monoclonal Antibody upon Freeze-Drying, Pharmaceutical Research, 30 (2013) 968–984. [DOI] [PubMed] [Google Scholar]
- [279].Arora A, Tamm LK, Biophysical approaches to membrane protein structure determination, Current Opinion in Structural Biology, 11 (2001) 540–547. [DOI] [PubMed] [Google Scholar]
- [280].Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG, High-Resolution Molecular Structure of a Peptide in an Amyloid Fibril Determined by Magic Angle Spinning NMR Spectroscopy, Proceedings of the National Academy of Sciences of the United States of America, 101 (2004) 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Tycko R, Solid-State NMR Studies of Amyloid Fibril Structure, Annual Review of Physical Chemistry, 62 (2011) 279–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Gelenter MD, Smith KJ, Liao S-Y, Mandala VS, Dregni AJ, Lamm MS, Tian Y, Xu W, Pochan DJ, Tucker TJ, The peptide hormone glucagon forms amyloid fibrils with two coexisting β-strand conformations, Nature structural & molecular biology, 26 (2019) 592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Telikepalli S, Kumru OS, Kim JH, Joshi SB, O’Berry KB, Blake-Haskins AW, Perkins MD, Russell Middaugh C, Volkin DB, Characterization of the Physical Stability of a Lyophilized IgG1 mAb after Accelerated Shipping-Like Stress, Journal of Pharmaceutical Sciences, 104 (2015) 495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Wi S, Pancoska P, Keiderling TA, Predictions of protein secondary structures using factor analysis on Fourier transform infrared spectra: Effect of Fourier self-deconvolution of the amide I and amide II bands, Biospectroscopy, 4 (1998) 93–106. [DOI] [PubMed] [Google Scholar]
- [285].Moorthy BS, Iyer LK, Topp EM, Characterizing Protein Structure, Dynamics and Conformation in Lyophilized Solids, Curr Pharm Des, 21 (2015) 5845–5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Byler DM, Susi H, Examination of the secondary structure of proteins by deconvolved FTIR spectra, Biopolymers, 25 (1986) 469–487. [DOI] [PubMed] [Google Scholar]
- [287].Zandomeneghi G, Krebs MRH, McCammon MG, Fändrich M, FTIR reveals structural differences between native beta-sheet proteins and amyloid fibrils, Protein science : a publication of the Protein Society, 13 (2004) 3314–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Kong J, Yu S, Fourier Transform Infrared Spectroscopic Analysis of Protein Secondary Structures, Acta Biochimica et Biophysica Sinica, 39 (2007) 549–559. [DOI] [PubMed] [Google Scholar]
- [289].Carpenter JF, Crowe JH, An infrared spectroscopic study of the interactions of carbohydrates with dried proteins, Biochemistry, 28 (1989) 3916–3922. [DOI] [PubMed] [Google Scholar]
- [290].Chang L, Shepherd D, Sun J, Ouellette D, Grant KL, Tang X, Pikal MJ, Mechanism of protein stabilization by sugars during freeze-drying and storage: Native structure preservation, specific interaction, and/or immobilization in a glassy matrix?, Journal of Pharmaceutical Sciences, 94 (2005) 1427–1444. [DOI] [PubMed] [Google Scholar]
- [291].Moussa EM, Singh SK, Kimmel M, Nema S, Topp EM, Probing the Conformation of an IgG1 Monoclonal Antibody in Lyophilized Solids Using Solid-State Hydrogen–Deuterium Exchange with Mass Spectrometric Analysis (ssHDX-MS), Molecular Pharmaceutics, 15 (2018) 356–368. [DOI] [PubMed] [Google Scholar]
- [292].Milani S, Faghihi H, Roulholamini Najafabadi A, Amini M, Montazeri H, Vatanara A, Hydroxypropyl beta cyclodextrin: a water-replacement agent or a surfactant upon spray freeze-drying of IgG with enhanced stability and aerosolization, Drug Development and Industrial Pharmacy, 46 (2020) 403–411. [DOI] [PubMed] [Google Scholar]
- [293].Costantino HR, Carrasquillo KG, Cordero RA, Mumenthaler M, Hsu CC, Griebenow K, Effect of excipients on the stability and structure of lyophilized recombinant human growth hormone, Journal of pharmaceutical sciences, 87 (1998) 1412–1420. [DOI] [PubMed] [Google Scholar]
- [294].Royer CA, Fluorescence spectroscopy, Protein stability and folding, Springer; 1995, pp. 65–89. [DOI] [PubMed] [Google Scholar]
- [295].Ramachander R, Jiang Y, Li C, Eris T, Young M, Dimitrova M, Narhi L, Solid state fluorescence of lyophilized proteins, Analytical Biochemistry, 376 (2008) 173–182. [DOI] [PubMed] [Google Scholar]
- [296].Sharma VK, Kalonia DS, Steady-state tryptophan fluorescence spectroscopy study to probe tertiary structure of proteins in solid powders, Journal of pharmaceutical sciences, 92 (2003) 890–899. [DOI] [PubMed] [Google Scholar]
- [297].Moussa EM, Wilson NE, Zhou QT, Singh SK, Nema S, Topp EM, Effects of Drying Process on an IgG1 Monoclonal Antibody Using Solid-State Hydrogen Deuterium Exchange with Mass Spectrometric Analysis (ssHDX-MS), Pharmaceutical Research, 35 (2018) 12. [DOI] [PubMed] [Google Scholar]
- [298].Huang RYC, Chen G, Higher order structure characterization of protein therapeutics by hydrogen/deuterium exchange mass spectrometry, Analytical and Bioanalytical Chemistry, 406 (2014) 6541–6558. [DOI] [PubMed] [Google Scholar]
- [299].Weis DD, Hydrogen exchange mass spectrometry of proteins: fundamentals, methods, and applications, John Wiley & Sons; 2016. [Google Scholar]
- [300].Trabjerg E, Nazari ZE, Rand KD, Conformational analysis of complex protein states by hydrogen/deuterium exchange mass spectrometry (HDX-MS): Challenges and emerging solutions, TrAC Trends in Analytical Chemistry, 106 (2018) 125–138. [Google Scholar]
- [301].Deng B, Lento C, Wilson DJ, Hydrogen deuterium exchange mass spectrometry in biopharmaceutical discovery and development – A review, Analytica Chimica Acta, 940 (2016) 8–20. [DOI] [PubMed] [Google Scholar]
- [302].Kumar L, Chandrababu KB, Balakrishnan SM, Allmendinger A, Walters B, Zarraga IE, Chang DP, Nayak P, Topp EM, Optimizing the Formulation and Lyophilization Process for a Fragment Antigen Binding (Fab) Protein Using Solid-State Hydrogen–Deuterium Exchange Mass Spectrometry (ssHDX-MS), Molecular Pharmaceutics, 16 (2019) 4485–4495. [DOI] [PubMed] [Google Scholar]
- [303].Kammari R, Topp EM, Solid-State Hydrogen–Deuterium Exchange Mass Spectrometry (ssHDX-MS) of Lyophilized Poly-d,l-Alanine, Molecular Pharmaceutics, 16 (2019) 2935–2946. [DOI] [PubMed] [Google Scholar]
- [304].Vadas O, Jenkins ML, Dornan GL, Burke JE, Chapter Seven - Using Hydrogen–Deuterium Exchange Mass Spectrometry to Examine Protein–Membrane Interactions, in: Gelb MH (Ed.) Methods in Enzymology, Academic Press; 2017, pp. 143–172. [DOI] [PubMed] [Google Scholar]
- [305].Pan LY, Salas-Solano O, Valliere-Douglass JF, Conformation and Dynamics of Interchain Cysteine-Linked Antibody-Drug Conjugates as Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry, Analytical Chemistry, 86 (2014) 2657–2664. [DOI] [PubMed] [Google Scholar]
- [306].Lampi KJ, Murray MR, Peterson MP, Eng BS, Yue E, Clark AR, Barbar E, David LL, Differences in solution dynamics between lens β-crystallin homodimers and heterodimers probed by hydrogen–deuterium exchange and deamidation, Biochimica et Biophysica Acta (BBA) - General Subjects, 1860 (2016) 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].AbouGhaly MHH, Du J, Patel SM, Topp EM, Effects of ionic interactions on protein stability prediction using solid-state hydrogen deuterium exchange with mass spectrometry (ssHDX-MS), International Journal of Pharmaceutics, 568 (2019) 118512. [DOI] [PubMed] [Google Scholar]
- [308].Iyer LK, Moorthy BS, Topp EM, Photolytic Cross-Linking to Probe Protein–Protein and Protein–Matrix Interactions in Lyophilized Powders, Molecular Pharmaceutics, 12 (2015) 3237–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Chen Y, Topp EM, Quantitative Analysis of Peptide–Matrix Interactions in Lyophilized Solids Using Photolytic Labeling, Molecular Pharmaceutics, 15 (2018) 2797–2806. [DOI] [PubMed] [Google Scholar]
- [310].Iyer LK, Sacha GA, Moorthy BS, Nail SL, Topp EM, Process and Formulation Effects on Protein Structure in Lyophilized Solids Using Mass Spectrometric Methods, Journal of Pharmaceutical Sciences, 105 (2016) 1684–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Chen Y, Topp EM, A Novel Photoreactive Excipient to Probe Peptide-Matrix Interactions in Lyophilized Solids, Journal of Pharmaceutical Sciences, 109 (2020) 709–718. [DOI] [PubMed] [Google Scholar]
- [312].Pieters S, De Beer T, Kasper JC, Boulpaep D, Waszkiewicz O, Goodarzi M, Tistaert C, Friess W, Remon J-P, Vervaet C, Vander Heyden Y, Near-Infrared Spectroscopy for In-Line Monitoring of Protein Unfolding and Its Interactions with Lyoprotectants during Freeze-Drying, Analytical Chemistry, 84 (2012) 947–955. [DOI] [PubMed] [Google Scholar]
- [313].Starciuc T, Malfait B, Danede F, Paccou L, Guinet Y, Correia NT, Hedoux A, Trehalose or Sucrose: Which of the Two Should be Used for Stabilizing Proteins in the Solid State? A Dilemma Investigated by In Situ Micro-Raman and Dielectric Relaxation Spectroscopies During and After Freeze-Drying, Journal of Pharmaceutical Sciences, 109 (2020) 496–504. [DOI] [PubMed] [Google Scholar]