

Abstract
Background
Leber congenital amaurosis (LCA) is a congenital retinal dystrophy that results in significant and often severe vision loss at an early age. Comprehensive analysis of the genetic mutations and phenotypic correlations in LCA patients has allowed for significant improvements in understanding molecular pathways of photoreceptor degeneration and dysfunction. The purpose of this article is to review the literature on the subject of retinal gene therapy for LCA, including historical descriptions, preclinical animal studies, and human clinical trials.
Methods
A literature search of peer-reviewed and indexed publications from 1996–2011 using the PubMed search engine was performed. Key terms included “Leber congenital amaurosis”, LCA, RPE65, ”cone-rod dystrophy”, “gene therapy”, and “human trials” in various combinations. Seminal articles prior to 1996 were selected from primary sources and reviews from the initial search. Articles were chosen based on pertinence to clinical, genetic, and therapeutic topics reviewed in this manuscript. Fundus photographs from LCA patients were obtained retrospectively from the clinical practice of one of the authors (R.A.S).
Results
Herein, we reviewed the literature on LCA as a genetic disease, the results of human gene therapy trials to date, and possible future directions towards treating inherited retinal diseases at the genetic level. Original descriptions of LCA by Theodor Leber and subsequent research demonstrate the severity of this disease with early-onset blindness. Discoveries of the causative heritable mutations revealed genes and protein products involved in photoreceptor development and visual transduction. Animal models have provided a means to test novel therapeutic strategies, namely gene therapy. Stemming from these experiments, three independent clinical trials tested the safety of subretinal delivery of viral gene therapy to patients with mutations in the RPE65 gene. More recently, efficacy studies have been conducted with encouraging results.
Conclusions
Initial safety studies indicated promising results of subretinal delivery of viral vector with subclinical immunologic or surgical sequelae. Overall, these initial studies demonstrate that viral vector gene therapy results are very promising, safe, and effective. Future studies measuring potential improvement in photoreceptor function may rely on recent advances in retinal imaging and electrophysiologic testing.
Keywords: Leber congenital amaurosis, LCA, Retinal dystrophy, Gene therapy, Adeno-associated virus, Human trials, RPE65
Introduction
Gene therapy has recently become a feasible approach to the treatment of inherited retinal disease in human patients [1–4]. Theoretically, in patients with a known mutation of the coding region of a single gene, a normal allele can be introduced and expressed to return cells to normal functioning. Multiple delivery systems have been developed in animal models to transfer the correct DNA sequence to the proper tissues and cell types and to specifically control its expression. Due to its accessibility, immunological advantages, and available modalities for physiologic testing and monitoring tissue pathology, the eye is an excellent model for testing the safety and efficacy of gene therapy. Over the last three years, multiple groups have reported their findings utilizing adeno-associated viral gene therapy for the treatment of Leber congenital amaurosis (LCA) resulting from mutation of the RPE65 gene (LCA2, OMIM#204100) [1–10]. These trials comprise low-dose safety studies in young adult patients, and dose-escalation studies including children and adolescents. Adeno-associated viral gene therapy is relatively safe, with acceptable sequelae, and improvements in light responses and functional vision noted in several patients are encouraging. In addition to reviewing their findings, the purpose of this article is to summarize our understanding of inherited LCA mutations, gene therapy results in animal models of LCA preceding the human trials, and new technologies that may improve the applicability of gene therapy to other diseases.
Clinical descriptions of LCA
The first report of LCA was published in 1869 by Theodor Leber [11]. In one young adult patient blind from birth, he described a constellation of low or absent vision with amaurotic pupils, wandering nystagmus, and pigmented retinopathy. Postmortem histologic examination revealed marked retinal atrophy accentuated in the outer layers and periphery, vascular thickening and sclerosis, retinal pigment epithelium (RPE) changes, and intraretinal pigmentation [11, 12]. His original description included LCA as a member of the family of retinitis pigmentosa (RP, RP1 OMIM #180100 and other subtypes), but a severe form with nearly complete vision loss by birth and a strong hereditary component. In 1954, Franceschetti and Dieterle described the absence or severe reduction of measured electroretinography (ERG) as a hallmark of LCA, thereby differentiating it from other retinal dystrophies [13].
Disease presentation and natural history
LCA is predicted to affect approximately 1/81,000 individuals [14] and up to 20 % of children attending schools for the blind worldwide [15]. Careful clinical observation and description of patients with LCA have revealed a spectrum of disease phenotypes and progression. All patients have early, severe visual deficits in childhood, highly attenuated or absent ERG recordings, and the absence of other ophthalmologic or systemic disease processes. The early onset and rapid rate of vision loss result in lifelong poor visual outcomes for LCA patients. Visual acuity loss can be quite severely progressive or profound from birth. One reported patient from a cohort described by Lambert et al. had decreasing visual acuity over a span of 12 years, from 20/120 to counting fingers [16]. Figure 1 illustrates different pigmentation patterns seen in the spectrum of LCA in human patients. Other associated ophthalmic signs include the oculodigital sign (habitual rubbing or poking the eyes), keratoconus/keratoglobus, cataracts, strabismus, intraretinal pigment migration, macular atrophy, and optic nerve pallor [15–17]. It is not uncommon for LCA patients to have associated intellectual disability [15,16].
Fig. 1.
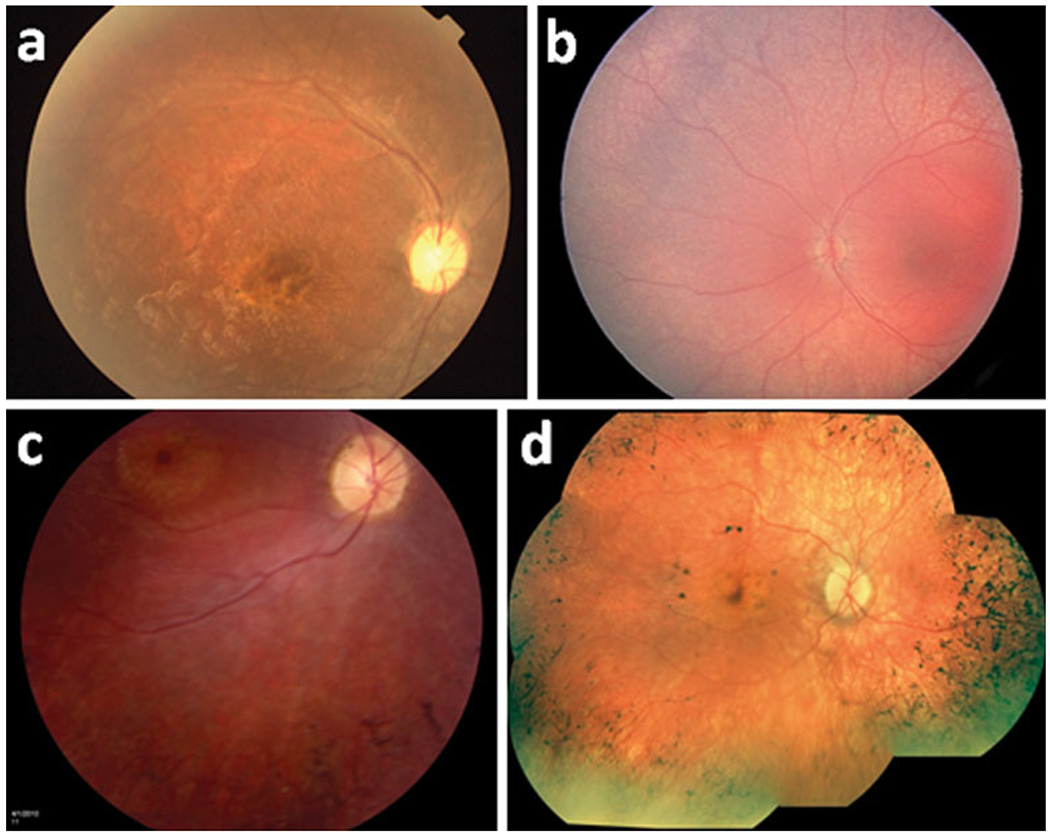
Various retinal phenotypes among LCA patients. a Focal perimacular atrophy and pigment accumulation. b Fundus hypopigmentation with diffuse RPE granularity. c Macular atrophy and bone spicule pigmentation. d Macular RPE clumping and atrophy accompanying peripheral bone spicule changes
While a severe form of congenital blindness, there is marked variability in disease presentation among LCA patients. Many patients are born with congenital blindness, and for those that are not, visual symptoms are noted within the first year of life by parents or caretakers. In one study following the progression of vision loss in infants and children affected by LCA, half never demonstrated grating visual acuity, and a third had no light perception whatsoever [18]. A recent study comparing LCA and early-childhood onset RP demonstrated a greater chance of retaining useful vision in the future when visual symptoms occur after infancy [19]. While some may have ambulatory vision initially and preserve reading visual acuity into adulthood, others are forced to read by Braille initially or after some years of reading by sight [16]. Very rarely will visual testing ever demonstrate improvement [18]. Before the advent of gene therapy, options available to LCA patients have been supportive only, including low vision aids, low vision social services, and genetic counseling of parents and patient.
Differential diagnoses and testing
A diagnosis of LCA must be differentiated from other retinal dystrophies that result in progressive or complete vision loss. Aside from LCA and other forms of RP, diseases with similar features include achromatopsia (ACHM2 OMIM #216900 and other subtypes), congenital stationary night blindness (CSNB, CSNB 1A OMIM #310500 and other subtypes), and forms of con-rod dystrophy (CORD, CORD1 OMIM #600624 and other subtypes). All are inherited in classic autosomal or sex-linked patterns. LCA represents 5 % of retinal dystrophies and is typically at the severe end of the spectrum, with nearly complete or total vision loss and severely reduced or absent ERGs. Lambert et al. evaluated 75 patients labeled as LCA, and found that only 45 met diagnostic criteria for LCA [16]. Of the remaining patients, five had CSNB, four had achromatopsia, five had other forms of RP or cone-rod dystrophy, and the remaining patients had miscellaneous syndromic conditions.
Of the nonsyndromic conditions, LCA can be distinguished in childhood by low or non-detectable cone and rod ERG responses [13, 20]. Achromatopsia affects only cone photoreceptors, and has a normal rod-mediated ERG. In complete CSNB, only rod photoreceptor responses are absent, probably caused by a defect in ON-bipolar cell signaling, and the cone-mediated responses are normal. Non-LCA forms of inherited RP present with rod-mediated changes first, including nyctalopia and peripheral vision loss, before slowly progressive reduction in ERG cone responses and central vision. Classically, intra-retinal pigment epithelial migration results in the classic fundoscopic appearance of bone spicule pigmentation, indicative but not universal for RP. Central visual changes rarely occur early in RP, and though forms of central RP have been reported [21], these are exceedingly rare and may represent cases of cone-rod dystrophy [22]. Early childhood-onset RP is very similar to LCA, except that visual symptoms are noted after 1 year of age [19]. Syndromic conditions that may present with congenital blindness include Joubert syndrome (cerebellar and corpus collosum hypoplasia, OMIM #213300), Senior–Loken syndrome (oculo-renal nephronophthisis, OMIM #266900), Alström’s syndrome (hearing loss and diabetes mellitus, OMIM #203800), peroxisomal disorders such as infantile Refsum disease (OMIM #266510) and Zellweger syndrome (OMIM #214100), Bardet–Biedl syndrome (polydactyly, obesity, and intellectual disability, OMIM #209900), and others.
Genetic mutations and phenotypic significance in LCA
LCA is typically inherited in an autosomal recessive fashion, though an autosomal dominant pattern has been reported [23]. Many gene mutations underlying the inheritance of LCA have been described and used to classify the disease into subtypes by the affected gene, including GUCY2D (LCA1), RPE65 (LCA2), , AIPL1 (LCA4), , RPGRIP1 (LCA6), CRX (LCA7), and CRB1 (LCA8). Other associated genes have been found and characterized, including LRAT, TULP1, and CEP290, among others. Please see Table 1 for a list of LCA-associated genes and function of the protein products. Figure 2 illustrates the location and presumed functions for the proteins affected in different subtypes of LCA.
Table 1.
Known LCA-associated genes and their function
| Disorder | Gene | Location | Protein | Function |
|---|---|---|---|---|
| LCA1 | GUCY2D[71] | 17p13.1 | Guanylate cyclase 2D | Produces RetGC-1, phototransduction |
| LCA2 | RPE65[72] | 1p31 | 65-kilodalton RPE-localized protein | Retinoid phototransduction cycle |
| LCA3 | SPATA7 | 14q31.3 | Spermatogenesis-associated protein | Unknown retinal function |
| LCA4 | AIPL1[73] | 17p13.1 | Aryl hydrocarbon interacting protein | cGMP-PDE folding, cell cycle progression |
| LCA5 | LCA5 | 6q14.1 | Lebercillin | Centrosomal or ciliary function |
| LCA6 | RPGRIP1[74] | 14q11 | RP GTPase regulator-interacting protein 1 | Outer segment formation |
| LCA7 | CRX | 9q13.3 | Homeodomain transcription factor | Photoreceptor development |
| LCA8 | CRB1[75] | 1q31 | Crumbs like protein 1 | Photoreceptor morphogenesis |
| LCA9 | Unknown | 1p36 | ––– | ––– |
| LCA10 | CEP290 | 12q21.3 | Centrosomal protein (Bardet-Biedel Syndrome) | Centrosome maintenance |
| LCA11 | IMPDH1 | 17q31.3 | Inosine monophosphate dehydrogenase | Cell growth |
| LCA12 | RD3 | 1q32.3 | Retinal degeneration protein | Nuclear PML bodies |
| LCA13 | RDH12 | 14q24.1 | Retinol dehydrogenase | Retinoid phototransduction cycle |
| LCA14 | LRAT | 4q32.1 | Lecithin retinol acyltransferase | Retinoid phototransduction cycle |
| LCA15 | TULP1 | 6p21.3 | Tubby-like protein | Phagocytosis |
Information collected from referenced works and Online Mendelian Inheritance in Man database (OMIM, http://www.ncbi.nlm.nih.gov/omim).
Fig. 2.
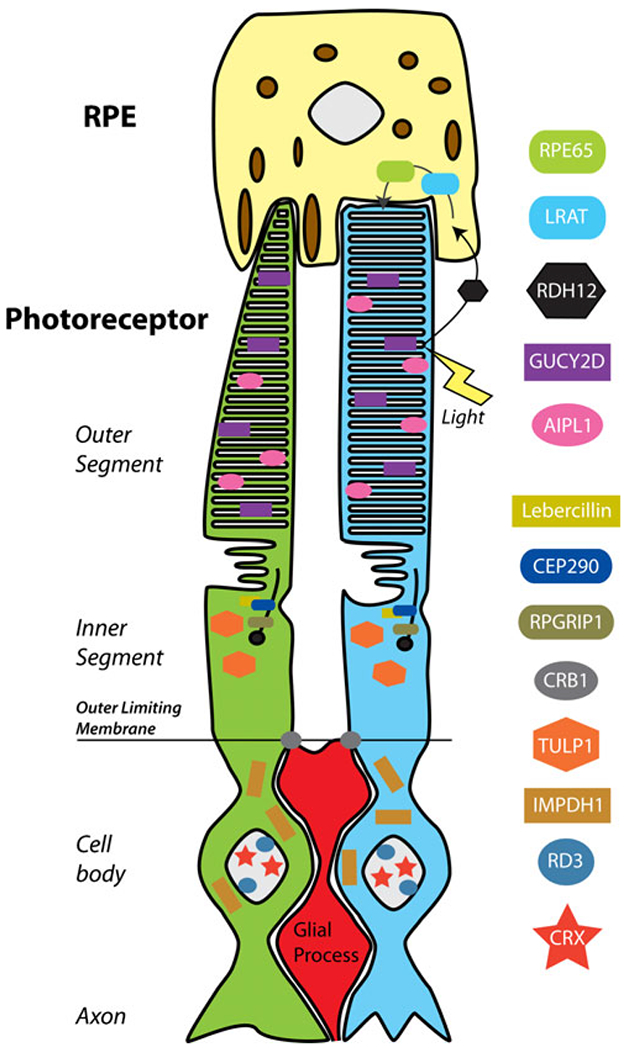
Schematic illustration of LCA-associated proteins and their location in the photoreceptor-RPE complex. Protein functions are outlined in Table 1. RPE cells (yellow) are in contact with the outer segments of cone (green) and rod (blue) photoreceptors. Proteins altered by LCA-causing mutations affect photoreceptor cells at several levels, including development, outer segment formation, protein trafficking, and photoreceptor-glia connections that form the outer limiting membrane. GUCY2D and AIPL1 participate in necessary machinery for transduction of light into hyperpolarization response. Additionally, genes expressed in RPE cells are affected, including RPE65 and LRAT, which participate in recycling vitamin A analogues in the visual transduction cycle (arrows; for further description, see corresponding text)
Dhamaraj et al. examined the prevalence of LCA1 (GUCY2D), LCA2 (RPE65), and LCA7 (CRX) in 100 consecutive LCA patients, and found that, overall, 11 % of these cases were attributed to mutations in GUCY2D (6 %), RPE65 (3 %), and CRX (2 %) [24]. In another study of Italian patients, approximately 28 % of affected patients had identifiable gene mutations, with 5.2 % due to GUCY2D, 8.4 % due to RPE65, 7.4 % due to CRB1, 4.2 % due to CEP290, and 3.2 % due to AIPL1 [25].
Genotype appears to play a role in the variability of phenotype and course in human LCA patients. In a study comparing LCA1 and LCA2 phenotypes, patients with GUCY2D mutations presented with normal appearing fundus, severe vision loss, hyperopia, photophobia, and a stable course, while patients with RPE65 mutations presented with a range of visual acuities, emmetropia or myopia, night blindness, and transient visual improvements through the second decade [26]. The study by Dhamaraj et al. noted similar findings in patients with RPE65 mutations, as well as poor pupillary reactions, nystagmus, and progressive decline in visual acuity during adulthood [24]. Prenatal retinal degenerative changes in the human fetus as early as 33 weeks gestation have been noted for LCA2 as well [27]. In the Italian population, keratoconus has been noted in patients with GUCY2D and AIPL1 mutations, subcapsular cataracts in patients with AIPL1, CRB1, and RPE65 mutations, and variability in fundus appearance in patients with CRB1 mutations, including one patient with preserved para-arteriole retinal pigment epithelium [25]. A recent report by Walia et al. suggests that patients with RPE65 or CRB1 mutations tend to lose central vision with age, and those with AIPL1 mutations remain stable across time [19]. Variations of macular morphology have been noted, in particular decreased macular thickness with RPE65 mutations and retention of the outer nuclear layer in foveae with CEP290 mutations, but these do not correlate with visual acuity [28].
Role of RPE65 in phototransduction and disease
This remainder of this article will focus on RPE65 (LCA2). Mutations in the RPE65 gene in humans have been linked to LCA2, autosomal recessive RP with early onset, early onset retinal dystrophy, and severe early childhood-onset retinal dystrophy (SECORD) [29–31]. The RPE65 gene has been very well studied in the phototransduction pathway, and is the therapeutic agent for the first gene therapy trials in LCA patients. RPE65 encodes a 65-kilodalton protein expressed in pigmented epithelial cells in all vertebrate models studied. The RPE65 protein is a carotenoid oxygenase required for the isomerization of all-trans to 11-cis-retinol in the visual transduction pathway [32]. During phototransduction in photoreceptors, all-trans-retinal is converted from the 11-cis form and transferred to the retinal pigment epithelium as all-trans-retinol, converted back to 11-cis-retinol by RPE65, and returned to the photoreceptor outer segment after conversion to the retinal form. Recently, RPE65 was shown to be translocated under light stimulation to the center of RPE cells by myosin VIIA, a protein which, when defective, causes Usher syndrome type 1 [33].
Mice lacking the Rpe65 gene have a slow retinal degeneration, primarily of rod photoreceptors, with reduced light sensitivity due to the lack of chromophore in the photoreceptors and the accumulation of vitamin A analogues in the RPE [32]. Loss of Rpe65 also affects cone photoreceptors in this mouse model, with slow cone photoreceptor degeneration and concomitant reduction of cone opsin expression [34]. Initially, however, photoreceptors are rendered nonfunctional by the deletion, without immediate retinal degeneration. Because Rpe65-deficient retinas are initially intact, with a latent period preceding degeneration, human LCA2 is potentially treatable by the restoration of RPE65 gene function.
Approaching LCA treatment with gene therapy
Gene therapy holds great potential for treating inheritable diseases. The principle of gene therapy is to replace nonfunctional or aberrant gene expression with an exogenous source of normal DNA capable of being transcribed and translated into functional protein. Thus, gene therapy requires the following: (1) a known genetic cause, (2) living cells with intact but malfunctioning cellular machinery, (3) access to tissue with little potential for vector dissemination or widespread inflammation, and (4) ability to accurately and reliably transfer DNA to the tissue and cell type of choice.
Determining appropriate candidate patients and diseases for gene therapy follows the above guidelines. Disease selection requires a known genetic alteration. Patient selection involves several levels of inclusion and exclusion criteria. Patients must first be genotyped to identify the gene involved. Second, in the case of retinal dystrophies, patients must be assessed to determine that ocular tissue is intact and photoreceptors are present. This can be accomplished by spectral-domain ocular coherence tomography (SD-OCT) to gauge the size and healthy appearance of the photoreceptor layer and fovea, or by VEP or ERG to determine responsiveness of photoreceptors to light [35]. Notably, variability in the photoreceptor layer architecture among human LCA2 patients has been demonstrated using OCT as a method for determining candidacy for gene therapy [35]. Third, in the case of viral delivery of DNA, risk of dissemination should be taken into account for immunosuppressed patients. Therefore, patients with unknown mutations, massive photoreceptor degeneration and death, and immunosuppression (in the case of viral vectors) are poor candidates for gene therapy.
Commonly used vectors in gene therapy studies
Vector selection can also play a role in the safety and efficacy of gene therapy. Genetic material can be delivered in a multitude of ways. Naked DNA can be electroporated into tissues, lipophilic vesicles can deliver DNA constructs, and viruses can infect and utilize host transcriptional machinery to deliver a DNA payload. Viruses, which commonly invade cells and subsume transcriptional machinery, are a rational and highly studied solution. Viral types being considered are adeno-associated virus (AAV), herpes simplex virus (HSV), and lentivirus. These viruses may have specific tropisms for different cell types and mechanisms for genetic expression, but only AAV infection of photoreceptors will be discussed here. Importantly, these viruses are engineered to lack integrative capacity, thereby preventing them from causing stable mutations in the human genome. Further, they must be able to infect non-dividing cell types, as is the case in most adult tissues. Finally, the expression construct, that is, the promoter and DNA regulatory elements controlling transcription, must express the gene reliably within cells. To date, all human trials have used a cytolomegalovirus (CMV) promoter and chicken β-actin enhancer that confer constitutive expression of the genetic construct.
Gene therapy trials of LCA in animal models: canine, mouse, and primate
Animal models have provided a wealth of knowledge about the pathophysiology of LCA, and the opportunity to test the safety and efficacy of viral gene therapy. The most celebrated of these is the Swedish Briard dog, which carries a natural four base pair deletion of the Rpe65 gene [36]. Similar to human patients, these animals are blind at birth and have absent ERG responses. Subretinal injections of AAV2 with CMV promoter and β-actin enhancer in 4-month-old dogs resulted in improved ERGs, VEPs, pupillary response, and vision-dependent behaviors [37]. Histologic analyses revealed detectable 11-cis retinal expression in photoreceptors of treated eyes, indicating functional return of the visual transduction pathway [38]. These studies also demonstrated that injections and reinjections are safe in large animals, and that subretinal injections are superior in efficacy to intravitreal injections [37–42].
Gene therapy has also been used to restore vision in Rpe65-deficient mice using viral vectors. Rpe65 homozygous mutant mice exhibit inherited retinal degeneration similar to LCA2. Injection of viral vector carrying mouse Rpe65 gene into the eyes of Rpe65−/− embryos in utero rescued Rhodopsin expression and visual function in these mice [43]. Results for gene therapy in adult mice have been mixed, however, with less dramatic results at older ages [44, 45]. Interestingly, one study examined photoreceptor layer thickness in these mice, and found that mice with better-preserved retinal thickness had greater improvement with viral gene therapy [35].
Prior to human trials, several studies testing the safety of AAV2 virus in primates had been performed. In the macaque, viral DNA was found in lacrimal and nasal fluids for up to 4 days and serum for up to 20 days [46]. Another study testing for transmission to other organs and to the germline demonstrated a lack of systemic distribution [47]. Importantly, viral transgene expression was measurable for up to 18 months, and no retinal toxicity was noted [47, 48].
Gene therapy trials in human LCA patients
The first human trials to test the safety of viral vector-mediated gene transfer to human LCA patients were reported in 2008 by three groups; from University College London, a collaboration between Children’s Hospital of Philadelphia (CHoP) and Italy, and a collaboration between the Universities of Pennsylvania and Florida [1, 3, 4]. Since then, follow-up reports and dose-escalation trials have been published. Here, we review these findings as an ongoing body of work from these three consortiums. The goals of the initial studies were primarily to test safety and secondarily to test efficacy of viral gene therapy. Each group injected AAV2 viral vectors driving RPE65 expression with a CMV promoter, into the subretinal space of three patients. Doses ranged from 1.5×1010–1.0×1011 viral particles, and the Bainbridge et al. [1] and Maguire et al. [4] groups placed patients on an oral course of corticosteroids to prevent inflammation. Table 2 outlines the studies, viral vectors and dosing used, patient demographic and mutation information, and visual outcomes. A comprehensive review of this clinical data in greater depth is available [49].
Table 2.
Summary of outcome measures for human clinical trials
| Trial | Dosing | Patient | Mutation | VA (treated) | Perimetry | Pupillometry | Nystagmus | Functional |
|---|---|---|---|---|---|---|---|---|
| Bainbridge, Smith et al (1 year[1]) | 2/2.hRPE65 [76] | 23yo M (OD) | Missense (homo.) |
20/286→20/145* 20/150→20/120* |
No Δ | ----- | ----- | No Δ |
| 1e11 particles (1.0mL inj.) | 17yo F (OD) | Missense |
No Δ No Δ |
No Δ | No Δ | No Δ | ||
| Added: AV5 helper virus; oral steroids | 18yo M (OD) | Missense |
No Δ No Δ |
Improved micro, dark | ----- | Improved time, errors | ||
| Maguire, Simonelli et al (6m[4], 1.5y[9]) | AAV2-hRPE65v2[77] | 26yo F (OD) | Missense (homo.) |
HM→20/640 20/1040→20/762 |
Improved (OD) | Improved (OD>OS) | Improved (OU) | Improved time, errors |
| 1.5e10 part. (0.1SmLinj.) | 26yo M (OD) | Missense (homo.) |
HM→20/459 20/500→20/126* |
Improved (OD) | Improved (OD>OS) | Improved (OU) | Improved time, errors | |
| Added: surfactant; oral steroids | 19yo F (OD) | Null (homo.) |
20/640→20/167 No Δ |
Improved (OD) | Improved (OD>OS) | Improved (OU) | Improved time, errors | |
| Maguire, High et al (2y[7]) | AAV2-hRPE65v2 | 17yo M (M-OS) | Missense | 20/112 → 20/69 20/300->20/104 |
Improved (OS) | No Δ | Improved | NA |
| Medium dose (M): 4.8e10 part. (0.15mL inj.) | 20yo F (M-OD) | Missense + Splice Mut |
20/448→20/1352 No Δ |
Improved (OD) | Improved | No Δ | No Δ | |
| 9yo M (M-OD) | Nonsense (homo.) |
No Δ No Δ |
Improved (OD) | Improved | Improved | Improved time, errors | ||
| 8yo M (M-OS) | Null + Deletion | No Δ No Δ |
Improved (OS) | Improved | No Δ | Improved time, errors | ||
| 10yo M (M-OD) | Splice Mut + Deletion |
20/564→20/390 No Δ |
Improved (OD) | Improved | Improved | Improved time, errors | ||
| 24yo F (M-OD) | Missense (homo.) |
20/209→20/80 No Δ |
Improved (OD) | No Δ | No Δ | No Δ | ||
| High dose (H): 1.5e11 part. (0.15mL inj.) | 44yo F (H-OD) | Null + Missense |
No Δ No Δ |
Improved (OD) | Improved | Improved | No Δ | |
| 35yo M (H-OD) | Splice Mut (homo.) |
No Δ No Δ |
Improved (OD) | Improved (amplitude) | No Δ | No Δ | ||
| 11yo M (H-OD) | Missense |
20/200→20/61.2 20/80→20/63 |
Improved (OD) | Improved (velocity) | No Δ | Improved time, errors | ||
| Hauswirth, Cideciyan et al (3m[2,3], 1y[5, 6]) | CBsb-hRPE65[42] | 24yo M (OS) | Missense (homo.) | No Δ No Δ |
Improved dark adapt | ----- | ----- | ----- |
| 5.96e10 part. (0.15ml inj.) | 23yo F (OD) | Missense |
No Δ No Δ |
Improved dark adapt | ||||
| No steroids | 21yo M (OD) | Missense (homo.) |
No Δ No Δ |
Improved dark adapt | ||||
| Banin et al (3m [8]) | CBSB-hRPE65 1.19e11 part. (0.30mL inj.) | 26yo M (OS) | Splice Mut (homo.) | ----- | Improved dark adapt (OD only) | ----- | ----- | ----- |
Visual acuity is underlined and italicized for treated eye.
Not statistically significant.
Abbreviations/symbols used: HM (hand motion), H (high dose), homo, (homozygous), inj. (injection), M (medium dose), Mut. (mutation), OD (right eye), OS (left eye), OU (both eyes), part, (particles), VA (visual acuity), Δ (change from baseline to post-therapy measures).
Summary of safety outcomes
The three groups demonstrated acceptable immunologic changes and ocular sequelae. In the Bainbridge et al. study [1], 1.0 ml was injected subretinally, by definition inducing a temporary retinal detachment that resolved within 24 hours. Similarly, smaller macular blebs occurred with 0.15 ml injections in the Maguire et al. [4] and Hauswirth et al. [3] groups, and spontaneously resolved. Generally, the groups detected no dissemination of the virus in other tissues following the injections. One patient in Maguire et al. [4] was found to have viral DNA in a tear sample on post-operative day 1 but not following, and another patient temporarily had detectable neutralizing antibodies to AAV2 capsid. A third patient in that study developed a macular hole that did not affect visual acuity, and both the macular hole and visual acuity remained unchanged through 1.5 years [4, 9]. Patients from Bainbridge et al. [1] were noted to have a self-limited intraocular inflammation following surgery, and two patients had a non-specific activation of T-cells, which may have been a result of corticosteroid withdrawal. One patient from the Hauswirth et al. study [1] also demonstrated antibodies to the AAV2 capsid that resolved after 90 days, and mildly increased lymphocyte stimulation index to the capsid protein.
Summary of efficacy data
Full-field ERG readings never changed from baseline in any of the three initial studies. However, even these initial studies demonstrate the occurrence of several promising visual changes. In Bainbridge et al. [1], none of the patients had improvements in best-corrected visual acuity. Though one patient did improve from 20/286 to 20/145 in the injected eye, it was not considered a significant change because the non-injected eye improved from 20/150 to 20/120. A different patient noted improvements in both dark-adapted perimetry and micro-perimety, unlike the others, and was the only patient to improve his performance on a mobility test, both in time and obstacle avoidance.
Maguire et al. [4] observed that patients had increased amplitude and velocity of the pupillary light response, as well as decreased nystagmus. At 3 months post-injection, all three patients noted an increase in best-corrected visual acuity, and improvements of time and obstacle avoidance in navigating a maze. However, it is unclear to what extent the improvement in nystagmus and the learning effect of repeated testing could contribute to the observed findings.
At 1.5 years, the pupillary responses remained significantly improved, amplitude of nystagmus diminished, best-corrected visual acuity continued to improve, and functional vision in the mobility test remained improved [9].
The same group also performed a dose-escalation trial [7]. In addition to the three original, low-dose patients (1.5×1010), six patients were given an increased dose at 4.8×1010, and another three patients given a dose at 1.5×1011 particles directly compared up to 2 years posttreatment. Improvements in pupillometry, nystagmus, mobility test, perimetry, and visual acuity did not correlate with dose, though all showed improvement on at least one measure. Interestingly, improvement in these tests was consistently greater overall in the younger patients. Multifocal ERGs also became detectable in three patients, which also may have been attributable to their reduced nystagmus.
The third study, published in separate parts by Hauswirth et al. [3] and Cideciyan et al. [2] also did not find visual acuity improvements, but showed improvement in dark-adapted full-field sensitivity testing (FST) in all three patients. Two of these patients had injection blebs superior and temporal to the macula respectively, which resolved. Light sensitivity, measured by visual thresholds in the dark, was increased in the areas of the retina where the injections were originally made [2]. This observation suggests that rod-photoreceptor driven dim light vision was improved specifically in those regions. At 1-year follow-up of the three patients, these changes remained stable [5]. One patient noted that she could read a digital clock in a family automobile, interpreted as visual gain due to fixation over the injection point, in this case the superotemporal retina. This phenomenon was termed a pseudofovea, a reorganization of visual fixation around a new point [6].
Jacobson et al. [10] also performed a dose-escalation trial compared to the original cohort. Doses ranged from 5.96×1010–1.79×1011. Their findings indicated that the improvement remained specific to treated areas, and the change in location of visual fixation was stable up to 3 years. As in the other dose-escalation study, younger patients generally showed greater improvement than older patients, while dose-dependent effects were not apparent [10]. Similarly, in a study of RPE65 mutations in Israel, one patient injected with the same RPE65 vector, though at a higher dose (1.19×1011), demonstrated an improvement in dark-adapted light sensitivity [8].
Long-term follow-up studies have shown other potential long-term effects of retinal gene therapy. Functional magnetic resonance imaging (fMRI), used to study activation of specific brain areas to discrete stimuli, was performed at 2 years on three patients in the CHOP–Italy cohort. Along with improvements in visual field, patients demonstrated a greater activation of cortical pathways in the treated eye, which had worse pretreatment visual acuity, compared to the control eye [50]. The activated cortical pathways correlate with the visual system, suggesting that these sight-specific pathways are intact and subject to potential improvement with treatment.
Most recently, Bennett et al. published efficacy data for subretinal injection of high dose AAV-RPE65 into the untreated eye of patients involved in the original clinical trial [51]. These patients showed modest improvements in visual acuity, pupillometry, and full-field sensitivity testing, with a similar safety profile to that of the original injection [51]. To date, no reports of reinjections into a previously injected eye have been published.
Successes and limitations of retinal gene therapy
These studies represent the first wave of human gene therapy trials for inherited retinal disease. Each showed, in its own way, the obstacles in quantifying visual improvement. Parsing out the effects of viral vector therapy on photoreceptor function is not simple. All three groups studied have in common an improvement in selected measures of vision, objective or subjective. While many measures of visual improvement correlated within patients, it is not clear how closely retinal changes can be measured by subjective visual acuity and mobility testing. Learning effects can result in improvement on repeat trials of the same test, and reduction in nystagmus can improve functional vision by enhancing stability of gaze.
For the majority of patients, full-field ERG responses did not improve, and for those that did have measurable ERGs, this finding is also characteristic of reduced nystagmus. This is perhaps not surprising, since a localized injection would not affect the entire retina, and modest improvements in a small portion of the retina may not be noted with full-field ERG. The use of dark-adapted perimetry by two of the groups to measure rod photoreceptor function in RPE65-deficient patients is an appropriate method for localizing changes in retinal sensitivity specifically to the affected cell type [1, 2, 5]. The method of measuring activity at the injection site with dark-adapted FST to monitor changes in retinal function helps refine the effects at the viral delivery site compared to neighboring tissue [2, 5]. Probably, a combination of both gross measures, such as refraction, nystagmus, and pupillometry, and fine measures including microperimetry and dark-adapted FSTs, will be required to understand the effects of viral gene therapy on the human visual system.
Further evaluation of viral vector safety and efficacy is required to move ahead to larger patient groups. Since these viruses do not integrate into the genome, they will be eventually cleared from the tissue by native inflammatory cells. Probably this will require repeated injections, which at least in canines has been shown to be safe [39]. One risk is that virus will spread inadvertently to the other eye, as shown in one canine study, or to the brain. In one report, RPE65 protein was noted in the uninjected control eye of several treated dogs, suggesting spread of the viral vector via retinal ganglion cell axons in the optic nerve [41]. This might represent trans-neuronal spread via retinal ganglion cells, or via communication in the optic nerve sheaths. Whether central nervous system or organism-wide spread occurs remains to be determined. A recent study on long-term effects of retinal gene therapy in a mouse model of LCA1 demonstrated viral genome present in the contralateral brain hemisphere from the injected eye in one of 20 injections, 11 months after injection [52]. While this may result from technical difficulties, this small but real effect may represent subclinical systemic effects of viral injection even in a small cohort of treated patients.
Future directions in gene therapy
RPE65 protein is thought to primarily function in the RPE. Alternative functions of RPE65 in cone and rod photoreceptors are still in question, and may play a role in the disease process and treatment effects. Other viral vectors may provide differential tropism for cone photoreceptors, rod photoreceptors, or RPE, which may affect the outcome. Recently, AAV2 and AAV8 viruses were compared for transfection efficiency in different cell types, demonstrating equal efficiency in RPE cells for both viruses, and enhanced efficiency for AAV8 in photoreceptor cells [53] Other viral vectors have been employed besides AAV, such as lentivirus, with similar results [54]. Aside from the CMV promoters currently used in human viral gene therapy, RPE- or photoreceptor-specific promoters can be used to conditionally activate viral gene expression only in the desired cell type. Additionally, antibiotic- and hormone-sensitive elements can control the timing of vector gene expression in order to delay activation until after initial post-operative inflammation [55–57].
The use of non-viral means for transferring gene constructs may prove effective as well. Gene delivery by protein-based nanoparticle carriers and by electrical transfer of naked DNA into retinal cells by electroporation has been used in animal models [58, 59]. Nanoparticles are easily manufactured, may have less immunologic responses, and readily pass through cell membranes. Nanoparticles also have the advantage of being modifiable to produce sustained-release compounds. However, these particles, unless specifically modified, may also readily pass through the blood-retina barrier and produce systemic effects [60]. Characterizing new vectors and genes for viral gene therapy may benefit from the use of postmortem human retinal cells and retinal explants, to reduce both animal use and bench-to-bedside timing [61].
Given the rare prevalence of inherited retinal dystrophies, expanding gene therapy efforts to other cone and rod dystrophies, and even other cell types, will greatly facilitate the advancement of the field. Gene therapy is evolving for other forms of LCA in animal models, including LCA1 and LCA4 [62–64]. Studies characterizing patients with LCA4-causing AIPL1 mutations indicate they may benefit from early intervention due to rapid macular and extra-macular photoreceptor loss [65–67]. RP is primarily a disease of rod photoreceptors, and initially affects the periphery then spreads centrally. Inherited diseases specific to cone photoreceptors, such as complete and incomplete achromatopsia and blue cone monochromatism, primarily affect central vision and are ideal for this strategy, since the macula is entirely populated by cone photoreceptors.
Treatment of other ocular diseases is also on the horizon. Leber hereditary optic neuropathy (LHON), originally described by Theodor Leber, is a mutation in mitochondrial DNA which produces failure of ATP generation within the optic nerve axons. Replacement of normal ND4 and ND1 gene transcript in fibroblasts of patients harboring mutations in these genes restores electron transport chain activity [68]. Further, viral delivery of normal gene activity to the rat vitreous can rescue vision in an animal model of LHON [69]. Patient screening and recruitment is currently underway to deliver AAV vector intravitreally to transfect retinal ganglion cells with a correct ND4 gene transcript [70].
As gene therapy evolves, genetic diagnostic testing will be paramount in identifying pre-phenotypic patients with retinal dystrophies. Custom microarrays can be used to detect a battery of specific known mutations without having to screen genes individually. Younger patients with greater populations of viable photoreceptors stand to gain the most from early intervention with gene therapy.
Conclusions
LCA has presented an excellent opportunity to intervene on the genetic level in an attempt to improve visual outcomes of patients with this disease. Representing 5 % of all retinal dystrophies, LCA has a strongly autosomal recessive inheritance pattern of single gene mutations that have been well-characterized. While far from common, patients are readily identified and genetically and phenotypically characterized by many groups around the world. These patients have severe visual loss, often without measurable visual acuity or light perception. The future of gene therapy rests with these and future studies characterizing the safety and efficacy of vector transfer of normal RPE65 expression to a degenerating retina in LCA patients. Careful documentation of all aspects of patient phenotype and response will yield an immense amount of data on the mechanism of visual improvement. With time, new vectors, expression constructs, genes, and tissues will be able to be employed to treat inherited diseases.
Acknowledgements
We thank Marcus Blum (Helios Klinikum Erfurt) for his generous gift of English translations of the original German descriptions of LCA by Theodor Leber, and Michael Gorin for his comments on this manuscript. We especially thank the patients and families of all LCA patients who have contributed and will contribute to human gene therapy trials. This work was partially supported by the National Institute on Deafness and Other Communication Disorders, National Institutes of Health (R00-DC009287-03), and a Career Development Award from Research to Prevent Blindness (Z.A.).
Footnotes
Conflict of interest None
Contributor Information
Robert B. Hufnagel, Department of Pediatrics, Division of Pediatric Ophthalmology, University of Cincinnati and Cincinnati Children’s Hospital, College of Medicine, 3333 Burnet Ave, ML 7003, Cincinnati, OH 45229, USA
Zubair M. Ahmed, Cincinnati Children’s Hospital, Cincinnati, OH 45229, USA
Zélia M. Corrêa, University of Cincinnati College of Medicine, Cincinnati, OH 45229, USA
Robert A. Sisk, Cincinnati Eye Institute, Cincinnati, OH 45242, USA
References
- 1.Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, Petersen-Jones S, Bhattacharya SS, Thrasher AJ, Fitzke FW, Carter BJ, Rubin GS, Moore AT, Ali RR (2008) Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 358:2231–2239 [DOI] [PubMed] [Google Scholar]
- 2.Cideciyan AV, Aleman TS, Boye SL, Schwartz SB, Kaushal S, Roman AJ, Pang JJ, Sumaroka A, Windsor EA, Wilson JM, Flotte TR, Fishman GA, Heon E, Stone EM, Byrne BJ, Jacobson SG, Hauswirth WW (2008) Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci U S A 105:15112–15117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, Conlon TJ, Boye SL, Flotte TR, Byrne BJ, Jacobson SG (2008) Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther 19:979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR, Konkle B, Stone E, Sun J, Jacobs J, Dell’Osso L, Hertle R, Ma JX, Redmond TM, Zhu X, Hauck B, Zelenaia O, Shindler KS, Maguire MG, Wright JF, Volpe NJ, McDonnell JW, Auricchio A, High KA, Bennett J (2008) Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 358:2240–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, Windsor EA, Conlon TJ, Sumaroka A, Pang JJ, Roman AJ, Byrne BJ, Jacobson SG (2009) Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum Gene Ther 20:999–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, Windsor EA, Conlon TJ, Sumaroka A, Roman AJ, Byrne BJ, Jacobson SG (2009) Vision 1 year after gene therapy for Leber’s congenital amaurosis. N Engl J Med 361:725–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maguire AM, High KA, Auricchio A, Wright JF, Pierce EA, Testa F, Mingozzi F, Bennicelli JL, Ying GS, Rossi S, Fulton A, Marshall KA, Banfi S, Chung DC, Morgan JI, Hauck B, Zelenaia O, Zhu X, Raffini L, Coppieters F, De Baere E, Shindler KS, Volpe NJ, Surace EM, Acerra C, Lyubarsky A, Redmond TM, Stone E, Sun J, McDonnell JW, Leroy BP, Simonelli F, Bennett J (2009) Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet 374:1597–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banin E, Bandah-Rozenfeld D, Obolensky A, Cideciyan AV, Aleman TS, Marks-Ohana D, Sela M, Boye S, Sumaroka A, Roman AJ, Schwartz SB, Hauswirth WW, Jacobson SG, Hemo I, Sharon D (2010) Molecular anthropology meets genetic medicine to treat blindness in the North African Jewish population: human gene therapy initiated in Israel. Hum Gene Ther 21:1749–1757 [DOI] [PubMed] [Google Scholar]
- 9.Simonelli F, Maguire AM, Testa F, Pierce EA, Mingozzi F, Bennicelli JL, Rossi S, Marshall K, Banfi S, Surace EM, Sun J, Redmond TM, Zhu X, Shindler KS, Ying GS, Ziviello C, Acerra C, Wright JF, McDonnell JW, High KA, Bennett J, Auricchio A (2010) Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol Ther 18:643–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, Roman AJ, Peden MC, Aleman TS, Boye SL, Sumaroka A, Conlon TJ, Calcedo R, Pang JJ, Erger KE, Olivares MB, Mullins CL, Swider M, Kaushal S, Feuer WJ, Iannaccone A, Fishman GA, Stone EM, Byrne BJ, Hauswirth WW (2012) Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 130 (1):9–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leber T (1869) Über Retinitis pigmentosa und angeborene Amaurose. Graefes Arch Clin Exp Ophthalmol 15:1–25 [Google Scholar]
- 12.Blum M, Hykin PG, Sanders M, Volcker HE (1992) Theodor Leber: a founder of ophthalmic research. Surv Ophthalmol 37:63–68 [DOI] [PubMed] [Google Scholar]
- 13.Franceschetti A, Dieterle P (1954) Importance diagnostique et prognostique de l’électrorétinogramme (ERG), dans les dégénérescences tapéto-rétiniennes avec rétrécissement du champ visuel et héméralopie. Confin Neurol 14:184–186 [PubMed] [Google Scholar]
- 14.Stone EM (2007) Leber congenital amaurosis — a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol 144:791–811 [DOI] [PubMed] [Google Scholar]
- 15.Schappert-Kimmijser J, Henkes HE, Van Den Bosch J (1959) Amaurosis congenita (Leber). AMA Arch Ophthalmol 61:211–218 [DOI] [PubMed] [Google Scholar]
- 16.Lambert SR, Kriss A, Taylor D, Coffey R, Pembrey M (1989) Follow-up and diagnostic reappraisal of 75 patients with Leber’s congenital amaurosis. Am J Ophthalmol 107:624–631 [DOI] [PubMed] [Google Scholar]
- 17.Heher KL, Traboulsi EI, Maumenee IH (1992) The natural history of Leber’s congenital amaurosis. Age-related findings in 35 patients. Ophthalmology 99:241–245 [DOI] [PubMed] [Google Scholar]
- 18.Fulton AB, Hansen RM, Mayer DL (1996) Vision in Leber congenital amaurosis. Arch Ophthalmol 114:698–703 [DOI] [PubMed] [Google Scholar]
- 19.Walia S, Fishman GA, Jacobson SG, Aleman TS, Koenekoop RK, Traboulsi EI, Weleber RG, Pennesi ME, Heon E, Drack A, Lam BL, Allikmets R, Stone EM (2010) Visual acuity in patients with Leber’s congenital amaurosis and early childhood-onset retinitis pigmentosa. Ophthalmology 117:1190–1198 [DOI] [PubMed] [Google Scholar]
- 20.Brecelj J, Stirn-Kranjc B (1999) ERG and VEP follow-up study in children with Leber’s congenital amaurosis. Eye 13(Pt 1):47–54 [DOI] [PubMed] [Google Scholar]
- 21.Godel V, Regenbogen L (1977) Functional evaluation in central retinitis pigmentosa. Ophthalmologica 174:121–128 [DOI] [PubMed] [Google Scholar]
- 22.Birch DG, Peters AY, Locke KL, Spencer R, Megarity CF, Travis GH (2001) Visual function in patients with cone-rod dystrophy (CRD) associated with mutations in the ABCA4(ABCR) gene. Exp Eye Res 73:877–886 [DOI] [PubMed] [Google Scholar]
- 23.Perrault I, Hanein S, Gerber S, Barbet F, Dufier JL, Munnich A, Rozet JM, Kaplan J (2003) Evidence of autosomal dominant Leber congenital amaurosis (LCA) underlain by a CRX heterozygous null allele. J Med Genet 40:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dharmaraj SR, Silva ER, Pina AL, Li YY, Yang JM, Carter CR, Loyer MK, El-Hilali HK, Traboulsi EK, Sundin OK, Zhu DK, Koenekoop RK, Maumenee IH (2000) Mutational analysis and clinical correlation in Leber congenital amaurosis. Ophthalmic Genet 21:135–150 [PubMed] [Google Scholar]
- 25.Simonelli F, Ziviello C, Testa F, Rossi S, Fazzi E, Bianchi PE, Fossarello M, Signorini S, Bertone C, Galantuomo S, Brancati F, Valente EM, Ciccodicola A, Rinaldi E, Auricchio A, Banfi S (2007) Clinical and molecular genetics of Leber’s congenital amaurosis: a multicenter study of Italian patients. Invest Ophthalmol Vis Sci 48:4284–4290 [DOI] [PubMed] [Google Scholar]
- 26.Perrault I, Rozet JM, Ghazi I, Leowski C, Bonnemaison M, Gerber S, Ducroq D, Cabot A, Souied E, Dufier JL, Munnich A, Kaplan J (1999) Different functional outcome of RetGC1 and RPE65 gene mutations in Leber congenital amaurosis. Am J Hum Genet 64:1225–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porto FB, Perrault I, Hicks D, Rozet JM, Hanoteau N, Hanein S, Kaplan J, Sahel JA (2002) Prenatal human ocular degeneration occurs in Leber’s congenital amaurosis (LCA2). J Gene Med 4:390–396 [DOI] [PubMed] [Google Scholar]
- 28.Pasadhika S, Fishman GA, Stone EM, Lindeman M, Zelkha R, Lopez I, Koenekoop RK, Shahidi M (2010) Differential macular morphology in patients with RPE65-, CEP290-, GUCY2D-, and AIPL1-related Leber congenital amaurosis. Invest Ophthalmol Vis Sci 51:2608–2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A (1997) Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet 17:194–197 [DOI] [PubMed] [Google Scholar]
- 30.Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP (1998) Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci U S A 95:3088–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weleber RG, Michaelides M, Trzupek KM, Stover NB, Stone EM (2011) The phenotype of Severe Early Childhood Onset Retinal Dystrophy (SECORD) from mutation of RPE65 and differentiation from Leber congenital amaurosis. Invest Ophthalmol Vis Sci 52:292–302 [DOI] [PubMed] [Google Scholar]
- 32.Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, Goletz P, Ma JX, Crouch RK, Pfeifer K (1998) Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet 20:344–351 [DOI] [PubMed] [Google Scholar]
- 33.Lopes VS, Gibbs D, Libby RT, Aleman TS, Welch DL, Lillo C, Jacobson SG, Radu RA, Steel KP, Williams DS(2011)The Usher 1B protein, MYO7A, is required for normal localization and function of the visual retinoid cycle enzyme, RPE65. Hum Mol Genet 20:2560–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Znoiko SL, Rohrer B, Lu K, Lohr HR, Crouch RK, Ma JX (2005) Downregulation of cone-specific gene expression and degeneration of cone photoreceptors in the Rpe65−/− mouse at early ages. Invest Ophthalmol Vis Sci 46:1473–1479 [DOI] [PubMed] [Google Scholar]
- 35.Jacobson SG, Aleman TS, Cideciyan AV, Sumaroka A, Schwartz SB, Windsor EA, Traboulsi EI, Heon E, Pittler SJ, Milam AH, Maguire AM, Palczewski K, Stone EM, Bennett J (2005) Identifying photoreceptors in blind eyes caused by RPE65 mutations: Prerequisite for human gene therapy success. Proc Natl Acad Sci U S A 102:6177–6182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Veske A, Nilsson SE, Narfstrom K, Gal A (1999) Retinal dystrophy of Swedish briard/briard-beagle dogs is due to a 4-bp deletion in RPE65. Genomics 57:57–61 [DOI] [PubMed] [Google Scholar]
- 37.Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, Pearce-Kelling SE, Anand V, Zeng Y, Maguire AM, Jacobson SG, Hauswirth WW, Bennett J (2001) Gene therapy restores vision in a canine model of childhood blindness. Nat Genet 28:92–95 [DOI] [PubMed] [Google Scholar]
- 38.Acland GM, Aguirre GD, Bennett J, Aleman TS, Cideciyan AV, Bennicelli J, Dejneka NS, Pearce-Kelling SE, Maguire AM, Palczewski K, Hauswirth WW, Jacobson SG (2005) Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther 12:1072–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amado D, Mingozzi F, Hui D, Bennicelli JL, Wei Z, Chen Y, Bote E, Grant RL, Golden JA, Narfstrom K, Syed NA, Orlin SE, High KA, Maguire AM, Bennett J (2010) Safety and efficacy of subretinal readministration of a viral vector in large animals to treat congenital blindness. Sci Transl Med 2:21ra16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barker SE, Broderick CA, Robbie SJ, Duran Y, Natkunarajah M, Buch P, Balaggan KS, MacLaren RE, Bainbridge JW, Smith AJ, Ali RR (2009) Subretinal delivery of adeno-associated virus serotype 2 results in minimal immune responses that allow repeat vector administration in immunocompetent mice. J Gene Med 11:486–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Narfstrom K, Katz ML, Bragadottir R, Seeliger M, Boulanger A, Redmond TM, Caro L, Lai CM, Rakoczy PE (2003) Functional and structural recovery of the retina after gene therapy in the RPE65 null mutation dog. Invest Ophthalmol Vis Sci 44:1663–1672 [DOI] [PubMed] [Google Scholar]
- 42.Jacobson SG, Acland GM, Aguirre GD, Aleman TS, Schwartz SB, Cideciyan AV, Zeiss CJ, Komaromy AM, Kaushal S, Roman AJ, Windsor EA, Sumaroka A, Pearce-Kelling SE, Conlon TJ, Chiodo VA, Boye SL, Flotte TR, Maguire AM, Bennett J, Hauswirth WW (2006) Safety of recombinant adeno-associated virus type 2-RPE65 vector delivered by ocular subretinal injection. Mol Ther 13:1074–1084 [DOI] [PubMed] [Google Scholar]
- 43.Dejneka NS, Surace EM, Aleman TS, Cideciyan AV, Lyubarsky A, Savchenko A, Redmond TM, Tang W, Wei Z, Rex TS, Glover E, Maguire AM, Pugh EN Jr, Jacobson SG, Bennett J (2004) In utero gene therapy rescues vision in a murine model of congenital blindness. Mol Ther 9:182–188 [DOI] [PubMed] [Google Scholar]
- 44.Lai CM, Yu MJ, Brankov M, Barnett NL, Zhou X, Redmond TM, Narfstrom K, Rakoczy PE (2004) Recombinant adeno-associated virus type 2-mediated gene delivery into the Rpe65−/− knockout mouse eye results in limited rescue. Genet Vaccines Ther 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pang JJ, Chang B, Kumar A, Nusinowitz S, Noorwez SM, Li J, Rani A, Foster TC, Chiodo VA, Doyle T, Li H, Malhotra R, Teusner JT, McDowell JH, Min SH, Li Q, Kaushal S, Hauswirth WW (2006) Gene therapy restores vision-dependent behavior as well as retinal structure and function in a mouse model of RPE65 Leber congenital amaurosis. Mol Ther 13:565–572 [DOI] [PubMed] [Google Scholar]
- 46.Weber M, Rabinowitz J, Provost N, Conrath H, Folliot S, Briot D, Cherel Y, Chenuaud P, Samulski J, Moullier P, Rolling F (2003) Recombinant adeno-associated virus serotype 4 mediates unique and exclusive long-term transduction of retinal pigmented epithelium in rat, dog, and nonhuman primate after subretinal delivery. Mol Ther 7:774–781 [DOI] [PubMed] [Google Scholar]
- 47.Jacobson SG, Boye SL, Aleman TS, Conlon TJ, Zeiss CJ, Roman AJ, Cideciyan AV, Schwartz SB, Komaromy AM, Doobrajh M, Cheung AY, Sumaroka A, Pearce-Kelling SE, Aguirre GD, Kaushal S, Maguire AM, Flotte TR, Hauswirth WW (2006) Safety in nonhuman primates of ocular AAV2-RPE65, a candidate treatment for blindness in Leber congenital amaurosis. Hum Gene Ther 17:845–858 [DOI] [PubMed] [Google Scholar]
- 48.Le Meur G, Weber M, Pereon Y, Mendes-Madeira A, Nivard D, Deschamps JY, Moullier P, Rolling F (2005) Postsurgical assessment and long-term safety of recombinant adeno-associated virus-mediated gene transfer into the retinas of dogs and primates. Arch Ophthalmol 123:500–506 [DOI] [PubMed] [Google Scholar]
- 49.Cideciyan AV (2010) Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog Retin Eye Res 29:398–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ashtari M, Cyckowski LL, Monroe JF, Marshall KA, Chung DC, Auricchio A, Simonelli F, Leroy BP, Maguire AM, Shindler KS, Bennett J (2011) The human visual cortex responds to gene therapy-mediated recovery of retinal function. J Clin Invest 121:2160–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bennett J, Ashtari M, Wellman J, Marshall KA, Cyckowski LL, Chung DC, McCague S, Pierce EA, Chen Y, Bennicelli JL, Zhu X, Ying GS, Sun J, Wright JF, Auricchio A, Simonelli F, Shindler KS, Mingozzi F, High KA, Maguire AM (2012) AAV2 gene therapy readministration in three adults with congenital blindness. Sci Transl Med 4:120ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boye SL, Conlon T, Erger K, Ryals R, Neeley A, Cossette T, Pang J, Dyka FM, Hauswirth WW, Boye SE (2011) Long-term preservation of cone photoreceptors and restoration of cone function by gene therapy in the guanylate cyclase-1 knockout (GC1KO) mouse. Invest Ophthalmol Vis Sci 52:7098–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vandenberghe LH, Bell P, Maguire AM, Cearley CN, Xiao R, Calcedo R, Wang L, Castle MJ, Maguire AC, Grant R, Wolfe JH, Wilson JM, Bennett J (2011) Dosage thresholds for AAV2 and AAV8 photoreceptor gene therapy in monkey. Sci Transl Med 3:88ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bemelmans AP, Kostic C, Crippa SV, Hauswirth WW, Lem J, Munier FL, Seeliger MW, Wenzel A, Arsenijevic Y (2006) Lentiviral gene transfer of RPE65 rescues survival and function of cones in a mouse model of Leber congenital amaurosis. PLoS Med 3:e347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lheriteau E, Libeau L, Mendes-Madeira A, Deschamps JY, Weber M, Le Meur G, Provost N, Guihal C, Moullier P, Rolling F (2010) Regulation of retinal function but nonrescue of vision in RPE65-deficient dogs treated with doxycycline-regulatable AAV vectors. Mol Ther 18:1085–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stieger K, Le Meur G, Lasne F, Weber M, Deschamps JY, Nivard D, Mendes-Madeira A, Provost N, Martin L, Moullier P, Rolling F (2006) Long-term doxycycline-regulated transgene expression in the retina of nonhuman primates following subretinal injection of recombinant AAV vectors. Mol Ther 13:967–975 [DOI] [PubMed] [Google Scholar]
- 57.Bemelmans AP, Kostic C, Hornfeld D, Jaquet M, Crippa SV, Hauswirth WW, Lem J, Wang Z, Schorderet DE, Munier FL, Wenzel A, Arsenijevic Y (2006) Lentiviral vectors containing a retinal pigment epithelium specific promoter for leber congenital amaurosis gene therapy. Lentiviral gene therapy for LCA. Adv Exp Med Biol 572:247–253 [DOI] [PubMed] [Google Scholar]
- 58.Conley SM, Naash MI (2010) Nanoparticles for retinal gene therapy. Prog Retin Eye Res 29:376–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson CJ, Berglin L, Chrenek MA, Redmond TM, Boatright JH, Nickerson JM (2008) Technical brief: subretinal injection and electroporation into adult mouse eyes. Mol Vis 14:2211–2226 [PMC free article] [PubMed] [Google Scholar]
- 60.Jo DH, Lee TG, Kim JH (2011) Nanotechnology and nanotoxicology in retinopathy. Int J Mol Sci 12:8288–8301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fradot M, Busskamp V, Forster V, Cronin T, Leveillard T, Bennett J, Sahel JA, Roska B, Picaud S (2011) Gene therapy in ophthalmology: validation on cultured retinal cells and explants from postmortem human eyes. Hum Gene Ther 22:587–593 [DOI] [PubMed] [Google Scholar]
- 62.Sun X, Pawlyk B, Xu X, Liu X, Bulgakov OV, Adamian M, Sandberg MA, Khani SC, Tan MH, Smith AJ, Ali RR, Li T (2010) Gene therapy with a promoter targeting both rods and cones rescues retinal degeneration caused by AIPL1 mutations. Gene Ther 17:117–131 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63.Tan MH, Smith AJ, Pawlyk B, Xu X, Liu X, Bainbridge JB, Basche M, McIntosh J, Tran HV, Nathwani A, Li T, Ali RR (2009) Gene therapy for retinitis pigmentosa and Leber congenital amaurosis caused by defects in AIPL1: effective rescue of mouse models of partial and complete Aipl1 deficiency using AAV2/2 and AAV2/8 vectors. Hum Mol Genet 18:2099–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boye SE, Boye SL, Pang J, Ryals R, Everhart D, Umino Y, Neeley AW, Besharse J, Barlow R, Hauswirth WW (2010) Functional and behavioral restoration of vision by gene therapy in the guanylate cyclase-1 (GC1) knockout mouse. PLoS One 5:e11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jacobson SG, Cideciyan AV, Aleman TS, Sumaroka A, Roman AJ, Swider M, Schwartz SB, Banin E, Stone EM (2011) Human retinal disease from AIPL1 gene mutations: foveal cone loss with minimal macular photoreceptors and rod function remaining. Invest Ophthalmol Vis Sci 52:70–79 [DOI] [PubMed] [Google Scholar]
- 66.Pennesi ME, Stover NB, Stone EM, Chiang PW, Weleber RG (2011) Residual Residual electroretinograms in young Leber congenital amaurosis patients with mutations of AIPL1. Invest Ophthalmol Vis Sci 52:8166–8173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Testa F, Surace EM, Rossi S, Marrocco E, Gargiulo A, Di Iorio V, Ziviello C, Nesti A, Fecarotta S, Bacci ML, Giunti M, Della Corte M, Banfi S, Auricchio A, Simonelli F (2011) Evaluation of Italian patients with Leber congenital amaurosis due to AIPL1 mutations highlights the potential applicability of gene therapy. Invest Ophthalmol Vis Sci 52:5618–5624 [DOI] [PubMed] [Google Scholar]
- 68.Bonnet C, Augustin S, Ellouze S, Benit P, Bouaita A, Rustin P, Sahel JA, Corral-Debrinski M (2008) The optimized allotopic expression of ND1 or ND4 genes restores respiratory chain complex I activity in fibroblasts harboring mutations in these genes. Biochim Biophys Acta 1783:1707–1717 [DOI] [PubMed] [Google Scholar]
- 69.Ellouze S, Augustin S, Bouaita A, Bonnet C, Simonutti M, Forster V, Picaud S, Sahel JA, Corral-Debrinski M (2008) Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction. Am J Hum Genet 83:373–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lam BL, Feuer WJ, Abukhalil F, Porciatti V, Hauswirth WW, Guy J (2010) Leber hereditary optic neuropathy gene therapy clinical trial recruitment: year 1. Arch Ophthalmol 128:1129–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Perrault I, Rozet JM, Calvas P, Gerber S, Camuzat A, Dollfus H, Chatelin S, Souied E, Ghazi I, Leowski C, Bonnemaison M, Le Paslier D, Frezal J, Dufier JL, Pittler S, Munnich A, Kaplan J (1996) Retinal-specific guanylate cyclase gene mutations in Leber’s congenital amaurosis. Nat Genet 14:461–464 [DOI] [PubMed] [Google Scholar]
- 72.Marlhens F, Bareil C, Griffoin JM, Zrenner E, Amalric P, Eliaou C, Liu SY, Harris E, Redmond TM, Arnaud B, Claustres M, Hamel CP (1997) Mutations in RPE65 cause Leber’s congenital amaurosis. Nat Genet 17:139–141 [DOI] [PubMed] [Google Scholar]
- 73.Dharmaraj S, Leroy BP, Sohocki MM, Koenekoop RK, Perrault I, Anwar K, Khaliq S, Devi RS, Birch DG, De Pool E, Izquierdo N, Van Maldergem L, Ismail M, Payne AM, Holder GE, Bhattacharya SS, Bird AC, Kaplan J, Maumenee IH (2004) The phenotype of Leber congenital amaurosis in patients with AIPL1 mutations. Arch Ophthalmol 122:1029–1037 [DOI] [PubMed] [Google Scholar]
- 74.Dryja TP, Adams SM, Grimsby JL, McGee TL, Hong DH, Li T, Andreasson S, Berson EL (2001) Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet 68:1295–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lotery AJ, Jacobson SG, Fishman GA, Weleber RG, Fulton AB, Namperumalsamy P, Heon E, Levin AV, Grover S, Rosenow JR, Kopp KK, Sheffield VC, Stone EM (2001) Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch Ophthalmol 119:415–420 [DOI] [PubMed] [Google Scholar]
- 76.Le Meur G, Stieger K, Smith AJ, Weber M, Deschamps JY, Nivard D, Mendes-Madeira A, Provost N, Pereon Y, Cherel Y, Ali RR, Hamel C, Moullier P, Rolling F (2007) Restoration of vision in RPE65-deficient Briard dogs using an AAV serotype 4 vector that specifically targets the retinal pigmented epithelium. Gene Ther 14:292–303 [DOI] [PubMed] [Google Scholar]
- 77.Bennicelli J, Wright JF, Komaromy A, Jacobs JB, Hauck B, Zelenaia O, Mingozzi F, Hui D, Chung D, Rex TS, Wei Z, Qu G, Zhou S, Zeiss C, Arruda VR, Acland GM, Dell’Osso LF, High KA, Maguire AM, Bennett J (2008) Reversal of blindness in animal models of leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol Ther 16:458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]