

Abstract
Lysosomal acid lipase (LAL) is essential for cholesteryl ester (CE) and triacylglycerol (TAG) hydrolysis in lysosomes. Clinically, an autosomal recessive LIPA mutation causes LAL deficiency (LAL-D), either Wolman Disease or Cholesterol Ester Storage Disease (CESD). LAL-D is associated with ectopic neutral lipid accumulation in the liver, small intestine, spleen, adrenal glands, and blood. Considering the importance of unesterified cholesterol and fatty acids in bone metabolism, we hypothesized that LAL is essential to bone formation, and ultimately, skeletal health. To investigate the role of LAL in skeletal homeostasis, we used LAL-deficient (−/−) mice and osteoblast cell cultures. Male LAL−/− mice had lower trabecular BV/TV (12%) compared to WT mice (21%), due to decreased trabecular number and increased trabecular separation; this change was not apparent in the females. While both sexes of LAL−/− mice displayed decreased cortical bone thickness and polar moment of inertia, only the female LAL−/− mice showed increased cortical porosity. Histological analyses revealed that LAL−/− mice tended to have less osteoblasts but no change in osteoclast numbers. In studying the cell-autonomous role of LAL, we observed impaired osteoblastogenesis of LAL−/− calvarial osteoblasts and in bone marrow stromal cells treated with the LAL inhibitor lalistat. Consistent with LAL’s role in other tissues, lalistat resulted in profound lipid puncta accumulation and an altered intracellular lipid profile. Finally, we analyzed a large de-identified national insurance database (i.e. 2016/2017 Optum Clinformatics®) which revealed that adults (≥18 years) with CESD (n=3,076) had a higher odds ratio (OR=1.21; 95% CI=1.03-1.41) of all-cause fracture at any location compared to adults without CESD (n=13.7 M) after adjusting for demographic variables and osteoporosis. These data demonstrate that alterations in LAL have significant clinical implications related to fracture risk and that LAL’s modulation of lipid metabolism is a critical for osteoblast function.
Keywords: Osteoblast, Skeleton, Bone, Lipid, Cholesterol, Lipophagy, Metabolism
1. Introduction
Rare genetic diseases provide an invaluable opportunity to learn about novel molecular mechanisms regulating bone turnover. Such conditions have led to the identification of critical proteins/ signaling pathways involved in maintaining bone health including bone morphogenetic protein (BMP)/ activin as in fibrodysplasia ossificans progressiva [1], WNT/ lipoprotein receptor-related protein (LRP)-5 in osteoporosis-pseudoglioma syndrome (OPPG) [2], and sclerostin from Sclerosteosis/ Van Buchem disease [3]. We have identified an additional rare disease, lysosomal acid lipase (LAL) deficiency, or LAL-D, that modulates bone formation via the newly discovered role of LAL in osteoblasts.
As a lipase, LAL’s predominant role involves two distinct mechanisms involving the hydrolysis of lipids, namely cholesteryl esters (CE) and triacylglycerols (TAGs). The first, more classically described role for LAL, involves modulating cholesterol homeostasis via low density lipoprotein (LDL)-receptor (LDL-R). Following LDL-LDLR binding and endocytosis, the late endosome fuses with the lysosome, whereby LAL can liberate free cholesterol from CE, and to a lesser extent, free fatty acids from TAG [4]. A second, more recently described mechanism involves LAL’s role in lipolysis via the lysosome during selective autophagy, termed lipophagy, primarily mobilizing free fatty acids from TAGs within the lipid droplet core [5]. Clinical conditions of LAL deficiency, LAL-D (OMIM 278000), results from mutations in the LIPA gene in an autosomal recessive pattern which occurs at a frequency of 1 in 40,000 to 100,000 varying by population. The disease presents in two distinct manners described as ‘early-onset’ (Wolman) disease, and ‘later onset’ previously termed cholesteryl ester storage disease (CESD), ranging in severity [6]. Early onset LAL-D is much more severe and includes hepatosplenomegaly, steatorrhea, abdominal distention, adrenal calcification, and results in death within the first year of age [7]. Conversely, the milder, late-onset LAL-D manifests with variable CE and TAG accumulation in the liver, adrenal gland, and small intestine [8]. Additionally, patients often display premature atherosclerosis and hypercholesterolemia, but skeletal phenotyping has not been reported. These pathologies have primarily been attributed to the dyslipidemia observed in individuals with LAL- D, characterized by elevated LDL-cholesterol and low HDL-cholesterol [8]. To date, there has been no documented skeletal phenotype associated with LAL-D. This is particularly interesting because epidemiologic data indicate that high concentrations of circulating cholesterol and accelerated atherosclerosis are linked to low bone mass and osteoporosis [9–12]. Moreover, our lab previously reported that intracellular lipid droplets are critical for osteoblast differentiation, and that peripheral adipose depots have the ability to contribute fatty acid energy sources to support osteoblast function [13, 14]. Hence, rare disorders of lipid metabolism provide the novel opportunities to study the effect of skeletal homeostasis.
Many mechanistic insights have been discovered relative to LAL-D using LAL-deficient (−/−) mice, which phenocopy the human disease as they suffer from severe abnormalities ascribed with early-onset LAL-D but a life expectancy of up to one year [15–18]. Quite striking, LAL−/− mice demonstrate the absence of inguinal and retroperitoneal white adipose tissue (WAT), as well as reduced interscapular brown adipose tissue (BAT) despite the lipid-loaded phenotypes observed in other tissues such as in the liver, spleen, and intestine [16]. These data demonstrate tissue-specific regulatory machineries and functions of LAL, which provides a particularly exciting opportunity to exploit in the context of bone biology. As such, the current manuscript identifies lysosomal acid lipase deficiency (LAL-D) as an additional rare disease associated with increased fracture risk and altered bone homeostasis.
2. Material and Methods
2.1. Animals
To identify potential mechanisms contributing to skeletal fragility in patients with LAL-D, we characterized male and female mice containing a global deletion of the Lipa gene (referred to hereafter as ‘LAL−/−’) and their respective littermate non-mutated, wild-type controls (WT) (Medical University of Graz, Austria). Mice were maintained in a clean environment with a regular light-dark (12 hr/12 hr) cycle and ad libitum access to food (chow diet; 4% fat and 9% protein; Altromin 1324, Lage, Germany) and water. At 16 weeks of age, mice were sacrificed, femora and tibiae were cleaned of soft-adhering tissue, placed in 10% neutral-buffered formalin (NBF) for 48 hrs, and then stored in 70% ethanol for future analyses. Ex vivo/ in vitro experiments were performed using 8- to 10-week-old male and female C57BL/6J or LAL−/− and WT mice, while LAL−/− and WT pups (5 days) were used to isolate calvaria osteoblast (cOBs). Plasma was isolated from whole blood for assessment of biochemical bone markers. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) and/ or the Austrian Federal Ministry of Science, Research, and Economy, Vienna, Austria, in accordance with the European Directive 2010/63/EU.
2.2. Micro-computed Tomography (μCT)
To assess trabecular architecture and cortical structure, the distal femur and mid-diaphysis were imaged using an ex vivo micro-computed tomography (μCT) scanner (μCT 50, Scanco Medical AG, Brüttisellen, Switzerland). With a peak X-ray tube intensity and current of 70 kVp and 114 mA, respectively, 1000 projections per full rotation of the sample, and an integration time of 300 ms, image stacks with an isotropic voxel size of 6 μm were acquired for the femur metaphysis (620 slices) and diaphysis (310 slices). Due to the length discrepancies between the WT and KO mice, the femur cancellous bone compartment was evaluated to account for these differences. As such, the male and female WT mice cancellous bone was analyzed from a 2.7 mm-long region (450 transverse slices) beginning 300 μm above the peak of the distal growth plate and extending proximally. Conversely, the cancellous bone from the KO females included 2.45 mm-long region beginning 270 μm above the peak of the distal growth plate and extending proximally to account for 9% reduction in femur length. Male KO femurs were 4% shorter compared to WT, and thus 2.6 mm-long region was included for analyses of cancellous bone starting 288 μm above the peak of the distal growth plate and extending proximally. Cortical bone architecture was evaluated in a 0.5 mm-long region (50 transverse slices) at the femoral mid-diaphysis. After defining the region of interest with contours for trabecular bone and cortical bone (tight fit to periosteal surface), a Gaussian filter (sigma = 0.2 and support =1) and global threshold of 220 per mille (344.9 mgHA/cm3 for trabeculae) or 420 per mille (828.1 mgHA/cm3 for cortices) were applied to segment bone from soft tissue and air. Standard Scanco evaluation scripts provided the morphological parameters of trabecular bone and structural parameters of cortical bone.
2.3. μCT Finite Element Analysis (μFEA)
The segmented image stack from each μCT scan of the distal femurs was converted to a finite element (FE) model in which each bone voxel was directly converted to 8-node hexahedral elements (fe_solveD, v1.16, Scanco Medical AG, Brüttisellen, Switzerland). Each element was assigned to 68-95 materials based on bins of tissue mineral density (TMD) with a bin size of 18.9 mg•HA/cm3 (e.g., 344.9.0-363.8 mg•HA/cm3, 363.8-382.7 mg•HA/cm3, etc.), and the modulus of each material was equal to 0.1127×TMD1.746 (MPa from mg•HA/cm3 where TMD is the median value of each bin). Using built-in boundary conditions that subjected the distal femur to uniaxial compression such that the nodes of the distal metaphyseal cross-section (Supplemental Figure 1) were rigidly fixed in the 3 orthogonal directions and the nodes of the distal diaphyseal cross-section were prescribed a vertical unit displacement in the z-direction but fixed in the orthogonal x- and y-directions, to impart an apparent strain of 1%. The Scanco FE-elastic solver determined the apparent stiffness (reaction force at the fixed distal nodes divided by vertical displacement of the proximal nodes) and the reaction force that caused 2% of the bone volume to exceed an equivalent strain of 0.007. The latter was the estimated failure load.
2.4. Static Bone Histomorphometry
Tibias were placed in Richard-Allan Scientific™ Decalcifying Solution (ThermoFisher Scientific, Waltham, MA) for 2 days, subjected to sequential ethanol dehydration, and embedded in paraffin. Embedded samples were then sectioned (5 μm) on a transverse plane, and sections were stained with hematoxylin and eosin (H&E) for subsequent analysis of static bone parameters [19]. Osteoblasts were identified as 3 or more cuboidal cells lining the trabecular bone surface, osteoclasts were identified as large, multinucleated cells on the bone surface (often adjacent to eroded surface), and bone marrow adipocytes were identified as white ‘ghost cells’ within the marrow area and excluding vasculature and/ or sectioning artifacts. All analyses were performed using BioQuant® Osteo 2018 version 18.2.6 (BioQuant® Image Analysis Corporation, Nashville, TN). The region of interest in the tibia was a 1 mm long region of the cancellous area starting 200 μm proximal to the distal growth plate.
2.5. In Vitro Cell Culture Experiments
Primary murine bone marrow stromal cells (BMSCs) and cOB were isolated as previously described [20]. Briefly, femur, tibia, and iliac crest were cleaned of soft adhering tissue, distal and proximal ends were opened, and bone marrow was isolated by centrifugation. Total bone marrow was plated in complete α-MEM (10% fetal bovine serum or FBS, 100 U/L penicillin G, and 100 mg/L streptomycin). The mesenchymal/ stromal population was isolated via plastic-adherence (48 hrs), trypsinized, counted, and plated for respective experiments. Primary cOBs were isolated from 5-day-old LAL−/− and WT pups. Calvariae were cleaned of soft-adhering tissue and osteoblasts were isolated using sequential 1.5 U/mg collagenase P incubations and collections. Following the final collection, cells were plated accordingly and cultured in a humidified incubator (5% CO2, 37°C). Upon confluency of both BMSCs and cOBs, osteoblast differentiation was induced by the addition of osteogenic medium: complete α-MEM supplemented with 5 mM β-glycerol phosphate (Sigma-Aldrich, St. Louis,MO), and 50 μg/mL ascorbic acid (Sigma-Aldrich, St. Louis, MO). Medium was changed every 2-3 days.
2.6. Alkaline Phosphatase and Von Kossa Staining
To determine how genetic deletion of LAL impacted osteoblast differentiation, cOBs (12 × 104 cells/ cm2) isolated from LAL−/− and WT mice were differentiated for 7 days and stained for alkaline phosphatase. Primary BMSCs (13.1 × 104 cells/ cm2) were cultured in the absence (control) or presence of the selective and specific LAL inhibitor lalistat 2 (100 μM, Tocris Bioscience, Boston, MA, referred to hereafter as ‘lalistat’) for 7 days and stained for ALP and Von Kossa. Staining was quantified using ImageJ (NIH; version 1.53h). TIF image files of the plates were uploaded into ImageJ. The freehand selection tool was used to indicate the whole well as the region of interest. The ‘Find Maxima’ analysis tool was used to measure weighted or unweighted average of colors. Maxima was presented as the number of maxima per unit area (in2). The dose of lalistat was based both on previously published reports [21] and from our data given cell viability and lipid droplet puncta (Supplemental Figure 1).
2.7. RNA Isolation and RNA Sequencing
Total RNA was extracted using ReliaPrep RNA Miniprep Systems (Promega, Madison, WI), according to the manufactures protocol. Each RNA sample was evaluated for integrity and quantitation using a Qubit RNA Assay and the Agilent Bioanalyzer. RNASeq libraries were prepared using 200 ng of RNA and the NEBNext® Ultra™ II RNA Library Prep (NEB, Cat: E7765S) per manufacturer’s instructions, with mRNA enriched via poly-A-selection using oligoDT beads. The RNA was then thermally fragmented and converted to cDNA, adenylated for adaptor ligation and PCR amplified. The libraries were sequenced using the NovaSeq 6000 with 150 bp paired end reads. RTA (version 2.4.11; Illumina) was used for base calling and analysis was completed using MultiQC v1.7. Adapters were trimmed by Cutadapt (v2.10). After trimming, reads were mapped to the mouse genome mm10 using STAR (v2.7.3a) and quantified by featureCounts (v2.0.0). DESeq2 (v.1.24.0) was used to detect differential expression between two groups. WebGestaltR (v0.4.4) was used to perform functional enrichment analysis.
2.8. Protein Extraction and Western Blot Analyses
To determine LAL protein expression, BMSCs were plated (10.5 × 10 cells/ cm2) and cell monolayers were lysed in radio-immune precipitation assay (RIPA) buffer (Cell Signaling, Danvers, MA) after 0 and 7 days in osteogenic medium. Additionally, protein was isolated from murine femur cortex (devoid of bone marrow). Protein samples were separated by SDS-PAGE and transferred to polyvinylidene-difluoride (PVDF) membranes using a Trans-Blot® Turbo™ (Bio-Rad, Hercules, CA). Blots were incubated with anti-LAL (1:1,000; Invitrogen, Carlsbad, CA) and β-actin antibodies (1:5,000; Santa Cruz Biotechnology, Inc., Dallas, TX). The protein bands were visualized by chemiluminescence using a ChemiDoc™ Tough Imaging System (Bio-Rad, Hercules, CA).
2.9. Confocal Microscopy
Primary murine BMSCs were cultured in osteogenic medium for 0 or 7 days in the absence (control) or presence of lalistat, 100 μM, 24 hr. Following treatment, cells were fixed using 4% paraformaldehyde (PFA) and stained with BODIPY 493/503 (1 μg/mL, Thermo Fisher Scientific, Waltham, MA) and Hoechst 33342 350/461 Solution (20 μg/mL, Thermo Fisher Scientific, Waltham, MA). A Leica SP8 confocal inverted microscope was used to visualize neutral lipids in the cells.
2.10. Lipidomics
Lipids were extracted from primary murine BMSC pellets (2.5 × 106 cells) following 0 or 7 days of incubation with osteogenic medium, in the absence (control) or presence of lalistat (100 μM, 24 hr) using a dichloromethane-methanol-water extraction protocol. Lipids were subsequently analyzed by direct infusion on a hybrid quadrupole time-of-flight (TOF) mass spectrometer (TripleTOF 5600, SCIEX, Framingham, MA) using the MS/MSALL workflow in both positive and negative modes. Lipid species were identified using LipidView™ software (SCIEX, Framingham, MA), and principal component analysis (PCA) (MarkerView™, SCIEX, Framingham, MA) was used to identify and group lipid species according to contribution to sample differences. Lipids were identified following the manuals from LipidView™ 1.2 software (ABSCIEX, Framingham, MA). Lipid annotation and assignment followed the metabolomics standards initiative (MSI) guidelines for metabolite (lipid) identification, i.e., Level 2: identification was based on spectral database (match factor > 80%) and Level 3: only compound groups were known, e.g. specific ions and RT regions of metabolites (lipids).
2.11. Clinical Data Analysis of Fracture Risk and LAL-D
To characterize the skeletal fragility phenotype among patients with LAL-D, we leveraged the Clinformatics® Data Mart Database (OptumInsight™, Eden Prairie, MN, USA) to compare prevalent all-cause fracture between adults with and without LAL-D. The Clinformatics® Data Mart Database is a U.S. nationwide single private payer administrative claims database [22, 23]. Claims data from 2016 and 2017 were extracted. We identified all medical conditions (e.g., LAL-D, fracture, chronic disease comorbidities) using the International Classification of Diseases, Tenth Revision (ICD-10), Clinical Modification codes (Supplemental Table 1). Information regarding how diagnoses were made or by whom (e.g., primary care physician) is not available in claims data.
Adults ≥18 years of age with at least one claim for fracture of the vertebral column, hip, upper extremities (not including wrist/hand), lower extremities (not including foot/toes), or unspecified site (i.e., stress fracture, osteoporosis with current pathological fracture, and pathological or non-traumatic fracture not classified elsewhere) were identified. The single claim-based definition has excellent accuracy for identifying fractures with up to 98% positive predictive value. Individuals were included if they had at least 12 full months of enrollment in a health plan in 2016 to 2017. Individuals with LAL-D were identified by at least one claim. The comparison group included individuals without any claims for LAL-D. Prevalent chronic diseases were selected based on their relevance to adults with LAL-D and fracture, as well as availability in the administrative claims database. Chronic diseases were identified by at least two different claim days (i.e., same claim over two or more different days). This claims-based definition improves accuracy for identifying non-event medical conditions (e.g., fracture is an “event” condition) over a single claim [24]. Chronic diseases included osteoporosis, ischemic heart disease, heart failure, cerebrovascular disease, hypertension, and type 2 diabetes. The data are de-identified and the University of Michigan Institutional Review Board approved this study as non-regulated.
2.12. Statistical Analyses
To determine the statistical significance (p ≤ 0.05) between groups, Student’s paired T-tests were performed. Variability, depicted as error bars, was calculated as the standard error of the mean (SEM) in the non-box plot figures. The figures using box plots represent median and interquartile range; the error bars represent 10th and 90th percentile data points[25, 26]. For analysis of the lipidomic data, the individual MS data extracted using LipidView™ 1.2 software (ABSCIEX, Framingham, MA) were assembled into a concatenated data set into a single .csv sheet in Microsoft Excel (Microsoft Corp., Seattle, WA). Statistical processing of the lipidomics data was performed using the statistical software R (Version 3.6.1) and some statistical tests such as T-test were performed using Prism 5 software (GraphPad Software, Inc. La Jolla, CA). Normalized, transformed, imputed, outlier removed, and scaled peak areas representative of relative lipid amounts were obtained using DeviumWeb and are represented. Hierarchical clustering analysis (HCA) was performed on Pearson distances using PermutMatrix, where the data were normalized using z-scores of the relative abundance of the lipids for a heat map display. Lipid fold changes with cut-offs of >1.2 and <0.8, and significant changes (p < 0.05) were calculated. Probability level a was set to 0.05, adjusted for multiple hypotheses testing using BH to allow for a maximum 5% probability (q = 0.05) in false positive detection (FDR). Correlations reported are Pearson correlations. PCA and partial least squared discriminant analyses (PLSDA) were performed using MetaboAnalyst where the output were displayed as score plots to visualize the sample groups. The data were scaled with unit variance without any transformation. MetaboAnalyst was also consulted for random forest (RF) analysis and outputs represented as mean decrease accuracies (MDA), whereas PLSDA driven VIP (variable importance in projection) are also reported.
To analyze the clinical data relative to fracture risk among adults with LAL-D, analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA). Descriptive characteristics were summarized for adults with and without LAL-D. Group differences between adults with and without LAL-D were examined using independent t-tests for continuous variables and Chi-square tests for categorical variables. Logistic regression models were developed with the outcome as any fracture and for each of the fracture subtypes (e.g., by location) and the primary exposure variable as LAL-D. To determine the effect of LAL-D on fracture prevalence beyond differences in sociodemographic variables and prevalent osteoporosis independent of age, sex, and race, model 1 was adjusted for age, sex, race, and U.S. region of residence and model 2 for the variables in model and osteoporosis. When significant, we performed additional analyses after stratifying by the appropriate variable and the main effect of the LAL-D group was interpreted. Effect estimates were reported as odds ratios (OR) with 95% confidence intervals (CI).
3. Results
3.1. LAL knockout mice display a low bone mass phenotype and reduced biomechanical properties.
Analysis of the distal femur metaphysis demonstrated lower trabecular bone (BV/TV) in the LAL−/− female and male mice compared to WT (Figure 1A). This reduction in BV/TV from LAL−/− mice was accompanied by decreased trabecular number (Figure 1B) and trabecular thickness (Figure 1C), with an increase in trabecular separation (Figure 1D). Elevated SMI suggested that LAL−/− mice had a more ‘rod-like’, weaker bone morphometry (Figure 1E). Connectivity density (Conn.Dn) was also significantly decreased in the both male and female LAL−/− mice compared with WT (Figure 1F). Both male and female LAL−/− mice displayed a mild reduced femur length (Figure 2A), which was accounted for in the μCT analyses. The cortical bone followed a similar pattern as the trabecular bone in that both male and female LAL−/− mice displayed reduced cortical parameters when compared to WT including cortical area (Figure 2B) and thickness (Figure 2C). Interestingly, female LAL−/− mice alone demonstrated increased cortical porosity (Figure 2D). Both female and male LAL−/− mice also demonstrated a lower polar moment of inertia (pMOI), which is a calculated assessment of bone strength, thus suggesting reduced biomechanical strength (Figure 2E), while no change was noted in bone marrow area (Figure 2F).
Figure 1. Male and female LAL−/− mice have low trabecular bone mass.
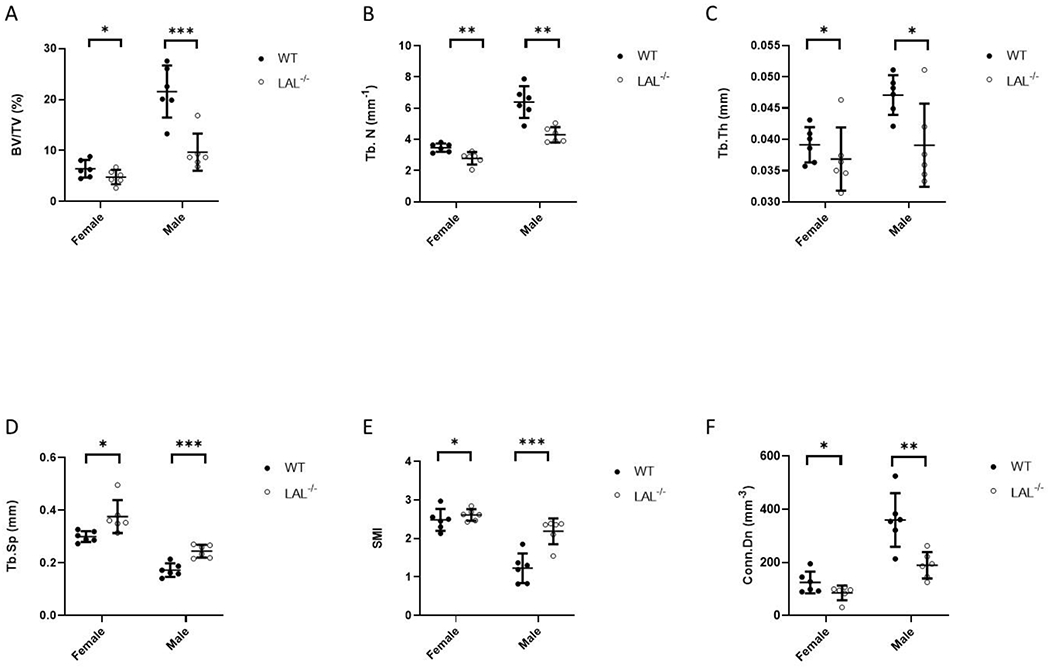
F1 Legend: Results from micro-computed tomography (μCT) analysis (A-F) of the trabecular bone in the distal femur metaphysis from 16-week-old WT and LAL−/− mice. A) Bone volume per total volume (BV/TV, %); B) Trabecular number (Tb.N); C) Trabecular thickness (Tb.Th.); D) Trabecular separation (Tb.Sp.); E) Structure model index (SMI); and F) Connectivity density (Conn. Dens., 1/mm3). Plots represent individual data points, mean, and standard deviation. Student’s T-Test was used to calculate significance (*; p < .05, **; p < .01, ***; p < .001).
Figure 2: LAL Deficiency reduced cortical bone parameters.
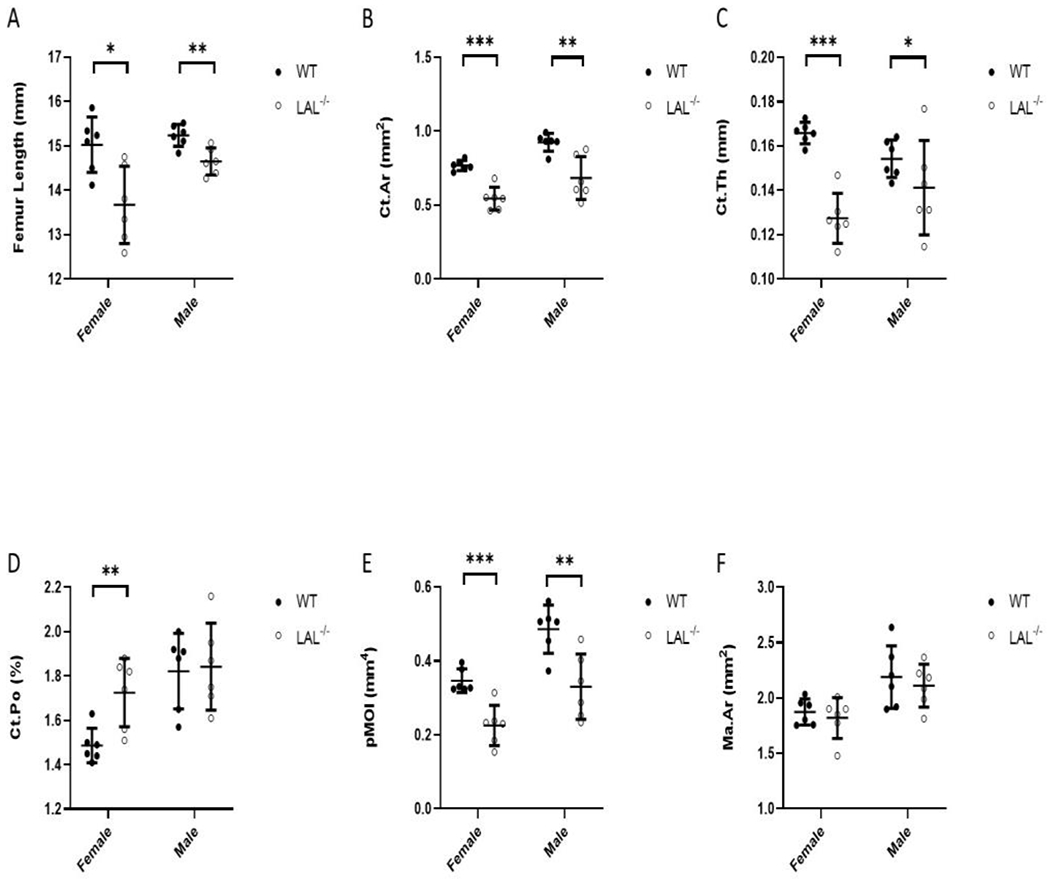
F2 Legend: Micro-computed tomography (μCT) analysis (A-F) of the cortical bone assessed at the femur diaphysis from 16-week-old WT and LA−/− mice. A) Femur length; B) Cortical bone area (Ct.A); C) Cortical thickness (Ct.Th.); D) Cortical Porosity (Ct.Po.); E) Polar moment of inertia (pMOI); and F) Bone marrow area (Ma.Ar). Plots represent individual data points, mean, and standard deviation. Student’s T-Test was used to calculate significance (*; p < .05, **; p < .01, ***; p < .001).
To gain further insight into the biomechanical alterations occurring in the bones from LAL−/− mice, we conducted μFEA to simulate compression testing. Consistent with reduced bone microarchitecture, both male and female LAL−/− mice displayed significantly lower apparent stiffness and failure load compare to WT (Table 1).
Table 1.
Genotype-related difference in compressive properties of the distal femur as determined by linear, elastic micro-finite element analysis (μFEA) using a heterogenous material model.
| Parameters | Unit | WT | Female LAL−/− | p-value | WT | Male LAL−/− | p-value |
|---|---|---|---|---|---|---|---|
| Apparent stiffness | N/m m | 6667.12 ± 619.35 | 4412.80 ± 709.90 | <0.001 | 6775.63 ± 864.65 | 5307.05 ± 1513.32 | 0.035 |
| Failure load | N | −75.65 ± 6.12 | −46.15 ± 8.81 | <0.001 | −79.82 ± 11.07 | −56.38 ± 22.27 | 0.044 |
Mean ± SD; WT-Wild type; LAL−/− – Lysosomal acid lipase knock out
Analysis of histological sections revealed that, within the region of interest, male LAL−/− mice had fewer number of osteoblasts compared to WT (Figure 3A). Importantly, once these values are expressed per bone perimeter (N.Ob/BS), the significance is reduced (p=0.008) to reflect only a trend in osteoblast reduction in both male and female LAL−/− mice compared to WT (Figure 3B), while expressing osteoblasts per total area (N.Ob/T.Ar) again confirmed a reduction in osteoblasts (Figure 3C). There was no change observed in osteoclast number (Osteoclasts, N.Oc/BS) (Figure 3D, 3E), average adipocyte volume (Ad.V) (Figure 3F), or number of adipocytes (N.Ad/Ma.Ar) (Figure 3G) in either sex. No change in bone marrow adipocytes was particularly striking as the cortex of the tibia shaft appear ‘white’ in color (Supplemental Figure 2A.) The growth plate also appeared similar to that of the WT in LAL−/− mice (Supplemental Figure 2B). Lastly, despite the low bone mass phenotype observed in LAL−/− mice, biochemical bone markers CTX-I and PINP were both increased in female and male LAL−/− mice compared to WT (Supplemental Figure 3).
Figure 3: Male LAL−/− mice have fewer osteoblasts.
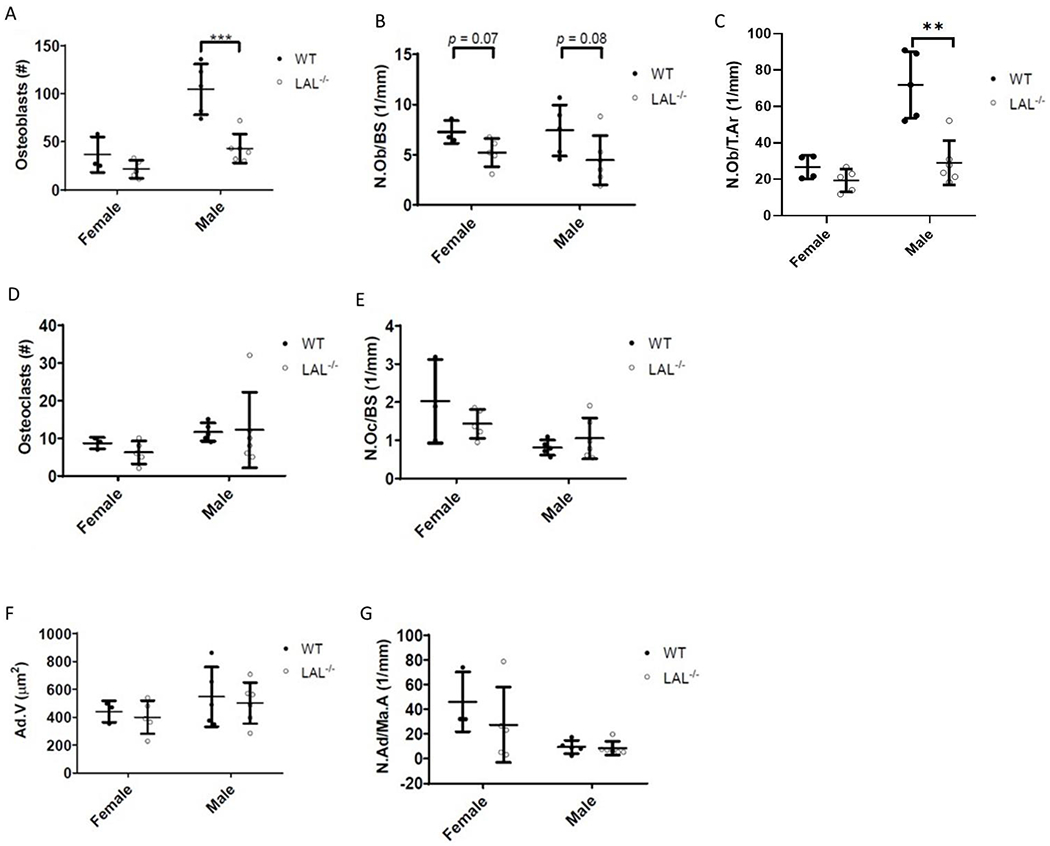
F3 Legend: Histological analysis of the trabecular bone of the proximal tibia of 16-week-old WT and LAL−/− mice. Number of A) osteoblasts (N.Ob.), B) osteoblasts per millimeter bone surface (N.Ob/BS, 1/mm), C) number of osteoblasts per total area (N.Ob./T.Ar); D) osteoclasts (N.Oc), and E) osteoclasts per millimeter of bone surface (N.Oc/BS, 1/mm). F) Average adipocyte volume (Ad.Vol, μm2). G) Adipocytes per bone marrow area (N.Ad/Ma.Ar, 1/mm). Plots represent individual data points, mean, and standard deviation. Student’s T Test was used to calculate significance (*; p < .05, **; p < .01, ***; p < .001).
3.2. LAL has a critical role in osteoblastogenesis and maintaining osteoblast function.
First, we indeed confirmed that LAL protein is expressed in osteoblasts and the flushed femoral cortex (Figure 4A). Next, we demonstrate that calvariae osteoblasts (cOBs) from LAL−/− mice exhibit impaired differentiation potential, as indicated by reduced ALP staining, 4.25 ± 0.9 units, compared to WT controls, 14.98 ± 0.6 units (Figure 4B). Despite this impairment, comparable hematoxylin staining was noted between LAL−/− and WT cells (Figure 4B), which suggests the reduction in ALP is not simply a function of reduced cell number. With this data in hand, we next targeted identifying the molecular role of LAL in osteoblasts by using an in vitro system. Importantly, given the potential systemic, secondary effects loss of LAL could be exerting on skeletal homeostasis, we utilized the commonly used potent and selective LAL inhibitor, lalistat, to study LAL’s role in an osteoblast-specific manner [21, 27–31]. Pharmacological inhibition of LAL using lalistat in BMSCs recapitulates the impaired osteoblast phenotype observed in LAL−/− cOB such that control cells displayed ALP/ VK staining of 44.13 ± 8.1 units whereas lalistat treatment resulted in 9.67 ± 7.3 units (Figure 4C). In addition, bright-field light microscopy revealed intracellular ‘vesicle’ accumulation during inhibition of LAL with lalistat (Figure 4C). We next performed RNA Sequencing on BMSCs harvested from WT and LAL−/− KO under osteogenic conditions for 7 days. These data demonstrate and distinct clustering of gene expression compared between WT and KO cells along PC1 along with a milder difference between sexes (PC2) (Figure 4D). Of the differentially expressed genes, female KO osteoblasts displayed 118 up-regulated genes and 456 down-regulated genes compared to WT, while osteoblasts cultured from male KO cells demonstrated 432 up-regulated genes and 869 down-regulated compared to WT (data not shown). Enrichment of significantly altered genes from osteoblasts, both male (Figure 4E) and female (Figure 4F) mice, demonstrated involvement of the WNT and NOTCH signaling, BMP pathways, mesenchymal lineage markers, osteogenesis, as well as phosphate, glucose, and fatty acid metabolism.
Figure 4. LAL is critical for proper osteoblast function.
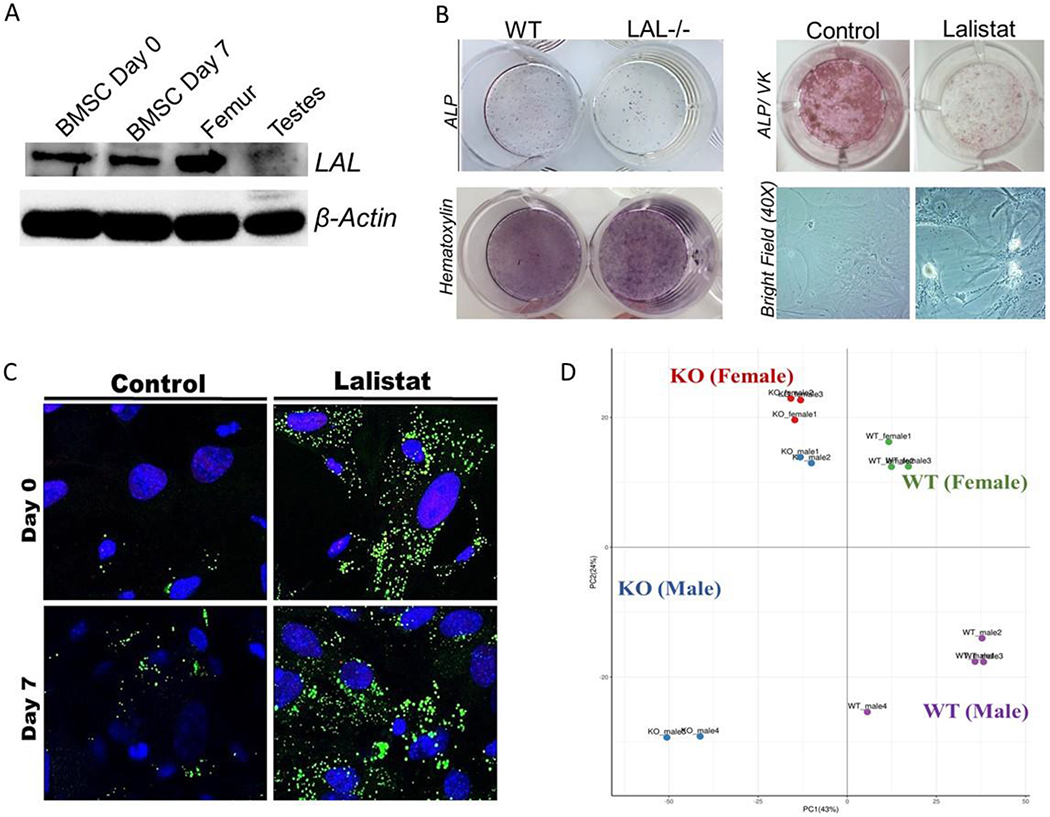
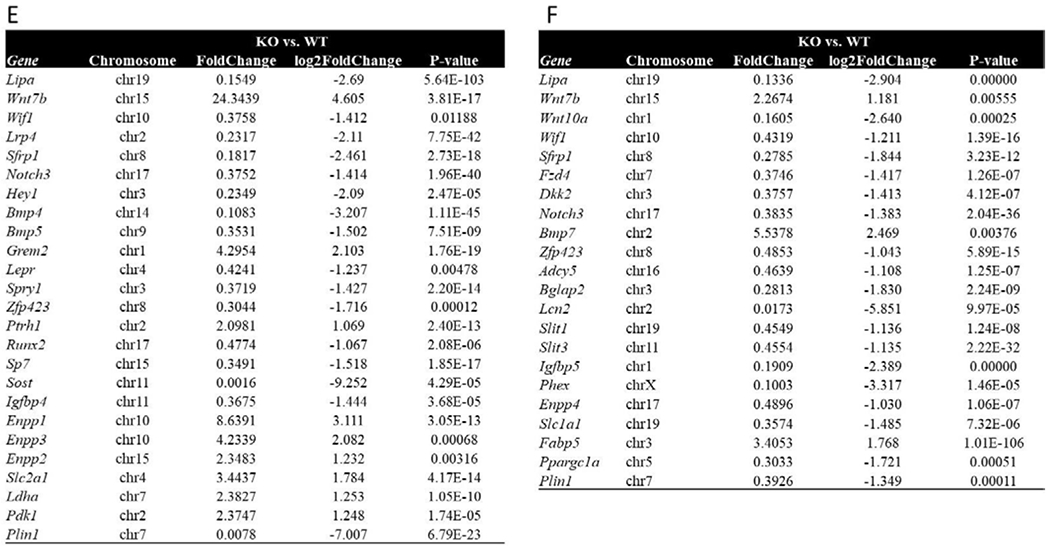
F4 Legend: A) LAL and β-actin protein expression in murine BMSCs at day 0 and day 7 of osteoblastogenesis, flushed femurs, and testes (n=3). B) Representative images of WT and LAL−/− calvarial osteoblasts (cOBs) or primary BMSCs treated with lalistat (100 μM) for 7 days during osteoblast differentiation. cOBs were stained for ALP and hematoxylin, while BMSCs were stained for ALP and Von Kossa (VK). Bright field images are also included at 20X magnification (n=3-4). C) BMSCs were cultured under osteogenic differentiation conditions for 0 or 7 days, in the absence (control) or presence of 24 hr treatment with lalistat (100 μM). A) Cells stained with BODIPY 493/503 (green; neutral lipids) and Hoechst 33342 Solution (blue; nuclear material) and visualized by confocal microscopy (n=5). D) Principle component analyses of RNA sequencing from BMSCs isolated from male and female WT or LAL−/− (KO) following 7 days in osteogenic medium (n=2-4). Further analyses of the significantly altered genes in (E) male and (F) female mice.
3.3. Inhibition of LAL in osteoblasts alters lipid metabolism throughout differentiation.
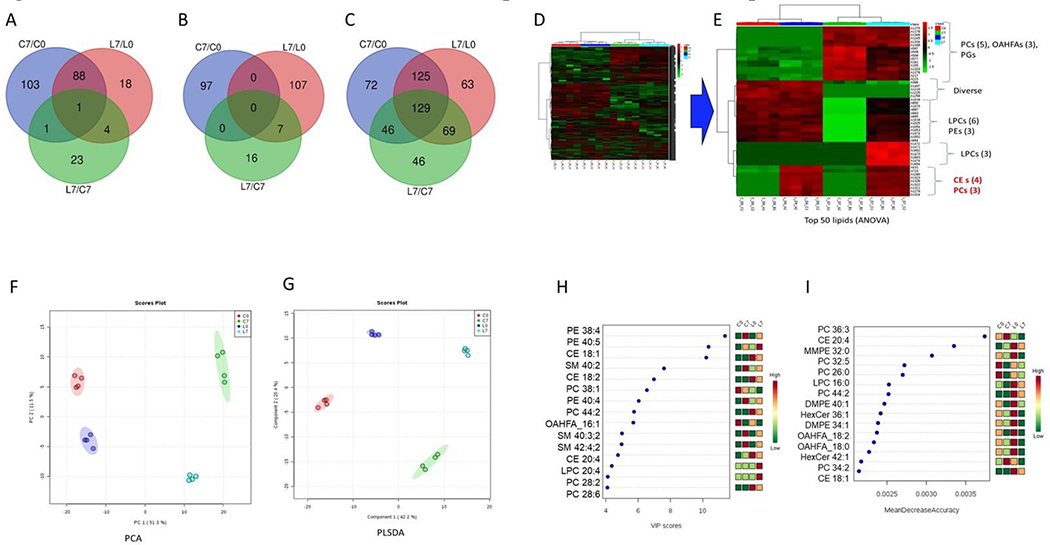
Using untargeted lipidomic analysis, we captured 1,264 and 1,006 unique lipid features in the 4 groups; control day 0 (C0), lalistat day 0 (L0), control day 7 (C7), and lalistat day 7 (L7). Upon further consolidation, 1,606 unique lipid species were revealed. However, with removal of missing values (with only 1 missing value allowed per group, n=4), the list of lipids was reduced to 822 lipids with quantitative abundance values. Analysis for the 4 groups revealed 651 lipids that were statistically significant, i.e., FDR corrected P-value of 0.05 (Figure 5A–C). These lipids were classified as 129 phosphatidylcholines (PCs), 82 phosphatidylethanolamines (PEs), 45 sphingomyelins (SMs), 39 (O-acyl)-ω-hydroxy fatty acids (OAHFAs), 40 dimethylphosphatidylethanolamines (DMPEs), 49 ceramides (CERs), 31 lysophosphatidylcholines (LPCs), and 10 cholesteryl esters (CEs). Interestingly, the highest number of increased accumulations of lipids were observed as a function of osteoblast differentiation, i.e. 193 lipids (C7-C0) (Figure 5A). The most pronounced decrease in lipid accumulation was observed for lalistat treatment (114 lipids), followed by that for 7 days of control cells (97 lipids) (Figure 5B). Among the total number of significant dysregulated lipids, time effect was highest followed by lalistat treatment and effect of time by lalistat treatment (Figure 5C).
Figure 5: Inhibition of LAL in osteoblasts results in profound alterations in lipid metabolism.
F5 Legend: A) Upregulated lipids (FC cut off >1.2) B) Downregulated lipids (FC cut off< 0.8) and C) Significantly dysregulated (both up and down) lipids (p-value < 0.05). Hierarchical clustering for D) all 823 lipids and E) top 50 statistically significant ANOVA resulted lipids. F) Principle component analysis (PCA) and G) partial least squared discriminant analyses (PLSDA) score plots. H) Variable importance in projection (VIP) scores from PLSDA models. I) Mean decrease accuracy from random forest (RF) analysis.
Hierarchical clustering indicated that there is a clear differential clustering of lipid species for the 4 groups (Figure 5D), as shown for the top 50 lipids (Figure 5E). From these data, the upper cluster represented lipid changes occurring as a function of osteoblast differentiation, such as PCs, OAHFAs, and PGs (Figure 5E). Whereas the bottom cluster corresponded to the lalistat treatment and as such, was heavily dominated by CEs and PCs (Figure 5E).
Principal component analysis (PCA) revealed that lipid abundances alone were able to discriminate the 4 groups and explain the variation in the dataset by virtue of the first 2 PCs (PC1, PC2) by 62.8% (Figure 5F). Similarly, using a supervised partial least squares discriminant analysis (PLS-DA), we observed that lipid abundances were able to discriminate the 4 groups (C0, C7, L0, L7) and explain the variation in the dataset by virtue of the first 2 PCs (PC1, PC2) by 62.6% (Figure 6G). The 1000-fold cross-validation measures for the model fitting were R2 cum= 0.999, and Q2 cum= 0.908, thereby discarding the chances of over-fit. We further identified the lipids responsible for the discrimination among the profiles, using VIP score to select those with the most significant contribution in the PLS-DA model [25]. These analyses revealed that PE (38:4), PE (40:5), CE (18:1), SM (40:2), and CE (18:2) were the top performers in summarizing the importance of the groups among other PEs, CEs, PCs and SMs (Figure 5H). Lastly, random forest analysis RF analysis we present the top 15 features based on mean importance of each feature. These were 5 PCs, 2 CEs, 2 OAHFAs and 2 hexosylceramides (HexCers) among others (Figure 5I).
3.4. Clinical manifestation of LAL-D is associated with increased incidence of osteoporosis and fracture.
Finally, to determine if patients with LAL-D suffer from impairments in skeletal health, we leveraged data from Clinformatics® Data Mart Database (a de-identified national insurance claims database) to compare prevalent fracture between adults with and without LAL-D. Descriptive characteristics for adults with (n = 3,076) and without (n = 13,683,481) LAL-D are presented in Table 1. Consistent with previous reports, both men and women with LAL-D had higher prevalence of cardiometabolic diseases. These data also demonstrate that adults affected by LAL-D had higher unadjusted prevalence of osteoporosis and fracture for all measures, except for stress fracture, compared to adults without LAL-D. After adjusting for age, sex, race, and U.S. region of residence (model 1), adults with LAL-D had higher odds of any fracture compared to adults without LAL-D (OR=1.23; 95% CI=1.05-1.43) (Table 2). After further adjustment for osteoporosis (model 2), which is a strong predictor of fracture, adults with LAL-D still had higher odds (OR=1.21; 95% CI=1.03-1.41), suggesting that adults with LAL-D may be vulnerable to fracture beyond the presence of osteoporosis. Age, sex, and race interactions were not significant (p > 0.100), indicating that the elevated fracture prevalence for individuals with LAL-D is present across the adult lifespan, for both men and women and for all races. Notably, when the same analyses were performed for each of the site-specific and site unspecified measures, adults with LAL-D had higher odds of fracture of the lower extremities (model 2, OR=1.49; 95% CI=1.21-1.85), suggesting a potential potent site-specific susceptibility to fracture.
Table 2.
Descriptive characteristics of adults with and without LAL-D.
| With LAL-D (n=3,076) | Without LAL-D (n=13,683,481) | p-value | |
|---|---|---|---|
| Age, mean (Gong et al.) | 64.0 (15.0) | 54.0 (18.8) | <0.001 |
| Sex, % (n) | |||
| Male | 45.5 (1,398) | 45.5 (6,228,465) | 0.938 |
| Female | 54.6 (1,678) | 54.5 (7,455,016) | |
| Race, % (n) | <0.001 | ||
| White | 56.5 (1,737) | 59.1 (8,091,034) | |
| Black | 10.9 (334) | 8.8 (1,207,472) | |
| Hispanic | 11.2 (344) | 10.3 (1,402,659) | |
| Asian | 3.4 (104) | 4.0 (553,039) | |
| Other/unknown | 18.1 (557) | 17.8 (2,429,277) | |
| U.S. region of residence, % (n) | <0.001 | ||
| West | 20.2 (620) | 21.6 (2,954,039) | |
| Midwest | 14.2 (437) | 24.1 (3,300,970) | |
| South | 51.5 (1,583) | 43.3 (5,930,863) | |
| Northeast | 14.2 (436) | 10.9 (1,497,609) | |
| Chronic diseases, % (n) | |||
| Osteoporosis | 7.1 (218) | 3.9 (530,749) | <0.001 |
| Ischemic heart disease | 21.4 (657) | 9.2 (1,254,273) | <0.001 |
| Cerebrovascular disease | 9.2 (284) | 4.0 (550,266) | <0.001 |
| Heart failure | 9.8 (301) | 4.5 (613,896) | <0.001 |
| Hypertension | 65.9 (2,028) | 38.3 (5,243,462) | <0.001 |
| Type 2 diabetes | 32.5 (1,000) | 16.1 (2,208,933) | <0.001 |
CI, confidence interval; SD, standard deviation.
4. Discussion
The current study establishes, for the first time, that LAL is essential for skeletal health as it supports osteoblast differentiation in a cell-autonomous manner via modulation of lipid metabolism. Moreover, our innovative clinical analyses provide strong evidence that adults with LAL-D suffer from heightened fracture susceptibility, especially in the lower extremities.
By utilizing the LAL−/− mice we were able to investigate the skeletal phenotype caused by loss of functional LAL, which previous characterizations have failed to mention. Interestingly, we observed that both male and female LAL−/− mice have lower bone mass in the trabecular and cortical bone compartment compared to WT controls. Both, pMOI and μFEA, demonstrate that LAL−/− mice have reduced bone biomechanical properties, indicative of a weaker bone more prone to fracture. While we are unable to determine dynamic bone histomorphometric indices in the current project, static parameters revealed no significant changes in osteoclast or bone marrow adipocytes. These findings were somewhat surprising given the critical role of the lysosome in osteoclast function along with the ectopic accumulation of lipids in various other tissues [16]. Nonetheless, osteoblasts, which express LAL, were reduced in LAL−/− compared to WT controls, which suggests reduced bone formation led to reduced bone mass. Despite a slight reduction in femur length in the LAL−/− mice, growth plate morphology appeared comparable to WT controls. We speculate that given the dramatic alterations in systemic metabolism occurring in these mice this reduction in longitudinal bone growth could be due to ‘failure to thrive’ (possible nutrient, vitamins, mineral, deficiencies) and/ or alterations from maternal health in utero during development. While we attempted to characterize bone turnover markers in the plasma to provide additional insights related to bone turnover, both CTx-1 and PINP are metabolized by the liver. As such, given the severe hepatic pathology that has been well described in LAL−/− mice starting at as early as 4 weeks of age and progressing throughout the animal’s life [15–17, 32–34], it is highly likely that the increase in both biomarkers is reflective of this liver dysfunction and not skeletal homeostasis.
Along these same lines, the multisystemic disorder of LAL-D, which is similarly observed in LAL−/− mouse, it is logical that the skeletal phenotype might be an indirect consequence of such confounders including vitamin deficiencies, chronic inflammation, altered mineral homeostasis, and/or the profound perturbations in whole-body metabolism. However, impaired osteoblast differentiation in both cOB and BMSCs treated with lalistat demonstrate that LAL is essential for osteoblastogenesis in a direct, cell-autonomous manner. While we did not observe profound alterations in hematoxylin staining of cOb, we did note reduced cell numbers during lalistat treatment using nuclear staining, which is a more sensitive method. Therefore, it is impossible to completely define the mechanism whereby LAL reduced osteoblast number, but this is an active area of research. In addition, our data provide evidence that LAL hydrolyzes neutral lipids within osteoblasts, thereby being critically involved in lipid metabolism [4], which although less understood, is of rising interests in bone biology [14, 35]. For example, essential cellular constituents, free cholesterol and energy dense fatty acids are presumed to remain entrapped in the lysosomes as CE and/ or TAGs, causing severe metabolic imbalance. To this point, our RNASeq data demonstrate that these impairments in osteoblastogenesis involve WNT, NOTCH, and BMP signaling pathways, as well as intracellular metabolism. Finally, our rigorous lipidomic analyses revealed that LAL inhibition in osteoblasts resulted in distinct alterations in intracellular lipid profiles. Particularly, lalistat increased CE in both undifferentiated stromal cells (day 0) and mature osteoblasts (day 7) in line with its well described hydrolytic activity [36, 37]. How free cholesterol status is altered in LAL−/− osteoblasts needs to be evaluated further in future studies. As such, aside from LDL-LDLR-LAL endocytosis, cells can endogenously synthesize cholesterol from acetyl-CoA precursors. Interestingly, inhibition of the rate-limiting enzyme in cholesterol biosynthesis by statins increased bone formation in vivo by increasing osteoblast activity [38]. Taken together, these suggest that cholesterol generation via the biosynthetic pathway may negatively impacts osteoblasts, which is somewhat counterintuitive; however, the explanation may involve cell bioenergetic alterations and cellular energy charge. For example, while cholesterol is important for osteoblast function, generating endogenous cholesterol from acetyl-CoA requires 99 adenosine triphosphate (ATP) molecules per cholesterol molecule synthesized, compared to 49 ATP mols for fatty acid synthesis or 10 ATP mols for glucose synthesis [39, 40]. Impaired exogenous cholesterol uptake from LDL-LDLR-LAL would initiate the endogenous cholesterol biosynthetic pathway, compromising the overall biogenetic capacity of osteoblasts during a time when ATP generation is critical, such as active bone formation. Therefore, these data related to LAL’s role in osteoblast function and reduced cell number underscore the need for future studies to examine how these differential cholesterol pathways can alter the biology of skeletal metabolism.
Finally, our novel clinical data demonstrating both elevated incidence of osteoporosis and increased fracture risk in individuals affected by LAL-D emphasize the translation potential of the animal and mechanistic data. While we recognize the limitations of such retrospective data (i.e., unable to examine cause of fracture, lifestyle confounding factors, overall health status, nutrition, and fall risk, among other factors) these data are the first of their kind and set the precedence for enhanced screening of osteoporosis in patients with LAL-D. While these data demonstrate the strong translational application for future scientific exploration, it is important to emphasize that these clinical data are from patients with late-onset LAL-D. Within the context of the current manuscript, the LAL−/− mouse model phenocopies a hybrid disease characterized with clinical manifestations observed in both early- and late-onset LAL-D. Therefore, while our data complement each other regarding reduced bone mass and skeletal health both in our mouse model and patient population, it is be possible that the disease is more severe in the LAL−/− mice compared to the patients late-onset LAL-D.
5. Conclusions
In conclusion, our data outline a previously unknown role for LAL relative to bone health, specifically as it relates to osteoblast function and bone formation. As the current manuscript utilized models with genetic ablation and pharmacological inhibition of LAL, these results are expected to have high impact applications. In fact, understanding the role of LAL in osteoblast differentiation is anticipated to provide a more comprehensive molecular appreciation of how conditions of dyslipidemia and cholesterol homeostasis impact skeletal health.
Supplementary Material
Table 3.
Prevalence and odds ratio (OR) of fracture measures for adults with and without LAL-D.
| With LAL-D (n=3,076) | Without LAL-D (n=13,683,481) | p-value | Model 1 Reference: without LAL-D | Model 2 Reference: without LAL-D | |
|---|---|---|---|---|---|
| Fracture | % (n) | % (n) | OR (95% CI) | OR (95% CI) | |
| Any fracture | 5.8 (179) | 3.6 (494,911) | <0.001 | 1.23 (1.05, 1.43) | 1.21 (1.03,1.41) |
| Site-specific fracture | |||||
| Cervical/thoracic vertebra | 0.8 (24) | 0.4 (60,612) | 0.005 | 1.23 (0.82, 1.84) | * |
| Lumbar spine/pelvis | 1.0 (31) | 0.7 (94,963) | 0.036 | 1.00 (0.70, 1.42) | 0.97 (0.68, 1.38) |
| Shoulder/arm/forearm | 1.7 (51) | 1.2 (160,553) | 0.013 | 1.10 (0.84, 1.46) | 1.09 (0.83, 1.44) |
| Femur/lower leg/ankle | 2.9 (89) | 1.4 (196,551) | <0.001 | 1.51 (1.22, 1.87) | 1.49 (1.21, 1.85) |
| Site unspecified | |||||
| Stress fracture | 0.5 (14) | 0.3 (41,163) | 0.118 | * | * |
| Pathological/non-traumatic fracture | 0.9 (28) | 0.5 (67,848) | 0.001 | 1.26 (0.87, 1.83) | 1.19 (0.81, 1.75) |
CI, confidence interval.
Model 1 adjusted for age, sex, race, and region of residence.
Model 2 adjusted for age, sex, race, region of residence, and osteoporosis.
Sample size insufficient for further adjustment.
Highlights:
Lysosomal acid lipase (LAL) deficiency results in reduced skeletal health and an increase in fracture incidence.
LAL regulates osteoblastogenesis and function via modulation of intracellular lipid metabolism.
Acknowledgements
The authors wish to acknowledge the expert technical support of the VANTAGE core and VANGARD core facilities, supported in part by the Vanderbilt-Ingram Cancer Center (P30 CA068485) and Vanderbilt Vision Center (P30 EY08126). Additional support was provided by the Maine Medical Center Research Institute Physiology Core Facility, Microscopy Core Facility, and the Proteomics and Lipidomics Analysis Core Facility. We also wish to express our gratitude to Sasi Uppuganti for his technical assistance on the μCT and μFEA analyses.
Funding
This work was supported by the National Institute of Health (NIH) National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) Grant AR072123 (to ERR), the University of Michigan Office of Health Equity and Inclusion Diversity Fund (to DGW), the Austrian Science Fund FWF (DK-MCD W1226, SFB F73, and P30882 to DK), the BioTechMed-Graz flagship project “Lipases and Lipid Signaling” (to DK), and the PhD program “Molecular Medicine” of the Medical University of Graz (to DK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
The authors declare no competing interests.
Bibliography
- [1].Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS, A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva, Nat Genet 38(5) (2006) 525–7. [DOI] [PubMed] [Google Scholar]
- [2].Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Juppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, Van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML, LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development, Cell 107(4) (2001) 513–23. [DOI] [PubMed] [Google Scholar]
- [3].Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, Van Hul W, Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST), Hum Mol Genet 10(5) (2001) 537–43. [DOI] [PubMed] [Google Scholar]
- [4].Li F, Zhang H, Lysosomal Acid Lipase in Lipid Metabolism and Beyond, Arterioscler Thromb Vasc Biol 39(5) (2019) 850–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zechner R, Madeo F, Kratky D, Cytosolic lipolysis and lipophagy: two sides of the same coin, Nat Rev Mol Cell Biol 18(11) (2017) 671–684. [DOI] [PubMed] [Google Scholar]
- [6].Hoffman EP, Barr ML, Giovanni MA, Murray MF, Lysosomal Acid Lipase Deficiency, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (Eds.), GeneReviews((R)), University of Washington, Seattle University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved., Seattle (WA), 1993. [Google Scholar]
- [7].Porto AF, Lysosomal acid lipase deficiency: diagnosis and treatment of Wolman and Cholesteryl Ester Storage Diseases, Pediatr Endocrinol Rev 12 Suppl 1 (2014) 125–32. [PubMed] [Google Scholar]
- [8].Chora JR, Alves AC, Medeiros AM, Mariano C, Lobarinhas G, Guerra A, Mansilha H, Cortez-Pinto H, Bourbon M, Lysosomal acid lipase deficiency: A hidden disease among cohorts of familial hypercholesterolemia?, J Clin Lipidol 11(2) (2017) 477–484.e2. [DOI] [PubMed] [Google Scholar]
- [9].Bagger YZ, Rasmussen HB, Alexandersen P, Werge T, Christiansen C, Tanko LB, Links between cardiovascular disease and osteoporosis in postmenopausal women: serum lipids or atherosclerosis per se?, Osteoporos Int 18(4) (2007) 505–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mandal CC, High Cholesterol Deteriorates Bone Health: New Insights into Molecular Mechanisms, Front Endocrinol (Lausanne) 6 (2015) 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Uyama O, Yoshimoto Y, Yamamoto Y, Kawai A, Bone changes and carotid atherosclerosis in postmenopausal women, Stroke 28(9) (1997) 1730–2. [DOI] [PubMed] [Google Scholar]
- [12].Yerges-Armstrong LM, Shen H, Ryan KA, Streeten EA, Shuldiner AR, Mitchell BD, Decreased bone mineral density in subjects carrying familial defective apolipoprotein B-100, J Clin Endocrinol Metab 98(12) (2013) E1999–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Maridas DE, Rendina-Ruedy E, Helderman RC, DeMambro VE, Brooks D, Guntur AR, Lanske B, Bouxsein ML, Rosen CJ, Progenitor recruitment and adipogenic lipolysis contribute to the anabolic actions of parathyroid hormone on the skeleton, Faseb j 33(2) (2019) 2885–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rendina-Ruedy E, Guntur AR, Rosen CJ, Intracellular lipid droplets support osteoblast function, Adipocyte 6(3) (2017) 250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Du H, Duanmu M, Witte D, Grabowski GA, Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage, Hum Mol Genet 7(9) (1998) 1347–54. [DOI] [PubMed] [Google Scholar]
- [16].Du H, Heur M, Duanmu M, Grabowski GA, Hui DY, Witte DP, Mishra J, Lysosomal acid lipase-deficient mice: depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span, J Lipid Res 42(4) (2001) 489–500. [PubMed] [Google Scholar]
- [17].Du H, Sheriff S, Bezerra J, Leonova T, Grabowski GA, Molecular and enzymatic analyses of lysosomal acid lipase in cholesteryl ester storage disease, Mol Genet Metab 64(2) (1998) 126–34. [DOI] [PubMed] [Google Scholar]
- [18].Sheriff S, Du H, Grabowski GA, Characterization of lysosomal acid lipase by site-directed mutagenesis and heterologous expression, J Biol Chem 270(46) (1995) 27766–72. [DOI] [PubMed] [Google Scholar]
- [19].Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM, Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee, J Bone Miner Res 28(1) (2013) 2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Maridas DE, Rendina-Ruedy E, Le PT, Rosen CJ, Isolation, Culture, and Differentiation of Bone Marrow Stromal Cells and Osteoclast Progenitors from Mice, J Vis Exp (131) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tuohetahuntila M, Molenaar MR, Spee B, Brouwers JF, Wubbolts R, Houweling M, Yan C, Du H, VanderVen BC, Vaandrager AB, Helms JB, Lysosome-mediated degradation of a distinct pool of lipid droplets during hepatic stellate cell activation, J Biol Chem 292(30) (2017) 12436–12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Whitney DG, Caird MS, Jepsen KJ, Kamdar NS, Marsack-Topolewski CN, Hurvitz EA, Peterson MD, Elevated fracture risk for adults with neurodevelopmental disabilities, Bone 130 (2020) 115080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].French ZP, Torres RV, Whitney DG, Elevated prevalence of osteoarthritis among adults with cerebral palsy, J Rehabil Med 51(8) (2019) 575–581. [DOI] [PubMed] [Google Scholar]
- [24].Chang HY, Weiner JP, Richards TM, Bleich SN, Segal JB, Validating the adapted Diabetes Complications Severity Index in claims data, Am J Manag Care 18(11) (2012) 721–6. [PubMed] [Google Scholar]
- [25].Grapov D, Wanichthanarak K, Fiehn O, MetaMapR: pathway independent metabolomic network analysis incorporating unknowns, Bioinformatics 31(16) (2015) 2757–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Misra BB, Fahrmann JF, Grapov D, Review of emerging metabolomic tools and resources: 2015-2016, Electrophoresis 38(18) (2017) 2257–2274. [DOI] [PubMed] [Google Scholar]
- [27].Schlager S, Vujic N, Korbelius M, Duta-Mare M, Dorow J, Leopold C, Rainer S, Wegscheider M, Reicher H, Ceglarek U, Sattler W, Radovic B, Kratky D, Lysosomal lipid hydrolysis provides substrates for lipid mediator synthesis in murine macrophages, Oncotarget 8(25) (2017) 40037–40051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Panda PK, Patra S, Naik PP, Praharaj PP, Mukhopadhyay S, Meher BR, Gupta PK, Verma RS, Maiti TK, Bhutia SK, Deacetylation of LAMP1 drives lipophagy-dependent generation of free fatty acids by Abrus agglutinin to promote senescence in prostate cancer, J Cell Physiol 235(3) (2020) 2776–2791. [DOI] [PubMed] [Google Scholar]
- [29].Masi S, Chennamaneni N, Turecek F, Scott CR, Gelb MH, Specific Substrate for the Assay of Lysosomal Acid Lipase, Clin Chem 64(4) (2018) 690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lim H, Lim YM, Kim KH, Jeon YE, Park K, Kim J, Hwang HY, Lee DJ, Pagire H, Kwon HJ, Ahn JH, Lee MS, A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes, Nat Commun 9(1) (2018) 1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hamilton J, Jones I, Srivastava R, Galloway P, A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2, Clin Chim Acta 413(15-16) (2012) 1207–10. [DOI] [PubMed] [Google Scholar]
- [32].Du H, Duanmu M, Rosa LR, Mouse lysosomal acid lipase: characterization of the gene and analysis of promoter activity, Gene 208(2) (1998) 285–95. [DOI] [PubMed] [Google Scholar]
- [33].Pajed L, Wagner C, Taschler U, Schreiber R, Kolleritsch S, Fawzy N, Pototschnig I, Schoiswohl G, Pusch LM, Wieser BI, Vesely P, Hoefler G, Eichmann TO, Zimmermann R, Lass A, Hepatocyte-specific deletion of lysosomal acid lipase leads to cholesteryl ester but not triglyceride or retinyl ester accumulation, J Biol Chem 294(23) (2019) 9118–9133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Leopold C, Duta-Mare M, Sachdev V, Goeritzer M, Maresch LK, Kolb D, Reicher H, Wagner B, Stojakovic T, Ruelicke T, Haemmerle G, Hoefler G, Sattler W, Kratky D, Hepatocyte-specific lysosomal acid lipase deficiency protects mice from diet-induced obesity but promotes hepatic inflammation, Biochim Biophys Acta Mol Cell Biol Lipids 1864(4) (2019) 500–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rendina-Ruedy E, Rosen CJ, Lipids in the Bone Marrow: An Evolving Perspective, Cell Metab (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Radovic B, Vujic N, Leopold C, Schlager S, Goeritzer M, Patankar JV, Korbelius M, Kolb D, Reindl J, Wegscheider M, Tomin T, Birner-Gruenberger R, Schittmayer M, Groschner L, Magnes C, Diwoky C, Frank S, Steyrer E, Du H, Graier WF, Madl T, Kratky D, Lysosomal acid lipase regulates VLDL synthesis and insulin sensitivity in mice, Diabetologia 59(8) (2016) 1743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang H, Lysosomal acid lipase and lipid metabolism: new mechanisms, new questions, and new therapies, Curr Opin Lipidol 29(3) (2018) 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Parhami F, Mody N, Gharavi N, Ballard AJ, Tintut Y, Demer LL, Role of the cholesterol biosynthetic pathway in osteoblastic differentiation of marrow stromal cells, J Bone Miner Res 17(11) (2002) 1997–2003. [DOI] [PubMed] [Google Scholar]
- [39].Luu W, Gelissen IC, Brown AJ, Manipulating Cholesterol Status Within Cells, Methods Mol Biol 1583 (2017) 41–52. [DOI] [PubMed] [Google Scholar]
- [40].Luu W, Sharpe LJ, Gelissen IC, Brown AJ, The role of signalling in cellular cholesterol homeostasis, IUBMB Life 65(8) (2013) 675–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.