

Abstract
Epigenetics is the study of heritable changes in DNA or its associated proteins except mutations in gene sequence. Epigenetic regulation plays fundamental roles in the processes of kidney cell biology through the action of DNA methylation, chromatin modifications via epigenetic regulators and interaction via transcription factors, and noncoding RNA species. Kidney diseases, including acute kidney injury, chronic kidney disease, nephritic and nephrotic syndromes, pyelonephritis and polycystic kidney diseases are driven by aberrant activity in numerous signaling pathways in even individual kidney cell. Epigenetic alterations, including DNA methylation, histone acetylation and methylation, noncoding RNAs, and protein posttranslational modifications, could disrupt essential pathways that protect the renal cells from uncontrolled growth, apoptosis and establishment of other renal associated syndromes, which have been recognized as one of the critical mechanisms for regulating functional changes that drive and maintain the kidney disease phenotype. In this chapter, we briefly summarize the epigenetic mechanisms in kidney cell biology and epigenetic basis of kidney development, and introduce epigenetic techniques that can be used in investigating the molecular mechanism of kidney cell biology and kidneys diseases, primarily focusing on the integration of DNA methylation and chromatin immunoprecipitation technologies into kidney disease associated studies. Future studies using these emerging technologies will elucidate how alterations in the renal cell epigenome cooperate with genetic aberrations for kidney disease initiation and progression. Incorporating epigenomic testing into the clinical research is essential to future studies with epigenetics biomarkers and precision medicine using emerging epigenetic therapies.
1. Introduction
Kidney diseases are multistep processes associated with the accumulation of numerous molecular alterations. These molecular changes impact cellular function within the kidney cells and its microenvironment. Numerous genetic alterations (mutations, loss of heterozygosity, deletions, insertions, aneuploidy, etc.) have been associated with different kidney diseases (Sadikovic et al., 2008), and can ultimately result in aberrant gene expression. However, the landscape of genetic alterations is insufficient to explain the pervasive gene expression changes and alterations to cellular function in some kinds of kidney diseases, such as autosomal dominant polycystic kidney disease (ADPKD). According to the two-hit hypothesis, deletions on both alleles of PKD1 and PKD2 genes block the mechanisms in renal epithelial cells that prevent aberrant cellular growth (Murcia, Sweeney, & Avner, 1999; Pei, 2001). One of these alterations (or “hits”) is a hereditary or somatic mutation in PKD gene and the second “hit” is an acquired mutation or copy number loss in the other PKD allele. ADPKD cannot be fully understood in terms of the constrained genetic mutations of PKD1 and PKD2, especially, in families with the same genetic mutations but variable disease severity (Harris & Rossetti, 2010). Instead, epigenetic drift can occur even in genetically identical humans (Shah et al., 2014), which may be an alternative means of explaining PKD-associated alterations.
Epigenetic alterations are heritable traits that impact the phenotype by interfering with gene expression independent of the DNA sequence (Egger et al., 2004). Epigenetic changes are as pervasive in kidney diseases as genetic alterations, and likely are responsible for the hidden source of variation in kidney diseases (Beckerman, Ko, & Susztak, 2014). The epigenetic mechanisms include DNA methylation, chromatin remodeling, noncoding RNAs (ncRNAs), binding of regulatory proteins (such as different epigenetic regulators to histone and DNA) and transcription factors (Dawson & Kouzarides, 2012; Gaykalova et al., 2012). Many of these mechanisms result in the changes of chromatin states.
Epigenetic events usually act together with other molecular processes in normal or disease states to have persistent gene expression and functional alterations (Luo & Lin, 2016; Zoghbi & Beaudet, 2016). For example, epigenetic alterations that impact transcription factor binding can explain genome-wide transcription dysregulation independent of genetic variation in diseases (Esteller, 2008; Hadnagy, Beaulieu, & Balicki, 2008; Simon & Lange, 2008). Recent studies have also found that PKD mutations are associated with the increase of DNA methylation in PKD1 gene body region (Woo et al., 2014), which may result in closed chromatin and is inaccessible to DNA repair genes during replication. Therefore, alterations to chromatin structure may be critical drivers of PKD (Li, 2015). Genome-wide characterization of epigenetic aberrations with functional impacts on gene expression in individual kidney disease remain key targets of interest, which is crucial for the development of new therapeutic agents to reverse specific alterations to the epigenetic landscape and to identify new biomarkers for different kidney diseases.
This chapter summarizes the epigenetic mechanisms in kidney cells and epigenetic basis of kidney development and focuses on describing experimental techniques to discover reversible epigenetic events, including DNA methylation and chromatin modification, in renal cells and kidney tissues. The detailed protocols for DNA methylation assay and chromatin immunoprecipitation (ChIP) assay are provided and discussed.
2. Epigenetic mechanisms in kidney cell biology
There are three basic epigenetic mechanisms related to kidney cell biology and kidney development, including DNA methylation, histone modification and microRNAs.
2.1. DNA methylation
The methylation of DNA at CpG dinucleotides was among the first genome modifications described and is an attractive mechanism for the regulation of kidney cell biology and kidney development (Dressler, 2008). DNA methylation involves the covalent transfer of a methyl radical (CH3) from S-adenyl methionine (SAM) to the 5-carbon on cytosine residues in CpG dinucleotides of DNA to form 5-methylcytosine (5mC) (Jin et al., 2011; Moore, Toomire, & Strauss, 2013). Normal DNA methylation or hypomethylation on specific CpG sites plays critical roles during different stages of normal kidney development, whereas aberrant methylation or hypermethylation on specific CpG sites can occur in kidneys under disease conditions (Bechtel-Walz & Huber, 2014). Abnormal genome-wide DNA methylation on regulatory DNA sequences (promoters, insulators and enhancers) with high CpG-concentrated regions called CpG islands, which are about 800–1000 nucleotides in average, have been a focus in epigenetic studies. The methylation patterns of CpG islands are highly tissue specific in normal human samples and may be changed in patient samples (Irizarry et al., 2009). The impact and function of the genome-wide alterations on DNA methylation remain poorly understood.
DNA methylation is catalyzed by a family of DNA methyltransferases (DNMTs) (Okano et al., 1999; Okano, Xie, & Li, 1998). In vertebrates, there are five known DNMTs that differ in structure and function. Apart from DNMT2, all DNMTs are comprised of an N-terminal regulatory domain in addition to a C-terminal catalytic domain. The ubiquitously expressed DNMT1, which displays a strong preference for hemimethylated CpG sites, functions to maintain the DNA methylation patterns established by the DNMT3 subfamily, comprising DNMT3a and DNMT3b, on unmethylated DNA (Kaneda et al., 2004; Okano et al., 1998, 1999) during DNA replication and DNA repair (Kaneda et al., 2004; Leonhardt et al., 1992; Mortusewicz et al., 2005; Okano et al., 1998, 1999). The expression of DNMT3a is also relatively ubiquitous and mice lacking Dnmt3a die at about 4 weeks of age, while the expression of DNMT3b is less in majority of differentiated tissues and knockout of Dnmt3b induces embryonic lethality (Okano et al., 1999; Xie et al., 1999), which suggest that DNMT3a is required for normal cellular differentiation, while DNMT3b is required during early development. The cofactor DNMT3L1 stimulates the activity of DNMT3A and DNMT3B, but by itself lacks enzymatic activity (Hata et al., 2002; Suetake et al., 2004). Recently, mouse studies show that DNMT3L is expressed during gametogenesis and is required for the establishment of maternal genomic imprints (Bourc’his et al., 2001; Hata et al., 2002) and mice lacking DNMT3L die early during development (Hata et al., 2002). The fifth member of the DNMT family, DNMT2, has very weak activity toward DNA (Goll et al., 2006; Hermann, Gowher, & Jeltsch, 2004).
DNA methylation as a gene-silencing mechanism plays an important role during organ and disease development. Under normal conditions, dynamic changes in DNA methylation, lead to stable and unique patterns that regulate tissue-specific gene transcription in somatic cells. In disease conditions, these methylation patterns have been known to change; either preceding the disease, or occurring as a consequence of it. During embryonic development, DNA methylation is a dynamic yet tightly controlled process, which contributes to the regulation of cell fate transitions (Bechtel-Walz & Huber, 2014; Holliday & Pugh, 1975). DNMT3a and DNMT3b were thought to establish the methylation patterns at the early development which is further maintained through somatic cell divisions by DNMT1, acting on the hemimethylated CpG sites generated by DNA replication (Ichiyanagi et al., 2013; Liao et al., 2015; Ramsahoye et al., 2000).
Recently, it has been showed that maintenance of DNA methylation plays a key role during nephron development (Wanner et al., 2019). DNMT1 and DNMT3a are highly enriched in the nephrogenic zone of the developing kidneys. Loss of DNMT1 in nephron progenitor cells led to a strong reduction of DNA methylation in all cells originating from the cap mesenchyme (CM). Deletion of DNMT1 in nephron progenitor cells (in contrast to deletion of DNMT3a or DNMT3b) mimics nutritional models of kidney growth restriction and results in a substantial reduction of nephron number as well as renal hypoplasia at birth (Wanner et al., 2019). This is in accordance with previous reports showing a global approximately 80% decrease of DNA methylation after deletion of DNMT1 (Lei et al., 1996). This study indicates that DNA demethylation results in transcriptional activation of genes in the CM, leading to reduced progenitor cell renewal and differentiation capacity by the nephrogenic niche. In addition, site-specific DNA methylation changes have been detected in patients with chronic kidney diseases (CKD) in general and diabetic nephropathy (Bell et al., 2010; Ko et al., 2013). Since DNA methylation patterns are known to be fairly stable and unique in differentiated cells, evaluating the differences in methylation status could extend our understanding of kidney development and the pathophysiology of kidney diseases.
2.2. Histone modifications
Histone modifications usually occur on critical amino acids of histone tails which extend out of the nucleosome (Iizuka & Smith, 2003). Histone modifications correlate with open or closed conformations of chromatin and drive different accessibility to transcription factors and regulatory proteins to specific genes (Bannister & Kouzarides, 2011). The most important histone modifications are acetylation and methylation of target lysine residues of the histone tails. Acetylation of histones strongly results in transcription activation, whereas deacetylation of histones mediated by histone deacetylases (HDACs) represses gene expression in almost all experimental systems (Haberland, Montgomery, & Olson, 2009; Sterner & Berger, 2000). Methylation of histones can either activate or repress gene transcription, depending on which specific residues, including H3K4, H3K9, H3K27, H3K36, etc., are modified. Many more histone modifications exist, including phosphorylation, ubiquitylation and glycosylation, which spread to other amino acid residues, such as arginine, serine and threonine (Bhaumik, Smith, & Shilatifard, 2007; Strahl & Allis, 2000; Sun & Allis, 2002; Turner, 2002).
Multiple epigenetic regulatory proteins write, erase and read histone marks to alter chromosomal structure by directly modifying and regulating DNA accessibility (Tarakhovsky, 2010). Histone acetyltransferases/deacetylases and methyltransferases/demethylases can either write or erase histone acetylation and methylation, respectively (El-Osta & Wolffe, 2000; Seto & Yoshida, 2014). The bromodomain-containing proteins (BRDs) selectively read and bind to acetylated histone lysine residues to regulate gene expression (Fujisawa & Filippakopoulos, 2017), while the chromatin remodeling factors, such as chromodomain protein, selectively read and recognize methylated lysine residues to promote heterochromatin formation and to suppress gene transcription (Tajul-Arifin et al., 2003). In addition to the histone modifications, epigenetic regulators also modify nonhistone substrates and regulates the activity of these proteins, such as histone methyltransferase, SMYD2, methylates histone H3K4 and H3K36 (Brown et al., 2006), histone H4K20 (Xu et al., 2018), but it also could methylate nonhistone proteins including HSP90, p53, Rb, STAT3 and p65 (Hamamoto et al., 2014; Huang et al., 2006; Li et al., 2017; Saddic et al., 2010). Many of these epigenetic regulators are dysregulated to affect cell homeostasis pathways in diseased cells and tissues (Rodriguez-Paredes & Esteller, 2011). Because histone marks have stable covalent structures, they can be inherited during cell division and DNA duplication and serve as disease markers (Judes et al., 2016). Genome-wide epigenetic analysis is essential to determine the source of alterations to the distribution of histone markers, which is critical to developing epigenetics therapies.
It is recognized that the methylation and acetylation of histones on lysine (K) or arginine (R) residues are key epigenetic mechanisms regulating gene expression during kidney development and disease (Table 1) (Hurtado Del Pozo et al., 2018). Studies have shown that the differentiation of metanephric mesenchyme cells to nephron progenitors largely depends on two regulators, Six2 and the Wnt pathway. In pluripotent stem cells (PSCs), the promoters of these genes are marked by tri-methylation H3K4 (H3K4me3; activating) and H3K27 (H3K27me3; repressive). Chromatin immunoprecipitation (ChIP)-DNA sequencing analysis with antibodies against H3K4me3 and H3K27me3 in immortalized metanephric mesenchyme mouse clonal cell lines indicted that Six2low/Wnthigh cells shows a loss of the H3K9me2 and H3K27me3 repressive histone marks and retained the H3K4me3 activating mark on the promoters of nephrogenic lineage genes, including Pax2, Pax8, Lef1, Jag1 or Lhx1 (McLaughlin et al., 2013). Moreover, a significant enrichment of H3K4me3, H3K9me3 and H3K27me3 on the Six2 gene was detected by immunolocalization and real-time quantitative PCR in mouse embryonic 15.5 kidneys. In contrast, nascent nephron cells showed high levels of H3K4me3, and low levels of both H3K9me3 and H3K27me3, in the Lhx1 gene. The increase of H3K79me2/3 marks and upregulation of histone H3K79 methyltransferase Dot1l is important for the generation of mature nephrons (McLaughlin et al., 2014). In addition, it has been reported that in differentiating mouse embryonic stem cells, MLL3/4-dependent deposition of H3K4me1 at enhancers correlates with increased levels of chromatin interactions, whereas loss of this histone modification leads to reduced levels of chromatin interactions and defects in gene activation during differentiation (Yan et al., 2018). Therefore, genome-wide epigenetic studies are essential to determine the source of alterations to the distribution of histone markers during kidney and disease development.
Table 1.
The key histone marks in kidney development and disease.
| Histone marks |
Effects on gene expression |
Writer | Eraser | Key gene promoters |
Roles in kidney development or diseases |
References |
|---|---|---|---|---|---|---|
| H3K27me3 | Repression | Ezh2 | Utx, Jmjd3 | Jag1 | Development in human and mouse | Majumder et al. (2018) |
| H3K4me3 | Activation | Mll | Pax2, Pax8, Lef1, Jag1, Lhx1 | Development in mouse | McLaughlin et al. (2013) | |
| H3k79me3 | Activation | Dot1l | Pax2, Pax8, Lef1, Jag1, Lhx1 | Development in mouse | McLaughlin et al. (2014) | |
| H3K9/14ac | Activation | CBP | Tgfβ1, Tgfβ3, Ctgf | Disease in Mouse | Deb et al. (2017) | |
| H3K9ac | Repression | Sirt6 (HDAC) | Notch1, Notch4 | Development and Disease in human and mouse | Liu et al. (2017) |
2.3. Non-coding RNA (ncRNA) and microRNAs
Chromatin changes also result from expression changes to types of non-coding RNA (ncRNA), a functional RNA molecule that is transcribed from DNA but not translated into proteins (Esteller, 2011; Peschansky & Wahlestedt, 2014). Epigenetic related ncRNAs include miRNA, siRNA, piRNA and lncRNA. In general, ncRNAs function to regulate gene expression at the transcriptional and post-transcriptional level (Sun, Hao, & Prasanth, 2018). miRNAs are highly conserved ncRNAs of ~22 nucleotides in length, acting as post-transcriptional gene regulators that canonically target 3′-UTR of many mRNAs through translational repression and/or mRNA degradation (Towler, Jones, & Newbury, 2015). Biosynthesis of miRNAs begins in the nucleus, where a primary miRNA (pri-miRNA) transcript is first transcribed by RNA polymerase II. Then, the Drosha-Dgcr8 complex processes pri-miRNAs into precursor miRNAs (pre-miRNAs). Subsequently, pre-miRNAs are exported into the cytoplasm and processed to mature miRNA by Dicer (Gonzalez-Duarte, Cazares-Ordonez, & Avila-Chavez, 2014; Ha & Kim, 2014; Sand, 2014). Details of the role of ncRNAs and miRNAs in gene expression regulation and kidney diseases are the subject of chapter 19 in this book. As an example, long noncoding RNAs (lncRNAs) can bind to DNA and to chromatin remodeling complexes, which are associated with complex alterations in the distribution of nucleosomes (Kawaguchi & Hirose, 2015). The functional role of specific ncRNAs on kidney epigenetics is an active area of research.
3. High-throughput platforms for epigenetic analysis in renal cells and tissues
3.1. DNA methylation analysis techniques and protocols
DNA methylation on specific gene(s) and the whole genome can be measured with different techniques. Most of these rely on an initial genomic fractionation and probe preparation step. These measurements include the use of antibodies [methylated DNA immunoprecipitation (MeDIP)] or conjugated methyl-CpG binding proteins (MBPs) to recognize methylated cytosines on native DNA (Judes et al., 2016), and the use of sodium bisulfite treatment to convert unmethylated cytosines to uracil residues with no effect on methylated cytosines. Sodium bisulfite conversion can be coupled with methylation specific PCR (MSP) and next-generation sequencing to evaluate the methylation on specific gene(s) and on the whole-genome DNA, respectively. Although incomplete binding of the antibodies to methylated CpG may result in false negatives, antibody associated techniques still have strong true-positive rates because of the nanomolar binding affinity to symmetrically methylated CpG. By comparison, sodium bisulfite conversion is more specific and can detect methylation changes at single nucleotide resolution (Kurdyukov & Bullock, 2016; Wreczycka et al., 2017). For this reason, we will mainly focus on the process of sodium bisulfite conversion and some of its related techniques. In addition, we will briefly describe the Methyl-CpG binding domain protein-enriched genome sequencing (MBD-seq) technique.
3.1.1. Sodium bisulfite conversion
Bisulfite treatment leads to the deamination of unmethylated cytosines which are converted to uracils at high pH, and then subsequently converted to thymidines during a PCR reaction by DNA polymerase (Fig. 1). Methylated cytosines (5mC) and 5-hydroxymethylcytosines (5hmC) are resistant to deamination and will not be converted. This selective deamination process of bisulfite conversion provides the basis for using different assays to distinguish the location and abundance of methylated and unmethylated CpG sites and general methylation patterns at particular regions of the genome. Detailed quantitative measures of percent methylation at individual CpG sites are obtained by Pyrosequencing. Alternative methods for broad-scale methylation analysis include methylation-specific PCR (MSP).
FIG. 1.

Sodium bisulfite conversion. (A) Bisulfite treatment converts unmethylated cytosine to uracil. (B) DNA polymerase substitutes dU for dT after bisulfite conversion.
So far, there are numbers of methods and commercial kits available for bisulfite conversion. The process of bisulfite conversion is harsh, resulting in the generation of different sizes of converted DNA fragments due to acidic conditions, temperature and reaction time. Although bisulfite conversion cannot distinguish between 5-mC and 5-hmC DNA, however, following bisulfite conversion, the DNA methylation status at specific locus or genome-wide can be further analyzed with methylation specific PCR (MSP) or whole-Genome Bisulfite Sequencing (WGBS), respectively.
3.1.2. Methylation specific PCR (MSP) and quantitative real-time MSP (QMSP)
Methylation-specific PCR is a highly specific and sensitive method that uses PCR to amplify methylated or unmethylated CpG sites on specific locus, and requires high specificity to discriminate between cytosine and thymine bases derived from methylated and unmethylated cytosines following bisulfite conversion. Two pairs of primers are designed for the amplification step; one pair specific for the methylated DNA and the other for the unmethylated DNA. To discriminate the methylated from the unmethylated alleles, at least one of the primer pairs, preferentially the methylated primers should include at least two CpG sites. Depending on the location of the primers, both methylated and unmethylated DNA would produce amplicons of different sizes.
Primer design for methylation-specific PCR is the key step for this assay due to the need to cover several CpG sites per primer, which does not allow the detection of a single CpG methylation in a CpG island. The primers used for methylated DNA (M pair) and for unmethylated DNA (U pair) in MSP assay should contain at least two CpG site within their sequence, and ideally be located at the far 3′-end of their sequence in order to discriminate maximally methylated DNA against unmethylated DNA. For example, if a forward primer in the M pair has this sequence: ATTAGTTTCGTTTAAGGTTCGA, the forward primer in the U pair must also contain the two CpG sites, e.g., ATTAGTTTTGTTTAAGGTTTGA; although they may differ in length and start position. The M pair and U pair should also include an adequate number of non-CpG Cs and ideally have a similar annealing temperature to amplify only the bisulfite-modified DNA.
It is important to note that standard MSP cannot quantify methylated alleles, when both methylated and unmethylated alleles are present in the DNA sample. Also, the use of low quality DNA such as the DNA extracted from formalin-fixed paraffin-embedded (FFPE) samples, limits MSP amplification to short amplicons since bisulfite treatment fragments the DNA. Due to these limitations of standard MSP, other advanced techniques such as pyrosequencing and quantitative real-time MSP (QMSP), also known as MethyLight, can be used.
Quantitative real-time MSP (QMSP) is based on standard MSP. Similar to standard MSP, primers are designed for either the methylated or unmethylated CpG sites. However, unlike standard MSP, QMSP is able to quantify methylated alleles using fluorescence-based quantitative PCR (qPCR) technology. Based on the nature of this method (qPCR amplification), high sensitivity and specificity at up to a single-nucleotide resolution may be achieved. Furthermore, since QMSP is more sensitive than standard MSP, it is capable of detecting low frequency hypermethylated alleles. Owing to its high sensitivity and specificity, QMSP is an ideal technique for the detection of DNA methylation biomarkers.
Described below is an established protocol for QMSP, adapted from MSP sequencing of urinary cells (Roupret et al., 2007).
Extract DNA from desired sample using Qiagen Tissue and Blood kits (Qiagen) or comparable method.
For methylation analysis, treat 1–2μg DNA with sodium bisulfite using the CpGenome kit (Chemicon) or comparable method according to the manufacturer’s instructions. The bisulfite-treated DNA is then used as a template for the quantitative fluorescence-based real-time methylation-specific PCR (QMSP).
Perform PCR to probe for the gene promoter regions of interest. Note: bisulfite-specific primers to actin should be used as the internal reference gene, and SssI (a CpG-specific methylase) treated DNA also known as a universally methylated DNA as positive control.
Use the QMSP Accuprime Taq polymerase (Invitrogen) if using the ABI Prism 7000 Sequences Detection System. Alternatively, the fluorescent probe can be adapted based on the detection system available.
For a 10μL PCR volume, add 1μL of Accuprime buffer, 0.25μL Accuprime Taq polymerase, 5pmol/L forward and reverse primers, 5pmol/L probe, 0.2μL Rox reference dye, and water. Sixty cycles of denaturation (95 °C for 45s), annealing (specific primer temperature for 2min), and extension (72 °C for 1min).
3.1.3. Whole genome bisulfite sequencing (WGBS)
Whole genome bisulfite sequencing (WGBS) is considered the “gold standard” for profiling DNA methylation, which allows the interrogation of methylation status of individual CpGs in a genome-wide scale (Wreczycka et al., 2017). Both bisulfite converted DNA and untreated input controls are sequenced with high throughput next generation sequencing technologies. This method permits the genome-wide evaluation of DNA methylation at single-base resolution (Liu et al., 2016; Wreczycka et al., 2017; Yong, Hsu, & Chen, 2016). Here, we describe the established protocols adapted from studies in tissues (Wreczycka et al., 2017; Yong et al., 2016).
Isolate genomic DNA from kidney tissues and renal cells, using the MasterPure™ DNA Purification Kit (Epicenter) or comparable method according to the manufacturer’s instructions. This kit employs a non-enzymatic approach to cell lysis, followed by protein precipitation and subsequent nucleic acid isolation.
Re-suspend the extracted DNA in TE buffer (1 × buffer: 10mM Tris-HCl, 1mM EDTA at pH 8.0) and quantify by fluorometry.
Perform bisulfite treatment of genomic DNA. Briefly, treat 50–100ng of purified genomic DNA with Zymo Lightning Conversion Reagent in a thermal cycle for 8min at 98°C, followed by 60min at 54°C. (Note: bisulfite treatment is known to fragment DNA.)
Purify bisulfite-treated DNA on a spin column, and then prepare the sequencing library using the EpiGnome™ Kit (Epicenter). In this procedure, bisulfite-treated single-stranded DNA is random primed using a polymerase able to read uracil nucleotides, to synthesize DNA containing a specific sequence tag.
Dilute and load the generated libraries onto the cBot DNA Cluster Generation System. After cluster generation is complete, transfer the flow cell to the HiSeq 2500 System for sequencing using 75bp paired-ends reads.
The advantages of WGBS is that (1) it typically covers over 90% of the CpG sites in the genome in an unbiased representation and (2) it also allows the identification of non-CG methylation as well as identification of partially methylated domains (PMDs). A major drawback of the WGBS is that it is expensive to run, with the library prep requiring relatively large quantities of DNA. In addition, analysis of sequencing data can be difficult and the development of new bioinformatics techniques for processing WGBS data remain a critical challenge (Stirzaker et al., 2014). In the situation where differential methylation occurs in only a small fraction of the genome, an alternative analysis, called reduced representation bisulfite sequencing (RRBS), can be used, in which only a fraction of the genome is sequenced. In addition, the method without bisulfite conversion has also been developed for the whole-genome DNA methylation detection which will be briefly described below.
3.1.4. Methyl-CpG binding domain protein-enriched genome sequencing (MBD-seq)
Just like WGBS, the MBD-Seq provides a probe-independent strategy to detect the genome-wide methylome landscape. Using an optimized protocol, the performance of MBD-seq can approximate the sensitivity/specificity obtained with WGBS, but at a fraction of the costs and time to complete the analysis. The methyl-binding domain of MBD2 protein has nanomolar affinity for a single symmetrically methylated CpG dinucleotide but has no affinity to unmethylated DNA oligonucleotides to any appreciable extent. Methylated sequence capturing using a MBD based enrichment followed by next-generation sequencing provides a great combination of sensitivity and cost-efficiency for genome-wide DNA-methylation profiling which allows estimating a single mCpG site (Ichiyanagi et al., 2013; Ramsahoye et al., 2000). The advantages for using MBD-Seq include: (1) it utilizes unconverted DNA, and therefore it does not depend on the efficiency of the intermediate steps, such as bisulfite conversion; (2) it has approximate basepair resolution; (3) it has greater accuracy of whole-genome DNA methylation detection, in which >95% of WGBS data is also found with MBD-Seq; and (4) it is applicable to all mammalian samples and tissue types. In contrast to WGBS which provides quantitative measurements of the percentage of methylation at each probe or genome coordinate, the disadvantage of MBD-seq is nonquantitative.
MBD sequencing workflow
Qualify and quantify the genomic DNA purified from kidney tissues
Perform a random fragmentation of the genomic DNA samples followed by size selection of the resulting fragments.
Separate the resulting DNA fragments by performing MBD-capture during which all fragments containing methylation are withheld, while fragments that do not contain methylation are discarded.
Prepare the library for sequencing which are then performed using the HiSeq 2500 System. The technical quality of the sequencing run is monitored in real time.
MBD-seq data provides two sets of reads, (1) methylated regions determined from pull-down and (2) input control. The number of reads corresponding to any given genomic DNA segment detected by MBD-Seq relative to the number of reads for an input control sample is proportional to the number of methylated CpG dinucleotides across all DNA fragments from that region. Therefore, this data is similar to ChIP-seq data. As with bisulfite sequencing, alignment of both sets of reads is a critical first step to analysis. Once aligned, regions of the genome that are methylated are determined from regions with large read counts in the methylated signal relative to input control. Peak calling algorithms such as MACS are a particularly popular set of enrichment-based methods for this problem, developed first for ChIP-seq data (Liao et al., 2015). Drawing from the well-established statistical approaches for differential analysis, many peak calling algorithms rely on models derived from negative controls to call enrichment.
Although there are a wide variety of techniques available to determine the DNA methylation status of kidney samples, choosing the best method to answer specific questions still proves difficult, as each assay has its advantages and disadvantages. The factors to be taken into consideration include the aim of the study, the quality and quantity of DNA available and the sensitivity and specificity of the method as well as the simplicity and the cost of the method (Kurdyukov & Bullock, 2016). Regardless of the technique being chosen, it is important that the data generated should be unbiased.
3.2. Histone modification and interactions analysis techniques and protocols
Chromatin is the DNA–protein complex that compacts and protects the genomic DNA within the cellular nucleus and the carrier of epigenetic information. Chromatin state can be mainly classified into two types: active (or open) and inactive (or condensed), which are often associated with different histone modifications. For example, the acetylation of histone H3 lysine 9 (H3K9ac) results in the open chromatin in promoters to promote gene transcription, while the tri-methylation of histone H3 lysine 9 (H3K9me3) results in the condensed chromatin in constitutively repressed genes. The combination of histone modifications, such as H3K4me and H3k27ac, is more precisely associated with active chromatin states. The binding of modified histones or other proteins, such as epigenetic regulators or transcription factors, to DNA can be evaluated by Chromatin immunoprecipitation (ChIP)-based methodologies, including chromatin immunoprecipitation (ChIP), Chip-PCR and ChIP-sequencing (ChIP-seq) analysis.
The chromatin immunoprecipitation (ChIP) assay is a powerful and versatile technique used for probing protein-DNA interactions within the natural chromatin context of the cell. The ChIP assay is designed to study the association of any proteins, including histones and their modified isoforms, epigenetic regulator(s) and transcriptional factor(s), with genomic DNA (Gade & Kalvakolanu, 2012; Wagner et al., 2016), in which ChIP isolates regions of DNA bound for the protein of interest, recognized by specific antibodies. Following that, PCR/qPCR or sequencing are used to determine the sequence of these DNA fragments bound by the studied proteins. For example, we can use ChIP grade antibodies against different histone marks, such as H3K4me2/3 or H3K9ac, to pull down either of these marks together with the bound DNA. If the research purpose is to identify whether these histone marks bind on specific gene, then purified DNA can be further analyzed by semi-quantitative PCR (ChIP) and quantitative PCR (ChIP-qPCR), whereas if the purpose is to identify the genome-wide targets of these histone marks, then purified DNA can be deeply analyzed by ChIP-sequencing, also known as ChIP-seq technique. ChIP-seq which combines ChIP with massively parallel DNA sequencing is used to map global binding sites precisely for any protein of interest, in which these proteins can be histone marks and others, such as epigenetic regulator(s) and transcriptional factor(s). Since the corresponding epigenetic regulators which are responsible for the modification of certain histone marks can easily be deduced, then ChIP-seq analysis can address if certain epigenetic regulators regulate gene expression indirectly through modification of histones. As an example, Li et al. performed ChIP-seq using antibodies against H3K9me2 and H4R3me2s, and revealed that histone demethylase JMJD1B distinctly mediates H3K9me2 and H4R3me2s demethylation at different loci in vivo, even though JMJD1B demethylates both H3K9me2 and H4R3me2s substrates with similar efficiency in vitro (Li et al., 2018). To identify the novel targets directly regulated by epigenetic regulators or other DNA-bound proteins, the antibodies against these proteins will be used to perform the immunoprecipitation assays instead of antibodies against histone marks. As an example, Saifudeen et al. reported a novel p53-Pax2 pathway for nephrogenesis based on the ChIP-seq data derived from p53 binding with antibodies against p53 other than histone marks (Saifudeen et al., 2012).
In general, ChIP and ChIP-qPCR are applied to confirm the binding of specific proteins to the predicted and limited target genes or to verify the potent novel protein targets identified by ChIP-seq, whereas ChIP-seq by itself is used to identify a mass of unknown targets of specific proteins and histone marks genome-wide. As an example, we performed ChIP and ChIP-qPCR to confirm that Ptpn13 was a novel target of Smyd2, a histone/lysine methyltransferase, according to the ChIP-seq analysis (Li et al., 2017). ChIP, ChIP-qPCR and ChIP-seq shared most of steps as below (Fig. 2). In this section, the protocol for the preparation of DNA from chromatin immunoprecipitation will be described in detail, which could be done in almost all the laboratories with no special equipment requirement. The primer design for ChIP and ChIP-qPCR will also be discussed. ChIP-qPCR is mainly dependent on the real-time PCR machine, while ChIP-seq needs more expensive and more professional equipment, such as HiSeq 3000/HiSeq 4000 Systems. Here, we describe the established protocols for preparation of DNA adapted from studies in cells and tissues (Gilchrist, Fargo, & Adelman, 2009; Schmidt et al., 2009).
FIG. 2.
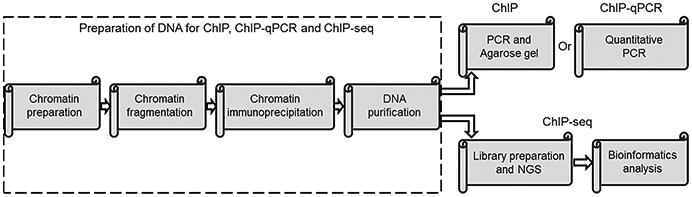
Workflow for ChIP, ChIP-qPCR and ChIP-seq.
3.2.1. Chromatin preparation
Weigh 30–40mg of fresh or frozen tissue (such as kidneys) in a Petri dish.
Chop tissue into small pieces (between 1 and 3mm3) using a scalpel blade in 1mL of ice-cold PBS with protease inhibitor cocktail.
Disaggregate the tissue using a homogenizer (pestle A) to get a homogeneous suspension.
Transfer the tissue suspension into a 1.5mL tube and centrifuge at 500 × rcf for 5min at 4°C. Gently discard the supernatant and keep the pellet.
Add 1mL (per 30–40mg tissue) of fresh 11% formaldehyde solution (50mM Hepes-KOH, 100mM NaCl, 1mM EDTA, 0.5mM EGTA, 11% formaldehyde) to resuspend the pellet. Swirl briefly and let sit at room temperature for 10 min. The formaldehyde crosslinked DNA and protein need be reversed at later step.
-
Add 1/20 volume of 2.5M glycine to quench formaldehyde. Rinse cells twice with ice cold PBS. Spin at 2000 × rcf for 4min. Proceed with cell lysis or freeze cells in liquid nitrogen and store pellets at −80°C.
Please note: The ChIP assay in cultured cells is started by adding 1/10 volume of fresh 11% formaldehyde solution as described at step 5 to culture medium for 10min at room temperature. Followed by adding 1/20 volume of 2.5M glycine to quench the reaction. Rinse the cells twice with ice cold PBS. Harvest and spin cells at 2000 × rcf for 5min at 4 °C to collect cell pellet for next steps.
Note: Samples should be kept on ice all the time and it also should minimize the time for sample manipulation to prevent protein degradation.
3.2.2. Chromatin fragmentation
Typically, there are two approaches for the chromatin fragmentation available. The first approach is to fragment native chromatin by standard micrococcal nuclease digestion of nuclei, which is referred to as nChIP (Hebbes, Thorne, & Crane-Robinson, 1988; O’Neill & Turner, 2003). This method is used for the study of proteins that bind to DNA with high affinity, such as histones, their modified isoforms and RNA polymerases. The second approach is to fragment the formaldehyde crosslinked chromatin to small sizes by sonication (Dedon et al., 1991; Solomon, Larsen, & Varshavsky, 1988), which is referred to as xChIP. This is the most common used method in ChIP assay, which works for proteins that bind to DNA with either higher or lower affinity. Thus, it will be detailed below.
Resuspend each pellet in 10mL of LB1 (50mM Hepes-KOH, pH 7.5; 140mM NaCl; 1mM EDTA; 10% Glycerol; 0.5% NP-40 or Igepal CA-630; 0.25% Triton X-100). Rock at 4°C for 10min. Spin at 2000 × rcf at 4°C for 4min in a tabletop centrifuge.
Resuspend each pellet in 10mL of LB2 (10mM Tris-HCl, pH 8.0; 200mMNaCl; 1mMEDTA; 0.5mM EGTA). Rock gently at 4°C for 5min. Pellet nuclei in tabletop centrifuge by spinning at 2000 × rcf at 4°C for 5min.
Resuspend each pellet in each tube in 3mL LB3 (10mMTris-HCl, pH 8; 100mM NaCl; 1mM EDTA; 0.5mM EGTA; 0.1% Na-Deoxycholate; 0.5% N-lauroylsarcosine).
Transfer cells to a homemade “sonication tube” (cut a polypropylene, 15mL conical tube into two pieces at the 7mL mark).
Sonicate suspension with a microtip attached to an Ultrasonic processor (120W, 20kHz, Fisher Scientific). Samples should be kept in an ice-water bath during sonication. Sonicate 12–18cycles of 30s ON and 60s OFF with 70% amplitude.
Add 300μL of 10% Triton X-100 to sonicated lysate. Split into 2mL centrifuge tubes. Spin at 20,000 × rcf at 4°C for 10min to pellet debris.
Combine supernatants from the 2mL centrifuge tubes in a new 15mL conical tube. The amount of LB3 and Triton X-100 is adjusted to the number of ChIPs to be performed, which depends on the starting population.
Save 50μL of cell lysate from each sonication as whole-cell extract (WCE) DNA. Store at −20°C.
Note: All lysis buffers should be supplemented with protease inhibitors (Complete, EDTA-free, Roche, #11873580001. The Bioruptor® Pico Sonication System is another option for researchers to shear DNA, which is an easier sonication protocol, just following the manufacture’s instruction.
3.2.3. Chromatin immunoprecipitation
Add 100μL magnetic beads (Invitrogen, Dynabeads) to a 1.5mL microfuge tube. Add 1mL block solution (0.5% BSA (w/v) in PBS). Set up one tube per IP.
Collect the beads using magnetic stand. Remove supernatant by aspiration. Wash beads in 1.0mL block solution two more times.
Resuspend beads in block solution and add 2–15μg of antibody in a final volume of 250μL
Incubate overnight or a minimum of 4h on a rotating platform at 4°C. Wash magnetic beads as described above (three times in 1mL block solution). Resuspend in 100μL block solution.
Add 100μL antibody/magnetic bead to cell lysates. Gently mix overnight on rotator or rocker at 4°C.
Let tubes sit in magnetic stand to collect the beads. Add 1mL RIPA Buffer (50mM Hepes-KOH, pH 7.5; 500mM LiCl; 1mM EDTA; 1% NP-40 or Igepal CA-630; 0.7% Na-Deoxycholate) to each tube. Remove tubes from magnetic stand and shake or agitate tube gently to resuspend beads. Replace tubes in magnetic stand to collect beads. Remove supernatant. Repeat this wash 4–6 more times.
Wash once with 1mL TBS (20mM Tris-HCl, pH 7.6; 150mM NaCl). Spin at 960 × rcf for 3min at 4°C and remove any residual TBS buffer using the magnetic stand. Add 200μL of elution buffer (50mM Tris-HCl, pH 8; 10mM EDTA; 1% SDS).
Elute and perform reverse crosslinking at 65°C for 6–18h. Resuspend beads in the first 15min with brief vortexing every 5 min.
Thaw 50μL of the WCE, add 150μL of elution buffer and mix. Reverse the formaldehyde crosslinking simultaneously with the ChIP samples.
Note: Step 1–3 could be done before chromatin fragmentation. All steps are performed at 4°C.
For each assay, in addition to antibody against the interested protein, the isotype control antibody, such as normal IgG, and positive control antibody, such as histone H3 and RNA polymerase II, are also necessary to either verify the specificity of binding or test the experimental operations.
3.2.4. Purification of DNA
Remove 200μL of supernatant and transfer to new tube. Add 200μL of TE to each tube of IP and WCE DNA to dilute SDS in elution buffer. Add 8μL of 1 mg/mL RNaseA (Ambion Cat # 2271). Mix and incubate at 37°C for 30min.
Add 4μL of 20mg/mL proteinase K (Invitrogen, 25530-049). Mix and incubate at 55°C for 1–2h.
Add 400μL phenol:chloroform:isoamyl alcohol (P:C:IA) and separate phases with 2mL Phase Lock Gel Light tubes FPR5101 Flowgen Bioscience and follow the instructions provided.
Transfer aqueous layer to new centrifuge tube containing 16μL of 5M NaCl (200mM final concentration) and 1μL of 20μg/μL GlycoBlue (Ambion, AM9516). Add 800μL 100% EtOH. Incubate for 30min at −80°C.
Spin at 20,000 × rcf for 10min at 4°C to pellet DNA. Wash pellets with 500μL of 80% EtOH and spin at 20,000 × rcf for 5min.
Dry pellets 10–20min in a speedvac at 45°C and resuspend each in 30μL of 10mM Tris-HCl, pH 8.0.
Measure DNA concentration of WCE with NanoDrop 2000 (Thermo Fisher Scientific).
Note: ChIP samples are too low in DNA concentration to give reliable results using a NanoDrop.
3.2.5. Primers design for ChIP and ChIP-qPCR
The primers for amplification of ChIP-DNA are crucial for the success of ChIP and ChIP-qPCR assay. In additional to basic principles for regular RT-PCR primers, primers design for ChIP-qPCR is more difficult than these RT-PCR primers, because of the unique sequence features of most gene promoter regions that are near the transcription start sites of genes and about 100–1000 base pairs long containing CpG islands (Deaton & Bird, 2011). Below are several guidelines for the primer design for ChIP-qPCR.
If it has been reported that the conserved binding sequence or motives of target gene by other transcriptional factors, then the primers could be designed according to binding sequence or sequence around that motif using common primers design tools, such as NCBI primer design and IDT, and then order primers from IDT.
If no reference, please refer the website: http://www.ensembl.org/index.html and then choose an organism, such as human or mouse http://www.ensembl.o…iens/Info/Index. Then search your gene and click the right hit on the search result page and it will bring you to the gene summary page. On the left, under “Gene Summary,” click “Sequence,” the sequence of the gene including 5′ flanking, exons, introns and flanking region will be displayed. (The exons are highlighted in pink background and red text, the sequence in front of the first exon is the promoter sequence.) By default, 600 bp 5′-flanking sequence (promoter) is displayed. Then put the sequence into common primers design tools, such as NCBI primer design and IDT, and then order primers from IDT.
In both case 1 and 2, multiple pairs of primers will be necessary to optimize the binding sites and specificity.
Negative primers are required for ChIP-qPCR assays. Follow the same routine to find the promoter region, then click “Configure this page” in the lower left column, a popup window opens allowing to input the size of 5’ Flanking sequence (upstream). You can put for example “5000″ or more and then save the configuration. Since promoters are usually up to 1000 base pairs. It is assumed that sequence beyond 5000 base pairs will be not related to the gene. Then these sequences could be used as templates for primers design.
Beware some genes have alternative promoters. To find those sequences, it requires extensive bioinformatics and experimental analysis.
3.2.6. Library preparation for ChIP-seq with next generation sequencing
Currently, lots of commercial kits are available for the preparation of the library, such as TruSeq ChIP Library Preparation Kit from Illumina, Inc., ChIP-Seq DNA Library Preparation from NEB, Inc., etc. By using these kits, all the researchers can construct the library according to the manufacturer’s instructions. And then the prepared library will be applied to the sequencing by the genomic core facility.
All the analysis steps described above define only significant regions in chromatin structure. Further analysis is essential to annotate the function of these genomic regions into various states, including active promoter, weak promoter, poised promoter, strong enhancer, weak/poised enhancers, insulator, transcriptional transition and heterochromatin. ChIP and ChIP-qPCR are applied to confirm the binding of specific proteins to the predicted and limited target genes. While ChIP-Seq analysis provides information about whole-genome distribution of individual proteins or their modifications—individual samples should be prepared for each study proteins, and therefore analysis of several proteins can be costly and require high material input.
4. Conclusion and discussion
Kidney disease is increasingly becoming a great epidemiologic concern worldwide. Despite the considerable advances in research, the pathophysiologic pathways involved in the progression of these kidney diseases remain to be elucidated. In recent years, multiple studies in kidney diseases have shown that mis-regulation of epigenetic modifiers contribute to kidney disease pathophysiology. It is becoming increasingly clear that DNA methylation and histone modifications contribute to kidney disease progression. Therefore, to better understand the pathophysiology of kidney diseases, it is important to identify the changes in DNA methylation and histone modifications in disease conditions compared to the health individuals, which is essential for finding hidden sources of variation in kidney diseases and therapeutic selection.
The newly developed high-throughput measurement technologies enable unprecedented, quantitative measurements of the epigenetic state in normal and disease kidneys. For DNA methylation, these techniques can be applied to kidney samples from human and model organisms, in which the functional impact of methylation alterations can be assessed bioinformatically in targeted experiments on model organisms and across sample population. In contrast, chromatin assays require higher-quality and quantity samples that are typical not feasible for preserved kidney samples or biopsies. As a result, chromatin measurements are typically limited to model organisms and cultured cell lines, which are essential to kidney epigenetics studies. Due to that DNA methylation and histone modifications are reversible, identifying these changes may potentially serve as therapeutic targets for kidney disease treatment in the future.
Acknowledgments
X.L. acknowledges support from National Institutes of Health grant R01 DK084097 and the PKD Foundation research grant as well as the Kansas Research and Translation Core Center (P30 DK106912) and Mayo Translation PKD Center (P30 DK090728).
References
- Bannister AJ, & Kouzarides T (2011). Regulation of chromatin by histone modifications. Cell Research, 21(3), 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtel-Walz W, & Huber TB (2014). Chromatin dynamics in kidney development and function. Cell and Tissue Research, 356(3), 601–608. [DOI] [PubMed] [Google Scholar]
- Beckerman P, Ko YA, & Susztak K (2014). Epigenetics: A new way to look at kidney diseases. Nephrology, Dialysis, Transplantation, 29(10), 1821–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CG, et al. (2010). Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Medical Genomics, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaumik SR, Smith E, & Shilatifard A (2007). Covalent modifications of histones during development and disease pathogenesis. Nature Structural & Molecular Biology, 14(11), 1008–1016. [DOI] [PubMed] [Google Scholar]
- Bourc’his D, et al. (2001). Dnmt3L and the establishment of maternal genomic imprints. Science, 294(5551), 2536–2539. [DOI] [PubMed] [Google Scholar]
- Brown MA, et al. (2006). Identification and characterization of Smyd2: A split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Molecular Cancer, 5, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, & Kouzarides T (2012). Cancer epigenetics: From mechanism to therapy. Cell, 150(1), 12–27. [DOI] [PubMed] [Google Scholar]
- Deaton AM, & Bird A (2011). CpG islands and the regulation of transcription. Genes & Development, 25(10), 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb DK, et al. (2017). Critical role of the cAMP-PKA pathway in hyperglycemia-induced epigenetic activation of fibrogenic program in the kidney. FASEB Journal., 31(5), 2065–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedon PC, et al. (1991). A simplified formaldehyde fixation and immunoprecipitation technique for studying protein-DNA interactions. Analytical Biochemistry, 197(1), 83–90. [DOI] [PubMed] [Google Scholar]
- Dressler GR (2008). Epigenetics, development, and the kidney. Journal of the American Society of Nephrology, 19(11), 2060–2067. [DOI] [PubMed] [Google Scholar]
- Egger G, et al. (2004). Epigenetics in human disease and prospects for epigenetic therapy. Nature, 429(6990), 457–463. [DOI] [PubMed] [Google Scholar]
- El-Osta A, & Wolffe AP (2000). DNA methylation and histone deacetylation in the control of gene expression: Basic biochemistry to human development and disease. Gene Expression, 9(1–2), 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M (2008). Epigenetics in cancer. The New England Journal of Medicine, 358(11), 1148–1159. [DOI] [PubMed] [Google Scholar]
- Esteller M (2011). Non-coding RNAs in human disease. Nature Reviews. Genetics, 12(12), 861–874. [DOI] [PubMed] [Google Scholar]
- Fujisawa T, & Filippakopoulos P (2017). Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nature Reviews. Molecular Cell Biology, 18(4), 246–262. [DOI] [PubMed] [Google Scholar]
- Gade P, & Kalvakolanu DV (2012). Chromatin immunoprecipitation assay as a tool for analyzing transcription factor activity. Methods in Molecular Biology, 809, 85–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaykalova D, et al. (2012). Dose-dependent activation of putative oncogene SBSN by BORIS. PLoS One, 7(7), e40389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist DA, Fargo DC, & Adelman K (2009). Using ChIP-chip and ChIP-seq to study the regulation of gene expression: Genome-wide localization studies reveal widespread regulation of transcription elongation. Methods, 48(4), 398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll MG, et al. (2006). Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science, 311(5759), 395–398. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Duarte RJ, Cazares-Ordonez V, & Avila-Chavez E (2014). The microRNA biogenesis machinery: Regulation by steroid hormones and alterations in cancer. Revista de Investigación Clínica, 66(5), 460–464. [PubMed] [Google Scholar]
- Ha M, & Kim VN (2014). Regulation of microRNA biogenesis. Nature Reviews. Molecular Cell Biology, 15(8), 509–524. [DOI] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, & Olson EN (2009). The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nature Reviews. Genetics, 10(1), 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadnagy A, Beaulieu R, & Balicki D (2008). Histone tail modifications and noncanonical functions of histones: Perspectives in cancer epigenetics. Molecular Cancer Therapeutics, 7(4), 740–748. [DOI] [PubMed] [Google Scholar]
- Hamamoto R, et al. (2014). SMYD2-dependent HSP90 methylation promotes cancer cell proliferation by regulating the chaperone complex formation. Cancer Letters, 351(1), 126–133. [DOI] [PubMed] [Google Scholar]
- Harris PC, & Rossetti S (2010). Determinants of renal disease variability in ADPKD. Advances in Chronic Kidney Disease, 17(2), 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata K, et al. (2002). Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development, 129(8), 1983–1993. [DOI] [PubMed] [Google Scholar]
- Hebbes TR, Thorne AW, & Crane-Robinson C (1988). A direct link between core histone acetylation and transcriptionally active chromatin. The EMBO Journal, 7(5), 1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann A, Gowher H, & Jeltsch A (2004). Biochemistry and biology of mammalian DNA methyltransferases. Cellular and Molecular Life Sciences, 61(19–20), 2571–2587. [DOI] [PubMed] [Google Scholar]
- Holliday R, & Pugh JE (1975). DNA modification mechanisms and gene activity during development. Science, 187(4173), 226–232. [PubMed] [Google Scholar]
- Huang J, et al. (2006). Repression of p53 activity by Smyd2-mediated methylation. Nature, 444(7119), 629–632. [DOI] [PubMed] [Google Scholar]
- Hurtado Del Pozo C, et al. (2018). Modeling epigenetic modifications in renal development and disease with organoids and genome editing. Disease Models & Mechanisms, 11(11), dmm035048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyanagi T, et al. (2013). Accumulation and loss of asymmetric non-CpG methylation during male germ-cell development. Nucleic Acids Research, 41(2), 738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka M, & Smith MM (2003). Functional consequences of histone modifications. Current Opinion in Genetics & Development, 13(2), 154–160. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, et al. (2009). The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics, 41(2), 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SG, et al. (2011). Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Research, 39(12), 5015–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judes G, et al. (2016). H3K4 acetylation, H3K9 acetylation and H3K27 methylation in breast tumor molecular subtypes. Epigenomics, 8(7), 909–924. [DOI] [PubMed] [Google Scholar]
- Kaneda M, et al. (2004). Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature, 429(6994), 900–903. [DOI] [PubMed] [Google Scholar]
- Kawaguchi T, & Hirose T (2015). Chromatin remodeling complexes in the assembly of long noncoding RNA-dependent nuclear bodies. Nucleus, 6(6), 462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko YA, et al. (2013). Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biology, 14(10), R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdyukov S, & Bullock M (2016). DNA methylation analysis: Choosing the right method. Biology (Basel), 5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei H, et al. (1996). De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development, 122(10), 3195–3205. [DOI] [PubMed] [Google Scholar]
- Leonhardt H, et al. (1992). A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell, 71(5), 865–873. [DOI] [PubMed] [Google Scholar]
- Li X (2015). Epigenetics in ADPKD: Understanding mechanisms and discovering treatment. In Li X (Ed.), Polycystic kidney disease. Brisbane, Australia: Codon Publications. The Journal of Clinical Investigation. [PubMed] [Google Scholar]
- Li LX, et al. (2017). Lysine methyltransferase SMYD2 promotes cyst growth in autosomal dominant polycystic kidney disease. The Journal of Clinical Investigation, 127(7), 2751–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, et al. (2018). JMJD1B demethylates H4R3me2s and H3K9me2 to facilitate gene expression for development of hematopoietic stem and progenitor cells. Cell Reports, 23(2), 389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, et al. (2015). Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nature Genetics, 47(5), 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, et al. (2016). Promoter methylation status of tumor suppressor genes and inhibition of expression of DNA methyltransferase 1 in non-small cell lung cancer. Experimental Biology and Medicine (Maywood, N.J.), 241(14), 1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, et al. (2017). Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nature Communication, 8(1), 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, & Lin C (2016). Enhancer, epigenetics, and human disease. Current Opinion in Genetics & Development, 36, 27–33. [DOI] [PubMed] [Google Scholar]
- McLaughlin N, et al. (2013). Histone signature of metanephric mesenchyme cell lines. Epigenetics, 8(9), 970–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S, et al. (2018). Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. Journal of Clinical Investigation, 128(1), 483–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin N, et al. (2014). In situ histone landscape of nephrogenesis. Epigenetics, 9(2), 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SP, Toomire KJ, & Strauss PR (2013). DNA modifications repaired by base excision repair are epigenetic. DNA Repair (Amst), 12(12), 1152–1158. [DOI] [PubMed] [Google Scholar]
- Mortusewicz O, et al. (2005). Recruitment of DNA methyltransferase I to DNA repair sites. Proceedings of the National Academy of Sciences of the United States of America, 102(25), 8905–8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murcia NS, Sweeney WE Jr., & Avner ED (1999). New insights into the molecular pathophysiology of polycystic kidney disease. Kidney International, 55(4), 1187–1197. [DOI] [PubMed] [Google Scholar]
- Okano M, Xie S, & Li E (1998). Dnmt2 is not required for de novo and maintenance methylation of viral DNA in embryonic stem cells. Nucleic Acids Research, 26(11), 2536–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, et al. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell, 99(3), 247–257. [DOI] [PubMed] [Google Scholar]
- O’Neill LP, & Turner BM (2003). Immunoprecipitation of native chromatin: NChIP. Methods, 31(1), 76–82. [DOI] [PubMed] [Google Scholar]
- Pei Y (2001). A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends in Molecular Medicine, 7(4), 151–156. [DOI] [PubMed] [Google Scholar]
- Peschansky VJ, & Wahlestedt C (2014). Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics, 9(1), 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsahoye BH, et al. (2000). Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proceedings of the National Academy of Sciences of the United States of America, 97(10), 5237–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Paredes M, & Esteller M (2011). Cancer epigenetics reaches mainstream oncology. Nature Medicine, 17(3), 330–339. [DOI] [PubMed] [Google Scholar]
- Roupret M, et al. (2007). Molecular detection of localized prostate cancer using quantitative methylation-specific PCR on urinary cells obtained following prostate massage. Clinical Cancer Research, 13(6), 1720–1725. [DOI] [PubMed] [Google Scholar]
- Saddic LA, et al. (2010). Methylation of the retinoblastoma tumor suppressor by SMYD2. The Journal of Biological Chemistry, 285(48), 37733–37740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadikovic B, et al. (2008). Cause and consequences of genetic and epigenetic alterations in human cancer. Current Genomics, 9(6), 394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifudeen Z, et al. (2012). A p53-Pax2 pathway in kidney development: Implications for nephrogenesis. PLoS One, 7(9), e44869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sand M (2014). The pathway of miRNA maturation. Methods in Molecular Biology, 1095, 3–10. [DOI] [PubMed] [Google Scholar]
- Schmidt D, et al. (2009). ChIP-seq: Using high-throughput sequencing to discover protein-DNA interactions. Methods, 48(3), 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto E, & Yoshida M (2014). Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harbor Perspectives in Biology, 6(4), a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, et al. (2014). Genetic and environmental exposures constrain epigenetic drift over the human life course. Genome Research, 24(11), 1725–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, & Lange CA (2008). Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutation Research, 647(1–2), 21–29. [DOI] [PubMed] [Google Scholar]
- Solomon MJ, Larsen PL, & Varshavsky A (1988). Mapping protein-DNA interactions in vivo with formaldehyde: Evidence that histone H4 is retained on a highly transcribed gene. Cell, 53(6), 937–947. [DOI] [PubMed] [Google Scholar]
- Sterner DE, & Berger SL (2000). Acetylation of histones and transcription-related factors. Microbiology and Molecular Biology Reviews, 64(2), 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirzaker C, et al. (2014). Mining cancer methylomes: Prospects and challenges. Trends in Genetics, 30(2), 75–84. [DOI] [PubMed] [Google Scholar]
- Strahl BD, & Allis CD (2000). The language of covalent histone modifications. Nature, 403(6765), 41–45. [DOI] [PubMed] [Google Scholar]
- Suetake I, et al. (2004). DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. The Journal of Biological Chemistry, 279(26), 27816–27823. [DOI] [PubMed] [Google Scholar]
- Sun ZW, & Allis CD (2002). Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature, 418(6893), 104–108. [DOI] [PubMed] [Google Scholar]
- Sun Q, Hao Q, & Prasanth KV (2018). Nuclear long noncoding RNAs: Key regulators of gene expression. Trends in Genetics, 34(2), 142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajul-Arifin K, et al. (2003). Identification and analysis of chromodomain-containing proteins encoded in the mouse transcriptome. Genome Research, 13(6B), 1416–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarakhovsky A (2010). Tools and landscapes of epigenetics. Nature Immunology, 11(7), 565–568. [DOI] [PubMed] [Google Scholar]
- Towler BP, Jones CI, & Newbury SF (2015). Mechanisms of regulation of mature miRNAs. Biochemical Society Transactions, 43(6), 1208–1214. [DOI] [PubMed] [Google Scholar]
- Turner BM (2002). Cellular memory and the histone code. Cell, 111(3), 285–291. [DOI] [PubMed] [Google Scholar]
- Wagner M, et al. (2016). Chromatin immunoprecipitation assay to identify genomic binding sites of regulatory factors. Methods in Molecular Biology, 1366, 53–65. [DOI] [PubMed] [Google Scholar]
- Wanner N, et al. (2019). DNA methyltransferase 1 controls nephron progenitor cell renewal and differentiation. Journal of the American Society of Nephrology, 30(1), 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo YM, et al. (2014). Genome-wide methylation profiling of ADPKD identified epigenetically regulated genes associated with renal cyst development. Human Genetics, 133(3), 281–297. [DOI] [PubMed] [Google Scholar]
- Wreczycka K, et al. (2017). Strategies for analyzing bisulfite sequencing data. Journal of Biotechnology, 261, 105–115. [DOI] [PubMed] [Google Scholar]
- Xie S, et al. (1999). Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene, 236(1), 87–95. [DOI] [PubMed] [Google Scholar]
- Xu W, et al. (2018). Overexpression of SET and MYND domain-containing protein 2 (SMYD2) is associated with tumor progression and poor prognosis in patients with papillary thyroid carcinoma. Medical Science Monitor, 24, 7357–7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, et al. (2018). Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Research, 28(3), 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong WS, Hsu FM, & Chen PY (2016). Profiling genome-wide DNA methylation. Epigenetics & Chromatin, 9, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, & Beaudet AL (2016). Epigenetics and human disease. Cold Spring Harbor Perspectives in Biology, 8(2), a019497. [DOI] [PMC free article] [PubMed] [Google Scholar]