

Abstract
Nitric oxide (nitrogen monoxide) (NO) plays an important role in a wide range of physiologic processes. A major mediator of endothelial function, NO regulates vasodilatory and antithrombotic actions in the vasculature and plays a role in reproductive functions, bronchodilation, bone formation, memory, insulin sensitivity, and gastrointestinal relaxation. NO is formed from NO synthase. Impaired NO bioactivity is strongly associated with endothelial dysfunction and cardiovascular disease, but is also implicated in a broad range of other disorders, including pulmonary hypertension, insulin resistance, erectile dysfunction, and preeclampsia. Numerous therapies designed to target NO are being investigated and developed, including NO donors and stimulants. The recent African‐American Heart Failure Trial (A‐HeFT) showed that the NO donor isosorbide dinitrate, combined with the vasodilator hydralazine, significantly reduced morbidity and mortality in black patients with moderate‐to‐severe heart failure. Antihypertensive drugs, including angiotensin‐converting enzyme inhibitors, calcium channel blockers, and third‐generation β‐blockers, are NO stimulants that have demonstrated significant improvement of endothelial function and NO bioactivity. Other cardiovascular therapies that may improve NO bioactivity include statins, L‐arginine, and hydralazine approaches such as exercise and dietary changes.
Nitric oxide (nitrogen monoxide) (NO) is a gas that is widely distributed throughout the human body and plays strikingly diverse roles in a broad spectrum of physiologic and pathophysiologic processes. 1 , 2 NO is a free radical and highly reactive molecule and is now recognized as an important signaling molecule or neurotransmitter, active in practically every major organ system, not only in mammals, but also in plant, fish, and insect species. 1 , 2
In humans, NO is an important mediator of functions related to the cardiovascular (CV) system. In the endothelium, NO has emerged as a major factor in mediating vascular tone and exerting an antithrombotic action. Protection of target organs (eg, heart, brain, and kidneys) is also a function of NO. 1 , 2 , 3 Other functions of NO related to CV function and related metabolic activity include peripheral nervous system functions in the respiratory tract, sexual function, insulin release, and immune responses in inflammation. 1 , 2 , 4 , 5 , 6 , 7 , 8 NO is also believed to potentiate the protective benefits of high‐density lipoprotein cholesterol in preventing atherosclerosis 7 and exercise in reducing the risks of CV disease. 8
Impairment of NO bioactivity is strongly associated with endothelial dysfunction, hypertension, atherosclerosis, and CV disease, 3 , 9 , 10 and has been linked to a wide range of other disorders, including asthma and pulmonary hypertension, 5 , 11 erectile dysfunction,12 preeclampsia,13 and insulin resistance.14
NO often plays a paradoxical role as both a protective and pathogenic factor in multiple diseases, depending on the environment and pathway of NO production and expression. 1 , 2 , 5 , 6 , 15 The multiple functions of NO and the potential methods for therapeutically maintaining or restoring its optimal production and function have been investigated over the past 25 years. 16 , 17 This article reviews some of the major developments in this rapidly advancing field, chiefly as it pertains to the CV system in general and hypertension in particular.
Historical Background
The CV effects of NO were first highlighted in experiments to determine the mechanism for the vasodilating effects of acetylcholine (Ach). 1 , 2 In animal studies, Ach was observed to cause dilation of arteries supplying blood to skeletal muscle in vivo; researchers hypothesized that this effect was due to diffusion of Ach to the vascular smooth muscle cells. 1 Furchgott and Zawadzki, 18 however, found in a landmark experiment that accidental rubbing of the intimal surface of an isolated rabbit aorta preparation abolished the vasodilatory effect of Ach. 18 The investigators hypothesized that rubbing of the intima caused loss of endothelial cells, which was supported by their observation that removal of the adventitia did not affect the vasodilatory action of Ach. 18 This vasodilatory action thus appeared to be dependent on the production by endothelial cells of an additional substance that diffused into the arterial smooth muscle cells and induced relaxation; this hypothetical substance was called endothelium‐derived relaxation factor, and was distinct from endothelium‐derived hyper‐polarizing factor.
In seeking to identify endothelium‐derived relaxation factor, Furchgott and Zawadzki eliminated several substances from their search then associated with endothelial function, including bradykinin, prostacyclin, 3′,5′‐cyclic guanosine monophosphate, and cyclic adenosine monophosphate.1, 18Subsequent studies identified endothelium‐derived relaxation factor as NO. 19 , 20
Synthesis and Mechanism of Action
The production of NO is catalyzed by NO synthases (NOS), which convert the amino acid l‐arginine to l‐citrulline and NO. 2 , 21 NOS are members of a family of cytochrome P450–like reductases linked to a nicotinamide adenine dinucleotide phosphate (reduced) (NAD[P]H) oxidase enzyme. At least 3 isoforms, specific to different organ systems, exist. Endothelial NOS (eNOS), a constitutive isoform, is primarily expressed in the endothelium and airway epithelium. Neuronal NOS (nNOS), a second constitutive isoform, is present in the central and peripheral nervous systems, as well as skeletal muscle. Both eNOS and nNOS are calcium‐calmodulin‐dependent. 2 , 22 The third known NOS isoform is inducible NOS (iNOS), which is primarily produced by the action of endotoxin inflammatory cytokines such as interleukin 1 or tumor necrosis factor ? in macrophages and other cell types. 2 , 22 All 3 of the isoforms, however, have been found in a variety of tissues, and the constitutive isoforms may also be inducible. 23
Pulsatile blood flow and shear stress are the primary physiologic stimuli of NO production by the endothelium. 9 These forces open calcium channels on endothelial cells, thus promoting the calcium‐dependent activation of eNOS, which, in turn, induces the release of NO. 13 Once synthesized, NO diffuses into the underlying vascular smooth muscle, where it activates soluble guanylate cyclase, causing an increase in 3′,5′‐cyclic guanosine monophosphate and smooth muscle relaxation. 2 , 9 The small size, lipophilic nature, and chemical lability of NO allow it to diffuse readily across cell membranes without need for channels or receptors. 13
It had previously been thought that after diffusing into the blood stream, NO was bound to the hemoglobin in red blood cells. 24 Hemoglobin, however, reacts with NO in such a way that facilitates the redelivery of NO to the vessel wall as a biologically active S‐nitrosothiol molecule.24, 25Furthermore, this mechanism of preserved NO bioactivity is upregulated in oxygen‐deprived tissues that are most in need of NO‐induced vasodilation and increased blood flow. 25
The mechanisms of stimulation of NO synthesis by nNOS are not well understood. 26 Study data suggest, however, that NO production by nNOS in the brain is increased in response to stress and hypoxia, where it is regulated by interacting proteins, and that 60% of nNOS activity in the brain occurs in the membrane fraction.27, 28, 29, 30
The iNOS enzyme produces NO in 1000‐fold greater quantities than the constitutive enzymes eNOS and nNOS in response to biologic inflammatory and immunologic factors, including cytokines, lipopolysaccharide, and interferon γ, 2 , 31 or possibly to counteract arteriosclerosis. 32 While inducible NO may kill invading organisms, the overproduction of NO from iNOS can be pathogenic and has been observed in many disorders, including septic shock, hemorrhagic shock, multiple sclerosis, neurodegenerative diseases, rheumatoid arthritis, ulcerative colitis, and cancer. 2 , 31 , 33 , 34 , 35 Reduced eNOS and increased iNOS have been observed concomitantly in disease states. 35 , 36
Endothelium, NO, and the CV System
The endothelium serves as a thromboresistant surface; a barrier to macromolecules; and a regulator of vascular tone, structure, and function by releasing multiple vasoactive substances, including NO, in response to physiologic stimuli. 3 , 37 Normal function of the endothelium relies on the balanced interaction of autocrine‐paracrine relaxing and contracting factors within the vessel wall. 3 , 37 In response to hemodynamic shear stress, the endothelium continuously releases NO and up‐regulates the gene that expresses NOS; NO provides vasodilation and maintains vascular tone. 37 In addition, the endothelium releases other interactive vasodilators. 38 , 39
NO also exhibits antithrombotic and anti‐inflammatory actions in the endothelium, including inhibition of vascular smooth muscle cell growth, platelet aggregation, cytokine expression, monocyte migration, leukocyte adhesion, and lipid oxidation. 38 , 40 NO down‐regulates synthesis of angiotensin‐converting enzyme (ACE) and angiotensin II type 1 receptors, thus suppressing the renin‐angiotensin system, which promotes vasoconstriction and atherosclerotic processes and inhibits activity of the potent vasoconstrictor endothelin‐1. 39 , 41 At the cellular level, NO regulates mitochondrial respiration and inhibits mitochondrial metabolism during cell stress, thereby reducing oxygen consumption and preventing the apoptosis related to oxidative damage. 1 , 42 Thus, NO plays a major role in maintaining endothelial homeostasis and function, regulating blood pressure (BP), and inhibiting atherosclerotic processes. 38 , 39 , 43
The endothelial functions of NO also play a role in promoting systemic vascular and arterial relaxation and distensibility, thereby reducing cardiac preload and afterload, myocardial hypertrophy, and left ventricular dysfunction, and protecting target organs. 39 , 44 , 45 , 46 Furthermore, the vascular and cardioprotective functions of NO extend beyond the endothelium to involve neuronal NO and its role as a neuromodulator of the autonomic nervous system, maintaining vagal tone and suppressing sympathetic nervous system overactivity through both central and peripheral nervous system signaling. 47 Acting in the nucleus tractus solitarius, NO plays a significant role in regulating baroreflex function and cardiopulmonary reflex responses. 48
Impaired NO in Endothelial Dysfunction
The importance of NO in promoting endothelial homeostasis is demonstrated by the association between impaired NO bioactivity and endothelial dysfunction, which is characterized by the imbalance of endothelium‐derived vasoconstrictive and vasodilatory substances, with a shift toward greater vasoconstriction, inflammation, and thrombosis formation. 3 , 49 Endothelial dysfunction is present during the early stages of atherosclerosis and is a significant predictor of CV morbidity and mortality (Figure 1). 3 , 40 , 44 , 50 , 51 , 52 This has prompted some researchers to conclude that the endothelium should be the focus of therapeutic strategies to reduce the risks of CV disease. 53 Reduced NO bioactivity is a major component of endothelial dysfunction. 3 , 36 , 54 , 55
Figure 1.
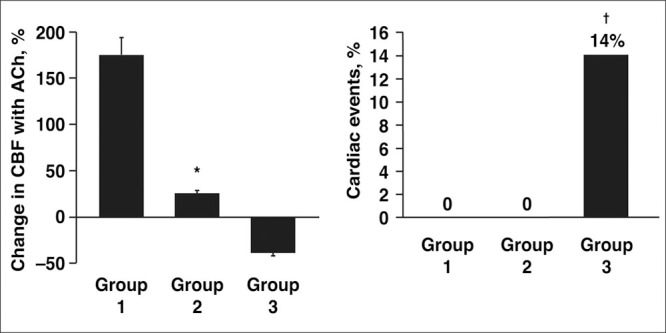
In 157 patients with mild atherosclerosis followed for an average of 28 months, severe endothelial dysfunction at baseline (group 3; n=42), identified as reduced coronary blood flow (CBF) response to acetylcholine (Ach), was associated with a significantly increased risk for cardiovascular events compared with patients with normal (group 1; n=83) or mildly impaired (group 2; n=32) endothelial function. *P<.0001 vs groups 1 and 3. †P<.05 vs groups 1 and 2. Reproduced with permission from Al Suwaidi et al. 51
One proposed mechanism of impaired NO availability and endothelial dysfunction is oxidative stress, which occurs when pro‐oxidant processes exceed the capacity of antioxidant mechanisms to maintain an appropriate balance. Oxidative stress is generated by increased production of reactive oxygen species, including superoxides, which are derived from xanthine oxidase, cyclooxygenase, and NAD[P]H oxidase. Reactive oxygen species react with NO, thereby decreasing its bioactivity. 10 , 49 Some well‐known CV risk factors such as hyperlipidemia, hypertension, diabetes, and cigarette smoking are associated with oxidative stress. 49 , 54 , 56
Impaired NO and Hypertension
Endothelial dysfunction is often described in individuals with hypertension. 3 Multiple studies of flow‐mediated dilatation of the brachial artery, usually examined by venous occlusion plethysmography of forearm blood flow, have shown that patients with hypertension exhibit blunted arterial vasodilation in response to endothelium‐dependent vasodilators, such as Ach, while vasodilatory response to endothelium‐independent vasodilators, such as sodium nitroprusside, is preserved (Figure 2). 43 , 57 , 58 Moreover, inhibition of NO with infusion of Ng‐monomethyl‐l‐arginine has significantly increased BP and total peripheral artery resistance (Figure 3). 59 It is unclear, however, whether endothelial dysfunction (with NO impairment) is a cause or consequence of increased BP. 3 Regardless of the mechanism, endothelial dysfunction may increase BP, and CV risk is positively and significantly correlated with the severity of endothelial dysfunction in hypertensive patients. 3 , 44 , 50
Figure 2.
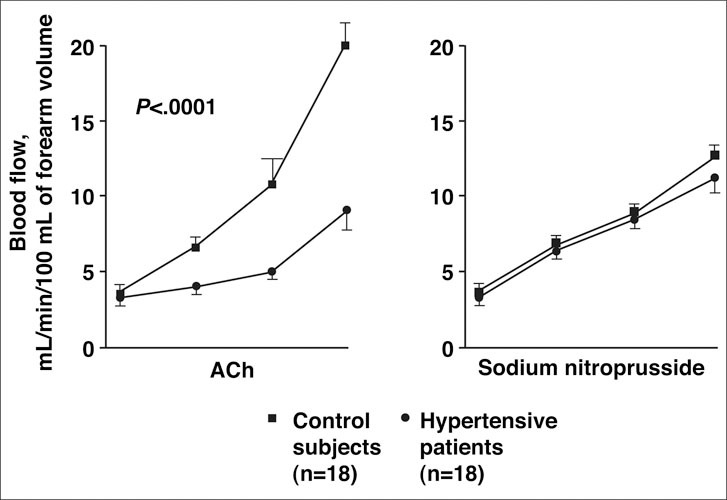
The effects of infusion of acetylcholine (Ach), an endothelium‐dependent vasodilator, and sodium nitroprusside, a direct endothelium‐independent vasodilator, on forearm blood flow (FBF) in the brachial artery were compared in 18 men and women with hypertension and 18 normotensive controls. As measured by strain‐gauge plethysmography, FBF Ach was significantly reduced in the hypertensive patients compared with the control patients (P<.0001), but no significant difference in FBF was observed between hypertensive and control patients in those infused with sodium nitroprusside. Reproduced with permission from Panza et al. 57
Figure 3.
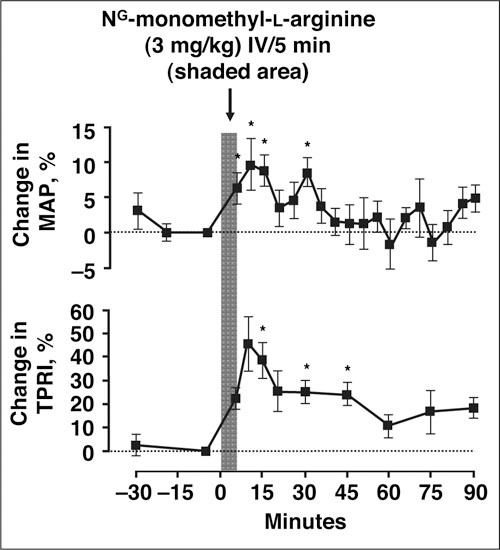
Intravenous (IV) infusion of 3 mg/kg Ng‐monomethyl‐l‐arginine, an inhibitor of nitric oxide synthase, significantly increased mean arterial pressure (MAP) by 10% (P<.05) and increased total peripheral resistance index (TPRI) by 46%, compared with saline placebo (P<.05), in 8 healthy subjects. Reproduced with permission from Haynes et al. 59
Impaired NO and CV Disease
Impairment of NO bioactivity has been viewed as a CV risk factor due to its strong association with endothelial dysfunction 3 , 60 ; however, studies have also identified the CV risks of reduced NO bioactivity per se. Studies have found, for example, that inhibition of NO with Ng‐monomethyl‐l‐arginine increases arterial stiffness. 45 , 61 Moreover, impaired platelet production of NO was shown to be a significant, independent risk factor, and a stronger predictor than atherosclerosis, for acute coronary syndromes in patients undergoing coronary angiography. 62 Another study showed that release of NO, as measured with a porphyrinic microsensor in atherosclerotic human carotid arteries and normal mammary arteries, was markedly reduced in the atherosclerotic arteries compared with normal arteries. 55 In patients with coronary heart disease (CHD), elevated C‐reactive protein level, an inflammatory marker, was shown to be significantly and independently associated with impaired NO bioavailability as measured by forearm blood flow vasodilatory response to Ng‐monomethyl‐l‐arginine; these findings further support the hypothesis that oxidative stress and inflammation are major causes of impaired NO. 63
Impaired NO in the Cardiometabolic Syndrome
Experimental data indicate that glucose stimulates islet activities of both constitutive and inducible forms of NOS, and NO may serve as a negative feedback inhibitor of inappropriate insulin release at hyperglycemic glucose levels. 64 Other preliminary data indicate that NO may stimulate insulin release. 65 Clinical studies have shown that the arterial vasodilating effect of insulin in skeletal muscle, a primary mechanism of insulin sensitivity, is dependent on endothelial NO release.14, 66 Overall, NO appears necessary for regulation of insulin secretion, normal β‐cell function, and insulin sensitivity. 14 , 67
Decreased NO bioactivity and endothelial dysfunction are associated with insulin resistance and diabetes mellitus. 14 , 68 , 69 Because the arterial vasodilating effect of insulin in skeletal muscle, which is a primary mechanism of insulin sensitivity, is dependent on endothelial NO release, NO impairment would theoretically reduce insulin sensitivity. 14 , 66 It is unclear, however, whether impaired insulin action causes decreased NO bioactivity, or vice versa, or whether both defects arise from a common biologic source. 66 One proposed pathogenic mechanism supporting this association is a genetic and/or acquired defect of NO, leading to the syndrome of reduced insulin‐mediated vasodilation, insulin resistance, and diabetes. 14 , 70
NON‐CV FUNCTIONS OF NO
Central and Peripheral Nervous System
In the brain, NO is a multifunctional messenger molecule involved in processes of synaptic plasticity, learning and memory, and possibly aging. 71 , 72 The antioxidant and antiapoptotic effects of NO may protect against neurodegeneration, 72 and vascular NO may promote cerebral perfusion, which improves memory function. 73 A study in an aging rat model of chronic brain hypoperfusion, which mimics human mild cognitive impairment, found that selective inhibition of eNOS impaired spatial memory function, suggesting that this brain function is dependent on NO‐regulated maintenance of cerebral blood flow and microvessel tone. 73 The effects of NO in neurodegenerative diseases are highly controversial, however, with evidence that NO may assume pathogenic as well as protective roles. 15 , 72 In the normal brain, for example, NO inhibits precursor brain cell proliferation; however, after brain damage, excessive NO production may promote pathologic neurogenesis. 15 , 74
In the peripheral nervous system, NO serves as a gastrointestinal neurotransmitter, mediating relaxation and tone of the stomach, colon, and internal anal sphincter. 2 Neuronal NO in the brain may also mediate the inhibition of gastric acid secretion, a stress‐induced defense mechanism. 27 , 30 In skeletal muscle, NO mediates excitation‐contraction coupling, mitochondrial energy production, glucose metabolism, and autoregulation of blood flow. 75 In addition, neuronal NO contributes to the regulation of renal function 76 and NO performs an important renoprotective role in opposing the effects of chronic renal renin‐angiotensin system overactivation, which contributes to renal hypertension and injury. 39 Inhibition of NO has also been associated with accelerated hypertensive renal disease in rats. 76
Reproductive System
Male Reproductive System. In men, NO regulates relaxation of smooth muscle cells of the corpus cavernosum and is a principal mediator of penile erection, speculatively through a combination of neuronal, neurohormonal, and vascular effects.2, 77 NO primarily acts as a neurotransmitter of erogenic stimuli, which diffuses to vascular and trabecular smooth muscle in the penis, thereby increasing blood flow and promoting erectile tumescence.77 Multiple animal studies have shown that decreased NOS is correlated with erectile dysfunction related to aging and diabetes. 78 , 79 , 80 Moreover, in clinical studies, reduced flow‐mediated dilation of the cavernous arteries 81 and of the brachial artery 82 were significantly correlated with erectile dysfunction.
Female Reproductive System. In women, NO increases during pregnancy and contributes to the reduction of peripheral vascular resistance through vascular smooth muscle relaxation. Although smooth muscle relaxation is observed in normal pregnancy, the role of NO bioactivity in this process appears to vary by species, vessel size, and vascular bed. 13 , 83 Additional data suggest that NO regulates uterine quiescence, cervical ripening, and labor. 84 , 85 , 86 Levels of NO in the uterus are elevated during pregnancy and significantly decreased during labor, whereas they are low in the cervix during pregnancy and increase dramatically before and during labor. 84 , 85 , 86 Production of NO in the uterus during pregnancy may be elevated to promote uterine relaxation and inhibit contractility, and the decrease of NO at term may help signal or promote labor. 84 Similarly, NO levels may be suppressed in the cervix during pregnancy to inhibit preterm labor, and may increase at term to enhance cervical ripening. 17
Reduced NO may play a role in preeclampsia.13 Several studies in isolated human resistance arteries from pregnant women in vitro demonstrated that flow‐mediated, endothelium/NO‐dependent vasodilatory responses were reduced in women with preeclampsia, compared with normotensive women. 87 , 88 , 89 Reduced flow‐mediated dilation of the brachial artery has also been observed in vivo in pregnant women with preeclampsia, compared with normotensive pregnant women, 90 and in pregnant women tested at 23–25 weeks′ gestation who later developed preeclampsia, suggesting that endothelial dysfunction precedes preeclampsia. 91 Other clinical studies, however, have failed to show an association between impaired NO and preeclampsia. 92 , 93
Respiratory System
NO is produced in the respiratory tract by multiple cell types, including epithelial cells, airway neurons, inflammatory cells, and vascular endothelial cells. 5 In the healthy lung, NO regulates pulmonary vascular reactivity and induces airway dilation in response to hypoxia‐induced vasoconstriction.5, 11 NO may be an important regulator of pulmonary vascular homeostasis because of its endothelial antithrombotic and anti‐inflammatory effects, and neuronal NO may also modulate bronchomotor tone. 11 In contrast, excessive production of NO induced by poorly controlled pulmonary inflammation is associated with asthma. 5 , 11 Overall, balanced production of NO appears to be essential for normal functioning of the lung. 11
Reduced expression of NO in the vascular endothelium of pulmonary arteries, as determined by histochemical and immunohistochemical analysis, has been significantly associated with primary and secondary pulmonary hypertension. The severity of pulmonary hypertension is proportional to the extent of NO reduction, although the pathophysiologic mechanism of this association is unclear. 11 , 94 Reduced endothelium‐dependent vasodilation also has been observed in patients with chronic obstructive pulmonary disease and cystic fibrosis, although the evidence of these associations is inconsistent. 11 , 95 In addition, a clinical study found that the immunohistochemical expression of eNOS in the arterial endothelium and protein content of lung tissue in smokers were lower than in control nonsmokers, suggesting that NO impairment from smoking is a pathogenic factor in respiratory disease associated with tobacco. 96 Preliminary data also suggest that airway hyper‐responsiveness, seen in asthma, may be associated with reduced endogenous NO in the lungs. 5 Paradoxically, however, asthma is associated with increased expression of iNOS and elevated levels of inducible NO, which have proinflammatory effects. 5
Bone Formation
Both constitutive and inducible forms of NO have been observed in a variety of bone cells, where they perform multiple functions. 6 , 97 At low levels, endothelial and neuronal constitutive NO mediate normal proliferation of bone‐forming osteoblasts and inhibit the activity of bone‐resorbing osteoclasts, promoting normal bone turnover and preventing osteoporosis. 97 , 98 A study in animal bone models found that NOS inhibition with N‐nitro‐l‐arginine methyl ester and aminoguanidine increased osteoclast resorptive activity, dramatically increasing the number and size of bone pits, compared with control bone cultures in vitro, and caused increased resorptive activity and a loss of bone density in an in vivo rat model of osteoporosis. 99 High levels of inducible NO, however, promoted by inflammatory states including rheumatoid arthritis or osteomyelitis, may exacerbate the osteoporotic processes caused by those conditions. 6 , 97 , 98 While controlled levels of constitutive NO are essential for normal bone growth, excessive inducible NO is pathogenic in osteoporosis. 6 , 97
Therapeutic Modulation of No
As the effects of impaired NO production and release have become increasingly defined in the pathophysiology of CV disease states, therapeutic strategies seeking to increase the availability of NO have been rapidly emerging. 16 , 17 , 100 These strategies include agents that donate or release NO, stimulate endogenous production of NO, or protect NO from oxidative stress and inactivation.
No Donors
Chemicals that release NO are being widely investigated for their potential in diverse therapeutic areas. 16 , 23 , 101 Nitrates such as nitroglycerin and nitroprusside provide acute vasodilatory relief in acute CV conditions such as angina, myocardial infarction, and hypertensive emergencies. The usefulness of these drugs is considered limited, however, because of adverse effects and the occurrence of nitrate tolerance with chronic administration. 23
The African‐American Heart Failure Trial (A‐HeFT), 33 conducted in 1050 black patients, supported the use of organic nitrate therapy to improve CV outcomes in patients with New York Heart Association class III or IV heart failure. Treatment with the NO donor isosorbide dinitrate plus hydralazine, added to standard heart failure therapies including diuretics, ACE inhibitors, and β‐blockers, achieved reductions of 43% for overall mortality (P=.01) and 33% for heart failure hospitalizations (P=.001), as well as improved quality‐of‐life scores (P=.02), compared with placebo (Figure 4). These results, which prompted early termination of the trial after a mean follow‐up of 10 months, provide the first substantial data indicating that NO modulation can improve CV outcomes in heart failure. 33
Figure 4.
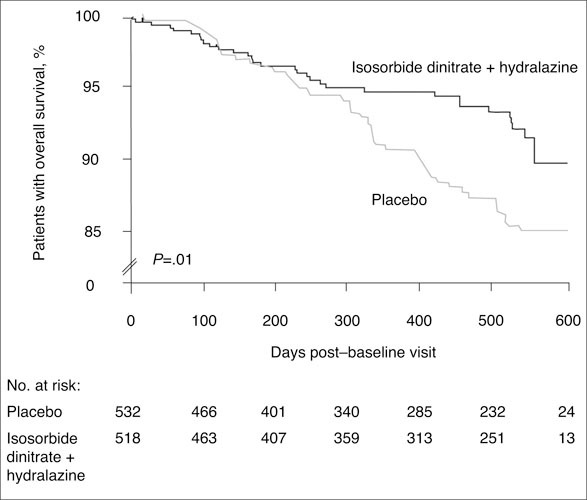
In the African‐American Heart Failure Trial (A‐HeFT), conducted in 1050 black patients with New York Heart Association class III or IV heart failure, treatment with a fixed dose of isosorbide dinitrate, a nitric oxide donor, plus hydralazine, when added to standard heart failure therapies, significantly reduced mortality by 43% (P=.01) compared with placebo. Reproduced with permission from Taylor et al.
The nitrate nicorandil has demonstrated not only relief of angina pectoris but also significant reductions of multiple CV disease outcomes in major randomized controlled clinical trials.102, 103 In the Impact of Nicorandil in Angina (IONA) study 103 (N=5126), addition of nicorandil to standard antianginal therapy significantly reduced the primary composite end point of CHD death, non‐fatal myocardial infarction, or unplanned hospitalization for angina pectoris (P=.01); acute coronary syndromes (P=.03); and all CV disease events (P=.03), compared with placebo.
In other disease areas, data from the Study of Osteoporotic Fractures 104 showed that intermittent use of nitrate supplements in 317 elderly women was associated with substantially increased bone mineral density of the hip and heel, compared with nonusers. Furthermore, topical nitroglycerine ointment was as effective as estrogen therapy in preventing bone loss in women who had undergone oophorectomy. 105 Several novel nitrates are also being investigated for their potential effects in providing neuroprotection in neurodegenerative diseases, including disorders such as dementia and Alzheimer's disease. 72
Inhaled NO has been used extensively for more than a decade in neonates and children for treatment of cardiorespiratory failure 106 and in adults for acute respiratory failure and pulmonary hypertension in critical care and perioperative settings.107 Inhaled NO in adults has reversed right ventricular dysfunction secondary to hypoxia and may alleviate other cardiopulmonary disorders, including idiopathic fibrosis. 11 In addition, S‐nitrosothiols are a new class of NO donor drugs, which include molecules derived from the reactions of endogenous NO with thiol groups; this class of drugs may avoid some of the drawbacks of the classic organic nitrates, and may enhance the anti‐inflammatory and antineoplastic functions of NO. 23
Hybrid NO Donors. New hybrid NO donors are being developed, including NO‐releasing nonsteroidal anti‐inflammatory drugs (NO‐NSAIDs), such as NO‐aspirin, NO‐naproxen, and NO‐ibuprofen. 16 These drugs are designed in part to avert the gastric injuries caused by NSAIDs; release of NO in animals has protected the stomach lining, perhaps by prevention of leukocyte adhesion on the endothelium, maintenance of mucosal blood flow, or stimulation of mucus secretion.16, 27, 101 Data from multiple animal studies suggest that NO‐NSAIDs provide enhanced efficacy and safety, compared with NSAIDs alone, in various inflammatory conditions, including rheumatoid arthritis, irritable bowel syndrome, colitis, and central nervous system inflammation. 16
NO Stimulants
Several therapies used to treat CV disease and hypertension have increased NO availability.100 These include antihypertensive drugs, such as β‐blockers, inhibitors of chronic renin‐angiotensin system activation, calcium channel blockers (CCBs), and 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase inhibitors (statins) and nonpharmacologic approaches. 100 , 108 , 109
β‐Blockers. The cardioselective β‐blocker nebivolol has demonstrated significant improvement in endo‐thelium/NO‐dependent vasodilation in healthy volunteers and patients with hypertension, compared with either placebo or atenolol, due to its ability to increase NO release (Figure 5). 110 , 111 The β‐blocker carvedilol may also improve endothelial function in high‐risk patients, possibly due to antioxidant activity. 112 , 113 Other β‐blockers that may improve endothelial/NO function, based on preliminary evidence, include bopindolol and celiprolol. 114 Interestingly, a clinical comparison study in 550 hypertensive patients found that nebivolol and celiprolol significantly reduced plasma levels of all 3 markers of thrombosis measured (homocystine, fibrinogen, and plasminogen activator inhibitor 1), thus suggesting promotion of antioxidant NO bioactivity, whereas carvedilol had no significant effect on any of these measures. 115
Figure 5.
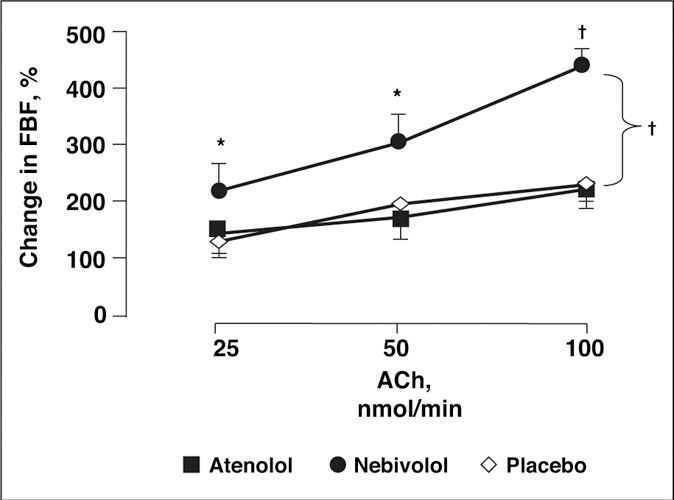
A double‐blind, randomized, 8‐week, crossover study in 12 hypertensive patients showed that nebivolol 5 mg + bendrofluazide 2.5 mg once daily significantly increased vasodilatory response to acetylcholine (Ach), as indicated by forearm blood flow (FBF), compared with baseline, while atenolol 50 mg + bendrofluazide 2.5 mg once daily and placebo had no effect on the vasodilatory response to Ach. *P<.05. †P<.001, compared with baseline. Reproduced with permission from Tzemos et al. 111
Renin‐Angiotensin System Modulating Drugs. Considerable data indicate that ACE inhibitors significantly improve endothelium‐dependent vasodilation and reverse endothelial dysfunction in normotensive patients and in hypertensive and CV disease patients (Figure 6). 100 , 116 , 117 , 118 , 119 In addition, ACE inhibitors have demonstrated improvement of endothelial function in high‐risk hypertensive patients with diabetes mellitus and nephropathy 120 , 121 and hypercholesterolemia and CHD. 122 , 123 These agents are hypothesized to promote NO bioactivity through preservation of bradykinin and stimulation of bradykinin β1 or β2 receptors, which promote NO release, and through antioxidant activity via suppression of angiotensin II, although the precise mechanisms of these effects remain unclear. 100 , 124 , 125
Figure 6.
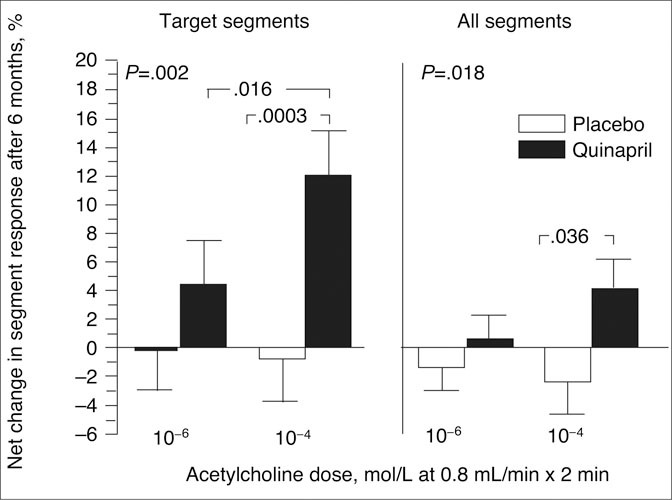
In the Trial on Reversing Endothelial Dysfunction (TREND), a double‐blind, randomized, placebo‐controlled trial, treatment with the angiotensin‐converting enzyme inhibitor quinapril for 6 months in 1 05 normotensive patients with coronary atherosclerosis significantly improved coronary artery vasomotor function as determined by the net percentage change in the acetylcholine‐provoked vasoconstriction response, compared with placebo, in target segments and in all segments. Reproduced with permission from Mancini et al. 116
Angiotensin II receptor blockers (ARBs) may also promote NO bioactivity, possibly through suppression of angiotensin II, leading to reduction of oxidative stress factors, and through activation of the angiotensin II type 2 receptor, which stimulates the bradykinin/β2 receptor to release NO. 126 , 127 In clinical studies, ARBs have demonstrated significant improvement of endothelial function and apparent promotion of NO bioactivity in patients with hypertension, hypercholesterolemia, and CV disease. 119 , 127 , 128 , 129 Other trials, however, have shown no effect of ARBs on endothelial function. 118 , 130 , 131
Calcium Channel Blockers. Substantial data also show that several CCBs may significantly improve endothelial function and restore NO bioactivity in various vascular beds, primarily through antioxidant activity, 100 , 132 , 133 , 134 although other studies have shown no improvement in endothelial function with CCBs in patients with hypertension and CV disease. 100 , 130 , 135
Statins. Statins have been shown to improve various indices of endothelial function, including increased coronary blood flow, reduced levels of adhesion molecules, and stimulation or restoration of eNOS. 136 , 137 In addition, statins inhibit the activity of angiotensin II and endothelin‐1, and reduce superoxide production and inflammatory processes associated with atherosclerosis, thus increasing NO in endothelial cells. 136 , 137 In clinical trials, several statins significantly improved endothelium‐dependent vasodilation in patients with hypercholesterolemia and other high‐risk populations. 138 , 139 , 140 , 141 Immunoassay studies found that statins up‐regulate the synthesis and bioactivity of endothelial NO through multiple pathways, including reduction of superoxide anion formation and decrease of caveolin‐1, a vascular inhibitor of eNOS activity (Figure 7). 137 , 142 , 143
Figure 7.
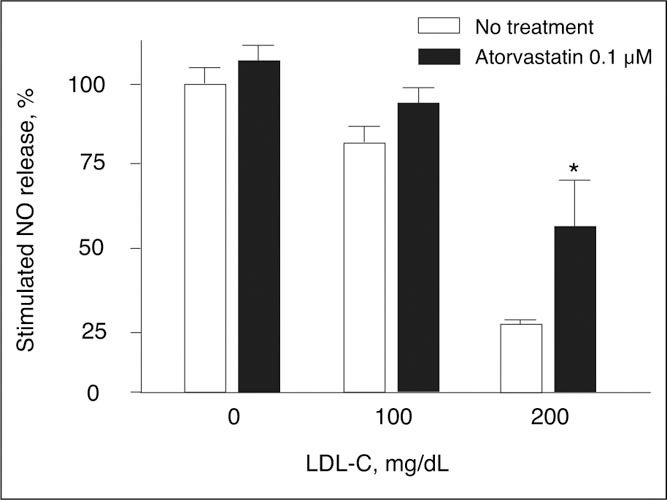
Three samples of intact endothelial cells were preincubated with or without low‐density lipoprotein cholesterol (LDL‐C), 100 mg/dL or 200 mg/dL, and the statin atorvastatin, 0.1 μmol/L, and then exposed to the calcium ionophore A23187, 5 μmol/L, a receptor‐independent agonist known to promote the binding of Ca2+‐activated calmodulin to endothelial nitric oxide synthase (eNOS). Exposure to LDL‐C without cotreatment with atorvastatin (open bars) substantially decreased nitric oxide (NO) release, while atorvastatin treatment (solid bars) significantly increased NO release from 5% in cells not exposed to LDL‐C to 17% and 107% in cells exposed to LDL‐C 100 mg/dL and 200 mg/dL, respectively. The greater effect of atorvastatin with higher exposure to LDL‐C is believed to derive from statin‐induced reduction of LDL‐C and caveolin‐1, which rises in concentration with increasing LDL‐C and potently inhibits eNOS function and NO release (see Figure 1 ). *P<.01 vs no treatment with atorvastatin. Reproduced with permission from Feron et al. 143
l‐Arginine. The sole substrate for NOS, l‐arginine, is an amino acid produced endogenously but mainly derived in humans through common dietary sources and as an oral supplement.144 Clinical studies have been inconsistent. A review of 17 small clinical studies of oral arginine supplement treatment found 12 studies showing reduced thrombotic activity and improved endothelium/ NO‐dependent vasodilation and 5 studies showing no CV benefit. 145 Two studies in women with pre‐eclampsia showed that l‐arginine treatment significantly reduced BP, possibly through increased NO bioactivity. 146 , 147 Another clinical study, however, found that oral l‐arginine treatment for 2 days did not reduce diastolic BP in preeclamptic women. 148
Nonpharmacologic Stimulants of NO
Several nonpharmacologic therapies used in the prevention of CV disease may promote increased NO production and release. Moderate and intense exercise significantly improved endothelium/NO‐dependent vasodilation in healthy and hypertensive subjects 149 , 150 and in patients with stable CHD. 151 In addition, studies have found that treatment with red wine from France in human endothelial cells in vitro resulted in significant increases in eNOS expression and NO release. 152 , 153 Incubation of platelets with purple grape juice and purple grape juice‐derived flavonoids‐substances that may account for the effects of red wine on NO‐also decreased platelet aggregation, increased platelet‐derived NO, and decreased superoxide production. 154 Other sources of anti‐oxidant flavonoids, such as black tea and cocoa, also improved endothelial function. 155 , 156 Ethanol also inhibits the activity of iNOS, an effect that may explain some of the CV benefits of moderate alcohol consumption. 157
Some studies suggest that nutritional supplements such as vitamins C and E, omega‐3 fatty acids, or folate may improve endothelial and NO function in patients with CV risk factors or disease, although these data are generally inconsistent. 109 A low‐calorie diet was also shown to significantly improve endothelium/NO‐dependent vasodilation in obese patients with hypertension. 158
Conclusions
NO is a signaling molecule fundamental to a wide array of physiologic processes. Impaired NO bio‐activity has been strongly associated with endothelial dysfunction and CV disease and is implicated to varying degrees in multiple disorders ranging from osteoporosis to erectile dysfunction. Various approaches to the therapeutic modulation of NO have been investigated, including the use of NO donors, antihypertensive agents, statins, l‐arginine, and nonpharmacologic CV therapies. Many of these therapies appear to significantly improve NO bioactivity, suggesting potential avenues for treatment. Data from the recent A‐HeFT trial showed that use of NO‐donor therapy significantly reduced morbidity and mortality in black patients with heart failure, providing the first substantial evidence that NO modulation may affect CV outcomes. Further data on the effects of NO modulation on outcomes in a range of disorders should help clarify the usefulness of this therapeutic approach.
Rererences
- 1. Stuart‐Smith K. Demystified…nitric oxide. Mol Pathol. 2002;55:360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moncada S, Higgs A. The L‐arginine‐nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. [DOI] [PubMed] [Google Scholar]
- 3. Brunner H, Cockcroft JR, Deanfield J, et al. Endothelial function and dysfunction. Part II: association with cardiovascular risk factors and diseases. A statement by the Working Group on Endothelins and Endothelial Factors of the European Society of Hypertension. J Hypertens. 2005;23:233–246. [DOI] [PubMed] [Google Scholar]
- 4. Tuteja N, Chandra M, Tuteja R, et al. Nitric oxide as a unique bioactive signaling messenger in physiology and pathophysiology. J Biomed Biotechnol. 2004;2004:227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ricciardolo FL. Multiple roles of nitric oxide in the air ways. Thorax. 2003;58:175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ralston SH. The Michael Mason Prize essay 1997. Nitric oxide and bone: what a gas! Br J Rheumatol. 1997;36:831–838. [DOI] [PubMed] [Google Scholar]
- 7. Spieker LE, Sudano I, Hürlmann D, et al. High‐density lipoprotein restores endothelial function in hypercholesterolemicmen. Circulation. 2002;105:1399–1402. [DOI] [PubMed] [Google Scholar]
- 8. Maiorana A, O'Driscoll G, Taylor R, et al. Exercise and the nitric oxide vasodilator system. Sports Med. 2003;33:1013–1035. [DOI] [PubMed] [Google Scholar]
- 9. Behrendt D, Ganz P. Endothelial function: from vascular biology to clinical applications. Am J Cardiol. 2002;90(suppl 3):40L–48L. [DOI] [PubMed] [Google Scholar]
- 10. Kawashima S, Yokoyama M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:998–1005. [DOI] [PubMed] [Google Scholar]
- 11. Hart CM. Nitric oxide in adult lung disease. Chest. 1999;115:1407–1417. [DOI] [PubMed] [Google Scholar]
- 12. Toda N, Ayajiki K, Okamura T. Nitric oxide and penile erectile function. Pharmacol Ther. 2005;106:233–266. [DOI] [PubMed] [Google Scholar]
- 13. Buhimschi IA, Saade GR, Chwalisz K, et al. The nitric oxide pathway in pre‐eclampsia: pathophysiological implications. Hum Reprod Update. 1998;4:25–42. [DOI] [PubMed] [Google Scholar]
- 14. Scherrer U, Sartori C. Defective nitric oxide synthesis: a link between metabolic insulin resistance, sympathetic overactivity and cardiovascular morbidity. Eur J Endocrinol. 2000;142:315–323. [DOI] [PubMed] [Google Scholar]
- 15. Contestabile A, Ciani E. Role of nitric oxide in the regulation of neuronal proliferation, survival and differentiation. Neurochem Int. 2004;45:903–914. [DOI] [PubMed] [Google Scholar]
- 16. Keeble JE, Moore PK. Pharmacology and potential therapeutic applications of nitric oxide‐releasing non‐steroidal anti‐inflammatory and related nitric oxide‐donating drugs. Br J Pharmacol. 2002;137:295–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grant MK, El‐Fakahany EE. Therapeutic interventions targeting the nitric oxide system: current and potential uses in obstetrics, bone disease and erectile dysfunction. Life Sci. 2004;74:1701–1721. [DOI] [PubMed] [Google Scholar]
- 18. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. [DOI] [PubMed] [Google Scholar]
- 19. Ignarro LJ, Buga GM, Wood KS, et al. Endothelium‐derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA. 1987;84:9265–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palmer RM, Ferrige AC, Moncada S. Nitric oxide release accounts for the biological activity of endothelium‐derived relaxing factor. Nature. 1987;327:524–526. [DOI] [PubMed] [Google Scholar]
- 21. Cylwik D, Mogielnicki A, Buczko W. L‐arginine and cardiovascular system. Pharmacol Rep. 2005;57:14–22. [PubMed] [Google Scholar]
- 22. Togashi H, Sasaki M, Frohman E, et al. Neuronal (type I) nitric oxide synthase regulates nuclear factor κB activity and immunologic (type II) nitric oxide synthase expression. Proc Natl Acad Sci USA. 1997;94:2676–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scatena R, Bottoni P, Martorana GE, et al. Nitric oxide donor drugs: an update on pathophysiology and therapeutic potential. Expert Opin Investig Drugs. 2005;14:835–846. [DOI] [PubMed] [Google Scholar]
- 24. Gow AJ, Luchsinger BP, Pawloski JR, et al. The oxyhemoglobin reaction of nitric oxide. Proc Natl Acad Sci USA. 1999;96:9027–9032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622–626. [DOI] [PubMed] [Google Scholar]
- 26. Dreyer J, Schleicher M, Tappe A, et al. Nitric oxide synthase (NOS)‐interacting protein interacts with neuronal NOS and regulates its distribution and activity. J Neurosci. 2004;24:10454–10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Esplugues JV, Barrachina MD, Beltrán B, et al. Inhibition of gastric acid secretion by stress: a protective reflex mediated by cerebral nitric oxide. Proc Natl Acad Sci USA. 1996;93:14839–14844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo Y, Ward ME, Beasjours S, et al. Regulation of cerebellar nitric oxide production in response to prolonged in vivo hypoxia. J Neurosci Res. 1997;49:89–97. [PubMed] [Google Scholar]
- 29. Saitoh F, Tian QB, Okano A, et al. NIDD, a novel DHHCcontaining protein, targets neuronal nitric‐oxide synthase (nNOS) to the synaptic membrane through a PDZ‐dependent interaction and regulates nNOS activity. J Biol Chem. 2004;279:29461–29468. [DOI] [PubMed] [Google Scholar]
- 30. Beltrán B, Barrachina MD, Méndez A, et al. Synthesis of nitric oxide in the dorsal motor nucleus of the vagus mediates the inhibition of gastric acid secretion by central bombesin. Br J Pharmacol. 1999;127:1603–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beck KF, Eberhardt W, Frank S, et al. Inducible NO synthase: role in cellular signalling. J Exp Biol. 1999;202:645–653. [DOI] [PubMed] [Google Scholar]
- 32. Ferrini MG, Davila HH, Valente EG, et al. Aging‐related induction of inducible nitric oxide synthase is vasculoprotective to the arterial media. Cardiovasc Res. 2004;61:796–805. [DOI] [PubMed] [Google Scholar]
- 33. Taylor AL, Ziesche S, Yancy C, et al., for the African‐American Heart Failure Trial Investigators . Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med. 2004;351:2049–2057. [DOI] [PubMed] [Google Scholar]
- 34. Boje KM. Nitric oxide neurotoxicity in neurodegenerative diseases. Front Biosci. 2004;9:763–776. [DOI] [PubMed] [Google Scholar]
- 35. Wang Y‐Z, Cao Y‐Q, Wu J‐N, et al. Expression of nitric oxide synthase in human gastric carcinoma and its relation to p53, PCNA. World J Gastroenterol. 2005;11:46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wilcox JN, Subramanian RR, Sundell CL, et al. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler Thromb Vasc Biol. 1997;17:2479–2488. [DOI] [PubMed] [Google Scholar]
- 37. Gibbons GH. Endothelial function as a determinant of vascular function and structure: a new therapeutic target. Am J Cardiol. 1997;79:3–8. [DOI] [PubMed] [Google Scholar]
- 38. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–III32. [DOI] [PubMed] [Google Scholar]
- 39. Raij L. Hypertension and cardiovascular risk factors: role of the angiotensin II‐nitric oxide interaction. Hypertension. 2001;37:767–773. [DOI] [PubMed] [Google Scholar]
- 40. Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 41. Ichiki T, Usui M, Kato M, et al. Downregulation of angiotensin II type 1 receptor gene transcription by nitric oxide. Hypertension. 1998;31:342–348. [DOI] [PubMed] [Google Scholar]
- 42. Beltrán B, Mathur A, Duchen MR, et al. The effect of nitric oxide on cell respiration: a key to understanding its role in cell survival or death. Proc Natl Acad Sci USA. 2000;97:14602–14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Panza JA. Endothelial dysfunction in essential hypertension. Clin Cardiol. 1997;20(suppl II):II26–II33. [PubMed] [Google Scholar]
- 44. Perticone F, Ceravolo R, Pujia A, et al. Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation. 2001;104:191–196. [DOI] [PubMed] [Google Scholar]
- 45. Wilkinson IB, Franklin SS, Cockcroft JR. Nitric oxide and the regulation of large artery stiffness: from physiology to pharmacology. Hypertension. 2004;44:112–116. [DOI] [PubMed] [Google Scholar]
- 46. Paulus WJ, Bronzwaer JG. Nitric oxide's role in the heart: control of beating or breathing? Am J Physiol Heart Circ Physiol. 2004;287:H8–H13. [DOI] [PubMed] [Google Scholar]
- 47. Chowdhary S, Townend JN. Nitric oxide and hypertension: not just an endothelium derived relaxing factor! J Hum Hypertens. 2001;15:219–227. [DOI] [PubMed] [Google Scholar]
- 48. Dias AC, Vitela M, Colombari E, et al. Nitric oxide modulation of glutamatergic, baroreflex, and cardiopulmonary transmission in the nucleus of the solitary tract. Am J Physiol Heart Circ Physiol. 2005;288:H256–H262. [DOI] [PubMed] [Google Scholar]
- 49. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. [DOI] [PubMed] [Google Scholar]
- 50. Felmeden DC, Spencer CG, Belgore FM, et al. Endothelial damage and angiogenesis in hypertensive patients: relationship to cardiovascular risk factors and risk factor management. Am J Hypertens. 2003;16:11–20. [DOI] [PubMed] [Google Scholar]
- 51. Al‐Suwaidi J, Hamasaki S, Higano ST, et al. Long‐term follow‐up of patients with mild coronary artery disease and endothelial dysfunction. Circulation. 2000;101:948–954. [DOI] [PubMed] [Google Scholar]
- 52. Heitzer T, Schlinzig T, Krohn K, et al. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104:2673–2678. [DOI] [PubMed] [Google Scholar]
- 53. Cohn JN. Arterial compliance to stratify cardiovascular risk: more precision in therapeutic decision making. Am J Hypertens. 2001;14(suppl 1):258S–263S. [DOI] [PubMed] [Google Scholar]
- 54. Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis? Circulation. 2004;109(suppl II):II27–II33. [DOI] [PubMed] [Google Scholar]
- 55. Oemar BS, Tschudi MR, Godoy N, et al. Reduced endothelial nitric oxide synthase expression and production in human atherosclerosis. Circulation. 1998;97:2494–2498. [DOI] [PubMed] [Google Scholar]
- 56. Higashi Y, Sasaki S, Nakagawa K, et al. Endothelial function and oxidative stress in renovascular hypertension. N Engl J Med. 2002;346:1954–1962. [DOI] [PubMed] [Google Scholar]
- 57. Panza JA, Quyyumi AA, Brush JE Jr, et al. Abnormal endothelium‐dependent vascular relaxation in patients with essential hypertension. N Engl J Med. 1990;323:22–27. [DOI] [PubMed] [Google Scholar]
- 58. Panza JA, Casino PR, Kilcoyne CM, et al. Role of endothelium‐derived nitric oxide in the abnormal endothelium‐dependent vascular relaxation of patients with essential hypertension. Circulation. 1993;87:1468–1474. [DOI] [PubMed] [Google Scholar]
- 59. Haynes WG, Noon JP, Walker BR, et al. Inhibition of nitric oxide synthesis increases blood pressure in healthy humans. J Hypertens. 1993;11:1375–1380. [DOI] [PubMed] [Google Scholar]
- 60. Deanfield J, Donald A, Ferri C, et al., for the Working Group on Endothelin and Endothelial Factors of the European Society of Hypertension . Endothelial function and dysfunction. Part I: methodological issues for assessment in the different vascular beds: a statement by the Working Group on Endothelin and Endothelial Factors of the European Society of Hypertension. J Hypertens. 2005;23:7–17. [DOI] [PubMed] [Google Scholar]
- 61. Kinlay S, Creager MA, Fukumoto M, et al. Endothelium‐derived nitric oxide regulates arterial elasticity in human arteries in vivo. Hypertension. 2001;38:1049–1053. [DOI] [PubMed] [Google Scholar]
- 62. Freedman JE, Ting B, Hankin B, et al. Impaired platelet production of nitric oxide predicts presence of acute coronary syndromes. Circulation. 1998;98:1481–1486. [DOI] [PubMed] [Google Scholar]
- 63. Fichtlscherer S, Breuer S, Schächinger V, et al. C‐reactive protein levels determine systemic nitric oxide bioavailability in patients with coronary artery disease. Eur Heart J. 2004;25:1412–1418. [DOI] [PubMed] [Google Scholar]
- 64. Henningsson R, Salehi A, Lundquist I. Role of nitric oxide synthase isoforms in glucose‐stimulated insulin release. Am J Physiol Cell Physiol. 2002;283:C296–C304. [DOI] [PubMed] [Google Scholar]
- 65. Smukler SR, Tang L, Wheeler MB, et al. Exogenous nitric oxide and endogenous glucose‐stimulated β‐cell nitric oxide augment insulin release. Diabetes. 2002;51:3450–3460. [DOI] [PubMed] [Google Scholar]
- 66. Petrie JR, Ueda S, Webb DJ, et al. Endothelial nitric oxide production and insulin sensitivity: a physiological link with implications for pathogenesis of cardiovascular disease. Circulation. 1996;93:1331–1333. [DOI] [PubMed] [Google Scholar]
- 67. Lajoix A‐D, Reggio H, Chardés T, et al. A neuronal isoform of nitric oxide synthase expressed in pancreatic β‐cells controls insulin secretion. Diabetes. 2001;50:1311–1323. [DOI] [PubMed] [Google Scholar]
- 68. Cleland SJ, Petrie JR, Small M, et al. Insulin action is associated with endothelial function in hypertension and type 2 diabetes. Hypertension. 2000;35:507–511. [DOI] [PubMed] [Google Scholar]
- 69. Calver A, Collier J, Vallance P. Inhibition and stimulation of nitric oxide synthesis in the human forearm arterial bed of patients with insulin‐dependent diabetes. J Clin Invest. 1992;90:2548–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cook S, Hugli O, Egli M, et al. Partial gene deletion of endothelial nitric oxide synthase predisposes to exaggerated high‐fat diet‐induced insulin resistance and arterial hypertension. Diabetes. 2004;53:2067–2072. [DOI] [PubMed] [Google Scholar]
- 71. Chien W‐L, Liang K‐C, Teng C‐M, et al. Enhancement of learning behaviour by a potent nitric oxide‐guanylate cyclase activator YC‐1. Eur J Neurosci. 2005;21:1679–1688. [DOI] [PubMed] [Google Scholar]
- 72. Thatcher GR, Bennett BM, Dringenberg HC, et al. Novel nitrates as NO mimetics directed at Alzheimer's disease. J Alzheimers Dis. 2004;6:S75–S84. [DOI] [PubMed] [Google Scholar]
- 73. Rauhala P, Andoh T, Chiueh CC. Neuroprotective properties of nitric oxide and S‐nitrosoglutathione. Toxicol Appl Pharmacol. 2005;207(suppl 2):91–95. [DOI] [PubMed] [Google Scholar]
- 74. De La Torre JC, Aliev G. Inhibition of vascular nitric oxide after rat chronic brain hypoperfusion: spatial memory and immunocytochemical changes. J Cereb Blood Flow Metab. 2005;25:663–672. [DOI] [PubMed] [Google Scholar]
- 75. Grozdanovic Z. NO message from muscle. Microsc Res Tech. 2001;55:148–153. [DOI] [PubMed] [Google Scholar]
- 76. Červenka L, Kramer HJ, Malý J, et al. Role of nNOS in regulation of renal function in hypertensive Ren‐2 transgenic rats. Physiol Res. 2002;52:571–580. [PubMed] [Google Scholar]
- 77. Burnett AL. Role of nitric oxide in the physiology of erection. Biol Reprod. 1995;52:485–489. [DOI] [PubMed] [Google Scholar]
- 78. Andersson KE. Pharmacology of penile erection. Pharmacol Rev. 2001;53:417–450. [PubMed] [Google Scholar]
- 79. Garban H, Vernet D, Freedman A, et al. Effect of aging on nitric oxide‐mediated penile erection in rats. Am J Physiol. 1995;268:H467–H475. [DOI] [PubMed] [Google Scholar]
- 80. Khan MA, Thompson CS, Jeremy JY, et al. The effect of superoxide dismutase on nitric oxide‐mediated and electrical field‐stimulated diabetic rabbit cavernosal smooth muscle relaxation. BJU Int. 2001;87:98–103. [DOI] [PubMed] [Google Scholar]
- 81. Virag R, Floresco J, Richard C. Impairment of shear‐stress‐mediated vasodilation of cavernous arteries in erectile dysfunction. Int J Impot Res. 2004;16:39–42. [DOI] [PubMed] [Google Scholar]
- 82. Kaya C, Uslu Z, Karaman I. Is endothelial function impaired in erectile dysfunction patients? Int J Impot Res. 2006;18:55–60. [DOI] [PubMed] [Google Scholar]
- 83. Sladek SM, Magness RR, Conrad KP. Nitric oxide and pregnancy. Am J Physiol. 1997;272:R441–R463. [DOI] [PubMed] [Google Scholar]
- 84. Ali M, Buhimschi I, Chwalisz K, et al. Changes in expression of the nitric oxide synthase isoforms in rat uterus and cervix during pregnancy and parturition. Mol Hum Reprod. 1997;3:995–1003. [DOI] [PubMed] [Google Scholar]
- 85. Norman JE, Thomson AJ, Telfer JF, et al. Myometrial constitutive nitric oxide synthase expression is increased during human pregnancy. Mol Hum Reprod. 1999;5:175–181. [DOI] [PubMed] [Google Scholar]
- 86. Bansal RK, Goldsmith PC, He Y, et al. A decline in myometrial nitric oxide synthase expression is associated with labor and delivery. J Clin Invest. 1997;99:2502–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Suzuki Y, Kajikuri J, Suzumori K, et al. Mechanisms underlying the reduced endothelium‐dependent relaxation in human omental resistance artery in pre‐eclampsia. J Physiol. 2000;527(pt 1):163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cockell AP, Poston L. Flow‐mediated vasodilatation is enhanced in normal pregnancy but reduced in preeclampsia. Hypertension. 1997;30:247–251. [DOI] [PubMed] [Google Scholar]
- 89. Pascoal IF, Lindheimer MD, Nalbantian‐Brandt C, et al. Preeclampsia selectively impairs endothelium‐dependent relaxation and leads to oscillatory activity in small omental arteries. J Clin Invest. 1998;101:464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yamamoto T, Susuki Y, Kojima K, et al. Reduced flow‐mediated vasodilation is not due to a decrease in production of nitric oxide in preeclampsia. Am J Obstet Gynecol. 2005;192:558–563. [DOI] [PubMed] [Google Scholar]
- 91. Savvidou MD, Hingorani AD, Tsikas D, et al. Endothelial dysfunction and raised plasma concentrations of asymmetric dimethylarginine in pregnant women who subsequently develop pre‐eclampsia. Lancet. 2003;361:1511–1517. [DOI] [PubMed] [Google Scholar]
- 92. Anumba DO, Ford GA, Boys RJ, et al. Stimulated nitric oxide release and nitric oxide sensitivity in forearm arterial vasculature during normotensive and preeclamptic pregnancy. Am J Obstet Gynecol. 1999;181:1479–1484. [DOI] [PubMed] [Google Scholar]
- 93. Diejomaoh FM, Omu AE, Al‐Busiri N, et al. Nitric oxide production is not altered in preeclampsia. Arch Gynecol Obstet. 2004;269:237–243. [DOI] [PubMed] [Google Scholar]
- 94. Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–221. [DOI] [PubMed] [Google Scholar]
- 95. Dinh‐Xuan AT, Higenbottam TW, Clelland CA, et al. Impairment of endothelium‐dependent artery relaxation in chronic obstructive lung disease. N Engl J Med. 1991;324:1539–1547. [DOI] [PubMed] [Google Scholar]
- 96. Barberà JA, Peinado VI, Santos S, et al. Reduced expression of endothelial nitric oxide synthase in pulmonary arteries of smokers. Am J Respir Crit Care Med. 2001;164:709–713. [DOI] [PubMed] [Google Scholar]
- 97. Das UN. Nitric oxide as the mediator of the antiosteoporotic actions of estrogen, statins, and essential fatty acids. Exp Biol Med (Maywood). 2002;227:88–93. [DOI] [PubMed] [Google Scholar]
- 98. Evans DM, Ralston SH. Nitric oxide and bone. J Bone Miner Res. 1996;11:300–305. [DOI] [PubMed] [Google Scholar]
- 99. Kasten TP, Collin‐Osdoby P, Patel N, et al. Potentiation of osteoclast bone‐resorption activity by inhibition of nitric oxide synthase. Proc Natl Acad Sci USA. 1994;91:3569–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Taddei S, Virdis A, Ghiadoni L, et al. Effects of anti‐hypertensive drugs on endothelial dysfunction. Drugs. 2002;62:265–284. [DOI] [PubMed] [Google Scholar]
- 101. Muscará MN, Wallace JL. Nitric oxide: V. Therapeutic potential of nitric oxide donors and inhibitors. Am J Physiol. 1999;276:G1313–G1316. [DOI] [PubMed] [Google Scholar]
- 102. Markham A, Plosker GL, Goa KL. Nicorandil. An updated review of its use in ischaemic heart disease with emphasis on its cardioprotective effects. Drugs. 2000;60:955–974. [DOI] [PubMed] [Google Scholar]
- 103. IONA Study Group. Effect of nicorandil on coronary events in patients with stable angina: the Impact Of Nicorandil in Angina (IONA) randomised trial. Lancet. 2002;359:1269–1275. [DOI] [PubMed] [Google Scholar]
- 104. Jamal SA, Browner WS, Bauer DC, et al. Intermittent use of nitrates increases bone mineral density: the Study of Osteoporotic Fractures. J Bone Miner Res. 1998;13:1755–1759. [DOI] [PubMed] [Google Scholar]
- 105. Wimalawansa SJ. Nitrogylcerin therapy is as efficacious as standard estrogen replacement therapy (Premarin) in pre vention of oophorectomy‐induced bone loss: a human pilot clinical study. J Bone Miner Res. 2000;15:2240–2244. [DOI] [PubMed] [Google Scholar]
- 106. Macrae DJ, Field D, Mercier JC, et al. Inhaled nitric oxide therapy in neonates and children: reaching a European consensus. Intensive Care Med. 2004;30:372–380. [DOI] [PubMed] [Google Scholar]
- 107. Germann P, Braschi A, Della Rocca G, et al. Inhaled nitric oxide therapy in adults: European expert recommendations. Intensive Care Med. 2005;31:1029–1041. [DOI] [PubMed] [Google Scholar]
- 108. Thuillez C, Richard V. Targeting endothelial dysfunction in hypertensive subjects. J Hum Hypertens. 2005;19(suppl 1):S21–S25. [DOI] [PubMed] [Google Scholar]
- 109. Brown AA, Hu FB. Dietary modulation of endothelial function: implications for cardiovascular disease. Am J Clin Nutr. 2001;73:673–686. [DOI] [PubMed] [Google Scholar]
- 110. Cockcroft JR, Chowienczyk PJ, Brett SE, et al. Nebivolol vasodilates human forearm vasculature: evidence for an L‐arginine/NO‐dependent mechanism. J Pharmacol Exp Ther. 1995;274:1067–1071. [PubMed] [Google Scholar]
- 111. Tzemos N, Lim PO, MacDonald TM. Nebivolol reverses endothelial dysfunction in essential hypertension: a randomized, double‐blind, crossover study. Circulation. 2001;104:511–514. [DOI] [PubMed] [Google Scholar]
- 112. Matsuda Y, Akita H, Terashima M, et al. Carvedilol improves endothelium‐dependent dilatation in patients with coronary artery disease. Am Heart J. 2000;140:753–759. [DOI] [PubMed] [Google Scholar]
- 113. Giugliano D, Marfella R, Acampora R, et al. Effects of perindopril and carvedilol on endothelium‐dependent vascular functions in patients with diabetes and hypertension. Diabetes Care. 1998;21:631–636. [DOI] [PubMed] [Google Scholar]
- 114. Kakoki M, Hirata Y, Hayakawa H, et al. Effects of vasodilatory β‐adrenoreceptor antagonists on endothelium‐derived nitric oxide release in rat kidney. Hypertension. 1999;33:467–471. [DOI] [PubMed] [Google Scholar]
- 115. Vyssoulis GP, Marinakis AG, Aznaouridis KA, et al. The impact of third‐generation beta‐blocker antihypertensive treatment on endothelial function and the prothrombotic state: effects of smoking. Am J Hypertens. 2004;17:582–589. [DOI] [PubMed] [Google Scholar]
- 116. Mancini GB, Henry GC, Macaya C, et al. Angiotensin‐converting enzyme inhibition with quinapril improves endothelial vasomotor dysfunction in patients with coronary artery disease. Circulation. 1996;94:258–265. [DOI] [PubMed] [Google Scholar]
- 117. Higashi Y, Oshima T, Sasaki S, et al. Angiotensin‐converting enzyme inhibition, but not calcium antagonism, improves a response of the renal vasculature to L‐arginine in patients with essential hypertension. Hypertension. 1998;32:16–24. [DOI] [PubMed] [Google Scholar]
- 118. Ghiadoni L, Magagna A, Versari D, et al. Different effect of antihypertensive drugs on conduit artery endothelial function. Hypertension. 2003;41:1281–1286. [DOI] [PubMed] [Google Scholar]
- 119. Hornig B, Landmesser U, Kohler C, et al. Comparative effect of ACE inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease: role of superoxide dis‐mutase. Circulation. 2001;103:799–805. [DOI] [PubMed] [Google Scholar]
- 120. O'Driscoll G, Green D, Maiorana A, et al. Improvement in endothelial function by angiotensin‐converting enzyme inhibition in non‐insulin‐dependent diabetes mellitus. J Am Coll Cardiol. 1999;33:1506–1511. [DOI] [PubMed] [Google Scholar]
- 121. Hermann TS, Li W, Dominguez H, et al. Quinapril treatment increases insulin‐stimulated endothelial function and adiponectin gene expression in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91:1001–1008. [DOI] [PubMed] [Google Scholar]
- 122. Esper RJ, Machado R, Vilariño J, et al. Endothelium‐dependent responses in patients with hypercholesterolemic coronary artery disease under the effects of simvastatin and enalapril, either separately or combined. Am Heart J. 2000;140:684–689. [DOI] [PubMed] [Google Scholar]
- 123. Lee AF, Dick JB, Bonnar CE, et al. Lisinopril improves arterial function in hyperlipidemia. Clin Sci (Lond). 1999;96:441–448. [PubMed] [Google Scholar]
- 124. Münzel T, Keaney JF Jr. Are ACE inhibitors a “magic bullet” against oxidative stress? Circulation. 2001;104:1571–1574. [DOI] [PubMed] [Google Scholar]
- 125. Ignjatovic T, Stanisavljevic S, Brovkovych V, et al. Kinin B1 receptors stimulate nitric oxide production in endothelial cells: signaling pathways activated by angiotensin‐1 converting enzyme inhibitors and peptide ligands. Mol Pharmacol. 2004;66:1310–1316. [DOI] [PubMed] [Google Scholar]
- 126. Landmesser U, Drexler H. Effect of angiotensin II type 1 receptor antagonism on endothelial function: role of bradykinin and nitric oxide. J Hypertens Suppl. 2006;24: S39–S43. [DOI] [PubMed] [Google Scholar]
- 127. Hornig B, Kohler C, Schlink D, et al. AT1‐receptor antagonism improves endothelial function in coronary artery dis ease by a bradykinin/B2‐receptor‐dependent mechanism. Hypertension. 2003;41:1092–1095. [DOI] [PubMed] [Google Scholar]
- 128. Ghiadoni L, Virdis A, Magagna A, et al. Effect of the angiotensin II type 1 receptor blocker candesartan on endothelial function in patients with essential hypertension. Hypertension. 2000;35(pt 2):501–506. [DOI] [PubMed] [Google Scholar]
- 129. Wassmann S, Hilgers S, Laufs U, et al. Angiotensin II type 1 receptor antagonism improves hypercholesterolemia‐associated endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2002;22:1208–1212. [DOI] [PubMed] [Google Scholar]
- 130. Anderson TJ, Elstein E, Haber H, et al. Comparative study of ACE‐inhibition, angiotensin II antagonism, and calcium channel blockade on flow‐mediated vasodilation in patients with coronary artery disease (BANFF study). J Am Coll Cardiol. 2000;35:60–66. [DOI] [PubMed] [Google Scholar]
- 131. Browne DL, Meeking DR, Allard SE, et al. Angiotensin II does not affect endothelial tone in type 1 diabetes—results of a double‐blind placebo controlled trial. Diabet Med. 2006;23:53–59. [DOI] [PubMed] [Google Scholar]
- 132. The ENCORE Investigators. Effect of nifedipine and cerivastatin on coronary endothelial function in patients with coronary artery disease. The ENCORE I Study (Evaluation of Nifedipine and Cerivastatin On Recovery of Coronary Endothelial function). Circulation. 2003;107:422–428. [DOI] [PubMed] [Google Scholar]
- 133. Taddei S, Virdis A, Ghiadoni L, et al. Calcium antagonist treatment by lercanidipine prevents hyperpolarization in essential hypertension. Hypertension. 2003;41:950–955. [DOI] [PubMed] [Google Scholar]
- 134. Verhaar MC, Honing ML, Van Dam T, et al. Nifedipine improves endothelial function in hypercholesterolemia, independently of an effect on blood pressure or plasma lipids. Cardiovasc Res. 1999;42:752–760. [DOI] [PubMed] [Google Scholar]
- 135. Higashi Y, Sasaki S, Nakagawa K, et al. A comparison of angiotensin‐converting enzyme inhibitors, calcium antagonists, beta‐blockers and diuretic agents on reactive hyperemia in patients with essential hypertension: a multicenter study. J Am Coll Cardiol. 2000;35:284–291. [DOI] [PubMed] [Google Scholar]
- 136. Wolfrum S, Jensen KS, Liao JK. Endothelium‐dependent effects of statins. Arterioscler Thromb Vasc Biol. 2003;23:729–736. [DOI] [PubMed] [Google Scholar]
- 137. Laufs U, La Fata V, Plutzky J, et al. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–1135. [DOI] [PubMed] [Google Scholar]
- 138. Perticone F, Ceravolo R, Maio R, et al. Effects of atorvastatin and vitamin C on endothelial function of hypercho lesterolemic patients. Atherosclerosis. 2000;152:511–518. [DOI] [PubMed] [Google Scholar]
- 139. Dogra GK, Watts GF, Chan DC, et al. Statin therapy improves brachial artery vasodilator function in patients with type 1 diabetes and microalbuminuria. Diabet Med. 2005;22:239–242. [DOI] [PubMed] [Google Scholar]
- 140. Beckman JA, Liao JK, Hurley S, et al. Atorvastatin restores endothelial function in normocholesterolemic smokers independent of changes in low‐density lipoprotein. Circ Res. 2004;95:217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Anderson TJ, Meredith IT, Yeung AC, et al. The effect of cholesterol‐lowering and antioxidant therapy on endothelium‐dependent coronary vasomotion. N Engl J Med. 1995;332:488–493. [DOI] [PubMed] [Google Scholar]
- 142. Mason RP, Walter MF, Jacob RF. Effects of HMG‐CoA reductase inhibitors on endothelial function: role of micro‐domains and oxidative stress. Circulation. 2004;109(suppl II):II34–II41. [DOI] [PubMed] [Google Scholar]
- 143. Feron O, Dessy C, Desager J‐P, et al. Hydroxy‐methylglutaryl‐coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation. 2001;103:113–118. [DOI] [PubMed] [Google Scholar]
- 144. Appleton J. Arginine: clinical potential of a semi‐essential amino acid. Altern Med Rev. 2002;7:512–522. [PubMed] [Google Scholar]
- 145. Preli RB, Klein KP, Herrington DM. Vascular effects of dietary L‐arginine supplementation. Atherosclerosis. 2002;162:1–15. [DOI] [PubMed] [Google Scholar]
- 146. Rytlewski K, Olszanecki R, Korbut R, et al. Effects of prolonged oral supplementation with L‐arginine on blood pressure and nitric oxide synthesis in preeclampsia. Eur J Clin Invest. 2005;35:32–37. [DOI] [PubMed] [Google Scholar]
- 147. Facchinetti F, Longo M, Piccinini F, et al. L‐arginine infusion reduces blood pressure in preeclamptic women through nitric oxide release. J Soc Gynecol Investig. 1999;6:202–207. [DOI] [PubMed] [Google Scholar]
- 148. Staff AC, Berge L, Haugen G, et al. Dietary supplementation with L‐arginine or placebo in women with pre‐eclampsia. Acta Obstet Gynecol Scand. 2004;83:103–107. [PubMed] [Google Scholar]
- 149. Higashi Y, Sasaki S, Kurisu S, et al. Regular aerobic exercise augments endothelium‐dependent vascular relaxation in normotensive as well as hypertensive subjects: role of endothelium‐derived nitric oxide. Circulation. 1999;100:1194–1202. [DOI] [PubMed] [Google Scholar]
- 150. Goto C, Higashi Y, Kimura M, et al. Effect of different intensities of exercise on endothelium‐dependent vasodilation in humans: role of endothelium‐dependent nitric oxide and oxidative stress. Circulation. 2003;108:530–535. [DOI] [PubMed] [Google Scholar]
- 151. Hambrecht R, Adams V, Erbs S, et al. Regular physical activity improves endothelial function in patients with coronary artery disease by increasing phosphorylation of endothelial nitric oxide synthase. Circulation. 2003;107:3152–3158. [DOI] [PubMed] [Google Scholar]
- 152. Leikert JF, Räthel TR, Wohlfart P, et al. Red wine polyphenols enhance endothelial nitric oxide synthase expression and subsequent nitric oxide release from endothelial cells. Circulation. 2002;106:1614–1617. [DOI] [PubMed] [Google Scholar]
- 153. Wallerath T, Poleo D, Li H, et al. Red wine increases the expression of human endothelial nitric oxide synthase: a mechanism that may contribute to its beneficial cardiovascular effects. J Am Coll Cardiol. 2003;41:471–478. [DOI] [PubMed] [Google Scholar]
- 154. Freedman JE, Parker C III, Li L, et al. Select flavonoids and whole juice from purple grapes inhibit platelet function and enhance nitric oxide release. Circulation. 2001;103:2792–2798. [DOI] [PubMed] [Google Scholar]
- 155. Duffy SJ, Keaney JF, Holbrook M, et al. Short‐and long‐term black tea consumption reverses endothelial dysfunction in patients with coronary artery disease. Circulation. 2001;104:151–156. [DOI] [PubMed] [Google Scholar]
- 156. Fisher ND, Hughes M, Gerhard‐Herman M, et al. Flavanolrich cocoa induces nitric oxide‐dependent vasodilation in healthy humans. J Hypertens. 2003;21:2281–2286. [DOI] [PubMed] [Google Scholar]
- 157. Lucas DL, Brown RA, Wassef M. Alcohol and the cardiovascular system: research challenges and opportunities. J Am Coll Cardiol. 2005;45:1916–1924. [DOI] [PubMed] [Google Scholar]
- 158. Sasaki S, Higashi Y, Nakagawa K, et al. A low‐calorie diet improves endothelium‐dependent vasodilation in obese patients with essential hypertension. Am J Hypertens. 2002;15:302–309. [DOI] [PubMed] [Google Scholar]