

Abstract
Objective:
Mitochondrial dysfunction plays a dominant role in the pathogenesis of alcoholic liver disease (ALD); however, the underlying mechanisms remain to be fully understood. Activating transcription factor 4 (ATF4) regulates genes involved in steatosis, oxidative stress, and apoptosis. This study aimed to investigate the function and mechanism of ATF4 in alcohol-induced hepatic mitochondrial dysfunction.
Design:
ATF4 activation was detected in the livers of patients with alcoholic hepatitis (AH). The role of ATF4 and mitochondrial transcription factor A (TFAM) in alcohol-induced liver damage was determined in hepatocyte-specific ATF4 knockout mice and liver-specific TFAM overexpression mice, respectively.
Results:
Hepatic PERK-eIF2α-ATF4 ER stress signaling was upregulated in the livers of patients with AH. Hepatocyte-specific ablation of ATF4 in mice ameliorated alcohol-induced hepatic steatosis, inflammation, and cell death. ATF4 ablation attenuated alcohol-impaired mitochondrial biogenesis and respiratory function along with the restoration of TFAM. Cell studies confirmed that TFAM expression was negatively regulated by ATF4. TFAM silencing in hepatoma cells abrogated the protective effects of ATF4 knockdown on ethanol-mediated mitochondrial dysfunction and cell death. Moreover, hepatocyte TFAM overexpression in mice attenuated alcohol-induced mitochondrial dysfunction and liver damage. Mechanistic studies revealed that ATF4 repressed the transcription activity of NRF1, a key regulator of TFAM, through binding to its promoter region. Clinical relevance among ATF4 activation, NRF1-TFAM pathway disruption, and mitochondrial dysfunction was validated in the livers of patients with AH.
Conclusion:
This study demonstrates that hepatic ATF4 plays a pathological role in alcohol-induced mitochondrial dysfunction and liver injury by disrupting the NRF1-TFAM pathway.
Keywords: Alcoholic liver disease, ER stress, mitochondrial biogenesis, OXPHOS
Introduction
Alcoholic liver disease (ALD), caused by long-term excessive alcohol consumption, represents one of the most prevalent types of chronic liver diseases worldwide [1, 2]. ALD encompasses a broad spectrum of clinical features ranging from simple fatty liver to more advanced steatohepatitis, fibrosis, cirrhosis, and, ultimately, hepatocellular carcinoma [3]. Hepatocyte is densely packed with mitochondria, which is the principal site of adenosine triphosphate (ATP) generation. Mitochondria integrate cell metabolism by regulating essential anabolic and catabolic pathways that ultimately depends on the proper function of oxidative phosphorylation (OXPHOS) [4, 5]. The OXPHOS system consists of 5 multisubunit complexes that are encoded by both nuclear DNA and mitochondrial DNA (mtDNA). The correct balance among mitochondrial complexes is essential to maintain cellular homeostasis and linked to cell fate decision [6, 7]. During the initiation and progression of ALD, hepatic mtDNA deletion/mutation and decreased respiratory chain complexes activities are intimately associated with liver damage in both human and mice [8–10].
Alcohol consumption induces accumulation of unfolded or misfolded proteins in the Endoplasmic reticulum (ER) lumen, which activates unfolded protein response (UPR) via the induction of protein kinase RNA-like endoplasmic reticulum kinase (PERK) [11]. As a result, the level of phosphorylated eukaryotic initiation factor 2 alpha (eIF2α) is distinctly elevated, resulting in the nuclear translation of activating transcription factor 4 (ATF4) [12]. ATF4 is a member of the cAMP-responsive element-binding protein family of basic zipper-containing proteins, regulates a variety of genes involved in various physiological processes, including apoptosis, lipid metabolism, and obesity [13–17]. Loss of ATF4 in HeLa cells led to enhanced ATP-dependent respiration, suggesting that ATF4 regulates mitochondrial functions [18]. Our previous studies have shown that alcohol-induced hepatic mitochondrial dysfunction was associated with ATF4 activation [19]. However, whether ATF4 regulates mitochondrial functions in the liver remains unclear. Considering ER stress is a pathological factor for mitochondrial dysfunction in multiple cell lines [20–22], and ATF4 activation plays a detrimental role in alcoholic steatosis [16], we hypothesized that ATF4 might be involved in the pathogenesis of alcohol-induced hepatic mitochondrial dysfunction.
Materials and Methods
Materials and methods used in this study were described in Supplementary Materials and Methods.
Results:
The hepatic PERK-eIF2α-ATF4 ER stress signaling pathway is upregulated in patients with alcoholic hepatitis.
Compared with healthy subjects, alcoholic hepatitis (AH) patients exhibited robustly enhanced phosphorylation of PERK and eIF2α proteins in the liver (Fig. 1A). The total protein levels of hepatic PERK and eIF2α were comparable between two groups (Supplementary Fig. 1A). Hepatic protein levels of ATF4 were markedly increased by 9-fold in AH patients, as evaluated by Western blot (Fig. 1A). Increased hepatic protein expression of ATF4 was also detected by IHC staining (Fig. 1A). In contrast to the cytoplasmic localization of PERK and eIF2α, ATF4 was primarily localized in the nuclei of hepatocytes (Fig. 1A). Hepatic mRNA levels of ATF4 were also upregulated by 5-fold in AH patients (Fig. 1B).
Fig. 1. Hepatic PERK-eIF2α-ATF4 signaling pathway is upregulated in ALD patients and hepatocyte-specific ablation of ATF4 protects alcohol-induced liver injury in mice.
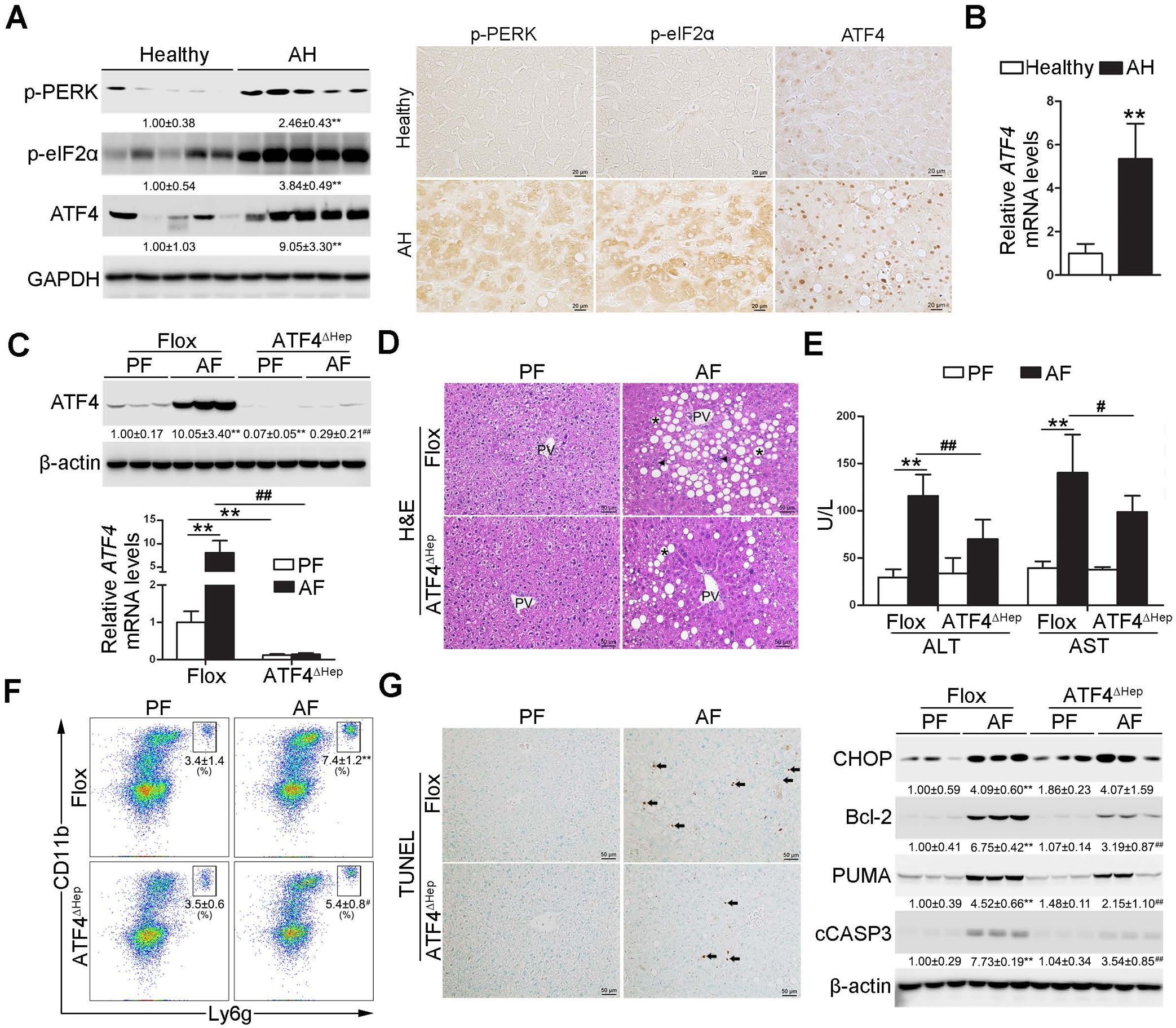
(A-B) Liver tissues were obtained from ALD patients and healthy controls. (A) Western bolt and immunohistochemistry analysis of hepatic PERK-eIF2α-ATF4 signaling expression (n=5/group). Scale bars: 20 μm. (B) Analysis of mRNA levels of ATF4 in the livers (n=5/group). (C-G) ATF4 floxed and ATF4ΔHep male mice were pair-fed or alcohol-fed for 8 weeks plus a single binge of ethanol (4 g/kg) before 4 hours of tissue collection. (C) Protein and mRNA levels of ATF4 in the liver (n=3–8). (D) Liver histopathological changes shown by H&E staining. Scale bars: 50 μm. Asterisks: lipid droplets. Arrowheads: Infiltrated immune cells. (E) Serum ALT and AST activities (n=8–11). (F) Representative dot plot and gating strategy for 7-AAD−CD45+CD11b+ Ly6g+ neutrophils (n=6). (G) Immunohistochemistry of TUNEL and Western bolt analysis of hepatic CHOP, Bcl-2, PUMA, and cleaved caspase-3. Scale bars: 50 μm. Arrow: TUNEL positive cells. Protein bands intensity was quantified by ImageJ (NIH). Data are presented as means ± SD. In panels A and B, statistical comparisons were made using Student’s t-test; **P<0.01 vs. healthy control. In panels C-G, statistical comparisons were made using one-way ANOVA with Tukey’s post hoc test; **P<0.01 vs. Flox/PF mice; #P<0.05, ##P<0.01 vs. Flox/AF mice. PF, pair-fed; AF, alcohol-fed.
Hepatocyte-specific deletion of ATF4 alleviates alcohol-induced liver injury.
To determine the role of ATF4 in the pathogenesis of ALD, hepatocyte-specific ATF4 knockout (ATF4ΔHep) mice were generated (Supplementary Fig. 1B). ATF4 floxed mice and ATF4ΔHep mice were subjected to the Lieber-DeCarli control diet (Flox/PF or ATF4ΔHep/PF) or alcohol diet (Flox/AF or ATF4ΔHep/AF) for eight weeks plus a single binge (4 g/kg) before four hours of tissue collection (modified NIAAA model). Chronic alcohol feeding activated the PERK-eIF2α-ATF4 signaling pathway in the liver of floxed mice (Fig. 1C and Supplementary Fig. 1B). Only trace amounts of ATF4 proteins were detected in the livers of ATF4ΔHep/AF mice despite the PERK-eIF2α signaling activation (Fig. 1C and Supplementary Fig. 1C). As shown in Fig. 1D, liver histopathological changes induced by chronic alcohol feeding, including lipid droplet accumulation and inflammatory cell infiltration, were alleviated in ATF4ΔHep mice. Alcohol-increased hepatic triglyceride (TG) and free fatty acid (FFA) contents were attenuated by ATF4 deletion (Supplementary Fig. 2A and B). The liver weight and body weight were comparable among all groups (Supplementary Fig. 2C). Alcohol-increased serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were also ameliorated by ATF4 deletion (Fig. 1E).
Neutrophils infiltration in the liver was analyzed by flow cytometry with a gating strategy showed in Supplementary Fig. 3A. Chronic alcohol feeding increased the frequency of neutrophils (CD45+CD11b+Ly6g+) in the livers of floxed mice, and this effect was ameliorated by ATF4 deletion (Fig. 1F). Correspondingly, ATF4ΔHep/AF mice displayed fewer hepatic myeloperoxidase (MPO) positive cells than Flox/AF mice (Supplementary Fig. 3B). Both the protein and mRNA levels of hepatic chemokine (C-X-C motif) ligand 1 (CXCL-1) were increased after alcohol exposure, whereas these effects were reversed by ATF4 ablation (Supplementary Fig. 3C). However, alcohol-increased number of hepatic F4/80 positive cells and abundance of CD45±CD11bintF4/80hi macrophage were not significantly affected by ATF4 ablation (Supplementary Fig. 3D and E).
In response to alcohol feeding, TUNEL positive cells were both detected in the livers of floxed mice and ATF4ΔHep mice compared with their corresponding controls; with ATF4 deletion, mice displayed fewer positive cells (Fig. 1G). Alcohol-increased hepatic protein levels of B-cell lymphoma 2 (Bcl-2), p53 upregulated modulator of apoptosis (PUMA), and cleaved caspase-3 (cCASP3) were all ameliorated in ATF4ΔHep mice (Fig. 1G). However, alcohol-induced hepatic C/EBP homologous protein (CHOP) activation was not affected by ATF4 deletion (Fig. 1G). Hepatic protein levels of alcohol metabolizing enzymes, including alcohol dehydrogenases (ADH) and cytochrome P450 2E1 (CYP2E1), and serum ethanol levels were comparable in Flox/AF mice and ATF4ΔHep/AF mice (Supplementary Fig. 4A and B).
Hepatocyte-specific deletion of ATF4 protects alcohol-induced mitochondrial dysfunction in mice.
We further determined if ATF4 deletion can improve alcohol-induced mitochondrial dysfunction. Primary hepatocytes from ATF4ΔHep mice displayed a markedly higher mitochondrial respiration than those from floxed mice fed with either control or alcohol diet (Fig. 2A). Alcohol-decreased basal oxygen consumption rate (OCR) was almost completely normalized by ATF4 knockout (Fig. 2A). We examined the effects of ATF4 and alcohol on mitochondrial fatty acid β-oxidation (FAO) in primary hepatocytes. Primary hepatocytes were incubated with fatty acids before flux analysis. In this condition, hepatocytes from ATF4ΔHep mice showed enhanced OXPHOS compared with those from floxed mice (Fig. 2A). Alcohol-decreased FAO was also largely preserved by hepatic ATF4 deletion (Fig. 2A). ATF4ΔHep/AF mice displayed significantly higher levels of hepatic ATP contents compared with Flox/AF mice (Fig. 2B).
Fig. 2. Ablation of ATF4 in hepatocyte protects alcohol-mediated mitochondrial dysfunction in the liver.
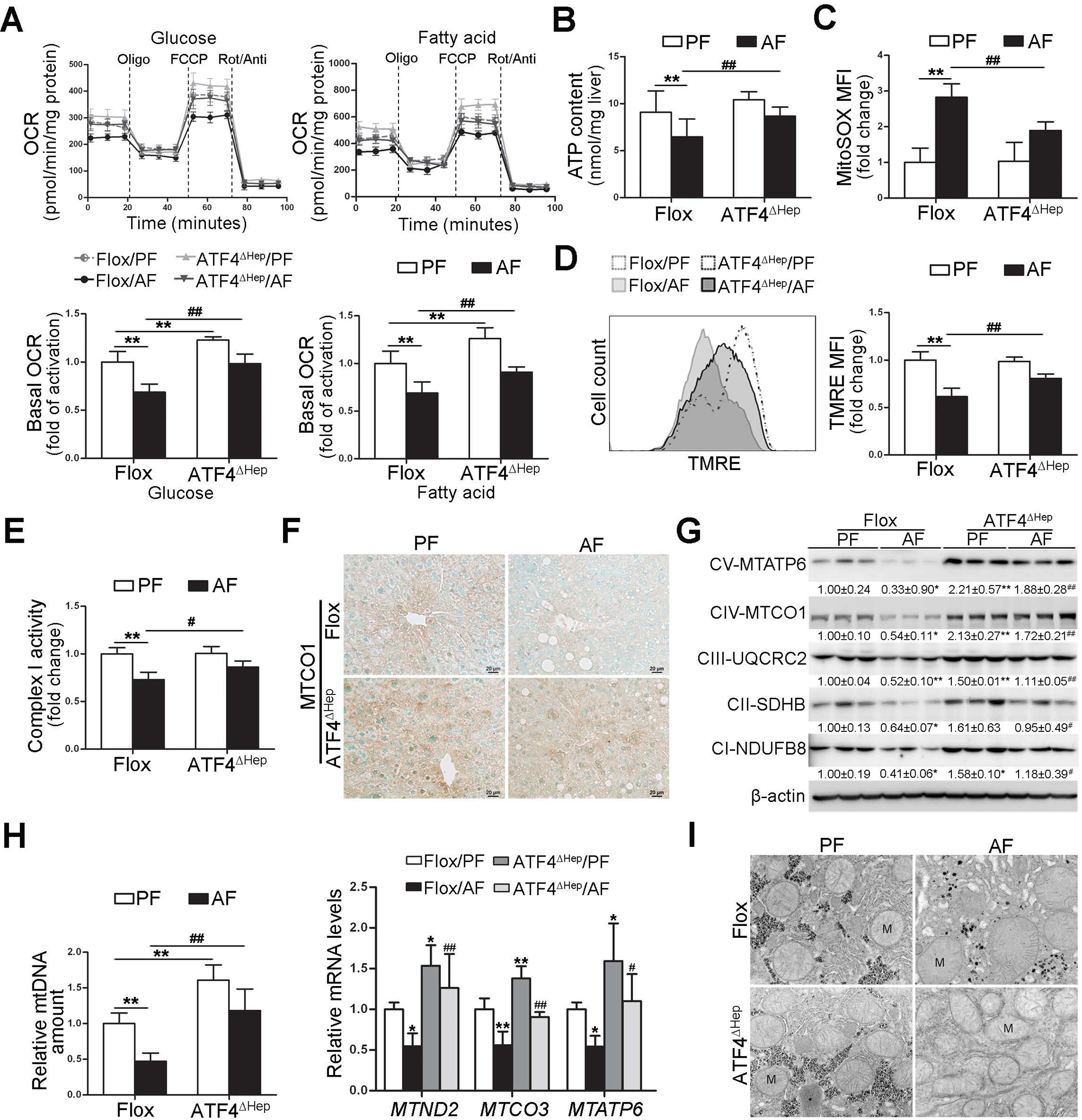
ATF4 floxed and ATF4ΔHep male mice were pair-fed or alcohol-fed for 8 weeks plus a single binge (4 g/kg) before 4 hours of tissue collection. (A) Representative oxygen consumption rate profile (OCR) and basal OCR in isolated primary hepatocytes. (B) ATP contents in the liver (n=5). (C) FACS analysis of mitochondrial ROS using MitoSOX dye (n=5). Data are the summary of the mean fluorescence intensity (MFI). (D) FACS analysis of mitochondrial membrane potential using TMRE probe (n=5). Data are the summary of the mean fluorescence intensity (MFI). (E) Hepatic complex I activity (n=6). (F) Immunohistochemistry of MTCO1 in the liver. Images were captured by light microscope. Scale bars: 20 μm. (G) Western blot analysis of mitochondrial respiratory complexes subunits (MTATP6, MTCO1, UQCRC2, SDHB, and NDUFB8). (H) mtDNA levels (MTND1) relative to nuclear DNA (SDHA) and the mRNA levels of mtDNA-encoded complexes subunits (n=5–6). (I) Mitochondrial morphology and ultrastructure. Protein bands intensity was quantified by ImageJ (NIH). Data are presented as means ± SD. Statistical comparisons were made using one-way ANOVA with Tukey’s post hoc test. *P<0.05, **P<0.01 vs. Flox/PF mice; #P<0.05, ##P<0.01 vs. Flox/AF mice. PF, pair-fed; AF, alcohol-fed; M, mitochondria.
We next measured the levels of mitochondrial reactive oxygen species (mtROS) and total ROS in primary hepatocytes. Compared with Flox/PF mice, higher mtROS and total ROS levels were detected in the hepatocytes from Flox/AF mice, whereas these effects were ameliorated by hepatic ATF4 knockout (Fig. 2C and Supplementary Fig. 5A). Alcohol-increased 4-hydroxynonenal (4-HNE) protein adducts formation in the liver was attenuated in ATF4ΔHep mice (Supplementary Fig. 5B). Hepatocytes isolated from Flox/AF mice exhibited significantly lower mitochondrial membrane potential than those from Flox/PF mice, whereas this effect was ameliorated in ATF4ΔHep mice (Fig. 2D). The enzymatic activity of mitochondria respiratory complex I, which control cellular NAD+/NADH redox balance, was decreased by chronic alcohol feeding in floxed mice (Fig. 2E). ATF4 deletion not only restored alcohol-reduced mitochondria respiratory complex I activity (Fig. 2E), but also corrected alcohol-decreased NAD+ contents and NAD+/NADH ratio in the liver (Supplementary Fig. 5C).
We next analyzed the effects of alcohol and ATF4 on hepatic mitochondrial respiratory chain (MRC) complexes, the proper function of which are essential for maintaining mitochondrial homeostasis. The protein levels of mtDNA-encoded mitochondrial complexes subunits, including cytochrome c oxidase subunit 1 (MTCO1) (Fig. 2F and G) and mitochondrial encoded ATP synthase membrane subunit 6 (MTATP6) (Fig. 2G), were remarkably higher in ATF4ΔHep mice than that in floxed mice, regardless of alcohol administration. Alcohol-decreased protein levels of nuclear genome-encoded complexes were also reversed by ATF4 knockout (Fig. 2G). Furthermore, hepatic relative mtDNA contents (MTND1 relative to SDHA) and mRNA levels of mtDNA-encoded complexes subunits were all significantly increased in ATF4ΔHep mice on either diet compared with floxed mice (Fig. 2H and Supplementary Fig. 6A and B). After chronic alcohol exposure, mitochondria in hepatocyte appear swollen with few cristae, whereas these changes were reversed by hepatic ATF4 knockout (Fig. 2I).
ATF4 negatively regulates TFAM expression in hepatocyte.
To understand the mechanisms underlying ATF4 deletion-restored hepatic mtDNA contents in ALD, major regulators involved in mtDNA replication, transcription, and maintenance were examined. Intriguingly, ATF4ΔHep mice displayed higher hepatic TFAM mRNA and protein levels on either diet compared with their genetic controls, as evaluated by RT-PCR (Fig. 3A), Western blot (Fig. 3B), and IHC staining (Fig. 3C). However, the mRNA levels of DNA polymerase subunit gamma (Plog), DNA-directed RNA polymerase, mitochondrial (Polrmt), and twinkle mtDNA helicase (Twinkle) were comparable between Flox/AF mice and ATF4ΔHep/AF mice (Fig. 3A). Furthermore, the association between ATF4 induction and TFAM reduction was observed in the liver from mice fed with ethanol for four weeks (Supplementary Fig. 7A).
Fig. 3. ATF4 negatively regulates TFAM in hepatocyte.
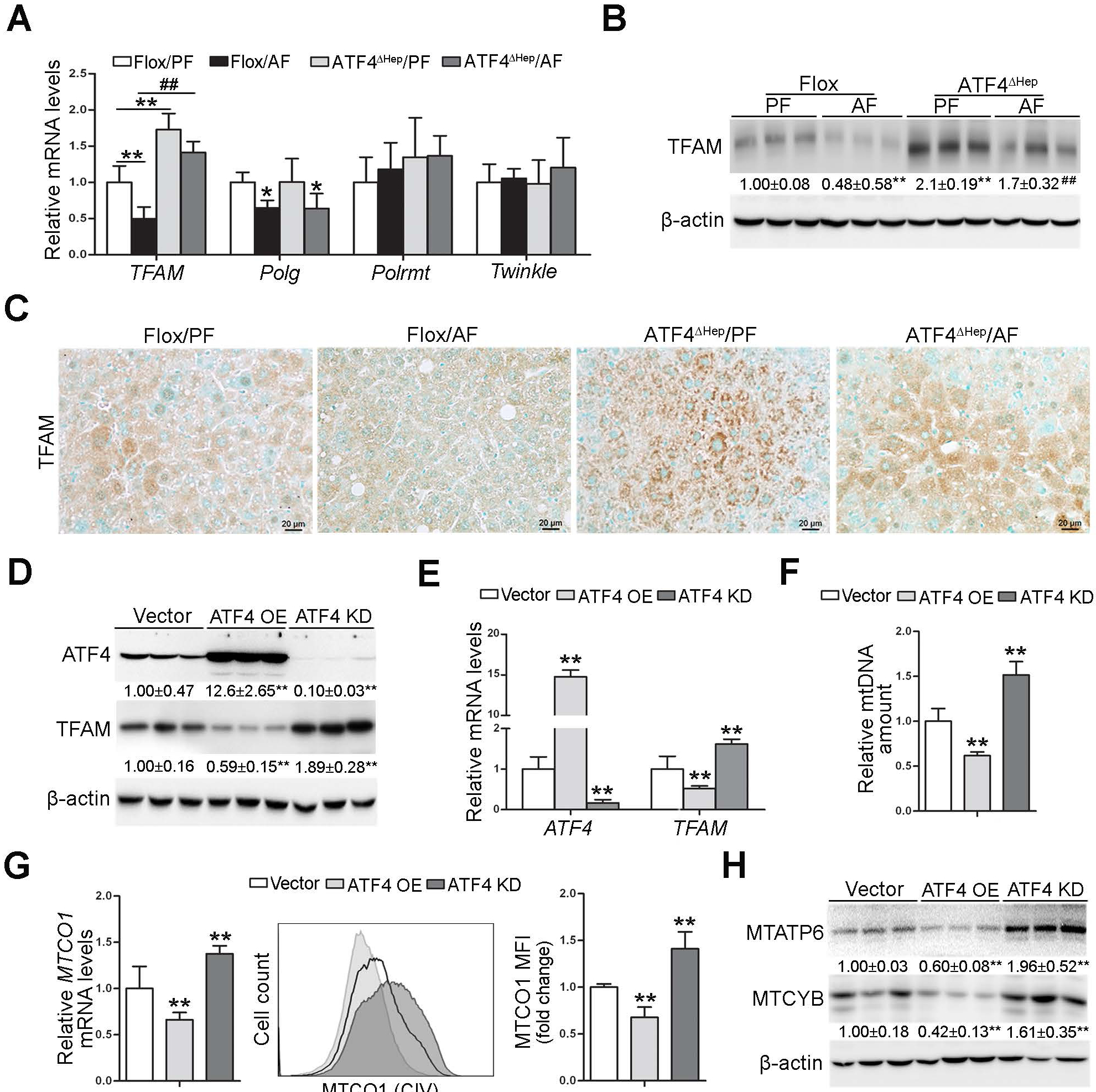
(A-C) ATF4 floxed and ATF4ΔHep male mice were pair-fed or alcohol-fed for 8 weeks plus a single binge of ethanol (4 g/kg) 4 hours before tissue collection. (A) Relative mRNA levels of hepatic TFAM, Polg, Polrmt, and Twinkle (n=5). (B) Western blot of hepatic TFAM. (C) Immunohistochemistry of hepatic TFAM. Images were captured by light microscope. Scale bars: 20 μm. (D-H) VL-17 cells were transfected with either ATF4 overexpression or ATF4 knockdown CRISPR plasmids. (D) Western blot analysis of ATF4 and TFAM in VL-17A cells. (E) Analysis of mRNA levels of ATF4 and TFAM in VL-17A cells (n=5–8). (F) mtDNA levels (MTND1) relative to nuclear DNA (SDHA) in VL-17A cells (n=6). (G) The mRNA levels and protein levels of MTCO1 were measured by RT-PCR and flow cytometry, respectively (n=4–6). (H) Western blot analysis of MTATP6 and MTCYB in VL-17A cells. Protein bands intensity was quantified by ImageJ (NIH). Data are presented as means ± SD. Statistical comparisons were made using one-way ANOVA with Tukey’s post hoc test. Panel A and B, *P<0.05, **P<0.01 vs. Flox/PF mice. ##P<0.01 vs. Flox/AF mice. Panel D-H, **P<0.01 vs. control cells. PF, pair-fed; AF, alcohol-fed.
To verify the animal data in vitro, we used VL-17A cells, a recombinant HepG2 cell line that constitutively expresses human ADH and CYP2E1 for efficient alcohol metabolism. Alcohol administration enhanced the expression of PERK-eIF2α-ATF4 signaling but decreased the expression of TFAM in a time- and dose-dependent manner in VL-17A cells (Supplementary Fig. 7B and C). We next created the stable ATF4 overexpression and knockdown cell lines, respectively, by using the CRISPR-Cas9 approach. The efficiency of ATF4 overexpression and knockdown in VL-17A cells was confirmed by Western blot (Fig. 3D) and RT-PCR (Fig. 3E). Both the protein and mRNA levels of TFAM were downregulated by ATF4 overexpression, while upregulated by ATF4 knockdown (Fig. 3D and E). Correlative to the changes in TFAM, the mtDNA contents (Fig. 3F), MTCO1 mRNA levels (Fig. 3G), and protein levels of the mtDNA-encoded genes, including Cytochrome b (MTCYB), MTCO1, and MTATP6, (Fig. 3G, H) were all markedly decreased by ATF4 overexpression but increased by ATF4 knockdown in VL-17A cells compared with the control cells.
TFAM reduction is involved in ATF4-mediated mitochondrial dysfunction in VL-17A cells with alcohol exposure.
To further determine whether TFAM is the key molecule mediating the effect of ATF4 on mitochondrial function under alcohol exposure, TFAM knockdown, and ATF4/TFAM double knockdown VL-17A cell lines were established and were treated with alcohol. As shown in Fig. 4A, cellular protein levels of ATF4 and TFAM were dramatically reduced by gene knockdown. ATF4 knockdown-increased protein levels of TFAM were entirely abrogated by TFAM knockdown in VL-17A cells (Fig. 4A). TFAM knockdown led to a severe reduction of mtDNA levels and exacerbated alcohol-decreased mtDNA levels in VL-17A cells (Fig. 4B). Moreover, the protective role of ATF4 knockdown in alcohol-mediated mtDNA depletion was completely abolished by TFAM knockdown (Fig. 4B). The same trend was also observed in the mRNA levels of MTCO1 in VL-17A cells (Supplementary Fig. 8A). Alcohol-decreased mitochondrial complex I activity was also ameliorated by ATF4 knockdown but exacerbated by TFAM knockdown (Supplementary Fig. 8B). ATF4 knockdown cells exhibited a higher respiration rate and attenuated ethanol-decreased basal OCR, whereas these effects were abrogated by TFAM knockdown (Fig. 4C). Although TFAM knockdown and ethanol exposure did not affect the ATP contents in VL-17A cells, reduction in ATP levels was observed when these two factors combined (Fig. 4D). ATF4 knockdown protected alcohol-perturbed mitochondrial membrane potential (Fig. 4E). Conversely, TFAM knockdown exacerbated the detrimental role of alcohol exposure in mitochondrial membrane potential disruption (Fig. 4E). As shown in Fig. 4F, alcohol exposure significantly increased the protein levels of PUMA and cCASP3, which were ameliorated by ATF4 knockdown but exacerbated by TFAM knockdown (Bar graph shown in Supplementary Fig. 8C). The frequency of Annexin V-positive cells was markedly elevated after alcohol exposure, which was protected by ATF4 knockdown but aggravated by TFAM knockdown (Fig. 4F). Moreover, the protective effects of ATF4 knockdown on alcohol-induced mitochondrial complex I activity reduction, mitochondrial membrane potential disruption, and apoptotic cell death were all abrogated by TFAM knockdown in VL-17A cells (Supplementary Fig. 8B and C and Fig. 4E, F).
Fig. 4. TFAM reduction is involved in ATF4-mediated mitochondrial dysfunction in VL-17A cells with alcohol exposure.
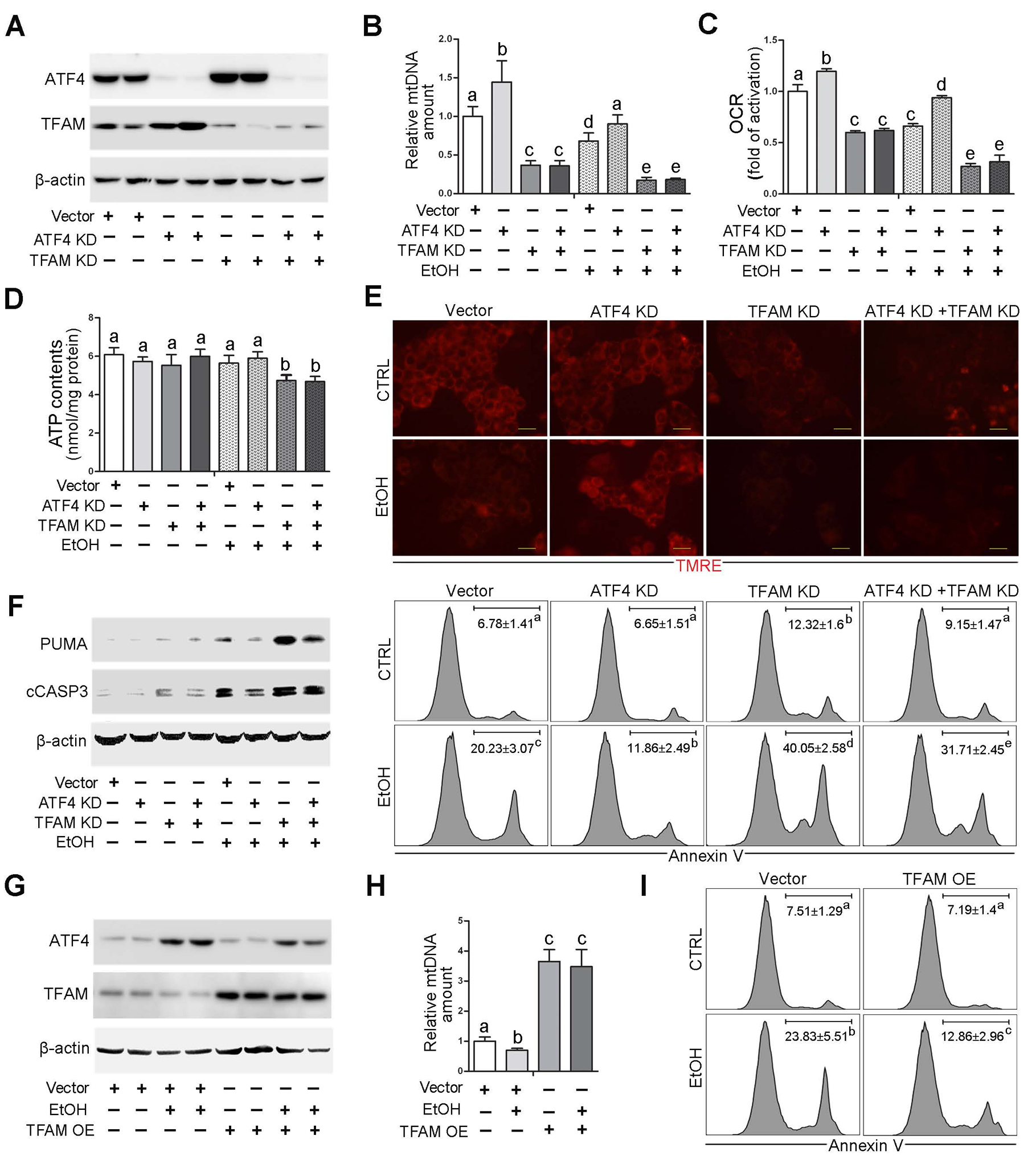
ATF4 knockdown, TFAM knockdown, ATF4/TFAM double knockdown VL-17A cells were generated and treated with 100 mM ethanol for 72 hours. (A) Western blot analysis of ATF4 and TFAM. (B) mtDNA levels (MTND1) relative to nuclear DNA (SDHA) in VL-17A cells (n=5). (C) Basal oxygen consumption rate of VL-17A cells (n=6). (D) ATP contents in VL-17A cells (n=5). (E) Immunofluorescent staining of mitochondrial membrane potential using TMRE probe. Scale bars: 20 μm. (F) Western blot analysis of PUMA and cleaved caspase-3, and FACS analysis of apoptotic cell death represented by Annexin V positive cells (n=4–6). (G-I) VL-17 cells were transfected with TFAM overexpression CRISPR plasmids and treated with 100 mM ethanol for 72 hours. (G) Western blot analysis of ATF4 and TFAM in VL-17A cells. (H) mtDNA levels (MTND1) relative to nuclear DNA (SDHA) in TFAM overexpression cells (n=5). (I) FACS analysis of apoptotic cell death represented by Annexin V positive cells. Protein bands intensity was quantified by ImageJ (NIH). Data are presented as means ± SD. Statistical comparisons were made using one-way ANOVA with Tukey’s post hoc test. Bars with different characters differ significantly (P < 0.05). EtOH, ethyl alcohol.
TFAM-overexpressing VL-17A cells were generated and subjected to alcohol. Alcohol-decreased TFAM protein levels were completely reversed by TFAM overexpression (Fig. 4G). Interestingly, alcohol-induced ATF4 activation was also reversed by TFAM overexpression (Fig. 4G). TFAM overexpression increased the cellular mtDNA levels and the mRNA levels of mtDNA-encoded complexes subunits regardless of alcohol exposure (Fig. 4H and Supplementary Fig. 9A). However, the mRNA levels of nuclear genome-encoded complexes subunits remain unchanged (Supplementary Fig. 9A). Alcohol-perturbed mitochondrial membrane potential was ameliorated by TFAM overexpression in VL-17A cells (Supplementary Fig. 9B). TFAM overexpression also reversed alcohol-increased frequencies of Annexin V-positive cells (Fig. 4I). This effect was accompanied by decreased protein levels of PUMA and cleaved caspase-3 (Supplementary Fig. 9C).
Hepatocyte-specific TFAM overexpression prevents alcohol-induced mitochondrial dysfunction in mice.
To further validate the protective function of TFAM in alcohol-induced hepatic mitochondrial dysfunction in vivo, hepatocyte-specific TFAM overexpressing (AAV8-TFAM) mice as well as control mice (AAV8-null) were generated. The mRNA and protein levels of TFAM in the liver of TFAM overexpressing mice were increased nearly 6-fold and 7-fold, respectively, at the time of starting alcohol feeding (Supplemental Fig. 10A and B). After eight weeks of alcohol feeding, hepatic TFAM protein levels in the AAV8-TFAM/AF mice were 5-fold higher than the AAV8-null/AF mice (Fig. 5A). Hepatic mtDNA contents and the mRNA levels of mtDNA-encoded mitochondrial complexes subunits were remarkably increased by TFAM overexpression (Fig. 5B). Accordingly, hepatic protein levels of mtDNA-encoded subunits were higher in AAV8-TFAM/AF mice than that in AAV8-null/AF mice (Fig. 5A and Supplemental Fig. 11A). The overexpression of TFAM did not affect the expression of nuclear DNA-encoded complexes subunits (Fig. 5A, 5B, and Supplemental Fig. 11A). Overexpression of TFAM ameliorated alcohol-impaired mitochondrial respiratory complex I activity (Fig. 5C) along with elevated NAD+ levels and NAD+ to NADH ratio in the liver (Supplementary Fig. 11B). Subsequent flow cytometry analysis showed that AAV8-TFAM/AF mice displayed higher levels of mitochondrial membrane potential (Fig. 5D) and lower levels of mtROS and total ROS levels in the liver than that in AAV8-null/AF mice (Fig. 5G). Alcohol-decreased hepatic ATP contents were reversed by TFAM overexpression (Fig. 5E). Primary hepatocytes from AF/AAV8-TFAM mice also displayed higher OCR values in response to either glucose or palmitate (Fig. 5F). Furthermore, alcohol-altered mitochondrial morphology, including swollen and loss of cristae, were reversed by TFAM overexpression (Fig. 5H).
Fig. 5. Hepatocyte-specific TFAM overexpression protects alcohol-induced mitochondrial dysfunction in mice.
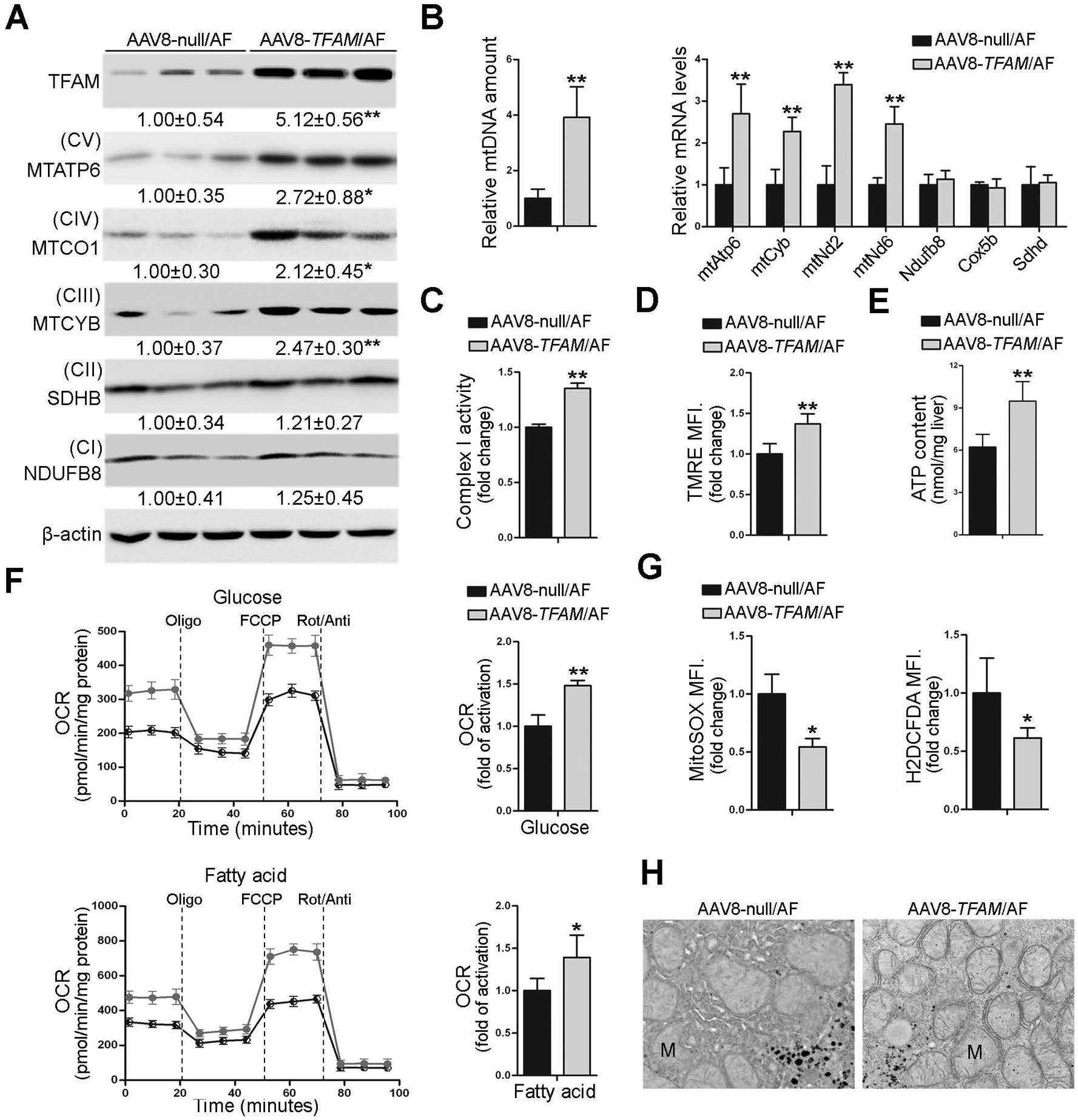
Hepatocyte-specific TFAM overexpression mice were generated by injected in the retrial orbital sinus with recombinant adeno-associated viral (AAV) serotype 8 gene transfer vectors bearing a liver-specific promoter combination (TBG) with mouse TFAM sequence. Mice injected with null-vector are served as control. These mice were fed with alcohol for 8 weeks plus a single binge of alcohol (4 g/kg) 4 hours before tissue collection. (A) Protein levels of TFAM, MTATP6, MTCO1, MTCYB, SDHB, and NDFUB8 in the liver. (B) Hepatic mtDNA contents (MTND1 relative to SDHA) and mtDNA- and nuDNA-encoded genes expression (n=4–6). (C) Hepatic mitochondrial complex I activity (n=5). (D) FACS analysis of mitochondrial membrane potential using TMRE probe (n=5). Data are the summary of the mean fluorescence intensity (MFI). (E) ATP contents in the liver (n=5). (F) Representative oxygen consumption rate profile (OCR) and basal OCR in isolated primary hepatocytes. (G) FACS analysis of mitochondrial ROS and total ROS using MitoSOX dye and H2DCFDA dye, respectively (n=5). Data are the summary of the mean fluorescence intensity (MFI). (H) Mitochondrial morphology and ultrastructure. Protein bands intensity was quantified by ImageJ (NIH). Data are presented as means ± SD. Statistical comparisons were made using one Student’s t-test. *P<0.05, **P<0.01 vs. AAV8-null/AF mice. AF, alcohol-fed; M, mitochondria.
Hepatocyte-specific TFAM overexpression attenuates alcoholic steatohepatitis.
We found that AAV8-TFAM/AF mice displayed markedly fewer hepatic lipid droplets and infiltrated immune cells in the liver compared with AAV8-null/AF mice (Fig. 6A). Lower levels of serum ALT and AST were observed in AAV8-TFAM/AF mice than that in AAV8-null/AF mice (Fig. 6B). TFAM overexpression in the hepatocytes also decreased neutral lipid accumulation and TG and FFA contents in the liver under alcohol exposure condition (Fig. 6C).
Fig. 6. Hepatocyte-specific TFAM overexpression ameliorates alcoholic steatohepatitis in mice.
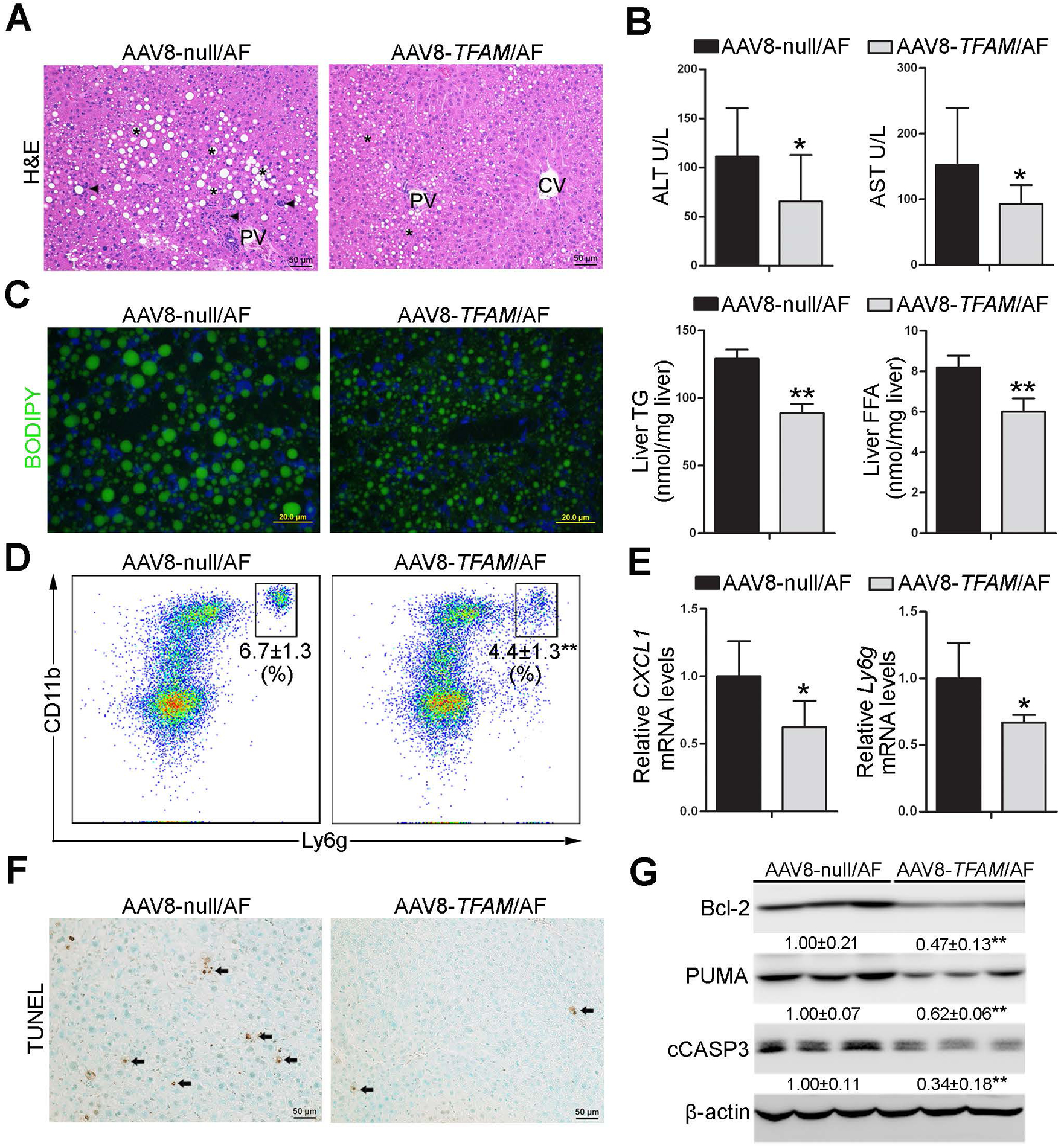
Hepatocyte-specific TFAM overexpression mice were generated by injected in the retrial orbital sinus with recombinant adeno-associated viral (AAV) serotype 8 gene transfer vectors bearing a liver-specific promoter combination (TBG) with mouse TFAM sequence. Mice injected with null-vector are served as control. These mice were fed with alcohol for 8 weeks plus a single binge of alcohol (4 g/kg) 4 hours before tissue collection. (A) H&E staining. Scale bars: 50 μm. Asterisks: lipid droplets. Arrowheads: Infiltrated immune cells. (B) Serum ALT and AST activities (n=6). (C) BODIPY staining of the neutral lipids in mouse liver, and hepatic TG and FFA contents (n=6). Scale bars: 20 μm. (D) Representative dot plot for CD11b+ and Ly6g+ neutrophils are displayed of singlet 7-AAD− CD45+ cells (n=5). (E) The mRNA levels of hepatic Cxcl-1 and Ly6g (n=5). (F) Immunohistochemistry of hepatic TUNEL. Scale bars: 50 μm. Arrow: TUNEL positive cells. (G) The protein levels of Bcl-2, PUMA, and cleaved caspase3 in the liver. Protein bands intensity was quantified by ImageJ (NIH). Data are presented as means ± SD. Statistical comparisons were made using one Student’s t-test. *P<0.05, **P<0.01 vs. AAV8-null/AF mice. AF, alcohol-fed.
TFAM overexpression ameliorated alcohol-mediated neutrophils infiltration in the liver (Fig. 6D and Supplementary Fig.12A). The mRNA levels of hepatic CXCL1 and Ly6g were also lower in AAV8-TFAM/AF (Fig. 6E). However, the number of F4/80 positive cells were not significant affected by TFAM overexpression (Supplementary Fig.12B). The number of TUNEL positive cells was reduced in the liver of AAV8-TFAM/AF mice (Fig. 6F). Furthermore, hepatic protein levels of Bcl-2, PUMA as well as cCASP3 were all lower in AAV8-TFAM/AF mice (Fig. 6G). TFAM overexpression did not affect the serum ethanol levels and hepatic protein levels of ADH and CYP2E1 (Supplementary Fig.12C).
ATF4 negatively regulates TFAM expression by suppressing NRF1 in the hepatocyte.
To explore the possible mechanism by which ATF4 negatively regulates TFAM, the effects of alcohol and ATF4 deletion on major signaling molecules related to mitochondrial biogenesis were measured. As shown in Fig. 7A, the mRNA levels of nuclear respiratory factor 1 (NRF1) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) but not NRF2 and PGC-1β were increased in ATF4ΔHep/PF mice compared with Flox/PF mice. However, lack of ATF4 in the hepatocytes completely reversed alcohol-decreased expression of NRF1 but not PGC-1α, suggesting that ATF4 may suppress TFAM expression through inhibiting NRF1 (Fig. 7A). Western blot analysis showed that alcohol-reduced hepatic NRF1 protein levels were reversed by ATF4 deletion (Fig. 7B). Furthermore, we found that ATF4 knockdown enhanced the expression of NRF1 in VL-17A cells (Fig. 7C). By contrast, ATF4 overexpression decreased the mRNA and protein levels of NRF1 (Fig. 7C).
Fig. 7. NRF1 is directly interact and inactivated by ATF4.
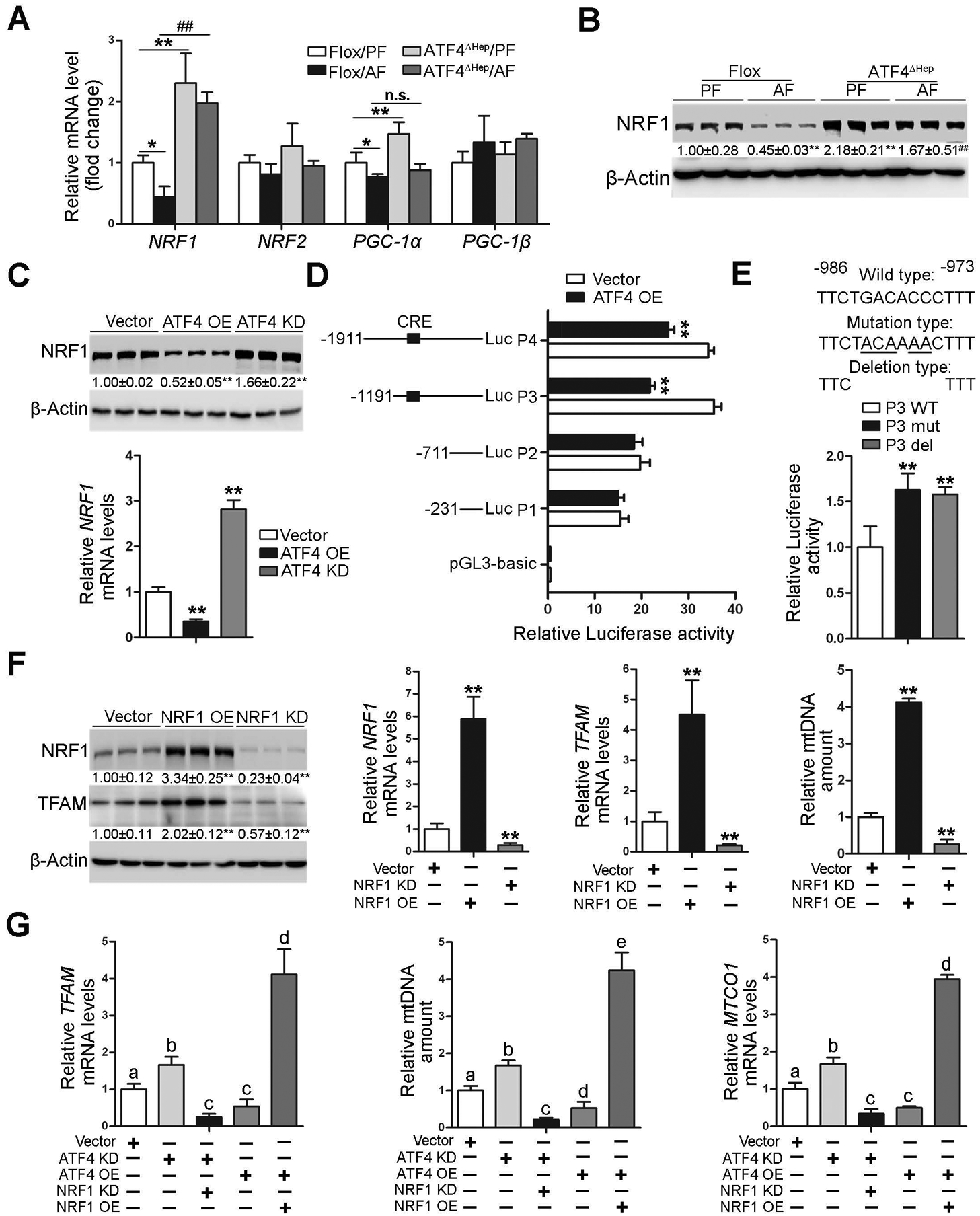
(A-B) ATF4 floxed and ATF4ΔHep male mice were pair-fed or alcohol-fed for 8 weeks plus a single binge of ethanol (4 g/kg) 4 hours before tissue collection. (A) Analysis of mRNA levels of NRF1, NRF2, PGC-1α, and PGC-1β in livers (n=5). (B) Western blot analysis of NRF1. (C) The expression of NRF1 in VL-17A cells transfected with either ATF4 overexpression CRISPR plasmid or ATF4 knockdown CRISPR plasmid. (D) The Relative luciferase activity of NRF1 promoters. (E) Relative promoter activity of wild-type of P3 (P3 WT) promoter and the mutated (P3 mut) and deleted (P3 del) constructs. The dual-luciferase activity was measured after 48 h transfection. (F) The protein levels and mRNA levels of NRF1 and TFAM after transfected either NRF1 overexpression CRISPR plasmid or NRF1 knockdown CRISPR plasmid. (G) Relative TFAM mRNA levels, mtDNA contents (MTND1 relative to SDHA), and mRNA levels of MTCO1 in ATF4/NRF1 double overexpression and ATF4/NRF1 double knockout VL-17A cell lines. Protein bands intensity was quantified by ImageJ (NIH). Data are presented as means ± SD. Statistical comparisons were made using one-way ANOVA with Tukey’s post hoc test (n=3–6). In panels A and B, *P<0.05, **P<0.01 vs. Flox/PF mice; # P<0.05, ##P<0.01 vs. Flox/AF mice. In panels C-F, **P < 0.05 versus corresponding control. In panel G, bars with different characters differ significantly (P < 0.05). PF, pair-fed; AF, alcohol-fed.
ATF4 positively or negatively regulates the transcription of its target genes via cAMP response element (CRE) sequences [23]. To further determine if ATF4 represses NRF1 transcription, a series of human NRF1 promoter constructs with (P3 and P4) or without (P1 and P2) CRE-binding sites were created and co-transfected into 293T cells with ATF4 overexpression CRISPR plasmids. As shown in Fig. 7D, overexpression of ATF4 significantly inhibited P3 and P4 promoter activity in reporter assays. However, no activity was observed when promoters P1 and P2 were used (Fig. 7D). Our bioinformatics analysis further identified conserved elements at the ATF4 binding site in the human and mouse NRF1 promoters (Supplementary Fig. 13A, B). When this cAMP response element was mutated or deleted in promoter P3, trans-inhibition of promoter activity by ATF4 was absent (Fig. 7E).
We next found that NRF1 overexpression significantly increased the mRNA and protein levels of TFAM in VL-17 cells, but NRF1 knockdown decreased the expression of TFAM (Fig. 7F). The cellular mtDNA levels were altered correlatively following TFAM expressions (Fig. 7F). Furthermore, ATF4 overexpression-decreased TFAM expression, mtDNA contents, and mtDNA-encoded complexes subunits expression were markedly reversed by NRF1 overexpression (Fig. 7G and Supplementary Fig. 13C). By contrast, ATF4 knockdown-increased TFAM expression, mtDNA levels, and MTCO1 expression were all abrogated by NRF1 knockdown (Fig. 7G). Alcohol-induced mitochondrial membrane potential disruption, OXPHOS dysfunction, and mitochondrial apoptotic pathway activation were ameliorated by NRF1 overexpression in VL-17A cells (Supplementary Fig. 14A–C).
The NRF1-TFAM signaling pathway is disrupted in the liver of patients with AH.
Finally, we examined the relevance of the identified ATF4-NRF1-TFAM pathway in patients with AH. In contrast to robust induction of ATF4, as described above, remarkable decreases in hepatic mRNA (Fig. 8A) and protein (Fig. 8B) levels of NRF1 and TFAM were observed in AH patients compared with healthy subjects. IHC staining further confirmed significant decreases in nuclear NRF1 and cytoplasmic TFAM as well as MTCO1 proteins in the liver of AH patients (Fig. 8C). The hepatic mtDNA contents and the protein levels of mtDNA-encoded complexes subunits were all significantly decreased in the liver of AH patients (Fig. 8D and E). Nuclear-encoded proteins involved in OXPHOS were also decreased in AH patients (Supplementary Fig. 15A). Decreased hepatic complex I activity (Fig. 8F) was observed in the livers of AH patients. Meanwhile, AH patients exhibited higher levels of hepatic oxidative damage than healthy controls, as indicated by elevated 4-HNE protein adducts formation in the liver (Supplementary Fig. 15B). The protein levels of hepatic PUMA and cleaved caspase-3 were also higher in AH patients than that in healthy subjects (Supplementary Fig. 15C). Collectively, these data indicate that ATF4-mediated NRF1-TFAM signaling pathway disruption may be involved in AH patients.
Fig. 8. NRF1-TFAM signaling pathway is disrupted in the liver of patients with alcoholic hepatitis.
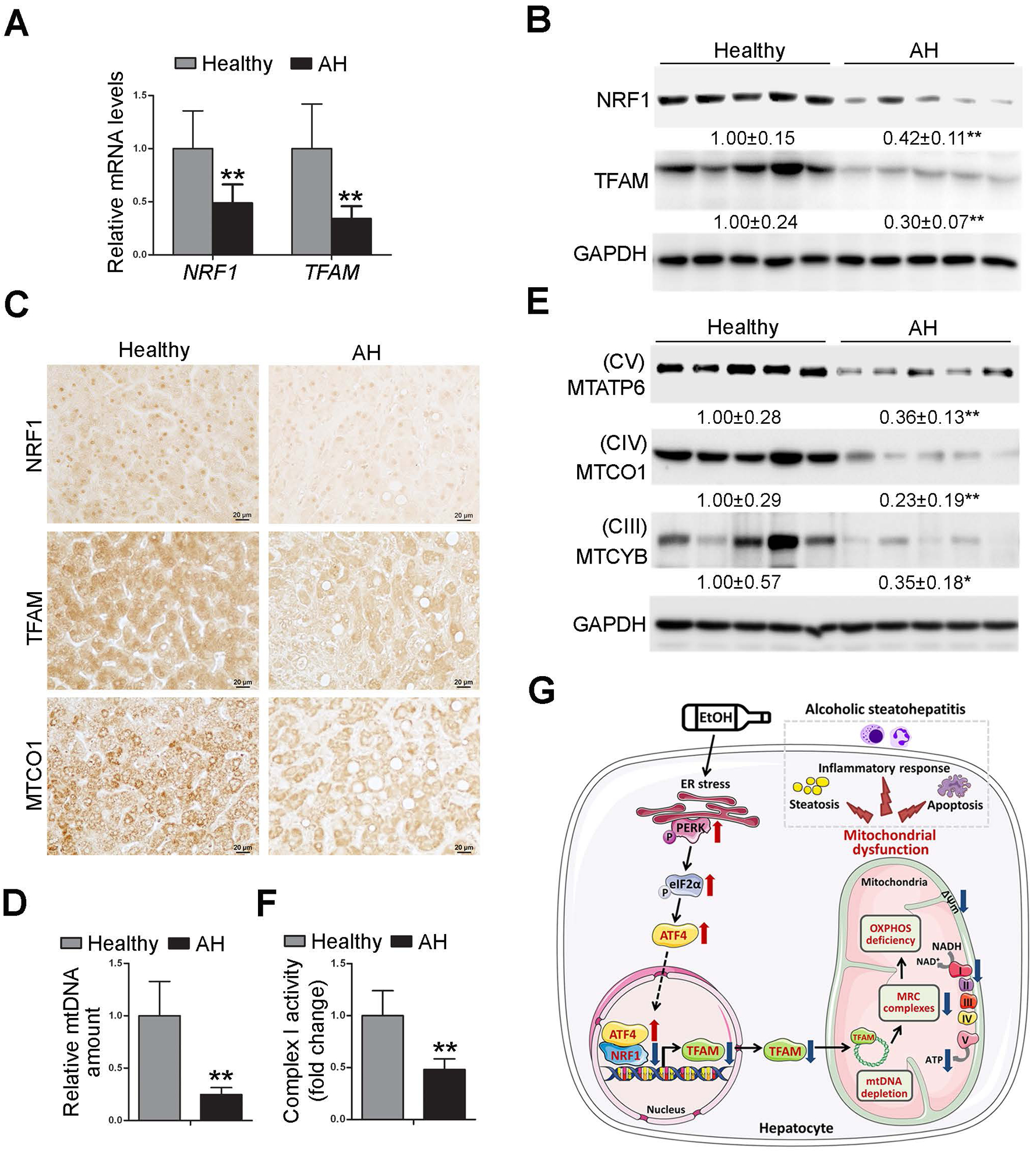
We obtained liver tissues from severe ALD patients and healthy donors. (A) The mRNA levels of hepatic NRF1 and TFAM (n=5). (B) Western blot analysis of hepatic NRF1 and TFAM. (C) Immunohistochemistry staining of NRF1, TFAM, and MTCO1 in the liver. Images were captured by light microscope. Scale bars: 20 μm. (D) mtDNA levels (MTND1) relative to nuclear DNA (SDHA) in the liver (n=5). (E) Western blot analysis of hepatic MTATP6, MTCO1, and MTCYB (n=5). (F) Hepatic complex I activity (n=5). Protein bands intensity was quantified by ImageJ (NIH). Data are presented as means ± SD. Statistical comparisons were made using Student’s t-test (n=5). *P<0.05, **P<0.01 vs. healthy controls.
Discussion
The present study demonstrated, for the first time, that ATF4 plays a critical role in the development of ALD by transmitting the cellular stress from ER to mitochondria. We observed that the PERK-eIF2α-ATF4 ER stress signaling pathway is activated in the livers of patients with severe AH. Using hepatocyte-specific ATF4 knockout mice and ATF4 deficiency VL-17A cell lines, we identified that hepatocyte ATF4 serves as a critical mediator in alcohol-impaired mitochondrial biogenesis and respiratory functions. Investigations of the underlying mechanisms revealed that hepatic TFAM inhibition, resulting from transcriptional suppression of NRF1, is mechanistically involved in ATF4-mediated mitochondrial dysfunction in the pathogenesis of ALD. The protective effects of hepatic ATF4 deletion and TFAM overexpression in alcohol-mediated mitochondrial dysfunction were linked to the amelioration of liver injury, which was evidenced by decreased hepatic steatosis, inflammatory response, and apoptosis (Fig. 8G).
Previous study have reported that hepatic knockout of ATF4 in mice alleviates alcoholic steatosis by inhibiting the expression of lipogenic genes [16], including fatty acid synthase (FAS) and Acetyl-CoA carboxylase (ACC). In this study, we confirmed the detrimental role of hepatic ATF4 in alcoholic steatosis. Our results also revealed that lack of ATF4 in the liver protects alcohol-induced oxidative stress, immune cells infiltration, and apoptosis. However, we found that the expression levels of FAS and ACC were significantly decreased after chronic ethanol feeding and not affected by ATF4 deletion (Supplementary Fig. 2D). One possible explanation for these discrepancies is that the different feeding method employed. In comparison with 4 weeks ethanol feeding model used by Li et al., our 8 weeks modified NIAAA model generates more damage to the liver and is therefore more clinically relevant. Indeed, the protein levels of hepatic FAS and ACC were also dramatically decreased in AH patients (Supplementary Fig. 2D).
Previous studies have reported that CHOP, a downstream target of ATF4, is activated after alcohol feeding and is involved in ER stress-induced cell death and mitochondrial dysfunction [24, 25]. Unexpectedly, hepatic abrogation of ATF4 did not affect CHOP protein levels in the liver regardless of alcohol exposure. Furthermore, the mRNA levels of hepatic Bcl-2-like protein 11 (Bim) and Death receptor 5 (DR5), two transcriptional downstream targets of CHOP [26, 27], were also not affected by ATF4 ablation (Supplementary Fig. 4C). These results suggested that AFT4 is dispensable for CHOP induction during alcohol-induced ER stress. In addition, lack of ATF4 in intestinal epithelial cells even leads to CHOP induction in ileum and colon [28]. It is noteworthy that a growing number of studies demonstrated that the ATF6 branch of the UPR is also a critical determinant of CHOP dynamics during ER stress [29–31]. Thus, further investigation is required to clarify the mechanisms of CHOP induction after ATF4 deletion in ALD.
TFAM is one of the most abundant mitochondria-localized DNA-binding proteins that control mtDNA replication, transcription, and packaging [32–34]. Whole-body TFAM deletion is embryonically lethal due to severe mtDNA deletion in mice [35], and tissue-specific lack of TFAM disrupts OXPHOS in different cell populations and generates a variety of alterations that recapitulate phenotypes of mitochondrial diseases in human and mice [6, 36–38]. In this study, we found that ATF4 is a negative regulator of TFAM in hepatocyte. Our results demonstrated that ATF4-mediated disruption of the TFAM-mtDNA signaling axis is a pathological factor for alcohol-injury mitochondrial dysfunction and liver injury. This notion is supported by several findings. Firstly, hepatic ATF4 ablation enhanced TFAM expression in vitro and in vivo. Lack of ATF4 protected against alcohol-decreased TFAM expression and mtDNA contents in hepatocyte. Conversely, forced ATF4 overexpressing in hepatocyte is sufficient leads to TFAM suppression and mtDNA content reduction. Secondly, the lack of TFAM abrogated the protective effects of ATF4 ablation on alcohol-induced mitochondrial dysfunction and cell death. Thirdly, hepatocyte-specific TFAM overexpression prevented alcohol-induced mitochondrial DNA deletion, complex I activity reduction, mitochondrial membrane potential disruption, and OXPHOS dysfunction. Finally, TFAM overexpression in hepatocyte markedly ameliorated alcohol-induced steatosis, apoptosis, and inflammatory cell infiltration in the liver. Collectively, these results suggest ATF4-mediated TFAM suppression mechanistically links to alcohol-induced mitochondrial dysfunction and liver damage.
NRF1 is a nuclear transcription factor that functions principally as a positive regulator of mitochondrial biogenesis-related genes, including TFAM [39, 40]. Specific and directly binding of NRF1 to TFAM promoter has been demonstrated by chromatin immunoprecipitation. NRF1 silencing produced a 1:1 knockdown of TFAM expression and decreased mtDNA contents [41, 42]. Sharing the same phenotype as TFAM, the genetic ablation of NRF1 also led to embryonically lethal [43]. These findings suggest that NRF1 is the master regulator in TFAM-mediated mtDNA homeostasis. In the present study, we found that both the protein and mRNA levels of NRF1 were remarkably higher in the liver of ATF4ΔHep mice than that in ATF4 floxed mice, regardless of alcohol administration. Our cell culture studies showed that ATF4 overexpression decreased NRF1 expression, but ATF4 knockdown enhanced NRF1 expression. We further found that ATF4 directly suppressed NRF1 transcription by binding at the CRE site in the NRF1 promoter. Moreover, NRF1 overexpression reversed ATF4-mediated TFAM inhibition and mtDNA depletion, suggesting the ATF4-NRF1 interaction and the subsequent TFAM suppression contribute to alcohol-induced mitochondrial dysfunction and liver injury. However, we could not rule out any other pathological factors that might be involved in ATF4-mediated mitochondrial dysfunction in ALD. Previous study reported that 8-week ethanol feeding plus-one binge strongly induced ER stress in the liver, which was associated with increased fat-specific protein 27 (FSP27) expression [44, 45]. Hepatic deletion of FSP27 protects alcohol-induced mitochondrial dysfunction but not ER stress [44]. Interestingly, ATF4 can induce the expression of peroxisome proliferator-activated receptor gamma (PPARγ) [46], a transcription factor known to promote the expression of FSP27α [47]. Therefore, further studies are necessary to test whether hepatic PPARγ-FSP27 axis upregulation is involved in ATF4-mediated mitochondrial dysfunction in ALD.
In summary, the present study uncovered a novel ATF4-NRF1-TFAM signaling pathway in the pathogenesis of alcoholic steatohepatitis. Our data demonstrate that hepatic ATF4 activation mediates alcohol-induced steatohepatitis by impairing mitochondrial biogenesis and respiratory function. Mechanistically, ATF4 negatively regulates TFAM via transcriptionally repressing NRF1 in the hepatocytes. Furthermore, the clinical relevance between ATF4 activation and NRF1-TFAM signaling pathway disruption was validated in the liver tissues from patients with alcoholic hepatitis. Therefore, targeting ATF4 or downstream NRF1-TFAM signals may be potential strategies for the prevention and treatment of alcoholic liver disease.
Supplementary Material
Significance of this study
What is already known on this subject?
Hepatic ATF4 is activated by chronic alcohol exposure in mice and liver-specific ATF4 deletion protects alcoholic steatosis.
Mitochondrial dysfunction contributes to alcohol-induced liver injury in mice.
What are the new findings?
Hepatocyte-specific ablation of ATF4 protects alcohol-induced mitochondrial dysfunction in mouse liver.
ATF4 acts as a negative regulator of the NRF1-TFAM signaling pathway in vivo and in vitro.
Overexpressing TFAM in hepatocyte protects against alcohol-induced mitochondrial dysfunction and steatohepatitis in mice.
The clinical relevance between ATF4 activation and NRF1-TFAM signaling pathway disruption is validated in the liver tissues from patients with severe alcoholic hepatitis.
How might it impact on clinical practice in the foreseeable future?
These findings suggest that expression of ATF4 in hepatocytes plays a critical role in the development of alcoholic liver disease. Targeting ATF4 and downstream NRF1-TFAM signals may be potential strategies for the prevention and treatment of alcoholic liver disease.
Acknowledgment:
We thank Dr. Christopher M. Adams at the University of Iowa for kindly providing the ATF4-floxed mice, and Dr. Dahn L. Clemens at the University of Nebraska for kindly providing the VL-17A cell line.
Financial support: This research was supported by the National Institutes of Health grants R01AA018844 (Zhanxiang Zhou), R01AA020212 (Zhanxiang Zhou), R24AA025017 (Zhaoli Sun), and Postdoctoral Fellowship Award from American Liver Foundation (Liuyi Hao).
Abbreviation:
- ALD
alcoholic liver disease
- PERK
protein kinase R (PKR)-like endoplasmic reticulum kinase
- AH
alcoholic hepatitis
- eIF2α
eukaryotic translation initiation factor 2A
- ATF4
activating transcription factor 4
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- H&E
hematoxylin and eosin
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- CHOP
C/EBP homologous protein
- PUMA
p53 upregulated modulator of apoptosis
- Bcl-2
B-cell lymphoma 2
- cCASP3
cleaved caspase-3
- NRF1
nuclear respiratory factor 1
- TFAM
mitochondrial transcription factor A
- OXPHOS
oxidative phosphorylation
- ROS
reactive oxygen species
- MFI
mean fluorescence intensity
- ATP
Adenosine triphosphate
- mtDNA
mitochondrial DNA
- MTCO1
cytochrome c oxidase subunit 1
- MTATP6
mitochondrial encoded ATP synthase membrane subunit 6
- SDH
succinate dehydrogenase
- TMRE
tetramethylrhodamine, ethyl ester
- ADH
alcohol dehydrogenases
- CYP2E1
cytochrome P450 2E1
- OCR
oxygen consumption rate
- NAD
nicotinamide adenine dinucleotide
- MTCYB
Cytochrome b
- NDUFB8
ubiquinone oxidoreductase subunit B8
- ND2
NADH dehydrogenase 2
- ND6
NADH dehydrogenase 6
- COX5b
Cytochrome c oxidase subunit 5B
- Bim
Bcl-2-like protein 11
- DR5
death receptor 5
- UQCRC2
ubiquinol-cytochrome C reductase core protein 2
- MPO
Myeloperoxidase
- F4/80
EGF-like module-containing mucin-like hormone receptor-like 1
- UPR
unfolded protein response
- ACC
acetyl-CoA carboxylase
- FAS
fatty acid synthase
- FSP27
fat-specific protein 27
- PPARγ
peroxisome proliferator-activated receptor gamma
- CRE
cAMP response element
Footnotes
Conflicts of Interest: No potential conflicts of interest relevant to this article are reported.
Patient consent: Obtained.
Reference
- [1].Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Seitz HK, Bataller R, Cortez-Pinto H, et al. Alcoholic liver disease. Nature reviews Disease primers 2018;4:16. [DOI] [PubMed] [Google Scholar]
- [3].Thursz M, Kamath PS, Mathurin P, et al. Alcohol-related liver disease: Areas of consensus, unmet needs and opportunities for further study. Journal of hepatology 2019;70:521–530. [DOI] [PubMed] [Google Scholar]
- [4].Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nature cell biology 2018;20:745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Koopman WJ, Distelmaier F, Smeitink JA, et al. OXPHOS mutations and neurodegeneration. The EMBO journal 2013;32:9–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baixauli F, Acin-Perez R, Villarroya-Beltri C, et al. Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell metabolism 2015;22:485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang J, Silva JP, Gustafsson CM, et al. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proceedings of the National Academy of Sciences of the United States of America 2001;98:4038–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mansouri A, Gattolliat CH, Asselah T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018;155:629–647. [DOI] [PubMed] [Google Scholar]
- [9].Fromenty B, Grimbert S, Mansouri A, et al. Hepatic mitochondrial DNA deletion in alcoholics: association with microvesicular steatosis. Gastroenterology 1995;108:193–200. [DOI] [PubMed] [Google Scholar]
- [10].Tang C, Liang X, Liu H, et al. Changes in mitochondrial DNA and its encoded products in alcoholic cirrhosis. International journal of clinical and experimental medicine 2012;5:245–250. [PMC free article] [PubMed] [Google Scholar]
- [11].Ji C New Insights into the Pathogenesis of Alcohol-Induced ER Stress and Liver Diseases. International journal of hepatology 2014;2014:513787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011;53:1752–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nishitoh H CHOP is a multifunctional transcription factor in the ER stress response. Journal of biochemistry 2012;151:217–219. [DOI] [PubMed] [Google Scholar]
- [14].Xiao Y, Deng Y, Yuan F, et al. An ATF4-ATG5 signaling in hypothalamic POMC neurons regulates obesity. Autophagy 2017;13:1088–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yoshizawa T, Hinoi E, Jung DY, et al. The transcription factor ATF4 regulates glucose metabolism in mice through its expression in osteoblasts. The Journal of clinical investigation 2009;119:2807–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li K, Xiao Y, Yu J, et al. Liver-specific Gene Inactivation of the Transcription Factor ATF4 Alleviates Alcoholic Liver Steatosis in Mice. The Journal of biological chemistry 2016;291:18536–18546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Seo J, Fortuno ES 3rd, Suh JM, et al. Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes 2009;58:2565–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Quiros PM, Prado MA, Zamboni N, et al. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. The Journal of cell biology 2017;216:2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hao L, Sun Q, Zhong W, et al. Mitochondria-targeted ubiquinone (MitoQ) enhances acetaldehyde clearance by reversing alcohol-induced posttranslational modification of aldehyde dehydrogenase 2: A molecular mechanism of protection against alcoholic liver disease. Redox biology 2018;14:626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hori O, Ichinoda F, Tamatani T, et al. Transmission of cell stress from endoplasmic reticulum to mitochondria: enhanced expression of Lon protease. The Journal of cell biology 2002;157:1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xie Y, He Y, Cai Z, et al. Tauroursodeoxycholic acid inhibits endoplasmic reticulum stress, blocks mitochondrial permeability transition pore opening, and suppresses reperfusion injury through GSK-3ss in cardiac H9c2 cells. American journal of translational research 2016;8:4586–4597. [PMC free article] [PubMed] [Google Scholar]
- [22].Chen Q, Thompson J, Hu Y, et al. Metformin attenuates ER stress-induced mitochondrial dysfunction. Translational research : the journal of laboratory and clinical medicine 2017;190:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Koyanagi S, Hamdan AM, Horiguchi M, et al. cAMP-response element (CRE)-mediated transcription by activating transcription factor-4 (ATF4) is essential for circadian expression of the Period2 gene. The Journal of biological chemistry 2011;286:32416–32423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang W, Zhong W, Sun Q, et al. Adipose-specific lipin1 overexpression in mice protects against alcohol-induced liver injury. Scientific reports 2018;8:408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen X, Zhong J, Dong D, et al. Endoplasmic Reticulum Stress-Induced CHOP Inhibits PGC-1alpha and Causes Mitochondrial Dysfunction in Diabetic Embryopathy. Toxicological sciences : an official journal of the Society of Toxicology 2017;158:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Puthalakath H, O’Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007;129:1337–1349. [DOI] [PubMed] [Google Scholar]
- [27].Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. The Journal of biological chemistry 2004;279:45495–45502. [DOI] [PubMed] [Google Scholar]
- [28].Hu X, Deng J, Yu T, et al. ATF4 Deficiency Promotes Intestinal Inflammation in Mice by Reducing Uptake of Glutamine and Expression of Antimicrobial Peptides. Gastroenterology 2019;156:1098–1111. [DOI] [PubMed] [Google Scholar]
- [29].Fusakio ME, Willy JA, Wang Y, et al. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Molecular biology of the cell 2016;27:1536–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yang H, Niemeijer M, van de Water B, et al. ATF6 Is a Critical Determinant of CHOP Dynamics during the Unfolded Protein Response. iScience 2020;23:100860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lebeaupin C, Vallee D, Hazari Y, et al. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. Journal of hepatology 2018;69:927–947. [DOI] [PubMed] [Google Scholar]
- [32].Kang I, Chu CT, Kaufman BA. The mitochondrial transcription factor TFAM in neurodegeneration: emerging evidence and mechanisms. FEBS letters 2018;592:793–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Matsushima Y, Goto Y, Kaguni LS. Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM). Proceedings of the National Academy of Sciences of the United States of America 2010;107:18410–18415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dolle C, Flones I, Nido GS, et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nature communications 2016;7:13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Larsson NG, Wang J, Wilhelmsson H, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nature genetics 1998;18:231–236. [DOI] [PubMed] [Google Scholar]
- [36].Vernochet C, Mourier A, Bezy O, et al. Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell metabolism 2012;16:765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Koh JH, Johnson ML, Dasari S, et al. TFAM Enhances Fat Oxidation and Attenuates High-Fat Diet-Induced Insulin Resistance in Skeletal Muscle. Diabetes 2019;68:1552–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li H, Wang J, Wilhelmsson H, et al. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America 2000;97:3467–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kiyama T, Chen CK, Wang SW, et al. Essential roles of mitochondrial biogenesis regulator Nrf1 in retinal development and homeostasis. Molecular neurodegeneration 2018;13:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays in biochemistry 2010;47:69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Piantadosi CA, Suliman HB. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. The Journal of biological chemistry 2006;281:324–333. [DOI] [PubMed] [Google Scholar]
- [42].Gao W, Wu M, Wang N, et al. Increased expression of mitochondrial transcription factor A and nuclear respiratory factor-1 predicts a poor clinical outcome of breast cancer. Oncology letters 2018;15:1449–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Huo L, Scarpulla RC. Mitochondrial DNA instability and peri-implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Molecular and cellular biology 2001;21:644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xu MJ, Cai Y, Wang H, et al. Fat-Specific Protein 27/CIDEC Promotes Development of Alcoholic Steatohepatitis in Mice and Humans. Gastroenterology 2015;149:1030–1041 e1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liangpunsakul S, Gao B. Alcohol and fat promote steatohepatitis: a critical role for fat-specific protein 27/CIDEC. Journal of investigative medicine : the official publication of the American Federation for Clinical Research 2016;64:1078–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yu K, Mo D, Wu M, et al. Activating transcription factor 4 regulates adipocyte differentiation via altering the coordinate expression of CCATT/enhancer binding protein beta and peroxisome proliferator-activated receptor gamma. The FEBS journal 2014;281:2399–2409. [DOI] [PubMed] [Google Scholar]
- [47].Matsusue K, Kusakabe T, Noguchi T, et al. Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell metabolism 2008;7:302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.