

Abstract
Why certain people relish healthy aging throughout their life span while others suffer pathological consequences? In this review, we focus on some of the dominant paradigms of pathological aging, such as amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), and Parkinson’s disease (PD), and predict that the antioxidant superoxide dismutase 1 (SOD1), when post-translationally modified by aging-associated oxidative stress, acts as a mechanism to accelerated aging in these age-related neurodegenerative diseases. Oxidative modifications of natively reduced SOD1 induce pathological confirmations such as misfolding, leading to a subsequent formation of monomeric, oligomeric, and multimeric aggregates. Misfolded SOD1 propagates like prions from cell to cell. These modified conformations are detected in brain tissues in ALS, AD, and PD, and are considered a contributing factor to their initial pathogenesis. We have also elaborated on oxidative stress-induced non-native modifications of SOD1 and offered a logistic argument on their global implication in accelerated or pathological aging in the context of ALS, AD, and PD.
Keywords: SOD1, Oxidative stress, Post-translational modification, Pathological aging, Amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), Parkinson’s disease (PD)
Introduction
The primeval pursuit of immortality is reflected in the extended life expectancy of the twenty-first century [1]. Unfortunately, the extra years of life are mostly burdened with age-related diseases. For instance, a demographic transformation, where the old age population outnumbers the children first ever in history, will be striking the USA by 2030 [2]. The senior and geriatric population will reach 2.1 billion by 2050. In recent years, this alarming shift of population aging is metaphorically termed as silver tsunami or gray tsunami [3]. The catastrophic tide of age-related chronic diseases threatens to overwhelm our health care system, finance, and quality of life. As a result, endeavors targeting the delayed onset of pathological aging have drawn significant attention in recent studies [1]. Pathological aging is an accelerated aging marked by the symptomatic existence of one or more age-related diseases, which may occur secondarily to genetic predisposition, infections, immunodeficiency, malnutrition, hormonal dysregulation, enzymatic malfunction, physical and mental stress, and trauma [4]. The functional deterioration in patients is variable depending upon the disease pathology. Externally, the onset of pathological aging is identified by the appearance of disease-specific symptoms, whereas, at the molecular level, the transition between healthy aging and pathological aging could be subtle and indistinguishable [4, 5]. However, a rational approach to increase the healthspan is to delay the onset of pathological aging by understanding global molecular changes associated with aging that might alter the internal homeostasis towards disease pathogenesis.
Whether aging is a disease or not is a long-run debate. We consider biological aging as an entity that includes both physiological and healthy aging. Healthy aging is constant, slowly progressive, and regulated by an active repair mechanism [6]. Contrary to this, pathological aging is selective and accelerated that features the incompetence of the intrinsic repair mechanisms and pre-clinical or clinical existence of one or more age-related diseases [7]. Comprehensively, in healthy aging, there is a constant balance between the damage accumulation and damage repair, known as a quasi-stable equilibrium state [6]. When this equilibrium is lost, the majority of cases end up suffering pathological aging. The universal criteria of age-related diseases are aging itself [8]. The commencement of pathological aging is marked by the onset of these debilitative conditions that potentially shortens our healthy lifespan. Hence, it is crucial to identify mechanistic aggravators of pathological aging.
Super oxide dismutase 1 (SOD1) is an endogenous protein that constitutes the earliest defense mechanism against bio-reactive free radicals. The efficiency of SOD1 in coping up with oxidative stress in the first place could be a major determinant of further consequences initiated by the free radicals [9]. In contrast to its universal role as an antioxidant, post-translational modifications (PTMs) associated with SOD1 such as phosphorylation, acetylation, oxidation, carbonylation, misfolding, aggregation, and its crosslinking with other pathological agents are currently implicated in the global aging process [10, 11]. Despite a considerable amount of study, the alleged toxic gain of function in SOD1 remains scattered and elusive [12]. The review aims to concise the argument of SOD1 post-translational modification in pathological aging by prioritizing the following crucial aspects: biological consequences of toxic conformations of SOD1, particularly in the aging population; toxic conformations of SOD1 as an aging factor in the disease pathogenesis in amyotrophic lateral sclerosis (ALS), and PTM SOD1 as a universal inducer of pathological aging.
Aging-associated oxidative modification of SOD1
Aging-associated oxidative stress induces alterations in SOD1 that are extensively studied and highly relevant to accelerated aging in ALS patients [13]. An earlier study has claimed that SOD1 can adopt more than 44 conformations depending upon the variety and extent of post-translational modifications, which exert physiological and pathological implications [14]. It is uncertain whether these modifications occur sequentially, one after another, until they reach the final toxic forms. Studies suggest that the pathogenicity of the intermediate conformations like misfolded or oligomeric SOD1 is more detrimental compared to the initial oxidized ones or the multimeric aggregates [15, 16]. Besides, oxidative stress may not be the only stimulus to provoke pathological modification. For example, irrespective of aging, protein may directly misfold by mutation, translational errors, or aberrant post-translational modifications and then initiate the oligomeric and multimeric transformations [17]. In contrast, oxidative modification of SOD1 appears more relevant and substantial during pathological aging [18]. Oxidation is one of the predominant non-enzymatic post-translational modifications associated with the aging process. It contributes to chemical modification at the susceptible side chains that shift the conformational stability towards the aggregation-prone state [19]. Oxidized proteins are associated with a number of age-related conditions such as ALS, Alzheimer’s disease, Parkinson’s disease, cataracts, rheumatoid arthritis, respiratory distress syndrome, and progeria [20, 21]. The thiol groups in cysteine residue are particularly susceptible to redox modification, and irreversible oxidation of a free single cysteine residue can substantially influence the native folding of a globular protein [22]. Cysteine residues in proteins can exist as different redox forms, among which thiol (-SH), disulfide forms (-S-S-) are the most common, but derivatives with a higher oxidation state such as sulfenic (RSOH), sulfinic (RSO2H), and sulfonic (RSO3H) acid are detected in a growing number of proteins. Sulfonic acid is the most highly oxidized thiol species, and its formation is irreversible [23]. Being an essential antioxidant, SOD1 is least expected to be damaged by oxidative stress, yet oxidative stress-induced post-translational modifications of SOD1 are studied extensively, mostly because of its structural conformation and close amalgamation with free radical homeostasis [24, 25]. A mass spectrometric analysis confirmed the oxidation of WT hSOD1 when exposed to H2O2. Electron capture dissociation (ECD) showed that the residue cysteine-111 in SOD1 was initially targeted for oxidative modification to form irreversible sulfonic acid [13]. It corresponds to our lab findings, where the pegylation reaction identified SOD1 oxidation in G93A transgenic mice [23]. Besides, point mutation of the cysteine residues in SOD1 recognized cysteine-111 as the primary target of PTM by oxidation.
Along with these, several other studies have addressed SOD1 oxidation at cysteine-111 residue as a preliminary stress response occurring to both wild type and mutant variants [26, 27]. It is understandable due to its natively reduced thiol state (-SH) and surface localization on the protein, offering more exposure to environmental stress [23]. Cysteine exhibits an extreme conservation pattern; for a strictly preserved cysteine residue in a protein, the degree of conservation is around 90%, for instance, the stable disulfide bond between cysteine 57 and cysteine 146. Whereas in a susceptible location, conservation efficiency is less than 10%, such as surface-exposed cysteine-111 in SOD1 [28].
Polarity-wise, cysteine is considered a hydrophobic residue because it prefers to stay buried within the protein, quite similar to the cysteine 6 residue in SOD1. Studies argue that this is likely evolutionary to avoid the existence of unpaired cysteine on the protein surface [29]. Nevertheless, it helps explain why cysteine-111 is not well tolerant of free radicals. Another notable feature of cysteine is its inclination towards cluster formation with other reduced cysteine residues nearby, a criterion of redox sensitivity. The burial tendency and the cluster forming nature imply that the cysteine-111, which is exposed and isolated/unpaired on the protein surface, is the minimally protected cysteine in SOD1 and a delicate target for oxidative modification [29].
Apart from cysteine-111, several other residues are described to be modified post-translationally. For instance, high molecular weight SOD1 aggregates that increased up to 40-fold in aged yeast cells consisted of oxidized cysteine 146, histone 120, and histone 71 residues as well [27]. Other forms of redox modification such as carbonylation, glutathionylation, palmitoylation, and nitration are detected at cysteine-111, cysteine 146, cysteine 57, and W32 residues of SOD1 [24]. Overall, oxidation of SOD1 is, so far, the most deliberate form of post-translational modification with potential consequences of misfolding and aggregate formation (Fig. 1).
Fig. 1.
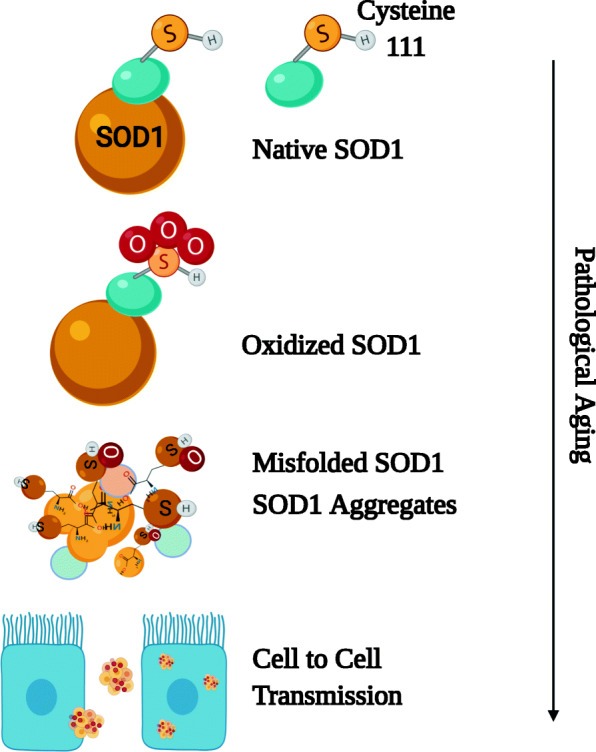
Post-translational modifications of SOD1. Unwarranted escalation of oxidative stress during aging could irreversibly oxidize the SOD1 protein at its cysteine-111 residue, culminating in protein misfolding and aggregate formation
Misfolding of SOD1
Protein misfolding refers to folding into an incorrect three-dimensional configuration that is typically non-functional and often resistant to breakdown [30]. Although the intramolecular disulfide bridge facilitates SOD1 stability, oxidative modifications could destabilize the protein and lead to misfolding before aggregate formation. These misfolded conformations are unusually stable, which is more often a feature of the natively folded protein [31].
However, biophysically, the SOD1 proteins are stuffed with beta-sheets, stabilized by H-bonds, linking the NH and CO groups of two distinct polypeptides pleated β strands [32]. While in α-helix, the H-bonds are formed between the NH group of one amino acid residue and the CO group of another amino acid residue within the same strand. Overall, in β sheets, the bonds are between 2 separate strands. Hence, the second strand could belong to different domains of the same protein or a separate molecule, giving rise to a stably crosslinked oligomeric or multimeric β sheet structure. Thus, the misfolded SOD1 self-associates or crosslinks with other proteins to deposit in various cells or tissues [30].
The proteins identified so far to be involved in conformational diseases have no specific sequence or structural resemblance [30, 33]. Specific epitopes in SOD1 are identified to be exposed by misfolding, which are utilized to raise conformation-specific antibodies [34]. Human SOD1 contains a masked Derlin-1 binding region (DBR) expanding over amino acid residues 6-16, which upon exposure, captures the essential Derlin protein of the EARD complex to induce ER stress [35]. The unveiled DBR region in misfolded SOD1 could be a potential therapeutic target for selective removal of aberrant SOD1 conformations without interrupting the native ones. Misfolded SOD1 may trigger detrimental consequences such as metal loss, disulfide bond reduction, monomerization, and further protein aggregation [36].
Aggregation and prion-like propagation of SOD1
Aggregates refer to insoluble pathological inclusions in the brain tissues predominantly featured in the end stage of neurodegenerative diseases [37]. Oxidatively modified SOD1 adopts a misfolded conformation that exposes the protein’s hydrophobic sequences to interact covalently with the exposed hydrophobic patches of other misfolded SOD1 in its vicinity. Such oligomers even offer covalent attachment to other native proteins and construct high molecular weight (HMW) aggregates [38]. The oligomers could be considered more toxic conformations as they resemble an active spreading state. In contrast, a relevant study has claimed that the hydrophobic residues tend to get concealed or dipped within the large aggregates, thereby acting in a neuroprotective manner by sequestering the toxic SOD1 species [15].
The term “prion-like propagation” is implicated conspicuously because prion diseases are infectious conditions, unlike neurodegenerative diseases such as ALS [39]. However, post-translationally modified SOD1 shares some of the mechanical properties of infectious prion aggregates, such as self-seeding and cross seeding with each other intracellularly and cell-to-cell transmissions [40]. It is hypothesized that misfolded SOD1 acts as a structural template that interacts with its natural counterpart and induces misfolding of the captured protein in a template-directed reaction [40]. Elongation of the misfolded SOD1 fibrils encounters spontaneous breakage that exposes new ends to allow prion-like replication of more native proteins and the spreading of the self-propagating core [38]. Also, the cell-to-cell transmission is another prion-like feature shared by modified SOD1 aggregates [35]. The dissemination into the extracellular space might occur, followed by cell death or exosome-mediated vesicular transport [41]. The large aggregates stimulate micropinocytosis in neurons to expedite their propagation [35]. Overall, once the protein is oxidatively modified to a critical limit, a chain reaction is instigated through subsequent misfolding, aggregation, and prion-like propagation, which may not precisely comply with the corresponding order.
The protein misfolding and aggregation theory is argued from different standpoints. Protein misfolding is autonomous of aggregation, which means the latter is an optional endpoint of conformational protein changes. Again, an opposite theory predicts aggregation as the primary inducer and stabilizer of the misfolded proteins [42]. It is wildly speculated that various mutant SOD1 variants acquire a toxic property to induce motor neuron death in ALS, although the gain of function is not yet well defined. Aging-related post-translational modifications of SOD1 offer a plausible explanation for the toxic gain of function. So far, the post-translational modifications stated above overlap each other and exhibit conformational plasticity, which means the potential to adopt multiple stable tertiary folds [43]. Besides, the molecular aging proceeds internally before the symptoms become evident [44]. In this regard, it is crucial to determine the initial appearance of these non-native conformations. It has been claimed that the aggregate formation in ALS is mostly an end-stage phenomenon [45], which indicates that oxidized or oligomeric SOD1 could be detected in a substantial amount at the presymptomatic phase in attainable samples like CSF or serum. Perhaps, the hypothesis might be tested initially in populations with a genetic predisposition to pathological aging.
SOD1 modification as a mechanism of global aging
Protein aggregation pathologies have gained importance concerning age-related neurodegenerative conditions since their topological distribution pattern and progression correlate with the clinical phenotypes of the patient [46]. Lately, aggregates composed of multiple pathological agents are being detected from patients phenotypically expressing one form of neurodegenerative diseases [47]. It raises the possibility of a potential primary inducer contributing to multiple neuropathological aging in diverse disease formats.
Amyotrophic lateral sclerosis and SOD1
Amyotrophic lateral sclerosis is an ideal example of pathological aging, a severely progressive and globally lethal age-related disease with underlying degeneration of upper and lower motor neurons (UMN & LMN) [48]. About 90% of the ALS cases are sporadic, while the remaining 10% are familial ALS linked to mutations in various genes, mostly chromosome-9 open reading frame 72 (C9ORF72), SOD1 (Superoxide dismutase 1), TAR DNA-binding protein (TARDBP), and fused in sarcoma (FUS) [48]. The peak age of disease onset is 58 to 63 years for the sporadic form and 47 to 52 years for familial cases. The average life expectancy with ALS is only 2-5 years after the disease onset [49], death occurring mostly due to the paralysis of respiratory muscles. The disease progression in ALS is fatal, involving neurons and glial cells, multiple organelles especially, ER and mitochondria, and initiates a cascade of inflammatory responses in the internal milieu [50]. A multifaceted disease pathology associated with ALS jeopardizes the therapeutic interventions due to which it is challenging to halt the disease progression. Instead, delaying the pathological aging in ALS by targeting the disease onset is a rational approach to increase the healthy life span in these patients.
Approximately 25% of familial ALS (fALS) are associated with genetic mutations in SOD1[10], whereas 5% of sporadic ALS (sALS) have shown alliance with the wild variety of SOD1. Transgenic mice overexpressing mutant forms of hSOD1 mimic the phenotypic trajectory of the human condition in ALS [11]. Studies with the G93A mouse model have shown that the onset of the disease is relative to the copy number of the hSOD1 transgene. For instance, G93A mice expressing 25, 18, 13, and 10 copies of human SOD1 have exhibited disease onset at around 90, 90-120, 200, and 300 days accordingly, which reflect a negative correlation between the levels of mutant SOD1 and the onset of pathological aging in fALS [51]. However, increasing evidence indicates that the toxic gain of function in SOD1 is the disease-causing mechanism in both fALS and sALS, where genetic predisposition could be considered an aggravating factor that accelerates the pathological aging further. It is speculated that both mutant or wild varieties of SOD1 form post-translationally modified non-native conformations that are adamant to breakdown by internal degradation machinery and exert their toxic effects like a cobweb manner over the native molecules and functioning apparatus of the cells [13].
Parkinson’s disease and SOD1
Histopathologically, Parkinson’s disease (PD) is characterized by the degeneration of dopaminergic neurons from the substantia nigra and the presence of intranuclear inclusions known as Lewy bodies. The Lewy bodies are misfolded, insoluble, fibrillated, and immuno-reactive alpha-synuclein inclusions along with ubiquitin deposition [52]. Alpha-synuclein is susceptible to PTM by phosphorylation in its serine 129 residues and contributes to abnormal cluster formation [53] that propagates toxic effects by synaptic vesicle impairment, mitochondrial dysfunction, ER stress, oxidative stress, autophagy, and lysosomal pathway dysfunction [54]. Compared to their widespread intra-neuronal and glial distribution [55], the susceptible regions are quite selective in PD, indicating the possible involvement of other influences in the disease pathology. Besides, therapies targeting alpha-synuclein brought disappointing results in clinical trials [56], supporting additional neurotoxic mechanisms in PD. It could be explained by the presence of a substantial amount of SOD1 aggregates in the vulnerable regions like substantia nigra (SN) and locus coeruleus (LC) in PD affected brain [56]. Similar regions in ALS affected brain also harbor these toxic conformations, indicating the universal incompetence of neurons in processing the aberrantly modified SOD1 [47]. Although direct physical interaction between alpha-synuclein and SOD1 is evident from earlier studies on PD, 50% of these SOD1 aggregates are independent of alpha-synuclein; they co-aggregate with other typical elements like ubiquitin [56]. The SOD1 proteinopathy in PD is still mostly speculative. Copper dysregulation and oxidative stress are potential catalysts in PD [57]. In this context, metal deficient and stress-induced modifications of SOD1 are likely relevant to PD pathogenesis. Besides, misfolded and copper deficit SOD1 appears in SN in the preliminary stage of the disease pathogenesis, even before the onset of neuronal loss [56], which substantiates the role of SOD1 as an instigator of pathological aging in PD (Fig. 2).
Fig. 2.
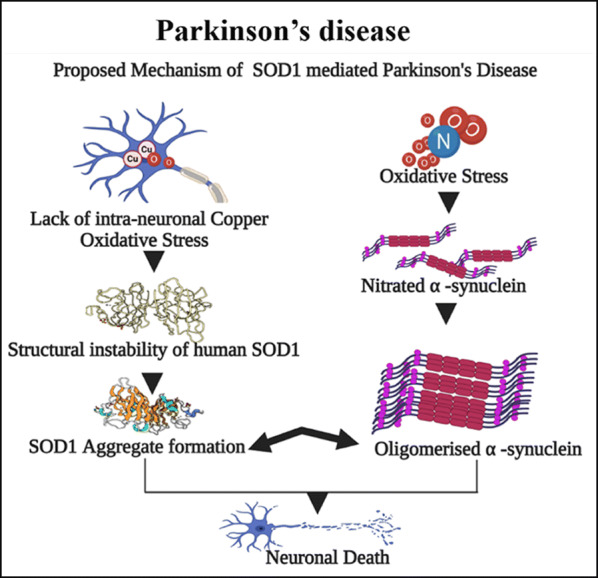
Direct physical interaction between SOD1 and alpha-synuclein promotes neuronal death in PD
Alzheimer’s disease and SOD1
Alzheimer’s disease, the most common form of dementia in the elderly [58], is characterized by extracellular beta-amyloid plaque (Aβ) and intracellular neurofibrillary tangles consisting of the microtubule-associated protein tau [59]. According to the amyloid hypothesis, the accumulation of Aβ is the pathogenesis associated with the onset and progression of the disease [60]. In contrast to Aβ, tau deposits correlate better with the disease course or the degree of cognitive impairment [58]. The primary prevention trials thus target the Aβ pathology [60]. However, at the incipient stage of the disease, the Aβ accumulates intracellularly. Hence, it is described as “a sign of worse things to come” [61]. What triggers the initial Aβ accumulation to progress into the advanced stages of extracellular plaque formation could persuade the process of pathological aging in Alzheimer’s disease (AD) [62]. Earlier, it was reported that SOD1 has a specific Aβ binding region with which it could interact with Aβ protein and initiate further aggregation and cell death [63]. Unpublished data from our lab shows that oxidized and misfolded SOD1 promotes intracellular Aβ aggregation, both in vivo, in vitro, and in the cell-free culture medium. Double transgenic mice (APP+/G37R+) have much aggressive expression of Aβ aggregates compared to APP+ transgenic mice. Besides, phenotypically, up to 50–60% of all ALS patients exhibit cognitive defects collectively known as the frontotemporal spectrum disorders of ALS (ALS-FTSD) [64], suggesting oxidatively modified SOD1 confirmations can induce Aβ aggregation (Fig. 3).
Fig. 3.
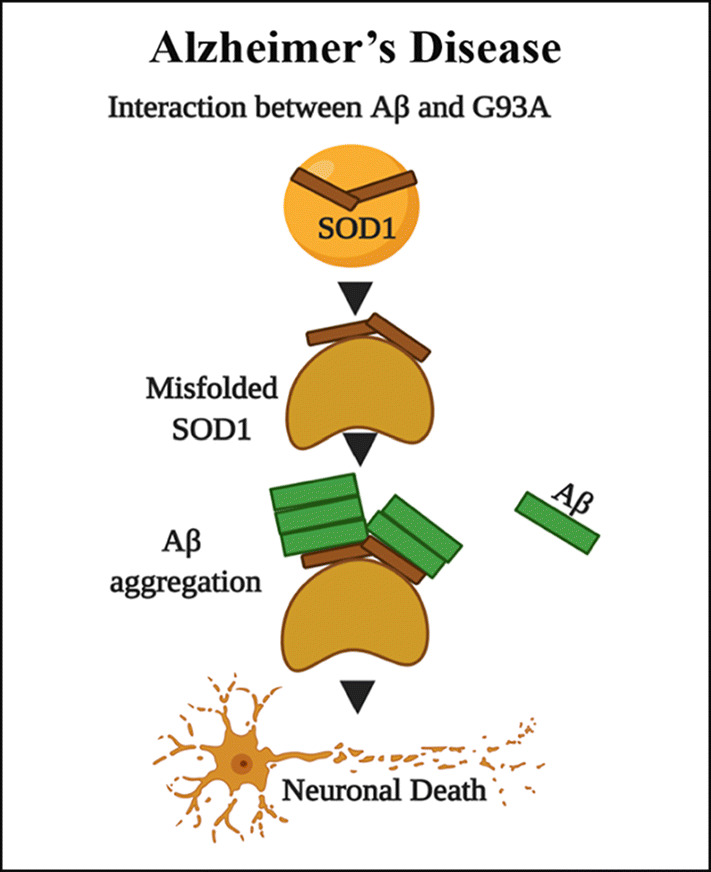
Mutant SOD1 increases intracellular Aβ aggregation
Conclusion
SOD1 is an essential antioxidant and directly relevant to aging-associated oxidative stress. The current review emphasizes the conformational changes of SOD1 implicated to accelerate the aging process, and is manifested in various aging-related neurodegenerative diseases like ALS, PD, and AD. It is imperative to investigate the specific structural alterations in the pathological forms of SOD1, which could further broaden our horizon to understand how the modified SOD1 is interacting with the other pathologically altered elements like alpha-synuclein and Aβ peptide. All pathogenic SOD1 conformations expose a short DBR, which can specifically bind to Derlin-1. Because DBR is a small and concealed segment in natively folded SOD1, it is relevant to expect any non-native conformation of SOD1 to expose the DBR. That means altered pathogenic conformations may share similar structural properties in SOD1. DBR could be a substantial experimental and therapeutic target to precisely identify the misfolded SOD1 in the context of pathological aging. In fact, meticulous knockdown strategies are required to identify and remove such aggregation-prone proteins. Therefore, more studies are warranted in this area to combat the universal aging process.
Funding
This work was supported by an Arthur J. Hudson Translation Team Grant from ALS Canada and Brain Canada and an operating grant from By-Health, China.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Crimmins EM. Lifespan and healthspan: past, present, and promise. Gerontologist. 2015;55(6):901–911. doi: 10.1093/geront/gnv130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ortman JM, Velkoff VA, Hogan H. An aging nation: the older population in the United States. United States Census Bureau, Economics and Statistics Administration, US; 2014.
- 3.Poirier TI, Butler LM, Devraj R, Gupchup GV, Santanello C, Lynch JC. A cultural competency course for pharmacy students. Am J Pharm Educ. 2009;73(5)81. 10.5688/aj730581. [DOI] [PMC free article] [PubMed]
- 4.Rajah M, Bastianetto S, Bromley-Brits K, Cools R, D’Esposito M, Grady C, et al. Biological changes associated with healthy versus pathological aging: a symposium review. Ageing Res Rev. 2009;8(2):140–146. doi: 10.1016/j.arr.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonker MJ, Melis JP, Kuiper RV, van der Hoeven TV, Wackers PF, Robinson J, et al. Life spanning murine gene expression profiles in relation to chronological and pathological aging in multiple organs. Aging Cell. 2013;12(5):901–909. doi: 10.1111/acel.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demetrius LA, Magistretti PJ, Pellerin L. Alzheimer’s disease: the amyloid hypothesis and the Inverse Warburg effect. Front Physiol. 2015;5:522. doi: 10.3389/fphys.2014.00522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13(1):179–189. doi: 10.1016/0197-4580(92)90027-U. [DOI] [PubMed] [Google Scholar]
- 8.Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012;22(17):R741–RR52. doi: 10.1016/j.cub.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 9.Medinas DB, Rozas P, Traub FM, Woehlbier U, Brown RH, Bosco DA, et al. Endoplasmic reticulum stress leads to accumulation of wild-type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2018;115(32):8209–8214. doi: 10.1073/pnas.1801109115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexander GM, Erwin KL, Byers N, Deitch JS, Augelli BJ, Blankenhorn EP, Heiman-Patterson TD. Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain Res Mol Brain Res. 2004;130(1-2):7–15. doi: 10.1016/j.molbrainres.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 11.De Giorgio F, Maduro C, Fisher EM, Acevedo-Arozena A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis Model Mech. 2019;12(1):dmm037424. 10.1242/dmm.037424. [DOI] [PMC free article] [PubMed]
- 12.Saccon RA, Bunton-Stasyshyn RK, Fisher EM, Fratta P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain. 2013;136(8):2342–2358. doi: 10.1093/brain/awt097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien JP, Brady ST, Brown RH., Jr Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13(11):1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huai J, Zhang Z. Structural properties and interaction partners of familial ALS-associated SOD1 mutants. Front Neurol. 2019;10:527. doi: 10.3389/fneur.2019.00527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gill C, Phelan JP, Hatzipetros T, Kidd JD, Tassinari VR, Levine B, et al. SOD1-positive aggregate accumulation in the CNS predicts slower disease progression and increased longevity in a mutant SOD1 mouse model of ALS. Sci Rep. 2019;9(1):1–13. doi: 10.1038/s41598-019-43164-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sangwan S, Zhao A, Adams KL, Jayson CK, Sawaya MR, Guenther EL, Pan AC, Ngo J, Moore DM, Soriaga AB, Do TD, Goldschmidt L, Nelson R, Bowers MT, Koehler CM, Shaw DE, Novitch BG, Eisenberg DS. Atomic structure of a toxic, oligomeric segment of SOD1 linked to amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2017;114(33):8770–8775. doi: 10.1073/pnas.1705091114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Díaz-Villanueva JF, Díaz-Molina R, García-González V. Protein folding and mechanisms of proteostasis. Int J Mol Sci. 2015;16(8):17193–17230. doi: 10.3390/ijms160817193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Migliore L, Coppedè F. Environmental-induced oxidative stress in neurodegenerative disorders and aging. Mutat Res. 2009;674(1-2):73–84. doi: 10.1016/j.mrgentox.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 19.Han Z-J, Feng Y-H, Gu B-H, Li Y-M, Chen H. The post-translational modification, SUMOylation, and cancer. Int J Oncol. 2018;52(4):1081–1094. doi: 10.3892/ijo.2018.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J, Li X, Li J-D. The roles of post-translational modifications on α-Synuclein in the pathogenesis of Parkinson’s diseases. Front Neurosci. 2019;13:381. doi: 10.3389/fnins.2019.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin MT, Beal MF. The oxidative damage theory of aging. Clin Neurosci Res. 2003;2(5-6):305–315. doi: 10.1016/S1566-2772(03)00007-0. [DOI] [Google Scholar]
- 22.Marinelli P, Navarro S, Graña-Montes R, Bañó-Polo M, Fernández MR, Papaleo E, Ventura S. A single cysteine post-translational oxidation suffices to compromise globular proteins kinetic stability and promote amyloid formation. Redox Biol. 2018;14:566–575. doi: 10.1016/j.redox.2017.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, Shang H, Qiu X, Fujiwara N, Cui L, Li X-M, Gao TM, Kong J. Oxidative modification of cysteine 111 promotes disulfide bond-independent aggregation of SOD1. Neurochem Res. 2012;37(4):835–845. doi: 10.1007/s11064-011-0679-8. [DOI] [PubMed] [Google Scholar]
- 24.Le Pecheur M, Bourdon E, Paly E, Farout L, Friguet B, London J. Oxidized SOD1 alters proteasome activities in vitro and in the cortex of SOD1 overexpressing mice. FEBS Lett. 2005;579(17):3613–3618. doi: 10.1016/j.febslet.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 25.Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi XF, Crow JP, Cashman NR, Kondejewski LH, Chakrabartty A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem. 2002;277(49):47551–47556. doi: 10.1074/jbc.M207356200. [DOI] [PubMed] [Google Scholar]
- 26.Martins D, English AM. SOD1 oxidation and formation of soluble aggregates in yeast: relevance to sporadic ALS development. Redox Biol. 2014;2:632–639. doi: 10.1016/j.redox.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilcox KC, Zhou L, Jordon JK, Huang Y, Yu Y, Redler RL, Chen X, Caplow M, Dokholyan NV. Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J Biol Chem. 2009;284(20):13940–13947. doi: 10.1074/jbc.M809687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marino SM, Gladyshev VN. Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J Mol Biol. 2010;404(5):902–916. doi: 10.1016/j.jmb.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagano N, Ota M, Nishikawa K. Strong hydrophobic nature of cysteine residues in proteins. FEBS Lett. 1999;458(1):69–71. doi: 10.1016/s0014-5793(99)01122-9. [DOI] [PubMed] [Google Scholar]
- 30.Dobson CM. Protein folding and misfolding. Nature. 2003;426(6968):884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 31.Soto C, Pritzkow S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat Neurosci. 2018;21(10):1332–1340. doi: 10.1038/s41593-018-0235-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ihara K, Fujiwara N, Yamaguchi Y, Torigoe H, Wakatsuki S, Taniguchi N, Suzuki K. Structural switching of Cu,Zn-superoxide dismutases at loop VI: insights from the crystal structure of 2-mercaptoethanol-modified enzyme. Biosci Rep. 2012;32(6):539–548. doi: 10.1042/bsr20120029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jahn TR, Radford SE. The Yin and Yang of protein folding. FEBS J. 2005;272(23):5962–5970. doi: 10.1111/j.1742-4658.2005.05021.x. [DOI] [PubMed] [Google Scholar]
- 34.Paré B, Lehmann M, Beaudin M, Nordström U, Saikali S, Julien JP, Gilthorpe JD, Marklund SL, Cashman NR, Andersen PM, Forsberg K, Dupré N, Gould P, Brännström T, Gros-Louis F. Misfolded SOD1 pathology in sporadic amyotrophic lateral sclerosis. Sci Rep. 2018;8(1):14223. doi: 10.1038/s41598-018-31773-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sundaramoorthy V, Walker AK, Yerbury J, Soo KY, Farg MA, Hoang V, Zeineddine R, Spencer D, Atkin JD. Extracellular wildtype and mutant SOD1 induces ER-Golgi pathology characteristic of amyotrophic lateral sclerosis in neuronal cells. Cell Mol Life Sci. 2013;70(21):4181–4195. doi: 10.1007/s00018-013-1385-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Banks CJ, Andersen JL. Mechanisms of SOD1 regulation by post-translational modifications. Redox Biol. 2019;26:101270. doi: 10.1016/j.redox.2019.101270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meiliana A, Dewi NM, Wijaya A. New insight in the molecular mechanisms of neurodegenerative disease. Indones Biomed J. 2018;10(1):16–34. doi: 10.18585/inabj.v10i1.448. [DOI] [Google Scholar]
- 38.Lindner AB, Demarez A. Protein aggregation as a paradigm of aging. Biochim Biophys Acta. 2009;1790(10):980–996. doi: 10.1016/j.bbagen.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 39.Liebman SW, Chernoff YO. Prions in yeast. Genetics. 2012;191(4):1041–1072. doi: 10.1534/genetics.111.137760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McAlary L, Plotkin SS, Yerbury JJ, Cashman NR. Prion-like propagation of protein misfolding and aggregation in amyotrophic lateral sclerosis. Front Mol Neurosci. 2019;12:262. doi: 10.3389/fnmol.2019.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider A, Simons M. Exosomes: vesicular carriers for intercellular communication in neurodegenerative disorders. Cell Tissue Res. 2013;352(1):33–47. doi: 10.1007/s00441-012-1428-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stroo E, Koopman M, Nollen EA, Mata-Cabana A. Cellular regulation of amyloid formation in aging and disease. Front Neurosci. 2017;11:64. doi: 10.3389/fnins.2017.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deller MC, Kong L, Rupp B. Protein stability: a crystallographer’s perspective. Acta Crystallogr F Struct Biol Commun. 2016;72(Pt 2):72–95. doi: 10.1107/s2053230x15024619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farage MA, Miller KW, Elsner P, Maibach HI. Characteristics of the aging skin. Adv Wound Care (New Rochelle) 2013;2(1):5–10. doi: 10.1089/wound.2011.0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chattopadhyay M, Valentine JS. Aggregation of copper-zinc superoxide dismutase in familial and sporadic ALS. Antioxid Redox Signal. 2009;11(7):1603–1614. doi: 10.1089/ars.2009.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeon GS, Shim YM, Lee DY, Kim JS, Kang M, Ahn SH, Shin JY, Geum D, Hong YH, Sung JJ. Pathological modification of TDP-43 in amyotrophic lateral sclerosis with SOD1 mutations. Mol Neurobiol. 2019;56(3):2007–2021. doi: 10.1007/s12035-018-1218-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trist BG, Hare DJ, Double KL. A Proposed mechanism for neurodegeneration in movement disorders characterized by metal dyshomeostasis and oxidative stress. Cell Chem Biol. 2018;25(7):807–816. doi: 10.1016/j.chembiol.2018.05.004. [DOI] [PubMed] [Google Scholar]
- 48.Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. 2015;6:171. doi: 10.4103/2152-7806.169561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bäumer D, Talbot K, Turner MR. Advances in motor neurone disease. J R Soc Med. 2014;107(1):14–21. doi: 10.1177/0141076813511451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pedrini S, Sau D, Guareschi S, Bogush M, Brown RH, Jr, Naniche N, Kia A, Trotti D, Pasinelli P. ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum Mol Genet. 2010;19(15):2974–2986. doi: 10.1093/hmg/ddq202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zwiegers P, Lee G, Shaw CA. Reduction in hSOD1 copy number significantly impacts ALS phenotype presentation in G37R (line 29) mice: implications for the assessment of putative therapeutic agents. J Negat Results Biomed. 2014;13:14. doi: 10.1186/1477-5751-13-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davie CA. A review of Parkinson’s disease. Br Med Bull. 2008;86:109–127. doi: 10.1093/bmb/ldn013. [DOI] [PubMed] [Google Scholar]
- 53.Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VMY, George JM, Rienstra CM. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat Struct Mol Biol. 2016;23(5):409–415. doi: 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fields CR, Bengoa-Vergniory N, Wade-Martins R. Targeting Alpha-synuclein as a therapy for Parkinson’s disease. Front Mol Neurosci. 2019;12:299. doi: 10.3389/fnmol.2019.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brück D, Wenning GK, Stefanova N, Fellner L. Glia and alpha-synuclein in neurodegeneration: a complex interaction. Neurobiol Dis. 2016;85:262–274. doi: 10.1016/j.nbd.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trist BG, Davies KM, Cottam V, Genoud S, Ortega R, Roudeau S, Carmona A, de Silva K, Wasinger V, Lewis SJG, Sachdev P, Smith B, Troakes C, Vance C, Shaw C, al-Sarraj S, Ball HJ, Halliday GM, Hare DJ, Double KL. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain. Acta Neuropathol. 2017;134(1):113–127. doi: 10.1007/s00401-017-1726-6. [DOI] [PubMed] [Google Scholar]
- 57.Cruces-Sande A, Méndez-Álvarez E, Soto-Otero R. Copper increases the ability of 6-hydroxydopamine to generate oxidative stress and the ability of ascorbate and glutathione to potentiate this effect: potential implications in Parkinson’s disease. J Neurochem. 2017;141(5):738–749. doi: 10.1111/jnc.14019. [DOI] [PubMed] [Google Scholar]
- 58.Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314(5800):777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 59.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/bf00308809. [DOI] [PubMed] [Google Scholar]
- 60.Kurfuerst S. Stop before it starts. Rehab Manag. 2003;16(8):42–44. [PubMed] [Google Scholar]
- 61.Cuello AC. Intracellular and extracellular Abeta, a tale of two neuropathologies. Brain Pathol. 2005;15(1):66–71. doi: 10.1111/j.1750-3639.2005.tb00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang X, Fu Z, Meng L, He M, Zhang Z. The early events that initiate β-amyloid aggregation in Alzheimer’s disease. Front Aging Neurosci. 2018;10:359. doi: 10.3389/fnagi.2018.00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jang JY, Cho H, Park HY, Rhim H, Kang S. ALS-linked mutant SOD1 proteins promote Aβ aggregates in ALS through direct interaction with Aβ. Biochem Biophys Res Commun. 2017;493(1):697–707. doi: 10.1016/j.bbrc.2017.08.127. [DOI] [PubMed] [Google Scholar]
- 64.Moszczynski AJ, Hintermayer MA, Strong MJ. Phosphorylation of threonine 175 tau in the induction of tau pathology in amyotrophic lateral sclerosis-frontotemporal spectrum disorder (ALS-FTSD). A review. Front Neurosci. 2018;12:259. doi: 10.3389/fnins.2018.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]