

Abstract
Nine bis(thiosemicarbazone) (BTSC) cobalt(III) complexes of the general formula [Co(BTSC)(L)2]NO3 were synthesized, where BTSC = diacetyl bis(thiosemicarbazone) (ATS), pyruvaldehyde bis(thiosemicarbazone) (PTS), or glyoxal bis(thiosemicarbazone) (GTS) and L = ammonia, imidazole (Im), or benzylamine (BnA). These compounds were characterized by multinuclear NMR spectroscopy, mass spectrometry, cyclic voltammetry, and X-ray crystallography. Their stability in phosphate-buffered saline was investigated and found to be highly dependent on the nature of the axial ligand L. These studies reveal that complex stability is primarily dictated by the axial ligand following the sequence NH3 > Im > BnA. The cellular uptake and cytotoxicity in cancer cells were also determined. Both the cellular uptake and cytotoxicity are significantly affected by the nature of the equatorial BTSC. Complexes of ATS are taken up much more effectively than those of PTS and GTS. The cytotoxicity of the complexes is correlated to that of the free ligand. Cell uptake and cytotoxicity was also determined under hypoxic conditions. Only minor differences in the hypoxia activity and uptake is observed. Treatment of the cancer cells with the copper-depleting agent tetrathiomolybdate decreases the cytotoxic potency of the complexes, indicating that they may operate via a copper-dependent mechanism. These results provide a structure-activity relationship for this class of compounds, which may be applied for the rational design of new cobalt(III) anticancer agents.
Graphical Abstract
Nine cobalt(III) bis(thiosemicarbazone) complexes were synthesized and characterized by mass spectrometry, X-ray crystallography, and multinuclear NMR spectroscopy. These compounds were evaluated as potential anticancer agents in normoxic and hypoxic conditions. Structure-activity relationships reveal that complex potency is correlated to that of the free bis(thiosemicarbazone) ligands, suggesting that the cobalt(III) complexes act as chaperones for these cytotoxic compounds.
Introduction
The platinum-based drugs cisplatin, carboplatin, and oxaliplatin comprise the first-line treatment for many cancer types.1–3 Despite their importance in the clinic, these drugs induce severe side effects, which hinder patient quality of life.4–8 These side effects arise from an inherent lack of selectivity of these cytotoxic drugs for cancer cells over normal cells. As such, there remains a need to develop anticancer complexes that actively target tumors. One approach to achieving this goal is to exploit the properties of the tumor microenvironment, which is characterized by leaky vasculature, low extracellular pH, and hypoxia.9–13 In this context, the use of metal ion redox chemistry is a strategy that has effectively been employed to target hypoxic cancer cells.14–17
Among the metal ion redox couples explored, those of the cobalt(III)/cobalt(II) and copper(II)/copper(I) couples are the most commonly exploited for the development of redox-activated prodrugs. Cobalt(III) complexes, which have been investigated as both hypoxia-targeting agents and as enzyme inhibitors,18–35 are kinetically inert due to their low-spin d6 electron configuration. Reduction to cobalt(II) yields a labile d7 complex, facilitating the loss of ligands within the coordination sphere. In this capacity, cobalt(III) complexes can acts as chaperones to deliver cytotoxic ligands selectively to hypoxic regions where metal reduction is favored.25,33,36
With regard to copper(II)-based hypoxia-targeting complexes, the most well characterized class of compounds are those of the bis(thiosemicarbazone) (BTSC) ligands.37–40 The radioactive BTSC 64Cu complex [Cu(ATSM)] where ATSM = diacetyl-bis(N4-methythiosemicarbazone) (Chart 1) is an established hypoxia imaging agent.41 With an appropriate copper(II)/copper(I) redox potential, [Cu(ATSM)] is reduced selectively in hypoxic cells.42–47 The resulting Cu(I) complex is substantially less stable, and the free ATSM ligand along with the radioactive 64Cu ion are released and trapped within the cell. The accumulation of radioactive 64Cu selectively in hypoxic cells gives rise to the hypoxia-imaging properties of this compound. In addition to diagnostic applications, Cu-BTSC complexes and free BTSC ligands possess well-documented anticancer activity.48 New drug candidates based on these ligands are currently under investigation.49
Chart 1.
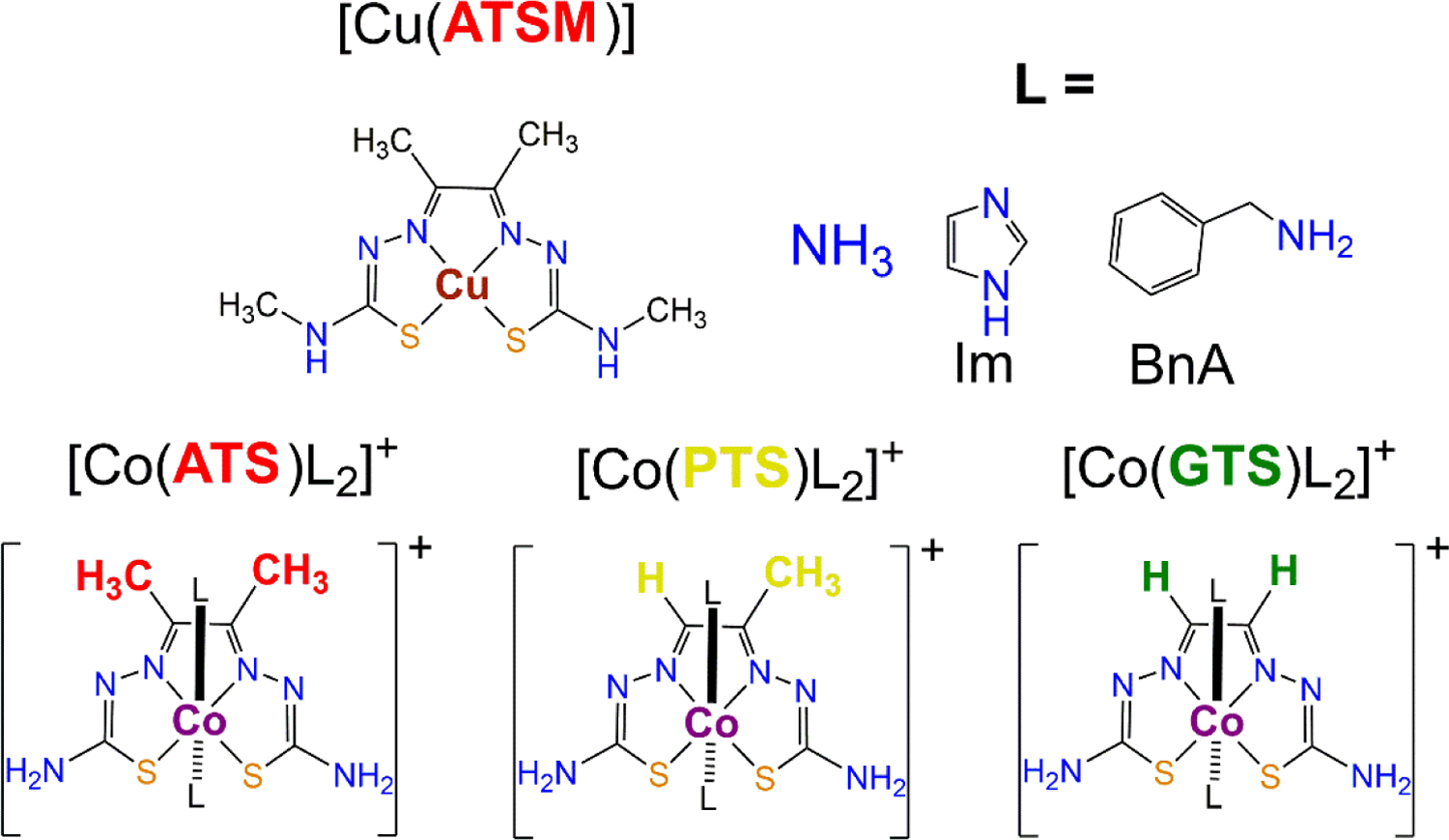
Structures of Cu[(ATSM)] and the [Co(BTSC)(L)2]+ Complexes Investigated in this Work.
In this study, we aimed to combine the hypoxia-targeting properties of the cobalt(III)/cobalt(II) redox couple with the anticancer potential of the BTSC ligands in the form of nine new cobalt(III) BTSC complexes (Chart 1). We hypothesized that the octahedral coordination geometry and increased kinetic inertness of cobalt(III), compared to the square planar coordination geometry and relative lability of copper(II), would confer key advantages to these complexes as hypoxia-targeting anticancer agents. The six-coordinate geometry of cobalt(III) allows for tuning of the physiochemical properties of the compounds for biological use via modification of axial ligands Furthermore, the greater kinetic inertness of cobalt(III) compared to copper(II) should give rise to more stable compounds before they are reduced to their active forms. Reduction of the cobalt(III) BTSC complexes in hypoxic environments may release both the axial ligands and the cytotoxic BTSC ligand. Although the current library of compounds includes only simple nitrogen donors, this work provides a foundation for development of complexes with bioactive axial ligands that may act synergistically with the BTSC ligand.. We have carried out detailed characterization of the cobalt(III) BTSC complexes, studied their stabilities, and investigated their potential as hypoxia-targeting anticancer agents. The results described herein detail a structure-activity relationship for this class of compounds and provide guiding principles for the next generation of cobalt(III)-based hypoxia-targeting agents.
Experimental
Materials and Methods.
Co(NO3)2·6H2O was obtained from Strem Chemicals (Newburyport, MA). Imidazole (Im) and benzylamine (BnA) were obtained from Alfa Aesar and used as received. The bis(thiosemicarbazone) ligands diacetyl bis(thiosemicarbazone) (ATS), pyruvaldehyde bis(thiosemicarbazone) (PTS), and glyoxal bis(thiosemicarbazone) (GTS) were synthesized as previously described.39 Solvents were of ACS grade or higher. CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate) lysis buffer was prepared using 1% CHAPS by mass, 5 mM ethylenediamine tetraacetic acid (EDTA), 50 mM tris(hydroxymethyl)aminomethane (Tris), and 110 mM NaCl; the pH was adjusted to 7.4 using dilute HCl or NaOH as necessary. All reactions were carried out under ambient atmospheric conditions without any efforts to exclude oxygen or water.
Physical Measurements.
NMR samples were prepared with DMSO-d6 as the solvent. Spectra were acquired on a 500 MHz Bruker AV 3HD spectrometer equipped with a broadband Prodigy cryoprobe. 1H and 13C{1H} NMR spectra were referenced to the residual solvent peak of DMSO at 2.50 and 39.52 ppm, respectively.50 59Co NMR spectra were referenced to K3[Co(CN)6] in D2O at 0 ppm.51 UV-Vis spectra were acquired using an Agilent Cary 8454 UV-Visible spectrophotometer. IR spectra were acquired on a Bruker Hyperion ATIR with ZnSe ATR attachment for solid powders. Graphite furnace atomic absorption spectroscopy (GFAAS) measurements were performed with a PerkinElmer PinAAcle 900z instrument. Electrochemical measurements were carried out using a Pine WaveNow potentiostat with a three-electrode setup consisting of a glassy carbon working electrode, a platinum counter electrode, and Ag wire quasi–reference electrode. Complexes were dissolved in anhydrous DMF with 0.10 M (Bu4N)(PF6) (TBAP) as the supporting electrolyte. Potentials were referenced using an internal standard of the ferrocene/ferricenium couple at 0.45 V vs the saturated calomel electrode (SCE).52,53 The sample cell was deoxygenated by bubbling nitrogen gas through the solution prior to analysis, and maintained under a blanket of nitrogen during the experiment. Analytical high-performance liquid chromatography (HPLC) was performed using a Shimadzu LC20–AT HPLC with an Ultra Aqueous C18 column, 100 Å, 5 μm, 250 mm x 4.6 mm (Restek, Bellefonte, PA) and an SPD-20AV UV/Vis detector monitoring at 220 and 260 nm. The flow rate for all HPLC analyses was 1 mL/min. Gradient elution was carried out as follows: 10% MeOH in water containing 0.1% TFA for 5 min, followed by linear gradient to 100% MeOH containing 0.1% TFA over 20 min. High-resolution mass spectra (HRMS) were recorded on an Exactive Orbitrap mass spectrometer in positive ESI mode (ThermoFisher Scientific, Waltham, MA). Elemental analyses (CHN) were performed by Atlantic Microlab Inc., Norcross, GA, USA.
[CoATS(NH3)2]NO3.
ATS (0.30 g, 1.5 mmol) was suspended in MeOH (10 mL) and added to a solution of CoNO3·6H2O (0.43 g, 1.5 mL) in MeOH (10 mL), resulting in an immediate color change to dark green. The mixture was heated under reflux for 3 h, and 30% aqueous NH4OH (3 mL) was added. The mixture was left stirring at room temperature for 16 h. The resulting bright red solid was isolated via vacuum filtration and washed with MeOH and Et2O. Yield 0.41 g (71%). 1H NMR (DMSO-d6, 500 MHz): δ 7.60 (s, 4H), 2.35 (s, 6H), 2.12 (s, 6H). 13C{1H}NMR (DMSO-d6, 125 MHz): δ 179.9, 155.4, 15.9. IR (ATR, cm−1): 3486 w, 3360 w, 3281 w, 3136 m, 2949 w, 2945 w, 2850 w, 1753 w, 1622 m, 1591 m, 1568 m, 1545 m, 1514 w, 1477 m, 1425 s, 1350 s, 1323 s, 1256 m, 1229 s, 1171 s, 1125 s, 1101 s, 1063 s, 998 w, 935 m, 918 m, 845 m, 825 s, 746 s, 708 s, 665 s, 655 s, 615 s. HR-ESI-MS (positive ion mode): m/z 323.0269 ([M]+, calcd. 323.0271). Anal. Calcd. for [Co(ATS)(NH3)2](NO3) (C6H16CoN9O3S2): C, 18.70; H, 4.19; N, 32.72. Found: C, 18.99: H, 4.07: N, 32.45.
[CoATS(Im)2]NO3.
ATS (0.31 g, 1.5 mmol) was suspended in MeOH (10 mL) and added to a solution of CoNO3·6H2O (0.42 g, 1.4 mmol) in MeOH (10 mL), resulting in an immediate color change to dark green. The mixture was heated under reflux for 2 h, and imidazole (1.00 g, 14.7 mmol) was added. After stirring at room temperature for 16 h, the resulting solution was concentrated to dryness under vacuum, and the remaining red residue was dissolved in approximately 2 mL of MeOH. The solution was cooled in an ice bath for 15 min, resulting in the precipitation of a bright red solid, which was collected by filtration and washed with Et2O. Yield 0.41 g (58%). 1H NMR (DMSO-d6, 500 MHz): δ 12.67 (br s, N-H), 7.69 (m, 6H), 7.14 (s, 2H), 6.75 (s, 2H), 2.46 (s, 6H). 13C{1H}NMR (DMSO-d6, 125 MHz): δ 181.4, 155.5, 137.8, 127.6, 117.5, 16.0. IR (ATR, cm−1): 3485 w, 3358 m, 3270 m, 3136 m, 3049 w, 2951 w, 2851 w, 2611 w, 2390 w, 1753 w, 1626 m, 1589 m, 1566 m, 1537 w, 1486 w, 1429 m, 1356 m, 1323 s, 1300 m, 1256 m, 1227 s, 1175 m, 1124 m, 1101 m, 1063 s, 999 w, 935 m, 916 m, 845 m, 826 m, 745 s, 665 s. HR-ESI-MS (positive ion mode): m/z 425.0490 ([M]+, calcd. 425.0489). Anal. Calcd. for [CoATS(Im)2]NO3·MeOH (C13H22CoN11O4S2): C, 30.06; H, 4.27; N, 29.66. Found: C, 30.04; H, 4.13; N, 29.59.
[CoATS(BnA)2]NO3.
ATS (0.16 g, 0.69 mmol) and CoNO3·6H2O (0.20 g, 0.69 mmol)) were suspended in 3 mL of MeOH. The resulting dark green solution was heated under reflux for 1 h, and benzylamine (0.50 mL, 4.7 mmol) was added. The mixture was allowed to stir at room temperature for 16 h. The resulting dark red solid was isolated via vacuum filtration and washed with MeOH and Et2O. Yield: 0.23 g (57%). 1H NMR (DMSO-d6, 500 MHz): δ 7.81 (s, 4H), 7.27 (m, 6H), 7.20 (m, 4H), 3.08 (s, 8H), 2.27 (s, 6H). 13C{1H} NMR (DMSO-d6, 125 MHz): δ 180.3, 157.1, 138.3, 128.3 (2C overlapping), 127.4, 46.5, 16.2. IR (ATR, cm−1): 3279 w, 3107 w, 1651 w, 1616 w, 1582 w, 1553 w, 1526 w, 1483 w, 1442 s, 1386 w, 1356 m, 1323 s, 1306 s, 1221 s, 1171 m, 1134 m, 1072 m, 1003 w, 972 s, 928 m, 846 m, 827 m, 758 s, 750 s, 706 m 694 s, 660 s, 619 s. HR-ESI-MS (positive ion mode): m/z 503.1204 ([M]+, calcd. 503.1210). Anal. Calcd. for [CoATS(BnA)2]NO3·H2O (C20H30CoN9O4S2): C, 41.16; H, 5.18; N, 21.60. Found: C, 41.31; H, 5.09; N, 21.83.
[CoPTS(NH3)2]NO3.
PTS (0.21 g, 0.96 mmol) was added as a suspension in MeOH (7 mL) to a red solution of CoNO3·6H2O (0.27 g, 0.92 mmol) in MeOH (7 mL), which immediately resulted in a dark green mixture. The mixture was heated under reflux for 1 h. An aqueous solution comprising 30% NH4OH (2 mL) was added, and the mixture was stirred at room temperature for 16 h. The resulting dark red solid was isolated via vacuum filtration and washed with MeOH and Et2O. Yield 0.25 g (73%). 1H NMR (DMSO-d6, 500 MHz): δ 7.80 (s, 2H), 7.65 (s, 1H), 7.63 (s, 2H), 2.32 (s, 3H), 2.22 (s, 6H). 13C{1H} NMR (DMSO-d6, 125 MHz): δ 182.3, 180.0, 154.6, 146.9, 15.7. IR (ATR, cm−1) : 3464 w, 3399 w, 3321 w, 3279 w, 3165 w, 3124 w, 1616 m, 1595 m, 1574 m, 1522 m, 1476 m, 1416 s, 1375 m, 1314 s, 1280 s, 1244 m, 1192 s, 1163 s, 1052 m, 932 w, 870 m, 824 w, 752 m, 700 s, 652 m. HR-ESI-MS (positive ion mode): m/z 309.0110 ([M]+, calcd. 309.0115). Anal. Calcd. for [CoPTS(NH3)2]NO3 (C5H14CoN9O3S2): C, 16.18; H, 3.80; N, 33.95. Found: C, 16.44; H, 3.65; N, 33.91.
[CoPTS(Im)2]NO3.
PTS (0.20 g, 0.92 mmol) was suspended in MeOH (4 mL) and added to a solution of CoNO3·6H2O (0.27 g, 0.92 mmol) in MeOH (4 mL), which immediately resulted in a dark green mixture. The mixture was heated under reflux for 1 h. Imidazole (0.63 g, 9.16 mmol) was added, and the mixture was stirred at room temperature for 16 h. The resulting dark red solid was isolated via vacuum filtration and washed with MeOH and Et2O. Yield 0.27 g (60%). 1H NMR (DMSO-d6, 500 MHz): δ 12.72 (br s, N-H), 7.90 (s, 2H), 7.80 (s, 1H), 7.77 (br s, 2H), 7.74 (s, 2H), 7.17 (s, 2H), 6.82 (s, 2H), 2.40 (s, 3H). 13C{1H} NMR (DMSO-d6, 125 MHz): δ 183.3, 181.5, 154.7, 147.2, 138.0, 127.8, 117.8, 15.8. IR (ATR cm−1): 3305 m, 3192 m, 3138 m, 3080 m, 2963 m, 1630 m, 1570 m, 1548 w, 1508 w, 1425 s, 1377 w, 1344 w, 1306 s, 1260 w, 1242 w, 1165 s, 1109 w, 1097 w, 1066 s, 1029 w, 1014 w, 926 s, 862 m, 825 m, 762 m, 735 s, 719 s, 687 m, 654 s, 606 s. HR-ESI-MS (positive ion mode): m/z 411.0334 ([M]+, calcd. 411.0333). Anal. Calcd. for [CoPTS(Im)2]NO3·1.1H2O (C11H18.2CoN11O4.1S2): C, 26.79; H, 3.72; N, 31.24. Found: C, 27.27; H, 3.81; N, 30.82.
[CoPTS(BnA)2]NO3.
PTS (0.15 g, 0.69 mmol) and CoNO3·6H2O (0.20 g, 0.69 mmol) were suspended in 1 mL of MeOH. The mixture was heated under reflux for 1 h to yield a dark green solution. Benzylamine (0.15 mL, 1.4 mmol) was added, and the mixture was stirred at room temperature for 16 h. The resulting dark red solid was isolated via vacuum filtration and washed with MeOH and Et2O. Yield 0.22 g (54%). 1H NMR (DMSO-d6, 500 MHz): δ 7.99 (s, 2H), 7.83 (s, 2H), 7.67 (s, 1H), 7.21–7.38 (m, 10H), 3.13–3.22 (m, 8H), 2.31 (s, 3H). 13C{1H} NMR (DMSO-d6, 125 MHz): δ 182.3, 180.0, 156.0, 148.0, 138.2, 128.5, 128.20, 127.5, 46.4, 15.8. IR (ATR, cm−1): 3420 w, 3281 w, 3107 m, 2945 w, 1622 m, 1574 m, 1439 s, 1312 s, 1244 w, 1159 s, 1070 w, 972 m, 928 m, 864 w, 827 w, 758 s, 698 s. HR-ESI-MS (positive ion mode): m/z 489.1051 ([M]+, calcd. 489.1054). Anal. Calcd. for [CoPTS(BnA)2]NO3·2H2O (C19H30CoN9O5S2): C, 38.84; H, 5.15; N, 21.46. Found: C, 38.55; H, 5.11; N, 21.65.
[CoGTS(NH3)2]NO3.
GTS (0.1 g, 0.50 mmol) was suspended in MeOH (3 mL) and added to a solution of CoNO3·6H2O (0.15 g, 0.50 mmol) in MeOH (3 mL), which immediately resulted in a dark brown mixture. The mixture was refluxed for 1 h, and 30% aqueous NH4OH (1.5 mL) was added. The mixture was left stirring at room temperature for 2 h. The resulting dark purple solid was isolated via vacuum filtration and washed with MeOH and Et2O. Yield: 0.12 g (64%). 1H NMR (DMSO-d6, 500 MHz): δ 7.82 (s, 4H), 7.56 (s, 2H), 2.28 (s, 6H). 13C{1H} NMR (DMSO-d6, 125 MHz): δ 182.0, 144.2. IR (ATR, cm−1): 3420 w, 3292 w, 3111 m, 1649 m, 1614 m, 1555 w, 1520 w, 1425 s, 1331 s, 1296 s, 1259 m, 1209 s, 1194 s, 1065 m, 970 w, 854 m, 816 s, 708 s, 675 m. HR-ESI-MS (positive ion mode): m/z 294.9960 ([M]+, calcd. 294.9958). Anal. Calcd. for [CoGTS(NH3)2]NO3·H2O (C4H14CoN9O4S2): C: 12.80, H, 3.76; N, 33.59. Found: C, 12.67; H, 3.70; N, 33.42.
[CoGTS(Im)2]NO3.
GTS (0.25 g, 1.2 mmol) was suspended in MeOH (5 mL) and added to a solution of CoNO3·6H2O (0.36 g, 1.3 mmol) in MeOH (3 mL), which immediately resulted in a dark brown mixture. The mixture was heated under reflux for 1.5 h. Imidazole (1.18 g, 19 mmol) in a solution of 3 mL MeOH was added, resulting in a color change to purple. The mixture was left stirring at room temperature for 16 h. The resulting dark purple solid was isolated via vacuum filtration and washed with MeOH and Et2O. Yield: 0.43 g (73%). 1H NMR (DMSO-d6, 300 MHz): δ 12.77 (s, 2H), 7.92 (s, 4H), 7.80 (s, 2H), 7.72 (s, 2H), 7.19 (s, 2H), 6.88 (s, 2H). 13C{1H} NMR (DMSO-d6, 125 MHz): δ 183.1, 144.8, 137.9, 127.8, 117.9. IR (ATR, cm−1): 3283 w, 3152 w, 3122 w, 2941 w, 1624 m, 1548 w, 1510 w, 1423 s, 1328 m, 1309 m, 1258 m, 1184 s, 1101 m, 1070 s, 1020 m, 1006 m, 902 m, 847 m, 810 s, 756 s, 710 s, 656 s, 634 m. HR-ESI-MS (positive ion mode): m/z 397.0179 ([M]+, calcd. 397.0176). Anal. Calcd. for [CoGTS(Im)2]NO3·0.75MeOH (C10.75H17CoN11O3.75S2): C, 26.71; H, 3.55; N, 31.87. Found: C, 26.79; H, 3.42; N, 32.29.
[CoGTS(BnA)2]NO3.
GTS (0.28 g, 1.37 mmol) and CoNO3·6H2O (0.40 g, 1.37 mmol)) were suspended in 4 mL of MeOH, which immediately resulted in formation of a dark brown solution. The mixture was heated under reflux for 2 h, and benzylamine (1.5 mL, 13.7 mmol) was added. The mixture was left stirring at room temperature for 16 h. The resulting dark red solid was collected by filtration and washed with MeOH and Et2O. Yield: 0.41 g (56%). 1H NMR (DMSO-d6, 500 MHz): δ 7.99 (s, 4H), 7.67 (s, 2H), 7.26 (m, 10H), 3.32 (m, obscured by H2O peak), 3.16 (m, 4H). 13C{1H} NMR (DMSO-d6 125 MHz): δ 181.9, 145.4, 138.1, 128.6, 128.2, 127.5, 46.3. IR (ATR, cm−1): 3389 w, 3310 w, 3284 w, 3144 m, 3065 w, 1643 m, 1628 m, 1587 w, 1551 w, 1522 w, 1495 w, 1435 s, 1410 s, 1393 s, 1310 s, 1192 s, 1052 m, 978 m, 922 m, 854 m, 812 s, 750 s, 698 s, 613 m. HR-ESI-MS (positive ion mode): m/z 475.0898 ([M]+, calcd. 475.0897). Anal. Calcd. for [CoGTS(BnA)2]NO3 (C18H28CoN9O5S2): C, 40.22; H, 4.50; N, 23.45. Found: C, 40.44; H, 4.54; N, 23.64.
X–Ray Crystallography
Single crystals were grown via the vapor diffusion of diethyl ether into a solution of the complexes in DMF. Low-temperature (223 K) X-ray diffraction data for [Co(GTS)(Im)2]NO3·DMF, [Co(ATS)(Im)2]NO3, and [Co(GTS)(BnA)2]NO3·DMF were collected on a Bruker X8 Kappa diffractometer coupled to an ApexII CCD detector with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The structures were solved through intrinsic phasing using SHELXT54 and refined against F2 on all data by full-matrix least squares with SHELXL55 following established refinement strategies56. All non-hydrogen atoms were refined anisotropically. Positions of hydrogen atoms bound to carbon atoms were geometrically calculated and refined using a riding model. Hydrogen atoms bound to nitrogen were located in the difference Fourier synthesis and subsequently refined semi-freely with the help of distance restraints. The isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the U value of the atoms they are linked to (1.5 times for methyl groups). In the structure of [Co(GTS)(Im)2]NO3·DMF, the DMF molecule and nitrate counterion exhibited disorder about their respective locations in the crystal lattice. The two disordered components of the DMF molecules were refined with appropriate similarity restraints, allowing the occupancy of each component to refine freely with net occupancy of both components summing to one. The nitrate counterion was handled in the same manner. Likewise, in the structure of [Co(ATS)(Im)2]NO3, the nitrate counterion exhibited similar disorder that was refined as described above. One of the axial imidazole ligands in [Co(ATS)(Im)2]NO3 was rotationally disordered about the Co–N axis. The two disordered components were refined as described above. Details of the data quality and a summary of the residual values of all the refinements are listed in Table 1.
Table 1.
X-ray Crystallographic Data Collection and Refinement Parameters
| [Co(GTS)(Im)2]NO3·DMF | [Co(ATS)(Im)2]NO3 | [Co(GTS)(BnA)2]NO3·DMF | |
|---|---|---|---|
| formula | C13H21CoN12O4S2 | C12H18CoN11O3S2 | C21H31CoN10O4S2 |
| fw | 532.47 | 487.42 | 610.61 |
| space group | P21/n | P21/n | P21/n |
| a, Å | 13.8070(7) | 8.4179(8) | 9.2715(5) |
| b, Å | 11.2235(6) | 9.3561(7) | 11.2627(6) |
| c, Å | 14.7344(7) | 24.287(2) | 26.5823(17) |
| β, deg | 104.089(2) | 90.303(6) | 98.548(3) |
| V, Å3 | 2214.6(2) | 1912.8(3) | 2744.9(3) |
| Z | 4 | 4 | 4 |
| ρcalcd g·cm−3 | 1.597 | 1.693 | 1.478 |
| T, K | 223(2) | 223(2) | 223(2) |
| μ(Mo Kα), mm−1 | 1.012 | 1.158 | 0.825 |
| θ range, deg | 1.813 to 26.372 | 2.333 to 26.369 | 1.967 to 25.349 |
| completeness to θ, % | 100.0 | 100.0 | 100.0 |
| total no. of data | 21018 | 17792 | 23249 |
| no. of unique data | 4524 | 3913 | 5029 |
| no. of param | 434 | 284 | 369 |
| no. of restraints | 533 | 94 | 8 |
| R1a, % | 4.25 | 6.21 | 7.77 |
| wR2b, % | 9.97 | 13.33 | 13.70 |
| GOFc | 1.055 | 1.035 | 1.039 |
| max, min peaks e·Å−1 | 0.840, −0.235 | 0.813, −0.623 | 0.735, −0.600 |
R1 = Σ||Fo| − |Fc||/Σ|Fo| for all data.
wR2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2 for all data.
GoF = {Σ[w(Fo2 − Fc2)2]/(n − p)}1/2, where n is the number of data and p is the number of refined parameters.
Stability and N-methylimidazole Challenge Studies
[Co(BTSC)(L)2]+ complexes were dissolved in pH 7.4 phosphate-buffer saline (PBS) to a final concentration of 1 mM in both the absence and presence of 5 mM N-methylimidazole as a competing ligand. Immediately after preparation, the solutions were analyzed via RP-HPLC. The solutions were then incubated at 37 °C for 24 h and analyzed again using the same HPLC conditions. The stability was characterized by the peak area remaining of the starting complex.
Cell Lines and Culture Conditions
HeLa (human cervical cancer), A549 (human lung cancer), and MRC-5 (human lung fibroblast) cells were obtained from American Type Culture Collection (ATCC). All cell lines were cultured as adherent monolayers in an incubator at 37 °C with a humidified atmosphere of 5% CO2. A549 and HeLa cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). MRC-5 cells were cultured in Minimum Essential Media (MEM) containing 10% FBS. Cells were checked for mycoplasma contamination monthly using the PlasmoTest™ mycoplasma detection kit from InvivoGen.
Cytotoxicity Assay
The colorimetric MTT assay was used to evaluate cytotoxicity.57 Trypsinized cells were plated at 2000 cells/well in 100 μL/well in a 96-well plate and incubated for 24 h. The media was removed and replaced with 200 μL of growth media containing varying concentrations of the complexes, ranging from 0 to 500 μM. For hypoxia/normoxia comparative studies, two plates were simultaneously dosed with the complex of interest; one plate was incubated under normoxic conditions, and the other was incubated in a hypoxia chamber (Billips-Rothenburg, Inc., Del Mar, CA, USA), which was purged with an atmosphere of 95% N2 and 5% CO2. After 24 h, the culture medium containing the complex was removed, 200 μL of fresh media was added, and both plates were allowed to incubate an additional 48 h under normoxic conditions. After this time, the media was removed again, and a solution of thiazolyl blue tetrazolium bromide (MTT) in DMEM (200 μL, 1 mg/mL) was added to each well. Upon incubation for 4 h, the DMEM/MTT solutions were aspirated, and the purple formazan crystals were dissolved in 200 μL of an 8:1 mixture of DMSO: pH 10 glycine buffer. The absorbance of each well at 570 nm was measured using a microplate reader. Absorbance values were normalized to the untreated wells and plotted as concentration of cobalt complex versus % viability. The resulting dose-response curves were analyzed using a logistic sigmoid function.58 Reported IC50 values represent the average of three independent experiments, each carried out with six replicates per concentration level. Stock solutions of the cobalt complexes were prepared fresh in 18.2 MΩ·cm H2O prior to serial dilution in the cell culture medium. Control experiments employing only the cobalt(III) complexes and MTT in the absence of cells were carried out to verify that the complexes do not reduce the MTT dye.
Uptake Experiments
Cellular uptake of cobalt was determined by implementing slight modifications to previously reported protocols.59,60 Trypsinized cells (2 × 106) were seeded in six 75 cm2 culture dishes and incubated for 24 h. The media was removed, and the cells were treated with either 0 or 100 μM cobalt complex and placed under normoxic or hypoxic conditions, as described above, for 24 h. Dishes containing no cells were also incubated with 100 μM of the cobalt complex to correct for non-specific adsorption of cobalt to the plastic. Media was removed from all dishes, and the cells were rinsed with 3 mL PBS and detached with trypsin (3 mL). The cells were centrifuged at 1000 rpm for 10 min. The supernatant was discarded, and the pellet was resuspended in 1 mL PBS. Samples were centrifuged and resuspended twice more using the same conditions to remove extracellular cobalt, and centrifuged a final time to pellet cells. The pellet was resuspended in ice-cold, ultrapure 1× CHAPS lysis buffer, and samples were gently agitated 30 min. The protein concentration in each sample was then determined using the Thermo Fisher Bicinchoninic Acid Protein Assay Kit according to the manufacturer’s instructions. The cobalt concentration in each sample was determined using GFAAS. Results were reported as the mass ratio of cobalt to protein (pg/μg) in each sample.
Results
Synthesis of Cobalt(III) Bis(thiosemicarbazone) Complexes.
Nine cobalt(III) bis(thiosemicarbazone) complexes were synthesized by the method shown in Scheme 1. Heating an equimolar mixture of the free BTSC ligand and Co(NO3)2 in MeOH under reflux for approximately 30 min afforded a green solid. This uncharacterized intermediate was treated with an excess of ammonia, imidazole, or benzylamine. A color change from green to red is accompanied by the addition of the axial ligands. The compounds were isolated by filtration. Characterization by NMR spectroscopy, mass spectrometry, IR spectroscopy, and elemental analysis is consistent with the proposed structures (Figures S1–S36, Supporting Information, SI). RP-HPLC was used to verify that the compounds were isolated in greater than 95% purity (Figures S37–S45, SI).
Scheme 1.
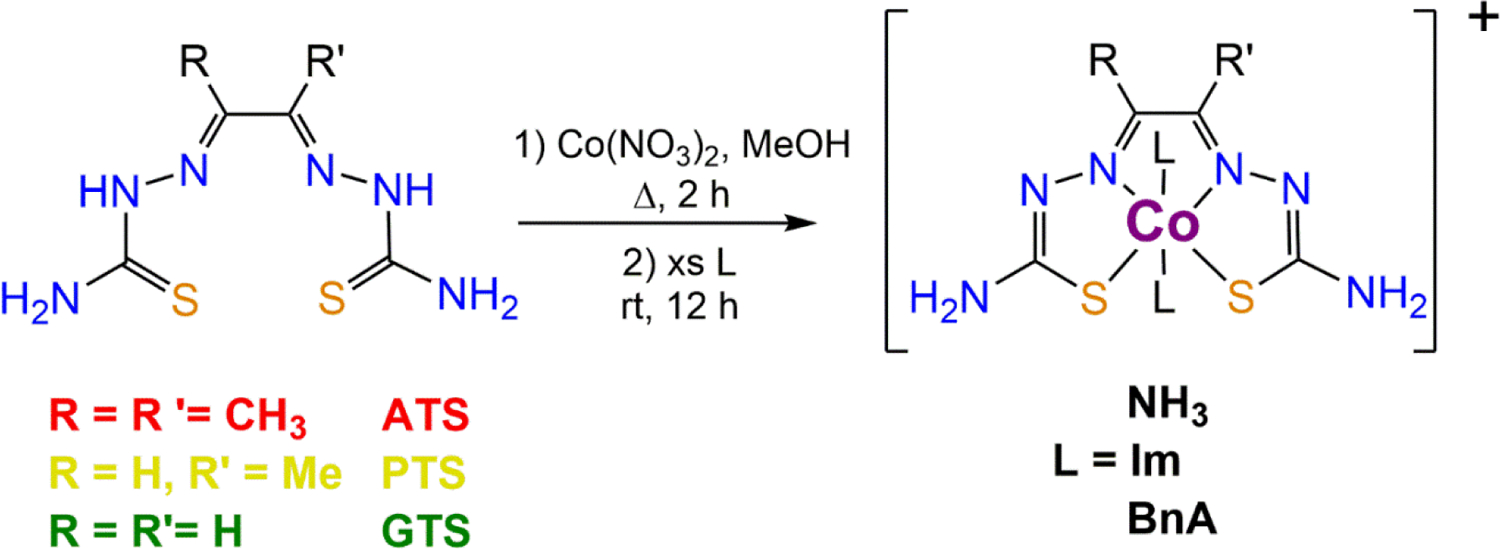
Synthesis of Cobalt(III) Bis(thiosemicarbazone) Complexes.
X–Ray Crystallography.
Single crystals, suitable for X-ray diffraction, were obtained for three [Co(BTSC)(L)2]NO3 complexes by the vapor diffusion of Et2O into DMF solutions. The crystal structures of the cations [CoGTS(Im)2]+, [CoATS(Im)2]+, and [CoGTS(BnA)2]+ are shown in Figure 1, and interatomic distances and angles are given in Table 2. All three complexes exhibit the expected octahedral coordination geometry for a low-spin d6 Co3+ ion. The BTSC ligands occupy four coordination sites in the equatorial plane, requiring the nitrogen donors to be arranged in a trans orientation. Despite the presence of two methyl groups in the backbone of ATS compared to GTS, the interatomic distances between cobalt and the sulfur and nitrogen donors of these ligands are indistinguishable between the three complexes. The Co–Im bond lengths of [CoGTS(Im)2]+ and [CoATS(Im)2]+ range from 1.943 to 1.9616 Å, similar to those reported for a related octahedral cobalt(III) Schiff base complex bearing axial imidazole ligands.61 Notably, the equatorial ligand has little effect on the axial ligand bond distances. For the complex [CoGTS(BnA)2]+, the benzylamine axial ligand Co–N bond distances are 2.003(3) and 2.006(3) Å, approximately 0.04 Å longer than the imidazole Co–N distances in [CoGTS(Im)2]+ and [CoATS(Im)2]+.
Figure 1.
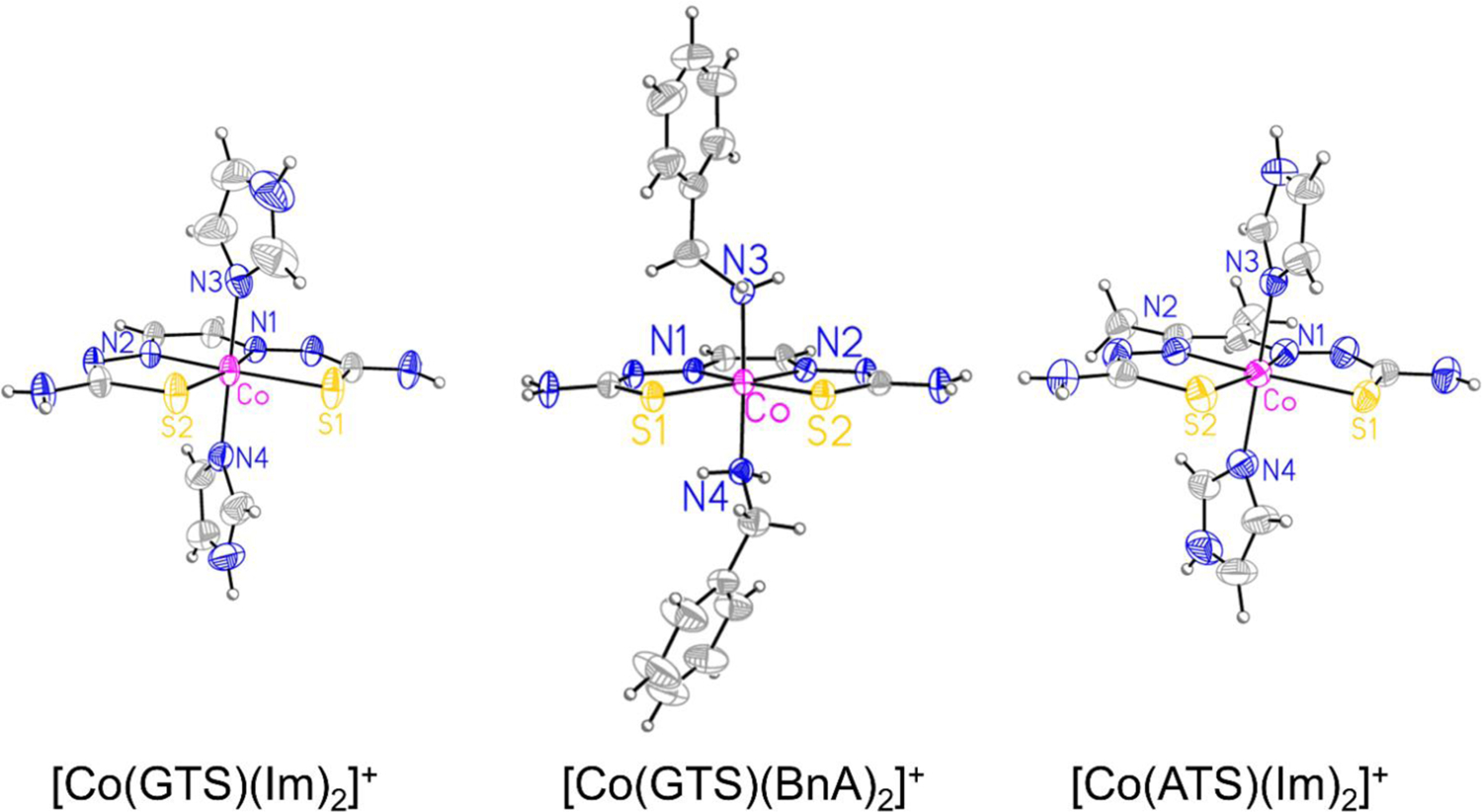
X-ray crystal structures of [Co(GTS)(Im)2]+, [Co(GTS)(BnA)2]+, and [Co(ATS)(Im)2]+. Ellipsoids are drawn at the 50% probability level. The disordered component of the imidazole ligand in [Co(ATS)(Im)2]+ is omitted for clarity.
Table 2.
Interatomic Distances (Å) and Angles (°) of [CoGTS(Im)2]+, [CoATS(Im)2]+, and [CoGTS(BnA)2]+.a
| Interatomic Distance | [CoGTS(Im)2]+ | [CoATS(Im)2]+ | [CoGTS(BnA)2]+ |
|---|---|---|---|
| Co–S1 | 2.2424(6) | 2.2472(9) | 2.2578(10) |
| Co–S2 | 2.2401(6) | 2.2563(9) | 2.2506(10) |
| Co–N1 | 1.8920(16) | 1.898(3) | 1.895(3) |
| Co–N2 | 1.8860(17) | 1.890(2) | 1.887(3) |
| Co–N3 | 1.9616(18) | 1.943(3) | 2.006(3) |
| Co–N4 | 1.9585(18) | 1.952(3) | 2.003(3) |
| Angle | [CoGTS(Im)2]+ | [CoATS(Im)2]+ | [CoGTS(BnA)2]+ |
| S1–Co–S2 | 105.01(2) | 106.56(3) | 106.59(4) |
| S1–Co–N1 | 85.85(5) | 85.38(8) | 85.17(9) |
| S1–Co–N2 | 169.65(5) | 168.00(9) | 168.38(9) |
| S1–Co–N3 | 90.32(6) | 89.47(8) | 90.99(10) |
| S1–Co–N4 | 89.59(6) | 90.13(9) | 87.66(10) |
| S2–Co–N1 | 169.13(5) | 168.06(8) | 168.23(9) |
| S2–Co–N2 | 85.29(5) | 87.75(9) | 85.00(9) |
| S2–Co–N3 | 90.23(6) | 89.39(8) | 89.02(9) |
| S2–Co–N4 | 90.96(5) | 87.75(9) | 93.13(10) |
| N1–Co–N2 | 83.86(7) | 82.64(12) | 83.23(12) |
| N1–Co–N3 | 89.35(7) | 90.49(11) | 91.31(13) |
| N1–Co–N4 | 89.43(7) | 92.58(12) | 86.74(13) |
| N2–Co–N3 | 90.76(7) | 91.28(11) | 89.90(13) |
| N2–Co–N4 | 89.10(7) | 89.76(12) | 91.05(13) |
| N3–Co–N4 | 178.78(7) | 176.87(11) | 177.71(13) |
Atoms are labeled as shown in Figure 1. Numbers in parentheses are the estimated standard deviations for the last significant figure.
59Co NMR Spectroscopy.
Cobalt-59 is an I = 7/2 nucleus with 100% natural abundance. Although linewidths may be broad due to quadrupolar relaxation, diamagnetic cobalt complexes are often amenable to characterization by 59Co NMR spectroscopy.51,62–64 All nine of the [Co(BTSC)(L)2]+ complexes were characterized by 59Co NMR spectroscopy. Representative spectra are shown in Figure 2, and chemical shifts (δ) and linewidths (ν1/2) are summarized in Table 3. The remaining 59Co NMR spectra are in the Supporting Information, Figures S19–S27.
Figure 2.
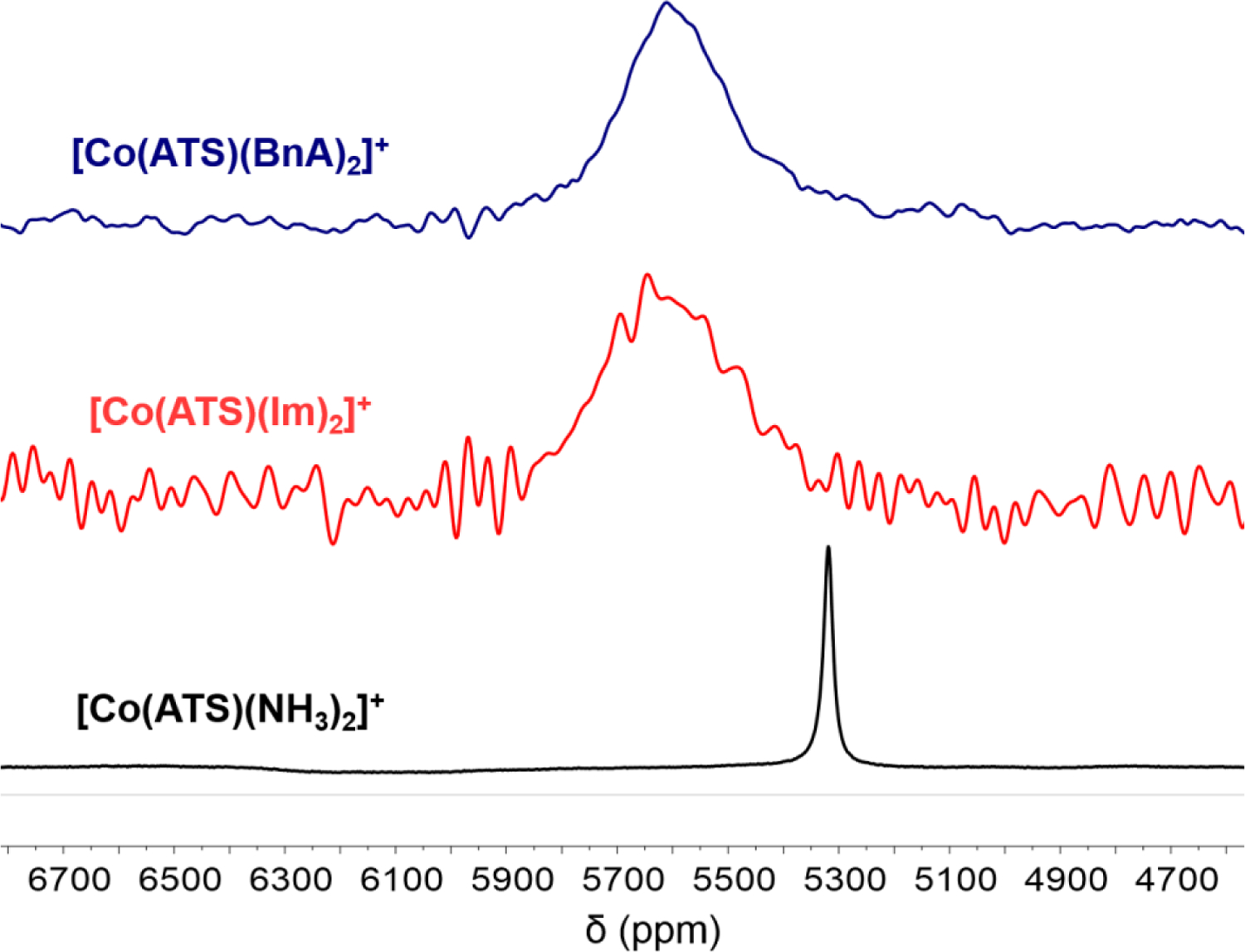
59Co NMR spectra of [Co(ATS)L2]+ complexes in DMSO-d6 at 25 °C obtained at a frequency of 119 MHz. Spectra are referenced to K3[Co(CN)6] in D2O at 0 ppm.
Table 3.
59Co NMR Chemical Shifts and Linewidthsa
| Compound | δ (ppm) | ν1/2 (Hz) |
|---|---|---|
| [Co(ATS)(NH3)2]+ | 5320 | 2600 |
| [Co(ATS)(Im)2]+ | 5640 | 33500 |
| [Co(ATS)(BnA)2]+ | 5600 | 27900 |
| [Co(PTS)(NH3)2]+ | 5320 | 1400 |
| [Co(PTS)(Im)2]+ | 5610 | 30700 |
| [Co(PTS)(BnA)2]+ | 5590 | 21100 |
| [Co(GTS)(NH3)2]+ | 5250 | 1200 |
| [Co(GTS)(Im)2]+ | 5570 | 30000 |
| [Co(GTS)(BnA)2]+ | 5510 | 20200 |
Chemical shifts are referenced to K3[Co(CN)6] in D2O set at δ = 0 ppm.
The chemical shift of the 59Co resonance exhibits a dependence on both the equatorial and axial ligands. For a given BTSC, the chemical shift moves downfield by approximately 300 ppm as the axial ligand is altered from NH3 to BnA or Im. Complexes of PTS and ATS exhibit chemical shifts that are within 30 ppm of each other. In contrast, the corresponding GTS complexes, which lack the electron-donating methyl groups on the ligand backbone, resonate approximately 80 ppm upfield from the PTS and ATS complexes. The linewidth of the resonance appears to correlate only with the nature of the axial ligand; complexes with axial ammonia ligands give rise to reasonably sharp signals with ν1/2 < 3000 Hz. In contrast, axial imidazole and benzylamine ligands give rise to very broad peaks with linewidths exceeding 20,000 Hz.
UV–Vis Spectroscopy.
The electronic structure of the cobalt bis(thiosemicarbazone) complexes was further characterized by UV-Vis spectroscopy. Representative UV–Vis spectra of the [Co(BTSC)(NH3)2]+ complexes are shown in Figure 3, and data for these and the remaining complexes are collected in Table 4. UV-Vis spectra for the other complexes are in the SI, Figures S46–S54. All of the complexes display two distinct absorption maxima: one highly intense feature between 340 and 390 nm and another, less intense feature ranging from 499 to 530 nm. A third less well-defined band is also apparent in the range of 440 to 450 nm. The low energy transitions are red-shifted as the equatorial ligand is changed from the most electron-donating ligand ATS to the least donating GTS. The influence of the axial ligand is substantially less pronounced.
Figure 3.
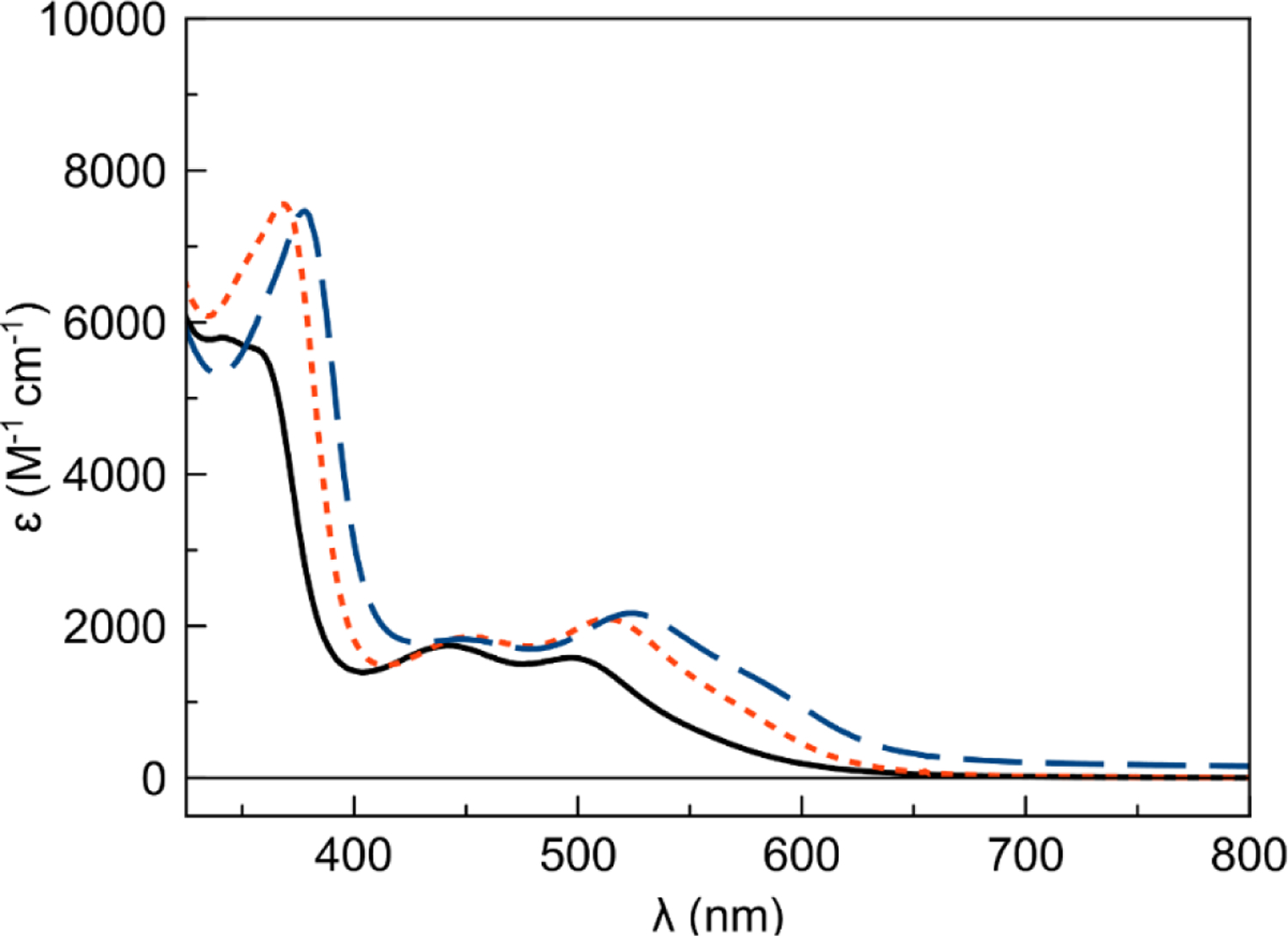
Electronic absorption spectra of [Co(ATS)(NH3)2]+ (solid black line), [Co(PTS)(NH3)2]+ (small orange dashes), and [Co(GTS)(NH3)2]+ (long blue dashes) complexes in pH 7.4 PBS at 25 °C.
Table 4.
Absorbance Maxima and Extinction Coefficients for the Complexes in pH 7.4 PBS
| Compound | λ, nm (ε, M−1 cm−1) | ||
|---|---|---|---|
| [Co(ATS)(NH3)2]+ | 341 (5800) | 443 (1708) | 499 (1566) |
| [Co(ATS)(Im)2]+ | 367 (5791) | 446 (1705) | 503 (1699) |
| [Co(ATS)(BnA)2]+ | 363 (7673) | 447 (1520) | 510 (1624) |
| [Co(PTS)(NH3)2]+ | 368 (7557) | 450 (1834) | 514 (2038) |
| [Co(PTS)(Im)2]+ | 374 (6339) | 449 (1576) | 516 (1842) |
| [Co(PTS)(BnA)2]+ | 375 (8016) | 450 (1700) | 524 (1870) |
| [Co(GTS)(NH3)2]+ | 377 (7456) | 446 (1961) | 523 (2015) |
| [Co(GTS)(Im)2]+ | 379 (7417) | 442 (1805) | 527 (2040) |
| [Co(GTS)(BnA)2]+ | 384 (8676) | 446 (1548) | 537 (1853) |
Electrochemistry.
The electrochemical properties of the [Co(BTSC)(L)2]+ complexes were analyzed by cyclic voltammetry in anhydrous DMF with 0.10 M NBu4PF6 as the supporting electrolyte. Results of the electrochemical studies are presented in Table 5, and representative voltammograms of the compound [Co(GTS)(Im)2]+ are displayed in Figure 4. Other voltammograms are deposited in the SI, Figures S55–S63.
Table 5.
Electrochemical Properties of [Co(BTSC)(L)2]+ Complexesa
| Compound | E1/2,Co(II/I) | Epc, Co(III/II) |
|---|---|---|
| [Co(ATS)(NH3)2]+ | −1.18 | −1.10 |
| [Co(ATS)(Im)2]+ | −1.19 | −0.86 |
| [Co(ATS)(BnA)2]+ | −1.18 | −0.83 |
| [Co(PTS)(NH3)2]+ | –1.11 | –0.92 |
| [Co(PTS)(Im)2]+ | –1.12 | –0.84 |
| [Co(PTS)(BnA)2]+ | –1.14 | –0.84 |
| [Co(GTS)(NH3)2]+ | –1.02 | –0.95 |
| [Co(GTS)(Im)2]+ | –1.00 | –0.72 |
| [Co(GTS)(BnA)2]+ | –1.00 | –0.69 |
Potentials are referenced to the SCE. Data were obtained at a glassy carbon working electrode in anhydrous DMF containing 0.10 M TBAP, using a scan rate of 100 mV/s at 25 °C.
Figure 4.
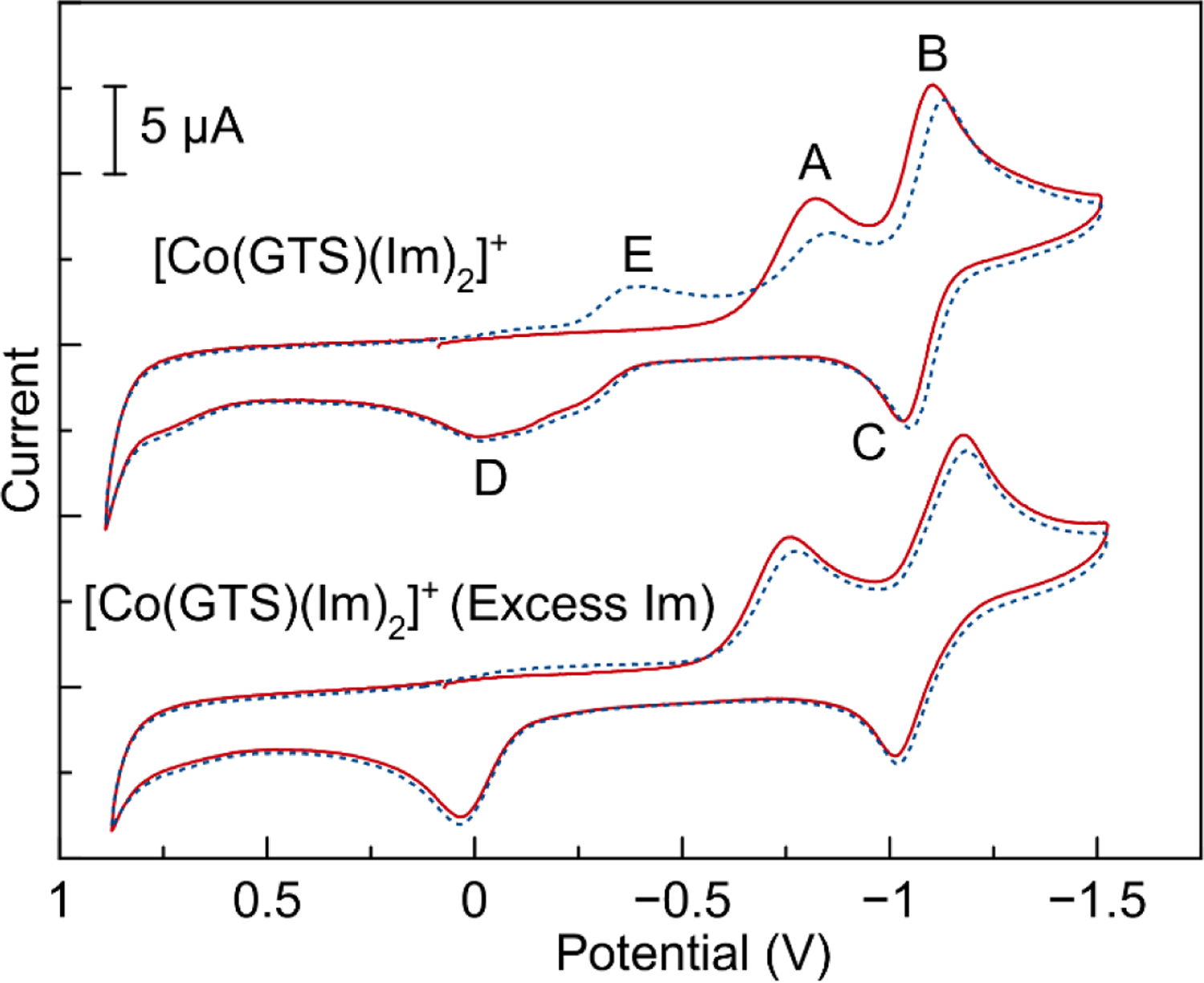
Cyclic voltammogram of [CoGTS(Im)2]+ before (top) and after (bottom) addition of excess imidazole. The first scan is shown as a solid red line, while the second scan is represented by small blue dashes. This experiment was performed at 25 °C in DMF solution with 0.10 M TBAP electrolyte at a scan rate of 100 mV/s. The potential is referenced to SCE, based on the position of the Fc/Fc+ couple as an internal standard.
All complexes exhibited the same general features: an initial, irreversible reduction (A) followed by a quasi–reversible reduction/oxidation (B/C), and then a broad oxidation feature (D) (Figure 4). Scans after the first sweep showed a new irreversible reduction feature (E) at a more positive potential than the initial irreversible reduction peak potential. To further probe the nature of this electrochemical reactivity, excess imidazole was added to the electrochemical cell. The addition of imidazole had three pronounced effects upon the voltammogram, as shown in Figure 4.
First, the irreversible reduction event (A) does not decrease in amplitude as dramatically upon the second cycle. Second, the broad irreversible oxidation feature (D) becomes much sharper, coalescing into what appears to be a single peak. Third, the reduction peak (E), a feature that is only apparent upon the second cycle, disappears altogether. These results indicate that axial ligand exchange plays an integral role in the complexes’ electrochemical behavior. We propose a mechanism in which the initial irreversible reduction peak (A) corresponds to the Co(III)/Co(II) reduction process. Population of eg* antibonding orbitals upon reduction of Co(III) leads to the dissociation of at least one of the axial ligands. The quasi-reversible reduction/oxidation event (B/C) is attributed to the Co(II)/Co(I) couple of the resulting complex, which lacks axial nitrogen donor ligands. The broad oxidation event (D) corresponds to the Co(II)/Co(III) oxidation of a mixture of complexes containing either nitrogen donor axial ligands or coordinated DMF axial ligands. The new reduction event (E) is the Co(III)/Co(II) couple of complexes bearing axial DMF ligands. When excess imidazole is added, recombination of this ligand with the cobalt complex to form [CoGTS(Im)2]+ occurs rapidly after feature (C). As such, feature (D) coalesces to a single oxidation event, corresponding to that of [CoGTS(Im)2]+ exclusively, and feature (E), which is attributed to the complex with axial DMF ligands, is not observed. Similar electrochemical mechanisms have been reported in other Co(III) complexes.18
Stability and N–Methylimidazole Challenge Studies.
The stability of the [Co(BTSC)(L)2]+ complexes at 37 °C in pH 7.4 PBS was evaluated by RP-HPLC. The peak area of the intact complex was measured after a 24 h incubation period. All complexes showed significant degradation over this time course; the amount of intact complex remaining ranges from 0–70% (Figure 5). Decomposition in PBS is accompanied by formation of a precipitate as well as the appearance of new peaks on the chromatogram. For the complexes bearing BnA, one of these peaks was conclusively identified to be free BnA, indicating that axial ligand loss is a key decomposition pathway for these complexes. Characterization of the precipitate by 1H NMR spectroscopy showed the presence of the intact complex, which may have precipitated as an insoluble phosphate or chloride salt. The nature of the axial ligand has the greatest impact on complex stability, whereas effects due to the equatorial ligand are subtler. Complexes with axial ammonia ligands, [Co(BTSC)(NH3)2]+, exhibit the highest stability, whereas those with benzylamine axial ligands are the least stable. For the benzylamine and ammonia complexes, the GTS ligand confers greater stability than the ATS ligand.
Figure 5.
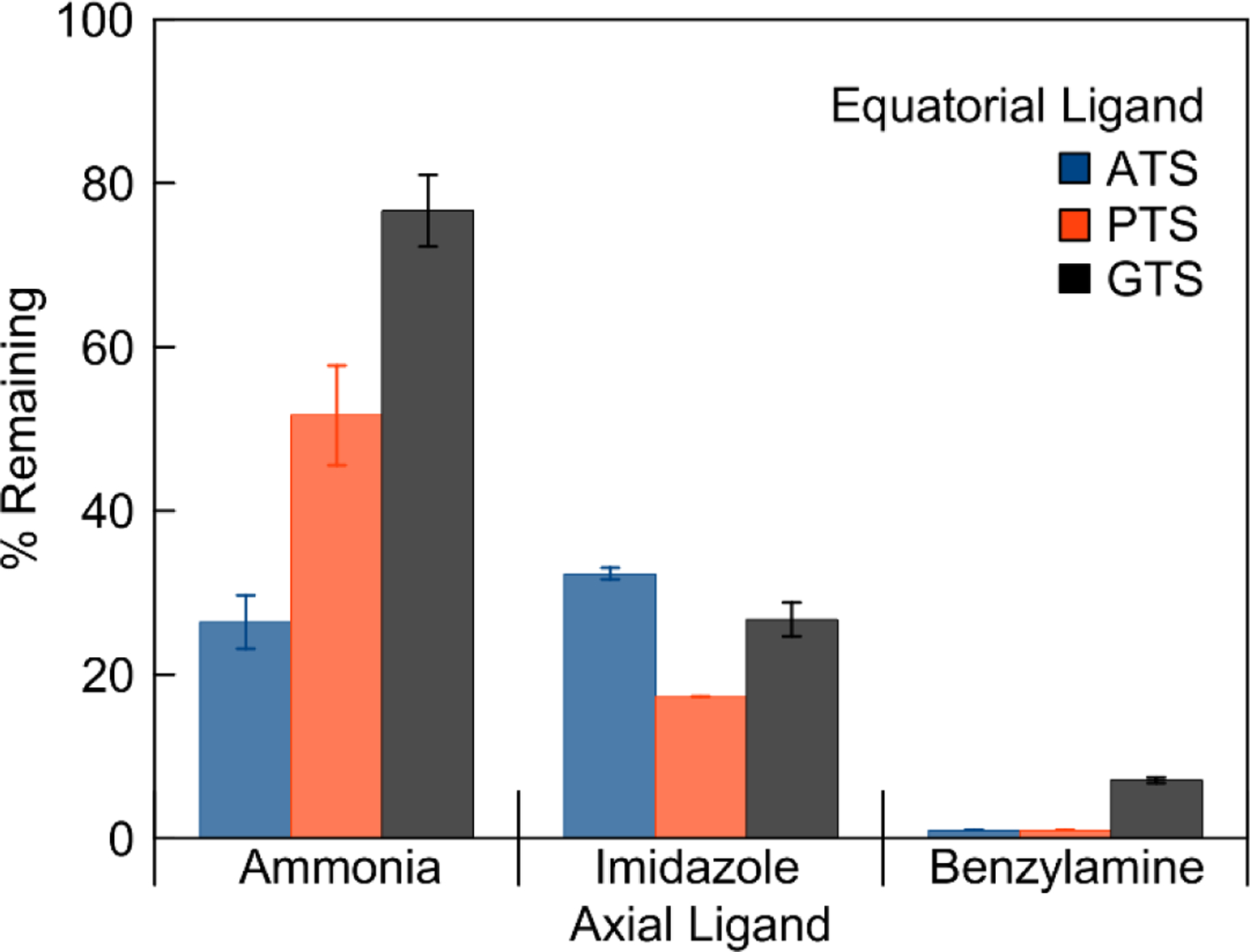
Percent of [Co(BTSC)(L)2]+ remaining in solution after 24 h incubation at 37 °C in PBS as measured by RP–HPLC. Values are the average of two independent experiments. Error bars represent the standard deviation.
The significant dependence on the nature of the axial ligand on the complex stability may indicate that these compounds are degrading via axial ligand substitution reactions, a phenomenon that is well studied for related cobalt-based Schiff-base enzyme inhibitors.18,29,61 This hypothesis was further explored by treating the [Co(BTSC)(L)2]+ complexes with 5 molar equivalents of N-methylimidazole (meIm) to qualitatively examine the susceptibility of these compounds to axial ligand substitution. The stability of the complexes with respect to the meIm challenge was investigated by RP-HPLC after 24 h. Decay of the peak corresponding to the intact complex was observed in conjunction with the appearance of two new peaks of higher retention times (Figure 6). We assign these new peaks as the monosubstituted and disubstituted complexes, [Co(BTSC)(L)(meIm)]+ and [Co(BTSC)(meIm)2]+ species based on their respective retention times on the column. The relative quantities of the starting complex, monosubstituted, and disubstituted complexes are shown in Table 6.
Figure 6.
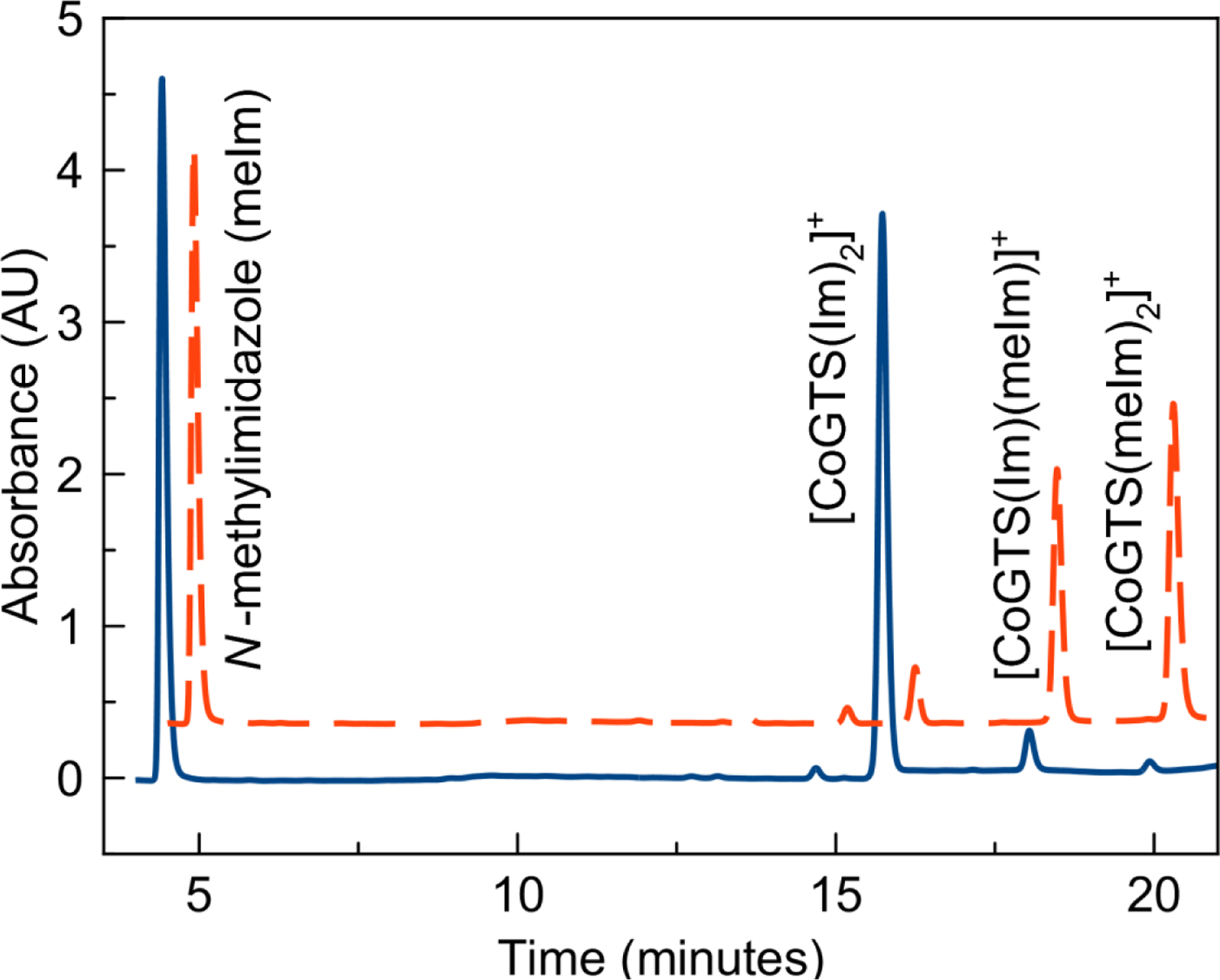
HPLC traces of 1 mM [CoGTS(Im)2]+ incubated with 5 mM N-methylimidazole immediately (solid blue) and 3 h (red dashes) after mixing. The 3-h trace is shifted up and to the right relative to the initial trace for clarity.
Table 6.
Axial ligand substitution of [Co(BTSC)(L)2]+ complexes by N–methylimidazole
| Compound | % Intact | % Monosubstituted | % Disubstituted |
|---|---|---|---|
| [Co(ATS)(NH3)2]+ | 38 | 27 | 35 |
| [Co(ATS)(Im)2]+ | 11 | 43 | 46 |
| [Co(ATS)(BnA)2]+ | 0 | 7 | 93 |
| [Co(PTS)(NH3)2]+ | 56 | 22 | 22 |
| [Co(PTS)(Im)2]+ | 7 | 39 | 54 |
| [Co(PTS)(BnA)2]+ | 1 | 6 | 93 |
| [Co(GTS)(NH3)2]+ | 65 | 15 | 20 |
| [Co(GTS)(Im)2]+ | 6 | 37 | 58 |
| [Co(GTS)(BnA)2]+ | 12 | 20 | 68 |
The stability trends for this meIm challenge study match those observed for the stability in PBS. Namely, the susceptibility of the complex to axial ligand substitution with meIm is mostly dependent on the axial ligand. The benzylamine and Im complexes exhibit the greatest amount of substitution by meIm after 24 h, whereas the complexes bearing axial ammonia ligands are resistant to meIm substitution. This trend is opposite to that observed for the axial ligand substitution of related cobalt(III) Schiff base compounds where axial ammonia ligands exhibited faster kinetics for meIm substitution compared to Im.61
Uptake Experiments.
To investigate the relationship between structure and intracellular accumulation, the uptake of the cobalt bis(thiosemicarbazone) complexes in A549 cells was quantified using GFAAS in conjunction with the Bradford protein assay. To test for hypoxia selectivity, these measurements were carried out under both normoxic and hypoxic conditions. The cells were treated with 100 μM of the complexes and incubated for 24 h prior to quantification of uptake. The results of these studies are shown in Figure 7. The ATS complexes are taken up much more efficiently than the PTS and GTS complexes. Notably, the GTS and PTS complexes exhibit less uptake than free Co(NO3). The efficient uptake of Co(NO3)2 by mammalian cells has been observed previously and was postulated to arise from either ion transporters or transferrin-mediated pathways.59 To investigate the generality of these trends among other cell lines, the uptake of [Co(BTSC)(Im)2]+ complexes was also evaluated in HeLa cells. These results verify that uptake follows the sequence GTS<PTS<ATS, as observed in A549 cells (Figure S64, SI).
Figure 7.
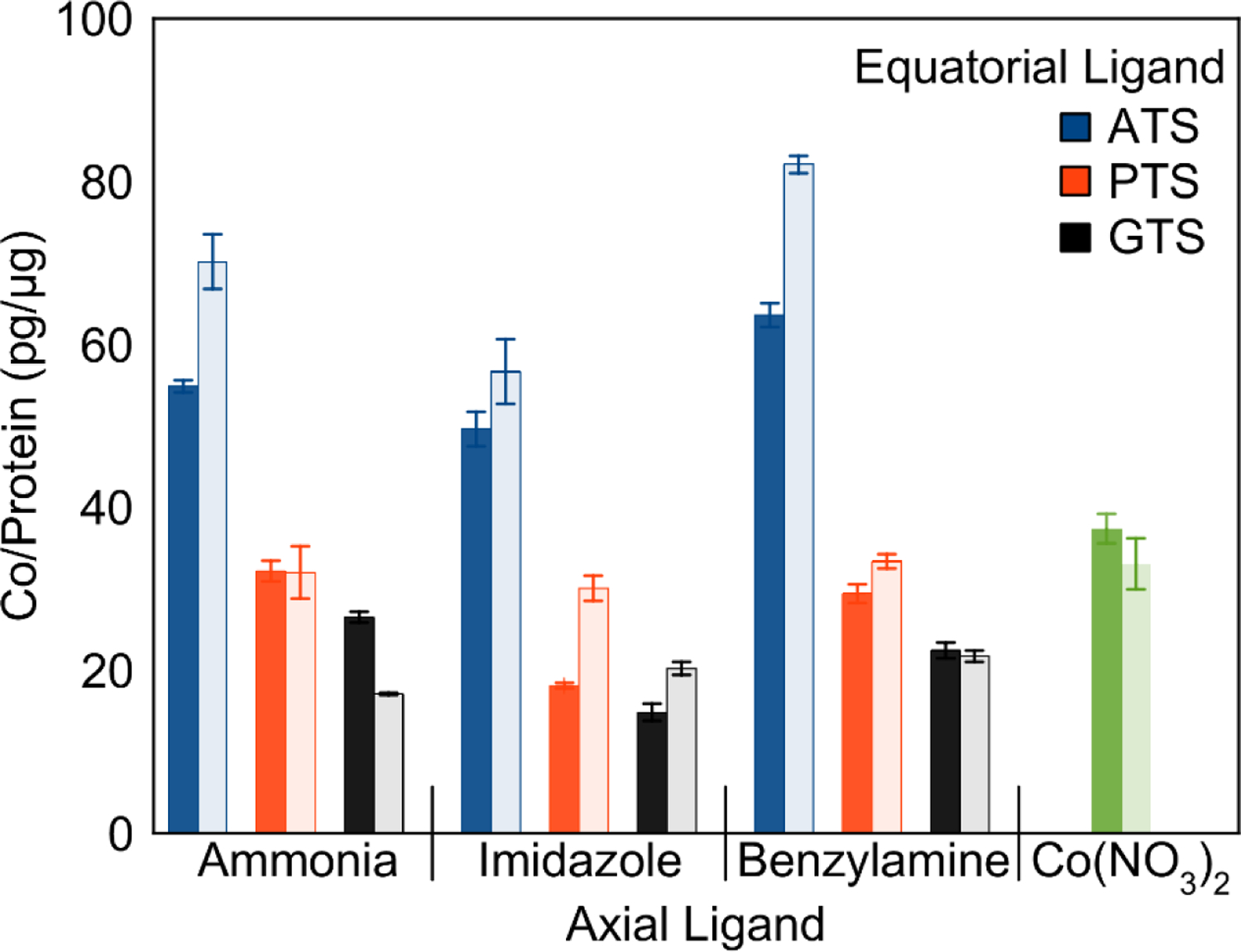
Uptake of [Co(BTSC)(L)2]+ complexes in normoxic (dark fill) and hypoxic (light fill) conditions. Bars indicate the mass ratio of cobalt to protein (pg/μg) in each sample after 24-h incubation with 100 μM Co complex. Results are the average of three samples for each complex.
Several complexes exhibited differential accumulation under hypoxic conditions. The ratio of uptake in normoxia and hypoxia is given in Table 7. Six out of the nine complexes showed >10% increase in uptake under hypoxic conditions. [CoGTS(NH3)2]+, in contrast, showed decreased uptake under hypoxic conditions. In general, the less stable, hydrophobic complexes containing imidazole and benzylamine demonstrated the highest hypoxia selectivity. Compounds containing the ATS and PTS equatorial ligands exhibit higher selectivity on average than GTS complexes. This trend in selectivity matches that reported for the [Cu(BTSC)] complexes, despite the fact that the copper complexes have a much more positive reduction potential relative to the cobalt(III) bis(thiosemicarbazones).65,66 The observed hypoxia selectivity is of these complexes is substantially lower than those reported for [Cu(ATSM)] (5× selectivity) and its derivatives.65,66
Table 7.
Cellular Uptake of Complexes in A549 Cells
| Compound | Co/Protein (pg/μg) (Normoxia) | Co/Protein (pg/μg) (Hypoxia) | Hypoxia/Normoxia Ratio |
|---|---|---|---|
| Co(NO3)2 | 37 ± 2 | 33 ± 3 | 0.88 ± 0.09 |
| [Co(ATS)(NH3)2]+ | 55 ± 1 | 70 ± 3 | 1.28 ± 0.05 |
| [Co(ATS)(Im)2]+ | 50 ± 2 | 57 ± 4 | 1.10 ± 0.09 |
| [Co(ATS)(BnA)2]+ | 64 ± 1 | 82 ± 1 | 1.30 ± 0.03 |
| [Co(PTS)(NH3)2]+ | 32 ± 1 | 32 ± 3 | 0.99 ± 0.09 |
| [Co(PTS)(Im)2]+ | 18.0 ± 0.3 | 30 ± 1 | 1.70 ± 0.07 |
| [Co(PTS)(BnA)2]+ | 29 ± 1 | 33 ± 1 | 1.10 ± 0.04 |
| [Co(GTS)(NH3)2]+ | 27 ± 1 | 17.0 ± 0.1 | 0.65 ± 0.01 |
| [Co(GTS)(Im)2]+ | 15 ± 1 | 20 ± 1 | 1.40 ± 0.09 |
| [Co(GTS)(BnA)2]+ | 22 ± 1 | 22 ± 1 | 0.97 ± 0.04 |
In Vitro Anticancer Activity.
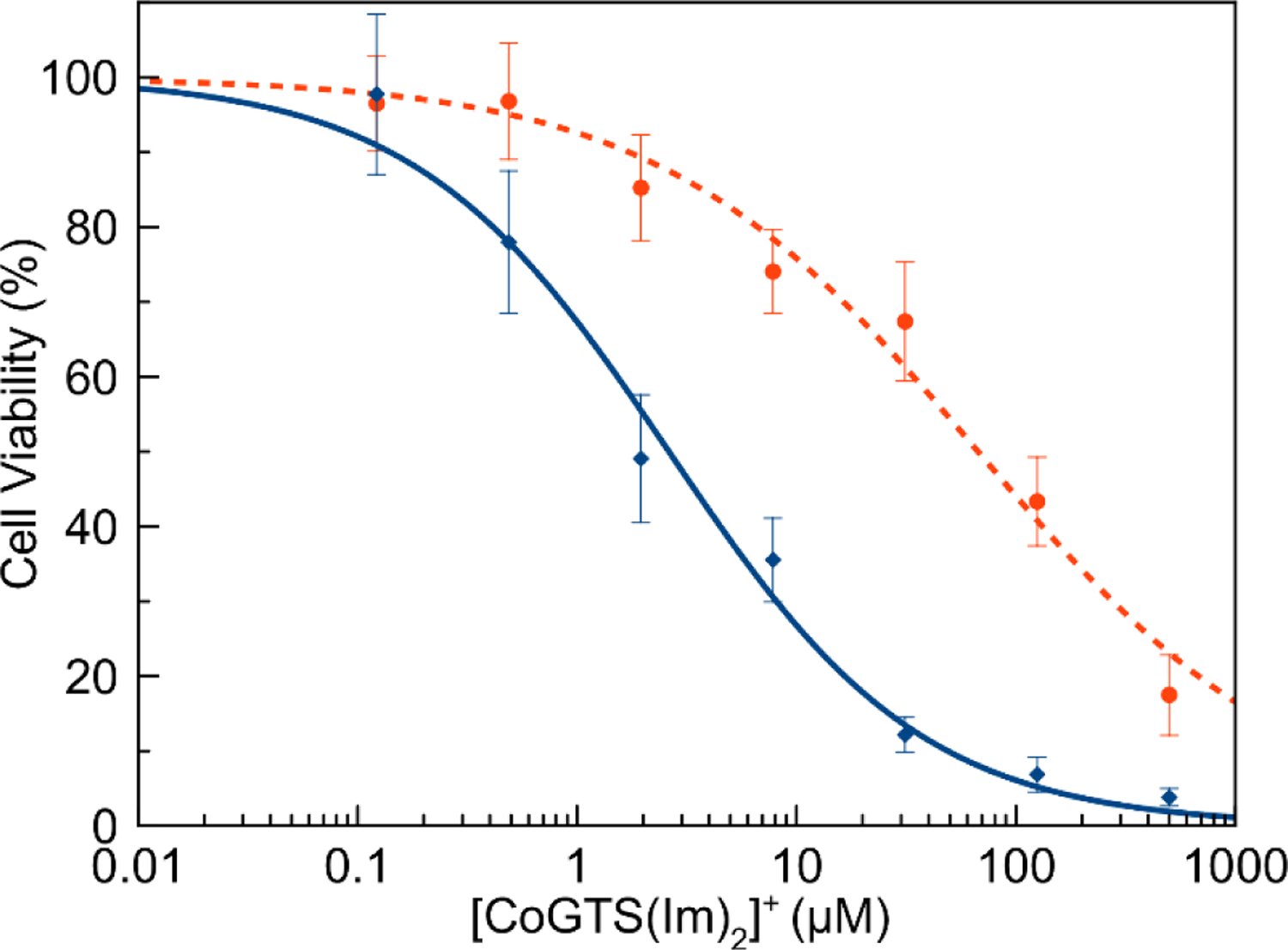
The cytotoxic effects of the complexes and free ligands were determined under both normoxic and hypoxic conditions in HeLa and A549 cell lines using the MTT assay. The cytotoxic activity of the complexes is tabulated as 50% growth inhibitory concentration (IC50) values in Table 8. Figure 8 shows the dose-response curves for the treatment of A549 with the three [Co(BTSC)(Im)2]+ complexes. Representative dose-response curves for the other compounds and cell lines are deposited in the SI, Figures S65–S73. Figure 8 reflects the general trends that are seen for all the complexes; complexes bearing the GTS ligand exhibit potent cytotoxicity, whereas those bearing the ATS ligand are inactive. The dose-response curves of complexes bearing the PTS ligand indicate a dose-dependent decrease in cell viability. However, these curves display a leveling out effect at 30–50% cell viability, where beyond a certain concentration threshold the cell viability remains constant. Even at the highest concentration screened (500 μM), the cell viability does not go to zero. This leveling out effect limited our ability to determine precise IC50 values, as the leveling out often occurred near 50% cell viability. The trends in cytotoxicity correspond with the activities of the free ligands, as the toxicity of the free ligands increases in the order ATS < PTS ≈ GTS. Notably, the activity of free PTS and GTS ligands exceeds that of the cobalt(III) complexes, indicating that coordination to cobalt(III) significantly attenuates their cytotoxicity. All the cobalt(III) complexes are also less active than the established metal-based anticancer drug cisplatin.
Table 8.
IC50 Values (μM) for [Co(BTSC)(L)2]NO3 Complexes
| HeLa | A549 | MRC-5 | |||
|---|---|---|---|---|---|
| Compound | Normoxia | Hypoxia | Normoxia | Hypoxia | Normoxia |
| Cisplatin | 1.7 ± 0.07 | 1.7 ± 0.21 | 5.5 ± 3.2 | 5.2 ± 2.4 | 3.5 ± 1.2 |
| Tirapazamine | 44 ± 19 | 13 ± 4.2 | 60 ± 5.4 | 11 ± 6.5 | n.d.b |
| ATS | >20 | >20 | 16.4 ± 0.87 | 17.5 ± 2.7 | n.d. b |
| PTS | 0.027 ± 0.002 | 0.055 ± 0.001 | 0.06 ± 0.03 | 0.09 ± 0.03 | n.d. b |
| GTS | 0.067 ± 0.008 | 0.094 ± 0.010 | 0.02 ± 0.01 | 0.04 ± 0.01 | >20 |
| Co(NO3)2 | >500 | >500 | >500 | >500 | >500 |
| [Co(ATS)(NH3)2]+ | >500 | >500 | >500 | >500 | n.d. b |
| [Co(ATS)(Im)2]+ | >500 | >500 | >500 | >500 | n.d. b |
| [Co(ATS)(BnA)2]+ | >500 | >500 | >500 | >500 | n.d. b |
| [Co(PTS)(NH3)2]+ | ≈22a | ≈15a | ≈16.9a | ≈21.8a | n.d. b |
| [Co(PTS)(Im)2]+ | ≈28a | ≈17a | ≈4.2a | ≈8.6a | n.d. b |
| [Co(PTS)(BnA)2]+ | ≈8.6a | ≈8.8a | ≈7.1a | ≈27a | n.d. b |
| [Co(GTS)(NH3)2]+ | 21 ± 2.9 | 15 ± 4.6 | 12 ± 1.4 | 9.2 ± 1.4 | >250 |
| [Co(GTS)(Im)2]+ | 7.4 ± 2.4 | 3.7 ± 2.3 | 2.6 ± 0.4 | 2.9 ± 0.7 | >250 |
| [Co(GTS)(BnA)2]+ | 7.3 ± 1.5 | 6.3 ± 0.76 | 5.8 ± 0.95 | 8.2 ± 1.9 | >250 |
Accurate IC50 values could not be obtained for these compounds because the dose-response curves level off near 50% cell viability. These values represent the average concentration at which the beginning of this leveling effect is observed in the dose-response curves.
n.d. = not determined.
Figure 8.
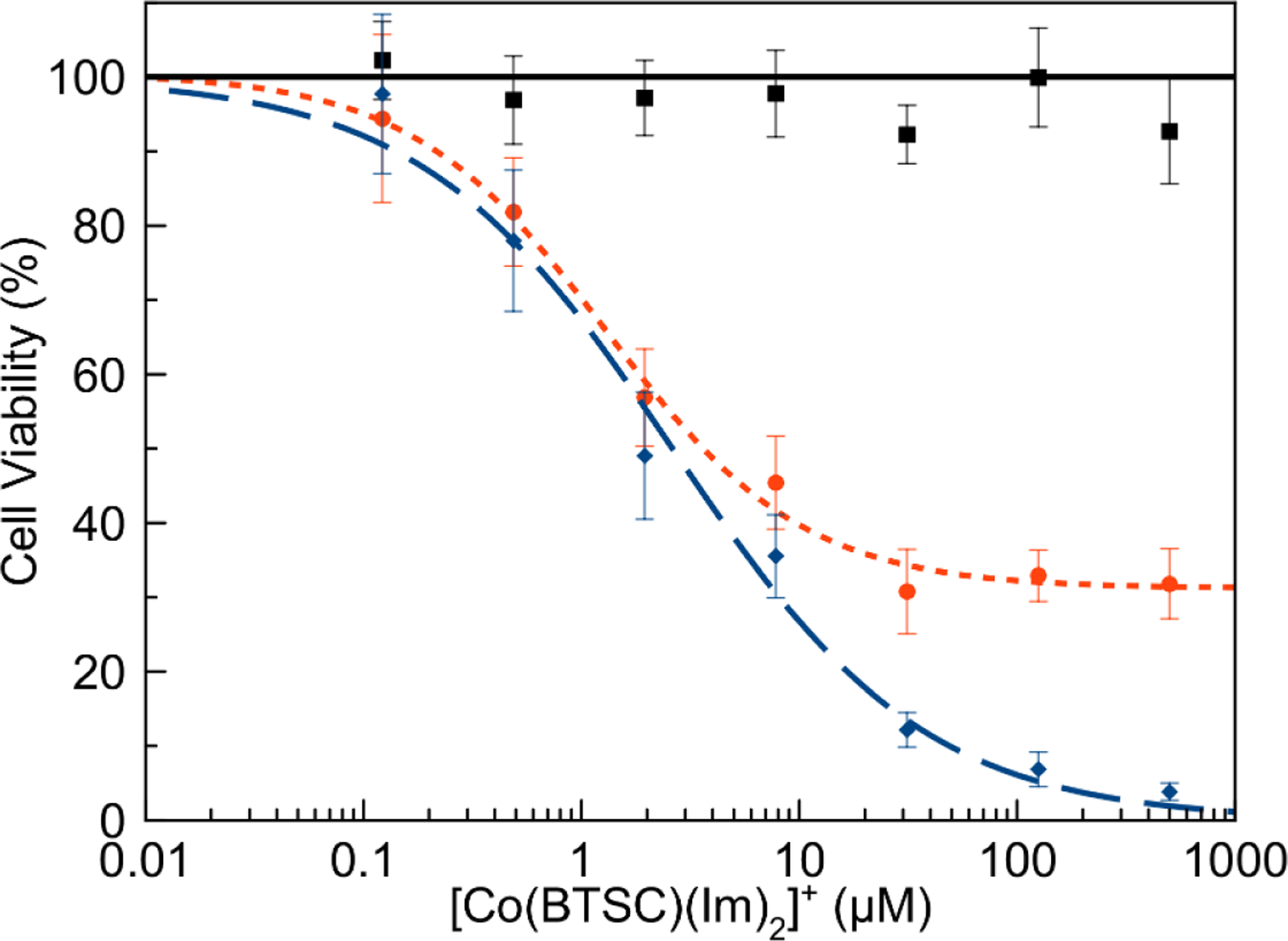
Effect of [Co(BTSC)(Im)2]+ complex concentration on A549 cell viability as measured by the MTT assay for [Co(ATS)(Im)2]+ (solid black line, square marker), [Co(PTS)(Im)2]+ (small-dashed orange line, circle marker), and [Co(GTS)(Im)2]+ (broad blue dashed line, diamond marker).
The axial ligands show less pronounced effects on the cytotoxicity. In general, complexes containing benzylamine and imidazole are slightly more potent than those with ammonia, but this trend does not hold for all nine complexes. The complexes were also tested in HeLa cells (Table 7). The activity of these complexes and the free BTSC ligands was greater than that observed in A549 cells, but the trends in activity remain the same. The cytotoxicity was also evaluated in MRC-5 normal lung fibroblasts as a model for non-cancerous cells (Figure 9, Table 7). The cytotoxicity of the complexes exhibits a distinct cell line dependence with the most potent activity in A549 cells, less activity in HeLa cells, and no substantial cytotoxicity in MRC-5 cells. Notably, cisplatin exhibits less cell-line dependence and is cytotoxic in the micromolar range against normal MRC-5 cells (Figure 9).
Figure 9.
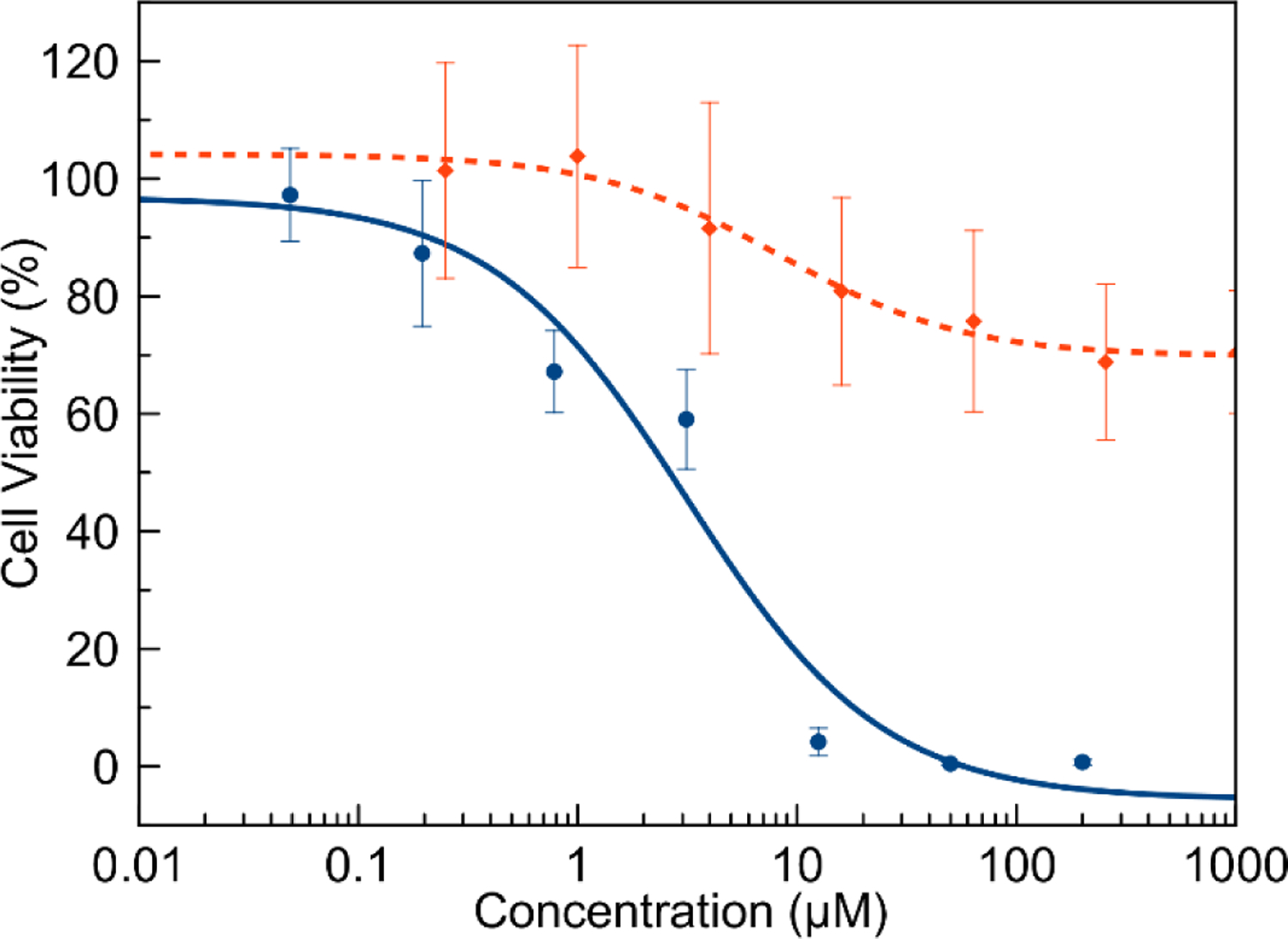
Dose-response curves of cisplatin (solid blue line, circular marker) and [Co(GTS)(NH3)2]+ (orange dashes, diamond marker) in MRC-5 (normal lung fibroblast) cells.
Most complexes showed no statistically significant increase in cytotoxicity under hypoxic conditions. Only a modest decrease in the IC50 values of the PTS and GTS complexes was observed under hypoxia. In contrast, the free PTS and GTS ligands exhibited decreased activity in hypoxia. Tirapazamine, a well-characterized hypoxia-selective cytotoxic agent, was used as a positive control.67,68 This compound displayed a hypoxia selectivity index of 3.3 in HeLa cells in our lab; this value is lower than the typical hypoxia selectivity index of 10 that is observed for this compound in other studies.69,70 Cisplatin exhibited no difference in activity under hypoxic versus normoxic conditions.
Cytotoxicity Experiments with Tetrathiomolybdate.
The correlation in activity of the complexes with their BTSC ligands suggests that they may be operating by similar mechanisms of action. The BTSC ligands induce their cytotoxic effects in a copper ion-dependent manner.48 These ligands act as ionophores for copper, shuttling [Cu(BTSC)] complexes into cells where they generate cytotoxic reactive species that induce cell death.71 Treatment of cells with 5 μM tetrathiomolybdate (TM) decreases the cytotoxic effects of these ligands by depleting the available copper pool.48,72,73 To test whether the [Co(GTS)(L)2]+ complexes operate via a similar mechanism, A549 cells were treated with both [Co(GTS)(L)2]+ complexes and TM. The IC50 values increased by a factor of 10 when cells were treated with TM, as shown in Table 9 and Figure 10. To confirm that TM does not react with the [Co(BTSC)(L)2]+ complexes, [Co(GTS)(NH3)2]NO3 and TM (1:1) were co-incubated in DMSO-d6 and monitored by NMR spectroscopy. Only intact [Co(GTS)(NH3)2]NO3 is detected, indicating that TM does not remove the cobalt(III) ion (SI, Figures S74–S75).
Table 9.
Effect of TM on IC50 values (μM) in A549 Cellsa
| Compound | Without TM | With 5 μM TM |
|---|---|---|
| GTS | 0.02 ± 0.01 | 3.93 ± 0.85 |
| [Co(GTS)(NH3)2]+ | 12 ± 1.4 | 290 ± 105 |
| [Co(GTS)(Im)2]+ | 2.6 ± 0.4 | 68 ± 23 |
| [Co(GTS)(BnA)2]+ | 5.8 ± 1.0 | 99 ± 20 |
TM does not affect the cell viability at 5 μM concentration.
Figure 10.
Effect of [CoGTS(Im)2]+ complex concentration on A549 cell viability in the presence (dashed line, circular marker) and absence (solid blue line, diamond marker) of TM.
Discussion
The anticancer potential of cobalt(III) complexes is currently a topic of significant interest, which has prompted the investigation of numerous different cobalt(III) coordination complexes.23,27,30 Additionally, the BTSC ligands are well-characterized, biomedically active agents.49,74,75 Although there are a few reports of cobalt(II) complexes of BTSCs,76,77 there is surprisingly no prior investigation on the cobalt(III) analogues. In this study, we were interested in combining the biological effects of the BTSC ligands with the hypoxia-targeting properties of cobalt(III) complexes, premised on the hypothesis that reductive activation of these complexes would give rise to hypoxia-selective anticancer agents. We synthesized a small library comprising nine [Co(BTSC)(L)2]+ complexes to elucidate structure-activity relationships for this class of compounds. The results of this study may be compared to related and well-established anticancer cobalt(III) Schiff base complexes29 and [Cu(BTSC)] compounds78 in order to provide a comprehensive picture regarding relationships between metal complex structure and biological activity.
The synthesis of the compounds proceeds by the sequential reaction of Co(NO3)2 with the BTSC followed by the addition of the axial ligand (Scheme 1). The reaction takes place with the aerobic oxidation of the cobalt(II) to cobalt(III). A green intermediate observed prior to addition of the axial ligands is hypothesized to be a cobalt(II) complex of the BTSC ligand. The cobalt(III) compounds were isolated as dark red solids. In contrast to the [Cu(BTSC)] complexes, the cobalt(III) analogues exhibit good water solubility; stock solutions of the compounds in mM concentrations in pure water could be readily made. These complexes were characterized and verified to be pure via NMR spectroscopy, X-ray crystallography, and HPLC. The NMR spectra of the nine complexes display no signs of paramagnetism, consistent with the expected diamagnetic nature of a low-spin cobalt(III) complex. Upon coordination to cobalt(III), the 1H NMR resonances of the BTSC ligands shift upfield, and the acidic azomethine proton resonance disappears. Analysis of these complexes by HR-ESI-MS revealed molecular ion peaks with masses matching those expected for the general formulae [Co(BTSC)(L)2]+. The crystal structures of [Co(GTS)(Im)2]+, [Co(GTS)(BnA)2]+, and [Co(ATS)(Im)2]+ (Figure 1) reveal the expected octahedral coordination geometry at the cobalt(III) center. The equatorial Co–BTSC ligand distances are relatively invariant in the three complexes. The axial Co–Im and Co–BnA distances are different; the Co–BnA distances are approximately 0.04 Å longer than the Co–Im distances, a feature that may be attributed to steric crowding at the cobalt(III) center by the large benzylamine ligands.
Further characterization of these compounds was accomplished by electronic absorption spectroscopy, 59Co NMR spectroscopy, and cyclic voltammetry. Absorbance bands in the UV-Vis spectra in the ranges of 340 to 390 nm and 499 to 530 nm shift depending on the nature of the equatorial ligand (Figure 3, Table 4). Both bands blue-shift following the sequence GTS < PTS < ATS. A smaller blue-shift is also apparent as the axial ligand is varied from either benzylamine or imidazole to ammonia. The low energy absorbance bands in the 499 to 530 nm range are assigned as a metal-to-ligand charge transfer (MLCT) transitions, and the higher energy feature in the 340 to 390 nm range is assigned as an intraligand π–π* transition. These assignments are made based on their large molar absorptivities and relative energies. The assignments are also consistent with the observed trends. As the equatorial ligand shifts from weakly electron donating (GTS) to strongly electron donating (ATS), the ligand-based π* orbital increases in energy leading to a concomitant increase in energy of both the MLCT and ligand-based transitions. These absorbance spectra are qualitatively similar to those of previously reported cobalt(III) Schiff base complexes, for which the major features were a low-energy MLCT and a high energy π –π* transition.18
The cyclic voltammograms show a clear relationship between ligand donor-capacity and the complex reduction potential. The initial reductive sweep in all complexes produces an irreversible reduction, which we assign to the Co(III)/Co(II) coupled with axial ligand loss, followed by a quasi-reversible Co(II)/Co(I) reduction of the remaining complex bearing only the equatorial BTSC ligand (Figure 4). As such, the reduction potential of the Co(II)/Co(I) couple is independent of the nature of the axial ligand (Table 5). As the equatorial ligands are altered from ATS to PTS to GTS the reduction potential of the Co(II/I) couple shifts by approximately +100 mV. This trend is consistent with that observed in the [Cu(BTSC)] complexes, which decrease in reduction potential by approximately 60 mV as methyl groups are added to the ligand backbone.78 ATS with two electron-donating methyl groups renders reduction of the cobalt center less favorable, giving rise to a more negative redox potential. The Co(III)/Co(II) redox couple, arguably the more important redox process for hypoxia targeting, is reflected by an irreversible peak potential. This peak potential is sensitive to both equatorial and axial ligands. The equatorial ligand effects follow the expected trend as for the Co(II)/Co(I) couple. For the same BTSC ligand, the benzylamine and imidazole complexes had more positive reduction potentials than the corresponding ammonia complexes, as expected based on the relative donating abilities of the ligands.79 The most positive reduction potential observed for the Co(III)/Co(II) couple is −0.69 V for [CoGTS(BnA)2]+, substantially more negative than the value reported for the established hypoxia-imaging agent CuATSM, −0.59 V.78 The reduction potentials of the cobalt(III) BTSC complexes lie on the edge of the range needed for hypoxia selectivity (−0.75 to −0.35 V vs SCE).15,23,53 Notably, irreversible reduction feature (E) (Figure 4) corresponds to the Co(III)/Co(II) couple of the complex with axial DMF ligands, which is generated electrochemically after the irreversible reduction of the intact complex. This reduction feature is 500 mV more positive than the Co(III)/Co(II) couples of the intact complexes, feature (A). This result suggests that ligand substitution of the axial ligands by an oxygen donor may shift the reduction potential to a region more suitable for reduction in the cellular environment.
The 59Co NMR spectra (Figure 2) corroborate the trends observed in UV-Vis spectra and cyclic voltammograms. The 59Co chemical shifts move upfield as the equatorial ligand is altered from ATS to PTS to GTS, and as the axial ligand is changed from Im to BnA to NH3 (Table 3). As discussed above, the linewidth of the resonances depends only on the axial ligand with the NH3 complexes displaying the sharpest signals. The linewidth of quadrupolar nuclei depends on the nuclear quadrupole coupling constant, the asymmetry of the electric field gradient, and on the rotational correlation time of the complex.62 The complexes are all roughly the same size; therefore their rotational correlation times should be similar. The difference in linewidth observed for the ammonia complex most likely arises from the smaller nuclear quadrupolar coupling constant and electric field gradient conferred by this ligand. The ammonia complexes give resonances that are shifted upfield by approximately 300 ppm relative to the corresponding benzylamine and imidazole complexes, signifying an increase in the shielding of the cobalt nucleus by ammonia.63,64 The chemical shifts of 59Co nuclei correlate linearly with the lowest energy d–d transition corrected for the nephelauxetic ratio of the ligands.51,62 In this case, the downfield chemical shift of the ammonia complexes is consistent with the stronger field nature of ammonia relative to imidazole.80 The weaker field nature of benzylamine relative to ammonia in this case may be a consequence of steric crowding of the metal center, as evidenced from the crystal structure, which minimizes metal-ligand orbital overlap and decreases the ligand field splitting energy.
The stability of the complexes in aqueous solution may be a key factor in their anticancer activity, and establishing trends in complex stability facilitates the design of improved analogues. We find that the axial ligand plays a large role in the stability of the complex. Complexes with axial ammonia ligands are more stable in pH 7.4 PBS than those with imidazole or benzylamine (Figure 5). This trend also holds when the complexes are challenged with N-methylimidazole as a competing ligand (Figure 6, Table 6). We hypothesize that the decrease in stability of the imidazole and benzylamine complexes arises due to weaker ligand donor strength and unfavorable steric interactions, respectively. Octahedral cobalt(III) complexes undergo ligand substitution reactions primarily via interchange dissociative-type mechanisms, processes that are accelerated by such factors.81 These results support the hypothesis that axial ligand identity is integral to complex stability, and the complexes are sufficiently labile to degrade even under normoxic conditions.
[Cu(BTSC)] complexes, when used in conjunction with the radionuclide 64Cu, can be employed for PET imaging of hypoxic tissue. The selective reduction of [Cu(BTSC)] complexes in hypoxic regions leads to the release and intracellular trapping of the copper ions. To determine the uptake profile and potential selectivity of the Co(BTSC) complexes for hypoxia based on the Co(III)/Co(II) redox couple, their uptake in both normoxic and hypoxic conditions was measured in A549 cells. These studies show a large dependence of the cellular uptake on the nature of the equatorial ligand (Figure 7, Table 7). Cellular uptake increases in the order GTS<PTS<ATS. This sequence follows the relative lipophilicity of the equatorial BTSC ligand. A similar trend in cellular uptake has been observed for cobalt(3,4-diarylsalen) complexes, whereby increased lipophilicity of the salen ligand facilitated cellular uptake of cobalt.82 The degree of uptake shows little correlation to the stability of the complexes. The uptake varies by a factor of approximately two for the different equatorial ligands, while the complex stabilities vary relatively little with respect to the equatorial ligand. The axial ligand has only a minor influence on the overall cell uptake. This result is somewhat surprising, especially for complexes bearing the axial benzylamine ligand. This lipophilic ligand should facilitate uptake of the complex via passive diffusion. The lack of significant axial ligand dependence may indicate that the axial ligands dissociate before the complex enters the cell or may suggest that more complex uptake pathways are operative. The presence of O2 influenced the uptake in some cases, as ATS complexes showed mildly increased uptake under hypoxic conditions, while PTS and GTS complexes generally showed no selectivity or normoxic selectivity. 82
Cytotoxicity of the complexes in cancer cells, like uptake, is primarily dependent on the nature of the equatorial ligand (Table 8). GTS complexes are the most cytotoxic, and ATS complexes are essentially inactive. This trend follows that observed for the free ligands and for the corresponding copper(II) complexes, where GTS and PTS are the most cytotoxic and ATS is the least.48,83 The IC50 values of the cobalt(III) complexes are at least a factor of 10 higher than those of the free ligand and copper complexes, indicating that coordination to this inert ion acts to attenuate the cytotoxicity of the ligand. [Cu(BTSC)] complexes, such as [Cu(ATSM)], are often more cytotoxic than the free ligands. Related Co(salen) complexes exhibit IC50 values in the range of 10 μM in different cancer cell lines, consistent with the activities of the Co(BTSC) complexes studied here.82 With respect to cytotoxic activity, the importance of the axial ligand is less than that of the equatorial ligand. In general, the complexes bearing axial ammonia ligands are less active than the corresponding imidazole and benzylamine complexes. As the axial ligands primarily affect the stabilities of the complexes, this trend suggests that there is an inverse relationship between complex stability and cytotoxicity in complexes containing the same equatorial ligand. The cell uptake studies indicate that the ATS complexes are taken up much more effectively than those of PTS and GTS. This inverse relationship between cell uptake and cytotoxicity indicates that different factors are important for these properties. The more lipophilic nature of ATS compared to GTS may lead to its increased uptake. However, the lack of cytotoxicity of the ATS complexes is consistent with the lack of activity of the free ligand. This result suggests that the cytotoxicity of the cobalt(III) complexes is mediated by that of the free ligand; the greater cell uptake of the [Co(ATS)(L)2]+ complexes cannot compensate for the lack of cytotoxic activity of the ATS ligand. As such, it is likely that the cobalt(III) complexes are acting as chaperones for the cytotoxic bis(thiosemicarbazones). Although the PTS complexes exhibit cytotoxic activity, they fail to kill 100% of the cells even at concentrations exceeding 100 μM. As such, the dose-response curves (Figure 8) level out at high concentrations. This leveling out effect has also been observed in the dose-response curves of related BTSC ligand and their corresponding copper complexes.84 This behavior for cytotoxic compounds is previously described and is attributed to cell-to-cell variability and is often observed in compounds that inhibit the cell cycle at a specific phase.58,85
With respect to hypoxic cytotoxicity, only moderate increases in activity––1.2 to 1.4-fold––are observed when the cells are incubated under conditions of hypoxia. The lack of significant increases in hypoxic cytotoxicity is consistent with the very negative reduction potentials of the cobalt(III) complexes that fall outside the window that is typically necessary for hypoxia targeting. Although not directly useful for therapeutic applications, the small increases in hypoxic activity suggest that further ligand tuning may give access to cobalt(III) BTSC complexes with suitable properties for hypoxia targeting. The observed lack of correlation between hypoxic uptake and reduction potential matches that observed for a class of cobalt(III)–cyclam complexes, which were proposed to be trapped intracellularly via ligand substitution rather than reduction.86
The GTS complexes exhibit minimal cytotoxic effects in the non-cancerous lung fibroblast cell line MRC-5 (Figure 9). The result indicates that such complexes may possess a beneficial therapeutic index for the selective killing of cancer cells. This property is also shared by the free BTSC ligands and the corresponding copper(II) complexes. The fact that this selectivity for cancerous over non-cancerous cell lines is maintained in the cobalt(III) complexes further supports the hypothesis that these complexes are acting as chaperones for the active BTSC ligand. The free BTSC ligands induce cytotoxicity by acting as copper(II) ionophores. These agents form copper(II) complexes which enter the cell and generate reactive-oxygen species, killing the cell.48,87 As such, treating cells with the copper-depleting agent TM protects them from BTSC-induced cytotoxicity. Treatment of A549 cells with 5 μM TM also inhibited the cytotoxic activity of [Co(GTS)(L)2]+ complexes (Figure 10). The IC50 values increased by a factor of 10 in the presence of TM (Table 9). This result suggests that these cobalt(III) complexes operate via a similar mechanism of action as the free BTSC ligand, which involves copper(II) ions, and is consistent with the fact that the cytotoxic activity of the cobalt(III) complexes correlates directly with that of the free ligands. As TM is known to induce a large number of effects on cancer cells, however, further studies are required to verify this hypothesis.
Based on the physical properties, uptake, and cytotoxicity of the complexes, we propose a mechanism in which the cobalt complex acts as a prodrug that breaks down upon a combination of reduction and ligand substitution to yield the cytotoxic BTSC ligand. The more labile imidazole and benzylamine complexes undergo substitution, yielding complexes with more positive reduction potentials in line with the cyclic voltammetric studies. Reduction to cobalt(II) may favor transmetalation of the BTSC ligand with copper(II), forming a cytotoxic agent. Transmetalation of a zinc(II) thiosemicarbazone complex with copper(II) has been observed,88 and the ability of copper(II) to transmetalate a cobalt(II) complex is expected thermodynamically on the basis of the Irving-Williams series.89 The more inert ammonia complexes are less cytotoxic, consistent with the proposed mechanism. These results indicate that compounds’ toxicity may be modified directly by changing the (BTSC) ligand, or indirectly by modifying the axial ligand, thus affecting the rate of release.
Summary and Conclusions
Nine [Co(BTSC)(L)2]NO3 complexes have been synthesized and characterized. A detailed investigation of their physical and biochemical properties enabled us to determine structure-activity relationships for this class of compounds. We find that the overall stability of the complex is modulated by the nature of the axial ligand, whereas the redox potential is primarily dictated by the nature of the equatorial ligand. The cytotoxicity of the complexes depends largely on the equatorial (BTSC) ligand, with toxicity increasing in the order ATS<PTS≈GTS, following the correlation expected for the free ligands and copper(II) complexes. The cellular uptake of these complexes, in contrast, follows the opposite trend where ATS complexes are taken up more efficiently than the corresponding PTS and GTS complexes. Poor hypoxia selectivity with respect to both cell uptake and cytotoxicity was observed, which we attribute to the highly negative redox potentials of the complexes. By using TM as a copper-depleting agent, we showed that the anticancer activity of the complexes may operate via a copper-dependent mechanism in a similar fashion as the free BTSC ligands, providing a feasible hypothesis for further mechanistic studies. Collectively these results suggest a mechanism where cobalt complexes enter the cell, engage in ligand substitution reactions that increase the redox potential, undergo reduction, and then transmetalate with copper(II) or release free cytotoxic BTSC ligands. Thus, structural modification of the axial ligands may be used to tune the kinetics of substitution of the prodrug complex. These results serve as a guide for the development of second generation anticancer [Co(BTSC)(L)2]+ prodrugs.
Supplementary Material
Acknowledgements
This research was supported by Cornell University. A. Paden King thanks the National Institute of Health, National Institute of General Medical Sciences, for a Chemical Biology Interface (CBI) Training Grant (grant number T32GM008500). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. Hendryck A. Gellineau thanks the Howard Hughes Medical Institute grant: Mentored Learning for Groups Underrepresented in Biomedical Research (Project ID 52008135), and the Frank L. and Lynnet Douglas Fellowship Award for summer research support. Dr. Ivan R. Keresztes is thanked for assistance with 59Co NMR spectroscopy. Mr. Liang Liang Feng is thanked for assistance with HPLC experiments and cobalt(III) complex stability studies. This work made use of the NMR facility and the Cornell Center for Material Research (CCMR) at Cornell University, which are supported by the NSF under award numbers CHE-1531632 and DMR-1120296, respectively.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS publications website at http://pubs.acs.org.
Complex characterization data (PDF)
Crystallographic data (CIF).
The authors declare no competing financial interests.
References
- (1).Abu-Surrah A; Kettunen M Platinum Group Antitumor Chemistry: Design and Development of New Anticancer Drugs Complementary to Cisplatin. Curr. Med. Chem 2006, 13, 1337–1357. [DOI] [PubMed] [Google Scholar]
- (2).Johnstone TC; Wilson JJ; Lippard SJ Monofunctional and Higher-Valent Platinum Anticancer Agents. Inorg. Chem 2013, 52, 12234–12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wilson JJ; Lippard SJ Synthetic Methods for the Preparation of Platinum Anticancer Complexes. Chem. Rev 2014, 114, 4470–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Daugaard G; Abildgaard U Cisplatin Nephrotoxicity. A Review. Cancer Chemother. Pharmacol 1989, 25, 1–9. [DOI] [PubMed] [Google Scholar]
- (5).Cvitkovic E Cumulative Toxicities from Cisplatin Therapy and Current Cytoprotective Measures. Cancer Treat. Rev 1998, 24, 265–281. [DOI] [PubMed] [Google Scholar]
- (6).Screnci D; McKeage MJ Platinum Neurotoxicity: Clinical Profiles, Experimental Models and Neuroprotective Approaches. J. Inorg. Biochem 1999, 77, 105–110. [DOI] [PubMed] [Google Scholar]
- (7).Yao X; Panichpisal K; Kurtzman N; Nugent K Cisplatin Nephrotoxicity: A Review. Am. J. Med. Sci 2007, 334, 115–124. [DOI] [PubMed] [Google Scholar]
- (8).Argyriou AA; Polychronopoulos P; Iconomou G; Chroni E; Kalofonos HP A Review on Oxaliplatin-Induced Peripheral Nerve Damage. Cancer Treat. Rev 2008, 34, 368–377. [DOI] [PubMed] [Google Scholar]
- (9).Joyce JA Therapeutic Targeting of the Tumor Microenvironment. Cancer Cell 2005, 7, 513–520. [DOI] [PubMed] [Google Scholar]
- (10).Hambley TW Physiological Targeting to Improve Anticancer Drug Selectivity. Aust. J. Chem 2008, 61, 647–653. [Google Scholar]
- (11).Le Q-T; Courter D Clinical Biomarkers for Hypoxia Targeting. Cancer Metastasis Rev 2008, 27, 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wilson WR; Hay MP Targeting Hypoxia in Cancer Therapy. Nat. Rev. Cancer 2011, 11, 393–410. [DOI] [PubMed] [Google Scholar]
- (13).Wojtkowiak JW; Verduzco D; Schramm KJ; Gillies RJ Drug Resistance and Cellular Adaptation to Tumor Acidic pH Microenvironment. Mol. Pharm 2011, 8, 2032–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hall MD; Failes TW; Yamamoto N; Hambley TW Bioreductive Activation and Drug Chaperoning in Cobalt Pharmaceuticals. Dalton Trans 2007, 3983–3990. [DOI] [PubMed]
- (15).Reisner E; Arion VB; Keppler BK; Pombeiro AJL Electron-Transfer Activated Metal-Based Anticancer Drugs. Inorg. Chim. Acta 2008, 361, 1569–1583. [Google Scholar]
- (16).Jungwirth U; Kowol CR; Keppler BK; Hartinger CG; Berger W; Heffeter P Anticancer Activity of Metal Complexes: Involvement of Redox Processes. Antioxid. Redox Signal 2011, 15, 1085–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Graf N; Lippard SJ Redox Activation of Metal-Based Prodrugs as a Strategy for Drug Delivery. Adv. Drug Deliv. Rev 2012, 64, 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Böttcher A; Takeuchi T; Hardcastle KI; Meade TJ; Gray HB; Cwikel D; Kapon M; Dori Z Spectroscopy and Electrochemistry of Cobalt(III) Schiff Base Complexes. Inorg. Chem 1997, 36, 2498–2504. [Google Scholar]
- (19).Takeuchi T; Böttcher A; Quezada CM; Simon MI; Meade TJ; Gray HB Selective Inhibition of Human α-Thrombin by Cobalt(III) Schiff Base Complexes. J. Am. Chem. Soc 1998, 120, 8555–8556. [Google Scholar]
- (20).Takeuchi T; Böttcher A; Quezada CM; Meade TJ; Gray HB Inhibition of Thermolysin and Human α-Thrombin by Cobalt(III) Schiff Base Complexes. Bioorg. Med. Chem 1999, 7, 815–819. [DOI] [PubMed] [Google Scholar]
- (21).Harney AS; Lee J; Manus LM; Wang P; Ballweg DM; LaBonne C; Meade TJ Targeted Inhibition of Snail Family Zinc Finger Transcription Factors by Oligonucleotide-Co(III) Schiff Base Conjugate. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 13667–13672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chang JY-C; Stevenson RJ; Lu G-L; Brothers PJ; Clark GR; Denny W. a; Ware DC Syntheses of 8-quinolinolatocobalt(III) Complexes Containing Cyclen Based Auxiliary Ligands as Models for Hypoxia-Activated Prodrugs. Dalton Trans 2010, 39, 11535–11550. [DOI] [PubMed] [Google Scholar]
- (23).Kim BJ; Hambley TW; Bryce NS Visualising the Hypoxia Selectivity of Cobalt(III) Prodrugs. Chem. Sci 2011, 2, 2135–2142. [Google Scholar]
- (24).Hurtado RR; Harney AS; Heffern MC; Holbrook RJ; Holmgren RA; Meade TJ Specific Inhibition of the Transcription Factor Ci by a Cobalt(III) Schiff Base–DNA Conjugate. Mol. Pharm 2012, 9, 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yamamoto N; Renfrew AK; Kim BJ; Bryce NS; Hambley TW Dual Targeting of Hypoxic and Acidic Tumor Environments with a Cobalt(III) Chaperone Complex. J. Med. Chem 2012, 55, 11013–11021. [DOI] [PubMed] [Google Scholar]
- (26).Chang JY-C; Lu G-L; Stevenson RJ; Brothers PJ; Clark GR; Botting KJ; Ferry DM; Tercel M; Wilson WR; Denny WA; Ware DC Cross-Bridged Cyclen or Cyclam Co(III) Complexes Containing Cytotoxic Ligands as Hypoxia-Activated Prodrugs. Inorg. Chem 2013, 52, 7688–7698. [DOI] [PubMed] [Google Scholar]
- (27).Heffern MC; Yamamoto N; Holbrook RJ; Eckermann AL; Meade TJ Cobalt Derivatives as Promising Therapeutic Agents. Curr. Opin. Chem. Biol 2013, 17, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Renfrew AK; Bryce NS; Hambley TW Delivery and Release of Curcumin by a Hypoxia-Activated Cobalt Chaperone: A XANES and FLIM Study. Chem. Sci 2013, 4, 3731–3739. [Google Scholar]
- (29).Heffern MC; Reichova V; Coomes JL; Harney AS; Bajema EA; Meade TJ Tuning Cobalt(III) Schiff Base Complexes as Activated Protein Inhibitors. Inorg. Chem 2015, 54, 9066–9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Munteanu CR; Suntharalingam K Advances in Cobalt Complexes as Anticancer Agents. Dalton Trans 2015, 44, 13796–13808. [DOI] [PubMed] [Google Scholar]
- (31).Renfrew AK; Bryce NS; Hambley T Cobalt(III) Chaperone Complexes of Curcumin: Photoreduction, Cellular Accumulation and Light-Selective Toxicity towards Tumour Cells. Chem. Eur. J 2015, 21, 15224–15234. [DOI] [PubMed] [Google Scholar]
- (32).Cressey PB; Eskandari A; Bruno PM; Lu C; Hemann MT; Suntharalingam K The Potent Inhibitory Effect of a Naproxen-Appended Cobalt(III)-Cyclam Complex on Cancer Stem Cells. ChemBioChem 2016, 17, 1713–1718. [DOI] [PubMed] [Google Scholar]
- (33).de Souza ICA; Faro LV; Pinheiro CB; Gonzaga DTG; da Silva F. de C.; Ferreira VF; Miranda F. da S.; Scarpellini M; Lanznaster M Investigation of Cobalt(III)-Triazole Systems as Prototypes for Hypoxia-Activated Drug Delivery. Dalton Trans 2016, 13671–13674. [DOI] [PubMed]
- (34).Garcia CV; Parrilha GL; Rodrigues BL; Barbeira PJS; Clarke RM; Storr T; Beraldo H Cobalt(III) Complexes with 2-Acetylpyridine-Derived Schiff Bases: Studies Investigating Ligand Release upon Reduction. Polyhedron 2017, 124, 86–95. [Google Scholar]
- (35).Todorović TR; Vukašinović J; Portalone G; Suleiman S; Gligorijević N; Bjelogrlić S; Jovanović K; Radulović S; Anđelković K; Cassar A; Filipović NR; Schembri-Wismayer P (Chalcogen)semicarbazones and Their Cobalt Complexes Differentiate HL-60 Myeloid Leukaemia Cells and Are Cytotoxic towards Tumor Cell Lines. Med. Chem. Commun 2017, 8, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Renfrew AK Transition Metal Complexes with Bioactive Ligands: Mechanisms for Selective Ligand Release and Applications for Drug Delivery. Metallomics 2014, 6, 1324–1335. [DOI] [PubMed] [Google Scholar]
- (37).French FA; Freedlander BL Carcinostatic Action of Polycarbonyl Carcinostatic Action of Polycarbonyl Compounds and Their Derivatives. IV. Glyoxal Bis (Thiosemicarbazone) and Derivatives. Cancer Res 1958, 18, 1290–1300. [PubMed] [Google Scholar]
- (38).French FA; Freedlander BL Chemotherapy Studies on Transplanted Mouse Tumors 1. Cancer Res 1960, 20, 505–538. [PubMed] [Google Scholar]
- (39).Gingras BA; Suprunchuk T; Bayley CH The Preparation of Some Thiosemicarbazones and Their Copper Complexes Part III. Can. J. Chem 1962, 40, 1053–1059. [Google Scholar]
- (40).Jansson PJ; Yamagishi T; Arvind A; Seebacher N; Gutierrez E; Stacy A; Maleki S; Sharp D; Sahni S; Richardson DR Di-2-Pyridylketone 4,4-Dimethyl-3-Thiosemicarbazone (Dp44mT) Overcomes Multidrug Resistance by a Novel Mechanism Involving the Hijacking of Lysosomal P-Glycoprotein (Pgp). J. Biol. Chem 2015, 290, 9588–9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vāvere AL; Lewis JS Cu-ATSM: A Radiopharmaceutical for the PET Imaging of Hypoxia. Dalton Trans 2007, 4893–4902. [DOI] [PubMed]
- (42).Obata A; Yoshimi E; Waki A; Lewis JS; Oyama N; Welch MJ; Saji H; Yonekura Y; Fujibayashi Y Retention Mechanism of Hypoxia Selective Nuclear Imaging/radiotherapeutic Agent Cu-Diacetyl-bis(N4-Methylthiosemicarbazone) (Cu-ATSM) in Tumor Cells. Ann. Nucl. Med 2001, 15, 499–504. [DOI] [PubMed] [Google Scholar]
- (43).Maurer RI; Blower PJ; Dilworth JR; Reynolds CA; Zheng Y; Mullen GED Studies on the Mechanism of Hypoxic Selectivity in Copper Bis(thiosemicarbazone) Radiopharmaceuticals. J. Med. Chem 2002, 45, 1420–1431. [DOI] [PubMed] [Google Scholar]
- (44).Xiao Z; Donnelly PS; Zimmermann M; Wedd AG Transfer of Copper between Bis(thiosemicarbazone) Ligands and Intracellular Copper-Binding Proteins. Insights into Mechanisms of Copper Uptake and Hypoxia Selectivity. Inorg. Chem 2008, 47, 4338–4347. [DOI] [PubMed] [Google Scholar]
- (45).Price KA; Crouch PJ; Volitakis I; Paterson BM; Lim S; Donnelly PS; White AR Mechanisms Controlling the Cellular Accumulation of Copper Bis(thiosemicarbazonato) Complexes. Inorg. Chem 2011, 50, 9594–9605. [DOI] [PubMed] [Google Scholar]
- (46).Donnelly PS; Liddell JR; Lim S; Paterson BM; Cater MA; Savva MS; Mot AI; James JL; Trounce IA; White AR; Crouch PJ An Impaired Mitochondrial Electron Transport Chain Increases Retention of the Hypoxia Imaging Agent Diacetylbis(4-Methylthiosemicarbazonato)copperII. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Xie D; King TL; Banerjee A; Kohli V; Que EL Exploiting Copper Redox for 19 F Magnetic Resonance-Based Detection of Cellular Hypoxia. J. Am. Chem. Soc 2016, 138, 2937–2940. [DOI] [PubMed] [Google Scholar]
- (48).Stefani C; Al-Eisawi Z; Jansson PJ; Kalinowski DS; Richardson DR Identification of Differential Anti-Neoplastic Activity of Copper Bis(thiosemicarbazones) That Is Mediated by Intracellular Reactive Oxygen Species Generation and Lysosomal Membrane Permeabilization. J. Inorg. Biochem 2015, 152, 20–37. [DOI] [PubMed] [Google Scholar]
- (49).Paterson BM; Donnelly PS Copper Complexes of Bis(thiosemicarbazones): From Chemotherapeutics to Diagnostic and Therapeutic Radiopharmaceuticals. Chem. Soc. Rev 2011, 40, 3005–3018. [DOI] [PubMed] [Google Scholar]
- (50).Fulmer GR; Miller AJM; Sherden NH; Gottlieb HE; Nudelman A; Stoltz BM; Bercaw JE; Goldberg KI NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar]
- (51).Bramley R; Brorson M; Sargeson AM; Schaeffer CE Cobalt-59 NMR Chemical Shifts of Cobalt(III) Complexes: Correlations with Parameters Calculated from Ligand-Field Spectra. J. Am. Chem. Soc 1985, 107, 2780–2787. [Google Scholar]
- (52).Connelly NG; Geiger WE Chemical Redox Agents for Organometallic Chemistry. Chem. Rev 1996, 96, 877–910. [DOI] [PubMed] [Google Scholar]
- (53).Pavlishchuk VV; Addison AW Conversion Constants for Redox Potentials Measured versus Different Reference Electrodes in Acetonitrile Solutions at 25°C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar]
- (54).Sheldrick GM SHELXT– Integrated Space-Group and Crystal-Structure Determination. Acta Cryst 2015, A71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Sheldrick GM A Short History of SHELX. Acta Cryst 2008, A64, 112–122. [DOI] [PubMed] [Google Scholar]
- (56).Müller P Practical Suggestions for Better Crystal Structures. Crystallogr. Rev 2009, 15, 57–83. [Google Scholar]
- (57).Freshney RI Culture of Animal Cells, 5th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- (58).Xia X; Owen MS; Lee REC; Gaudet S Cell-to-Cell Variability in Cell Death: Can Systems Biology Help Us Make Sense of It All? Cell Death Dis 2014, 5, e1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Kirin SI; Ott I; Gust R; Mier W; Weyhermüller T; Metzler-Nolte N Cellular Uptake Quantification of Metalated Peptide and Peptide Nucleic Acid Bioconjugates by Atomic Absorption Spectroscopy. Angew. Chem. Int. Ed 2008, 47, 955–959. [DOI] [PubMed] [Google Scholar]
- (60).Egger AE; Rappel C; Jakupec MA; Hartinger CG; Heffeter P; Keppler BK Development of an Experimental Protocol for Uptake Studies of Metal Compounds in Adherent Tumor Cells. J. Anal. At. Spectrom 2009, 24, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Manus LM; Holbrook RJ; Atesin TA; Heffern MC; Harney AS; Eckermann AL; Meade TJ Axial Ligand Exchange of N-Heterocyclic Cobalt(III) Schiff Base Complexes: Molecular Structure and NMR Solution Dynamics. Inorg. Chem 2013, 52, 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Yamasaki A Cobalt-59 Nuclear Magentic Resonance Spectroscopy in Coordination Chemistry. J. Coord. Chem 1991, 24, 211–260. [Google Scholar]
- (63).Mason J Patterns of Nuclear Magnetic Shielding of Transition-Metal Nuclei. Chem. Rev 1987, 87, 1299–1312. [Google Scholar]
- (64).Medek A; Frydman V; Frydman L Solid and Liquid Phase 59Co NMR Studies of Cobalamins and Their Derivatives. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 14237–14242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Dearling JL; Lewis JS; McCarthy DW; Welch MJ; Blower PJ Redox-Active Metal Complexes for Imaging Hypoxic Tissues: Structure–activity Relationships in Copper(II) Bis(thiosemicarbazone) Complexes. Chem. Commun 1998, 2531–2532.
- (66).Burgman P; O’Donoghue JA; Lewis JS; Welch MJ; Humm JL; Ling CC Cell Line-Dependent Differences in Uptake and Retention of the Hypoxia-Selective Nuclear Imaging Agent Cu-ATSM. Nucl. Med. Biol 2005, 32, 623–630. [DOI] [PubMed] [Google Scholar]
- (67).Zeman EM; Brown JM; Lemmon MJ; Hirst VK; Lee WW SR-4233: A New Bioreductive Agent with High Selective Toxicity for Hypoxic Mammalian Cells. Int. J. Radiat. Oncol. Biol. Phys 1986, 12, 1239–1242. [DOI] [PubMed] [Google Scholar]
- (68).Reddy SB; Williamson SK Tirapazamine: A Novel Agent Targeting Hypoxic Tumor Cells. Expert Opin. Investig. Drugs 2009, 18, 77–87. [DOI] [PubMed] [Google Scholar]
- (69).Ahmadi M; Ahmadihosseini Z; Allison SJ; Begum S; Rockley K; Sadiq M; Chintamaneni S; Lokwani R; Hughes N; Phillips RM Hypoxia Modulates the Activity of a Series of Clinically Approved Tyrosine Kinase Inhibitors. Br. J. Pharmacol 2014, 171, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Strese S; Fryknäs M; Larsson R; Gullbo J Effects of Hypoxia on Human Cancer Cell Line Chemosensitivity. BMC Cancer 2013, 13, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Djoko KY; Donnelly PS; Mcewan AG Inhibition of Respiratory Complex I by Copper(II)-Bis(thiosemicarbazonato) Complexes. Metallomics 2014, 6, 2250–2259. [DOI] [PubMed] [Google Scholar]
- (72).Lovejoy DB; Jansson PJ; Brunk UT; Wong J; Ponka P; Richardson DR Antitumor Activity of Metal-Chelating Compound Dp44mT Is Mediated by Formation of a Redox-Active Copper Complex That Accumulates in Lysosomes. Cancer Res 2011, 71, 5871–5880. [DOI] [PubMed] [Google Scholar]
- (73).Brewer GJ; Dick RD; Grover DK; LeClaire V; Tseng M; Wicha M; Pienta K; Redman BG; Jahan T; Sondak VK; Strawderman M; LeCarpentier G; Merajver SD Treatment of Metastatic Cancer with Tetrathiomolybdate, an Anticopper, Antiangiogenic Agent: Phase I Study. Clin. Cancer Res 2000, 6, 1–10. [PubMed] [Google Scholar]
- (74).Kalinowski DS; Quach P; Richardson DR Thiosemicarbazones: The New Wave in Cancer Treatment. Future Med. Chem 2009, 1, 1143–1151. [DOI] [PubMed] [Google Scholar]
- (75).Dilworth JR; Hueting R Metal Complexes of Thiosemicarbazones for Imaging and Therapy. Inorg. Chim. Acta 2012, 389, 3–15. [Google Scholar]
- (76).Zhong X; Yi J; Sun J; Wei H-L; Liu W-S; Yu K-B Synthesis and Crystal Structure of Some Transition Metal Complexes with a Novel Bis-Schiff Base Ligand and Their Antitumor Activities. Eur. J. Med. Chem 2006, 41, 1090–1092. [DOI] [PubMed] [Google Scholar]
- (77).El-Tabl AS; El-wahed MMA; Rezk AMSM Cytotoxic Behavior and Spectroscopic Characterization of Metal Complexes of Ethylacetoacetate Bis(thiosemicarbazone) Ligand. Spectrochim. Acta Part A Mol. Biomol. Spectrosc 2014, 117, 772–788. [DOI] [PubMed] [Google Scholar]
- (78).Dearling JL; Lewis JS; Mullen GE; Welch MJ; Blower PJ Copper Bis(thiosemicarbazone) Complexes as Hypoxia Imaging Agents: Structure-Activity Relationships. J. Biol. Inorg. Chem 2002, 7, 249–259. [DOI] [PubMed] [Google Scholar]
- (79).Lever ABP Electrochemical Parametrization of Metal Complex Redox Potentials, Using the Ruthenium(III)/Ruthenium(II) Couple to Generate a Ligand Electrochemical Series. Inorg. Chem 1990, 29, 1271–1285. [Google Scholar]
- (80).Navon G; Panigel R Spectroscopic and Solution Properties of the Cobalt(III) Hexaimidazole Ion. Inorg. Chem 1989, 28, 1405–1407. [Google Scholar]
- (81).Helm L; Merbach AE Inorganic and Bioinorganic Solvent Exchange Mechanisms. Chem. Rev 2005, 105, 1923–1960. [DOI] [PubMed] [Google Scholar]
- (82).Gust R; Ott I; Posselt D; Sommer K Development of Cobalt(3,4-Diarylsalen) Complexes as Tumor Therapeutics. J. Med. Chem 2004, 47, 5837–5846. [DOI] [PubMed] [Google Scholar]
- (83).Palanimuthu D; Vijay Shinde S; Somasundaram K; Samuelson AG In Vitro and in Vivo Anticancer Activity of Copper Bis(thiosemicarbazone) Complexes. J. Med. Chem 2013, 56, 722–734. [DOI] [PubMed] [Google Scholar]
- (84).Akladios FN; Andrew SD; Parkinson CJ Cytotoxic Activity of Expanded Coordination Bis-Thiosemicarbazones and Copper Complexes Thereof. J. Biol. Inorg. Chem 2016, 21, 931–944. [DOI] [PubMed] [Google Scholar]
- (85).Fallahi-Sichani M; Honarnejad S; Heiser LM; Gray JW; Sorger PK Metrics Other than Potency Reveal Systematic Variation in Responses to Cancer Drugs. Nat. Chem. Biol 2013, 9, 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Yamamoto N; Danos S; Bonnitcha PD; Failes TW; New EJ; Hambley TW Cellular Uptake and Distribution of Cobalt Complexes of Fluorescent Ligands. J. Biol. Inorg. Chem 2008, 13, 861–871. [DOI] [PubMed] [Google Scholar]
- (87).Richardson DR; Sharpe PC; Lovejoy DB; Senaratne D; Kalinowski DS; Islam M; Bernhardt PV Dipyridyl Thiosemicarbazone Chelators with Potent and Selective Antitumor Activity Form Iron Complexes with Redox Activity. J. Med. Chem 2006, 49, 6510–6521. [DOI] [PubMed] [Google Scholar]
- (88).Stacy AE; Palanimuthu D; Bernhardt PV; Kalinowski DS; Jansson PJ; Richardson DR Zinc(II)–Thiosemicarbazone Complexes Are Localized to the Lysosomal Compartment Where They Transmetallate with Copper Ions to Induce Cytotoxicity. J. Med. Chem 2016, 59, 4965–4984. [DOI] [PubMed] [Google Scholar]
- (89).Irving H; Williams RJP The Stability of Transition-Metal Complexes. J. Chem. Soc 1953, 3192–3210.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.