

Summary
Acute pancreatitis (AP), an inflammatory disorder of the pancreas with a high hospitalization rate, frequently leads to systemic inflammatory response syndrome (SIRS) and multiple organ dysfunction syndrome (MODS). However, therapeutic targets for effective treatment and early intervention of AP are still urgently required to be identified. Here, we have observed that the expression of pancreatic lincRNA‐EPS, a long intergenic non‐coding RNA, is dynamically changed during both caerulein‐induced AP (Cer‐AP) and sodium taurocholate‐induced severe AP (NaTc‐SAP). The expression pattern of lincRNA‐EPS is negatively correlated with the typical inflammatory genes such as IL‐6, IL‐1β, CXCL1, and CXCL2. Further studies indicate that knockout of lincRNA‐EPS aggravates the pathological symptoms of AP including more induction of serum amylase and lipase, severe edema, inflammatory cells infiltration and acinar necrosis in both experimental AP mouse models. Besides these intrapancreatic effects, lincRNA‐EPS also protects against tissue damages in the extra‐pancreatic organs such as lung, liver, and gut in the NaTc‐SAP mouse model. In addition, we have observed more serum pro‐inflammatory cytokines TNF‐α and IL‐6 in the lincRNA‐EPS‐/‐ NaTc‐SAP mice and more extracellular HMGB1 around injured acinar cells in the pancreas from lincRNA‐EPS‐/‐ NaTc‐SAP mice, compared with their respective controls. Pharmacological inhibition of NF‐B activity by BAY11‐7082 significantly abolishes the suppressive effect of lincRNA‐EPS on TLR4 ligand‐induced inflammatory genes in macrophages. Our study has described a protective role of lincRNA‐EPS in alleviating AP and SAP, outlined a novel pathway that lincRNA‐EPS suppresses HMGB1‐NF‐B‐dependent inflammatory response in pancreatic macrophages and provided a potential therapeutic target for SAP.
Keywords: lncRNA, lincRNA‐EPS, HMGB1, severe acute pancreatitis, NF‐B, inflammation
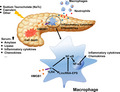
Injured acinar cells release a large amount of HMGB1 during Cer‐AP and NaTc‐SAP. HMGB1 activates the TLR4‐dependent downstream signalling in the pancreatic macrophages to produce pro‐inflammatory cytokines and chemokines, and thus leads to neutrophils and macrophages infiltration, and local and systemic inflammation. lincRNA‐EPS, downregulated at the initial stage of Cer‐AP and NaTc‐SAP, plays an important role in restricting the production of pro‐inflammatory cytokines and chemokines in pancreatic macrophages by targeting the HMGB1‐TLR4‐NF‐κB signalling pathway.
Abbreviations
- H&E
Hematoxylin and Eosin
- Cer‐AP
Caerulein‐induced AP
- CXCL
Chemokine (C‐X‐C motif) ligand
- DAMPs
Damage‐associated molecular patterns
- HMGB1
High‐mobility group box 1
- iBMM
Immortalized bone marrow‐derived macrophage
- IHC
Immunohistochemistry
- IL‐1β
Interleukin‐1β
- IL‐6
Interleukin‐6
- lincRNA
long intergenic non‐coding RNA
- MCP‐1
Monocyte chemo‐attractant protein‐1
- MODS
Multiple organ dysfunction syndrome
- MPO
Myeloperoxidase
- NaTc‐SAP
Sodium taurocholate‐induced severe AP
- PM
Peritoneal macrophage
- SAP
Severe acute pancreatitis
- SIRS
Systemic inflammatory response syndrome
- TLR
Toll‐like receptor
- TNF‐α
Tumour necrosis factor‐α
- ALT
Alanine aminotransferase
- AST
Aspartate aminotransferase
Introduction
Acute pancreatitis (AP) is the leading cause of admission to hospital for gastrointestinal disorders worldwide, with multiple potential aetiologies such as alcohol consumption, bile reflex, pancreatic duct obstruction, autoimmunity and genetic mutations. 1 , 2 AP is an inflammatory disorder of the pancreas, frequently leading to systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS) and even death unless early intervention. 1 As there is no effective treatment, the mortality of the patients with AP‐MODS is over 20%, and the life quality of those who experienced the devastating inflammatory diseases is significantly worse than the general population. 2 , 3
During AP, particularly the severe AP (SAP) progression, initial injury of acinar cells leads to local tissue damage and release of damage‐associated molecular patterns (DAMPs). 4 , 5 High‐mobility group box 1 (HMGB1), a key member of DAMPs family, activates pancreatic macrophages to induce inflammatory genes including TNF‐α, IL‐6, IL‐1β and MCP‐1 by binding to MD‐2 and activating Toll‐like receptor‐4 (TLR4) complex. 6 , 7 These inflammatory mediators further amplify the pancreatic inflammation and tissue damage by recruiting neutrophils and activating more macrophages, leading to pancreas oedema, intrapancreatic haemorrhage, acinar necrosis, and upregulation of serum inflammatory cytokines, chemokines, amylase and lipase. 5 , 8 , 9 Local pancreatic injuries induce SIRS and inflammation in extra‐pancreatic organs including lung, liver and gut, which may develop to AP‐MODS. 10 , 11 , 12 Hence, a novel strategy targeting the inflammatory responses in the pancreatic macrophage at the initial phase is a promising early intervention for AP and SAP.
Long intergenic non‐coding RNAs (lincRNAs) are long non‐coding transcripts (>200 nt) from the intergenic regions of annotated protein‐coding genes. 13 lincRNAs play important roles in controlling the dynamic transcriptional programmes that are a hallmark of immune cell activation. The importance of these molecules is underscored by their newly recognized roles in the regulation of inflammatory responses and related inflammatory diseases. For example, lincRNA‐Cox2 is identified as a dynamically regulated gene induced by TLR ligands and that, in turn, acts to both promote and repress inflammatory genes expression. 14 lincRNA‐Tnfaip3 also appears to mediate both the activation and repression of distinct classes of inflammatory genes in macrophages. 15 lincRNA‐Tnfaip3 physically interacts with HMGB1, assembling a NF‐B/HMGB1/lincRNA‐Tnfaip3 complex in the LPS‐stimulated macrophages. Therefore, induction of lincRNA‐Tnfaip3 is required for the transactivation of NF‐B‐regulated inflammatory genes in response to TLR4 activation. 15 lincRNA‐EPS (also known as Ttc39aos1) is considered as an important transcriptional brake that suppresses inflammatory gene expression in macrophages and protects the mice in the endotoxin‐shock mouse model. 16 Myeloid cells including macrophages and dendritic cells express a high level of lincRNA‐EPS, while it is significantly downregulated in these cells stimulated with LPS. 16 Several additional lncRNAs including THRIL, 17 lnc13, 18 and an antisense lncRNA, AS‐IL‐1α, 19 also regulate inflammatory gene expression in myeloid cells. However, it is unclear whether these lincRNAs (e.g. lincRNA‐EPS) that control inflammatory responses also play crucial roles in ameliorating inflammatory diseases such as SAP.
In this study, we find that the expression level of lincRNA‐EPS is dynamically changed during AP progression in both caerulein‐induced AP (Cer‐AP) and sodium taurocholate‐induced severe AP (NaTc‐SAP) mouse models. By comparing wild‐type (WT) and lincRNA‐EPS‐/‐ mice, our genetic evidence shows that lincRNA‐EPS protects mice against AP in both experimental AP models. Meanwhile, our current study has outlined a novel pathway that lincRNA‐EPS inhibits HMGB1‐triggered NF‐B activation and NF‐B‐dependent inflammatory genes expression in the pancreatic macrophages during AP.
Materials and methods
Mice
The C57BL/6N background lincRNA‐EPS‐/‐ mice were generated by Biocytogen (Beijing, China). Briefly, a pair of sgRNAs were designed to generate a ~ 4kb chromosomal deletion on gene Ttc39aos1 (Gene ID: 102635290). The targeting sequences of the paired sgRNAs were 5’‐CCGCCCGCTTTCCCGCCTTCTGG‐3’ and 5’‐GCATTACTTGGACAGCCCCTTGG‐3’. The cleavage activity of the paired sgRNAs was tested by UCATM (Universal CRISPR Activity Assay) in vitro, which was developed by Biocytogen. In vitro‐transcribed sgRNAs and purified Cas9 protein were co‐microinjected into 170 zygotes (C57BL/6N strain). Seven founder mice from 18 pups were generated and verified by PCR genotyping. Mice genotyping primer sequences were 5’‐GCAGACAGGCGTGGACATTCATTCT‐3’ (lincRNA‐EPS‐F1), 5’‐TCACTGAATACACAGGCTGCTGCAA‐3’ (lincRNA‐EPS‐F2) and 5’‐GCTTGTACTCGCCTCTTCTCTGCAA‐3’ (lincRNA‐EPS‐R). PCR products were 456 bp (WT mice) and 417 bp (KO mice). Other WT C57BL/6N mice used in this study were ordered from Vital River Laboratory Animal Technology (Beijing, China). All the mice were maintained in the specific pathogen‐free (SPF) environment at Suzhou Institute of Systems Medicine (ISM) under a controlled temperature (25°) and a 12 hr day–night cycle. All animal experiments were conducted according to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Service Center of ISM (ISM‐IACUC‐0009‐R).
Reagents
LPS (Escherichia coli 0111: B4) and anti‐α‐tubulin antibody were purchased from Sigma‐Aldrich (St. Louis, MO). Primary antibodies against GAPDH (#5174), p‐AKT (#4060S), AKT (#9272S), p‐ERK1/2 (#9101S), ERK1/2(#4695S), p‐JNK (#4668S), JNK (#9252S), p‐p38 (#9215S), p38 (#9212S), p‐p65 (#3033S) and p65 (#6956S) were from Cell Signaling Technology (Danvers, MA). All the fluorescence secondary antibodies were purchased from LI‐COR (Lincoln, NE). Fluor® 647 anti‐mouse F4/80 (#123122) was purchased from BioLegend (San Diego, CA). Anti‐HMGB1 (#ab79823) antibody was from Abcam (San Francisco, CA). Caerulein was ordered from MCE (Monmouth Junction, NJ). Sodium taurocholate was from Solarbio (Beijing, China). NF‐B inhibitor BAY11‐7082 was purchased from Selleckchem (Houston, TX) and prepared as 50 mmol/L stock solution in DMSO. PD98059, a potent and selective cell‐permeable MEK inhibitor, was ordered from InvivoGen (San Diego, CA). Lipofectamine 2000 was from Invitrogen (Carlsbad, CA). Fluid thioglycollate medium was purchased from Merck KGaA (Darmstadt, Germany).
Cell culture and treatment
HEK293T and RAW264·7 were obtained from American Type Culture Collection (Manassas, VA) and cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. M‐CSF conditioned medium was collected from the supernatant of GMG14‐12 cells (gifted by Genhong Cheng Laboratory, University of California, Los Angeles, CA). For mouse bone marrow‐derived macrophage (BMM) differentiation, bone marrow cells were harvested from WT or lincRNA‐EPS‐/‐ mice and differentiated in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin and 1% M‐CSF conditioned medium for 7 days. The media were replaced on day 3 and day 6, and the cells were used for experiments as BMMs on day 7. For pancreatic macrophages preparation, we sorted the CD11b+F4/80+ cells from pancreases as pancreatic macrophage preparation. These macrophages were cultured on the slides and stained with DAPI and p‐p65 antibody for immunofluorescence detection. For J2 virus‐immortalized macrophages (iBMMs), a cell line transformed by retrovirus expressing v‐raf and c‐myc was established (called GG2EE cells) and grown in RPMI‐1640 (10 mm HEPES, pH 7·8, 10% FBS, 1% penicillin/streptomycin). The supernatant containing J2 viruses was harvested and filtered through 0·22‐µm filter. Bone marrow cells from WT or lincRNA‐EPS‐/‐ mice were infected with the J2 virus and immortalized as described previously. 20 , 21 For peritoneal macrophage (PM) preparation, peritoneal lavage was collected from mice injected with 2 ml of 3% fluid thioglycollate medium for 3 days, followed by centrifugation for 5 min at 1200 rpm, and the peritoneal cells were cultured in RPMI‐1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin. After 1‐hr culture, the medium was replaced and most of the attached cells are PMs. For all experiments, cells were plated overnight and the medium was replaced before stimulation. Cells were stimulated or pretreated at following concentrations unless indicated elsewhere: LPS (100 ng/ml), HMGB1 (500 ng/ml), PD98059 (10 µm) and BAY11‐7082 (10 µm).
Experimental AP mouse models
Before the induction of experimental AP, the mice fasted for 8 hr. The experimental Cer‐AP mice were intraperitoneally administered with caerulein (200 µg/kg or 100 µg/kg) 8 or 10 times hourly. 8 The experimental NaTc‐SAP mice were induced by pancreatic ductal retrograde infusion of sodium taurocholate (NaTc) as previously described. 22 Briefly, after the mice were anaesthetized with pentobarbital, laparotomy was performed and the duodenum was taken out gently from the depths of the wound in the middle of the abdomen. The biliopancreatic duct could be clearly observed by rotating the duodenum. The needle passed through the duodenal wall directly opposite to the papilla. Once the needle was placed within the duct, it was fixed in the desired position with a tied ligature. Retrograde infusion of 5% NaTc was performed at a flow rate of 40 µl/min, 2 µl/g according to mouse weight. After the retrograde infusion, the needle was removed and the opened duodenum was sutured. Mice were killed at 24 hr post the initial induction of the Cer‐AP or NaTc‐SAP, and the serum and tissues, including pancreas, lung and liver, were collected for further analysis. The tissues were placed in 4% paraformaldehyde for histology and immunohistochemistry analysis or stored in RNAlater RNA Stabilization Reagent (Qiagen, Düsseldorf, Germany) for RNA extraction and gene expression analysis. In addition, fresh pancreases were isolated for analysing inflammatory immune cell infiltration via flow cytometry or snap‐frozen for protein extraction for Western blot analysis. Peritoneal lavage fluid from the sham and NaTc‐SAP mice was collected by injection with 5 mL saline. 100 µl of the diluted lavage fluid was spread in the antibiotics‐free LB plates and cultured for 24 hr.
Measurement of serum amylase, lipase, ALT and AST
Blood from experimental AP mouse models was centrifuged at 1500 g for 10 min at 4° to separate the serum. Serum samples were diluted to the appropriate concentration and incubated with corresponding reagents from commercial kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Serum amylase and lipase activities were measured by α‐Amylase Assay Kit (C016‐1) and Lipase Activity Kit (A054‐2) according to the manufacturer’s instructions, respectively. AST and ALT activities were measured by Alanine Aminotransferase Assay Kit (C009‐2‐1) and Aspartate Aminotransferase Assay Kit (C010‐2‐1) according to the manufacturer’s instructions, respectively.
Flow cytometry analysis
To analyse the infiltrated innate immune cells in the pancreases from NaTc‐SAP mice, freshly isolated pancreases were incubated with 1 mg/ml collagenase D (Thermo Fisher Scientific, Waltham, MA) and minced into small pieces on ice. Single pancreatic cell suspensions were firstly stained with Fc blocking antibody, then immune‐labelled with fluorochrome‐conjugated antibodies in PBS supplemented with 2% heat‐inactivated FBS (Gibco, Thermo Fisher Scientific). Isotype controls were also included. Antibodies Alexa Fluor 647‐conjugated anti‐CD11b, PE‐conjugated anti‐F4/80 and PerCP/Cy5·5‐conjugated anti‐Ly6G were purchased from BioLegend (San Diego, CA). Flow cytometry analysis was performed on a Life Launch Attune NxT Flow Cytometer (Thermo Fisher Scientific, Waltham, MA) after gating the living cells. Data were analysed using FlowJo software (version 10·0).
RNA isolation and real‐time quantitative PCR (RT‐qPCR)
RNA was isolated from macrophages or pancreatic tissue stored in RNAlater using TRIzol (Thermo Fisher Scientific) according to the manufacturer’s instructions. Following RNA concentration, 1 µg of RNA was used to make cDNA using PrimeScript TM RT Reagent Kit for RT‐qPCR (Takara, Tokyo, Japan). RT‐qPCR analysis was performed using cDNA in the Roche 480 instrument using SYBR from Toneker Biotech (Suzhou, Jiangsu). The relative mRNA expression level of genes was normalized to the internal control ribosomal protein gene Rpl32 by using 2−∆∆ C t cycle threshold method. 23 The primer sequences for RT‐qPCR were from the primer bank, 24 and all the sequences are available upon request.
RNA sequencing, data acquisition and processing
Total pancreas RNA was extracted from Cer‐AP and saline‐treated mice. RNA concentration was quantified using a Qubit 2·0 Fluorometer (Thermo Fisher Scientific). The integrity of extracted RNA was evaluated using an Agilent Technology 2100 Bioanalyzer. RNA libraries were constructed using a TruSeq Stranded mRNA Sample Prep Kit (Illumina, San Diego, CA) according to the manufacturer’s guidelines. The quantity and quality of the libraries were also assessed by Qubit and Agilent 2100 Bioanalyzer, respectively, and their molar concentration was validated by RT‐qPCR for library pooling (Table S1). Libraries were sequenced on the HiSeq X10 using the paired end 2 × 150 bp, dual‐index format. For RNA‐Seq data analysis, Trimmomatic was used to remove Illumina sequencing adapters within raw reads of every sample, trim low‐quality bases of both read ends (with parameters LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15) and drop one read if its length was < 36 bp. Secondly, the clean reads were mapped to mouse mm 10 reference genome with CLC Genomics Workbench (Qiagen) and the alignment bam files were used as htseq‐count (command of python package HTSeq) input to get read counts of genes. Finally, CLC was used to identify DEGs (P ≤ 0·05, FC ≥ 2) based on raw read counts. For DEGs, Ingenuity Pathway Analysis (IPA) and GO biological process were performed by Fisher’s exact test, the enrichment P‐values of which were corrected by the Bonferroni method. RNA sequencing raw data have been deposited in the Genome Sequence Archive of the National Genomics Data Center (NGDC) under the accession numbers CRA002365 (publicly accessible website: http://bigd.big.ac.cn/gsa).
Histological examination, immunohistochemistry and immunofluorescence
Pancreas, lung and liver tissues were collected 24 hr post the initial induction of AP and were fixed in 4% paraformaldehyde in PBS. Paraffin‐embedded tissues from each tissue were sectioned at 5 µm, and the slice was followed by H&E staining. The degree of pancreatic injury was evaluated by light microscopy in 200X magnification over five separate fields. Histology scores of lung injury were evaluated based on the description in the previous study. 25 The severity of pancreatitis was scored mainly based on the description in the previous study. 26 Histology scores of liver injury were evaluated via counting neutrophils in 200X magnification over seven separate fields. Immunohistochemistry for neutrophil marker MPO (#ab9535; Abcam) was performed on the pancreas of mice. Briefly, 5‐µm‐thick paraffin sections of formalin‐fixed paraffin‐embedded pancreatic tissue were fixed in dimethylbenzene, quenched with 3% H2O2 and blocked with goat serum. After three washes with PBS, the sections were treated with anti‐MPO primary antibody (1:50) overnight. Then, the sections were incubated with horseradish peroxidase‐conjugated secondary antibody for 1 hr. Finally, the colour was developed using DAB as a peroxidase substrate, and the slides were counterstained with haematoxylin for bright‐field microscopy. Immunofluorescence for HMGB1, F4/80 and p‐p65 was performed on snap‐frozen sections of pancreas from mice. Frozen sections were obtained from OCT‐embedded pancreatic tissue, and snap‐frozen tissue was sectioned at 5‐µm thickness. After blocking Fc receptor with 10% goat serum in PBS, frozen sections were incubated overnight with antibodies Alexa Fluor® 647 anti‐mouse F4/80 (1:100), anti‐HMGB1 (1:250) and anti‐p‐p65 (1:100). HMGB1 and p‐p65 colocalization in macrophages are analysed by SP8 LIGHTNING confocal microscope (Leica, Wetzlar, Germany).
Protein extraction and Western blotting
Fresh pancreatic tissues were frozen in liquid nitrogen and ground into powder. The ground tissue powder or cultured cells were lysed in cold RIPA lysis buffer (50 mm Tris–Cl, pH 7·5, 150 mm NaCl, 1 mm EDTA, 1% Triton‐X‐100 and 5% glycerol) containing complete protease inhibitor (Merk Limited, Darmstadt, Germany ) in the presence or absence of phosphatase inhibitor (Roche). Protein concentrations of the extracts were measured with a BCA assay (Thermo Fisher Scientific) and equalized with the lysis buffer. Equal amounts of the extracts were loaded and subjected to SDS‐PAGE, transferred onto PVDF membranes (Millipore Corporation, Billerica, MA, USA) and then blotted with enhanced chemiluminescence (Millipore Corporation, Billerica, MA, USA) or Odyssey Imaging Systems (LI‐COR Biosciences, Lincoln, Nebraska, USA).
Preparation of recombinant mouse HMGB1 (rmHMGB1) protein
Full‐length mouse HMGB1 gene was amplified from the cDNA of RAW264·7 cells. The PCR products were cloned into pcDNA3·1(‐) plasmid to make an mHMGB1‐His fusion protein expression vector, which was confirmed by Sanger DNA sequencing. HEK293T cells were seeded into 10‐cm dishes and transfected with pcDNA3·1(‐)‐mHMGB1‐6xHis vector to express rmHMGB1 protein by using Lipofectamine 2000 according to the manufacturer’s instructions. Extraction and purification of rmHMGB1 were performed by using the HisTrapTM column from GE Healthcare (Uppsala, Sweden), according to the manufacturer’s instructions. Purified rmHMGB1 was verified by Western blot and Coomassie blue staining. The concentration of purified rmHMGB1 was evaluated via BCA assay. ToxinSensor™ Chromogenic LAL Endotoxin Assay Kit (GenScript, Piscataway, NJ) was used to measure the concentration of the endotoxin.
Statistical analysis
The data represent the mean of at least three independent experiments, and error bars represent SEM or SD of the mean. Statistical analysis was performed by unpaired 2‐tailed Student's t‐test using the GraphPad Prism (version 6; GraphPad Software Inc, San Diego, CA, USA). P < 0.05 was considered as a statistically significant difference.
Results
Dynamic alternation of lincRNA‐EPS expression during AP and SAP
To investigate the genes potentially regulating AP progression, we analysed the transcriptome of pancreases from the saline‐treated and Cer‐AP mice by using high‐quality total pancreas RNA (Fig. 1A and Table S1). 162 inflammatory genes were significantly induced in the pancreases from the Cer‐AP mice compared with the saline‐treated mice (Table S2). These inflammatory genes were induced at Cer‐AP‐10h and fell back at Cer‐AP‐22h (Fig. S1A and Table S2). Among them, several well‐known inflammatory genes related to AP progressions such as IL‐6, IL‐1β, CXCL1 and CXCL2 were significantly induced at the early stage and downregulated at the later stage (Fig. S1B, C). Interestingly, more than 100 lncRNAs were shown the completely opposite expression pattern to the above inflammatory genes, which were suppressed at Cer‐AP‐10h and upregulated at Cer‐AP‐22h (Fig. 1B and Table S3). LincRNA‐EPS, which has been reported as a transcriptional brake to restrain inflammation, 16 was one of the representative lncRNAs, which were reversely correlated with the expression of the above inflammatory genes (Fig. 1B).
Figure 1.
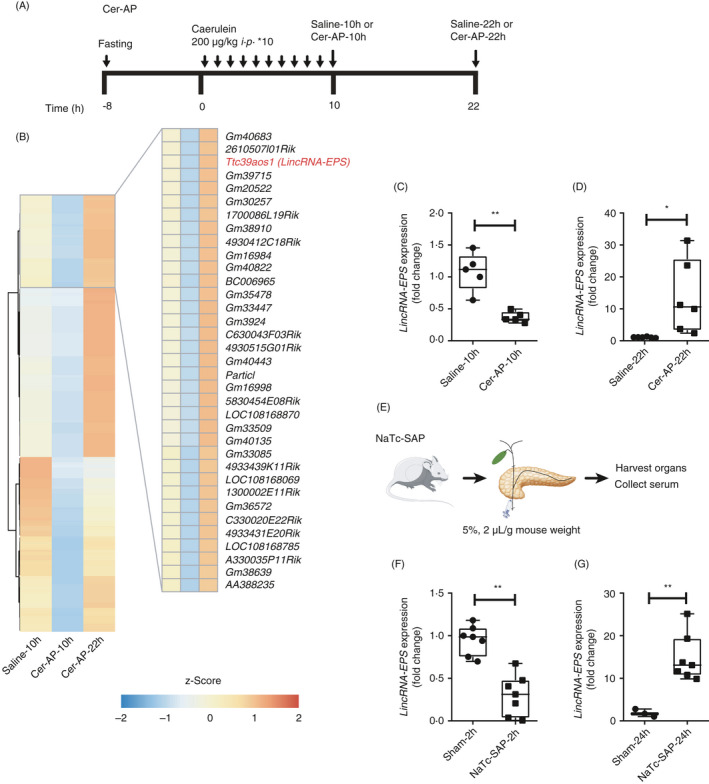
Dynamic alternation of lincRNA‐EPS expression during AP progression. (A) Schematic diagram of the Cer‐AP mouse model. (B) Transcriptomes of pancreases from the saline‐10h, Cer‐AP‐10h and Cer‐AP‐22h mice were analysed by RNA‐Seq. Heatmap of the representative lncRNAs (Cer‐AP‐10h/saline‐10h ≤ 0·5‐fold and Cer‐AP‐22h/Cer‐AP‐10h ≥ 2 fold) that were downregulated at the early stage and recovered expression at the later stage are shown. (C) RT‐qPCR analysis of lincRNA‐EPS transcripts in the pancreases from saline‐10h (n = 5) and Cer‐AP‐10h (n = 5) mice. (D) RT‐qPCR analysis of lincRNA‐EPS transcripts in the pancreases from saline‐22h (n = 6) and Cer‐AP‐22h (n = 6) mice. (E) Schematic diagram of the experimental SAP mouse models by pancreatic ductal retrograde infusion of NaTc. (F) RT‐qPCR analysis of lincRNA‐EPS transcripts in the pancreases from sham‐2h (n = 7) and NaTc‐SAP‐2h (n = 7) mice. (G) RT‐qPCR analysis of lincRNA‐EPS transcripts in the pancreases from sham‐24h (n = 3) and NaTc‐SAP‐24h (n = 7) mice. Data of (B) are shown as the mean of the biological triplicates. Data of (C), (D), (F) And (G) are shown as the mean ± SD of one representative experiment from three independent experiments. *P < 0·05 and **P < 0·01 by unpaired Student's t‐test.
We verified the dynamic expression changes of lincRNA‐EPS in a lower dose and better controlled Cer‐AP mouse model by RT‐qPCR. Comparing with the respective controls, lincRNA‐EPS was significantly downregulated in the pancreases from Cer‐AP‐10h mice, while it was dramatically upregulated in the pancreases from Cer‐AP‐22h mice (Fig. 1C, D and Fig. S1D). In addition, we confirmed the dynamic alternation of lincRNA‐EPS expression in the NaTc‐SAP mouse model, which mimics the clinical biliary (i.e. gallstone) SAP patients (Fig. 1E). Consistent with Cer‐AP, lincRNA‐EPS was significantly downregulated in the pancreases from NaTc‐SAP‐2h mice, while it was dramatically upregulated in the pancreases from NaTc‐SAP‐24h and NaTc‐SAP‐36h mice (Fig. 1F, G and Fig. S1E).
Together, we find that the expression of lincRNA‐EPS, known as an anti‐inflammation lncRNA, dynamically changed its expression during the AP progression and reversely correlated with the typical inflammatory genes in both Cer‐AP and NaTc‐SAP mouse models. These results suggest a potential role of lincRNA‐EPS in alleviating pancreas injury during AP.
More severe symptoms in the Cer‐AP and NaTc‐SAP lincRNA‐EPS‐/‐ mice
To determine whether lincRNA‐EPS plays a protective role during AP, we constructed a lincRNA‐EPS‐/‐ mouse strain using CRISPR/Cas9 genome editing technology and verified the founder mice by PCR genotyping and RT‐qPCR (Fig. S2). The lincRNA‐EPS‐/‐ mice did not show any defects and spontaneous diseases during development and breeding. In the Cer‐AP mouse model, serum amylase and lipase activities were significantly elevated in the Cer‐AP WT and lincRNA‐EPS‐/‐ mice, while higher activities were observed in the lincRNA‐EPS‐/‐ mice than the WT mice (Fig. 2A). More induction of inflammatory cytokine genes including IL‐1β and IL‐6 was also detected in the Cer‐AP lincRNA‐EPS‐/‐ mice than the WT mice (Fig. 2B). Consistently, histological analysis showed more severe oedema and leucocyte infiltration in the Cer‐AP lincRNA‐EPS‐/‐ mice than the WT mice (upper panel of Fig. 2C, D). Immunohistochemical analysis confirmed the increased infiltration of neutrophils marked by myeloperoxidase (MPO) in pancreas tissue sections from the Cer‐AP lincRNA‐EPS‐/‐ mice compared with the WT mice (middle panel of Fig. 2C and E). In addition, more TUNEL+ apoptotic acinar cells were detected in the pancreases from the Cer‐AP lincRNA‐EPS‐/‐ mice than the WT mice (bottom panel of Fig. 2C and F).
Figure 2.
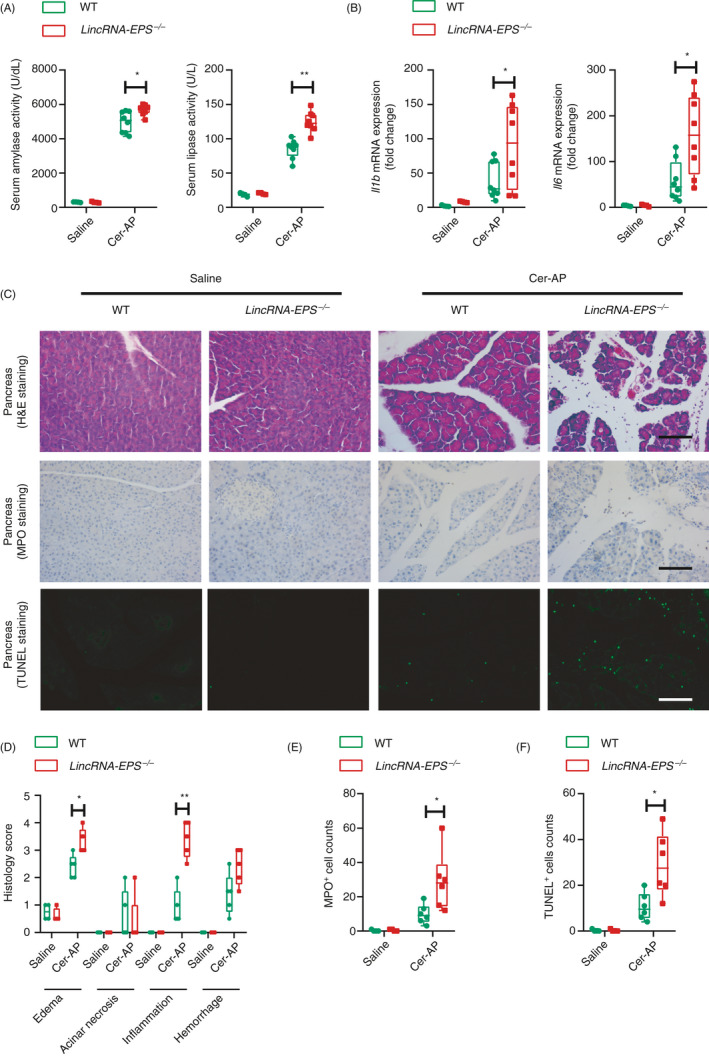
lincRNA‐EPS protects mice against Cer‐AP. (A) 24 hr post the initial injection of saline or caerulein, activities of the serum amylase (left panel) and lipase (right panel) of saline WT (n = 4), saline lincRNA‐EPS‐/‐ (n = 4), Cer‐AP WT (n = 8) and Cer‐AP lincRNA‐EPS‐/‐ (n = 8) mice were measured via enzymatic methods. (B) RT‐qPCR analysis of the Il1b and Il6 mRNA expression in the pancreases from the mice described in (A). (C) Histological examination of the pancreas sections from the indicated mice by H&E staining (upper panel). Neutrophils infiltration in the pancreases from the indicated mice were measured by MPO staining (middle panel). Necrotic cells in the pancreases from the indicated mice were measured by TUNEL staining (bottom panel). Data are shown as representative pancreas sections from the indicated mice; scale bar, 100 µm. Saline WT (n = 4), Saline lincRNA‐EPS‐/‐ (n = 4), Cer‐AP‐WT (n = 5) and Cer‐AP‐lincRNA‐EPS‐/‐ (n = 5). (D–F) Histology scores of pancreatitis (D), MPO+ cells (E) and TUNEL+ cells (F) were calculated based on the representative field of the pancreas sections from the indicated mice, group size was described in (C). Data of (A), (B), (D), (E) And (F) are shown as mean ± SD of one representative experiment from three independent experiments. *P < 0·05 and **p < 0·01 by unpaired Student's t‐test.
Similarly, higher serum amylase and lipase activities were detected in the lincRNA‐EPS‐/‐ mice than the WT mice in the experimental NaTc‐SAP mouse model (Fig. 3A). Much more severe pancreas damage such as inflammatory cell infiltration and tissue necrosis foci could be observed in the NaTc‐SAP lincRNA‐EPS‐/‐ mice than the WT mice (upper panel of Fig. 3B, C). Consistently, more acinar cell apoptosis was detected in the pancreases from the NaTc‐SAP lincRNA‐EPS‐/‐ mice (bottom panel of Fig. 3B and D). Taken together, the results from Cer‐AP and NaTc‐SAP mouse models indicate that knockout of lincRNA‐EPS aggravates the pancreas injury, which suggests the protective role of lincRNA‐EPS during both AP and SAP progression.
Figure 3.
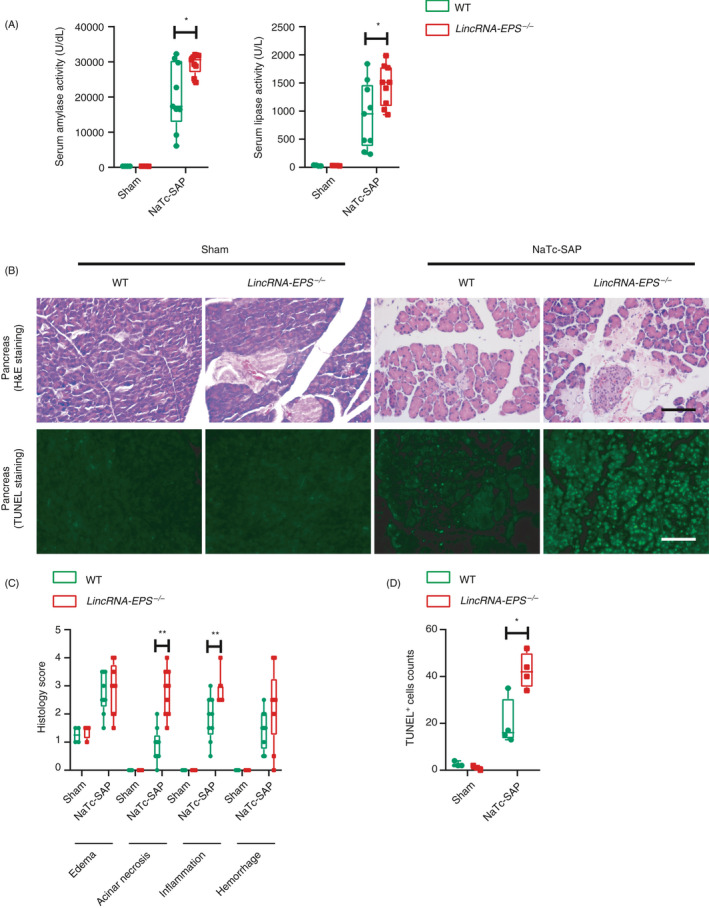
lincRNA‐EPS protects mice against NaTc‐SAP. (A) 24 hr post the retrograde infusion of injection of NaTc, activities of the serum amylase (left panel) and lipase (right panel) of sham WT (n = 4), sham lincRNA‐EPS‐/‐ (n = 4), NaTc‐SAP WT (n = 9) and NaTc‐SAP lincRNA‐EPS‐/‐ (n = 9) mice were measured via enzymatic methods. (B) Histological examination of the pancreas sections from the indicated mice by H&E staining (upper panel). Acinar cell apoptosis in the pancreases from the indicated mice was measured by TUNEL staining (bottom panel). (C) Histology scores of pancreatitis were evaluated based on the representative field of the pancreas sections from the indicated mice, group sizes were described in (A). (D) TUNEL+ cells were calculated based on the representative field of the pancreas sections from the indicated mice, sham WT (n = 3), sham lincRNA‐EPS‐/‐ (n = 3), NaTc‐SAP WT (n = 4) and NaTc‐SAP lincRNA‐EPS‐/‐ (n = 4). Data of (B) are shown as representative pancreas sections from the indicated mice; scale bar, 100 µm. Data of (A), (C) And (D) are shown as the mean ± SD of one representative experiment from three independent experiments. *P < 0·05 and **P < 0·01 by unpaired Student's t‐test.
More inflammatory cytokine production and more infiltration of neutrophils and macrophages in the NaTc‐SAP lincRNA‐EPS‐/‐ mice
We further compared the inflammatory responses between the NaTc‐SAP WT and lincRNA‐EPS‐/‐ mice. Knockout of lincRNA‐EPS significantly promoted the induction of inflammatory cytokine and chemokine genes including IL‐1β, IL‐6 and MCP1 in the injured pancreases from the NaTc‐SAP mice (Fig. 4A). In addition to the differential expression of the inflammatory genes in pancreases, we also found that more serum IL‐6 and TNF‐α proteins were induced in the NaTc‐SAP lincRNA‐EPS‐/‐ mice than the WT mice (Fig. 4B). Consistent with the inflammatory cytokines, a large number of neutrophils were recruited into the injured pancreases during AP progression, while more infiltrated neutrophils with the marker MPO were observed in the injured pancreases from the NaTc‐SAP lincRNA‐EPS‐/‐ mice than the WT mice (Fig. 4C, D). These immunohistochemistry (IHC) results were verified by flow cytometry assay, which indicated that more CD11b+Ly6G+ cells were detected in the pancreases from the NaTc‐SAP lincRNA‐EPS‐/‐ mice than the WT mice (Fig. 4E and Fig. S3A). Meanwhile, more CD11b+F4/80+ macrophages were detected in the pancreases from the NaTc‐SAP lincRNA‐EPS‐/‐ mice than the WT mice (Fig. 4F and Fig. S3B). Together, these results suggest that lincRNA‐EPS potentially protects the mice against SAP and SAP‐SIRS by suppressing intrapancreatic inflammatory responses.
Figure 4.
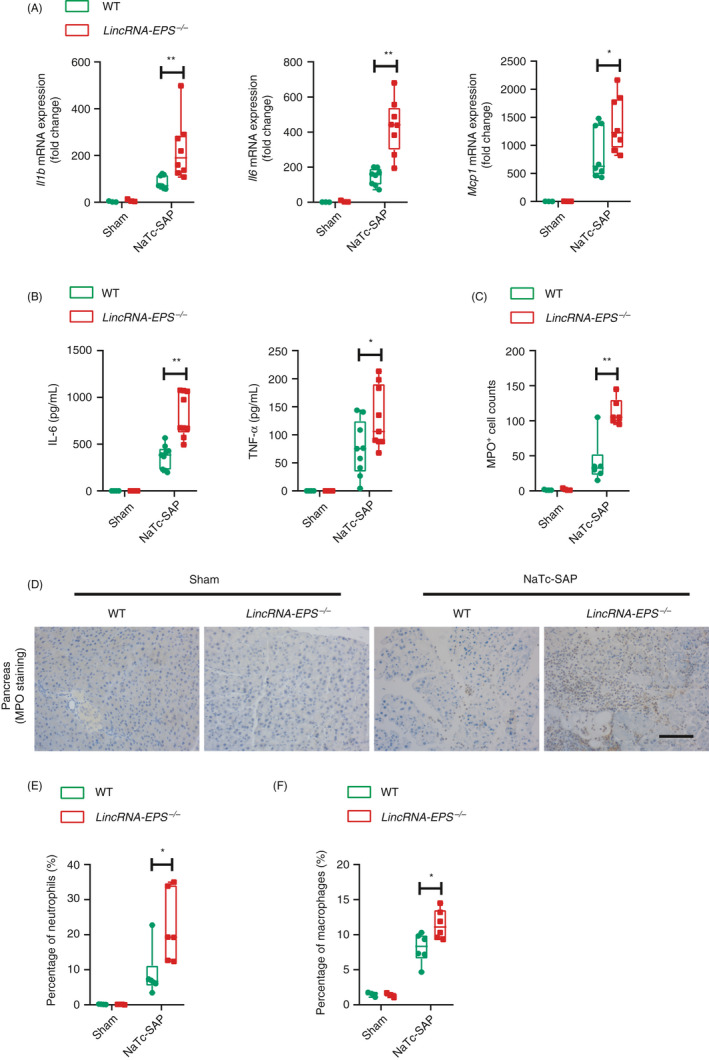
lincRNA‐EPS inhibits pro‐inflammatory cytokine production and neutrophil infiltrations in the NaTc‐SAP mice. (A) 24 hr post the retrograde infusion of injection of NaTc, RT‐qPCR analysis of Il1b, Il6 and Mcp1 mRNA expression was performed in the pancreases from the indicated mice. Sham WT (n = 3), sham lincRNA‐EPS‐/‐ (n = 3), NaTc‐SAP WT (n = 8) and NaTc lincRNA‐EPS‐/‐ (n = 8) mice. (B) ELISA detection of the serum IL‐6 and TNF‐α from indicated mice, group sizes were described in (A). (C, D) Neutrophil infiltrations in the pancreases from the indicated mice were measured by MPO staining (C), and MPO+ cells were calculated based on the representative field of the pancreas sections from the indicated mice (D), sham WT (n = 4), sham lincRNA‐EPS‐/‐ (n = 4), NaTc‐SAP WT (n = 6) and NaTc‐SAP lincRNA‐EPS‐/‐ (n = 6). (E, F) Pancreatic neutrophils (E) and macrophages (F) from indicated mice were analysed by flow cytometry, and the percentage were compared. Group sizes were described in (C). Data of (C) are shown as representative pancreas sections from the indicated mice; scale bar, 100 µm. Data of (A), (B), (D), (E) And (F) are shown as the mean ± SD of one representative experiment from three independent experiments. *P < 0·05 and **P < 0·01 by unpaired Student's t‐test.
More severe SIRS in the NaTc‐SAP lincRNA‐EPS‐/‐ mice
As higher serum IL‐6 and TNF‐α, two main pro‐inflammatory mediators of SIRS, were detected in the NaTc‐SAP lincRNA‐EPS‐/‐ mice than the WT mice, we were wondering whether lincRNA‐EPS also protected other organs such as lung, liver and gut in the NaTc‐SAP mouse model. Lung is one of the extra‐pancreatic organs that is severely affected by SAP in humans. H&E staining results indicated that the thickness of the alveolar wall was substantially increased in the NaTc‐SAP mice, while thicker alveolar walls were observed in the NaTc‐SAP lincRNA‐EPS‐/‐ mice than the WT mice (Fig. 5A, B). Experimental NaTc‐SAP led to severe liver damage, and more hepatocyte death, haemorrhage and neutrophil infiltration were observed in the NaTc‐SAP lincRNA‐EPS‐/‐ mice than the WT mice (Fig. 5C, D). Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST), the markers of hepatocyte injury, were also higher in the NaTc‐SAP lincRNA‐EPS‐/‐ mice, compared with the WT mice. (Fig. 5E, F). Meanwhile, gut permeability was increased significantly in the NaTc‐SAP mice, followed by translocation of the Gram‐negative bacteria across the mucosal barrier to enter the abdominal cavity. 27 Much more bacteria were detected in the abdominal cavity of the NaTc‐SAP lincRNA‐EPS‐/‐ mice than the WT mice (Fig. 5G). Taken together, these data demonstrate that lincRNA‐EPS also has a protective effect on the other organ in addition to pancreases during SAP progression.
Figure 5.
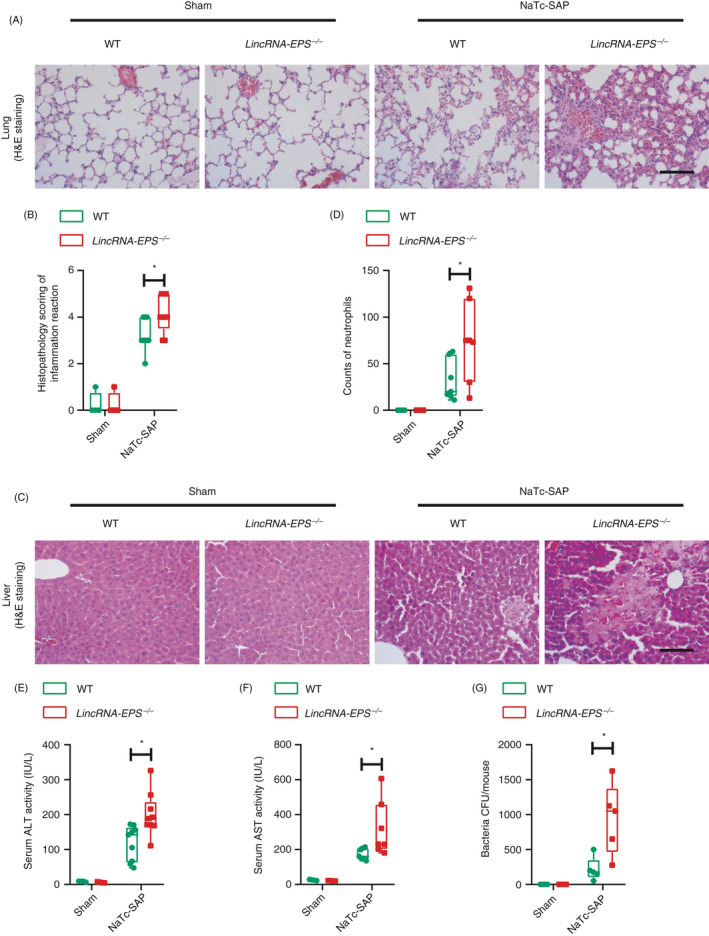
LincRNA‐EPS prevents systemic inflammatory responses in the NaTc‐SAP mice. (A, B) Twenty‐four hours post the retrograde infusion of injection of NaTc, histological examination of the lung sections from the indicated mice was performed by H&E staining (A). Histopathology scores of lung injury were evaluated by measuring the thickness of the alveolar walls in the representative fields of the lung sections from the indicated mice (B). Sham WT (n = 4), sham lincRNA‐EPS‐/‐ (n = 4), NaTc‐SAP‐WT (n = 9) and NaTc‐SAP‐lincRNA‐EPS‐/‐ (n = 9). (C, D) Twenty‐four hours post the retrograde infusion of injection of NaTc, histological examination of the liver sections from the indicated mice was performed by H&E staining (C), infiltrated neutrophils were counted in the representative field of the liver sections from the indicated mice (D). Group size was described in (A). (E, F) Serum ALT (E) and AST (F) activities were measured via enzymatic methods. Group sizes were described in (A). (G) Bacteria in the peritoneal lavage fluid from indicated mice were cultured in the antibiotics‐free LB plates, and the colony‐forming unit (CFU) was counted. Sham WT (n = 4), sham lincRNA‐EPS‐/‐ (n = 4), NaTc‐SAP WT (n = 5) and NaTc‐SAP lincRNA‐EPS‐/‐ (n = 5). Data of (A) and (C) are shown as a representative pancreas sections from the indicated mice; scale bar, 100 µm. Data of (B), (D) and (E–G) are shown as mean ± SD of one representative experiment from three independent experiments. *P < 0·05 and **P < 0·01 by unpaired Student t‐test.
More release of HMGB1 from acinar cells and higher activation of NF‐κB in the pancreatic macrophages from the NaTc‐SAP lincRNA‐EPS‐/‐ mice
To explore the molecular mechanism for the lincRNA‐EPS‐mediated protection on the injured organs during NaTc‐SAP, we focused on the pancreatic macrophages, which played critical roles in the initiation and progression of SAP. Necrotic acinar cells initiate the inflammatory responses by releasing HMGB1, and the extracellular HMGB1 activates pancreatic macrophages by binding with MD2 and triggering TLR4 signalling. 10 , 28 TLR4‐triggered activations of key transcription factor NF‐B drive the induction of inflammatory genes including IL‐1β, TNF‐α and IL‐6 in pancreases. In the pancreases from the NaTc‐SAP mice, acinar cells were damaged and HMGB1 was secreted (Fig. 6A). More extracellular localized HMGB1 was observed in the pancreases from the NaTc‐SAP lincRNA‐EPS‐/‐ mice than the WT mice (Fig. 6A, B). Isolated and cultured acinar cells were damaged upon NaTc treatment in vitro, and a large amount of HMGB1 was released into supernatant (Fig. S3C, D). NF‐B p65 phosphorylation (p‐p65) was clearly increased in the pancreatic F4/80+ cells during NaTc‐SAP (Fig. 6C), which suggests that the pancreatic macrophages were activated by the extracellular HMGB1 from damaged acinar cells. More activation of NF‐B was observed in the pancreatic macrophages from the NaTc‐SAP lincRNA‐EPS‐/‐ mice than the WT mice (Fig. 6C, D). Furthermore, we checked the p‐p65 level in the whole pancreases from the Cer‐AP mice and confirmed that knockout of lincRNA‐EPS promoted NF‐B activation in the pancreases (Fig. 6E, F). Collectively, the above results suggest that lincRNA‐EPS inhibits pancreatic macrophage activation triggered by the HMGB1 from the damaged acinar cells during SAP progression.
Figure 6.
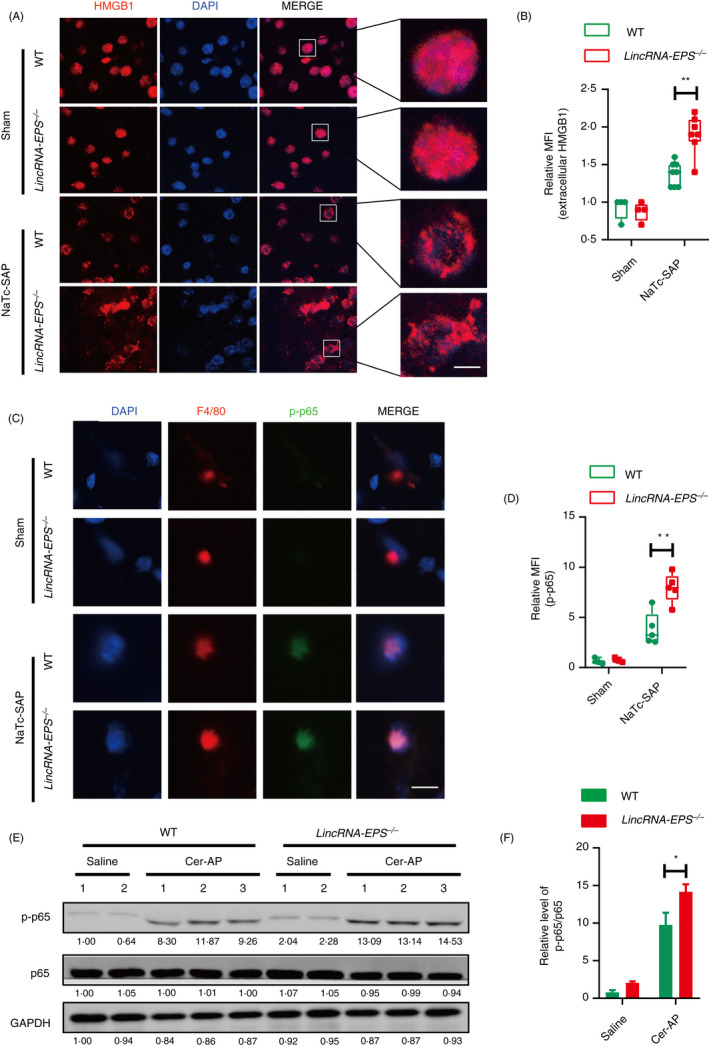
LincRNA‐EPS reduces the release of HMGB1 from acinar cells and the activation of NF‐B in the pancreatic macrophages from NaTc‐SAP mice. (A, B) Immunofluorescence of the subcellular localization of HMGB1 was measured in the snap‐frozen pancreas sections from indicated mice (A), and the mean fluorescent intensity (MFI) of extracellular HMGB1 was calculated in the representative field of the sections (B); sham WT (n = 4), sham lincRNA‐EPS‐/‐ (n = 4), NaTc‐SAP WT (n = 8) and NaTc‐SAP lincRNA‐EPS‐/‐ (n = 8). (C, D) Immunofluorescence of the p‐p65 in the sorted pancreatic macrophages was measured (C), and the MFI of p‐p65 was calculated in the representative field of the sections. Sham WT (n = 4), Sham lincRNA‐EPS‐/‐ (n = 4), NaTc‐SAP WT (n = 5) and NaTc‐SAP lincRNA‐EPS‐/‐ (n = 5). (E, F) Western blot analysis of p‐p65 and total p65 in the pancreas lysate from the indicated mice (E), and relative p‐p65 protein level in (E) were quantified using the ImageJ software (F). GAPDH was shown as a loading control. Data of (A) and (C) are shown as representative pancreas sections from the indicated mice; scale bar, 5 µm (A) and 10 µm (C). Data of (E) are shown as representative of three independent experiments. Data of (B) and (D) are shown as the mean ± SD of one representative experiment from three independent experiments. *P < 0·05 and**P < 0·01 by unpaired Student's t‐test.
Knockout of lincRNA‐EPS facilitates inflammatory cytokine production in response to HMGB1/LPS stimulation on macrophages
To evaluate the impact of lincRNA‐EPS on inflammatory cytokines expression in macrophages, we generated WT and lincRNA‐EPS‐/‐ immortalized bone marrow‐derived macrophages (iBMMs) for further experiments. First, freshly isolated pancreatic acinar cells were stimulated with a high dose of NaTc to induce cell injury in vitro, which mimics the NaTc‐SAP in vivo. Next, these NaTc‐stimulated acinar cells were co‐incubated with iBMMs. Much more induction of IL‐6 mRNA was detected in the lincRNA‐EPS‐/‐ iBMMs than in the WT cells (Fig. 7A). Sonicated 266‐6, a mouse pancreatic acinar cell line, was used to stimulate iBMMs. Similarly, the expression of IL‐1β was higher in the lincRNA‐EPS‐/‐ iBMMs than in the WT cells (Fig. 7B). To test whether HMGB1 mediated the effect of damaged acinar cells as an inflammatory stimulus, we prepared the high purity and endotoxin‐free recombinant mouse HMGB1 (rmHMGB1) (Fig. S4). rmHMGB1 stimulation led to higher induction of IL‐1β and IL‐6 transcription in the lincRNA‐EPS‐/‐ iBMMs than the WT cells (Fig. 7C), as both LPS and HMGB1 trigger inflammatory response through the TLR4‐dependent signalling pathway. Therefore, TLR4 ligand LPS was used to stimulate macrophages as an in vitro model in the following experiments. lincRNA‐EPS was sharply downregulated in response to LPS in all the tested macrophages, including RAW264·7, BMMs, peritoneal macrophages (PMs) and iBMMs (Fig. 7D and Fig. S5A–C). Comparing with their relative controls, more transcripts of IL‐1β and IL‐6 were detected in the LPS‐stimulated lincRNA‐EPS‐/‐ iBMMs, BMMs and PMs (Fig. 7E and Fig. S5D, E). Moreover, the production of IL‐6 and TNF‐α was also significantly higher in the LPS‐stimulated lincRNA‐EPS‐/‐ iBMMs than in the control cells (Fig. 7F). However, overexpression of lincRNA‐EPS suppressed transcription of both IL‐1β and TNF‐α in iBMMs (Fig. 7G). Taken together, lincRNA‐EPS inhibits the TLR4 ligands, HMGB1‐ and LPS‐triggered inflammatory cytokine production in macrophages, and thus potentially alleviates the pancreas injury during SAP progression.
Figure 7.
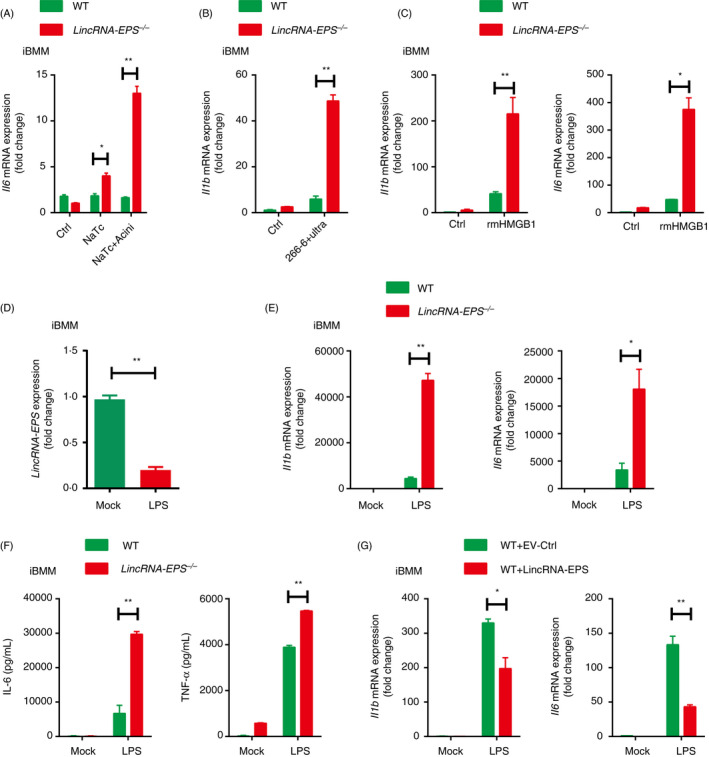
LincRNA‐EPS inhibits inflammatory cytokine production in response to HMGB1 and LPS stimulation in macrophages. (A) RT‐qPCR analysis of Il6 mRNA expression in the WT or lincRNA‐EPS‐/‐ iBMMs stimulated with 200 µM NaTc, or stimulated with the injured mouse acinar cells for 6 hr. The acinar cells were freshly isolated and pretreated with 200 µM NaTc for 2 hr. (B) RT‐qPCR analysis of Il1b mRNA expression in the WT or lincRNA‐EPS‐/‐ iBMMs cocultured with the ultrasonic damaged 266‐6 cells for 6 hr. (C) RT‐qPCR analysis of Il1b and Il6 expression in the WT or lincRNA‐EPS‐/‐ iBMMs stimulated with rmHMGB1 (500 ng/ml) for 6 hr. (D) RT‐qPCR analysis of lincRNA‐EPS transcripts in the WT or lincRNA‐EPS‐/‐ iBMMs stimulated with LPS (100 ng/ml) for 6 hr. (E) RT‐qPCR analysis of Il1b and Il6 mRNA expression in the WT or lincRNA‐EPS‐/‐ iBMMs stimulated with LPS (100 ng/ml) for 6 hr. (F) ELISA measurement of IL‐6 and TNF‐α in the supernatant of WT or lincRNA‐EPS‐/‐ iBMMs stimulated with LPS(100 ng/ml) for 24 hr. (G) RT‐qPCR analysis of Il1b and Il6 mRNA expression in the empty vector control (EV‐Ctrl) or lincRNA‐EPS rescued lincRNA‐EPS‐/‐ iBMMs stimulated with LPS (100 ng/ml) for 6 hr. Data are shown as mean ± SEM from at least three independent experiments. *P < 0·05 and **P < 0·01 by unpaired Student's t‐test.
LincRNA‐EPS suppresses the induction of inflammatory genes in the TLR4‐activated macrophage mainly via the NF‐ B‐dependent pathway
To gain insight into how lincRNA‐EPS suppressed IL‐1β and IL‐6 production in macrophages, we examined their upstream signalling pathways including NF‐B, MAPK and PI3K/AKT pathways. We found that more phosphorylation of p65 and ERK was detected in the LPS‐stimulated lincRNA‐EPS‐/‐ iBMMs than the WT cells (Fig. 8A, B). Overexpression of lincRNA‐EPS in WT iBMMs showed decreased phosphorylation of p65 and ERK after stimulation with LPS (Fig. 8C, D). BAY11‐7082, an inhibitor of NF‐B pathway, was used to test whether lincRNA‐EPS suppressed IL‐6 via the NF‐B pathway in macrophages (Fig. S6A, B). We found more induction of IL‐6 mRNA in the LPS‐stimulated lincRNA‐EPS‐/‐ iBMMs than WT cells, while pharmacological inhibition of NF‐B significantly impaired the upregulation of IL‐6 mRNA in the lincRNA‐EPS‐/‐ iBMMs (Fig. 8E). However, pretreatment with PD98059, an effective ERK inhibitor, did not affect the upregulation of IL‐6 mRNA in the lincRNA‐EPS‐/‐ iBMMs (Fig. 8F and Fig. S6C, D). These results indicated that lincRNA‐EPS suppresses TLR4 ligand‐triggered inflammatory response mainly via the NF‐B signalling pathway.
Figure 8.
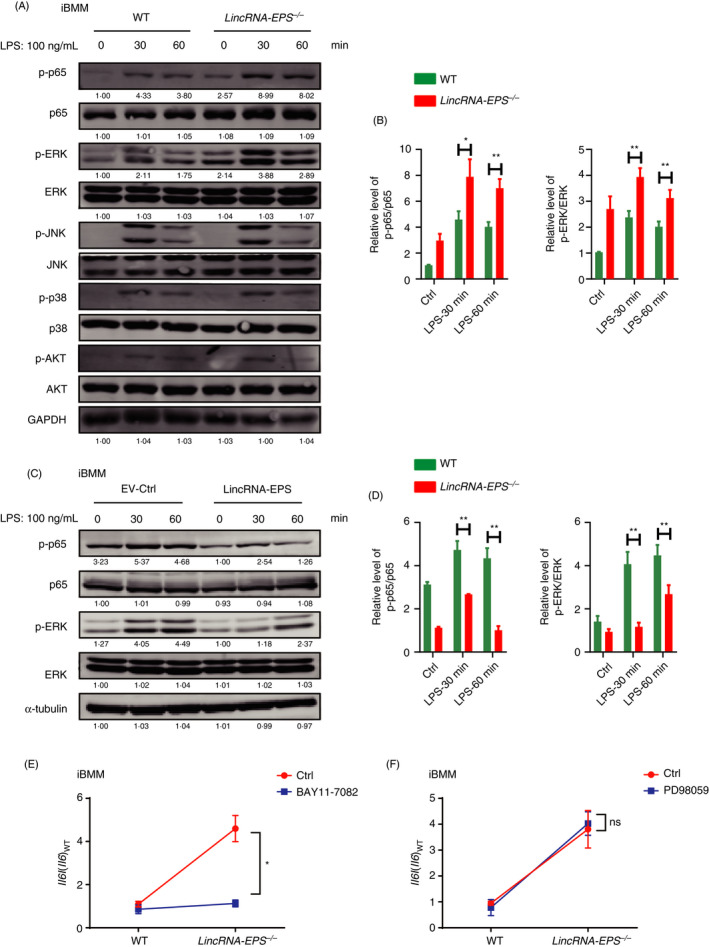
lincRNA‐EPS suppresses TLR4‐triggered inflammatory responses in macrophages via the NF‐B‐dependent signalling. (A, B) Western blot analysis of NF‐B, MAPK and PI3K/AKT signalling activation in the WT and lincRNA‐EPS‐/‐ iBMMs stimulated with LPS (100 ng/ml) for the indicated time (A), and relative p‐p65 and p‐ERK protein levels in (A) were quantified using the ImageJ software (B). GAPDH was shown as a loading control. (C, D) Western blot analysis of p‐p65, p‐ERK and their relative total proteins in the stably transfected with EV‐Ctrl or lincRNA‐EPS iBMMs stimulated with LPS (100 ng/ml) for the indicated time (C), and relative p‐p65 and p‐ERK protein level in (C) were quantified using the ImageJ software (D). α‐Tubulin was shown as a loading control. (E, F) RT‐qPCR analysis of Il6 mRNA expression in the WT and lincRNA‐EPS‐/‐ iBMMs pretreated with 10 µM NF‐B inhibitor BAY11‐7082 (E) or ERK inhibitor PD98059 (F) for 1 hr and then stimulated with LPS (100 ng/ml) for 6 hr. Data of (A) and (C) are shown as representative of three independent experiments. Data of (B) and (D–F) are shown as mean ± SEM from three independent experiments. ns, not significant; *P < 0·05 by unpaired Student's t‐test.
Discussion
lncRNAs are differentially expressed in the cells or organs with healthy and pathological conditions, implying that they may have important biological functions. In this study, we have analysed the expression profile of all transcripts in pancreases during Cer‐AP, and have identified a negative correlation of the expression pattern between lincRNA‐EPS and typical inflammatory genes that are related to AP. lincRNA‐EPS, initially identified as an erythroid‐specific lncRNA with anti‐apoptotic activity, is required for erythroid differentiation. 29 Recent study shows lincRNA‐EPS also restrains inflammation responses and protects the mice in the endotoxin‐shock models. 16 During bacterial infection, lincRNA‐EPS suppresses host protective NO expression and impairs the host defence against the lethal dose of L. monocytogenes infection. 30 In addition to the roles of lincRNA‐EPS in controlling inflammatory and bacterial infectious disease, here we show that lincRNA‐EPS alleviates the local and systemic inflammation in AP and SAP mouse models by suppressing HMGB1‐triggered inflammatory responses. Our current study has not only described a novel function of lincRNA‐EPS in the regulation of HMGB1‐NF‐B‐dependent inflammation but also provided a potential therapeutic target for AP and SAP.
Numerous lncRNAs have been shown to involve in the progression of pancreatic cancer (PC) and pancreatic ductal adenocarcinoma (PDAC). For example, XLOC_000647, which is downregulated in PC tissues, inhibits cancer cell proliferation, invasion and epithelial–mesenchymal transition by downregulating NLRP3· 31 Hypoxia‐induced lncRNA‐MTA2TR significantly promotes PC cell proliferation and invasion via driving the deacetylation‐dependent accumulation of HIF‐1α. 32 LOC389641 is upregulated in PDAC tissues and promotes PDAC progression by suppressing E‐cadherin. 33 However, it is almost unknown whether any lncRNAs regulate the progression of pancreatitis, which is a high‐risk factor for both PC and PDAC. We have shown that the dynamic alteration of lincRNA‐EPS expression is correlated with the AP progression, which suggests a potential role of lincRNA‐EPS in the regulation of chronic pancreatitis (CP), PC and PDAC. Further studies are required to investigate whether lincRNA‐EPS also regulates CP and CP‐associated cancers.
lincRNA‐EPS expression is suppressed in macrophages exposed to TLR ligands including Pam3CSK4 (TLR2/1), LPS (TLR4) and polyI:C (TLR3). 16 Microbes such as L. monocytogenes and Sendai virus also inhibit lincRNA‐EPS expression. 16 In addition, the lincRNA‐EPS expression is downregulated in the monocytes from patients with active pulmonary tuberculosis (PTB), compared with healthy individuals. 34 Blocking of activation of NF‐B impairs the suppression of lincRNA‐EPS in the TLR4‐activated macrophages. 16 In our study, the lincRNA‐EPS expression is downregulated at the early stage of Cer‐AP and NaTc‐SAP compared with the control groups, while it is significantly upregulated at the later stage of both AP models. It is interesting to further investigate the molecular mechanism for modulating lincRNA‐EPS expression, particularly the signalling pathway that induces lincRNA‐EPS transcription at the later stage of AP. Multiple lncRNAs are potentially used as diagnostic biomarkers and therapeutic targets in human diseases. 35 , 36 , 37 Given the dramatic and dynamic changes in lincRNA‐EPS expression during AP progression, it is promising that lincRNA‐EPS serves as a sensitive diagnosis biomarker for AP and SAP.
Pro‐inflammatory cytokines TNF‐α and IL‐6 are the key mediators to induce systemic inflammation and cytokine storm. 38 , 39 , 40 lincRNA‐EPS effectively suppresses the induction of serum amylase and lipase, alleviating pancreas injuries including oedema, haemorrhage, inflammatory cell infiltration and acinar necrosis in both experimental AP mice. Besides these intrapancreatic effects, lincRNA‐EPS also protects the extra‐pancreatic organs such as lung, liver and gut in the NaTc‐SAP mouse model mainly by inhibiting the production of serum pro‐inflammatory cytokines and chemokines. SAP elicited by gallstone, alcohol misuse and hyperlipidaemia always accompanies by higher serum TNF‐α and IL‐6. Therefore, intrapancreatic delivery or systemic administration of lincRNA‐EPS is a potential treatment for most SAP patients with different aetiologies. Severe influenza and coronavirus disease 2019 (COVID‐19) are always with cytokine storm syndrome. 41 , 42 lincRNA‐EPS may also have protective effects on lungs infected by influenza virus and SARS‐CoV‐2 by suppressing cytokine storm.
Our data collectively indicate that lincRNA‐EPS inhibits the activation of NF‐B in the pancreatic macrophage stimulated by HMGB1, which is released from the damaged acinar cells. Inside the cells, HMGB1 binds DNA, regulating transcription and inhibiting inflammatory nucleosome release, and thus limits AP. 43 However, extracellular HMGB1 is the initial mediator to trigger local inflammation. Excessive amounts of extracellular HMGB1 may cause external tissue injury and dysfunction in the pathogenesis of many diseases of both sterile and infectious origin. 7 , 44 , 45 Pancreas‐resident and pancreas‐recruited macrophages play an active role in inflammation progression of AP. 46 , 47 Early intervention by lincRNA‐EPS suppresses the HMGB1‐NF‐B‐dependent inflammation gene expression in intrapancreatic macrophages, and thus effectively limits AP severity.
In conclusion, injured acinar cells release large amounts of DAMPs including HMGB1 during AP or SAP, particularly during biliary pancreatitis. HMGB1 activates the TLR4‐dependent downstream signalling in pancreatic macrophages to produce pro‐inflammatory cytokines and chemokines and thus leads to neutrophil infiltration, and local and systemic inflammation. lincRNA‐EPS is downregulated at the initial stage of Cer‐AP and NaTc‐SAP and sharply upregulated at the later stage, and plays a role in inhibiting the pro‐inflammatory cytokine and chemokine production in pancreatic macrophages by targeting the TLR4‐NF‐B signalling pathway (Fig. S7). lincRNA‐EPS negatively regulates inflammatory responses during AP and SAP progression and thus alleviates the symptoms of these inflammatory diseases. Therefore, lincRNA‐EPS, a lncRNA highly expressed in myeloid immune cells, is a promising therapeutic target for effective treatment and early intervention of AP and SAP, owing to its ability to suppress inflammatory response via modulation of NF‐B pathway in pancreatic macrophages.
Disclosures
The authors have no financial conflict of interest.
Author contributions
F. M. and Q. Z. conceived the idea and designed the experiments. S. C., J. Z., LQ. S., S. L., T. Z., C. H., D. L. and H. Y. performed all the experiments. Y. J., J. H., Y. Q., M. Z. and G. C. provided the reagents and suggestions. F. M. and S. C. analysed the data and wrote the manuscript.
Supporting information
Fig. S1. Dynamic alternation of inflammatory gene expression during Cer‐AP progression.
Fig. S2. Construction and verification of lincRNA‐EPS‐/‐ mice.
Fig. S3. Neutrophils and macrophages infiltration in the pancreases and the source of HMGB1.
Fig. S4. Expression and purification of recombinant mouse HMGB1.
Fig. S5. Downregulation of lincRNA‐EPS facilitates IL‐1β and IL‐6 expression in the TLR4‐triggered macrophages.
Fig. S6. Verification of the effect of the NF‐B and ERK inhibitors.
Fig. S7. Working model for the role of lincRNA‐EPS in the regulation of inflammatory responses during Cer‐AP and NaTc‐SAP. Injured acinar cells release a large amount of HMGB1 during Cer‐AP and NaTc‐SAP.
Table S1. Differentially expressed inflammatory genes in pancreases during AP analyzed by RNA‐Seq.
Table S2. Differentially expressed lncRNAs in pancreases during AP analyzed by RNA‐Seq.
Table S3. The quantity and quality of pancreas RNA used for RNA‐Seq.
Acknowledgements
We appreciate the technical supports from the RNA technology platform of Suzhou Institute of Systems Medicine. This work was supported by the National Key Research and Development Program of China (2018YFA0900803), National Natural Science Foundation of China (81900583, 31800760, 31801825, 31771560, 81471606, 31670883 and 31771560), Fundamental Research Funds for the Central Universities (KJQN201924), CAMS Initiative for Innovative Medicine (2016‐I2 M‐1‐005), Non‐profit Central Research Institute Fund of CAMS (2016ZX310189, 2016ZX310194, 2017NL31004 and 2019PT310028), Innovation Fund for Graduate Students of Peking Union Medical College (2019‐1001‐10) and Natural Science Foundation of Jiangsu Province (BK20200004 and BK20170408).
Shengchuan Chen, Jingfei Zhu and Li‐Qiong Sun contributed equally.
Senior author: Feng Ma, Suzhou Institute of Systems Medicine, Office 810, No 100 Chongwen Road, Suzhou Industrial Park, Suzhou215123, China
Contributor Information
Qiyu Zhang, Email: qiyuz@126.com.
Feng Ma, Email: maf@ism.pumc.edu.cn.
Data availability statement
The data support the finding of this study are available from the corresponding author upon reasonable request.
References
- 1. Lankisch PG, Apte M, Banks PA. Acute pancreatitis. Lancet 2015; 386:85–96. [DOI] [PubMed] [Google Scholar]
- 2. Lerch MM, Gorelick FS. Models of acute and chronic pancreatitis. Gastroenterology 2013; 144:1180–93. [DOI] [PubMed] [Google Scholar]
- 3. Skouras C, Hayes AJ, Williams L, Garden OJ, Parks RW, Mole DJ. Early organ dysfunction affects long‐term survival in acute pancreatitis patients. HPB (Oxford) 2014; 16:789–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Habtezion A. Inflammation in acute and chronic pancreatitis. Curr Opin Gastroenterol 2015; 31:395–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hoque R, Malik AF, Gorelick F, Mehal WZ. Sterile inflammatory response in acute pancreatitis. Pancreas 2012; 41:353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol 2012; 8:195–202. [DOI] [PubMed] [Google Scholar]
- 7. Yang H, Wang H, Andersson U. Targeting inflammation driven by HMGB1. Front Immunol 2020; 11:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang C, Chen S, Zhang T, Li D, Huang Z, Huang J, et al. TLR3 ligand polyi: C prevents acute pancreatitis through the interferon‐beta/interferon‐alpha/beta receptor signaling pathway in a caerulein‐induced pancreatitis mouse model. Front Immunol 2019; 10:980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Awla D, Zetterqvist AV, Abdulla A, Camello C, Berglund LM, Spegel P, et al. NFATc3 regulates trypsinogen activation, neutrophil recruitment, and tissue damage in acute pancreatitis in mice. Gastroenterology 2012; 143:1352–60.e7. [DOI] [PubMed] [Google Scholar]
- 10. Sharif R, Dawra R, Wasiluk K, Phillips P, Dudeja V, Kurt‐Jones E, et al. Impact of toll‐like receptor 4 on the severity of acute pancreatitis and pancreatitis‐associated lung injury in mice. Gut 2009; 58:813–9. [DOI] [PubMed] [Google Scholar]
- 11. Lundberg AH, Eubanks JW 3rd, Henry J, Sabek O, Kotb M, Gaber L, et al. Trypsin stimulates production of cytokines from peritoneal macrophages in vitro and in vivo. Pancreas 2000; 21:41–51. [DOI] [PubMed] [Google Scholar]
- 12. Closa D, Sabater L, Fernandez‐Cruz L, Prats N, Gelpi E, Rosello‐Catafau J. Activation of alveolar macrophages in lung injury associated with experimental acute pancreatitis is mediated by the liver. Ann Surg 1999; 229:230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cabili MN, Trapnell C, Goff L, Koziol M, Tazon‐Vega B, Regev A, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011; 25:1915–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science 2013; 341:789–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ma S, Ming Z, Gong AY, Wang Y, Chen X, Hu G, et al. A long noncoding RNA, lincRNA‐Tnfaip3, acts as a coregulator of NF‐kappaB to modulate inflammatory gene transcription in mouse macrophages. FASEB J 2017; 31:1215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Atianand MK, Hu W, Satpathy AT, Shen Y, Ricci EP, Alvarez‐Dominguez JR, et al. A long noncoding RNA lincRNA‐EPS acts as a transcriptional brake to restrain inflammation. Cell 2016; 165:1672–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Z, Chao TC, Chang KY, Lin N, Patil VS, Shimizu C, et al. The long noncoding RNA THRIL regulates TNFalpha expression through its interaction with hnRNPL. Proc Natl Acad Sci USA 2014; 111:1002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Castellanos‐Rubio A, Fernandez‐Jimenez N, Kratchmarov R, Luo X, Bhagat G, Green PH, et al. A long noncoding RNA associated with susceptibility to celiac disease. Science 2016; 352:91–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan J, Atianand M, Jiang Z, Carpenter S, Aiello D, Elling R, et al. Cutting edge: a natural antisense transcript, AS‐IL‐1alpha, controls inducible transcription of the proinflammatory cytokine IL‐1alpha. J Immunol 2015; 195:1359–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palleroni AV, Varesio L, Wright RB, Brunda MJ. Tumoricidal alveolar macrophage and tumor infiltrating macrophage cell lines. Int J Cancer 1991; 49:296–302. [DOI] [PubMed] [Google Scholar]
- 21. Ma F, Liu SY, Razani B, Arora N, Li B, Kagechika H, et al. Retinoid X receptor alpha attenuates host antiviral response by suppressing type I interferon. Nat Commun 2014; 5:5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perides G, van Acker GJ, Laukkarinen JM, Steer ML. Experimental acute biliary pancreatitis induced by retrograde infusion of bile acids into the mouse pancreatic duct. Nat Protoc 2010; 5:335–41. [DOI] [PubMed] [Google Scholar]
- 23. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc 2008; 3:1101–8. [DOI] [PubMed] [Google Scholar]
- 24. Wang X, Spandidos A, Wang H, Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res 2012; 40:D1144–D1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szarka RJ, Wang N, Gordon L, Nation PN, Smith RH. A murine model of pulmonary damage induced by lipopolysaccharide via intranasal instillation. J Immunol Methods 1997; 202:49–57. [DOI] [PubMed] [Google Scholar]
- 26. Wang Y, Kayoumu A, Lu G, Xu P, Qiu X, Chen L, et al. Experimental models in syrian golden hamster replicate human acute pancreatitis. Sci Rep 2016; 6:28014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Juvonen PO, Alhava EM, Takala JA. Gut permeability in patients with acute pancreatitis. Scand J Gastroenterol 2000; 35:1314–8. [DOI] [PubMed] [Google Scholar]
- 28. Yang H, Wang H, Ju Z, Ragab AA, Lundback P, Long W, et al. MD‐2 is required for disulfide HMGB1‐dependent TLR4 signaling. J Exp Med 2015; 212:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hu W, Yuan B, Flygare J, Lodish HF. Long noncoding RNA‐mediated anti‐apoptotic activity in murine erythroid terminal differentiation. Genes Dev 2011; 25:2573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Agliano F, Fitzgerald KA, Vella AT, Rathinam VA, Medvedev AE. Long non‐coding RNA LincRNA‐EPS inhibits host defense against listeria monocytogenes infection. Front Cell Infect Microbiol 2019; 9:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hu H, Wang Y, Ding X, He Y, Lu Z, Wu P, et al. Long non‐coding RNA XLOC_000647 suppresses progression of pancreatic cancer and decreases epithelial‐mesenchymal transition‐induced cell invasion by down‐regulating NLRP3. Mol Cancer 2018; 17:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zeng Z, Xu FY, Zheng H, Cheng P, Chen QY, Ye Z, et al. LncRNA‐MTA2TR functions as a promoter in pancreatic cancer via driving deacetylation‐dependent accumulation of HIF‐1alpha. Theranostics 2019; 9:5298–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zheng S, Chen H, Wang Y, Gao W, Fu Z, Zhou Q, et al. Long non‐coding RNA LOC389641 promotes progression of pancreatic ductal adenocarcinoma and increases cell invasion by regulating E‐cadherin in a TNFRSF10A‐related manner. Cancer Lett 2016; 371:354–65. [DOI] [PubMed] [Google Scholar]
- 34. Ke Z, Lu J, Zhu J, Yang Z, Jin Z, Yuan L. Down‐regulation of lincRNA‐EPS regulates apoptosis and autophagy in BCG‐infected RAW264.7 macrophages via JNK/MAPK signaling pathway. Infect Genet Evol 2020; 77:104077. [DOI] [PubMed] [Google Scholar]
- 35. Sanchez Y, Huarte M. Long non‐coding RNAs: challenges for diagnosis and therapies. Nucleic Acid Ther 2013; 23:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dong P, Xiong Y, Yue J, Hanley SJB, Kobayashi N, Todo Y, et al. Long non‐coding RNA NEAT1: a novel target for diagnosis and therapy in human tumors. Front Genet 2018; 9:471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pandya G, Kirtonia A, Sethi G, Pandey AK, Garg M. The implication of long non‐coding RNAs in the diagnosis, pathogenesis and drug resistance of pancreatic ductal adenocarcinoma and their possible therapeutic potential. Biochim Biophys Acta Rev Cancer 2020; 1874:188423. [DOI] [PubMed] [Google Scholar]
- 38. Makhija R, Kingsnorth AN. Cytokine storm in acute pancreatitis. J Hepatobiliary Pancreat Surg 2002; 9:401–10. [DOI] [PubMed] [Google Scholar]
- 39. Tanaka T, Narazaki M, Kishimoto T. IL‐6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 2014; 6:a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev 2012; 76:16–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet 2020; 395:1033–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu Q, Zhou YH, Yang ZQ. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol 2016; 13:3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kang R, Zhang Q, Hou W, Yan Z, Chen R, Bonaroti J, et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 2014; 146:1097–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 2011; 29:139–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Andersson U, Yang H, Harris H. Extracellular HMGB1 as a therapeutic target in inflammatory diseases. Expert Opin Ther Targets 2018; 22:263–77. [DOI] [PubMed] [Google Scholar]
- 46. Habtezion A, Gukovskaya AS, Pandol SJ. Acute pancreatitis: a multifaceted set of organelle and cellular interactions. Gastroenterology 2019; 156:1941–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fan HN, Chen W, Fan LN, Wu JT, Zhu JS, Zhang J. Macrophages‐derived p38alpha promotes the experimental severe acute pancreatitis by regulating inflammation and autophagy. Int Immunopharmacol 2019; 77:105940. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Dynamic alternation of inflammatory gene expression during Cer‐AP progression.
Fig. S2. Construction and verification of lincRNA‐EPS‐/‐ mice.
Fig. S3. Neutrophils and macrophages infiltration in the pancreases and the source of HMGB1.
Fig. S4. Expression and purification of recombinant mouse HMGB1.
Fig. S5. Downregulation of lincRNA‐EPS facilitates IL‐1β and IL‐6 expression in the TLR4‐triggered macrophages.
Fig. S6. Verification of the effect of the NF‐B and ERK inhibitors.
Fig. S7. Working model for the role of lincRNA‐EPS in the regulation of inflammatory responses during Cer‐AP and NaTc‐SAP. Injured acinar cells release a large amount of HMGB1 during Cer‐AP and NaTc‐SAP.
Table S1. Differentially expressed inflammatory genes in pancreases during AP analyzed by RNA‐Seq.
Table S2. Differentially expressed lncRNAs in pancreases during AP analyzed by RNA‐Seq.
Table S3. The quantity and quality of pancreas RNA used for RNA‐Seq.
Data Availability Statement
The data support the finding of this study are available from the corresponding author upon reasonable request.