

Abstract
BACKGROUND AND AIMS:
The proinflammatory cytokine IL-1β has been implicated in the pathophysiology of nonalcoholic and alcoholic steatohepatitis. How IL-1β promotes liver injury in these diseases is unclear, as no IL-1β receptor-linked death pathway has been identified. Autophagy functions in hepatocyte resistance to injury and death, and findings of decreased hepatic autophagy in many liver diseases suggest a role for impaired autophagy in disease pathogenesis. Recent findings that autophagy blocks mouse liver injury from lipopolysaccharide led to an examination of autophagy’s function in hepatotoxicity from proinflammatory cytokines.
APPROACH AND RESULTS:
AML12 cells with decreased autophagy from a lentiviral autophagy-related 5 (Atg5) knockdown were resistant to toxicity from TNF, but sensitized to death from IL-1β, which was markedly amplified by TNF co-treatment. IL-1β/TNF death was necrosis by trypan blue and propidium iodide positivity, absence of mitochondrial death pathway and caspase activation, and failure of a caspase inhibitor or necrostatin-1s to prevent death. IL-1β/TNF depleted autophagy-deficient cells of ATP, and ATP depletion and cell death were prevented by supplementation with the energy substrate pyruvate or oleate. Pharmacological inhibitors and genetic knockdown studies demonstrated that IL-1β/TNF-induced necrosis resulted from lysosomal permeabilization and release of cathepsins B and L in autophagy-deficient cells. Mice with a tamoxifen-inducible, hepatocyte-specific Atg5 knockout were similarly sensitized to cathepsin-dependent hepatocellular injury and death from IL-1β/TNF in combination, but neither IL-1β nor TNF alone. Knockout mice had increased hepatic inflammation, and IL-1β/TNF-treated, autophagy-deficient AML12 cells secreted exosomes with proinflammatory damage–associated molecular patterns.
CONCLUSIONS:
The findings delineate mechanisms by which decreased hepatocyte autophagy promotes IL-1β/TNF-induced necrosis from impaired energy homeostasis and lysosomal permeabilization and inflammation through the secretion of exosomal damage–associated molecular patterns.
Liver injury triggers the death of hepatocytes, which leads to the development of liver failure, hepatic fibrosis, and hepatocellular carcinoma. Hepatocyte death occurs by necrosis, apoptosis, necroptosis or pyroptosis, but in many liver diseases—including nonalcoholic and alcoholic steatohepatitis—multiple forms of death coexist.(1) The mechanisms underlying hepatocyte injury and death remain unclear, and there are currently no established therapies that target these pathways. A better understanding of the regulatory pathways of hepatocyte death has the potential to lead to new treatments for liver disease.
Integral to hepatocyte death in most liver diseases is an overactive inflammatory response.(2) Self-limited inflammation after liver injury mediates tissue repair, but excessive inflammation promotes further hepatocyte injury and death. An innate immune response is triggered by pattern recognition receptor stimulation from gut-derived pathogen-associated molecular patterns, such as lipopolysaccharide (LPS),(3) which promote liver injury.(4) LPS-activated hepatic macrophages release proinflammatory cytokines, prominent among which are TNF and IL-1β. Cytokine production is further amplified by the release from injured hepatocytes of damage-associated molecular patterns (DAMPs).(5) TNF can induce hepatocellular apoptosis through a well-defined TNF receptor 1 (TNFR1) death pathway.(6) Recently IL-1β cytotoxicity has also been implicated in liver injury in steatohepatitis.(7–10) The IL-1 receptor (IL-1R) is not linked to a death signaling pathway, however, making it unclear how IL-1β promotes steatohepatitis. Delineating the mechanisms of IL-1β’s toxic and inflammatory effects is essential to understanding not only the molecular basis of hepatic injury, but also the mechanisms of many other human diseases that have been linked to this cytokine.(11)
Macroautophagy (hereafter referred to as autophagy) performs multiple functions with the potential of regulating hepatocyte death.(12,13) We and others have demonstrated that factors that sensitize the liver to injury, including toxins, steatosis and aging, decrease hepatocyte and macrophage autophagy.(14–18) Decreased autophagy may therefore have an important role in liver injury, but the mechanisms by which impaired autophagy promotes liver disease remain unclear.(12) Studies have demonstrated that hepatocyte autophagy mediates resistance to toxin-induced injury from alcohol, galactosamine (GalN), and acetaminophen.(14,19,20) Decreased hepatocyte autophagy amplifies GalN/LPS-induced apoptosis through the mechanism of increased caspase-8 cleavage, demonstrating that autophagy regulates TNF-mediated apoptosis.(14) Subsequent studies have shown that autophagy is hepatoprotective against LPS in the absence of hepatotoxin co-treatment,(21) suggesting that autophagy protects hepatocytes from cytokine-induced injury by other mechanisms.
The present study examined the effect of decreased autophagy on hepatocyte death from TNF and IL-1β. Decreased autophagy sensitized AML12 hepatocytes to TNF-dependent IL-1β toxicity. Death occurred through cathepsin-dependent necrosis triggered by impaired energy homeostasis. Mice with decreased hepatocyte autophagy were similarly sensitized to IL-1β/TNF-induced liver injury and inflammation. Decreased autophagy led to AML12 cell proinflammatory exosomal DAMP secretion in response to IL-1β/TNF, indicating that autophagy regulates hepatocyte inflammation. These findings identify a critical role for autophagy in blocking IL-1β-induced hepatocyte death and inflammation, as well as a mechanism of synergistic IL-1β/TNF hepatotoxicity.
Materials and Methods
AML12 CELL CULTURE
The nontransformed mouse cell line AML12 (American Type Culture Collection, Manassas, VA)(22) was cultured in Dulbecco’s modified Eagle’s medium/F12 medium with glutamine and Hepes (Corning, Corning, NY) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin (Gemini Bio Products, West Sacramento, CA), 1% insulin-transferrin-selenium (Thermo Fisher Scientific, Waltham, MA), and 1 μM dexamethasone (MilliporeSigma, St. Louis, MO) in a 5% CO2, 37°C incubator. Cells were treated with mouse-recombinant IL-1β (5 ng/mL) and/or TNF (10 ng/mL) (R&D Systems, Minneapolis, MN). Some cells were pretreated for 1 hour with 10 μM QVD-Oph, 50 μM necrostatin-1s (Abcam, Cambridge, MA), 10 mM pyruvate, or 250 μM oleate conjugated to bovine serum albumin (MilliporeSigma), as previously described.(15) To induce apoptosis, cells were pretreated with 0.25 ug/mL actinomycin D (MilliporeSigma) 1 hour before 10 ng/mL of TNF. To determine autophagic flux, cells were treated with 20 mM ammonium chloride/100 μM leupeptin or 0.2 μM bafilomycin (MilliporeSigma). Cathepsins were inhibited by 1-hour pretreatment with 10 μM CA-074Me ([L-3-trans-(propylcarbamoyl)oxirane-2-carbonyl]-L-isoleucyl-L-proline methyl ester), Cathepsin Inhibitor I (Z-Phe-Gly-NHO-Bz), Cathepsin L Inhibitor I (Z-Phe-Phe-CH2F), Cathepsin L Inhibitor III (Z-Phe-Tyr(t-Bu)-diazomethylketone), or Cathepsin L Inhibitor IV (1-Naphthalenesulfonyl-Ile-Trp-CHO) (MilliporeSigma).
LENTIVIRAL SMALL HAIRPIN RNA CONSTRUCTION AND INFECTION
To generate cells with decreased autophagy, a short hairpin RNA (shRNA) for the autophagy gene autophagy-related 5 (Atg5) 5′-GATCCCCGTCAG GTGATCAACGAAATTTCAAGAGAATTTC GTTGATCACCTGACTTTTTC-3′ was cloned into the BglII-XhoI sites of pSUPER (Oligoengine, Seattle, WA). After SmaI-XhoI digestion, the H1 promoter-shRNA cassette was subcloned into the EcoRV-XhoI sites of the vector pCCL.sin.PPT.hPGK.GFPWpre.(23) Lentiviral stocks were prepared by calcium phosphate transfection of the vector and the packaging vectors pMDLg/pRRE, pRSV-Rev, and pMD2.VSVG into HEK-293T cells. Stably infected, polyclonal knockdown AML12 cells (siAtg5 cells) and control cells with empty vector (Vec cells) were generated.
siAtg5 cells with a cathepsin B (Ctsb) knockdown were generated with the shRNA 5′-CCGGCGAACC TGCATTCACACCAATCTCGAGATTGGTGT GAATGCAGGTTCGTTTTTGA-3′. Similarly, a cathepsin L (Ctsl) knockdown was generated with the shRNA 5′-CCGGGCTTTCCAGTACAT TAAGGAACTCGAGTTCC TTAATGTACTGG AAAGCTTTTTG-3′. Polyclonal, doubly infected lines were selected in 10 μg/mL and maintained in 3 μg/mL of puromycin (MilliporeSigma). Control doubly infected siAtg5 cells were created by a second infection with the empty vector lentivirus (siAtg5-Vec cells).
ANIMALS
Mice were maintained under 12-hour light/dark cycles with automatic water feeding and unlimited access to food (PicoLab Rodent Diet 20 #5053; LabDiet, St. Louis, MO). Mice were housed in static cages with 1/4-inch corncob bedding and cotton nestlets. Results in 10–14-week old male and female mice were equivalent and combined for all experiments. Hepatocyte-specific autophagy knockout mice were generated by crossing Atg5F/F(24) and ERt-albumin-Cre mice with a tamoxifen-inducible, albumin promoter–driven Cre recombinase(25) to generate Atg5Δhep mice. Cre expression was induced by a single subcutaneous injection of 3 mg of tamoxifen (MilliporeSigma) 4 days before the study. Tamoxifen-injected littermate Atg5F/F mice lacking the Cre transgene served as controls. Mice were injected intravenously with 0.25 μg of TNF and/or 2.5 μg of IL-1β (PeproTech, Rocky Hill, NJ). To inhibit cathepsin activity, mice were injected intraperitoneally with Cathepsin L Inhibitor IV (20 μg/gm dissolved in DMSO) intraperitoneally at 12 hours and 1 hour before IL-1β/TNF administration. C57BL/6J male mice (Jackson Laboratory, Bar Harbor, ME) were injected with GalN/LPS as described previously.(14) Animal studies were approved by the Animal Care and Use Committee of the Emory University School of Medicine and followed the National Institutes of Health guidelines for animal care.
STATISTICAL ANALYSIS
Numerical results are reported as means ± SEM from three or more independent experiments. Statistical significance between control and treated groups was determined by one-way analysis of variance. Statistical significance was defined as P < 0.05.
ADDITIONAL METHODS
Additional detailed methods can be found in the Supporting Information.
Results
DECREASED AUTOPHAGY SENSITIZES AML12 CELLS TO DEATH FROM IL-1β/TNF
In light of findings that decreased hepatocyte autophagy sensitizes to LPS-induced liver injury,(21) toxicity of the LPS-inducible cytokines TNF and IL-1β was examined in cultured hepatocytes. Stably infected, polyclonal lines of the nontransformed murine hepatocyte cell line AML12 were established with a lentivirus containing empty vector as a control (Vec cells) or a shRNA knockdown of the autophagy gene Atg5 (siAtg5 cells). ATG5 but not ATG7 protein content is decreased in siAtg5 cells (Fig. 1A). Consistent with decreased autophagy, siAtg5 cells have increased microtubule-associated protein 1 light chain 3 (LC3)-I and decreased LC3-II protein levels, and reduced autophagic flux as indicated by the failure of lysosomal inhibition with ammonium chloride/leupeptin to increase LC3-II levels (Fig. 1A).
FIG. 1.
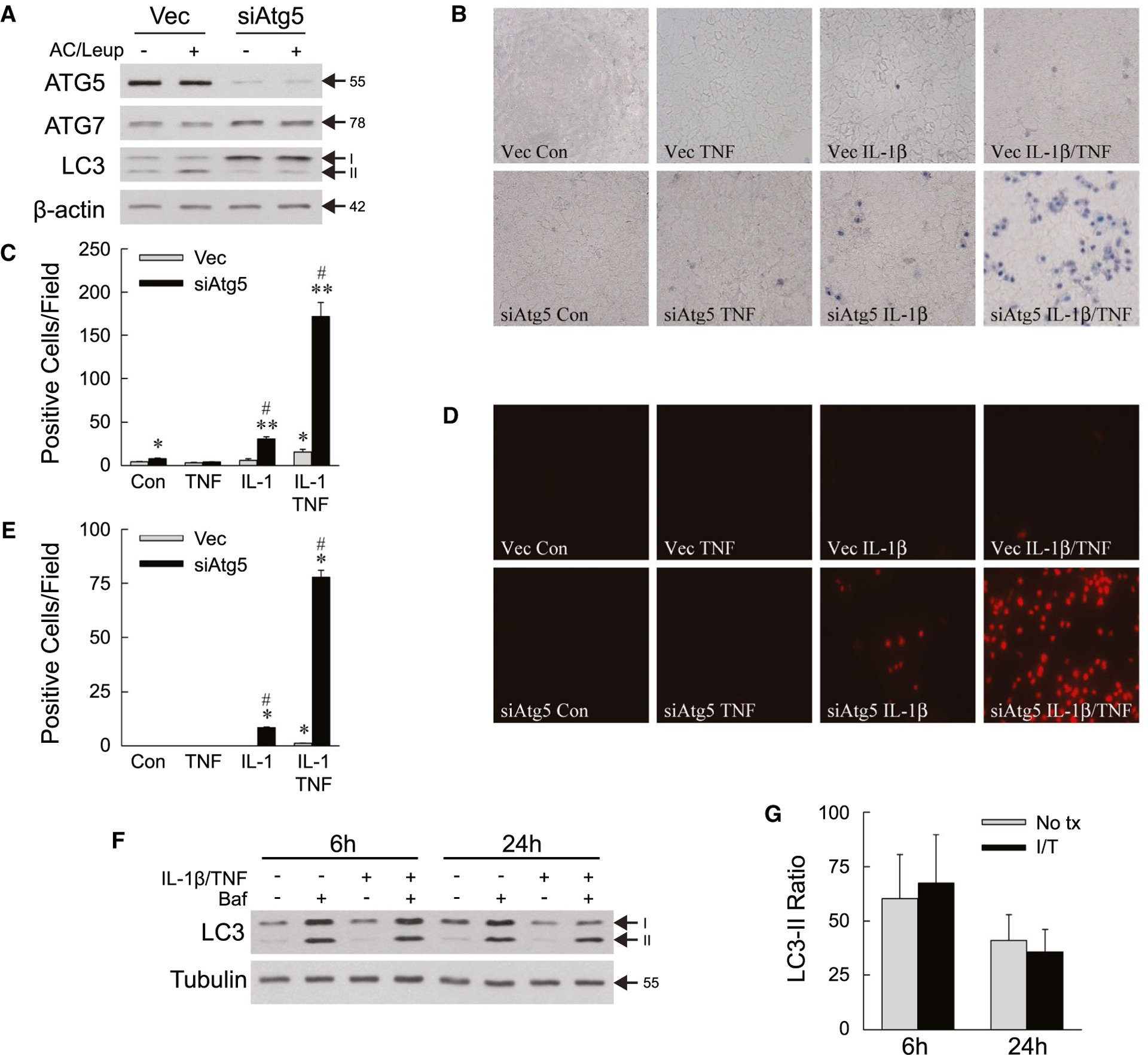
Decreased autophagy sensitizes AML12 cells to death from IL-1β/TNF. (A) Immunoblots of total protein from Vec and siAtg5 cells untreated or treated with ammonium chloride/leupeptin. ATG5 band is the ATG5-ATG12 conjugate. (B) Representative images of trypan blue–stained cells at 24 hours (×200). (C) Numbers of trypan blue–positive cells untreated (control) or treated with TNF (10 ng/mL) and/or IL-1β (5 ng/mL) for 24 hours (*P < 0.01 and **P < 0.0001 compared with untreated Vec cells; #P < 0.0001 compared with Vec cells with the same treatment; n = 4–7). (D) Representative images of propidium iodide–stained cells with the same treatments (×200). (E) Numbers of propidium iodide–positive cells untreated or treated with TNF and/or IL-1β for 24 hours (*P < 0.000001 compared with untreated Vec cells; #P < 0.000001 compared with Vec cells with the same treatment; n = 8). (F) Immunoblots of wild-type AML12 cells untreated or treated with IL-1β/TNF for 6 hours or 24 hours with some cells receiving bafilomycin. (G) Ratios of LC3-II with bafilomycin treatment to LC3-II without bafilomycin in AML12 cells untreated or treated with IL-1β/TNF for 6 hours or 24 hours obtained from densitometric scanning of immunoblots from three independent experiments. Arrows in (A) and (F) indicate molecular weights (in kD) and the LC3-I and LC3-II forms. Abbreviations: AC/Leup, ammonium chloride/leupeptin; Baf, bafilomycin; Con, control; I/T, IL-1β/TNF; No tx, untreated.
By trypan blue staining at 24 hours, untreated Vec and siAtg5 cells had a very low level of cell death that was slightly higher in siAtg5 cells (Fig. 1B,C). Neither cell type underwent cell death from TNF alone (Fig. 1B,C). Vec cells were resistant to death from IL-1β alone and had a small increase in death from IL-1β/TNF combined (Fig. 1B,C). In contrast, siAtg5 cells underwent a modest but significant increase in death from IL-1β alone, which was markedly amplified by TNF co-treatment (Fig. 1B,C). Propidium iodide staining confirmed that mild cell death from IL-1β alone and high levels of cell death from IL-1β/TNF occurred exclusively in siAtg5 cells with decreased autophagy (Fig. 1D,E). IL-1β/TNF treatment did not increase autophagic flux by LC3 immunoblots of untreated and bafilomycin-treated wild-type cells (Fig. 1F), and densitometric determinations of the ratio of LC3-II levels in cells treated with bafilomycin to that in untreated cells (Fig. 1G). The lack of a cytokine effect on autophagic flux indicates that basal levels of autophagy mediate hepatocyte resistance to IL-1β/TNF cytotoxicity.
Increased death in siAtg5 cells was not secondary to differences in receptor levels, as IL-1R1 and TNFR1 levels were unaffected by a decrease in autophagy (Fig. 2A). The effects of IL-1β or TNF alone were equivalent for cytokine concentrations of 5–50 ng/mL. IL-1β/TNF death was equivalent over the same IL-1β concentrations, and maximal at 10 ng/mL of TNF (data not shown). Subsequent experiments were therefore performed with 5 ng/mL IL-1β and/or 10 ng/mL TNF.
FIG. 2.
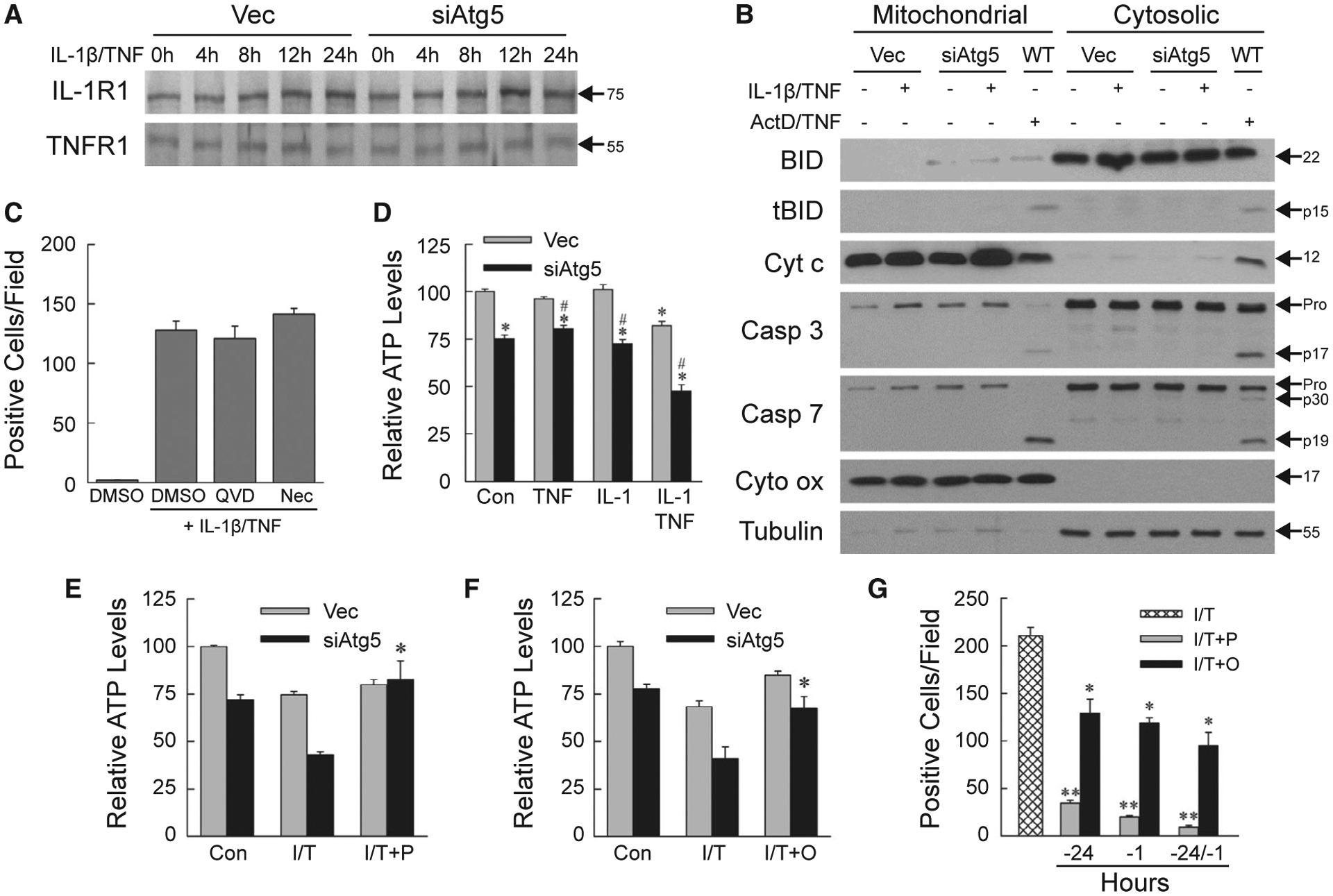
IL-1β/TNF-induced death is necrosis secondary to ATP depletion. (A) Immunoblots of total protein from Vec and siAtg5 cells treated with IL-1β/TNF. (B) Immunoblots of mitochondrial and cytosolic proteins from Vec, siAtg5, and wild-type AML12 cells untreated, or treated with IL-1β/TNF or actinomycin D + TNF. Blots were probed for BID and truncated BID, cytochrome c, caspase 3, caspase 7, and cytochrome oxidase, and tubulin as mitochondrial and cytosolic protein loading and purity controls, respectively. Immunoblots in (A) and (B) are representative of four independent experiments, and protein molecular weights (in kD) are indicated by arrows. (C) Numbers of trypan blue–positive siAtg5 cells treated with DMSO vehicle alone, or IL-1β/TNF after pretreatment with DMSO, QVD-OPh or necrostatin-1s (n = 4). (D) Relative ATP levels in untreated control and 24-hour TNF and/or IL-1β treated cells (*P < 0.000002 compared with untreated Vec cells; #P < 0.00001 compared with Vec cells with the same treatment; n = 6–8). (E) ATP content in cells untreated or treated for 24 hours with IL-1β/TNF alone or with a 1-hour pretreatment with pyruvate (*P < 0.0002 compared with siAtg5 cells treated with IL-1β/TNF; n = 4–8). (F) ATP levels in cells untreated or treated for 24 hours with IL-1β/TNF alone or with oleate pretreatment at 24 hours and 1 hour (*P < 0.01 compared with siAtg5 cells treated with IL-1β/TNF; n = 4–8). (G) Numbers of trypan blue–positive siAtg5 cells treated with IL-1β/TNF alone, or with pyruvate or oleate pretreatment at 24 hours and/or 1 hour (*P < 0.0005 and **P < 0.0000001 compared with IL-1β/TNF; n = 4–8). Abbreviations: ActD/TNF, actinomycin D + TNF; Casp 3, caspase 3; Casp 7, caspase 7; Cyt c, cytochrome c; Cyt ox, cytochrome oxidase; DMSO, DMSO vehicle alone; I/T/, IL-1β/TNF; I/T+O, IL-1β/TNF + oleate; I/T+P, IL-1β/TNF + pyruvate; Nec, necrostatin-1s; QVD, QVD-OPh; tBID, truncated BID; WT, wild-type.
IL-1β/TNF DEATH IS NECROSIS
Cellular trypan blue and propidium iodide positivity reflects primary or secondary necrosis. To exclude secondary necrosis from apoptosis, mitochondrial death pathway activation and downstream caspase cleavage were examined. In IL-1β/TNF-treated siAtg5 cells, BH3-interacting domain death agonist (BID) cleavage and mitochondrial cytochrome c release into the cytoplasm did not occur as in wild-type cells undergoing apoptosis from actinomycin D/TNF (Fig. 2B). Effector caspase 3 and 7 cleavage was absent in IL-1β/TNF-treated siAtg5 cells but present after actinomycin D/TNF along with decreased procaspase levels (Fig. 2B). Total cellular protein levels of the apoptotic factors B cell lymphoma 2–associated X protein (BAX), B cell lymphoma extra large (BCL-XL), Bcl-2-interacting mediator of cell death (BIM), myeloid cell leukemia 1 (MCL1), and tumor protein 53 (p53) were unaltered by the knockdown of autophagy or IL-1β/TNF treatment (Supporting Fig. S1). IL-1β/TNF death did not occur through the apoptotic mitochondrial death pathway characteristic of hepatocyte death from TNF.
To further confirm that IL-1β/TNF death was necrosis, the effects of caspase and necroptosis inhibition were examined. Death was not blocked by the caspase inhibitor QVD-OPh, excluding apoptosis or pyroptosis, or the necroptosis inhibitor necrostatin-1s (Fig. 2C). IL-1β/TNF death in AML12 cells with defective autophagy is necrosis.
DEATH RESULTS FROM IMPAIRED ENERGY HOMEOSTASIS
Autophagy maintains hepatocyte energy homeostasis, and necrosis often results from ATP depletion, suggesting that decreased energy stores might be the mechanism underlying siAtg5 cell death from IL-1β/TNF. Compared with Vec cells, untreated siAtg5 cells had a modest but significant decrease in ATP that was unaltered by treatment with TNF or IL-1β alone (Fig. 2D). IL-1β/TNF in combination induced a significant decrease in ATP in both cell types, but levels were 42% less in siAtg5 cells (Fig. 2D), demonstrating that autophagy-defective hepatocytes undergo energy depletion from IL-1β/TNF in combination.
Autophagy degrades cellular components into energy substrates.(26) To determine whether decreased siAtg5 cell ATP was secondary to an inadequate supply of autophagic products or defective mitochondrial function, we examined the effects of supplementation with the energy substrate pyruvate or oleate. Pyruvate and oleate had no significant effect on ATP levels in autophagy-intact Vec cells but prevented the IL-1β/TNF-induced ATP decrease in siAtg5 cells (Fig. 2E,F). These data indicate that siAtg5 cells have an intact ATP-generating capacity but lack sufficient autophagy-supplied substrates. ATP depletion is the mechanism of necrosis, as pyruvate and oleate significantly reduced siAtg5 cell death from IL-1β/TNF (Fig. 2G).
IL-1β/TNF-INDUCED NECROSIS IS CATHEPSIN-DEPENDENT
ATP depletion triggers lysosomal permeabilization, causing necrosis from the release of hydrolytic enzymes such as cathepsins.(27,28) AML12 cell medium contained equivalent CTSB levels, but increased amounts of CTSL from IL-1β/TNF-treated siAtg5 cells (Fig. 3A). Levels of the secreted protein albumin were equivalent and decreased by IL-1β/TNF (Fig. 3A). Increased CTSL activity was detected in the medium (Fig. 3B). Cathepsins promoted necrosis, as pharmacological CTSB and CTSL inhibitors blocked siAtg5 cell death, although the effect was only significant for CTSL inhibitors (Fig. 3C).
FIG. 3.
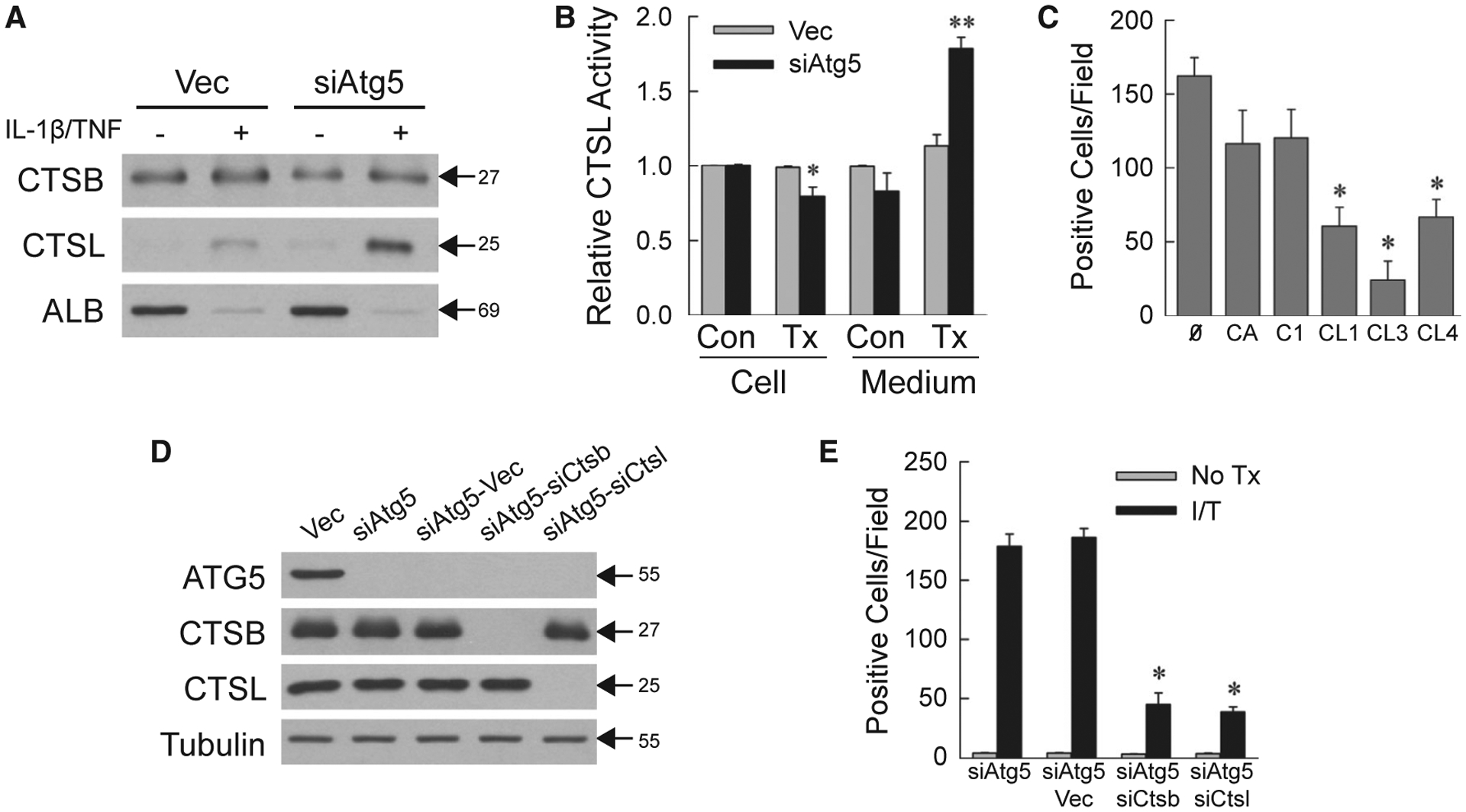
Necrosis is cathepsin-dependent. (A) Immunoblots of protein isolated from the medium of cells untreated or 24-hour IL-1β/TNF treated and probed for CTSB, CTSL, and albumin. (B) Relative CTSL activity in cells and medium from untreated control and 24-hour IL-1β/TNF-treated cells (*P < 0.01 compared with untreated Vec cells; **P < 0.000001 compared with untreated Vec cell medium; n = 5–6). (C) Numbers of trypan blue–positive siAtg5 cells that received no inhibitor or CA-074Me, Cathepsin Inhibitor I, Cathepsin L Inhibitor I, Cathepsin L Inhibitor III, or Cathepsin L Inhibitor IV for 1 hour before IL-1β/TNF (*P < 0.001 compared with no inhibitor; n = 4–6). (D) Immunoblots of total protein from the indicated cells. (E) Numbers of trypan blue–positive cells in untreated and IL-1β/TNF-treated cells at 24 hours (*P < 0.0000001 compared with IL-1β/TNF-treated siAtg5-Vec cells; n = 6–12). Molecular weights (in kD) are indicated by arrows on western blots, which are representative of three independent experiments. Abbreviations: Ø, no inhibitor; ALB, albumin; C1, Cathepsin Inhibitor I; CA, CA-074Me; CL1, Cathepsin L Inhibitor I; CL3, Cathepsin L Inhibitor III; CL4, Cathepsin L Inhibitor IV; I/T, IL-1β/TNF-treated; No Tx, untreated; Tx, IL-1β/TNF-treated.
To further establish cathepsin involvement, lentiviral double knockdowns of Atg5 and Ctsb (siAtg5-siCtsb cells) or Ctsl (siAtg5-siCtsl cells) were generated along with empty vector–infected siAtg5 cells (siAtg5-Vec cells). CTSB or CTSL was selectively decreased in the respective knockdown cells (Fig. 3D). IL-1β/TNF death was equivalent in siAtg5 and siAtg5-Vec cells, but significantly decreased in cells with a CTSB or CTSL knockdown (Fig. 3E). These findings demonstrate that lysosomal cathepsin release occurs from impaired energy homeostasis in IL-1β/TNF-treated siAtg5 cells and triggers necrosis.
HEPATOCYTE-SPECIFIC Atg5 KNOCKOUT MICE UNDERGO IL-1β/TNF LIVER INJURY
To determine the effect of decreased autophagy on cytokine-induced hepatocellular injury in vivo, IL-1β/TNF toxicity was examined in tamoxifen-inducible, hepatocyte-specific Atg5 knockout Atg5Δhep mice. Four days after tamoxifen injection, ATG5 but not ATG7 was decreased in Cre+ mouse livers (Fig. 4A). Control and Atg5Δhep mice were resistant to TNF injury with only minor increases in serum ALT or TUNEL (terminal deoxynucleotide transferase-mediated deoxyuridine triphosphate nick end-labeling)–positive hepatocytes at 6 hours after injection (Fig. 4B,F). Mice had normal ALTs after IL-1β administration, and knockout mice had a slight increase in TUNEL-positive cells at 6 hours (Fig. 4C,G). IL-1β/TNF co-administration led to slight increases in ALT and TUNEL positivity at 6 hours and 12 hours in control mice (Fig. 4D,E,H). In contrast, Atg5Δhep mice had markedly increased serum ALTs and TUNEL positivity from 6 to 24 hours after IL-1β/TNF treatment (Fig. 4D,E,H). Areas of hepatic necrosis were detected by histology exclusively in IL-1β/TNF-treated knockout mice (Fig. 4I).
FIG. 4.
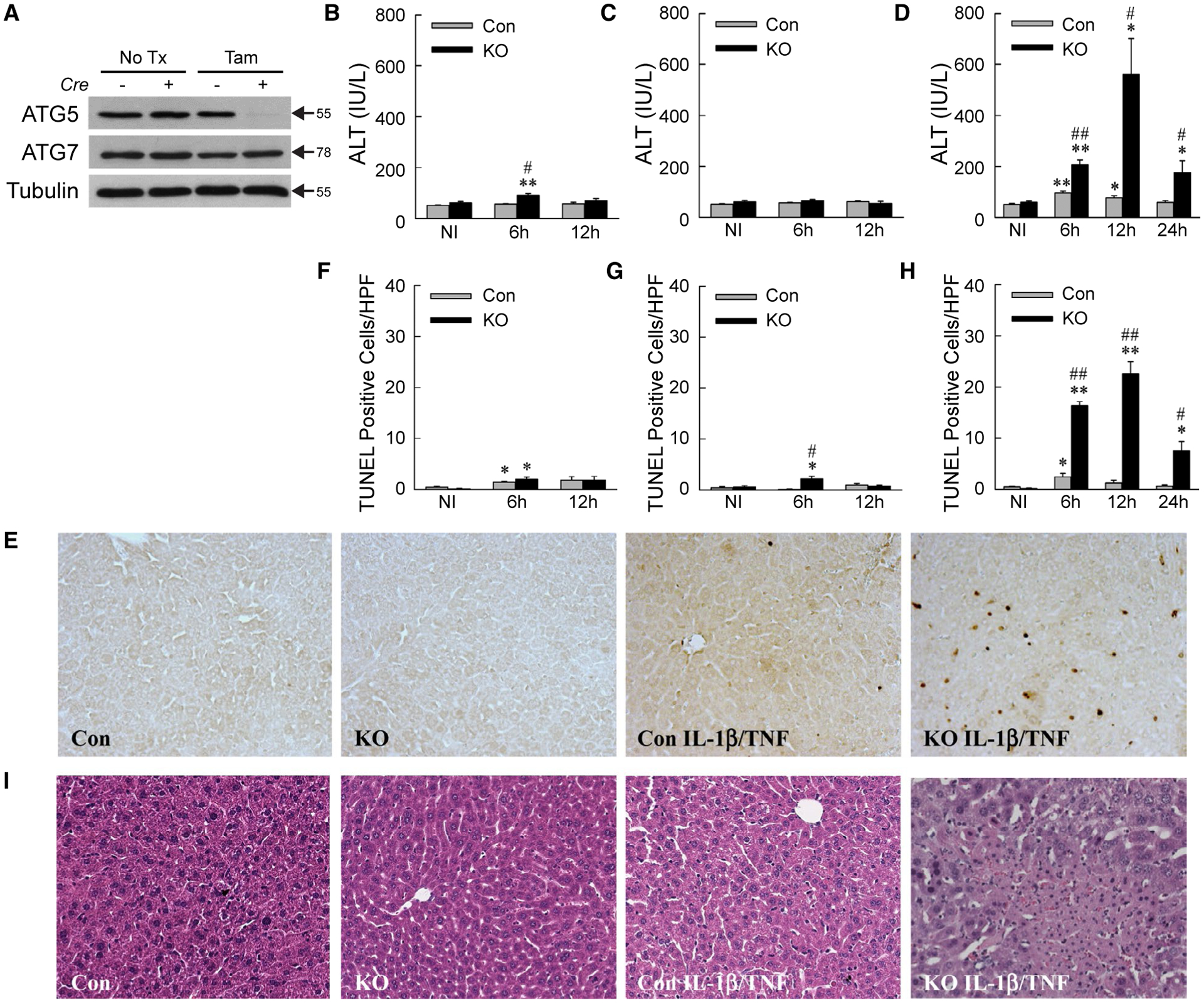
IL-1β/TNF induces liver injury in Atg5Δhep mice. (A) Representative immunoblots from three independent experiments of total liver protein from Cre− and Cre+ mice that received no treatment or tamoxifen. ATG5 band represents the ATG5-ATG12 conjugate. (B-D) Serum ALT levels in control and knockout mice not injected or administered TNF (B), IL-1β (C), or IL-1β (D) for the times indicated (*P < 0.01 and **P < 0.0001 compared with untreated control mice; #P < 0.01 and ##P < 0.0001 compared with treated control mice; n = 5–20). (E) Representative TUNEL images in mice untreated or 12-hour IL-1β/TNF-treated. (F-H) Numbers of TUNEL-positive cells per high power field (×200) in the same mice treated with TNF (F), IL-1β (G), or IL-1β/TNF (H) (*P < 0.01 and **P < 0.0001 compared with untreated control mice; #P < 0.01 and ##P < 0.001 compared with treated control mice; n = 5–9). (I) Representative hematoxylin and eosin stained images of livers from the same mice (×200). Abbreviations: HPF, high power field; KO, knockout; NI, not injected; No Tx, no treatment; Tam, tamoxifen.
TUNEL positivity can represent apoptosis or necrosis.(29) Apoptosis was excluded by the absence of mitochondrial cytochrome c release and BID and caspase cleavage in IL-1β/TNF-injured knockout livers in contrast to wild-type mice undergoing GalN/LPS-induced apoptosis (Fig. 5A). In addition, total cellular protein levels of the apoptotic factors BAX, BCL-XL, BIM, MCL-1, and p53 were unaffected by decreased autophagy or IL-1β/TNF (Supporting Fig. S2). Lysosomal permeabilization occurred in IL-1β/TNF-treated knockout mouse livers, as cathepsins B and L were detected in the cytosol of these mice in parallel with decreased lysosomal levels (Fig. 5B). Lysosomal cathepsin release led to liver injury as Cathepsin L Inhibitor IV significantly inhibited the IL-1β/TNF-induced increases in serum ALT (Fig. 5C) and numbers of TUNEL-positive cells (Fig. 5D) in knockout mice. Identical to AML12 cells, decreased hepatocyte autophagy in vivo led to hepatocyte lysosomal permeabilization and cathepsin-dependent necrosis from IL-1β/TNF.
FIG. 5.
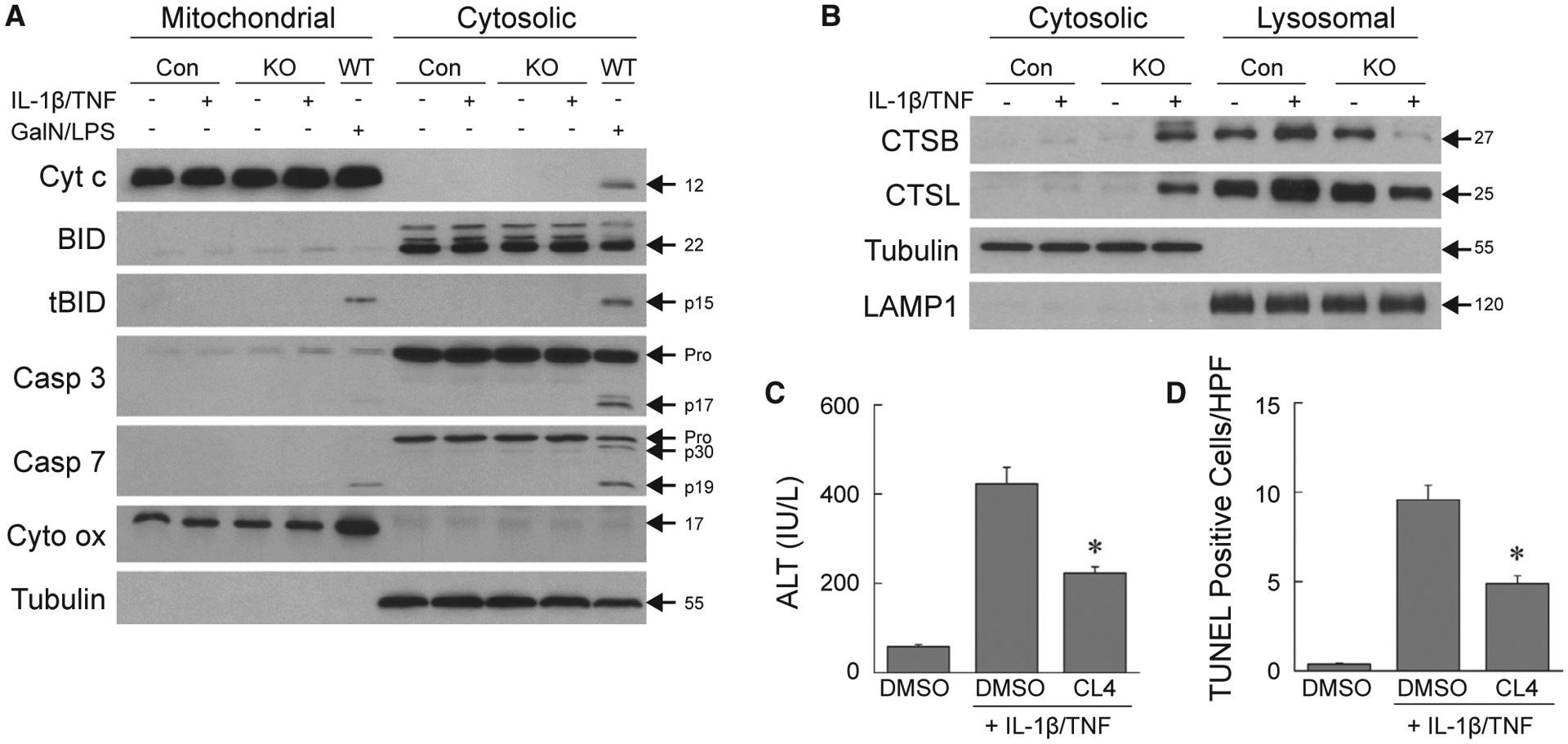
Mouse hepatocyte death is cathepsin-dependent necrosis. (A) Immunoblots of hepatic mitochondrial and cytosolic protein from control, knockout, or wild-type mice treated with IL-1β/TNF or GalN/LPS for 6 hours. Blots were probed for cytochrome c, BID and truncated Bid, caspase 3, caspase 7, and cytochrome oxidase and tubulin as loading and purity controls. (B) Western blots of cytosolic and lysosomal protein from the same mice probed for CTSB, CTSL, and tubulin and lysosomal membrane associated protein 1 as loading and purity controls. Images are representative of three independent experiments, and molecular weights are indicated by arrows. (C) Serum ALT levels in knockout mice injected with DMSO vehicle alone, or DMSO or Cathepsin L Inhibitor IV with IL-1β/TNF (*P < 0.00003 compared to mice with DMSO + IL-1β/TNF; n = 9–14). (D) Numbers of TUNEL-positive cells in the livers of the same mice (*P < 0.00006 compared to mice with DMSO + IL-1β/TNF; n = 9–11). Abbreviations: Casp 3, caspase 3; Casp 7, caspase 7; CL4, Cathepsin L Inhibitor IV; Cyt c, cytochrome c; Cyt ox, cytochrome oxidase; DMSO, DMSO vehicle alone; LAMP1, lysosomal membrane associated protein 1; tBID, truncated BID; WT, wild-type.
Atg5Δhep MICE HAVE INCREASED INFLAMMATION
Examination of hepatic gene expression revealed that levels of the proinflammatory cytokines Il6 and interferon-γ (Ifng) (Fig. 6A,B), and markers cyclooxygenase 2 (Cox2) and nitric oxide synthase 2 (Fig. 6C,D), were significantly increased in IL-1β/TNF-treated Atg5Δhep mice. In contrast, there were no changes in mRNA levels for macrophage galactose N-acetyl-galactosamine specific lectin 2 and resistin like alpha, which are genes expressed in anti-inflammatory M2-polarized macrophages (Fig. 6E,F). Greater numbers of inflammatory cells were present in IL-1β/TNF-treated Atg5Δhep mice, as mRNA levels (Fig. 6G,H) and immunofluorescence staining for the macrophage marker clusters of differentiation 68 (CD68) (Fig. 7A,B) and neutrophil marker lymphocyte antigen 6 complex, locus G (LY6G) (Fig. 7C,D) were increased in these mice. Decreased hepatocyte autophagy magnified the hepatic inflammatory response to IL-1β/TNF.
FIG. 6.
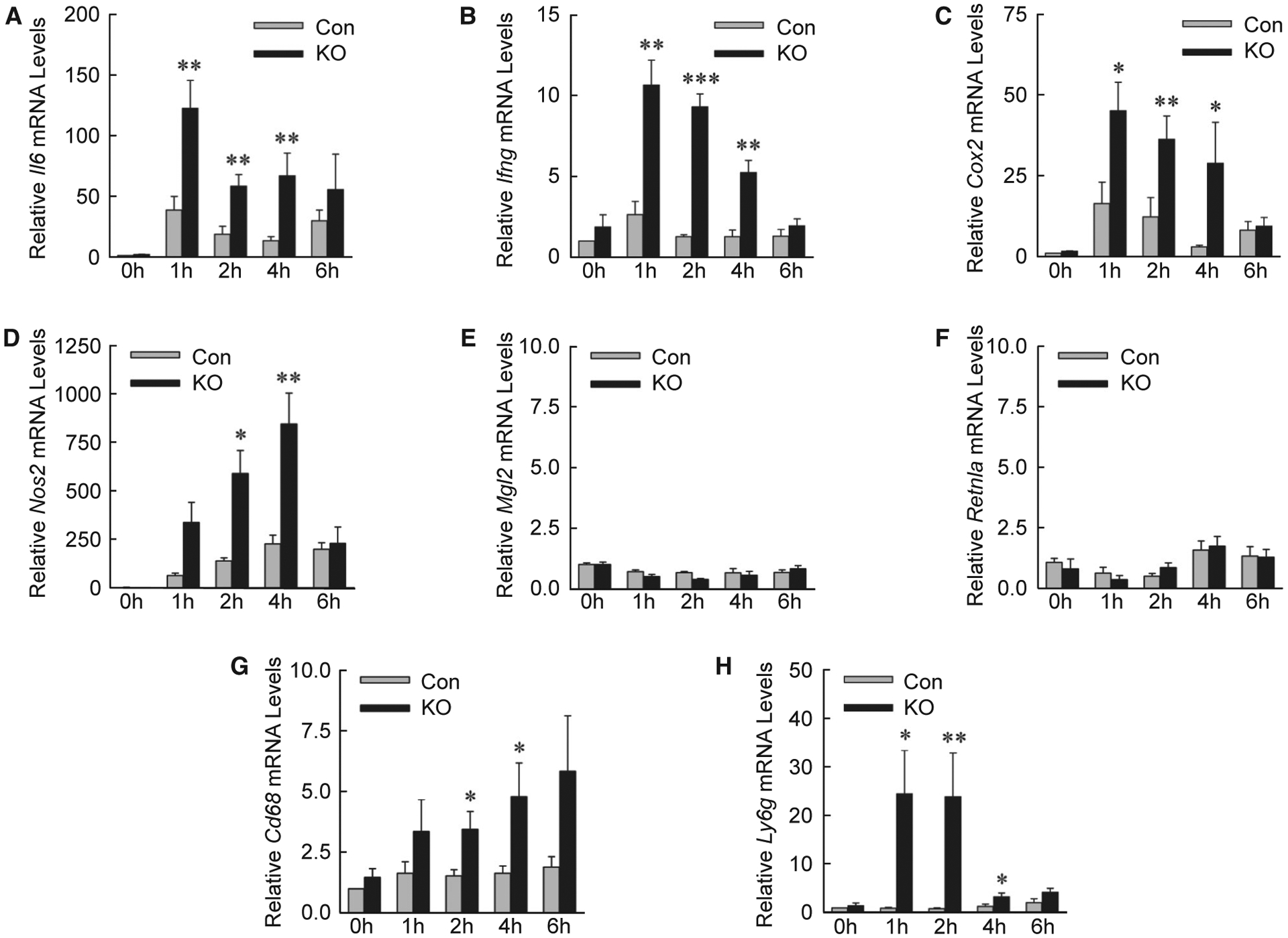
Knockout mice have increased proinflammatory gene expression. (A-H) Relative hepatic mRNA levels in control and knockout mice for the genes Il6, Ifng, cyclooxygenase 2, nitric oxide synthase 2, macrophage galactose N-acetyl-galactosamine specific lectin 2, resistin like alpha, Cd68 and Ly6g (*P < 0.05, **P < 0.01, and ***P < 0.001 compared with control mice at the same time; n = 4–7). Abbreviations: Cox2, cyclooxygenase 2; Ifng, interferon-γ; Mgl2, macrophage galactose N-acetyl-galactosamine specific lectin 2; Nos2, nitric oxide synthase 2; Retnla, resistin like alpha.
FIG. 7.
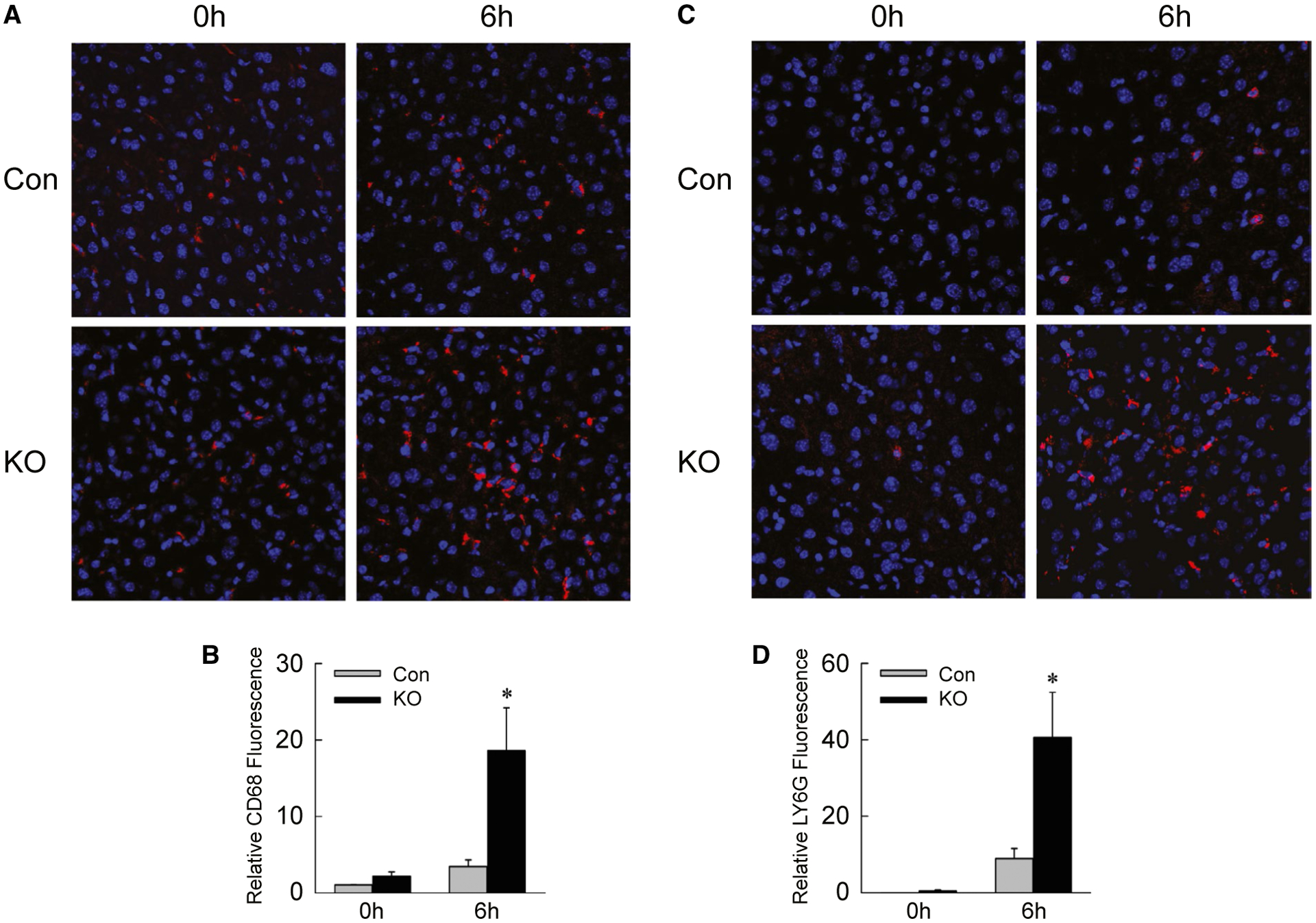
Inflammatory infiltration is increased in knockout mice. (A) Immunofluorescence staining of livers from control and knockout mice untreated or 6 hours after IL-1β/TNF (×200). (B) Quantification of CD68 fluorescence (*P < 0.01 compared with untreated control; n = 4–10). (C) LY6G immunofluorescence in the same livers. (D) LY6G fluorescence quantification (*P < 0.01 compared with untreated control; n = 4–10).
DECREASED AUTOPHAGY PROMOTES PROINFLAMMATORY EXOSOMAL DAMP SECRETION
Findings of increased inflammation in Atg5Δhep mice suggested that DAMP release from IL-1β/TNF-injured hepatocytes amplified the innate immune response. Autophagy sequesters and degrades multivesicular bodies containing proteins that other-wise are secreted in exosomes.(30) Extracellular vesicles (EVs) isolated from AML12 cell medium consisted largely of exosomes, as indicated by a characteristic mean diameter of approximately 65 nm (Fig. 8A). EV diameter and quantity were unchanged by decreased autophagy or IL-1β/TNF treatment (Fig. 8A,B). Exosome isolation was further confirmed by enrichment of the exosome-specific protein ALIX in the EV isolate (Fig. 8C). EVs from IL-1β/TNF-treated siAtg5 cells contained the DAMPs high-mobility group box 1 (HMGB1) and heat shock protein 90 (HSP90), which were absent in EVs from Vec cells (Fig. 8C).
FIG. 8.
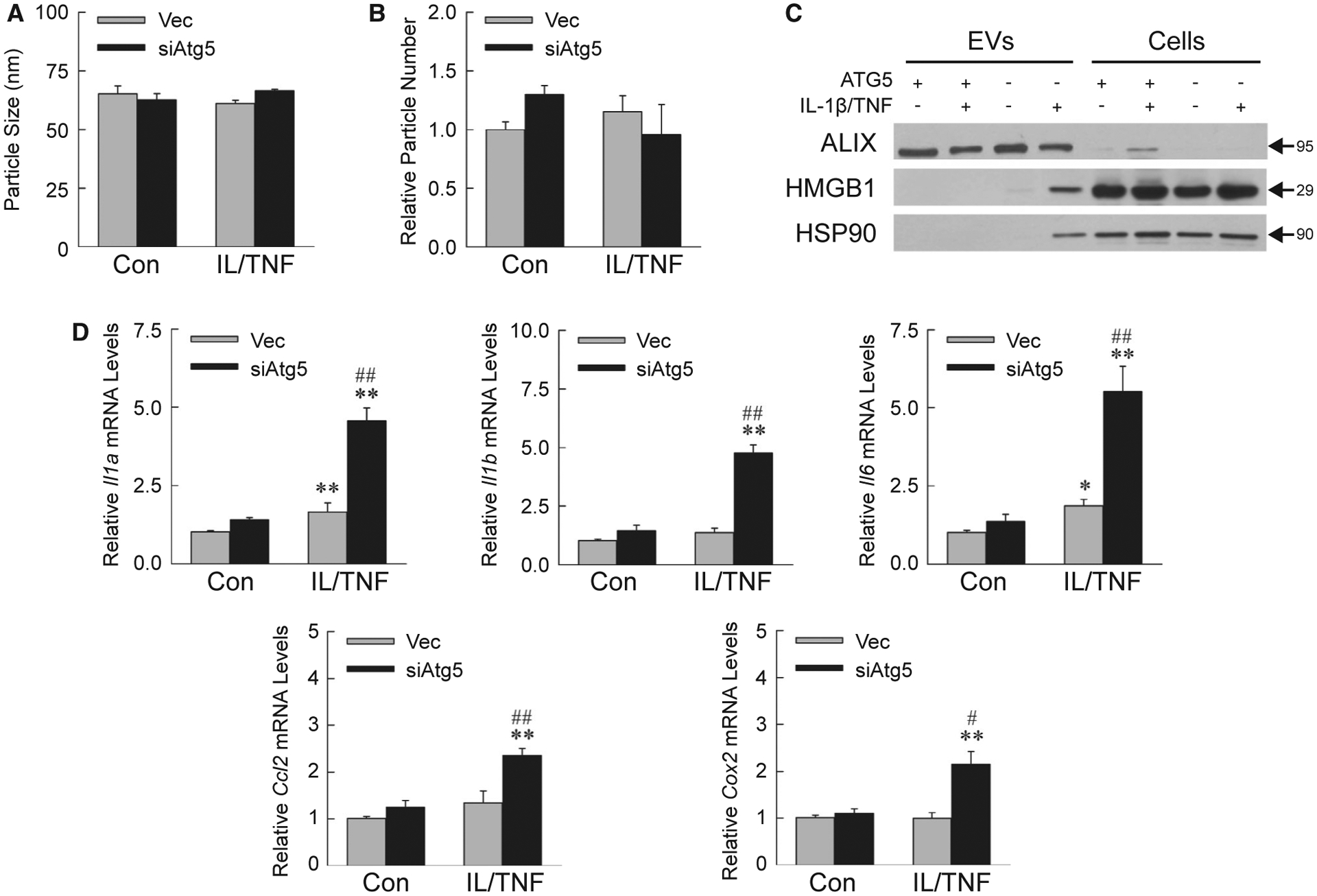
IL-1β/TNF-treated siAtg5 cells release proinflammatory exosomes. Mean size (A) and number (B) of EVs isolated from AML12 cell medium (n = 3). (C) Immunoblots of EV and cellular protein from Vec (ATG5+) or siAtg5 (ATG5−) cells untreated or IL-1β/TNF-treated for 24 hours. Images are representative of the results of three independent experiments, and molecular weights are indicated by arrows. (D) Kupffer cell mRNA levels after 6 hours of exposure to EVs from control or IL-1β/TNF-treated Vec and siAtg5 cells (*P < 0.01 and **P < 0.001 compared with untreated Vec cells; #P < 0.01 and ##P < 0.001 compared with IL-1β/TNF-treated Vec cells; n = 6–7).
To prove the proinflammatory nature of siAtg5 cell exosomes, wild-type Kupffer cells were exposed to AML12 cell–derived EVs for 6 hours. mRNA levels of proinflammatory Kupffer cell genes Il1a, Il1b, Il6, Ccl2 (chemokine [C-C motif] ligand 2), and Cox2 were significantly increased by EVs from IL-1β/TNF-treated siAtg5 cells as compared with Vec cell EVs (Fig. 8D). Decreased autophagy therefore promotes IL-1β/TNF-treated hepatocyte secretion of exosomal DAMPs that activate macrophages to cause inflammation.
Discussion
Recent studies have implicated IL-1β in the pathophysiology of nonalcoholic and alcoholic steatohepatitis.(7–10) These findings suggest that IL-1β induces hepatocyte injury and death, but how IL-1β causes such an effect is unclear as this cytokine lacks a defined receptor-linked death pathway. The present study delineates a molecular mechanism for IL-1β-induced liver injury by finding that with decreased autophagy, IL-1β becomes cytotoxic and proinflammatory to hepatocytes (summarized in Supporting Fig. S3).
Although IL-1β has been reported to induce death in normal, cultured hepatocytes,(31) other investigations have failed to detect IL-1β cytotoxicity.(9,32) Our findings demonstrate a lack of significant IL-1β-induced death in normal mice, but sensitization to IL-1β toxicity in the presence of TNF and decreased autophagy. Interestingly, previous studies in which IL-1β was nontoxic to normal hepatocytes did demonstrate toxicity in steatotic and GalN/LPS-treated cells,(9,32) two conditions that we have previously demonstrated impaired hepatocyte autophagy.(14,15) Decreased autophagy, which is found in many liver diseases including nonalcoholic and alcoholic steatohepatitis,(33) may be a mechanism for IL-1β-dependent hepatic injury in these diseases. IL-1β/TNF toxicity may also be a mechanism underlying our previous findings that hepatocyte-specific autophagy knockout mice undergo increased liver injury and mortality from high-dose LPS,(21) and that mice with decreased autophagy from aging develop increased steatohepatitis from high-fat diet feeding.(34)
IL-1β hepatotoxicity is due to necrosis rather than apoptosis. Necrosis is dependent on the co-presence of TNF, which like IL-1β is itself nontoxic to normal hepatocytes. Death from IL-1β/TNF results from compromised energy homeostasis, which only occurs in autophagy-defective hepatocytes. The required presence of both cytokines is consistent with findings in rat and rabbit liver that IL-1β and TNF in combination create a synergistic metabolic demand on the liver.(35,36) Hepatocytes are able to meet this metabolic demand when autophagy supplies energy substrates. Hepatocytes with impaired autophagy lack these substrates, resulting in ATP depletion that triggers lysosomal permeabilization and release of necrosis-inducing cathepsins.
Cathepsin-dependent cytotoxicity from TNF has been reported previously due to caspase-dependent apoptosis.(37) Our findings define a distinct, IL-1β-induced, caspase-independent necrotic death pathway. Death was CTSB-dependent and CTSL-dependent, as knockdown of either cathepsin effectively blocked death. Differential levels of CTSL, but not CTSB, were detected in the medium of dying cells, possibly explaining the better effectiveness of CTSL pharmacological inhibitors despite equal efficacy of a genetic knockdown of either form. Consistent with the involvement of both cathepsins was the finding of lysosomal release into the cytosol of CTSB and CSTL in the livers of IL-1β/TNF-treated autophagy knockout mice.
In addition to sensitizing to IL-1β cytotoxicity, decreased autophagy promoted hepatocyte inflammation. Recent studies have emphasized a role for hepatocytes as immune mediators in liver disease through the release of cytokines and proinflammatory DAMPs.(38) In IL-1β-dependent murine alcoholic liver injury, the DAMPs HMGB1 and HSP90 promote inflammation and HSP90 is detected in exosomes.(39,40) We describe how decreased hepatocyte autophagy is a mechanism that promotes IL-1β-dependent exosomal release of the DAMPs HMGB1 and HSP90. These hepatocyte findings, together with the ability of macrophage autophagy to down-regulate inflammation,(18,41,42) demonstrate a significant role for decreased autophagy in the promotion of liver injury–associated inflammation. Based on nonhepatic cell studies, the mechanism of hepatocyte exosome DAMP release is likely the shunting of undegraded multivesicular body DAMPs into exosomes for secretion. However, no increase in exosome number was detected in cytokine-stimulated, autophagy-deficient AML12 hepatocytes, as would be expected from this mechanism. Rather, selective elimination by autophagy of multivesicular bodies with DAMPs may occur. Additional studies will be required to further clarify the mechanism of exosomal DAMP release from autophagy-defective hepatocytes.
These findings define a mechanism by which IL-1β toxicity occurs in liver and potentially other organs. That decreased autophagy promotes IL-1β-induced hepatic injury, and inflammation suggests an additional mechanism by which therapies targeted to increase autophagy may be an effective treatment for liver diseases such as steatohepatitis.
Supplementary Material
Acknowledgments
Supported by National Institutes of Health grants R01DK044234, R01AA022601 (to M.J.C.), and T32DK108735 (to M.A.). Core resources were supported by G12MD007602.
Abbreviations:
- ALT
alanine aminotransferase
- Atg5
autophagy-related 5
- BID
BH3-interacting domain death agonist
- CD68
clusters of differentiation 68
- Con
control
- CTSB
cathepsin B
- CTSL
cathepsin L
- DAMP
damage-associated molecular pattern
- EV
extracellular vesicle
- GalN
galactosamine
- HMGB1
high-mobility group box 1
- HSP90
heat shock protein 90
- KO
knockout
- LC3
microtubule-associated protein 1 light chain 3
- LPS
lipopolysaccharide
- LY6G
lymphocyte antigen 6 complex, locus G
- shRNA
small hairpin RNA
- TUNEL
terminal deoxynucleotide transferase-mediated deoxyuridine triphosphate nick end-labeling
Footnotes
Potential conflict of interest: Nothing to report.
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.31209/suppinfo.
REFERENCES
- 1).Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014;147:765–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol 2013;59:583–594. [DOI] [PubMed] [Google Scholar]
- 3).Zannetti C, Roblot G, Charrier E, Ainouze M, Tout I, Briat F, et al. Characterization of the inflammasome in human Kupffer cells in response to synthetic agonists and pathogens. J Immunol 2016;197:356–367. [DOI] [PubMed] [Google Scholar]
- 4).Czaja MJ, Xu J, Ju Y, Alt E, Schmiedeberg P. Lipopolysaccharide-neutralizing antibody reduces hepatocyte injury from acute hepatotoxin administration. Hepatology 1994;19:1282–1289. [PubMed] [Google Scholar]
- 5).McDonald B, Kubes P. Innate immune cell trafficking and function during sterile inflammation of the liver. Gastroenterology 2016;151:1087–1095. [DOI] [PubMed] [Google Scholar]
- 6).Mann A, Czaja MJ, Schattenberg JM. TNF signaling. In: Dufour JF, Clavien PA, eds. Signaling Pathways in Liver Diseases, 3rd Edition. Hoboken, NJ: John Wiley & Sons; 2015:186–202. [Google Scholar]
- 7).Tilg H, Moschen AR, Szabo G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016;64:955–965. [DOI] [PubMed] [Google Scholar]
- 8).Cui K, Yan G, Xu C, Chen Y, Wang J, Zhou R, et al. Invariant NKT cells promote alcohol-induced steatohepatitis through inter-leukin-1β in mice. J Hepatol 2015;62:1311–1318. [DOI] [PubMed] [Google Scholar]
- 9).Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology 2010;139:323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest 2012;122:3476–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Furman D, Chang J, Lartigue L, Bolen CR, Haddad F, Gaudilliere B, et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med 2017;23:174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Czaja MJ, Ding WX, Donohue TM, Friedman SL, Kim JS, Komatsu M, et al. Functions of autophagy in normal and diseased liver. Autophagy 2013;9:1131–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Czaja MJ. Function of autophagy in nonalcoholic fatty liver disease. Dig Dis Sci 2016;61:1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Amir M, Zhao E, Fontana L, Rosenberg H, Tanaka K, Gao G, et al. Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation. Cell Death Differ 2013;20:878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010;11:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell 2011;146:682–695. [DOI] [PubMed] [Google Scholar]
- 18).Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015;11:271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010;139:1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012;55:222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Lalazar G, Ilyas G, Malik SA, Liu K, Zhao E, Amir M, et al. Autophagy confers resistance to lipopolysaccharide-induced mouse hepatocyte injury. Am J Physiol Gastrointest Liver Physiol 2016;311:G377–G386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Wu JC, Merlino G, Fausto N. Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc Natl Acad Sci USA 1994;91:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Piva R, Chiarle R, Manazza AD, Taulli R, Simmons W, Ambrogio C, et al. Ablation of oncogenic ALK is a viable therapeutic approach for anaplastic large-cell lymphomas. Blood 2006;107:689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006;441:885–889. [DOI] [PubMed] [Google Scholar]
- 25).Schuler M, Dierich A, Chambon P, Metzger D. Efficient temporally controlled targeted somatic mutagenesis in hepatocytes of the mouse. Genesis 2004;39:167–172. [DOI] [PubMed] [Google Scholar]
- 26).Cingolani F, Czaja MJ. Regulation and functions of autophagic lipolysis. Trends Endocrinol Metab 2016;27:696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene 2008;27:6434–6451. [DOI] [PubMed] [Google Scholar]
- 28).Kosic M, Arsikin-Csordas K, Paunovic V, Firestone RA, Ristic B, Mircic A, et al. Synergistic anticancer action of lysosomal membrane permeabilization and glycolysis inhibition. J Biol Chem 2016;291:22936–22948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 1995;21:1465–1468. [DOI] [PubMed] [Google Scholar]
- 30).Fader CM, Sanchez D, Furlan M, Colombo MI. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 2008;9:230–250. [DOI] [PubMed] [Google Scholar]
- 31).Petrasek J, Dolganiuc A, Csak T, Kurt-Jones EA, Szabo G. Type I interferons protect from toll-like receptor 9-associated liver injury and regulate IL-1 receptor antagonist in mice. Gastroenterology 2011;140:697–708.e694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Gehrke N, Hovelmeyer N, Waisman A, Straub BK, Weinmann-Menke J, Worns MA, et al. Hepatocyte-specific deletion of IL1-RI attenuates liver injury by blocking IL-1 driven autoinflammation. J Hepatol 2018;68:986–995. [DOI] [PubMed] [Google Scholar]
- 33).Czaja MJ. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology 2011;140:1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Fontana L, Zhao E, Amir M, Dong H, Tanaka K, Czaja MJ. Aging promotes the development of diet-induced murine steatohepatitis but not steatosis. Hepatology 2013;57:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Jin MB, Shimahara Y, Yamaguchi T, Ichimiya M, Kinoshita K, Oka T, et al. The effect of a bolus injection of TNF-α and IL-1β on hepatic energy metabolism in rats. J Surg Res 1995;58:509–515. [DOI] [PubMed] [Google Scholar]
- 36).Tredget EE, Yu YM, Zhong S, Burini R, Okusawa S, Gelfand JA, et al. Role of interleukin 1 and tumor necrosis factor on energy metabolism in rabbits. Am J Physiol 1988;255:E760–E768. [DOI] [PubMed] [Google Scholar]
- 37).Werneburg N, Guicciardi ME, Yin XM, Gores GJ. TNF-α-mediated lysosomal permeabilization is FAN and caspase 8/Bid dependent. Am J Physiol Gastrointest Liver Physiol 2004;287:G436–G443. [DOI] [PubMed] [Google Scholar]
- 38).Jia L, Vianna CR, Fukuda M, Berglund ED, Liu C, Tao C, et al. Hepatocyte toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat Commun 2014;5:3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Saha B, Momen-Heravi F, Furi I, Kodys K, Catalano D, Gangopadhyay A, et al. Extracellular vesicles from mice with alcoholic liver disease carry a distinct protein cargo and induce macrophage activation through heat shock protein 90. Hepatology 2018;67:1986–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Ge X, Antoine DJ, Lu Y, Arriazu E, Leung TM, Klepper AL, et al. High mobility group box-1 (HMGB1) participates in the pathogenesis of alcoholic liver disease (ALD). J Biol Chem 2014;289:22672–22691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41).Ilyas G, Cingolani F, Zhao E, Tanaka K, Czaja MJ. Decreased macrophage autophagy promotes liver injury and inflammation from alcohol. Alcohol Clin Exp Res 2019;43:1403–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Ilyas G, Zhao E, Liu K, Lin Y, Tesfa L, Tanaka KE, et al. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1β. J Hepatol 2016;64:118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.