

Abstract
Although the dinuclear center (DNC) of the resting oxidized “as-isolated” cytochrome c oxidase (CcO) is not a catalytically active state, its detailed structure, especially the nature of the bridging species between the Fea33+ and CuB metal sites, is still both relevant and unsolved. Recent crystallographic work has shown an extended electron density for a peroxide type dioxygen species (O1–O2) bridging the Fea3 and CuB centers. In this paper, our density functional theory (DFT) calculations show that the observed peroxide type electron density between the two metal centers is most likely a mistaken analysis due to overlap of the electron density of a water molecule located at different positions between apparent O1 and O2 sites in DNCs of different CcO molecules with almost the same energy. Because the diffraction pattern and the resulting electron density map represent the effective long-range order averaged over many molecules and unit cells in the X-ray structure, this averaging can lead to an apparent observed superposition of different water positions between the Fea33+ and CuB metal sites.
Short abstract
X-ray crystal structures have shown an extended electron density for a dioxygen species (O1−O2) bridging the Fea3 and CuB centers of the oxidized resting state of cytochrome c oxidase (CcO). Our current DFT calculations show that the observed peroxide type electron density is most likely an overlap of the electron density of a water molecule located at different positions between O1 and O2 in different CcO molecules with almost the same energy.
1. Introduction
Cytochrome c oxidase (CcO), located in the inner mitochondrial or bacterial membrane, is the terminal enzyme in the respiratory chain that reduces O2 to H2O and pumps protons across the membrane to create the chemiosmotic proton gradient used by ATP synthase to synthesize ATP.1−5 The catalytic site of CcO that binds and reduces O2 by 4e–/4H+ transfer contains a heme a3 (Fea3) and a Cu (CuB) ion. Fea3 and CuB are close to each other (∼5 Å). This Fea3-CuB active site is usually called the dinuclear (or binuclear) center/complex (DNC or BNC). In all types of CcO molecules, the iron in the Fea3 site is coordinated to heme and an axial histidine ligand (His384; residue numbers in this paper are by default for ba3 CcO from Thermus thermophilus (Tt)), while the copper in the CuB site is coordinated to three histidine ligands: His233, His282, and His283. His233 covalently links to the Tyr237 side chain. This unique cross-linked tyrosine residue takes an important role in the processes of electron/proton transfer in CcO. There are two other redox centers also present in CcO. One is a homodinuclear Cu dimer (2CuA), which serves as the initial site of electron entry to CcO,6,7 and the other is also a heme, which is heme A (Fea) in the case of the aa3 type of CcO or heme B (Feb) in the ba3 type of CcO. Electrons transfer from cytochrome c to CuA, then on to heme A/B, and from there to the DNC Fea3-CuB.8,9 The DNC structures of aa3 and ba3 oxidases are nearly identical.
The oxidation, spin, and ligation states of the Fea3 and CuB sites change during the catalytic cycle. Starting from the binding of O2 with the reduced (R) DNC, several catalytic intermediates (see Figure 1) have been well characterized by resonance Raman (rR) studies.10−14 Very recently, we have calculated the Fea3–O/O–O stretching frequencies for several DNC intermediate states and compared them with the available rR data.15 When molecular O2 binds with Fea32+, state A[Fea3–O2•–···CuB+] is formed,10−14,16,17 where Fea3–O2•− is likely in a similar bent end-on geometry as in oxymyoglobin.15,18−20 The following characterized states are P, F, and OH. State P is not a peroxide-containing compound (as implied in the notation), but one in which the dioxygen O–O bond has already been cleaved.21−25 It can be represented as P[Fea34+=O2–···OH––CuB2+]. State F[Fea3=O2–···H2O–CuB2+] is then formed when the OH– ligand of CuB in P receives a proton and becomes a water ligand. Our density functional calculations reproduced the rR observed ∼20 cm–1 shift of the Fea3–O stretching mode from state P[Fea34+=O2–···OH––CuB] to F[Fea34+=O2–···H2O–CuB].15 Therefore, it is highly likely that the H2O ligand is still on the CuB2+ site in state F. On the basis of our energy calculations,26 an FH[Fea3=O2–···CuB2+] state, where the H2O ligand has dissociated from the CuB site and the O2– on Fea34+ also weakly binds with CuB, although not observed, may exist before the catalytically active form of the oxidized state OH is formed by 1e–/1H+ reduction of Fea34+=O2– to Fea3–OH–. A very low frequency Fea33+–OH– stretching mode at 450 cm–1 was observed in OH, and our calculations have shown that this low Fe–O stretching mode can be produced by a nearly symmetrically bridged OH[Fea3–OH––CuB2+] structure with a relatively long Fea3–O distance near 2 Å.15
Figure 1.

A, P, F, and OH are the catalytic intermediates that are identified by resonance Raman (rR) experiments after O2 binding with Fea32+ in the reduced (R) state. Their DNCs are likely in the forms presented above. However, the resting “as-isolated” oxidized O state is different from the active OH state.
It is interesting to note that the DNC structure of the resting “as-isolated” oxidized (O) state is different from that of the active OH state. OH is a metastable catalytically active state, which decays to the relaxed lower-energy O state when a one-electron (1e–) transfer to the DNC in OH is delayed (or absent) during redox cycling, just after cycling from R → OH. Given sufficient time, OH decays to O, which is the same as the isolated oxidized (“resting”) state. The differences between O and OH were first seen in kinetics studies using optical difference spectroscopy and electrometry to examine the 1e– redox transitions OH + 1e– → EH in comparison to O + 1e– → E.9,27 In the next step to regenerate R, one more electron is added to the active site, and a proton may move into the active site region to protonate an OH– (if needed). The observed kinetics of OH → EH is very different from that of O → E, and the first gives more efficient electron transfer, ending at CuB, and more effective proton transfer, for both ba3 and aa3 type enzymes. In the kinetic experiments, to generate state EH from OH, the 1e– added is injected after a timed laser flash, and the prior reaction cycle ending in state OH is also started by a laser flash in the CO-inhibited R state,28 synchronized with a pulse of molecular oxygen.27 Overall, the full 2e– reduction of O → R is much slower than that of OH → R.29−31 It has also been found in aa3 that the reduction of OH is coupled to proton translocation, while the reduction of O is not.29−31 From these variations in kinetics, some significant structural differences between OH and O are expected, but these specific differences are not at all evident. It has been a hot topic in the past 30 years and is still under debate which species bridges the Fea33+ and CuB sites in the resting “as-isolated” O state, despite various spectral and structural analyses.32−40
The electron density between Fea3 and CuB in the as-isolated oxidized aa3 type CcOs from Paracoccus denitrificans (Pd) and Rhodobacter sphaeroides (Rs) was originally interpreted as a H2O and an OH– ligand.33,34 Later, on the basis of the high-resolution X-ray crystal structures of the oxidized CcO’s from Pd (PDB code 3HB3, 2.25 Å resolution)35 and from bovine heart (PDB code 2ZXW, 1.95 Å resolution),36 two research groups have independently proposed that a peroxide dianion (O22–) bridges the Fea3 and CuB in the DNC. Similarly, strong electron density for a peroxide type dioxygen species (O1–O2) bridging the Fea3 and CuB DNC was also observed in the high-resolution (1.8 Å) X-ray crystal structures (PDB entries 3S8G and 3S8F) of ba3 CcO from Tt (see Figure 2).37 Further, the peroxide type species in the resting oxidized DNC was also observed by the X-ray free-electron laser (XFEL) experiment (1.9 Å resolution).38 Different groups reported slightly different O1–O2 distances of around 1.5–1.7 Å. Recently, Andersson et al. reported a radiation-damage-free oxidized ba3 CcO structure (2.3 Å resolution) at room temperature, in which a single hydroxide or a water molecule resides between the Fea3 and CuB sites.39 However, the very recent low-dose high-energy X-ray data analysis on the oxidized-resting bovine heart CcO again showed the peroxide-type electron density between the Fea3 and CuB sites.40
Figure 2.
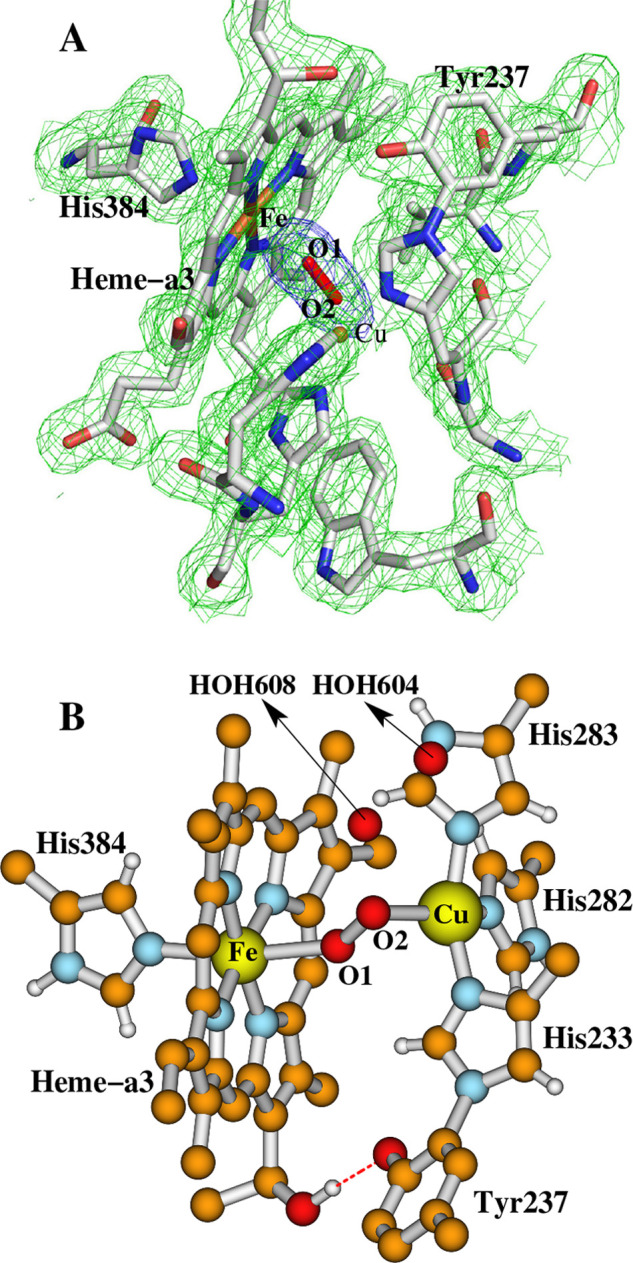
Dinuclear center (DNC) of the X-ray crystal structure 3S8G of ba3 CcO from Thermus thermophilus (Tt).37 (A) The electron density map adapted with permission from Figure 5 of ref (37). Copyright © 2011 Tiefenbrunn et al. (B) The ball and stick structure of this DNC in a different orientation.
Theoretically, Kaila et al. proposed through their quantum chemical calculations that this bridging ligand in the resting oxidized DNC of bovine heart CcO is dioxygen (O2), which may be reduced to superoxide (O2•–) in the X-ray beam.41 However, their calculated O–O distances of 1.29–1.32 Å for O2 or O2•– bridges are much shorter than the apparent O–O distances observed in the X-ray structures. Further, we are not aware of any experimental evidence favoring the presence of molecular oxygen (O2) between the oxidized Fe3+ and Cu2+ metal centers. If the affinity of O2 binding with Fe3+ and Cu2+ is low, the O–O species with high occupancy observed in the X-ray crystal structures cannot be molecular O2. We have therefore also performed density functional theory (DFT) calculations42 on the ba3 CcO from Tt on the basis of the 3S8G(37) X-ray crystal structure with O22–, O2•–, HO2–, or H2O2 in the bridging position. These calculations have shown that the geometry optimized DNC structures with either O22– or O2•– as the bridging species would have large structural discrepancies in comparison with the X-ray crystal structure.42 When H2O2 was put in between the Fea32+ and CuB sites (Fea33+ and CuB metal sites were assumed to be reduced in the X-ray beam),37 the O–O bond broke during the geometry optimizations regardless of the spin state of Fea32+.42 Also, it is well-known experimentally that, in the oxidized resting O state in ba3, the dinuclear center is closed and reacts only very slowly with Fea3 ligands, including H2O2. (The aa3 enzyme shows more access to H2O2 in state O.) The dinuclear site in ba3 only opens up for binding of H2O2 and reaction after a single-electron photoactivated injection into the enzyme.43,44 The presence then of H2O2 or related species within the active site of the oxidized resting O state in ba3 would be unexplained, aside from possible radiation-induced reactions.
Finally, our previous calculations indicated that the O1–O2 species observed in the DNC of the X-ray crystal structure was best represented as HO2–, which may be a product of the photoreaction of the H2O/OH– ligands with 2e– transfer to the adjacent oxidized Fea33+ and CuB sites in the X-ray beam.42 However, in evaluating the most likely and unlikely physical explanation for the observed electron density map, we need to take into account that the operating physical mechanism will produce atomic structures that represent both a spatial and time average over billions of enzyme molecules. While some effects due to the X-ray irradiation may be observable with careful attention to the time course, these effects are not likely to be dominant when they are averaged over billions of structural sites. Indeed, as we now show in the Results and Discussion, our current calculations demonstrate that the Fea33+–H2O···OH––CuB/Fea33+−OH–···H2O–CuB type structures are also unlikely to represent the resting state of the DNC, and the observed O1–O2 peroxide type electron density between the two metal centers is most likely the overlap of electron densities of single water molecules positioned somewhere between O1 and O2 in different DNCs with almost the same energy. The effective electron density averaging occurs because of the long-range averaging over molecular subunits in Fourier (reciprocal) space converted back to real coordinate space (Cartesian space).
2. Calculation Methods
The starting geometries of the DNC model clusters studied in this paper were established on the basis of the Cartesian coordinates of the ba3 CcO X-ray crystal structure 3S8G.37 All calculations were performed using the DFT dispersion-corrected OLYP-D3(BJ)45 functional implemented within the ADF2017 software package.46−48 Goerigk et al. assessed 217 variations of dispersion-corrected and -uncorrected density functional approximations and carried out a detailed analysis to identify reliable approaches based on the analysis of the GMTKN55 benchmark database for general main-group thermochemistry, kinetics, and noncovalent interactions.49 Their studies have shown that OLYP-D3(BJ) is one of the best three GGA functionals overall, outperforming all dispersion-corrected meta-GGAs except for SCAN-D3(BJ).49
Our geometry optimization calculations were performed using the broken-symmetry50−52/OLYP-D3(BJ)45/TZP plus COSMO15,26,53−56 solvation model methodology. The inner cores of C(1s), N(1s), and O(1s) were treated by the frozen core approximation.
Starting from the active OH[Fea33+–OH––CuB] state model, we will examine whether there are H2O/OH– ligands or a H2O molecule between the Fea33+ and CuB sites in the resting as-isolated O state through energetic and pKa calculations. The whole structure of our DNC model for the OH state is shown in Figure 3. For a clearer view, the top portion of this cluster is shown in Figure 4I, and the central Fea33+–OH––CuB part of the model is given in Figure 4II. In our OH model, the bridging OH– is H-bonding with a water molecule, which originates from the H2O ligand of CuB in the prior state F.
Figure 3.
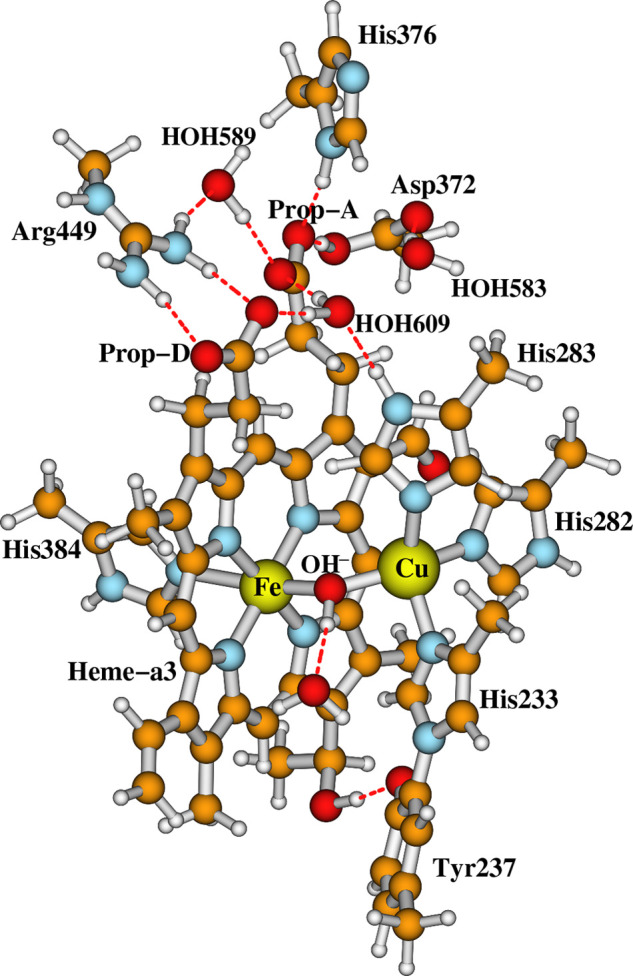
Our whole DNC model for state OH. Clearer views for both the top cluster and the central Fea33+–OH––CuB portion of the model are given in Figure 4I,II, respectively.
Figure 4.

A closer look at the top (I) and the central (II–VIII) portions of the DNC model clusters studied here (see Table 1). The rest of each model is similar to what was shown in Figure 3. Legend: (II) OH[Fea33+–OH––CuB] state; (III) Fea33+–H2O···OH––CuB model state; (IV) Fea33+–OH–···H2O–CuB model state; (V) Fea33+–H2O–CuB(a); (VI) Fea33+–H2O–CuB(b); (VII) Fea33+–H2O–CuB(c); (VIII) Fea33+–H2O–CuB(d). Note that structures V–VIII are four major optimized structures (see the Results and Discussion and Table 1) with very similar energies obtained by protonating the bridging OH– ligand in OH[Fea33+–OH––CuB] (model II).
When each model is constructed from the X-ray crystal structure 3S8G,37 the Cα atoms of Tyr237, His233, His282, His283, Asp372, His376, and His384 are each replaced with a link H atom along the original Cβ–Cα direction with a Cβ–Hlink distance of 1.09 Å. The Cβ of Arg449 and C228 of the geranyl side chain of the a3-heme are also replaced with a Hlink atom. During the geometry optimizations, except for the Hlink atom on the geranyl side chain of the a3-heme, the positions of all other Hlink atoms are fixed. Note that, in order to avoid the energy difference caused by the different H-bonding patterns, the two water molecules HOH604 and HOH608 above the O1–O2 species found in the 3S8G X-ray crystal structure are not included in our current models. We have geometry-optimized our DNC model clusters with the Fea33+ site in different spin states (low spin, intermediate spin, and high spin). Our calculations show that the intermediate-spin-Fea33+ state is generally lower in energy than the corresponding high-spin-Fea3 state for each model cluster studied here. However, the rR and EPR experimental data suggest that the Fea33+ is high spin in both the active OH and the resting O states.5,57 One needs more experimental data and higher-level theoretical calculations to determine whether different spin states of Fea3 can coexist in the OH and the resting O states. In fact, the existence of intermediate-/low-spin-Fea3 states in the resting O DNCs may be temperature-dependent, since Mössbauer spectroscopy experiments on Tt ba3(58) and c1aa3(59) have shown the coexistence of different high-spin and “low-spin” (which might also be intermediate-spin according to the isomer shift and quadrupole splitting values) Fea33+ species at very low temperature (4.2 K), and the “low-spin” Fea3 species changes to high spin as the temperature is increased above 190 K.58 These spin states may exhibit spin crossover, which depends on a sensitive balance between the respective energies and entropies. For simplicity, we will present our calculated results in which the high-spin (HS) Fea33+ site is antiferromagnetically (AF) coupled with the CuB site and put the results of the intermediate- and low-spin-Fea33+ states in the Supporting Information. Note that all conclusions we find for the HS-Fea3 states in the following section will remain the same if the Fea33+ is in the intermediate-spin state.
3. Results and Discussion
Our calculated energies and the main geometric and Mulliken net spin population properties of the geometry-optimized OH[Fea3HS,3+–OH–CuB] state and the possible O[Fea3HS,3+–H2O···OH––CuB] and O[Fea3HS,3+–OH–···H2O–CuB] states are given in the first three rows of Table 1. The central portions of the Fea3HS,3+–H2O···OH––CuB and Fea3HS,3+–OH–···H2O–CuB DNC clusters are given in Figure 4III,IV, respectively. The net spin populations from Mulliken population analysis are the main indication of the spin state for the Fea33+ site. In the ideal ionic limit, the net unpaired spin populations for HS-Fea3 and CuB2+ are 5 and 1, respectively. However, because of the metal–ligand covalency, the calculated net spin magnitudes for the Fea3 and CuB2+ sites are smaller than their corresponding ionic limits. The opposite signs for the spin densities of Fea3 and CuB2+ sites indicate the AF coupling. Our calculated net spins on Fea3 and CuB2+ show that we obtained the HS-Fea3 AF-coupled to the CuB2+ state for each cluster.
Table 1. OLYP-D3(BJ) Calculated Geometrical, Energetic, and Net Spin Properties of the Optimized DNC Clusters in Fea3HS,3+–OH––CuB, Fea3HS,3+–H2O···OH––CuB, Fea3HS,3+–OH–···H2O–CuB, and Fea3HS,3+–H2O–CuB Statesa.
| geometry
(Å) |
net spinf |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| structureb | Fe–N(H384) | Fe–O | Cu–O | O···O | Fe···Cu | Ec | Qd | pKa(H2O)e | Fea3 | O(Fe)/O(Cu) | CuB | Y237 |
| Fea3HS,3+–OH––CuB | 2.50 | 1.99 | 2.04 | 3.73 | 0.0 | 0 | 4.08 | 0.12 | –0.39 | –0.25 | ||
| Fea3HS,3+–H2O···OH––CuB | 2.18 | 2.17 | 1.95 | 2.50 | 4.78 | 6.3 | 0 | 4.08 | 0.08/–0.16 | –0.50 | 0.11 | |
| Fea3HS,3+–OH–···H2O–CuB | 2.28 | 1.98 | 2.13 | 2.45 | 4.64 | 6.6 | 0 | 4.12 | 0.20/–0.03 | –0.39 | –0.36 | |
| Fea3HS,3+–H2O–CuB(a) | 2.11 | 2.39 | 2.94 | 4.98 | –1.5 | 1 | 12.1 | 4.11 | 0.04 | –0.24 | –0.62 | |
| Fea3HS,3+–H2O–CuB(b) | 2.12 | 2.47 | 2.77 | 4.90 | –1.6 | 1 | 12.2 | 4.10 | 0.04 | –0.25 | –0.61 | |
| Fea3HS,3+–H2O–CuB(c) | 2.06 | 3.55 | 2.21 | 4.73 | –1.2 | 1 | 11.9 | 4.06 | –0.01 | –0.36 | –0.36 | |
| Fea3HS,3+–H2O–CuB(d) | 2.09 | 3.26 | 2.20 | 4.73 | –0.9 | 1 | 11.7 | 4.09 | –0.02 | –0.35 | –0.44 | |
| 3S8Gg | 2.22 | 2.39 | 2.25 | 1.52 | 4.92 | |||||||
Geometries were optimized in the broken-symmetry state with high-spin (HS) Fea33+ AF-coupled to the CuB site.
Calculated broken-symmetry state energies (offset by −26336.6 kcal mol–1).
The net charge of the model clusters.
The pKa(H2O) values were calculated for the process Fea3HS,3+–OH––CuB → Fea3HS,3+–H2O–CuB(a–d).
The Mulliken net spin populations on Fea33+, O of the bridging OH–/H2O, CuB, and the heavy atoms of the Tyr237–O– side chain (the sum total).
The X-ray crystal structure.37
OH is a metastable catalytically active state, which decays to the relaxed O state when no electron transfers to the DNC. Therefore, state O is lower in energy than OH. However, our calculations show that both the Fea3HS,3+–H2O···OH––CuB and Fea3HS,3+–OH–···H2O–CuB states are higher in energy (by >6 kcal mol–1) than the Fea3HS,3+–OH–CuB state. Further, the O···O distances (∼2.5 Å) in Fea3HS,3+–H2O···OH––CuB and Fea3HS,3+–OH–···H2O–CuB structures are ∼1 Å longer than the apparent O1–O2 distance found from the 3S8G X-ray crystal structure (see the last row of Table 1). Therefore, the DNC of the resting as-isolated O state is not likely in the Fea3HS,3+–H2O···OH––CuB or the Fea3HS,3+–OH–···H2O–CuB form.
Next, we examine whether a water molecule is preferred residing between the Fea33+ and CuB sites in the as-isolated O state. We then protonated the bridging OH– in the state OH[Fea33+–OH––CuB], kept the H-bonding H2O molecule, and optimized the structure. We find that, by modification of the position of the proton added to the bridging OH– or the orientation of the H-bonding H2O molecule, the geometry optimizations will end in different minima with different Fe–O, Cu–O, and Fe···Cu distances, but with very similar energies. The central Fea3HS,3+–H2O–CuB structures of four major optimized geometries (a–d) are shown in Figure 4V–VIII, and their main bond distances and energies are given in rows 4–7 of Table 1.
In the optimized structure Fea3HS,3+–H2O–CuB(a) (Figure 4.V), the H2O molecule binds with the Fea33+ site with Fe-O distance of 2.39 Å. In Fea3–H2O–CuB2+(b) (Figure 4VI), the H2O molecule is in between Fea3 and CuB2+ and does not bind with either site (with Fe–O and Cu–O distances of 2.47 and 2.77 Å, respectively). Although structurally different, both structures are essentially at the same energy. In structures Fea3–H2O–CuB2+(c) (Figure 4VII) and Fea3–H2O–CuB2+(d) (Figure 4VIII), the H2O molecule binds with the CuB site (with Cu–O distances of 2.21 and 2.20 Å, respectively) in different orientations. In Fea3–H2O–CuB2+(c), the H2O molecule tilts up and has an H-bonding interaction with one of the N atoms of the heme ring; in Fea3–H2O–CuB2+(d), however, the H2O simply moves closer to CuB starting from the bridging position. Fea3HS,3+–H2O–CuB(c) and Fea3HS,3+–H2O–CuB(d) also have nearly the same energy. In fact, all four Fea3HS,3+–H2O–CuB(a–d) structures are close in energy, with a maximum difference of 0.7 kcal mol–1. Since these four structures have one more proton than the OH[Fea3HS,3+–OH––CuB] state, in order to compare the energetic stabilities, we calculated the pKa of the H2O molecules in the four Fea3HS,3+–H2O–CuB(a–d) structures using the equation26,42,60,61
| 1 |
where E(A–) and E(AH) are the calculated total energies of the deprotonated (A–, here OH[Fea3HS,3+–OH––CuB]) and protonated (AH, here the four Fea3HS,3+–H2O–CuB(a–d)) states. In ADF, the “total energy” of the system is defined relative to a sum of atomic fragments (spherical spin-restricted atoms). The calculated gas-phase energy of a proton E(H+) is therefore relative to a spin-restricted hydrogen atom. ΔGsol(H+,1 atm) is the solvation free energy of a proton at 1 atm pressure. We use the “best available” experimental value of –264.0 kcal mol–1 for this term, on the basis of an analysis of cluster-ion solvation data.62−65 For E(H+), here we take the empirically corrected values 293.1 kcal mol–1 (i.e., 12.71 eV) for OLYP based on experimental standard hydrogen electrode energy and the proton solvation free energy (see Appendix in ref (60)). The translational entropy contribution to the gas-phase free energy of a proton is taken as –TΔSgas(H+) = –7.8 kcal mol–1 at 298 K and 1 atm pressure.66 (5/2)RT = 1.5 kcal mol–1 includes the proton translational energy (3/2)RT and PV = RT.66 The term ΔZPE is the zero-point energy difference for the deprotonated state (A–) minus the protonated state (AH), and it was estimated as ΔZPE = −7.7 kcal mol–1 for OH–/H2O by only optimizing the geometries (and then performing frequency calculations) of an OH– and an H2O molecule within the COSMO solvation model.
The calculated pKas of the H2O molecule in each of the Fea3HS,3+–H2O–CuB(a–d) states are also given in Table 1. They are between 11.7 and 12.2, indicating that the Fea3HS,3+–H2O–CuB(a–d) states are energetically much more stable than the OH[Fea3HS,3+–OH––CuB] state. We therefore speculate that it is a water molecule between the Fea33+ and CuB sites in the resting as-isolated O state. However, an extended electron density, which was interpreted as a dioxygen molecule, was observed between Fea33+ and CuB in the DNC of the X-ray crystal structures.35−37 Since our initial geometries of our DNC model structures are constructed on the basis of the Cartesian coordinates of the X-ray crystal structure 3S8G,37 to compare our optimized structures Fea3HS,3+–H2O–CuB(a–d) with 3S8G, we superimpose the Cartesian coordinates of the central portions of the five structures together. The overlapped structures are shown in Figure 5 with two orientations: A and B. The colors of the atoms in the different structures are as follows: 3S8G, silver; Fea3HS,3+–H2O–CuB(a), orange; Fea3HS,3+–H2O–CuB(b), purple; Fea3HS,3+–H2O–CuB(c), blue; Fea3HS,3+–H2O–CuB(d), green.
Figure 5.
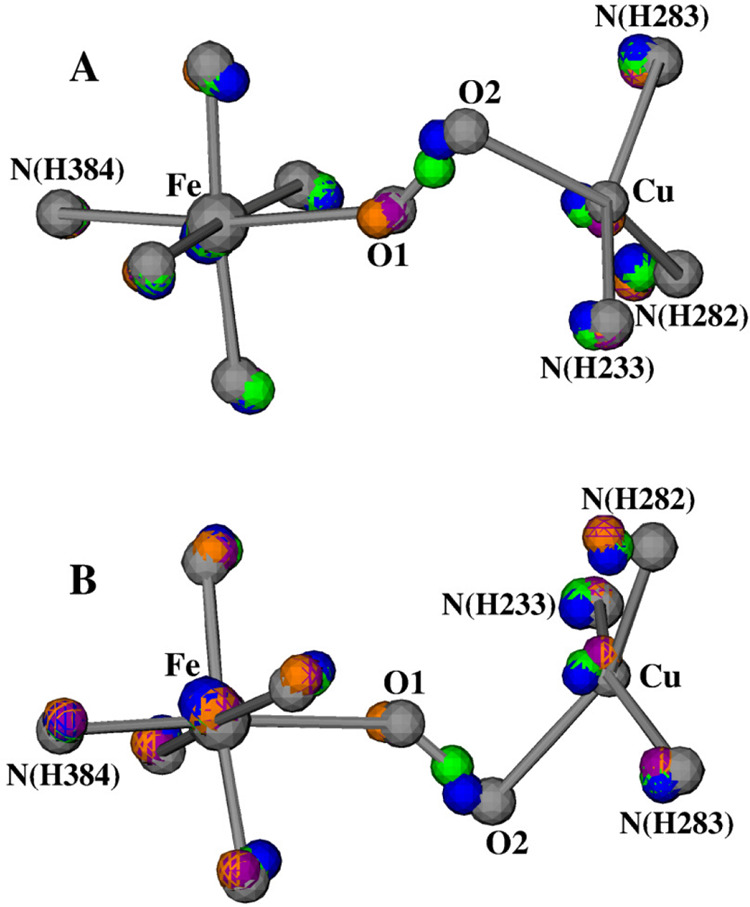
Overlap of the central portions of the following DNC structures (see Table 1): silver, 3S8G X-ray crystal structure; orange, Fea3HS,3+–H2O–CuB(a); purple, Fea3HS,3+–H2O–CuB(b); blue, Fea3HS,3+–H2O–CuB(c); and green, Fea3HS,3+–H2O–CuB(d). (A) and (B) are the views of this overlap from two different angles. For clarity, the H-bonding water molecules and the hydrogen atoms are not shown.
During our geometry optimizations, the CuB site and its ligands move slightly more than the heme-Fea3 site. Now focusing on the positions of the central oxygen atoms, we do see that the four O atoms in the optimized structures align with the 3S8G O1–O2 directions very well. The orange and purple O atoms are close to the O1 position in 3S8G, the blue O atom is near O2, and the green O atom is in about the middle between O1 and O2. This supports our proposal that the electron density around O1–O2 in the X-ray crystal structures represents the overlap of the electron density of one water molecule that is in different positions along O1–O2 in different CcO molecules in a crystal.
For clarity, the water molecule that has an H-bonding interaction with the ligand H2O in each of the four geometry-optimized structures is not shown in Figure 5. In 3S8G, a water molecule (HOH608) was seen 3.01 Å above the position of O2. Therefore, HOH608 may have an H-bonding interaction with the H2O ligand in a position as in Fea33+–H2O–CuB(c) (Figure 4VII). No other H2O molecules were identified within the H-bonding distances around O1–O2 in 3S8G. This means that not all CcO molecules in the crystal have an H2O molecule H-bonding to the H2O ligand, and even if there is an H-bonding H2O molecule in some of the CcO DNCs, the H-bonding patterns and the positions of the H-bonding H2O molecules may differ; therefore, they may not be identified in the X-ray crystal structure.
Since it is not clear whether there is an H-bonding H2O molecule in the DNC of the O state, we removed the H-bonding H2O from the Fea3HS,3+–H2O–CuB(a–d) structures and optimized the geometries again. The central portions of the corresponding optimized structures (S) are given as S1–S4 in Figure 6, respectively. Their main bond distances and the calculated energies are given in Table 2.
Figure 6.

Structures (S) S1–S4 (see Table 2): the central portions of the optimized geometries after removing the H-bonding H2O molecule from the Fea3HS,3+–H2O–CuB(a–d) structures (see Figure 4V–VIII). S5 is a constrained geometry optimized structure with a fixed Fe–O distance at 2.39 Å. (A) and (B) show the overlap of S1–S5 with the X-ray crystal structure 3S8G in two different viewing angles (see also Figure 7).
Table 2. Geometry Optimized Fea3HS,3+–H2O–CuB DNC Structures and Energies, in Which No Water Molecule H-Bonding to the H2O Liganda.
| distance (Å) |
||||
|---|---|---|---|---|
| structureb | Fe–O | Cu–O | Fe···Cu | Ec (kcal/mol) |
| S1 | 2.61 | 2.82 | 5.03 | 1.4 |
| S2 | 2.97 | 2.52 | 4.98 | 0.3 |
| S3 | 3.54 | 2.29 | 4.76 | 0.0 |
| S4 | 3.07 | 2.41 | 4.87 | 0.6 |
| S5d | 2.39 | 2.81 | 4.91 | 0.8 |
Geometries (S1–S5) were optimized in the broken-symmetry state with high-spin Fea33+ AF-coupled to the CuB site.
Calculated broken-symmetry state energies (offset by –26004.7 kcal mol–1). Note that Fea33+ and CuB are very weakly coupled. The F-coupled calculations on these structures yield essentially the same energy as the corresponding broken-symmetry state.
The Fe–O distance was fixed at 2.39 Å during this geometry optimization.
Without the H-bonding H2O molecule, the binding of the H2O ligand with the two metal sites, especially with the Fea33+ site, are weakened. For instance, from Fea3–H2O–CuB2+(a) to S1, the H2O dissociates from the Fea3 site (from an Fe–O distance of 2.39 Å) and moves to about the middle between Fea33+ and CuB (Fe–O, 2.61 Å; Cu–O, 2.82 Å). S1 has the longest calculated Fea33+ and CuB distance of 5.03 Å. S3 differs from the Fea3HS,3+–H2O–CuB(c) structure with slightly elongated Cu–O and Fe–Cu distances. S2 and S4 look very similar and have almost the same energy, but with different Fe–O, Cu–O, and Fe–Cu distances. The calculated energies among S2–S4 are within 1 kcal mol–1. Although S1 has the highest calculated energy, it is only 1.4 kcal mol–1 higher than S3. Further, to see how high the energy would be if the H2O molecule is close to the Fea33+ site as in the structure Fea3–H2O–CuB2+(a), we reoptimized the geometry of S1 with a fixed Fe–O distance at 2.39 Å. The constraint optimized structure is also shown in Figure 6 as S5, and its geometric and energetic properties are also given in Table 2. It turned out that the calculated energy of S5 is very close to those of S2 and S4 and is only 0.8 kcal mol–1 higher than that of S3. Because of the very similar energies, in principle, the structures S1–S5 may coexist in the DNCs of different CcO molecules.
Similar to the case for Figure 5, in Figure 6A,B, we have also shown the overlap of the DNCs of S1–S5 and the X-ray crystal structure 3S8G in two different viewing angles. The colors of the atoms in different structures are as follows: 3S8G, silver; S1, orange; S2, purple; S3, blue; S4, green; S5, red. In the overlap structure, the H2O molecules in S1 and S5 are in the vicinity of the O1 position in 3S8G, the H2O in S3 is close to the position of O2, and the H2O molecules in S2 and S4 are located between O1 and O2. The same overlap structure is also shown in Figure 7, where the electron density map is reconstructed from the 3S8G data file. Again, this supports the proposal that in the resting O state the H2O molecule may reside at different positions along the O1–O2 direction between the Fea33+ and CuB sites in different enzyme molecules.
Figure 7.
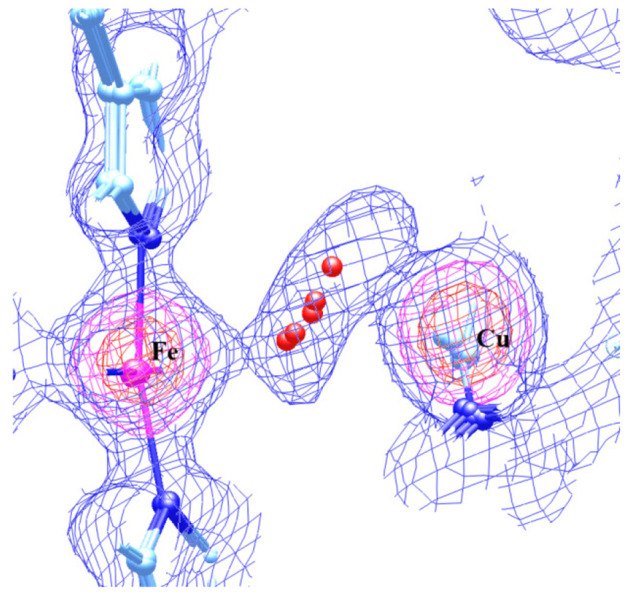
Overlap of the structures S1–S5 (also see Table 2 and Figure 6) with the electron density map that was reconstructed from the X-ray crystal structure 3S8G data file.
Further, although the structures and energies for Fea3HS,3+–H2O–CuB(a–d) and S1–S5 given in Tables 1 and 2 are obtained from broken-symmetry state calculations in which the HS-Fea33+ is AF-coupled to CuB, the coupling between the HS-Fea33+ and CuB sites appears to be very weak. Our Fea3HS,3+-CuB ferromagnetically coupled (F-coupled) calculations show that each of these structures also represents an optimization-converged geometry in the F-coupled state with essentially the same energy as the corresponding broken-symmetry state. Therefore, both Fea3HS,3+-CuB AF-coupled and F-coupled configurations of the DNCs may coexist in the resting O state.
4. Conclusions
Starting from the OH[Fea33+–OH––CuB] DNC structure of the active oxidized state CcO, where a hydroxo bridges the Fea33+ and the CuB sites, we have studied the feasible DNC structures of the resting as-isolated oxidized O state. Our calculations show that the O state is not likely in the Fea33+–H2O···OH––CuB or the Fea33+–OH–···H2O–CuB forms, where an H2O/OH– binds with Fea33+/CuB and the H2O and OH– ligands H-bond with each other, since both the Fea33+–H2O···OH––CuB and the Fea33+–OH–···H2O–CuB structures are higher in energy than the OH[Fea33+–OH––CuB] state. Also, the structures of these metal bridging OH–···H2O systems show O···O distances in poor agreement with the X-ray structures of ba3 and aa3 CcOs. Further, our pKa calculations show that the bridging OH– ligand in the OH[Fea33+–OH––CuB] state energetically prefers to be protonated at neutral pH. We therefore propose that a water molecule is between the Fea33+ and CuB sites in the resting O state of CcO. Our calculations further suggest that the H2O molecule can bind with either the Fea33+ or the CuB site or it can stay at various positions between the Fea33+ and CuB sites with very similar energies, depending on the Fea33+–CuB distance and whether or not this H2O molecule has an H-bonding interaction with another H2O molecule.
The X-ray crystal structures of the oxidized CcOs from Pd (3HB3),35 from bovine heart (2ZXW),36 and from Tt (3S8G and 3S8F)37 all show that there is strong electron density for a dioxygen type species (O1–O2) bridging the Fea33+ and CuB2+ in the O state. In a crystal structure with ∼1.9 Å resolution, it is actually not possible to tell if the ∼1.5 Å apart O–O species is due to overlapping water molecules or a peroxide, as both models would produce very similar overlapping densities in comparison to the experimental electron density map. Since our initial DNC model structures were constructed on the basis of the Cartesian coordinates of the X-ray crystal structure 3S8G, we then superimposed the 3S8G DNC with several of our geometry-optimized O[Fea33+–H2O–CuB] structures that have different H2O positions between Fea33+ and CuB and have very similar calculated energies. The overlap structures show that the H2O molecules lie between and along the O1–O2 direction in 3S8G. There are billions of CcO molecules in the crystal, and the water molecule may occupy one equilibrium position at a given time in a given DNC but different ones at other times or in other DNCs. The positions of the different water molecules may be “locked” after the crystal is rapidly frozen (normally at 100 K). However, even at 100 K, the atoms and the water molecules vibrate around their equilibrium positions. The atomic positions obtained from X-ray diffraction analysis are the averages of billions of unit cells over both time and space. Both dynamic and static disorder comparing DNCs in different CcO molecules will contribute to the X-ray structure. We therefore propose that the extended electron density between Fea33+ and CuB observed in the X-ray crystal structures is the overlap of the electron density of a water molecule located at different positions in different CcO molecules in the crystals.
The change in protonation state and structure from state OH to O leads to large differences in the electron and proton transfer kinetics for the two different subsequent reaction pathways, OH → EH → R in comparison to OH → O → E → R, with the sequence of 1e– and 1H+ transfers to the reactive oxidized Fea33+–OH––CuB complex switched. The new structural analysis of state O in comparison to OH provides a foundation for further exploration of these differences in kinetics.
Acknowledgments
We thank Tzanko Doukov and Aina Cohen for very helpful discussions. We thank the NIH for financial support (R01 GM100934) and thank The Scripps Research Institute for computational resources. This work also used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (grant number ACI-1053575, resources at the San Diego Supercomputer Center through award TG-CHE130010 to A.W.G.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c00724.
The authors declare no competing financial interest.
Supplementary Material
References
- Wikström M. Active Site Intermediates in the Reduction of O2 by Cytochrome Oxidase, and Their Derivatives. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 468–475. 10.1016/j.bbabio.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Kaila V. R. I.; Verkhovsky M. I.; Wikström M. Proton-Coupled Electron Transfer in Cytochrome Oxidase. Chem. Rev. 2010, 110, 7062–7081. 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- Konstantinov A. A. Cytochrome c Oxidase: Intermediates of the Catalytic Cycle and Their Energy-Coupled Interconversion. FEBS Lett. 2012, 586, 630–639. 10.1016/j.febslet.2011.08.037. [DOI] [PubMed] [Google Scholar]
- von Ballmoos C.; Adelroth P.; Gennis R. B.; Brzezinski P. Proton Transfer in ba3 Cytochrome c Oxidase from Thermus thermophilus. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 650–657. 10.1016/j.bbabio.2011.11.015. [DOI] [PubMed] [Google Scholar]
- Wikström M.; Krab K.; Sharma V. Oxygen Activation and Energy Conservation by Cytochrome c Oxidase. Chem. Rev. 2018, 118, 2469–2490. 10.1021/acs.chemrev.7b00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farver O.; Chen Y.; Fee J. A.; Pecht I. Electron Transfer among the CuA, Heme-b and a3 Centers of Thermus thermophilus Cytochrome ba3. FEBS Lett. 2006, 580, 3417–3421. 10.1016/j.febslet.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Fee J. A.; Case D. A.; Noodleman L. Toward a Chemical Mechanism of Proton Pumping by the B-Type Cytochrome c Oxidases: Application of Density Functional Theory to Cytochrome ba3 of Thermus thermophilus. J. Am. Chem. Soc. 2008, 130, 15002–15021. 10.1021/ja803112w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siletsky S. A.; Belevich I.; Jasaitis A.; Konstantinov A. A.; Wikström M.; Soulimane T.; Verkhovsky M. I. Time-Resolved Single-Turnover of ba3 Oxidase from Thermus thermophilus. Biochim. Biophys. Acta, Bioenerg. 2007, 1767, 1383–1392. 10.1016/j.bbabio.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Verkhovsky M. I.; Jasaitis A.; Verkhovskaya M. L.; Morgan J. E.; Wikstrom M. Proton Translocation by Cytochrome c Oxidase. Nature 1999, 400, 480–483. 10.1038/22813. [DOI] [PubMed] [Google Scholar]
- Han S.; Ching Y. C.; Rousseau D. L. Primary Intermediate in the Reaction of Oxygen with Fully Reduced Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 2491–2495. 10.1073/pnas.87.7.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S. H.; Ching Y. C.; Rousseau D. L. Primary Intermediate in the Reaction of Mixed-Valence Cytochrome c Oxidase with Oxygen. Biochemistry 1990, 29, 1380–1384. 10.1021/bi00458a006. [DOI] [PubMed] [Google Scholar]
- Ogura T.; Takahashi S.; Shinzawa-Itoh K.; Yoshikawa S.; Kitagawa T. Observation of the FeII-O2 Stretching Raman Band for Cytochrome Oxidase Compound A at Ambient Temperature. J. Am. Chem. Soc. 1990, 112, 5630–5631. 10.1021/ja00170a032. [DOI] [PubMed] [Google Scholar]
- Ogura T.; Takahashi S.; Hirota S.; Shinzawa-Itoh K.; Yoshikawa S.; Appelman E. H.; Kitagawa T. Time-Resolved Resonance Raman Elucidation of the Pathway for Dioxygen Reduction by Cytochrome c Oxidase. J. Am. Chem. Soc. 1993, 115, 8527–8536. 10.1021/ja00072a002. [DOI] [Google Scholar]
- Varotsis C.; Woodruff W. H.; Babcock G. T. Time-Resolved Raman Detection of ν(Fe-O) in an Early Intermediate in the Reduction of O2 by Cytochrome-Oxidase. J. Am. Chem. Soc. 1989, 111, 6439–6440. 10.1021/ja00198a075. [DOI] [Google Scholar]
- Han Du W.-G.; Gotz A. W.; Noodleman L. DFT Fea3-O/O-O Vibrational Frequency Calculations over Catalytic Reaction Cycle States in the Dinuclear Center of Cytochrome c Oxidase. Inorg. Chem. 2019, 58, 13933–13944. 10.1021/acs.inorgchem.9b01840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigami I.; Hikita M.; Egawa T.; Yeh S. R.; Rousseau D. L. Proton Translocation in Cytochrome c Oxidase: Insights from Proton Exchange Kinetics and Vibrational Spectroscopy. Biochim. Biophys. Acta, Bioenerg. 2015, 1847, 98–108. 10.1016/j.bbabio.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa S.; Shimada A. Reaction Mechanism of Cytochrome c Oxidase. Chem. Rev. 2015, 115, 1936–1989. 10.1021/cr500266a. [DOI] [PubMed] [Google Scholar]
- Van Wart H. E.; Zimmer J. Resonance Raman Evidence for the Activation of Dioxygen in Horseradish Oxyperoxidase. J. Biol. Chem. 1985, 260, 8372–8377. [PubMed] [Google Scholar]
- Hirota S.; Li T. S.; Phillips G. N.; Olson J. S.; Mukai M.; Kitagawa T. Perturbation of the Fe-O2 Bond by Nearby Residues in Heme Pocket: Observation of νFe-O2 Raman Bands for Oxymyoglobin Mutants. J. Am. Chem. Soc. 1996, 118, 7845–7846. 10.1021/ja9608297. [DOI] [Google Scholar]
- Tsubaki M.; Nagai K.; Kitagawa T. Resonance Raman Spectra of Myoglobins Reconstituted with Spirographis and Isospirographis Hemes and Iron 2,4-Diformylprotoporphyrin IX. Effect of Formyl Substitution at the Heme Periphery. Biochemistry 1980, 19, 379–385. 10.1021/bi00543a020. [DOI] [PubMed] [Google Scholar]
- Wikström M. Energy-Dependent Reversal of the Cytochrome-Oxidase Reaction. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 4051–4054. 10.1073/pnas.78.7.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng L. C.; Baker G. M. Reaction of Hydrogen-Peroxide with the Rapid Form of Resting Cytochrome-Oxidase. Biochemistry 1991, 30, 5727–5733. 10.1021/bi00237a014. [DOI] [PubMed] [Google Scholar]
- Morgan J. E.; Verkhovsky M. I.; Wikstrom M. Observation and Assignment of Peroxy and Ferryl Intermediates in the Reduction of Dioxygen to Water by Cytochrome c Oxidase. Biochemistry 1996, 35, 12235–12240. 10.1021/bi961634e. [DOI] [PubMed] [Google Scholar]
- Proshlyakov D. A.; Pressler M. A.; Babcock G. T. Dioxygen Activation and Bond Cleavage by Mixed-Valence Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 8020–8025. 10.1073/pnas.95.14.8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian M.; Wong W. W.; Gennis R. B.; Palmer G. Mass Spectrometric Determination of Dioxygen Bond Splitting in the ″Peroxy″ Intermediate of Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 13114–13117. 10.1073/pnas.96.23.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Du W.-G.; Götz A. W.; Noodleman L. A Water Dimer Shift Activates a Proton Pumping Pathway in the PR → F Transition of ba3 Cytochrome c Oxidase. Inorg. Chem. 2018, 57, 1048–1059. 10.1021/acs.inorgchem.7b02461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siletsky S. A.; Belevich I.; Belevich N. P.; Soulimane T.; Wikstrom M. Time-Resolved Generation of Membrane Potential by ba3 Cytochrome c Oxidase from Thermus thermophilus Coupled to Single Electron Injection Into the O and OH States. Biochim. Biophys. Acta, Bioenerg. 2017, 1858, 915–926. 10.1016/j.bbabio.2017.08.007. [DOI] [PubMed] [Google Scholar]
- Szundi I.; Funatogawa C.; Fee J. A.; Soulimane T.; Einarsdottir O. CO Impedes Superfast O2 Binding in ba3 Cytochrome Oxidase from Thermus thermophilus. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 21010–21015. 10.1073/pnas.1008603107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch D.; Belevich I.; Jasaitis A.; Ribacka C.; Puustinen A.; Verkhovsky M. I.; Wikström M. The Catalytic Cycle of Cytochrome c Oxidase Is Not the Sum of Its Two Halves. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 529–533. 10.1073/pnas.0306036101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevich I.; Bloch D. A.; Belevich N.; Wikström M.; Verkhovsky M. I. Exploring the Proton Pump Mechanism of Cytochrome c Oxidase In Real Time. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 2685–2690. 10.1073/pnas.0608794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siletsky S. A.; Belevich I.; Wikström M.; Soulimane T.; Verkhovsky M. I. Time-Resolved OH→EH Transition of the Aberrant ba3 Oxidase from Thermus thermophilus. Biochim. Biophys. Acta, Bioenerg. 2009, 1787, 201–205. 10.1016/j.bbabio.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Moody A. J. ’As Prepared’ Forms of Fully Oxidised Haem/Cu Terminal Oxidases. Biochim. Biophys. Acta, Bioenerg. 1996, 1276, 6–20. 10.1016/0005-2728(96)00035-7. [DOI] [PubMed] [Google Scholar]
- Ostermeier C.; Harrenga A.; Ermler U.; Michel H. Structure at 2.7 Å Resolution of the Paracoccus denitrificans Two-Subunit Cytochrome c Oxidase Complexed with an Antibody FV Fragment. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 10547–10553. 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L.; Hiser C.; Mulichak A.; Garavito R. M.; Ferguson-Miller S. Identification of Conserved Lipid/Detergent-Binding Sites in A High-Resolution Structure of the Membrane Protein Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 16117–16122. 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepke J.; Olkhova E.; Angerer H.; Muller H.; Peng G. H.; Michel H. High Resolution Crystal Structure of Paracoccus denitrificans Cytochrome c Oxidase: New Insights into the Active Site and the Proton Transfer Pathways. Biochim. Biophys. Acta, Bioenerg. 2009, 1787, 635–645. 10.1016/j.bbabio.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Aoyama H.; Muramoto K.; Shinzawa-Itoh K.; Hirata K.; Yamashita E.; Tsukihara T.; Ogura T.; Yoshikawa S. A Peroxide Bridge Between Fe and Cu Ions in the O2 Reduction Site of Fully Oxidized Cytochrome c Oxidase Could Suppress the Proton Pump. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2165–2169. 10.1073/pnas.0806391106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiefenbrunn T.; Liu W.; Chen Y.; Katritch V.; Stout C. D.; Fee J. A.; Cherezov V. High Resolution Structure of the ba3 Cytochrome c Oxidase from Thermus thermophilus in a Lipidic Environment. PLoS One 2011, 6, e22348. 10.1371/journal.pone.0022348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata K.; Shinzawa-Itoh K.; Yano N.; Takemura S.; Kato K.; Hatanaka M.; Muramoto K.; Kawahara T.; Tsukihara T.; Yamashita E.; Tono K.; Ueno G.; Hikima T.; Murakami H.; Inubushi Y.; Yabashi M.; Ishikawa T.; Yamamoto M.; Ogura T.; Sugimoto H.; Shen J. R.; Yoshikawa S.; Ago H. Determination of Damage-Free Crystal Structure of An X-Ray-Sensitive Protein Using An XFEL. Nat. Methods 2014, 11, 734–U174. 10.1038/nmeth.2962. [DOI] [PubMed] [Google Scholar]
- Andersson R.; Safari C.; Dods R.; Nango E.; Tanaka R.; Yamashita A.; Nakane T.; Tono K.; Joti Y.; Bath P.; Dunevall E.; Bosman R.; Nureki O.; Iwata S.; Neutze R.; Branden G. Serial Femtosecond Crystallography Structure of Cytochrome c Oxidase at Room Temperature. Sci. Rep. 2017, 7, 4518. 10.1038/s41598-017-04817-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno G.; Shimada A.; Yamashita E.; Hasegawa K.; Kumasaka T.; Shinzawa-Itoh K.; Yoshikawa S.; Tsukihara T.; Yamamoto M. Low-Dose X-Ray Structure Analysis of Cytochrome c Oxidase Utilizing High-Energy X-Rays. J. Synchrotron Radiat. 2019, 26, 912–921. 10.1107/S1600577519006805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila V. R. I.; Oksanen E.; Goldman A.; Bloch D. A.; Verkhovsky M. I.; Sundholm D.; Wikström M. A Combined Quantum Chemical and Crystallographic Study on the Oxidized Binuclear Center of Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg. 2011, 1807, 769–778. 10.1016/j.bbabio.2010.12.016. [DOI] [PubMed] [Google Scholar]
- Han Du W.-G.; Noodleman L. Density Functional Study for the Bridged Dinuclear Center Based on a High-Resolution X-Ray Crystal Structure of ba3 Cytochrome c Oxidase from Thermus thermophilus. Inorg. Chem. 2013, 52, 14072–14088. 10.1021/ic401858s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siletskiy S.; Soulimane T.; Azarkina N.; Vygodina T. V.; Buse G.; Kaulen A.; Konstantinov A. Time-Resolved Generation of A Membrane Potential by ba3 Cytochrome c Oxidase from Thermus thermophilus. Evidence for Reduction-Induced Opening of the Binuclear Center. FEBS Lett. 1999, 457, 98–102. 10.1016/S0014-5793(99)01019-4. [DOI] [PubMed] [Google Scholar]
- Siletsky S. A. Steps of the Coupled Charge Translocation in the Catalytic Cycle of Cytochrome c Oxidase. Front. Biosci., Landmark Ed. 2013, 18, 36–57. 10.2741/4086. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- ADF, Amsterdam Density Functional Software; SCM, Theoretical Chemistry, Vrije Universiteit: Amsterdam, The Netherlands; http://www.scm.com.
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Guerra C. F.; Van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- Guerra C. F.; Visser O.; Snijders J. G.; te Velde G.; Baerends E. J., Parallelisation of the Amsterdam Density Functional Program. In Methods and techniques for computational chemistry; Clementi E., Corongiu C., Eds.; STEF: Cagliari, 1995; pp 303–395. [Google Scholar]
- Goerigk L.; Hansen A.; Bauer C.; Ehrlich S.; Najibi A.; Grimme S. A Look at the Density Functional Theory Zoo with the Advanced GMTKN55 Database for General Main Group Thermochemistry, Kinetics and Noncovalent Interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. 10.1039/C7CP04913G. [DOI] [PubMed] [Google Scholar]
- Noodleman L. Valence Bond Description of Anti-Ferromagnetic Coupling in Transition-Metal Dimers. J. Chem. Phys. 1981, 74, 5737–5743. 10.1063/1.440939. [DOI] [Google Scholar]
- Noodleman L.; Case D. A. Density-Functional Theory of Spin Polarization and Spin Coupling in Iron-Sulfur Clusters. Adv. Adv. Inorg. Chem. 1992, 38, 423–470. 10.1016/S0898-8838(08)60070-7. [DOI] [Google Scholar]
- Noodleman L.; Lovell T.; Han W.-G.; Liu T.; Torres R. A.; Himo F.. Density Functional Theory. In Comprehensive Coordination Chemistry II, From Biology to Nanotechnology; Lever A. B., Ed.; Elsevier: 2003; Vol. 2, pp 491–510. [Google Scholar]
- Klamt A.; Schüürmann G. COSMO - A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc., Perkin Trans. 2 1993, 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Klamt A. Conductor-Like Screening Model for Real Solvents - A New Approach to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 1995, 99, 2224–2235. 10.1021/j100007a062. [DOI] [Google Scholar]
- Klamt A.; Jonas V. Treatment of the Outlying Charge in Continuum Solvation Models. J. Chem. Phys. 1996, 105, 9972–9981. 10.1063/1.472829. [DOI] [Google Scholar]
- Pye C. C.; Ziegler T. An Implementation of the Conductor-Like Screening Model of Solvation within the Amsterdam Density Functional Package. Theor. Chem. Acc. 1999, 101, 396–408. 10.1007/s002140050457. [DOI] [Google Scholar]
- Han S.; Takahashi S.; Rousseau D. L. Time Dependence of the Catalytic Intermediates in Cytochrome c Oxidase. J. Biol. Chem. 2000, 275, 1910–1919. 10.1074/jbc.275.3.1910. [DOI] [PubMed] [Google Scholar]
- Zimmermann B. H.; Nitsche C. I.; Fee J. A.; Rusnak F.; Munck E. Properties of A Copper-Containing Cytochrome ba3: A Second Terminal Oxidase from the Extreme Thermophile Thermus thermophilus. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 5779–5783. 10.1073/pnas.85.16.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusnak F. M.; Munck E.; Nitsche C. I.; Zimmermann B. H.; Fee J. A. Evidence for Structural Heterogeneities and A Study of Exchange Coupling. Mössbauer Studies of Cytochrome c1aa3 from Thermus thermophilus. J. Biol. Chem. 1987, 262, 16328–16332. [PubMed] [Google Scholar]
- Noodleman L.; Han Du W.-G.; Fee J. A.; Götz A. W.; Walker R. C. Linking Chemical Electron-Proton Transfer to Proton Pumping in Cytochrome c Oxidase: Broken-Symmetry DFT Exploration of Intermediates along the Catalytic Reaction Pathway of the Iron-Copper Dinuclear Complex. Inorg. Chem. 2014, 53, 6458–6472. 10.1021/ic500363h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Du W.-G.; Noodleman L. Broken Symmetry DFT Calculations/Analysis for Oxidized and Reduced Dinuclear Center in Cytochrome c Oxidase: Relating Structures, Protonation States, Energies, and Mössbauer Properties in ba3Thermus thermophilus. Inorg. Chem. 2015, 54, 7272–7290. 10.1021/acs.inorgchem.5b00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissandier M. D.; Cowen K. A.; Feng W. Y.; Gundlach E.; Cohen M. H.; Earhart A. D.; Coe J. V.; Tuttle T. R. The proton’s absolute aqueous enthalpy and Gibbs free energy of solvation from cluster-ion solvation data. J. Phys. Chem. A 1998, 102, 7787–7794. 10.1021/jp982638r. [DOI] [Google Scholar]
- Truhlar D. G.; Cramer C. J.; Lewis A.; Bumpus J. A. Molecular modeling of environmentally important processes: Reduction potentials. J. Chem. Educ. 2004, 81, 596–604. 10.1021/ed081p596. [DOI] [Google Scholar]
- Truhlar D. G.; Cramer C. J.; Lewis A.; Bumpus J. A. Molecular modeling of environmentally important processes: Reduction potentials (vol 81, pg 596, 2004). J. Chem. Educ. 2007, 84, 934. 10.1021/ed084p934.1. [DOI] [Google Scholar]
- Marenich A. V.; Ho J. M.; Coote M. L.; Cramer C. J.; Truhlar D. G. Computational electrochemistry: prediction of liquid-phase reduction potentials. Phys. Chem. Chem. Phys. 2014, 16, 15068–15106. 10.1039/C4CP01572J. [DOI] [PubMed] [Google Scholar]
- Tawa G. J.; Topol I. A.; Burt S. K.; Caldwell R. A.; Rashin A. A. Calculation of the aqueous solvation free energy of the proton. J. Chem. Phys. 1998, 109, 4852–4863. 10.1063/1.477096. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.