

Summary
Immune checkpoint blockade (ICB) results in durable disease control in a subset of patients with advanced renal cell carcinoma (RCC), but mechanisms driving resistance are poorly understood. We characterize the single-cell transcriptomes of cancer and immune cells from metastatic RCC patients before or after ICB exposure. In responders, subsets of cytotoxic T cells express higher levels of co-inhibitory receptors and effector molecules. Macrophages from treated biopsies shift toward pro-inflammatory states in response to an interferon-rich microenvironment but also upregulate immunosuppressive markers. In cancer cells, we identify bifurcation into two subpopulations differing in angiogenic signaling and upregulation of immunosuppressive programs after ICB. Expression signatures for cancer cell subpopulations and immune evasion are associated with PBRM1 mutation and survival in primary and ICB-treated advanced RCC. Our findings demonstrate that ICB remodels the RCC microenvironment and modifies the interplay between cancer and immune cell populations critical for understanding response and resistance to ICB.
Keywords: cancer, single cell, immunotherapy, resistance, kidney
Graphical abstract
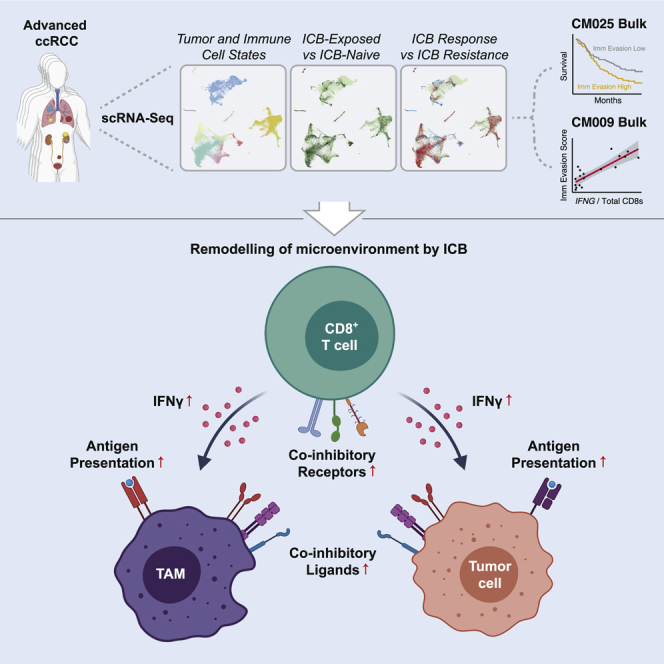
Highlights
-
•
Distinct CD8+ T cell phenotypes are enriched after immune checkpoint blockade (ICB)
-
•
Immune checkpoint ligands are upregulated in macrophages and tumor cells after ICB
-
•
Two cancer cell subpopulations are conserved across heterogeneous RCC tumors
-
•
Cancer cell programs drive distinct immune interactions and predict patient outcomes
Bi et al. dissect the cancer cell and immune microenvironmental transcriptional programs in immune checkpoint blockade-exposed metastatic renal cell carcinoma, revealing immune subpopulation reprogramming and interactions with distinct cancer cell populations in the context of clinical resistance.
Introduction
The tumor microenvironment plays critical roles in renal cell carcinoma (RCC) carcinogenesis, pathophysiology, and therapy. The most common RCC histologic subtype, clear cell RCC (ccRCC), frequently harbors characteristic second-hit loss-of-function mutations in VHL on a background of loss of chromosome 3p, where VHL resides (Creighton et al., 2013). These events result in decreased degradation of hypoxia-inducible factors (HIFs), causing mutant cells to shift to using glycolysis and secreting VEGF, which promotes angiogenesis (Hsieh et al., 2017). Moreover, ccRCC tumors are highly infiltrated by immune cells, particularly by T cells (Şenbabaoğlu et al., 2016). Leveraging these features of the microenvironment, treatment has evolved significantly over the past decade, with now widespread use of VEGF tyrosine kinase inhibitors (TKIs), immune checkpoint blockade (ICB), and combinations of these approaches (Choueiri and Motzer, 2017; Motzer et al., 2014, 2019; Rini et al., 2019).
ICB can provide durable benefit in responders, but resistance occurs in most patients, and the causal cancer and immune cellular processes are poorly understood. Genomic characterization of ICB response has focused on bulk-sequenced pre-treatment samples, where lower baseline myeloid inflammation and PBRM1 loss of function are associated with response in certain patient populations (Braun et al., 2019, 2020; McDermott et al., 2018; Miao et al., 2018). Observations of both intra- and intertumoral heterogeneity complicate mechanistic understanding, with multiple molecular subtypes of ccRCC defined by mutations in a set of chromatin remodelers that exhibit distinct patterns of evolution and metastasis (Creighton et al., 2013; Gerlinger et al., 2012; Turajlic et al., 2018).
Bulk sequencing is poorly suited to investigating cell-type-specific patterns of response to therapy. A lack of representative pre-clinical immune-competent RCC models poses further challenges to investigating interactions and signaling between cell types (Hsieh et al., 2017). To address these challenges, recent studies utilize single-cell analyses of patient samples to pinpoint cancer cell of origin and describe immune cells in untreated RCC (Chevrier et al., 2017; Young et al., 2018). We therefore hypothesized that single-cell transcriptomic dissection of biopsies from before or after ICB exposure would reveal immune and cancer cell programs that determine treatment response and nominate targets to combat resistance.
Results
Characterizing the tumor microenvironment of advanced RCC during therapy
We collected fresh biopsies or surgical resections from eight patients, seven with metastatic RCC and one with localized disease (Table S1). Dissociated cells were processed for single-cell RNA sequencing (scRNA-seq) using the 10× platform, and additional tissue was used for bulk whole-exome sequencing where available (Figure 1A). At time of biopsy, five patients had been exposed to ICB (including four to both TKIs and ICB), and three patients had not been exposed to systemic therapy (Figure 1B). Specimens were derived from the kidney in cases of nephrectomy and from lymph node and visceral metastases otherwise. Seven patients had ccRCC, while one tumor had a type 2 papillary RCC histology with PBRM1 and SETD2 mutations. As expected, chromosome 3p loss was universally detected in ccRCC patients for whom genomic characterization was available, with additional second-hit mutations in VHL and/or genes encoding chromatin remodelers. Broadly, our cohort recapitulated typical characteristics of advanced ccRCC in both genomic and clinical features (Creighton et al., 2013).
Figure 1.

Characterizing the tumor microenvironment of advanced RCC during therapy
(A) Study overview.
(B) Summary of treatment histories at time of biopsy, clinicopathological features, and genomic features across profiled RCC lesions. ICB Response: PR, partial response; SD, stable disease; PD, progressive disease; NE, not evaluable. For some samples without successful whole-exome sequencing, genomic characterization is incomplete or missing.
(C) Uniform manifold approximation and projection (UMAP) of malignant and non-malignant cells captured across all lesions, colored by broad cell type. Granular cell types and states were discerned through iterative reprojection and unsupervised clustering of lymphoid, myeloid, and tumor compartments, and merged into broader cell-type categories for this visualization. DC, dendritic cell; NK, natural killer cell; NKT, natural killer T cell; TAM, tumor-associated macrophage; T-Reg, regulatory T cell.
(D) UMAP of malignant and non-malignant cells captured across all lesions, colored by patient, biopsy site, ICB treatment history, and ICB response.
After quality control, our dataset included 34,326 total cells covering various cancer and non-malignant cell types. To mitigate possible batch effects and allow for joint analysis of cancer cells, which have patient-specific expression patterns, we used Seurat's CCA-based alignment to aggregate cells across samples and jointly cluster them in an unsupervised manner (Figures 1C and S1A; Table S1) (Butler et al., 2018). We labeled clusters based on marker gene expression, identifying cancer and non-cancer cell clusters shared across patients, biopsy site, and systemic treatment received (Figures 1C, 1D, S1B, and S1C; Table S1).
CD8+ T cell remodeling by ICB
All ICB-exposed patients in our cohort received a therapy targeting the PD-1 axis, so we hypothesized that treatment may enact cell-(sub)type-specific remodeling of the lymphoid compartment. To address this, we began by performing unsupervised clustering within broad lymphoid cell types to identify cell states conserved across patients, biopsy sites, and treatment histories. We identified 12 lymphoid cell subsets, including B cells, plasma cells, natural killer T cells, FGFBP2+ and FGFBP2− natural killer cells, regulatory T cells, and memory and effector T helper cells (Figures 2A, 2B, S2A, and S2B; Table S2).
Figure 2.

CD8+ T cell exhaustion states are differentially remodeled by ICB
(A) UMAP of lymphoid cells captured across all lesions, colored and labeled by cell type. Bar plots show cell-type proportions grouped by ICB treatment history.
(B) Heatmap of scaled normalized expression for cell-type-defining genes as determined by two-sided Wilcoxon rank-sum test with Bonferroni FDR correction (q < 0.01).
(C) UMAP of CD8+ T cell subtypes, followed by expression heatmaps in UMAP space of co-inhibitory receptors, exhaustion markers, and memory-associated genes.
(D) Heatmaps in UMAP space of VISION signature scores for terminally exhausted and progenitor exhausted CD8+ T cells.
(E) Signature score distributions for terminally exhausted and progenitor exhausted CD8+ T cell signatures within each CD8+ T cell subtype. Significance of differential signature enrichment (p value) between subtypes was determined by two-sided Wilcoxon rank-sum test. Boxplots include centerline, median; box limits, upper and lower quartiles; and whiskers extending at most 1.5× the interquartile range past upper and lower quartiles.
(F) Heatmap of differential gene expression q values (two-sided Wilcoxon rank-sum test with Bonferroni FDR correction) for comparisons of cells within each CD8+ T cell cluster from ICB-exposed versus ICB-naive patients.
(G) Gene set enrichment analysis (GSEA) of terminally exhausted and progenitor exhausted signatures in 4-1BB-Lo CD8+ T cells from ICB PR patients compared with ICB SD/PD patients.
(H) Signature scores for an ICB-exposed 4-1BB-Lo CD8+ T cell signature and expression values for individual genes in paired pre-/on-ICB bulk RNA-seq samples in the Checkmate 009 RCC cohort. Expression values for individual genes were normalized against total CD8 fraction per sample as inferred by CIBERSORTx. Significance of differential signature enrichment or expression (p value) was determined by paired two-sided Wilcoxon rank-sum test. Boxplots include centerline, median; box limits, upper and lower quartiles; and whiskers extending at most 1.5× the interquartile range past upper and lower quartiles.
∗∗∗p < 0.001, two-sided Wilcoxon rank-sum test. See also Figures S2 and S3, Table S2.
CD8+ T cells formed four distinct clusters, including a population progressing through the cell cycle, a small population marked by expression of the interferon-stimulated gene MX1, and two populations differentiated by their expression of a set of genes including TNFRSF9 (encoding the activation marker 4-1BB) (Figures 2A–2C and S2C; Table S2) (Horisberger et al., 1983; Wolfl et al., 2007). Co-inhibitory receptors PDCD1 (encoding PD-1), HAVCR2 (encoding TIM-3), and LAG3 were expressed across all CD8+ T cell subsets but were detected in a larger proportion of 4-1BB-Hi cells (Figure S2C) (Thommen and Schumacher, 2018). TOX and ENTPD1 (encoding CD39), two exhaustion markers upregulated in tumor-specific T cells, were also elevated in 4-1BB-Hi and cycling cells (Figures 2C and S2C) (Canale et al., 2018; Duhen et al., 2018; Scott et al., 2019). In a pattern opposite that of ENTPD1, we observed expression, in a subset of cells, of IL7R and STAT4, two genes linked with T cell longevity and memory that are upregulated in a population of cells associated with ICB response in melanoma (Figure 2C) (Sade-Feldman et al., 2018).
Studies in melanoma models have shown that tumor-infiltrating CD8+ T cells include a progenitor exhausted population, which persists long term, responds to anti-PD-1 therapy, and ultimately differentiates into terminally exhausted cells. Frequency of this CD8+ T cell population in pre-treatment biopsies predicts positive clinical outcomes in melanoma treated with ICB (Miller et al., 2019). To determine whether any of the CD8+ T cell subsets we recovered in RCC resemble this progenitor exhausted phenotype, we scored individual cells for progenitor and terminally exhausted gene signatures (Table S2) (DeTomaso et al., 2019; Sade-Feldman et al., 2018). We found that the progenitor exhausted signature was strongly enriched in a subset of the cells within the 4-1BB-Lo cluster (Figures 2D, 2E, and S3A–S3C). Conversely, the terminally exhausted signature was elevated in the other CD8+ T cell populations.
Having identified a candidate subset of CD8+ T cells within RCC that bears some similarities to the previously described progenitor exhausted population, we next examined expression differences based on exposure to ICB (Figure 2F). Among CD8+ T cell subsets, we found that PDCD1, TIGIT, and HAVCR2 were significantly upregulated in ICB-exposed biopsies only within the 4-1BB-Lo cluster, an effect driven by responders (Figure S3D). Of effector molecules, we observed elevated expression in ICB-exposed biopsies of GZMB, PRF1, and IFNG in both 4-1BB-Lo and 4-1BB-Hi populations, but only 4-1BB-Lo cells upregulated GZMA and FASLG. Likewise, only 4-1BB-Lo cells upregulated a host of co-stimulatory molecules and chemokines. Conversely, 4-1BB-Lo cells from ICB-exposed biopsies downregulated several genes associated with the progenitor exhausted phenotype, hinting at possible differentiation toward a more terminally exhausted state. In melanoma models, polyfunctional progenitor exhausted CD8+ T cells differentiate to a more terminally exhausted phenotype during response to anti-PD1, and the latter population is primarily responsible for tumor killing despite being short lived (Miller et al., 2019). Within 4-1BB-Lo cells from ICB-exposed patients in our cohort, we observed significant enrichment of the terminally exhausted gene signature in cells from responders compared with cells from non-responders (Figures 2G and S3E–S3G; Table S2).
To validate these phenomena seen in unpaired biopsies, we examined a larger cohort of patients (Checkmate 009) undergoing monotherapy with the anti-PD-1 antibody nivolumab for whom longitudinal pre- and on-therapy bulk transcriptomes were available (Choueiri et al., 2016; Miao et al., 2018). In these paired biopsies, we observed significantly increased levels in the on-therapy samples of an expression signature derived from comparing ICB-exposed with ICB-naive 4-1BB-Lo cells from our single-cell cohort (Figure 2H; Table S2). Concordantly, on-treatment samples exhibited higher levels of co-inhibitory (e.g., PDCD1, LAG3) and effector molecules (e.g., IFNG, PRF1). These findings are consistent with, but do not unequivocally establish, the possible existence in RCC of a subpopulation of 4-1BB-Lo CD8+ T cells that respond to anti-PD1 therapy in a mechanistic fashion similar to that observed in models based on melanoma and chronic viral infection. Longitudinal single-cell studies would be necessary to definitively detect subpopulation-specific treatment-associated differentiation within individual patients.
Shift toward inflammation in tumor-associated macrophages
In addition to adaptive immunity, the innate immune system plays critical roles in tumor survival and resistance to therapy. In particular, tumor-associated macrophages (TAMs) are reported to play diverse and opposing roles within the tumor microenvironment, with different phenotypes promoting inflammation, tumor growth, angiogenesis, and metastasis, as well as cancer cell killing (Guerriero, 2018; Mantovani et al., 2017). TAMs are diverse in ccRCC, with mass cytometry identifying 17 phenotypes and high myeloid inflammation at baseline associated with worse outcomes on anti-PD-1 therapy (Chevrier et al., 2017; McDermott et al., 2018). Consequently, we sought to characterize myeloid cells within our cohort and their response to therapy.
We extracted myeloid cells identified from the full dataset based on marker expression and sub-clustered them, identifying two clusters of dendritic cells (CLEC9A+ and CD1c+ classical dendritic cells), two clusters of monocytes (classical CD16− and CD16+ monocytes), and five clusters of TAMs, which expressed a diverse host of immunomodulatory genes (Figures 3A–3C; Table S3) (Collin and Bigley, 2018). The proportions of these cell populations were stable between patients and across ICB exposure status (Figures 3A and S4A). A simple M1/M2 dichotomization of macrophages into phenotypes that promote inflammation and wound healing/tissue repair, respectively, is increasingly understood to imperfectly capture the diversity of TAM phenotypes, and indeed, we did not observe a clean binarization of expression of M1 and M2 marker genes (Figure 3D; Table S3) (Mantovani et al., 2017). A small population of CXCL10-Hi TAMs specifically expressed a set of genes associated with a classical M1-activated phenotype (Wang et al., 2014). However, the other three noncycling TAM populations expressed different M2-associated genes to varying degrees. Multiple M2-associated genes were robustly detected in a population of TAMs highly expressing GPNMB, previously reported to promote M2 polarization in a mouse model of acute kidney injury (Zhou et al., 2017). A separate population of TAMs with elevated expression of FOLR2, canonically described as marking M2 macrophages, expressed a distinct subset of M2-associated genes (Puig-Kröger et al., 2009). A last population of TAMs with mixed expression of both M1- and M2-associated genes was marked by high expression of VSIR, which encodes VISTA, a PD-1 homolog that serves as a negative checkpoint that inhibits T cell activation (Lines et al., 2014).
Figure 3.

Tumor-associated macrophages shift toward inflammation during checkpoint blockade
(A) UMAP of myeloid cells captured across all lesions, colored and labeled by cell type. Bar plots show cell-type proportions grouped by ICB treatment history.
(B) Heatmap of scaled normalized expression for cell-type-defining genes as determined by two-sided Wilcoxon rank-sum test with Bonferroni FDR correction (q < 0.01).
(C) Dotplot showing percentage of cells in each cell type expressing immune checkpoint and evasion genes.
(D) Heatmap of scaled normalized expression for curated M1- and M2-associated genes within TAM subtypes.
(E) Heatmap of normalized enrichment scores (NES) for gene sets significantly enriched (p < 0.05, q < 0.25) in TAM subtypes from ICB PR patients compared with ICB SD/PD patients.
(F) Violin and boxplots comparing expression distributions of immune checkpoint and evasion genes between all TAM from ICB-exposed versus ICB-naive patients. Significance of differential expression (q value) was determined by two-sided Wilcoxon rank-sum test with Bonferroni FDR correction. Boxplots include centerline, median; box limits, upper and lower quartiles; and whiskers extending at most 1.5× the interquartile range past upper and lower quartiles.
(G) Violin and boxplots comparing expression distributions of immune checkpoint and evasion genes between all TAM from ICB PR versus ICB SD/PD patients. Significance of differential expression (q value) was determined by two-sided Wilcoxon rank-sum test with Bonferroni FDR correction. Boxplots include centerline, median; box limits, upper and lower quartiles; and whiskers extending at most 1.5× the interquartile range past upper and lower quartiles.
∗∗∗q < 0.001, ns: not significant, two-sided Wilcoxon rank-sum test with Bonferroni FDR correction. See also Figure S4, Table S3.
To better understand the roles these diverse TAM populations play in the course of ICB, we examined their expression of immune checkpoint genes and immune-related transcriptional programs. PD-L2 and, to a lesser extent, PD-L1, both ligands for PD-1 signaling mediating immune checkpoint in T cells, were detected sparsely across the TAM populations (Figure 3C). LGALS9, VSIG4, and VSIR, whose protein products promote the T cell immune checkpoint via other pathways, and SIGLEC10, which can inhibit inflammatory responses by macrophages, were more broadly detected (Barkal et al., 2019; Das et al., 2017; Lines et al., 2014; Vogt et al., 2006). Comparing ICB-exposed patients with and without response, we noted broad and significant changes in expression patterns indicating a shift toward an M1-like phenotype in response to interferon signaling in all TAM and monocyte populations, except in the CXCL10-Hi TAMs, which already showed evidence of M1 polarization (Figures 3E and S4B). In parallel, we observed systematic increases in expression signatures associated with aspects of macrophage activity, including antigen presentation and proteasome function (Figures 3E and S4C). Collectively, these patterns pointed to a widespread shift to a pro-inflammatory phenotype across TAMs in ICB responders, potentially induced by interferon-γ produced by CD8+ T cells (Figures S4D and S4E). At the same time, we also observed systematic and dramatic upregulation of CD8+ T cell immune checkpoint and macrophage anti-inflammatory signaling genes in ICB-exposed versus naive TAMs (Figure 3F). Much of this effect was driven by TAMs from patients with ICB response, which had further increases in expression levels of VSIR, VSIG4, PD-L2, and SIGLEC10 compared with cells from non-responders (all assessed post-ICB) (Figure 3G). Thus, even as TAMs in responders exhibited a more pro-inflammatory phenotype, they also exhibited expression changes that may suppress an inflammatory immune microenvironment, thereby potentially promoting eventual resistance to ICB.
Two distinct cancer cell subpopulations
To better understand cellular programs active in cancer cells that may drive interactions with the immune system, we next sought to identify shared patterns of expression across cancer cells. We sub-clustered cells identified as malignant via gene expression and copy number alterations (Figure S1B). We aligned sets of cancer cells derived from different biopsies to focus on shared variation in cellular programs across the cohort. Across biopsies, cells formed two major clusters, denoted tumor program 1 (TP1) and tumor program 2 (TP2), with a small number of cells distinguished by cell-cycle progression (Figures 4A and 4B; Table S4). Both TP1 and TP2 cells were recovered from all samples except one with few cancer cells (P912), independent of biopsy site or ICB exposure (Figure S5A).
Figure 4.

Two malignant cell programs with distinct metabolic and immune-reactive characteristics are conserved across biopsy sites
(A) UMAP of malignant cells captured across all lesions, colored and labeled by cluster. Bar plots show cluster proportions grouped by ICB treatment history.
(B) Heatmap of scaled normalized expression for cluster-defining genes as determined by two-sided Wilcoxon rank-sum test with Bonferroni FDR correction (q < 0.01).
(C) Violin and boxplots comparing single-cell signature score distributions between the two dominant malignant cell clusters, partitioned by biopsy site. Significance of differential signature enrichment (p value) was determined by two-sided Wilcoxon rank-sum test. Boxplots include centerline, median; box limits, upper and lower quartiles; and whiskers extending at most 1.5× the interquartile range past upper and lower quartiles.
(D) GSEA of hallmark interferon-γ response and gene ontology antigen presentation and processing via MHC class I signatures in TP1 cells from ICB PR patients compared with ICB SD/PD patients (top) and TP2 cells from ICB PR patients compared with ICB SD/PD patients (bottom).
(E) Heatmap of differential expression q values (two-sided Wilcoxon rank-sum test with Bonferroni FDR correction) for immune checkpoint and evasion genes in comparisons of cells within each cluster from ICB-exposed versus ICB-naive patients.
(F) Heatmap of differential expression q values (two-sided Wilcoxon rank-sum test with Bonferroni FDR correction) for immune checkpoint and evasion genes in comparisons of cells within each cluster from ICB PR versus ICB SD/PD patients.
∗∗p < 0.01, ∗∗∗p < 0.001, two-sided Wilcoxon rank-sum test. See also Figure S5, Table S4.
We scored cells from the two major clusters for activity of various gene signatures to identify the cellular programs underlying their transcriptional differences. Across different metastatic sites, TP1 cells scored higher for gene sets for kidney morphogenesis, adherens junction assembly, and angiogenesis (Figure 4C). TP2 cells were characterized by large differences in metabolic programs, with increases in both glycolysis and oxidative phosphorylation, along with concomitant increases in fatty acid metabolism. The simultaneous upregulation of both oxidative phosphorylation and glycolysis genes in TP2 cells was unexpected and points to elevated metabolism in this tumor subpopulation, which may indicate metabolic plasticity permitting opportunistic use of different energetic pathways. The presence of cells in this hybrid metabolic state is consistent with theoretical models that predict such a cell state under high HIF-1 activity, which is well established in RCC, with observations in cell lines linking such plasticity to metastasis (Jia et al., 2019; Wettersten et al., 2017; Yu et al., 2017).
We next examined the potential effects of these cancer cell programs on immune cells in the context of ICB therapy. Comparing ICB responders with non-responders, both cancer cell populations upregulated gene sets associated with interferon-γ response and antigen processing on major histocompatibility complex (MHC) class I, indicating a response to the pro-inflammatory microenvironment in responders (Figures 4D and S5B–S5E; Table S4). However, ICB-treated cancer cells also upregulated several T cell checkpoint molecules, with different patterns in the two populations (Figure 4E). TP1 cells robustly upregulated nectin-2 after therapy, while TP2 cells had more moderate increases in expression of several checkpoint molecules. Comparing ICB responders with non-responders, TP2 cells exhibited a more dramatic difference, broadly upregulating both a host of T cell checkpoint molecules and molecules involved in decreasing macrophage inflammation and evading phagocytosis (Figure 4F). We recapitulated our findings in a more homogeneous subset of our cohort (Figures S5F–S5L). In sum, our results suggest complex interactions between the many cell populations in the microenvironment, with the two cancer cell populations presenting different immunosuppressive signals to both CD8+ T cells and TAMs during ICB therapy.
TP1 cancer expression program associated with improved survival
The differences between TP1 and TP2 cancer cells with respect to kidney differentiation, metabolism, and immunosuppressive signaling prompted us to investigate in a large prospective cohort whether they play different roles in disease progression and resistance to therapy. We generated gene signatures describing TP1 and TP2 and restricted these signatures to cancer-cell-specific genes (Table S5). Having leveraged our scRNA-seq data to generate cancer-cell-specific signatures, we then scored bulk RNA-seq of pre-treatment samples from advanced ccRCC in a randomized clinical trial (Checkmate 025) comparing the mTOR inhibitor everolimus with nivolumab (Table S5) (Braun et al., 2020). We observed an association between pre-treatment TP1 score and significantly improved overall survival only within patients receiving nivolumab in multivariate analysis, although score levels between radiographic response categories were not significantly different (Figures 5A and S6A–S6C). In a joint analysis including patients from both arms modeling treatment arm, TP1 score, and a treatment-TP1 interaction term, we observed a significant interaction between TP1 score and treatment with nivolumab (p = 0.04, Cox proportional hazards). These results indicate that the relationship between TP1 score and survival is driven by cancer-immune interactions. There was no significant association between TP2 scores and survival (Figure S6D). However, in nivolumab-treated patients, decreased survival was associated with higher expression of a set of immune checkpoint and evasion genes, almost all of which were significantly upregulated in TP2 cells from ICB responders versus non-responders, albeit these genes are not cancer cell specific (Figures 4F and 5B) (pCox = 0.017).
Figure 5.

Tumor program signatures are prognostic in the Checkmate 025 RCC cohort and associated with distinct genomic features
(A) Kaplan-Meier analysis of overall survival (OS) in the Checkmate 025 RCC cohort, with patients separated by high and low TP1 score in bulk RNA-seq. plog-rank, log-rank test p value; pCox, p value determined via a multivariate Cox proportional hazard model using TP1 score dichotomized within treatment arm and incorporating age, sex, MSKCC risk group, prior lines of therapy (≤1 or ≥2), and days between biopsy collection and start of trial therapy as covariates.
(B) Kaplan-Meier analysis of OS in the Checkmate 025 RCC cohort, with patients separated by high and low immune checkpoint/evasion score in bulk RNA-seq. plog-rank, log-rank test p value; pCox, p value determined via a multivariate Cox proportional hazard model using immune checkpoint/evasion score dichotomized within treatment arm and incorporating age, sex, MSKCC risk group, prior lines of therapy (≤1 or ≥2), and days between biopsy collection and start of trial therapy as covariates.
(C) Bar plots comparing TP1 and TP2 score between mutant and wild-type samples for commonly mutated genes in the Checkmate 025 RCC cohort. Significance of differential score enrichment (q value) determined by two-sided Wilcoxon rank-sum test with Benjamini-Hochberg FDR correction. Gray dotted line corresponds to q = 0.05.
(D) Bar plots comparing TP1 and TP2 score between mutant and wild-type samples for common copy number alterations in the Checkmate 025 RCC cohort. Significance of differential score enrichment (q value) determined by two-sided Wilcoxon rank-sum test with Benjamini-Hochberg FDR correction. Gray dotted line corresponds to q = 0.05.
To better understand the etiology of these two cancer cell programs, we evaluated whether they were associated with underlying genomic features. In the Checkmate 025 cohort, we noted an increase in TP1 score in PBRM1 and KDM5C mutant tumors and an increase in TP2 score in tumors harboring 9p21.3 deletions, which were previously associated with ICB resistance (Figures 5C and 5D) (Braun et al., 2020). The association of TP1 score with PBRM1 mutations and TP2 score with 9p21.3 deletions was corroborated in the TCGA KIRC cohort (Figures S6E and S6F). In ICB-treated Checkmate 025 patients, TP1 score remained significantly associated with increased survival when incorporating PBRM1 mutation status as a covariate (Figure S6G). In conjunction with the observation of associations with KDM5C and copy number alterations, this suggested that TP1 captures transcriptional patterns associated with genomic alterations beyond single gene mutations.
The survival benefit in the Checkmate 025 ICB arm prompted further investigation of interactions between cancer cell subpopulations and immune cells. We examined CIBERSORTx immune cell population abundances inferred in Checkmate 025 (nivolumab arm) using cell-type expression signatures derived from our dataset (Figures S6H and S6I; Table S5) (Newman et al., 2019). In tumors with higher TP1 score, we observed increases in inferred abundance of 4-1BB-Lo CD8+ T cells, noted earlier for dramatic upregulation of effector molecule expression during ICB therapy and resemblance to progenitor exhausted cells promoting ICB response in melanoma (Figure 2F) (Miller et al., 2019). Although computational inference of abundances of related immune cell populations in bulk cohorts is challenging, these findings support a relationship between TP1 expression programs in cancer cells and survival mediated by cancer-immune cell interactions. Collectively, our observations of distinct tumor subpopulations with different immune cell interactions may help explain the observation in bulk RNA-seq of distinct clusters of RCC, including heterogeneously infiltrated highly angiogenic tumors and non-infiltrated highly metabolic tumors (Şenbabaoğlu et al., 2016).
Cell-cell interactions within the tumor microenvironment
Given observations of IFNG upregulation in ICB-exposed 4-1BB-Lo CD8+ T cells, immune checkpoint and evasion gene upregulation in TAMs from ICB responders, and differentially immunomodulatory cancer cell programs, we hypothesized that the different cell populations participate in a complex cross talk. To identify possible non-cell-autonomous effects, we used CellPhoneDB to identify putative signaling between different cell populations via known receptor-ligand pairs (Figure 6A) (Efremova et al., 2020). Both cancer cell populations were inferred to signal to CD8+ T cells expressing CD44 via osteopontin (encoded by SPP1), an interaction that suppresses T cell activation in mouse models (Klement et al., 2018). Likewise, both cancer cell populations expressed MIF and CD47, signaling to CD74 and SIRPα expressed on TAMs, interactions that promote TAM secretion of growth factors and that inhibit phagocytosis, respectively. However, only TP2 cells were inferred to engage TIM-3 (encoded by HAVCR2) expressed on both CD8+ T cells and TAMs via galectin-9 (encoded by LGALS9). Signaling through TIM-3 inhibits CD8+ cytotoxic cell activity and, in other human cancers, induces TAM secretion of growth factors (Das et al., 2017). In addition, TP2 cells expressed nectin-2, which binds to the CD8+ T cell co-inhibitory receptor TIGIT (Yu et al., 2009). Although such an in silico approach cannot definitively establish an interaction, it suggested the possibility of complex regulation of both T cells and TAMs by cancer cells.
Figure 6.

IFNG is associated with pro-inflammatory TAM and immune checkpoint/evasion phenotypes in the Checkmate 009 RCC cohort
(A) Heatmap of cell-type-specific receptor-ligand interactions inferred by CellPhoneDB. Shown are inferred interactions between malignant cell clusters and all CD8+ T cells, malignant cell clusters and all TAMs, and all CD8+ T cells and all TAMs. Circle size indicates significance of interaction and circle color indicates mean expression of receptor and ligand genes for each pair.
(B) Scatterplots of CXCL10-Hi TAM fraction (left) and ICB-exposed TAM signature score (right) versus IFNG gene expression normalized by total CD8+ T cell fraction in bulk RNA-seq of on-ICB samples in the Checkmate 009 RCC cohort. Cell-type fractions were inferred using CIBERSORTx. Pearson coefficient (R) and associated p value are reported for each correlation.
(C) Scatterplots of immune checkpoint/evasion signature score versus IFNG expression normalized by total CD8+ T cell fraction (left) and immune checkpoint/evasion score versus hallmark interferon-γ response signature score (right) in bulk RNA-seq of on-ICB samples in the Checkmate 009 RCC cohort. Cell-type fractions were inferred using CIBERSORTx. Pearson coefficient (R) and associated p value are reported for each correlation.
(D) Signature scores for the immune checkpoint/evasion signature and expression values for individual genes in paired pre-/on-ICB bulk RNA-seq samples in the Checkmate 009 RCC cohort. Significance of differential signature enrichment or expression (p value) was determined by paired two-sided Wilcoxon rank-sum test. Boxplots include centerline, median; box limits, upper and lower quartiles; and whiskers extending at most 1.5× the interquartile range past upper and lower quartiles.
We additionally observed evidence of interactions between CD8+ T cells and TAMs. Consistent with upregulation of interferon-response genes in TAMs from ICB responders versus non-responders, CellPhoneDB inferred regulation of TAMs by interferon-γ produced by CD8+ T cells (Figures 3E and 6A). Within on-ICB biopsies from Checkmate 009, normalized IFNG levels were robustly correlated with CIBERSORTx-estimated CXCL10-Hi TAM fractions and with an expression signature derived from comparing ICB-exposed to ICB-naive TAMs from our single-cell cohort (Figure 6B; Tables S2 and S3). Within the same samples, we observed concomitant increases in expression of immune checkpoint and evasion genes in samples with higher normalized IFNG expression, consistent with immunosuppressive negative feedback loops reported in other cancer types (Figure 6C; Tables S2 and S5) (Abiko et al., 2015; Bellucci et al., 2015; Garcia-Diaz et al., 2017). Indeed, comparing pre- and on-ICB biopsies from the same patients, we observed increases in expression levels of the immune checkpoint and evasion signature and of member genes, including VSIG4, PD-L2, and SIGLEC10 (Figure 6D; Tables S2 and S5). Several of these genes are expressed by both cancer cells and TAMs, and their origin cannot be deciphered in bulk transcriptomes, but these comparisons suggest that the tumor microenvironment may adapt to ICB treatment by increasing immunosuppressive gene expression.
Discussion
To investigate immune system and cancer responses to therapy, we examined single-cell transcriptomes from advanced RCC before or after ICB. A subpopulation of CD8+ T cells is robustly activated and differentiated toward a terminally exhausted phenotype in responders. These cells have similarities to those described as mediating ICB response in melanoma and may correspond to a population described as the progeny of stem-like CD8+ T cells in RCC (Jansen et al., 2019; Miller et al., 2019). Among TAMs, we identified a small pro-inflammatory population and multiple others that coherently shift toward a pro-inflammatory phenotype in response to interferon-γ signaling in ICB responders. ICB exposure was associated with increases in T cell checkpoint molecule expression and anti-inflammatory signaling, suggesting immune system adaptation that may underlie treatment resistance. Within cancer cells, we uncovered two populations differentiated by expression of pro-angiogenic, kidney differentiation, and metabolic programs. In response to ICB, metabolically plastic and less differentiated TP2 cancer cells upregulated a host of immunosuppressive genes. Whereas cells from both populations were observed across biopsies in our cohort, an increase in the TP1 cancer program was associated with increased survival with ICB, suggesting that differences in cancer-immune cross talk operate between the two populations.
Clinical heterogeneity poses a challenge to our study given the modest biopsy numbers. Almost all ICB-exposed patients had exposure to TKIs, so observed effects may be driven by the combination. Nonetheless, as TKI and ICB combination or sequential therapy is the standard of care, our findings remain relevant for future work. RCC most commonly manifests as ccRCC, and our dataset reflects this. Whereas we have explicitly focused on expression programs found across patients, detailed studies of other histologies are needed. We validated our findings in uniformly treated bulk-sequenced clinical trial cohorts, including one with longitudinal pre- and on-ICB biopsies. Nonetheless, bulk RNA-seq is limited by unknown cell-type admixture, so future work should focus on large cohorts of paired ICB-naive and ICB-resistant biopsies using single-cell approaches. Multiplex imaging analyses may elucidate mechanisms of cross talk, which appear pervasive in RCC (Braun et al., 2021). This is particularly pressing given the upregulation of immunosuppressive genes in both TAMs and cancer cells, which may ultimately precipitate resistance.
Our findings highlight the importance of studying immunomodulatory pathways away from the PD-1 axis, including T cell inhibition by VISTA expressed by both TAMs and cancer cells. Targeting the CD47/SIRPα axis with anti-CD47 antibodies, perhaps in conjunction with anti-PD-1 therapy, may promote macrophage phagocytosis of TP2 cells, which upregulate a broad range of immunosuppressive genes in ICB responders. These investigations are urgent now that ICB is a standard of care, and our findings provide a path toward identifying therapeutic targets and combinations to combat treatment resistance.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eliezer M. Van Allen (eliezerm_vanallen@dfci.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Raw single cell RNA-seq and bulk whole exome sequencing data are deposited to dbGaP: phs002065.v1.p1. Processed single cell RNA-Seq data can also be accessed via the Single Cell Portal (https://singlecell.broadinstitute.org/single_cell/study/SCP1288/tumor-and-immune-reprogramming-during-immunotherapy-in-advanced-renal-cell-carcinoma#study-summary). Code used for all processing and analysis is available upon request.
Transcriptional signatures for progenitor and terminally exhausted CD8+ T cells (CD8_G and CD8_B, respectively) were obtained from published results (Sade-Feldman et al., 2018) (GEO: GSE120575). For the Checkmate 009 cohort, raw bulk RNA-seq data were obtained from published results (Miao et al., 2018) (dbGaP: phs001493). For the Checkmate 025 cohort, normalized bulk RNA-seq, clinical, and mutational data were obtained from published results (Braun et al., 2020) (European Genome-Phenome Archive: EGAS00001004290, EGAS00001004291, EGAS00001004292). For the TCGA clear cell renal cell carcinoma (KIRC) cohort, normalized bulk RNA-seq data were obtained from Firebrowse (http://firebrowse.org/, illuminahiseq_rnaseqv2-RSEM_genes_normalized). Clinical data were downloaded from the cBioPortal (https://www.cbioportal.org/study/clinicalData?id=kirc_tcga). Somatic mutation data were obtained from published results (Ricketts et al., 2018) (https://gdc.cancer.gov/). GISTIC2 results describing amplification and deletion events in TCGA KIRC were downloaded from Firebrowse (http://firebrowse.org/, gdac.broadinstitute.org_KIRC-TP.CopyNumber_Gistic2.Level_4.2016012800.0.0).
Experimental model and subject details
Human patient samples were collected with written informed consent and ethics approval by the Dana-Farber Cancer Institute Institutional Review Board under protocol no. 15-349. Patient metadata, including age at diagnosis, sex, treatment regimen, clinical response, sample biopsy site, histological grade, and disease stage, are provided as Table S1.
Method details
Somatic mutation and copy number alteration calls
Where available, additional tissue was used for whole exome sequencing (samples P55, P915, P913, P906, P76) or panel sequencing via OncoPanel (sample P90), with downstream bioinformatic analyses as previously described (He et al., 2021). Briefly, exome capture was performed using Illumina’s Rapid Capture Exome Kit, reads were aligned using BWA v0.5.9 (Li and Durbin, 2009), somatic mutation calling was performed using MuTect (Cibulskis et al., 2013), and copy number alterations were called using FACETS (Shen and Seshan, 2016).
Sample collection and dissociation for scRNA-seq
Single-cell suspensions for single-cell RNA-seq were obtained by mechanical and enzymatic dissociation. Tumor tissue was were collected and transported in Medium 199 with Hank’s salts (ThermoFisher Scientific #12350039) (samples P912, P913, P915, P916) or Medium 199 with Earle′s salts (Sigma #M4530) (other samples) on ice. Enzymatic dissociation mix consisted of calcium- and magnesium-free Hank’s Balanced Salt Solution (ThermoFisher Scientific #14170120) with 5 mM CaCl2 (Sigma #10043524), 100 μg/mL Collagenase Type 4 (Worthington Biochemical Corporation #LS004186), 0.75 u/mL Dispase (StemCell Technologies #7913), and 10 μg/mL DNAse I (StemCell Technologies #07900) for most samples (P906, P912, P913, P915, P916). For samples P90 and P55, dissociation mix composition was similar with the exception that Dispase was used at a concentration of 1.125 u/mL. For sample P76, Accumax was used instead (Sigma-Aldrich #A7089). In all cases, the sample was first minced into pieces smaller than 0.5 mm in cold enzymatic dissociation mix using scalpels or spring scissors. The resulting tissue suspension was then incubated for a total of 20 min at 37°C with rotation, interrupted briefly by additional mincing or pipetting after 10 min (samples P906, P912, P913, P915, P916), or for 10 min at 37°C (P90, P55) or room temperature (P76), whereas subsequent steps were carried out at 4°C or on ice. When necessary, additional pipetting was used to complete the dissociation and residual tissue fragments were strained using a 100 μm cell strainer.
Cells were then centrifuged at 580g for 5 min, or using a brief centrifugation (8 sec with centrifugal force ramping up to but not exceeding 11,000 g) to minimize cell loss, and the resulting cell pellet was resuspended in ACK Lysing Buffer (ThermoFisher Scientific #A1049201) to lyse red blood cells. After 1 min incubation on ice, red blood cell lysis was stopped by addition of calcium- and magnesium-free PBS (ThermoFisher Scientific #10010-23) followed by centrifugation. If the resulting pellet appeared bloody, red blood cell lysis was repeated without exceeding 3 min incubation in total. Following red blood cell removal, cells were resuspended in calcium- and magnesium-free PBS with 0.4% BSA (Ambion #AM2616). Cell viability and the presence of cell clumps or debris were then assessed using a hemocytometer after mixing cells with Trypan blue (Sigma #T8154). Based on this assessment, samples P90 and P55 were centrifuged at low speed (200g for 5min) to remove debris, and dead cells were removed from samples P76 and P55 using Viahance (BioPAL #CP-50VQ02). All samples had viability greater than 58%, and 5,000-8,000 viable cells in PBS with 0.4% BSA were loaded per channel of the 10x Genomics Chromium platform using Single Cell 3' (v2 Chemistry) reagents (10x Genomics #PN-120237 and #PN-120236).
scRNA-seq data preprocessing
scRNA-seq samples were sequenced on an Illumina HiSeq X sequencer. Demultiplexing of sequencing results, barcode processing, read alignment, and UMI counting were performed using the 10x Cell Ranger analysis pipeline v3.1. Reads were aligned to the human genome reference b37 with the GENCODE 30 annotation lifted over to GRCh37 (Frankish et al., 2019). Confidently mapped, non-PCR duplicate reads were counted to generate a gene-barcode matrix for each sample with barcodes filtered to exclude low RNA content droplets.
To exclude data from droplets containing more than one cell, doublet detection and removal were performed on gene-barcode matrices using Scrublet (Wolock et al., 2019). An expected doublet rate parameter of 0.06 was used, and doublet score thresholds were chosen manually to divide putative singlet and neotypic doublet modes in the score distribution. Predicted doublets were then removed from gene-barcode matrices.
To abrogate sample-to-sample differences driven by the ambient RNA profile present in all droplets prepared from a given single-cell suspension, we performed an ambient RNA decontamination step using R package SoupX v1.2.2 (Young and Behjati, 2020). For each sample, an ambient RNA profile was determined from an unfiltered gene-barcode matrix provided by the Cell Ranger pipeline which included low RNA containing droplets. Three classes of genes exhibiting high cell type specificity and bimodality within the samples of our cohort (immunoglobulin, hemoglobin, and macrophage-associated genes such as APOE and C1QA) were used to estimate per-cell contamination fractions. The ambient RNA profile was then subtracted proportionately from cells of the original gene-barcode matrix. These cleaned gene-barcode matrices were used for all downstream analysis. Further QC, feature selection, dimension reduction, unsupervised clustering, and differential expression analyses were performed using the Seurat R package v3.1.5 (Butler et al., 2018).
Across-sample integration
Prior to across-sample integration, cells with fewer than 200 genes detected or more than 25% of counts attributed to mitochondrially-encoded transcripts were removed. Genes detected in fewer than three cells across all samples were also excluded. Each gene expression measurement was normalized by total expression in the corresponding cell and multiplied by a scaling factor of 10,000, then log2-transformed (default Seurat approach). To adjust for technical and biological sources of variation between samples, the standard anchor-based workflow for dataset integration in Seurat was used (Stuart et al., 2019). 10,000 variable features for CCA (canonical correlation analysis) were chosen based on stabilized variance, and integration anchors were identified using the first 30 reduced dimensions. The resulting merged dataset included 34,326 cells and 32,718 detected genes across the eight samples of the cohort. Integration-transformed expression values were used only for dimension reduction and clustering. The original log-normalized expression values were used for all differential expression and gene set level analyses.
Visualization, clustering, and identification of cell types
Principal components analysis (PCA) was performed on the integration-transformed expression matrix, and the first 50 principal components were used for Louvain clustering of cells with a resolution parameter of 0.5. Uniform manifold approximation and projection (UMAP) was performed on the same PCs with 100 nearest neighbors for visualization in two dimensions. To classify cells into broad cell types, differential expression analysis was performed comparing cells of each Louvain cluster to all other clusters using a two-sided Wilcoxon rank-sum test with Bonferroni FDR correction (q < 0.01 considered significant) (Figure S1A). These initial clusters were then merged into lymphoid, myeloid, normal tissue, and putative tumor lineages based on expression of differential marker genes. To identify immune cell types at a high resolution, lymphoid and myeloid cells were re-clustered and re-projected separately using 30 PCs for Louvain clustering and UMAP, with the same differential expression analysis applied to annotate cell type. Further iterations of re-clustering (30 PCs, resolution parameters ranging from 0.15-0.5) were performed within larger cell types, including CD8+ T cells, T-Helper cells, NK cells, and tumor associated macrophages to identify cell subtypes and states with clearly distinct differential expression profiles.
Identification of cancer cells
Malignant cells were identified using inferred CNV profiles and cluster-level marker gene expression. We first projected and clustered all cells from each sample separately to identify PTPRC+ and PTPRC− clusters. We then performed InferCNV (https://github.com/broadinstitute/inferCNV) within each sample, using PTPRC+ clusters as reference groups and PTPRC- clusters as observation groups (Figure S1B). Malignant clusters were identified based on established copy number features of RCC histologies. For clear cell RCC samples, clusters with chromosome 3p loss were called as malignant; for the single papillary RCC sample profiled in the cohort, clusters with gains in chromosomes 7 and 17 were called as malignant. Differential expression analysis showed that observation clusters without distinct CNAs exhibited clear expression patterns associated with fibroblasts or endothelial cells.
These InferCNV results were then cross-referenced with our gene expression marker-based lineage calls. In the fully integrated dataset, Louvain clusters 3, 4, 6, 17, and 21 were identified as putatively malignant based on significant differential expression of CA9, CA12, NDUFA4L2, VCAM1, or VEGFA. Cells in these clusters constituted 99.38% of cells labelled via InferCNV as malignant. Cancer cells were definitively called as the overlap between these clusters and the set of cells identified as malignant based on copy number profile with InferCNV.
Differential expression and gene set level analysis
Differential expression analysis comparing cells from treatment exposure or response groups was performed using a two-sided Wilcoxon rank-sum test with Bonferroni FDR correction. Preranked gene set enrichment analysis (GSEA) was performed using the fgsea v1.10.1 R package (Sergushichev, 2016; Subramanian et al., 2005). For a given pairwise GSEA comparison, the log2(fold-change) of the average expression between groups was used as a ranking metric. GSEA was performed using Hallmark, KEGG, and GO Biological Process gene sets from v6.2 of the MSigDB repository or select gene sets curated from literature (Bell et al., 2016; Liberzon et al., 2015; McDermott et al., 2018). Gene sets with p < 0.05 and FDR < 0.25 were considered significant.
Single cell signature scoring was performed using the VISION v2.0.0 R package (DeTomaso et al., 2019). Differential signature score enrichment between groups was determined using a two-sided Wilcoxon rank-sum test with Benjamini-Hochberg FDR correction (Benjamini and Hochberg, 1995). VISION scoring was performed using the same MSigDB gene sets cited above. CD8+ T cells with a progenitor exhausted signature score > 0.75 and a terminally exhausted signature score < 2.5 were called as Progenitor Exhausted Cells, and CD8+ T cells with a progenitor exhausted signature score < 0.75 and a terminally exhausted signature score > 2.5 were called as Terminally Exhausted Cells.
Receptor-ligand interaction inference
Inference of receptor-ligand interactions was performed using the CellPhoneDB Python library (Efremova et al., 2020). The algorithm was run on log-normalized expression values for tumor cells, tumor-associated macrophages, and CD8+ T cells with default parameters and no subsampling.
Bulk RNA-seq and survival analysis
To obtain an ICB-exposed 41BB-Lo CD8+ T cell signature for projection into bulk RNA-Seq, genes significantly upregulated in 41BB-Lo CD8+ T cells from ICB-exposed patients relative to those from ICB-naive patients (with q < 0.01, log2(fold-change) > 0.25) were restricted to lymphoid-specific genes. Lymphoid-specific genes were defined as the intersection between genes expressed in <5% of tumor, myeloid, or normal tissue cells and genes with at least 10% greater expression in lymphoid cells.
To obtain an ICB-exposed TAM signature, genes significantly upregulated in all ICB-exposed TAM (aggregated across the five subsets described) relative to all ICB-naive TAM (with q < 0.01, log2(fold-change) > 0.25) were restricted to myeloid-specific genes. Myeloid-specific genes were defined as the intersection between genes expressed in <5% of tumor, lymphoid, or normal tissue cells and genes with at least 10% greater expression in myeloid cells.
To obtain TP1 and TP2 signatures, genes differentially expressed between TP1 and TP2 cells (with q < 0.01, log2(fold-change) > 0.5 or < -0.5) were restricted to tumor-specific genes. Tumor-specific genes were defined as the intersection between genes expressed in <5% of myeloid, lymphoid, or normal tissue cells and genes with at least 10% greater expression in tumor cells.
We defined a curated Immune Checkpoint/Evasion signature as a set of putatively immunoregulatory genes observed as upregulated in TAM and/or tumor cells from ICB-exposed and ICB-responder lesions. This signature comprised the following genes: VSIR, VSIG4, LGALS9, CD274 (PD-L1), PDCD1LG2, SIGLEC10, NECTIN2, CD47, and MIF.
Bulk RNA-seq from the Checkmate 009 cohort were aligned to human reference genome b37 and the GENCODE 30 GRCh37 liftover transcriptome reference using STAR v2.7.0, and TPM quantification was performed with salmon v0.14.1 (Choueiri et al., 2016; Dobin et al., 2013; Frankish et al., 2019; Miao et al., 2018; Patro et al., 2017).
Signature scoring of all bulk RNA-seq samples was performed using the VISION R package (DeTomaso et al., 2019). Significance of differences in OS between samples stratified by signature score were determined via Kaplan-Meier analysis, log-rank tests, and multivariate Cox regression analysis using the survminer v0.4.6 and survival v3.1.7 R packages and the lifelines v0.23.9 Python package. In the Checkmate 025 cohort, multivariate analyses of each arm separately incorporated TP1, TP2, or Immune Checkpoint/Evasion score dichotomized within each arm in addition to patient age, sex, MSKCC risk group, prior lines of therapy (≤1 or ≥2), and days between biopsy collection and start of trial therapy as covariates. For the joint analysis of both arms, the model included continuous TP1 score, treatment arm (1 in nivolumab, 0 if everolimus), treatment arm∗TP1 score, patient age, sex, MSKCC risk group, prior lines of therapy (≤1 or ≥2), and days between biopsy collection and start of trial therapy. In TCGA KIRC and Checkmate 025 cohorts, comparisons of signature scores between mutant and wild-type tumors, or between ICB response groups, were performed using a two-sided two-sided Wilcoxon rank-sum test with Benjamini-Hochberg FDR correction (Benjamini and Hochberg, 1995). For comparisons of signature scores between mutant and wild-type samples, only mutations with predicted high impact Ensembl Sequence Ontology were considered. In the Checkmate 009 cohort, comparisons of signature scores or TPM between matched pre/on-ICB samples were performed using a paired two-sided two-sided Wilcoxon rank-sum test.
To generate a CIBERSORTx signature matrix using immune cell types from our single cell cohort, we ran the Create Signature Matrix module (https://cibersortx.stanford.edu/runcibersortx.php) with the log-normalized expression matrix from our dataset restricted to immune populations of interest supplied as a reference matrix (Newman et al., 2019). We used a Min. Expression parameter of 0.05 and a Min No. Barcode Genes parameter of 100, with all other parameters set to default. Using the resulting signature matrix, CIBERSORTx deconvolution was performed on the bulk RNA-seq cohorts in relative mode with S-mode batch correction and quantile normalization disabled. Comparisons of inferred immune population frequencies between samples grouped by signature score were performed using a two-sided Wilcoxon rank-sum test with Benjamini-Hochberg FDR correction (Benjamini and Hochberg, 1995).
Quantification and statistical analysis
All statistical analyses were performed using R (version 3.6.0). For differential expression analysis between groups of cells, a two-sided Wilcoxon rank-sum test with Bonferroni FDR correction was used. For comparisons of signature scores or CIBERSORTx-inferred immune fractions between groups of cells or bulk RNA-Seq samples, a two-sided Wilcoxon rank-sum test with Benjamini-Hochberg FDR correction was used. For survival analysis of bulk RNA-Seq samples stratified by signature score, significance of OS difference was determined using a log-rank test and multivariate Cox proportional hazards model. Descriptions of statistical tests performed for each individual analysis are provided in Figure legends and Method Details.
Acknowledgments
We thank the patients who participated in this study. We thank B.C. Miller for thoughtful discussions of this work. This work was supported by the National Institutes of Health (U01 CA233100 [E.M.V.A.], R01 CA227388 [E.M.V.A.], U2C CA233195 [K.B., A. Regev, E.M.V.A.], T32 GM008313 [M.X.H.], T32 CA009172 [A.K.T.]), the National Science Foundation (GRFP DGE1144152 [M.X.H.]), a Novartis-DDP grant (R.H., E.M.V.A., T.K.C.), a Kure It-AACR grant (E.M.V.A, T.C.), a Dunkin’ Donuts Breakthrough Grant (E.M.V.A.), the DF/HCC Kidney Cancer SPORE (P50CA101942-15 [D.A.B.], T.K.C.), the DOD CDMRP (KC170216 and KC190130 [D.A.B.]), and the DOD Academy of Kidney Cancer Investigators (KC190128 [D.A.B.]). T.K.C. is supported in part by the Kohlberg Chair at Harvard Medical School and the Trust Family, Michael Brigham, and Loker Pinard Funds for Kidney Cancer Research at DFCI. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Author contributions
Conceptualization, K.B., M.X.H., O.R.-R., A. Rotem, A. Regev, T.K.C., and E.M.V.A.; validation, K.B. and M.X.H.; formal analysis, K.B. and M.X.H.; investigation, A.K., S.N., J.W., G.G., M.S.C., T.D., S.V., and A. Rotem; resources, S.L.C., B.A.M., R.H., S.S., and J.L.G.; data curation, Z.B., A.M., L.D., G.B., J.S., A.K.T., and N.I.V.; writing – original draft, K.B., M.X.H., S.V., and E.M.V.A.; writing – reviewing & editing, Z.B., D.A.B., J.L.G., A. Regev, and T.K.C.; visualization, K.B.; supervision, S.S., S.V., O.R.-R., A. Rotem, A. Regev, T.K.C., and E.M.V.A; project administration, M.X.H, A.M., L.D., G.B., J.S., E.S., M.S., and J.P.; funding acquisition, R.H., A. Regev, T.K.C., and E.M.V.A.
Declaration of interests
M.X.H. has consulted for Amplify Medicines and Ikena Oncology. Z.B. reports research support from Bristol-Meyers Squibb (BMS) and Genentech/imCORE unrelated to this study. D.A.B. reports non-financial support from BMS, honoraria from LM Education/Exchange Services, and personal fees from Octane Global, Defined Health, Dedham Group, Adept Field Solutions, Slingshot Insights, Blueprint Partnerships, Charles River Associates, Trinity Group, and Insight Strategy, outside of this work. B.A.M. has consulted for Bayer, Astellas, AstraZeneca, Seattle Genetics, Exelixis, Nektar, Pfizer, Janssen, Genentech, Eisai, and EMD Serono. He received research support to Dana-Farber Cancer Institute from BMS, Calithera, Exelixis, and Seattle Genetics. J.L.G. is a consultant for and/or receives sponsored research support from GlaxoSmithKline (GSK), Array BioPharma, Codagenix, Verseau, Kymera, and Eli Lilly. A. Rotem is an employee of AstraZeneca and an equity holder in NucleAI and Celsius Therapeutics. A. Regev is a founder and equity holder of Celsius Therapeutics, holds equity in Immunitas Therapeutics, and, until August 31, 2020, was an SAB member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov, and Thermo Fisher Scientific; since August 1, 2020, she has been an employee of Genentech. E.M.V.A. reports advisory/consulting with Tango Therapeutics, Genome Medical, Invitae, Monte Rosa, Enara Bio, Manifold Bio, and Janssen; research support from Novartis and BMS; equity in Tango Therapeutics, Genome Medical, Syapse, Manifold Bio, Monte Rosa, and Enara Bio. T.K.C. reports research support/honoraria from, consulting/advisory relationships with, and/or equity stakes in Alexion, Analysis Group, AstraZeneca, Aveo, Bayer, BMS, Calithera, Cerulean, Corvus, Eisai, EMD Serono, Exelixis, F. Hoffmann-La Roche, Foundation Management, Genentech, GSK, Heron Therapeutics, Infinity Pharma, Janssen Oncology, IQVIA, Ipsen, Lilly, Merck, NCCN, Novartis, Peloton, Pfizer, Pionyr, Prometheus Labs, Roche, Roche Products Limited, Sanofi/Aventis, Takeda, Tracon, Surface Oncology, Tempest, Up-to-Date, OncLIve, PVI, and MJH Life Sciences; patents filed, royalties, or other intellectual properties related to biomarkers of immune checkpoint blockers; and support from the Dana-Farber/Harvard Cancer Center Kidney SPORE and Program, the Kohlberg Chair at Harvard Medical School and the Trust Family, Michael Brigham, and Loker Pinard Funds for Kidney Cancer Research at DFCI.
Published: March 11, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ccell.2021.02.015.
Supplemental information
TPM expression matrix, patient-level metadata, and sample-level metadata for Checkmate 009 bulk RNA-seq samples.
References
- Abiko K., Matsumura N., Hamanishi J., Horikawa N., Murakami R., Yamaguchi K., Yoshioka Y., Baba T., Konishi I., Mandai M. IFN- γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br. J. Cancer. 2015;112:1501–1509. doi: 10.1038/bjc.2015.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkal A.A., Brewer R.E., Markovic M., Kowarsky M., Barkal S.A., Zaro B.W., Krishnan V., Hatakeyama J., Dorigo O., Barkal L.J. CD24 signalling through macrophage Siglec-10 is a new target for cancer immunotherapy. Nature. 2019;572:392–396. doi: 10.1038/s41586-019-1456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell L.C.K., Pollara G., Pascoe M., Tomlinson G.S., Lehloenya R.J., Roe J., Meldau R., Miller R.F., Ramsay A., Chain B.M. In vivo molecular dissection of the effects of HIV-1 in active Tuberculosis. PLoS Pathog. 2016;12:e1005469. doi: 10.1371/journal.ppat.1005469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci R., Martin A., Bommarito D., Wang K., Hansen S.H., Freeman G.J., Ritz J. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. OncoImmunology. 2015;4:e1008824. doi: 10.1080/2162402X.2015.1008824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- Braun D.A., Ishii Y., Walsh A.M., Van Allen E.M., Wu C.J., Shukla S.A., Choueiri T.K. Clinical validation of PBRM1 alterations as a marker of immune checkpoint inhibitor response in renal cell carcinoma. JAMA Oncol. 2019;5:1631–1633. doi: 10.1001/jamaoncol.2019.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun D.A., Hou Y., Bakouny Z., Ficial M., Sant’ Angelo M., Forman J., Ross-Macdonald P., Berger A.C., Jegede O.A., Elagina L. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020;26:909–918. doi: 10.1038/s41591-020-0839-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun D.A., Street K., Burke K.P., Cookmeyer D.L., Denize T., Pedersen C.B., Gohil S.H., Schindler N., Pomerance L., Hirsch L. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell. 2021:632–648. doi: 10.1016/j.ccell.2021.02.013. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canale F.P., Ramello M.C., Núñez N., Furlan C.L.A., Bossio S.N., Serrán M.G., Boari J.T., Castillo A.del, Ledesma M., Sedlik C. CD39 expression defines cell exhaustion in tumor-infiltrating CD8+ T cells. Cancer Res. 2018;78:115–128. doi: 10.1158/0008-5472.CAN-16-2684. [DOI] [PubMed] [Google Scholar]
- Chevrier S., Levine J.H., Zanotelli V.R.T., Silina K., Schulz D., Bacac M., Ries C.H., Ailles L., Jewett M.A.S., Moch H. An immune atlas of clear cell renal cell carcinoma. Cell. 2017;169:736–749.e18. doi: 10.1016/j.cell.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choueiri T.K., Fishman M.N., Escudier B., McDermott D.F., Drake C.G., Kluger H., Stadler W.M., Perez-Gracia J.L., McNeel D.G., Curti B. Immunomodulatory activity of nivolumab in metastatic renal cell carcinoma. Clin. Cancer Res. 2016;22:5461–5471. doi: 10.1158/1078-0432.CCR-15-2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choueiri T.K., Motzer R.J. Systemic therapy for metastatic renal-cell carcinoma. N Engl J Med. 2017;376:354–366. doi: 10.1056/NEJMra1601333. [DOI] [PubMed] [Google Scholar]
- Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin M., Bigley V. Human dendritic cell subsets: an update. Immunology. 2018;154:3–20. doi: 10.1111/imm.12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton C.J., Morgan M., Gunaratne P.H., Wheeler D.A., Gibbs R.A., Gordon Robertson A., Chu A., Beroukhim R., Cibulskis K., Signoretti S. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M., Zhu C., Kuchroo V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017;276:97–111. doi: 10.1111/imr.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeTomaso D., Jones M.G., Subramaniam M., Ashuach T., Ye C.J., Yosef N. Functional interpretation of single cell similarity maps. Nat. Commun. 2019;10:4376. doi: 10.1038/s41467-019-12235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhen T., Duhen R., Montler R., Moses J., Moudgil T., de Miranda N.F., Goodall C.P., Blair T.C., Fox B.A., McDermott J.E. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018;9:2724. doi: 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- Frankish A., Diekhans M., Ferreira A.-M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz A., Shin D.S., Moreno B.H., Saco J., Escuin-Ordinas H., Rodriguez G.A., Zaretsky J.M., Sun L., Hugo W., Wang X. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–1201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M., Rowan A.J., Horswell S., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A., Tarpey P. Massachusetts Medical Society; 2012. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero J.L. Macrophages: the road less traveled, changing anticancer therapy. Trends Mol. Med. 2018;24:472–489. doi: 10.1016/j.molmed.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M.X., Cuoco M.S., Crowdis J., Bosma-Moody A., Zhang Z., Bi K., Kanodia A., Su M.-J., Ku S.-Y., Garcia M.M. Transcriptional mediators of treatment resistance in lethal prostate cancer. Nat. Med. 2021 doi: 10.1038/s41591-021-01244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horisberger M.A., Staeheli P., Haller O. Interferon induces a unique protein in mouse cells bearing a gene for resistance to influenza virus. PNAS. 1983;80:1910–1914. doi: 10.1073/pnas.80.7.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J.J., Purdue M.P., Signoretti S., Swanton C., Albiges L., Schmidinger M., Heng D.Y., Larkin J., Ficarra V. Renal cell carcinoma. Nat. Rev. Dis. Primers. 2017;3:1–19. doi: 10.1038/nrdp.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen C.S., Prokhnevska N., Master V.A., Sanda M.G., Carlisle J.W., Bilen M.A., Cardenas M., Wilkinson S., Lake R., Sowalsky A.G. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. 2019;576:465–470. doi: 10.1038/s41586-019-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D., Lu M., Jung K.H., Park J.H., Yu L., Onuchic J.N., Kaipparettu B.A., Levine H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. U S A. 2019;116:3909–3918. doi: 10.1073/pnas.1816391116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klement J.D., Paschall A.V., Redd P.S., Ibrahim M.L., Lu C., Yang D., Celis E., Abrams S.I., Ozato K., Liu K. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Invest. 2018;128:5549–5560. doi: 10.1172/JCI123360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The molecular signatures database Hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lines J.L., Sempere L.F., Wang L., Pantazi E., Mak J., O’Connell S., Ceeraz S., Suriawinata A.A., Yan S., Ernstoff M.S. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014;74:1924–1932. doi: 10.1158/0008-5472.CAN-13-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A., Marchesi F., Malesci A., Laghi L., Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017;14:399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott D.F., Huseni M.A., Atkins M.B., Motzer R.J., Rini B.I., Escudier B., Fong L., Joseph R.W., Pal S.K., Reeves J.A. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018;24:749–757. doi: 10.1038/s41591-018-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao D., Margolis C.A., Gao W., Voss M.H., Li W., Martini D.J., Norton C., Bossé D., Wankowicz S.M., Cullen D. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359:801–806. doi: 10.1126/science.aan5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B.C., Sen D.R., Al Abosy R., Bi K., Virkud Y.V., LaFleur M.W., Yates K.B., Lako A., Felt K., Naik G.S. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019;20:326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer R.J., Hutson T.E., McCann L., Deen K., Choueiri T.K. Overall survival in renal-cell carcinoma with pazopanib versus sunitinib. N Engl J Med. 2014;370(18):1769–1770. doi: 10.1056/NEJMc1400731. [DOI] [PubMed] [Google Scholar]
- Motzer R.J., Penkov K., Haanen J., Rini B., Albiges L., Campbell M.T., Venugopal B., Kollmannsberger C., Negrier S., Uemura M. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. 2019;380:1103–1115. doi: 10.1056/NEJMoa1816047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon: fast and bias-aware quantification of transcript expression using dual-phase inference. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Kröger A., Sierra-Filardi E., Domínguez-Soto A., Samaniego R., Corcuera M.T., Gómez-Aguado F., Ratnam M., Sánchez-Mateos P., Corbí A.L. Folate receptor β is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009;69:9395–9403. doi: 10.1158/0008-5472.CAN-09-2050. [DOI] [PubMed] [Google Scholar]
- Ricketts C.J., De Cubas A.A., Fan H., Smith C.C., Lang M., Reznik E., Bowlby R., Gibb E.A., Akbani R., Beroukhim R. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018;23:313–326.e5. doi: 10.1016/j.celrep.2018.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rini B.I., Plimack E.R., Stus V., Gafanov R., Hawkins R., Nosov D., Pouliot F., Alekseev B., Soulières D., Melichar B. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380:1116–1127. doi: 10.1056/NEJMoa1816714. [DOI] [PubMed] [Google Scholar]
- Sade-Feldman M., Yizhak K., Bjorgaard S.L., Ray J.P., de Boer C.G., Jenkins R.W., Lieb D.J., Chen J.H., Frederick D.T., Barzily-Rokni M. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018;175:998–1013.e20. doi: 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A.C., Dündar F., Zumbo P., Chandran S.S., Klebanoff C.A., Shakiba M., Trivedi P., Menocal L., Appleby H., Camara S. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571:270–274. doi: 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Şenbabaoğlu Y., Gejman R.S., Winer A.G., Liu M., Van Allen E.M., de Velasco G., Miao D., Ostrovnaya I., Drill E., Luna A. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016;17:231. doi: 10.1186/s13059-016-1092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergushichev A.A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. BioRxiv. 2016 060012. [Google Scholar]
- Shen R., Seshan V.E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommen D.S., Schumacher T.N. T cell dysfunction in cancer. Cancer Cell. 2018;33:547–562. doi: 10.1016/j.ccell.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S., Xu H., Litchfield K., Rowan A., Chambers T., Lopez J.I., Nicol D., O’Brien T., Larkin J., Horswell S. Tracking cancer evolution reveals constrained routes to metastases: TRACERx renal. Cell. 2018;173:581–594.e12. doi: 10.1016/j.cell.2018.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt L., Schmitz N., Kurrer M.O., Bauer M., Hinton H.I., Behnke S., Gatto D., Sebbel P., Beerli R.R., Sonderegger I. VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J. Clin. Invest. 2006;116:2817–2826. doi: 10.1172/JCI25673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N., Liang H., Zen K. Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front Immunol. 2014;5:614. doi: 10.3389/fimmu.2014.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettersten H.I., Aboud O.A., Lara P.N., Weiss R.H. Metabolic reprogramming in clear cell renal cell carcinoma. Nat. Rev. Nephrol. 2017;13:410–419. doi: 10.1038/nrneph.2017.59. [DOI] [PubMed] [Google Scholar]
- Wolfl M., Kuball J., Ho W.Y., Nguyen H., Manley T.J., Bleakley M., Greenberg P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood. 2007;110:201–210. doi: 10.1182/blood-2006-11-056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock S.L., Lopez R., Klein A.M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M.D., Behjati S. SoupX removes ambient RNA contamination from droplet based single-cell RNA sequencing data. BioRxiv. 2020 doi: 10.1093/gigascience/giaa151. 303727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M.D., Mitchell T.J., Braga F.A.V., Tran M.G.B., Stewart B.J., Ferdinand J.R., Collord G., Botting R.A., Popescu D.-M., Loudon K.W. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science. 2018;361:594–599. doi: 10.1126/science.aat1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., Lu M., Jia D., Ma J., Ben-Jacob E., Levine H., Kaipparettu B.A., Onuchic J.N. Modeling the genetic regulation of cancer metabolism: interplay between glycolysis and oxidative phosphorylation. Cancer Res. 2017;77:1564–1574. doi: 10.1158/0008-5472.CAN-16-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Harden K., Gonzalez L.C., Francesco M., Chiang E., Irving B., Tom I., Ivelja S., Refino C.J., Clark H. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009;10:48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
- Zhou L., Zhuo H., Ouyang H., Liu Y., Yuan F., Sun L., Liu F., Liu H. Glycoprotein non-metastatic melanoma protein b (Gpnmb) is highly expressed in macrophages of acute injured kidney and promotes M2 macrophages polarization. Cell. Immunol. 2017;316:53–60. doi: 10.1016/j.cellimm.2017.03.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TPM expression matrix, patient-level metadata, and sample-level metadata for Checkmate 009 bulk RNA-seq samples.
Data Availability Statement
Raw single cell RNA-seq and bulk whole exome sequencing data are deposited to dbGaP: phs002065.v1.p1. Processed single cell RNA-Seq data can also be accessed via the Single Cell Portal (https://singlecell.broadinstitute.org/single_cell/study/SCP1288/tumor-and-immune-reprogramming-during-immunotherapy-in-advanced-renal-cell-carcinoma#study-summary). Code used for all processing and analysis is available upon request.
Transcriptional signatures for progenitor and terminally exhausted CD8+ T cells (CD8_G and CD8_B, respectively) were obtained from published results (Sade-Feldman et al., 2018) (GEO: GSE120575). For the Checkmate 009 cohort, raw bulk RNA-seq data were obtained from published results (Miao et al., 2018) (dbGaP: phs001493). For the Checkmate 025 cohort, normalized bulk RNA-seq, clinical, and mutational data were obtained from published results (Braun et al., 2020) (European Genome-Phenome Archive: EGAS00001004290, EGAS00001004291, EGAS00001004292). For the TCGA clear cell renal cell carcinoma (KIRC) cohort, normalized bulk RNA-seq data were obtained from Firebrowse (http://firebrowse.org/, illuminahiseq_rnaseqv2-RSEM_genes_normalized). Clinical data were downloaded from the cBioPortal (https://www.cbioportal.org/study/clinicalData?id=kirc_tcga). Somatic mutation data were obtained from published results (Ricketts et al., 2018) (https://gdc.cancer.gov/). GISTIC2 results describing amplification and deletion events in TCGA KIRC were downloaded from Firebrowse (http://firebrowse.org/, gdac.broadinstitute.org_KIRC-TP.CopyNumber_Gistic2.Level_4.2016012800.0.0).