

Abstract
Mining of plant-derived antimicrobials is the major focus at current to counter antibiotic resistance. This study was conducted to characterize the antimicrobial activity and mode of action of linalyl anthranilate (LNA) against carbapenemase-producing Klebsiella pneumoniae (KPC-KP). LNA alone exhibited bactericidal activity at 2.5% (V/V), and in combination with meropenem (MPM) at 1.25% (V/V). Comparative proteomic analysis showed a significant reduction in the number of cytoplasmic and membrane proteins, indicating membrane damage in LNA-treated KPC-KP cells. Up-regulation of oxidative stress regulator proteins and down-regulation of oxidative stress-sensitive proteins indicated oxidative stress. Zeta potential measurement and outer membrane permeability assay revealed that LNA increases both bacterial surface charge and membrane permeability. Ethidium bromide influx/efflux assay showed increased uptake of ethidium bromide in LNA-treated cells, inferring membrane damage. Furthermore, intracellular leakage of nucleic acid and proteins was detected upon LNA treatment. Scanning and transmission electron microscopies again revealed the breakage of bacterial membrane and loss of intracellular materials. LNA was found to induce oxidative stress by generating reactive oxygen species (ROS) that initiate lipid peroxidation and damage the bacterial membrane. In conclusion, LNA generates ROS, initiates lipid peroxidation, and damages the bacterial membrane, resulting in intracellular leakage and eventually killing the KPC-KP cells.
Keywords: Comparative proteomic, KPC-KP, Linalyl anthranilate, Membrane damage, ROS
Graphical abstract
Highlights
-
•
Novel antimicrobial, LNA kills KPC-KP cells effectively at 2.5%.
-
•
LNA disrupts bacterial membrane by generating reactive oxygen species.
-
•
LNA causes intracellular leakage of nucleic acids and proteins, killing the cells.
1. Introduction
The emergence of carbapenemase-producing Klebsiella pneumoniae (KPC-KP) has been a major setback in the patient’s treatment outcomes since its first appearance in 1996 [1]. KPC-KP was prioritized by WHO in 2017 as one of the few antibiotic-resistant pathogens that urgently require novel treatment [2]. KPC-KP expresses the carbapenemase (KPC) gene that deactivates all existing β-lactam antibiotics including carbapenem, one of the last resort antibiotics used for severe bacterial infections. Within the next few years, antibiotic exposure pressures expedited the emergence of a more robust form of carbapenemase, the NDM-1 metallo-β-lactamase and VIM metallo-β-lactamase [3,4]. Emergence of these carbapenemases increases the mortality rate of patients infected with KPC-KP cells due to the lack of treatment options. The situation was exacerbated as the rates of novel antibiotic discovery have slowed significantly and the timelines for antibiotic approval for clinical use have been extended. A total of eleven antibiotics were discovered between 1995 and 1999, while only six were discovered between 2010 and 2014 [5].
To address the above issue on antibiotic resistance, the mining of novel antimicrobials is crucial; researchers have now diverted their attention to antimicrobial mining from the traditional microorganism to plants. This is because plants produce a plethora of complex secondary metabolites that have potential applications as novel antimicrobials. Plant secondary metabolites can be categorized into four classes, namely terpenes and terpenoids, phenolic compounds, alkaloids, and sulfur-containing compounds [6]. Terpenes and terpenoids, one of the major classes of plant secondary metabolites, have gained much attention due to their importance in physiological and ecological roles of plants, including industrial applications ranging from flavors to fragrances and pharmaceutical applications, particularly in aromatherapy [7]. Terpenes have a base unit of five carbons known as an isoprene unit and can be categorized into different groups such as the monoterpenes (C10), sesquiterpenes (C15), diterpenes (C20) and triterpenes (C30) [8]. Terpenoids, on the other hand, are oxygen-containing terpenes units [8]. Numerous terpenes and terpenoids have been reported in previous studies for antimicrobial activity against pathogenic bacteria such as Escherichia coli, Staphylococcus aureus, and Pseudomonas aeruginosa [[9], [10], [11]]. For instance, Ultee et al. [12] reported the antibacterial activity of the monoterpene, cymene, and monoterpenoid, carvacrol against Bacillus cereus. Further investigation has shown that both compounds may have disrupted the cellular plasma membrane, resulting in the alteration of the membrane potential as well as the pH gradient and intracellular ATP levels [12]. Later on, Zuniga et al. [13] related the DNA damage observed in terpene catechols-treated bacteria with the generation of reactive oxygen species (ROS). Another study by Rodenak-Kladniew et al. [14] has shown that terpene alcohol—linalool induced cell cycle arrest and apoptosis in HepG2 cells by generating ROS.
Oxidative stress is essentially the imbalance between the production of ROS during cellular respiration and metabolism, and the ability of cells to neutralize their harmful effects. ROS consists of radical and non-radical oxygen species such as hydrogen peroxide (H2O2), hydroxyl ion (OH−), hydroxyl radical (OH·), peroxide (O22−), singlet oxygen (1O2), and superoxide anion (O2−); these generally affect cellular components, including lipids, proteins, and nucleic acids [15,16]. Lipid peroxidation is a self-propagating chain reaction that involves reactions between ROS and membrane fatty acids [17,18]. At the cellular level, such reaction will cause membrane damage and eventually kill the cells. Interaction between ROS and proteins often results in covalent modification, which generally destabilizes and inactivates a particular protein [19]. Such modifications include carbonylation of arginine, lysine, proline, and threonine; conversion of histidine to oxo-histidine; and oxidation of cysteine and methionine, which impairs protein function [19]. Furthermore, the nucleic acid is also the common target of ROS, and oxidizing nucleotides, such as guanine, cause lesion, and DNA breakage [20]. This leads to the production of non-functional proteins that eventually kill the cells [20]. Antibiotics such as aminoglycosides, chloramphenicol, quinolones, and rifampicin, regardless of their modes of action, have been found to generate ROS during antimicrobial action [21]. For example, aminoglycosides, which disrupt protein synthesis, produce large amounts of hydroxyl radicals when proteins are mistranslated and misfolded [22]. Thus, the ROS inducer may serve as a potential therapeutic agent in the fight against antibiotic resistance.
Linalyl anthranilate (LNA) is an arene and terpene found in a variety of plants such as marjoram, Mexican giant hyssop, lavender, and thyme [[23], [24], [25], [26]]. Currently, LNA is commonly used in the food and beverage industry as a food flavoring agent. Our previous study involving the antimicrobial activity of lavender essential oil revealed that LNA is one of the major compounds in lavender essential oil with the highest composition at 49.5% [27]. This is similar to the study conducted by Yap et al. [25] which reported that LNA is the major compound in lavender essential oil with a total composition of 38.42%. This led us to believe that LNA could be the compound responsible for antimicrobial activity. To date, no study has been reported on the antimicrobial activity of this compound against any microorganism. However, LNA may play a role in inducing oxidative stress and killing the bacterial cell due to its nature and structure similarity to other members of a terpene. Therefore, this study aims to evaluate the antimicrobial activity of LNA alone and in combination with meropenem (MPM) against KPC-KP cells. This study also intends to elucidate the mode of action of LNA against KPC-KP cells using a comparative proteomic profiling analysis. The postulated mode of action is further validated by various qualitative and quantitative experiments.
2. Materials and methods
2.1. LNA and MPM
LNA (>99% purity, fragrance grade) and MPM (>98% purity, HPLC grade) used throughout the study were purchased from Penta Manufacturing Company (NJ, USA) and Sigma-Aldrich Corporation (MO, USA). MPM was dissolved in water to prepare a 10 mg/mL stock solution.
2.2. Bacterial strains and culture condition
The bacterial strains used in this study were KPC-KP BAA-1705 and Escherichia coli ATCC 25922 (EC25922), both obtained from American Type Culture Collection (Manassas, VA, USA). The bacterial strains were grown and maintained on Mueller-Hinton agar (MHA; Sigma-Aldrich, Missouri, USA). Subsequently, a single colony was inoculated into Mueller-Hinton broth (MHB; Sigma-Aldrich, Missouri, USA) at 37 °C and shaken at 250 rpm for 16 h according to the experimental procedures, unless otherwise specified.
2.3. Minimum inhibitory concentration (MIC) assay
The MIC assay was performed as described in the previous study [28]. Two-fold dilutions were performed in each test well to yield final well volumes consisting of 50 μL of LNA, 40 μL of a bacterial suspension at approximately 1 × 105 CFU/mL, and 10 μL of resazurin (7-hydroxy-3H-phenoxazin-3-one-10-oxide) at a final concentration of 0.02% (m/V). Apart from negative and growth controls, EC25922 was treated with MPM (1000 ng/mL to 0.5 ng/mL) as a positive control in this assay. All assays were performed in triplicates and incubated for 20 h at 37 °C with shaking at 200 rpm. The MICs of LNA and MPM were determined qualitatively from color change of resazurin and the assay was completed in triplicate.
2.4. Checkerboard assay
The checkerboard assay was performed as detailed in the previous study [28]. Ten serial, two-fold dilutions of MPM and five serial, two-fold dilutions of LNA were prepared to determine the combinatory effects of LNA and MPM against KPC-KP cells. Each well contained 25 μL of MPM and 25 μL of LNA, inoculated with 40 μL of bacterial suspension and 10 μL of resazurin to make a final concentration of approximately 1 × 105 CFU/mL and 0.02% (m/V). The 96-well plates were then incubated for 20 h at 37 °C with shaking at 200 rpm and the assay was completed in triplicate. The combinatory relationship between LNA and MPM was expressed in terms of the combined fractional inhibitory concentration index (FICIc) using the following formulas [29,30]:
| FICIc ≤ 0.5, synergistic; FICIc > 0.5–4.0, additive; FICIc > 4.0, antagonistic. |
2.5. Time kill assay
A standard inoculum of 1 × 105 CFU/mL was used for the time kill assay. The test concentrations of LNA and MPM used were determined from the checkerboard assay. The time kill analysis consisted of untreated KPC-KP cells (inoculum with MHB supplemented with 10% (m/V) Tween 80 at final concentration), KPC-KP cells treated with LNA (1.25%, V/V) and MPM (16 μg/mL), alone and in combination. Each treatment had a final volume of 20 mL with Tween 80 incorporated at a final concentration of 10% (m/V) to improve the solubility of LNA. The cells were incubated at 37 °C with shaking at 200 rpm. Immediately after inoculation, viable counting was performed every half-hourly until 4 h. About 50 μL of cells from each treatment group were obtained and subjected to a hundred-fold dilution with 0.85% (m/V) saline. The diluted samples were then plated onto MHA and incubated at 37 °C for 16 h, followed by colony counting. The assay was completed in triplicate.
2.6. Comparative proteomic analysis
2.6.1. Protein extraction
Protein extraction was carried out as described previously [31]. The KPC-KP cell treatment is detailed in Section 2.5 (LNA, 1.25% (V/V); MPM, 16 μg/mL). The LNA-treated and untreated cultures were supplemented with Tween 80 at a final concentration of 10% (m/V) to improve the solubility of LNA. A standard inoculum of 1 × 105 CFU/mL KPC-KP cells was used for both treated and untreated culture preparations. The cultures were incubated for 4 h at 37 °C with shaking at 200 rpm. The cell pellets from both treatment groups were obtained by centrifugation at 9,000 rpm for 10 min; pellets were washed at least three times and resuspended in 500 μL of cold protein extraction buffer (50 mM ammonium bicarbonate, 10 mM phenylmethylsulfonyl fluoride). The samples were then ultra-sonicated on the ice at 20 amplitude for 10 cycles; each cycle consisted of 10 s of sonication, followed by a 20 s cooling period (Qsonica Sonicator Q55, Fischer Scientific, USA). The ultra-sonicated samples were then centrifuged at 4 and 10,000 rpm for 1 h; the supernatant was then collected and the protein concentration was quantified by Bradford assay. The protein concentration of each sample was standardized to 1 mg/mL for the subsequent proteomic analysis. The assay was completed in triplicate.
2.6.2. Protein sample preparation
Approximately 100 μg of total protein was resuspended in 100 μL of 50 mM ammonium bicarbonate (pH 8.0). RapiGest surfactant (Waters Corporation, USA) was added to the extracted protein in equal parts at a final concentration of 0.05% (V/V). The proteins from each sample were then concentrated to a volume of 100 μL using a Vivaspin™ column (GE Healthcare, USA) with a molecular weight cut-off (MWCO) of 3000 Da and incubated at 80 for 15 min. The protein was reduced using 5 mM dithiothreitol (DTT) at 60 for 30 min and then alkylated in the dark using 10 mM iodoacetamide at room temperature for 45 min. Proteolytic digestion was performed using Trypsin Gold (Promega, USA) at a ratio by parts of 100:0.5 protein to trypsin, followed by overnight incubation at 37 . Tryptic digestion and RapiGest activity were terminated by the addition of 1 μL of concentrated trifluoroacetic acid and then the samples were incubated at 37 for 20 min. The tryptic peptide solution from each sample was centrifuged at 14,000 rpm for 20 min and the resulting supernatants were collected and stored at −80 until subsequent analysis.
2.6.3. Nano liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis
Nano LC-MS/MS analysis was performed as described in Ref. [31] using an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific, USA). The samples (2 μL containing 2 μg peptides) were injected and separated on an EASY-nLC 1000 (Dionex, Thermo Scientific, USA) equipped with an Easy-Spray Column Acclaim PepMap™ C18 100 Å (2 μm, 50 μm × 15 cm, Thermo Scientific, USA). The samples were then separated by a gradient of 5%–40% (V/V) acetonitrile (ACN) with 0.1% (V/V) formic acid (FA) for 91 min, followed by a wash gradient of 85% ACN with 0.1% FA for 6 min and then equilibrated back to 5% (V/V) ACN with 0.1% (V/V) FA for 1 min and maintained until subsequent sample injection. The flow rate was set at 250 nL/min. The mass spectrometer was operated in positive ion mode with a nanospray voltage of 1.5 kV and a source temperature of 250 . The instrument was operated in data-dependent acquisition mode with a survey MS scan by Orbitrap MS using the following parameters: a mass range of m/z 310–1800 with resolving power of 120,000, automatic gain control (AGC) of 400,000 and maximum injection time of 50 ms. The top speed mode of 3 s was used in the selection of precursors with a monoisotopic charge state of 2–7. These precursors were further analyzed in the MS/MS scan. All precursors were filtered using a 20-s dynamic exclusion window and an intensity threshold of 5,000. The MS/MS spectra were analyzed by ion trap MS with the following parameters: rapid scan rate with resolving power of 60,000, AGC of 100, isolation window of 1.6 m/z and maximum injection time of 250 ms. Precursors were then fragmented by collision-induced dissociation and high-energy collision dissociation at a normalized collision energy of 30% to 28%.
2.6.4. Protein identification and comparative analysis
Raw data were processed using Thermo ScientificTM Proteome DiscovererTM Software v2.1 with the SEQUEST® HT search engine. The MS ion intensities were calculated based on the accurate mass and time tag strategy. The accurate alignment of detected LC retention time and m/z value across different analyses and the area under chromatographic elution profiles of the identified peptides can be compared between different samples. For protein identification, the peptide identification data were compared with the Uniprot® K. pneumoniae database with a 1% strict FDR and 5% relax FDR criteria using Percolator®. Search parameters were set up to two mis-cleavages with fixed amino acid modification through carbamidomethylation and variable modification through methionine oxidation, together with asparagine and glutamine deamidation. A fragment tolerance of 0.6 Da and a precursor tolerance of 10 ppm were used with trypsin as a digestion enzyme. Identified proteins with at least two unique peptides suggested greater confidence of protein identity. Protein quantification and statistical analyses were performed using Perseus Software v1.6.0.7 (Max Planck Institute of Biochemistry). Each control and the treated sample consisted of three biological replicates with three technical replicates, each analyzed by LC-MS/MS. A protein file with three technical replicates in txt format from Proteome Discoverer™ was uploaded to the Perseus system for further comparative analysis between samples. The data were log2-transformed to stabilize the variance and scale-normalized to the same mean intensity across the technical replicates. The mean value for all three technical replicates of the same biological samples was grouped in the same matrix and the valid values were obtained by filtering with ‘at least two’, eliminating proteins that existed only in one of the technical replicates. Finally, all the biological replicates of the same treatment group were consolidated under the same matrix, with the missing values input to random numbers derived from the normal distribution. The histograms were plotted to obtain a similarity comparison of the ratio for all samples. The differential expressed protein between control and treatment was detected using a t-test, the P-value was also adjusted for multiple-tests using a permutation-based false discovery rate with a randomization number of 250. Proteins were considered to be significantly differentiated between treatment groups with adjusted q-value <0.05 and fold change ≤−1 or ≥1.
2.7. Bacterial membrane disruption assays
2.7.1. Zeta potential measurement
KPC-KP cell treatment is described in Section 2.5 (LNA, 1.25% (V/V); MPM, 16 μg/mL). The zeta potential of untreated, LNA-treated, and MPM-treated KPC-KP cells was determined as detailed in Ref. [28] using the Zetasizer Nano ZS instrument (Malvern Instruments, Malvern, UK). The treatment time for all treatment groups was determined in the time kill analysis, whereas the concentration of LNA and MPM used was determined by the checkerboard assay. Treated cells were washed with 0.85% (m/V) saline at least five times before zeta potential measurement. The experiment was performed in triplicate.
2.7.2. Outer membrane permeability assay
The outer membrane permeability assay was performed as detailed in previous studies [25,28]. Overnight culture of KPC-KP cells was washed, adjusted to an optical density (OD600nm) 0.3 and followed by treatment with LNA with concentration and treatment time as determined by the checkerboard assay and time kill analysis. After treatment completion, the samples were first washed with 0.85% (m/V) saline (five times) to remove the treatment and then divided into two equal portions of 10 mL. Next, sodium dodecyl sulfate (SDS) solution at a final concentration of 0.1% (m/V) was added to one portion, whereas 0.85% (m/V) saline was added to the other. SDS acts as a permeabilizing probe that causes cell death when a sudden influx has occurred. This can be measured in terms of OD600nm at intervals of 0, 5, 10, 30, and 60 min using a spectrophotometer. The assay was completed in triplicates.
2.7.3. Ethidium bromide influx/efflux assay
The assay was performed as described previously with slight modification [32]. Overnight cultures of KPC-KP were washed and adjusted to OD600nm of 0.5 with MHB. The cells were then distributed into three groups; one group consisted of untreated KPC-KP cells and two groups were LNA-treated KPC-KP cells. Untreated KPC-KP cells were supplemented with ethidium bromide at a final concentration of 1 μg/mL, whereas LNA-treated KPC-KP cells were supplemented with LNA and ethidium bromide at a final concentration of 1.25% (V/V) and 0.1% (m/V), respectively. The accumulation of ethidium bromide was measured using an EnSight multimode plate reader (PerkinElmer, Massachusetts, USA) with an excitation wavelength of 530 nm and an emission wavelength of 585 nm every 5 min up to 1 h at 37 . Subsequently, the cells were harvested and washed with MHB at least 2 times. Fresh MHB supplemented with 0.5% (m/V) glucose was added to the untreated KPC-KP cells. One of the LNA-treated KPC-KP groups was reconstituted with MHB supplemented with 0.5% (m/V) glucose and 1.25% (V/V) LNA, whereas the second group of LNA-treated KPC-KP cells were reconstituted with MHB supplemented with 0.5% (m/V) glucose, 1.25% (V/V) LNA and 0.1% (m/V) ethidium bromide. Ethidium bromide efflux was again measured at the wavelength mentioned above for another 1 h every 5 min at 37 . The assay was completed in triplicate.
2.7.4. Nucleic acid leakage measurement
The nucleic acid leakage was measured as detailed in Ref. [33]. KPC-KP cell treatment is detailed in Section 2.5 (LNA, 1.25% (V/V); MPM, 16 μg/mL). The cell pellets of untreated and LNA-treated KPC-KP cells were collected at 10,000 rpm centrifugation for 5 min. The supernatant was collected and measured for nucleic acid at a ultraviolet (UV) absorption wavelength of 260 nm using a UV–Vis spectrophotometer (Thermo Fischer Scientific, Massachusetts, USA). The assay was completed in triplicate.
2.7.5. Protein leakage measurement
The protein leakage was measured as detailed in Ref. [33]. KPC-KP cell treatment is detailed in Section 2.5 (LNA, 1.25% (V/V); MPM, 16 μg/mL). The cell pellets of untreated and LNA-treated KPC-KP cells were collected by centrifugation at 10,000 rpm for 5 min. The supernatant was collected and measured at 595 nm for protein content using the Bradford solution. The assay was completed in triplicate.
2.7.6. Scanning electron microscopy
Scanning electron microscopy was performed as detailed previously [28]. KPC-KP cell treatment is detailed in Section 2.5 (LNA, 1.25% (V/V); MPM, 16 μg/mL). The cells were harvested and the pellet was washed with 0.85% (m/V) saline for five times. Following this, the samples were then fixed with 4% (V/V) glutaraldehyde for 5 h and 1% (m/V) osmium tetroxide for 2 h at 4 °C. Sodium cacodylate buffer at 0.1 M was used in all the washing steps. Then, the samples were further dehydrated by sequential exposure to increased acetone concentrations (35%–95%, V/V) for 10 min, followed by 100% (V/V) acetone for 15 min for three times. After dehydration, the samples were subjected to critical point drying for 30 min (BalTec CPD 030, Bal-Tec, Balzers, Liechtenstein). The samples were then secured onto the specimen stub using double-sided tape. Finally, the samples were sputter-coated with gold using a cool sputter coater (BalTec SCD 005) and observed via a JEOL JSM-6400 instrument (JEOL, Tokyo, Japan) at 15 kV.
2.7.7. Transmission electron microscopy
KPC-KP cell treatment is detailed in Section 2.5 (LNA, 1.25% (V/V); MPM, 16 μg/mL). Untreated and LNA-treated KPC-KP cells were harvested and the pellet was washed with 0.85% (m/V) saline for five times, followed by fixation in 4% (V/V) glutaraldehyde for 2 days at 4 °C. Then, the specimen was washed with 0.1 M sodium cacodylate buffer for 30 min three times. Subsequently, the specimen was post-fixed in 1% (m/V) osmium tetroxide for 2 h at 4 °C. The specimen was then washed again three times with 0.1 M sodium cacodylate buffer for 30 min. Next, the specimen was subjected to a series of acetone washes for dehydration purposes. The specimen was washed with 35% (V/V) acetone, followed by 50% (V/V), 75% (V/V) and 95% (V/V) acetone in each wash for 45 min. Finally, 100% (V/V) acetone was used in the final washing step, which took 1 h and was repeated for three times. Next, the specimens were subjected to infiltration procedure with various ratios of acetone and resin mixture (1:1 for 12 h, 1:3 for 12 h and 0:1 for 16 h), before beam capsulation. The capsulated specimen was polymerized in an oven at 60 °C for 48 h. Upon completion of polymerization, the specimens block was cut into 1 μm thick sections and stained with toluidine blue and dried on a hot plate. Excess stains were removed by washing under tap water. The specimens were then viewed under a light microscope and the areas of interest were selected for ultrathin sectioning. The resin block was then trimmed according to the selected area of interest by shaving the resin blocks into a trapezoid having dimensions not exceeding 0.5 mm across the base, 0.4 mm across the top, and 0.3 mm along the sides. Ultrathin sectioning was performed via i-Ultramicrotome EM UC6 (Leica, Germany), and samples were collected from the surface of the water bath and placed on the copper mesh. The samples were then stained with uranyl acetate for 15 min and then with lead for 10 min. Each sample was washed before the next stain. The samples were then viewed under transmission electron microscope Leo Libra-120 (ZEISS, Germany) at 120 kV.
2.8. Oxidative stress assays
2.8.1. Lipid peroxidation assay
Lipid peroxidation assay was performed as detailed in Ref. [34]. KPC-KP cell treatment is detailed in Section 2.5 (LNA, 1.25% (V/V); MPM, 16 μg/mL). The cell pellets were collected using centrifugation at 10,000 rpm for 5 min and washed with phosphate-buffered saline (PBS). The supernatant collected was referred to as treatment media. The resulting cell pellet was then sonicated as detailed in Section 2.7.1 with the supernatant collected and termed as cell lysate. Both treatment media and cell lysate were subjected to malondialdehyde (MDA) measurement by mixing 500 μL of either treatment media and cell lysate to 400 μL of 15% (V/V) trichloroacetic acid and 800 μL of 0.67% (m/V) thiobarbituric acid (TBA) in 0.01% (V/V) butylated hydroxytoluene. The samples were then vortexed and incubated at 95 for 20 min in a water bath. A volume of 3 mL of butanol was added to the sample, followed by gentle mixing. A volume of 200 μL of the butanol phase was collected from each sample with absorbance measured at 532 nm. The amount of MDA present was estimated using the MDA standard curve and normalized based on the protein concentration of each sample. The assay was completed in triplicate.
2.8.2. ROS measurement
ROS measurement was performed as detailed in Ref. [34]. KPC-KP cell treatment is detailed in Section 2.5 (LNA, 1.25% (V/V); MPM, 16 μg/mL). The cell pellets were collected by centrifugation at 10,000 rpm for 5 min and washed with PBS. Untreated and LNA-treated KPC-KP cells were treated with 20 μM of 2′,7′–dichlorofluorescein diacetate (DCF-DA) for 30 min at 37 °C. After incubation, the cell pellet was collected by centrifugation at 10,000 rpm for 5 min with a supernatant consisting of DCF-DA removed. The cell pellets were resuspended in PBS and the fluorescence intensity was measured at an excitation and emission wavelengths of 485 and 528 nm using the Tecan microplate reader (Tecan Trading AG, Switzerland). The assay was completed in triplicate.
3. Results and discussion
3.1. Antimicrobial activity and killing kinetics of LNA against KPC-KP cells
In recent years, antibiotic resistance in human infections has been a major challenge in the clinical setting, preventing patients from recovering and even increasing mortality during infection. This is further complicated by the emergence of carbapenem-resistant bacteria, such as KPC-KP, in the last decade. This resistance phenomenon is particularly worrying in the medical field, as carbapenem is the last-resort antibiotic, which has the widest range of activity and highest potency amongst all β-lactam antibiotics [35]. Thus, the discovery of novel antimicrobials is crucial, spurring many research groups to look at plant secondary metabolites with the hope of solving the problem, especially for terpenes and terpenoids, which are known to be bactericidal. Additionally, the discovery of novel antimicrobials will revive the efficacy of older generation antibiotics through combinatory antimicrobial treatment. As detailed in Table 1, LNA demonstrated bactericidal activity against KPC-KP cells at MIC of 2.5% (V/V), 9.2 μmol/L, while the combination with MPM resulted in an additive interaction that further reduces the MIC to 1.25% (V/V), 4.6 μmol/L. MPM had a MIC value of 32 μg/mL (0.00008 μmol/L) to 16 μg/mL (0.00004 μmol/L), indicating the potential of LNA as an antibiotic resistance modifier.
Table 1.
MIC, FIC and FIC indices of LNA and MPM against KPC-KP cells.
| Antimicrobial agents | KPC-KP |
Type of interaction | |||
|---|---|---|---|---|---|
| MICO | FIC | FICI | FICIc | ||
| LNA (%, V/V) | 2.5 | 1.25 | 0.50 | 1.00 | Additive |
| MPM (μg/mL) | 32 | 16 | 0.50 | ||
MIC: minimum inhibitory concentration; FIC: fractional inhibitory concentration; LNA: linalyl anthranilate; MPM: meropenem; KPC-KP: carbapenemase-producing Klebsiella pneumoniae. MICO: MIC of one component alone; FIC: MIC of one component in the most effective combination; FICIC: total FICI of the combination of both samples. FICIc ≤0.5: synergistic; FICIc >0.5–4.0: additive; FICIc >4.0: antagonistic.
Time kill analysis was first performed to determine the efficiency of LNA in killing KPC-KP cells, alone and in combination with MPM. As shown in Fig. 1, the combination of LNA and MPM at their respective sub-inhibitor concentration successfully killed all KPC-KP cells within 1.5 h. Hence, 1.5 h was used as the standard treatment time for subsequent assays.
Fig. 1.

Time kill kinetics of untreated carbapenemase-producing Klebsiella pneumoniae (KPC-KP) cells and KPC-KP cells treated with linalyl anthranilate (LNA), meropenem (MPM), and LNA-MPM combination.
3.2. Comparative proteomic analysis revealed loss of membrane and cytoplasmic proteins and signs of oxidative stress in LNA-treated KPC-KP cells
The untreated and LNA-treated KPC-KP cells were subjected to whole protein extraction for comparative proteomic analysis. The Pearson correlation value and principal component analysis showed good separation between the three treatment groups, indicating significant changes in proteomic abundance between the groups (Figs. S1 and S2). As shown in Fig. 2A-i, we have successfully identified 368 proteins from untreated KPC-KP cells and 361 proteins from LNA-treated KPC-KP cells. A total of 296 similar proteins were shared by both groups. From the analysis, 67 proteins were up-regulated and 85 proteins were down-regulated following the exposure to LNA (Fig. 2A-ii & iii).
Fig. 2.

Comparative proteomic analysis between untreated and LNA-treated KPC-KP cells. (A-i) Venn diagram of the total protein obtained from untreated and LNA-treated KPC-KP cells; (A-ii) The total numbers of exclusive, up-regulated, and down-regulated proteins; (A-iii) Volcano plot showing up-regulated (designated green square) and down-regulated (designated red square) proteins of the LNA-treated KPC-KP cells. GO analysis for LNA-treated KPC-KP cells in terms of biological processes (B-i), cellular components (B-ii) and molecular functions (B-iii) of identified proteins and their relative abundance in terms of the mentioned GO categories (B-iv). (C) KEGG pathway analysis of differentially expressed proteins in LNA-treated KPC-KP cells. The proteomic analysis is detailed in the Tables S1–S3. LNA: linalyl anthranilate; KPC-KP: carbapenemase-producing Klebsiella pneumoniae; GO: gene ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes.
Furthermore, 65 proteins were found to be exclusive towards the LNA-treated cells and 72 proteins were exclusive to untreated cells (Fig. 2A-ii). All identified proteins were subjected to gene ontology analysis and classified into three categories, namely, biological processes, cellular components, and molecular functions together with total protein abundance in each group, followed by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis to determine the overall effect of LNA on the proteome of the KPC-KP cells (Figs. 2B and C). In terms of biological processes of KPC-KP cells exposed to LNA, the majority of the proteins identified were categorized under cellular and metabolic processes (36.21% and 34.22%), followed by cellular component organization and response to a stimulus (9.68% and 8.09%) as shown in Fig. 2B-i. Cellular component-wise, the majority of proteins identified were categorized under cytoplasm and membrane (61.11% and 16.67%) followed by protein-containing complex and ribosome (10.61% and 8.84%; Fig. 2B-ii).
Subsequently, the molecular function categorizations of the identified proteins showed that the majority of proteins were involved in binding and catalytic activity (43.60% and 41.09%; Fig. 2B-iii). According to the KEGG pathway analysis shown in Fig. 2C, proteins related to protein biosynthesis were most affected by the exposure towards LNA, with a total of 57 proteins. This was followed by stress response and amino acid biosynthesis proteins, with 44 and 32 proteins, respectively. As seen in Fig. 2B-iv, LNA-treated KPC-KP cells reduced the abundance of cytoplasmic proteins compared to untreated cells. Additionally, after exposure to LNA, a large amount of membrane-related proteins had reduced abundance. For instance, after exposure to LNA, 132 cytoplasmic proteins and 36 membrane-related proteins had reduced abundance compared to untreated KPC-KP cells, suggesting a disrupted bacterial membrane that could lead to intracellular leakage, explaining the loss of cytoplasmic proteins.
From the proteomic KEGG pathway analysis, 19 stress-related proteins had increased abundance following exposure to LNA. Of the 19 up-regulated proteins, 4 were oxidative stress-related proteins involved in the repair of genetic material and proteins (Table 2). Additionally, we also found that the majority of proteins involved in ribosome biogenesis, and DNA and RNA processing in LNA-treated cells had reduced abundance compared to untreated KPC-KP cells (Table 2). Ribosomal and genetic material processing proteins are known to be relatively sensitive to oxidative stress [36,37]. For instance, oxidative damage can induce base substitution, addition, deletion, and other mutations in nucleic acids, leading to the formation of non-functioning proteins [38]. Besides, oxidative damage also affects proteins, especially ribosomal proteins due to the affinity of the ROS to RNA [39]. Studies have reported that oxidation of RNA causes indirect damages to ribosomal RNA through covalent modification, leading to defective protein synthesis [36,39]. Evidence from the proteomic analysis suggested that LNA induces oxidative stress in the KPC-KP cells. Induction of oxidative stress is also known to affect the integrity of the eukaryotic cell membranes. This is termed as lipid peroxidation, a self-propagating chain reaction that involves reactions between ROS and membrane fatty acid, eventually destroying the cell membrane [17,18].
Table 2.
List of up-regulated oxidative stress regulator proteins and down-regulated oxidative stress-sensitive proteins identified form LNA-treated KPC-KP cells.
| Upregulated/downregulated protein | Accession no. | Protein | Fold change |
|---|---|---|---|
| Upregulated protein | A6TCW1 | Protein RecA | 3.31 |
| A6TC47 | DNA ligase | 1.14 | |
| A6TCJ1 | Autonomous glycyl radical cofactor | 1.05 | |
| B5XQ04 | Holliday junction ATP-dependent DNA helicase RuvA | aa | |
| Downregulated protein | B5Y305 | 50S ribosomal protein L9 | −2.24 |
| B5XNB4 | 30S ribosomal protein S13 | −1.52 | |
| B5XN86 | 30S ribosomal protein S7 | ba | |
| B5XQC8 | 50S ribosomal protein L35 | ba | |
| A6TES6 | Ribosomal protein L11 methyltransferase | ba | |
| A6T6T1 | Ribosomal protein S12 methylthiotransferase RimO | ba | |
| A6TEC2 | Ribosomal RNA large subunit methyltransferase G | ba | |
| A6T4I7 | Ribosomal RNA small subunit methyltransferase A | ba | |
| A6TEU2 | Ribosomal RNA small subunit methyltransferase B | ba | |
| A6TG44 | Ribosomal RNA small subunit methyltransferase G | ba | |
| A6T4M5 | Ribosomal RNA small subunit methyltransferase H | ba | |
| A6TCL6 | Ribosome maturation factor RimM | ba | |
| B5XSX5 | Ribosome-binding factor A | ba |
a refers to protein exclusive to LNA-treated KPC-KP cells; b refers to protein exclusive to untreated KPC-KP cells. LNA: linalyl anthranilate; KPC-KP: carbapenemase-producing Klebsiella pneumoniae.
3.3. LNA kills KPC-KP cells by generating ROS, which disrupts the bacterial membrane via lipid peroxidation
As demonstrated by the concept, membrane-related assays and electron microscopy techniques were employed to demonstrate the ability of LNA to disrupt the bacterial membrane of KPC-KP cells. Lipid peroxidation assay and ROS measurement were also performed to validate its role in inducing oxidative stress, which has cascaded into lipid peroxidation and membrane disruption.
Lipopolysaccharide (LPS) is the major component in the outer membrane of Gram-negative bacteria, which grants the bacterial membrane high negative charges [40]. Thus, the disrupted outer membrane would have a more positive value indicated by an increase in the zeta potential measurement. According to Fig. 3A, the zeta potential of untreated KPC-KP cells has a negative value of −12.1 mV. KPC-KP cells exposed to several concentrations of LNA had significantly more positive zeta potential, from −7.53 mV to −8.60 mV, than untreated KPC-KP cells. Additionally, we also found that the membrane disruption ability of LNA was concentration-independent.
Fig. 3.

LNA disrupts the bacterial membrane of KPC-KP cells by inducing oxidative stress. (A) Membrane zeta potential of untreated and LNA-treated KPC-KP cells at several concentrations; (B) Outer membrane permeability of KPC-KP cells exposed to 0.1% (m/V) SDS or saline after treatment with LNA at 1.25% (V/V); (C) Ethidium bromide influx (i) and efflux (ii) activity of untreated and LNA-treated KPC-KP cells; (D) Intracellular leakage of UV-absorbing material: nucleic acid (i) and protein (ii) of KPC-KP cells exposed to LNA; (E) Oxidative stress assessment via ROS measurement (i) and lipid peroxidation assay (ii); and (F) Scanning and transmission electron micrographs of untreated (i and ii) and LNA-treated (iii and iv) KPC-KP cells (Δ indicates membrane damage). LNA: linalyl anthranilate; KPC-KP: carbapenemase-producing Klebsiella pneumoniae; SDS: sodium dodecyl sulfate; UV: ultraviolet; ROS: reactive oxygen species.
Moreover, the outer membrane permeability assay was performed to understand the bacterial membrane permeabilization role by LNA. A low concentration of SDS was used as a permeabilization probe, which shows bacterial membrane disruption. Under normal conditions, the outer membrane of Gram-negative bacteria is negatively charged due to the presence of LPS and generally repels other negatively-charged molecules, such as SDS. However, the disrupted bacterial membrane will enable the influx of SDS, eventually killing the cell. The concentration of SDS was optimized and fixed at 0.1% (m/V) as this concentration does not cause significant damages to bacterial cells with a healthy outer membrane. Also, the duration of this experiment was optimized and fixed at 1 h to prevent cell damages due to prolonged exposure to SDS. Fig. 3B compares the growth of the untreated and LNA-treated KPC-KP cells in terms of absorbance at 600 nm between pre- and post-exposure to 0.1% (m/V) SDS solution. The untreated group showed normal growth in the absence or presence of an SDS solution. However, in the absence or presence of 0.1% (m/V) SDS, bacterial cells treated with LNA had lower absorbance than that of untreated cells, indicating that the sub-inhibitory concentration of LNA slowed the growth of KPC-KP cells. A significant drop in absorbance could be detected when LNA treated KPC-KP cells were exposed to 0.1% (m/V) SDS. This indicated the ability of LNA to cause the influx of SDS into the cell and eventually kill it.
Subsequently, ethidium bromide influx and efflux activity were performed to determine the effect of LNA on the influx and efflux activity of KPC-KP cells (Fig. 3C). According to Fig. 3C-i, untreated cells demonstrated an active efflux activity upon exposure towards ethidium bromide over time, whereas LNA-treated cells gradually increased the uptake of ethidium bromide as indicated by the relative fluorescence unit (Fig. 3C-i). This suggested the ability of LNA to increase the membrane permeability of KPC-KP cells. Upon removal of LNA from the LNA-treated group in the efflux assay, the efflux activity of KPC-KP cells returned to normal with the reduction of fluorescent signal over time (Fig. 3C-ii). This suggested that LNA does not play a role in inhibiting the efflux system of KPC-KP cells.
Next, the presence of oxidative stress indicated by the comparative proteomic profiling analysis in Section 3.2 suggests the presence of ROS, which leads to lipid peroxidation. To validate these phenomena, lipid peroxidation assay and measurement of ROS were performed. Lipid peroxidation assay measures the end product of lipid peroxidation, MDA, while the level of ROS generation was quantified by the reaction involving DCF-DA, esterase enzyme, and ROS species [25]. Fig. 3E-ii shows that the concentration of MDA quantified in LNA-treated KPC-KP cells is significantly higher than that in the untreated cells, indicating the presence of lipid peroxidation of cells when exposed to LNA. The level of ROS quantified in LNA-treated KPC-KP cells was also significantly higher than that in untreated cells (Fig. 3E-i), indicating a high level of ROS generated in KPC-KP cells after exposure to LNA.
In conclusion, our study reported antimicrobial activity and mode of action of LNA that had not been previously studied. In future clinical applications, LNA has demonstrated a tremendous potential as novel antimicrobial as it exhibits antimicrobial activity against the robust KPC-KP cells with a low MIC value of 2.5% (V/V), 9.2 μmol/L. The combination between LNA and MPM further reduces the MIC value of both by 2-fold; MIC of LNA to 1.25%, 4.6 μmol/L, and MPM to 16 μg/mL, 0.00004 μmol/L. This suggests that LNA has the potential in combinatory antibiotic therapy that will revive the efficacy of older generation antibiotics in the clinical setting. Besides, our study elucidated the antimicrobial mode of action of LNA, which involved the generation of ROS that resulted in lipid peroxidation in KPC-KP cells and membrane damages. Subsequently, intracellular materials were lost, eventually killing the cell. We also found that LNA did not affect the efflux system of KPC-KP cells. The proposed mode of action of LNA is illustrated in Fig. 4. Compared to commercialized antibiotics currently in use in clinical settings, the antibacterial activity of LNA is lower in terms of effective concentration. However, LNA has a different approach in killing bacterial cells involving the induction of ROS, which eventually disrupts the bacterial membrane and kills the cell. This makes LNA a promising antibiotic adjuvant by which LNA would facilitate the uptake of antibiotics, thereby enhancing the activity of existing antibiotics. Shortly, an in vivo study may be performed to investigate the toxicity of LNA. Additionally, an effective delivery system can also be established for efficient delivery of LNA to the site of infection, enhancing the efficiency of this compound in the fight against infection. Taken together, this would eventually pave the way for the clinical use of LNA as an antimicrobial agent.
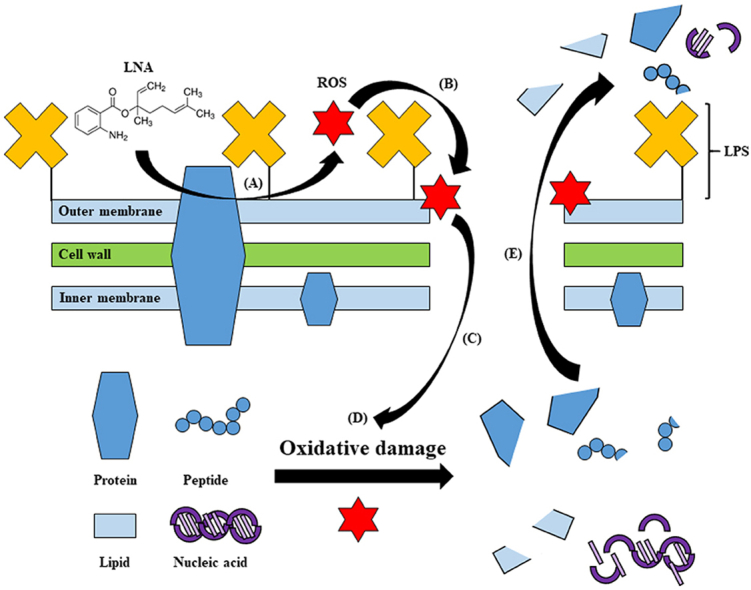
Fig. 4.

Proposed mode of action of LNA against KPC-KP cells. (a) LNA reacted with bacterial membrane components to form ROS. (b) ROS initiates lipid peroxidation by attacking membrane lipid, causing a chain reaction, which disrupts the bacterial membrane. (c) Influx of ROS into the KPC-KP intracellular region. (d) ROS degrades nucleic acids, lipids, and proteins. (e) Intracellular materials leakage due to bacterial membrane disruption. LNA: linalyl anthranilate; KPC-KP: carbapenemase-producing Klebsiella pneumoniae; ROS: reactive oxygen species; LPS: lipopolysaccharide.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This study was supported by the Higher College of Technology (HCT) Interdisciplinary Research Grant (Grant No. 113118), the Malaysian Medical Association Grant, and the UCSI PSIF Grant (Grant No. Proj-2019-In-Fas-062). The authors would like to acknowledge all members of the Floral Biotechnology Laboratory (FBL), Universiti Putra Malaysia, and Malaysia Genome Institute (MGI) for providing the necessary assistance and facilities throughout this study.
Footnotes
Peer review under responsibility of Xi’an Jiaotong University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2020.05.014.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Yigit H., Queenan A.M., Anderson G.J. Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2001;45:1151–1161. doi: 10.1128/AAC.45.4.1151-1161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization, Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. http://remed.org/wp-content/uploads/2017/03/lobal-priority-list-of-antibiotic-resistant-bacteria-2017.pdf. (accessed on 29 March 2020).
- 3.van der Bij A.K., Pitout J.D. The role of international travel in the worldwide spread of multiresistant Enterobacteriaceae. J. Antimicrob. Chemother. 2012;67:2090–2100. doi: 10.1093/jac/dks214. [DOI] [PubMed] [Google Scholar]
- 4.Moo C.L., Yang S.K., Yusoff K. Mechanisms of antimicrobial resistance (AMR) and alternative approaches to overcome AMR. Curr. Drug Discov. Technol. 2020;17:430–447. doi: 10.2174/1570163816666190304122219. [DOI] [PubMed] [Google Scholar]
- 5.Ventola C.L. The antibiotic resistance crisis: part 1: causes and threats. P&T. 2015;40:277–283. [PMC free article] [PubMed] [Google Scholar]
- 6.Yang S.K., Low L.Y., Yap P.S.X. Plant-derived antimicrobials: insights into mitigation of antimicrobial resistance. Record Nat. Prod. 2018;12:295–396. [Google Scholar]
- 7.Jiang Z., Kempinski C., Chappell J. Extraction and analysis of terpenes/terpenoids. Curr. Protoc. Plant Biol. 2016;1:345–358. doi: 10.1002/cppb.20024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahizan N.A., Yang S.K., Moo C.L. Terpene derivatives as a potential agent against antimicrobial resistant (AMR) pathogens. Molecules. 2019;24 doi: 10.3390/molecules24142631. 2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trombetta D., Castelli F., Sarpietro M.G. Mechanisms of antibacterial action of three monoterpenes. Antimicrob. Agents Chemother. 2005;49:2474–2478. doi: 10.1128/AAC.49.6.2474-2478.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez-Romero J.C., González-Ríos H., Borges A. Antibacterial effects and mode of action of selected essential oils components against Escherichia coli and Staphylococcus aureus, Evid. Based Complement. Alternative Med. 2015;2015 doi: 10.1155/2015/795435. 795435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haraguchi H., Oike S., Muroi H. Mode of antibacterial action of totarol, a diterpene from Podocarpus nagi. Planta Med. 1996;62:122–125. doi: 10.1055/s-2006-957832. [DOI] [PubMed] [Google Scholar]
- 12.Ultee A., Bennik M.H.J., Moezelaar R. The phenolic hydroxyl group of carvacrol is essential for action against the food-borne pathogen Bacillus cereus. Appl. Environ. Microbiol. 2002;68:1561–1568. doi: 10.1128/AEM.68.4.1561-1568.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuniga M.A., Dai J., Wehunt M.P. DNA oxidative damage by terpene catechols as analogues of natural terpene quinone methide precursors in the presence of Cu(II) and/or NADH. Chem. Res. Toxicol. 2006;19:828–836. doi: 10.1021/tx060021s. [DOI] [PubMed] [Google Scholar]
- 14.Rodenak-Kladniew B., Castro A., Stärkel P. Linalool induces cell cycle arrest and apoptosis in HepG2 cells through oxidative stress generation and modulation of Ras/MAPK and Akt/mTOR pathways. Life Sci. 2018;199:48–59. doi: 10.1016/j.lfs.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Memar M.Y., Ghotaslou R., Samiei M. Antimicrobial use of reactive oxygen therapy: current insights. Infect. Drug Resist. 2018;11:567–576. doi: 10.2147/IDR.S142397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pizzino G., Irrera N., Cucinotta M. Oxidative stress: harms and benefits for human health. Oxid. Med. Cell. Longev. 2017;2017 doi: 10.1155/2017/8416763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van der Paal J., Netys E.C., Verlackt C.C.W. Effect of lipid peroxidation on membrane permeability of cancer and normal cells subjected to oxidative stress. Chem. Sci. 2016;7:489–498. doi: 10.1039/c5sc02311d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong-Ekkabut J., Xu Z., Triampo W. Effect of lipid peroxidation on the properties of lipid bilayers: a molecular dynamics study. Biophys. J. 2007;93:4225–4236. doi: 10.1529/biophysj.107.112565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ezraty B., Gennaris A., Barras F. Oxidative stress, protein damage and repair in bacteria. Nat. Rev. Microbiol. 2017;15:385–396. doi: 10.1038/nrmicro.2017.26. [DOI] [PubMed] [Google Scholar]
- 20.Cadet J., Wagner J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013;5 doi: 10.1101/cshperspect.a012559. a012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vatansever F., de Melo W.C., Avci P. Antimicrobial strategies centered around reactive oxygen species--bactericidal antibiotics, photodynamic therapy, and beyond. FEMS Microbiol. Rev. 2013;37:955–989. doi: 10.1111/1574-6976.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kohanski M.A., Dwyer D.J., Wierzbowski J. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell. 2008;135:679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hashmi L.S., Hossain M.A., Weli A.M. Gas chromatography-mass spectrometry analysis of different organic crude extracts from the local medicinal plant of Thymus vulgaris L. Asian Pac. J. Trop. Biomed. 2013;3:69–73. doi: 10.1016/S2221-1691(13)60026-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Navarrete A., Avila-Rosas N., Majín-León M. Mechanism of action of relaxant effect of Agastache mexicana ssp. mexicana essential oil in Guinea-pig trachea smooth muscle. Pharm. Biol. 2017;55:96–100. doi: 10.1080/13880209.2016.1230140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yap P.S.X., Krishnan T., Yiap B.C. Membrane disruption and anti-quorum sensing effects of synergistic interaction between Lavandula angustifolia (lavender oil) in combination with antibiotic against plasmid-conferred multi-drug-resistant Escherichia coli. J. Appl. Microbiol. 2014;116:1119–1128. doi: 10.1111/jam.12444. [DOI] [PubMed] [Google Scholar]
- 26.Bialon M., Krzyśko-Łupicka T., Pik A. Chemical composition of herbal macerates and corresponding commercial essential oils and their effect on bacteria Escherichia coli. Molecules. 2017;22 doi: 10.3390/molecules22111887. 1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang S.K., Yusoff K., Thomas W. Lavender essential oil induces oxidative stress which modifies the bacterial membrane permeability of carbapenemase producing Klebsiella pneumoniae. Sci. Rep. 2020;10 doi: 10.1038/s41598-019-55601-0. 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang S.K., Yusoff K., Mai C.W. Additivity vs synergism: investigation of the additive interaction of cinnamon bark oil and meropenem in combinatory therapy. Molecules. 2017;22 doi: 10.3390/molecules22111733. 1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yap P.S.X., Lim S.H.E., Hu C.P. Combination of essential oils and antibiotics reduce antibiotic resistance in plasmid-conferred multidrug resistant bacteria. Phytomedicine. 2013;20:710–713. doi: 10.1016/j.phymed.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Meletiadis J., Pournaras S., Roilides E. Defining fractional inhibitory concentration index cutoffs for additive interactions based on self-drug additive combinations, Monte Carlo simulation analysis, and in vitro-in vivo correlation data for antifungal drug combinations against Aspergillus fumigatus. Antimicrob. Agents Chemother. 2010;54:602–609. doi: 10.1128/AAC.00999-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang S.K., Yusoff K., Ajat M. Disruption of KPC-producing Klebsiella pneumoniae membrane via induction of oxidative stress by cinnamon bark (Cinnamomum verum J. Presl) essential oil. PloS One. 2019;14 doi: 10.1371/journal.pone.0214326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viveiros M., Rodrigues L., Martins M. Evaluation of efflux activity of bacteria by a semi-automated fluorometric system. Methods Mol. Biol. 2010;642:159–172. doi: 10.1007/978-1-60327-279-7_12. [DOI] [PubMed] [Google Scholar]
- 33.Yao C., Li X., Bi W. Relationship between membrane damage, leakage of intracellular compounds, and inactivation of Escherichia coli treated by pressurized CO2. J. Basic Microbiol. 2014;54:858–865. doi: 10.1002/jobm.201200640. [DOI] [PubMed] [Google Scholar]
- 34.Kumar A., Sharma V., Dhawan A. Methods for detection of oxidative stress and genotoxicity of engineered nanoparticles. Methods Mol. Biol. 2013;1028:231–246. doi: 10.1007/978-1-62703-475-3_15. [DOI] [PubMed] [Google Scholar]
- 35.Papp-Wallace K.M., Endimiani A., Taracila M.A. Carbapenems: past, present, and future. Antimicrob. Agents Chemother. 2011;55:4943–4960. doi: 10.1128/AAC.00296-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willi J., Küpfer P., Evéquoz D. Oxidative stress damages rRNA inside the ribosome and differentially affects the catalytic center. Nucleic Acids Res. 2018;46:1945–1957. doi: 10.1093/nar/gkx1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cabiscol E., Tamarit J., Ros J. Oxidative stress in bacteria and protein damage by reactive oxygen species. Int. Microbiol. 2000;3:3–8. [PubMed] [Google Scholar]
- 38.Guo C., Ding P., Xie C. Potential application of the oxidative nucleic acid damage biomarkers in detection of diseases. Oncotarget. 2017;8:75767–75777. doi: 10.18632/oncotarget.20801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kong Q., Lin C.L. Oxidative damage to RNA: mechanisms, consequences, and diseases. Cell. Mol. Life Sci. 2010;67:1817–1829. doi: 10.1007/s00018-010-0277-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bononi I., Balatti V., Gaeta S. Gram-negative bacterial lipopolysaccharide retention by a positively charged new-generation filter. Appl. Environ. Microbiol. 2008;74:6470–6472. doi: 10.1128/AEM.00552-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.