

Abstract
As the underlying pathophysiology of progressive forms of multiple sclerosis (MS) remains unclear, current treatment strategies are inadequate. Progressive MS is associated with increased oxidative stress and neuronal damage in lesions along with an extensive representation of activated microglia/macrophages. To target these disease mechanisms, we tested the novel combination of generic medications, hydroxychloroquine (HCQ), and indapamide, in tissue culture and in mice. HCQ is an anti-malarial medication found to inhibit microglial activation and to ameliorate disease activity in experimental autoimmune encephalomyelitis. We are currently completing a phase II trial of HCQ in primary progressive MS (ClinicalTrials.gov Identifier: NCT02913157). Indapamide is an antihypertensive previously discovered in our laboratory drug screen to be an anti-oxidant. As these medications have a different spectrum of activities on disease mechanisms relevant to progressive MS, their use in combination may be more effective than either alone. We thus sought preclinical data for the effectiveness of this combination. In vitro, indapamide had robust hydroxyl scavenging activity, while HCQ and indapamide alone and in combination protected against iron-induced neuronal killing; TNF-α levels in activated microglia were reduced by either drug alone, without additional combination effects. In mice with a lysolecithin lesion that manifests demyelination and axonal loss in the spinal cord, the combination but not individual treatment of HCQ and indapamide reduced CD68+ microglia/macrophage representation in lesions, attenuated axonal injury, and lowered levels of lipid peroxidation. Our study supports the combination of indapamide and HCQ as a new treatment strategy targeting multiple facets of progressive MS.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13311-020-01002-5.
Key Words: Multiple sclerosis, oxidative stress, neurodegeneration, axonal loss, hydroxychloroquine, indapamide, microglia, neuroprotection, anti-oxidant
Introduction
Multiple sclerosis (MS) is an inflammatory and neurodegenerative disease of the central nervous system (CNS) involving demyelination and axonal injury [1]. It is characterized by an increased density of activated microglia/macrophages within lesions [2]. The microglia/macrophages often contain intracellular myelin breakdown products, suggesting their active role in demyelination and debris clearance [3]. These phagocytic cells are found in close proximity to damaged axons [4] and there is a positive correlation between microglia/macrophage accumulation and acute axonal damage in active MS lesions [5]. In this regard, microglia/macrophages may contribute to injury through mechanisms such as their production of reactive oxygen species (ROS) [6]. The amount of oxidative damage associated markers, including elevated NADPH oxidase, E06-immunoreactive oxidized phospholipids, and malondialdehyde for lipid peroxides, along with markers for oxidized DNA, such as 8-hydroxy-2′-deoxyguanosine, is correlated to lesion activity in post-mortem MS tissue [7–9]. Prior to white matter lesion formation, the normal-appearing white matter (NAWM) shows clusters of activated microglia/macrophages, termed microglia nodules [10], suggesting the critical involvement of these cells in lesion initiation.
Current pharmacological disease-modifying therapies (DMTs) in MS reduce leukocyte activation and their entry into the CNS that contribute to clinical relapses [11]. These DMTs are useful in the relapsing-remitting form of MS (RRMS) but do not meaningfully halt the evolving disability in the primary and secondary progressive forms of MS [12]. Besides the greater axonal loss in progressive relative to RRMS cases [1], reasons for the failure of current DMTs in progressive MS could be that they largely lack the capacity to enter the CNS to inhibit microglia/macrophages within, or to detoxify the ROS produced by activated microglia/macrophages, or by pro-oxidative iron deposited in the extracellular space [13].
In many lesions of progressive MS patients, microglia and macrophages are abundant in the border of demyelinated chronic active lesions, consistent with the hypothesis that microglia/macrophages are contributing to the expanding injury [1, 14]. Furthermore, engulfment of apoptotic neurons by microglia may result in their polarization to a disease-associated phenotype (e.g., elevation of Apoe, Axl, Ccl2), and a downregulation of homeostatic (e.g., tgfb1, P2ry12, Tmem119) markers [15].
Thus far, only one medication, ocrelizumab, an anti-CD20 monoclonal antibody, is approved for primary progressive MS [12, 16]; being a monoclonal antibody, its large size would likely preclude its entry into the CNS to act locally within. One medication, siponimod, a sphingosine-1-phosphate receptor modulator, is approved for a subset of secondary progressive MS patients that still has active inflammatory disease [17]. The capacity of siponimod to detoxify ROS or microglia is unclear; however, other sphingosine-1-phosphate receptor modulators such as fingolimod are able to inhibit microglia activation [18]. This lack of available therapies for progressive MS emphasizes the need to focus research efforts on developing new treatments, including the repurposing of well-tolerated generic medications.
We previously described the generic medication, hydroxychloroquine (HCQ), to inhibit microglia activation and attenuate the severity of mice afflicted with experimental autoimmune encephalomyelitis (EAE), an inflammatory model of MS [19]. We are currently completing a phase II trial of HCQ in primary progressive MS (ClinicalTrials.gov Identifier: NCT02913157). Indapamide is a generic antihypertensive medication which our group found in a previous drug screen to scavenge hydroxyl radicals and to protect neurons from iron-induced killing in culture [20]. Moreover, following lysolecithin injury to the spinal cord of middle-aged mice, indapamide mitigated the extent of axonal loss, NADPH oxidase–mediated oxidative stress, and lesion expansion [21]. Both hydroxychloroquine and indapamide cross into the brain, and their low cost and high tolerability make them promising candidates for the treatment of progressive MS, either singly or in combination.
In this manuscript, we used in vitro cell culture assays and the mouse lysolecithin spinal cord model, which produces demyelination and axonal injury/loss [22] as relevant models of MS. We tested the hypothesis that HCQ and indapamide are a potent combination to reduce oxidative stress, iron-induced killing of neurons, and microglia/macrophage activation in vitro, and that their combination in the lysolecithin demyelination model will have additive axonal protective effects.
Methods
Mice
All experiments conducted with animals were approved by the University of Calgary Animal Care Committee following regulations set forth by the Canadian Council of Animal Care. Microglia cultures used mouse pups at postnatal P0-P2 delivered from a pregnant female CD1 mouse from Charles River (Montreal) in our facility. Female C57BL/6 mice 6–8 weeks of age from Charles River were used for all in vivo experiments.
Human Neurons
This assay followed the protocol described previously [20]. Briefly, human fetal neurons were isolated from brain tissues from therapeutically aborted 14–20 week old human fetuses [23]. Consent was provided by each donor for the use of these specimens and has been approved by the University of Calgary Research Ethics Board. Cells were maintained in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS; Sigma, F1051). Prior to experimentation, 96-well plates were coated in poly-l-ornithine (Sigma, P4957) for at least 1 h. Neurons were plated at a density of 100,000 cells/well in AIM-V® Serum Free Medium (Gibco, 12055-091) and incubated at 37 °C and 5% CO2 for 24 h. Indapamide (2 mM stock prepared in 10% dimethylsulfoxide (DMSO; Sigma, D2650) in PBS, with final DMSO concentration exposed to cells at 0.05%; Sigma-Aldrich, I1887) and/or HCQ (1.5 mM stock prepared in PBS; Sigma-Aldrich, PHR1782) was then added for 1 h preceding addition of 50 μM of FeSO4 (Sigma-Aldrich, F8633). After 24 h of incubation, cultures were fixed with 4% paraformaldehyde (PFA; Electron Microscopy Sciences, 19210) for 15 min and stained for Tubulin β3 to visualize neurons.
Mouse Microglia
Neonatal mouse microglia of over 95% purity were cultured as previously described [24]. P0-P2 CD1 mouse pups (Charles River) were decapitated and the cortex of brains were extracted. Following meningeal removal, the brains were placed into complete Dulbecco’s Modified Eagle Medium (DMEM; Gibco, 11960-044) supplemented with 10% FBS, 1% GlutaMAX™ (Gibco, 35050-061), 1% penicillin/streptomycin (Gibco, 15070063), and 1% MEM non-essential amino acids (Gibco, 11140050) and minced with a scalpel. These cells were then vortexed for 1 min to kill neurons. Cells were then seeded at a density of one brain/flask in T75 flasks precoated with 3.125 μg/ml poly-d-lysine (Sigma, P7280). Medium was changed on days 1, 3, 6, and 8. When cultures were confluent, flasks were placed on an orbital shaker at 250 rpm for 20 min to detach the loosely adherent microglia from the astrocyte monolayer, and medium was collected from the flasks. This cell suspension was centrifuged at 1200 rpm for 10 min and subsequently counted on a hemocytometer and assessed for viability with Trypan Blue Stain (Gibco, 15250-061). Cells were plated on a 96-well plate at a density of 30,000 cells/well in serum-free supplemented DMEM. Cells were incubated at 37 °C and 5% CO2 for 1 h to allow their adherence to the plate, followed by treatment with HCQ and/or indapamide for another hour. The cells were then treated with 10 ng/ml lipopolysaccharide (LPS; Sigma, L2630) and incubated for 6 h. Supernatant was collected and stored at − 20 °C until TNF-α analysis.
Mouse TNF-α ELISA
The Mouse TNF-α Platinum ELISA kit (Invitrogen, BMS607-3) was used to measure TNF-α cytokine levels from mouse microglia. Cell-conditioned media following thawing was diluted 1:50. The ELISA was performed according to the protocol provided in the kit.
HORAC Assay
The hydroxyl radical anti-oxidant capacity (HORAC) assay method was followed as described [25]. Briefly, hydroxyl radicals are generated by a Co(II)-mediated Fenton-like reaction to oxidize fluorescein causing a loss of fluorescence [25]. When an anti-oxidant is present, the reaction forming the hydroxyl radical is attenuated and there is reduction in the loss of fluorescence. Gallic acid (Sigma, G7384) was used as a positive anti-oxidant control, and various concentrations of HCQ and/or indapamide were used. Absorbance of fluorescein (Sigma, F2456) was measured every 5 min for 60 min on a SPECTRAMax® GEMINI-XS Spectrofluorometer (Molecular Devices). The decay of absorbance was recorded as relative fluorescent units, and an area under the curve (AUC) analysis was calculated for each experiment. AUC was calculated by summing the quotient of each sequential time point to the initial time point of each respective group.
Lysolecithin Model of Demyelination
The lysolecithin demyelination surgeries were completed as previously described [26]. Briefly, mice were anesthetized with a ketamine (100 mg/kg) and xylazine (10 mg/kg) solution intraperitoneally. Buprenorphine (0.05 mg/kg) was injected subcutaneously immediately prior to surgery and 12 to 16 h post-surgery as an analgesic agent. Ophthalmic gel was applied to both eyes prior to and immediately after surgery to prevent corneal drying. Roughly 6 cm of the dorsal side of the mouse was shaved caudally from the ears and disinfected with 70% ethanol and an iodine solution. The mouse was then secured and positioned in a stereotactic surgical frame. A midline incision was made from the cervical to lower thoracic region on the dorsal side of the mouse. A retractor was placed to separate the underlying adipose and muscle tissue to expose the T2 vertebrae, which is an easily recognizable anatomical landmark in the C57BL/6 mice. Surgical spring scissors were used to expose the dorsal spinal cord between T3 and T4 vertebrae, followed by a 30G needle to remove the spinal cord meninges in this same region. A 10-μl Hamilton syringe attached to a 34G needle was positioned in between the T3 and T4 vertebrae, and 0.5 μl of 5 μg lysolecithin (Sigma, L1381) was injected 1.3 mm into the spinal cord ventral column between T3 and T4 at a flow rate of 0.25 μl/min. The needle remained in the spinal cord for an additional 2 min to limit backflow. The incision was sutured, and the mouse was monitored in a thermally controlled environment to promote recovery.
Quantitative Fluorescence Microscopy Analysis
Human neurons were stained in 96-well flat bottom black/clear plates and imaged using the 10 ×/0.5 NA air objective on the ImageXpress Micro XLS High-Content Analysis System (Molecular Devices). The following filters were used for detection: Nuclear yellow excitation 387/11 nm—emission 440/40 nm and FITC excitation 485/20 nm—emission 525/30 nm. For each well, 9 fields of view were imaged to be used for quantitative analysis. Multiwavelength cell scoring analysis in the MetaXpress High-Content Image Acquisition and Analysis Software (Molecular Devices) was used to measure the number of cells from images obtained from the ImageXpress. For representative images, the image was created by merging each channel with pseudo-colors. Only brightness and contrast were adjusted and kept consistent between samples to produce a clearer image.
In Vivo Treatment Paradigm
Six–eight-week-old young female C57BL/6 mice (Charles River) were used for this experiment. Four experimental groups (n ≥ 5 each) were used for each experiment, which was repeated 3 times. We used a pretreatment paradigm as the lysolecithin demyelination is very severe and produces myelin disruption, and death of Olig2+ oligodendroglial lineage cells occurs by 4 h of the insult; the Olig2+ cells are almost completely lost in lesions at 24 h [22]. The HCQ group began a 10-day pretreatment, receiving 300 μl intraperitoneal (IP) injection of 100 mg/kg HCQ once a day dissolved in sterile PBS, as described previously in the experimental autoimmune encephalomyelitis model [19]. The combination group also began receiving HCQ 10 days prior to surgery. All other groups (vehicle, indapamide) received vehicle injections (300 μl of PBS) at the same time. Indapamide treatment was a 3-day pretreatment. Mice were injected IP with 300 μl of 20 mg/kg of indapamide, dissolved in sterile PBS with 10% DMSO twice daily, 12 h apart, as described previously [21]. The HCQ and vehicle groups received a vehicle injection of 10% DMSO in PBS in the evening while the indapamide and combination groups received indapamide treatment. On the day of lysolecithin surgery (considered day 0), mice were injected in the morning and then immediately underwent surgery. The injections continued for another 3 days following surgery, when the mice were then sacrificed.
Spinal Cord Harvest and Sectioning
Mice were sacrificed through overdose of ketamine/xylazine, followed by intracardiac perfusion of ~ 15 mL of PBS, followed by ~ 15 mL of 4% PFA to fix the tissue. Spinal cords were then dissected from the dorsal side of the mouse. The cervical spinal cord to the lower thoracic region was taken and placed into microtubes containing 1 mL of 4% PFA and were stored at 4 °C for 24 h. After this fixation, the spinal cords were transferred to a 30% sucrose solution for 72 h. Spinal cords were then placed in Frozen Section Compound 22 Clear (Leica, 3801480) and frozen over dry ice and 2-methylbutane (Sigma, 78-78-4). The cords were then cut in 20-μm sections on a cryostat (ThermoFisher), and sections were collected on Superfrost® Plus microscope slides (VWR, 48311-703) and stored at − 20 °C for subsequent analysis.
Mouse Spinal Cord Histology
To visualize demyelination for calculation of total lesion volume and lesion epicenter area, spinal cords were stained with eriochrome cyanine and neutral red solution. Briefly, spinal cord sections on microscope slides were removed from − 20 °C storage and left to air dry at RT for 30 min. The slides were then subjected to citrisolv (Fischer Scientific, 22-143975), isopropanol (Sigma, 279544), 100% ethanol, 95% ethanol, 90% ethanol, 70% ethanol, and 50% ethanol in succession for 1 min each. Slides were then washed in distilled water for 1 min, followed by 15-min treatment with an eriochrome cyanine solution (10% FeCl3) and an immediate treatment with 0.5% NH4OH for 5 s preceding another 1 min wash with distilled water. Finally, samples were dehydrated with successive treatments of 50%, 70%, 90%, 95%, and 100% ethanol each for 1 min, followed by 2-min washes in isopropanol and 4 min in citrisolv. Coverslips were then mounted on slides with Acrytol Mounting Medium (Electron Microscopy Science, 13518) and left to dry at RT for 30 min. Bright-field images were acquired at 20 × 0.75 NA air objective using the Olympus VS110 Slidescanner.
Lesion Volume Analysis
Spinal cords were sectioned at 20 μm over a series of 10 sets of slides; thus, each section was 200 μm away from the adjacent section on a given slide. Sections were then stained with eriochrome cyanine and neutral red, and images were captured. Using cellSens software (Olympus), each lesion was traced and the area was recorded. The lesion area that was the largest for a given mouse was designated the epicenter lesion. The volume of lesion per mouse was estimated by taking the sum of the area of myelin disruption of all eriochrome cyanine sections of a single animal’s spinal cord and multiplying that by 200 μm to account for the distance between each section. This was then converted from cubic micrometer to cubic millimeter.
Antibodies
The following antibodies were used for immunofluorescence microscopy: mouse anti-mouse/human tubulin β3 (1:500; Biolegend, 801202), rat anti-mouse CD68 (1:500; Biolegend), rat anti-mouse MBP (1:100; Abcam, ab40390), goat anti-mouse Olig2 (1:250; R&D Systems, AF2418), rabbit anti-mouse NF-H (1:1000; EnCor Biotechnology, RPCA-NF-H), goat anti-mouse amyloid precursor protein (1:400; Abcam, ab2084), and mouse anti-4-hydroxynonenal (1:100; R&D Systems, MAB3249). The following secondary antibodies from Jackson ImmunoResearch were used at 1:400 dilution: Alexa Fluor 488 donkey anti-mouse IgG, Alexa Fluor 488 donkey anti-rabbit IgG, Alexa Fluor 488 donkey anti-goat IgG, Cyanine Cy3 donkey anti-goat IgG, Cyanine Cy3 donkey anti-rat IgG, Alexa Fluor donkey anti-rabbit 647, Alexa Fluor donkey anti-rat 647. Hoescht S769121 (Nuclear yellow; 1:800; Abcam, ab138903) was used for nuclear staining in all experiments.
Confocal Immunofluorescence Microscopy
Microscope slides with mouse spinal cord samples were removed from − 20 °C storage and left at RT to air dry for 30 min. For MBP staining, slides underwent delipidation with successive washes of 1 minute each with 50%, 70%, 90%, 95%, 100%, 95%, 90%, 70%, and 50% ethanol. Horse blocking solution (PBS, 10% horse serum, 1% BSA, 0.1% cold fish stain gelation, 0.1% Triton X-100, 0.05% Tween-20) was used to block all samples for 1 h at RT. After blocking, slides were incubated with primary antibodies in antibody dilution buffer (PBS, 1% BSA, 0.1% cold fish stain gelation, 0.1% Triton X-100) overnight at 4 °C. Following this, slides were washed three times using a 0.05% Tween-20 in PBS solution for 5 min, then incubated with secondary antibodies at RT for 1 h, protected from light. Slides were then washed three times with 0.05% Tween-20 in PBS for 5 min and coverslips were mounted with Fluoromount-G solution (SouthernBiotech, 0100-01).
Laser confocal immunofluorescence images were acquired using the Lecia TCS Sp8 laser confocal microscope, using the 25 × 0.5 NA water objective for all samples and experiments. The 405 nm, 488 nm, 552 nm, and 640 nm lasers were used to excite the fluorophores from antibodies bound to samples and detected by two low dark current Hamamatsu PMT detectors and two high sensitivity hybrid detectors on the Sp8. Images of spinal cords were acquired in 8-bits in a z-stack using bi-directional scanning at 2048 × 2048 resolution with the pinhole set to 1 airy unit, 0.75 × zoom, and 0.57 μm per optical section with 30 sections. Laser power, gain, and offset were maintained consistent for all samples within each experiment. A slide of secondary controls (i.e., containing nuclear yellow and only secondary antibodies) was used in each experiment to determine optimal settings to minimize and account for non-specific fluorescence from the secondary antibodies. The lesion epicenter on each slide sampled was imaged for analysis. Leica Application Suite X was used for image acquisition and ImageJ was used for image threshold and particle analysis.
We had predetermined exclusion criteria for analysis of spinal cord sections. Tissues and thus mice were excluded if the sections had folds or tears in the area of the lesion, or if the lesion was too small or ended up outside the ventral or ventrolateral white matter. This led to our sample sizes varying slightly between groups and stains.
Confocal Microscopy Image Analysis
All images were analyzed with ImageJ (National Institute of Health). For each z-stack, the maximum intensity projections were created for each channel (i.e., marker) and converted from 8-bit to RGB. The region of interest (ROI; i.e., the demyelinated lesion) was traced using MBP or CD68 and was pasted onto each channel, similar to that described previously [27]. To determine the positive signal, the color threshold was set using the contralateral uninjured white matter. The analyze particles function was used to create a mask of positive signals and quantify the particles within each ROI. For APP, the mask of NF-H was pasted onto the APP channel, then the outside was cleared so APP+ signal was only analyzed within NF-H+ axons. The same color threshold values and settings for positive signals, size, and circularity inclusion for particle analysis were maintained across all samples for each experiment. The values quantified were divided by the mean from the vehicle or control samples to provide a fold change value. The fold change values from all experiments were then plotted for subsequent statistical analysis.
For all representative images shown, the maximum intensity projection of each channel in the respective z-stack was merged with pseudo-colors in ImageJ. Only brightness and contrast settings were adjusted, and done consistently, between all samples for clearer image display.
Statistical Analysis
All statistical analysis was performed in GraphPad Prism 8.0. All experimental groups in vitro and in vivo included a minimum of four replicates, with each experiment being completed three times at minimum. Given the number of groups, all experiments were analyzed using a one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison test to be compared to the vehicle group. Graphs represent the mean and error bars depict the standard error of the mean.
Results
Indapamide Displays Robust Anti-oxidant Ability Against Hydroxyl Radical Generated by H2O2
Utilizing the HORAC assay that generates the hydroxyl radical from the Fenton-like reaction of a divalent cation with hydrogen peroxide, and where the hydroxyl radical then destroys fluorescein in solution, we evaluated the anti-oxidant capacity of the drugs in isolation and in combination. The presence of an anti-oxidant would attenuate the degradation of fluorescein and therefore the loss of fluorescence. Thus, a steep slope occurs in the absence of any anti-oxidant while a less steep slope represents the presence of anti-oxidant activity (Fig. 1(A–C)).
Fig. 1.

Anti-oxidant activity of treatments against the hydroxyl radical. Representative relative fluorescence unit (RFU) graphs from HORAC assay of (A) indapamide, (B) HCQ, and (C) indapamide and HCQ alone or in combination. Gallic acid was used as a positive control anti-oxidant. Each data point is represented in RFU, taken every 5 min for 60 min. The greater magnitude of decay shown in the no anti-oxidant and 5 μM HCQ groups demonstrates an inability to counter the hydroxyl radical degradation of fluorescein. (D) AUC analysis from HORAC assay with various drug treatments. Bar graphs display the mean with error bars representing the SEM. Data were analyzed using a one-way ANOVA with Dunnett’s post hoc multiple comparison test. Each data point in (D) represents one experiment. ****p < 0.0001 compared to no anti-oxidant control. IND: indapamide; HCQ: hydroxychloroquine
Figure 1(A) shows that indapamide at increasing concentrations from 1 to 10 μM progressively decreased the slope of the degradation of fluorescein, indicative of anti-oxidant activity. HCQ was without effect at concentrations of 1 to 10 μM (Fig. 1(B)). To evaluate whether there was a combination effect of HCQ or indapamide over multiple experiments, we analyzed indapamide at 1 μM and HCQ at 5 μM (Fig. 1(C)), as these represent concentrations likely to be achieved in patients taking these medications [28, 29]. From an experiment, area under the curve (AUC) analysis was then calculated for each reaction and pooled per condition from multiple experiments. A higher AUC represented greater anti-oxidant capacity. The AUC for HCQ was similar to that of no anti-oxidant control, indicating the HCQ had no direct anti-oxidant activity in this assay. In combination of the drugs, the AUC reflected the activity of indapamide alone (Fig. 1(D)). Thus, HCQ did not have direct anti-oxidant activity against the hydroxyl radical, unlike indapamide. Moreover, there was no combination effect as the activity of HCQ and indapamide together was not greater than either drug in isolation.
Drug Treatments Are Neuroprotective Against FeSO4 Challenge In Vitro
Ferrous iron is toxic to neurons in culture [30]. The mechanisms of iron-mediated neurotoxicity are not entirely clear; however, it may involve direct cytolytic mechanisms or through the formation of hydroxyl radicals such as by the Fenton reaction of ferrous ion and peroxides in cell membranes or solution [31]. Within 24 h of exposure to 50 μM of FeSO4, few neurons identified through tubulin β3 staining were detected in culture compared to DMSO-treated control (Fig. 2(A, B)).
Fig. 2.

Drug treatments are neuroprotective against FeSO4 treatment in vitro on human fetal neurons. Representative images of human fetal neurons in vitro of (A) healthy DMSO control; (B) FeSO4 control; (C–E) 50 μM FeSO4 with drug treatments of (C) 1 μM indapamide, (D) 5 μM HCQ, and (E) combination. (F) Quantitation of surviving neurons with 1 μM indapamide, 5 μM HCQ, and their combination after 24-h incubation with 50 μM FeSO4. Scale bar represents 50 μm. Bar graphs represent the mean with error bars depicting SEM. Data were analyzed using a one-way ANOVA with Dunnett’s post hoc multiple comparison test to 50 μM FeSO4. Each data point represents one replicate well. The protective trend observed from HCQ and indapamide treatment was reproduced in two additional experiments. ****p < 0.0001 compared to iron. IND: indapamide, HCQ: hydroxychloroquine
As previous work showed that neurons promptly began to die after exposure to FeSO4 [20], we utilized a 1 h pretreatment paradigm with HCQ and indapamide alone and in combination preceding FeSO4 administration. After 24 h of exposure, the survival of tubulin β3+ neurons was quantified. Indapamide protected human fetal neurons in vitro from iron-induced toxicity at the therapeutically relevant concentration of 1 μM (Fig. 2(C)) as did HCQ at 5 μM (Fig. 2(D)). The combination of 1 μM indapamide and 5 μM HCQ produced a strong protective effect (Fig. 2(E)); however, since the number of neurons remaining alive in the presence of individual drugs was already similar to that found in control, non-iron-exposed cultures (i.e., near complete protection), the additional protective effect of the combination could not be ascertained (Fig. 2(F)). Taken together, indapamide and HCQ alone and in combination mitigated the neuronal death induced by ferrous iron in culture.
Stimulated Microglia TNF-α Levels Are Attenuated Following Drug Treatments In Vitro
To assess the direct effect of the drug treatments on microglia activation, we isolated and cultured microglia from P0-P2 CD1 mouse pups. Using an ELISA, we assessed TNF-α secretion by LPS-activated microglia pretreated for 1 h with or without indapamide and/or HCQ. We found that HCQ (5 μM) and indapamide (1 μM) both reduced the LPS-enhanced level of TNF-α by about 50%, and no further decrease was elicited by the combination (Fig. 3).
Fig. 3.
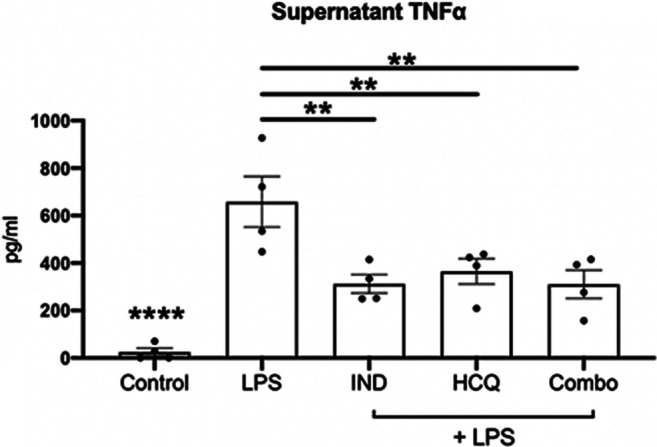
Drug treatments alone and in combination reduce TNF-α levels in vitro. Representative data from supernatant collected from cultured mouse microglia which were stimulated with 10 ng/ml LPS and subsequently subjected to drug treatments of 5 μM HCQ, 1 μM indapamide, and their combination. Supernatant was analyzed for TNF-α with an ELISA. Bar graphs display the mean with error bars representing the SEM. Data were analyzed using a one-way ANOVA with Dunnett’s post hoc multiple comparison test against the 10 ng/ml LPS group. Data has been reproduced with similar trends in two other experiments. Each data point represents one replicate well. **p < 0.01, ****p < 0.0001 compared to LPS. IND: indapamide, HCQ: hydroxychloroquine
Drug Treatments Do Not Affect the Size of a Day 3 Lysolecithin Lesion in Mice
The lysolecithin model of demyelination involves administration of lysolecithin, a lipid disrupting detergent, into the spinal cord white matter to cause demyelination, axonal damage, oligodendrocyte loss, and microglia/macrophage recruitment to the lesion site [21]. This model has rapid-onset and well-validated temporal characteristics, with a demyelinated area present as early as 4 h following lysolecithin injection [22]. Figure 4(A) depicts a schematic of the timeline of the in vivo paradigm followed in this study. Through imaging eriochrome cyanine-stained (Fig. 4(B)) coronal sections and measuring the area of lesion per section (see Methods), the area of the epicenter of the lesion was obtained for each mouse and expressed as a ratio of the mean epicenter area for the vehicle control groups for that experiment; this enabled us to pool data across 3 experiments. Figure 4(C) shows that lesion epicenter area was similar across drug alone or combination groups compared to vehicle control, and this was also the case for lesion volume (Fig. 4(D)).
Fig. 4.

Treatment paradigm and effect of drug treatments on day 3 lysolecithin lesion size. (A) Timeline of pretreatment paradigm. (B) Representative image of the lysolecithin lesion, visualized with eriochrome cyanine staining for lipids (blue) and neutral red for nuclei (red). Closed arrowhead depicts lesion area. (C) Lesion epicenter area quantitation of each treatment group. (D) Quantitation of the lesion volume of each treatment group. Pooled data from the three separate experiments (n ≥ 5 each), normalized and represented as fold change of the vehicle group within a particular experiment. Graphs represented are the mean with error bars depicting SEM. Data were analyzed using a one-way ANOVA with Dunnett’s post hoc multiple comparison test, compared to vehicle treatment. Each data point represents one animal combined from 3 separate experiments. Horizontal dashed line represents fold change of 1 (the mean of vehicle group). IND: indapamide, HCQ: hydroxychloroquine
Together, these data show that neither the drug treatments alone nor in combination at the tested dosages were able to reduce the size of the catastrophic lysolecithin lesion 3 days following lysolecithin injection.
Drug Treatments Do Not Alter Oligodendrocyte Lineage Cell Numbers or Extent of MBP Immunoreactivity in Lesions
Following lysolecithin deposition, myelin disruption and death of Olig2+ oligodendroglial lineage cells occur by 4 h of the insult; the Olig2+ cells are almost completely lost in lesions at 24 h and they begin to repopulate by 3 days [22]. Thus, we investigated the effects of individual or combination treatments on Olig2+ cell numbers at 3 days. Our analysis found similar content across all groups compared to vehicle controls (Fig. 5).
Fig. 5.

Drug treatments have no effect on myelin degradation or oligodendrocytes in the lesion. (A–D) Representative images of treatment groups from the day 3 lysolecithin lesion, visualizing Olig2+ cells (white), NFH+ axons (green; see Fig. 7), MBP+ myelin (red), and the overlay image. (E) Number of oligodendrocyte lineage cells in the lesion. (F) Comparisons of MBP+ staining within the lesion, indicative of myelin debris. Pooled data from the three separate experiments (n ≥ 5 each), normalized and represented as fold change of the vehicle group. Graphs represented are the mean with error bars depicting SEM. Data were analyzed using a one-way ANOVA with Dunnett’s post hoc multiple comparison test, compared to vehicle treatment. Each data point represents one animal. Horizontal dashed line represents fold change of 1 (the mean of vehicle group). IND: indapamide, HCQ: hydroxychloroquine
Subsequent to lysolecithin injection, MBP immunoreactivity is initially intense within the lesion compared to spared areas, presumably due to exposure of epitopes of MBP in fragmented myelin to the staining antibody [22] (Fig. 5(A–D)). We determined that HCQ or indapamide alone, and in combination, did not alter the total amount of MBP+ myelin debris within the lesion 3 days following lysolecithin (Fig. 5(F)).
Thus, at 3 days following lysolecithin administration in the spinal cord, the drug treatments tested did not protect against oligodendrocyte loss or influence the amount of MBP+ myelin debris within the lesion.
Reduced CD68 Expression on Microglia/Macrophages by Combination Treatment In Vivo
To measure the representation of microglia/macrophages in lysolecithin spinal cord lesions, we utilized CD68+ immunostaining. CD68 is expressed by monocyte-derived macrophages and CNS resident microglia. Furthermore, immunoreactivity of CD68 has been positively associated with microglia/macrophage activation [32]. We first measured the total CD68+ area with lesions (Fig. 6). We did not find any significant difference across any treatment group for the area occupied by CD68+ microglia/macrophages with lesions. However, the mean fluorescence intensity (MFI) of CD68+ stains showed that the drugs in combination, but not alone, significantly reduced the CD68+ immunoreactivity (Fig. 6(C)), suggestive of a reduction of their activation state.
Fig. 6.

Effect of drug treatments on CD68+ microglia/macrophages in vivo. (A) Representative images of the day 3 lysolecithin lesion, visualizing DAPI for nuclei (blue) and CD68 for microglia/macrophages within the lesion (red). (B) Number of CD68+ cells within the lesion, expressed as percent coverage of the lesion area. (C) Mean fluorescence intensity of CD68+ microglia/macrophages within the lesion. Lower immunoreactivity represents reduced cell activation. Pooled data from the three separate experiments (n ≥ 5 each), normalized and represented as fold change of the vehicle group. White dashed line depicts lesion area. Graphs represented are the mean with SEM. Data were analyzed using a one-way ANOVA with Dunnett’s post hoc multiple comparison test, compared to vehicle treatment. Each data point represents one animal. Horizontal dashed line represents fold change of 1 (the mean of vehicle group). ***p < 0.001 compared to vehicle control. IND: indapamide, HCQ: hydroxychloroquine
Combination Treatment of HCQ and Indapamide Decreases Axonal APP Accumulation In Vivo
Given the robust neuroprotection by HCQ, indapamide, and the combination treatment against FeSO4 toxicity in vitro (Fig. 2), and the reduced CD68+ immunoreactivity in lesions of combination-treated lysolecithin-demyelinated mice (Fig. 6), we determined whether the axonal injury occurring in lysolecithin lesions [22] could be alleviated. First, we stained and counted the total number of NFH+ axons remaining 3 days following lysolecithin injection (Fig. 7(A–E)). This analysis did not reveal any differences between any of the treatment groups and vehicle-treated mice (Fig. 7(F)), perhaps because lysolecithin causes extensive early injury where axonal and myelin spheroids occur after an hour, and immunoreactivity for APP in axons appear by 4 h, the earliest time point investigated [22]. The accumulation of APP is an indicator of disrupted axonal axoplasmic flow [33, 34] and is commonly used as a marker of neuronal injury [7]. Thus, we evaluated APP immunoreactivity in NFH+ axons that have survived the injury at 72 h post-lysolecithin. The treatment groups of HCQ and indapamide in isolation did not significantly alter the number APP+NFH+ axons within the lesion compared to vehicle control. However, the combination treatment reduced the number of APP+NFH+ axons, indicative of a neuroprotective effect (Fig. 7(G)).
Fig. 7.

Combination treatment reduces the magnitude of axonal damage in vivo. (A–E) Representative images of normal-appearing white matter (NAWM), or of lysolecithin lesions in mice treated with vehicle, IND, HCQ, and combination. Each image includes NFH for axons (green), APP for compromised axons (magenta), and an overlay image. The insets of the overlay image depict APP accumulation on NFH+ axons (white). (F) Total number of NFH+ axons in the lesion. (G) Quantitation of number of NFH+ axons with accumulated levels of APP, represented as fold change of vehicle group. Graphs represent the mean with error bars depicting SEM. Data were analyzed using a one-way ANOVA with Dunnett’s post hoc multiple comparison test; *p < 0.05 compared to vehicle treatment. Each data point represents one animal. Pooled data from the three separate experiments (n ≥ 5 each), normalized and represented as fold change of the vehicle group. Horizontal dashed line represents fold change of 1 (the mean of vehicle group). IND: indapamide, HCQ: hydroxychloroquine
HCQ and Combination Treatment Reduce Lipid Peroxidation In Vivo
We evaluated oxidative injury within the lysolecithin injury at 72 h by determining the magnitude of the lipid peroxidation end product, 4-hydroxynonenal (4-HNE) [35]. We observed 4-HNE in lesions of all groups (Fig. 8(A)). Normalized density shows a robust and statistically significant reduction of 4-HNE present within the lesion in mice treated with the combination of HCQ and indapamide (Fig. 8(B)). Surprisingly, there was a near equivalent reduction in the HCQ alone group, yet no effect observed following indapamide treatment (Fig. 8(B)).
Fig. 8.

HCQ and combination treatment reduce lipid peroxidation in vivo. (A) Representative images of the day 3 lysolecithin lesion for all treatment groups. Images are overlays of 4-HNE (green) for lipid peroxidation and DAPI (blue) for nuclei; the DAPI signal has been reduced to illuminate the 4-HNE better. White dashed line depicts lesion area. (B) Percent of the lesion containing 4-HNE+ immunofluorescence. Graph represents the mean with error bars depicting SEM. Data were analyzed using a one-way ANOVA with Dunnett’s post hoc multiple comparison test, compared to vehicle treatment; **p < 0.01; ***p < 0.001. Each data point represents one animal, normalized and represented as fold change of the vehicle group. Horizontal dashed line represents fold change of 1 (the mean of vehicle group). IND: indapamide, HCQ: hydroxychloroquine
Discussion
We sought to establish a combination drug treatment in mice with the goal of clinical translation to prevent disease progression in MS. Currently, there are few treatment options available for those living with progressive MS, and we aspire to fill this unmet need. The mechanisms of progression in MS are related to neuronal/axonal loss and chronic microglia/macrophage activation including their elevation of oxidative stress [13], so we focused on mitigating these mechanisms with the combination of HCQ and indapamide. Our main findings are summarized in Table 1. There was no combination effect in the fluorimeter-based HORAC assay as indapamide was active but HCQ was not. No additional inhibition of microglial production of TNF-α was found in the combination over the extent exerted by either alone. We could not determine the potential combination effect on iron-mediated killing in culture as both drugs were already preventing toxicity at a high magnitude. In vivo, however, likely because lysolecithin acts through a detergent-like mechanism that creates a prominent demyelinated lesion by 4 h of deposition [22], HCQ and indapamide alone did not reduce CD68+ immunoreactivity or extent of APP accumulation in NFH+ axons. However, their combination lowered the CD68+ manifestation of macrophage/microglia activity, and had a neuroprotective outcome as informed by the alleviation of APP+NFH+ axons. In this regard, APP is transported down axons by fast axoplasmic flow in healthy axons and is normally not detected by immunohistochemistry; with neuronal compromise, axoplasmic flow is perturbed and APP accumulates to levels that become detectable by an APP antibody in axons [7, 33, 34].
Table 1.
Summary of treatment effects. Summary of in vitro and in vivo effects observed following administration of HCQ and indapamide alone and in combination. ++: marginal effect; +++: modest effect; ++++: robust effect
| HCQ | Indapamide | Combination Effect? | |
|---|---|---|---|
|
Protection against neurotoxicity in vitro |
+++ | +++ | Unable to assess since each drug by itself was already very potent |
|
Anti-oxidant capacity (HORAC) in vitro |
No effect | ++++ | No |
|
Inhibition of microglial TNF-α in vitro |
++ | ++ | No |
| CD68+ microglia/macrophage in vivo | No effect | No effect | Yes. Reduces CD68+ immunoreactivity within lesion |
| Axonal health (APP accumulation) in vivo | No effect | No effect | Yes. Reduces number of APP+ axons in lesion |
| Marker of oxidative injury (4-HNE) in vivo | ++++ | No effect | The magnitude of 4-HNE reduction by HCQ is marked, so it is not possible to determine any additional effect |
Recently, we reported that in middle-aged 8–10-month-old mice, indapamide decreased the extent of axonal loss occurring within 72 h of lysolecithin injury [21]. These aging mice had a more severe loss of axons compared to young 6-8-week-old mice that was associated with elevated NADPH oxidase activation in microglia [21], so it appears that the additional components (e.g., that linked to NADPH oxidase activation in aging microglia) of aging-enhanced lysolecithin-induced axonal loss are amendable to indapamide therapy while the neurodegeneration occurring in young mice is not. As age is associated with progression in MS [36], it would be important in future work to test the combination effect of HCQ and indapamide in aging mice, as this would provide guides that are more informative for progressive MS.
In correspondence with the lack of effect of indapamide by itself to reduce APP+ axons in young mice in vivo, we determined that indapamide did not lower the amount of the lipid peroxidation end product, 4-HNE, when compared to vehicle treatment. Thus, the strong ability of indapamide to antagonize hydroxyl radical degradation of substrate in vitro did not manifest as reduced lipid peroxidation in vivo. Although the hydroxyl radical is often considered the most reactive of oxygen radicals [37], other radicals such as superoxide anion are also associated with peroxidation of lipids [38]. The lack of indapamide effect on lowering 4-HNE in young mice in the current study may thus be associated with its inadequacy in scavenging and protecting against non-hydroxyl radicals.
That HCQ potently attenuated 4-HNE by itself, and accounting for the magnitude in the combination treatment, was initially unexpected. However, HCQ inhibits NADPH oxidase activity [39] and is a microglia inhibitor [19], and activated microglia are a major source of many toxins including superoxide through NADPH oxidase as discussed above [21]. Activated microglia containing iron are thought to produce the oxidative stress that drives the slowly expanding lesions that are prominent in the brains of patients with progressive MS [40, 41]. Recently, a sophisticated single-cell RNA sequencing of CNS immune cells and focusing on oxidative stress (Tox-seq) found a core signature shared among a microglia cluster and subsets of infiltrating myeloid cells in lesions of MS and its animal model, EAE [42]. Inhibiting the degradation of the anti-oxidant glutathione through blocking ɣ-glutamyl transferase was shown to reduce markers of oxidative damage, including 4-HNE, in the spinal cord of EAE-inflicted mice along with attenuation of clinical severity [42]. Thus, we contend that by reducing several aspects of the activity of microglia/macrophages including their propensity for oxidative stress, treatment with hydroxychloroquine led to lowering of 4-HNE in vivo.
Taken together, our data support the use of a combination of HCQ and indapamide to mitigate the substrates of progression in MS, particularly via the inhibition of microglia/macrophage activity and neuronal injury in vivo. While a combination neuroprotective effect could not be clearly demonstrated in culture because of the potency of individual agents, the alleviation of CD68+ immunoreactivity, APP+ axonal profiles, and lipid peroxidation in the combination group against the substantial lysolecithin injury in mice provides the rationale to consider the co-administration of HCQ and indapamide for MS. With HCQ already in a clinical trial in primary progressive MS, we hope for these findings to lay the preclinical evidence to encourage a future clinical trial of the combination of HCQ and indapamide in progressive MS.
Supplementary Information
(PDF 516 kb)
Acknowledgments
We thank the Multiple Sclerosis Society of Canada and the Canadian Institutes of Health Research (CIHR) for the support of operating grants. DB was supported by a CIHR Canada Graduate Scholarships Master’s Award while YD received the Alberta MS Collaboration postdoctoral fellowship; YD is the current recipient of a fellowship from CIHR. VWY acknowledges support from Canada Research Chair (Tier 1) program.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018;378:169–180. doi: 10.1056/NEJMra1401483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lassmann H. Mechanisms of white matter damage in multiple sclerosis. Glia. 2014;62:1816–1830. doi: 10.1002/glia.22597. [DOI] [PubMed] [Google Scholar]
- 3.Kuhlmann T, Ludwin S, Prat A, et al. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017;133:13–24. doi: 10.1007/s00401-016-1653-y. [DOI] [PubMed] [Google Scholar]
- 4.Trapp BD, Peterson J, Ransohoff RM, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 5.Bitsch A, Schuchardt J, Bunkowski S, et al. Acute axonal injury in multiple sclerosis: Correlation with demyelination and inflammation. Brain. 2000;123:1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- 6.O’Loughlin E, Madore C, Lassmann H, et al. Microglial phenotypes and functions in multiple sclerosis. Cold Spring Harb Perspect Med. 2018;8:a028993. doi: 10.1101/cshperspect.a028993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haider L, Fischer MT, Frischer JM, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134:1914–1924. doi: 10.1093/brain/awr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witte ME, Mahad DJ, Lassmann H, et al. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med. 2014;20:179–187. doi: 10.1016/j.molmed.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14:183–193. doi: 10.1016/S1474-4422(14)70256-X. [DOI] [PubMed] [Google Scholar]
- 10.Singh S, Metz I, Amor S, et al. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol. 2013;125:595–608. doi: 10.1007/s00401-013-1082-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rommer PS, Milo R, Han MH, et al. Immunological aspects of approved MS therapeutics. Front Immunol. 2019;10:1564. doi: 10.3389/fimmu.2019.01564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciccarelli O. Multiple sclerosis in 2018: new therapies and biomarkers. Lancet Neurol. 2019;18:10–12. doi: 10.1016/S1474-4422(18)30455-1. [DOI] [PubMed] [Google Scholar]
- 13.Faissner S, Plemel JR, Gold R, et al. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat Rev Drug Discov. 2019;18:905–922. doi: 10.1038/s41573-019-0035-2. [DOI] [PubMed] [Google Scholar]
- 14.Frischer JM, Weigand SD, Guo Y, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol. 2015;78:710–721. doi: 10.1002/ana.24497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017;47:566–581. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376:209–220. doi: 10.1056/NEJMoa1606468. [DOI] [PubMed] [Google Scholar]
- 17.Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018;391:1263–1273. doi: 10.1016/S0140-6736(18)30475-6. [DOI] [PubMed] [Google Scholar]
- 18.O’Sullivan S, Dev KK. Sphingosine-1-phosphate receptor therapies: Advances in clinical trials for CNS-related diseases. Neuropharmacology. 2017;113:597–607. doi: 10.1016/j.neuropharm.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 19.Koch MW, Zabad R, Giuliani F, et al. Hydroxychloroquine reduces microglial activity and attenuates experimental autoimmune encephalomyelitis. J Neurol Sci. 2015;358:131–137. doi: 10.1016/j.jns.2015.08.1525. [DOI] [PubMed] [Google Scholar]
- 20.Faissner S, Mishra M, Kaushik DK, et al. Systematic screening of generic drugs for progressive multiple sclerosis identifies clomipramine as a promising therapeutic. Nat Commun. 2017;8:1990. doi: 10.1038/s41467-017-02119-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michaels NJ, Lemmon K, Plemel JR, et al. Aging-exacerbated demyelinating injury is associated with microglia-derived reactive oxygen species: alleviation by indapamide. J Neurosci. 2020;44:8587–8600. doi: 10.1523/JNEUROSCI.1098-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plemel JR, Michaels NJ, Weishaupt N, et al. Mechanisms of lysophosphatidylcholine-induced demyelination: A primary lipid disrupting myelinopathy. Glia. 2018;66:327–347. doi: 10.1002/glia.23245. [DOI] [PubMed] [Google Scholar]
- 23.Vecil GG, Larsen PH, Corley SM, et al. Interleukin-1 is a key regulator of matrix metalloproteinase-9 expression in human neurons in culture and following mouse brain trauma in vivo. J Neurosci Res. 2000;61:212–224. doi: 10.1002/1097-4547(20000715)61:2<212::AID-JNR12>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 24.Mishra MK, Wang J, Keough MB, et al. Laquinimod reduces neuroaxonal injury through inhibiting microglial activation. Ann Clin Transl Neurol 2014;6:409-422. [DOI] [PMC free article] [PubMed]
- 25.Ou B, Hampsch-Woodill M, Flanagan J, et al. Novel fluorometric assay for hydroxyl radical prevention capacity using fluorescein as the probe. J Agric Food Chem. 2002;50:2772–2777. doi: 10.1021/jf011480w. [DOI] [PubMed] [Google Scholar]
- 26.Keough MB, Jensen SK, Wee Yong V. Experimental demyelination and remyelination of murine spinal cord by focal injection of lysolecithin. J Vis Exp 2015:52679. [DOI] [PMC free article] [PubMed]
- 27.Pu A, Mishra MK, Dong Y, et al. The glycosyltransferase EXTL2 promotes proteoglycan deposition and injurious neuroinflammation following demyelination. J Neuroinflammation. 2020;17:220. doi: 10.1186/s12974-020-01895-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mackenzie AH. Pharmacologic actions of 4-aminoquinoline compounds. Am J Med. 1983;75:5–10. doi: 10.1016/0002-9343(83)91264-0. [DOI] [PubMed] [Google Scholar]
- 29.Caruso FS, Szabadi RR, Vukovich RA. Pharmacokinetics and clinical pharmacology of indapamide. Am Heart J. 1983;106:212–220. doi: 10.1016/0002-8703(83)90119-9. [DOI] [PubMed] [Google Scholar]
- 30.Xue M, Mikliaeva EI, Casha S, et al. Improving outcomes of neuroprotection by minocycline: Guides from cell culture and intracerebral hemorrhage in mice. Am J Pathol. 2010;176:1193–1202. doi: 10.2353/ajpath.2010.090361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braughler JM, Duncan LA, Chase RL. The involvement of iron in lipid peroxidation. Importance of ferric to ferrous ratios in initiation. J Biol Chem. 1986;261:10282–10289. [PubMed] [Google Scholar]
- 32.Chistiakov DA, Killingsworth MC, Myasoedova VA, et al. CD68/macrosialin: Not just a histochemical marker. Lab Investig. 2017;97:4–13. doi: 10.1038/labinvest.2016.116. [DOI] [PubMed] [Google Scholar]
- 33.Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stone JR, Okonkwo DO, Dialo AO, et al. Impaired axonal transport and altered axolemmal permeability occur in distinct populations of damaged axons following traumatic brain injury. Exp Neurol. 2004;190:59–69. doi: 10.1016/j.expneurol.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 35.Shoeb M, Ansari N, Srivastava S, et al. 4-Hydroxynonenal in the Pathogenesis and Progression of Human Diseases. Curr Med Chem. 2014;21:230–237. doi: 10.2174/09298673113209990181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koch M, Mostert J, Heersema D, et al. Progression in multiple sclerosis: Further evidence of an age dependent process. J Neurol Sci. 2007;255:35–41. doi: 10.1016/j.jns.2007.01.067. [DOI] [PubMed] [Google Scholar]
- 37.Singh E, Devasahayam G. Neurodegeneration by oxidative stress: a review on prospective use of small molecules for neuroprotection. Mol Biol Rep. 2020;47:3133–3140. doi: 10.1007/s11033-020-05354-1. [DOI] [PubMed] [Google Scholar]
- 38.Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med Cell Longev. 2014;2014:360438. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Müller-Calleja N, Manukyan D, Canisius A, et al. Hydroxychloroquine inhibits proinflammatory signalling pathways by targeting endosomal NADPH oxidase. Ann Rheum Dis. 2017;76:891–897. doi: 10.1136/annrheumdis-2016-210012. [DOI] [PubMed] [Google Scholar]
- 40.Dal-Bianco A, Grabner G, Kronnerwetter C, et al. Slow expansion of multiple sclerosis iron rim lesions: pathology and 7 T magnetic resonance imaging. Acta Neuropathol. 2017;133:25–42. doi: 10.1007/s00401-016-1636-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hametner S, Wimmer I, Haider L, et al. Iron and neurodegeneration in the multiple sclerosis brain. Ann Neurol. 2013;74:848–861. doi: 10.1002/ana.23974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mendiola AS, Ryu JK, Bardehle S, et al. Transcriptional profiling and therapeutic targeting of oxidative stress in neuroinflammation. Nat Immunol. 2020;21:513–524. doi: 10.1038/s41590-020-0654-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 516 kb)