

Abstract
IL-10+ regulatory B cells (Bregs) inhibit immune responses in various settings. While Bregs appear to inhibit inflammatory cytokine expression by CD4+ T cells and innate immune cells, their impact on CD8+ T cells is contradictory. Moreover, it remains unclear, which effects of Bregs are direct. The subanatomical localization of Breg suppressive function and nature of their intercellular interactions remains unknown. Using novel tamoxifen-inducible B cell-specific IL-10 knockout mice, we found that Bregs inhibit CD8+ T cell proliferation and inhibit inflammatory cytokine expression by both CD4+ and CD8+ T cells. Sort-purified Bregs from IL-10-reporter mice were adoptively transferred into wild type hosts and examined by live-cell imaging. Bregs localized to the T:B border, specifically entered the T cell zone, and made more frequent and longer contacts with both CD4+ and CD8+ T cells, than non-Bregs. These Breg:T cell interactions were antigen-specific and reduced subsequent T:DC contacts. Thus, Bregs inhibit T cells through direct cognate interactions that subsequently reduces DC:T cell interactions.
1. Introduction
Regulatory B cells (Bregs) play an important role modulating immune responsiveness to autoimmune, alloimmune, tumor and infectious challenges(1–3). Their regulatory function is widely attributed to their expression of IL-10, as demonstrated in numerous transfer studies and in several studies where mice constitutively lacked B cell IL-10 expression(3–9). Although in certain models, B cells can inhibit the immune response using other mechanisms, such Bregs are less well defined and their relationship to IL-10-expressing Bregs is unclear(10–14). Thus, herein, the term Bregs refers to IL-10+ B cells.
Definitive study of Bregs has been challenging because they are rare (~1% of B cells) and lack a specific marker(3). The spleen contains the highest frequency and number of Bregs and has been the primary site from which various subpopulations, including marginal zone (MZ), T2-marginal zone precursor (T2-MZP), and CD1dhiCD5+ (B10) B cells has been isolated(3). These Breg populations are sufficiently enriched in IL-10+ B cells (e.g. 10–15%) to transfer regulatory activity in various models(6, 8, 14, 15). As such, emphasis has been placed on IL-10 frequency rather than an unbiased assessment of IL-10 distribution amongst all B cell subsets, and this has been compounded by the near universal use of ex vivo stimulation of B cells to enhance detection of weak IL-10 expression(3). In fact, each of these subsets contains a minority (e.g. 10–20%) of all IL-10+ B cells(9, 16). In contrast, TIM-1 and CD9 are more inclusive markers, encompassing the majority of all IL-10+ B cells from most of the canonical B cell subsets(9, 17). Recently, two groups examined IL-10 expression by B cells without ex vivo stimulation utilizing transcriptional analysis or IL-10 reporter mice and surprisingly, reported that plasma cells (PCs) represented the vast majority of IL-10+ cells of B lineage(6, 14). Thus, it remains uncertain which B cell subsets carry out Breg function in vivo.
Numerous studies have shown that Bregs inhibit Th1 and Th17 cells, while promoting Th2 cells and Foxp3+ Tregs(5, 18, 19). In contrast, in vivo studies examining the effect of Bregs per se on CD8+ T cells are limited and conflicting. After infection with different pathogens, constitutive loss of B cell IL-10 in CD19-CreXIL-10fl/fl mice increased CD8+ T cell IFNγ expression, while specific loss of B cell IL-10 expression in mixed BM chimeras had no effect on either CD8+ T cell expansion or cytokine expression(20, 21). Moreover, the effects of B cells on CD8 cell effector or memory function may be indirectly mediated through CD4 or innate cells(20, 22). In this regard, Bregs have also been shown to modulate the function of innate immune cells including monocytes, DCs, neutrophils and NK T cells(5, 6, 9, 14, 18, 23, 24).
It remains unclear how Bregs actually modulate immune responses in vivo and the subanatomical localization of their suppressive function remains unknown. Antigen specificity, and the requirement for MHCII and CD40 suggest direct cognate interactions, at least with CD4+ T cells(5, 7, 9, 23, 25). As noted above, there is evidence that Bregs may affect CD8 cells through CD4+ T cells(20, 22). However, other interpretations are possible. First, B cells maturing from transitional B cells in the absence of conventional (MHCII and CD40-dependent) T:B interactions may not be entirely normal(26, 27). Second, Bregs may indirectly suppress T cell function by inhibiting antigen presenting cell (APC) function, similar to Tregs. In this case, apparent antigen (Ag) specificity may result from a requirement for cell activation in close proximity to DCs involved in presenting antigen to reactive T cells. Indeed, the true mechanism of Treg activity was not revealed until in vivo live cell imaging was performed(28).
Here we set out to identify the subanatomical localization and cellular interactions of Bregs in vivo and clarify the effect of IL-10 expression by Bregs on CD4 and CD8 T cells. Using novel Tamoxifen (TAM)-inducible hCD20-ERT2.Cre-IL-10fl/fl (B-IL-10KO) mice, which are developmentally normal, to acutely delete B cell IL-10 expression, we show that B cell IL-10 expression regulates cytokine expression by CD4 cells and both proliferation and cytokine expression by CD8 cells. Utilizing IL-10-GFP reporter mice, GFP+ Bregs could be directly sorted without further stimulation. This allowed us to examine Breg dynamic interactions with antigen-specific CD4 and CD8 T cells in mouse spleen using 2-photon microscopy for the first time. These imaging studies showed that Bregs specifically enter the T cell zone and make dynamic cellular interactions with OT-1 and OT-2 cells in vivo in an antigen-dependent manner. In contrast to Tregs, Bregs interact directly with T cells and this limits subsequent T cell interactions with DCs.
2. Materials and Methods
2.1. Mice
Wild type mice were obtained from Jackson Laboratories. The following mice (all on the C57BL/6 (B6) background) were kindly provided as follows: OT-1 Rag1−/− DsRed (Fadi Lakkis, University of Pittsburgh); IL-10-GFP (Richard Flavell, Yale University (29)); hCD20-ERT2.Cre (Mark Shlomchik, University of Pittsburgh) and IL-10fl/fl (Werner Muller, University of Manchester (30)) were crossed to generate hCD20-ERT2.Cre-IL-10fl/fl (B-IL-10KO) mice. OT-2 and eGFP mice (Jackson Laboratories) were crossed in our laboratory. The University of Pittsburgh Animal Care and Use committee approved all animal studies.
2.2. Islet isolation and transplantation
400 islets isolated from BALB/c pancreata were placed under the left kidney capsule of hCD20-ERT-2.Cre+ Control B6 or B-IL-10KO sex-matched chemically-diabetic recipients, as we previously described(9). All recipients had glycemia <150 mg/dl within 2 days post-transplant. Blood glucose >250 mg/dl for 2 consecutive days after engraftment was defined as rejection.
2.3. Immunization and flow cytometry
IL-10-GFP reporter (Tiger) or control B6 mice were immunized i.p with 30×106 mitomycin C treated fully allogeneic BALB/c splenocytes, or nitrophenyl chicken gamma globulin (NP-CGG) or NP-ovalbumin (NP-OVA) precipitated in alum (50μg/mouse; BIOSEARCH Technologies, Petaluma CA). IL-10 expression was examined in total splenocytes left unstimulated or stimulated with “LPIM” (10μg/mg LPS, 50ng/ml PMA, 0.50μg/ml Ionomycin (Sigma Aldrich) and monensin (eBioscience Inc.,) for 5h(5, 7, 9, 23, 25). B cell subsets were identified by staining cells with AA4.1,CD19, CD21, CD23, CD138, CD5, B220, IgD and IgM (Thermo Fisher Scientific) and evaluated by the gating strategy described by Allman et al. (Fig. S1). B cells were classified as MZ cells (IgMhi, CD21hi, CD23lo/-), MZP cells (IgMhi, CD21hi, CD23hi), follicular I (FOI: IgDhi, IgMlo, CD23hi), follicular II (FOII: IgDhi, IgMInt, CD23hi), transitional 1 (T1: IgMhi, IgD−, CD21−, CD23−, B220+, AA4.1+), Transitional 2(T2: IgMhi, IgD−, CD21−, CD23+, B220+, AA4.1+), plasma cell/plasmablast (PC: IgMhi, IgD−, CD21−, B220−, CD138+), and splenic B1a (IgMhi, IgD−, CD21−, CD5+) (31). T cell IFN-γ and IL-17 were examined by stimulating total splenocytes with 50ng/ml PMA, 0.50μg/ml ionomycin and monensin for 5h in vitro. Intracellular staining was carried out using BD Fixation/Permeabilization kit (BD Biosciences). Flow cytometry was performed with BD LSRII or Fortessa cytometers.
hCD20-ERT2.Cre-IL-10fl/fl mice or Cre-negative littermate controls were treated with 2mg of tamoxifen (in corn oil) i.p for 3 consecutive days. Mice were immunized 3–5d later with allogeneic-splenocytes as above and endogenous T cell cytokines were examined on day 14. Alternatively, mice received 2×106 naïve OT-1 or OT-2 T cells stained with 2.5μM CFSE, and were then immunized with NP-OVA (50μg, i.p.). T cell proliferation and cytokine production were examined on day 5.
2.4. Bone marrow derived dendritic cell (BMDC) generation
BMDCs were generated by culturing bone marrow cells in RPMI media containing 10ng/ml of GM-CSF and IL-4 (Peprotech Inc.)(32, 33). Media was changed every 2 days. On day 6, DCs were treated with LPS (100ng/ml, 6h) and pulsed with 10μg/ml NP-OVA for 1 hour.
2.5. Ex vivo imaging
IL-10-GFP spleens were harvested 6–8d following immunization and Bregs or Control cells (GFP+/−CD19+ CD86+CD44+CD3−) were sorted using a BD FACSARIA. The cells were pulsed with NP-OVA or NP-CGG (10μg/ml for 1h at 37°C), washed, then stained with 1.5μM CFSE for 20 minutes and 1–2×106 cells transferred into naïve congenic CD45.1 C57BL/6 mice. Antigen-pulsed BMDCs (as above) were stained with 2.5μM Cell tracker Violet (Thermo Fisher Scientific) and mice received 1–2×106 cells. The same mice also received 2×106 naïve DsRED OT-1 T cells one day before Breg/DC transfer. Separately, we transferred eGFP-OT-2 cells and Breg or Control B cells stained with 5μM SNARF. Spleens were harvested 18h after Breg/DC transfer, glued to a coverslip and cut with a vibratome longitudinally. Spleen sections were kept in a warmed chamber at 37°C in 95% oxygenated and 5% CO2 in plain phenol free RMPI media with 10mM HEPES for the duration of imaging. Images were acquired with Olympus FVE1000 multiphoton microscope (Olympus) with a Mai Tai DeepSee femtosecond pulsed laser (Spectral Physics, Santa Clara CA) using a 25x water immersion objective (NA = 1.05). Time-lapsed image acquisition was carried out with Fluoview software (Olympus). Excitation wavelength between 850–875nm was used for imaging. The data were analyzed with Bitplane Imaris software. Cellular properties of B cells, T cells and DCs were examined by creating cell surfaces and tracks for each individual channel as we previously described(34). Tracks were inspected manually in order to ensure accuracy. Subsequently, cellular speed, displacement, meandering index and sphericity were obtained from built in modules. Cell contact duration was determined by manually examining each track for the entirety of the movie and scoring the duration of signal overlap between contacting cells.
2.6. Immunofluorescence
Spleens were frozen in OCT. Sections (10μm) and blocked with 5% goat serum for 1h. Rabbit anti-GFP (Abcam Plc. Cambridge MA), anti-rabbit Alexa-555, anti-IgM-Alexa488 or Alexa647, anti-B220-biotin, streptavidin-Alexa488 (or Alexa647), anti-CD3-Alexa647 (Thermo Fisher Scientific) and anti-SIGNR-1-Alexa488 (eBioscience Inc) were used as indicated. Whole spleen images were acquired with Nikon 90i and A1 immunofluorescence or confocal microscopes connected to a motorized stage. Analysis was carried out with Nikon NIS Elements software.
2.7. Statistical analyses
GraphPad Prism or Microsoft Excel were used to perform Student’s t tests (unpaired, two tailed) or ANOVA , as indicated. Results were considered significant if p ≤0.05.
3. Results
3.1. IL-10 is a key Breg inhibitor of CD4 and CD8 cells
To directly assess the role of IL-10 expression by Bregs, we used B-IL-10KO mice, which are entirely normal until specific deletion of IL-10 expression in B cells is induced by TAM treatment (~90% reduction; Fig. 1a). Alloimmunization of TAM-treated B-IL-10KO mice led to a 1.5–2 fold increase in IFN-γ and IL-17 expression by endogenous CD4 and a 1.5 fold increase in IFN-γ by CD8 cells compared to TAM-treated Cre-negative littermate controls (Fig. 1b,c). Moreover, transfer of OT-1 TCR-tg (CD8+) T cells followed by OVA immunization resulted in increased proliferation and ~2 fold higher levels of IFN-γ expression by splenic OT-1 cells in TAM-treated B-IL-10KO mice compared to Cre-negative controls (Fig. 1d,e).
Figure 1. T cell produce increased levels of inflammatory cytokines and proliferate more in mice with B cells specific IL-10 deficiency.
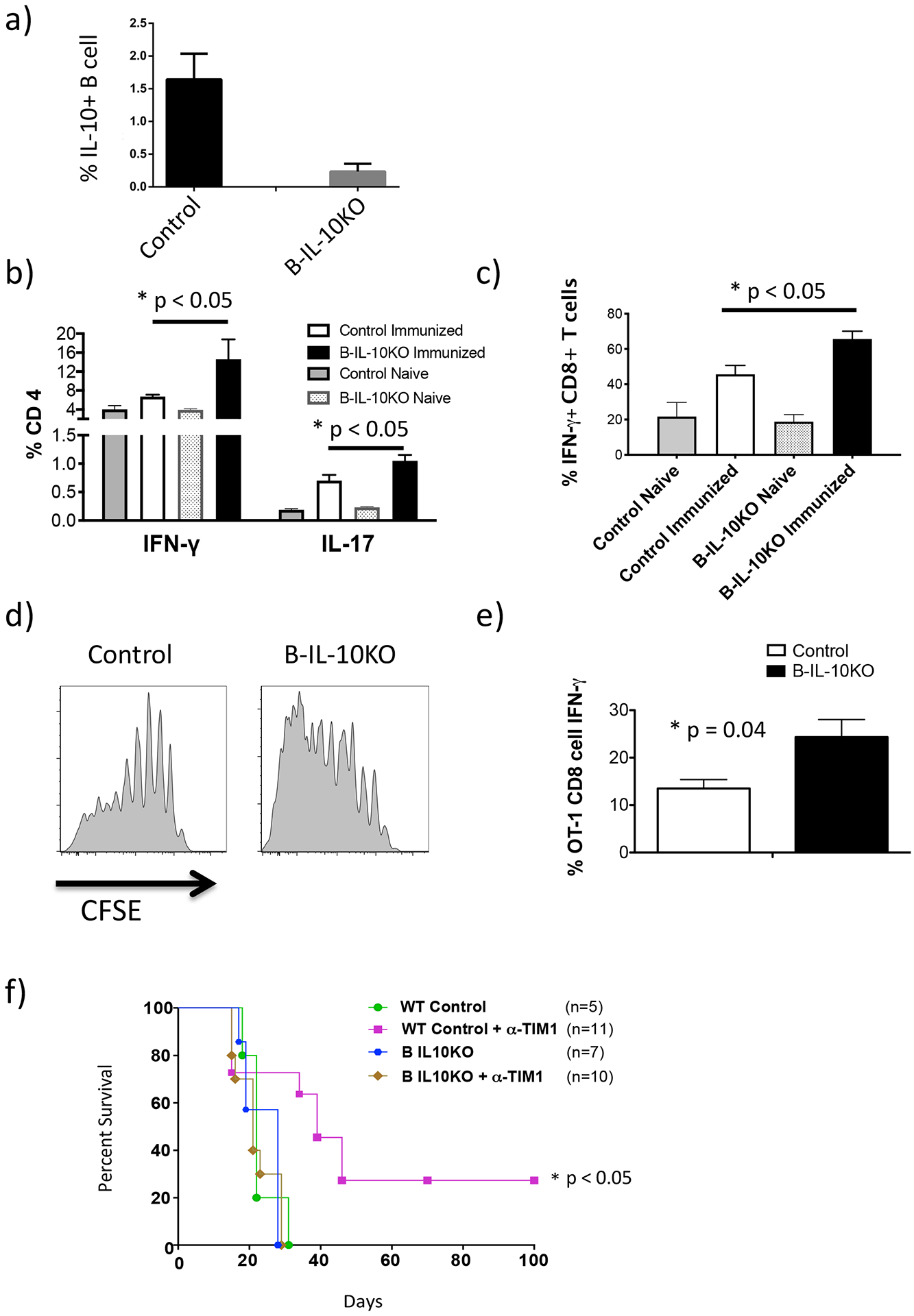
hCD20-ERT2.Cre-IL-10fl/fl mice (B-IL-10KO) or Control (IL-10fl/fl) mice were treated with tamoxifen on days −3, −2 and −1 to specifically knockout the IL-10 gene in B cells. All mice were immunized with BALB/c splenocytes on day 0 and spleens harvested 14 days later to examine T cell cytokines. a) Bar graph showing IL-10 expression by B cells from B-IL-10KO vs. Control mice (n = 3). b) IFN-γ and IL-17 production by splenic CD4 T cells (n = 5). c) IFN-γ production by splenic CD8 T cells (n = 5). d) CFSE-stained OT-1 T cells were transferred into tamoxifen treated B-IL-10KO or Control mice, as described above, that were subsequently immunized with NP-OVA-alum i.p. and spleens were harvested on day 5. Histograms show OT-1 CFSE dilution for indicated treatment groups (n = 4). e) IFN-γ production by transferred OT-1 T cells on day 5 (n = 4). Graphs (a, b, c, e) show mean ± SD with Student’s t tests (unpaired, two tailed) to analyze data. f) Kaplan-Meir plots showing allograft survival of Cre+ Control (B6) or B-IL-10KO BALB/c islet recipient with and without anti-TIM-1 treatment. Log-rank test was carried out the analyze survival curve.
We previously showed that prolonged allograft survival mediated by tolerogenic anti-TIM-1 (RMT1–10) is B cell dependent and associated with an increase in IL-10+ TIM-1+ B cells (9). To directly demonstrate that B cell IL-10 is essential for anti-TIM-1-mediated tolerance, we examined allograft survival (GS) in diabetic TAM-treated B-IL-10KO or Cre-neg controls. Anti-TIM-1 prolonged GS in control mice, with ~30% achieving long-term (>100d) engraftment (Fig. 1f). However, prolonged GS mediated by anti-TIM-1 was entirely dependent on B cell IL-10. These results substantiate the IL-10-mediated inhibition of T cells by Bregs, and specifically establish that Bregs can suppress CD8+ T cells in vivo.
3.2. IL-10-GFP reporter mice allow direct identification of Bregs
Consistent with other reports, we observed a small population of splenic Bregs in naïve IL-10-GFP reporter mice, even in the absence of in vitro stimulation (Fig. 2a,b)(6, 29, 35, 36). The frequency of GFP+ B cells increased significantly after mice were immunized with either alloantigen or NP-CGG-Alum (Fig. 2a and not shown). By comparison, typical ex vivo stimulation of splenocytes from immunized mice with LPS, PMA, ionomycin, plus monensin (LPIM) resulted in a further 3–5 fold increase in frequency of GFP+ B cells compared to unstimulated cells (monensin alone) (Fig. 2a,b)(9, 23).
Figure 2. Immunized Tiger-IL-10eGFP reporter mice allow for Breg detection by flow cytometry without ex-vivo stimulation.
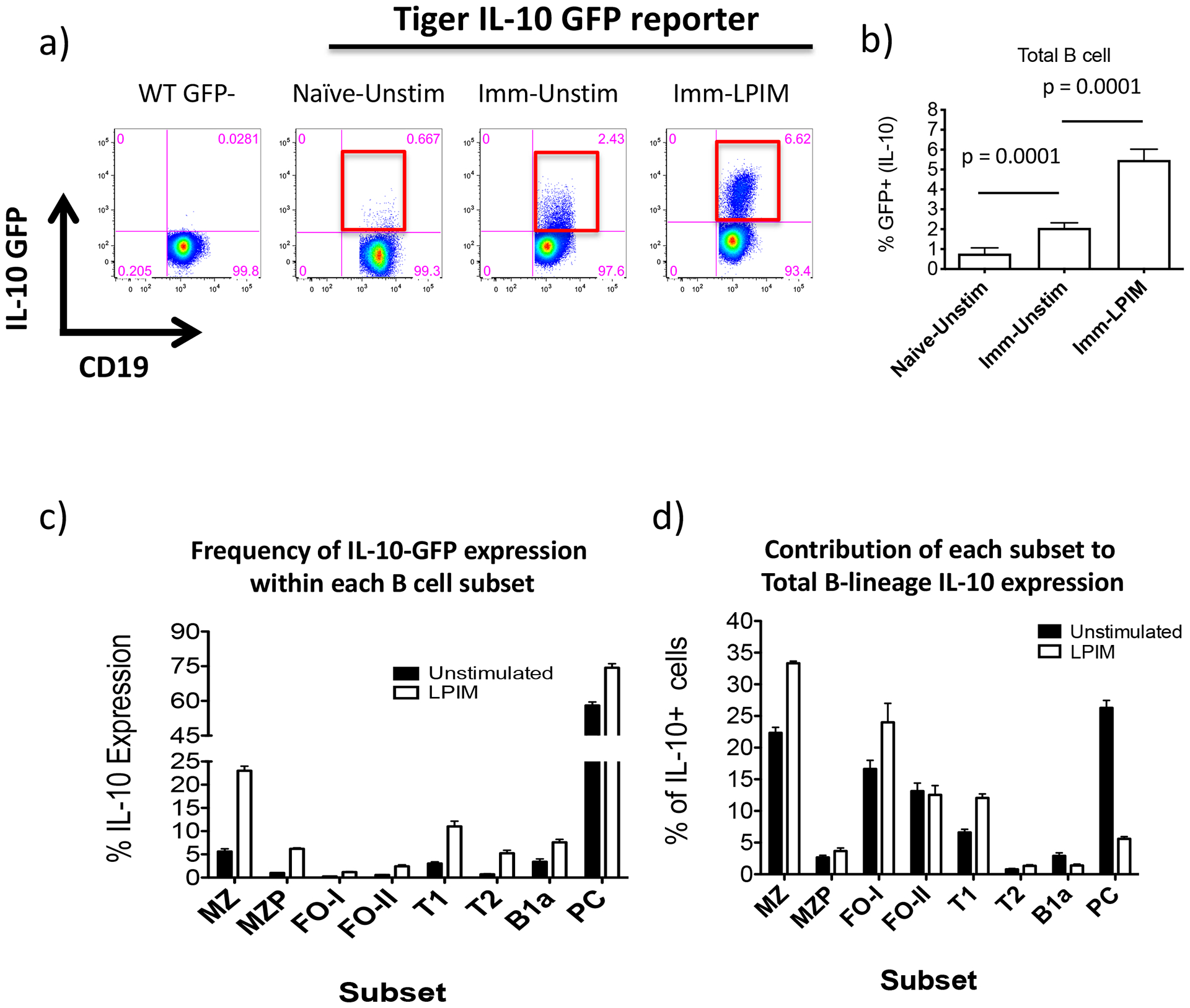
a) Splenocytes were isolated from IL-10-reporter mice that were Naïve or immunized (Imm) with allo-antigen (d3). Splenocytes were left unstimulated (Unstim) or stimulated with LPS, PMA, ionomycin and monensin (LPIM) for 5 hours. Subsequently, B cells were examined for IL-10-GFP expression by flow cytometry. Red boxes highlight GFP expression within each plot. b) Cumulative data showing B cell IL-10-GFP expression in LPIM stimulated vs. unstimulated cells. Student’s t tests (unpaired, two tailed) was used to assess statistical differences between groups (n = 3–6/group). c) Percent of B cells expressing IL-10-GFP within each B cell subset from unstimulated vs. LPIM-stimulated splenocytes (MZ, marginal zone; MZP, marginal zone precursor; FOI, follicular I; FOII, follicular II; T1, transitional 1; T2, transitional 2; PC, plasma cells; B1a) (n = 6). d) Percent contribution to total B cell IL-10-GFP expression by each subset. Data are representative of 4–7 mice. Graphs show mean ± SD.
Bregs have typically been described in terms of their frequency in various B cell subsets after in vitro stimulation. Using IL-10-reporter mice and established markers(31), we compared the distribution of GFP+ B cells amongst canonical B cells subsets, with/without in vitro stimulation (Fig. 2c and S1). IL-10-GFP expression by both unstimulated and stimulated splenic B cells was most enriched within MZ B cells (5.6% ±1.0% vs. 23% ±1.7%, respectively, p=0.0001) and was least enriched amongst follicular I (FO-I) B cells (0.26% ±0.050% vs. 1.2% ±0.15%, respectively, p=0.0006; Fig. 2c). Thus, stimulation led to a 4–7 -fold increase in the frequency of IL-10-GFP expression in most B cell subsets (e.g. 4x MZ; 6x MZP; 4.5x FO; 7x T2) compared to unstimulated cells, however, the rank order of subset distribution and overall frequency remained similar (Fig. S1g). Notably, unstimulated PCs, were highly enriched for IL-10-GFP, yet exhibited relatively little increase (1.3x) after ex vivo stimulation (from 58% ±2.6% to 74% ±3.1%, p=0.002). Similar results were obtained when mice were immunized with NP-CGG (data not shown).
Because B cell subsets differ markedly in size, we examined the contribution of each subset to the total number of IL-10+ B cells. Although FO B cells are the least enriched (~0.25%), they are the largest splenic B cell population (~55%), and as a result, make up a larger than expected contribution to the total Breg population (Fig. 2c,d and S1). These results show that MZ (~23%) and FO B cells (~30%) and PCs (~27%), comprise ~80% of all IL-10+ B cells – contrasting with transfer studies that emphasize IL-10 frequency within small subsets. The fact that IL-10+ cells are rare amongst FO-I B cells may explain why they lack regulatory activity in most transfer studies and their role as Bregs has been overlooked. Because IL-10 expression of B cells, but not PCs, markedly increases with in vitro stimulation, the contribution of PCs (<1% of B cells) to overall IL-10 falls from 27% to ~5% after in vitro stimulation. This may help explain why PCs were missed as a major source of B cell IL-10 for so long. Importantly, our findings open the door to sort-purify GFP+ Bregs directly from IL-10-reporter mice to examine their function in vivo.
3.3. Live-cell imaging of splenic Bregs Reveals Breg:T cell interactions at the T:B border
While IL-10 is a key mediator of Breg activity, essentially nothing is known about the intercellular interactions between Bregs and other immune cells in vivo nor the subanatomic localization of these events. To examine the interactions between Bregs, T cells, and DCs, we utilized 2-photon microscopy. IL-10 reporter mice were immunized with NP-CGG, to enrich for hapten-specific NP-reactive B cells(37). 7 days later, GFP+ B cells (Bregs) and GFP- control B cells, were sort-purified from spleens, CFSE-stained (to allow visualization), pulsed with NP-OVA and transferred into separate, otherwise naïve, congenic (CD45.1) mice along with naive TCR-transgenic DsRed-OT-1 (CD8+) and NP-OVA-pulsed BMDCs (Fig. S2a). In this manner, NP-reactive B cells and DCs can present processed Ova peptide antigens to both OT-1 and OT-2 cells. 18–24h after cell transfer, recipient spleens were removed and cut with a vibratome to facilitate imaging of the white pulp (Fig. S2a)(34). To determine localization of Bregs and T cells in spleen, we took advantage of the congenic difference between transferred cells (CD45.2) and host mice (CD45.1) (Fig. S2a). Immunofluorescence staining revealed that, as expected, most OT-1 trafficked to the T cell zone (TCZ), forming clusters (see imaging movies below and Fig. 3a). Transferred B cells were readily observed in B cell follicles and at the T:B border around these T cell clusters (Fig. 3a,b,c).
Figure 3. Localization and interactions of Bregs and Control B cells with OT-1 T cells in the spleen.
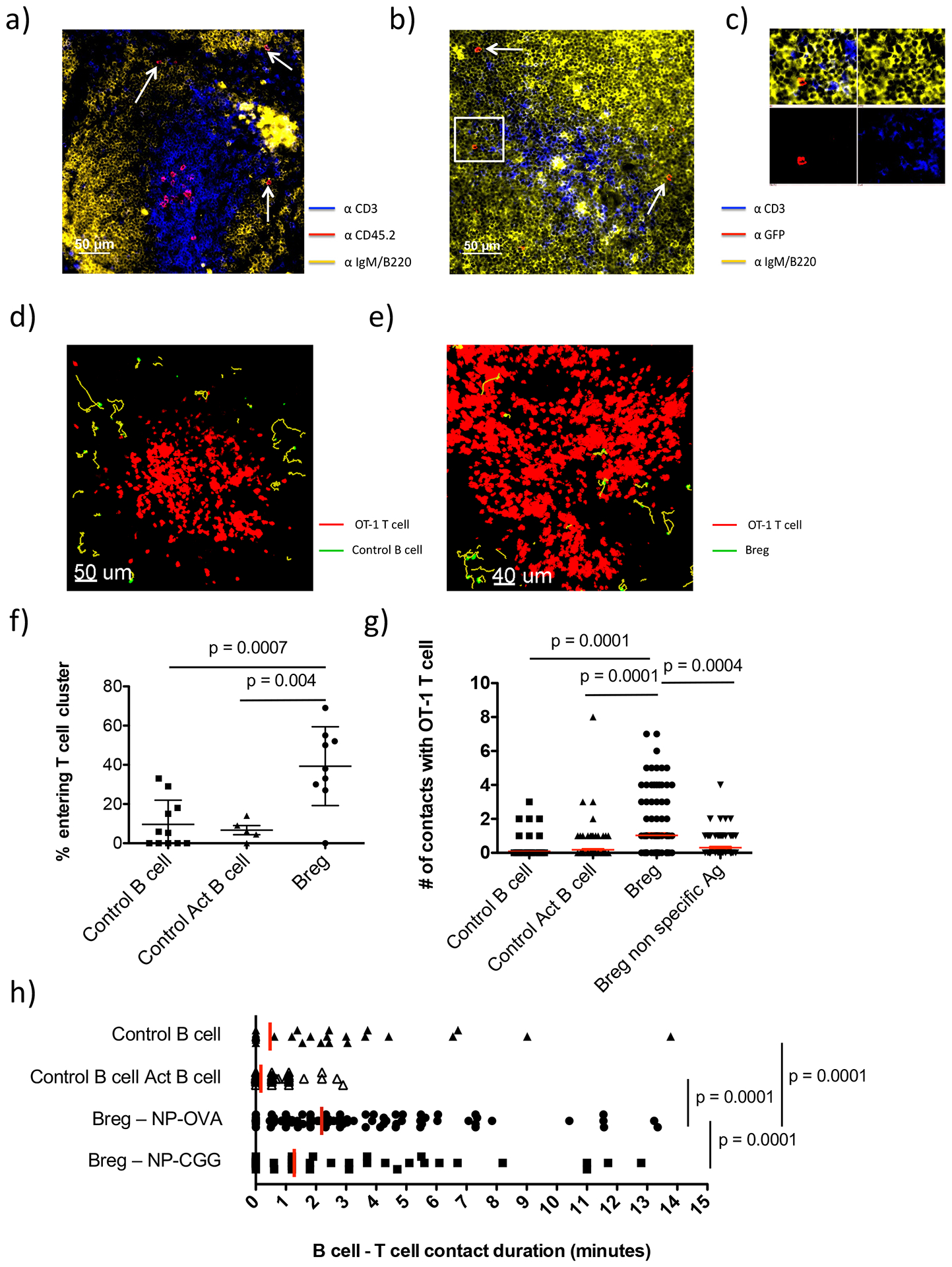
Experiments were carried out as described in Fig. S2a. (a-c): Immunofluorescence microscopic images of spleen sections of CD45.1 congenic mice 24h after transfer of CD45.2 Bregs and OT-1 cells. a) Section stained with anti-CD3 (T cells), anti-CD45.2 (transferred cells), and anti-IgM/B220 (B cells: same fluorochrome). b) Section stained with anti-CD3 (T cells), anti-IgM/B220 (B cells) and anti-GFP to specifically detect transferred Bregs. c) Inset from (b) zoomed in on a Breg cell showing separated fluorescent signals from each channel. d-e). 2-photon imaging snapshots showing transferred CFSE-stained Control B cells (GFP-, green) (d), or Bregs (GFP+, green) (e), around the periphery or within DS-red OT-1 T cell (red) clusters. f) Graph showing the frequency of Control B cell vs. Control activated B cell (Control Act B cell) vs. Breg cells entering T cell cluster. Each dot represents one individual 20–35 minute recording. Mean ± SD is shown. g) Graph showing the number of contacts between individual Control B cells, Control Act B cells, Bregs pulsed with NP-OVA or Bregs pulsed with NP-CGG with OT-1 T cells. Mean ± SEM is shown. h) Graph showing the duration (minutes) of each contact made by Control B cells, Control Act B cells, Bregs pulsed with NP-OVA or Bregs pulsed with NP-CGG with OT-1 T cells. Red line shows mean contact duration. Imaging results were obtained from 3–4 separate mice for each group with 131 Control B cells, 157 Control Act B cell, 135 Breg-NP-OVA and 90 Breg-NP-CGG cells tracked. Student’s t tests (unpaired, two tailed) was used to assess significant differences between groups.
Analysis of imaging movies revealed that although there were no clear differences in the number of GFP+ vs. GFP- control B cells that localized to B cell follicles or T:B border, GFP+ Bregs actually entered these OT-1 T cell clusters at a 4-fold higher frequency than GFP- control B cells sorted from the same mice (Bregs, 39% ±6.7% vs. Control, 9.6% ±3.7%, p=0.0007; Fig. 3d,e,f and Movies S1–S2). We also sorted GFP- control B cells that expressed the same levels of CD44 and CD86 as the GFP+ B cells to ensure the observed differences were not due to the activation state of the B cells (see Fig. S2a). As above, these “activated” GFP- controls (Control-Act) tended to localize in the follicle and T:B border (Fig. 3a) and were much less frequently observed within the T cell clusters than Bregs (6.7% ±5.2% vs. 39% ±6.7%, respectively p=0.004; Fig. 3d,e,f and Movies S1–S2). Bregs made contacts with T cells both within and around the periphery of these T cell clusters. In fact, Bregs made >5-fold more contacts with OT-1 T cells than did control B cells (Bregs, 1.0 ±0.14 vs. Control, 0.091 ±0.037 vs. Control Act 0.18 ±0.061 contacts/cell, p=0.0001; Fig. 3g and Movie S3). These Breg:T cell contacts tended to be relatively brief with only a minority of the cells making contacts greater than 5 minutes (Fig. 3h). Nonetheless, Bregs still made significantly longer contacts with OT-1 T cells than did Control B cells. We also compared Bregs and Control B cells using other motility parameters including velocity and displacement, but found no notable differences (Fig. S2b–e). Interestingly, we did find that OT-1 T cells moved faster in spleens of mice that had received Bregs compared to control B cells (Fig. S2f), suggesting that Bregs may decrease T cell:DC interactions(28, 38). However, the magnitude of these differences was small.
Similar results were obtained when the same experiment was repeated using OT-2 CD4+ T cells rather than OT-1 (CD8+) T cells (Fig. S2g,h). Again, compared to Control-Act B cells, Bregs exhibited increased entry into the T cell clusters, made more contacts (0.40 ±0.068 vs. 1.1 ±0.20 contacts/cell, respectively; p=0.007), and more prolonged contacts (2.9 ±5.8 min vs. 6.0 ±9.8, respectively; p=0.002) with OT-2 cells. Thus, Bregs exhibit increased interactions with both CD4+ and CD8+ T cells than do non-Bregs.
Several studies have demonstrated that Breg activity is dependent on antigen-specific priming (5, 9, 25). As such, we asked whether the contacts made between Bregs and OT-1 T cells were antigen-specific. Experiments were carried out in which Bregs were pulsed with NP-CGG (non-specific antigen) instead of NP-OVA (specific antigen) prior to transfer. Bregs pulsed with NP-CGG, and those pulsed with NP-OVA, entered the T cell clusters with similar frequency. However, compared to Bregs pulsed with specific antigen (NP-OVA), those pulsed with NP-CGG made fewer contacts with OT-1 T cells, and their interactions with OT-1 T cells were shorter in duration, similar to GFP- control B cells (Fig. 3g,h). Thus, Breg interaction with T cells, but not localization, was affected by cognate antigen.
3.4. Breg:T cell interactions inhibit subsequent T cell:DC interactions
To determine whether Bregs also interact with DCs, experiments similar to above were performed, using fluorescent-labeled NP-OVA-pulsed BMDCs to enable their detection. We found that neither transferred control B cells nor Bregs interacted with the co-transferred BMDCs (Fig. 4a,b and c, and movies S4–S5). However, further analysis revealed that in the presence of Bregs, OT-1 T cells made 25% fewer contacts with BMDCs than they did in the presence of control B cells (Breg; 1.4 ±0.17/contacts per cell vs. Control; 1.9 ±0.18/contacts/per cell, , p=0.01), and moreover, T cell:DC contact duration was almost 40% shorter in the presence of Bregs (Breg; 10 ±11 min vs. Control; 16 ±12 min, , p=0.0001) (Fig. 4d,e). Reduced T:DC contact in the presence of Bregs could result from ongoing effects of previous cognate T cell:Breg encounters, or could be the result of Breg:DC interactions not visualized in this experimental set-up. We reasoned that while T:Breg interactions are cognate, Breg:DC interactions would be non-cognate. Thus, similar to the experiments described above, Bregs were pulsed with non-cognate NP-CGG vs. cognate NP-OVA antigen and interactions between OT-1 T cells and NP-OVA pulsed DCs were examined. When Bregs were pulsed with NP-CGG, DCs made more T cell contacts with T cells, than when Bregs were pulsed with NP-OVA (Breg NP-CGG; , 2.0 ±0.15/T:DC contacts per cell vs. Breg-NP-OVA; , 1.4 ±0.17/T:DC contacts/per cell, ; p=0.01) and the T:DC contacts lasted significantly longer (Breg NP-CGG; , 24 ±12 min vs. Breg-NP-OVA; , 10 ±11 min, p=0.0001) (; Fig. 4e). Thus, Bregs reduce the number and duration of contacts between T cells and DC’s – only when they present specific antigen. This indicates that cognate Breg:T cell interactions are necessary for altered T cell behavior. This provides an additional mechanism by which Ag-specific Bregs may restrain T cell responses.
Figure 4. Breg, DC, and OT-1 T cell interactions in the spleen.
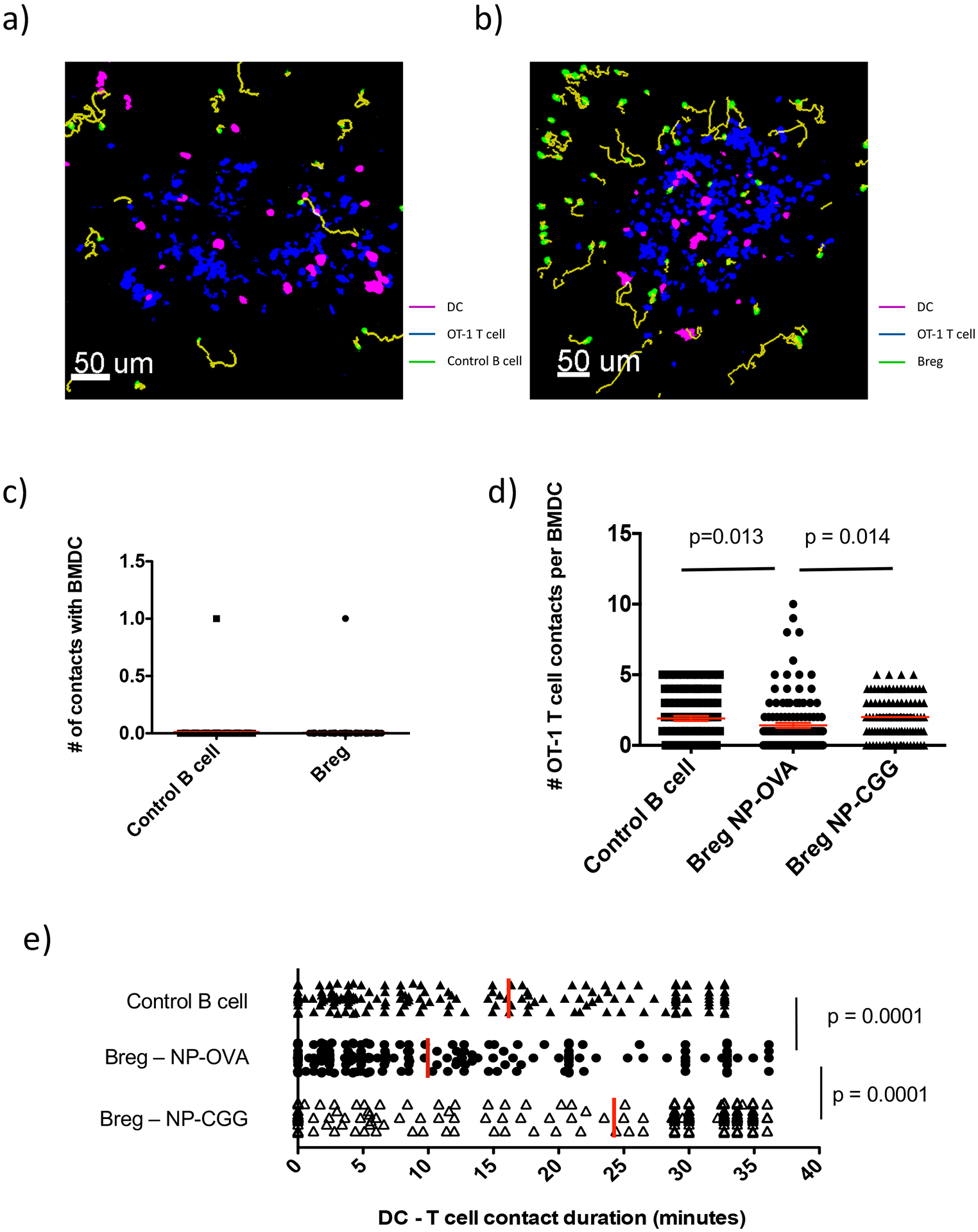
Experiments were carried out as described in Fig. S2a with minor modification. Mice received 2–3 million BMDC, 2 million Control B cells or Bregs and 1 million DsRed naïve OT-1 T cells. a) 2-photon microscopy imaging snapshot showing transferred Control B cells (green) with OT-1 T cells (blue) and BMDC (pink). Yellow lines represent track path and length for each B cell. b) 2-photon microscopy imaging snapshot showing Bregs (green) with OT-1 T cells (blue) and BMDC (pink). Yellow lines represent track path and length for each B cell. c) Graph showing the number of contacts individual Control B cells or Bregs made with BMDC. d) Graph showing the number of contacts each BMDC made with OT-1 T cells in the presence of Control B cells (98 BMDCs tracked) versus Bregs pulsed with NP-OVA (123 BMDCs tracked) versus Bregs pulsed with NP-CGG (102 BMDCs tracked). Mean (red line) ± SEM is shown. n = 2–4 mice per group. Student’s t tests (unpaired, two tailed) was used to assess significant differences between groups.
3.5. FO Bregs exhibit increased CCR7 expression
Based on the striking difference in the ability of Bregs to enter the T cell clusters noted above, we examined CCR7 expression, which would facilitate B cell migration to the T-B border and entry into the TCZ. Indeed, Bregs expressed significantly higher levels of CCR7 compared to control B cells (2.7-fold increase in mean fluorescent intensity (MFI); Fig. 5a,b). Moreover, FO-Bregs express higher levels CCR7 than MZ-Bregs (Fig. 5c,d). To confirm that FO-Bregs preferentially localized to B cell follicles where they can access the T:B border, we sort-purified FO (GFP+CD19+IgDhi IgMloCD21lo) vs. MZ (GFP+CD19+IgDlo IgMhiCD21hi) Bregs from immunized IL-10-GFP reporter mice and transferred them into WT recipients. Examination of whole spleen section images revealed that transferred MZ Bregs localized mostly to the MZ amongst SIGNR1+ macrophages (36% ±11) and red pulp (RP) (44% ±17), with 20% ±6 migrating to the follicles. In contrast, transferred FO Bregs localized mostly to the follicles (46% ±11) and RP (40% ±5) with only 13% ±3.5 locating to the MZ (Fig. 5e–i). This is consistent with conventional MZ and FO B cells which, after initial activation, can migrate to the RP to generate extrafollicular responses that ultimately give rise to memory B cells and short-lived PCs (39, 40). In this regard, Bregs can develop into antibody-producing PCs (35).
Figure 5. Breg chemokine expression and splenic localization.
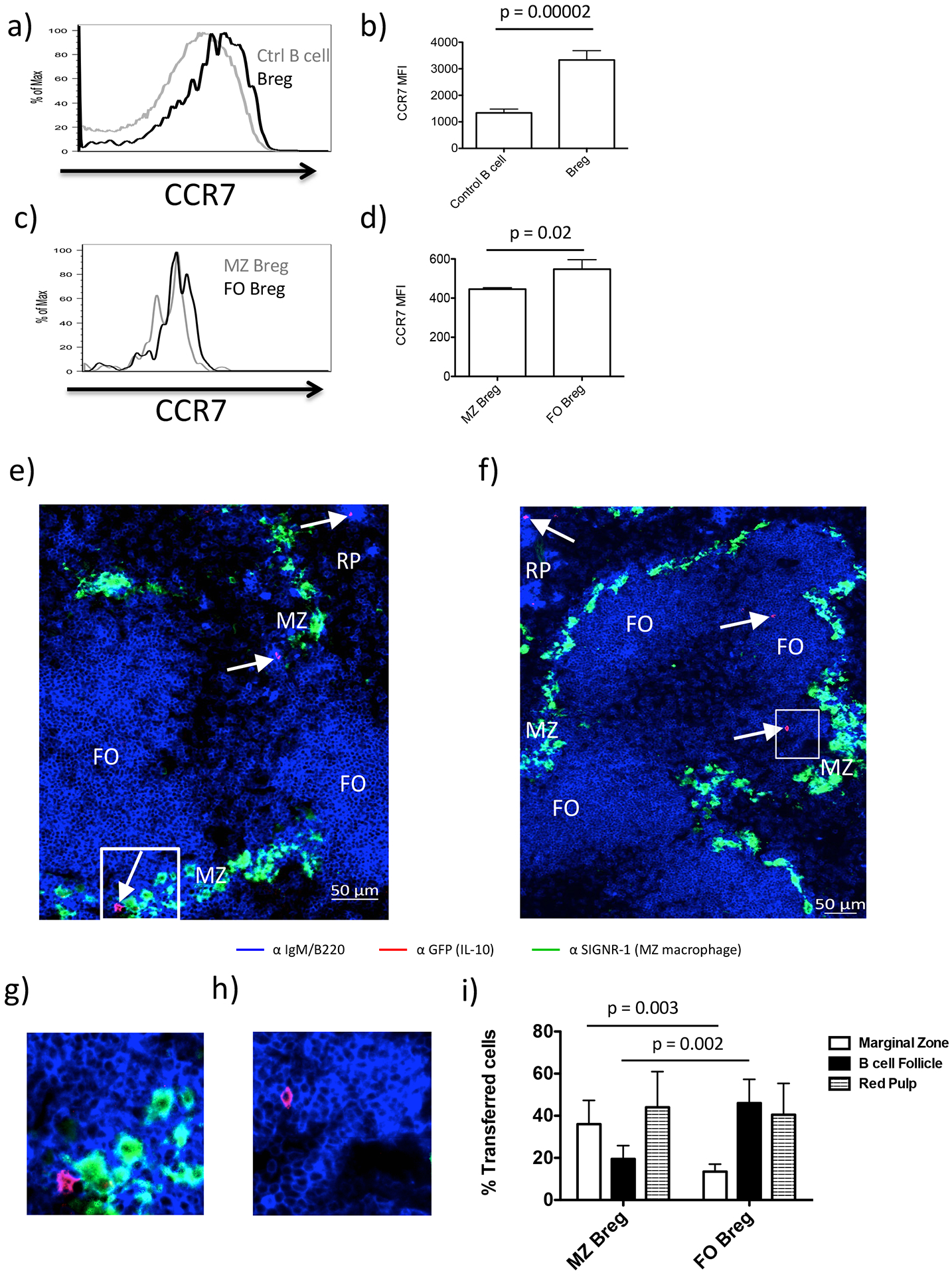
Representative histogram overlays showing relative expression of: a) CCR7 expression on Bregs (black) vs. Control B cells (grey). b) Bar graph showing mean MFI ± SD for each cell type in (a). Data is representative of 4 mice. c) CCR7 expression on FO-Bregs (black) vs. MZ-Breg (grey). d) Bar graph showing mean MFI ± SD for each cell type in (c). e) MZ-Bregs were sorted from previously immunized Tiger mice based on established phenotypic markers (Fig. S1b) and transferred into B6 WT mice. To simultaneously detect endogenous FO B cells (B220hi IgMlo) and endogenous B cells localizing to the MZ and red pulp (B220lo IgMhi), we stained spleen sections with anti-B220 and anti-IgM conjugated to the same fluorophore (blue). The sections were also stained with anti SIGNR-1 (green), which labels MZ macrophages, demarcating the MZ as a narrow band that separates the follicles from the red pulp (RP) and anti-GFP (red) to detect the transferred cells. f) FO-Bregs were sorted from previously immunized Tiger mice based on established phenotypic markers (Fig. S1c) and transferred into B6 WT mice. Sections were stained as in described in (e). Arrows indicate staining for transferred cells. g) Inset from (e) showing zoomed in area of GFP+ cell located in MZ. h) Inset from (f) showing zoomed in area of GFP+ cell located in B cell follicle. i) Summary graph showing quantitation of transferred MZ and FO Breg migration to indicated splenic niche. Mean ± SD is shown for each graph. n = 3–5 mice per group. Student’s t tests (unpaired, two tailed) was used to assess significant differences between groups.
Finally, we examined localization of endogenous splenic B cells in IL-10-GFP reporter mice. 79% ±6.9 of all IL-10+ B cells localized to the RP, 16% ±10 localized to the MZ, and only 4.7% ±3.5% localized to the follicles (Fig. S3a–d, Movie S6). However, since immunofluorescence staining is both less sensitive and has lower dynamic range than flow cytometry, this analysis likely underestimates the frequency of follicular Bregs, which exhibit dimmer IL-10 expression (Fig. S3e). Thus, Bregs are localized in all three major splenic niches including the follicles.
4. Discussion
Our understanding of Bregs has been complicated by the lack of a specific marker and resulting focus on B cell subpopulations sufficiently enriched in IL-10 expression that they can transfer immunomodulatory activity to B cell deficient hosts (1–3, 9, 19). It remains unclear which B cell subsets express IL-10 and/or perform Breg function in vivo, and essentially nothing was known about how and where Bregs carry out their regulatory function. Breg function has primarily been examined by transfer into B cell deficient mice, which lack normal splenic and lymph node architecture. Few groups have utilized more physiologically relevant models where endogenous B cells specifically lack IL-10 expression(5, 20, 21, 41). To address these fundamental questions we used novel hCD20-ERT2.Cre.IL-10fl/fl mice with an inducible B cell-specific deletion of IL-10. This was important because the only two studies to have used mice with (congenitally) floxed B cell IL-10, exhibited more modest regulatory defects than studies using mixed BM chimeras or Breg transfers into μMT mice(21, 41). We also utilized IL-10-reporter mice to definitively identify distribution and localization of IL-10+ B cell subsets, and study their intercellular interactions.
Bregs appear to skew CD4 T cells towards Th2 and Treg responses,(6, 14). However, it is was unknown whether Bregs interact directly with CD4+ T cells and/or inhibit their inflammatory activity indirectly by suppressing DC function, as seen with Tregs (6, 14). While Bregs from MHC II-deficient and CD40-deficent mice are defective, T cell interactions might be required for proper Breg development and activation (25, 42). By comparison, studies examining the in vivo effects of Bregs on CD8 T cells are limited. Mice lacking B cells, or B cell-IL-10, exhibit increased CD8 effector/memory responses after CMV or L. donovani infection compared to WT mice(43–45). However, B cell-mediated suppression of CD8 cells was not related to IL-10. Moreover, B cells can present L. donovani to CD4+ but not CD8+ cells in vitro, suggesting that regulation of CD8 responses by B cells may be indirect.
Here, we definitively demonstrated that acute loss of B cell IL-10 resulted in a significant increase in proinflammatory cytokines by both CD4 and CD8 cells, and increased the proliferation of antigen-specific CD8 cells. Thus, Bregs dampen both CD4+ and CD8+ T effector responses in vivo. Moreover, such mice were unresponsive to anti-TIM-1-mediated inhibition of allograft rejection, confirming that the tolerogenic effects of this mAb are truly dependent on induction of IL-10+ Bregs in vivo(9).
Using IL-10-reporter mice, we confirmed that PCs are highly enriched in IL-10 compared to canonical B cell subsets. However, when adjusted for subpopulation size, PCs comprise only ~25% of all B lineage IL-10, while MZ B cells comprised 25% and FO B cells, the least enriched but largest subset, made up ~30%. Thus, MZ and FO B cells and PCs comprise ~80% of all IL-10+ Bregs. The differential localization and interactions with distinct T cells amongst these three subsets suggests distinct regulatory functions, an issue now amenable to study.
Examination of bona fide Bregs by live-cell imaging allowed us to determine that a fraction of transferred Bregs localize to the T-B border, where their regulatory function may be maximized by encountering recently activated T cells. Bregs actually made forays into clusters of Ag-specific T cells in the TCZ, compared to controls B cells, which may be facilitated by expressing higher levels of CCR7(Fig. 5). Moreover, FO Bregs express higher levels of CCR7 than MZ Bregs, and in cell transfer experiments more than twice as many FO Bregs as MZ Bregs migrated to B cell follicles, further suggesting that FO Bregs are most likely to interact with T cells at the T:B border and in the nearby TCZ. Bregs exhibited an increased number and duration of contacts with both OT1 and OT2 cells compared to non-Bregs, and these were specific-antigen dependent (Fig. 3 and S2). Thus, our data directly demonstrate that Bregs interact in a cognate manner with both naïve CD4 and CD8 cells. Breg:T cell Ag-specific vs. non Ag-specific interactions are likely to be purposeful, since T cells moved more rapidly and made significantly fewer and briefer contacts with DCs (Fig. 4e and S2f).
Our data show that Bregs do not make contacts with transferred BMDCs. It remains possible that Bregs could interact with specific subsets of DCs in certain niches to impact T cell activation. In our model, Bregs might inhibit DC function through elaboration of cytokines without requiring direct cell-cell contact. However, reduced T:DC interactions required cognate Breg:T cell interactions. This provides mechanistic insight into previous reports that Bregs inhibit innate immune cell function (6, 24, 46). Decreased T cell inflammatory cytokines and increased Tregs resulting from Breg activity might indirectly reduce APC activation, further limiting T cell activation (9, 18). Importantly, our data indicates that Ag-specific Breg and Treg activity occur through biologically distinct mechanisms. Tregs are activated by specific Ag, but such cognate interactions suppress the ability of DCs to subsequently present Ags to other nearby T cells that may actually respond to distinct antigenic specificities. In contrast, Breg activation through the BCR (NP-CGG) is not sufficient to induce Breg:Tcell, or suppress DC:T cell, interactions. Rather, Bregs must also capture Ag through their BCR (NP-OVA) and after processing, present this Ag to relevant Ag-specific T cells.
In summary, we firmly establish that Bregs have a significant impact on both CD4 and CD8 T cell effector function in vivo. We demonstrate that the T-B border, an important junction for cognate interaction between T cells and B cells, is also a likely primary site for antigen-specific Breg-mediated T cell regulation through direct cognate interactions, and that FO B cell derived Bregs are the most likely Breg subset carrying out these interactions. Activated FO and MZ Bregs also migrate to the RP, where their role remains to be determined. It is possible that Bregs belonging to other subsets, or found in other locations, exhibit different interactions with innate cells and/or T cells in different settings. This might explain why Bregs exist in various B cell subsets. Elucidation of such functional distinctions is now feasible.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01AI114587 (DMR). KM was supported by an AST-Basic Science Fellowship Grant. We would like to thank Dr. Fadi Lakkis for critical reading of the manuscript. We also thank Dr. Mark Shlomchik, Dr. Richard Flavell and Dr. Werner Muller for providing us with genetically altered mice developed in their laboratories.
Abbreviations
- Ag
antigen
- APC
antigen presenting cell
- B-IL-10KO
hCD20-ERT2.Cre-IL-10fl/fl mice
- BMDC
bone marrow derived dendritic cells
- Breg
regulatory B cells
- Control-Act
GFP- activated control B cell
- FO-I
follicular I
- FO-II
follicular II
- LPIM
LPS, PMA, Ionomycin, Monnesin
- MZ
marginal zone
- NP-CGG
nitrophenyl chicken gamma globulin
- NP-CGG
nitrophenyl ovalbumin
- PC
plasma cell
- RP
Red pulp
- T2-MZP
T2 marginal zone precursor
- TAM
tamoxifen
- TCZ
T cell zone
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- 1.Mauri C Regulation of immunity and autoimmunity by B cells. Current opinion in immunology 2010;22(6):761–767. [DOI] [PubMed] [Google Scholar]
- 2.Lino AC, Dorner T, Bar-Or A, Fillatreau S. Cytokine-producing B cells: a translational view on their roles in human and mouse autoimmune diseases. Immunological reviews 2016;269(1):130–144. [DOI] [PubMed] [Google Scholar]
- 3.DiLillo DJ, Matsushita T, Tedder TF. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Annals of the New York Academy of Sciences 2010;1183:38–57. [DOI] [PubMed] [Google Scholar]
- 4.Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002;16(2):219–230. [DOI] [PubMed] [Google Scholar]
- 5.Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nature immunology 2002;3(10):944–950. [DOI] [PubMed] [Google Scholar]
- 6.Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity 2014;41(6):1040–1051. [DOI] [PubMed] [Google Scholar]
- 7.Mauri C, Gray D, Mushtaq N, Londei M. Prevention of arthritis by interleukin 10-producing B cells. The Journal of experimental medicine 2003;197(4):489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. The Journal of clinical investigation 2008;118(10):3420–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding Q, Yeung M, Camirand G, Zeng Q, Akiba H, Yagita H et al. Regulatory B cells are identified by expression of TIM-1 and can be induced through TIM-1 ligation to promote tolerance in mice. The Journal of clinical investigation 2011;121(9):3645–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lundy SK, Boros DL. Fas ligand-expressing B-1a lymphocytes mediate CD4(+)-T-cell apoptosis during schistosomal infection: induction by interleukin 4 (IL-4) and IL-10. Infection and immunity 2002;70(2):812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khan AR, Hams E, Floudas A, Sparwasser T, Weaver CT, Fallon PG. PD-L1hi B cells are critical regulators of humoral immunity. Nature communications 2015;6:5997. [DOI] [PubMed] [Google Scholar]
- 12.Nouel A, Pochard P, Simon Q, Segalen I, Le Meur Y, Pers JO et al. B-Cells induce regulatory T cells through TGF-beta/IDO production in A CTLA-4 dependent manner. Journal of autoimmunity 2015;59:53–60. [DOI] [PubMed] [Google Scholar]
- 13.Lindner S, Dahlke K, Sontheimer K, Hagn M, Kaltenmeier C, Barth TF et al. Interleukin 21-induced granzyme B-expressing B cells infiltrate tumors and regulate T cells. Cancer research 2013;73(8):2468–2479. [DOI] [PubMed] [Google Scholar]
- 14.Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014;507(7492):366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans JG, Chavez-Rueda KA, Eddaoudi A, Meyer-Bahlburg A, Rawlings DJ, Ehrenstein MR et al. Novel suppressive function of transitional 2 B cells in experimental arthritis. Journal of immunology 2007;178(12):7868–7878. [DOI] [PubMed] [Google Scholar]
- 16.Mohib K, Cherukuri A, Rothstein DM. Regulatory B cells and transplantation: almost prime time? Current opinion in organ transplantation 2018;23(5):524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun J, Wang J, Pefanis E, Chao J, Rothschild G, Tachibana I et al. Transcriptomics Identify CD9 as a Marker of Murine IL-10-Competent Regulatory B Cells. Cell reports 2015;13(6):1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carter NA, Rosser EC, Mauri C. Interleukin-10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen-induced arthritis. Arthritis research & therapy 2012;14(1):R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding Q, Mohib K, Kuchroo VK, Rothstein DM. TIM-4 Identifies IFN-gamma-Expressing Proinflammatory B Effector 1 Cells That Promote Tumor and Allograft Rejection. Journal of immunology 2017;199(7):2585–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bankoti R, Gupta K, Levchenko A, Stager S. Marginal zone B cells regulate antigen-specific T cell responses during infection. Journal of immunology 2012;188(8):3961–3971. [DOI] [PubMed] [Google Scholar]
- 21.Madan R, Demircik F, Surianarayanan S, Allen JL, Divanovic S, Trompette A et al. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. Journal of immunology 2009;183(4):2312–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watt V, Ronchese F, Ritchie D. Resting B cells suppress tumor immunity via an MHC class-II dependent mechanism. Journal of immunotherapy 2007;30(3):323–332. [DOI] [PubMed] [Google Scholar]
- 23.Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 2008;28(5):639–650. [DOI] [PubMed] [Google Scholar]
- 24.Iwata Y, Matsushita T, Horikawa M, Dilillo DJ, Yanaba K, Venturi GM et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 2011;117(2):530–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature 2012;491(7423):264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golovkina TV, Shlomchik M, Hannum L, Chervonsky A. Organogenic role of B lymphocytes in mucosal immunity. Science 1999;286(5446):1965–1968. [DOI] [PubMed] [Google Scholar]
- 27.Barbaric I, Miller G, Dear TN. Appearances can be deceiving: phenotypes of knockout mice. Briefings in functional genomics & proteomics 2007;6(2):91–103. [DOI] [PubMed] [Google Scholar]
- 28.Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P et al. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nature immunology 2006;7(1):83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara-Tejero M, Galan JE et al. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity 2006;25(6):941–952. [DOI] [PubMed] [Google Scholar]
- 30.Roers A, Siewe L, Strittmatter E, Deckert M, Schluter D, Stenzel W et al. T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. The Journal of experimental medicine 2004;200(10):1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allman D, Pillai S. Peripheral B cell subsets. Current opinion in immunology 2008;20(2):149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. The Journal of experimental medicine 1992;176(6):1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Labeur MS, Roters B, Pers B, Mehling A, Luger TA, Schwarz T et al. Generation of tumor immunity by bone marrow-derived dendritic cells correlates with dendritic cell maturation stage. Journal of immunology 1999;162(1):168–175. [PubMed] [Google Scholar]
- 34.Camirand G, Wang Y, Lu Y, Wan YY, Lin Y, Deng S et al. CD45 ligation expands Tregs by promoting interactions with DCs. The Journal of clinical investigation 2014;124(10):4603–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maseda D, Smith SH, DiLillo DJ, Bryant JM, Candando KM, Weaver CT et al. Regulatory B10 cells differentiate into antibody-secreting cells after transient IL-10 production in vivo. Journal of immunology 2012;188(3):1036–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neves P, Lampropoulou V, Calderon-Gomez E, Roch T, Stervbo U, Shen P et al. Signaling via the MyD88 adaptor protein in B cells suppresses protective immunity during Salmonella typhimurium infection. Immunity 2010;33(5):777–790. [DOI] [PubMed] [Google Scholar]
- 37.Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC et al. T follicular helper cell dynamics in germinal centers. Science 2013;341(6146):673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004;427(6970):154–159. [DOI] [PubMed] [Google Scholar]
- 39.Smith KG, Hewitson TD, Nossal GJ, Tarlinton DM. The phenotype and fate of the antibody-forming cells of the splenic foci. European journal of immunology 1996;26(2):444–448. [DOI] [PubMed] [Google Scholar]
- 40.William J, Euler C, Shlomchik MJ. Short-lived plasmablasts dominate the early spontaneous rheumatoid factor response: differentiation pathways, hypermutating cell types, and affinity maturation outside the germinal center. Journal of immunology 2005;174(11):6879–6887. [DOI] [PubMed] [Google Scholar]
- 41.Teichmann LL, Kashgarian M, Weaver CT, Roers A, Muller W, Shlomchik MJ. B cell-derived IL-10 does not regulate spontaneous systemic autoimmunity in MRL.Fas(lpr) mice. Journal of immunology 2012;188(2):678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blair PA, Chavez-Rueda KA, Evans JG, Shlomchik MJ, Eddaoudi A, Isenberg DA et al. Selective targeting of B cells with agonistic anti-CD40 is an efficacious strategy for the generation of induced regulatory T2-like B cells and for the suppression of lupus in MRL/lpr mice. Journal of immunology 2009;182(6):3492–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hon H, Oran A, Brocker T, Jacob J. B lymphocytes participate in cross-presentation of antigen following gene gun vaccination. Journal of immunology 2005;174(9):5233–5242. [DOI] [PubMed] [Google Scholar]
- 44.Marino E, Tan B, Binge L, Mackay CR, Grey ST. B-cell cross-presentation of autologous antigen precipitates diabetes. Diabetes 2012;61(11):2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heit A, Huster KM, Schmitz F, Schiemann M, Busch DH, Wagner H. CpG-DNA aided cross-priming by cross-presenting B cells. Journal of immunology 2004;172(3):1501–1507. [DOI] [PubMed] [Google Scholar]
- 46.Horikawa M, Weimer ET, DiLillo DJ, Venturi GM, Spolski R, Leonard WJ et al. Regulatory B cell (B10 Cell) expansion during Listeria infection governs innate and cellular immune responses in mice. Journal of immunology 2013;190(3):1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.