

Building evidence implicates the gut microbiome in critical animal functions such as regulating digestion, nutrition, immune regulation, and development. Simplified, phylogenetically diverse models for hypotheses testing are necessary because of the difficulty of assigning causative relationships in complex gut microbiomes.
KEYWORDS: Aeromonas, Bacteroidetes, bladder, conservation, gut, Hirudo, leech, Macrobdella, microbiome
ABSTRACT
Leeches are found in terrestrial, aquatic, and marine habitats on all continents. Sanguivorous leeches have been used in medicine for millennia. Modern scientific uses include studies of neurons, anticoagulants, and gut microbial symbioses. Hirudo verbana, the European medicinal leech, maintains a gut community dominated by two bacterial symbionts, Aeromonas veronii and Mucinivorans hirudinis, which sometimes account for as much as 97% of the total crop microbiota. The highly simplified gut anatomy and microbiome of H. verbana make it an excellent model organism for studying gut microbial dynamics. The North American medicinal leech, Macrobdella decora, is a hirudinid leech native to Canada and the northern United States. In this study, we show that M. decora symbiont communities are very similar to those in H. verbana. We performed an extensive study using field-caught M. decora and purchased H. verbana from two suppliers. Deep sequencing of the V4 region of the 16S rRNA gene allowed us to determine that the core microbiome of M. decora consists of Bacteroides, Aeromonas, Proteocatella, and Butyricicoccus. The analysis revealed that the compositions of the gut microbiomes of the two leech species were significantly different at all taxonomic levels. The R2 value was highest at the genus and amplicon sequence variant (ASV) levels and much lower at the phylum, class, and order levels. The gut and bladder microbial communities were distinct. We propose that M. decora is an alternative to H. verbana for studies of wild-caught animals and provide evidence for the conservation of digestive-tract and bladder symbionts in annelid models.
IMPORTANCE Building evidence implicates the gut microbiome in critical animal functions such as regulating digestion, nutrition, immune regulation, and development. Simplified, phylogenetically diverse models for hypothesis testing are necessary because of the difficulty of assigning causative relationships in complex gut microbiomes. Previous research used Hirudo verbana as a tractable animal model of digestive-tract symbioses. Our data show that Macrobdella decora may work just as well without the drawback of being an endangered organism and with the added advantage of easy access to field-caught specimens. The similarity of the microbial community structures of species from two different continents reveals the highly conserved nature of the microbial symbionts in sanguivorous leeches.
INTRODUCTION
Leeches are a diverse animal group native to freshwater, marine, and terrestrial environments. They are found on all continents and oceans on planet Earth (1, 2). Records of humans applying leeches medicinally survive from civilizations as far back as ancient Egypt (3, 4), resulting in the name medicinal leech, Hirudo medicinalis Linnaeus 1758. As the understanding of hirundinid taxonomy improved, Hirudo medicinalis was subdivided into additional species, including Hirudo verbana Carena 1820 and Hirudo orientalis Utevsky & Trontelj 2005 (5, 6). Since 2004, only H. medicinalis and H. verbana are approved for use as a medical device in the United States and must be shipped from approved suppliers in Europe (5). Although it shares the same common name, the North American medicinal leech, Macrobdella decora, was rarely used for bloodletting (7). No mechanical or pharmaceutical product has yet been able to replicate the reduction of venous congestion achieved by the medical application of Hirudo leeches (8, 9). This results in a continued need for medicinal leeches and a better understanding of their biology.
As approved medical devices, the natural feeding habits of leeches are utilized to reduce venous congestion and improve blood circulation in affected patients. To make the most of unpredictable encounters with prey, hirudinid leeches consume up to five times their body weight in one feeding and can go 6 to 12 months between feedings (1). After initial attachment, the leech stimulates blood flow in the prey by secreting vasodilators and a number of anticoagulant peptides, including hirudin, antistasins, and apyrases (10–14). The gastrointestinal system of H. verbana is highly simplified and consists of a pharynx, crop, and intestinum (1, 8). Once the blood is consumed, excess ions and water are rapidly removed from the blood meal to form a highly viscous intraluminal fluid (ILF) in the crop (8, 15). The ILF remains in the crop over long periods of time before it slowly passes into the intestinum, where it is digested (8).
The gut microbiome of medicinal leeches is simple compared to common mammalian gastrointestinal models. Previous research described an ILF microbiota in H. verbana dominated by Aeromonas veronii and Mucinivorans hirudinis (16–21). Subsequent studies revealed the presence of additional Aeromonas spp. in H. verbana and other hirudinid leeches (22–24). Clostridial species have also been detected in culture-independent studies of the ILF of H. verbana (17, 24) and H. orientalis (24). Several functions for the dominant crop microbiota have been proposed, including providing essential nutrients to the host (25–27), preventing other bacteria from colonizing, inhibiting putrefaction of the ILF (28), aiding the host’s immune responses (29, 30), or initiating the digestion of erythrocytes (31).
In most animals, the diversity of the microbiome increases along the length of the digestive tract, and similar findings have been reported for Hirudo spp. The H. verbana intestinum contains Alphaproteobacteria, Gammaproteobacteria, Deltaproteobacteria, Fusobacteria, Firmicutes, and Bacteroidetes as well as Aeromonas and Mucinivorans (17). A number of closely related hirudiniform leech species have also tested positive for Aeromonas and Bacteroidetes from the digestive tract (10, 23, 24). The composition of the microbial community among the different Hirudo species studied is similar in both crop and intestinum (17, 21, 23, 24).
Unlike humans but similar to other annelids (32, 33), H. verbana has multiple paired bladders colonized by a number of microbial species (34). Sequence analysis and fluorescence in situ hybridization micrographs of the H. verbana bladder show a stratified community consisting of Ochrobactrum, Bdellovibrio, Niabella, and Sphingobacterium (34). The difference between the bladder microbiome and that of the ILF or intestinum indicates that H. verbana microbiomes are body site specific and suggests a selection process that regulates the composition of these communities.
Much less is known about the gut microbiome of M. decora. Prior studies attempting to characterize the M. decora gut microbiome relied on aerobic culturing methods or sequencing total ILF DNA with primers specific for Aeromonas or Bacteroidetes symbionts. These studies revealed that the Aeromonas species associated with M. decora was Aeromonas jandaei (35) and the Bacteroidetes species was most similar to uncultured and unidentified species in a clade with Rikenella, Mucinivorans, and Alistipes (10). In this study, we strove for a more complete understanding of the microbiomes associated with the M. decora gut and bladders.
In this work, we describe the microbiotas of the ILF, bladder, and intestinum in wild-caught and laboratory-fed specimens of the North American medicinal leech, Macrobdella decora, and H. verbana purchased from two different suppliers. Culture-independent deep sequencing of the V4 region of the16S rRNA gene was confirmed by fluorescence in situ hybridization (FISH). Comparison of core and common microbial amplicon sequence variants (ASVs) from M. decora to those of the well-described H. verbana provides insights about the level of conservation of the microbiomes between distantly related and geographically isolated sanguivorous leeches.
RESULTS
The microbial communities of three organs from two leech species (H. verbana and M. decora) were analyzed in this study: the intestinum, crop, and bladder (see Table S1 in the supplemental material). Due to their small size, the entire organ was homogenized for the intestinum and bladder. Only the ILF was collected from the crop. Total DNA was extracted from the samples, and the V4 hypervariable region of the 16S rRNA gene was sequenced using an Illumina MiSeq instrument to determine the composition of the microbiome. After processing (see Materials and Methods), the rarefied data set consisted of 182 ASVs in 210 unique samples from 174 leeches sampling 3 organ sites in 2 leech species (Table S1).
ILF, intestinum, and bladder microbiomes differ between leech species.
An initial analysis was performed to determine if the composition of the microbiotas of the three organs analyzed differed between H. verbana and M. decora. Of all parameters tested, the leech host species had the greatest effect on microbiome composition (Fig. 1A). A permutational multivariate analysis of variance (PERMANOVA) using the Bray-Curtis and binary Ochiai metrics indicated that at all taxonomic levels, the microbiotas differed significantly between the two leech species. At the low taxonomic levels (ASV, genus, and family), the R2 values for the Bray-Curtis metric ranged between 47 and 54% and at higher taxonomic levels (order, class, or phylum), this R2 decreased to 20 to 22% (Table S2 and Fig. 1B). The remaining variation between samples was not accounted for by host leech species alone and indicates that further factors are important in determining microbial community composition.
FIG 1.

Bray-Curtis NMDS plot of leech-associated microbiotas showing that the microbiota is host species specific at the genus level but not at higher taxonomic levels. Two leech species were sampled (Hirudo verbana [orange] and Macrobdella decora [cyan]) at three organ sites (ILF [circles], intestinum [triangles], and bladder [plus signs]). (A) At the ASV level, 47% of the total variation between samples is described by separating the two leech host species. (B) When taxa were grouped at 88% sequence similarity (i.e., order level), only 20% of the total variation between samples was described by separating the two leech host species. Ellipses were calculated at 95% confidence. Stress was <0.14.
Other variables affect the microbiome to a smaller extent.
After samples were examined as a group, the data were split into smaller host species-specific groups to determine additional variables that influenced the microbiome. In H. verbana, the sampled organ had the greatest effect (accounting for 33% of variation; Bray-Curtis PERMANOVA) on microbiome composition, with the supplier, feeding, and shipment each accounting for an additional 5 to 10% of variation (Table 1). When the binary Ochiai metric was used, a similar pattern was observed (Table S3). For both dissimilarity metrics, the lot number of blood fed to the animals did not produce significant variation between samples (P ≥ 0.279) (Table 1 and Table S3).
TABLE 1.
PERMANOVA between leech-derived samples based on Bray-Curtis dissimilaritiesa
| Organism and characteristic | df | Pseudo-F | R2 | Pb |
|---|---|---|---|---|
| Hirudo verbana | ||||
| Organ | 2 | 55.81 | 0.33 | 0.001 |
| Supplier | 1 | 34.94 | 0.10 | 0.001 |
| Feeding | 6 | 3.71 | 0.07 | 0.001 |
| Shipment lot | 3 | 6.08 | 0.05 | 0.001 |
| Blood meal lotc | 5 | 1.03 | 0.02 | 0.439 |
| Macrobdella decora | ||||
| Organ | 2 | 12.92 | 0.17 | 0.001 |
| Feeding | 6 | 3.86 | 0.15 | 0.001 |
| Mo collected | 5 | 3.19 | 0.11 | 0.002 |
| Collection site | 3 | 3.10 | 0.06 | 0.006 |
| Collection/blood lotd | 3 | 1.14 | 0.02 | 0.357 |
Variables were tested and listed in order of decreasing R2.
Bold values indicate significant differences (P ≤ 0.01). P values are based on 999 permutations.
The blood meal lot is the fresh shipment of blood on which the animals fed in the laboratory.
Because of the extended time between collection events and the short shelf life of blood products, each blood lot was fed to only one collection lot of M. decora. The variation caused by either of these metadata variables therefore cannot be separated for M. decora samples.
In M. decora, the sampled organ also had the greatest effect on microbiome composition (accounting for 17% of variation; Bray-Curtis PERMANOVA), with feeding, month of animal collection, and collection site accounting for an additional 6 to 15% of variation (Table 1). A similar pattern was observed when the binary Ochiai metric was used (Table S3). Only for the binary Ochiai dissimilarity metric did the collection lot (when animals were collected from the same site two or more times in 1 month) and lot number of blood fed to the animals account for significant variation between samples (P = 0.045) (Table 1 and Table S3). In both leech species, the organ sampled had the greatest effect on variation between samples, with collection/shipment and feeding also having significant effects.
Deep sequencing of the H. verbana ILF microbiome confirms a simple core.
The first organ site to be examined in-depth was the ILF. The data from 16S rRNA gene V4 deep sequencing of the H. verbana ILF were analyzed to identify members of the core (ASVs present in ≥ 90% of samples), common (70 to 90% of animals), and transient (<70% of samples) microbiomes. For taxonomic identification of ASVs, we used the following cutoffs: ASVs with >97% identity to a genus in RDP were identified as that genus, and those with an 80 to 97% match were identified as genus-like, while those with a match that was <80% were identified with the prefix “unk” before the closest taxonomic level identified with >97% percent identity. The core consisted of two taxa, Aeromonas and Mucinivorans (Table 2 and Table S4). In the 39 H. verbana ILF samples tested, Mucinivorans and Aeromonas together accounted for 58.7 to 100% (median = 93.3%) of all the 16S rRNA gene V4 sequences from the ILF and were detected in 94.9% and 100% of the ILF samples, respectively (Table 2 and Table S4).
TABLE 2.
Median abundance (percent of total 16S rRNA gene V4 sequences) of core and common ASVs in ILF and intestinum samples from Hirudo verbana and Macrobdella decora
| Class | Genus or familya | ASVb | % abundance inc: |
||||
|---|---|---|---|---|---|---|---|
|
H. verbana |
M. decora |
||||||
| S1 ILF (n = 37) | S2 ILF (n = 75) | Intestinum (n = 13) | ILF (n = 30) | Intestinum (n = 22) | |||
| Bacteroidia | Bacteroides-like | ASV3 | ND | ND | 0.0 | 27.9 | 25.7 |
| Bacteroides-like | ASV5 | ND | ND | ND | 26.1 | 17.5 | |
| Mucinivorans | ASV1 | 84.2 | 68.1 | 8.0 | ND | ND | |
| Rikenella-like | ASV15 | ND | 0.1 | 15.6 | ND | ND | |
| Clostridia | Butyricicoccus-like | ASV6 | ND | ND | ND | 6.3 | 4.8 |
| Proteocatella | ASV4 | 0.0 | ND | 0.1 | 23.3 | 21.3 | |
| Sporobacter-like | ASV13 | ND | ND | ND | 3.6 | 0.1 | |
| Tyzzerella | ASV20 | ND | ND | ND | 1.5 | 0.2 | |
| Alphaproteobacteria | Rhodospirillaceae | ASV11 | 0.0 | ND | 26.7 | ND | ND |
| Deltaproteobacteria | Desulfovibrio | ASV33 | 0.0 | 0.4 | 1.4 | ND | 0.0 |
| Gammaproteobacteria | Aeromonas | ASV2 | 14.0d | 9.6 | 18.5 | 4.2 | 5.6 |
ASVs were determined at the 97% confidence level. The suffix “-like” indicates the most closely related genus listed in RDP.
ASV ID specific to this study. See Table S4 for GenBank accession numbers.
The relative abundances of ASVs belonging to the core (present in ≥ 90% of samples) are highlighted in gray. Common (present in 70 to 90% of samples) ASVs are in bold. Core and common ASVS were determined for each species of leech and sampled organ individually. ILF samples are grouped to show slight differences between suppliers (S1, supplier 1; S2, supplier 2). H. verbana intestinum samples are not divided by supplier due to insufficient sampling (supplier 1 = two animals) to confirm the nonsignificant difference between the intestinum microbiome from the two suppliers (P = 0.02). ND, none detected in sample group (abundance to determine presence = 0.1%). ASVs present in <70% of samples are considered transient. Median abundance values of 0.0% indicate an ASV present at a low level in a low number of samples.
The prevalence of Aeromonas ASV2 was 89.5% in the ILF of animals sampled from supplier 1 and thus just below the cutoff of ≥90% (see Table S4).
The H. verbana organisms used in this study were obtained from two suppliers, a factor that affected the gut microbiome composition. The core ILF microbiome of H. verbana from supplier 1 contains only Mucinivorans, while Aeromonas is a member of the common microbiome (present in 89.5% of samples). In contrast, the core ILF microbiome of H. verbana from supplier 2 consisted of ASVs associated with Mucinivorans, Aeromonas, and a Desulfovibrio-like taxon. The common ILF microbiome of H. verbana from supplier 2 contains an ASV associated with Bacteroides and a second ASV associated with Mucinivorans, which are both completely absent in H. verbana from supplier 1 (Tables S4, S5, and S6). Low-abundant, transient genera also showed a slight difference in specific ASVs between suppliers. Mucinivorans ASV35 and ASV93 are present only in animals from supplier 1. Proteocatella ASV22 is present only in animals from supplier 2, while Proteocatella ASV4 is present only in animals from supplier 1 (Tables S4 and S6). This suggests that the H. verbana supplier has an effect on several members of the ILF microbiome.
Characterization of the M. decora ILF microbiome.
The M. decora ILF core microbiome consisted of Bacteroidales (Bacteroides-like taxon) and Aeromonas. But unlike the H. verbana ILF, the M. decora ILF core microbiome also contained two species belonging to the Clostridiales (Table 2 and Table S4). Aeromonas and Bacteroides-like taxon sequences made up 40.2 to 74.8% (median = 60.4%) of the M. decora ILF microbiota, while Clostridiales species accounted for another 17.3 to 46.6% (median = 29.5%). Sequences from only one of these Clostridiales species (Proteocatella) had been reported previously from the H. verbana intestinum but not from the crop (17) (Table 2 and Table S4).
In the M. decora ILF, one ASV met the requirements for the common microbiome and was identified as a Sporobacter-like taxon (Table 2 and Table S4), which has not previously been found to be associated with a leech. The transient microbiome in M. decora consisted of 12 ASVs, including some that were not identified from H. verbana samples in this study or other previous studies (Table S4). Overall, the farm-housed H. verbana ILF samples (30 total ASVs) contained more ASVs than field-caught M. decora (18 total ASVs). The number of ASVs per animal was slightly different between the two groups (7 ± 5 ASVs in H. verbana versus 9 ± 3 ASVs in M. decora), resulting in a significant difference in Shannon diversity (P = 1.5 × 10−8) between ILF microbiome samples from the two leech species (Fig. 2).
FIG 2.
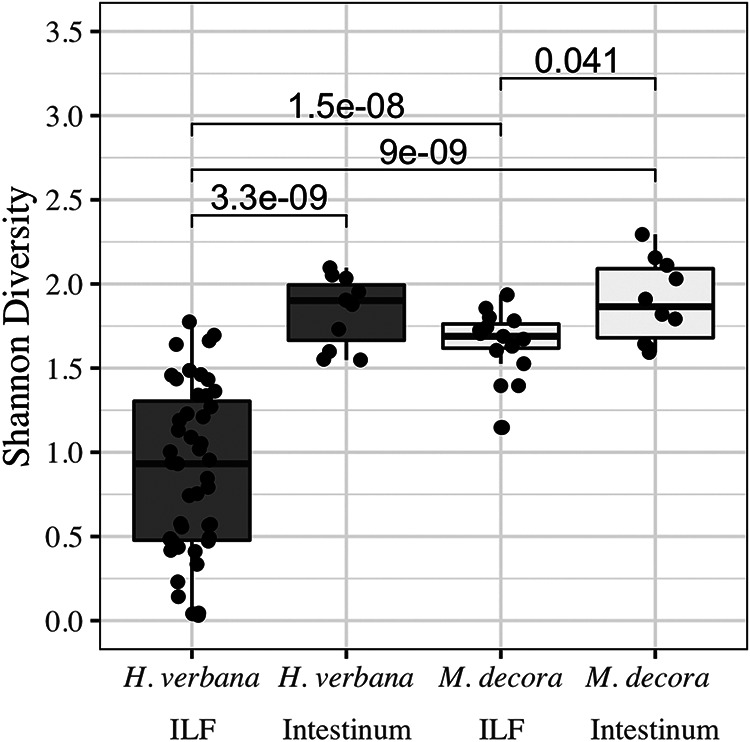
Shannon alpha diversity showing that the microbiome of Macrobdella decora ILF is more diverse than that of Hirudo verbana but that this is not true of the intestinum microbiomes. All samples were collected before feeding or more than 90 days after feeding. Alpha diversity does not change between the ILF and intestinum of wild-caught M. decora (white) but increases in farm-raised H. verbana (gray). A Wilcoxon rank-sum analysis with Bonferroni correction was used to compare sample groups. Significant P values (<0.05) are given above lines indicating comparisons.
Characterization of the intestinum microbiome.
Based on previous descriptions of intestinal microbiomes in other animals, we expected the leech intestinum to harbor a more complex microbiome than the crop. The intestinum microbiota from H. verbana was dominated by sequences from three genera: Aeromonas, Mucinivorans, and an unknown Rhodospirillaceae ASV that had been previously identified in the shed mucus castings of H. verbana (36) (Table 2 and Table S4). The difference in microbial community between the unfed H. verbana ILF and intestinum was marked by changes in the relative abundance of sequences from both dominant and common ASVs (Table 3) as well as an increase in alpha diversity, as measured by Shannon (P = 3.3 × 10−9) (Fig. 2) and inverse Simpson (data not shown) metrics. The increase in alpha diversity between the H. verbana ILF and intestinum was not detected with observed richness or Chao-1 calculations (data not shown). This analysis may be affected by a smaller sample size for the intestinum than for the ILF (12 and 39 individual samples, respectively) (Table S1).
TABLE 3.
Changes in abundance of bacterial ASVs in the ILF and intestinum of Hirudo verbana and Macrobdella decora leeches
| Host species and bacterial class | Genus or familya | ASVb | Base meanc | Log2 fold changed | Adjusted Pe | Location with higher abundancef |
|---|---|---|---|---|---|---|
| Hirudo verbana | ||||||
| Bacteroidia | Bacteroides | ASV34 | 29 | −9 | 3.8 × 10−10 | ILF |
| Bacteroides | ASV51 | 11 | −8 | 5.0 × 10−6 | ILF | |
| Mucinivorans | ASV1 | 4,520 | −5 | 2.8 × 10−44 | ILF | |
| Mucinivorans | ASV10 | 187 | −27 | 1.0 × 10−62 | ILF | |
| Mucinivorans | ASV23 | 10 | 8 | 0.008 | Intestinum | |
| Rikenella | ASV31 | 15 | 28 | 3.2 × 10−8 | Intestinum | |
| Rikenella-like | ASV15 | 25 | 5 | 1.1 × 10−7 | Intestinum | |
| Clostridia | Proteiniclasticum-like | ASV7 | 239 | −27 | 5.6 × 10−77 | ILF |
| Proteocatella | ASV22 | 39 | −26 | 7.9 × 10−44 | ILF | |
| Fusobacteriia | Fusobacterium | ASV8 | 182 | −28 | 1.6 × 10−40 | ILF |
| Alphaproteobacteria | Rhodospirillaceae | ASV11 | 45 | 10 | 6.4 × 10−7 | Intestinum |
| Gammaproteobacteria | Aeromonas | ASV2 | 820 | −2 | 0.03 | ILF |
| Morganella | ASV55 | 17 | −6 | 2.1 × 10−5 | ILF | |
| Proteus | ASV40 | 13 | −8 | 2.6 × 10−6 | ILF | |
| Macrobdella decora | ||||||
| Bacteroidia | Bacteroides-like | ASV28 | 41 | 3 | 0.03 | Intestinum |
| Bacteroides-like | ASV3 | 1,327 | 0 | 0.99 | — | |
| Bacteroides-like | ASV5 | 1,110 | 0 | 0.99 | — | |
| Mucinivorans | ASV49 | 28 | 11 | 2.5 × 10−9 | Intestinum | |
| Clostridia | Butyricicoccus-like | ASV6 | 763 | −1 | 0.12 | ILF |
| Proteocatella | ASV4 | 1,124 | 0 | 0.99 | — | |
| Sporobacter-like | ASV13 | 96 | −1 | 0.99 | ILF | |
| Tyzzerella | ASV20 | 93 | −3 | 2.1 × 10−5 | ILF | |
| Alphaproteobacteria | Insolitispirillum | ASV52 | 13 | 7 | 1.1 × 10−4 | Intestinum |
| unk_Rhodospirillaceae | ASV38 | 20 | 8 | 9.1 × 10−5 | Intestinum | |
| Gammaproteobacteria | Aeromonas | ASV2 | 486 | 0 | 0.99 | — |
ASVs belonging to the core (present in ≥90% of samples) are highlighted in gray. Common (present in 70 to 90% of samples) ASVs are in bold. Core and common ASVS were determined for each species of leech and sampled organ individually. The ASV labeled “unk_” has no closely related genus listed in RDP.
ASV ID specific to this study. See Table S4 for GenBank accession numbers.
Mean number of reads per sample of 4,990 total reads, calculated using both ILF and intestinum samples. Median reads per sample type are reported in Table S4. The minimum read count to be considered present in a sample was 7 (see Materials and Methods).
Calculated with abundance in intestinum samples as the numerator and abundance in ILF samples as the denominator.
P values were adjusted for multiple comparisons. Values in bold are significant (P < 0.001).
Gastrointestinal location with higher abundance of specified ASV. —, log2 fold change of <1 between gastrointestinal locations.
In M. decora, the Shannon (Fig. 2) and inverse Simpson alpha diversity between ILF and intestinum samples changed only slightly (P ≥ 0.04), but changes in observed richness and Chao-1 measurements were not significant (data not shown). The slight difference in microbial community between the unfed M. decora ILF and intestinum was marked by changes in the relative abundance of sequences from transient ASVs with a base mean of <100/4,990 reads (Table 3). The four core genera of both ILF and intestinum from M. decora were Aeromonas, a Bacteroides-like taxon, a Butyricicoccus-like taxon, and Proteocatella (Table 2 and Table S4). In the intestinum, these four genera comprised five core ASVs accounting for 66.9 to 98.6% (median = 81.6%) of the total 16S rRNA gene V4 sequences, while in the ILF they accounted for 77.7 to 99.9% (median = 86.4%) of sequences (Table 2 and Table S4).
Some of the ASVs identified in M. decora intestinum were also detected in H. verbana or were previously reported from H. verbana samples. The dominant Proteocatella-related ASV from M. decora was previously found in the H. verbana intestinum (17) but was detected in only three ILF, four intestinum, and two bladder samples from H. verbana processed in this study. The Desulfovibrio-associated ASV that was common in H. verbana intestinum was detected in only two intestinum and one ILF samples from M. decora. Twenty-five ASVs in the M. decora intestinum were not found in H. verbana samples, while 10 ASVs in the H. verbana intestinum were not found in the M. decora intestinum (Tables S4 and S5). In the intestinum, like in the ILF, the microbial community observed at the order level is conserved between leech species, while at the ASV level, it may be leech species specific (Table S5).
The H. verbana bladder community contains three core ASVs.
The final organ site to be analyzed was the bladder. The 16S rRNA gene V4 deep sequencing of H. verbana bladders identified 21 total ASVs (Table S4). The core microbiome comprised Nubsella, Ochrobactrum, and an unknown Comamonadaceae taxon (Table 4 and Table S4). The common microbiome additionally contained an ASV associated with a Bdellovibrio-like taxon. The remaining 17 ASVs were present only transiently (Table S4).
TABLE 4.
Presence of core and common bladder ASVs in total 16S rRNA gene V4 sequences from Macrobdella decora and Hirudo verbana bladder samples
| Class | Genusa | ASVb | % of 16S rRNA gene V4 sequences inc: |
|||
|---|---|---|---|---|---|---|
|
H. verbana |
M. decora |
|||||
| Median | Max | Median | Max | |||
| Bacteroidia | Nubsella | ASV17 | 18.2 | 40.0 | 0.0 | 0.2 |
| Clostridia | Butyricicoccus-like | ASV6 | 0.0 | 0.3 | 6.3 | 81.5 |
| Proteocatella | ASV4 | 0.0 | 3.2 | 15.9 | 63.7 | |
| Sporobacter-like | ASV13 | ND | ND | 1.2 | 7.9 | |
| Tyzzerella | ASV20 | ND | ND | 2.1 | 10.2 | |
| Alphaproteobacteria | Azospirillum-like | ASV18 | 0.0 | 0.2 | 1.7 | 55.1 |
| Ochrobactrum | ASV9 | 3.9 | 93.6 | 0.0 | 0.2 | |
| Betaproteobacteria | unk_Comamonadaceae | ASV12 | 19.7 | 73.8 | ND | ND |
| unk_Comamonadaceae | ASV21 | ND | N.D. | 3.6 | 39.4 | |
| unk_Methylophilaceae | ASV30 | 0.0 | 0.0 | 2.2 | 19.4 | |
| Deltaproteobacteria | Bdellovibrio-like | ASV41 | 2.1 | 21.2 | ND | ND |
ASVs were determined at the 97% confidence level. ASVs labeled “-like” reflect the most closely related genus listed in RDP. ASVs labeled “unk_” have no closely related genus listed in RDP.
ASV ID specific to this study. See Table S4 for GenBank accession numbers.
Percentage of total 16S rRNA gene V4 sequences. Values for core (present in ≥90% of samples) ASVs are highlighted in gray; those for common (present in ≥70% of samples) ASVs are in bold. ND, none detected in sample group (abundance to determine presence = 0.1%). ASVs present in <70% of samples are considered transient. Median abundance values of 0.0% indicate ASVs present at a low level in a low number of samples.
The M. decora bladder community contains conserved ASVs.
The 16S rRNA gene V4 deep sequencing of M. decora bladders identified 25 total ASVs (Table S4). Of these identified taxa, only Proteocatella was recovered frequently enough to be considered part of the core (Table 4 and Table S4). Six ASVs were identified as common (present in 69 to 90% of bladder samples) (Table 4). The remaining 18 ASVs were present only transiently.
The localization of bacterial taxa within the bladder also appears to be conserved between H. verbana and M. decora. FISH imaging of M. decora bladders suggested that Rhizobiaceae occur in the matrix of the bladder contents while other Alphaproteobacteria occur intracellularly in the epithelial cells lining the interior of the bladder. Betaproteobacteria occur in close association with the M. decora bladder epithelial cells (Fig. 3). This layered community is consistent with localizations reported previously in H. verbana bladders (34).
FIG 3.

False-color FISH micrographs of Macrobdella decora bladder samples. From left to right, columns contain images obtained with DAPI (blue, eukaryotic DNA), Eub338 (green, Eubacteria), CF319a (white, Bacteroidetes [Bacteroides]), and additional probes (red) and composite images. From top to bottom, the fourth column contains images obtained with probes Alf968 (Alphaproteobacteria [Azospirillum-like taxa, Elstera-like taxa, Ensifer, Phreatobacter-like taxa, and Shinella]), Bet42a (Betaproteobacteria [unk_Comamonadaceae taxa and unk_Methylophilaceae taxa]), Rhiz1244 (Rhizobiales [Ensifer Phreatobacter-like taxa, and Shinella]), and Aer66 (Aeromonas). The sixth column contains high-resolution images of the areas marked with white squares in the composite image. Dashed lines in the DAPI and high-resolution composite images indicate the bladder epithelial layer, with large dashes indicating external (toward body tissue) and small dashes indicating internal (toward the bladder matrix) epithelial layers. The layer is mostly one cell layer thick. The bladder matrix begins immediately adjacent to the internal epithelial layer. The M. decora bladder is colonized in a stratified manner with intracellular Alphaproteobacteria (Azospirillum-like taxa and Elstera-like taxa), epithelium-associated Betaproteobacteria, and Rhizobiales (Ensifer, Phreatobacter-like taxa, and Shinella) in the matrix. Aeromonas (red arrows) is associated with eukaryotic cells, suggesting that it was carried here by leech hemocytes and is not a normal member of the bladder microbiota. Bars = 10 μm.
Feeding affects the composition of the gut microbiome.
PERMANOVA results indicated that feeding has the second greatest influence on microbiome composition after organ site. A Bray-Curtis PERMANOVA and Bonferroni-adjusted Wilcoxon rank-sum analysis found that H. verbana ILF microbiota at 1 to 7 days after feeding (DaF) are significantly different from those of unfed animals (0 DaF) (P ≤ 0.05) (Table S7). The perturbed H. verbana ILF microbial community rebounds by 30 to 90 DaF to a state undifferentiable from that of an unfed animal (Table S7). The M. decora ILF microbiome follows a similar pattern, with the community at 4 DaF being significantly different from that of unfed animals (0 DaF) (P ≤ 0.022) (Table S7). The perturbed M. decora ILF microbial community rebounds by 30 DaF to a state undifferentiable from that of an unfed animal (P > 0.29) (Fig. 4 and Table S7).
FIG 4.
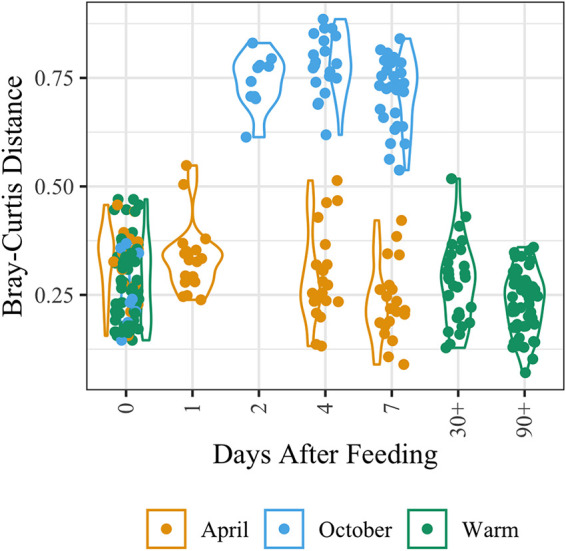
Bray-Curtis distance calculation shows that Macrobdella decora ILF microbiomes at 2 to 7 DaF are significantly different from those of unfed animals (0 DaF) and that the community rebounds at 30 to 90 DaF. Warm months are June, July, August, and September. Note the appearance of two subpopulations of M. decora ILF samples (related to season of collection) that (i) appear to rebound by ∼4 DaF and (ii) appear to require >7 DaF to rebound. PERMANOVA distance and Bonferroni-corrected Wilcoxon rank-sum calculations for all comparisons are available in Table S7.
In the ILF microbial community, significant changes occur in the relative abundance of transient but not core ASVs after feeding (Fig. 5, Fig. 6, and Table S8). In H. verbana, Desulfovibrio and Pseudomonas ASVs are most significantly different in abundance with time after feeding (P ≤ 0.03) (Table S8). In M. decora ILF, the Clostridiales-associated ASVs are most significantly different in abundance after feeding (P ≤ 0.002) (Fig. 5, Fig. 6, and Table S8). These data suggest that the overall composition of the ILF microbial community is relatively stable after feeding.
FIG 5.

Box-and-whisker plot of leech-associated gut microbiotas showing that the prevalence of taxa in the ILF appears to be greatly affected within ∼48 h of a blood meal, while the prevalence of taxa in the intestinum maintains relative stability. The prevalences of eight core and common gut taxa from deep-sequenced microbiomes of Hirudo verbana ILF, Macrobdella decora ILF, and M. decora intestinum were assessed at 7 time points after a blood meal (0, 1, 2, 4, 7, 30+, and 90+ days after feeding). See Table S1 for number of samples analyzed at each time point. See Table S8 for Wald test P values.
FIG 6.

Heat map of Macrobdella decora ILF ASVs as a function of days after feeding (DaF) and collection site.
The abundances of the same ASVs were examined in the intestinum. Although slight changes in relative abundances were measured after feeding, the changes in composition of the community in the M. decora intestinum after feeding were not significant (P ≥ 0.15) (Fig. 5). This suggests that the intestinum microbial community is more stable than that of the ILF.
We further assessed the composition and abundance of the ILF microbiome after feeding in M. decora using FISH. Immediately after capture and before feeding, the microbial population in the M. decora crop was below the limit of detection for FISH, although occasional Aeromonas cells were found (Fig. 7). After a blood meal, the microbial population in the M. decora crop increased at 4 and 7 DaF, with Bacteroidetes species forming microcolonies by 4 DaF and individual Aeromonas cells spread throughout the crop by 7 DaF (Fig. 7). A similar pattern was previously observed and quantified in H. verbana (18). This confirms that although the relative abundance of Aeromonas and Bacteroides-like taxon 16S rRNA gene V4 sequences do not significantly change after feeding in M. decora, both species are actively proliferating in the crop at different rates.
FIG 7.

False-color FISH micrograph of Macrobdella decora ILF. From left to right, columns contain images obtained with the probes DAPI (blue, eukaryotic DNA), Eub338 (green, Eubacteria), CF319a (white, Bacteroidetes [Bacteroides]), and Aer66 (red, Aeromonas) and composite and high-resolution composite (enlarged areas are indicated with white squares in the composites) images. From top to bottom, the rows contain images from animals sacrificed 0 days (wild-caught animals), 4 days, and 7 days after a laboratory-administered sterile blood meal (DaF). Blue arrows indicate eukaryotic cells (most likely leech hemocytes), green arrows indicate notable bacteria not labeled by the two specific probes, white arrows indicate Bacteroidetes microcolonies, and red arrows indicate Aeromonas. Background fluorescence is from crop contents, especially the blood meal at 4 and 7 DaF. Few bacteria are present in the crop of wild-caught, unfed animals. Bacteroidetes colony expansion occurs by 4 DaF, while Aeromonas prevalence increases by 7 DaF. Other bacteria are present at 4 DaF; however, their numbers appear to be overwhelmed by Bacteroidetes and Aeromonas growth at 7 DaF. In animals at 7 DaF, Aeromonas organisms are occasionally found associated with hemocytes. Bars = 10 μm.
Collection events may affect the composition of the M. decora microbiome.
PERMANOVA indicated that the collection date and site also correlated with the Bray-Curtis distances in M. decora. Our data suggested that the M. decora ILF microbial community composition changed after feeding in M. decora collected in October and to a much smaller extent in those collected in April (Fig. 4). This analysis is confounded by differences in the location where the animals were collected (Fig. S1 to S3), and because of the small sample size, a more comprehensive study is required to draw firm conclusions about the effects of collection date and site on the gut microbiome composition in M. decora.
DISCUSSION
In this study, we used deep sequencing of the V4 region of the 16S rRNA gene and FISH imaging to characterize the microbiomes from three organs in the North American medicinal leech, Macrobdella decora, and the European medicinal leech, Hirudo verbana. Our analysis revealed that the ILF, intestinum, and bladder microbiomes are similar at the order level between the two leech species but differ at lower taxonomic levels. Especially at the ASV level, the bacteria present are specific to the two host leech species. This result is similar to observations of other animal models in which researchers have noted host-specific genera showing an evolutionary pattern that mimics that of the host’s evolution (37–41) and has been suggested previously for leeches (10).
In both leech species, the gut microbial communities are dominated by Aeromonas and Bacteroidales species, which is consistent with previous studies performing 454 pyrosequencing of the 16S rRNA gene V6 region (42), 16S rRNA gene V3-V4 deep sequencing (21), and 16S rRNA gene clone library analysis (17). Aeromonas and Mucinivorans constitute the core microbiome in H. verbana. In M. decora ILF, we also found Clostridiales to be very abundant, and one ASV was associated with a Desulfovibrio-like taxon which was previously identified associated with H. verbana (17, 21, 34). The core microbiome in M. decora consisted of ASVs associated with Aeromonas, Bacteroides, Proteocatella, and a Butyricicoccus-like taxon. This is consistent with previous findings that Bacteroidetes and Aeromonas are commonly (but not universally) found in the gut of animals and insects fed on blood (43–48). Siddall et al. showed that many sanguivorous leeches maintain a gut symbiosis with Aeromonas and Bacteroidetes species (10), and we therefore hypothesize that the conserved taxonomic orders have been evolutionarily maintained in hirudinid leeches despite host species diversification and geographic separation. Our study shows that while the Bacteroidales and Clostridiales genera changed between leech hosts, the genus Aeromonas is found in both leech species, suggesting that the capabilities for colonizing hosts and performing the required physiological functions are conserved at different taxonomic levels in these bacteria.
Previous genetic studies of Aeromonas veronii provide mechanistic insight into key physiological functions that symbionts need to possess for successful colonization of the ILF. A number of colonization factors, such as type III secretion systems (49), carbon starvation response (50), oxidative stress response inhibition (8), and heme acquisition processes (51), are critical to enabling leech gut colonization. Interestingly, not all members of this genus possess these colonization factors (30), and this leads to strain specificity that our 16S rRNA survey cannot address. Such strain specificity has been noted in several other bacterium-animal symbioses (52–55). New studies comparing genomic differences between Aeromonas strains isolated from sanguivorous leech species and those from diseased veterinary samples or non-host-associated environmental samples may be used to further identify genes critical for host colonization and persistence (56). Comparison of genomes of Bacteroidales and Clostridiales strains isolated from these leech hosts may identify additional factors critical for symbiosis in a wide range of taxa.
In addition to confirming a simple core microbiome, our study also confirmed the presence of a rich, low-abundance microbiome in both the H. verbana and M. decora ILF, as was recently suggested to occur in the gut of wild H. verbana (21). We found that ASVs from low-abundance microbes in the ILF of both H. verbana and M. decora were present transiently. Because of the terminal nature of our sample collection methods, we defined transient ASVs as those present in <70% of samples, but we cannot determine if these ASVs are present consistently in <70% of animals or rather if they may occur in all animals but only temporarily. Although three Bacteroides and a number of Rikenellaceae ASVs identified in the transient microbiome have not been described before in H. verbana, other researchers have previously noted that Mucinivorans may not be the only Bacteroidales taxon associated with hirudinid leeches (17, 23, 36). Sequences belonging to other taxa noted by previous researchers were found in the H. verbana ILF samples at median abundances below 1% of the total 16S rRNA gene V4 sequences (17, 24, 42) (Table S4). The list of transient bacteria identified in this study is likely incomplete due to the limitations of 16S V4 rRNA gene sequencing. In order to reduce the identification of false positives, ASVs present in a sample at <0.14% were eliminated. In this process, bacteria that were only ever present as an especially low fraction of the community may have been lost. Previous research in a variety of environments has concluded that although a relatively small number of ASVs may dominate samples, low-abundance populations may be responsible for driving changes in observed community diversity between treatment groups (57, 58). While many transient microbes simply pass through the digestive tract, other transient species may have a significant role in metabolic cycles and thus may be hidden drivers of ecosystem functioning (59).
Transient microbiotas may also allow specific identification of leech source populations. We compared the microbiomes of H. verbana specimens obtained from two suppliers: one that sold field-caught animals and one that sold farm-bred animals. A number of ASVs sequenced from the ILF were identified as specific to the supplier (Table S6). Different treatments before shipping, including different starvation times (to reduce abundance of human pathogens) (60), potentially feeding animals antibiotic-contaminated blood (22, 61), and supplementing farmed animals with animals captured in different geographic regions are factors that could have influenced the microbiome but are not discernible in our study. The low concentrations and intermittent presence of many previously identified bacteria highlight the importance of greater sequencing depth and a greater number of specimens tested in observation of wild populations.
Not only was the core gut microbiome similar at the order level between M. decora and H. verbana, but also the dynamic response of these symbionts in response to feeding a blood meal followed a similar trend. Transient bacteria exhibited the most significant changes in abundance between time points after feeding. In the most extreme cases, bacteria present at abundances below the limit of detection were observed at only certain time points after population expansion. FISH imaging revealed that the nonsignificant changes in relative abundances of core symbionts were due to proliferation over similar time scales. Other researchers have observed clostridial populations increasing rapidly in response to feeding, while Bacteroidales numbers increase at a lower rate, and high levels of Bacteroidales or Rikenellaceae are indicative of long periods between feedings in avian, reptilian, and mammalian hosts (62–64). This consistency with community dynamics observed in other animal models suggests that leeches may be good models to predict microbial changes due to intermittent feedings or extended periods of starvation.
Although part of the decrease in alpha diversity in the M. decora ILF microbiome (Fig. S1) after blood meal consumption is possibly due to properties of the innate immunity of the ingested blood meal (65), in a number of vertebrate species it has also been shown that extreme feeding or fasting events do result in decreased alpha diversity (62, 63). It has been suggested that in H. verbana, the Aeromonas symbiont may release antimicrobial peptides responsible for restricting bacterial colonization in addition to host-produced antimicrobial peptides (66). Because the ILF core community and postfeeding community dynamics are similar in M. decora, it is reasonable to assume that a similar process may also occur in this host species. Thus, the observation of wild M. decora provides a new ability to observe community dynamics of taxa that appear to have been eliminated in farm-housed H. verbana.
In contrast to the observed temporal changes in the ILF microbiome, the microbiome in the intestinum of H. verbana and M. decora is much more stable. This might be largely due to a more consistent flow of nutrients resulting in a more constant environment that is less affected by sporadic feeding events than the ILF. It is also possible that the intestinum serves as a high-abundance reservoir of core symbionts from which new blood meals may be seeded after ingestion.
We also examined the microbiome within the leech bladders. The 17 pairs of bladders are adjacent to the lateral ceca of the crop. Microbes in the bladder are thought to recycle nitrogenous waste (67). The core microbiome in H. verbana is consistent with previous studies and composed of Nubsella (previously Sphingobacterium), Ochrobactrum, and an unknown Comamonadaceae taxon (previously Comamonas) (34). However, the Bdellovibrio-like taxon we detected was not the same ASV that was sequenced by Kikuchi et al. (34). In M. decora, only Proteocatella met the criteria for being in the core microbiota. Four taxa sequenced from bladder samples exhibited conservation at the genus level between leech species. ASVs associated with Bdellovibrio, Comamonadaceae, Phreatobacter, and Ensifer were present in bladders from both leeches; however, the ASVs present were unique to the leech host species. In contrast, ASVs associated with Bacteroidia, Clostridia, Alphaproteobacteria, Betaproteobacteria, Deltaproteobacteria, and Gammaproteobacteria were found in bladders from both leech species. The ASVs associated with Flavobacterium, Proteocatella, an unknown Rhodospirillaceae, an unknown Comamonadaceae, and a Desulfovibrio-like taxon detected within M. decora are very similar to those identified from H. verbana bladders in this and other studies (Table S4) (34). The taxa we detected in the bladders of both H. verbana and M. decora were also similar to those found in other annelids (32, 33); i.e., a transient ASV associated with an Elstera-like taxon (ASV29) was most closely related to a bacterium sequenced from the earthworm bladder (32). These data suggest a conservation of leech bladder symbionts at the genus level despite geographic and evolutionary separation that is greater than for the digestive-tract microbiome. Further research is needed to determine if the transmission of leech bladder symbionts is similar to that reported in earthworms, where the transmission occurs within the cocoon (33).
Our results indicate that there are distinct bladder and intestinal microbiota in hirudinid leeches. Based on their absence in ILF and intestinum samples, 15 ASVs were identified as specific to the leech bladder. In addition, the very low abundance of Ochrobactrum and unknown Comamonadaceae taxon sequences in ILF samples (maximum = 0.6%) and the absence of any ALF968-, Rhiz1244-, or Bet42a-binding cells in FISH images (data not shown) lead us to hypothesize that these genera are specific to the bladder microbiome. The presence of these ASVs at a low abundance in an ILF sample would therefore possibly be due to index hopping during sequencing (68, 69) (analysis of positive controls indicates that in rare instances, core and common ASVs were detected in abundances of up to 1.8% of total sequences in a sample) or contamination during dissection of the animal. In M. decora and H. verbana, bilateral pairs of bladders are present in most body segments and found in close proximity to the crop or intestinum. The close proximity of these organs leads to a high chance of puncture during dissection, especially after feeding, when the bladders and crop are both filled with fluid and stretched tissue structure becomes incredibly friable. This may have occurred in previous studies where bladder-specific symbionts were found in the ILF and intestinum of 3 wild H. verbana (21) and Pedobacter and Ochrobactrum were cultured from intestinum samples (17, 70). Such contamination can be difficult to overcome, especially when one is working with small animals, and this highlights the importance of using FISH to confirm the presence of taxa predicted by 16S rRNA gene surveys.
This work revealed a conservation of gut and bladder symbioses in medicinal leech species from Europe and North America. While the community structures of H. verbana and M. decora microbiomes have many similarities, the differences at the lower taxonomic levels can be exploited in the future to gain a better understanding of the coevolution of hosts and symbionts. From our data we can hypothesize that the gut microbiome composition is affected by periods of satiated feeding, starvation, or torpidity. In H. verbana, the simple gut community helps to digest the blood meal (8) and protect the host from invading bacteria (66). It is likely that the microbial communities in the gut of M. decora perform very similar functions. Future work should focus on isolating dominant members of both symbioses to compare conserved metabolic and colonization abilities and to test for host species specialization. Through understanding molecular and mechanistic studies enabled by simple systems, especially in evolutionarily conserved biological processes and metabolic capabilities, one can test predictions that would be challenging to test in more complex communities (71, 72).
MATERIALS AND METHODS
Animals.
Macrobdella decora organisms were collected from ponds located in Storrs, CT (41°49′3.074″N, 72°15′32.704″W), Groton, MA (42°35′26.993″N, 71°32′25.63″W), Caroga, NY (43°11′28.684″N, 74°28′10.026″W), and Mount Snow, VT (42°58′25.3″N, 72°55′47.9″W). Macrobdella decora organisms were identified through distinctive body coloration and by counting 4 gonopores. The only known confounding species that would overlap in this range is Macrobdella sestertia, which has 24 gonopores (73, 74). If sacrificed within 1 week of collection, animals were maintained in native pond water.
Hirudo verbana organisms were purchased from Leeches U.S.A. (Westbury, NY, USA) and BBEZ (Biebertal, Germany) and maintained as described below.
Leeches were maintained in sterile circular tanks (up to 10 animals/tank) in an environmental chamber with 12-h/12-h day/night cycles at 25/23°C, respectively. Between uses, tanks were scrubbed with distilled water, autoclaved for 30 min, baked at 80°C overnight, and then allowed to come to room temperature for storage. Tanks contained groups of animals from a single shipment or collection lot. Dilute Instant Ocean (DIO) water, sterilized twice with 0.2-μm filters and consisting of 34 mg/liter Instant Ocean salts (Aquarium Systems, Inc., Mentor, OH, USA) in Nanopure water, was changed weekly or when the animals vomited or an animal in the tank died. Tanks contained a minimum of one autoclaved rock.
Tanks were cleaned by completely emptying the old water and performing multiple complete, small-volume water changes until water remained clear for at least 20 min after the last change. Tanks were then filled to previous level and returned to environmental chamber.
Feeding.
Animals were fed as described previously (16). Sheep blood with heparin as an anticoagulant was purchased from Lampire Biological Laboratories (Pipersville, PA, USA). Animals were fed in groups of 1 to 3 animals on sterile 50-ml Falcon tubes containing 30 ml sheep blood that had been warmed to 37°C and covered with Parafilm (American National Can, Greenwich, CT, USA). All animals were allowed to feed to satiety and then let sit in sterile DIO for ∼30 min before handling after feeding.
Study design and dissections.
The microbial communities of three organs from two leech species (H. verbana and M. decora) were analyzed in this study: the intestinum, crop, and bladder (Table S1).
Animals were removed from the tank, and the anterior end was tied with string to inhibit regurgitation before narcotizing in 70% ethanol (16). Each animal was then treated with RNase Away and rinsed with molecular biology-grade water to remove external microbiota and nucleic acids. An additional string was tied around the animal to bisect it into anterior and posterior fractions. Sterile technique and instruments were used for all further dissection.
(i) Bladder.
A small primary incision was made immediately lateral to the dorsal lateral dextral stripe and immediately posterior to the second string. Bladders were identified and carefully bluntly dissected so as to minimize contamination with ILF. Bladders were briefly rinsed in sterile phosphate-buffered saline (PBS) and then placed in a sterile bead-beating tube. Each sample consisted of 1 to 3 bladders from the same leech. In this paper, the term “bladders” includes bladders and the attached nephridia.
(ii) ILF.
To access the crop contents, a secondary central incision was made with a fresh blade immediately below the second string. Approximately 100 μl ILF was collected as it was released and placed in a sterile bead-beating tube. In animals with especially low volumes of ILF, a sterile pipette tip was used to gently scrape the inside of the crop.
(iii) Intestinum.
To access the intestinum, a second incision was made immediately lateral to the anus. The intestinum was carefully dissected from the anus and consisted of ≥1 cm intestinum with contents. Samples were placed in bead-beating tubes or 1.5-ml microcentrifuge tubes and snap-frozen in liquid nitrogen before storage at −80°C.
Tissue fixation and embedding.
After bladder, ILF, and intestinum sample were collected, a fully bisecting incision was made immediately posterior to the second string. The anterior portion of the animal was placed in methacarn (6:3:1 methanol-chloroform-acetic acid) (75). Tissues in methacarn were stored at 4°C with rocking. Regular fixative changes were made when the fixative was no longer clear. Tissues were dissected, and a final fixative change was performed with fresh anhydrous methacarn before incubating for one additional week at 4°C with rocking. The anhydrous methacarn was replaced with anhydrous methanol, and the tissues were stored at 4°C until embedding. Tissues were cleared with a decreasing methanol-xylene series and then embedded in Paraplast Plus (Millipore Sigma, St. Louis, MO, USA).
FISH.
Paraffin-embedded tissues were sectioned at 6 to 8 μm using a microtome and placed on poly-l-lysine-coated slides. Sections on slides were cleared with xylene and then rehydrated with an ethanol-water series. Slides were bleached for 8 to 12 h using 2% hydrogen peroxide and a standard fluorescent light bulb (with concomitant cooling over an ice bath). Bleached slides were incubated in distilled water at 90°C for 5 min, then hybridized at 55°C for 20 min in a hybridization chamber, and washed in 50 ml wash buffer at 50°C for 20 min on a rotating incubator (76). Hybridization solutions contained 0.54 M NaCl, 12 mM Tris-Cl, 30% formamide, and 1.2% sodium dodecyl sulfate with 1 μM Cy3 probe, 1 μM Cy5 probe, and 3 μM Eub338. Slides were briefly rinsed with distilled water and allowed to dry before mounting with SlowFade Gold. Negative no-probe controls were subjected to the same conditions and used to estimate autofluorescence of the samples (data not shown). Probe sequences and targets are listed in Table 5. Slides were observed using a Nikon A1R confocal microscope (laser wavelengths, 405 nm [DAPI {4′,6-diamidino-2-phenylindole}], 488 nm [Alexa Fluor 488], 558 nm [Cy3], and 640 nm [Cy5]), and images were processed using ImageJ 1.51s (77).
TABLE 5.
FISH probes used in this study
| Probe | Nucleotide sequence (5′–3′) | Reference | Target organism(s) |
|---|---|---|---|
| ALF968 | GGTAAGGTTCTGCGCGTT | 88 | Aminobacter, Elstera, Insolitispirillum-like, Ochrobactrum, Phreatobacter-like, Sphingomonas |
| BET42a | GCCTTCCCACWTCGTTT | 89 | Achromobacter, Acidovorax, Pelomonas, Ramlibacter, Variovorax |
| CF319a | TGGTCCGTGTCTCAGTAC | 90 | Bacteroides, Flavobacterium, Pedobacter, Mucinivorans, (some) Phreatobacter-like, (some) Bdellovibrio |
| AER66 | CTACTTTCCCGCTGCCGC | 91 | Aeromonas |
| Rhiz1244 | TCGCTGCCCACTGTCACC | 92 | Aminobacter, Ensifer, Phreatobacter-like, (some) Ochrobactrum |
| Eub338 | GCTGCCTCCCGTAGGAGT | 93 | (Most) Eubacteria |
Bead-beating DNA extraction.
A modified version of a protocol published by Yu and Morrison was used (78). Cell lysis buffer (300 μl) was added to sample in a bead tube containing 0.1-mm and 0.5-mm zirconia/silica beads (BioSpec Products, OK, USA) beads. Samples were beaten for 90 s at ∼3,500 oscillations per min and briefly centrifuged before supernatant was transferred to clean 1.5-ml microcentrifuge tubes. An additional 150 μl lysis buffer was added to the bead tubes, and the sample was beaten again for 90 s. The supernatant previously removed was returned to bead tube and incubated for 15 min at 56°C with 5 s vortexing every 4 min. The sample was briefly centrifuged before addition of 100 μl 10 M ammonium acetate at 4°C and vortexed for 15 s before being incubated for 10 min on ice. The sample was centrifuged for 10 min at 16,000 × g. The supernatant was transferred to a clean 1.5-ml microcentrifuge tube to which 350 μl ethanol at 4°C was added before vortexing for 10 s and transferring the supernatant to a QIAamp Mini spin column (Qiagen, Germantown, MD, USA). The sample was processed as recommended in the QIAamp DNA Mini handbook and eluted with 10 mM Tris-Cl, pH 8.5. The eluted DNA concentration was measured using a Qubit double-stranded DNA (dsDNA) high-sensitivity (HS) assay kit (Thermo Fisher Scientific, Carlsbad, CA, USA). Eluted DNA samples were stored at −20°C until sequencing.
MasterPure DNA extraction.
MasterPure complete DNA extraction without column purification into 50 μl Tris-EDTA (TE) was performed according to the manufacturer’s protocol (Epicentre, Madison, WI, USA). No significant difference was found between the two extraction methods (PERMANOVA; P ≥ 0.359) (Fig. S4).
Sequencing and initial ASV taxonomy assignment.
Extracted DNA samples were analyzed by amplifying the V4 hypervariable region of the 16S rRNA gene using primers designed in a previous study (79). PCRs were prepared as described in reference 80 and sequenced using an Illumina MiSeq instrument (Illumina, San Diego, CA, USA) with custom sequencing primers added to the reagent cartridge (79) and sequenced at 2 × 250 bp.
Resulting community sequences were processed using R as outlined below. Complete coding is available in supplemental material and via GitHub: https://github.com/joerggraflab/McClureE_Md2019/.
Initial ASV assignments were processed using the standard DADA2 pipeline (81). Sequences were filtered using the recommended standard filtering parameters: 0 Ns, truncated quality score less than 2, and maximum expected error rate of 2. Forward and reverse sequences were then merged, chimeras were removed, and amplicon size was limited to 240 to 263 bp to reach a data set containing 2,191 unique ASVs. ASVs were assigned taxonomy by comparing to the SILVA SSU 132 data set (82). Any ASVs that were not identified to the genus level and were considered significant in later analysis were compared to the RDP data set for taxonomic assignment to the lowest possible taxonomic level (83).
(i) Negative controls.
Two types of negative controls were prepared and sequenced. (i) Reagent controls were prepared by performing the DNA extraction procedure using the same reagents without any sample. The resulting DNA yields for these reagent controls after extraction were always below the limit of detection for the Qubit dsDNA HS assay. (ii) Negative-PCR controls were prepared by performing V4-specific PCR amplification using molecular biology-grade water. The resulting reactions produced no bands when analyzed with the QIAxcel DNA fast analysis cartridge (Qiagen, Germantown, MD, USA).
After sequencing, each negative control contained fewer than 2,050 reads. Furthermore, contaminating ASVs were identified through the use of the decontam package (84). After removal of contaminants identified by decontam, the data set consisted of 2,288 ASVs. Samples contained 4,992 to 308,479 reads, with a median of 49,250. A rarefaction curve was calculated for all samples and revealed that a rarefaction level of 2,000 reads was sufficient to describe the composition of the microbiome. Rarefaction to 4,990 reads/sample without replacement resulted in the removal of 1,339 ASVs from the data set. Any ASV not present in at least two samples after rarefaction was also removed from the data set. The final trimmed and rarefied data set consisted of 209 samples, 182 ASVs, and 4,990 reads per sample. The trimmed data set was then transferred to phyloseq (85) for further analysis.
(ii) Positive controls.
Two types of positive control were prepared and sequenced. (i) A dilution series of a ZymoBIOMICS microbial community DNA standard (Zymo Research, Irvine, CA, USA) was amplified. (ii) Some samples were amplified multiple times with different PCR primers and on different runs to confirm the reproducibility of the data (data not shown). For samples amplified and sequenced multiple times, the sample with the most reads was used for analysis.
The maximum abundance of any spurious ASV in a positive-control sample was 1.8%, with only three ASVs at >1% abundance (Table S4). The majority of spurious ASVs occurring in both positive and negative controls were those identified as core or common in at least one sample group. Together, the control sample results suggest that ASVs with measured abundances of ≤1.8% in a sample must be viewed with some skepticism until confirmed in future research.
Core and common ASV determination.
The median read count of a single ASV in any negative control (medNeg) was calculated to be 7. This number was then used to calculate a conservative estimate for the minimum fraction of a sample that an ASV must constitute to be considered present in that sample. It was assumed that any true ASV would contain a read count greater than or equal to the medNeg, so an ASV must contain at least 7/4,990 (0.1%) of the total community in order to be considered present. The core community was defined as only those ASVs present in at least 90% of samples in a group. The common community was defined as only those ASVs present in 70 to 90% of samples in a group.
Comparison to previous sequences.
16S sequences from previously published research were downloaded from the NCBI SRA database. Published sequences were trimmed to the appropriate primer region and aligned with sequences from this study using CLC Genomics Workbench (Qiagen, Aarhus, Denmark). Sequences from regions outside the V4 hypervariable region of the 16S rRNA gene could not be directly compared to the sequences in this study and so were compared only qualitatively at the identified genus level. Aligned sequences were identified as identical to those produced by this study or were labeled as unique ASVs.
Effect of extraction method.
Sample collection and processing were conducted over a period of 6 years. Over this time, two DNA extraction methods were used, as described above. A PERMANOVA using the Bray-Curtis metric was performed through the adonis function of the vegan package (86) to determine if the two extraction methods affected results. No significant difference was found between the two extraction methods (PERMANOVA: pseudo-F = 0.695; R2 = 0.06; P ≥ 0.586, Bray-Curtis) (Fig. S4).
Beta diversity calculations.
PERMANOVA using the Bray-Curtis, Jaccard, Chao1, or binary Ochiai metrics were performed using the adonis function of the vegan package (86). Nonmetric multidimensional scaling (NMDS) plots were prepared using the distance, ordinate, and plot_ordination function of the phyloseq package (85). Distances were calculated with the Bray-Curtis, Jaccard, Chao1, or binary Ochiai metrics, as indicated (87).
Alpha diversity calculations.
Alpha diversity was calculated using the Shannon, inverse Simpson, Chao-1, and abundance-based coverage estimator (ACE) metrics in the plot_richness function of the phyloseq package (85). Alpha diversity was calculated for ILF and intestinum samples separately.
Data availability.
The sequence data were deposited in the NCBI SRA under project ID PRJNA544194. Individual ASV sequences are available in Table S4 as well as in the NCBI SRA under the accession numbers listed in Table S4.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by NIH R01 GM095390 to J.G., P. Visscher, and H. Morrison, NSF 1710511 to J.G. and V. Cooper, and the University of Connecticut.
We thank R. Rubinstein and M. T. McClure for help with Python coding and the Microbial Resources and Services Facility of the University of Connecticut for sequencing the samples.
J.G. is a leech microbiology consultant for the German leech farm Biebertaler Blutegelzucht GmbH, Biebertal, Germany, but the company does not direct or approve J.G.’s research and publications.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Sawyer RT. 1986. Leech biology and behavior. Clarendon Press, Oxford, United Kingdom. [Google Scholar]
- 2.Brinkmann A, Jr. 1947. Two new Antarctic leeches. Nature 160:756. 10.1038/160756a0. [DOI] [PubMed] [Google Scholar]
- 3.Whitaker IS, Rao J, Izadi D, Butler PE. 2004. Historical article: Hirudo medicinalis: ancient origins of, and trends in the use of medicinal leeches throughout history. Br J Oral Maxillofac Surg 42:133–137. 10.1016/S0266-4356(03)00242-0. [DOI] [PubMed] [Google Scholar]
- 4.Graf J. 2000. The symbiosis of Aeromonas and Hirudo medicinalis, the medicinal leech. ASM News 66:147–153. [Google Scholar]
- 5.Siddall ME, Trontelj P, Utevsky SY, Nkamany M, Macdonald KS. 2007. Diverse molecular data demonstrate that commercially available medicinal leeches are not Hirudo medicinalis. Proc Biol Sci 274:1481–1487. 10.1098/rspb.2007.0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Utevsky SY, Trontelj P. 2005. A new species of the medicinal leech (Oligochaeta, Hirudinida, Hirudo) from Transcaucasia and an identification key for the genus Hirudo. Parasitol Res 98:61–66. 10.1007/s00436-005-0017-7. [DOI] [PubMed] [Google Scholar]
- 7.Poly WJ. 2018. Range extension for the elusive New England medicinal leech, Macrobdella sestertia Whitman, 1886 (Hirudinida: Macrobdellidae), in South Carolina, U.S.A., with notes on morphology, coloration, and biology. Proc Cal Acad Sci 64:347–359. [Google Scholar]
- 8.Marden JN, McClure EA, Beka L, Graf J. 2016. Host matters: medicinal leech digestive-tract symbionts and their pathogenic potential. Front Microbiol 7:1569. 10.3389/fmicb.2016.01569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Müller IW. 2000. Handbuch der Blutegeltherapie. HAUG, Heidelberg, Germany. [Google Scholar]
- 10.Siddall ME, Min GS, Fontanella FM, Phillips AJ, Watson SC. 2011. Bacterial symbiont and salivary peptide evolution in the context of leech phylogeny. Parasitology 138:1815–1827. 10.1017/S0031182011000539. [DOI] [PubMed] [Google Scholar]
- 11.Min GS, Sarkar IN, Siddall ME. 2010. Salivary transcriptome of the North American medicinal leech, Macrobdella decora. J Parasitol 96:1211–1221. 10.1645/GE-2496.1. [DOI] [PubMed] [Google Scholar]
- 12.Whitaker IS, Oboumarzouk O, Rozen WM, Naderi N, Balasubramanian SP, Azzopardi EA, Kon M. 2012. The efficacy of medicinal leeches in plastic and reconstructive surgery: a systematic review of 277 reported clinical cases. Microsurgery 32:240–250. 10.1002/micr.20971. [DOI] [PubMed] [Google Scholar]
- 13.Kvist S, Manzano-Marin A, de Carle D, Trontelj P, Siddall ME. 2020. Draft genome of the European medicinal leech Hirudo medicinalis (Annelida, Clitellata, Hirudiniformes) with emphasis on anticoagulants. Sci Rep 10:9885. 10.1038/s41598-020-66749-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Babenko VV, Podgorny OV, Manuvera VA, Kasianov AS, Manolov AI, Grafskaia EN, Shirokov DA, Kurdyumov AS, Vinogradov DV, Nikitina AS, Kovalchuk SI, Anikanov NA, Butenko IO, Pobeguts OV, Matyushkina DS, Rakitina DV, Kostryukova ES, Zgoda VG, Baskova IP, Trukhan VM, Gelfand MS, Govorun VM, Schioth HB, Lazarev VN. 2020. Draft genome sequences of Hirudo medicinalis and salivary transcriptome of three closely related medicinal leeches. BMC Genomics 21:331. 10.1186/s12864-020-6748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wenning A, Zerbst-Boroffka I, Bazin B. 1980. Water and salt excretion in the leech. J Comp Physiol B 139:97–102. 10.1007/BF00691022. [DOI] [Google Scholar]
- 16.Graf J. 1999. Symbiosis of Aeromonas veronii biovar sobria and Hirudo medicinalis, the medicinal leech: a novel model for digestive tract associations. Infect Immun 67:1–7. 10.1128/IAI.67.1.1-7.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Worthen PL, Gode CJ, Graf J. 2006. Culture-independent characterization of the digestive-tract microbiota of the medicinal leech reveals a tripartite symbiosis. Appl Environ Microbiol 72:4775–4781. 10.1128/AEM.00356-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kikuchi Y, Graf J. 2007. Spatial and temporal population dynamics of a naturally occurring two-species microbial community inside the digestive tract of the medicinal leech. Appl Environ Microbiol 73:1984–1991. 10.1128/AEM.01833-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ott BM, Dacks AM, Ryan KJ, Rio RV. 2016. A tale of transmission: Aeromonas veronii activity within leech-exuded mucus. Appl Environ Microbiol 82:2644–2655. 10.1128/AEM.00185-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson M, Bomar L, Maltz M, Graf J. 2015. Mucinivorans hirudinis gen. nov., sp. nov., an anaerobic, mucin-degrading bacterium isolated from the digestive tract of the medicinal leech, Hirudo verbana. Int J Syst Evol Microbiol 65:990–995. 10.1099/ijs.0.000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neupane S, Modry D, Pafčo B, Zurek L. 2019. Bacterial community of the digestive tract of the European medicinal leech (Hirudo verbana) from the Danube River. Microb Ecol 77:1082–1090. 10.1007/s00248-019-01349-z. [DOI] [PubMed] [Google Scholar]
- 22.Beka L, Fullmer MS, Colston SM, Nelson MC, Talagrand-Reboul E, Walker P, Ford B, Whitaker IS, Lamy B, Gogarten JP, Graf J. 2018. Low-level antimicrobials in the medicinal leech select for resistant pathogens that spread to patients. mBio 9:e01328-18. 10.1128/mBio.01328-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laufer AS, Siddall ME, Graf J. 2008. Characterization of the digestive-tract microbiota of Hirudo orientalis, a european medicinal leech. Appl Environ Microbiol 74:6151–6154. 10.1128/AEM.00795-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitaker IS, Maltz M, Siddall ME, Graf J. 2014. Characterization of the digestive tract microbiota of Hirudo orientalis (medicinal leech) and antibiotic resistance profile. Plast Reconstr Surg 133:408e–418e. 10.1097/01.prs.0000438461.06217.bb. [DOI] [PubMed] [Google Scholar]
- 25.Rio RVM, Attardo GM, Weiss BL. 2016. Grandeur alliances: symbiont metabolic integration and obligate arthropod hematophagy. Trends Parasitol 32:739–749. 10.1016/j.pt.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nogge G. 1981. Significance of symbionts for the maintenance of an optimal nutritional state for successful reproduction in hematophagous arthropods. Parasitology 82:101–104. [Google Scholar]
- 27.Manzano-Marin A, Oceguera-Figueroa A, Latorre A, Jimenez-Garcia LF, Moya A. 2015. Solving a bloody mess: B-vitamin independent metabolic convergence among gammaproteobacterial obligate endosymbionts from blood-feeding arthropods and the leech Haementeria officinalis. Genome Biol Evol 7:2871–2884. 10.1093/gbe/evv188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rio RVM, Anderegg M, Graf J. 2007. Characterization of a catalase gene from Aeromonas veronii, the digestive-tract symbiont of the medicinal leech. Microbiology (Reading) 153:1897–1906. 10.1099/mic.0.2006/003020-0. [DOI] [PubMed] [Google Scholar]
- 29.Husnik F. 2018. Host–symbiont–pathogen interactions in blood-feeding parasites: nutrition, immune cross-talk and gene exchange. Parasitology 145:1294–1303. 10.1017/S0031182018000574. [DOI] [PubMed] [Google Scholar]
- 30.Silver AC, Williams D, Faucher J, Horneman AJ, Gogarten JP, Graf J. 2011. Complex evolutionary history of the Aeromonas veronii group revealed by host interaction and DNA sequence data. PLoS One 6:e16751. 10.1371/journal.pone.0016751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maltz M, Graf J. 2011. The type II secretion system is essential for erythrocyte lysis and gut colonization by the leech digestive tract symbiont Aeromonas veronii. Appl Environ Microbiol 77:597–603. 10.1128/AEM.01621-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidson SK, Powell R, James S. 2013. A global survey of the bacteria within earthworm nephridia. Mol Phylogenet Evol 67:188–200. 10.1016/j.ympev.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 33.Viana F, Paz LC, Methling K, Damgaard CF, Lalk M, Schramm A, Lund MB. 2018. Distinct effects of the nephridial symbionts Verminephrobacter and Candidatus Nephrothrix on reproduction and maturation of its earthworm host Eisenia andrei. FEMS Microbiol Ecol 94. 10.1093/femsec/fix178. [DOI] [PubMed] [Google Scholar]
- 34.Kikuchi Y, Bomar L, Graf J. 2009. Stratified bacterial community in the bladder of the medicinal leech, Hirudo verbana. Environ Microbiol 11:2758–2770. 10.1111/j.1462-2920.2009.02004.x. [DOI] [PubMed] [Google Scholar]
- 35.Siddall ME, Worthen PL, Johnson M, Graf J. 2007. Novel role for Aeromonas jandaei as a digestive tract symbiont of the North American medicinal leech. Appl Environ Microbiol 73:655–658. 10.1128/AEM.01282-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ott BM, Rickards A, Gehrke L, Rio RV. 2014. Characterization of shed medicinal leech mucus reveals a diverse microbiota. Front Microbiol 5:757. 10.3389/fmicb.2014.00757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clark MA, Moran NA, Baumann P, Wernegreen JJ. 2000. Cospeciation between bacterial endosymbionts (Buchnera) and a recent radiation of aphids (Uroleucon) and pitfalls of testing for phylogenetic congruence. Evolution 54:517–525. 10.1111/j.0014-3820.2000.tb00054.x. [DOI] [PubMed] [Google Scholar]
- 38.Moeller AH, Li Y, Mpoudi Ngole E, Ahuka-Mundeke S, Lonsdorf EV, Pusey AE, Peeters M, Hahn BH, Ochman H. 2014. Rapid changes in the gut microbiome during human evolution. Proc Natl Acad Sci U S A 111:16431–16435. 10.1073/pnas.1419136111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chandler JA, Morgan Lang J, Bhatnagar S, Eisen JA, Kopp A. 2011. Bacterial communities of diverse Drosophila species: ecological context of a host–microbe model system. PLoS Genet 7:e1002272. 10.1371/journal.pgen.1002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brune A, Dietrich C. 2015. The gut microbiota of termites: digesting the diversity in the light of ecology and evolution. Annu Rev Microbiol 69:145–166. 10.1146/annurev-micro-092412-155715. [DOI] [PubMed] [Google Scholar]
- 41.Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K, Rawls JF. 2011. Evidence for a core gut microbiota in the zebrafish. ISME J 5:1595–1608. 10.1038/ismej.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maltz MA, Bomar L, Lapierre P, Morrison HG, McClure EA, Sogin ML, Graf J. 2014. Metagenomic analysis of the medicinal leech gut microbiota. Front Microbiol 5:151. 10.3389/fmicb.2014.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Gilbreath TM, Kukutla P, Yan G, Xu J. 2011. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One 6:e24767. 10.1371/journal.pone.0024767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aksoy S. 2000. Tsetse—a haven for microorganisms. Parasitol Today 16:114–118. 10.1016/s0169-4758(99)01606-3. [DOI] [PubMed] [Google Scholar]
- 45.Strand MR. 2018. Composition and functional roles of the gut microbiota in mosquitoes. Curr Opin Insect Sci 28:59–65. 10.1016/j.cois.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zepeda Mendoza ML, Xiong Z, Escalera-Zamudio M, Runge AK, Thézé J, Streicker D, Frank HK, Loza-Rubio E, Liu S, Ryder OA, Samaniego Castruita JA, Katzourakis A, Pacheco G, Taboada B, Löber U, Pybus OG, Li Y, Rojas-Anaya E, Bohmann K, Carmona Baez A, Arias CF, Liu S, Greenwood AD, Bertelsen MF, White NE, Bunce M, Zhang G, Sicheritz-Pontén T, Gilbert MPT. 2018. Hologenomic adaptations underlying the evolution of sanguivory in the common vampire bat. Nat Ecol Evol 2:659–668. 10.1038/s41559-018-0476-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michel AJ, Ward LM, Goffredi SK, Dawson KS, Baldassarre DT, Brenner A, Gotanda KM, McCormack JE, Mullin SW, O’Neill A, Tender GS, Uy JAC, Yu K, Orphan VJ, Chaves JA. 2018. The gut of the finch: uniqueness of the gut microbiome of the Galápagos vampire finch. Microbiome 6:167–167. 10.1186/s40168-018-0555-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siddall ME, Barkdull M, Tessler M, Brugler MR, Borda E, Hekkala E. 2019. Ideating iDNA: lessons and limitations from leeches in legacy collections. PLoS One 14:e0212226. 10.1371/journal.pone.0212226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silver AC, Kikuchi Y, Fadl AA, Sha J, Chopra AK, Graf J. 2007. Interaction between innate immune cells and a bacterial type III secretion system in mutualistic and pathogenic associations. Proc Natl Acad Sci U S A 104:9481–9486. 10.1073/pnas.0700286104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bomar L, Graf J. 2012. Investigation into the physiologies of Aeromonas veronii in vitro and inside the digestive tract of the medicinal leech using RNA-seq. Biol Bull 223:155–166. 10.1086/BBLv223n1p155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maltz M, LeVarge B, Graf J. 2015. Identification of iron and heme utilization genes in Aeromonas and their role in the colonization of the leech digestive tract. Front Microbiol 6:763. 10.3389/fmicb.2015.00763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mandel MJ, Wollenberg MS, Stabb EV, Visick KL, Ruby EG. 2009. A single regulatory gene is sufficient to alter bacterial host range. Nature 458:215–218. 10.1038/nature07660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yip ES, Grublesky BT, Hussa EA, Visick KL. 2005. A novel, conserved cluster of genes promotes symbiotic colonization and sigma-dependent biofilm formation by Vibrio fischeri. Mol Microbiol 57:1485–1498. 10.1111/j.1365-2958.2005.04784.x. [DOI] [PubMed] [Google Scholar]
- 54.Murfin KE, Lee MM, Klassen JL, McDonald BR, Larget B, Forst S, Stock SP, Currie CR, Goodrich-Blair H. 2015. Xenorhabdus bovienii strain diversity impacts coevolution and symbiotic maintenance with Steinernema spp. nematode hosts. mBio 6:e00076-15. 10.1128/mBio.00076-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng H, Perreau J, Powell JE, Han B, Zhang Z, Kwong WK, Tringe SG, Moran NA. 2019. Division of labor in honey bee gut microbiota for plant polysaccharide digestion. Proc Natl Acad Sci U S A 116:25909–25916. 10.1073/pnas.1916224116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rangel LT, Marden J, Colston S, Setubal JC, Graf J, Gogarten JP. 2019. Identification and characterization of putative Aeromonas spp. T3SS effectors. PLoS One 14:e0214035. 10.1371/journal.pone.0214035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci U S A 103:12115–12120. 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benjamino J, Lincoln S, Srivastava R, Graf J. 2018. Low-abundant bacteria drive compositional changes in the gut microbiota after dietary alteration. Microbiome 6:86. 10.1186/s40168-018-0469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jousset A, Bienhold C, Chatzinotas A, Gallien L, Gobet A, Kurm V, Küsel K, Rillig MC, Rivett DW, Salles JF, van der Heijden MGA, Youssef NH, Zhang X, Wei Z, Hol WHG. 2017. Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J 11:853–862. 10.1038/ismej.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roth M. 2009. Zur Biologie der Blutegel, p 10–43. In Michalsen A, Roth M (ed), Blutegeltherapie. Haug, Stuttgart, Germany. [Google Scholar]
- 61.Sartor C, Bornet C, Guinard D, Fournier PE. 2013. Transmission of Aeromonas hydrophila by leeches. Lancet 381:1686. 10.1016/S0140-6736(13)60316-5. [DOI] [PubMed] [Google Scholar]
- 62.Vasai F, Brugirard Ricaud K, Bernadet MD, Cauquil L, Bouchez O, Combes S, Davail S. 2014. Overfeeding and genetics affect the composition of intestinal microbiota in Anas platyrhynchos (Pekin) and Cairina moschata (Muscovy) ducks. FEMS Microbiol Ecol 87:204–216. 10.1111/1574-6941.12217. [DOI] [PubMed] [Google Scholar]
- 63.Costello EK, Gordon JI, Secor SM, Knight R. 2010. Postprandial remodeling of the gut microbiota in Burmese pythons. ISME J 4:1375–1385. 10.1038/ismej.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carey HV, Walters WA, Knight R. 2013. Seasonal restructuring of the ground squirrel gut microbiota over the annual hibernation cycle. Am J Physiol Regul Integr Comp Physiol 304:R33–R42. 10.1152/ajpregu.00387.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Indergand S, Graf J. 2000. Ingested blood contributes to the specificity of the symbiosis of Aeromonas veronii biovar sobria and Hirudo medicinalis, the medicinal leech. Appl Environ Microbiol 66:4735–4741. 10.1128/aem.66.11.4735-4741.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tasiemski A, Massol F, Cuvillier-Hot V, Boidin-Wichlacz C, Roger E, Rodet F, Fournier I, Thomas F, Salzet M. 2015. Reciprocal immune benefit based on complementary production of antibiotics by the leech Hirudo verbana and its gut symbiont Aeromonas veronii. Sci Rep 5:17498. 10.1038/srep17498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nelson MC, Graf J. 2012. Bacterial symbioses of the medicinal leech Hirudo verbana. Gut Microbes 3:322–331. 10.4161/gmic.20227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nelson MC, Morrison HG, Benjamino J, Grim SL, Graf J. 2014. Analysis, optimization and verification of Illumina-generated 16S rRNA gene amplicon surveys. PLoS One 9:e94249. 10.1371/journal.pone.0094249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Farouni R, Djambazian H, Ferri LE, Ragoussis J, Najafabadi HS. 2020. Model-based analysis of sample index hopping reveals its widespread artifacts in multiplexed single-cell RNA-sequencing. Nat Commun 11:2704. 10.1038/s41467-020-16522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mumcuoglu KY, Huberman L, Cohen R, Temper V, Adler A, Galun R, Block C. 2010. Elimination of symbiotic Aeromonas spp. from the intestinal tract of the medicinal leech, Hirudo medicinalis, using ciprofloxacin feeding. Clin Microbiol Infect 16:563–567. 10.1111/j.1469-0691.2009.02868.x. [DOI] [PubMed] [Google Scholar]
- 71.Graf J. 2016. Lessons from digestive-tract symbiosis between bacteria and invertebrates. Annu Rev Microbiol 70:375–393. 10.1146/annurev-micro-091014-104258. [DOI] [PubMed] [Google Scholar]
- 72.McFall-Ngai M, Hadfield MG, Bosch TC, Carey HV, Domazet-Loso T, Douglas AE, Dubilier N, Eberl G, Fukami T, Gilbert SF, Hentschel U, King N, Kjelleberg S, Knoll AH, Kremer N, Mazmanian SK, Metcalf JL, Nealson K, Pierce NE, Rawls JF, Reid A, Ruby EG, Rumpho M, Sanders JG, Tautz D, Wernegreen JJ. 2013. Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci U S A 110:3229–3236. 10.1073/pnas.1218525110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith DG. 1977. The rediscovery of Macrobdella sestertia Whitman (Hirudinea: Hirudinidae). J Parasitol 63:759–760. 10.2307/3279592. [DOI] [PubMed] [Google Scholar]
- 74.Phillips AJ, Salas-Montiel R, Oceguera-Figueroa A. 2016. Distribution of the New England medicinal leech, Macrobdella sestertia Whitman, 1886 and redeterminations of specimens of Macrobdella (Annelida: Clitellata: Macrobdellidae) at the National Museum of Natural History, Smithsonian Institution. Proc Biol Soc Washington 129:103–113. 10.2988/0006-324X-129.Q2.103. [DOI] [Google Scholar]
- 75.Puchtler H, Waldrop FS, Meloan SN, Terry MS, Conner HM. 1970. Methacarn (methanol-Carnoy) fixation. Practical and theoretical considerations. Histochemie 21:97–116. 10.1007/BF00306176. [DOI] [PubMed] [Google Scholar]
- 76.Tang YZ, Gin KY, Lim TH. 2005. High-temperature fluorescent in situ hybridization for detecting Escherichia coli in seawater samples, using rRNA-targeted oligonucleotide probes and flow cytometry. Appl Environ Microbiol 71:8157–8164. 10.1128/AEM.71.12.8157-8164.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu Z, Morrison M. 2004. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 36:808–812. 10.2144/04365ST04. [DOI] [PubMed] [Google Scholar]
- 79.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Benjamino J, Beka L, Graf J. 2018. Microbiome analyses for toxicological studies. Curr Protoc Toxicol 79:e53. 10.1002/cptx.53. [DOI] [PubMed] [Google Scholar]
- 81.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pruesse E, Peplies J, Glöckner FO. 2012. SINA: accurate high throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829. 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, Bandela AM, Cardenas E, Garrity GM, Tiedje JM. 2007. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res 35:D169–D172. 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. 2018. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6:226. 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H. 2019. vegan: community ecology package. R Package version 2.5-5.
- 87.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Neef A. 1997. Anwendung der in situ Einzelzell‐Identifizierung von Bakterien zur Populationsanalyse in komplexen mikrobiellen Biozönosen. Technical University of Munich, Munich, Germany. [Google Scholar]
- 89.Manz W, Amann R, Ludwig W, Wagner M, Schleifer KH. 1992. Phylogenetic oligodeoxynucleotide probes for the major subclasses of Proteobacteria: problems and solutions. Syst Appl Microbiol 15:593–600. 10.1016/S0723-2020(11)80121-9. [DOI] [Google Scholar]
- 90.Manz W, Amann R, Ludwig W, Vancanneyt M, Schleifer KH. 1996. Application of a suite of 16S rRNA-specific oligonucleotide probes designed to investigate bacteria of the phylum cytophaga-flavobacter-bacteroides in the natural environment. Microbiology 142:1097–1106. 10.1099/13500872-142-5-1097. [DOI] [PubMed] [Google Scholar]
- 91.Kämpfer P, Erhart R, Beimfohr C, Böhringer J, Wagner M, Amann R. 1996. Characterization of bacterial communities from activated sludge: culture-dependent numerical identification versus in situ identification using group- and genus-specific rRNA-targeted oligonucleotide probes. Microb Ecol 32:101–121. 10.1007/BF00185883. [DOI] [PubMed] [Google Scholar]
- 92.Thayanukul P, Zang K, Janhom T, Kurisu F, Kasuga I, Furumai H. 2010. Concentration-dependent response of estrone-degrading bacterial community in activated sludge analyzed by microautoradiography-fluorescence in situ hybridization. Water Res 44:4878–4887. 10.1016/j.watres.2010.07.031. [DOI] [PubMed] [Google Scholar]
- 93.Amann RI, Krumholz L, Stahl DA. 1990. Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J Bacteriol 172:762–770. 10.1128/jb.172.2.762-770.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequence data were deposited in the NCBI SRA under project ID PRJNA544194. Individual ASV sequences are available in Table S4 as well as in the NCBI SRA under the accession numbers listed in Table S4.