

Abstract
Several lines of evidence implicate the protein tau in the pathogenesis of multiple brain disorders, including Alzheimer’s disease, other neurodegenerative conditions, autism, and epilepsy. Tau is abundant in neurons and interacts with microtubules, but its main functions in the brain remain to be defined. These functions may involve the regulation of signaling pathways relevant to diverse biological processes. Informative disease models have revealed a plethora of abnormal tau species and mechanisms that might contribute to neuronal dysfunction and loss, but the relative importance of their respective contributions is uncertain. This knowledge gap poses major obstacles to the development of truly impactful therapeutic strategies. The current expansion and intensification of efforts to translate mechanistic insights into tau-related therapeutics should address this issue and could deliver better treatments for a host of devastating conditions.
Graphical Abstract
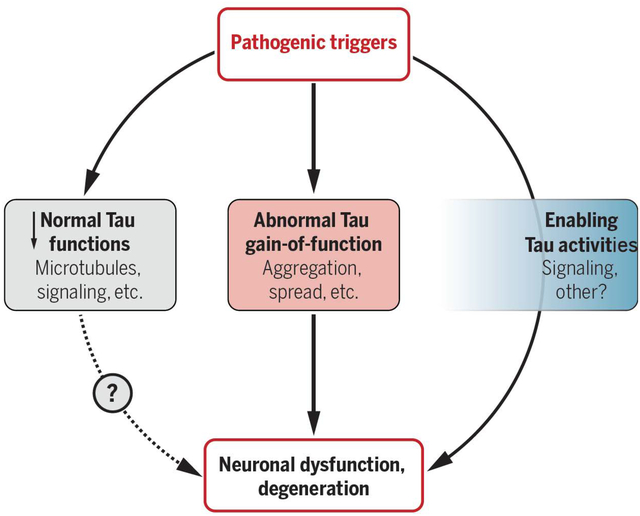
Summary Figure: Potential tau pathomechanisms. Developing effective tau-targeting therapeutics will require a better understanding of how exactly tau contributes to Alzheimer’s disease and other disorders of the central nervous system. Potential mechanisms likely fall into the three broad categories shown. However, the relative pathogenic impact and overall importance of individual mechanisms have yet to be defined in truly disease-relevant contexts and may differ among diseases and even patients. The blue box on the right indicates tau activities that do not directly mediate but indirectly promote or facilitate pathogenic processes.
Summary sentence:
Expanding efforts to develop tau-targeting therapeutics will shed light on this enigmatic protein and could yield better treatments for multiple brain disorders.
REVIEW SUMMARY
Background:
The microtubule-associated protein tau has been implicated in the pathogenesis of Alzheimer’s disease and a range of other neurodegenerative disorders (called “tauopathies”). As the number of people with tauopathies is rising in aging populations across the world, interest in the fundamental biology of this protein and in the development of tau-targeting treatments have been expanding rapidly. Recent insights into the complexity of this intrinsically disordered protein suggest that tau is a worthy but challenging target whose multifaceted nature will likely require a multipronged therapeutic approach. Derived from a single gene by alternative splicing, six major isoforms of tau have been identified in the human brain. In addition, tau is subject to many different posttranslational modifications, further indicating that it may be regulated by multiple processes and may participate in diverse functions.
Advances:
Tau is widely presumed to stabilize microtubules. However, the experimental reduction or ablation of tau in vivo does not alter many neural properties and processes that likely depend on microtubules, including neuronal integrity, axonal transport, synapse formation, and complex brain functions. Although tau reduction seems to have minimal effects on otherwise unmanipulated brains, it can prevent or diminish aberrant cell signaling, neural network dysfunctions (e.g., epileptic activity) and behavioral alterations caused by diverse disease processes, which suggests that tau activities are needed for other pathogenic triggers to cause these derangements. In addition to this “enabling bystander” role, tau’s interactions with a large number of other proteins can cause adverse gains of function, which are associated with—and possibly caused by— the formation of abnormal tau structures and assemblies. Because abnormal forms of tau trigger a plethora of pathomechanisms, targeting individual downstream mechanisms may have limited therapeutic impact, unless the relative pathogenic importance of the specific mechanism has been well established in experimental models that allow for conclusive validation of cause-and-effect relationships. Although much attention has focused on the abnormal aggregation of tau in tauopathies and on the ability of tau “seeds” to spread from neuron to neuron, internalization of propagating tau does not appear to impair neuronal survival or brain functions. Moreover, tau reduction prevents or diminishes neural network dysfunction and behavioral abnormalities also in disease models that do not have abnormal tau inclusions, which suggests that there is more to tau than aggregation and propagation. A promising diversification of tau-targeting therapeutic strategies is beginning to address this complexity. Lowering overall tau levels may have the greatest potential, as this strategy bypasses the unresolved questions of which forms of tau and which downstream mechanisms are most detrimental in any given condition.
Outlook:
Many efforts to develop better treatments for neurodegenerative diseases have failed, in large part because of an inadequate understanding of disease mechanisms and, perhaps, because too many fundamental knowledge gaps, alternative interpretations of data, and methodological complexities did not receive the attention they deserved. This Review highlights important gaps in the understanding of tau and the methodological advances needed to fill them. It also pinpoints obstacles that could complicate the translation of tau-related scientific discoveries into better therapeutics and offers pragmatic strategies to overcome these challenges. Despite the extraordinary progress that has been made to date, the main physiological functions that tau fulfills in the adult and aging brain remain to be defined. Another critical objective is to develop better experimental models and technologies to rigorously compare different tau species and pathomechanisms, particularly their relative impacts on neuronal functions and survival in vivo. For the development of truly informative biomarkers and effective therapeutics, it will be critical to rigorously differentiate between associations and cause-and-effect relationships. Until the main drivers of neuronal dysfunction and demise have been identified for Alzheimer’s disease and other conditions in which tau has a causal or enabling role, it seems prudent to focus on pragmatic strategies, such as overall tau reduction, while also expanding efforts to further validate the importance of more specific targets and approaches. Investigational approaches to lower overall tau levels include tau-targeting antisense oligonucleotides, which have advanced into a clinical trial for early Alzheimer’s disease, and the development of small-molecule drugs that can modulate the production or degradation of tau. The most desirable tau-targeting therapeutics would be efficacious across diverse tauopathies, as well as affordable, easy to access, and well tolerated when administered over long periods of time to fragile groups of people who likely take multiple other medications.
Introduction
The microtubule-associated protein tau (MAPT) is an intrinsically disordered protein that is most abundant in neuronal axons (1). Its interaction with microtubules is intermittent and rather short lived and has been characterized as a rapid “kiss-and-hop” process (2). Neurons can release tau and tau fragments into the extracellular milieu, a process that is enhanced by increased neuronal activity (3). Tau is also present, but less abundant, in neuronal cell bodies, nuclei and synaptic specializations, as well as in glia and other cell types in the brain and peripheral organs (1, 4). In humans, six major tau isoforms are generated in brain cells from a single MAPT gene by alternative splicing of exons 2, 3 and 10 (Fig. 1A). These isoforms differ in their number of N-terminal inserts (zero, one, or two) and microtubule-binding repeat domains [three (3R) or four (4R)] (1, 5), as well as in their subcellular distributions (6, 7) and protein interactomes (8), suggesting involvement in distinct functions. Tau undergoes various posttranslational modifications (Fig. 1B) (1, 9–12), indicating that it may be regulated by multiple pathways and may participate in diverse biological processes. Low body temperature (13) and high dietary salt intake (14) increase tau phosphorylation. The biological purpose of these and other tau modifications is uncertain.
Fig. 1. Tau isoforms and posttranslational modifications.
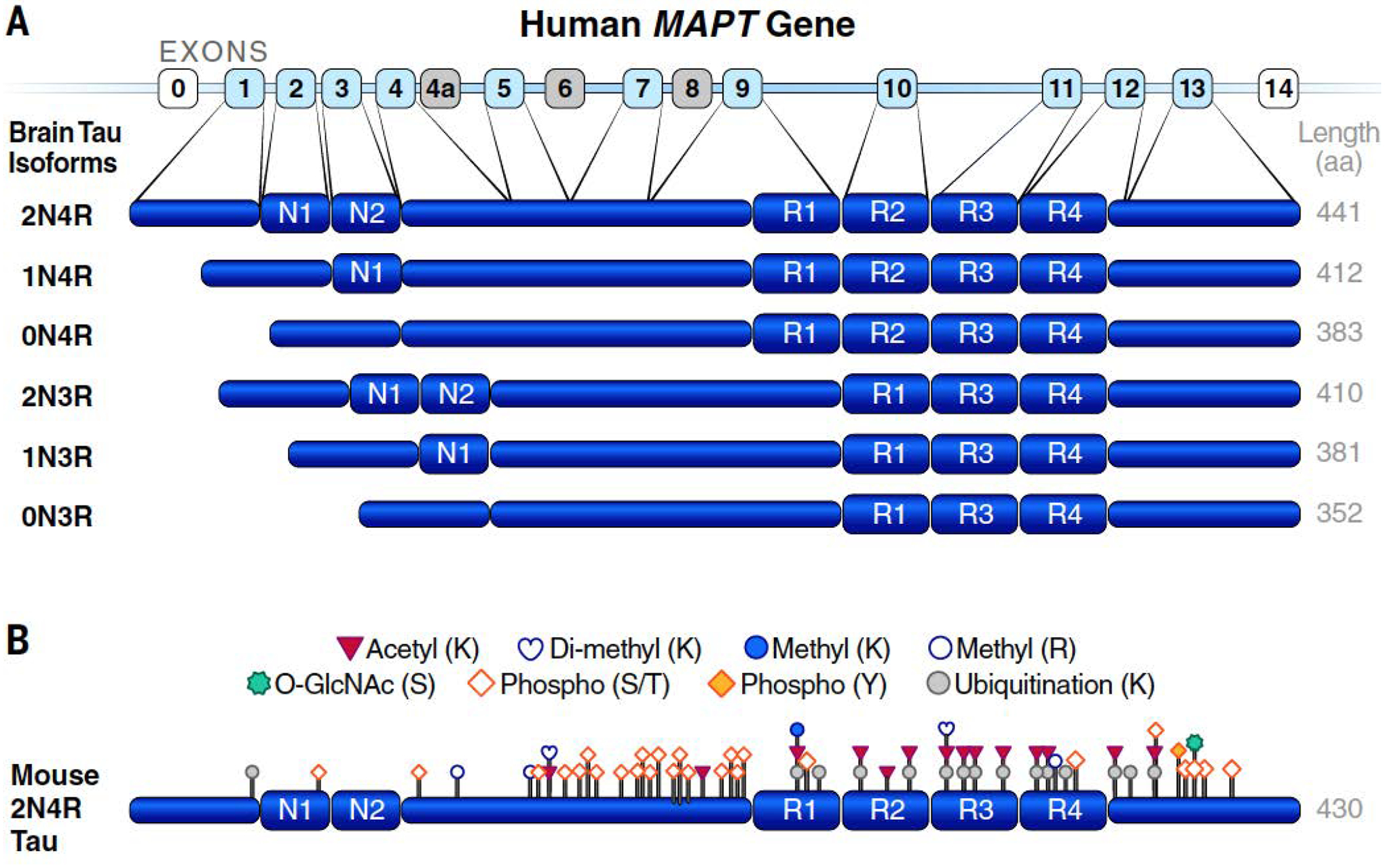
(A) Six major isoforms of tau are produced in brain from the MAPT gene through alternative splicing. N1 and N2 denote N-terminal inserts; R1 to R4 indicate microtubule-binding repeat domains, whose differential inclusion results in the generation of 3R versus 4R tau. Noncoding exons are shown in white; coding exons not expressed in brains or humans are in light grey; coding exons in human brain are in light blue. aa, amino acids. (B) Physiological posttranslational modifications identified by mass spectrometry of tau isolated from brain tissues of healthy adult WT mice. Note that some amino acid modifications could compete with each other (e.g., ubiquitination and acetylation). K, Lys; R, Arg; S, Ser; T, Thr; and Y, Tyr. [Adapted from figure 1a of (9)]
Although the main functions of tau also remain to be defined, there is no question that it is involved in a variety of common and disabling brain diseases. Intraneuronal accumulations of insoluble, filamentous tau aggregates underlie the formation of neurofibrillary tangles, a pathological hallmark of Alzheimer’s disease (AD) and multiple other neurodegenerative disorders (“tauopathies”) (1, 15). Besides these highly organized tau assemblies, nonfibrillar tau species, including tau oligomers and fragments, may also contribute to neurological disease (1, 16, 17). A causal role of tau in brain diseases is suggested by positive correlations between neurofibrillary tangle burden and brain dysfunctions, autosomal dominant mutations in MAPT that cause neurodegenerative disorders, and neuronal deficits caused or enabled by tau in experimental models (1, 11, 15, 18–22).
Intraneuronal accumulation of abnormal tau species is required for the postmortem diagnosis of AD. Although the heterogenous group of less common non-AD tauopathies by definition also have “tau pathology,” they differ from AD and from each other with respect to many other pathological and clinical features. Notably, a large number of very rare MAPT mutations (mostly within or adjacent to the microtubule-binding repeat domain of tau) can each cause non-AD tauopathies—in particular, frontotemporal lobar degeneration (FTLD)—but they never cause AD (1, 15, 23, 24). By contrast, the rare Ala152→Thr (A152T) variant of tau seems to increase the risk for both AD and non-AD tauopathies (25, 26). These associations between specific changes in the amino acid sequence of tau and distinct neurodegenerative disorders merit further exploration, especially because animal and cell culture models expressing human tau with any one of several MAPT mutations that cause FTLD in humans (henceforth referred to as “FTLD-tau”) are widely used to study the pathogenic role of tau in AD and to develop better treatments for this condition. What if the pathogenic roles of tau in AD and FTLD differed fundamentally? Therapeutically targeting FTLD-tau might fail in AD, the most frequent cause of dementia, unless the targeted mechanism is at work in both types of disorders.
A particular challenge in this regard is the plethora of potential pathomechanisms that have been identified for (mostly FTLD-associated) tau. Identifying the mechanism that is most critical in AD has been made difficult by the lack of unquestionably AD-relevant experimental models that allow rigorous comparison of different tau species and pathomechanisms, particularly their effects on neuronal functions and survival. Notably, reducing levels of endogenous wild-type (WT) tau has beneficial effects in experimental models of autism (18), depression (19), epilepsy (21, 22, 27), and stroke (20), even though these models do not develop intraneuronal accumulations of abnormal tau aggregates, indicating that there is much more to tau than aggregation.
Putative physiological functions
Tau’s interaction with microtubules raises at least two possible roles: that tau supports the functions of microtubules and that microtubules support the functions of tau. It is widely presumed that tau’s main function is to stabilize microtubules. However, this dogma has been challenged, if not refuted, by a growing body of evidence. Early experiments carried out in cell-free conditions or using microinjection of tau into non-neuronal cells lacking endogenous tau suggested that tau stabilizes microtubules and regulates their dynamics (28–30). More recent studies refuted the hypothesis that tau stabilizes microtubules in primary rat neurons while yielding additional evidence that tau regulates microtubule dynamics (31). Whether genetic ablation of tau has a long-term impact on the neuronal expression of other microtubule-associated proteins (MAPs) under otherwise normal circumstances is uncertain. Western blot analysis of brain tissue from a small number of mice suggested increased brain levels of MAP1A in Mapt−/− mice during early postnatal stages compared with WT controls (32). In mice maintained on a high-fat diet throughout life, hippocampal levels of MAP1A, MAP1B and MAP2 were increased in adult Mapt−/− mice but either unchanged or decreased in aged Mapt−/− mice (33). In contrast to tau ablation, MAP1B ablation causes defects in the formation of neuronal layers and axon tracts during development and this phenotype is markedly exacerbated by concomitant tau ablation (34), suggesting that tau can partially fulfill or assume developmental MAP1B functions in the absence of the latter molecule. Microtubule-stabilizing drugs reduced tau pathology, behavioral deficits, and neurodegeneration in transgenic mice overexpressing FTLD-tau (1, 35, 36), but whether these agents did so by compensating for a loss of tau function, counteracting an adverse gain of tau function, or another mechanism is uncertain. Moreover, such drugs have failed in clinical trials for AD and other tauopathies (37).
Notably, genetic ablation and acute knockdown of tau did not change axonal transport, which critically depends on microtubules, in cultured primary neurons (38, 39) or in mouse optic nerve axons in vivo (40). These findings seem difficult to reconcile with the notion that tau has a major role in stabilizing microtubules, at least in axons. Furthermore, with the possible exceptions discussed below, genetic ablation of tau during early development is well tolerated in rodents (18, 21, 22, 41–44), and so is acute knockdown of tau in the brains of young adult rodents and non-human primates (27, 45). No compelling evidence could be found to show that tau reduction causes derangements of microtubules or alterations in microtubule dynamics that affect neuronal functions or integrity in young, adult, or aging mammalian brains.
By contrast, mounting evidence suggests that tau helps regulate multiple signaling pathways. Tau promotes insulin-induced tyrosine phosphorylation of the insulin receptor substrate 1 (IRS-1) and inhibits activation of phosphatase and tensin homolog (PTEN) (46). Ablation of these functions in Mapt−/− mice reduces anorexigenic effects of insulin in the hypothalamus (46), which may contribute to the moderate age-dependent obesity of these mice (42, 46). Conversely, neuronal accumulation of FTLD-tau enhanced insulin responsiveness in transgenic mice (47), a process that might contribute to weight loss observed in people who have or are at risk for AD (48). Yet, tau ablation prevented synaptic and cognitive deficits in a streptozotocin-induced model of type 1 diabetes (49).
Several studies have explored whether tau has roles in synaptic physiology and plasticity. Deficits in long-term potentiation were identified in the hippocampal CA1 region of one tau-deficient (Mapt−/−) model (50) but not in the dentate gyrus of another (51). Similarly, some groups observed reductions in long-term depression in Mapt−/− mice (46, 52), whereas others did not (50), possibly because of differences in mouse ages and experimental protocols. Reductions in long-term depression might counteract processes that abnormally increase this activity in AD (53).
Mapt−/− mice have normal excitatory postsynaptic transmission in the hippocampal CA1 region and dentate gyrus (44, 51). However, they have an increased frequency of inhibitory postsynaptic currents in dentate granule cells (51), which may contribute to their increased resistance to epilepsy of diverse causes (18, 21, 22, 27, 44, 51). The potential roles of tau in the activity of excitatory versus inhibitory neurons and synapses merit further investigation.
Tau also seems to be involved in the generation, maintenance or regulation of brain rhythms. Compared with WT controls, Mapt−/− mice have alterations in various neural network oscillations (41, 54, 55). Some of these alterations may be related to the abnormal sleep structure that has been observed in these mice, which includes decreased non-rapid eye movement (NREM) sleep time, a higher number of state transitions between NREM sleep and wakefulness, and shortened sleep bouts (55). Sleep deprivation increases tau levels in cerebrospinal fluid (CSF) and brain interstitial fluid (56). Additional studies are needed to determine whether this process reflects a role of tau in sleep regulation and whether it promotes tau pathology in AD and other conditions associated with sleep disturbances (57).
The changes in brain rhythms and the resistance of Mapt−/− mice to epilepsy may reflect, at least in part, roles of tau in neurogenesis (58) or in neuronal migration and maturation (59). However, although Mapt−/− mice have slightly smaller brains than WT controls (18), there appear to be no studies demonstrating obvious changes in the overall brain anatomy and cytoarchitecture of these mice. It could be interesting to further explore the effects of tau reduction on neurogenesis and connectivities among brain regions and specific neuronal populations, especially in models of autism in which tau reduction prevented excessive brain growth (megalencephaly) and behavioral abnormalities (18).
Reports of marked behavioral deficits in Mapt−/− mice suggesting an essential role of tau in locomotor functions and in learning and memory (33, 60) could not be replicated by others (18, 21, 22, 42, 44, 51, 61). In humans, the chromosome 17q21.31 microdeletion syndrome causes intellectual disability, epilepsy, and reduced muscle tone (62). Because MAPT resides at this location, it could be tempting to speculate that tau reduction contributes to the impairments in these patients. However, this deletion includes other genes, and mutations in one of them (KANSL1, encoding KAT8 regulatory NSL complex subunit 1) are sufficient to cause the syndrome (62), making a causal role of tau reduction unlikely. Indeed, tau reduction has beneficial effects in a variety of disease models, as described further below.
Taken together, these findings suggest that tau’s most important physiological functions in the central nervous system (CNS) have yet to be defined. In light of tau’s multiple isoforms and many posttranslational modifications, it is tempting to speculate that these functions may differ among cell types, subcellular compartments, and perhaps even life stages.
Roles in neurological and psychiatric disorders
In principle, tau may be involved in the pathogenesis of CNS diseases in at least three ways. A disease process might (i) block a critical tau function (loss of function), (ii) change tau in ways that turn it into a pathogenic effector or mediator (gain of function), or (iii) require the presence of tau in essentially unaltered form (enablement) (Fig. 2; also see the summary figure).
Fig. 2. Physiological but disease-enabling functions of tau.
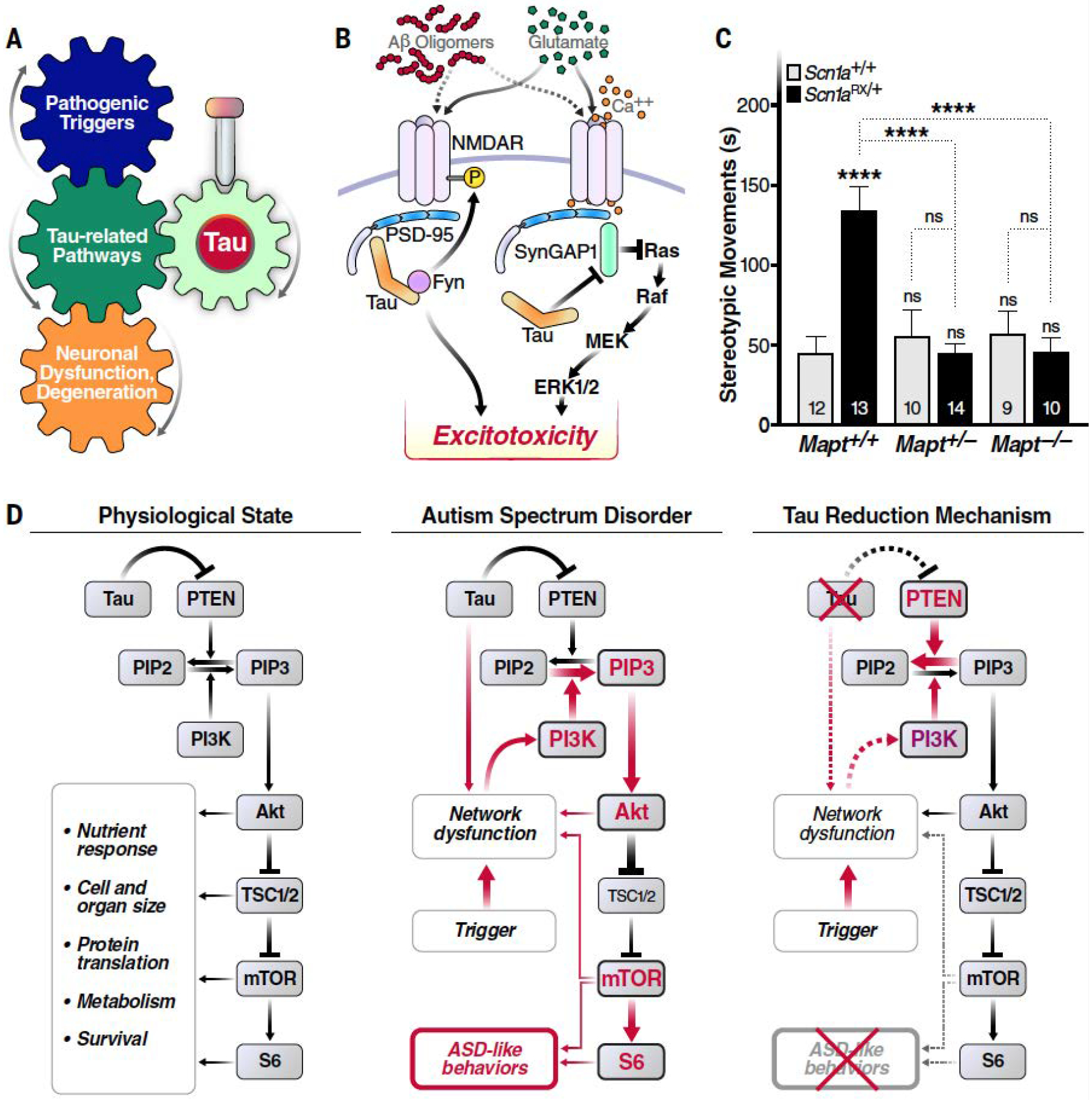
(A) Simplified conceptual diagram highlighting how targeting tau could block the development or progression of disease, even when tau is neither a driver nor a direct mediator of the pathogenic cascade. (B) Tau regulates interactions among the tyrosine kinase Fyn, N-methyl-D aspartate receptors (NMDARs), and postsynaptic density protein 95 (PSD-95) (44), as well as between PSD-95 and synaptic Ras GTPase-activating protein 1 (SynGAP1), which negatively regulates synaptic extracellular signal-regulated kinase (ERK) activation (20). Although these tau activities may normally form part of the complex network of regulators that support synaptic and neuronal activities, they may also allow pathological overstimulation of glutamate receptors to result in “excitotoxicity,” a process tau reduction has been shown to counteract (4, 20). P, phosphorylation; MEK, mitogen-activated protein kinase kinase. [Adapted from figure 3 of (4) with permission from Elsevier] (C and D) In mouse models, the presence of endogenous WT tau allows other processes to overactivate the PI3K-Akt-mTOR signaling pathway, which in turn promotes the development of autism-like behaviors, including excessive stereotypic movements, social interaction deficits and cognitive inflexibility (18). (C) Partial or complete genetic reduction of tau prevented excessive stereotypic movements (time spent engaged in such movements in seconds) in Scn1aRX/+ knockin mice, which carry a sodium channel mutation that causes autism and epilepsy in humans. By contrast, tau reduction did not alter this behavior in mice without this mutation. Numbers of mice per group are indicated in bars. ****P < 0.0001 vs. WT (Scn1a+/+Mapt+/+) controls or as indicated by brackets [two-way analysis of variance (ANOVA) with Holm-Sidak test]. ns, not significant. Error bars indicate SEM. [Reprinted from figure 1A of (18) with permission from Elsevier] (D) Because PTEN counteracts PI3K, tau’s ability to inhibit PTEN activation (46) may enable normal activity of the PI3K-Akt-mTOR pathway under physiological conditions. However, when pathological processes overactivate PI3K, as in some autism spectrum disorders, tau’s constraint of PTEN becomes maladaptive and allows for overactivation of the downstream signaling cascade, promoting the development of autism symptoms through diverse anatomical and pathophysiological mechanisms. Tau reduction counteracts this process by releasing the activity of PTEN and possibly also by blocking neural network dysfunctions. PIP2, phosphatidylinositol 4,5-biphosphate; PIP3, phosphatidylinositol 3,4,5-triphosphate; PI3K, phosphatidylinositol 3-kinase; Akt, protein kinase B; mTOR, mammalian target of rapamycin; TSC1/2, tuberous sclerosis 1 and 2; S6, ribosomal protein S6; ASD, autism spectrum disorder. [Reprinted from figure 3 of (18) with permission from Elsevier]
Early studies raised the then-plausible possibility that sequestration of tau into intraneuronal aggregates might prevent tau from stabilizing microtubules, leading to neurodegeneration (63). In light of the more-recent studies discussed above, this and other loss-of-function mechanisms now seem unlikely. By contrast, evidence continues to mount for pathogenic gains of function of tau. Intraneuronal accumulation of aggregated tau correlates well with clinical disease manifestations in AD, FTLD, and other tauopathies (1, 11, 15, 64). In addition to this “guilt by association”, interventions that block neuronal tau accumulation reduce functional deficits and neuronal loss in experimental models (1, 11, 45, 65, 66).
Another possibility is that physiological tau functions enable brain diseases by allowing other factors and processes to alter signaling pathways (Fig. 2). An analogy that might help illustrate this concept in general is the role of the C-C chemokine receptor type 5 (CCR5) in the pathogenesis of AIDS. Because HIV-1 uses CCR5 to enter T lymphocytes, genetic loss of this co-receptor prevents the development of HIV-1 disease (67). Thus, although CCR5 is not an active effector or mediator of HIV-1-dependent immunopathogenesis, it clearly enables this process in critical ways. In a similar vein, physiological functions of tau may enable neural network dysfunction and behavioral abnormalities caused by other pathogenic drivers in models of autism, depression, epilepsy, and stroke (18–22, 27, 44). Consistent with this notion, there is no evidence for abnormal tau aggregation or propagation in these models. Tau reduction also prevents epileptiform activity and some other functional abnormalities in human amyloid precursor protein (hAPP) transgenic mice that show no evidence of abnormal tau accumulation but simulate several other aspects of AD (9, 22, 51). In primary neuronal cultures, tau reduction does not alter axonal transport under baseline conditions but effectively prevents axonal transport deficits caused by oligomeric assemblies of amyloid-β (Aβ) peptides (38, 39), which have also been causally implicated in the pathogenesis of AD (53).
The analogy to CCR5, genetic deficiency of which is well tolerated (67), may also help explain why tau reduction has such limited adverse effects under physiological conditions and such profound protective effects under pathological conditions. Under baseline conditions, redundancies in the distinct biological processes in which CCR5 and tau participate may minimize the consequences of their ablation. By contrast, pathological processes that more specifically depend on the presence of these molecules such as HIV-1 entry into T cells or Aβ-induced deficits in axonal transport, can be blocked by the ablation or reduction of CCR5 or tau, respectively.
The “enablement” and “adverse gain-of-function” mechanisms are not mutually exclusive and, indeed, may form vicious cycles that drive progression of AD and possibly other disorders. For example, tau enables epileptogenesis in models of diverse diseases (18, 20–22, 27, 44), and chronic epileptic activity in humans is associated with the development of tau pathology, which may contribute to cognitive decline in these patients (68). Furthermore, overexpression of tau can cause epileptiform activity in transgenic mice (69–71). Possibly related to this modulation of network hypersynchrony, tau reduction lowers, whereas tau overexpression increases, the power of brain oscillations in the low frequency range (41). Such changes in brain rhythms could alter cognitive functions (72). In addition, increased power of low frequency oscillations predicts progression to AD in people with subjective cognitive impairments (73), whereas increasing the power of higher-frequency oscillations reduces cognitive deficits and counteracts AD pathology in animal models (74, 75), suggesting that changes in brain rhythms could have symptomatic as well as disease-modifying consequences.
Pathogenic mechanisms of abnormal tau
Because tauopathies are defined by the accumulation of abnormal tau aggregates, many studies have focused on tau misfolding and aggregation (1, 15, 76–78). However, the most pathogenic forms of abnormal tau have not yet been conclusively identified and may differ across tauopathies (15, 77, 78) and among individual patients with AD (76). Notably, even when specific tau alterations are tightly associated with disease states, the biological meaning of such alterations is difficult to discern without further experimental analysis. For example, tau that accumulates in brains of people with distinct tauopathies has disease-specific structural features and can be enriched for distinct isoforms of tau (15, 78). However, whether these tau structures and isoform differences causally contribute to the respective tauopathies or result from specific disease processes that occur in one condition but not others remains to be determined. Similar uncertainties about causality apply to many posttranslational modifications of tau. For example, hyperphosphorylation of tau can be pathogenic or protective, depending on the specific sites targeted (79). These complexities are relevant not only to the development of tau-related therapeutics, but also to the interpretation of tau-related biomarkers, an important and rapidly evolving aspect of this field.
Increased binding of radioligands to detect tau aggregates in brains of live people by positron emission tomography (64, 80) and increased levels of phospho-tau species in CSF (81) and blood (82, 83) are becoming increasingly useful in the differential diagnosis of tauopathies. In AD, these biomarkers also correlate with disease progression (64, 81, 83). How well they can predict clinically meaningful benefits in trials of tau-related therapeutic strategies remains to be determined. Notably, preclinical evaluations of therapeutic interventions in FTLD-tau-overexpressing mouse models have revealed thought provoking dissociations between the intraneuronal accumulation of tau aggregates on one hand and neural dysfunctions and neurodegeneration on the other. For example, reducing overall tau levels decreased behavioral deficits despite the persistence of intraneuronal tau aggregates (66), and reducing the RNA binding protein T cell intracellular antigen 1 (TIA1) increased the formation of neurofibrillary tangles while reducing neurodegeneration, behavioral deficits, premature mortality, and tau oligomer formation (84). These results are consistent with other findings, indicating that oligomeric assemblies and fragments of tau are more bioactive than highly organized aggregates such as tau fibrils and that small, nonfibrillar tau species can contribute to functional neural impairments both within neurons and from the extracellular space (12, 16, 17, 69, 85, 86).
Most putative pathomechanisms of tau have been identified in experimental models in which individual isoforms or fragments of tau are overexpressed at rather high levels and studied in isolation from other factors that tau interacts with in the aging human brain (1, 11, 12, 87). Genomic alterations caused by transgene insertions may contribute to tauopathy-like phenotypes (88) but have been excluded in only some of the most widely used animal models in this field (89). Further highlighting that no individual experimental model is perfect, the half-life of tau is much shorter in cultured human and rodent neurons than in human and rodent brains (90), and the relative abundance of tau isoforms differs markedly between immature neurons in vitro and mature neurons in the adult or aging brain (91, 92). Thus, it is critical to scrutinize all available evidence, ideally from human studies and multiple experimental models, before embarking on the costly development of treatments that target individual forms of abnormal tau or specific pathomechanisms. The following discussion focuses on potentially targetable pathomechanisms that are supported by in vivo evidence and whose manipulation in experimental models reduced not only putative disease-relevant morphological or biochemical alterations but also neuronal dysfunctions or loss.
In experimental models, abnormal tau can impair diverse subcellular compartments, including neuronal axons, presynaptic terminals, and postsynaptic specializations (1, 4, 11, 12, 93). It seems unlikely that a single pathomechanism accounts for all of these changes. For example, cleavage of tau at Asp314 by caspase-2 causes an enrichment of FTLD-tau in dendritic spines and disrupts synaptic and cognitive functions, presumably by reducing the trafficking or anchoring of AMPA-type glutamate receptors (17), the main transducers of rapid excitatory transmission in the mammalian brain (94). Whether this cleavage is also critical for tau-dependent impairments of other subcellular compartments remains to be determined.
Diverse lines of evidence suggest that abnormal tau can disrupt proteostasis mechanisms (25, 95–97), which are critical for neuronal survival and communication (98). For example, in transgenic mice overexpressing FTLD-tau, the reduction of cognitive deficits by activation of the cyclic adenosine monophosphate-protein kinase A pathway has been related to the prevention of tau-induced impairments of the 26S proteasome (99). Enhancing autophagy by pharmacological or genetic manipulations reduced tau pathology and neuronal loss in zebrafish that express A152T-variant human tau (25).
Another intriguing possibility is that abnormal tau promotes the activation and increased expression of transposable elements (100, 101), a process that may involve the intranuclear depletion of piwi-interacting RNAs (100) and could contribute to neuronal dysfunction and degeneration through a variety of mechanisms, including formation of DNA double-strand breaks and aberrant changes in gene expression (100–103). The suppression of neurodegeneration and locomotor abnormalities in a fruit fly tauopathy model by treatment with a reverse transcriptase inhibitor (100) provides additional support for the potential importance of this mechanism.
Acetylation of tau can promote the formation, accumulation and cell-to-cell propagation of abnormal tau species (104, 105). Acetylated, hyperphosphorylated FTLD-tau may contribute to neural dysfunctions by altering the neuronal axon initial segment, a process that may involve interactions between tau and the microtubule end-binding protein 3, and by changing neuronal excitability and neural network activity (106, 107). Acetylated tau can also disrupt kidney and brain expressed protein (KIBRA)-mediated signaling at synapses (12, 108).
Abnormal tau may also cause neuronal dysfunction and degeneration through aberrant activities of the innate immune system (109). Intraneuronal accumulation of FTLD-tau triggers neuronal phagocytosis by microglia through a process that involves secretion of an opsonin, a molecule that binds to and enhances phagocytosis of a target antigen or cell, and production of nitric oxide (110). Deletion of the complement C3a receptor reduced neuroinflammation, synaptic deficits, and neurodegeneration in transgenic mice overexpressing FTLD-tau (111). In similar models, ablation of inflammasome components reduced tau pathology as well as learning and memory deficits (112), whereas overexpression of the cytokine interleukin 1β increased neuroinflammation and tau pathology (113). Coexpression of apolipoprotein (apo) E4, the most common genetic risk factor that strongly increases AD risk (114), enhanced neurodegeneration in transgenic mice overexpressing FTLD-tau, and pharmacological deletion of microglia reduced both tau pathology and neurodegeneration in this model (115).
Considerable attention has been paid to the transcellular propagation (or “spread”) of tau aggregates from neuron to neuron (15, 65, 77, 109, 116, 117), which is enhanced by neuronal overexcitation (118, 119). However, there does not appear to be any conclusive published demonstration that the propagating tau impairs brain functions or the survival of neurons that take it up from the extracellular space, even in transgenic mice whose neurons overexpress an abundance of abnormal tau and, thus, might be prone to the seed-dependent formation of toxic tau aggregates. Indeed, injection of various tau species promoted tau aggregation and propagation in mouse models overexpressing human WT or FTLD-tau, but no neuronal loss or degeneration was observed even 15 months after injection (120–122). In primary cultures, aggregates of a fluorescently tagged phosphomimetic of tau efficiently propagated from neuron to neuron but did not impair the survival, presynaptic terminals, basic electrophysiological properties, or activity of neurons (123). Whether the axonal transport deficits observed in this study resulted from neuronal overproduction of the engineered tau species or uptake of its propagating aggregates is unclear.
Notwithstanding these caveats, extracellular tau may impair neural functions independently of cell-to-cell propagation. For example, when added to the medium of cultured human neurons, N-terminal fragments of tau released from neurons of AD patients caused neuronal hyperactivity and increased production of Aβ (124). Moreover, even brief exposure of mouse brain slices to extracellular human tau oligomers impaired long-term potentiation (86).
Despite these multiple potential pathomechanisms, it is far from clear how exactly abnormal tau causes or contributes to neuronal dysfunction and degeneration, in large part because available methods and experimental models have not yet been optimized to (i) rigorously and efficiently compare the relative pathogenicity of specific forms of tau in truly disease-relevant contexts and (ii) selectively modulate these tau species in vitro and in vivo.
Interactions with other pathogenic factors
Tau aggregation in transgenic mice overexpressing FTLD-tau is enhanced by coexpression of α-synuclein (125), a protein that accumulates in neurons in Parkinson’s disease, dementia with Lewy bodies, and many cases with AD (126, 127). In FTLD-tau transgenic mice, tau accumulation is also increased by injecting Aβ peptides into the brain (128) and by neuronal overexpression of APP bearing autosomal dominant mutations that cause early onset familial AD and elevate cerebral Aβ levels (129). By contrast, overexpression of tau does not increase Aβ accumulation in such models, as compared to singly transgenic APP mice (129). However, ablation or reduction of endogenous, non-aggregated WT tau prevents or diminishes Aβ toxicity in cultures of rodent primary neurons (130), as well as several (but not all) synaptic, network, and cognitive alterations in different lines of APP transgenic mice (22, 44, 51, 131, 132). These results raise the possibility that even physiological forms of tau can enable or promote disease processes under otherwise pathological circumstances, possibly by allowing other factors such as APP or some of its metabolites to alter signaling pathways that tau normally enhances or constrains (Fig. 2).
Tau also has bidirectional relations with apoE4. Compared with the more common apoE3 isoform, apoE4 increases AD risk, lowers age of disease onset, and promotes hyperphosphorylation of endogenous WT tau in induced pluripotent stem cell (iPSC)-derived human neurons and in transgenic mice (114, 133–135). Conversely, ablation of endogenous tau protects transgenic mice expressing human apoE4 fragments against age-dependent cognitive decline (136), which suggests a mutual interdependence between these pathogenic factors. Neurons of apoE4 carriers produce more apoE fragments than those of apoE3 carriers and these fragments impair mitochondria as well as the cytoskeleton (114, 133, 134). Compared with apoE2 and apoE3, apoE4 also exacerbates tau pathology and neurodegeneration in transgenic mice overexpressing FTLD-tau, whereas ablation of apoE or of microglia reduces these abnormalities (115, 137). Thus, apoE may be required for the development of these tau-dependent abnormalities, and microglia may have a key role in this copathogenic interaction.
Therapeutic approaches
Most tau-related therapeutic strategies that have advanced into clinical trials were designed to target select tau assemblies or posttranslational modifications presumed to be pathogenic, as judged from clinicopathological associations and, in some instances, additional evidence for causal contributions in preclinical models (11, 37, 138). Many of these trials were discontinued because of toxicity or lack of efficacy (37). As with other drug trials that have yielded negative results in AD or other neurodegenerative disorders, it is possible that they were initiated too late in the course of the disease, that target engagement was inadequate in the most relevant sites in the brain, or that the targeted form of tau does not actually contribute much to clinically meaningful functional impairments.
In light of these challenges, it is encouraging that the range and number of tau-targeting approaches have been steadily increasing after many anti-Aβ treatments failed in late-stage clinical trials for AD. More than 20 tau-related therapeutic strategies are undergoing clinical trials, including small-molecule inhibitors of tau acetylation, aggregation, O-GlcNAcylation (GlcNAc, N-acetylglucosamine), or phosphorylation, as well as active and passive immunizations against tau, and microtubule stabilizers (37).
Predicting the outcome of these trials is made difficult by the lack of conclusive answers to the following key questions. Are the most pathogenic forms of tau misfolded monomers, fragments, or multimers, and are they fibrillar or nonfibrillar? Do these pathogenic tau species act intracellularly or extracellularly? What are the roles of tau’s many posttranslational modifications and isoforms in health and disease? Does cell-to-cell spreading of tau impair neuronal survival or brain functions? What are the most impactful downstream mechanisms that medicate tau’s contributions to the development and progression of brain diseases? Could they be blocked without substantial side effects?
The importance of these questions is highlighted by the following hypothetical examples. If tau species that cause the greatest amount of neuronal dysfunction were produced by the affected neurons themselves and acted primarily within their presynaptic terminals or dendritic spines, tau-targeting antibodies that remain in the extracellular space and cannot reach these intracellular compartments would probably not be able to reduce or prevent functional deficits in clinical trials (139). In a similar vein, if the formation and propagation of tau aggregates in human tauopathies were merely coincidental with or a parallel consequence of an unidentified upstream process that is the main driver of neuronal degeneration and brain dysfunction, inhibiting tau aggregation or propagation might yield negative outcomes in clinical trials.
Among therapeutic strategies that are in preclinical development or undergoing clinical evaluation, those that could lower overall tau levels may, thus, have the greatest potential, as this approach bypasses the unresolved questions of which forms of tau and pathomechanisms are most critical in any given condition (Fig. 3). Preclinical proof-of-concept for the benefits of overall tau reduction comes from the suppression of regulatable tau-encoding transgenes, which diminished neural dysfunctions putatively caused by FTLD-tau (66) or A152T-variant tau (41), as well as from the genetic ablation or partial reduction of endogenous mouse tau, which reduced neural network dysfunctions and behavioral abnormalities in mouse models of AD, autism, depression, epilepsy, and stroke (18–22, 27, 44). Notably, even partial (~50%) reduction of tau was effective in several of these studies, and there is no evidence that this level of tau reduction impairs CNS functions in animal models.
Fig. 3. Tau-focused therapeutic strategies.

Approaches that target the upstream components (bold) of the depicted cascade may have the greatest therapeutic potential because the downstream components fan out into a variety of potential targets whose relative pathogenic impacts are uncertain.
Although gene editing approaches are rapidly advancing toward clinical applications, a more readily translatable approach to reducing overall tau levels in humans is the intrathecal injection (via lumbar puncture) of antisense oligonucleotides ASOs that promote the degradation of MAPT mRNAs. This approach, or intracerebroventricular injection of such ASOs, effectively lowers overall tau levels in brains of rodents and nonhuman primates and reduces chemically induced epilepsy in WT mice (27) as well as behavioral abnormalities and neurodegeneration in transgenic mice overexpressing FTLD-tau (45). Intrathecal injections of tau-targeting ASOs were well tolerated in nonhuman primates (45) and are undergoing a phase 1 safety assessment in people with early AD (ClinicalTrials.gov identifier NCT03186989). ASOs can also be used to modulate alternative splicing, a strategy that could help lower the abundance of the most pathogenic isoforms of tau (140), which may differ across tauopathies (1, 15).
An ASO that targets survival of motor neuron2, centromeric (SMN2) precursor mRNA (pre-mRNA) has shown substantial clinical benefits in spinal muscular atrophy (141), a neurodegenerative disease of childhood, and encouraging results seem to be emerging from a trial of an ASO that targets mutant huntingtin for Huntington’s disease (142). However, because ASOs are large, negatively charged molecules that are subject to degradation, bringing ASOs across the blood-brain barrier and maintaining efficacious levels in brain tissues require repeated lumbar punctures, which are suboptimal for the prolonged treatment of chronic brain diseases in older people. Thus, developing effective tau-lowering small-molecule drugs that can be given orally is an important objective. So far, this has been a difficult feat to accomplish, at least in part because decreases in tau levels are a sensitive and early indicator of neurotoxic off-target effects. Consequently, neurotoxic compounds can be false-positive confounders of screens for tau-lowering drugs (143).
In principle, overall tau reducers might act by directly or indirectly reducing the transcription of MAPT or the translation of MAPT mRNA or enhancing the degradation of MAPT (pre-)mRNA or tau protein (Fig. 3). Several types of drugs were selected or developed to reduce specific forms of tau but may also be able to lower overall tau levels. A relevant strategy at preclinical stages of development is the identification of small molecules that specifically target MAPT pre-mRNA (144).
In cultures of primary rodent neurons or iPSC-derived human neurons, tau levels can be reduced by treatment with statins at concentrations that are not toxic in vitro (145, 146). However, it is unknown whether high enough brain concentrations of these drugs could be achieved to adequately reduce tau levels in vivo and whether such concentrations could be tolerated.
Acetylation can compete with ubiquitination and thereby counteract tau degradation (147). Genetic ablation or pharmacological inhibition of the lysine acetyltransferases p300 and CREB-binding protein (CBP) reduces tau acetylation and total tau levels, as well as tau pathology, neurodegeneration and behavioral deficits in transgenic mice (12, 105). Salsalate, a non-acetylated dimer of salicylic acid used to treat arthritis, reduces tau acetylation in the brain (12) and is undergoing a phase 1 trial for mild to moderate AD (NCT03277573).
Enhancing autophagy by pharmacological or genetic manipulations reduced tau pathology and neuronal loss in zebrafish that express A152T-variant human tau (25). A recent study showed that tau degradation is regulated by the farnesylated protein Rhes, a guanosine triphosphate (GTPase) in the Ras family (148). Treatment with lonafarnib, an inhibitor of farnesyltransferase used in cancer treatment, reduced brain atrophy, tau pathology, neuroinflammation, and behavioral deficits in transgenic mice overexpressing FTLD-tau (148).
Another strategy to reduce tau levels involves the generation of proteolysis-targeting chimeras (PROTACs)—molecules linking a target recognition domain with a ubiquitin ligase that helps shuttle the target molecule into protein degradation pathways, particularly the proteasome (149). Such compounds also hold promise for the targeted degradation of tau and are undergoing preclinical evaluation (149, 150).
These strategies highlight an encouraging diversification of the tau-related drug development pipeline, which will hopefully yield better therapeutics for tauopathies and other disorders in which tau has a causal or enabling role. The most important needs (Fig. 4) in this exciting area of translational neurobiology include: (i) affordable anti-tau therapeutics that can be administered safely and easily for long periods of time to fragile populations who are middle-aged or older and likely to take other medications for aging-related comorbidities; (ii) drugs that effectively and safely reduce as many disease-promoting tau species as possible in the most vulnerable cell types and subcellular compartments; and (iii) biomarkers that reliably detect target engagement in the brain of live people and, ideally, correlate well with clinically meaningful functional outcomes.
Fig. 4. Tau-related challenges and needs.
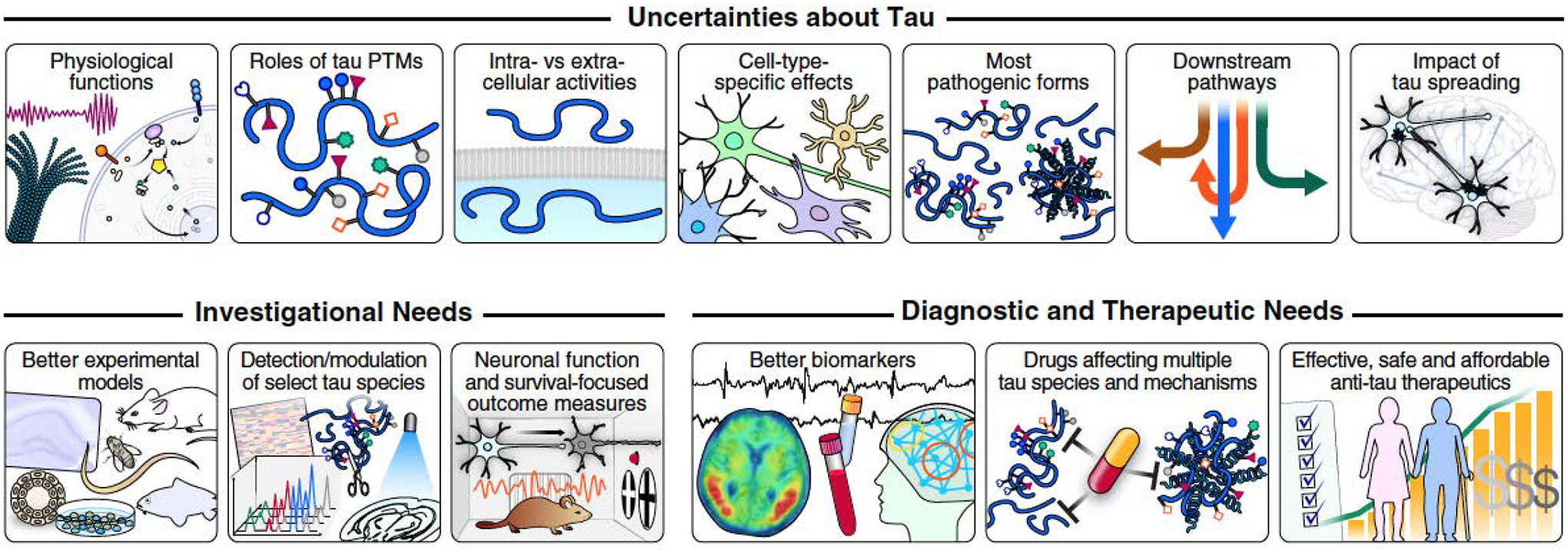
In principle, several of the trailblazing discoveries reviewed here could lead to the development of better treatments for AD and other tauopathies. However, many efforts to develop treatments for brain diseases have failed, in large part because of an inadequate understanding of the pathobiology of these complex disorders and, perhaps, because too many fundamental knowledge gaps, alternative interpretations of data, and methodological complexities did not receive the attention they deserved. Addressing the scientific knowledge gaps and technological challenges highlighted in this Review could accelerate and enable the development of truly impactful tau-related diagnostics and therapeutics. PTM, posttranslational modification.
Conclusion
The multifaceted protein tau raises fascinating scientific questions, and its importance from a public health perspective is increasing as the prevalence of tauopathies continues to surge around the world. Tau may fulfill different functions in different cell types, subcellular locations and circumstances, and may contribute to different diseases through diverse mechanisms. However, its most important functions and pathomechanisms remain to be defined. Adverse gains of function may turn tau into a direct mediator of neurotoxic effects, and even some of its physiological activities may allow disease processes to destabilize neural networks in ways that contribute to behavioral abnormalities and cognitive decline. Developing more effective treatments for tauopathies and other conditions involving tau will likely require a more definitive understanding of its pathobiology. Until this important goal has been reached, lowering overall tau levels would seem to be the most pragmatic and promising therapeutic approach. This strategy has shown beneficial effects in diverse disease models and a safety profile that has allowed related therapeutics to advance into clinical trials.
ACKNOWLEDGMENTS
We regret that many important papers with direct relevance to this topic could not be mentioned because of space constraints. We thank members of the Mucke Lab for helpful comments on the manuscript, S. Ordway for editorial review, J. Carroll for preparation of graphics, and R. Mott and J. Rosendorf for administrative assistance.
Funding:
Tau-related research in the Mucke Lab is supported by the National Institutes of Health (grants R01 MH115679, R56 AG060631 and UH3 AG054304), the Tau Consortium, and Cure Network Dolby Acceleration Partners. C.-W.C. is supported by a Research Fellowship from the Alzheimer’s Association.
Footnotes
Competing interests: L.M. is a co-inventor on patents held by the Gladstone Institutes that focus on tau reduction as a strategy to block neural network dysfunction. He has served on the scientific advisory boards of Arvinas, Biogen, and Dolby Family Ventures, and has provided consulting services to Sangamo Therapeutics.
REFERENCES
- 1.Wang Y, Mandelkow E, Tau in physiology and pathology. Nat. Rev. Neurosci 17, 5–21 (2016). doi: 10.1038/nrn.2015.1; [DOI] [PubMed] [Google Scholar]
- 2.Janning D et al. , Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons. Mol Biol Cell 25, 3541–3551 (2014). doi: 10.1091/mbc.e14-06-1099; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamada K et al. , Neuronal activity regulates extracellular tau in vivo. J Exp Med 211, 387–393 (2014). doi: 10.1084/jem.20131685; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ittner A, Ittner LM, Dendritic Tau in Alzheimer’s disease. Neuron 99, 13–27 (2018). doi: 10.1016/j.neuron.2018.06.003; [DOI] [PubMed] [Google Scholar]
- 5.Caillet-Boudin ML, Buee L, Sergeant N, Lefebvre B, Regulation of human MAPT gene expression. Mol Neurodegener 10, 1–14 (2015). doi: 10.1186/s13024-015-0025-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zempel H et al. , Axodendritic sorting and pathological missorting of Tau is isoform specific and determined by axon initial segment architecture. J. Biol. Chem 292, 12192–12207 (2017). doi: 10.1074/jbc.M117.784702; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu C, Götz J, Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS One 8, e84849 (2013). doi: 10.1371/journal.pone.0084849; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu C, Song X, Nisbet R, Götz J, Co-immunoprecipitation with Tau isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N Tau in disease. J. Biol. Chem 291, 8173–8188 (2016). doi: 10.1074/jbc.M115.641902; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morris M et al. , Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci 18, 1183–1189 (2015). doi: 10.1038/nn.4067; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mair W et al. , FLEXITau: Quantifying post-translational modifications of Tau protein in vitro and in human disease. Anal Chem 88, 3704–3714 (2016). doi: 10.1021/acs.analchem.5b04509; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Götz JJ, Götz J, “Experimental models of tauopathy - From mechanisms to therapies” in Tau Biology, Advances in Experimental Medicine and Biology, Takashima A, Wolozin B, Buee L, Eds. (Springer, 2019), vol. 1184, chap. 28, pp. 381–391. doi: 10.1007/978-981-32-9358-8_28 [DOI] [PubMed] [Google Scholar]
- 12.Tracy TE, Gan L, Tau-mediated synaptic and neuronal dysfunction in neurodegenerative disease. Curr Opin Neurobiol 51, 134–138 (2018). doi: 10.1016/j.conb.2018.04.027; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arendt T et al. , Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J. Neurosci 23, 6972–6981 (2003). doi: 10.1523/JNEUROSCI.23-18-06972.2003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faraco G et al. , Dietary salt promotes cognitive impairment through tau phosphorylation. Nature 574, 686–690 (2019). doi: 10.1038/s41586-019-1688-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goedert M, Spillantini MG, Ordered assembly of tau protein and neurodegeneration. Tau Biology, Advances in Experimental Medicine and Biology, vol 1184 1184, 3–21 (2019). doi: 10.1007/978-981-32-9358-8_1 [DOI] [PubMed] [Google Scholar]
- 16.Cowan CM, Mudher A, Are tau aggregates toxic or protective in tauopathies? Front Neurol 4, 1–13 (2013). doi: 10.3389/fneur.2013.00114; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao X et al. , Caspase-2 cleavage of tau reversibly impairs memory. Nat. Med 22, 1268–1276 (2016). doi: 10.1038/nm.4199; [DOI] [PubMed] [Google Scholar]
- 18.Tai C et al. , Tau reduction prevents key features of autism in mouse models. Neuron 106, 421–437 (2020). doi: 10.1016/j.neuron.2020.01.038; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sotiropoulos I et al. , Atypical, non-standard functions of the microtubule associated Tau protein. Acta. Neuropathol. Commun 5, 1–11 (2017). doi: 10.1186/s40478-017-0489-6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bi M et al. , Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat. Commun 8, 1–15 (2017). doi: 10.1038/s41467-017-00618-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gheyara AL et al. , Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol 76, 443–456 (2014). doi: 10.1002/ana.24230; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberson ED et al. , Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754 (2007). doi: 10.1126/science.1141736; [DOI] [PubMed] [Google Scholar]
- 23.Kunkle BW et al. , Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 51, 414–430 (2019). doi: 10.1038/s41588-019-0358-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jansen IE et al. , Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51, 404–413 (2019). doi: 10.1038/s41588-018-0311-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopez A et al. , A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain 140, 1128–1146 (2017). doi: 10.1093/brain/awx005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coppola G et al. , Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum. Mol. Genet 21, 3500–3512 (2012). doi: 10.1093/hmg/dds161; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devos SL et al. , Antisense reduction of tau in adult mice protects against seizures. J. Neurosci 33, 12887–12897 (2013). doi: 10.1523/JNEUROSCI.2107-13.2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trinczek B, Biernat J, Baumann K, Mandelkow EM, Mandelkow E, Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol Biol Cell 6, 1887–1902 (1995). doi: 10.1091/mbc.6.12.1887; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panda D, Goode BL, Feinstein SC, Wilson L, Kinetic stabilization of microtubule dynamics at steady state by tau and microtubule-binding domains of tau. Biochemistry 34, 11117–11127 (1995). doi: 10.1021/bi00035a017; [DOI] [PubMed] [Google Scholar]
- 30.Drubin DG, Kirschner MW, Tau protein function in living cells. J. Cell Biol 103, 2739–2746 (1986). doi: 10.1083/jcb.103.6.2739; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiang L et al. , Tau does not stabilize axonal microtubules but rather enables them to have long labile domains. Curr Biol 28, 1–9 (2018). doi: 10.1016/j.cub.2018.05.045; [DOI] [PubMed] [Google Scholar]
- 32.Harada A et al. , Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 369, 488–491 (1994). doi: 10.1038/369488a0; [DOI] [PubMed] [Google Scholar]
- 33.Ma QL et al. , Loss of MAP function leads to hippocampal synapse loss and deficits in the Morris Water Maze with aging. J. Neurosci 34, 7124–7136 (2014). doi: 10.1523/JNEUROSCI.3439-13.2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takei Y, Teng J, Harada A, Hirokawa N, Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J. Cell Biol 150, 989–1000 (2000). doi: 10.1083/jcb.150.5.989; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barten DM et al. , Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027. J Neurosci 32, 7137–7145 (2012). doi: 10.1523/JNEUROSCI.0188-12.2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang B et al. , The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci 32, 3601–3611 (2012). doi: 10.1523/JNEUROSCI.4922-11.2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.VandeVrede L, Boxer AL, Polydoro M, Targeting tau: Clinical trials and novel therapeutic approaches. Neurosci Lett 731, 1–13 (2020). doi: 10.1016/j.neulet.2020.134919; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vossel KA et al. , Tau reduction prevents Aβ-induced axonal transport deficits by blocking activation of GSK3β. J. Cell Biol 209, 419–433 (2015). doi: 10.1083/jcb.201407065; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vossel KA et al. , Tau reduction prevents Aβ-induced defects in axonal transport. Science 330, 198–198a (2010). doi: 10.1126/science.1194653; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan A, Kumar A, Peterhoff C, Duff K, Nixon R-A, Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J. Neurosci 28, 1682–1687 (2008). doi: 10.1523/JNEUROSCI.5242-07.2008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Das M et al. , Neuronal levels and sequence of tau modulate the power of brain rhythms. Neurobiol. Dis 117, 181–188 (2018). doi: 10.1016/j.nbd.2018.05.020; [DOI] [PubMed] [Google Scholar]
- 42.Morris M et al. , Age-appropriate cognition and subtle dopamine-independent motor deficits in aged tau knockout mice. Neurobiol. Aging 34, 1523–1529 (2013). doi: 10.1016/j.neurobiolaging.2012.12.003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ke YD et al. , Lessons from tau-deficient mice. Int J Alzheimers Dis 2012, 1–9 (2012). doi: 10.1155/2012/873270; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ittner LM et al. , Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397 (2010). doi: 10.1016/j.cell.2010.06.036; [DOI] [PubMed] [Google Scholar]
- 45.DeVos SL et al. , Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med 9, 1–14 (2017). doi: 10.1126/scitranslmed.aag0481; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marciniak E et al. , Tau deletion promotes brain insulin resistance. J. Exp. Med 214, 2257–2269 (2017). doi: 10.1084/jem.20161731; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leboucher A et al. , Brain insulin response and peripheral metabolic changes in a Tau transgenic mouse model. Neurobiol Dis 125, 14–22 (2019). doi: 10.1016/j.nbd.2019.01.008; [DOI] [PubMed] [Google Scholar]
- 48.Johnson DK, Wilkins CH, Morris JC, Accelerated weight loss may precede diagnosis in Alzheimer disease. Arch Neurol 63, 1312–1317 (2006). doi: 10.1001/archneur.63.9.1312; [DOI] [PubMed] [Google Scholar]
- 49.Abbondante S et al. , Genetic ablation of tau mitigates cognitive impairment induced by type 1 diabetes. Am J Pathol 184, 819–826 (2014). doi: 10.1016/j.ajpath.2013.11.021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahmed T et al. , Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol Aging 35, 2474–2478 (2014). doi: 10.1016/j.neurobiolaging.2014.05.005; [DOI] [PubMed] [Google Scholar]
- 51.Roberson ED et al. , Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci 31, 700–711 (2011). doi: 10.1523/JNEUROSCI.4152-10.2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimura T et al. , Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. B. Biol. Sci 369, 1–8 (2014). doi: 10.1098/rstb.2013.0144; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li S et al. , Soluble oligomers of amyloid β-protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801 (2009). doi: 10.1016/j.neuron.2009.05.012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cantero JL et al. , Role of tau protein on neocortical and hippocampal oscillatory patterns. Hippocampus 21, 827–834 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Cantero JL et al. , Tau protein role in sleep-wake cycle. J. Alzheimers Dis 21, 411–421 (2010). doi: 10.3233/JAD-2010-100285; [DOI] [PubMed] [Google Scholar]
- 56.Holth JK et al. , The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 363, 880–884 (2019). doi: 10.1126/science.aav2546; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peter-Derex L, Yammine P, Bastuji H, Croisile B, Sleep and Alzheimer’s disease. Sleep Med Rev 19, 29–38 (2015). doi: 10.1016/j.smrv.2014.03.007; [DOI] [PubMed] [Google Scholar]
- 58.Fuster-Matanzo A, Llorens-Martin M, Jurado-Arjona J, Avila J, Hernandez F, Tau protein and adult hippocampal neurogenesis. Front Neurosci 6, 1–6 (2012). doi: 10.3389/fnins.2012.00104; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sapir T, Frotscher M, Levy T, Mandelkow EM, Reiner O, Tau’s role in the developing brain: Implications for intellectual disability. Hum. Mol. Genet 21, 1681–1692 (2012). doi: 10.1093/hmg/ddr603; [DOI] [PubMed] [Google Scholar]
- 60.Lei P et al. , Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med 18, 291–295 (2012). doi: 10.1038/nm.2613; [DOI] [PubMed] [Google Scholar]
- 61.Li Z, Hall AM, Kelinske M, Roberson ED, Seizure resistance without parkinsonism in aged mice after tau reduction. Neurobiol. Aging 35, 2617–2624 (2014). doi: 10.1016/j.neurobiolaging.2014.05.001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zollino M et al. , Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype. Nat. Genet 44, 636–638 (2012). doi: 10.1038/ng.2257; [DOI] [PubMed] [Google Scholar]
- 63.Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K, Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A 91, 5562–5566 (1994). doi: 10.1073/pnas.91.12.5562; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.La Joie R et al. , Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci Transl Med 12, 1–12 (2020). doi: 10.1126/scitranslmed.aau5732; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DeVos SL et al. , Tau reduction in the presence of amyloid-beta prevents tau pathology and neuronal death in vivo. Brain 141, 2194–2212 (2018). doi: 10.1093/brain/awy117; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.SantaCruz K et al. , Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481 (2005). doi: 10.1126/science.1113694; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gupta RK et al. , Evidence for HIV-1 cure after CCR5Delta32/Delta32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: a case report. Lancet HIV 7, e340–e347 (2020). doi: 10.1016/S2352-3018(20)30069-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tai XY et al. , Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain 139, 2441–2455 (2016). doi: 10.1093/brain/aww187; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maeda S et al. , Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep 17, 530–551 (2016). doi: 10.15252/embr.201541438; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wheeler JM et al. , High copy wildtype human 1N4R tau expression promotes early pathological tauopathy accompanied by cognitive deficits without progressive neurofibrillary degeneration. Acta Neuropathol. Commun 3, 1–13 (2015). doi: 10.1186/s40478-015-0210-6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garcia-Cabrero AM et al. , Hyperexcitability and epileptic seizures in a model of frontotemporal dementia. Neurobiol. Dis 58, 200–208 (2013). doi: 10.1016/j.nbd.2013.06.005; [DOI] [PubMed] [Google Scholar]
- 72.Buzsaki G, Watson BO, Brain rhythms and neural syntax: implications for efficient coding of cognitive content and neuropsychiatric disease. Dialogues Clin Neurosci 14, 345–367 (2012). doi: 10.31887/DCNS.2012.14.4/gbuzsaki; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gouw AA et al. , EEG spectral analysis as a putative early prognostic biomarker in nondemented, amyloid positive subjects. Neurobiol Aging 57, 133–142 (2017). doi: 10.1016/j.neurobiolaging.2017.05.017; [DOI] [PubMed] [Google Scholar]
- 74.Verret L et al. , Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721 (2012). doi: 10.1016/j.cell.2012.02.046; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Iaccarino HF et al. , Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540, 230–235 (2016). doi: 10.1038/nature20587; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dujardin S et al. , Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat Med 26, 1256–1263 (2020). doi: 10.1038/s41591-020-0938-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaufman SK et al. , Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron 92, 796–812 (2016). doi: 10.1016/j.neuron.2016.09.055; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Scheres SH, Zhang W, Falcon B, Goedert M, Cryo-EM structures of tau filaments. Curr Opin Struct Biol 64, 17–25 (2020). doi: 10.1016/j.sbi.2020.05.011; [DOI] [PubMed] [Google Scholar]
- 79.Ittner A et al. , Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science 354, 904–908 (2016). doi: 10.1126/science.aah6205; [DOI] [PubMed] [Google Scholar]
- 80.Leuzy A et al. , Diagnostic performance of RO948 F 18 tau positron emission tomography in the differentiation of Alzheimer disease from other neurodegenerative disorders. JAMA Neurol 77, 955–965 (2020). doi: 10.1001/jamaneurol.2020.0989; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barthelemy NR et al. , A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat Med 26, 398–407 (2020). doi: 10.1038/s41591-020-0781-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Palmqvist S et al. , Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA Neurol 324, 772–781 (2020). doi: 10.1001/jama.2020.12134; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ashton NJ et al. , An update on blood-based biomarkers for non-Alzheimer neurodegenerative disorders. Nat Rev Neurol 16, 265–284 (2020). doi: 10.1038/s41582-020-0348-0; [DOI] [PubMed] [Google Scholar]
- 84.Apicco DJ et al. , Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat Neurosci 21, 72–80 (2018). doi: 10.1038/s41593-017-0022-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen XQ, Mobley WC, Alzheimer disease pathogenesis: insights from molecular and cellular biology studies of oligomeric abeta and tau species. Front Neurosci 13, 1–21 (2019). doi: 10.3389/fnins.2019.00659; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fa M et al. , Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci Rep 6, 1–15 (2016). doi: 10.1038/srep19393; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jankowsky JL, Zheng H, Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol Neurodegener 12, 1–22 (2017). doi: 10.1186/s13024-017-0231-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gamache J et al. , Factors other than hTau overexpression that contribute to tauopathy-like phenotype in rTg4510 mice. Nat Commun 10, 1–12 (2019). doi: 10.1038/s41467-019-10428-1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Goodwin LO et al. , Large-scale discovery of mouse transgenic integration sites reveals frequent structural variation and insertional mutagenesis. Genome Res 29, 494–505 (2019). doi: 10.1101/gr.233866.117; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sato C et al. , Tau kinetics in neurons and the human central nervous system. Neuron 97, 1284–1298 (2018). doi: 10.1016/j.neuron.2018.02.015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Beevers JE et al. , MAPT genetic variation and neuronal maturity alter isoform expression affecting axonal transport in iPSC-derived dopamine neurons. Stem Cell Reports 9, 587–599 (2017). doi: 10.1016/j.stemcr.2017.06.005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Iovino M et al. , Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 138, 3345–3359 (2015). doi: 10.1093/brain/awv222; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McInnes J et al. , Synaptogyrin-3 mediates presynaptic dysfunction induced by tau. Neuron 97, 823–835 e828 (2018). doi: 10.1016/j.neuron.2018.01.022; [DOI] [PubMed] [Google Scholar]
- 94.Derkach VA, Oh MC, Guire ES, Soderling TR, Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci 8, 101–113 (2007). doi: 10.1038/nrn2055; [DOI] [PubMed] [Google Scholar]
- 95.Yu A et al. , Tau protein aggregates inhibit the protein-folding and vesicular trafficking arms of the cellular proteostasis network. J Biol Chem 294, 7917–7930 (2019). doi: 10.1074/jbc.RA119.007527; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fu H et al. , A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat Neurosci 22, 47–56 (2019). doi: 10.1038/s41593-018-0298-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Caballero B et al. , Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 17, 1–17 (2018). doi: 10.1111/acel.12692; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hipp MS, Kasturi P, Hartl FU, The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20, 421–435 (2019). doi: 10.1038/s41580-019-0101-y; [DOI] [PubMed] [Google Scholar]
- 99.Myeku N et al. , Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med 22, 46–53 (2016). doi: 10.1038/nm.4011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sun W, Samimi H, Gamez M, Zare H, Frost B, Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat Neurosci 21, 1038–1048 (2018). doi: 10.1038/s41593-018-0194-1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Guo C et al. , Tau activates transposable elements in Alzheimer’s disease. Cell Rep 23, 2874–2880 (2018). doi: 10.1016/j.celrep.2018.05.004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shanbhag NM et al. , Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol Commun 7, 1–18 (2019). doi: 10.1186/s40478-019-0723-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tam OH, Ostrow LW, Gale Hammell M, Diseases of the nERVous system: retrotransposon activity in neurodegenerative disease. Mob DNA 10, 1–14 (2019). doi: 10.1186/s13100-019-0176-1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Min SW et al. , SIRT1 deacetylates tau and reduces pathogenic tau spread in a mouse model of tauopathy. J Neurosci 38, 3680–3688 (2018). doi: 10.1523/JNEUROSCI.2369-17.2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen X et al. , Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol Neurodegener 15, 1–19 (2020). doi: 10.1186/s13024-019-0354-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hatch RJ, Wei Y, Xia D, Götz J, Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta. Neuropathol 133, 717–730 (2017). doi: 10.1007/s00401-017-1674-1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sohn PD et al. , Pathogenic tau impairs axon initial segment plasticity and excitability homeostasis. Neuron 104, 458–470.e455 (2019). doi: 10.1016/j.neuron.2019.08.008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tracy TE et al. , Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron 90, 245–260 (2016). doi: 10.1016/j.neuron.2016.03.005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vogels T, Murgoci AN, Hromadka T, Intersection of pathological tau and microglia at the synapse. Acta Neuropathol Commun 7, 1–25 (2019). doi: 10.1186/s40478-019-0754-y; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brelstaff J, Tolkovsky AM, Ghetti B, Goedert M, Spillantini MG, Living neurons with tau filaments aberrantly expose phosphatidylserine and are phagocytosed by microglia. Cell Rep 24, 1939–1948 (2018). doi: 10.1016/j.celrep.2018.07.072; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Litvinchuk A et al. , Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer’s disease. Neuron 100, 1337–1353 (2018). doi: 10.1016/j.neuron.2018.10.031; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ising C et al. , NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673 (2019). doi: 10.1038/s41586-019-1769-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ghosh S et al. , Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci 33, 5053–5064 (2013). doi: 10.1523/JNEUROSCI.4361-12.2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Huang Y, Mucke L, Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222 (2012). doi: 10.1016/j.cell.2012.02.040; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shi Y et al. , Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med 216, 2546–2561 (2019). doi: 10.1084/jem.20190980; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rauch JN et al. , LRP1 is a master regulator of tau uptake and spread. Nature 580, 381–385 (2020). doi: 10.1038/s41586-020-2156-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gibbons GS, Lee VMY, Trojanowski JQ, Mechanisms of cell-to-cell transmission of pathological tau: A review. JAMA Neurol 76, 101–108 (2019). doi: 10.1001/jamaneurol.2018.2505; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP, Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep 14, 389–394 (2013). doi: 10.1038/embor.2013.15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wu JW et al. , Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19, 1085–1092 (2016). doi: 10.1038/nn.4328; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ahmed Z et al. , A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol 127, 667–683 (2014). doi: 10.1007/s00401-014-1254-6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Iba M et al. , Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 33, 1024–1037 (2013). doi: 10.1523/JNEUROSCI.2642-12.2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Clavaguera F et al. , Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11, 909–913 (2009). doi: 10.1038/ncb1901; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hallinan GI, Vargas-Caballero M, West J, Deinhardt K, Tau misfolding efficiently propagates between individual intact hippocampal neurons. J Neurosci 39, 9623–9632 (2019). doi: 10.1523/JNEUROSCI.1590-19.2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bright J et al. , Human secreted tau increases amyloid-beta (Aβ) production. Neurobiol Aging 36, 693–709 (2015). doi: 10.1016/j.neurobiolaging.2014.09.007; [DOI] [PubMed] [Google Scholar]
- 125.Moussaud S et al. , Alpha-synuclein and tau: teammates in neurodegeneration? Mol Neurodegener 9, 1–14 (2014). doi: 10.1186/1750-1326-9-43; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kotzbauer PT, Trojanowski JQ, Lee VMY, Lewy body pathology in Alzheimer’s disease. J.Mol.Neurosci 17, 225–232 (2001). doi: 10.1385/JMN:17:2:225; [DOI] [PubMed] [Google Scholar]
- 127.Outeiro TF et al. , Dementia with Lewy bodies: an update and outlook. Mol Neurodegener 14, 1–18 (2019). doi: 10.1186/s13024-019-0306-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Götz J, Chen F, van Dorpe J, Nitsch RM, Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science 293, 1491–1495 (2001). doi: 10.1126/science.1062097; [DOI] [PubMed] [Google Scholar]
- 129.Lewis J et al. , Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491 (2001). doi: 10.1126/science.1058189; [DOI] [PubMed] [Google Scholar]
- 130.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A, Tau is essential to β-amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 99, 6364–6369 (2002). doi: 10.1073/pnas.092136199; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Puzzo D et al. , Tau is not necessary for amyloid-beta-induced synaptic and memory impairments. J Clin Invest 130, 4831–4844 (2020). doi: 10.1172/JCI137040; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hall AM et al. , Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer’s disease. J. Neurosci 35, 6221–6230 (2015). doi: 10.1523/JNEUROSCI.2552-14.2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang C et al. , Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med 24, 647–657 (2018). doi: 10.1038/s41591-018-0004-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Brecht WJ et al. , Neuron-specific apolipoprotein E4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci 24, 2527–2534 (2004). doi: 10.1523/JNEUROSCI.4315-03.2004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tesseur I et al. , Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am. J. Pathol 156, 951–964 (2000). doi: 10.1016/S0002-9440(10)64963-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Andrews-Zwilling Y et al. , Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci 30, 13707–13717 (2010). doi: 10.1523/JNEUROSCI.4040-10.2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shi Y et al. , ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527 (2017). doi: 10.1038/nature24016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jadhav S et al. , A walk through tau therapeutic strategies. Acta Neuropathol Commun 7, 1–31 (2019). doi: 10.1186/s40478-019-0664-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Congdon EE, Sigurdsson EM, Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 14, 399–415 (2018). doi: 10.1038/s41582-018-0013-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schoch KM, Miller TM, Antisense oligonucleotides: Translation from mouse models to human neurodegenerative diseases. Neuron 94, 1056–1070 (2017). doi: 10.1016/j.neuron.2017.04.010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mercuri E et al. , Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med 378, 625–635 (2018). doi: 10.1056/NEJMoa1710504; [DOI] [PubMed] [Google Scholar]
- 142.Tabrizi SJ et al. , Targeting huntingtin expression in patients with Huntington’s disease. N Engl J Med 380, 2307–2316 (2019). doi: 10.1056/NEJMoa1900907; [DOI] [PubMed] [Google Scholar]
- 143.Dehdashti SJ et al. , A high-throughput screening assay for determining cellular levels of total tau protein. Curr Alzheimer Res 10, 679–687 (2013). doi: 10.2174/15672050113109990143; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chen JL et al. , Design, optimization, and study of small molecules that target tau pre-mRNA and affect splicing. J Am Chem Soc 142, 8706–8727 (2020). doi: 10.1021/jacs.0c00768; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.van der Kant R et al. , Cholesterol metabolism is a druggable axis that independently regulates tau and amyloid-beta in iPSC-derived Alzheimer’s disease neurons. Cell Stem Cell 24, 363–375 e369 (2019). doi: 10.1016/j.stem.2018.12.013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hamano T, Yen SH, Gendron T, Ko LW, Kuriyama M, Pitavastatin decreases tau levels via the inactivation of Rho/ROCK. Neurobiol. Aging 33, 2306–2320 (2012). doi: 10.1016/j.neurobiolaging.2011.10.020; [DOI] [PubMed] [Google Scholar]
- 147.Min SW et al. , Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966 (2010). doi: 10.1016/j.neuron.2010.08.044; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hernandez I et al. , A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci Transl Med 11, 1–17 (2019). doi: 10.1126/scitranslmed.aat3005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Burslem GM, Crews CM, Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 181, 102–114 (2020). doi: 10.1016/j.cell.2019.11.031; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Silva MC et al. , Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 8, 1–31 (2019). doi: 10.7554/eLife.45457; [DOI] [PMC free article] [PubMed] [Google Scholar]