

Abstract
Multidrug resistant (MDR) bacteria is a global threat with many common infections becoming increasingly difficult to eliminate. While significant effort has gone into the development of potent biocides, the effectiveness of many first-line antibiotics has been diminished due to adaptive resistance mechanisms. Bacterial membrane proteins belonging to the resistance-nodulation-cell division (RND) superfamily play significant roles in mediating bacterial resistance to antimicrobials. They participate in multidrug efflux and cell wall biogenesis to transform bacterial pathogens into “superbugs” that are resistant even to last resort antibiotics. In this review, we summarize the RND superfamily of efflux transporters, with a primary focus on the assembly and function of the inner membrane pumps. These pumps are critical for extrusion of antibiotics from the cell as well as the transport of lipid moieties to the outer membrane to establish membrane rigidity and stability. We analyze recently solved structures of bacterial inner membrane efflux pumps as to how they bind and transport their substrates. Our cumulative data indicate that these RND membrane proteins area able to utilize different oligomerization states to achieve particular activities, including forming MDR pumps and cell wall remodeling machineries, to ensure bacterial survival. This mechanistic insight, combined with simulated docking techniques, allows for the design and optimization of new efflux pump inhibitors to more effectively treat infections that today are difficult or impossible to cure.
Graphical Abstract

1. Introduction
According to the World Health Organization, antibiotic resistance is one of the biggest threats to global health, food security and development today.1 The Center of Disease Control reports that there are more than 2.8 million new cases of antibiotic resistant infections each year in the United States alone2 with significantly more worldwide. While resistance mechanisms are a natural phenomenon in bacteria,3 there is an accelerated timeline for resistance due to the misuse of antibiotics in both humans and animals. In order to combat this, current guidelines suggest to minimize exposure to these compounds, take only when necessary, and only for the recommended duration. Newer data suggests that for many infections, shorter courses of treatments can be just as effective as following the older guidelines, thereby reducing the exposure of these bacteria to antibiotics thus slowing down the speed in which they develop resistance.1 Unfortunately, in many countries, distribution of these compounds is unregulated, and thus their usage is widely abused. In addition, as of today, there is extensive use of antibiotics and biocides in plants and animals for human consumption, further exacerbating the resistance problem. As a result, many of the first-line antibiotics routinely used to treat bacterial infections have been rendered ineffective. Common infections, such as pneumonia, tuberculosis, gonorrhea and chlamydia are not as easily treated, as multidrug resistant (MDR) strains of bacteria that cause these infections have emerged. This forces the use of more expensive medicines, longer treatment durations, higher mortality rates and an overall increase in healthcare costs. While it is encouraging that the number of reported deaths caused by antibiotic-resistant infections has significantly decreased since 2013,2 continued improvement in prevention and treatment strategies are still necessary. A small number of new antibiotics are in development but none of them are expected to be effective against the most dangerous forms of antibiotic-resistant bacteria. Therefore, it is imperative that we continue to invest in research and development of new antibiotics, vaccines, diagnostics and other tools.1
2. How do bacteria confer resistance?
Resistance mechanisms are widespread in bacteria.4,5 Some mechanisms are innate to the bacterium while many are acquired over time. These can be either permanent attributes of the cell or reversible when the harmful stimulus is removed. Favorable mutations within the bacterial genome can occur when exposed to sub lethal amounts of antibiotic. These mutations, as well as mobile genetic elements, such as plasmids, transposons and phages that can transfer genetic material from one strain to another, greatly increase the rate of acquired resistance. These genetic alterations allow the bacterium to resist the threat of antibiotics in a variety of ways. The simplest way is through changes in the lipid composition of the outer membrane to create an impermeable barrier.6,7 In many Gram-negative bacteria, the composition of lipopolysaccharide (LPS) on the outer leaflet and glycerophospholipids on the inner leaflet can be modified to effectively block the penetration of harmful agents.8,9 The reduced expression of nonspecific diffusion channels (porins) expressed when exposed to antibiotics also aids in the ability of the bacterium to confer resistance.10,11 The permeability properties of Gram-positive bacteria are due to, in part, the glycan chain length and cross linking properties of its peptidoglycan layer,12 although most compounds are able to traverse this layer unhindered and other resistance mechanisms are more effective. In the case of organisms belonging to the Corynebacteria–Mycobacteria–Nocardia group, however, an outer membrane that consists of lipid, such as trehalose dimycolate (TDM), covalently linked to the arabinogalactan layer significantly hinders biocides from entering the cell.13–15
Additional methods bacteria use to combat antibiotics include: antibiotic inactivation – either by degradation (several strains of bacteria synthesize beta-lactamase, an enzyme that breaks down beta-lactam based antibiotics, such as penicillin. Klebsiella pneumonia synthesizes carbapenemases to break down carbapenem drugs and most beta-lactam compounds) or modification (such as acetylation, phosphorylation and adenylation, all of which decrease the affinity of the antibiotic toward its intended cellular receptor; resistance mutations (in the presence of quinolones, many strains of Escherichia coli acquire mutations in the quinolone resistance-determining regions.6,16 which allows continued gyrase function and DNA duplication) Other mutations affect resistance by alteration of the antimicrobial target receptor, upregulation of the expression of important efflux systems17 and the modulation of important regulatory networks18; specific target modification Lipid A is the lipid component of lipopolysaccharide (LPS), the major component of the outer leaflet of the outer membrane of Gram-negative bacteria. LPS is immunogenic and is recognized by the host to elicit an immune response. To counteract this, several different mechanisms have evolved to circumvent this response, with the most common being neutralization of the negative charge of lipid A. The mobilized colistin resistance gene mcr-1 has been found in several strains of bacteria, including: E. coli, Salmonella enterica, K. pneumoniae, Enterobacter aerogenes, and Enterobacter cloacae). The mcr-1 enzyme transfers a phosphoethanolamine to the lipid A present on the outer membrane. This results in alteration of the lipid A topology and, as a result, reduces the affinity of several classes of drugs including colistin and polymyxins.19,20 In a similar fashion, ethanolamine phosphotransferase A, EptA, and 4-amino-4-deoxy-L-arabinose transferase, ArnT mediate the modification of Lipid A by the addition of phosphoethanolamine and 4-amino-4-deoxy-L-arabinose, respectively21. These modifications result in a decrease of the negatively charged surface area of the cell membrane which inhibits binding of polymyxin B22); development of new processes to avoid antibiotic targets (some Staphylococcus aureus bacteria can bypass the drug effects of trimethoprim)2, and the extrusion of antibiotic through efflux transporters. Efflux transporters can either be dedicated systems designed to extrude a single class of drugs, such as the Tet(A) and Tet(B) which recognize tetracycline and its derivatives, or multidrug efflux pumps that can transport several classes of unrelated compounds.
3. Efflux transporter classifications
Efflux transporters can either be primary transporters or secondary transporters. Primary transporters use the energy generated from ATP hydrolysis to pump their substrates out from the cells. This is in contrast to secondary transporters that utilize the biological energy from ion gradients to transport their substrates across the membrane. Based on the classification system of the Saier group23–25 efflux transporters can be further classified into distinct families: 1) The ATP-binding cassette superfamily (ABC); 2) the major facilitator superfamily (MFS)26; 3) the multidrug and toxic compound extrusion family (MATE)27,28; 4) the small multidrug resistance family (SMR)29,30; 5) the resistance-nodulation-cell division family (RND)31; 6) the p-aminobenzoyl-glutamate transporter (AbgT) family32–34; and 7) the recently described proteobacterial antimicrobial compound efflux (PACE) family.35 Each family shares common attributes and yet has unique features that define their classifications.
3.1. The ABC superfamily.
The ABC superfamily is the only family of the seven listed to be classified as primary transporters. ABC transporters classically have two domains, a highly conserved ATP-hydrolyzing domain and a predominantly transmembrane helical domain that is unique to the ABC transport family. These transporters use the energy created by ATP hydrolysis to transport their substrates across their cell membranes. Known substrates include hemolysins, macrolides,36 fluoroquinolones37 and fusaric acid,38 among many others. This family of transporters has been extensively studied and reviewed in-depth elsewhere.39–41
3.2. The MFS superfamily.
The MFS superfamily of transporters can be classified into symporters (substrate import along with a coupling ion), antiporters (substrate and co-substrate in opposite directions) and uniporters (single substrate transport),42 although all three types are thought to go through a similar conformational change upon substrate transport. They share a common 12 transmembrane helical architecture with known deviations of up to 16 transmembrane segments, N- and C-bundle domains, and a twofold pseudo-symmetry.43 In bacteria, their substrate specificity is determined by the aqueous pocket at the center of the membrane. For symporters, substrates include lactose and fucose during normal cellular function and for antiporters, compounds such as tetracycline44 and bile salts are recognized during cellular challenge.45–47 MFS transporters are also important for biofilm formation.48–50 Common substrates for this superfamily include tetracycline, norfloxacin, trimethoprim, acriflavine, and ethidium bromide.51–53
3.3. The MATE superfamily.
The MATE family of transporters are involved in the extrusion of cationic and lipophilic compounds from the cell. Structurally, they are similar to other transporter families with 12 transmembrane helices and a pseudo two-fold symmetry. Substrates bind via a monovalent cationic binding site with a proposed mechanism of transport similar to that of the MFS superfamily with either H+ or Na+-coupled antiport as their primary energy source.54 The MATE transporters appear to have a broad substrate profile. Examples of MATE substrates include tigecycline55 and fluoroquinolone-based antimicrobials such as norfloxacin and ciprofloxacin.56
3.4. The SMR superfamily.
The SMR family is composed of small proteins (100–140 amino acids), each with four transmembrane helices. These can function as either homo or heterodimeric proteins in the native membrane-embedded state. They confer resistance to quaternary ammonium compounds (QAC) such as tetraphenylphosphonium (TPP) and methyl viologen in addition to a variety of antibiotics, antiseptics and detergents (30 and references therein). SMR substrate export is driven by the energy derived from the electrochemical gradient of the cell.
3.5. The AbgT superfamily.
Even though over 13,000 putative transporters of the AbgT family have been identified, very little structural or functional information exists. The first two structures, YdaH from Alcanivorax borkumensis, and MtrF from Neisseria gonorrhoeae were recently solved.33,34,57,58 Both YdaH and MtrF assemble as dimers and share nearly identical overall topology. Each protomer contains nine transmembrane helices and two helical hairpins with small periplasmic and cytoplasmic domains, with the overall structure organized in a two-fold pseudosymmetry. This inward-open conformation displays a concave aqueous basin that penetrates the inner leaflet of the cytoplasmic membrane. While its canonical role within the cell is to export p-aminobenzoic acid, it also acts as an antibiotic efflux transporter, with sulfonamide drugs as a known substrate.57 As is common with other efflux systems, the electrochemical gradient of the cell is the primary energy source used to power substrate transport.
3.6. The PACE superfamily.
Recently classified, very little structural information is known about the PACE family of transporters. It is predicted to have four transmembrane segments that span the inner membrane of Gram-negative bacteria with both the N- and C-termini located within the cytoplasm.35 Studies have shown that these PACE transporters can confer resistance to various biocides and pesticides, such as chlorhexidine, acriflavine and benzalkonium.59 Acinetobacter baumannii AceI, the seminal member of this superfamily, was shown to accumulate chlorhexidine in the presence of the protonophore carbonyl cyanide m-chlorophenylhydrazone (CCCP), suggesting that the proton motive force is required for substrate transport within this superfamily.60
3.7. The RND superfamily.
Among various types of efflux transporters, members of the resistance-nodulation-cell division (RND) superfamily are the most powerful in mediating antibiotic resistance in Gram-negative bacteria.61 It is known that many of these RND efflux transporters function as trimers. However, our recent studies indicate that these transporters can also oligomerize as dimers or monomers. Owing to the significant functions of these RND transporters with their intriguing assemblies and action mechanisms, this review will focus on the RND family, with a detailed analysis of the structure, function and substrate/antibiotic binding within the integral membrane RND efflux pump.
4. RND transporters
The RND superfamily of transporters were discovered almost 30 years ago with the identification of MDR pumps in E. coli and Pseudomonas aeruginosa.62–64 Since then, thousands of this class of transporters have been identified in many different organisms. Current phylogenetic classification lists nine separate but related branches (Figure 1) (in-depth information on transporter superfamilies can be found in the Transporter Classification Database23,25,65 and review66).
Figure 1.

Phylogenetic Tree of RND transporters. Simplified diagram based on the SuperfamilyTree2program that analyzes proteins and generates a tree showing phylogenetic positions of members within the superfamily.298 Black boxes show the RND pumps that are highlighted in this review. AAPE – aryl-polyene pigments, EST – eukaryotic sterol transporter, HAE - hydrophobe/amphiphile efflux, HME, heavy metal efflux, NFE – nodulation factor exporter.
In bacteria, the majority of RND transporters can be classified into one of two subfamilies based upon their substrates, 1) the hydrophobe/amphiphile efflux (HAE) and the heavy metal efflux (HME) families. In most cases these transporters function as a tripartite system: 1) the RND inner membrane pump (IMP) that resides in the inner membrane that grabs the substrate from the cell, 2) a periplasmic membrane fusion protein (MFP) that aids in RND transporter assembly and/or substrate transfer to the 3) outer membrane channel protein (OMP) that is located in the outer membrane. The OMP then shuttles the substrate outside of the cell. This method of substrate extrusion is almost exclusively driven by the proton motive force as their prime source of energy.67 The ability of these transport systems to extrude the substrate directly into the extracellular space makes it an extremely important process for cell viability and antibiotic resistance, and thus an attractive drug target. The ability of RND transporters to remove toxic compounds, coupled with outer membrane impermeability, creates a formidable challenge to introduce bactericides into the cell. This is in contrast to other transport systems that only transfer their substrates into the periplasm where many times it is not fully released into the extracellular space. The RND efflux systems, however, extrude toxic compounds out of the cell, bypassing the periplasm and increasing the overall efficacy of the cellular efflux system.68,69
4.1. RND transport assembly.
The canonical assembly of the RND transport system is perhaps best understood through structural and biochemical information obtained from the E. coli AcrAB-TolC transporter. This type of tripartite efflux system has been identified as the most critical HAE transporters for the extrusion of toxic compounds.70 They consist of an IMP, an MFP and an OMP (Figure 2) with a stoichiometry of 3:6:3, although alternate stoichiometries have been reported.71–73 While the main function of RND transporters is to use the proton motive force as the energy source to shuttle cellular components, antibiotics and other toxic agents to the outer membrane, the structural determinants by which they achieve this are varied. This is, in large part, due to different specificities of the RND inner membrane pumps. Therefore, a detailed understanding of these pumps is necessary to aid in the design of new inhibitors to effectively block the transport of antibiotics from the cell.
Figure 2.
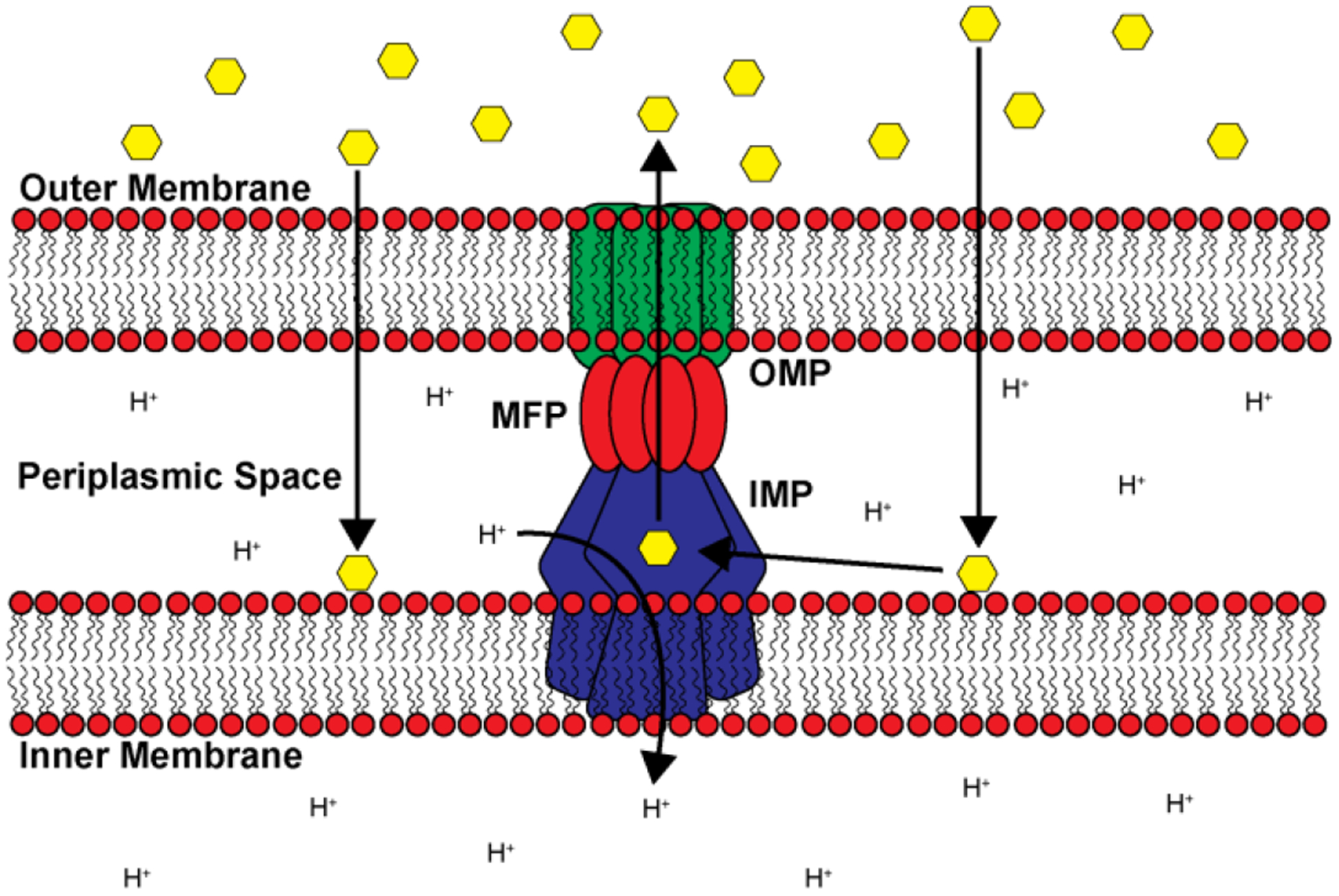
RND transport assembly. Cartoon representation depicting a trimeric RND transport system that spans both the inner and outer membranes of Gram-negative bacteria. Substrates such as antibiotics and other harmful biocides (yellow hexagons) that have entered the cell are shuttled from the periplasmic space to the exterior through a channel created by the coordinated assembly between the IMP (blue), MFP (red) and OMP (green) subunits.
4.2. Inner membrane RND pump characteristics.
Most RND pumps are known to contain 12 transmembrane helices (TM) with large periplasmic domains between TM1-TM2 and TM7-TM8. These large periplasmic domains, perhaps the most conserved feature of RND inner membrane pumps, are flexible in nature and are important for substrate recognition, translocation and delivery to the MFP and OMP for extrusion. These domains, along with the periplasmic adaptor proteins, are also important for association with the OMP. RND pumps traditionally associate as a trimer, with the three protomers aligning to form a central funnel where substrates can, upon binding one of the multiple binding pockets, be shuttled through until it is transferred to the OMP and released. This is accompanied by a concurrent shuttling of a proton from the periplasm to the cell interior (Figure 2) in a 1:1 ratio. While most RND pumps assemble as a trimeric complex, there are several examples where alternative configurations have been discovered.
Recently, a new subfamily of the RND superfamily of transporters, termed hopanoid biosynthesis-associated RND (HpnN) transporters74 has been characterized. HpnN is involved in the shuttling of hopanoids from the cytoplasm to the outer membrane of Gram-negative bacteria for cell wall remodeling.75 This, in turn, is critical for mediating multidrug resistance. It is unique because structural data of HpnN from Albican Burkholderia supports it as assembling as a dimer without the association of periplasmic adaptor proteins. It shuttles hopanoids to the outer membrane through a rigid body swinging motion, where the flexible periplasmic domains open and close the tunnel within each subunit of the transporter.76
In 2019, the crystal structure of mycobacterium membrane protein Large 3 (MmpL3) was solved.77,78 MmpL3 is an essential transporter in mycobacteria, most notably mycobacterium tuberculosis. Its main function is to shuttle trehalose monomycolate (TMM) from the outer leaflet of the cytoplasmic membrane into the periplasmic space79 where it is either converted into trehalose dimycolate (TDM) or attached to arabinogalactan.80 This results in a waxy outer layer difficult for many of the current antibiotics to penetrate. Inactivation or knockout of MmpL3 renders the mycobacterium unviable. Therefore, MmpL3 is an attractive inhibitory target. It is unique from the other RND transporters in that it is thought to perform its transport function as a monomer although alternative assembly configurations have been suggested.81
Therefore, while structural similarities exist between all RND pumps, their stoichiometries and mechanisms of substrate transport varies greatly. This implies that the design of efflux pump inhibitors must be specifically tailored to each individual structure. In-depth knowledge of the critical determinants for substrate recognition and mechanism of transport for each type will greatly aid in the intelligent design of future inhibitors.
5. RND pumps with trimeric architecture
The trimeric RND efflux systems are found in many species of bacteria. While they are important for the removal of a diverse array of toxic metabolites and other compounds detrimental to cells, those found in Gram-negative bacteria are of special interest. In most cases, bacterial efflux systems contribute to resistance for a broad spectrum of substrates such as toxic compounds, antibiotics, detergents, antimicrobials and metal ions.82,83 A phylogenetic analysis of the bacterial genome divides the trimeric RND transporters into either the hydrophobe/amphiphile efflux (HAE) class or the heavy metal efflux (HME) class (Figure 1).84 The dynamic expression of these HAE-RND efflux proteins play a significant role in the intrinsic and acquired resistance of antibiotics and toxic compounds as they are essential for transporting drugs directly into the extracellular space.85,86 Generally, HAE-RND efflux pumps have flexible, considerably hydrophobic binding pockets that selectively and actively transport a broad range of structurally unrelated toxic compounds out of the bacterial cell, such as antibacterial peptides, hormones, long-chain fatty acids and many important last resort antibiotics.87 The larger, bulky ligands are often stabilized by additional interactions with polar or charged residues in combination with the hydrophobic interactions that are also involved in binding nonpolar ligands within the binding cleft. In contrast to the broad-spectrum HAE-RND efflux pumps, the HME-RND efflux pumps possess more selective mechanisms of recognizing and binding substrates. Substrate recognition of these transmembrane pumps depends heavily on the residues of the binding site that contain asparagine, aspartic acid, glutamic acid and methionine to form channels throughout the components of the efflux system.88 The binding sites of the HME-RND efflux pumps possess unique coordination geometries to specifically recognize and transport monovalent and divalent metal ion out of the cell.89
A prototypical trimeric HAE class of RND efflux system is shown in Figure 3(a–d). The IMP:MFP:OMP proteins form the tripartite complex in the ratio of 3:6:3 to create the periplasmic-spanning RND efflux system and this oligomerization is necessary for the transport of substrates.90,91 IMPs are functional as trimers within the inner membrane of the bacterium and typically contain 12 transmembrane segments (TM1-TM12). Two large loops of each protomer protrude into the periplasmic space to form a large periplasmic domain that is further separated into six subdomains.91 The MFPs form a hexamer of flexible elongated adaptor proteins that connect and interact with both IMPs and OMPs in the periplasmic space to form a channel to transport substrate (Figure 3a). The flexible MFP contains an α-hairpin domain, a lipoyl domain, a β-barrel domain and a membrane proximal domain, where the α-hairpin domain contacts the OMP and the β-barrel and membrane proximal domains interact with the IMP to assemble the complete tripartite efflux complex. The occurrence of the MFP assembling into a hexamer within the RND efflux systems is required for the extrusion of substrates across the periplasm.92 Extrusion of the substrate across both membranes requires the association of the MFP with the IMP and OMP to form the three component RND efflux system.
Figure 3.

General structure of trimeric RND efflux systems. (a) The assembled components of a tripartite efflux system (adapted from PDB ID 5O66) visualized in side view with the inner membrane pump IMP (dark blue), membrane fusion protein MFP (dark red) and outer membrane protein OMP (dark green). Subunits are color-coded by slight variations of the indicated color accordingly. (b) Top view of trimeric OMP visualized from above the outer membrane suface (green). (c) Top view through the MFP hexamer within the periplasm (red). (d) The trimeric IMP as viewed from the periplasmic inner membrane surface (blue).
The OMPs are embedded in the outer membrane and function as a channel comprised of three protomers.93 Each OMP protomer contains three structurally significant domains: a transmembrane β-barrel domain, the periplasmic extending α-helical domain and an equatorial domain.
5.1. Conformational states of the trimeric efflux IMP.
Within the hydrophobe-amphiphile efflux (HAE) subfamily, these RND proteins largely form trimeric efflux pumps as evidenced by the first crystal structure of the E. coli AcrB pump.94 It was later found that the IMP of this system, AcrB, is capable of forming an asymmetric trimer, where the three protomers are unique and take different conformational states. These three conformations correspond to the “access” (L), “binding” (T) and “extrusion” (O) states.95–97 Because of these asymmetric crystal structures, the drug efflux process of HAE-RND pumps has been proposed to entail a synchronized motion between subunits of the trimer to advance the transport cycle from “access” to “binding” and then “extrusion” states to export drug molecules. However, a recent study of the Campylobacter jejuni CmeB HAE-RND-type multidrug efflux pump using x-ray crystallography and single-molecule fluorescence energy transfer (sm-FRET) indicated that individual protomers of the CmeB trimer can have various conformational states, not necessarily following the “access”, “binding” and “extrusion” pattern of the conformation, which led to a postulation that individual protomers of these trimeric RND pumps could bind and export substrates independently instead of operating in a synchronized fashion. A direct observation of the CmeB transport dynamics of the CmeB trimer embedded in membrane vesicles via sm-FRET depicted that each CmeB subunit undergoes conformational transitions uncoordinated and independent of each other and that there are at least four different transient states within individual protomers during the transport cycle. Based on these new findings, a proposed model for transport mechanism where the three protomers within the trimer function independently and uncoordinated was developed.98 Nonetheless, the study suggests that more efforts may be needed to elucidate the detailed mechanism of these fascinating efflux systems.
In the active export of substrate, an IMP protomer can allosterically shift between three distinct active transport conformations: the access state, the binding state and the extrusion state within each of the three protomers.90,99 These allosteric changes are transmitted through the MFP adaptor proteins to open the periplasmic channel and subsequently to the OMP to extrude the substrate through the outer membrane. After substrate export, the transporter returns to its resting state with the OMP again in its closed conformation. The completely assembled tripartite efflux system crosses the inner membrane, periplasm and outer membrane of Gram-negative bacteria to export substrates from the cytoplasm and periplasm out of the cell.100
5.2. Substrate transport energetics.
Typically, the trimeric RND efflux systems undergo a stepwise mechanism of substrate extrusion fueled by the proton-motive force.101,102 The transfer of these protons from the periplasm to the cytoplasm is carried out by a relay of conserved protonated residues within the transmembrane domain of the IMP.103,104 For example, the proton-relay network of the E. coli IMP, AcrB, allows for the transfer of protons across the cytoplasmic membrane and generation of the PMF. At least four residues (D407, D408, K940 and T978) within the transmembrane region are involved in this process.105 It is interesting to note that a single point mutation on these corresponding residues in homologues MexB106, AcrB107, CmeB98 and CusA104 has been found to impair the function of these pumps, suggesting the important functional role of these proton-relay residues. Crystal structures of the AcrB proton-relay network mutants D407A, D408A, K940A and T978A have been reported.105 These mutant proteins revealed remarkably similar conformations, which show distinct conformational differences compared with the conformations of the wild-type protein.105 These conformations may mimic one of the transient states of the pump during the transport cycle. In both CmeB98 and CusA,104 the corresponding proton-relay mutants has been used to embed in proteoliposomes. In vitro studies using these proteoliposomes, either by sm-FRET or stopped-flow transport assay, indicated that these mutant transporters are completely nonfunctional. These proteoliposome studies indeed highlight the importance of these residues for energy coupling.
5.3. E. coli AcrAB-TolC.
The AcrAB-TolC efflux system is the best studied efflux system in E. coli and whose broad specificity governs its intrinsic resistance to numerous antibiotics, solvents, detergents, dyes and organic compounds.108 The periplasmic MFP of this efflux system AcrA (Figure 4a – red), mediates the interactions between the IMP AcrB (Figure 4a – blue) and the OMP TolC (Figure 4a - green) of this system.109,110 The extrusion of substrates through the AcrAB-TolC efflux system is powered by the proton-motive force that synchronously fuels the conformational changes within AcrB, the pump of the efflux system.111 Similar to other HAE-RND efflux pumps, AcrB is able to bind and transport substrates from both the outer leaflet of the inner membrane and periplasm.112 The first experiment to successfully quantify the ligand-transporter interaction for an RND IMP was reported for AcrB in 2007.113 This study showed that AcrB binds drugs such as rhodamine 6G, ethidium, proflavine and ciprofloxacin in the micromolar range.113 The PMF is critical for this process and the transport capacity of AcrB was completely abolished when residues important for the proton-relay network (D407, D408, K490 and T978) were individually mutated to alanine.114 TolC has been shown in previous studies to not only be essential to the extrusion of antimicrobial substrates, but also to play a major role in the transport of large polypeptides.115 The loss of TolC function results in the incomplete transport of substrates and causes an increased susceptibility to antimicrobial drugs.93 The importance of TolC is highlighted by the fact that it appears to be a promiscuous OMP. For example, TolC serves as the OMP for AcrAB, AcrAD, AcrEF and others.116
Figure 4.
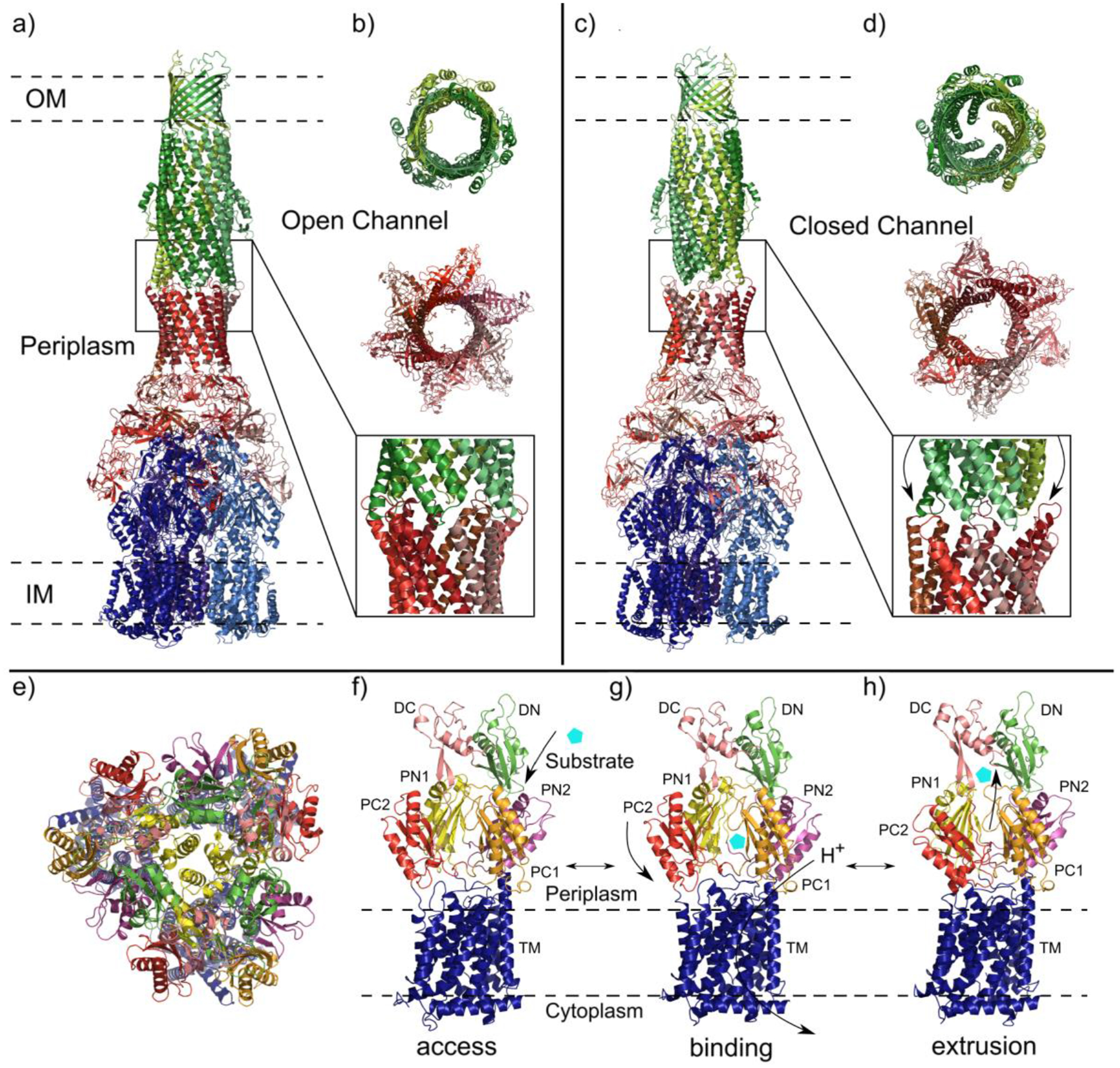
Comparison of the AcrAB-TolC efflux system components in open and closed channel conformations. Subunits are color-coded by slight variations of the indicated color accordingly: AcrA (red), AcrB (blue) and TolC (green). (a) Structure of AcrAB-TolC efflux system in the open channel state visualized in side view (adapted from PDB ID 5O66). (b) Top view (top and middle panels) and side view (bottom panel) of AcrA-TolC interface in the ligand bound open channel state. (c) Structure of the apo closed channel state of the AcrAB-TolC pump visualized in side view (adapted from PDB ID 5V5S). (d) Top view (top and middle panels) and side view (bottom panel) of AcrA-TolC interface in apo closed channel state. (e-h) AcrB protomers viewed in their access, binding and extrusion states. Separate colors depict distinct subdomains. (e) Ribbon diagram of the AcrB protomers viewed from the inner membrane plane (adapted from PDB ID 5NC5). (f) Side view of the access state of the AcrB protomer with substrate (cyan pentagon) entering the periplasmic cleft. (g) Side view of the binding state with arrows indicating the closing of the periplasmic cleft and the transport of protons through the transmembrane domain. (h) Side view of the extrusion state with closed PN1/PC2 and PN2/PC1 subdomains and exported substrate.
Studies examining the interactions between AcrA, AcrB and TolC provide the basis for the extrusion model of efflux systems in which allosteric changes in the IMP are communicated through the adaptor protein MFP to open the periplasmic channel of the OMP. In addition to the E. coli AcrAB-TolC system, other HAE-RND efflux systems such as Neisseria gonorrhoeae MtrCDE, Campylobacter jejuni CmeABC and Acinetobacter baumannii AdeABC have comparable proposed extrusion mechanisms. The repeated recruitment of TolC occurs after the AcrAB complex has formed and is only able to self-assemble the efflux system in the presence of AcrA.100 Studies on the α-helical residues near the end of the periplasmic domain of TolC suggests that the conversion between the closed and open states of OMPs is synchronously linked to the transition of two α-helices from an inward position to an outward position (Figure 4a and 4c – magnified).
With the use of cryo-electron microscopy (cryo-EM), two structures of the full AcrAB-TolC efflux pump were determined: 1) an open channel in the presence of inhibitor or antibiotic (Figure 4a and 4b) and 2) a closed channel in the apo-state (Figure 4c and 4d). These structures provide insight into the conformational states of this transporter and possibly serves as a model for many other trimeric efflux systems. In these structures, the AcrB protomers are able to adopt three distinct conformational states during the substrate transport process: access, binding and extrusion (Figure 4e – 4h).117 The structures of the apo state of the AcrB pump and a closed TolC represent the resting state of the efflux system while the structures of an opened TolC in the presence of transferrable substrates represent numerous transport states. The AcrB trimer in the apo structures can assume asymmetric conformational states (access:access:binding), (access:binding:binding) or (access:binding:extrusion).118,119 The AcrB protomers undergo substantial allosteric changes when the efflux system shifts from the apo state to the multiple transport states. In the transport states, large allosteric changes occur in subdomains PN1/PC2 and PN2/PC1 upon the binding of substrate.111,120 In addition, molecular dynamics simulations in AcrB suggest that the G-loop of AcrB is very flexible, with the phenylalanine residue, F617, within the G-loop 614GFGFAGRG621 necessary for the substrate transfer process and stabilization of substrate binding.121 Recently, a cryo-EM study of the A. baumannii AdeB multidrug efflux pump depicts that a flexible F-loop 661PAIDELGT668, which forms part of the substrate binding site within the periplasmic cleft, may be critical for substrate recognition and transport.122 In AcrB, the composition of this F-loop is 669PAIVELGT676, where residue I671 has been revealed to be significant for substrate specificity.123
A structure of erythromycin-bound AcrB revealed that this macrolide was anchored within the proximal drug binding site (located in periplasmic domain PN2) buried within the periplasmic cleft of the multidrug efflux pump, where six distinct amino acids (one Phe, one Leu, three Ser, one Thr, one Lys and one Asp) at the proximal site are liable for the binding.124 The entrance of the AcrB periplasmic cleft is surrounded by residues F664, F666, L668, R717 and L828 and these residues have been shown to be essential for substrate recognition.105,119,120 A substrate bound crystal structure of AcrB with minocycline shows that the residues Q176, F178, N274, F615 and F617 directly interact the bound minocycline to stabilize the binding of substrates at the distal site within the periplasmic cleft.111 In addition, MD simulations suggest that the distal site of AcrB also contains a hydrophobic patch that influences drug binding.121 Interestingly, the connections between TolC and AcrA and between AcrA and AcrB undergo no significant structural changes with the different conformational transport states of the AcrB pump; rather the MFP repacks to communicate the allostery between IMP and OMP.125
Studies on the AcrAB-TolC system have also investigated how substrate binding in the apo state initiates quaternary structural alterations in AcrB that are interconnected to AcrA. This triggers the AcrA monomers to repack and subsequently generates tertiary allosteric changes in TolC that open the channel from a closed apo state. The rearrangement of AcrA is essential to prevent the substrate from leaking into the periplasm by forming a sealed channel within the pump. This repacking of AcrA allosterically couples the transport state of AcrB to TolC to guarantee that the opening of the TolC channel is synchronized with the closing of the efflux system to the periplasm.126 When the substrate has been removed or is absent, the efflux system returns to the apo state with TolC adopting its closed conformation.118 Comparison of these conformational states indicates that the efflux system constricts along the long axis of the pump by approximately 10 Å when shifting to the ligand bound transport state.100 The transition from the apo state to an active transport state likely causes a contraction of the local periplasm to accommodate the axial constriction in the efflux system and the change in the curvature of the surrounding membrane near the AcrB transmembrane domain. It is likely that these changes in the structures surrounding the efflux system impact the energetics and kinetics of the substrate transporting process. This structural data may be important for the development of inhibitory drugs that target the interfaces between the components of this efflux system or the conformational switching of the tripartite pumps.
Multiple homologs of the AcrAB-TolC efflux system are also expressed in Gram-negative bacteria.116 One such IMP homolog is AcrD, which features an overall high level of sequence and folding identity and resemblance to AcrB and also possesses some corresponding substrate specificities. While AcrD is capable of expelling some of the same compounds as AcrB, only the AcrD is capable of recognizing and expelling aminoglycosides.115 This ability of Gram-negative bacteria to express and essentially switch out some components of the AcrAB-TolC efflux system further broadens its range of substrate resistance. This interchangeable capability allows other homologs combinations to be compensate in E. coli in the event one or more of the AcrAB-TolC components are inhibited. As an example, the MFP AcrE and IMP AcrF interconnect with TolC to form the AcrEF-TolC tripartite efflux structure.116 AcrEF and AcrAB have similar substrate selectivity and the MFPs are interchangeable with one another such that AcrA can also operate with AcrF. Earlier studies have revealed that resistance to antimicrobials in AcrAB-impaired cells can be maintained due to the expression of AcrEF. Overexpression of AcrEF complements AcrAB function and contributes to the antimicrobial resistance of mutants when AcrA or AcrB was incapacitated.127 TolC is also a component of the MdtABC and MdtEF efflux systems that expel nitrosyl indole derivatives generated from anaerobic respiration. The MdtABC-TolC efflux system is unusual in that the IMP pump exists as a heteromultimeric pump formed by 2:1 ratio of MdtB/MdtC proteins.128,129 Therefore, drugs targeting TolC may allow inhibition of the major AcrAB-TolC efflux pump in E. coli, as well as all homologous systems relying on the OMP TolC, thus unable to transport bactericides from within the cell.
5.4. N. gonorrhoeae MtrCDE.
The Gram-negative Diplococcus N. gonorrhoeae is a bacterium that causes the sexually transmitted disease gonorrhea only found in humans.130 Despite being well studied, gonorrhea remains a major concern with more than 100 million cases reported annually worldwide.131 In order to overcome the host’s innate immune system, N. gonorrhoeae has developed mechanisms to overwhelm antimicrobial systems.132 The most clinically significant HAE-RND efflux system in N. gonorrhoeae is the MtrCDE tripartite multidrug efflux system.133 Similar to other RND multidrug pumps, this efflux system recognizes and actively transfers a broad spectrum of structurally distinct toxic compounds from the bacterial cell, including the host’s antibacterial peptides, long-chain fatty acids, hormones and several clinically significant antibiotics.134 For the MtrCDE tripartite efflux system, MtrD is a large proton-motive force dependent IMP trimer pump with 1,067 residues per protomer (Figure 5a).135 MtrE is a 447 residues protein that forms the homotrimeric OMP tunnel.136 The MFP is MtrC, containing 412 residues per monomer and oligomerizes in order to interconnect MtrD and MtrE forming the tripartite efflux system MtrCDE.136 Like the other trimeric HAE-RND efflux systems, MtrCDE extends through the entire cellular envelope of N. gonorrhoeae. MtrCDE shares the ability to transport hydrophobic antimicrobial compounds, such as antibiotics, nonionic detergents and antibacterial peptides, but can also uniquely export gonadal steroidal hormones and bile salts present in the mucosal environment.137
Figure 5.
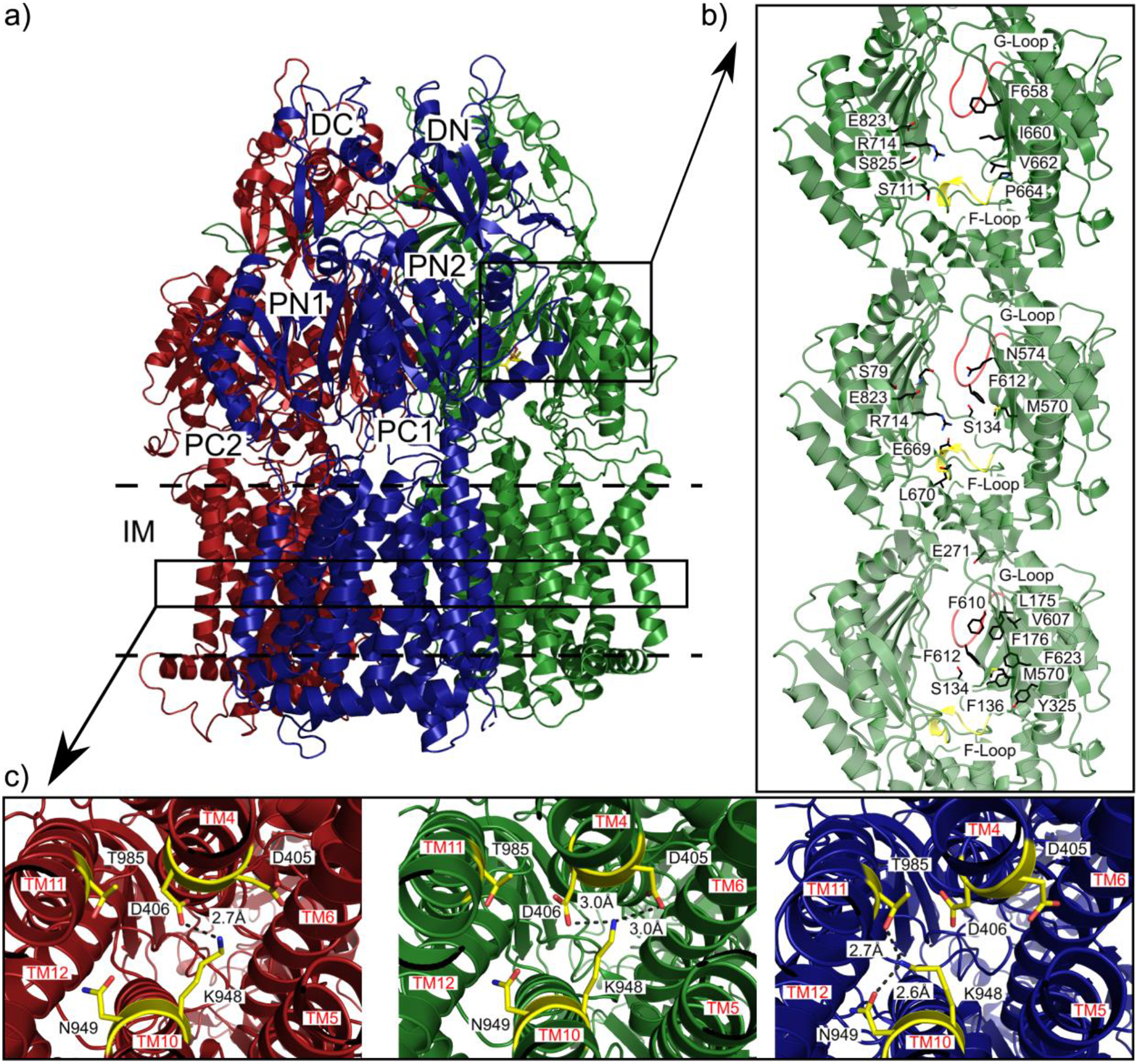
Substrate binding sites and proton-relay network of the MtrDCR103 pump (adapted from PDB ID 6VKT). (a) Ribbon diagram of the trimeric MtrD CR103 pump viewed from the membrane plane with the access, binding and extrusion protomers colored dark red, dark green and dark blue, respectively. (b) The periplasmic multidrug binding sites of MtrD CR103 with the periplasmic cleft entrance site, proximal drug binding site and distal drug binding site in the first, second and third panels. Residues that are important for selectivity are represent as sticks (black). The F loop and G loop are colored green and yellow, respectively, in each panel. (c) The proton-relay network of the MtrD CR103 multidrug efflux pump with the residue involved in the proton transport process (yellow sticks). In the access state K948 forms a hydrogen bond with D406 in the first panel. In the second panel the binding state of MtrD CR103 protomer K948 forms hydrogen bonds with both D405 and D406. In the extrusion state the K948 forms hydrogen bonds with N949 and T985 passing the proton through the transmembrane domain.103
A new, exceedingly drug resistant mutant strain of N. gonorrhoeae was discovered in the United Kingdom in 2018.132 This strain overexpresses the MtrCDE efflux pump along with a single K823E mutation in MtrD that bestows increased antimicrobial resistance. A cryo-EM structure of a mutant MtrD from the gonococcal strain CR.103 (designated MtrDCR103), which carries a mosaic-like sequence and significantly elevates drug resistance, revealed that this IMP adopts the overall fold of an HAE-RND pump and forms a homotrimer.125,126,134 Each protomer comprises 12 transmembrane α-helices (TM1-TM12) and an expansive periplasmic domain which can be separated into six subdomains (PN1, PN2, PC1, PC2, DN and DC), similar to what is seen from E. coli AcrB (Figure 4a).135 In the AcrB multidrug pump, approximately 22 residues form the proximal binding site and 11 of these residues are conserved in MtrD. The conserved residues S79, S134, M570, Q574, F612, E669, L670, G671, R714, G717 and E823 of MtrD are implicated in the substrate recognition of the pump. The periplasmic cleft of MtrD also contains the flexible gate G-loop (609GFSFSGS615). The glycines of this loop are critical for protein function and flexibility and allow the G-loop to swing the bound substrate from proximal to distal binding sites. In the distal binding site, 11 residues, S134, F136, L175, F176, E271, Y325, M570, V607, F610, F612 and F623, are conserved between MtrD and AcrB.120 The residues F176, V607 and F610 form part of a hydrophobic patch of the distal binding site and are potentially important for the binding of substrates. The subdomains PC1 and PC2 create the periplasmic cleft that allows substrates to enter the pump through the periplasm.135 Deep inside the periplasmic cleft the gate G-loop separates the proximal and distal multidrug recognition sites. Substrates from the periplasm are likely captured in the cleft and bind to one or both of these sites prior to extrusion. The entry to the MtrD periplasmic cleft is enclosed by residues F658, I660, V662, P664, S711, R714, E823 and S825 (Figure 5a and 5b). Most of these residues are not conserved amongst the HAE-RND efflux pumps and likely account for the substrate specificity and selectivity of MtrD. However, the conserved residue R714 of MtrD corresponds to R717 of the E. coli pump AcrB, with both of these residues located within substrate binding periplasmic clefts. R717 of AcrB has been implicated in substrate specificity and the corresponding arginine R714 of MtrD is likely contributes to the selectivity of the pump.121 Similar to other IMP pumps there is a flexible F-loop (665PPILELGN672) in MtrD to link the periplasmic cleft entrance and proximal multidrug binding sites. In the AcrB F-loop, a conserved isoleucine I671 is essential for drug selectivity and would suggest that the corresponding residue I667 of MtrD aids in the substrate selectivity of the IMP.
In a separate report, the analysis of 4852 N. gonorrhoeae genomes from a global meta-analysis collection identified mutations at positions 714 and 823 are able to cause the N. gonorrhoeae to be clinically non-susceptible to azithromycin.138 This agrees well with the structural data from cryo-EM experiments that suggest that these two residues are important for substrate selectivity and entrance into the MtrD periplasmic cleft, respectively.103
The cryo-EM structure of MtrDCR103 shows that this multidrug efflux pump forms an asymmetric trimer and the protomers show distinct access, binding and extrusion conformations (Figure 5a).103 This is unique within the crystal structure of MtrD, where the protomers are identical in conformation and form a homotrimer.103 The cryo-EM structure also has bound ampicillin which allows identification of the residues important for drug interaction within the binding protomer of the MtrD trimer (see Figure 4b for periplasmic, proximal and distal views of the antibiotic binding site. The bound ampicillin forms hydrophobic interactions with F136, I139, F176, V607 and F610 at the distal binding site and is anchored by electrostatic interactions from N135, S275 and T277. A second, reported cryo-EM structure has been solved with bound erythromycin, elucidating how MtrDCR103 recognizes macrolide antibiotics.103 The large erythromycin molecule is bound deep within the periplasmic cleft at the distal drug-binding site, suggesting that the protein is more flexible in solution to accommodate larger drugs. Erythromycin forms extensive contacts with seven aromatic, two hydrophobic, one positively charged and two polar residues. These form aromatic stacking, hydrophobic and electrostatic interactions with the bound drug having direct contact with residues F136, I139, R174, F176, S275, T277, Y325, F568, V607, F610, F612 and F623 of MtrDCR103.103 In AcrB, the conserved residues have been observed to be important for the binding of substrates and suggest that these residues are also important for MtrD to bind similar substrates such as antibiotics. Understanding the dynamics of these interactions will help improved drug design to better inhibit the both critical MtrD multidrug efflux pump in N. gonorrhoeae as well as efflux pumps from other organisms to help combat the increasing threat of antibiotic resistance.
It is well established that the proton motive force powers RND efflux pumps to extrude drugs from their periplasmic domain.90 In the transmembrane domain of MtrDCR103, residues D405, D406, K948, N949 and T985 are conserved and form the proton relay network (Figure 5c).103 The efflux of drugs from the cell is coupled and synchronized to the influx of protons to the cytosol. The residues K948 and D406 are conserved in multiple RND efflux pumps and closely interact with each other to funnel protons to the cytosol.103 The side-chain nitrogen of K948 forms a single hydrogen bond to the carboxylate oxygen of the side-chain of D406 (Figure 5c). When a drug binds to the protomer of MtrDCR103 a shift in conformation to the binding state occurs. The side chain of D405 moves closer to K948 to form an additional hydrogen bond. When MtrDCR103 shifts to the extrusion state K948 swings away from D405 breaking the hydrogen bond and offers the proton released from D405 to form a hydrogen bond to N949.103 The dynamic change of this hydrogen bonding network may promote the transfer of protons from the periplasm to the cytosol which creates the proton motive force to energize the drug efflux process.101 It appears that K948 plays a major role in sweeping protons from the periplasm to the cytoplasm by transferring protons from D405 to either N949 or T985.103 D406 may also be important by helping to stabilize different transient states by interacting with the critical K948 residue.
The OMP crystal structure of the MtrCDE multidrug efflux system has also been determined.134 MtrE forms a trimeric channel that extends down vertically from the outer membrane surface to the periplasm. MtrE is a homotrimer that forms an α-β barrel tunnel that is approximately 130 Å long to span the outer membrane, similar to what is observed in TolC. Each protomer of MtrE is composed of four β-strands and eight α-helices that contribute to the 12-stranded outer membrane β-barrel and the extended periplasmic α-barrel domains. The four β-strands of each protomer arrange in an antiparallel orientation to form the outer membrane spanning β-barrel domain. The α-helices of the MtrE protomers arrange to form the extended periplasmic α-helical tunnel that interacts with the MFP. Within the periplasmic α-helical tunnel two long α-helices reach through the center of the tunnel.108 In addition, this tunnel also contains two pairs of shorter α-helices that stack end-to-end to form semi-continuous helices to form coiled-coil interactions with the two long α-helices. The equatorial domain of MtrE contains two additional helices and many unstructured coils that may assist in the transfer of substrates through the MtrE tunnel.136 The channel of MtrE is capable of opening without the other components of the efflux system, however constriction and dilation of the MtrE channel has been linked to the reorganization of MtrC.91 MtrC contains β-barrel domains that interact with the periplasmic domain of the MtrD pump to bridge the periplasmic space in-between the MtrD and MtrE membrane proteins. Similar to the other RND efflux pumps, MtrC repacks to transfer the changes in allosteric states of the MtrD pump to the MtrE channel to regulate the opening and closing of MtrE.96,139
5.5. C. jejuni CmeABC.
Enterocolitis is an inflammation of the digestive tract that can be caused by C. jejuni. Genomic sequence analyses show that C. jejuni expresses multiple drug efflux transporters to facilitate antibiotic resistance.140 The most characterized efflux system in C. jejuni is the tripartite HAE-RND CmeABC efflux system that functions as the primary expeller of antimicrobials.61,141 The CmeABC drug efflux system comprises the inner membrane transporter CmeB, the periplasmic fusion protein CmeA and the outer membrane channel CmeC.142 C. jejuni has acquired resistance to several clinically relevant antibiotics, with the CmeABC efflux system critical for fluoroquinolone-resistant campylobacters. The Cme locus contains three tandemly linked genes (cmeABC) that encode for the CmeA the MFP, CmeB the IMP and CmeC the OMP.143
The CmeB efflux pump crystal structures show that CmeB forms a homotrimer and assumes a similar protein folding seen with the other HAE-RND family members (Figure 6a). The CmeB is composed of three protomers, each with 12 transmembrane helices (TM1-TM12) and a large periplasmic domain that is divided into the six subdomains: PN1, PN2, PC1, PC2, DN and DC (Figure 6b).99 Similar to the other HAE-RND efflux systems, a pore in the center of the CmeB pump is formed by PN1, PN2, PC1 and PC2 of each protomer that stabilizes the trimeric system. As with other HAE-RND efflux pumps, PC1 and PC2 combine to form the periplasmic cleft of CmeB. This cleft contains the binding sites for substrate recognition and forms the docking domain to anchor substrates prior to extrusion.99 Select mutations within CmeB have been determined to increase C. jejuni drug resistance, with 22 distinct amino acid residues identified. Many of these residues are localized at or near the substrate binding site, with mutations conferring resistance to a broader range of antibiotics, including chloramphenicol, ciprofloxacin, erythromycin and tetracycline.144
Figure 6.
The closed/open states of CmeB and the gates of CmeC. (a) Ribbon diagram of the CmeB homotrimer viewed in the membrane plane (adapted from PDB ID 5T0O). Each subunit of CmeB is labeled with a different color. (b) The closed “resting” state protomer of CmeB (slate) superimposed onto the open binding state protomer (light green). Subdomains DN, DC, PN1, PN2, PC1 and PC2 are labeled on the front of the protomers. In the “resting” state the periplasmic cleft between PC1 and PC2 is closed in each protomer. The “binding” state of the CmeB protomer creates a channel through the opening of the periplasmic cleft and is exposed to solvent. (c) Ribbon diagram of the CmeC trimer viewed in the outer membrane plane (adapted from PDB ID 4MT4). Each subunit of CmeC is labeled with a different color. (d) Top view of the CmeC trimer looking down from the extracellular space. The three R104 residues (colored sticks) from each protomer are found to interact and form the first gate of the channel. (e) Periplasmic view from the top of the CmeC trimer showing the charged and polar residues Q412, D413, E416 and N420 form each protomer forming the second gate to block the channel.
Two distinct structures of CmeB were determined using x-ray crystallography. Both structures arrange to form conformational asymmetric trimers similar to AcrB and MtrD.125,135,145 In the first crystal structure, the protomers of the CmeB trimer are all in the extrusion form, similar to the extrusion protomers of AcrB and 4MtrD, where the periplasmic cleft is closed (Figure 6b - slate). In a second crystal structure one of the protomers is in the binding state with the periplasmic cleft open, similar to the substrate-bound state of MtrD.(Figure 6b – light green)135 The other two protomers possess closed conformations of the periplasmic cleft, where one protomer adopts the extrusion state and the other protomer displays the resting state, similar to what in observed for apo-CusA.104 These various structures likely reflect different transient conformational states that the CmeB pump must go through during the transport cycle. Taken together, these crystal structures of CmeB show protomers with a different conformation cycle than the other HAE-RND asymmetric trimer pumps that have two periplasmic clefts open and one cleft closed.99 Individual protomers of CmeB also show conformations of various transient states within the transport cycle. The CmeB protomers are arranged so that only one out of the three periplasmic clefts in one crystal structure are open and in the other crystal structure all of these clefts are closed.99 Single-molecule FRET experiments on CmeB also indicate that the individual protomers are able to independently transition to the binding and then extrusion conformations in order to bind and expel substrates.99 The transport dynamics and conformational changes of the periplasmic domain movements were observed using total internal reflection sm-FRET imaging. In this experiment a functional mutant CmeB pump with its three native cysteines removed was derivatized with a mixture of maleimide-activated Alexa Fluor 546 (AF546) and Alexa Fluor 647 (AF647) serving as a molecular ruler to measure the relative change in distance between two inter-protomer cysteines.99 Using this method the opening and closing of periplasmic cleft can be monitored. The data showed that in the absence of a proton gradient multiple CmeB protomers prefer to assume the access conformation with occasional reversible transitions to the binding conformations. In the presence of a proton gradient more transitions were observed indicating that multiple protomers can advance from the binding to the extrusion conformation independently of the state of the adjacent protomers. Additionally, D409, a residue critical in the proton relay pathway, abolished the transport activity of CmeB when changed to an alanine. Single molecule-FRET experiments suggested that CmeB D409A pump reconstituted into proteoliposomes is inactive. It prefers to stay in the “resting” form and is incapable of changing its conformational state to advance the transport cycle.99 Combined, this data suggests that each CmeB protomer can independently transition through at least four different conformations to complete the transport cycle and that a singular disruption of the electron transport pathway is sufficient to abolish all transport activity.
The crystal structure of the OMP component of the CmeABC efflux system shows that CmeC arranges as a homotrimer with 492 residues per protomer, similar to the other identified structures of OMP channels such as TolC and MtrE.126,146,147 The CmeC component forms a tunnel 130 Å long spanning the outer membrane.146 Similar to TolC and MtrE, the structure of CmeC is comprised of four β-strands that form the transmembrane β-barrel and six α-helices to form the periplasmic α-helical domains (Figure 6c), with the interior surface of the tunnel is extensively occupied by negatively charged residues.93 CmeC is distinct from other OMPs in that its exterior surface is predominately populated with acidic residues. The N-terminals of the CmeC protomers each contain an elongated loop that extends from the membrane surface that continues down to the equatorial domain in the periplasm.124,146 In this N-terminal loop there is a conserved cysteine residue that interacts with a lipid of the inner leaflet via a thioester bond. This cysteine is necessary for insertion of CmeC into the outer membrane and mutation of this residue in other OMPs disrupts the ability for the protein to unfold the transmembrane β-barrel.99 The channel of the CmeC outer membrane β-barrel is partially obstructed by a flexible loop suggesting that this structure is in the closed conformation. This loop functions as a cap to open and close the transmembrane channel of CmeC. The residue R104 from each of the protomers of CmeC associate with one another through hydrogen bond interactions forming a gateway to regulate the passage of compounds across the membrane (Figure 6d). On the periplasmic side of the α-helical domains a second gateway is created by a layer of charged and polar residues Q412, D413, E416 and N420 with hydrogens bonds linking Q412 to E417 on the subsequent protomer (Figure 6e).146 In order to actively transport substrates across the outer membrane and out of the cell the opening and closing of these gates must be synchronized to the repacking of CmeA and conformational states of CmeB. CmeA likely interacts with CmeC through grooves that are located on the outermost surface of the periplasmic domain of the trimer. Like TolC and MtrE, the protomers align to form three intra-protomer and three inter-protomer grooves.147,148 MFPs such as CmeA contain an α-helical coiled-coil domain that that are proposed to fit into these grooves and contact CmeC, forming the transport channel.147 This is also in agreement with the 3:6:3 ratio of IMP:MFP:OMP since there are a total of 6 grooves in the CmeC trimer and 6 monomers of CmeA.91 This synchronized repacking of CmeA and interaction with CmeC regulates the opening and closing of the CmeC channel, which coincides with what has been observed in the Neisseria gonorrhoeae MtrCDE efflux system.134,146
5.6. A. baumannii AdeABC.
A. baumannii is an opportunistic pathogen that was determined as the root cause of peculiar infections that produced symptoms such as pneumonia, bloodstream infections, meningitis and necrotizing fasciitis.149 A. baumannii has become one of the most dangerous threats in hospitals due to its ability to quickly mutate and upregulate efflux pumps which allows the bacterium to acquire resistance to a broad spectrum of antimicrobial compounds.150 Many strains are now resistant to carbapenems, colistin and tigecycline and has become a top priority pathogen for the research and development of new inhibitors and antibiotics.151 One of the main mechanism contributing to this resistance is tripartite AdeABC efflux system that mediates resistance to several classes of antimicrobial agents, such as β-lactams, tetracyclines, aminoglycosides, chloramphenicol, cefepime, fluoroquinolones, trimethoprim, tigecycline and novobiocin.150,152 The AdeABC locus consists of three tandemly connected genes that encode for the protein components of this tripartite efflux pump: AdeA, AdeB and AdeC where all three components are required for transporting substrates out of the cell.153 The tertiary structure of AdeB is a trimer of protomers that consist of 1,035 amino acids each, similar to other IMPs in the HAE-RND efflux family such as AcrB, MtrD and CmeB (Figure 7a).99,135,154 The unbound conformation of the three AdeB protomers shows that the periplasmic cleft formed by the subdomains PC1 and PC2 is closed and no channel is found within the periplasmic domain in this state. This is typical of IMP protomers lacking substrate such as the apo conformation seen in CmeB and CusA efflux pumps.104,142
Figure 7.
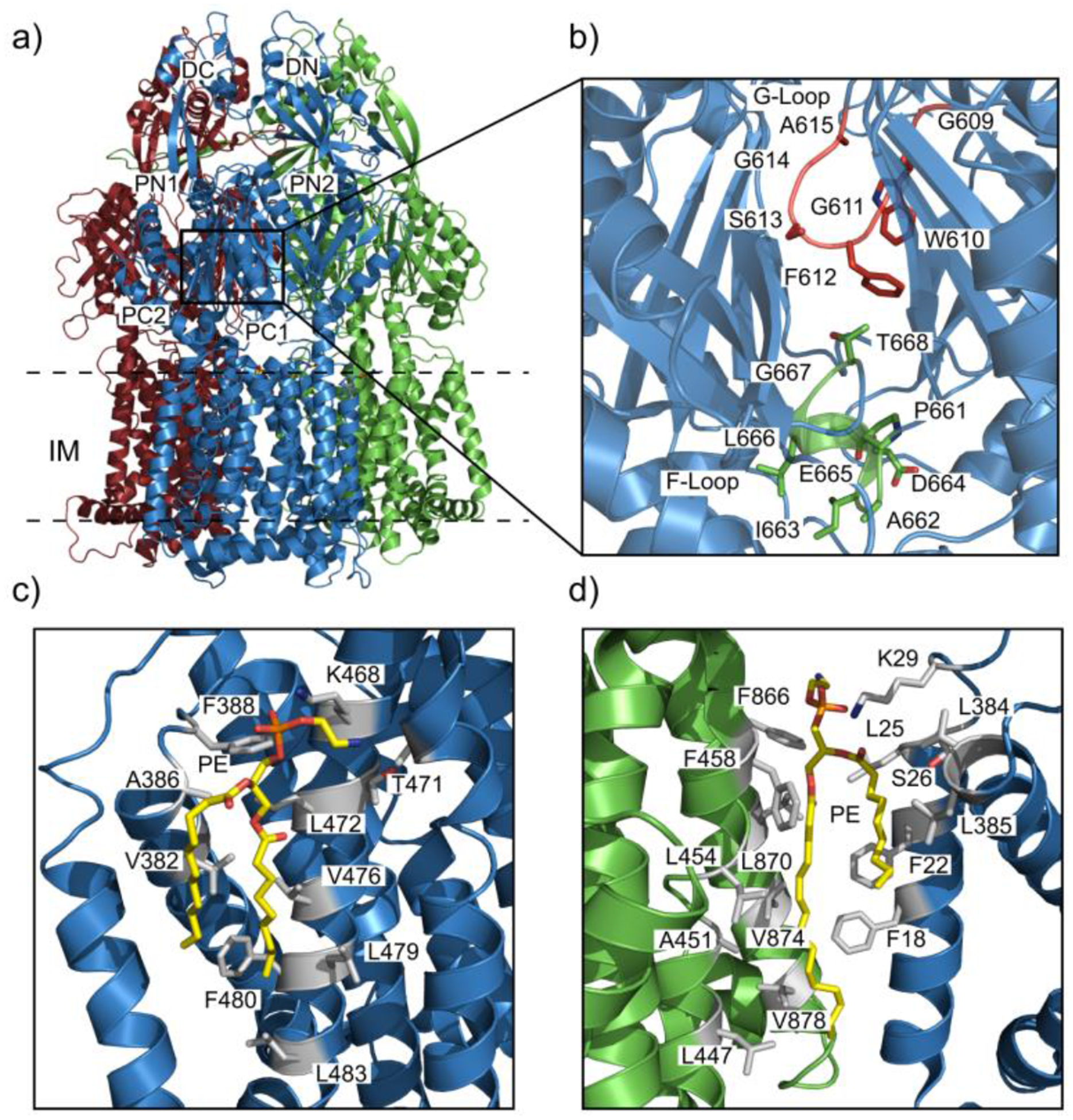
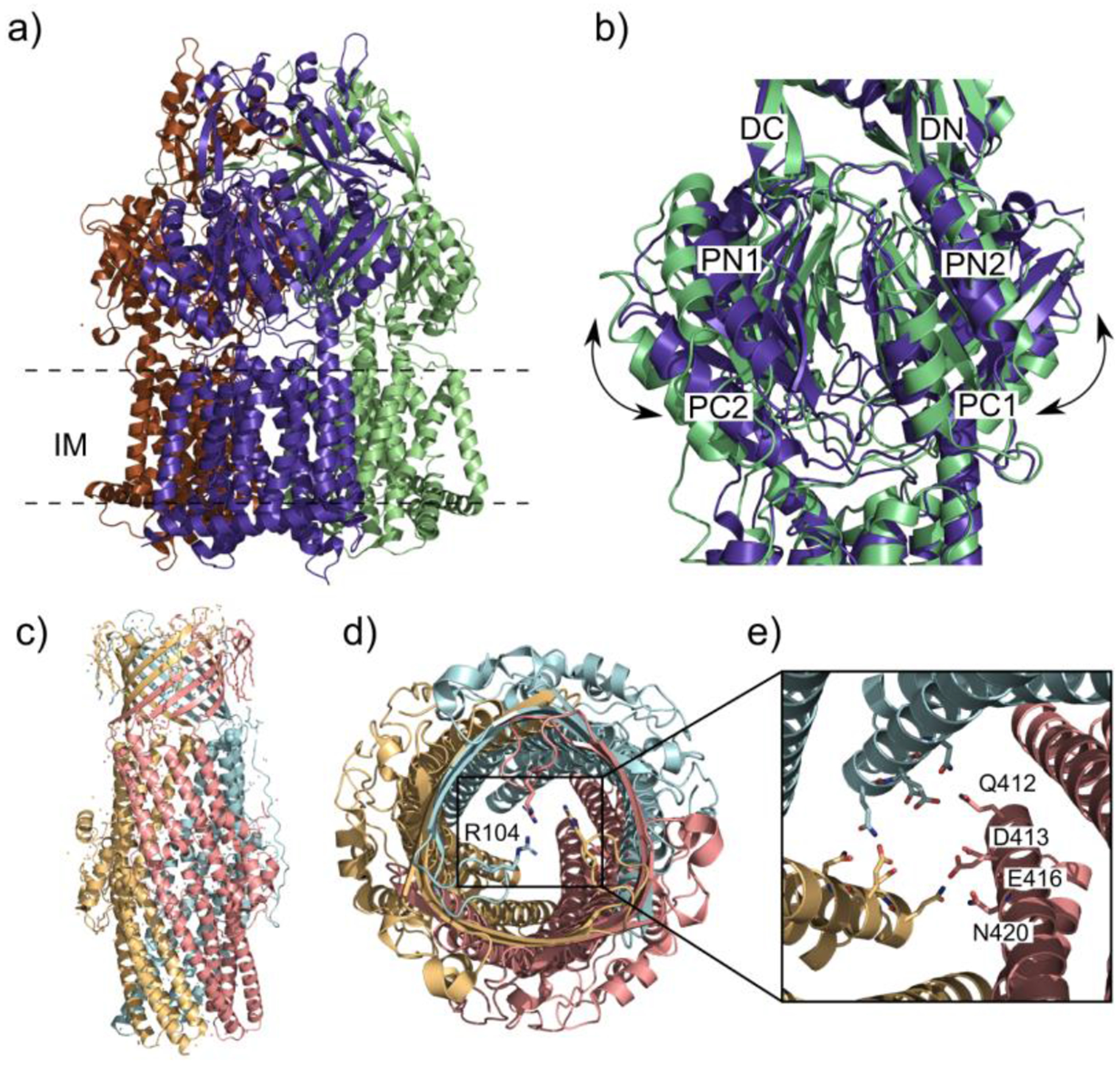
Drug and lipid binding sites of the AdeB pump. (a) Ribbon diagram of the cryo-EM structure of the trimeric AdeB multidrug efflux pump viewed in the inner membrane plane. Each protomer of AdeB is labeled with a different color (dark red, green, light blue). (b) The F-loop that forms part of the proximal multidrug binding site with residues (dark red sticks) that are important (dark red) for drug binding in this F-loop. The G-loop (green) with residues (green sticks) important for delivering drug molecules to the distal multidrug binding site. (c) The PE lipid (yellow sticks) binding site at the interior surface of the central cavity of an AdeB protomer. The residues (grey sticks) of the AdeB protomer that are involved in binding the lipid. (d) The PE lipid binding site at the interface between two AdeB protomers (green and light blue) with the residues (grey sticks) involved in binding the lipid.
Based on the ligand-bound structures of MtrD, AcrB, or CmeB, we anticipate that the substrate specificity of AdeB would also be geared toward binding hydrophobic compounds. In AdeB, a flexible loop designated the F-loop (Figure 7b) contains residues 661PAIDELGT668 in which E665, L666 and G667 are conserved among the other pumps MtrD, CmeB and AcrB.99,120,121,133 This F-loop is situated close to the inner membrane plane may play a role in the proximal substrate binding site. The second loop in the proximal binding site is the G-loop (Figure 7b) with residues that are positioned in the motif of GXGFXGX, containing three glycines and single phenylalanine that are conserved in the other pumps CmeB, MtrD and AcrB. In AdeB the G-loop consists of residues 609GWGFSGA615 where G616 and G619 are anticipated to be necessary for providing flexibility to the G-loop in order to pass the bound substrate from the proximal to distal binding site (Figure 7b) based on the mutation studies on the corresponding residues in other IMPs. Mutation of the corresponding glycines in AcrB disrupts the flexibility of the G-loop to transport the substrate from proximal to the distal binding site and reduces the resistance to erythromycin and doxorubicin.155 Sequence alignment and docking studies suggest that the distal substrate binding pocket of AdeB contains a hydrophobic patch comprised of residues F178, F277, I607 and W610 that likely plays a significant role in AdeB-substrate interactions.156 Docking simulations of the substrate gentamicin into the AdeB binding sites shows that the residues Y77, T91, S134, F277 and W610 of AdeB are important for the distal binding site residues to interact with the gentamicin molecule. Docking studies also show that E563 at the periplasmic entrance of the AdeB binding site may be an essential residue for anchoring gentamicin.154
Each of the AdeB protomers are found to interact with phosphoethanolamine lipids at distinct binding sites located on the interior surface of a transmembrane central cavity created by the joining of the trimeric AdeB efflux pump protomers (Figure 6c and 6d). In order to stabilize the lipids at these positions the residues V382, A386, F388, L454, A457, T471, L472, S475, V476, L479, F480 and L483 form hydrophobic interactions with the acyl chains of the lipid and the positively charged residue K468 to form a hydrogen bond with the phosphate headgroup anchor the lipid to the protomer (Figure 7c). It has been proposed that there are corresponding sites in AcrB and MtrD that are capable of interacting with lipids and may also be able to bind substrates.112,155 A second lipid binding site is located at the protomer-protomer interface. The lipid phosphate headgroup is oriented toward outer leaflet of the cytoplasmic membrane with the acyl chains entrenched in the hydrophobic region of the inner membrane. The residues L447, A451, L454, F458, F866, L870, V874 and V878 from one protomer interact with one of the acyl chains. The residues F18, F22, L25, S26, L384 and L385 from the subsequent protomer form favorable hydrophobic interactions with the hydrocarbon chains and the phosphate headgroup is anchored by a hydrogen bond with the K29 side chain of the adjacent protomer (Figure 7d).154 It is likely that these well-defined lipid binding sites are important for the oligomerization and stabilization of the AdeB trimer as well as the other efflux pumps MtrD, AcrB and CmeB.157
As with other RND efflux pumps, drug extrusion via AdeABC is driven by the proton-motive force with the influx of protons synchronized and coupled to the efflux of substrates. In AdeB the proton relay network is buried within the transmembrane domain of each protomer comprised of the residues D407, D408, K931, N932 and T968 that are conserved amongst the various RND efflux pump family members.105,114 Without the presence of substrates, the RND pumps remain in an apo state with no apparent transfer of protons. In the presence of drugs, AdeB shifts to the binding conformation which allows the transfer of protons and export of substrates from the cell. The opening of the periplasmic cleft causes the reorientation of the transmembrane helices of AdeB similar to the shifts in the same transmembrane helices seen in MtrD, AcrB and CmeB.121,135,144 In these HAE-RND pumps TM8 and TM5 shift away from the cluster of the transmembrane helices in order to open the periplasmic cleft and allow the entrance of substrate into the proximal binding site.158 This shift of the transmembrane helices weakens the hydrogens bonds D408 and T446 and triggers the reorganization of hydrogen bonds within the proton relay network. Similar to the proton relay mechanism in MtrD, the side chain of the proton sweeper K973 flips to hydrogen bond to D407 and D408. In this orientation a proton is able to be transferred from the carboxylate oxygen of the side chain of D407 and subsequently passed to the side-chain nitrogen atom of K973. Upon transition to the extrusion state, the K973 side-chain swings toward T968 and N932 to form hydrogen bonds and pass the proton toward the cytosol. During the allosteric transitions from access, binding and extrusion states the side-chain of K973 sweeps toward the cytosol breaking and forming hydrogen bond networks to translocate the periplasmic protons.154 This mechanism allows efficient extrusion of substrate from the bacterium and disruption of this process, in concert or separate from direct substrate inhibition, is a viable strategy to counteract the acquired antibiotic resistance of the cell.
5.7. Heavy metal efflux (HME) class of RND transporters.
In order to regulate the levels of transition metals bacteria use efflux pumps to transport metals such as calcium, cobalt, copper, iron, manganese, magnesium, zinc that are common in enzymatic cofactors.159 Transition metals are incorporated into approximately 40% of enzymes and are essential for cellular functions such as bacterial Cu/Zn superoxide dismutase and the prokaryotic electron transport chain. While these transition metals are needed for the functions of the cell, in excess they can become toxic. Nonessential metals such as silver can be utilized as effective biocides. The main cause of this cellular toxicity is the high reactivity of many transition metals. Cationic metals are especially toxic and react with many abundant biomolecules such as thiols, imidazoles, nucleic acids and sulfides.160 Copper also has a high toxicity due to the ability of the metal ion to displace other metals from enzymatic complexes as well as its high redox potential which can generate extremely reactive hydroxyl radicals that can damage proteins, lipids and DNA.161 The intentional use of transition metals as biocides has been an effective method to reduce bacterial growth on common objects. Silver, copper and zinc have been used in a number of manmade items to prevent bacterial growth such as brass doorknobs, silverware, paint, water filters and medical instruments.162,163 In order to combat the use of these metal biocides, bacteria have adapted to use efflux pumps to acquire resistance to metals similar to the acquired resistance to antibiotics.88,164
Transcription of the HME RND transporter genes is strongly dependent on the concentration of toxic metal ions in the cellular environment.165 CusCFBA from E. coli is one of the most studied efflux systems among the HME-RND family members. CusCFBA is selective in its metal extrusion capabilities and confers resistance of the cell to Ag+ and Cu+ ions.166 Other, less understood HME-RND efflux system family members include Cd2+, Zn2+ and Co2+ transporter CzcABC in various Ralstonia metallidurans and Zn2+-specific ZneABC from Cupriavidus metallidurans.167,168.
The canonical HME-RND efflux systems consist of three proteins that work in conjunction: CusCFBA, ZneBAC and CzcCBA.165,168,169 The IMPs of each efflux system, CusA, ZneA and CzcA, are inner membrane proteins that function as a trimeric pump to bind and transport metals to the next component of the efflux system, the MFP.104,168,170 The corresponding efflux system MFPs, CusB, ZneB and CzcB are the periplasmic adaptor proteins that rearrange to form a hexameric tunnel and allosterically communicate conformational changes to the OMP.171–173 The OMPs, CusC, ZneC and CzcC each form a trimeric gated channel to transport substrate through the outer membrane and out of the cell.169,171 An additional small, periplasmic, metal-binding protein CusF functions as a metal ion chaperone and has no analog amongst other HAE-RND family members.174 While the HAE-RND efflux systems transport a broad spectrum of substrates, the HME-RND family members are extremely selective and possess the ability to differentiate between monovalent and divalent ions.61
The CusCFBA operon encodes CusC, the OMP, followed by CusF, a chaperone, the MFP CusB and finally the IMP pump CusA.175 The CzcCBA operon is encoded with genes for the IMP CzcA, the MFP CzcB and OMP CzcC. This operon is transcribed tricistronically and is bordered by multiple genes that are involved in regulation of metal dependent proteins and expression of efflux system proteins.176 The ZneBAC operon structure encodes genes in the BAC order instead of the more common CBA structure and encodes the genes for the integral membrane pump protein belonging to the RND superfamily ZneA, a membrane fusion protein ZneB and an outer membrane pore protein ZneC.177
5.7.1. Heavy metal inner membrane pumps.
The first reported structure in the HME subfamily, CusA exhibits the conserved tertiary structure of RND efflux pumps forming a homotrimer with each protomer containing six periplasmic subdomains PN1, PN2, PC1, PC2, DN and DC and twelve transmembrane α-helices, TM1-TM12 (Figure 8a).104,178 The subdomains PC1 and PC2 form the periplasmic cleft characteristic of RND pumps; however, in CusA a single α-helix branches across the bottom of the cleft separating the periplasmic and transmembrane domains.104,179 This horizontal α-helix contains an important methionine residue M672 that couples with M573 and M623 to form a central metal binding site coordinated by the three methionine thioesters within the CusA periplasmic cleft (Figure 8c). Mutation of these residues to isoleucines increases cell susceptibility to copper indicating the importance of these residues to the function of CusA.180–182 Crystal structures acquired by soaking CusA in Cu+ and Ag+ were determined to be coordinated by the thioesters of M573, M623 and M672 in both structures.104 The binding of Cu+ and Ag+ causes a large conformation shift in the position of the horizontal α-helix, swinging the α-helix and prying open the periplasmic cleft by about 30° to expose the binding site to the periplasm. Comparison of the metal-bound and apo CusA structures reveal that the transmembrane α-helices TM1, TM2, TM3, TM6, TM8 and pore subdomain PN1 shift upwards away from the inner membrane upon metal binding. There is also a channel made up of four distinct methionine pairs that shuttle metal ions in a stepwise mechanism within each of the CusA protomers.104 One conserved pair, M271-M755, located in the periplasmic domain above the three methionine central metal binding site, is likely involved in passing metal ions up the pump to the next component in the efflux system (Figure 8c).104,182 Below the main metal binding site, three pairs, M410-M501, M403-M486 and M391-M1009 form the channel through the transmembrane domain (Figure 8d). In vitro transport assays and in vivo susceptibility assays show that mutation of these methionine pairs hinder the ability of the bacterium to expel copper and silver further supporting the importance of these methionine residues for the transport of metal ions.58,182 CusA mutants M391I, M486I, M573I, M623I, M672I and M755I were incorporated into liposomes in order to investigate the importance of these residues for the transfer of copper and silver across lipid membrane. Metal ion transport assays of these CusA mutants reveal that the methionine deficient mutants are unable to transport Ag+ ions across the liposome membranes.104 The proton motive force fuels the allostery of the IMP protomers to shift between the access, binding, and extrusion states. The crystal structure of CusA reveals that the conserved charged residues D405, E939 and K984 contribute to a proton relay network of residues necessary for proton translocation across the inner membrane. The importance of these residues was further investigated using metal ion transport assays. CusA mutants D405A, E939A and K984A were reconstituted into liposomes and are unable to transfer Ag+ ions from the extravesicular fluid and into the liposome illuminating the requisite of these residues in the proton relay network.104
Figure 8.
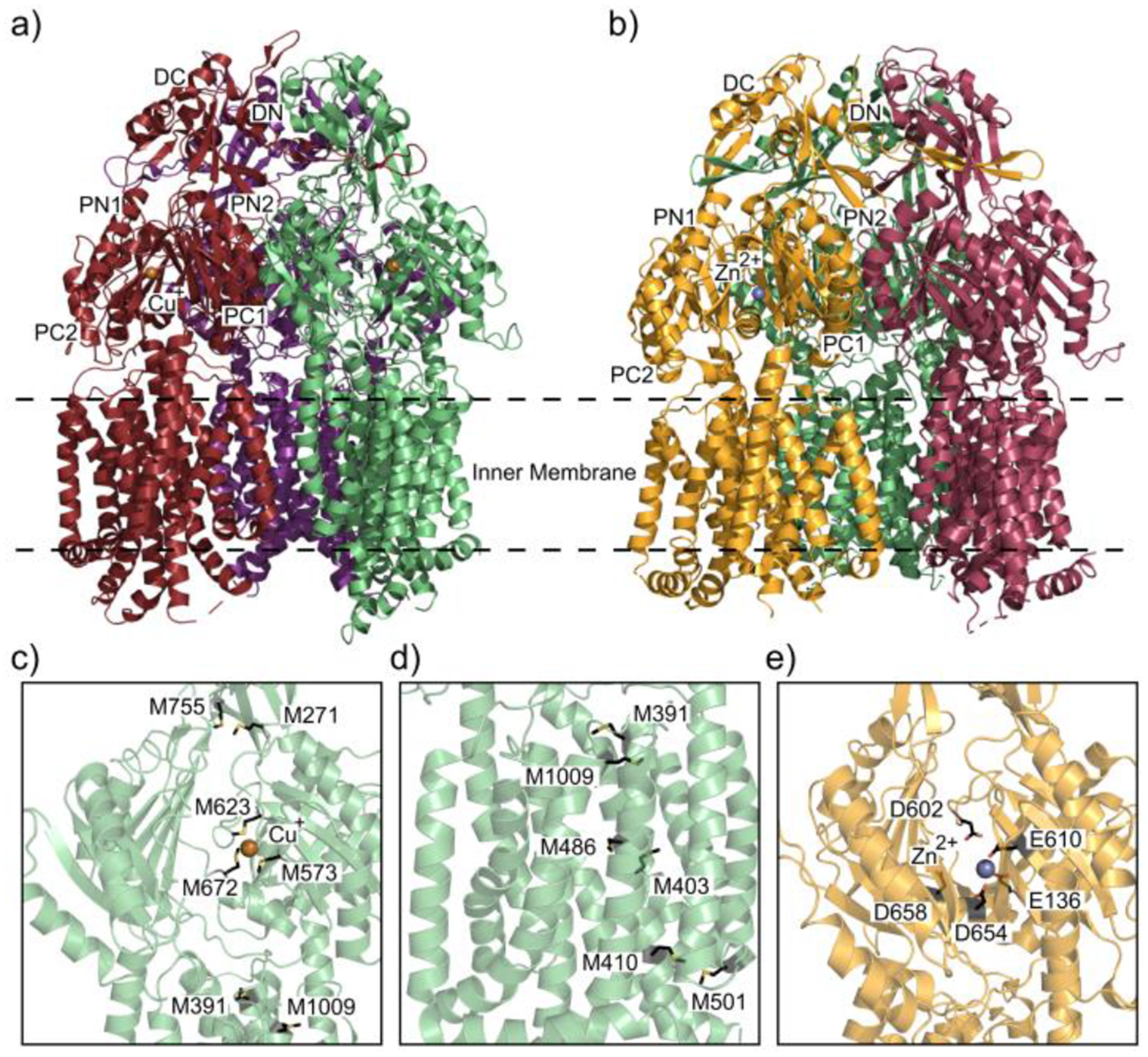
Heavy metal binding sites of CusA and ZneA. (a) The ribbon diagram of the Cu+ ion (dark orange sphere) bound CusA trimer pump (adapted from PDB ID 3KSS) viewed parallel to the inner membrane plane. Each protomer of CusA is labeled with a different color (ruby, purple and green). (b) Ribbon diagram of the Zn2+ ion (slate sphere) bound structure of ZneA (adapted from PDB ID 4K0J), viewed parallel to the inner membrane plane with colored protomers (orange, sand and dark magenta). (c) Metal extrusion pathway of CusA from the periplasm. The Cu+ ion (dark orange sphere) is coordinated by the three methionine binding site M573, M623 and M672 (black sticks) The methionine pair M271-M755 is where the metal ion could then be released for extrusion through CusB. (d) Metal extrusion pathway of CusA from the cytoplasm. Metal ions are passed through the transmembrane via the residues (black sticks) that form the methionine pair channel. (e) The Zn2+ ion binding site within the ZneA periplasmic cleft with a Zn2+ ion (slate sphere) coordinated by acidic residues (black sticks).
A crystal structure of ZneA, the efflux pump of the Zn2+ specific ZneABC efflux system, shows structural similarities to CusA (Figure 8b).104,168. ZneA exhibits the same general structure as efflux pump CusA, both possess a short horizontal α-helix within the periplasmic cleft unique to HME-RND efflux pumps. The crystal structure of ZneA shows that the protomers all adopt an apo conformational state with the periplasmic cleft closed that is indicative of efflux pumps that are not in the presence of substrate.168,183 This conformation likely reflects a transient state before ligand binding. Near the short horizontal α-helix within the periplasmic cleft an anionic pocket is formed by the side chains E136, D602, E610, D654 and D658 (Figure 8e). The structure of ZneA with bound Zn2+ reveals that the carboxylate sidechains of these residues form the proximal binding site in ZneA, which is conserved in another divalent metal binding efflux pump CzcA.168,169 Structural and sequence comparisons indicate that this anionic pocket is partly conserved in the monovalent central metal binding site of CusA but is entirely absent in the HAE-RND efflux pumps. The Zn2+ proximal metal binding site resides approximately 5 Å away from the three methionine main binding site in CusA suggesting a differing allostery to transport substrate. The residue D602 of ZneA is conserved in both D617 of CusA and D619 of CzcA; in addition, this residue has also been shown to be necessary for the transport of metal ions. While the carboxylate sidechain of D617 is only adjacent to the Cu+ binding site of CusA and doesn’t directly interact with Cu+, the mutant D617A exhibits no measurable Cu+ transport activity suggesting an important role of this residue in HME-RND efflux pumps.168,170,179 Loss of this residue in ZneA or CzcA is likely to reduce the affinity for Zn2+ and hinder the ability of HME-RND efflux systems to transport metals.
5.7.2. Heavy metal channel proteins.
Similar to other OMPs in the RND efflux family, CusC is arranged as a cannon shaped homotrimer with each protomer composed of four β-strands and nine α-helices that form a ~130 Å long α/β barrel protein (Figure 9a and 9b).184 The structure of CusC also has a conserved N-terminal cysteine residue that has been shown in other OMPs to covalently link each protomer to an outer membrane lipid and is necessary for the correct insertion of OMPs into the outer membrane.93,124,184 CusC possesses a channel opening of approximately 30 Å. This is much larger than most HAE-RND outer membrane channels such as TolC93, MtrE134 and CmeC146 that only have a diameter of roughly 12Å. In comparison, the periplasmic domains of CusC are in a closed conformation suggesting that a large conformational change is needed to for the extrusion of substrates. The interior surface of the channel is markedly populated by electronegative residues that may work in conjunction with the electronegative channel of CusB to form a pathway for ion extrusion.184 Mutation studies that target or delete the N-terminal cysteine of CusC resulted in the disruption of the trimeric channel and the assembly of the 12 stranded transmembrane β-barrel.124 The crystal structures of the CusC mutants revealed that the loss of the N-terminal cysteine disrupted the formation of the four β-strands that contribute to the 12 stranded transmembrane β-barrel and instead this domain formed highly disordered coils. In comparison to wild-type CusC the cysteine deficient mutants possess periplasmic α-helices that are swung outward so that the periplasmic region of these CusC mutants are in the open channel state.124 This would suggest that interactions with the transmembrane β-barrel are necessary for wild-type CusC for closing the periplasmic channel.
Figure 9.
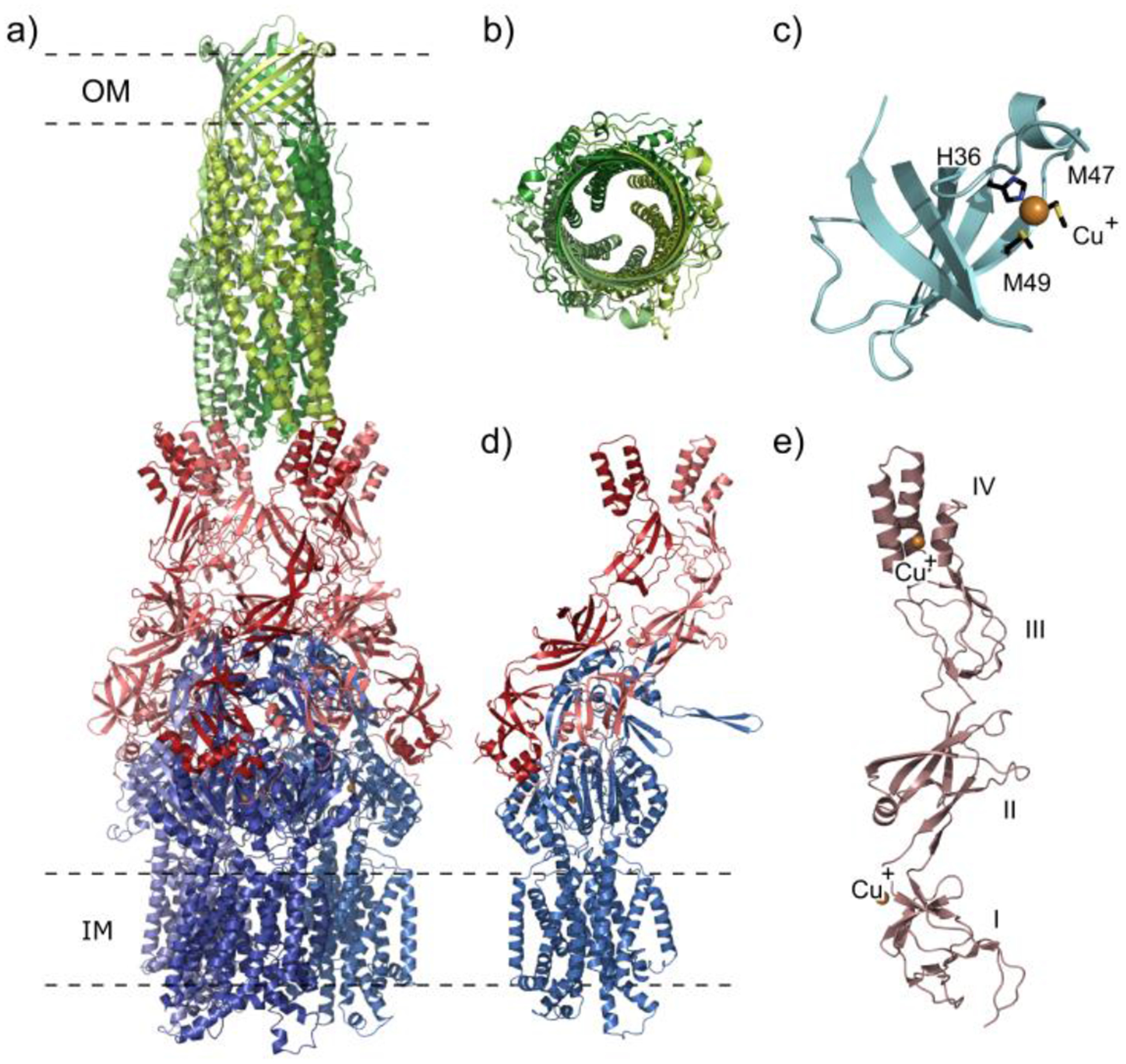
Components of the CusCFBA efflux system. (a) Model of the CusCBA tripartite efflux system. The system contains of the inner membrane pump CusA protomers (shades of blue), the membrane fusion adaptor CusB subunits (shades of red), and the outer membrane channel CusC monomers (shades of green) adapted from PDB ID 4DNT and PDB ID 4K7R. (b) Extracellular top view of the tripartite CusC monomers (shades of green) by a ribbon diagram (adapted from PDB ID 4K7R). (c) Ribbon structure of the Cu+ ion (dark orange sphere) bound CusF metallochaperone (adapted from PDB ID 3E6Z). The binding of the Cu+ ion is coordinated by the residues H36, M47 and M49 (black sticks). (d) The ribbon diagram of the CusBA subunit with each subunit of CusBA containing one CusA molecule (blue) and two CusB molecules (red and light pink) adapted from PDB ID 4DNT. (e) Ribbon diagram of single flexible CusB subunit coordinating two Cu+ ions (dark orange sphere) adapted from PDB ID 3OW7.
5.7.3. Heavy metal IMP-MFP Interactions.
CusB (Figure 9a, 9d and 9e) is an HME-RND family MFP that differs greatly in identity and sequence in comparison to the MFPs of the HAE-RND efflux systems.92,171 CusB is highly flexible and can be separated into one α-helical domain and three β-domains that can assemble into multiple conformations.171 The first β-domain (membrane proximal domain) is formed by six β-strands and contains both the N-terminal and C-terminal ends of the protein. The second β-domain (β-barrel domain) can combine with the first to form one of two different spatial conformations: 1) four β-sheets, two β-strands and one short α-helix, or 2) six β-strands and two α-helices. These conformations allow this domain acts a hinge shifting the other domains by approximately 20°. The third β-domain (lipoyl domain) consists of eight β-strands while the fourth domain (α-hairpin domain) is the α-helical domain; this domain is comprised of three α-helices that are capable of interacting with the periplasmic grooves of the CusC channel.124,171,185 Binding studies indicate that CusB is capable of binding Ag+ and Cu+ ions and x-ray absorption spectroscopy revealed that the CusB-Cu+ complex has a three sulfur coordinate bonding system.186 Mutant binding studies and in vivo susceptibility assays examining the M21I, M36I and M38I CusB mutant show a reduction in Cu+ ion affinity and increased cell susceptibility to Cu+ ions suggesting that these residues form a key Cu+ binding site in CusB.182
It is likely that Cu+ or Ag+ ions are shuttled to CusB from the metal binding chaperone CusF.174,187 CusF is a small, five-stranded β-barrel protein comprised of a pair of three stranded antiparallel β-sheets with residues H36, M47 and M49 coordinating a metal ion (Figure 9c).188 A CusF homolog has also been discovered in the CzcCBA efflux system of R. metallidurans.188 CusF likely scavenges the surrounding periplasm and has been shown to be important to the transfer of Cu+ and Ag+ ions prior to being expelled out of the cell by the CusCBA efflux system. The interaction between CusF and CusB could be the initial step before metal ion binding to the CusA efflux pump.
The co-crystal structures of CusBA provide the first structural information for adaptor-transporter interaction.185 The structures reveal that one protomer of CusA interacts with two monomers of CusB (Figure 9d), providing the first structural information of a protein ratio of 3:6:3 (Figure 9a) for the three component RND efflux systems.58,179,185 Six CusB proteins form an inverted funnel atop of the trimeric CusA pump with the first β-domain of CusB attached to CusA, with the flexible second β-domain acting as a hinge, while the other domains form the central channel. The interior surface of the central channel is populated with negatively charged residues suggesting the capability to bind and transport positively charged metals ions. Binding studies indicate that CusB forms a tight interaction with CusA and multiple co-crystal structures reveal two differing models of the interactions between CusA and CusB. In one model four salt bridges form between D155, R771, R777 and E584 of CusA to the corresponding residues K95, D386, E388 and R397 of CusB. The second model consists of charge-dipole and dipole-dipole interactions from residues Q785, Q194, D800 and Q198 of CusA forming hydrogen bonding to Q108, S109, S253 and N312 of CusB.185
6. Structural data as a guide for the generation of new efflux pump inhibitors (EPIs)
There has been a rapid evolution of scientific technologies since the first structure-based drug design experiments 30 years ago. Advances in cloning, protein expression and purification, proteomics, structural genomics and information technology have greatly accelerated the timeline in which new inhibitors can be discovered.189 Nonetheless, the process from idea to clinic is still a long and arduous process that can take years before a compound becomes available for patient care.190 The information that structural biology provides is essential for the rational design of inhibitory compounds in order to streamline the bench to bedside process.191
The threat of a worldwide outbreak of an antibiotic resistant strain of bacteria requires the requisite effort and resources to develop effective treatments that are currently unavailable. Structural data of RND multidrug efflux pumps from several bacterial species as well as cell-based inhibitory assays have confirmed the idea that these pumps are promising targets.192,193 Their inhibition halts substrate export, thus improving the efficacy of current antibiotics. The determination of high-resolution structures has provided a wealth of information toward the understanding of how RND pumps transport their substrates as well as how current EPIs bind and inhibit this transport process.120,194,195 A number of computational techniques can be utilized to accelerate the discovery of new RND pump inhibitors while significantly reducing costs of screening when compared to cell-based high-throughput screens.196,197 As the number of solved RND pump-EPI-bound structures grows, so does our overall understanding of how to improve upon these compounds for better efficacy and increased pharmacokinetic and pharmacologic properties.
6.1. Small molecule targeting of HAE pumps.
While trimeric RND proteins serve as excellent inhibitory targets due to their essential role in the export of antibiotics, no compounds are currently approved for treatment in patients. Efflux pumps are generally regulated at the level of transcription6, and there are currently no known small molecule EPIs found in bacteria for efflux pump regulation. It is encouraging, however, that several different compound classes significantly decrease the resistance of multidrug resistant bacteria by inhibiting the activities of these pumps.198 Disruption of RND transport activity, in turn, increases the effective concentration of antibiotic within the cell and potentially revives the usefulness of “old” drugs.199 This method of efficacy recovery is exemplified in plants, where species such as Berberis vulgaris and Dalea versicolor produce EPIs in conjunction with antibacterial compounds to effectively restore bactericidal activities against resistant organisms.200–202 Therefore, the inhibition of RND efflux pumps is an excellent strategy to combat multidrug resistance and serves as a high-priority target for the development of new drugs to be used as standalone treatments or, most likely, in conjunction with current antibiotics.
6.2. Types of HAE inhibitors.
To date, to study efflux pump inhibition in a laboratory setting there are two types of molecules currently used as Efflux Pump inhibitors (EPIs): 1) proton conductors and 2) direct inhibitors. Proton conductors, such as carbonyl cyanide m-chlorophenylhydrazine (CCCP), alter the proton motive force used by the RND pumps for substrate export.192 CCCP and other proton gradient disruptors have seen significant use in the laboratory,203–205 but are not specific for the study of efflux pumps. These molecules work by affecting both ΔpH and Δψ across the cellular membrane, altering the proton-motive-force (PMF) available to fuel the pumps.198,206 They are also quite toxic and are seen as having little application clinically due to their non-specific nature and, therefore, are only loosely considered EPIs in this context.
The second class of EPI molecules is direct inhibitors. Direct inhibitors can bind either competitively or non-competitively to the RND pump.192 EPIs can alter the overall affinities for the pumps towards their respective substrates by binding at or near the substrate recognition site (competitive binding), or at a distant site that causes a conformational change within the protein, thus eliminating the substrate recognition site (non-competitive binding). Due to the direct interaction of EPIs with the pumps, they are attractive candidates for future clinical use. A number of classes have been found to act as effective EPIs through small molecule screens of both synthetic and natural compounds, such as pyranopyridines, quinolines and arylpiperazines.202 To date, only three EPI-bound efflux pump structures have been solved: 1) the widely used inhibitor PAβN120, 2) the pyridopyrimidine inhibitor D13–9001207 and 3) pyranopyridine inhibitors derived from the pyranopyridine MBX-2319.195 These inhibitor-bound structures have provided significant understanding of RND transporter function and has laid the groundwork toward future generations of EPI drug design.
6.2.1. PAβN.
In 2001, a family of peptidomimetic compounds were discovered that targeted multidrug efflux pumps in Gram-negative bacteria by screening synthetic compounds and fermentation extracts.208,209 This family of molecules includes the compound PAβN, the most widely utilized and studied EPI to date.192 PAβN is a modified dipeptide-amide that exhibits potent inhibition of drug efflux in the three major RND efflux pumps from P. aeruginosa (MexAB-OprM, MexCD-OprJ and MexEF-OprN) as well as the AcrAB-TolC system in E. coli, showing broad specificity for these different pump systems.209 This made PAβN an exciting molecule for initial EPI development on a clinical level. A crystal structure of PAβN bound to AcrB illuminates how this EPI functions to inhibit drug export (Figure 10a).120 Coupling this data with mutation studies, the critical site was found to be in the periplasmic domain. A number of favorable interactions stabilize the binding of this drug, namely hydrophobic contacts between Phe664 and Arg717 and the naphthylamide and phenylalanine moieties, respectively.120 Participation of backbone oxygen atoms from Pro718 and Leu828 also appeared to stabilize the binding of the ligand, while the amino nitrogen from the Phe moiety of PAβN interacts with the sidechain oxygen of Ser715 (Figure 10b). A number of structural rearrangements are present in the co-crystal structure when compared to the apo structure of AcrB. To accommodate binding of PAβN, Cα3 shifts significantly (3.6 Å shift in the α-carbon of Gln657) along with sidechain rearrangements of up to 8 Å. Despite this structural information it was still unclear how this periplasmic binding site of PAβN inhibits drug export. It was later observed that proximal substrate binding site of AcrB overlaps with its PAβN periplasmic binding site.192 This site is surrounded by residues Asp566, Phe664, Phe666, Glu673, Arg717 and Asn719, most which are also important for PAβN binding. This site plays an important role in the export of large antibiotics such as macrolides, doxorubicin and rifampin,210,211 and explains how PAβN can inhibit macrolides and levofloxacin while export of other antibiotics such as carbenicillin and tetracycline remain largely unaffected.192
Figure 10.
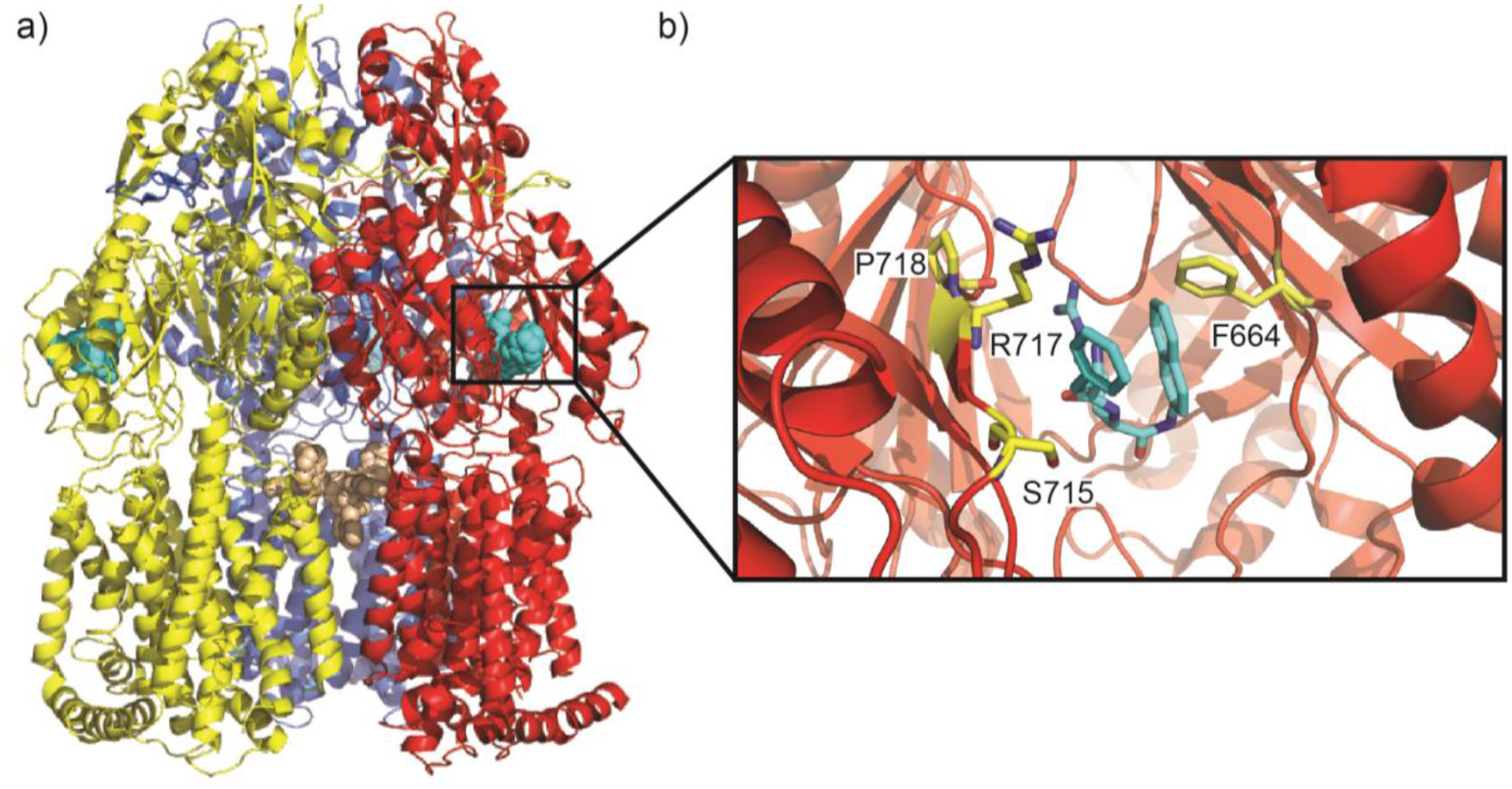
PAβN binds to AcrB (a) Trimeric architecture of AcrB (adapted from PDB ID 1T9Y). AcrB crystallized with three-fold symmetry with PAβN bound at two sites, a periplasmic site (cyan spheres) and the central cavity (wheat spheres). (b) Magnified view of PAβN bound at the periplasmic site. Residues F664, S715, R717 and P718 (yellow sticks) contribute electrostatic interactions to stabilize PAβN (cyan sticks).
Unfortunately, while a potent inhibitor, the development of PAβN was halted due to significant nephrotoxicity in vivo.193,212 This may be due to a secondary effect of membrane dysfunction caused by PAβN.213 Despite the halting of development, the molecule has been widely used as a laboratory benchmark due to its broad specificity for RND multidrug efflux pumps. Moreover, determination of the AcrB-PAβN and its overlap with the proximal binding site provides a starting point for the development of less toxic inhibitors.
6.2.2. D13–9001.
As the search for a clinically-viable EPIs continued, another major class to be discovered was pyridopyrimidines.214 This class was developed specifically to lower resistance to the fluoroquinolone levofloxacin and the β-lactam aztreonam in P. aeruginosa by targeting the MexAB-OprM pump system.215 It was designed to be specific for MexB and not to interact with the other major multidrug exporter of P. aeruginosa MexY, leading to specific targeting. From this class of pyridopyrimidine, D13–9001, was found to be potent in vivo while maintaining high solubility and a good safety profile.192,215 D13–9001 was later found to interact with and inhibit the AcrAB-TolC pump system from E. coli, giving it the potential to perform as a wide-ranging multidrug-efflux inhibitor.
The structure of D13–9001 was solved bound to both AcrB and MexB in 2013.194 Differing from PAβN, the structure showed that D13–9001 binds to the distal binding site of each RND pump. The tert-butyl thiazolyl aminocarboxyl pyridopyrimidine (TAP) moiety is surrounded by a number of Phe residues on both AcrB (F136, F178, F610, F615 and F628) (Figure 11a) and MexB (F136, F178, F610, F573, F615 and F628) (Figure 11b). The tetrazole ring also interacts with similar residues in both AcrB (Q176, S180, N274 and R620) and MexB (K151, D274 and R620). The piperidine aceto-amino ethylene ammonio-acetate (PAEA) moiety takes a slightly different conformation in the structures though, where PAEA is extended in the MexB structure compared to the AcrB structure.
Figure 11.
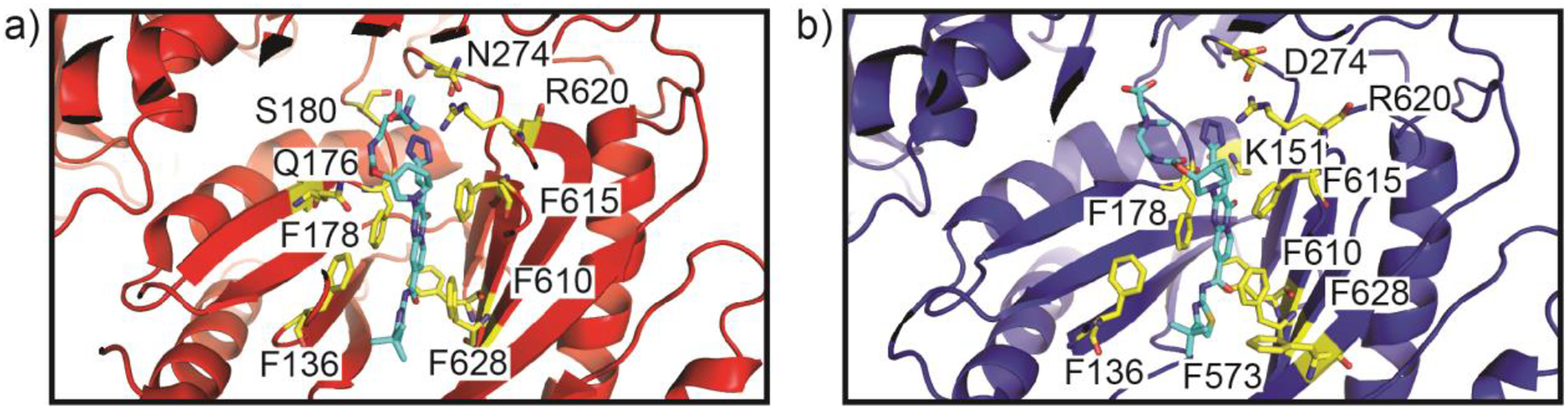
D13–9001 binds to the hydrophobic trap. (a) D13–9001 binds to AcrB (PDB ID 3W9H). The hydrophobic trap is lined with Phe residues while hydrophilic residues contribute important electrostatic interactions. F136, F178, F610, F615 and F628, Q176, S180, N274 and R620 (yellow sticks) stabilize D13–9001 (cyan sticks) at this site. (b) D13–9001 binds to MexB (PDB ID 3W9J). The hydrophobic trap of MexB is similar to that of AcrB. Residues F136, F178, F573, F610, F615, F628, K151, D274 and R620 (yellow sticks) create favorable electrostatic interactions with D13–9001 (cyan sticks).
Based off of this structural information, the binding pocket of D13–9001 was divided into two parts; the substrate export channel and the hydrophobic trap.194 The strong hydrophobic interactions between the TAP moiety of D13–9001 and the string of Phe residues in the hydrophobic trap captures D13–9001 and inhibits export, while the tetrazole and PAEA moieties extent into the substrate export channel and block export of other substrates. The trapping of D13–9001 was also hypothesized to halt conformational changes. While these results provided important information for the continued development of new EPIs, the development of this small molecule for clinical applications was abandoned, likely due to its specificity to only MexB and not MexY in P. aeruginosa193 due to structural clashes in the hydrophobic trap.194
6.2.3. MBX-2319.
More recently, the pyranopyridine MBX-2319 was discovered to inhibited the AcrAB-TolC pump system using a high-throughput screen.216 This molecule represents a new class of EPI molecules. MBX-2319 was found to potentiate antibiotics that are substrates of AcrB such as chloramphenicol, minocycline and erythromycins well as inhibit RND pumps in organisms other than E. coli such as Klebisella pneumoniae and Enterobacter cloacae, along with P. aeruginosa when combined with PMBN, a molecule that permeabilizes the outer membrane.193 Structural studies using a truncated AcrB protein, which only contains the periplasmic domain, found that MBX-2319 interacts in a manner similar to that of D13–9001.195 D13–9001 binds tightly to the hydrophobic trap of AcrB, with favorable interactions between the pyridine ring and F628 and the phenyl and morpholinyl groups and F176 and F615, respectively. A portion of the inhibitor also extends out into the substrate export channel, similar to D13–9001.194 The structural similarities between MBX-2319-AcrB and D13–9001-AcrB substantiate the conclusion that the binding of these molecules by the pump effectively locks the pump into a single conformation and sterically hinders the movement of substrates through the transport channel. Optimizations of MBX-2319 have added acetamide (MBX-3132) and acrylamide (MBX-3135) groups, which have increased the inhibitory activity by 10- and 20-fold, respectively, when compared to MBX-2319.193,195,217 Structural studies show that these MBX-2319 derivatives interact with AcrB in a similar manner as their parent compound. The effectiveness of this pyranopyridine class of inhibitors shows promise for a future combinatorial therapy where specific RND pumps are inoperative, thereby increasing the potency of antibiotics against drug-resistant bacteria. These pyranopyridines are currently under development.193
6.3. Simulated docking to the RND pump.
In order to effectively target RND efflux pumps for antibacterial treatment, new classes of inhibitors must be discovered. A number of computational methods are available to identify and characterize potential EPIs in a rapid, cost-effective manner. After identification of the substrate binding site through structural and mechanistic studies, such as the proximal binding site or hydrophobic trap of the HAE pumps, a large chemical library can docked using a number of docking tools such as Autodock Vina218, Schrodinger GLiDE219,220 and UCSF Dock.221 These programs fit each molecule to the specified binding site and calculate an energy that is then used to rank the overall affinities for each site. Large chemical libraries are available for download through the Zinc database, which contains a large number of subsets such as FDA approved drugs and commercially available molecules.222 After docking, the short list of molecules can be further refined using a final step of simulation. A common approach is the utilization of a molecular mechanic’s force field with the generalized born surface area implicit solvation model (MM-GBSA) to calculate a more accurate binding energy for the final molecules.223,224 While these results must be confirmed in vivo, this is an effective method to generate a small number of candidate molecules from a large subset of data.
A number of studies using computational techniques have been attempted to identify and characterize inhibitors of RND pumps. One example of this is by Mangiaterra et. al., where they identified two new EPIs, morelloflavone and pregnan-20-one, using virtual screening and verified the hits in vitro against MexAB.225 Another example is from Vargiu et. al., where they characterized known EPIs and their interactions with AcrB using a combinatorial docking and molecular dynamics approach,121,192 providing details on small molecule binding in the distal pocket of AcrB. Their simulated results for the inhibitor MBX-2319 were later confirmed through the determination of the bound crystal structure.195 These studies show the potential for EPIs to both be identified quickly and characterized through the combined use of structural and computational techniques.
7. RND inner membrane pump with dimeric architecture
7.1. HpnN: The dimeric butterfly pump.
The Burkholderia Cepacia Complex (Bcc) is a group of naturally occurring bacteria that can severely affect individuals with underlying conditions such as cancer and other diseases where the immune system is compromised.226 These pathogens are especially dangerous to those with cystic fibrosis (CF).227 Bcc in CF patients can cause a rapid deterioration of lung function with increased mortality.228 Interestingly, the severity of Bcc infections of those with CF varies greatly, underlying the fact that little mechanistic information is known about these bacteria. Research thus far has focused on the differences between the spread and infectivity of individual B. cepacia species as well as the discovery of new treatment options. B. multivorans is one of the most commonly isolated Bcc species that poses a threat for a widespread outbreak, and has been found to be deadly even for patients without CF.229,230 This, combined with the discovery of a ceftazidime-resistant mutant strain,231 makes B. multivorans, and other highly infectious Bcc pathogens, a high priority for the development of new antibiotics.
One strategy to combat the spread of Bcc bacteria is to increase the permeability of the outer membrane of the cell, thus allowing for the increased influx of drugs and other biocides. The outer membrane of B. multivorans is stabilized by the insertion of hopanoids. Hopanoids are pentacyclic triterpenoid lipids that are the sterol analogy in prokaryotes.232–234 Incorporation of hopanoids into the outer membrane helps cells mitigate stressful conditions including low pH and high temperature.235 They provide stability and barrier function to the cell,236 and thus are important for the conferment of multidrug resistance.237,238 Incorporation of hopanoids into the outer membrane leads to antibiotic resistance for polymyxin B, colistin (also known as polymyxin E), chloramphenicol and novobiocin.76,237 The hopanoid biosynthesis-associated RND transporter, HpnN, is important for hopanoid trafficking in B. multivorans and other Gram-negative opportunistic human pathogens that are capable of producing hopanoids.74,75 This integral membrane protein shuttles hopanoids from the cytoplasmic to the outer membrane of the organism. Hypersensitivity to certain antibiotics is achieved upon knockdown of the HpnN gene which makes HpnN an attractive target for the development of small molecule inhibitors.76
Contrary to transport pumps with trimeric architecture, HpnN assembles and functions as a homodimer.76 It is currently the only bacterial protein of this class known to function in a homo-dimeric assembly.239 HpnN also appears to function without coordination of a periplasmic membrane fusion protein unlike other RND efflux pumps.240,241 These traits make HpnN unique in the RND transporter family.
7.1.1. Structural characteristics of HpnN.
The first structural information of the HpnN-family transporters was obtained from the B. multivorans HpnN membrane protein. A monomer of B. multivorans HpnN consists of 877 amino acids. In crystallographic structural studies, the protein crystallized as a dimer, and this assembly was confirmed in solution through size exclusion chromatography.76 Similar to other RND-family pumps,154 each monomeric subunit contains 12 α-helices that form the transmembrane domain (TMs 1–12).76 The periplasmic region is made up of two separate loops found between TM 1 and 2 (loop 1) and TM 7 and 8 (loop 2), where loop 1 is made up of 11 α-helices and 4 β-strands, while loop 2 consists of 10 α-helices and 3 β-strands (Figure 12a). The TM domain of HpnN is structurally similar to members of the HAE-HME trimeric class of RND efflux pumps.76 One notable difference is that TM8 is significantly longer than other proteins of this class and extends into the periplasmic domain. Despite this difference, backbone root mean square deviation values of 2.8 Å and 2.7 Å are observed when compared to the TM domains of AcrB210 and CusA,104 respectively. Therefore, even though HpnN assembles in dimeric form, it still displays significant similarities in arrangement and orientation between the trimeric RND protein family of inner membrane efflux pumps.
Figure 12.
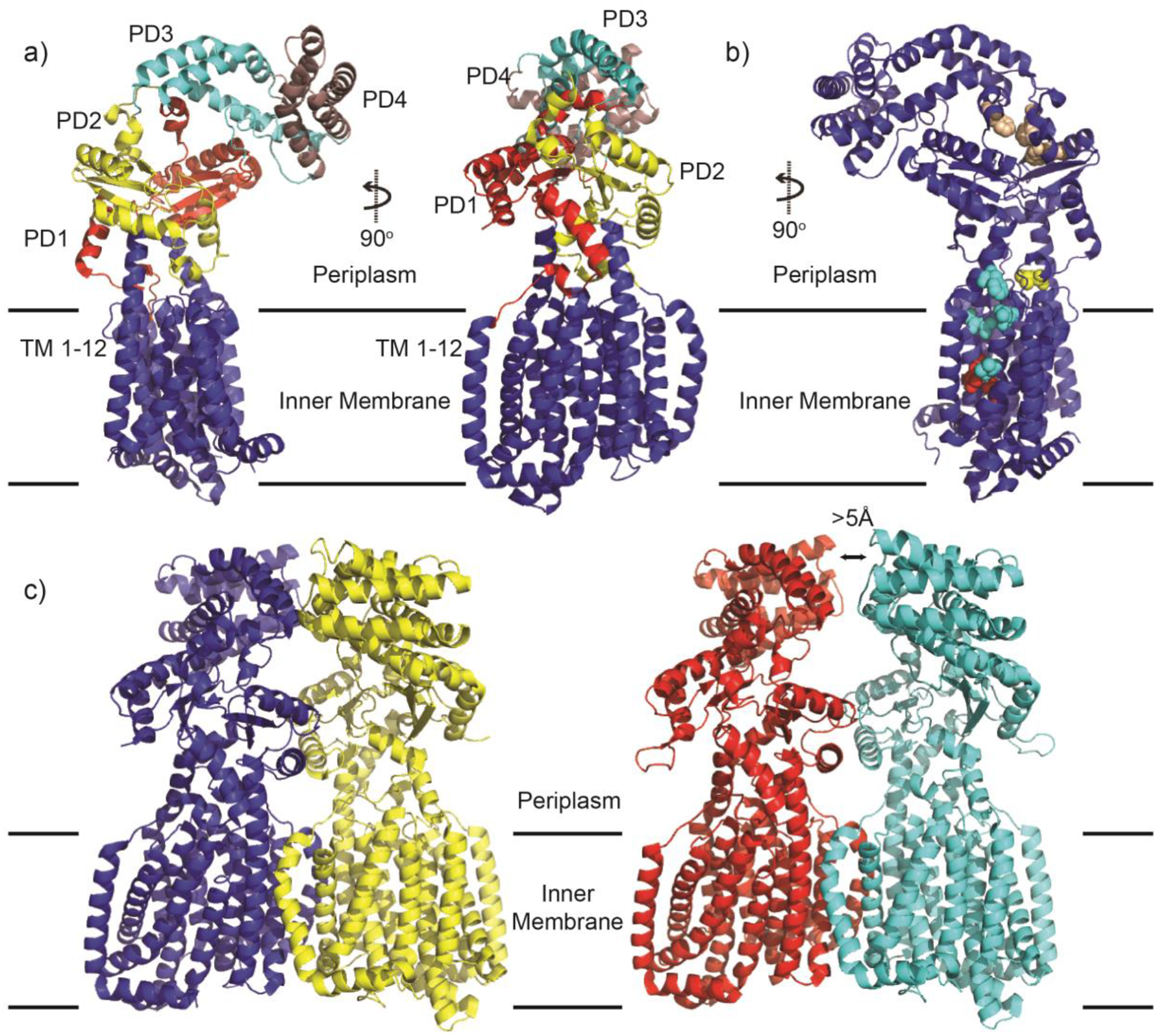
The structure of B. multivorans HpnN. (a) HpnN structural organization. A monomer of HpnN consists of 12 transmembrane domains (TMs 1–12; blue) and four periplasmic domains: PD1 (red), PD2 (yellow), PD3 (cyan) and PD4 (magenta). (b) Important residues in HpnN. Important conserved residues have been identified in HpnN through a combination of structural and mutational studies. Residues F270, V332, V339 and L826 (cyan spheres) form the tunnel entrance and are important for hopanoid recognition in the inner membrane. Residue L48 (yellow spheres) constricts the tunnel and controls the flow of hopanoids through the pump. Residues F117, F541 and W661 (Wheat spheres) are conserved residues that form the exit site of the tunnel. Residues D344, T818 and T819 (red spheres) participate in the proton transport chain (PTC) to fuel the pump. (c) Dimeric forms of HpnN. Form I (left) and Form II (right) differ by a >5 Å swinging motion of the periplasmic domains. This motion constricts the tunnel at L48 and is believed to regulate hopanoid shuttling by HpnN. All figures adapted from PDB ID 5KHN (Form I) and ID 5KHN (Form II).
Despite the TM domain similarities, the periplasmic domains of HpnN are unique to this class RND transporters.76 HpnN contains four distinct periplasmic domains, PDs 1–4 (Figure 12a). PD1 has eight α-helices (α1–5 and α9–11) and four-β sheets (β1–4), PD2 is made up of six α-helices (α12–15, α20–21) and three β-sheets (β5–7), PD3 is make up of three α-helices (α6, α16, α19) and PD4 is made up of four α-helices (α7–8 and α17–18). These periplasmic domains are joined by several flexible loops which suggests that the total periplasmic region of HpnN is elastic in nature, facilitating the movement and release of hopanoids for outer membrane insertion.76 Each protomer is able to form a channel from the outer leaflet of the inner membrane up through the periplasmic domain, and thus, is able to shuttle hopanoids to the cell surface. Whether both protomers are necessary for function is not clear.
7.1.2. The HpnN-hopanoid binding channel.
The hopanoid binding channel begins near the inner membrane surface with a cavity formed between TMs 2,4 and 11, and ends in the periplasm between PDs 1 and 2 (Figure 12b). The initial cavity contains four conserved hydrophobic residues F270, V332, V339 and L826, which likely play an important role in the recognition of hopanoids.76 The tunnel is constricted by the conserved residue L48, located at the narrowest region of the tunnel, which likely controls flow of the hopanoids as they travel the tunnel. Lastly, three conserved hydrophobic residues, F117, F541 and W661, are found at the exit site of the tunnel. Two distinct conformations of the HpnN dimer allow insight into the mechanism of hopanoid transport.76 Significant differences between the two forms are observed, notably within the periplasmic region. Compared to Form I, Form II experiences a significant swinging motion in the periplasmic domain, swinging more than 5 Å and breaking contacts between α6 at the dimeric interface of each monomer (Figure 12c). This swinging motion is believed to facilitate the movement of HpnN between the two separate conformations. Differences within the hopanoid-binding tunnel is seen between the two forms of the HpnN as visualized by unique crystal structures. In Form I, the tunnel is open in both units of the dimer, allowing hopanoids to be transported from the inner membrane to the periplasm. In Form II, the swinging of the periplasmic domains relative to the transmembrane regions effectively constricts the tunnel at L48, inhibiting the transport of hopanoids. Therefore, the two crystal structures are thought to represent biologically relevant conformations and display how this dimeric assembly might function.
7.1.3. The HpnN dimer interface.
Oligomerization of HpnN is created largely through the interactions between TMs 7–9 of each monomer, with smaller contributions from periplasmic helices α6, α14, α18, α21, a single β-sheet, β6, from PD2 and helix α6 of PD3. This region of HpnN is largely hydrophobic in nature, therefore, dimerization is likely to be stabilized through van der Waals interactions.76
7.1.4. The proton relay network of HpnN.
As with all of the RND transporters described in this review, HpnN utilizes the proton motive force (PMF) in order to provide energy for hopanoid transport from the inner membrane to the periplasm.76 A triad of conserved residues that are proton donors/acceptors less than four Å away from each other are found in the transmembrane region of HpnN. These residues are D344, T818 and T819. Aspartate and threonine residues have both been shown to take part in PMF pathways previously,242 and the triad is organized in a manner that would allow the flow of protons from T819 to D344 and, finally, T818 (Figure 11b). The remainder of the pathway is likely facilitated by nearby residues or water molecules.
7.2. HpnN as a drug target.
Although HpnN plays an important role in the overall antibiotic resistance of B. multivorans, it may also serve other functions within the cell such as assembly of protein-lipid microdomains important for recruitment of the cell division machinery.74 Phylogenic analysis reveals that HpnN and eukaryotic steroid transporters evolved from a common ancestor.74 Understanding the mechanisms of hopanoid binding and transport in bacteria may offer insight into sterol trafficking in multicellular organisms and allow the design of more efficient compounds to combat abnormalities in steroid hormone production, metabolism and mechanisms of action.243
Currently, the development of inhibitors for HpnN has been underexplored. This may be due to challenges brought on by this unique dimeric pump system. Trimeric HAE and HME pumps (AcrB, MexB, CusA, etc.) typically have three channels that allow for the binding of compounds located at or near the inner-membrane as well as substrates within the periplasm.244 The presence of multiple binding sites coupled with the promiscuous nature of the trimeric pumps has allowed several substrate-bound structures to be determined. These structures have advanced our structural understanding of RND-bound transporters and have aided in the rational design and development of new inhibitors. Contrary to this, only a single entrance site for the hopanoid tunnel has been found for HpnN, beginning in the outer-leaflet of the inner membrane.76 This site would likely require inhibitors to intercalate into the inner-membrane in order to enter to tunnel, thus limiting the ability to capture HpnN in its substrate-bound conformation. Therefore, strategies to develop small molecule inhibitors of HpnN will likely have to differ from the trimeric RND inhibitor development, where a number of small molecules have been discovered that enter the channel but cannot be pumped, halting conformational changes and “clogging” the pump to inhibit substrate export.195,207 For HpnN, it may be possible to exploit the differences between the two forms of the HpnN dimer, where the channel is either open (Form I) or closed (Form II). The two flexible loops connecting TM1 to TM2 appear to be flexible and form a “hinge” to facilitate dynamic changes between these two forms.76 Targeting this region with small molecule inhibitors could restrict conformational changes between the two forms and alter the biological function of this important protein.
To date, there has yet to be a structure of the hopanoid-bound form of HpnN. Determining the structure of hopanoid bound to HpnN at one or multiple stages of transport (at the inner membrane, in the middle of the channel or upon exit at the or near the top of the periplasmic domain) would identify additional important HpnN-hopanoid interactions that can be targeted through structure-based drug design. Nonetheless, the crystal structure of HpnN has allowed for the identification of important residues for cell growth and hopanoid binding.76 Residues identified include those important for conformational flexibility (L48), the docking of hopanoids (L826), the hydrophobic hopanoid transport channel (F117, F541, W661) and proton transport (D344, T818, T819) (Figure 12b). As hopanoids and their transporters are not essential in certain strains such as B. thailandensis and Rhodopseudomonas palustris TIE-1, these strains allow for experimental verification of structural-based hypotheses. Engineered mutations at these important residues severely inhibit growth of B. thailandensis. This suggests that inhibition of HpnN is a viable strategy to combat Bcc infections and other bacteria where hopanoid deposition in the outer membrane is important for membrane rigidity and antibiotic exclusion from the cell.
8. Monomeric RND inner membrane pumps
8.1. MmpL3: The single-handed mycobacterial pump.
In the previous sections, we have examined how RND efflux pumps can function as a trimer in coordination with other periplasmic adaptor proteins and OMPs (AcrB, MtrD, CmeB, AdeB, CusA) or as a dimer (HpnN). Both types of pumps have distinct yet similar methods for substrate transport. Currently, there are only a few RND pumps that have been shown to transport their substrates in monomeric form. In eukaryotic systems, examples include Niemann-Pick C1 (NPC1) and human patched1 protein. NPC1 functions as a monomer that transports cholesterol out of endosomes. Mutations that inhibit this process result in the human Niemann-Pick Type C disease.245,246 Human patched1 protein is a member of the well-studied hedgehog signaling pathway. The binding of Sonic Hedgehog to Patched1 turns on signaling important for embryogenesis and tissue regeneration.247,248 In bacteria, the only known RND pump to function as a monomer is MmpL3. MmpL3 belongs to the mycobacterial membrane protein Large family and is essential for mycobacterial growth and survival. Its main function is to transport trehalose monocycolate, TMM, from the inner membrane to the cell envelope where it is either converted into trehalose dimycolate, TDM, or attached onto arabinogalactan. MmpL3 is classified as an RND transporter categorized into the hydrophobe/amphiphile efflux 2 (HAE2) subfamily (Figure 1).61,249
8.1.1. Structural characteristics of MmpL3.
Recently, multiple x-ray crystallographic structures of MmpL3 from Mycobacterium smegmatis have been reported.77,78 M. smegmatis is a benign strain of mycobacteria that allows experiments to be performed without increased biosafety precautions as needed when working with Mycobacterium tuberculosis (Mtb). Additionally, M. smegmatis MmpL3 is a viable substitute for Mtb MmpL3 as these two proteins share significant sequence homology and Mtb MmpL3 is able to rescue the viability of an M. smegmatis MmpL3 null strain, suggesting that these two orthologs can substitute for each other in vivo.79 MmpL3 is an RND transporter with 1,013 amino acids. An attempt to determine the full length MmpL3 structure resulted in a 3.3 Å resolution structure. However, a detailed inspection indicated that more than 60 residues at the C-terminal end of this membrane protein was missing. It has been found that this C-terminal domain (amino acids 773–1013) is unimportant for the overall transport function of the protein250, although it has been suggested that this region may be responsible for its oligomerization state in vivo.81 Since this region contains 52 proline residues, it is likely to be easily proteolytically cleaved and unstable in solution. Indeed, when purified, the full length protein is preferentially cleaved to remove this domain.77,78 Therefore, MmpL3 constructs consisting of amino acids 1–748 fused with T4 lysozyme at the C-terminal end78 or amino acids 1–776 alone77 were used for structural determination. The MmpL3 structures reported represent the bound form of MmpL3, as they either contain lipid substrates or one of several antimicrobial compounds previously shown to inhibit mycobacterial cell growth and replication. The apo-MmpL3 form has not yet been determined, perhaps because of the difficulty in removing all lipids during large scale MmpL3 protein preparations.
MmpL3 crystallizes as a monomer with a two-fold pseudo symmetry. The overall architecture of this molecule consists of 12 TM and two large periplasmic loops, as predicted (see Figure 13a and 13c). These loops assemble into two periplasmic domains, PD1 and PD2, which are located between TM1 and TM2 and TM7 and TM8 respectively. PD1 is composed of four α-helices and three β-strands. Most of PD1 is formed from amino acids within the first periplasmic loop between TM1 and TM2, however, residues 437 to 448 from periplasmic loop two form a helix that intertwines in PD1 to help shape its overall structure. PD2 consists of three α-helices and three β-strands, mainly from amino acids of periplasmic loop two between TM7 and TM8. Similar to PD1, an additional α-helix from residues 49–60 is located within PD2. The cross-over of residues from each loop help to define the structural characteristics of the PDs. This, combined with the elongated TM helices 2 and 8 as well as long flexible linkers at the C-terminal ends of TM1 and TM7 form the basis of the spatial orientation of PD1 and PD2 with respect to each other as well as their proximity to the inner membrane.
Figure 13.
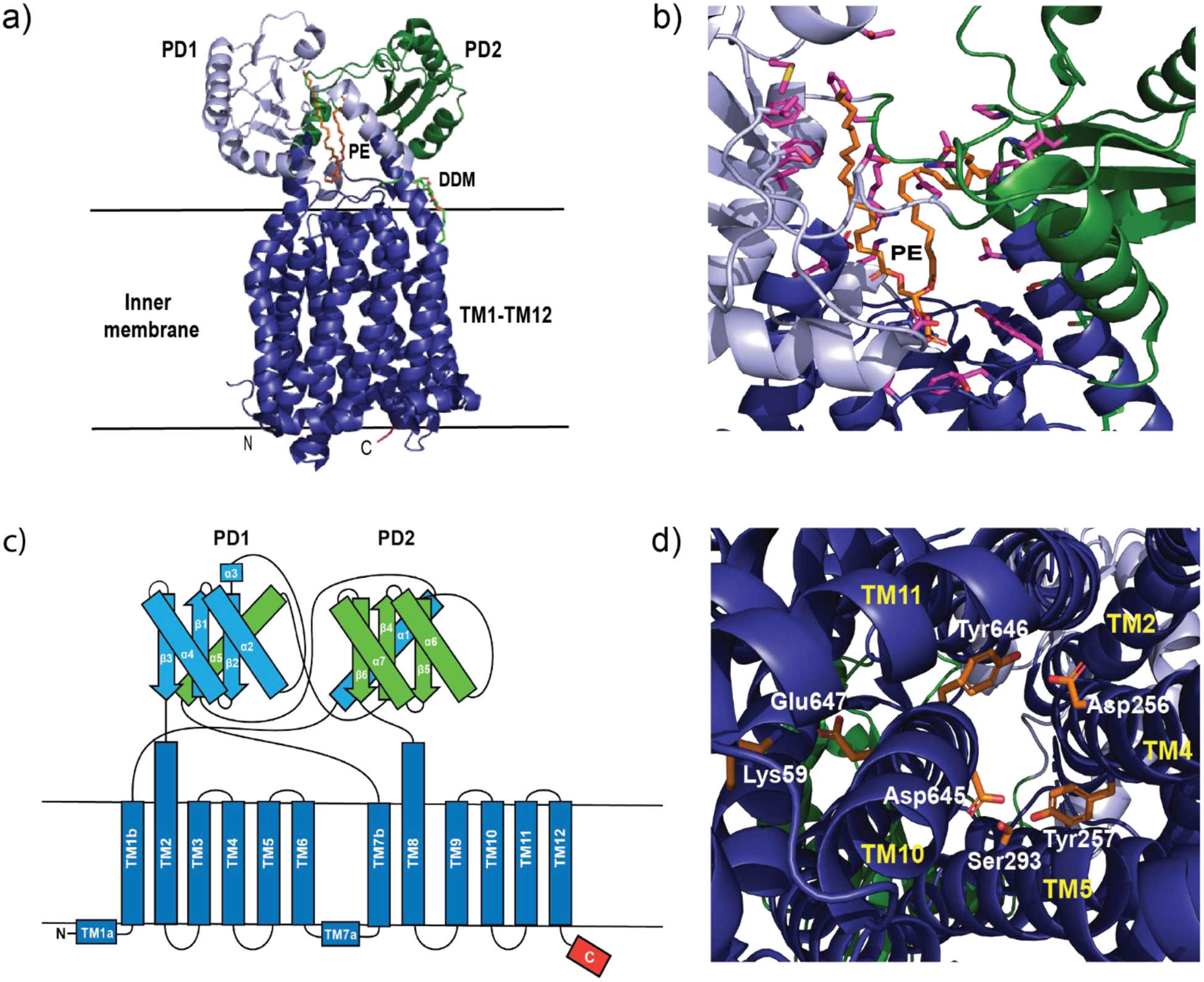
The structure of M. smegmatis MmpL3. (a) Ribbon diagram depicting the crystal structure of MmpL3. Colors are consistent for all panels. Dark blue represents the 12 α-helices that constitute the transmembrane region of the protein as well as two small α-helices (TM1a and TM7a) that are located on the inner leaflet of the inner membrane. The TM helices are mostly embedded within the inner membrane with TM2 and TM8 extending into the periplasm. PD1 and PD2 are tethered to the membrane through flexible loops, allowing significant mobility and interaction between the two periplasmic domains. PD1 is light blue along with the helix α5 (green). PD2 is green with the helix α1 (light blue). Other features include the start of the cytoplasmic c-terminal domain (red), DDM detergent molecule (green and red) and PE located in the cavity formed by PD1 and PD2 (orange). (b) magnified view of the PE binding pocket. The side chains of MmpL3 that can stabilize PE binding are shown in purple. (c) Cartoon schematic identifying each secondary structural component. (d) Magnified view of important side chains for proton translocation (orange) TMs are labeled in yellow. All figures adapted from PDB ID 6OR2.
8.1.2. The MmpL3-TMM binding channel.
PD1 and PD2 orient to form a central channel originating from the outer leaflet of the inner membrane up to the periplasmic domain. A large hydrophobic pocket created by TMs 7–10 opens up to this central channel. This hydrophobic center is postulated to contain one or more TMM binding sites. Overall, there appears to be three openings in the periplasmic domain.78 Lauryl-6-Trehaloside (6LT), a TMM mimic, was seen bound at multiple locations, suggesting a possible path for substrate transport. Additionally, a separate distinct ligand, co-purified and co-crystallized with MmpL3 was identified.77 Within the central cavity created by PD1 and PD2 of the periplasmic domain was a ligand of approximately 700 Da. Liquid chromatography coupled with mass spectrometry identified this ligand as phosphatidyl ethanolamine (PE). The binding of PE is extensive with conserved amino acids lining the central cavity participating in stabilizing this molecule (Figure 13b). Further analysis revealed that other lipids similar to PE, such as phosphatidyl glycerol (PG), phosphatidyl inositol (PI) diacylglycerol (DAG) and cardiolipin (CDL) can also bind MmpL377, suggesting that this transporter is capable of binding and shuttling a variety of lipids synthesized within the cell to the periplasm and/or the outer membrane. MmpL3 has also been suggested to be involved in heme uptake through transfer from Mtb protein Rv0203251. This allows faster heme transport into the cell than from simple dissociation from Rv0203 itself. The heme is then degraded in the cytoplasm and iron released.251,252 Since siderophore-driven iron acquisition is critical for Mtb virulence,253 it suggests that iron deprivation is a viable strategy to help eliminate Mtb infection. Indeed, limiting iron has been reported to increase the effectiveness of well-established Mtb antibiotics.254 The absence of iron results in decreased MmpL3 synthesis, decreased surface TMM deposition, and as a result, increased membrane permeability and fluidity.254 Thus, pharmacologic or genetic depletion of iron creates a synergistic effect when combined with Mtb-targeting drugs. To date, little has been delineated about the influx function of this protein. It is possible that limiting heme (iron) uptake may result in a negative feedback loop that limits MmpL3 expression and consequently, lipid transport. Further experiments are required to fully understand the scope and mechanism of heme transport.
8.1.3. The proton relay network of MmpL3.
MmpL3 has conserved hydrophilic residues interspersed within the TM helices. Certain residues on adjacent TMs are positioned to form hydrogen-bonded pairs. TM4 and TM10 are connected through Asp256-Tyr646 and Tyr257-Asp645, residues conserved throughout almost the entire MmpL family and critical for the growth of mycobacteria in culture.255 Additionally, these pairs are located in similar regions to other Asp-Asp-Lys and Asp-Asp-Tyr triads from other transporters confirmed to be critical for proton translocation.125,256,257 Conserved residues Ser293 and Glu647-Lys591 may also contribute to the network for proton transfer of MmpL3, as most of them are deemed important for the function of this transporter (Figure 13d).250 This proton relay network is critical for the function of MmpL3, as it supplies the energy required for lipid transport to the outer membrane.
8.2. MmpL3 mechanism of action.
The structural and biochemical information of MmpL3 has provided a preliminary view of a unique RND transporter that is proposed to function as a monomer, although it cannot be ruled out of assembling in higher oligomeric states when located in the inner membrane of intact mycobacteria. Previous studies indicate that MmpL3 can act as a flippase.258 Through a spheroplast assay, MmpL3 was found to be critical for the translocation of TMM across the membrane of intact spheroplasts to reside on its outer leaflet. This process was effectively blocked upon the addition of known MmpL3 inhibitors. The structures of MmpL3 indicate that there are up to three openings within the periplasmic domain. TMM that is positioned in the outer leaflet can be extracted from the membrane and transported from the entrance of the channel (formed by TMs 7–10) up through the central cavity of the periplasmic domain formed by PD1 and PD2. TMM is then released to the inner leaflet of the outer membrane to be used for TDM and mAGP synthesis. Substrate transport is powered by the PMF of the cell with protons being shuttled into the cell through a series of conserved hydrogen-bonded hydrophilic residues. Whether the flippase activity is coupled with its MmpL3 lipid transport and release is still unknown. It is possible that there are helper proteins that aid in one or both of these functions. Presently, whether it acts alone or in conjunction with other factors is still unclear.
8.3. MmpL3-protein interactions.
Little is known about how MmpL3 assembles within the cell. As a monomer, it is already distinct from all other known RND transporters. As described above, RND inner membrane pumps mostly assemble as trimers, with periplasmic adaptor proteins and outer membrane factors forming the complete efflux complex in 3:6:3 stoichiometric ratios. While it is possible that MmpL3 can function as a singular protein within the membrane, it is more likely that other adaptor proteins aid in this process. A bacterial two-hybrid system using MmpL3 as bait identified 19 potential interacting proteins.81 Surprisingly, a low number of proteins involved in mycolic acid biosynthesis or transport were found. Only TmaT, a mycolic acid acyltransferase required for TMM export and cell viability was identified. Instead, known functions of potential MmpL3-interacting proteins include the synthesis and export of peptidoglycan, arabinogalactan and lipoglycans, the cell regulatory protein, CrgA, the elongation-specific regulatory factor Wag31 as well as several with unknown functions. Taken together, these data suggest that, in addition to its import/export functions, MmpL3 may serve as an anchor in the coordination of new cell wall deposition with cell elongation and division at the pole and septum of growing cells.81
A separate report identified the secreted lipoprotein LpqN/Rv0583c (Mtb gene designation) through a yeast-2 hybrid assay that interacted with PD2 but not PD1 of MmpL3. LpqN is a small, 228 amino acid-interacting partner of both MmpL3 and MmpL11. LpqN interacts with lipid biosynthetic enzymes, notably mycolyltransferase Ag85A, and is thought to interact as a periplasmic adaptor protein to link synthesized lipid (TMM) to proteins near the cell envelope. To help confirm its in vivo role, the crystal structures of LpqN were solved to high resolution, with and without bound lipid substrates. The apo structure of LpqN contains three α-helices and seven β-strands with the interior oriented to form a shallow cavity, a potential ligand binding site (Figure 14a). The interior wall of this cavity consists mainly of hydrophobic and polar residues including Leu-87, Ile-121, Val-183, Ala-184, Leu-201, Ala-203 and Met-214, which would allow amphiphilic lipid substrates to bind. The crystal structures of LpqN with dodecyl trehalose (6LT) and trehalose 6-decanoate (T6D) show a pronounced shift in both secondary and overall structure to further enhance lipid binding (see Figure 14b and 14c). This structural shift provides a deeper binding pocket to better accommodate the long hydrophobic acyl chain. Since the water-soluble 6LT and T6D are structurally similar to the insoluble lipid molecule TMM, they are ideal mimetics to help elucidate the role of LpqN in lipid transfer. This combined data gives insight into the mechanism of TMM transfer from export from the inner membrane through MmpL3, with either direct or indirect transfer of TMM to LpqN and delivered to the cell envelope. Attempts to establish a binding between LpqN and MmpL3 were unsuccessful. Since a lpqN-deficient Mtb strain displays normal growth characteristics, it is likely that additional proteins with redundant functions to LpqN exist. Whether other membrane associated or periplasmic adaptor proteins are involved to solidify the link between these two proteins is yet to be established.
Figure 14.
Structures of MmpL3-associated proteins. (a) Crystal structure of LpqN (adapted from PDB ID 6E5D). The central ribbon is depicted in green with α-helices in blue and loop regions in pink. (b) and (c) Overlay of substrate bound LpqN with the apo-form shown in (a). Arrows point to areas of significant shift upon substrate binding, within the central ribbon (compare light green (bound) to dark green (unbound)) and the proximal α-helix (compare light blue (bound) to dark blue (unbound)). L6T (b) is shown as orange (carbon atoms) and red (oxygen atoms) and T6D (c) is shown as light grey (carbon atoms) and red (oxygen atoms). L6T-bound LpqN ane T6D-bound LpqN structures adapted from PDB ID 6E5F and PDB ID 6MNA, respectively. (d) Crystal structure of TftA (adapted from PDB ID 6T84). The central antiparallel β-sheet region (tan) is surrounded by α-helices (light blue) and flexible loops (grey).
Recently, additional proteins thought to interact with MmpL3 were identified using a co-purification assay.259 In M. smegmatis, MmpL3 fused with monomeric superfolder GST (msfGST) was inserted into the genome to replace native MmpL3 at its original chromosomal location. Cell membranes were solubilized and MmpL3-GST plus copurified proteins were isolated with anti-GFP nanobodies covalently bound to magnetic beads. These proteins were then identified by mass spectrometry and their effects on mycobacterial viability were tested in both M. smegmatis and M. tuberculosis. The most abundant proteins were MSMEG_0736 and MSMEG_5308. MSMEG_0736 and its Mtb homolog, Rv0383C, were found to be essential for TMM transport and named TMM transport factor A, TftA. MSMEG_0736 and its Mtb homolog, Rv1057, appear to be nonessential for growth and TMM transport and postulated to be stress response proteins that can stabilizes the MmpL3-TftA complex. TftA is a 278-residue protein that co-localizes with MmpL3 at the poles and septa. Its C-terminal domain (amino acids 206–278) is not essential for MmpL3 association or TMM transport. TftA is thought to aid in TMM translocation either by interacting with MmpL3 directly (linking the TMM biosynthetic machinery directly to MmpL3 through TMM binding), or being a scaffold protein that recruits other as yet unidentified complex members. The crystal structure of TftA from M smegmatis (residues 24–205) shows a unique fold comprised of a central antiparallel β-sheet flanked by α-helices.260 (Figure 14d). No known structural similarities have been reported. Structural analysis suggests that there is no obvious binding pocket for TMM or other lipids. Whether TftA interacts with the C-terminal cytoplasmic domain of MmpL3 or via its N-terminal transmembrane helix with the transmembrane domains of MmpL3 is yet to be determined.
8.4. MmpL3 as an anti-mycobacterial drug target.
Despite considerable efforts to develop treatments for tuberculosis (TB), there are an estimated 1.5 million people that still die each year from active TB infection.261 This is in part due the emergence of Mtb strains resistant to some or all of the currently available antibiotics. The standard treatment for TB consists of the first-line anti-TB drugs isoniazid, rifampin, pyrazinamide and ethambutol. Unfortunately, multidrug resistant (MDR) strains have emerged to some or all of these antibiotics.262 Treatment for MDR-TB consists of compounds derived from fluoroquinolone, aminoglycosides, cyclic polypeptides, thioamides, serine analogs and salicylic acid derivatives.263 Occasionally, these second-line antibiotics fail to cure TB infection as extensively drug-resistant (XDR) and total drug-resistant (TDR) strains have emerged. Thus, there is a critical need for the development of unique and effective targets to help battle these difficult or impossible to treat infections. For a list of commonly used anti-tuberculosis drugs with the genes associated with their respective resistance and major mechanism see264. A primary focus has been on the MMPL family of transporters. The Mtb genome encodes 13 mmpL genes whose functions are critical to the survival of the mycobacteria. They have been implicated in the transport of lipids, biofilm formation and drug efflux.265 It is well established that while other members of the MmpL3 family are important for cellular homeostasis only MmpL3 is deemed essential for Mtb survival.
MmpL3 has also been investigated as an antimicrobial target for nontuberculosis mycobacterial infections NTM),266 specifically, from Mycobacteium avium complex, MAC, and Mycobacterium abscessus complex species, MABSC. MAC and MABSC account for up to 95% of all pulmonary NTM infections which are often fatal to people with underlying lung disease. Upon screening compound libraries against M. abscessus and M. avium in culture, several hits were identified that appear to target MmpL3. Therefore, given its central role in cell wall biogenesis combined with its inhibition bactericidal for both Mtb and NTM infections makes MmpL3 an attractive target for mycobacterial antibiotic development.
8.5. Identified inhibitors of MmpL3.
MmpL3 appears to be a very promiscuous target with inhibitors having a variety of diverse chemical scaffolds. The most common approach to find potential MmpL3 inhibitors is to screen small molecule libraries and select those compounds that show bacteriostatic and/or bactericidal properties against mycobacteria. Resistant mutants to these select compounds are then isolated and mutations that confer resistance identified through whole genome sequencing. In many cases, MmpL3 is the target of genetic modification that allows resistance. An interesting approach tried recently was to use a pool of inhibitor-resistant mmpl3 mutants to identify new potential MmpL3 inhibitors through cross-resistance.267 Inhibitors of MmpL3 include: SQ109, BM212, AU1235, ICA38, Rimonabant, NITD-304, NITD-349, E11, HC2060, HC2091, HC2149, HC2169, HC2184, C215, TBL-140, PIPD1 and indole-2 carboxamides among others (Table 1).78,79,276–279,268–275 Most of these compounds have been validated through a variety of in vitro and in vivo techniques (competitive binding, mycobacterial growth and lipid accumulation assays).280 Structure-activity relationship studies are then performed to optimize its chemical structure. Despite the wide range of structural characteristics, many of the effective MmpL3 inhibitors share common traits. They are generally highly soluble with low toxicity and are able to inhibit mycobacterial cell growth and viability through a defect in mycolic acid synthesis/transport. As Mtb typically resides in macrophages, these inhibitors also need to be active in and tolerant to the macrophage environment. Ideally, an MmpL3 inhibitor should be effective in both active TB and in states of non-replicating persistence (NRP). Finding an MmpL3 inhibitor that satisfies all of the above optimal criteria has been a challenge. Because of this, only a few of these inhibitors, notably SQ109, has made it past Stage I clinical trials. Therefore, a detailed knowledge of how these inhibitors bind MmpL3, and how these interaction affects MmpL3 function is required. Additionally, mutations within MmpL3 that confer resistance need to be identified and characterized.
Table 1.
Chemical Structures of MmpL3 Inhibitors.
| SQ109 | |
| BM212 |  |
| AU1235 |  |
| ICA38 | |
| Rimonabant |  |
| NITD-304 |  |
| NITD-349 |  |
| E11 |  |
| HC2060 |  |
| HC2091 |  |
| HC2149 |  |
| HC2169 | |
| HC2184 |  |
| C15 |  |
| TBL-140 | |
| PIPD1 |  |
| THPP1 |  |
| Indole-2 carboxamide |
 backbone backbone |
8.5.1. How MmpL3 inhibitors function.
An important unanswered question is how do MmpL3 inhibitors cause their bacteriostatic/bactericidal effects. Despite modeling efforts, it remained unclear whether these compounds inhibit MmpL3 function by binding MmpL3 directly or indirectly through disruption of the proton motive force or inhibition of a secondary target.281 Inhibition of the PMF by non-specific PMF disruptors may affect the transport function of MmpL3 and cell viability without any direct interaction. Previously, the best evidence for a direct inhibitor-MmpL3 interaction was through competition experiments such as the use of MmpL3-specific labeled dyes that were displaced when increasing amounts of antibiotic were added. Before structural information of MmpL3 became available, important amino acid residue required for mycobacterial growth and TMM transport were determined through site-directed mutagenesis. As a starting point, the entire class of Mtb MmpL proteins was analyzed for areas of significant sequence homology. Those residues that were conserved across all or most MmpLs were individually mutated in MmpL3 to cysteine in a Δcys-MmpL3 background. Those that failed to rescue viability in an mmpl3 null strain were deemed essential. These included D251C (D256), S288C (S293), G543C (G548), D640C (D645), Y641C (Y646), D710C (D715)and R715C (R720)250 (residues in parentheses correspond to the M. smegmatis nomenclature – a non-virulent mycobacterium whose MmpL3 ortholog can rescue cell viability and cell wall integrity of an Mtb mmpl3-null strain). All of these residues are predicted to map to the central core of the protein within the TM domain. Many resistance mutations to well-established inhibitors such as SQ109271, BM212269,282 and indoleamides272 also map to this region. It was posited that these inhibitors diminish MmpL3 function due to disruption of the proton translocation pathway rather than inhibit lipid (TMM) binding directly. The possibility was also suggested that these compounds do not bind to MmpL3, rather disruption of the proton motive force of the cell indirectly inhibits MmpL3-guided TMM transport, and as a consequence, promotes cell death. As the number of identified MmpL3 inhibitors grows in the literature, so does the number of identified MmpL3 resistance mutations. Mutations have been mapped to positions Q40 (Q40), V240 (V245), I244 (I249), Y252 (Y257), G253 (G258), V285 (V290), L299 (L304), T311 (T316), R373 (R378), L566 (L571), I585 (I590), S591 (S596), V643 (V648), F644 (F649), A662 (A667), L693 (L698) and L699 (L704) (Figure 14a – red and cream colored spheres), among others and are located not only within the central transmembrane region but also in the periplasmic domain which forms the central cavity for lipid transport79,271,273–275,277,278,283,284 This diversity of mutations suggests that compounds can exert their inhibitory effects in a variety of ways. Therefore, the link between MmpL3 and drug resistance has not yet been fully elucidated.
8.5.2. Structural characteristics of MmpL3-inhibitor binding.
Recently, the crystal structure of M. smegmatis MmpL3 was solved with bound inhibitors: the 1,2-ethylenediamine SQ109, the adamantly urea AU1235, the indolcarboxamide IC38 and rimonabant, an analog of the 1,5-diarylpyrrole derivative BM212.78 This was the first direct evidence of an MmpL3-inhibitor structural interaction. SQ109 is perhaps the most promising MmpL3 inhibitor to date. It has been shown to successfully inhibit growth of both Mtb and non-replicating mycobacteria in a dose-dependent manner.78,266 Structural determination of the MmpL3-SQ109 complex shows the inhibitor is bound in an extended conformation near the center of the transmembrane region (Figure 15b). There are noticeable shifts observed in six of the 12 TM helices (TM4, TM5, TM6, TM10 and TM11). This allows the narrow cavity within the TMs to open up and accommodate SQ109, with two phenylalanine side chains (F640 and F649) swinging out to make room for the bulky adamantine group. There are total of 18 amino acid residues in direct contact with SQ109, most being conserved across all mycobacterial species. These are I249, I253, D256, Y257, F260, S293, I297, S301, I319, A637, V638, G641, L642, D645, Y646, F649, A682 and L686 (Figure 15a – red spheres). Of special note, both of the Asp-Tyr hydrogen bonded pairs (D256-Y646 and Y257-D645) (Figure 15b – magnified insert), implicated in MmpL3 proton translocation, are disrupted upon SQ109 binding. Previously, site-directed mutagenesis studies have confirmed the importance of these Asp-Tyr pairs.250
Figure 15.
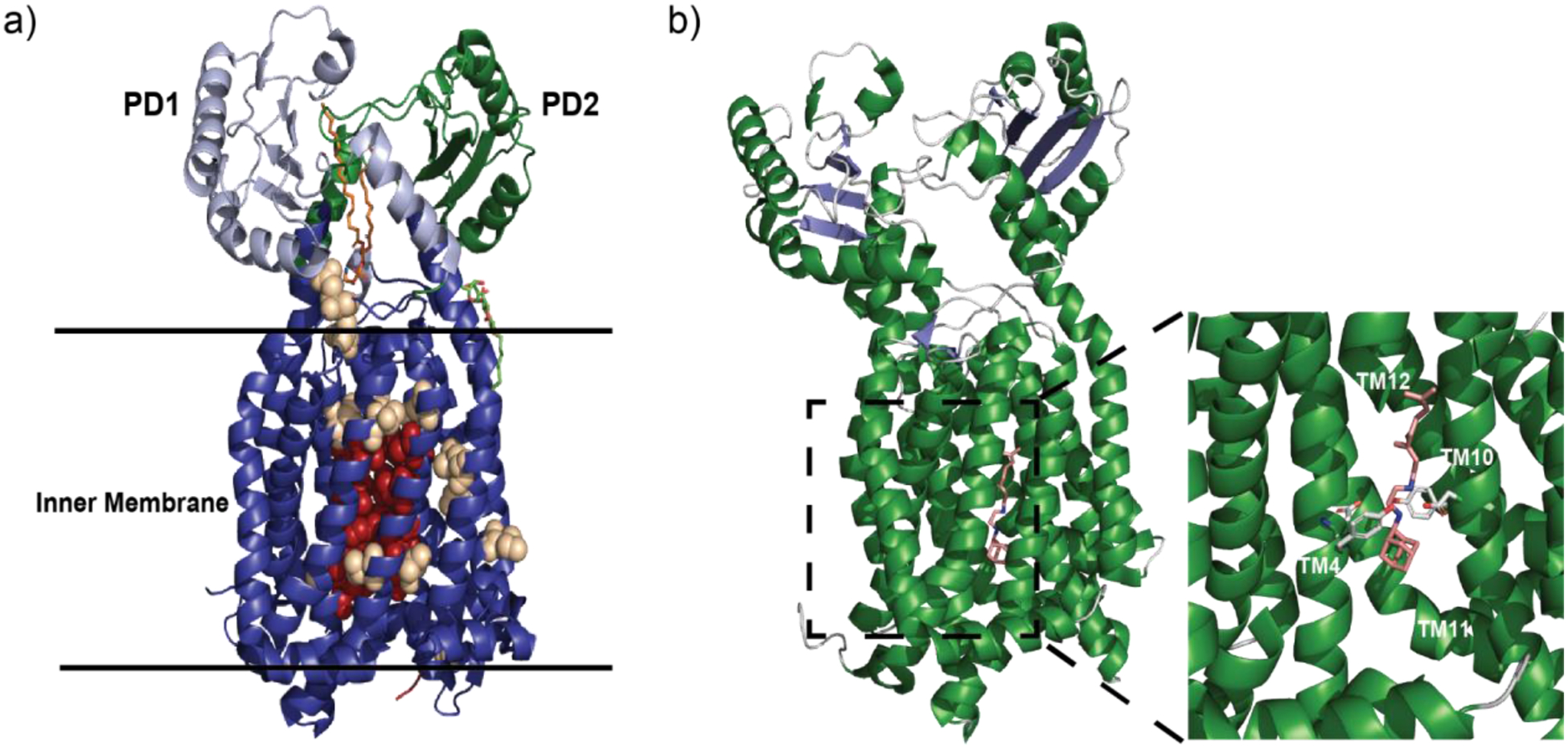
MmpL3 as a drug target. (a) Mutations within MmpL3 are able to confer resistance to a variety of antibiotics. Red spheres represent residues whose side chains can directly affect the putative binding site of SQ109 and other MmpL3 inhibitors. Tan spheres represent residues that are outside of the inhibitor binding pocket (adapted from PDB ID 6OR2). (b) SQ109 directly interrupts the proton translocation chain in MmpL3. α-helices are depicted in green while β-strands are blue. SQ109 is shown in tan (carbon atoms) and blue (nitrogen atoms) (adapted from PDB ID 6AJH). A magnified view of the SQ109 binding pocket shows how the critical Asp-Tyr pairs are disrupted when SQ109 is present. Side chains are shown in grey (carbon atoms) and red (oxygen atoms). Parts of TM5 and TM6 are removed for clarity.
Both AU1235 and ICA38 bind in similar fashion as SQ109 with small differences based on the size of the head group and the overall length of each compound. All three disrupt the putative proton translocation channel by disruption of the Asp-Tyr pairs. Rimonabant is structurally distinct from the other three inhibitors (Table 1). Although it targets the same general region within the transmembrane domain, its Rimonabant has three arms that protrude into unique pockets within the protein. The pyrazole ring core inserts at the center of the bundle and interacts with I253 (TM4), G641 and L642 (TM10). The three “arms” protruding from this central core insert into separate pockets, with the 2,4-dichlorophenyl ring located in a distinct pocket from those occupied by the other inhibitors. L248, V689, A690 and L708 all form unique interactions due to the branched structural design. As with SQ109, AU12135 and ICA38, Rimonabant disrupts the critical Asp-Tyr pairs important for proton translocation. There have been reported 13 mutations that confer resistance to one or more of SQ109, AU1235 and ICA38 derivatives. Of those, nine are in residues that directly contact the inhibitor in the structure. These mutations all have significant effects on inhibitor binding78 which validates the structural data. Four known resistant mutations, Q40R, I194R, I572P and A705T are not associated with the putative inhibitor binding pocket. In a competition assay these mutations do not alter the KD significantly therefore, it is unclear how these MmpL3 variants confer resistance. Nonetheless, these structural and functional studies have provided the framework in which to understand the binding characteristics of some of the well-studied MmpL3 inhibitors and, as a result, a beginning strategy for structure-based drug design.
8.6. Future design and use of MmpL3-targeted therapeutics.
The fundamental understanding of how select inhibitors interact with MmpL3 has been greatly increased with recent structural and functional data. These data allow for the intelligent design of new chemical scaffolds as well as improvement upon existing ones to provide increased MmpL3 inhibition, and as a result, increased bactericidal ability. Many of the current MmpL3 inhibitors are hydrophobic in nature with low solubility. This hydrophobicity allows them to bury with the transmembrane helices and disrupt the proton translocation pathway. However, this low solubility may preclude effective dose administration. Therefore, inhibitors need to be optimized for both solubility and potency. Determination of the crystal structure allows new areas of MmpL3 to be considered for inhibition, such as within the periplasmic domain, where compounds with lower hydrophobicity/increased solubility could be utilized due to the aqueous environment of the periplasm. The well-established resistance mutations at Q40 (located in a loop within the periplasm) among others, resistant to several MmpL3 inhibitors, represents alternative targets separate from those that target the TM region. While most of the characterized inhibitors disrupt the PMF of the mycobacteria these compounds may cause deleterious effects within the host. Compounds that target alternative means of MmpL3 inhibition separate from disruption of the inner membrane proton motive force may increase overall treatment tolerability.276 Future efforts to identify compounds with alternative scaffolds that preferentially target the PN domains and/or the TMM flippase/translocation pathway exclusively may prove beneficial.
Current resistance mutation data suggests that approximately 80% of the mutations lie within 10 Å of the combined binding pocket described for SQ109, Au1235, IC38 and rimonabant, while the rest reside in other areas of the protein.78 It has been suggested that the strength of resistance of these mutant strains depends upon their proximity to essential residues required for inhibitor binding. Mutagenesis studies have shown that while small changes in side chain density and charge may be tolerated, the effectiveness of the side chain mutation correlates with how they disrupt the binding pocket. It seems that the tolerance of side chain substitutions is stricter within the transmembrane region than it is at the outer regions of the periplasmic domain, most likely because of the increased flexibility in this area. This is important to consider as the spontaneous mutation rate is likely to increase with a compound that targets a flexible area of the protein. In this regard, it may be beneficial to use a combination of inhibitors that give rise to the lowest spontaneous mutation rate. It has also been established that MmpL3 inhibitors such as SQ109 and BM212 have increased effectiveness when combined with some of the currently used antibiotics. This combinatory therapy is especially important for multidrug and extensively drug resistant Mtb infections. Evaluation of genes affected by MmpL3 depletion shows an up-regulation of genes involved in osmoprotection and metal homeostasis while genes important for energy production and mycolic acids biosynthesis genes are repressed.285 This information provides new targets/pathways than can be exploited in combination with direct MmpL3 inhibition to further enhance the effectiveness of treatment. With any new inhibitor design it is important to consider all of the important factors outlined above. They must have sufficient solubility to deliver adequate potency, low toxicity, optimized for optimal delivery and used in combination with other drugs that would minimize the frequency of spontaneous resistance. Ideally it would also be bactericidal in cases of both Mtb and non-tuberculosis mycobacterial infections. While it is promising that analogs of BM212 Indole-2-carboxamide-based MmpL3 inhibitors have been shown to be effective in MDR, and XDR M. tuberculosis strains in murine TB infection models282,286,287, the fact that few compounds have made it to clinical trials suggests that continued development of novel compounds is warranted. With the amount of research done thus far, the successful design of an Mtb or pan-mycobacterial antibiotic treatment regimen including MmpL3 inhibition is a possible, if not probable, advancement for mitigation of mycobacterial infections.
9. Conclusion
It was thought that the RND superfamily of transporters largely function as trimers as evidenced by the oligomerization states of HAE-subfamily efflux pumps, including AcrB,119 MexB,288–290 MtrD135 and CmeB,99 as well as HME-subfamily proteins, such as CusA104,179,185 and ZneA.168 However, our recent studies have shown that the B. multivorans HpnN membrane protein, belonging to the HpnN (HAE3) subfamily of RND transporters, was crystallized as a dimer.76 We also have shown that the M. smegmatis MmpL3 transporter, which is affiliated with the MmpL subfamily of RND proteins, functions as a monomer77. These observations are in line with the eukaryotic RND-type transporters NPC1246 and PTCH1,247,291,292 where these human proteins also assemble as monomers. Our studies indeed pose a strong likelihood that different subfamilies of these RND membrane proteins are able to assemble into various oligomerization states to function.
The RND transporters represent very important drug targets. However, it is always a challenging process to develop new inhibitory compounds. To date, most compounds that target these systems with high efficacy have either low solubility, high toxicity or both. To successfully advance in structure-based inhibitor design there exist both 1) technical and 2) technological innovations that will shape the future of RND transporter-bases therapeutics.
9.1. Technical Innovations.
Membrane proteins are historically difficult to purify in sufficient quantity for structural analysis. This is especially the case for eukaryotic membrane proteins, where the traditional expression systems, such as E. coli and insect cells, often produce low yields of unstable product prone to aggregation. These difficulties often lead to modification of the construct, with domains shortened or exchanged, thus changing its fundamental nature. In addition, the native lipid bilayer, imperative for the overall function of membrane proteins and protein complexes, is missing. The absence of this lipid bilayer also creates instability as lipid and lipid-associated molecules are critical for stabilization of the overall structure. Nonetheless, detergent extraction has led to successful structural determination of a majority of the RND transporters and other membrane proteins currently found in the protein data bank. To facilitate successful removal of these proteins from their cellular location, however, it is often a laborious process to identify a detergent that both solubilizes the membrane protein and keeps it in its functional state. Common detergents such as n-dodecyl-β-D-maltoside, (DDM) or 6-cyclohexyl-1-hexyl-β-D-maltoside (Cymal-6) historically have been used with good results, but the absence of native lipid may exclude important structural features critical to obtain a complete mechanistic view of substrate transport. Thus, alternatives to detergent-based extraction, such as lipid bilayer mimetics which include bicelles, lipid cubic phase and nanodiscs derived from detergent-free systems (native cell membrane nanoparticle system (NCMNS)157 and styrene–maleic acid lipid particles (SMALPs)293 may result in structures of RND transporters, with and without bound substrate, in their native lipid-associated state. This can be achieved not only with standard x-ray crystallography, but also with cryo-EM and other techniques that focus on determining structures of these important membrane proteins.
9.2. Technological Innovations.
Structure-based inhibitor design for targeting RND transporters has been predominantly based on x-ray crystallographic data, which has its inherent drawbacks. X-ray structures typically only display a singular static conformational state. This provides limited insight with regards to the dynamic nature of these proteins, a critical component necessary for drug discovery, although single-molecule fluorescence resonance energy transfer (sm-FRET)99 and molecular dynamics (MD) simulations14 can allow for unfolding the important functional dynamics of these membrane proteins. Recently, the technological advances in electron microscope optics, sensors and data analysis software294 for the techniques of cryo-EM, cryo-electron tomography (cryo-ET), x-ray free electron laser (XFEL) and x-ray tomography have greatly facilitated our capabilities to quickly generate structural data outside of the typical method of x-ray crystallography. Cryo-EM is an imaging technique that collects single-particle images at random orientation in a frozen-hydrated state. Theoretically, as long as we can obtain a sufficient number of single-particle images, cryo-EM should be able to allow us to obtain structures of relatively large biomacromolecules of greater than 100 kDa to a near atomic resolution. This technique also offers the capacity of solving various structures at different conformational states of a biomacromolecule to high resolution from a single cryo-EM sample122,295. XFEL allows for the serial determination of structures and dynamics of membrane proteins at room temperature, which is more physiologically relevant. This exciting technique can allow us to observe the detailed action and motion of target membrane proteins during a biological process at high resolution. Both cryo-ET and x-ray tomography are novel cell imaging technologies that allow for imaging intact cell structure and morphology at physiological conditions, although tomographic data are still taken in a frozen-hydrated state. Owing to recent technological advancements, cryo-ET can now allow us to elucidate structures of membrane proteins and membrane protein complexes in intact cells to near atomic resolution. It is our hope that we will be able to study cell biology and critically important membrane proteins in intact cells at a near atomic resolution.
In many cases, structures determined from these methods will more accurately display how RND inner membrane pumps are assembled within the cell membrane and allow optimization of inhibitory compounds based on the conformations of these proteins in the solution state. Additionally, it will not be unusual to detect multiple conformations of an RND pump or unique assembly states of entire RND transport systems in a single cryo-EM grid. Historically, as referenced above, structural determination requires homogenous and pure samples. Sample heterogeneity often hampers the progress of drug design and development, especially those that target membrane proteins and complexes, where sample preparation and crystallization tend to be more challenging and time consuming. We recently developed an iterative methodology, designated “Build and Retrieve” (BaR), that allows us to identify and solve cryo-EM structures of a variety of inner and outer membrane proteins, including membrane protein complexes of different sizes and dimensions, from a heterogeneous, impure protein sample. In addition, the BaR methodology is capable of leading us to elucidate structural information from E. coli K12 crude membrane and raw lysate. Importantly, the work highlights the potential of cryo-EM for systems structural proteomics. In the near future, the techniques of cryo-EM may allow us to obtain important structural information, possibly at the atomic level, of how various membrane proteins interact to assemble as a functional complex or form an enzymatic chain in native membranes. Coupled with the technique of native mass spectrometry (nMS),296 it is expected that cryo-EM will soon be able to facilitate the study of membrane proteome and elucidate how membrane proteins contact each other within native membrane.
Alternatives to detergent-based membrane protein extraction may also be useful for these emerging technologies to produce meaningful data of RND transporters with native lipid, thus allowing insight into the protein-lipid stability and dynamics of these systems. The heterogeneity of membrane samples, a common experimental problem, may no longer be a significant roadblock for meaningful data generation, as it is already feasible to generate several complete structures of membrane proteins and other membrane associated factors, from a single membrane extraction. Thus, with cryo-EM, it is no longer imperative, and perhaps even beneficial, to have a heterogenous, impure sample.
The ability to determine the structure of RND pumps in their solution state, alongside cofactors, membrane-associated proteins and lipid will greatly advance our understanding of their protein dynamics as well as substrate/inhibitor/membrane interactions. A recent example shows how structure-guided modification of mefloquine to piperidine resulted in the 2-fold potency enhancement towards the Plasmodium falciparum 80S ribosome.189,297 This, combined with the robust chemical libraries available and advanced docking tools will rapidly advance the identification and validation of new inhibitors for RND-efflux pumps as well as many other proteins that are potential targets for drug development.
Acknowledgments
E.W.Y. would like to acknowledge the past and present members of the Yu lab whose contributions were critical in solving the structures of several of the RND-type inner membrane pumps described in this review, especially Chih-Chia Su, Feng Long, Jani Reddy Bolla and Hsiang-Ting Lei. This work is supported by U.S. NIH Grant R01 AI145069 (E.W.Y.) and based upon research conducted at the Northeastern Collaborative Access Team beamlines of the Advanced Photon Source, supported by award GM103403 from the National Institutes of General Medical Sciences. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Additionally, data acquisition and analysis for structures solved by cryo-electron microscopy was supported by the Cryo-Electron Microscopy Core at Case Western Reserve University as well as the National Cancer Institute’s National Cryo-EM Facility at the Frederick National Laboratory for Cancer Research under contract HSSN261200800001E
We would also like to thank the reviews for their thorough critique and many helpful comments which significantly strengthened the content and presentation of this review.
Abbreviations
- 6-DDTre
lauryl-6-trehaloside
- 6LT
dodecyl trehalose
- ABC
ATP-binding cassette
- Acr
acriflavine
- AbgT
p-aminobenzoyl-glutamate transporter
- Bcc
Burkholderia Cepacia Complex
- CCCP
carbonyl cyanide m-chlorophenylhydrazine
- CDL
cardiolipin
- CF
cystic fibrosis
- DAG
diacyl glycerol
- EPI
efflux pump inhibitors
- EptA
ethanolamine phosphotranserase A
- EM
electron microscopy
- HAE
hydrophobic and amphiphilic efflux
- HME
heavy metal efflux
- HpnN
hopanoid biosynthesis-associated RND transporter
- IMP
inner membrane efflux pump
- LPS
lipopolysaccharide
- mAGP
mycolyl-arabinogalactan-peptidoglycan
- MABSC
mycobacterium abscessus complex
- MAC
mycobacterium avium complex
- MATE
multidrug and toxic compound extrusion
- MFS
major facilitator superfamily
- MDR
multidrug resistant
- MFP
membrane fusion protein
- MmpL
mycobacterial membrane protein large
- Mtb
mycobacterium tuberculosis
- NRP
nonreplicating persistence
- NTM
nontuberculosis mycobacterial
- OMP
outer membrane protein
- PaβN
phenylalanine-arginine β-naphthylamide
- PACE
proteobacterial antimicrobial compound efflux
- PD
periplasmic domain
- PE
phosphatidyl ethanolamine
- PEAE
piperidine aceto-amino ethylene ammonio-acetate
- PI
phosphatidyl inositol
- PG
phosphatidyl glycerol
- PMF
proton motive force
- QAC
quaternary ammonium compounds
- RND
resistance-nodulation-cell division
- SMR
small multidrug resistance
- T6D
trehalose 6-decanoate
- TB
tuberculosis
- TDR
total drug resistant
- TM
transmembrane
- TDM
trehalose dimycolate
- TMM
trehalose monomycolate
- XDR
extensively drug resistant
Biographies
Philip A. Klenotic received his Ph.D in Chemistry from The Ohio State University studying the structure and function of E.coli Signal Peptidase I. He has since held research positions in Ophthalmology and Cell Biology at the Cleveland Clinic and, Dermatology, Cardiovascular Medicine and Pharmacology at Case Western Reserve University. His current research interest as lab manager of the Yu lab is how membrane proteins recognize and transport their substrates from the cell into the extracellular space. A better understanding of this process will help in the design of more effective inhibitors to help treat antibiotic resistance in bacteria.
Mitchell A. Moseng obtained his degree in Biochemistry from Indiana University South Bend in 2012 and his Ph.D. in Chemistry from Miami University in 2019. In 2019 he joined the Yu research group as a postdoctoral scholar in the department of Pharmacology at Case Western Reserve University. Currently, he is conducting research using cryo-EM, x-ray crystallography, and other biophysical/biochemical techniques to elucidate the molecular mechanisms of human and bacterial membrane transport proteins.
Christopher E. Morgan graduated with a B.A. in Chemistry from Youngstown State University in 2013 and received his Ph.D. in Chemistry from Case Western Reserve University in 2018. Following graduation, he joined the Yu research group in 2018 as a postdoctoral scholar. His current research focuses on utilizing Cryo-EM, computational and biophysical techniques to study the mechanisms of bacterial membrane transport proteins and their interactions with antibiotics along with ribosome structure and function.
Edward W. Yu is Interim Chair and Professor of Pharmacology at Case Western Reserve University School of Medicine. He is also Co-Leader of the Molecular Mechanisms of Resistance Research Group of the Case VA CARES Center. His research program broadly focuses on the structure, assembly, and mechanisms of the resistance-nodulation-cell division (RND)-superfamily of efflux pumps as well as their regulation. These efflux systems are key components for Gram-negative pathogens to ensure their survival in toxic environments by extruding a variety of antimicrobial agents from bacterial cells. His lab has elucidated the structures of several of these important RND-type membrane proteins with the overall goal to discover novel ways to effectively inhibit their export functions.
Footnotes
The authors declare no conflicts of interest.
References
- (1).WHO. https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance. Accessed May 4–6, 2020.
- (2).Antibiotic Resistance Threats in the United States;2019. https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf Accessed May 4–6, 2020.
- (3).D’Costa VM; King CE; Kalan L; Morar M; Sung WWL; Schwarz C; Froese D; Zazula G; Calmels F; Debruyne R; Golding GB; Poinar HN; Wright GD Antibiotic Resistance Is Ancient. Nature 2011, 477, 457–461. [DOI] [PubMed] [Google Scholar]
- (4).Allen HK; Donato J; Wang HH; Cloud-Hansen KA; Davies J; Handelsman J Call of the Wild: Antibiotic Resistance Genes in Natural Environments. Nat. Rev. Microbiol 2010, 8, 251–259. [DOI] [PubMed] [Google Scholar]
- (5).Forsberg KJ; Patel S; Gibson MK; Lauber CL; Knight R; Fierer N; Dantas G Bacterial Phylogeny Structures Soil Resistomes across Habitats. Nature 2014, 509, 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Anes J; McCusker MP; Fanning S; Martins M The Ins and Outs of RND Efflux Pumps in Escherichia Coli. Front. Microbiol 2015, 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gunn JS Bacterial Modification of LPS and Resistance to Antimicrobial Peptides. J. Endotoxin Res 2001, 7, 57–62. [PubMed] [Google Scholar]
- (8).Nikaido H Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev 2003, 67, 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Krishnamoorthy G; Leus IV; Weeks JW; Wolloscheck D; Rybenkov VV; Zgurskaya HI Synergy between Active Efflux and Outer Membrane Diffusion Defines Rules of Antibiotic Permeation into Gram-Negative Bacteria. MBio 2017, 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cohen SP; McMurry LM; Levy SB MarA Locus Causes Decreased Expression of OmpF Porin in Multiple-Antibiotic-Resistant (Mar) Mutants of Escherichia Coli. J. Bacteriol 1988, 170, 5416–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Liu X; Ferenci T Regulation of Porin-Mediated Outer Membrane Permeability by Nutrient Limitation in Escherichia Coli; J. Bacteriol 1998, 180, 3917–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lambert PA Cellular Impermeability and Uptake of Biocides and Antibiotics in Gram-positive Bacteria and Mycobacteria. J. Appl. Microbiol 2002, 92, 46S–54S. [PubMed] [Google Scholar]
- (13).Minnikin DE Complex Lipids, Their Chemistry Biosynthesis and Roles. Biol. Mycobact 1982, 1, 95–184. [Google Scholar]
- (14).Bansal-Mutalik R; Nikaido H Mycobacterial Outer Membrane Is a Lipid Bilayer and the Inner Membrane Is Unusually Rich in Diacyl Phosphatidylinositol Dimannosides. Proc. Natl. Acad. Sci 2014, 111, 4958–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Brennan PJ; Nikaido H The Envelope of Mycobacteria. Annu. Rev. Biochem 1995, 64, 29–63. [DOI] [PubMed] [Google Scholar]
- (16).Moon DC; Seol SY; Gurung M; Jin JS; Choi CH; Kim J; Lee YC; Cho DT; Lee JC Emergence of a New Mutation and Its Accumulation in the Topoisomerase IV Gene Confers High Levels of Resistance to Fluoroquinolones in Escherichia Coli Isolates. Int. J. Antimicrob. Agents 2010, 35, 76–79. [DOI] [PubMed] [Google Scholar]
- (17).Maslov DA; Shur KV; Vatlin AA; Danilenko VN MmpS5-MmpL5 Transporters Provide Mycobacterium Smegmatis Resistance to Imidazo [1, 2-b][1, 2, 4, 5] Tetrazines. Pathogens 2020, 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Munita JM; Arias CA Mechanisms of Antibiotic Resistance. Virulence Mech. Bact. Pathog 2016, 481–511. [Google Scholar]
- (19).Pristovšek P; Kidric J Solution Structure of Polymyxins B and E and Effect of Binding to Lipopolysaccharide: An NMR and Molecular Modeling Study. J. Med. Chem 1999, 42, 4604–4613. [DOI] [PubMed] [Google Scholar]
- (20).Liu Y-Y; Wang Y; Walsh TR; Yi L-X; Zhang R; Spencer J; Doi Y; Tian G; Dong B; Huang X Emergence of Plasmid-Mediated Colistin Resistance Mechanism MCR-1 in Animals and Human Beings in China: A Microbiological and Molecular Biological Study. Lancet Infect. Dis 2016, 16, 161–168. [DOI] [PubMed] [Google Scholar]
- (21).Anandan A; Vrielink A Structure and Function of Lipid A–Modifying Enzymes. Ann. N. Y. Acad. Sci 2020, 1459, 19–37. [DOI] [PubMed] [Google Scholar]
- (22).Lee H; Hsu F-F; Turk J; Groisman EA The PmrA-Regulated PmrC Gene Mediates Phosphoethanolamine Modification of Lipid A and Polymyxin Resistance in Salmonella Enterica. J. Bacteriol 2004, 186, 4124–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Saier MH Jr; Tran CV; Barabote RD TCDB: The Transporter Classification Database for Membrane Transport Protein Analyses and Information. Nucleic Acids Res 2006, 34 (suppl_1), D181–D186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Saier MH Jr; Yen MR; Noto K; Tamang DG; Elkan C The Transporter Classification Database: Recent Advances. Nucleic Acids Res. 2008, 37 (suppl_1), D274–D278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Saier MH Jr; Reddy VS; Tamang DG; Västermark Å The Transporter Classification Database. Nucleic Acids Res. 2013, 42, D251–D258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Saier MH Jr; Beatty JT; Goffeau A; Harley KT; Heijne WH; Huang S-C; Jack DL; Jahn PS; Lew K; Liu J The Major Facilitator Superfamily. J Mol Microbiol Biotechnol 1999, 1, 257–279. [PubMed] [Google Scholar]
- (27).Brown MH; Paulsen IT; Skurray RA The Multidrug Efflux Protein NorM Is a Prototype of a New Family of Transporters. Mol. Microbiol 1999, 31, 394–395. [DOI] [PubMed] [Google Scholar]
- (28).Kuroda T; Tsuchiya T Multidrug Efflux Transporters in the MATE Family. Biochim. Biophys. Acta (BBA)-Proteins Proteomics 2009, 1794, 763–768. [DOI] [PubMed] [Google Scholar]
- (29).Paulsen IT; Skurray RA; Tam R; Saier MH Jr; Turner RJ; Weiner JH; Goldberg EB; Grinius LL The SMR Family: A Novel Family of Multidrug Efflux Proteins Involved with the Efflux of Lipophilic Drugs. Mol. Microbiol 1996, 19, 1167–1175. [DOI] [PubMed] [Google Scholar]
- (30).Bay DC; Rommens KL; Turner RJ Small Multidrug Resistance Proteins: A Multidrug Transporter Family That Continues to Grow. Biochim. Biophys. Acta (BBA)-Biomembranes 2008, 1778, 1814–1838. [DOI] [PubMed] [Google Scholar]
- (31).Saier MH Jr; Tam R; Reizer A; Reizer J Two Novel Families of Bacterial Membrane Proteins Concerned with Nodulation, Cell Division and Transport. Mol. Microbiol 1994, 11, 841–847. [DOI] [PubMed] [Google Scholar]
- (32).Veal WL; Shafer WM Identification of a Cell Envelope Protein (MtrF) Involved in Hydrophobic Antimicrobial Resistance in Neisseria Gonorrhoeae. J. Antimicrob. Chemother 2003, 51 (1), 27–37. 10.1093/jac/dkg031. [DOI] [PubMed] [Google Scholar]
- (33).Su CC; Bolla JR; Kumar N; Radhakrishnan A; Long F; Delmar JA; Chou TH; Rajashankar KR; Shafer WM; Yu EW Structure and Function of Neisseria Gonorrhoeae MtrF Illuminates a Class of Antimetabolite Efflux Pumps. Cell Rep. 2015, 11, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Delmar JA; Yu EW The A Bg T Family: A Novel Class of Antimetabolite Transporters. Protein Sci. 2016, 25, 322–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hassan KA; Liu Q; Elbourne LDH; Ahmad I; Sharples D; Naidu V; Chan CL; Li L; Harborne SPD; Pokhrel A Pacing across the Membrane: The Novel PACE Family of Efflux Pumps Is Widespread in Gram-Negative Pathogens. Res. Microbiol 2018, 169, 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kobayashi N; Nishino K; Yamaguchi A Novel Macrolide-Specific ABC-Type Efflux Transporter InEscherichia Coli. J. Bacteriol 2001, 183, 5639–5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Baylay AJ; Ivens A; Piddock LJV A Novel Gene Amplification Causes Upregulation of the PatAB ABC Transporter and Fluoroquinolone Resistance in Streptococcus Pneumoniae. Antimicrob. Agents Chemother 2015, 59, 3098–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hu R-M; Liao S-T; Huang C-C; Huang Y-W; Yang T-C An Inducible Fusaric Acid Tripartite Efflux Pump Contributes to the Fusaric Acid Resistance in Stenotrophomonas Maltophilia. PLoS One 2012, 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Davidson AL; Dassa E; Orelle C; Chen J Structure, Function, and Evolution of Bacterial ATP-Binding Cassette Systems. Microbiol. Mol. Biol. Rev 2008, 72, 317–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Davidson AL; Chen J ATP-Binding Cassette Transporters in Bacteria. Annu. Rev. Biochem 2004, 73, 241–268. [DOI] [PubMed] [Google Scholar]
- (41).Oswald C; Holland IB; Schmitt L The Motor Domains of ABC-Transporters. Naunyn. Schmiedebergs. Arch. Pharmacol 2006, 372, 385–399. [DOI] [PubMed] [Google Scholar]
- (42).Quistgaard EM; Löw C; Guettou F; Nordlund P Understanding Transport by the Major Facilitator Superfamily (MFS): Structures Pave the Way. Nat. Rev. Mol. Cell Biol 2016, 17, 123–132. [DOI] [PubMed] [Google Scholar]
- (43).Yan N Structural Advances for the Major Facilitator Superfamily (MFS) Transporters. Trends Biochem. Sci 2013, 38, 151–159. [DOI] [PubMed] [Google Scholar]
- (44).Levy SB Active Efflux, a Common Mechanism for Biocide and Antibiotic Resistance. J. Appl. Microbiol 2002, 92, 65S–71S. [PubMed] [Google Scholar]
- (45).Lomovskaya O; Lewis KIM; Matin A EmrR Is a Negative Regulator of the Escherichia Coli Multidrug Resistance Pump EmrAB. J. Bacteriol 1995, 177, 2328–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Woolley RC; Vediyappan G; Anderson M; Lackey M; Ramasubramanian B; Jiangping B; Borisova T; Colmer JA; Hamood AN; McVay CS Characterization of the Vibrio Cholerae VceCAB Multiple-Drug Resistance Efflux Operon in Escherichia Coli. J. Bacteriol 2005, 187, 5500–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Pasqua M; Grossi M; Zennaro A; Fanelli G; Micheli G; Barras F; Colonna B; Prosseda G The Varied Role of Efflux Pumps of the MFS Family in the Interplay of Bacteria with Animal and Plant Cells. Microorganisms 2019, 7, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Pérez-Varela M; Corral J; Vallejo JA; Rumbo-Feal S; Bou G; Aranda J; Barbé J Mutations in the β-Subunit of the RNA Polymerase Impair the Surface-Associated Motility and Virulence of Acinetobacter Baumannii. Infect. Immun 2017, 85, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sahu PK; Iyer PS; Gaikwad MB; Talreja SC; Pardesi KR; Chopade BA An MFS Transporter-like ORF from MDR Acinetobacter Baumannii AIIMS 7 Is Associated with Adherence and Biofilm Formation on Biotic/Abiotic Surface. Int. J. Microbiol 2012, 2012. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Liu JY; Miller PF; Willard J; Olson ER Functional and Biochemical Characterization of Escherichia Coli Sugar Efflux Transporters. J. Biol. Chem 1999, 274, 22977–22984. [DOI] [PubMed] [Google Scholar]
- (51).Kobylka J; Kuth MS; Müller RT; Geertsma ER; Pos KM AcrB: A Mean, Keen, Drug Efflux Machine. Ann. N. Y. Acad. Sci 2020, 1459, 38–68. [DOI] [PubMed] [Google Scholar]
- (52).Zomot E; Yardeni EH; Vargiu AV; Tam H-K; Malloci G; Ramaswamy VK; Perach M; Ruggerone P; Pos KM; Bibi E A New Critical Conformational Determinant of Multidrug Efflux by an MFS Transporter. J. Mol. Biol 2018, 430, 1368–1385. [DOI] [PubMed] [Google Scholar]
- (53).Lewinson O; Adler J; Sigal N; Bibi E Promiscuity in Multidrug Recognition and Transport: The Bacterial MFS Mdr Transporters. Mol. Microbiol 2006, 61, 277–284. [DOI] [PubMed] [Google Scholar]
- (54).Tanaka Y; Hipolito CJ; Maturana AD; Ito K; Kuroda T; Higuchi T; Katoh T; Kato HE; Hattori M; Kumazaki K Structural Basis for the Drug Extrusion Mechanism by a MATE Multidrug Transporter. Nature 2013, 496, 247–251. [DOI] [PubMed] [Google Scholar]
- (55).McAleese F; Petersen P; Ruzin A; Dunman PM; Murphy E; Projan SJ; Bradford PA A Novel MATE Family Efflux Pump Contributes to the Reduced Susceptibility of Laboratory-Derived Staphylococcus Aureus Mutants to Tigecycline. Antimicrob. Agents Chemother 2005, 49, 1865–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kaatz GW; McAleese F; Seo SM Multidrug Resistance in Staphylococcus Aureus Due to Overexpression of a Novel Multidrug and Toxin Extrusion (MATE) Transport Protein. Antimicrob. Agents Chemother 2005, 49, 1857–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Bolla JR; Su C-C; Delmar JA; Radhakrishnan A; Kumar N; Chou T-H; Long F; Rajashankar KR; Edward WY Crystal Structure of the Alcanivorax Borkumensis YdaH Transporter Reveals an Unusual Topology. Nat. Commun 2015, 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Su CC; Long F; Lei HT; Bolla JR; Do SV; Rajashankar KR; Yu EW Charged Amino Acids (R83, E567, D617, E625, R669, and K678) of CusA Are Required for Metal Ion Transport in the Cus Efflux System. J. Mol. Biol 2012, 422, 429–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Hassan KA; Liu Q; Henderson PJF; Paulsen IT Homologs of the Acinetobacter Baumannii AceI Transporter Represent a New Family of Bacterial Multidrug Efflux Systems. MBio 2015, 6, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Hassan KA; Jackson SM; Penesyan A; Patching SG; Tetu SG; Eijkelkamp BA; Brown MH; Henderson PJF; Paulsen IT Transcriptomic and Biochemical Analyses Identify a Family of Chlorhexidine Efflux Proteins. Proc. Natl. Acad. Sci 2013, 110, 20254–20259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Tseng T-T; Gratwick KS; Kollman J; Park D; Nies DH; Goffeau A; Saier MH Jr The RND Permease Superfamily: An Ancient, Ubiquitous and Diverse Family That Includes Human Disease and Development Proteins. J. Mol. Microbiol. Biotechnol 1999, 1, 107–125. [PubMed] [Google Scholar]
- (62).Ma D; Cook DN; Alberti M; Pon NG; Nikaido H; Hearst JE Molecular Cloning and Characterization of AcrA and AcrE Genes of Escherichia Coli. J. Bacteriol 1993, 175, 6299–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Poole K; Krebes K; McNally C; Neshat S Multiple Antibiotic Resistance in Pseudomonas Aeruginosa: Evidence for Involvement of an Efflux Operon. J. Bacteriol 1993, 175, 7363–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Li X-Z; Nikaido H; Poole K Role of MexA-MexB-OprM in Antibiotic Efflux in Pseudomonas Aeruginosa. Antimicrob. Agents Chemother 1995, 39, 1948–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Saier MH Jr; Tran CV; Barabote RD TCDB: The Transporter Classification Database for Membrane Transport Protein Analyses and Information. Nucleic Acids Res. 2006, 34 (suppl_1), D181–D186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Nikaido H RND Transporters in the Living World. Res. Microbiol 2018, 169, 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Nikaido H; Takatsuka Y Mechanisms of RND Multidrug Efflux Pumps. Biochim. Biophys. Acta (BBA)-Proteins Proteomics 2009, 1794, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Lee A; Mao W; Warren MS; Mistry A; Hoshino K; Okumura R; Ishida H; Lomovskaya O Interplay between Efflux Pumps May Provide Either Additive or Multiplicative Effects on Drug Resistance. J. Bacteriol 2000, 182, 3142–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Tal N; Schuldiner S A Coordinated Network of Transporters with Overlapping Specificities Provides a Robust Survival Strategy. Proc. Natl. Acad. Sci 2009, 106, 9051–9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Sulavik MC; Houseweart C; Cramer C; Jiwani N; Murgolo N; Greene J; DiDomenico B; Shaw KJ; Miller GH; Hare R; Shimer G Antibiotic Susceptibility Profiles Of Escherichia Coli Strains Lacking Multidrug Efflux Pump Genes. Antimicrob. Agents Chemother 2001, 45, 1126–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Zgurskaya HI; Nikaido H Cross-Linked Complex between Oligomeric Periplasmic Lipoprotein AcrA and the Inner-Membrane-Associated Multidrug Efflux Pump AcrB from Escherichia Coli. J. Bacteriol 2000, 182, 4264–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Fernandez-Recio J; Walas F; Federici L; Venkatesh Pratap J; Bavro VN; Miguel RN; Mizuguchi K; Luisi B A Model of a Transmembrane Drug-efflux Pump from Gram-negative Bacteria. FEBS Lett. 2004, 578, 5–9. [DOI] [PubMed] [Google Scholar]
- (73).Symmons MF; Bokma E; Koronakis E; Hughes C; Koronakis V The Assembled Structure of a Complete Tripartite Bacterial Multidrug Efflux Pump. Proc. Natl. Acad. Sci 2009, 106, 7173–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Doughty DM; Coleman ML; Hunter RC; Sessions AL; Summons RE; Newman DK The RND-Family Transporter, HpnN, Is Required for Hopanoid Localization to the Outer Membrane of Rhodopseudomonas Palustris TIE-1. Proc. Natl. Acad. Sci 2011, 108, E1045–E1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Daligault HE; Davenport KW; Minogue TD; Bishop-Lilly KA; Broomall SM; Bruce DC; Chain PS; Coyne SR; Frey KG; Gibbons HS Whole-Genome Assemblies of 56 Burkholderia Species. Genome Announc. 2014, 2, e01106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Kumar N; Su C-C; Chou T-H; Radhakrishnan A; Delmar JA; Rajashankar KR; Edward WY Crystal Structures of the Burkholderia Multivorans Hopanoid Transporter HpnN. Proc. Natl. Acad. Sci 2017, 114, 6557–6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Su C-C; Klenotic PA; Bolla JR; Purdy GE; Robinson CV; Yu EW MmpL3 Is a Lipid Transporter That Binds Trehalose Monomycolate and Phosphatidylethanolamine. Proc. Natl. Acad. Sci 2019, 116, 11241–11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Zhang B; Li J; Yang X; Wu L; Zhang J; Yang Y; Zhao Y; Zhang L; Yang X; Yang X; Cheng X; Liu Z; Jiang B; Jiang H; Guddat LW; Yang H; Rao Z Crystal Structures of Membrane Transporter MmpL3, an Anti-TB Drug Target. Cell 2019, 176, 636–648. [DOI] [PubMed] [Google Scholar]
- (79).Grzegorzewicz AE; Pham H; Gundi VAKB; Scherman MS; North EJ; Hess T; Jones V; Gruppo V; Born SEM; Korduláková J Inhibition of Mycolic Acid Transport across the Mycobacterium Tuberculosis Plasma Membrane. Nat. Chem. Biol 2012, 8, 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Jankute M; Cox JAG; Harrison J; Besra GS Assembly of the Mycobacterial Cell Wall. Annu. Rev. Microbiol 2015, 69, 405–423. [DOI] [PubMed] [Google Scholar]
- (81).Belardinelli JM; Stevens CM; Li W; Tan YZ; Jones V; Mancia F; Zgurskaya HI; Jackson M The MmpL3 Interactome Reveals a Complex Crosstalk between Cell Envelope Biosynthesis and Cell Elongation and Division in Mycobacteria. Sci. Rep 2019, 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Nikaido H; Pagès JM Broad-Specificity Efflux Pumps and Their Role in Multidrug Resistance of Gram-Negative Bacteria. FEMS Microbiology Reviews. 2012, 36, 340–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Butler MS; Cooper MA Antibiotics in the Clinical Pipeline in 2011. Journal of Antibiotics. 2011, 64, 413–425. [DOI] [PubMed] [Google Scholar]
- (84).Meguro N; Kodama Y; Gallegos MT; Watanabe K Molecular Characterization of Resistance-Nodulation-Division Transporters from Solvent- and Drug-Resistant Bacteria in Petroleum-Contaminated Soil. Appl. Environ. Microbiol 2005, 71, 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Nikaido H Multidrug Resistance in Bacteria. Annu. Rev. Biochem 2009, 78, 119–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Mobarki N; Almerabi B; Hattan A Antibiotic Resistance Crisis. Int. J. Med. Dev. Ctries 2019, 40, 561–564. [Google Scholar]
- (87).Yu EW; Aires JR; Nikaido H AcrB Multidrug Efflux Pump of Escherichia Coli: Composite Substrate-Binding Cavity of Exceptional Flexibility Generates Its Extremely Wide Substrate Specificity. Journal of Bacteriology. 2003, 185, 5657–5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Nies DH Efflux-Mediated Heavy Metal Resistance in Prokaryotes. FEMS Microbiology Reviews. Elsevier 2003, 27, 313–339. [DOI] [PubMed] [Google Scholar]
- (89).Ma Z; Jacobsen FE; Giedroc DP Coordination Chemistry of Bacterial Metal Transport and Sensing. Chem. Rev 2009, 109, 4644–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Misra R; Bavro VN Assembly and Transport Mechanism of Tripartite Drug Efflux Systems. Biochimica et Biophysica Acta - Proteins and Proteomics. 2009, 1794, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Janganan TK; Bavro VN; Zhang L; Matak-Vinkovic D; Barrera NP; Venien-Bryan C; Robinson CV; Borges-Walmsley MI; Walmsley AR Evidence for the Assembly of a Bacterial Tripartite Multidrug Pump with a Stoichiometry of 3:6:3. J. Biol. Chem 2011, 286, 26900–26912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Stegmeier JF; Polleichtner G; Brandes N; Hotz C; Andersen C Importance of the Adaptor (Membrane Fusion) Protein Hairpin Domain for the Functionality of Multidrug Efflux Pumps. Biochemistry 2006, 45, 10303–10312. [DOI] [PubMed] [Google Scholar]
- (93).Luisi B; Koronakis V; Hughes C; Sharff A; Koronakis E Crystal Structure of the Bacterial Membrane Protein TolC Central to Multidrug Efflux and Protein Export. Nature 2002, 405, 914–919. [DOI] [PubMed] [Google Scholar]
- (94).Murakami S; Nakashima R; Yamashita E; Yamaguchi A Crystal Structure of Bacterial Multidrug Efflux Transporter AcrB. Nature 2002, 419, 587–593. [DOI] [PubMed] [Google Scholar]
- (95).Murakami S; Nakashima R; Yamashita E; Matsumoto T; Yamaguchi A Crystal Structures of a Multidrug Transporter Reveal a Functionally Rotating Mechanism. Nature 2006, 443, 173–179. [DOI] [PubMed] [Google Scholar]
- (96).Seeger MA; Schiefner A; Eicher T; Ois Verrey F$; Diederichs K; Pos KM Structural Asymmetry of AcrB Trimer Suggests a Peristaltic Pump Mechanism. Science 2006, 313, 1295–1298. [DOI] [PubMed] [Google Scholar]
- (97).Sennhauser G; Amstutz P; Briand C; Storchenegger O; Grütter MG Drug Export Pathway of Multidrug Exporter AcrB Revealed by DARPin Inhibitors. PLoS Biol 2006, 5, 0106–0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Su C-C; Yin L; Kumar N; Dai L; Radhakrishnan A; Bolla JR; Lei H-T; Chou T-H; Delmar JA; Rajashankar KR Structures and Transport Dynamics of a Campylobacter Jejuni Multidrug Efflux Pump. Nat. Commun 2017, 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Su CC; Yin L; Kumar N; Dai L; Radhakrishnan A; Bolla JR; Lei HT; Chou TH; Delmar JA; Rajashankar KR; Zhang Q; Shin YK; Yu EW Structures and Transport Dynamics of a Campylobacter Jejuni Multidrug Efflux Pump. Nat. Commun 2017, 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Ramaswamy VK; Vargiu AV; Malloci G; Dreier J; Ruggerone P Molecular Rationale behind the Differential Substrate Specificity of Bacterial RND Multi-Drug Transporters. Sci. Rep 2017, 7, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Paulsen IT; Brown MH; Skurray RA Proton-Dependent Multidrug Efflux Systems. 1996, 60, 575–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Tseng T-T; Gratwick KS; Kollman J; Park D; Nies DH; Goffeau A; Saier MH Jr; Saier MH The RND Permease Superfamily: An Ancient, Ubiquitous and Diverse Family That Includes Human Disease and Development Proteins. J. Mol. Microbiol. Biotechnol 1999, 1, 107–125. [PubMed] [Google Scholar]
- (103).Lyu M; Moseng MA; Reimche JL; Holley CL; Dhulipala V; Su C Crossm Cryo-EM Structures of a Gonococcal Multidrug Efflux Pump Illuminate a Mechanism of Drug Recognition and Resistance. 2020, 11, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Long F; Su C-CC; Zimmermann MT; Boyken SE; Rajashankar KR; Jernigan RL; Yu EW; Edward WY Crystal Structures of the CusA Efflux Pump Suggest Methionine-Mediated Metal Transport. Nature 2010, 467, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Su CC; Li M; Gu R; Takatsuka Y; McDermott G; Nikaido H; Yu EW Conformation of the AcrB Multidrug Efflux Pump in Mutants of the Putative Proton Relay Pathway. J. Bacteriol 2006, 188, 7290–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Guan L; Nakae T Identification of Essential Charged Residues in Transmembrane Segments of the Multidrug Transporter MexB OfPseudomonas Aeruginosa. J. Bacteriol 2001, 183, 1734–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Takatsuka Y; Nikaido H Threonine-978 in the Transmembrane Segment of the Multidrug Efflux Pump AcrB of Escherichia Coli Is Crucial for Drug Transport as a Probable Component of the Proton Relay Network. J. Bacteriol 2006, 188, 7284–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Pu Y; Zhao Z; Li Y; Zou J; Ma Q; Zhao Y; Ke Y; Zhu Y; Chen H; Baker MAB; Ge H; Sun Y; Xie XS; Bai F Enhanced Efflux Activity Facilitates Drug Tolerance in Dormant Bacterial Cells. Mol. Cell 2016, 62, 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Du D; van Veen HW; Luisi BF Assembly and Operation of Bacterial Tripartite Multidrug Efflux Pumps. Trends in Microbiology. 2015, 23, 311–319. [DOI] [PubMed] [Google Scholar]
- (110).Ge Q; Yamadah Y; Zgurskaya E The C-Terminal Domain of AcrA Is Essential for the Assembly and Function of the Multidrug Efflux Pump AcrAB-TolC. J. Bacteriol 2009, 191, 4365–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Eicher T; Seeger MA; Anselmi C; Zhou W; Brandstätter L; Verrey F; Diederichs K; Faraldo-Gómez JD; Pos KM Coupling of Remote Alternating-Access Transport Mechanisms for Protons and Substrates in the Multidrug Efflux Pump AcrB. Elife 2014, 3. 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Zgurskaya HI; Nikaido H Bypassing the Periplasm: Reconstitution of the AcrAB Multidrug Efflux Pump of Escherichia Coli. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 7190–7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Su C-C; Nikaido H; Edward WY Ligand-Transporter Interaction in the AcrB Multidrug Efflux Pump Determined by Fluorescence Polarization Assay. FEBS Lett. 2007, 581, 4972–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Takatsuka Y; Nikaido H Threonine-978 in the Transmembrane Segment of the Multidrug Efflux Pump AcrB of Escherichia Coli Is Crucial for Drug Transport as a Probable Component of the Proton Relay Network. J. Bacteriol 2006, 188, 7284–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Aires JR; Nikaido H Aminoglycosides Are Captured from Both Periplasm and Cytoplasm by the AcrD Multidrug Efflux Transporter of Escherichia Coli. J. Bacteriol 2005, 187, 1923–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Horiyama T; Nishino K AcrB, AcrD, and MdtABC Multidrug Efflux Systems Are Involved in Enterobactin Export in Escherichia Coli. PLoS One 2014, 9, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Du D; van Veen HW; Murakami S; Pos KM; Luisi BF Structure, Mechanism and Cooperation of Bacterial Multidrug Transporters. Current Opinion in Structural Biology. 2015, 33, 76–91. [DOI] [PubMed] [Google Scholar]
- (118).Wang Z; Fan G; Hryc CF; Blaza JN; Serysheva II; Schmid MF; Chiu W; Luisi BF; Du D An Allosteric Transport Mechanism for the AcrAB-TolC Multidrug Efflux Pump. 2017, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Yu EW; McDermott G; Zgurskaya HI; Nikaido H; Koshland DE Structural Basis of Multiple Drug-Binding Capacity of the AcrB Multidrug Efflux Pump. Science 2003, 300, 976–980. [DOI] [PubMed] [Google Scholar]
- (120).Yu EW; Aires JR; McDermott G; Nikaido H A Periplasmic Drug-Binding Site of the AcrB Multidrug Efflux Pump: A Crystallographic and Site-Directed Mutagenesis Study. J. Bacteriol 2005, 187, 6804–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Vargiu AV; Nikaido H Multidrug Binding Properties of the AcrB Efflux Pump Characterized by Molecular Dynamics Simulations. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 20637–20642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Su C-C; Morgan CE; Kambakam S; Rajavel M; Scott H; Huang W; Emerson CC; Taylor DJ; Stewart PL; Bonomo RA; Yu EW; Kambakan S; Rajavel M; Scott H; Huang W; Emerson CC; Taylor DJ; Stewart PL; Bonomo RA; Yu EW Cryo-Electron Microscopy Structure of an Acinetobacter Baumannii Multidrug Efflux Pump. MBio 2019, 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Schuster S; Vavra M; Kern WV Evidence of a Substrate-Discriminating Entrance Channel in the Lower Porter Domain of the Multidrug Resistance Efflux Pump AcrB. Antimicrob. Agents Chemother 2016, 60, 4315–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Lei HT; Bolla JR; Bishop NR; Su CC; Yu EW Crystal Structures of CusC Review Conformational Changes Accompanying Folding and Transmembrane Channel Formation. J. Mol. Biol 2014, 426, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Murakami S; Nakashima R; Yamashita E; Matsumoto T; Yamaguchi A Crystal Structures of a Multidrug Transporter Reveal a Functionally Rotating Mechanism. Nature 2006, 443, 173–179. [DOI] [PubMed] [Google Scholar]
- (126).Du D; Wang Z; James NR; Voss JE; Klimont E; Ohene-Agyei T; Venter H; Chiu W; Luisi BF Structure of the AcrAB-TolC Multidrug Efflux Pump. Nature 2014, 509, 512–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Zhang CZ; Chang MX; Yang L; Liu YY; Chen PX; Jiang HX Upregulation of AcrEF in Quinolone Resistance Development in Escherichia Coli When AcrAB-TolC Function Is Impaired. Microb. Drug Resist 2018, 24, 18–23. [DOI] [PubMed] [Google Scholar]
- (128).Novoa D; Conroy-Ben O The Anaerobic Efflux Pump MdtEF-TolC Confers Resistance to Cationic Biocides bioRxiv 2019, 1–16 [Google Scholar]
- (129).Kim HS; Nagore D; Nikaido H Multidrug Efflux Pump MdtBC of Escherichia Coli Is Active Only as a B 2C Heterotrimer. J. Bacteriol 2010, 192, 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Quillin SJ; Seifert HS Neisseria Gonorrhoeae Host Adaptation and Pathogenesis. Nature Reviews Microbiology. 2018, 16, 226–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Unemo M; Shafer WM Antimicrobial Resistance in Neisseria Gonorrhoeae in the 21st Century: Past, Evolution, and Future. Clin. Microbiol. Rev 2014, 27, 587–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Smolarchuk C; Wensley A; Padfield S; Fifer H; Lee A; Hughes G Persistence of an Outbreak of Gonorrhoea with High-Level Resistance to Azithromycin in England. Eurosurveillance 2018, 23, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Warner DM; Shafer WM; Jerse AE Clinically Relevant Mutations That Cause Derepression of the Neisseria Gonorrhoeae MtrC-MtrD-MtrE Efflux Pump System Confer Different Levels of Antimicrobial Resistance and in Vivo Fitness. Mol. Microbiol 2008, 70, 462–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Lei HT; Chou TH; Su CC; Bolla JR; Kumar N; Radhakrishnan A; Long F; Delmar JA; Do SV; Rajashankar KR; Shafer WM; Yu EW Crystal Structure of the Open State of the Neisseria Gonorrhoeae MtrE Outer Membrane Channel. PLoS One 2014, 9, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Bolla JR; Su CC; Do SV; Radhakrishnan A; Kumar N; Long F; Chou TH; Delmar JA; Lei HT; Rajashankar KR; Shafer WM; Yu EW Crystal Structure of the Neisseria Gonorrhoeae MtrD Inner Membrane Multidrug Efflux Pump. PLoS One 2014, 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Delahay RM; Robertson BD; Balthazar JT; Shafer WM; Ison CA Involvement of the Gonococcal MtrE Protein in the Resistance of Neisseria Gonorrhoeae to Toxic Hydrophobic Agents. Microbiology 1997, 143, 2127–2133. [DOI] [PubMed] [Google Scholar]
- (137).Rouquette-Loughlin CE; Reimche JL; Balthazar JT; Dhulipala V; Gernert KM; Kersh EN; Pham CD; Pettus K; Abrams AJ; Trees DL; Cyr SS; Shafer WM Mechanistic Basis for Decreased Antimicrobial Susceptibility in a Clinical Isolate of Neisseria Gonorrhoeae Possessing a Mosaic-like Mtr Efflux Pump Locus. MBio 2018, 9, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Ma KC; Mortimer TD; Grad YH Efflux Pump Antibiotic Binding Site Mutations Are Associated with Azithromycin Nonsusceptibility in Clinical Neisseria Gonorrhoeae Isolates. BioRxiv 2020. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Booth L; Blyth MT; Mara MLO; Brown MH Of Functionally Important Residues in the MtrD Efflux Protein. 2019, 10, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Ruiz-Palacios GM The Health Burden of Campylobacter Infection and the Impact of Antimicrobial Resistance: Playing Chicken. Clin. Infect. Dis 2007, 44, 701–703. [DOI] [PubMed] [Google Scholar]
- (141).Engberg J; Aarestrup FM; Taylor DE; Gerner-smidt P; Nachamkin I Engberg-J_Resistance Mechanisms Campylobacter_Emerg Infect 2001. Emerg. Infect. Dis 2001, 7, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Lin J; Overbye Michel L; Zhang Q CmeABC Functions as a Multidrug Efflux System in Campylobacter Jejuni. Antimicrob. Agents Chemother 2002, 46, 2124–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Yan M; Sahin O; Lin J; Zhang Q Role of the CmeABC Efflux Pump in the Emergence of Fluoroquinolone-Resistant Campylobacter under Selection Pressure. J. Antimicrob. Chemother 2006, 58, 1154–1159. [DOI] [PubMed] [Google Scholar]
- (144).Pumbwe L; Piddock LJV Identification and Molecular Characterisation of CmeB, a Campylobacter Jejuni Multidrug Efflux Pump. FEMS Microbiol. Lett 2002, 206, 185–189. [DOI] [PubMed] [Google Scholar]
- (145).Tikhonova EB; Yamada Y; Zgurskaya HI Sequential Mechanism of Assembly of Multidrug Efflux Pump AcrAB-TolC. Chem. Biol 2011, 18, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Su CC; Radhakrishnan A; Kumar N; Long F; Bolla JR; Lei HT; Delmar JA; Do SV; Chou TH; Rajashankar KR; Zhang Q; Yu EW Crystal Structure of the Campylobacter Jejuni CmeC Outer Membrane Channel. Protein Sci. 2014, 23, 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Janganan TK; Zhang L; Bavro VN; Matak-Vinkovic D; Barrera NP; Burton MF; Steel PG; Robinson CV; Borges-Walmsley MI; Walmsley AR Opening of the Outer Membrane Protein Channel in Tripartite Efflux Pumps Is Induced by Interaction with the Membrane Fusion Partner. J. Biol. Chem 2011, 286, 5484–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Neuberger A; Du D; Luisi BF Structure and Mechanism of Bacterial Tripartite Efflux Pumps. Res. Microbiol 2018, 169, 401–413. [DOI] [PubMed] [Google Scholar]
- (149).Lee CR; Lee JH; Park M; Park KS; Bae IK; Kim YB; Cha CJ; Jeong BC; Lee SH Biology of Acinetobacter Baumannii: Pathogenesis, Antibiotic Resistance Mechanisms, and Prospective Treatment Options. Front. Cell. Infect. Microbiol 2017, 7, 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Doi Y; Murray GL; Peleg AY Acinetobacter Baumannii: Evolution of Antimicrobial Resistance-Treatment Options. Semin. Respir. Crit. Care Med 2015, 36, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Kim YJ; Kim SI; Kim YR; Hong KW; Wie SH; Park YJ; Jeong H; Kang MW Carbapenem-Resistant Acinetobacter Baumannii: Diversity of Resistant Mechanisms and Risk Factors for Infection. Epidemiol. Infect 2012, 140, 137–145. [DOI] [PubMed] [Google Scholar]
- (152).Navon-Venezia S; Leavitt A; Carmeli Y High Tigecycline Resistance in Multidrug-Resistant Acinetobacter Baumannii-Authors’ Response. Journal of Antimicrobial Chemotherapy. 2007, 60, 178–179. [DOI] [PubMed] [Google Scholar]
- (153).Magnet S; Courvalin P; Lambert T Resistance-Nodulation-Cell Division-Type Ef Ux Pump Involved in Aminoglycoside Resistance In. Society 2001, 45, 3375–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Su C-C; Morgan CE; Kambakam S; Rajavel M; Scott H; Huang W; Emerson CC; Taylor DJ; Stewart PL; Bonomo RA; Yu EW Cryo-Electron Microscopy Structure of an Acinetobacter Baumannii Multidrug Efflux Pump.mBio 2019, 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Nakashima R; Sakurai K; Yamasaki S; Nishino K; Yamaguchi A Structures of the Multidrug Exporter AcrB Reveal a Proximal Multisite Drug-Binding Pocket. Nature 2011, 480, 565–569. [DOI] [PubMed] [Google Scholar]
- (156).Sciences H Properties of AdeABC and AdeIJK Efflux Systems of Acinetobacter Baumannii Compared with Those of the AcrAB-TolC System of Escherichia Coli. 2014, 58, 7250–7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Qiu W; Fu Z; Xu GG; Grassucci RA; Zhang Y; Frank J; Hendrickson WA; Guo Y Structure and Activity of Lipid Bilayer within a Membrane-Protein Transporter. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 12985–12990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Takatsuka Y; Nikaido H Covalently Linked Trimer of the AcrB Multidrug Efflux Pump Provides Support for the Functional Rotating Mechanism. J. Bacteriol 2009, 191, 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Waldron KJ; Rutherford JC; Ford D; Robinson NJ Metalloproteins and Metal Sensing. Nature. 2009, 460, 823–830. [DOI] [PubMed] [Google Scholar]
- (160).Silver S; Misra TK Plasmid-Mediated Heavy Metal Resistances. Ann. Rev. Microbiol 1988, 42, 717–743. [DOI] [PubMed] [Google Scholar]
- (161).Macomber L; Imlay JA The Iron-Sulfur Clusters of Dehydratases Are Primary Intracellular Targets of Copper Toxicity. PNAS 2009, 20, 8344–8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Grass G; Rensing C; Solioz M Metallic Copper as an Antimicrobial Surface. Applied and Environmental Microbiology. 2011, 77, 1541–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Chopra I The Increasing Use of Silver-Based Products as Antimicrobial Agents: A Useful Development or a Cause for Concern? Journal of Antimicrobial Chemotherapy. 2007, 59, 587–590. [DOI] [PubMed] [Google Scholar]
- (164).Alvarez-Ortega C; Olivares J; Martínez JL RND Multidrug Efflux Pumps: What Are They Good For? Frontiers in Microbiology. 2013, 4, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Delmar JA; Su CC; Yu EW Heavy Metal Transport by the CusCFBA Efflux System. Protein Science. 2015, 24,1720–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Franke S; Grass G; Nies DH The Product of the YbdE Gene of the Escherichia Coli Chromosome Is Involved in Detoxification of Silver Ions. Microbiology 2001, 147, 965–972. [DOI] [PubMed] [Google Scholar]
- (167).Nies DH; Nies A; Chu L; Silver S Expression and Nucleotide Sequence of a Plasmid-Determined Divalent Cation Efflux System from Alcaligenes Eutrophus. Proc. Natl. Acad. Sci. U. S. A 1989, 86, 7351–7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Pak JE; Ekendé EN; Kifle EG; O’Connell JD; De Angelis F; Tessema MB; Derfoufi KM; Robles-Colmenares Y; Robbins RA; Goormaghtigh E; Vandenbussche G; Stroud RM Structures of Intermediate Transport States of ZneA, a Zn(II)/Proton Antiporter. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 18484–18489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Goldberg M; Pribyl T; Juhnke S; Nies DH Energetics and Topology of CzcA, a Cation/Proton Antiporter of the Resistance-Nodulation-Cell Division Protein Family. J. Biol. Chem 1999, 274, 26065–26070. [DOI] [PubMed] [Google Scholar]
- (170).Rensing C; Pribyl T; Nies DH New Functions for the Three Subunits of the CzcCBA Cation-Proton Antiporter 1997, 179, 6871–6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Su CC; Yang F; Long F; Reyon D; Routh MD; Kuo DW; Mokhtari AK; Van Ornam JD; Rabe KL; Hoy JA; Lee YJ; Rajashankar KR; Yu EW Crystal Structure of the Membrane Fusion Protein CusB from Escherichia Coli. J. Mol. Biol 2009, 393, 342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).De Angelis F; Lee JK; O’Connell JD; Miercke LJW; Verschueren KH; Srinivasan V; Bauvois C; Govaerts C; Robbins RA; Ruysschaert JM; Stroud RM; Vandenbussche G Metal-Induced Conformational Changes in ZneB Suggest an Active Role of Membrane Fusion Proteins in Efflux Resistance Systems. PNAS 2010, 107, 11038–11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Silver S; Phung LT BACTERIAL HEAVY METAL RESISTANCE: New Surprises. 1996, 50, 753–789. [DOI] [PubMed] [Google Scholar]
- (174).Xue Y; Davis AV; Balakrishnan G; Stasser JP; Staehlin BM; Focia P; Spiro TG; Penner-Hahn JE; O’Halloran TV Cu(I) Recognition via Cation-π and Methionine Interactions in CusF. Nat. Chem. Biol 2008, 4, 107–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Franke S; Grass G; Rensing C; Nies DH Molecular Analysis of the Copper-Transporting Efflux System CusCFBA of Escherichia Coli. J. Bacteriol 2003, 185, 3804–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Stahl A; Pletzer D; Mehmood A; Ullrich MS Marinobacter Adhaerens HP15 Harbors Two CzcCBA Efflux Pumps Involved in Zinc Detoxification. Antonie van Leeuwenhoek, Int. J. Gen. Mol. Microbiol 2015, 108, 649–658. [DOI] [PubMed] [Google Scholar]
- (177).Degrade HB Biodegradative Bacteria. 2014. Spinger; Japan. [Google Scholar]
- (178).Delmar JA; Su C-C; Yu EW Bacterial Multidrug Efflux Transporters. Annu. Rev. Biophys 2014, 43, 93–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Long F; Su CC; Lei HT; Bolla JR; Do SV; Yu EW Structure and Mechanism of the Tripartite CusCBA Heavy-Metal Efflux Complex. Philosophical Transactions of the Royal Society B: Biological Sciences. Royal Society 2012, 367, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Rensing C; Grass G Escherichia Coli Mechanisms of Copper Homeostasis in a Changing Environment. FEMS Microbiology Reviews. Elsevier 2003, 27, 197–213. [DOI] [PubMed] [Google Scholar]
- (181).Helbig K; Bleuel C; Krauss GJ; Nies DH Glutathione and Transition-Metal Homeostasis in Escherichia Coli. J. Bacteriol 2008, 190, 5431–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Su CC; Long F; Yu EW The Cus Efflux System Removes Toxic Ions via a Methionine Shuttle. Protein Science. 2011, 20, 6–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Hung LW; Kim HB; Murakami S; Gupta G; Kim CY; Terwilliger TC Crystal Structure of AcrB Complexed with Linezolid at 3.5 Å Resolution. J. Struct. Funct. Genomics 2013, 14, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Kulathila R; Kulathila R; Indic M; van den Berg B Crystal Structure of Escherichia Coli CusC, the Outer Membrane Component of a Heavy Metal Efflux Pump. PLoS One 2011, 6, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Su CC; Long F; Zimmermann MT; Rajashankar KR; Jernigan RL; Yu EW Crystal Structure of the CusBA Heavy-Metal Efflux Complex of Escherichia Coli. Nature 2011, 470, 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Bagai I; Liu W; Rensing C; Blackburn NJ; McEvoy MM Substrate-Linked Conformational Change in the Periplasmic Component of a Cu(I)/Ag(I) Efflux System. J. Biol. Chem 2007, 282, 35695–35702. [DOI] [PubMed] [Google Scholar]
- (187).Loftin IR; Blackburn NJ; McEvoy MM Tryptophan Cu(I)-π Interaction Fine-Tunes the Metal Binding Properties of the Bacterial Metallochaperone CusF. J. Biol. Inorg. Chem 2009, 14, 905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Loftin IR; Franke S; Roberts SA; Weichsel A; Héroux A; Montfort WR; Rensing C; McEvoy MM A Novel Copper-Binding Fold for the Periplasmic Copper Resistance Protein CusF. Biochemistry 2005, 44, 10533–10540. [DOI] [PubMed] [Google Scholar]
- (189).Scapin G; Potter CS; Carragher B Cryo-EM for Small Molecules Discovery, Design, Understanding, and Application. Cell Chem. Biol 2018, 25, 1318–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Morgan S; Grootendorst P; Lexchin J; Cunningham C; Greyson D The Cost of Drug Development: A Systematic Review. Health Policy 2011, 100, 4–17. [DOI] [PubMed] [Google Scholar]
- (191).Anderson AC The Process of Structure-Based Drug Design. Chem. Biol 2003, 10, 787–797. [DOI] [PubMed] [Google Scholar]
- (192).Li XZ; Plésiat P; Nikaido H The Challenge of Efflux-Mediated Antibiotic Resistance in Gram-Negative Bacteria. Clin. Microbiol. Rev 2015, 28, 337–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Aron Z; Opperman TJ Optimization of a Novel Series of Pyranopyridine RND Efflux Pump Inhibitors. Curr. Opin. Microbiol 2016, 33, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Nakashima R; Sakurai K; Yamasaki S; Hayashi K; Nagata C; Hoshino K; Onodera Y; Nishino K; Yamaguchi A Structural Basis for the Inhibition of Bacterial Multidrug Exporters. Nature 2013, 500, 102–107. [DOI] [PubMed] [Google Scholar]
- (195).Sjuts H; Vargiu AV; Kwasny SM; Nguyen ST; Kim H-S; Ding X; Ornik AR; Ruggerone P; Bowlin TL; Nikaido H; others. Molecular Basis for Inhibition of AcrB Multidrug Efflux Pump by Novel and Powerful Pyranopyridine Derivatives. Proc. Natl. Acad. Sci 2016, 113, 3509–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Wang Z; Sun H; Shen C; Hu X; Gao J; Li D; Cao D; Hou T Combined Strategies in Structure-Based Virtual Screening. Phys. Chem. Chem. Phys 2020. 22, 3149–3159. [DOI] [PubMed] [Google Scholar]
- (197).Wang Z; Sun H; Yao X; Li D; Xu L; Li Y; Tian S; Hou T Comprehensive Evaluation of Ten Docking Programs on a Diverse Set of Protein-Ligand Complexes: The Prediction Accuracy of Sampling Power and Scoring Power. Phys. Chem. Chem. Phys 2016, 18, 12964–12975. [DOI] [PubMed] [Google Scholar]
- (198).Sharma A; Gupta VK; Pathania R Efflux Pump Inhibitors for Bacterial Pathogens: From Bench to Bedside. Indian J. Med. Res 2019, 149, 129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Stavri M; Piddock LJV; Gibbons S Bacterial Efflux Pump Inhibitors from Natural Sources. J. Antimicrob. Chemother 2007, 59, 1247–1260. [DOI] [PubMed] [Google Scholar]
- (200).Stermitz FR; Lorenz P; Tawara JN; Zenewicz LA; Lewis K Synergy in a Medicinal Plant: Antimicrobial Action of Berberine Potentiated by 5′-ethoxyhydnocarpin, a Multidrug Pump Inhibitor. Proc. Natl. Acad. Sci 2000, 97, 1433–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Belofsky G; Percivill D; Lewis K; Tegos GP; Ekart J Phenolic Metabolites of Dalea v Ersicolor That Enhance Antibiotic Activity against Model Pathogenic Bacteria. J. Nat. Prod 2004, 67, 481–484. [DOI] [PubMed] [Google Scholar]
- (202).Lamut A; Peterlin Mašič L; Kikelj D; Tomašič T Efflux Pump Inhibitors of Clinically Relevant Multidrug Resistant Bacteria. Med. Res. Rev 2019, 39, 2460–2504. [DOI] [PubMed] [Google Scholar]
- (203).Ni W; Li Y; Guan J; Zhao J; Cui J; Wang R; Liu Y Effects of Efflux Pump Inhibitors on Colistin Resistance in Multidrug-Resistant Gram-Negative Bacteria. Antimicrob. Agents Chemother 2016, 60, 3215–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Park YK; Ko KS Effect of Carbonyl Cyanide 3-Chlorophenylhydrazone (CCCP) on Killing Acinetobacter Baumannii by Colistin. J. Microbiol 2015, 53, 53–59. [DOI] [PubMed] [Google Scholar]
- (205).Baron SA; Rolain J-M Efflux Pump Inhibitor CCCP to Rescue Colistin Susceptibility in Mcr-1 Plasmid-Mediated Colistin-Resistant Strains and Gram-Negative Bacteria. J. Antimicrob. Chemother 2018, 73, 1862–1871. [DOI] [PubMed] [Google Scholar]
- (206).Bhattacharyya T; Sharma A; Akhter J; Pathania R The Small Molecule IITR08027 Restores the Antibacterial Activity of Fluoroquinolones against Multidrug-Resistant Acinetobacter Baumannii by Efflux Inhibition. Int. J. Antimicrob. Agents 2017, 50, 219–226. [DOI] [PubMed] [Google Scholar]
- (207).Nakashima R; Sakurai K; Yamasaki S; Hayashi K; Nagata C; Hoshino K; Onodera Y; Nishino K; Yamaguchi A Structural Basis for the Inhibition of Bacterial Multidrug Exporters. Nature 2013, 500, 102–106. [DOI] [PubMed] [Google Scholar]
- (208).Renau TE; Léger R; Flamme EM; Sangalang J; She MW; Yen R; Gannon CL; Griffith D; Chamberland S; Lomovskaya O; others. Inhibitors of Efflux Pumps in Pseudomonas Aeruginosa Potentiate the Activity of the Fluoroquinolone Antibacterial Levofloxacin. J. Med. Chem 1999, 42, 4928–4931. [DOI] [PubMed] [Google Scholar]
- (209).Lomovskaya O; Warren MS; Lee A; Galazzo J; Fronko R; Lee MAY; Blais J; Cho D; Chamberland S; Renau T; others. Identification and Characterization of Inhibitors of Multidrug Resistance Efflux Pumps in Pseudomonas Aeruginosa: Novel Agents for Combination Therapy. Antimicrob. Agents Chemother 2001, 45, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Eicher T; Cha H; Seeger MA; Brandstätter L; El-Delik J; Bohnert JA; Kern WV; Verrey F; Grütter MG; Diederichs K; others. Transport of Drugs by the Multidrug Transporter AcrB Involves an Access and a Deep Binding Pocket That Are Separated by a Switch-Loop. Proc. Natl. Acad. Sci 2012, 109, 5687–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Nakashima R; Sakurai K; Yamasaki S; Nishino K; Yamaguchi A Structures of the Multidrug Exporter AcrB Reveal a Proximal Multisite Drug-Binding Pocket. Nature 2011, 480, 565–569. [DOI] [PubMed] [Google Scholar]
- (212).Lomovskaya O; Bostian KA Practical Applications and Feasibility of Efflux Pump Inhibitors in the Clinic—a Vision for Applied Use. Biochem. Pharmacol 2006, 71, 910–918. [DOI] [PubMed] [Google Scholar]
- (213).Matsumoto Y; Hayama K; Sakakihara S; Nishino K; Noji H; Iino R; Yamaguchi A Evaluation of Multidrug Efflux Pump Inhibitors by a New Method Using Microfluidic Channels. PLoS One 2011, 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Nakayama K; Ishida Y; Ohtsuka M; Kawato H; Yoshida K; Yokomizo Y; Hosono S; Ohta T; Hoshino K; Ishida H; others. MexAB-OprM-Specific Efflux Pump Inhibitors in Pseudomonas Aeruginosa. Part 1: Discovery and Early Strategies for Lead Optimization. Bioorg. Med. Chem. Lett 2003, 13, 4201–4204. [DOI] [PubMed] [Google Scholar]
- (215).Yoshida K; Nakayama K; Ohtsuka M; Kuru N; Yokomizo Y; Sakamoto A; Takemura M; Hoshino K; Kanda H; Nitanai H; others. MexAB-OprM Specific Efflux Pump Inhibitors in Pseudomonas Aeruginosa. Part 7: Highly Soluble and in Vivo Active Quaternary Ammonium Analogue D13–9001, a Potential Preclinical Candidate. Bioorg. Med. Chem 2007, 15, 7087–7097. [DOI] [PubMed] [Google Scholar]
- (216).Opperman TJ; Kwasny SM; Kim H-S; Nguyen ST; Houseweart C; D’Souza S; Walker GC; Peet NP; Nikaido H; Bowlin TL Characterization of a Novel Pyranopyridine Inhibitor of the AcrAB Efflux Pump of Escherichia Coli. Antimicrob. Agents Chemother 2014, 58, 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Nguyen ST; Kwasny SM; Ding X; Cardinale SC; McCarthy CT; Kim H-S; Nikaido H; Peet NP; Williams JD; Bowlin TL; others. Structure--Activity Relationships of a Novel Pyranopyridine Series of Gram-Negative Bacterial Efflux Pump Inhibitors. Bioorg. Med. Chem 2015, 23, 2024–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Trott O; Olson AJ AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; others. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (220).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- (221).Allen WJ; Balius TE; Mukherjee S; Brozell SR; Moustakas DT; Lang PT; Case DA; Kuntz ID; Rizzo RC DOCK 6: Impact of New Features and Current Docking Performance. J. Comput. Chem 2015, 36, 1132–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Irwin JJ; Shoichet BK ZINC- a Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model 2005, 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Lyne PD; Lamb ML; Saeh JC Accurate Prediction of the Relative Potencies of Members of a Series of Kinase Inhibitors Using Molecular Docking and MM-GBSA Scoring. J. Med. Chem 2006, 49, 4805–4808. [DOI] [PubMed] [Google Scholar]
- (224).Shriram V; Khare T; Bhagwat R; Shukla R; Kumar V Inhibiting Bacterial Drug Efflux Pumps via Phyto-Therapeutics to Combat Threatening Antimicrobial Resistance. Front. Microbiol 2018, 9, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Mangiaterra G; Laudadio E; Cometti M; Mobbili G; Minnelli C; Massaccesi L; Citterio B; Biavasco F; Galeazzi R Inhibitors of Multidrug Efflux Pumps of Pseudomonas Aeruginosa from Natural Sources: An in Silico High-Throughput Virtual Screening and in Vitro Validation. Med. Chem. Res 2017, 26, 414–430. [Google Scholar]
- (226).Mahenthiralingam E; Urban TA; Goldberg JB The Multifarious, Multireplicon Burkholderia Cepacia Complex. Nat. Rev. Microbiol 2005, 3, 144–156. [DOI] [PubMed] [Google Scholar]
- (227).Sajjan U; Corey M; Humar A; Tullis E; Cutz E; Ackerley C; Forstner J Immunolocalisation of Burkholderia Cepacia in the Lungs of Cystic Fibrosis Patients. J. Med. Microbiol 2001, 50, 535–546. [DOI] [PubMed] [Google Scholar]
- (228).Whiteford ML; Wilkinson JD; McColl JH; Conlon FM; Michie JR; Evans TJ; Paton JY Outcome of Burkholderia (Pseudomonas) Cepacia Colonisation in Children with Cystic Fibrosis Following a Hospital Outbreak. Thorax 1995, 50, 1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Hanulik V; Webber MA; Chroma M; Uvizl R; Holy O; Whitehead RN; Baugh S; Matouskova I; Kolar M An Outbreak of Burkholderia Multivorans beyond Cystic Fibrosis Patients. J. Hosp. Infect 2013, 84, 248–251. [DOI] [PubMed] [Google Scholar]
- (230).Hanulik V; Webber MA; Holy O; Roterva M; Kolar M Epidemiology of Burkholderia Multivorans Strains Obtained from Non-Cystic Fibrosis Patients Isolated in Large Hospitals across the Czech Republic. J. Hosp. Infect 2014, 86, 74–75. [DOI] [PubMed] [Google Scholar]
- (231).Stokell JR; Gharaibeh RZ; Steck TR Rapid Emergence of a Ceftazidime-Resistant Burkholderia Multivorans Strain in a Cystic Fibrosis Patient. J. Cyst. Fibros 2013, 12, 812–816. [DOI] [PubMed] [Google Scholar]
- (232).Ourisson G; Rohmer M; Poralla K Prokaryotic Hopanoids and Other Polyterpenoid Sterol Surrogates. Annu. Rev. Microbiol 1987, 41, 301–333. [DOI] [PubMed] [Google Scholar]
- (233).Mahato SB; Sen S Advances in Triterpenoid Research, 1990–1994. Phytochemistry 1997, 44, 1185–1236. [DOI] [PubMed] [Google Scholar]
- (234).Sáenz JP; Grosser D; Bradley AS; Lagny TJ; Lavrynenko O; Broda M; Simons K Hopanoids as Functional Analogues of Cholesterol in Bacterial Membranes. Proc. Natl. Acad. Sci 2015, 112, 11971–11976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Schmerk CL; Bernards MA; Valvano MA Hopanoid Production Is Required for Low-PH Tolerance, Antimicrobial Resistance, and Motility in Burkholderia Cenocepacia. J. Bacteriol 2011, 193, 6712–6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Welander PV; Hunter RC; Zhang L; Sessions AL; Summons RE; Newman DK Hopanoids Play a Role in Membrane Integrity and PH Homeostasis in Rhodopseudomonas Palustris TIE-1. J. Bacteriol 2009, 191, 6145–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Malott RJ; Steen-Kinnaird BR; Lee TD; Speert DP Identification of Hopanoid Biosynthesis Genes Involved in Polymyxin Resistance in Burkholderia Multivorans. Antimicrob. Agents Chemother 2012, 56, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Malott RJ; Wu C-H; Lee TD; Hird TJ; Dalleska NF; Zlosnik JEA; Newman DK; Speert DP Fosmidomycin Decreases Membrane Hopanoids and Potentiates the Effects of Colistin on Burkholderia Multivorans Clinical Isolates. Antimicrob. Agents Chemother 2014, 58, 5211–5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Du D; Wang-Kan X; Neuberger A; van Veen HW; Pos KM; Piddock LJV; Luisi BF Multidrug Efflux Pumps: Structure, Function and Regulation. Nat. Rev. Microbiol 2018, 16, 523–539. [DOI] [PubMed] [Google Scholar]
- (240).Su C-C; Long F; Zimmermann MT; Rajashankar KR; Jernigan RL; Edward WY Crystal Structure of the CusBA Heavy-Metal Efflux Complex of Escherichia Coli. Nature 2011, 470, 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Shi X; Chen M; Yu Z; Bell JM; Wang H; Forrester I; Villarreal H; Jakana J; Du D; Luisi BF; others. In Situ Structure and Assembly of the Multidrug Efflux Pump AcrAB-TolC. Nat. Commun 2019, 10, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Takatsuka Y; Nikaido H Threonine-978 in the Transmembrane Segment of the Multidrug Efflux Pump AcrB of Escherichia Coli Is Crucial for Drug Transport as a Probable Component of the Proton Relay Network. J. Bacteriol 2006, 188, 7284–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Honour JW Diagnosis of Diseases of Steroid Hormone Production, Metabolism and Action. J. Clin. Res. Pediatr. Endocrinol 2009, 1, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Zwama M; Yamasaki S; Nakashima R; Sakurai K; Nishino K; Yamaguchi A Multiple Entry Pathways within the Efflux Transporter AcrB Contribute to Multidrug Recognition. Nat. Commun 2018, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Gong X; Qian H; Zhou X; Wu J; Wan T; Cao P; Huang W; Zhao X; Wang X; Wang P Structural Insights into the Niemann-Pick C1 (NPC1)-Mediated Cholesterol Transfer and Ebola Infection. Cell 2016, 165, 1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Li X; Lu F; Trinh MN; Schmiege P; Seemann J; Wang J; Blobel G 3.3 Å Structure of Niemann–Pick C1 Protein Reveals Insights into the Function of the C-Terminal Luminal Domain in Cholesterol Transport. PNAS 2017, 114, 9116–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Gong X; Qian H; Cao P; Zhao X; Zhou Q; Lei J; Yan N Structural Basis for the Recognition of Sonic Hedgehog by Human Patched1. 2018, 361, 1–18. [DOI] [PubMed] [Google Scholar]
- (248).Qian H; Cao P; Hu M; Gao S; Yan N; Gong X Inhibition of Tetrameric Patched1 by Sonic Hedgehog through an Asymmetric Paradigm. Nat. Commun 2019, 10, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Sandhu P; Akhter Y Siderophore Transport by MmpL5-MmpS5 Protein Complex in Mycobacterium Tuberculosis. J. Inorg. Biochem 2017, 170, 75–84. [DOI] [PubMed] [Google Scholar]
- (250).Belardinelli JM; Yazidi A; Yang L; Fabre L; Li W; Jacques B; Angala SK; Rouiller I; Zgurskaya HI; Sygusch J Structure-Function Profile of MmpL3, the Essential Mycolic Acid Transporter from Mycobacterium Tuberculosis. ACS Infect Dis 2016, 2, 2702–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Owens CP; Chim N; Graves AB; Harmston CA; Iniguez A; Contreras H; Liptak MD; Goulding CW The Mycobacterium Tuberculosis Secreted Protein Rv0203 Transfers Heme to Membrane Proteins MmpL3 and MmpL11. J. Biol. Chem 2013, 288, 21714–21728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Owens CP; Chim N; Goulding CW Insights on How the Mycobacterium Tuberculosis Heme Uptake Pathway Can Be Used as a Drug Target. Future Med. Chem 2013, 5, 1391–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Doherty CP Host-Pathogen Interactions: The Role of Iron. J. Nutr 2007, 137, 1341–1344. [DOI] [PubMed] [Google Scholar]
- (254).Pal R; Hameed S; Fatima Z Altered Drug Efflux under Iron Deprivation Unveils Abrogated MmpL3 Driven Mycolic Acid Transport and Fluidity in Mycobacteria. BioMetals 2019, 32, 49–63. [DOI] [PubMed] [Google Scholar]
- (255).Bernut A; Viljoen A; Dupont C; Sapriel G; Blaise M; Bouchier C; Brosch R; de Chastellier C; Herrmann J; Kremer L Insights into the Smooth-to-rough Transitioning in Mycobacterium Bolletii Unravels a Functional Tyr Residue Conserved in All Mycobacterial MmpL Family Members. Mol. Microbiol 2016, 99, 866–883. [DOI] [PubMed] [Google Scholar]
- (256).Eicher T; Seeger MA; Anselmi C; Zhou W; Brandstätter L; Verrey F; Diederichs K; Faraldo-Gómez JD; Pos KM Coupling of Remote Alternating-Access Transport Mechanisms for Protons and Substrates in the Multidrug Efflux Pump AcrB. Elife 2014, 3, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Tsukazaki T; Mori H; Echizen Y; Ishitani R; Fukai S; Tanaka T; Perederina A; Vassylyev DG; Kohno T; Maturana AD Structure and Function of a Membrane Component SecDF That Enhances Protein Export. Nature 2011, 474, 235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Xu Z; Meshcheryakov VA; Poce G; Chng S-S MmpL3 Is the Flippase for Mycolic Acids in Mycobacteria. Proc. Natl. Acad. Sci 2017, 114, 7993–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Fay A; Czudnochowski N; Rock JM; Johnson JR; Krogan NJ; Rosenberg O; Glickman MS Two Accessory Proteins Govern MmpL3 Mycolic Acid Transport in Mycobacteria. MBio 2019, 10, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Ung KL; Alsarraf HMAB; Kremer L; Blaise M The Crystal Structure of the Mycobacterial Trehalose Monomycolate Transport Factor A, TtfA, Reveals an Atypical Fold. Proteins Struct. Funct. Bioinforma 2019, 88, 809–815. [DOI] [PubMed] [Google Scholar]
- (261).Rapid Communication: Key changes to treatment of multidrug- and rifampicin-resistant tuberculosis. 2018, https://www.who.int/tb/publications/2018/rapid_communications_MDR/en/ accessed June 23, 2020.
- (262).Frieden TR; Sterling T; Pablos-Mendez A; Kilburn JO; Cauthen GM; Dooley SW The Emergence of Drug-Resistant Tuberculosis in New York City. N. Engl. J. Med 1993, 328, 521–526. [DOI] [PubMed] [Google Scholar]
- (263).Velayati AA; Masjedi MR; Farnia P; Tabarsi P; Ghanavi J; ZiaZarifi AH; Hoffner SE Emergence of New Forms of Totally Drug-Resistant Tuberculosis Bacilli: Super Extensively Drug-Resistant Tuberculosis or Totally Drug-Resistant Strains in Iran. Chest 2009, 136, 420–425. [DOI] [PubMed] [Google Scholar]
- (264).Bailo R; Bhatt A; Aínsa JA Lipid Transport in Mycobacterium Tuberculosis and Its Implications in Virulence and Drug Development. Biochem. Pharmacol 2015, 96, 159–167. [DOI] [PubMed] [Google Scholar]
- (265).Sandhu P; Akhter Y Evolution of Structural Fitness and Multifunctional Aspects of Mycobacterial RND Family Transporters. Arch. Microbiol 2018, 200, 19–31. [DOI] [PubMed] [Google Scholar]
- (266).Li W; Yazidi A; Pandya AN; Hegde P; Tong W; Calado Nogueira de Moura V; North EJ; Sygusch J; Jackson M MmpL3 as a Target for the Treatment of Drug-Resistant Nontuberculous Mycobacterial Infections. Front. Microbiol 2018, 9, 1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Williams JT; Haiderer ER; Coulson GB; Conner KN; Ellsworth E; Chen C; Alvarez-Cabrera N; Li W; Jackson M; Dick T Identification of New MmpL3 Inhibitors by Untargeted and Targeted Mutant Screens Defines MmpL3 Domains with Differential Resistance. Antimicrob. Agents Chemother 2019, 63 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Tahlan K; Wilson R; Kastrinsky DB; Arora K; Nair V; Fischer E; Barnes SW; Walker JR; Alland D; Barry III CE; Boshoff HI SQ109 Targets MmpL3, a Membrane Transporter of Trehalose Monomycolate Involved in Mycolic Acid Donation to the Cell Wall Core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother 2012. 56, 1797–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).La Rosa V; Poce G; Canseco JO; Buroni S; Pasca MR; Biava M; Raju RM; Porretta GC; Alfonso S; Battilocchio C MmpL3 Is the Cellular Target of the Antitubercular Pyrrole Derivative BM212. Antimicrob. Agents Chemother 2012, 56 (1), 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Stanley SA; Grant SS; Kawate T; Iwase N; Shimizu M; Wivagg C; Silvis M; Kazyanskaya E; Aquadro J; Golas A Identification of Novel Inhibitors of M. Tuberculosis Growth Using Whole Cell Based High-Throughput Screening. ACS Chem. Biol 2012, 7, 1377–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Tahlan K; Wilson R; Kastrinsky DB; Arora K; Nair V; Fischer E; Barnes SW; Walker JR; Alland D; Barry CE SQ109 Targets MmpL3, a Membrane Transporter of Trehalose Monomycolate Involved in Mycolic Acid Donation to the Cell Wall Core of Mycobacterium Tuberculosis. Antimicrob. Agents Chemother 2012, 56, 1797–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Lun S; Guo H; Onajole OK; Pieroni M; Gunosewoyo H; Chen G; Tipparaju SK; Ammerman NC; Kozikowski AP; Bishai WR Indoleamides Are Active against Drug-Resistant Mycobacterium Tuberculosis. Nat. Commun 2013, 4, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Rao SPS; Lakshminarayana SB; Kondreddi RR; Herve M; Camacho LR; Bifani P; Kalapala SK; Jiricek J; Ma NL; Tan BH Indolcarboxamide Is a Preclinical Candidate for Treating Multidrug-Resistant Tuberculosis. Sci. Transl. Med 2013, 5, 1–12. [DOI] [PubMed] [Google Scholar]
- (274).Remuiñán MJ; Pérez-Herrán E; Rullás J; Alemparte C; Martínez-Hoyos M; Dow DJ; Afari J; Mehta N; Esquivias J; Jiménez E Tetrahydropyrazolo [1, 5-a] Pyrimidine-3-Carboxamide and N-Benzyl-6′, 7′-Dihydrospiro [Piperidine-4, 4′-Thieno [3, 2-c] Pyran] Analogues with Bactericidal Efficacy against Mycobacterium Tuberculosis Targeting MmpL3. PLoS One 2013, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Foss MH; Pou S; Davidson PM; Dunaj JL; Winter RW; Pou S; Licon MH; Doh JK; Li Y; Kelly JX Diphenylether-Modified 1, 2-Diamines with Improved Drug Properties for Development against Mycobacterium Tuberculosis. ACS Infect. Dis 2016, 2, 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Dupont C; Chen Y; Xu Z; Roquet-Banères F; Blaise M; Witt A-K; Dubar F; Biot C; Guérardel Y; Maurer FP A Piperidinol-Containing Molecule Is Active against Mycobacterium Tuberculosis by Inhibiting the Mycolic Acid Flippase Activity of MmpL3. J. Biol. Chem 2019, 294, 17512–17523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Shetty A; Xu Z; Lakshmanan U; Hill J; Choong ML; Chng S-S; Yamada Y; Poulsen A; Dick T; Gengenbacher M Novel Acetamide Indirectly Targets Mycobacterial Transporter MmpL3 by Proton Motive Force Disruption. Front. Microbiol 2018, 9, 2960–2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Zheng H; Williams JT; Coulson GB; Haiderer ER; Abramovitch RB HC2091 Kills Mycobacterium Tuberculosis by Targeting the MmpL3 Mycolic Acid Transporter. Antimicrob. Agents Chemother 2018, 62, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Shao M; McNeil M; Cook GM; Lu X MmpL3 Inhibitors as Antituberculosis Drugs. Eur. J. Med. Chem 2020, 200, 112390 1–15. [DOI] [PubMed] [Google Scholar]
- (280).Li W; Stevens CM; Pandya AN; Darzynkiewicz Z; Bhattarai P; Tong W; Gonzalez-Juarrero M; North EJ; Zgurskaya HI; Jackson M Direct Inhibition of MmpL3 by Novel Antitubercular Compounds. ACS Infect. Dis 2019, 5, 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Li W; Upadhyay A; Fontes FL; North EJ; Wang Y; Crans DC; Grzegorzewicz AE; Jones V; Franzblau SG; Lee RE Novel Insights into the Mechanism of Inhibition of MmpL3, a Target of Multiple Pharmacophores in Mycobacterium Tuberculosis. Antimicrob. Agents Chemother 2014, 58, 6413–6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Poce G; Bates RH; Alfonso S; Cocozza M; Porretta GC; Ballell L; Rullas J; Ortega F; De Logu A; Agus E Improved BM212 MmpL3 Inhibitor Analogue Shows Efficacy in Acute Murine Model of Tuberculosis Infection. PLoS One 2013, 8, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).McNeil MB; Dennison D; Parish T Mutations in MmpL3 Alter Membrane Potential, Hydrophobicity and Antibiotic Susceptibility in Mycobacterium Smegmatis. Microbiology 2017, 163, 1065–1070. [DOI] [PubMed] [Google Scholar]
- (284).Ioerger TR; O’Malley T; Liao R; Guinn KM; Hickey MJ; Mohaideen N; Murphy KC; Boshoff HIM; Mizrahi V; Rubin EJ Identification of New Drug Targets and Resistance Mechanisms in Mycobacterium Tuberculosis. PLoS One 2013, 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).McNeil MB; Cook GM Utilization of CRISPR Interference to Validate MmpL3 as a Drug Target in Mycobacterium Tuberculosis. Antimicrob. Agents Chemother 2019, 63, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Stec J; Onajole OK; Lun S; Guo H; Merenbloom B; Vistoli G; Bishai WR; Kozikowski AP Indole-2-Carboxamide-Based MmpL3 Inhibitors Show Exceptional Antitubercular Activity in an Animal Model of Tuberculosis Infection. J. Med. Chem 2016, 59, 6232–6247. [DOI] [PubMed] [Google Scholar]
- (287).Trofimov V; Kicka S; Mucaria S; Hanna N; Ramon-Olayo F; Del Peral LV-G; Lelièvre J; Ballell L; Scapozza L; Besra GS Antimycobacterial Drug Discovery Using Mycobacteria-Infected Amoebae Identifies Anti-Infectives and New Molecular Targets. Sci. Rep 2018, 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Sakurai K; Yamasaki S; Nakao K; Nishino K; Yamaguchi A; Nakashima R Crystal Structures of Multidrug Efflux Pump MexB Bound with High-Molecular-Mass Compounds. Sci. Rep 2019, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Akama H; Kanemaki M; Yoshimura M; Tsukihara T; Kashiwagi T; Yoneyama H; Narita S; Nakagawa A; Nakae T Crystal Structure of the Drug Discharge Outer Membrane Protein, OprM, of Pseudomonas Aeruginosa Dual Modes of Membrane Anchoring and Occluded Cavity End. J. Biol. Chem 2004, 279, 52816–52819. [DOI] [PubMed] [Google Scholar]
- (290).Tsutsumi K; Yonehara R; Ishizaka-Ikeda E; Miyazaki N; Maeda S; Iwasaki K; Nakagawa A; Yamashita E Structures of the Wild-Type MexAB–OprM Tripartite Pump Reveal Its Complex Formation and Drug Efflux Mechanism. Nat. Commun 2019, 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Qi X; Schmiege P; Coutavas E; Wang J; Li X Structures of Human Patched and Its Complex with Native Palmitoylated Sonic Hedgehog. Nature 2018, 560, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Rudolf AF; Kinnebrew M; Kowatsch C; Ansell TB; El Omari K; Bishop B; Pardon E; Schwab RA; Malinauskas T; Qian M The Morphogen Sonic Hedgehog Inhibits Its Receptor Patched by a Pincer Grasp Mechanism. Nat. Chem. Biol 2019, 15, 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Parmar M; Rawson S; Scarff CA; Goldman A; Dafforn TR; Muench SP; Postis VLG Using a SMALP Platform to Determine a Sub-Nm Single Particle Cryo-EM Membrane Protein Structure. Biochim. Biophys. Acta (BBA)-Biomembranes 2018, 1860, 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Rivera-Calzada A; Carroni M Technical Advances in Cryo-Electron Microscopy. Front. Mol. Biosci 2019, 6, 72. 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Ho C-M; Beck JR; Lai M; Cui Y; Goldberg DE; Egea PF; Zhou ZH Malaria Parasite Translocon Structure and Mechanism of Effector Export. Nature 2018, 561, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Chorev DS; Baker LA; Wu D; Beilsten-Edmands V; Rouse SL; Zeev-Ben-Mordehai T; Jiko C; Samsudin F; Gerle C; Khalid S Protein Assemblies Ejected Directly from Native Membranes Yield Complexes for Mass Spectrometry. Science (80-.) 2018, 362, 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Wong W; Bai X-C; Sleebs BE; Triglia T; Brown A; Thompson JK; Jackson KE; Hanssen E; Marapana DS; Fernandez IS Mefloquine Targets the Plasmodium Falciparum 80S Ribosome to Inhibit Protein Synthesis. Nat. Microbiol 2017, 2, 17031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Yen MR; Chen JS; Marquez JL; Sun EI; Saier MH Multidrug Resistance: Phylogenetic Characterization of Superfamilies of Secondary Carriers That Include Drug Exporters. In Membrane Transporters in Drug Discovery and Development; Springer, 2010; pp 47–64. [DOI] [PubMed] [Google Scholar]