

Abstract
Modulation of chromatin structure and/or modification by Polycomb repressive complexes (PRCs) provides an important means to partition the genome into functionally distinct subdomains and to regulate the activity of the underlying genes. Both the enzymatic activity of PRC2 and its chromatin recruitment, spreading, and eviction are exquisitely regulated via interactions with cofactors and DNA elements (such as unmethylated CpG islands), histones, RNA (nascent mRNA and long noncoding RNA), and R-loops. PRC2-catalyzed histone H3 lysine 27 trimethylation (H3K27me3) is recognized by distinct classes of effectors such as canonical PRC1 and BAH module-containing proteins (notably BAHCC1 in human). These effectors mediate gene silencing by different mechanisms including phase separation-related chromatin compaction and histone deacetylation. We discuss recent advances in understanding the structural architecture of PRC2, the regulation of its activity and chromatin recruitment, and the molecular mechanisms underlying Polycomb-mediated gene silencing. Because PRC deregulation is intimately associated with the development of diseases, a better appreciation of Polycomb-based (epi)genomic regulation will have far-reaching implications in biology and medicine.
Keywords: Polycomb, PRC2, PRC1, Chromatin, Histone, H3K27me3, methylation, RNA, Transcription, CpG island, R-loop, Allosteric activation, Phase separation, BAH, chromodomain
Introduction
Pioneering studies on the genetics of Drosophila led to the discovery of the multi-subunit Polycomb repressive complexes, PRC2 and PRC1. Polycomb proteins EZH2/1, EED, SUZ12, and RBBP4/7 (Table 1) jointly establish a PRC2 ‘core’ (Box 1) which exhibits an exquisite molecular architecture that permits functional modulation. This topic has been reviewed elsewhere [1–5]; our aim here is to overview recent advances in understanding PRC-mediated (epi)genomic and transcriptomic regulation at the interface of chromatin, RNA, and regulatory machineries. In the following we focus on (i) modulation of core PRC2 enzymatic activity, (ii) chromatin targeting and spreading of PRC2 versus its eviction and functional restriction, and (iii) Polycomb-mediated gene-silencing mechanisms, with a focus on effectors for H3K27me3, a PRC2-catalyzed histone post-translational modification (PTM).
Table 1.
Function of the core subunit, accessory cofactor and modulatory factor of mammalian PRC2.
| Protein ID | Function | |
|---|---|---|
| “core” PRC2 | Enhancer of Zeste Homolog 2 (EZH2) or Enhancer of Zeste Homolog 1 (EZH1) | (i) Sole methyltransferase class in cells for ‘writing’ H3K27me3/2 on chromatin, with the catalytic activity residing in a domain termed Su(var)3–9, Enhancer-of-zeste and Trithorax (SET); (ii) Provides protein-protein interaction interfaces for various partners; (iii) EZH2 is subjected to allosteric activation and potentially, autoinhibition whereas EED-mediated allosteric activation cannot be applied for EZH1, contributing to a functional distinction between the two EZHs |
| Embryonic Ectoderm Development (EED) | (i) Mediates allosteric activation and on-chromatin spreading of PRC2 via binding H3K27me3 (or methylated cofactor such as JARID2 and PALI1) with a ‘reader’ module of WD40 repeat; (ii) Preferentially activates EZH2:PRC2 over EZH1:PRC2 due to a variation at their interaction interfaces | |
| Suppressor Of Zeste 12 Homolog (SUZ12) | Exhibits a dual modular organization, with (i) its C-terminal VEFS domain mediating assembly of PRC2 core and (ii) an N-terminal domain extension serving as a platform or “hub” for interaction with various cofactors (at least six) including PCL1/2/3, JARID2, EPOP and ABEP2, which are involved in the de novo targeting or the enhanced binding of PRC2 “core” onto the chromatin. | |
| Retinoblastoma Binding Protein 4 or 7 (RBBP4, also known as RbAp48; or RBBP7, also known as RbAp46) | Bridges PRC2 and chromatin together by directly binding to nucleosome; and such binding is suppressed when histone H3 Lys4 (H3K4) is methylated | |
| Accessory or “non-core” factor unique to the PRC2.1 sub-complex | Polycomb-like (PCL), including PCL1/PHF1, PCL2/MTF2 and PCL3/PHF19 | (i) One PCL and “core” PRC2 form the PRC2.1 subcomplex, in which the C-terminal chromo-like region of PCL forming direct interaction with SUZ12; (ii) Targets PRC2.1 to chromatin; (iii) the N-terminal domains of PCL harbor various chromatin-binding activities, such as binding to DNA (notably, CpG island, GC- and/or CpG- rich elements) by the EH module and to H3K36me3/2 (or testis-specific H3tK27me3) by the Tudor domain, which mediate PRC2.1’s de novo recruitment and/or enhance its residence time on chromatin |
| Elongin BC and Polycomb Repressive Complex 2 associated protein (EPOP; also known as C17ORF96 or esPRC2p48) | (i) EPOP:PRC2 interaction is established through a C-terminal region of EPOP and the ZnB-Zn domain of SUZ12; (ii) EPOP connects PRC2.1 to a functionally antagonizing complex, Elongin, and suppresses transcriptional elongation by the latter; this action is potentially through the EPOP:PRC2.1-mediated sequestration of the cofactor EloB/C and/or direct methylation of EloA, as well as via PRC2- independent effect | |
| PRC2-associated LCOR isoform 1 (PALI1; also known as C10ORF12); PALI2 | (i) PALI1/2 positively modulates the catalytic activity of PRC2 during mouse embryogenesis but its regulatory role merits additional study; (ii) EPOP and PALI1/2 are present in the PRC2.1 complex variant in a mutually exclusive manner | |
| Accessory factor for PRC2.2 subcomplex | Jumonji and AT-rich interaction domain containing 2 (JARID2) | (i) Specifies the PRC2.2 sub-complex and directly binds to ZnB-Zn of SUZ12; (ii) Modulates chromatin recruitment and enzymatic activity of PRC2 via various mechanisms; (iii) JARID2-K116me3, catalyzed by PRC2, mimics H3K27me3 for allosterically stimulating PRC2.2; (iv) JARID2 binds H2AK119ub1, lncRNAs and DNA via its various embedded domains; (v) JARID2 binding to H2AK119ub1 is essential for establishing a pathway of Polycomb domain establishment, which involves ncPRC1->H2AK119ub1->JAR1D2:PRC2.2-> H3K27me3 deposition |
| AE binding protein 2 (AEBP2) | (i) AEBP2 interacts with SUZ12 to stabilize PRC2 binding to genomic targets; (ii) the Zn figures of AEBP2 also bind DNA; (iii) AEBP2 may further promote allosteric stimulation of PRC2; however, its PRC2-regulatory function requires additional study. | |
| Other modulator of PRC2 | Chromosome X open-reading-frame 67 (CXORF67) also known as EZH Inhibitory Protein (EZHIP; aka CATACOMB) | (i) Uses a conserved, H3-like peptide sequence to form direct interaction with the SET domain of EZH2 and acts as a competitive inhibitor of PRC2; (ii) when overexpressed in cells, it alters global patterns of H3K27me3. |
| “oncohistone” H3K27M (cancer-related mutant) | (i) Acts as inhibitor of PRC2 via direct binding to the SET domain of EZH2; (ii) Dramatically changes the global patterning of H3K27me3. |
Box 1. Molecular Architecture of the PRC2 Core.
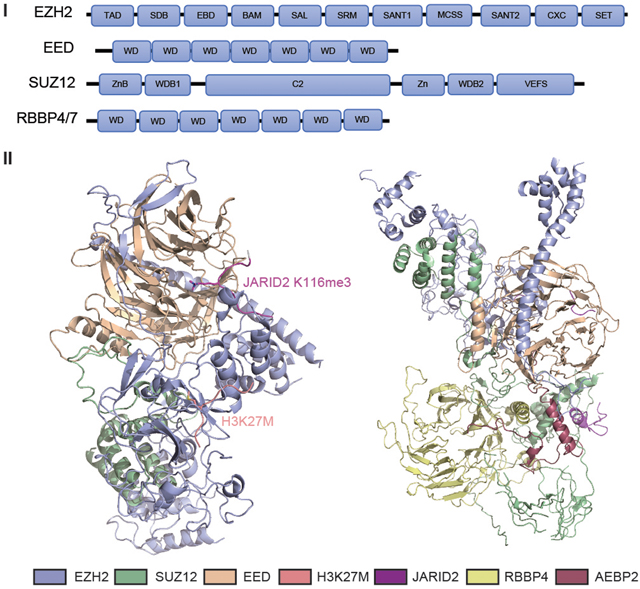
In mammals, a functional PRC2 core complex is formed by EZH2 (or a related EZH1), EED, SUZ12, and RBBP4/RbAp48 (or RBBP7/RbAp46), establishing an exquisite architecture and molecular structure. In essence, EZH2/1 acts as catalytic subunit for ‘writing’ the H3K27me1/2/3 marks. The EZH2/1 methyltransferase domain, termed Su(var)3–9, Enhancer of zeste, and Trithorax (SET) (Figure I), directly binds to the H3K27 substrate (indicated in Figure II) by H3K27M, a substrate mutant and mimic that has stronger binding to SET) and to the methyl donor, S-adenosyl methionine (SAM). EED is essential for PRC2 allosteric activation, upon binding to H3K27me3 (indicated in Figure III) by JARID2-K116me3, an H3K27me3 mimic). SUZ12 can be split into two functional modules – the SUZ12 C-terminal VEFS domain, together with EZH2 and EED, makes a stable PRC2 [19,21], and its long N-terminal extension directly binds to PRC2 cofactors such as Polycomb-like (PCLs) and JARID2 that mediate PRC2 targeting to nucleation sites [28]. RBBP4 or RBBP7 (also known as Nurf55 in Drosophila), by contrast, is not required for PRC2 methyltransferase activity but assists in chromatin recruitment by directly binding to histones, a process that is greatly suppressed by histone H3 lysine 4 (H3K4) methylation [36]. In addition to four ‘core’ components, we include in the illustration accessory cofactors of PRC2 (such as AEBP2 and JARID2) to demonstrate their interactions with PRC2 core that contribute to complex stability, activity, and/or chromatin binding.
Regulation of Core PRC2 Activity
The catalytic function of PRC2 is subject to multilevel regulation (Figure 1A) by conformational changes (such as allosteric activation, see Glossary), competitive inhibition, and influences from the chromatin environment.
Figure 1. Inhibition and Allosteric Activation of the PRC2 Core.
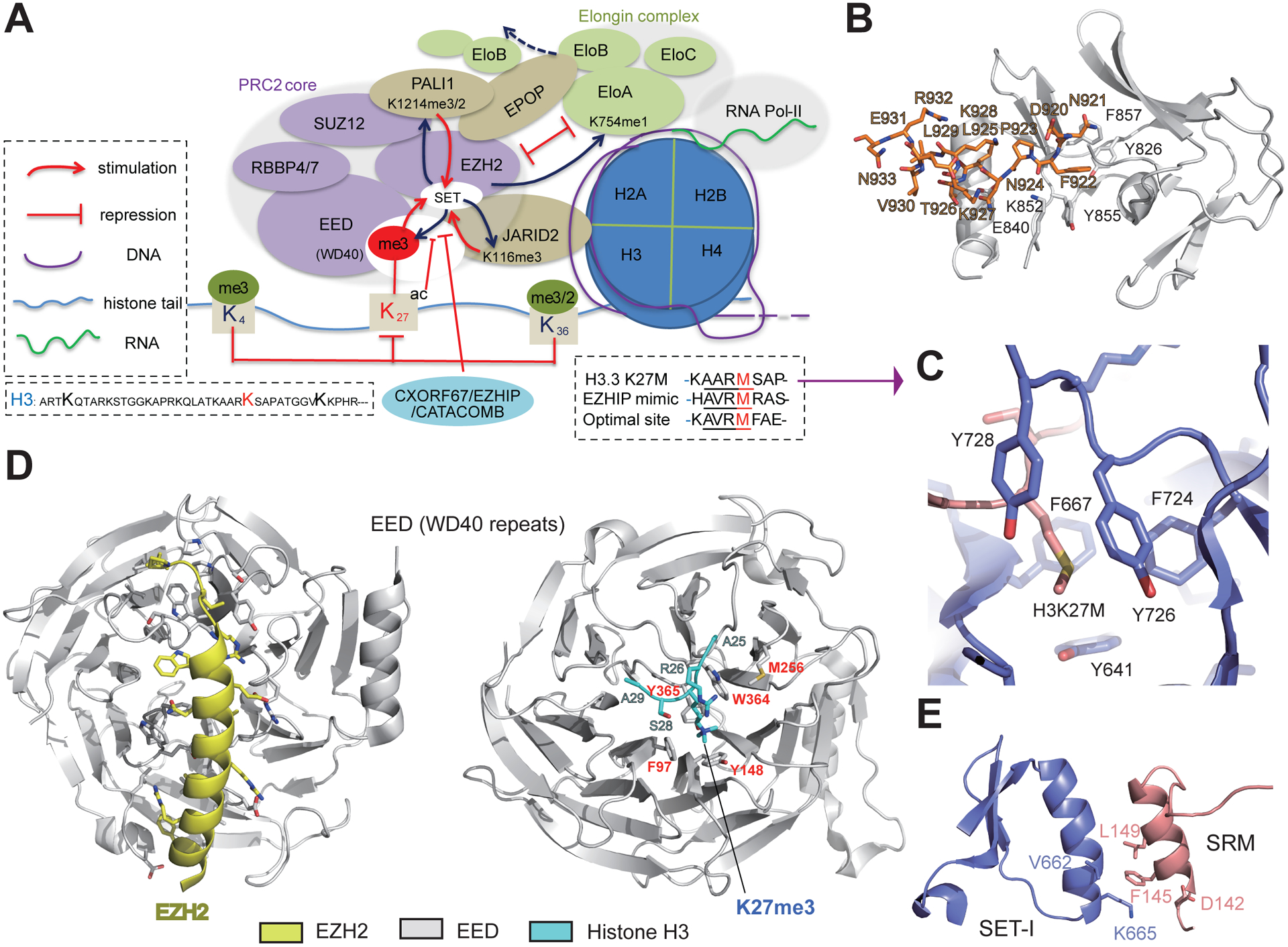
(A) Inhibition and activation of PRC2 ‘core’ activity are mediated by both ‘core’ and ‘non-core’ subunits, as well as by the chromatin environment (e.g., histone post-translational modifications and nascent RNA). EZHIP/CATACOMB mimics the K27M ‘oncohistone’ mutant of histone H3 (box on the right) and suppresses PRC2 catalytic activity via competitive inhibition. Conversely, EED binding to H3 trimethylated on Lys27 (H3K27me3) via its WD40 repeat leads to allosteric activation of PRC2:EZH2. In addition to H3K27, PRC2 also methylates the cofactors JARID2 and PALI1, and the trimethylated cofactors can mimic H3K27me3 and be read by EED for PRC2 allosteric activation. PRC2 also methylates EloA, a subunit of the Elongin complex, to inhibit Elongin-mediated transcriptional elongation. In addition, EPOP, a ‘non-core’ subunit of PRC2.1, can sequester EloB/C away from the Elongin complex, thereby downregulating target gene expression. (B) The autoinhibited conformation of the catalytic center of the fungus Chaetomium thermophilum PRC2 (PDB code 5BJS). The catalytic domain is shown as a cartoon in white, and the post-SET subdomain is shown as orange sticks. (C) Interaction between the oncohistone H3K27M and the active site of EZH2. (D) Structures showing EED in complex with the N terminus of EZH2 (left; PDB code 2QXV) and EED in complex with H3K27me3 peptide (right; PDB 3IIW). The N terminus of EZH2 is shown in yellow (left) and EED in gray, with its H3K27me3-recognizing residues labeled in red (right). (E) Binding interface between EZH2 SRM and SETI motifs (PDB 5HYN). Residues involving in interactions for allosteric activation are shown as sticks.
Autoinhibition and Competitive Inhibition of EZH2
In mammals, EZH2 adopts an autoinhibitory state and has little methyltransferase activity [6, 7]. The formation of the multi-subunit PRC2 generally makes EZH2 enzymatically competent. However, a crystallography study of fungal PRC2 captured an autoinhibited conformation in the absence of substrate and a methyl donor, a post-SET EZH2 motif binds to its catalytic center, excluding substrate from binding (Figure 1B) [8]. It is not known whether such autoinhibition applies to mammalian PRC2.
Recently, CXorf67/EZHIP/CATACOMB was found to be a negative regulator of PRC2. CXorf67 was initially identified as a gene involved in aberrant reciprocal chromosomal translocation of t(X;17)(p11.2; q21.33) that generates a fusion gene, termed MBTD1–CXorf67, in a subset of patients with endometrial stromal sarcoma [9]. A flurry of recent papers showed that CXorf67 directly inhibits PRC2, and was thus renamed EZHIP or CATACOMB (Table 1) [10–13]. Mechanistically, a conserved H3-like peptide of CXorf67/EZHIP/CATACOMB binds to the EZH2 SET domain, interfering with EZH2:H3 engagement [10–12] (Figure 1A). Thus, it acts as a competitive inhibitor of EZH2:PRC2 and suppresses global H3K27me3 in cells [10,11]. Such competitive inhibition of the active site of EZH2(SET) was previously demonstrated in cancer cells. In particular, a deadly pediatric glioma subtype is characterized by a lysine-to-methionine missense mutant of H3K27 (H3K27M), generating a so-called oncohistone that dramatically suppresses PRC2 activity [14–16] (Figure 1C). Likewise, EZHIP is frequently overexpressed in posterior fossa type A ependymoma, resulting in H3K27me3 loss in affected tumors [10,11]. In sarcoma, MBTD1–CXorf67 likely inhibits PRC2 by recruiting the antagonizing NuA4/TIP60 acetyltransferase complex via its MBTD1 segment [12]. Competitive inhibitors of PRC2:EZH2, CXorf67/EZHIP, and H3K27M impede H3K27me3 spreading, leading to perturbations in Polycomb-mediated gene silencing and altered cellular states during pathogenesis [10,13]. The function of EZHIP/CATACOMB during normal development, however, requires further investigation.
Allosteric Activation of PRC2 Coupled with Spreading on Chromatin
The binding affinity of EZH2 for histone substrates is inversely related to the degree of H3K27 methylation [17]. Therefore, elevated EZH2 activity is necessary to complete multimethylation and generate H3K27me3. The multi-subunit PRC2 core establishes a platform that permits enzymatic stimulation of EZH2:PRC2. A mechanism involving a series of EZH2:EED:H3K27me3 interactions modulates PRC2 conformation, inducing allosteric activation – once the EZH2 N terminus binds to a well-defined region of EED, it forms an α-helix (Figure 1D, left/yellow) [18]; meanwhile, the WD40 repeat of EED (for domain names, see Table S1 in the supplemental information online) reads H3K27me3 deposited by PRC2:EZH2 on the same or an adjacent nucleosome (Figure 1D, right), leading to stepwise allosteric activation of EZH2 [19]. The initial H3K27me3 reading involves an aromatic ‘cage’ formed by the EED WD40 repeat (Figure 1D, right/red) [20]. This binding then leads to a conformational change of EZH2 in which its stimulus-responsive motif (SRM; Box 1) becomes ordered and forms an interaction with SET domain (Figure 1E) [21]. Ultimately, the catalytic SET is stabilized and its methyltransferase activity is enhanced [19]. Disruption of such allosteric activation is recurrently seen in cancer via somatic mutations of the EED H3K27me3-reading motif (i.e., I363M, Y365A; Figure 1D, right/red) or the EZH2 SRM (P132S, D142V and F145L; Figure 1E), resulting in PRC2 loss of function (LOF) [22–24]. Generally, allosteric activation disrupting mutations do not affect PRC2 complex formation or its binding to nucleation sites, supporting the notion that PRC2 allosteric activation is independent of chromatin recruitment [24].
PRC2 also trimethylates cofactors JARID2 and PALI1 [25,26] (Table 1). Methylation of cofactors, such as JARID2-K116me3 and PALI1-K1241me2/3, can mimic H3K27me3 and be bound by EED, allosterically stimulating PRC2 [25,26] (Figure 1A).
EED binding to H3K27me3 and subsequent stimulation of PRC2:EZH2 immediately suggest a model (Figure 2A) in which PRC2:EZH2 spreads on chromatin to generate broad Polycomb domains, which was recently experimentally confirmed [27–29]. A cryogenic electron microscopy (cryo-EM) structure of PRC2 bound to a dinucleosome provides further support [30]. Essentially, one PRC2 simultaneously binds to two adjacent mononucleosomes, a process that is insensitive to the length of the linker DNA. The EZH2 CXC domain (Box 1) binds to DNA, positioning the H3 tail towards the EZH2 catalytic center [30]. If H3K27me3 is present on both adjacent nucleosomes, EED is then able to recognize it, causing further activation of PRC2 and deposition/spreading of H3K27me3 [30].
Figure 2. De Novo Targeting and Spreading of PRC2 on Chromatin versus its Eviction and Functional Restriction in Mammalian Cells.
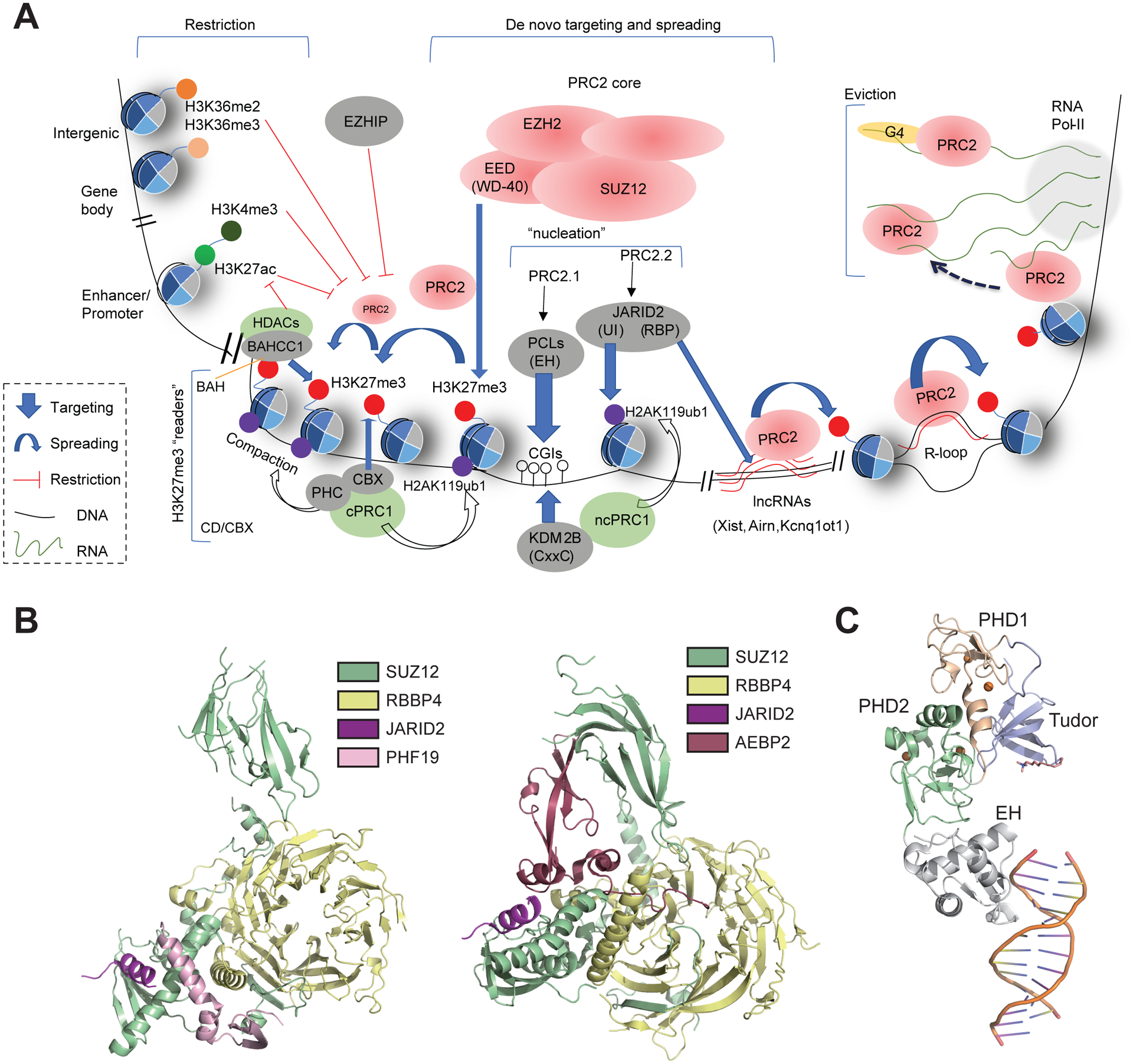
(A) (Top left) Both PRC2 enzymatic activity and Polycomb domain spreading are suppressed by gene activation-related histone post-translational modifications (PTMs), such as active promoter/enhancer-associated histone H3 Lys27 acetylation (H3K27ac)/Lys4 trimethylation (H3K4me3) and transcription-associated Lys36 trimethylation (H3K36me3; located within gene bodies); H3 Lys36 dimethylation (H3K36me2), that is deposited at intergenic regions or promoters, also inhibits PRC2-mediated H3K27me3 deposition/spreading. Likewise, EZHIP, which binds to EZH2 SET domain, impedes H3K27me3 deposition/spreading. (Middle) Reciprocal recruitment between PRC1 and PRC2 to establish Polycomb domains in the genome. On the one hand, the chromatin binding by PCLs [notably via its EH module that binds to unmethylated CpG-rich islands (CGIs) and/or G+C-rich elements] mediates de novo targeting of cPRC1, which deposits H3K27me3; this H3K27me3 can be recognized by EED, which ‘kicks off’ PRC2.1 spreading, and can in addition be ‘read’ by CBX within the canonical PRC1 complex (cPRC1) for deposition of histone H2A ubiquitinated on Lys119 (H2AK119ub1). On the other hand, a noncanonical PRC1 complex (ncPRC1) is recruited independently by a DNA-binding factor, such as KDM2B (which binds to unmethylated CGIs via its CxxC motif), and catalyzes H2AK119ub1; this H2AK119ub1 is subsequently bound by JARID2 (via its UI motif) to recruit/target PRC2.2 to chromatin. In addition, JARID2 binds to long noncoding RNAs (lncRNAs) via its RBP motif, and contributes to PRC2 spreading/deposition and Polycomb domain formation at silenced loci. R-loop is also relevant for PRC2 chromatin binding/recruitment. (Top right) PRC2 interactions with nascent RNAs. At active sites of transcription, PRC2 is evicted from chromatin via interactions with nascent precursor mRNAs, notably G-quadruplex (G4) RNA structures that have a higher affinity for PRC2 binding. Note that a different ‘RNA bridge’ model has also been proposed [79]. (Bottom left) At least two different classes of H3K27me3-specific readers (Figure 3), namely chromodomains (CDs) within CBXs in cPRC1 and BAH-containing proteins such as BAHCC1, induce chromatin compaction and histone decetylation that contribute to Polycomb-mediated gene silencing in mammalian cells. (B) Structures showing interactions between the N-terminal region of SUZ12 and a set of non-core subunits that are unique to PRC2.1 (PHF19/PCL3; left, PDB code 6NQ3) or PRC2.2 (JARID2 and AEBP2; right, PDB 5WAI). Subunits are colored as labeled. (C) Structure illustrating binding of the CG-rich double-stranded (ds)DNA by the conserved EH motif of PCLs (PHF1; PDB 5XFQ). Different domains are colored as labeled. The dsDNA is shown as a cartoon. Abbreviation: Pol II, polymerase II.
Overall, the molecular architecture of PRC2:EZH2 combines allosteric activation and chromatin spreading, which does not apply to PRC2:EZH1. Based on these findings, small molecules have been developed that either inhibit or promote allosteric activation of PRC2:EZH2. The allosteric inhibitor EED-226 causes a conformational change of the EED H3K27me3 reading cage (W364 and Y365; Figure 1D), thus suppressing EED:H3K27me3 engagement [33]. By contrast, the small molecules UNC5635 and UNC5636 serve as EED activators and are potentially useful for selectively correcting phenotypes caused by EED LOF mutations (i.e., I363M that is found in patients with myeloid disorders), without affecting wild-type EED [34].
Modulation of PRC2 Activity by the Chromatin Environment
Histone density enhances PRC2 function, and PRC2 enzymatic activity is much higher on dinucleosomes than on mononucleosomes [35]. Histone PTMs, including H3K27ac [a single lysine can be acetylated (ac) or methylated, but not both], H3K4me3, and H3K36me3/2, directly suppress PRC2 catalytic function [36] (Figures 1A and 2A).
De Novo Recruitment and Spreading of PRC2, versus Its Eviction and Polycomb Domain Restriction
PRC proteins are markedly dynamic in cells, and the majority rapidly diffuse throughout the nucleus [37,38]. It is widely held (Figure 2A) that de novo recruitment of PRC2 establishes initial H3K27me2/3 at ‘nucleation’ sites, followed by spreading of PRC2 and H3K27me3. Studies have also uncovered both stimulatory and antagonizing factors that affect PRC2 binding/residence on chromatin and its eviction or functional restriction. In contrast to Polycomb response elements (PREs), defined in Drosophila as cis-elements that mediate PRC recruitment or antagonize Trithorax group proteins, their mammalian counterparts have been unclear, and increasing evidence indicates that unmethylated CG-rich sequences such as CpG islands (CGIs) [1,27,28,39–42] and other chromatin features such as R-loops [43,44] are also involved.
Vertebrate SUZ12 Uses Separate Protein Modules To Coordinate PRC2 Complex Assembly and De Novo Chromatin Targeting via Cofactor Binding
Chromatin targeting by PRC2 can be independent of H3K27me3 catalysis, although the two synergize in forming broad Polycomb domains. In support, mammalian SUZ12 (Table 1), a core component of PRC2, can be split to two functional modules – its C-terminal VEFS (Box 1), together with EZH2 and EED, generate a stable minimally active PRC2 [19,21], while its long N-terminal extension directly binds to various ‘non-core’ PRC2 cofactors of PRC2 (Figure 2B), PCLs, JARID2, and AEBP2 [45,46], that have distinct chromatin targeting/binding activities (Table 1) and have no requirement for pre-existing H3K27me3 or other PRC2 core components [28]. In mESCs, PRC2 assembled with SUZ12-VEFS cannot deposit H3K27me3 at well defined Polycomb sites, suggesting a chromatin-targeting defect [28]. The SUZ12 homolog (UniProt D6NKI3) in Tetrahymena, a unicellular ciliate, only contains VEFS, indicating progressive evolution of the multifaceted functions of SUZ12.
Two Distinctive Non-Core Cofactor Classes, PCLs and JARID2, Mediate De Novo PRC2 Establishment via Different Pathways
Following inducible EED re-expression in Eed−/− mESCs, Oksuz et al. measured initial seeding of EED and H3K27me3 in the genome, termed nucleation, and demonstrated that MTF2/PCL2 (a PCL member that is abundantly expressed in mESCs), and to a lesser extent JARID2 (but not AEBP2), mediate de novo PRC2 establishment at nucleation sites enriched in unmethylated CGIs [27]. Consistent findings were shown by separate studies in mESCs [40,41,47]. Based on composition variation, a subcomplex variant of PRC2.1 was defined by its unique cofactors –PCLs together with EPOP or PALI1/2 – whereas JARID2, together with AEBP2, defines PRC2.2 [48–53] (Box 2 and Table 1).
Box 2. PRC2 variants PRC2.1 and PRC2.2.
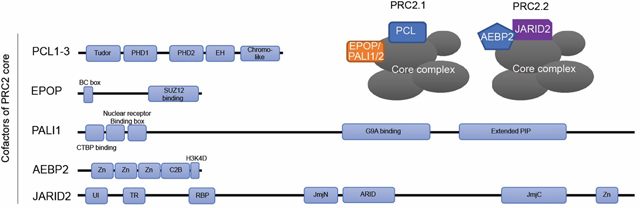
Mammalian PRC2 core can associate with a set of non-core cofactors – Polycomb-like (PCL), EPOP, and PALI1/2 – that define a variant complex termed PRC2.1 [48–53]. PCL was first identified in Drosophila, and there are three mammalian PCL family genes, PCL1/PHF1, PCL2/MTF2, and PCL3/PHF19. Each PRC2.1 complex contains only one PCL [53]. Whereas the PCL:PRC2.1 interaction is mediated through the chromo-like domain of PCL and the C2 domain of SUZ12 (Box 1), the EPOP:PRC2 interaction is established through a C-terminal region of EPOP and the ZnB-Zn domain of SUZ12 [45,52]. Via its PIP-box, PALI1/2 interacts with PRC2.1, and PALI1/2 and EPOP are present in PRC2.1 in a mutually exclusive manner, but both can colocalize with PCL [51,143]. Another set of cofactors, JARID2 and AEBP2, define the PRC2.2 subcomplex variant [50,51,53]. In PRC2.2, ZnB-Zn of SUZ12 directly binds to JARID2 [45,46]. AEBP2:SUZ12 interaction interfaces are mapped to SUZ12 C2 and AEBP2 C2-binding (C2B) domains, as well as AEBP2 zinc fingers and the SUZ12 Zn finger-binding domain (ZnB), which is in structurally proximity to the SET domain of EZH2.
PRC2.1 chromatin targeting is mediated mainly by evolutionarily conserved PCLs that ‘kick off’ the EED-mediated feedforward loop for PRC2.1 to establish broad Polycomb domains in mESCs [27,40,41,47]. Biophysical and crystallographic studies of the EH domain found in all PCLs (Box 2) have revealed a winged-helix structure (Figure 2C) that binds to unmethylated CGIs in vitro and is essential for PRC2.1 binding to CGI-enriched sites in mESCs [40]. Independent investigation with the mapped MTF2 binding sites further suggested the involvement of unmethylated GCG trinucleotides in generating a specific DNA shape feature in the context of CGIs that distinguishes MTF2-targeted CGIs from non-targeted CGIs [41]. Furthermore, Choi et al. performed in vitro characterization of EH in human PCL1/PHF1 or Drosophila Pcl and revealed that it enhances PRC2-mediated H3K27me3, which is likely due to EH direct binding to unmethylated DNA, thereby increasing PRC2 residence time on chromatin [54]; however, they also showed that DNA binding by these EHs was not sequence-specific [54]. In multiple myeloma, a plasma cell malignancy exhibiting PCL3/PHF19 overexpression and dependency, point mutations in the PCL3/PHF19 EH motif that abolish CGI-binding [40] phenocopied PHF19 depletion or deletion of its PRC2-interacting domain (chromo-like; Figure 2B), which all resulted in derepression of PRC2 target genes and delayed tumor growth [55]. These findings collectively support an essential requirement of PCLs, and most likely their DNA-binding EH modules, for PRC2.1 chromatin targeting (Figure 2A, middle). In cases of PCL2/MTF2 and PCL3/PHF19, EH binds to CGI- and/or G+C-rich DNA [27,40,41].
Operating independently of the above mechanism, JARID2 also directly binds to the N-terminal module of SUZ12 (Figure 2B) and mediates de novo recruitment of PRC2.2 in mESCs [27,41,47]. The ubiquitin-interacting (UI) motif of JARID2 (Box 2) binds to mono-ubiquitinated H2A Lys119 (H2AK119ub1) [56], a catalytic product of PRC1 (Figure 2A, middle and Box 3), and depletion of H2AK119ub1 preferentially affects overall genomic binding of PRC2.2 with respect to PRC2.1 [57,58], thus demonstrating a pathway the differs from the canonical axis and that involves PRC2→H3K27me3→PRC1(CBX)→H2AK119ub1 (reviewed in [1,3]). Initially, Tavares et al. reported H3K27me3-independent recruitment of a RYBP-containing PRC1 variant, termed ncPRC1 (Box 3), in mESCs [59], which was further demonstrated by an artificial targeting system wherein ncPRC1-dependent H2AK119ub1 leads to the de novo recruitment of PRC2, followed by the formation of a Polycomb domain [42]. ncPRC1 chromatin targeting relies on its interaction with DNA/chromatin-binding factors such as KDM2B/FBXL10 (which harbors a CxxC zinc finger that recognizes unmethylated CGIs [42]), E2F6, and L3MBTL2 [60–62]. This noncanonical axis was further verified during X-inactivation wherein PRC2 recruitment onto inactive X-chromosome is preceded by ncPRC1-induced deposition of H2AK119ub1 [63,64], and can also be applied to genome-wide where knockout or catalytic inactivation of PRC1 (by I53A/D56K of RING1B) significantly reduced binding of PRC2, especially of PRC2.2, at H2AK119ub1-marked sites in mESCs [57,58].
Box 3. PRC1 subcomplex variants.
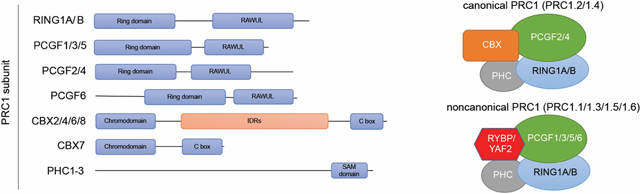
Mammalian PRC1 is composed of a ubiquitin ligase (either RING1A or RING1B), one of six PCGF family members (PCGF1–6), and one of three PHC family members [42,59,97,98]. Depending on which PCGF is included, PRC1 can be subdivided into six different subcomplexes, PRC1.1 to PRC1.6, with each containing a single PCGF member [97]. Another way to classify PRC1 subcomplexes is based on the presence of a chromobox (CBX) subunit generate canonical PRC1 (cPRC1) and noncanonical PRC1 (ncPRC1), respectively. In addition to RING1A/B, cPRC1s contain a CBX subunit and a PCGF member, either PCGF2 (also known as MEL18) or PCGF4 (also known as BMI1), which are also termed PRC1.2 and PRC1.4, respectively [97]. Conversely, ncPRC1s include a unique subunit, either RING1- and YY1-binding protein (RYBP) or YY1-associated factor 2 (YAF2), and one of the four PCGF1/3/5/6 proteins [97], also termed PRC1.1, PRC1.3, PRC1.5, and PRC1.6, respectively. In addition to RING1, both cPRC1 and ncPRC1 contain a Polyhomeotic homolog 1–3 subunit (PHC1/2/3).
Altogether, distinct cofactor-based recruitment mechanisms not only operate interdependently but, importantly, synergize in the establishment of broad Polycomb domains (Figure 2A, middle).
Cis-Acting lncRNAs Mediate/Stabilize PRC2 Chromatin Recruitment
Cis-acting long noncoding RNAs (lncRNAs) enhance PRC2 function at PRCs-silenced regions (Figure 2A, bottom/right), exemplified by Xist-dependent X-inactivation, Kcnq1ot1-induced genetic imprinting, and HOX repression by HOTAIR [65–67]. Furthermore, Schertzer et al. show that, in trophoblast stem cells, lncRNAs (Xist, Airn, and Kcnq1ot1) direct PRC2 chromatin occupancy and mediate H3K27me3 spreading [68]. The effects of these lncRNAs are determined by spatial proximity to their targeted loci (thus being restricted to local chromatin), the lncRNA copy number, the presence of PRC2 nucleation sites, and assistance from the RNA-binding protein HNRNPK [68]. Because PRC2 nucleation is required [68], lncRNAs likely act in concert with other PRC2-recruitment mechanisms. Indeed, in addition to H2AK119ub1, JARID2 also binds to lncRNAs through its RBP motif (Box 2), an event that stimulates the JARID2:PRC2 interaction and chromatin targeting [69,70] (Figure 2A, middle).
R-Loops Emerge as a Chromatin Feature for PRC Targeting
Chromatin binding by PRCs involves R-loops (Figure 2A, bottom/right) [43,44], a DNA:RNA hybrid that often forms cotranscriptionally near gene promoters [71]. In Drosophila and mESCs, PRCs including PRC2 recognize targets where the promoters form R-loops and expression is low [43,44]. Resolution of R-loops by degrading RNAs with RNase H1 specifically reduced local PRC-binding levels and corresponding PTMs (H3K27me3 and H2AK118/119ub1), leading to derepression of R-loop-associated, but not R-loop-negative, targets [43,44]. Interestingly, R-loop removal in Ezh2−/− mESCs further derepressed the R-loop-positive targets, indicating PRC2-independent repression by R-loops [44]. Although PRC2 was reported to induce RNA:DNA exchange and promote R-loop formation in vitro [43], another study showed that EZH2 depletion had no effect on R-loops in mESCs [44]. Further study is warranted to dissect interplays among R-loops, transcription, and PRC targeting/recruitment.
Nascent RNAs Potentially Mediate PRC2 Chromatin Eviction via a Contentious ‘RNA Bridge’ Model of PRC2-Binding Enhancement
Although PRC2 cannot efficiently bind to chromatin at actively transcribed genes, it associates directly with nascent RNAs [72,73]. Although the PRC2-interacting regions of primary mRNAs have been mapped to exon–intron boundaries and 3′ untranslated regions (3′-UTRs) [74], the RNA-interacting interfaces within PRC2 seem to be clustered within the same sites used for allosteric regulation (Figure 1E), thereby inhibiting PRC2 activation [73,75]. Furthermore, both SUZ12 [74] and JARID2 [69,70,75,76] bind to RNAs. It was proposed that, following rapid transcription, PRC2 binding to nascent RNAs causes its chromatin dissociation (Figure 2A, right/top) because RNA degradation triggers PRC2 recruitment to CGIs associated with transcriptionally active genes, consistent with a finding that transcription inhibition is sufficient to enhance genome-wide recruitment of PRC2 [74,77]. In addition, nascent precursor mRNAs harboring a G-quadruplex (G4; Figure 2A, right/top) bind to PRC2 with high affinity, and induce PRC2 eviction from substrate nucleosomes in vitro and in vivo [73,78]. CRISPR/dCas9-mediated recruitment of G4-containing RNAs to CDKN2A causes reactivation of this Polycomb-repressed tumor suppressor in malignant rhabdoid tumor cells and induces senescence [78].
In contrast to a model that nascent RNAs act in cis to evict PRC from active sites, recent findings also indicate that they can induce PRC recruitment to Polycomb-repressed targets [43,44,79]. In human induced pluripotent stem cells (iPSCs), perturbation of RNA:PRC2 interactions by RNase A or inhibition of transcription, or by using an RNA binding-defective form of EZH2, all disrupted PRC2 chromatin occupancy and localization genome-wide [79]. The genes affected are predominantly developmental, leading to stem cell differentiation defects, indicating that PRC2 requires RNA binding for appropriate chromatin localization [79]. Such an RNA binding defective PRC2 maintains its ability to form complexes, DNA/nucleosome binding, and methyltransferase activity, and an ‘RNA bridge’ model was proposed in which RNA serves as a linker for PRC2 to ‘scan’ for chromatin status [79].
The Chromatin Context Restricts the Formation/Spreading of Polycomb Domains
Whereas EED-mediated reading of H3K27me3 underlies PRC2 spreading, DNA binding by the PCL EH motif increases the PRC2 residence time on chromatin [54]. Likewise, other modules within PCLs (i.e., Tudor [80,81]), JARID2 (ARID and zinc-finger domains), AEBP (zinc-finger), and RBBP4/7 can bind to DNA, histones, and/or nucleosomes [80]. Conceivably, these chromatin-engaging activities further enhance/stabilize PRC2 recruitment and/or spreading under various contexts.
H3K27ac, H3K4me3, and H3K36me3/2 all directly suppress PRC2 catalytic activity [36], thereby preventing H3K27me3 formation at these marked regions [81,82] (Figure 2A, left). Although H3K27ac, H3K4me3, and H3K36me3 mark the enhancer, promoter, and body regions of actively transcribed genes, respectively, H3K36me2 forms large intergenic domains [81]. In addition, H3K36me3/2 are read by the PWWP domains of DNMT3A and DNMT3B [81] that establish de novo DNA methylation at CpG sites, and thus affect PRC2 recruitment and/or H3K27me3 patterns [83,84]. Thus, chromatin modifications provide various toolkits for restricting Polycomb domains from establishment/spreading in different chromatin contexts.
Overall, a combination of protein:DNA, protein:RNA, protein:R-loop, and protein:protein interactions, as well as local chromatin environment, collectively determine genomic seeding and spreading of PRC2 versus its eviction and domain restriction (Figure 2A). It is worth noting that, because parental H3K27me3 is twofold diluted following DNA replication [85,86], recent studies have also explored mitotic inheritance of H3K27me3 [85–90] (Box 4); a local redeposition model was postulated in which, during DNA replication, parental H3K27me3 is locally recycled to newly synthesized DNA where PRC2 propagates/restores the H3K27me3 pattern at the newly incorporated histones, possibly via allosteric activation [88,89].
Box 4. Mitotic Inheritance of H3K27me3 Marks.
During DNA replication, parental histone PTMs such as H3K27me3 are twofold diluted, which influences cell fate decision making during differentiation [85,86]. For cells to maintain a defined identity, histone PTM patterns must be fully and accurately restored in daughter cells within one cell cycle [85,88]. To illustrate how the H3K27me3 mark is inherited, Alabert et al. proposed a model in which H3K27me3 propagates through continuous modification of both new and old histones, because restoration of this mark is much slower than for gene-active histone marks [85]. Based on the profiles of chromatin occupancy after replication (ChOR-seq), which tracks the occupancy of chromatin modifiers and histone PTMs after replication-fork passage genome-wide, Reveron-Gomez et al. further postulated a model in which parental H3K27me3 is locally ‘recycled’ onto newly synthesized DNA during replication [88]. The speed of H3K27me3 restoration correlates positively with PRC2 occupancy, suggesting that PRC2 propagates H3K27me3 at newly incorporated histones, possibly through the existing H3K27me3-induced allosteric activation [3,88]. In the absence of EZH2 activity, H3K27me3 levels in parental H3K27me3 domains are stable across the cell cycle, in agreement with the local redeposition model [88]. Furthermore, Escobar et al. used a CRISPR-biotinylation system to biotin-label parental H3.1/H3.2 histones at individual genomic sites (demarcated by either gene activation- or gene repression-associated histone PTMs), which then allows tracking their redeposition by ChIP-seq of cells during and after DNA replication [89]; they observed that the local redeposition model applies to repressive but not to active histone PTMs, suggesting that only repressive epigenomic patterns are inherited by the daughter cells [89].
Molecular Underpinnings of Polycomb-Mediated Gene Silencing
Since the discovery that Polycomb factors are repressors of Hox expression in Drosophila, the field has speculated about the underlying mechanisms. Recent studies have unveiled the multilevel involvement of PRC2 and its enzymatic product (H3K27me3) in the recruitment of readers/effectors (Figure 2A, bottom/left; and Figure 3A) that comprise distinct classes that may mediate gene silencing via different molecular mechanisms. We also touch on gene-silencing mechanisms that are independent of H3K27me3 (notably, nonhistone methylation) and of PRC2 methyltransferase activity.
Figure 3. Distinct Classes of Readers of H3K27me3 (Histone H3 Trimethylated on Lys27) Mediate Gene Silencing by Polycomb via Different Molecular Mechanisms.
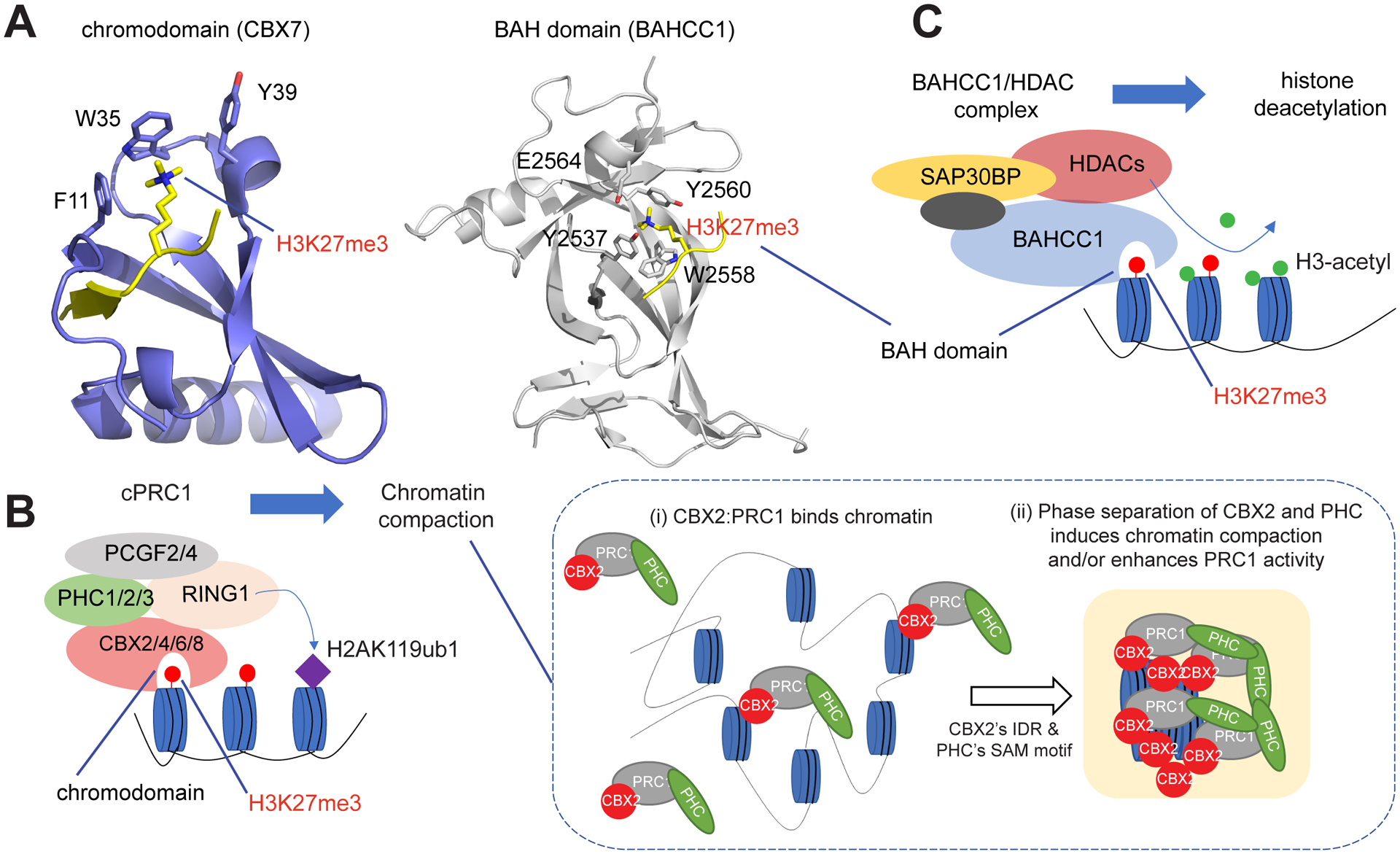
(A) Structure showing recognition of H3K27me3 by the chromodomain of CBX7 (left) or by the BAH domain of BAHCC1 (right); the key H3K27me3 reading cage residues are labeled. H3 peptide is shown in yellow. (B) Scheme illustrating that, cPRC1 reads H3K27me3 via the chromodomain of its CBX subunit, and then mediates deposition of histone 2A ubiquitinated on Lys119 (H2AK119ub1) and chromatin compaction (left). For the latter (right), intrinsically disordered regions (IDRs) that are present in CBX2/4/6/8 (but not in CBX7), as well as the SAM domain of the PHC subunit, both phase-separate, inducing cPRC1 condensation to enhance chromatin compaction/folding. (C) A model that specific readout of H3K27me3 by a conserved BAH module in BAHCC1 leads to recruitment of BAHCC1, HDAC1/2, and corepressors to deacetylate local histones and silence H3K27me3-demarcated genes.
H3K27me3 Is Read by cPRC1, Inducing Chromatin Compaction via Phase Separation
cPRC1 (Box 3) is defined by inclusion of a CBX subunit (CBX2/4/6/7/8) that uses a chromodomain (CD) to read H3K27me3 (Figure 3A, left). Acting downstream of PRC2/H3K27me3, cPRC1 induces chromatin compaction [91–95]. Importantly, chromatin compaction by cPRC1 does not require RING1 catalytic activity in mESCs [91,96]. Owing to inclusion of a RYBP or YAF2 subunit (Box 3), which enhances RING1 enzymatic activity, ncPRC1 serves as the primary writer of H2AK119ub1. Because of the absence of CBX, ncPRC1 is recruited independently of H3K27me3, and in fact acts upstream of PRC2.2 for the de novo establishment of H3K27me3 [42,59,97–99] (Figure 2A, bottom/middle).
In mESCs, cPRC1-induced chromatin compaction was found to rely mainly on CBX2 and PHC, and compaction was independent of its ubiquitination activity [91–95] – the SAM domain of PHC1/2 (Box 3), that mediates head-to-tail oligomerization of PRC1, and intrinsically disordered regions (IDRs) of CBX2, are both essential for repressing Polycomb gene targets [93,94,100], supporting a model involving phase separation or liquid–liquid phase separation (LLPS). Indeed, whereas cPRC1 resides in membraneless Polycomb bodies in cells [101], recombinant CBX2–RING1B heterodimers form phase-separated condensates in vitro, independently of H3K27me3 [102,103]. In addition to the H3K27me3-binding CD, CBX2 contains IDRs (Box 3), including a lysine(K)/arginine(R)-rich cluster and a serine-rich region (SRR; a 15 residue region containing 14 serines) [102,103]. Electrostatic interactions among positively charged K/R-rich clusters and phosphorylated SRRs [104,105] mediate LLPS formation. Serine-to-alanine (S→A) mutations in SRR or K/R-to-alanine mutations of the K/R-rich cluster (K/R→A for all 23 K/Rs) abolished CBX2 condensate formation in vitro and in cells, establishing that IDS are essential for LLPS formation [102]. Interestingly, CBX2 K/R-rich cluster was also shown to be essential for nucleosomal compaction in vitro [92], and for axial patterning in genetically modified mice [95]. Studies using severe versus intermediate mutations of the K/R-rich cluster (such as 23Å~, 13Å~, or 10Å~ KR→A) revealed a positive correlation between the extent of the phase-separation defect and the severity of nucleosomal decompaction or axial patterning defects [95,102]. Because the E3 ligase activity of RING1B is not essential for chromatin compaction by cPRC1, a phase separation mechanism likely underlies the gene-silencing function of cPRC1 [91,106]. PHC SAM domains also phase-separate in vitro, enhancing PRC1 activity [107]. Such a cPRC1-mediated chromatin compaction/folding mechanism is evolutionarily conserved, and loss of Polycomb/Pc or Polyhomeotic/Ph (the sole CBX or PHC homolog in Drosophila), resulted in opened chromatin at Hox cluster genes before their reactivation [94,108]. Collectively, phase separated CBXs/PHCs drive the formation of cPRC1 condensates, thereby enhancing chromatin compaction and/or PRC1 ubiquitination activity (Figure 3B, right).
Genomic profiling conducted in mESCs and Drosophila has demonstrated that PRC1-mediated compaction at both local (~100 kb) and distal levels (>10 Mb or interchromosomal) operates independently of anchor proteins (cohesin and CTCF) because disruption of cPRC1 does not affect topologically associated domains (TADs) [94,109–111]. However, such chromatin compaction does alter general chromatin accessibility, gene expression, and nuclear organization, and long-range loops (often poised enhancer–promoter and promoter–promoter contacts) generated by PRC1/2 keep Polycomb targets in a repressed state [94,109–113].
It is notable that cPRC1-mediated compaction is cell type-specific owing to differences between CBX components in different cell lineages. Self-renewing ESCs preferentially express CBX7 over other CBXs [114]. CBX7 (Box 3), unlike CBX2/4/6/8, lacks IDRs and thus lacks the IDRassociated chromatin-compaction function of CBX2 (Figure 3B, right). H3K27me3-marked cPRC1 targets in ESCs are transcriptionally poised [112,115], and active histone marks (H3K4me3) coexist at bivalent domain gene promoters and H3K4me1 at poised enhancers that restrict the formation of a fully repressed chromatin structure [116–118]. In support, depletion of KMT2B/MLL2, an H3K4me3 writer, in ESCs results in a tighter chromatin structure [117]. Thus, PRC targets in ESCs, predominantly developmental genes, maintain flexibility and are subject to differential regulation in response to developmental cues [112,119,120]. Intriguingly, upon differentiation, CBX7 is replaced by IDR-containing CBX2/4 in specific lineages, and CBX2/4-containing cPRC1s are likely to lead to more stable repression of gene expression, setting cellular identity towards a differentiation trajectory [114].
BAH Module-Based Readout of H3K27me3 Mediates Gene Silencing by Polycomb across Different Species
A conserved motif, termed Bromo-adjacent homology (BAH), that is present in plant [121–123], animal [124], and filamentous fungus (Neurospora crassa) [125] chromatin-associated proteins, was recently shown to be a new class of H3K27me3-specific readers that induce Polycomb mediated target repression. BAH-based readout of H3K27me3 is not only evolutionarily conserved but also seems to be more prevalent than the PRC1-based mechanism because the latter does not exist in plants [121–123], whereas BAH-based H3K27me3 readers are present in fungi, plants, and mammals. Fan et al. recently demonstrated that the BAH module in BAH-containing protein 1 (BAHCC1), a chromatin regulator that is overexpressed in acute leukemias, specifically recognizes H3K27me3 (Figure 3A, right) and that, in leukemia lines and 293 cells, depletion of BAHCC1, or disruption of its BAH-mediated H3K27me3 binding via point mutagenesis, significantly derepressed H3K27me3-demarcated genes [124]. In mice, disruption of H3K27me3 reading by the BAHCC1 BAH caused partial postnatal lethality and dwarfism, indicating that the BAHCC1 pathway is a crucial determinant of development [124]. BAHCC1 interacts with histone deacetylases (HDACs) and associated corepressors, thus connecting H3K27me3 to histone deacetylation, an integral step in gene silencing [124] (Figure 3C). Interestingly, another mammalian protein, BAHD1, that also contains an H3K27me3-binding BAH domain [126], was previously suggested to mediate heterochromatin formation via its multifunctional scaffolding role [127] and also interacts with HDACs [128]; however, the exact involvement of the BAHD1 BAH module in regulating Polycomb-mediated gene expression remains to be fully defined. Furthermore, the gene-silencing mechanisms utilized by the protein complexes assembled by non-vertebrate BAH-based H3K27me3 readers, including EARLY BOLTING IN SHORT DAYS (EBS) and homologous SHORT LIFE (SHL) in plants [121–123] and Effector of Polycomb Repression 1 (EPR-1) in Neurospora crassa [125], are likely to be diverse based on recent reports, which merits additional characterizations.
H3K27me3-Independent and/or Methyltransferase-Independent Mechanisms also Contribute to Gene Silencing by PRC2
PRC2 also monomethylates GATA4 at Lys299, an event that interferes with GATA4 interaction with coactivator p300 [129]. Likewise, PRC2 monomethylates Lys754 of EloA (Figure 1A), a subunit of the Elongin complex that is known to promote transcriptional elongation [130]. Compared to wild-type EloA, the Lys754 methylation-defective Elo mutant induced upregulation of a subset of PRC2 targets in mESCs and interfered with cell differentiation, indicative that EloA-mediated methylation is necessary for optimal repression of Polycomb targets [130]. These findings point to a nonhistone methylation-dependent pathway for gene silencing wherein PRC2 directly methylates (co)activators to suppress their gene activation function.
EPOP and PALI1/2, cofactors that are exclusively associated with the PCL-containing PRC2.1 [48–52] (Table 1 and Box 2), can contribute to PRC2 target repression in an H3K27me3-independent manner. First, through its blocks complex formation (BC-box)b, EPOP interacts with EloB/C, which, together with EloA, make up the Elongin complex. EPOP:EloB/C and EloB/C:EloA interactions are mutually exclusive, and EPOP may thus sequester EloB/C, thereby preventing Elongin complex formation and inducing gene downregulation [52] (Figure 1A). However, it is also noteworthy to mention that EPOP often acts in a PRC2-independent context [48] – independently of PRC2, EPOP:EloB/C cooperates with USP7 to remove gene activation-related H2B ubiquitination, thus inhibiting transcription [131]. Targets co-bound by EloA:EloB/C, EPOP, and PRC2.1 are generally poorly expressed, reminiscent of ‘bivalent domain’ genes. In support, H3K27me3 and PRC2 levels at these targets were found to be enhanced in EPOP-null mESCs [48,52]. In addition to interacting with PRC2.1, PALI/2 (Box 2) also binds to G9A, an H3K9 mono- and dimethyltransferase, indicating crosstalk between PRC2 and H3K9me2/1, a potent gene silencing histone PTM [26,50]. PALI1 was previously shown to promote PRC2 methyltransferase activity during mouse embryogenesis [50,132]; however, such an effect was not verified in other systems [26], meriting further study.
Concluding Remarks and Future Perspectives
Recent understanding of PRC2 architecture, its chromatin recruitment versus eviction, and Polycomb gene-silencing mechanisms, has greatly changed current views regarding how PRC2 exerts its (epi)genome- and transcriptome-regulatory effects. However, several key questions remain to be addressed (see Outstanding Questions). First, the dissection of the roles of individual PRC2/1 subcomplexes/cofactors was mainly performed with mESCs [27,40,41,47]. Although this cell model has many advantages, the relative contributions of PRC variants to H3K27me3 deposition/spreading may vary among differentiated cell types and cancers where PRC2 cofactors tend to be differentially or aberrantly expressed [80,133]. Højfeldt et al. recently showed that the non-core subunits, PCLs, JARID2, and AEBP2, collectively mediate targeting of the PRC2 core and thus the deposition of H3K27me3 [134]. Although discrepancies between the findings of this work [134] and other studies [27,40,41,47] require further investigation, there is consensus in the essential involvement of cofactors, and that different cellular states alter PRC binding upon differentiation [112] or during transition of ground-to-primed pluripotency [110]. In addition, interactions between RNAs and PRCs appear to be complex, and somewhat contradictory results have been reported, meriting further study. Second, PRCs-mediated (epi)genomic and transcriptomic regulation is executed in a context of 3D chromatin. Chromatin looping in 3D helps to spread PRC-catalyzed H3K27me3/H2AK199ub1 marks to distal but spatially close sites. Phase separation-mediated PRC condensation is also relevant. Conceivably, phase-separated assemblies (PRC2/1) and genome 3D organization may be coordinated. Phase-separated PRC condensates could both promote chromatin compaction and exclude transcriptional (co)activators. Furthermore, PRC-generated chromatin loops may bring in cis-elements, termed silencers, that inhibit gene expression via their increased local concentration [119]. The interplay between these different factors remains to be dissected.
Outstanding Questions.
It was reported that nascent RNAs function in cis to evict PRCs’ binding at actively transcribed sites whereas RNAs induce PRCs’ recruitment to the repressed Polycomb target sites. It remains unclear how RNAs achieve context-dependent regulation of PRC2.
Can Polycomb proteins, other than CBX and PHC, phase separate, mediating the assembly of PRCs’ condensates seen in cells?
What is the causal relationship between chromatin compaction and gene silencing mediated by PRCs?
A class of conserved BAH modules are shown to be H3K27me3 “readers”, mediating Polycomb gene repression. Cooperation, synergistic and/or competition effects potentially exist among them and those previously known H3K27me3 “readers” (cPRC1), either at same or differential sets of targets, which remains to be defined among various cell and tissue types.
Increasing amount of evidence demonstrates the PRC-unrelated functions of Polycomb proteins. For example, EZH2 and PRC1 were shown to induce gene activation under certain biological contexts. How are these distinct activities of Polycomb proteins regulated?
We favor the view that chromodomain- and BAH-based readout of H3K27me3 collectively mediates gene silencing by Polycomb proteins in mammals (Figure 3). The expression patterns of BAHCC1 and PRC1 factors may vary during development, and their respective contributions to target gene repression by Polycomb are likely to differ accordingly, but the details will require more investigation.
Lastly, PRC2 methylates a range of nonhistone substrates in vitro [130]. Future study is warranted to determine the PRC2 methylome under physiological conditions, thereby allowing functional dissection of nonhistone methylation by PRC2. In addition, although predominantly held to be repressive, Polycomb proteins can be associated with active transcription. In addition to being involved in PRC2, EZH2 has noncanonical PRC2-independent functions that may operate in a methyltransferase-dependent fashion [135]. For instance, EZH2 activates particular NF-κB target genes in breast cancer [136,137] and harbors gene activation associated protein motifs [138]; similarly, PRC1 positively regulates the expression of specific targets in epidermal progenitors, cardiac mesoderm precursors, and cancer [139–141]; in Drosophila, PRC1 was recently reported to activate developmental genes by promoting enhancer–promoter looping [142]. However, few studies have addressed the regulation of the PRC-dependent versus PRC-independent activities of Polycomb proteins, or of their gene-repression versus -activation roles.
Supplementary Material
Highlights:
Catalytic activity of the PRC2 “core” is delicately regulated by conformational change (auto-inhibition versus allosteric activation), its “non-core” cofactors and the chromatin context.
A collection of factors, at least including DNA cis-elements (such as unmethylated CpG island), RNA, R-loop and chromatin modification, are involved in the regulation of PRC2 targeting and spreading in the chromatin, as well as its eviction or functional restriction.
A “local redeposition” model is postulated that, during DNA replication, local “recycling” of parental H3K27me3 assists in the PRC2-mediated propagation of H3K27me3 at newly-synthesized histones.
Liquid-liquid phase separation establishes protein condensates of the H3K27me3-”reading” cPRC1 complex, leading to chromatin compaction and Polycomb gene silencing.
Conserved BAH modules emerge as a new class of H3K27me3-specific “readers”, mediating Polycomb gene silencing among species of fungi, plant and mammal.
Critical questions remain to be addressed regarding complicated cross-talks among PRC variants, chromatin, transcription (RNA) and H3K27me3 “readers” in a range of tissue and cell types; nonhistone- or non-PRC-related regulatory activities of Polycomb proteins also exist, bringing in additional layers of complexity.
GLOSSARY
- Allosteric activation
Regulatory site (other than the active site) of an enzyme binds a modulator for activating/enhancing the enzyme function, which often involves a conformational change of the protein
- Bivalent domain genes
Genes that harbor both active and repressive histone modifications (usually H3K4me3 and H3K27me3)
- Competitive inhibition
inhibition or interruption of a chemical pathway through competing off an involved chemical substance, such as blockade of the substrate binding via a competitor
- CpG island (CGI)
interspersed stretches of DNA, usually 500–1500 bp long, display an elevated G+C base composition (GC-rich and CpG-rich) that are normally found at gene promoters and predominantly nonmethylated
- cryo-EM
Cryogenic electron microscopy. It is a method for imaging frozen-hydrated specimens at cryogenic temperatures where specimens remain in their native state without the need for dyes or fixatives, allowing the study at molecular resolution
- Genetic imprinting
a gene-regulatory mechanism that depends upon the sex of the parent who passed on the gene alleles, which affects expression only from the paternal or maternal allele by altering the chromatin structure and/or chromatin modification
- G-quadruplex (G4)
Guanine-rich DNA or RNA sequences folded into four-stranded, noncanonical secondary structures called G-quadruplexes (G4s)
- lncRNAs
Long non-coding RNAs, which are transcribed RNA molecules with a length of more than 200 nucleotides that do not encode proteins
- iPSC
Induced pluripotent stem cells. iPSCs are derived from adult somatic cells by genetic reprograming back into an embryonic-like pluripotent state that enables the development of any type of cells
- mESC
Murine embryonic stem cells. It is derived from the inner cell mass of early mouse embryos (at blastocyst stage) that can be differentiated to all tissue types
- Onco-histone
cancer-associated recurrent mutations identified within histones that can dominantly drive or significantly contribute to oncogenesis
- Phase-separation
Phase-separation is the creation of two distinct phases from a single homogeneous mixture
- Polycomb bodies
nuclear foci where Polycomb group proteins are concentrated in
- Polycomb domains
genomic regions bound by Polycomb proteins and marked by their respective histone marks (such as H3K27me3 and H2AK119ub1), which often extend many tens and even hundreds of kilobases to create the silenced region
- Polycomb responsive elements (PREs)
are regulatory sites that contain high-affinity DNA-binding sites for Polycomb proteins such as PRC2
- R-loop
a nucleic acid structure including two antiparallel DNA strands and one RNA strand. In this structure, the RNA is base-paired to one of the DNA strands whereas the other DNA strand is unpaired
- Trithorax group proteins
typically function in large complexes whose main action is to maintain gene expression. depending on their functions, they can be categorized into histone-modifying complexes and ATP-dependent chromatin-remodeling complexes
- X-inactivation
in female mammalian cells, one of the two copies of X chromosomes is silenced by packaged into a transcriptionally inactive structure
Footnotes
Supplemental Information
Supplemental information associated with this article can be found, in the online version.
Reference:
- 1.Blackledge NP et al. (2015) Targeting Polycomb systems to regulate gene expression: modifications to a complex story. Nat Rev Mol Cell Biol 16 (11), 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuettengruber B et al. (2017) Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 171 (1), 34–57. [DOI] [PubMed] [Google Scholar]
- 3.Yu JR et al. (2019) PRC2 is high maintenance. Genes Dev 33 (15–16), 903–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Mierlo G et al. (2019) The Complexity of PRC2 Subcomplexes. Trends Cell Biol 29 (8), 660–671. [DOI] [PubMed] [Google Scholar]
- 5.Chammas P et al. (2020) Engaging chromatin: PRC2 structure meets function. Br J Cancer 122 (3), 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu H et al. (2013) Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PLoS ONE 8 (12), e83737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antonysamy S et al. (2013) Structural Context of Disease-Associated Mutations and Putative Mechanism of Autoinhibition Revealed by X-Ray Crystallographic Analysis of the EZH2-SET Domain. PLoS ONE 8 (12), e84147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bratkowski M et al. (2017) Polycomb repressive complex 2 in an autoinhibited state. J Biol Chem 292 (32), 13323–13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dewaele B et al. (2014) Identification of a novel, recurrent MBTD1-CXorf67 fusion in low-grade endometrial stromal sarcoma. Int J Cancer 134 (5), 1112–22. [DOI] [PubMed] [Google Scholar]
- 10.Jain SU et al. (2019) PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat Commun 10 (1), 2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hubner JM et al. (2019) EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol 21 (7), 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piunti A et al. (2019) CATACOMB: An endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism. Sci Adv 5 (7), eaax2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain SU et al. (2020) H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol Cell 80 (4), 726–735 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartzentruber J et al. (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482 (7384), 226–31. [DOI] [PubMed] [Google Scholar]
- 15.Lewis PW et al. (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340 (6134), 857–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stafford JM et al. (2018) Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 4 (10), eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng Y et al. (2012) Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc Natl Acad Sci U S A 109 (34), 13549–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han Z et al. (2007) Structural basis of EZH2 recognition by EED. Structure 15 (10), 1306–15. [DOI] [PubMed] [Google Scholar]
- 19.Jiao L and Liu X (2015) Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 350 (6258), aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Margueron R et al. (2009) Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461 (7265), 762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Justin N et al. (2016) Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun 7, 11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ueda T et al. (2016) Propagation of trimethylated H3K27 regulated by polycomb protein EED is required for embryogenesis, hematopoietic maintenance, and tumor suppression. Proc Natl Acad Sci U S A 113 (37), 10370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee CH et al. (2018) Distinct Stimulatory Mechanisms Regulate the Catalytic Activity of Polycomb Repressive Complex 2. Mol Cell 70 (3), 435–448 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CH et al. (2018) Allosteric Activation Dictates PRC2 Activity Independent of Its Recruitment to Chromatin. Mol Cell 70 (3), 422–434 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanulli S et al. (2015) Jarid2 Methylation via the PRC2 Complex Regulates H3K27me3 Deposition during Cell Differentiation. Mol Cell 57 (5), 769–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q et al. (2020) Convergent evolution between PALI1 and JARID2 for the allosteric activation of PRC2. bioRxiv, 2020.05.28.122556. [Google Scholar]
- 27.Oksuz O et al. (2018) Capturing the Onset of PRC2-Mediated Repressive Domain Formation. Mol Cell 70 (6), 1149–1162 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hojfeldt JW et al. (2018) Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat Struct Mol Biol 25 (3), 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge EJ et al. (2019) Nucleation and Propagation of Heterochromatin by the Histone Methyltransferase PRC2: Geometric Constraints and Impact of the Regulatory Subunit JARID2. J Am Chem Soc 141 (38), 15029–15039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poepsel S et al. (2018) Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat Struct Mol Biol 25 (2), 154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Margueron R et al. (2008) Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 32 (4), 503–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lavarone E et al. (2019) Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. Nat Commun 10 (1), 1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qi W et al. (2017) An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol 13 (4), 381–388. [DOI] [PubMed] [Google Scholar]
- 34.Suh JL et al. (2019) Discovery of selective activators of PRC2 mutant EED-I363M. Sci Rep 9 (1), 6524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan W et al. (2012) Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science 337 (6097), 971–5. [DOI] [PubMed] [Google Scholar]
- 36.Schmitges FW et al. (2011) Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell 42 (3), 330–41. [DOI] [PubMed] [Google Scholar]
- 37.Tatavosian R et al. (2018) Live-cell single-molecule dynamics of PcG proteins imposed by the DIPG H3.3K27M mutation. Nat Commun 9 (1), 2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Youmans DT et al. (2018) Live-cell imaging reveals the dynamics of PRC2 and recruitment to chromatin by SUZ12-associated subunits. Genes Dev 32 (11–12), 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rickels R et al. (2016) An Evolutionary Conserved Epigenetic Mark of Polycomb Response Elements Implemented by Trx/MLL/COMPASS. Mol Cell 63 (2), 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H et al. (2017) Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 549 (7671), 287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perino M et al. (2018) MTF2 recruits Polycomb Repressive Complex 2 by helical-shape-selective DNA binding. Nat Genet 50 (7), 1002–1010. [DOI] [PubMed] [Google Scholar]
- 42.Blackledge NP et al. (2014) Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 157 (6), 1445–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alecki C et al. (2020) RNA-DNA strand exchange by the Drosophila Polycomb complex PRC2. Nat Commun 11 (1), 1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skourti-Stathaki K et al. (2019) R-Loops Enhance Polycomb Repression at a Subset of Developmental Regulator Genes. Mol Cell 73 (5), 930–945 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen S et al. (2018) Unique Structural Platforms of Suz12 Dictate Distinct Classes of PRC2 for Chromatin Binding. Mol Cell 69 (5), 840–852 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kasinath V et al. (2018) Structures of human PRC2 with its cofactors AEBP2 and JARID2. Science 359 (6378), 940–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Healy E et al. (2019) PRC2.1 and PRC2.2 Synergize to Coordinate H3K27 Trimethylation. Mol Cell 76 (3), 437–452 e6. [DOI] [PubMed] [Google Scholar]
- 48.Liefke R and Shi Y (2015) The PRC2-associated factor C17orf96 is a novel CpG island regulator in mouse ES cells. Cell Discov 1, 15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Z et al. (2011) PRC2 complexes with JARID2, MTF2, and esPRC2p48 in ES cells to modulate ES cell pluripotency and somatic cell reprogramming. Stem cells 29 (2), 229–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Conway E et al. (2018) A Family of Vertebrate-Specific Polycombs Encoded by the LCOR/LCORL Genes Balance PRC2 Subtype Activities. Mol Cell 70 (3), 408–421 e8. [DOI] [PubMed] [Google Scholar]
- 51.Alekseyenko AA et al. (2014) Reciprocal interactions of human C10orf12 and C17orf96 with PRC2 revealed by BioTAP-XL cross-linking and affinity purification. Proc Natl Acad Sci U S A 111 (7), 2488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beringer M et al. (2016) EPOP Functionally Links Elongin and Polycomb in Pluripotent Stem Cells. Mol Cell 64 (4), 645–658. [DOI] [PubMed] [Google Scholar]
- 53.Hauri S et al. (2016) A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep 17 (2), 583–595. [DOI] [PubMed] [Google Scholar]
- 54.Choi J et al. (2017) DNA binding by PHF1 prolongs PRC2 residence time on chromatin and thereby promotes H3K27 methylation. Nat Struct Mol Biol 24 (12), 1039–1047. [DOI] [PubMed] [Google Scholar]
- 55.Ren Z et al. (2019) PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation. Blood 134 (14), 1176–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cooper S et al. (2016) Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat Commun 7, 13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tamburri S et al. (2020) Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol Cell 77 (4), 840–856 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blackledge NP et al. (2020) PRC1 Catalytic Activity Is Central to Polycomb System Function. Mol Cell 77 (4), 857–874 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tavares L et al. (2012) RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 148 (4), 664–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qin J et al. (2012) The polycomb group protein L3mbtl2 assembles an atypical PRC1-family complex that is essential in pluripotent stem cells and early development. Cell Stem Cell 11 (3), 319–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stielow B et al. (2018) MGA, L3MBTL2 and E2F6 determine genomic binding of the non-canonical Polycomb repressive complex PRC1.6. PLoS Genet 14 (1), e1007193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang Y et al. (2018) Combinatorial Control of Recruitment of a Variant PRC1.6 Complex in Embryonic Stem Cells. Cell Rep 22 (11), 3032–3043. [DOI] [PubMed] [Google Scholar]
- 63.Brockdorff N (2017) Polycomb complexes in X chromosome inactivation. Philos Trans R Soc Lond B Biol Sci 372 (1733). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zylicz JJ et al. (2019) The Implication of Early Chromatin Changes in X Chromosome Inactivation. Cell 176 (1–2), 182–197 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mager J et al. (2003) Genome imprinting regulated by the mouse Polycomb group protein Eed. Nat Genet 33 (4), 502–7. [DOI] [PubMed] [Google Scholar]
- 66.Silva J et al. (2003) Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell 4 (4), 481–95. [DOI] [PubMed] [Google Scholar]
- 67.Rinn JL et al. (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129 (7), 1311–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schertzer MD et al. (2019) lncRNA-Induced Spread of Polycomb Controlled by Genome Architecture, RNA Abundance, and CpG Island DNA. Mol Cell 75 (3), 523–537 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaneko S et al. (2014) Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell 53 (2), 290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.da Rocha ST et al. (2014) Jarid2 Is Implicated in the Initial Xist-Induced Targeting of PRC2 to the Inactive X Chromosome. Mol Cell 53 (2), 301–16. [DOI] [PubMed] [Google Scholar]
- 71.Allison DF and Wang GG (2019) R-loops: formation, function, and relevance to cell stress. Cell Stress 3 (2), 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang X et al. (2017) Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat Struct Mol Biol 24 (12), 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Q et al. (2019) RNA exploits an exposed regulatory site to inhibit the enzymatic activity of PRC2. Nat Struct Mol Biol 26 (3), 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beltran M et al. (2016) The interaction of PRC2 with RNA or chromatin is mutually antagonistic. Genome Res 26 (7), 896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Long Y et al. (2017) Conserved RNA-binding specificity of polycomb repressive complex 2 is achieved by dispersed amino acid patches in EZH2. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaneko S et al. (2014) Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev 28 (18), 1983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Riising EM et al. (2014) Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell 55 (3), 347–60. [DOI] [PubMed] [Google Scholar]
- 78.Beltran M et al. (2019) G-tract RNA removes Polycomb repressive complex 2 from genes. Nat Struct Mol Biol 26 (10), 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Long Y et al. (2020) RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells. Nat Genet 52 (9), 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vizan P et al. (2015) Role of PRC2-associated factors in stem cells and disease. FEBS J 282 (9), 1723–35. [DOI] [PubMed] [Google Scholar]
- 81.Li J et al. (2019) Understanding histone H3 lysine 36 methylation and its deregulation in disease. Cell Mol Life Sci 76 (15), 2899–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Streubel G et al. (2018) The H3K36me2 Methyltransferase Nsd1 Demarcates PRC2-Mediated H3K27me2 and H3K27me3 Domains in Embryonic Stem Cells. Mol Cell 70 (2), 371–379 e5. [DOI] [PubMed] [Google Scholar]
- 83.Baubec T et al. (2015) Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520 (7546), 243–7. [DOI] [PubMed] [Google Scholar]
- 84.Weinberg DN et al. (2019) The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573 (7773), 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alabert C et al. (2015) Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev 29 (6), 585–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jadhav U et al. (2020) Replicational Dilution of H3K27me3 in Mammalian Cells and the Role of Poised Promoters. Mol Cell 78 (1), 141–151 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hansen KH et al. (2008) A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol 10 (11), 1291–300. [DOI] [PubMed] [Google Scholar]
- 88.Reveron-Gomez N et al. (2018) Accurate Recycling of Parental Histones Reproduces the Histone Modification Landscape during DNA Replication. Mol Cell 72 (2), 239–249 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Escobar TM et al. (2019) Active and Repressed Chromatin Domains Exhibit Distinct Nucleosome Segregation during DNA Replication. Cell 179 (4), 953–963 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hugues A et al. (2020) Mitotic Inheritance of PRC2-Mediated Silencing: Mechanistic Insights and Developmental Perspectives. Front Plant Sci 11, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eskeland R et al. (2010) Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol Cell 38 (3), 452–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grau DJ et al. (2011) Compaction of chromatin by diverse Polycomb group proteins requires localized regions of high charge. Genes Dev 25 (20), 2210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Isono K et al. (2013) SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev Cell 26 (6), 565–77. [DOI] [PubMed] [Google Scholar]
- 94.Wani AH et al. (2016) Chromatin topology is coupled to Polycomb group protein subnuclear organization. Nat Commun 7, 10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lau MS et al. (2017) Mutation of a nucleosome compaction region disrupts Polycomb-mediated axial patterning. Science 355 (6329), 1081–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Endoh M et al. (2012) Histone H2A mono-ubiquitination is a crucial step to mediate PRC1-dependent repression of developmental genes to maintain ES cell identity. PLoS Genet 8 (7), e1002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gao Z et al. (2012) PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell 45 (3), 344–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fursova NA et al. (2019) Synergy between Variant PRC1 Complexes Defines Polycomb-Mediated Gene Repression. Mol Cell 74 (5), 1020–1036 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rose NR et al. (2016) RYBP stimulates PRC1 to shape chromatin-based communication between Polycomb repressive complexes. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kundu S et al. (2017) Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation. Mol Cell 65 (3), 432–446 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saurin AJ et al. (1998) The human polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J Cell Biol 142 (4), 887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Plys AJ et al. (2019) Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev 33 (13–14), 799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tatavosian R et al. (2019) Nuclear condensates of the Polycomb protein chromobox 2 (CBX2) assemble through phase separation. J Biol Chem 294 (5), 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hatano A et al. (2010) Phosphorylation of the chromodomain changes the binding specificity of Cbx2 for methylated histone H3. Biochem Biophys Res Commun 397 (1), 93–9. [DOI] [PubMed] [Google Scholar]
- 105.Kawaguchi T et al. (2017) Phosphorylation of CBX2 controls its nucleosome-binding specificity. J Biochem 162 (5), 343–355. [DOI] [PubMed] [Google Scholar]
- 106.Illingworth RS et al. (2015) The E3 ubiquitin ligase activity of RING1B is not essential for early mouse development. Genes Dev 29 (18), 1897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Seif E et al. (2020) Phase separation by the polyhomeotic sterile alpha motif compartmentalizes Polycomb Group proteins and enhances their activity. Nat Commun 11 (1), 5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cheutin T and Cavalli G (2018) Loss of PRC1 induces higher-order opening of Hox loci independently of transcription during Drosophila embryogenesis. Nat Commun 9 (1), 3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schoenfelder S et al. (2015) Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat Genet 47 (10), 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Joshi O et al. (2015) Dynamic Reorganization of Extremely Long-Range Promoter-Promoter Interactions between Two States of Pluripotency. Cell Stem Cell 17 (6), 748–757. [DOI] [PubMed] [Google Scholar]
- 111.Eagen KP et al. (2017) Polycomb-mediated chromatin loops revealed by a subkilobase-resolution chromatin interaction map. Proc Natl Acad Sci U S A 114 (33), 8764–8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cruz-Molina S et al. (2017) PRC2 Facilitates the Regulatory Topology Required for Poised Enhancer Function during Pluripotent Stem Cell Differentiation. Cell Stem Cell 20 (5), 689–705 e9. [DOI] [PubMed] [Google Scholar]
- 113.Ogiyama Y et al. (2018) Polycomb-Dependent Chromatin Looping Contributes to Gene Silencing during Drosophila Development. Mol Cell 71 (1), 73–88 e5. [DOI] [PubMed] [Google Scholar]
- 114.Morey L et al. (2012) Nonoverlapping functions of the Polycomb group Cbx family of proteins in embryonic stem cells. Cell Stem Cell 10 (1), 47–62. [DOI] [PubMed] [Google Scholar]
- 115.Ku M et al. (2008) Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4 (10), e1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rada-Iglesias A et al. (2011) A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470 (7333), 279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mas G et al. (2018) Promoter bivalency favors an open chromatin architecture in embryonic stem cells. Nat Genet 50 (10), 1452–1462. [DOI] [PubMed] [Google Scholar]
- 118.King HW et al. (2018) Polycomb repressive complex 1 shapes the nucleosome landscape but not accessibility at target genes. Genome Res 28 (10), 1494–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ngan CY et al. (2020) Chromatin interaction analyses elucidate the roles of PRC2-bound silencers in mouse development. Nat Genet 52 (3), 264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Taberlay PC et al. (2011) Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell 147 (6), 1283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang Z et al. (2018) EBS is a bivalent histone reader that regulates floral phase transition in Arabidopsis. Nat Genet 50 (9), 1247–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Qian S et al. (2018) Dual recognition of H3K4me3 and H3K27me3 by a plant histone reader SHL. Nat Commun 9 (1), 2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li Z et al. (2018) Polycomb-mediated gene silencing by the BAH-EMF1 complex in plants. Nat Genet 50 (9), 1254–1261. [DOI] [PubMed] [Google Scholar]
- 124.Fan H et al. (2020) BAHCC1 binds H3K27me3 via a conserved BAH module to mediate gene silencing and oncogenesis. Nat Genet 52 (12), 1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wiles ET et al. (2020) Evolutionarily ancient BAH-PHD protein mediates Polycomb silencing. Proc Natl Acad Sci U S A 117 (21), 11614–11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhao D et al. (2016) The BAH domain of BAHD1 is a histone H3K27me3 reader. Protein Cell 7 (3), 222–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bierne H et al. (2009) Human BAHD1 promotes heterochromatic gene silencing. Proc Natl Acad Sci U S A 106 (33), 13826–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lakisic G et al. (2016) Role of the BAHD1 Chromatin-Repressive Complex in Placental Development and Regulation of Steroid Metabolism. PLoS Genet 12 (3), e1005898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.He A et al. (2012) PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev 26 (1), 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ardehali MB et al. (2017) Polycomb Repressive Complex 2 Methylates Elongin A to Regulate Transcription. Mol Cell 68 (5), 872–884 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liefke R et al. (2016) EPOP Interacts with Elongin BC and USP7 to Modulate the Chromatin Landscape. Mol Cell 64 (4), 659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shi Y et al. (2019) C10ORF12 modulates PRC2 histone methyltransferase activity and H3K27me3 levels. Acta Pharmacol Sin 40 (11), 1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xu B et al. (2015) Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp Hematol 43 (8), 698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hojfeldt JW et al. (2019) Non-core Subunits of the PRC2 Complex Are Collectively Required for Its Target-Site Specificity. Mol Cell 76 (3), 423–436 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang J and Wang GG (2020) No Easy Way Out for EZH2: Its Pleiotropic, Noncanonical Effects on Gene Regulation and Cellular Function. Int J Mol Sci. 21(24), E9501. doi: 10.3390/ijms21249501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee ST et al. (2011) Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell 43 (5), 798–810. [DOI] [PubMed] [Google Scholar]
- 137.Lawrence CL and Baldwin AS (2016) Non-Canonical EZH2 Transcriptionally Activates RelB in Triple Negative Breast Cancer. PLoS One 11 (10), e0165005.27764181 [Google Scholar]
- 138.Jiao L et al. (2020) A partially disordered region connects gene repression and activation functions of EZH2. Proc Natl Acad Sci U S A 117 (29), 16992–17002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Morey L et al. (2015) Polycomb Regulates Mesoderm Cell Fate-Specification in Embryonic Stem Cells through Activation and Repression Mechanisms. Cell Stem Cell 17 (3), 300–15. [DOI] [PubMed] [Google Scholar]
- 140.Cohen I et al. (2018) PRC1 Fine-tunes Gene Repression and Activation to Safeguard Skin Development and Stem Cell Specification. Cell Stem Cell 22 (5), 726–739 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chan HL et al. (2018) Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms. Nat Commun 9 (1), 3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Loubiere V et al. (2020) Widespread activation of developmental gene expression characterized by PRC1-dependent chromatin looping. Sci Adv 6 (2), eaax4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kloet SL et al. (2016) The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat Struct Mol Biol 23 (7), 682–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.