

SUMMARY
Cellular processes are largely carried out by macromolecular assemblies, most of which are dynamic, having components that are in constant flux. One such assembly is the nuclear pore complex (NPC), an ~50 MDa assembly comprised of ~30 different proteins called Nups that mediates selective macromolecular transport between the nucleus and cytoplasm. We developed a proteomics method to provide a comprehensive picture of the yeast NPC component dynamics. We discovered that, although all Nups display uniformly slow turnover, their exchange rates vary considerably. Surprisingly, this exchange rate was relatively unrelated to each Nup’s position, accessibility, or role in transport but correlated with its structural role; scaffold-forming Nups exchange slowly, whereas flexible connector Nups threading throughout the NPC architecture exchange more rapidly. Targeted perturbations in the NPC structure revealed a dynamic resilience to damage. Our approach opens a new window into macromolecular assembly dynamics.
In Brief
The nuclear pore complex (NPC) is commonly represented as a rather static assembly. Using a quantitative proteomics approach, Hakhverdyan et al. open a window into the dynamics of NPC components and show how constant exchange of key components governs maintenance and resilience to damage of this molecular machine.
Graphical Abstract
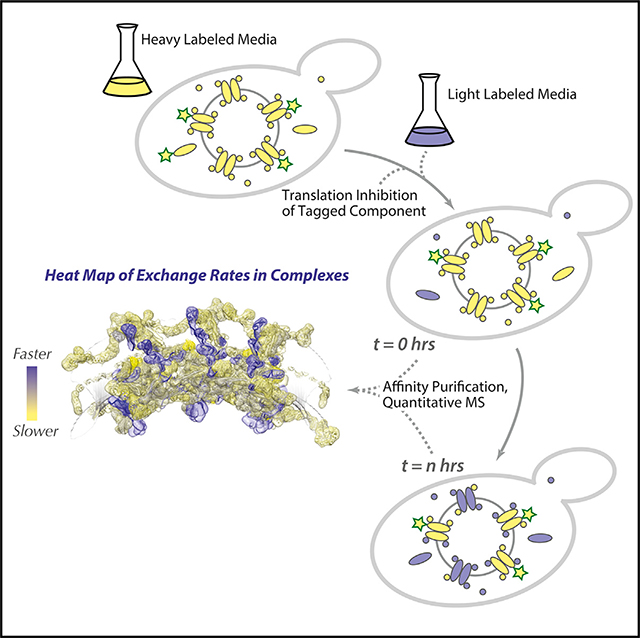
INTRODUCTION
Cellular processes are carried out by macromolecular assemblies largely comprised of protein subunits. Some assemblies, when formed, appear to be relatively unchanged for long periods of time and are considered stable, but many are much more dynamic. We can consider two processes contributing to the dynamics of assemblies in vivo: turnover of their subunits, being the sum of subunit synthesis and degradation (Garlick and Millward, 1972), and subunit exchange, the process by which assembly-bound subunits are replaced with subunits from a free pool (Figure 1A). Modulation of the equilibrium between turnover and exchange can alter the cellular amounts of macromolecular assemblies and can also produce assemblies with different compositions, conformations, and functions (De Souza et al., 2004; Niño et al., 2016; Williamson, 2008). However, many of the principles determining and regulating the dynamic organization of macromolecular assemblies in living cells remain poorly understood because of a paucity of methodologies that can reliably and comprehensively quantify the dynamics of subunits (Aitchison and Rout, 2015). The nuclear pore complex (NPC) is an excellent system to study the dynamic organization of macromolecular assemblies in vivo because it has long-lived and rapidly exchanging components (Knockenhauer and Schwartz, 2016; Rabut et al., 2004).
Figure 1. Analysis of Nucleoporin Turnover and Exchange.

(A) Schematic representation of protein dynamics in cells.
(B and C) Schematic representation of turnover (B) and exchange (C) measurement workflows. See STAR Methods for details.
(D) Heavy label decay time course for Nups (blue) and transport factors (purple), indicating the turnover rate for each protein. Measured data and fit are presented; error bars, standard error. The turnover rate fits were calculated for other abundant cytoplasmic proteins with a known low turnover rate to establish their turnover rate distribution (stable protein average, solid black line; 2 standard deviations, dashed black lines). For a comparison, fast turnover proteins from Christiano et al. (2014) were also plotted (but not observed as part of this dataset) (red). An average of turnover rates for GFP-Nup84- and GFP-Nup157-captured NPCs is shown; tagged Nups are indicated by an asterisk.
(E) Heavy label decay time course for Nups and transport factors. Measured data and fit are presented; error bars, standard error. The exchange rate fits were also calculated for other abundant cytoplasmic proteins, which represent nonspecific ultrafast exchanging proteins, to establish their distribution of exchange rates (ultrafast protein average, solid black line; 2 standard deviations, dashed black lines). An average of exchange rates for GFP-Nup84- and GFP-Nup157-captured NPCs is shown; tagged Nups are indicated by an asterisk.
(F) Plot comparing turnover rates with exchange rates.
(G) Plot comparing the exchange rates inferred in vivo (Figure 1E) from those measured on isolated NPCs (STAR Methods).
In terms of structure, each NPC is an annular proteinaceous machine, embedded in the nuclear envelope, responsible for active gated transport of macromolecular cargoes between the nucleoplasm and cytoplasm. In the model yeast (Saccharomyces sp.), the 52-MDa NPC is composed of 30 different proteins, called nucleoporins or Nups, present as a total of ~550 subunits. Its functions, constituent protein subunits, and three-dimensional architecture have been characterized in high detail, and its overall bauplan and functions are conserved with that of NPCs in other eukaryotes (Kim et al., 2018). All NPCs possess an overall 8-fold radial symmetry through their cylindrical axis so that there are 8 repeating units, called spokes, making up the structure (Beck and Hurt, 2017; Kosinski et al., 2016). Coaxial outer and inner rings form a symmetric core scaffold that is connected to a membrane ring, a nuclear basket, and a cytoplasmic export platform; these discrete and relatively rigid assemblies are connected by flexible cables, a combination imbuing the structure with strength and flexibility (Fischer et al., 2015; Kim et al., 2018; Teimer et al., 2017) (Figure 2). The scaffold surrounds a central channel that is formed in part by nucleoporins called Phe-Gly (FG) Nups, from which domains with multiple intrinsically disordered FG repeat motifs project. These FG motifs mediate selective nucleocytoplasmic transport through specific interactions with nuclear transport factors that carry their cognate macromolecular cargoes through the NPC (Baade and Kehlenbach, 2019; Holzer and Antonin, 2018; Knockenhauer and Schwartz, 2016).
Figure 2. Heatmap of Nup Exchange Rates on an NPC Structure.

(A-E) Structure of an NPC and its components in different views; a model of the pore membrane is shown in gray. For each Nup, the localization probability density of the protein is shown with a representative structure embedded within it (Kim et al., 2018). The remaining rows show a heatmap of Nup exchange rates on an NPC structure (blue, rapid exchange; yellow, slow exchange). The second row shows a heatmap on an entire NPC, and subsequent rows show heatmapping on major NPC substructures, with remaining NPC shown in faded gray.
In terms of dynamics, it has been suggested that some proteins in the core scaffold (at least in vertebrates) can remain associated with NPCs for years (Savas et al., 2012; Toyama et al., 2013), whereas transport factors and their cargoes represent a huge transient flux, exchanging up to 1,000 macromolecules per second per NPC (Ribbeck and Görlich, 2001). The NPC also interfaces with the nucleoplasm, cytoplasm, and nuclear envelope and thus interacts directly with an enormous diversity of macromolecules, including cytoplasmic and nuclear proteins, transmembrane and luminal nuclear envelope proteins, ribonucleoproteins, and chromatin (Knockenhauer and Schwartz, 2016; Ptak et al., 2014). Significantly, unlike their metazoan counterparts, yeast NPCs do not coordinately disassemble and reassemble during mitosis, greatly simplifying dynamics assessment over many cell divisions.
Nucleoporin dynamics is an area of great interest, and studies have already provided valuable insights (Boisvert et al., 2012; Christiano et al., 2014; Dilworth et al., 2001; Griffis et al., 2004; Mathieson et al., 2018; Rabut et al., 2004; Savas et al., 2012; Toyama et al., 2013, 2018). Importantly, there has been no way to systematically, simultaneously, and comprehensively quantify turnover and exchange rates in the normal state or in response to specific subunit perturbations for any assembly. Here we developed a proteomics-based method to distinguish between old and newly synthesized proteins in an assembly that avoids potential issues introduced by exogenous expression or tagging the measured proteins. The method can determine and distinguish subunit turnover and exchange specifically in an assembly to provide a comprehensive picture of assembly dynamics. Applying it to the NPC, we discovered striking correlations between a protein’s structural role and its dynamics.
RESULTS AND DISCUSSION
Nups Have Uniformly Low Turnover Rates
We first established the turnover rate of Nups (Figure 1A) in logarithmically growing yeast cells. Every cell division, one daughter cell’s worth of new protein must be synthesized, diluting the old mother protein pool by one-half. This synthesis is the only source of turnover for a non-degraded protein; however, if a protein is also degraded, then its turnover is more rapid, making its half-life shorter than the cell doubling time. To distinguish old from new protein, we used a strategy that combines pulse-chase stable isotope labeling (Ong et al., 2002a; Pratt et al., 2002) with our method for affinity purification of native, intact assemblies (Hakhverdyan et al., 2015) applied to the NPC (Kim et al., 2018; Subbotin and Chait, 2014; Figure 1B). With this strategy, we simultaneously determined the turnover rates and, hence, degradation rates (Tables S1, S2, and S8), of all Nups as well as those of numerous NPC-associated nucleocytoplasmic transport factors. These rates are for native untagged proteins, avoiding possible issues with tagging altering a protein’s dynamics. As an important internal standard, we also determined the turnover and degradation rates of a set of abundant contaminating cytoplasmic proteins (Hakhverdyan et al., 2015; Mellacheruvu et al., 2013). The turnover rates of all Nups were very similar and gave essentially negligible degradation rates within the range of those seen for the abundant cytoplasmic proteins (Figure 1D; Table S2). Such low degradation rates are entirely consistent with previous studies and are indeed typical for the great majority of proteins in yeast (Christiano et al., 2014); the turnover rate of these proteins equals the doubling rate of the cells; because there is essentially no degradation, the heavy label is only diluted by cell growth and division. Notably, these rates differ sharply from the small fraction of yeast proteins that are known to have high degradation rates (Figure 1D; Boisvert et al., 2012; Christiano et al., 2014). Our data are also consistent with degradation rates for homologous Nups in dividing mammalian cells, with half-lives of a few hours to a few days (Boisvert et al., 2012; Cambridge et al., 2011; Mathieson et al., 2018), although degradation rates in non-dividing cells are much slower, with half-lives from days to months (Savas et al., 2012; Toyama et al., 2013).
Nups Show a Broad Range of Exchange Rates with a Surprising Structural Correlation
After the turnover analysis, we sought to quantify the other process that contributes to NPC dynamics: the exchange rate of Nups. The design of the exchange experiments is a straightforward adaptation of that for the turnover experiments with an additional capacity to also track “old” NPCs over time, allowing us to assess whether any Nups have been replaced with new ones (Figure 1C). This is accomplished by conditional expression of the tagged Nup with which we affinity capture the NPCs. During the heavy labeling step, the tagged Nup is expressed. When cells are then switched to light medium, a tetracycline-binding riboswitch (Hanson et al., 2003) is used to stop translation of the tagged Nup from its transcript to ensure extremely rapid cessation of its synthesis (Note S5; Figure S4). To avoid pulling out newly formed NPCs, a non-exchanging NPC constituent is required (Figure S1B). We chose Nup84 (a representative of the outer ring) and Nup157 (a representative of the inner ring) because homologs of these Nups have been reported to be relatively stable components of the vertebrate NPC (Mathieson et al., 2018; Rabut et al., 2004; Savas et al., 2012; Toyama et al., 2013), and our own data confirm that these two yeast Nups are indeed very slow exchangers (below). During affinity capture, post-lysis exchange of proteins is blocked (Figure S2). The affinity capture step thus generates only mature (i.e., “old”), initially heavy-labeled NPCs; the subsequent appearance of light-labeled Nups in this fraction from cells collected over time must result from the exchange between soluble Nups and those bound to mature NPCs (Figure 1E; Figure S2). Because Nups have negligible degradation rates (above), their light labeling rate closely tracks cell growth (Figures 1B and 1D), which also sets the upper limit of the exchange rate we can measure (Figure S1B). We estimated this upper rate experimentally from measurements of abundant contaminating proteins, whose “exchange rates” (because they have no affinity for NPCs in vivo) should be ultrarapid; as expected, most Nups have much slower exchange rates (Figure 1E).
In contrast to their tightly clustered turnover rates, Nups displayed a broad range of apparent exchange rates (residence half-lives), as estimated from our measurements (Figure 1E; Tables S3, S4, and S8), which fall into three broad categories: less than 2.5 h (Gle1, Dyn2, Nup2, Ndc1, Nup1, and Nup60), ~3–7 h (Nup53, Mlp1, Mlp2, Nup100, Nup145N, Gle2, Nup42, and Nup116), and more than 7 h (all remaining Nups); moreover, there is no obvious correlation between the Nup turnover rates and their exchange rates (Figure 1F) or between a Nup’s essentiality and its exchange rate. The fact that the majority of Nups were found to have half-lives significantly longer than the cell division time (~2 h) rationalizes the relatively constant stoichiometry measured for almost all NPC Nups (Kim et al., 2018; Rajoo et al., 2018). The exchange rates for several mammalian Nups have also been determined previously. These exchange rates also exhibit a wide range (Griffis et al., 2002; Mathieson et al., 2018; Nakielny et al., 1999; Rabut et al., 2004), with their relative values showing a mild correlation with the exchange rates we determined for their yeast homologs (R = 0.63; Rabut et al., 2004; Table S8), perhaps reflecting conservation of certain structural elements between yeast and mammalian NPCs. Also, as anticipated (and in validation of the method), transport factors, on average, displayed the fastest exchange rates (Figure 1E).
The limitations of our assay when measuring the upper limit of exchange rates tends to compress the faster-exchanging values (Figure 1E; Figure S1B). To explore this limitation, we took an orthogonal approach and measured the exchange rate between proteins in the lysate and affinity-captured isolated NPCs using an adaptation of the I-DIRT method (Tackett et al., 2005). Despite the obvious differences between measuring protein exchange between a soluble pool and NPC in vivo versus in vitro and the limitations of each approach, we found that the exchange rate in vitro correlated quite well (R ~0.8) with that in vivo (Figure 1G), implying that a significant contribution to a protein’s association with the NPC in vivo may be its passive on and off rates. Moreover, this method “spread out” the fastest-exchanging proteins, showing that transport factors exchanged extremely rapidly and that even the fastest-exchanging Nups were exchanging at significantly lower rates, which we believe reflects the situation in vivo.
To gain insights into functional relevance, we heatmapped the in vivo exchange rates of all yeast Nups onto a recently determined structure of the yeast NPC (Figure 2; Kim et al., 2018). It has been suggested that the more accessible peripheral Nups have higher exchange rates than the more inaccessible core Nups (D’Angelo et al., 2009; Daigle, 2001; Lyman and Gerace, 2001; Rabut et al., 2004). It was therefore surprising that we found no statistically significant correlation between a Nup’s exchange rate and the position of its center of mass relative to its distance from the center of mass of each spoke (i.e, proportional to how “peripheral” the Nup is). It has also been suggested that FG repeat-containing Nups tend to exchange relatively rapidly (D’Angelo et al., 2009; Daigle, 2001; Lyman and Gerace, 2001; Rabut et al., 2004). Again, surprisingly we observed several instances of very slowly exchanging FG repeat-containing Nups (e.g., Nsp1, Nup57, and Nup49; Figure 2; Table S4). However, we did find a remarkable correlation between the exchange rate of a Nup and its structural role in the NPC.
Components of the inner and outer rings had the slowest exchange rates; most of the mass comprising the membrane ring (Pom152 and Pom34) also displayed very slow exchange rates, with exception of the integral membrane Nup Ndc1, which shows a surprising and significant degree of exchange (Figures 1E and 2). Ndc1 is known to be shared by the NPC and the yeast spindle pole body, and its faster exchange may reflect how it is being shared structurally and functionally between these two nuclear envelope-associated assemblies (Chial et al., 1998; Rüthnick et al., 2017).
The outer, inner, and membrane rings thus represent extremely stable modules in the NPC even though some of their components are far from the spoke’s center of mass (Figure 2). The core components of the cytoplasmic export platform (Nup82, Nup159, and Nsp1) displayed similarly slow exchange rates, indicating that this module, despite being peripheral, is also very stable (Figure 2). We suggest that one reason for these slow exchange rates is the generally high surface area of contact each scaffold Nup forms with its neighbors (Kim et al., 2018). However, in striking contrast, all Nups comprising the connector cables that thread throughout the NPC’s architecture exchange relatively rapidly (Figure 2). These cables are comprised of intrinsically disordered regions (IDRs) and run between the stable outer rings, inner rings, membrane rings, and export platform. They connect to the Nups in these modules via conserved arrays of short linear motifs (SLiMs) in the IDRs that bind specific regions of Nups in the modules (Fischer et al., 2015; Kim et al., 2018; Lin et al., 2016; Teimer et al., 2017). We suggest that the high exchange rates revealed here reflect the inherently dynamic nature of the connector cables and an ability of the SLiMs to detach and re-bind, allowing the IDR connector cables to constantly seek optimum interactions for the NPC to adjust for changes in transport flux and NE morphology. The folded domains of all connector cable-containing Nups and their surface areas of contact with other Nups are small, likely aiding their faster exchange rate (Fischer et al., 2015; Kim et al., 2018). The NPC configuration is thus fixed in neither space nor time, with the dynamic association of the connections between all modules and spokes allowing the NPC to reconfigure or adjust its structure. In a similar vein, numerous NPC components involved in RNA export—the nuclear basket (Mlp1, Mlp2, Nup2, Nup1, and Nup60 (Niepel et al., 2013) and RNA remodeling factors associated with the cytoplasmic export platform (such as Gle1, Gle2, and Nup42) (Folkmann et al., 2011)—also appear among the more rapidly exchanging proteins (Figure 2). We suggest that at least some of these proteins represent a cloud of accessory factors surrounding and constantly exchanging with the NPC in vivo.
Having determined the dynamics of Nups in unperturbed NPCs, we sought to interrogate the main driving forces behind Nup dynamics, taking advantage of the capability of genetics in yeast. We targeted a select set of mutants, each of which could address key questions regarding the mechanisms underlying NPC dynamics.
The NPC’s Modular Construction Imparts Resilience to Damage
The yeast NPC is remarkably robust to structural damage; for example, roughly two-thirds of its components can be individually deleted without loss of viability (Doye and Hurt, 1995; Strawn et al., 2004b). To test the nature of this robustness, we removed Nup120, a major component of the core scaffold and member of the outer ring (Nup84) complex (Figure 2; Siniossoglou et al., 2000). Deletion of the gene encoding Nup120 results in slower growth and a temperature-sensitive mRNA export phenotype and causes NPCs to cluster in the plane of the nuclear envelope (Aitchison et al., 1995a; Heath et al., 1995). Surprisingly, despite this multitude of phenotypic effects caused by deletion of Nup120, the dynamics of most Nups were left largely unaffected (Figure 3A; Figure S3A; Table S5), except for components found in just two structural modules (Figure 3B). First, every component of the outer ring complex displayed a significant increase in its exchange rate, indicating that removal of Nup120 had destabilized the other components of the outer ring but did not cause them to completely dissociate from the NPC; furthermore, the exchange rate of these outer ring components are affected to a similar degree, indicating a strong structural interdependence within the outer ring module of the NPC. Second, the normally slow-exchanging Nup82 and Nup159 in the export platform displayed a similar increase in exchange rate upon Nup120 deletion. An increase in the exchange rate of the third slowly exchanging export platform component Nsp1 was also noted (but represented a much smaller apparent change, likely because a larger number of copies of this Nup also exists in the unaffected inner ring module). The outer ring complex is directly attached to the export platform (Figure 2); this attachment is essential for the latter’s stable anchoring to the NPC (Fernandez-Martinez et al., 2016; Gaik et al., 2015; Kim et al., 2018; Teimer et al., 2017). Thus, it is easy to rationalize why destabilization of the outer ring complex leads to a concomitant destabilization of the export platform. Moreover, the structural interdependence of the outer ring components indicated here can explain why deletion of any of the Nup84 complex’s Nups leads to similar mRNA export defects (Fabre and Hurt, 1997). In contrast, the exchange rates of the adjacent inner ring proteins generally showed only a minor increase, and the exchange rates of other Nups that interact with the export platform or the outer ring (Nup116, Nup100, Nup145N, Nup1, Nup60, Mlp1, and Mlp2) were relatively unaffected by deletion of Nup120 (Figure 3A; Figure S3A). However, these components also make numerous interactions with other structural modules (Figure 2; Fischer et al., 2015; Kim et al., 2018; Mészáros et al., 2015), providing a significant level of NPC attachment redundancy and likely alleviating the effects of the increased exchange rates for their outer ring or export platform attachment sites. Our results support the view that the NPC is made of modules whose components are structurally co-dependent but with more limited structural co-dependence between many of these modules. Such an architecture, as we also show below, allows the assembly as a whole to remain remarkably unperturbed by damage to any of its individual components.
Figure 3. Characterization of Nucleoporin Exchange Rates in the Nup120-Null Strain.

(A) Graph showing comparison of Nup exchange rates in the Nup120 wild-type and null strains. A doubling rate normalization line (solid black line; 3 standard deviations, dashed black lines) indicates the difference in labeling rates between the strains, calculated from other abundant cytoplasmic proteins. Proteins with the greatest deviation are outlined in red. The average of two experiments is shown; error bars are combined standard error of the parameter fits. Tagged Nup157 is indicated by an asterisk.
(B) Heatmap on the molecular structure of an NPC (Figure 2; Kim et al., 2018) of differences in exchange rates (in percent; white, 0%; red, +200% [faster]) between a wild-type and Nup120-null strains. Scale bar, 100 Å.
Probing Factors that Determine the Exchange Rate of Nups
What are the dominant factors determining a Nup’s exchange rate? One clear possibility is that some Nups exchange slowly because they are firmly anchored to the NPC’s scaffold, whereas faster exchangers are less strongly associated. Conversely, Nups also participate in ATP- and guanosine triphosphate (GTP)-driven transport and remodeling processes, and these might actively drive these components to reversibly associate with the NPC. We therefore wanted to find out whether it is possible to alter the exchange rate of a Nup by strongly associating with another Nup with a distinctly different rate and determine which exchange rate dominated. The strongest association is protein fusion, but in almost all cases, it is difficult to design such a fusion while ensuring no deleterious structural alterations within or steric hindrance between the two Nups. However, there is one particularly suitable test case in the NPC: Nup145N and Nup145C. Nup145N consists of an N-terminal FG repeat region, a middle flexible connector domain, and a C-terminal folded domain, whereas Nup145C consists almost entirely of an alpha-solenoid domain. These two Nups are normally generated by autocatalytic cleavage of a single precursor Nup145 protein; upon cleavage and release, Nup145N can re-bind Nup145C, largely maintaining its prior form. This cleavage is neither essential for growth nor necessary for localization of these Nups to the NPC (Emtage et al., 1997; Teixeira et al., 1997, 1999). Thus, elimination of the cleavage site effectively fuses the two proteins (Nup145NC) with no gross phenotypic effects. Nup145N normally has an exchange rate of ~3 h, whereas that of Nup145C is considerably slower (~9 h). Nup145N, by virtue of its FG repeat region, interacts with many active transport processes, whereas Nup145C is a core scaffold component. Conceivably, fusion could confer the faster exchange rate upon Nup145C, the slower rate upon Nup145N, or an intermediate rate between the two. In fact, we found that the exchange rate of the entire Nup145NC fusion protein matched that of Nup145C (Figures 4A and 4B; Table S6; Figure S3B). Thus, the more rapid exchange process here does not appear to be the dominant one. Instead, we suggest that the dominant factor is the strength of interaction each Nup makes with its surrounding Nups and that, as a core scaffold component, Nup145C forms stronger interactions than Nup145N that prevail in the Nup145NC fusion protein.
Figure 4. Characterization of Nucleoporin Exchange Rates in Nup145 Mutant Strains.

(A) Graph showing comparison of Nup exchange rates in wild-type and Nup145NC fusion strains. A doubling rate normalization line (solid black line; 3 standard deviations, dashed black lines) indicates the difference in labeling rates between strains, calculated from other abundant cytoplasmic proteins. Proteins with the greatest deviation are outlined in red. Tagged Nup84 is indicated by an asterisk.
(B) Heatmap on the molecular structure of an NPC (Figure 2; Kim et al., 2018) of differences in exchange rates (in percent; cyan, −60% [slower]; white, 0%; red, +70% [faster]) between wild-type and Nup145NC fusion strains (from Kim et al., 2018). Top: 3 NPC spokes. Bottom: single spoke with Nups dissected aside. Scale bars, 100 Å.
(C) Graph showing comparison of Nup exchange rates in wild-type and Nup145NFG fusion strains. A doubling rate normalization line (solid black line; 3 standard deviations, dashed black lines) indicates the difference in labeling rates between strains, calculated from other abundant cytoplasmic proteins.
(D) Plots showing the ODELAY!-measured phenotypic effect of temperature-induced (top) or benzyl alcohol-induced (bottom) stress in selected strains (see STAR Methods for details).
For (A) and (C), the average of two experiments is shown; error bars are combined standard error of the parameter fits.
Interestingly, fusion of Nup145N to Nup145C also significantly altered the exchange rate of several other Nups (Figure 4A). Although the absolute change in exchange rate seems to be modest (Figure 4A), the percentage change is actually quite dramatic (Figure S3B), a more than 50% increase in exchange rate in some cases. Strikingly, when heatmapping the change into the NPC structure, it can be seen that all of the affected Nups are immediately adjacent to the folded domain of Nup145N, these being most of the outer ring Nups; the basket proteins Mlp1 and Mlp2; and Nup192, which is found next to the folded domain Nup145N in the inner ring (Kim et al., 2018; Figure 4B). Distal Nups, even in the same structural modules, were essentially unaffected. These effects can be rationalized by noting that Nup145N actually has two main binding sites in the NPC; the first is on Nup145C copies found predominantly on the nucleus-facing outer ring adjacent to the nuclear basket, and the second is on the nucleus-facing inner ring (Figures 2 and 4B; Kim et al., 2018). We suggest that, by stabilizing Nup145N at its sites in the NPC and bringing a large Nup145C domain into its second binding site, we sterically compete all proteins surrounding Nup145N’s C-terminal folded domain. Notably, however, Nups adjacent to Nup145N’s connector region are unaffected in their exchange rates (Figure 4B), as might be expected from such a small and flexible domain (Fischer et al., 2015), which presents little steric hindrance.
A significant increase in exchange rate was also seen for Nup100 and Nup116 and its associated Gle2 (Figure 4A; Figure S3B). Nup100 and Nup116 are paralogs of Nup145N that normally occupy Nup145N’s two binding sites on the cytoplasmic side (Kim et al., 2018; Ratner et al., 2007; Yoshida et al., 2011). We propose that, by covalently attaching Nup145N to Nup145C, we anchored it to the normal cytoplasmic binding sites for Nup100 and Nup116, again sterically competing for space with these proteins and destabilizing Nup100 and Nup116 at the same sites. We again see evidence of the modular construction of the NPC in that the affected components are within the same structural module and immediately adjacent to the altered component, whereas distal modules are relatively unaffected.
FG Repeat Regions Are Not Major Contributors to FG Nup Exchange Rates
A significant proportion of the NPC’s mass is comprised of FG repeat domains, interacting with ~1,000 transport factors and their cargos every second (Grünwald and Singer, 2010; Smith et al., 2002; Yang et al., 2004). In addition, interactions of FG repeat regions with other Nups have been proposed to contribute to the structure of the NPC (Onischenko et al., 2017). To begin to probe the influence of such interactions on the exchange dynamics of FG Nups with the NPC, we deleted the non-essential FG repeat domain of Nup145N (a Nup with a medium range exchange rate; Figure 1E), representing approximately one-third of the mass of the protein; although considerable, this alteration results in a truncated protein that localizes correctly to the NPC in a strain that does not exhibit fitness defects (Strawn et al., 2004a). Deletion of this FG repeat region resulted in no substantial change in the exchange rate of Nup145N or any other Nup (Figure 4C; Table S6). Had interaction with the rapid transport flux been the dominant factor in the exchange rate measured for Nup145N, removal of the FG repeats would have decoupled this interaction and decreased the exchange rate. Lack of this effect indicates that the transport flux does not significantly affect the exchange rate of FG Nups; i.e., the transport flux does not necessarily “pull” FG Nups into a more exchanging regime. Conversely, we would expect that loss of any dominant interactions between this FG repeat and other FG repeats or Nups because of deletion of the Nup145N FG repeat region would have resulted in an increase in Nup145N’s exchange rate because of loss of these interactions. Because no such increase was detected, we conclude that interactions of Nup145N’s FG repeat region with other FG repeat regions or other Nups are not key factors that stabilize Nup145N’s association with the NPC. Taken together with the Nup145NC experiments, these findings suggest that it is a protein’s interaction with other Nups, and not with the transport flux or its associated active processes, that dictate its exchange rate.
Functional Consequences of Changes to Nup Dynamics
Although we found that the assembly as a whole can remain remarkably unperturbed by damage to any of its individual components, functionality is nonetheless compromised because removal of Nup120 deleteriously affects nuclear transport (Aitchison et al., 1995a). We therefore sought to test whether the NPC is functionally robust to minor alterations in its integrity and its components’ dynamics or whether even small changes lead to measurable functional issues. For this, we used growth rate under stress (temperature and benzyl alcohol, shown to be excellent proxies for alterations in NPC function; Fernandez-Martinez et al., 2012) using an extremely sensitive single-cell living yeast array growth assay, ODELAY! (Herricks et al., 2017b). As we show (Figure 4D), of the perturbations tested above, only the most dramatic, Nup120 deletion, gave altered growth phenotypes, indicating a degree of robustness for the NPC to minor structural perturbation, likely reflecting its strong, flexible, and resilient “suspension bridge”-like architecture (Kim et al., 2018).
Even Slowly Exchanging Nucleoporins Can Be Removed Rapidly upon Damage
In postmitotic mammalian tissues, Nups in the core scaffold have been identified as being among the longest-lived proteins in the organism, suggesting that NPCs are among the most stable and long-lived cellular assemblies (D’Angelo et al., 2009; Savas et al., 2012; Toyama et al., 2013). Although, in dividing cells, old NPCs are diluted out by new NPC synthesis, such a mechanism is absent in quiescent or post-mitotic cells. Damage or loss of individual Nup molecules would thus appear to be a major potential challenge faced by NPCs during the long lifetime of these cells, and indeed, accumulated Nup damage leading to deterioration of the transport barrier function has been proposed to contribute to cellular aging of post-mitotic cells (D’Angelo et al., 2009; Savas et al., 2012; Toyama et al., 2013). The more rapid exchange we observed for the connector Nups (Figures 1E and 2) should allow a damaged version to be replaced, but members of the core scaffold exchange so slowly that their damage could have deleterious effects to the cell if they are not repaired. However, as with most macromolecular assemblies, it is currently unknown whether damaged components of the NPC can be exchanged for new undamaged copies (D’Angelo et al., 2009; Daigle, 2001; Rabut et al., 2004; Savas et al., 2012; Toyama et al., 2013).
Now, at least in yeast, we can question whether this measured slow exchange in fact correlates with an inability to replace damaged Nups; that is, can a truly damaged component be removed and replaced, restoring the NPC? We sought to address this question using an adaptation of our turnover and exchange assays. To simulate nucleoporin damage, we used a diploid strain in which we tagged one copy of Nup170 with an inducible degron tag and left the other copy unaltered (wild type) to supply undamaged protein copies for potential replacement (Nishimura et al., 2009; Nishimura and Kanemaki, 2014). Nup170 was chosen because it is among the most slowly exchanging and most inaccessible Nups, and its deletion cannot be compensated by copies of its paralog Nup157 (Aitchison et al., 1995b; Kim et al., 2018; Rajoo et al., 2018). We first confirmed that the degron-tagged Nup170 (Nup170-AID) localized correctly to and was stoichiometrically incorporated within, NPCs (Figure S4A). Next we showed that, upon induction of the degron, Nup170-AID was degraded rapidly in cells (~30-min half-life); as a control, we also measured the degradation time course of the more accessible Nup116 and observed a similar decay rate (Figure S4B). We then tested whether it was the Nup170-AID already localized inside assembled NPCs that was being degraded (rather than degradation of just the pool of newly synthesized Nup170-AID in these growing cells). We did this by using metabolic labeling to compare the amount of Nup170, the AID tag, and other Nups in affinity-captured “old” NPCs (Figure 1C) after AID induction versus no induction (Figure 5A; Table S7; (Deng et al., 2019; Oda et al., 1999; Ong et al., 2002b). After 2 h of induction, ~80% of the mass spectrometry (MS) signature for the AID tag was lost, and based on this, the expected ~40% loss of Nup170-AID signal was also seen (after correction; STAR Methods; Figure 5A). This is a remarkable finding. Despite being buried deep in the NPC’s scaffold, the entire Skp, Cullin, F-box-containing (SCF) ubiquitin ligase machinery and megadalton-sized proteasome (Wolf and Menssen, 2018) are able to access the AID-tagged Nup170 within minutes (Figure S4B), extract it from the NPC, and degrade it.
Figure 5. NPC Dynamics after Induced Degradation of AID-Nup170 Protein.

(A) Fraction of each Nup in mature NPCs after degron induction (wild-type versus Nup170-AID strains). A doubling rate normalization line (solid black line; 1 standard deviation, dashed black lines) indicates the difference in labeling rates between the strains, calculated from other abundant cytoplasmic proteins. Proteins with significant deviation (p < 0.05) are indicated in red. The average of 2 experiments is shown; error bars show the range of the two values. Tagged Nup84 is indicated by an asterisk.
(B) Fraction of newly synthesized Nup incorporated into mature NPCs after degron induction (wild-type versus Nup170-AID strains) and new protein incorporation difference for Nups in AID-Nup170-degraded and -undegraded (mock-treated) cells; error bars, standard deviation. The fractional changes were also calculated for other abundant cytoplasmic proteins, which represent nonspecific ultrafast-exchanging proteins (stable protein average, solid black line; 3 standard deviations, dashed black lines). Proteins with significant deviation (p < 0.01) from the normal are indicated in red. The average of three experiments is shown; error bars are combined standard error of the mean. Tagged Nup84 is indicated by an asterisk.
(C) Heatmap on the molecular structure of NPC (Figure 2; Kim et al., 2018) of Nup exchange increase after AID-Nup170 degradation (white, no new protein; red, 0.06 [6%] new protein). Scale bar, 100 Å.
Turnover experiments show that there is no compensatory up-regulation of Nup170 synthesis in response to loss of Nup170-AID or for any other Nup (Figure 5). We therefore measured the degree of new protein exchange in NPCs after 2 h, comparing degron-induced with uninduced cells (Figure 5B; Table S7). The degree of exchange was similar for most Nups. However, we noted an increase for Nup170, consistent with restoration of new and undamaged Nup170 in the scaffold void left by the degraded Nup170-AID. This restoration was minor but consistent with the slow exchange rate measured for Nup170 in wild-type cells (Figure 1E). That is, it appears that repair of Nup170 proceeds through the same slow exchange process that replaces it in wild-type cells over time. Two other Nups similarly increased their degree of exchange. When we map these differences in the degree of exchange to the NPC structure (Figure 5C), we see that Nups with an increased exchange rate are Nup170 itself and neighbors of Nup170, Nup157 and Nup53. Our results are consistent with the idea that removal and degradation of copies of Nup170 (approximately half of the total Nup170 in the NPC) generates voids in the NPC structure (Rajoo et al., 2018) that allow neighbors flanking those voids to exchange as well. Again the NPC’s modularity is evidenced in that only components within the same inner ring module whose exchange rates are altered, and the remaining overall structure of the NPC remains remarkably unaltered and resilient to removal of this large core component.
Therefore, it would seem that NPCs do have a way to replace their damaged components, one we suggest is likely shared by many of a cell’s macromolecular assemblies. If undamaged, protein exchange (albeit slow in some cases) maintains the assembly. However, the proteolytic degradation machinery has the capacity to efficiently destroy a damaged component, seemingly regardless of apparent inaccessibility. We suggest that the fact that all proteins in the NPC can exchange, even slowly, reflects a significant flexibility to this structure (and likely many macromolecular assemblies), permitting access to SCF ubiquitin ligases and proteasomes. Upon efficient removal, the assembly’s damaged component is replaced by the same simple and ceaseless exchange processes present in the intact NPC. Thus, although all of our data agree with previous results showing that Nups are turned over slowly and that long-lived post-mitotic cells may retain a significant portion of “old” Nups, we suggest that a cell can remove and replace damaged Nups in the NPC. However, this process may not always keep up with the damage rate or errors during new complex assembly, and it may be the balance between cumulative Nup damage versus Nup replacement that dictates the contribution of NPCs to the aging process in each cell type.
Conclusions
Although we studied NPCs from one cell type, their conserved overall bauplan would suggest similar behavior in many other cells. More generally, given that the NPC typifies many macromolecular assemblies, the principles of maintenance and restoration we discussed here are likely common. Most cell proteins are very stable in terms of their turnover, being degraded and replaced at a slow rate in the range of tens of hours or longer. This is seen for Nups and, understandably, has given the impression that the NPC is in large part a very stable, permanent structure. However, upon measurement of the exchange of Nups to and from the NPC, a very different picture emerges. As well as dynamic flux of NPC-associating proteins, such as transport factors and RNA remodeling proteins, many Nups also constantly exchange in and out of the NPC. Even the core scaffold is an impermanent structure, its components exchanging slowly but constantly, and interconnected by proteins exchanging more rapidly; thus, the term “scaffold” in the dictionary sense of “temporary platform” is appropriate. The interaction strengths between adjacent Nups, rather than their apparent accessibility per se, seem to be the overriding factor in determining their exchange rates; we suggest that this is true for the components of many complexes. This constant exchange for even the largest assemblies and the most inaccessible of their components may also help solve the problem of their maintenance. Although very low turnover may seem to imply an inability to respond to damage, we demonstrated that a constant exchange of all components in an assembly ensures that, should any component suffer damage, it could potentially be swapped out for an undamaged copy provided by residual synthesis of that component, even in quiescent cells. Exchange is also the most extreme consequence of the normal molecular motions undertaken by an assembly’s components; thus, although structural work over preceding years has provided an increasingly detailed but static three-dimensional view of the macromolecular assemblies comprising the cell, our work underscores that all assemblies in cells are four-dimensional and in constant motion, their constituents constantly in flux to adjust, reconfigure, replace, and respond to their ever-changing environment.
The methods described here add a dynamic dimension to affinity capture-based interactomics studies. Previously, we could only read out an isolated macromolecular assembly as the sum of its components, with no temporal data embedded in it beyond the cellular conditions at the time of isolation. Now, with this approach, we can embed isotopic ratio information within the isolated assembly that has measured the rate of turnover and exchange of the assembly’s components in the living cell. If an assembly can be tagged and isolated, then the method should apply. The dynamics of complexes such as proteasomes, spindle organizers, and even organelles such as peroxisomes and mitochondria could potentially be studied in a manner similar to that used here for the NPC. Similarly, cell culture systems from a variety of model organisms can be isotopically labeled and are appropriate for study. This approach permits comprehensive quantitative determination and accurate temporal modeling of macromolecular assembly dynamics in many cellular structures.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact author Michael P. Rout (rout@rockefeller.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
Files containing the input data and custom Python scripts are available upon request. Mass spectrometry raw datasets used in this study are publicly available in Zenodo (https://zenodo.org/) under DOI #: 10.5281/zenodo.4062150.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All Saccharomyces cerevisiae strains used in this study are listed in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-HA (Y-11) rabbit polyclonal | Santa Cruz Biotechnoloy | Cat.# sc-805 |
| anti-GFP llama polyclonal IgG | Fridy et al. 2014 | anti-GFP antibody |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Chlortetracycline hydrochloride | Sigma-Aldrich | Cat.# 26430 |
| 3-Indoleacetic acid (auxin) | Sigma-Aldrich | Cat.# I2886 |
| StuI | New England Biolabs | Cat.# R0187S |
| NcoI | New England Biolabs | Cat.# R0193S |
| SacI | New England Biolabs | Cat.#R0156S |
| BamHI | New England Biolabs | Cat.#R0136S |
| HindIII | New England Biolabs | Cat.#R0104S |
| Calf Intestinal Alkaline Phosphatase (CIP) | New England Biolabs | Cat.# M0290 |
| Coomassie Blue R250 | MP Biomedicals | Cat.# 190682 |
| Glutaraldehyde | Sigma-Aldrich | Cat.# 340855 |
| Trypsin Sequencing Grade, modified | Roche | Cat.# 11418033001 |
| Iodoacetamide | Sigma | Cat.# I6125-10g |
| L-lysine:2HCl 13C6 | Cambridge Isotope Laboratories Inc. | Cat.# CNLM-291-H |
| Nupage LDS Sample buffer | Thermo Fisher Scientific | Cat.# NP0007 |
| Ultrapure Salmon Sperm DNA Solution | Invitrogen | Cat.# 15632011 |
| Critical Commercial Assays | ||
| Dynabeads M270 Epoxy | Thermo Fisher Scientific | Cat # 143.02D |
| Deposited Data | ||
| Mass spectrometry raw datasets | Zenodo (https://zenodo.org/) | DOI #: 10.5281/zenodo.4062150 |
| Experimental Models: Saccharomyces cerevisiae Strains | ||
| MATa/α his3-Δ200/his3-Δ200 leu2-3,112/leu2-3,112 lys2-801/lys2-801 trp1-1(am)/trp1-1(am) ura3-52ura3-52 | Rout et al, 2000 | DF5 |
| MATa/α ade2-1::ADE2/ade2-1::ADE2 trp1-1/TRP1 LYS2/lys2 ura3-1/ura3-1 leu2-3,112/leu2-3,112 his3-11,15/his3-11,15 | Strawn et al, 2004 | SWY2870 |
| can1-100/can1-100 myc-LoxP-nup145ΔGLFG/myc-LoxP-nup145ΔGLFG | ||
| DF5, NUP84/pTDH3-tc3-GFP-Nup84::SpHIS5 | This study | Tc3-GFP-Nup84 |
| DF5, NUP157/pTDH3-tc3-GFP-Nup157::SpHIS5 | This study | Tc3-GFP-Nup157 |
| DF5, NUP84/pTDH3-tc3-GFP-Nup84::SpHIS5, Nup145N-LoxP-6xHA-Nup 145C/Nup145N-LoxP-6xHA-Nup145C | This study | Nup145NC-fusion |
| SWY2870, pTDH3-tc3-GFP-Nup84::SpHIS5, lys2::KanR/lys2 | This study | nup145NΔFG |
| DF5, nup120::URA3/nup120::URA3 NUP157/ pTDH3-tc3-GFP-Nup157::SpHIS5 | This study | nup120null |
| DF5, NUP84/pTDH3-tc3-GFP-Nup84::SpHIS5, NUP170/Nup170-AID::KanR, OsTIR::URA3/ura3-52 | This study | Nup170-AID |
| DF5, pTDH3-tc3-GFP-Nup84::SpHIS5, NUP116/ Nup116-AID::KanR, OsTIR::URA3/ura3-52 | This study | Nup116-AID |
| Recombinant DNA: List of Plasmids Used in this Study | ||
| TDH3 promoter replacement and N-terminal tagging with tetracycline binding aptamers and 3HA tag, Amp, KanR | Euroscarf | pTDH3-tc3-3xHA |
| Gene deletion, Amp, HIS5 | Euroscarf | pUG27 |
| KanR marker in pTDH3-tc3-3xHA replaced with HIS5 in pTDH3-tc3-3xHA, Amp | This study | Ttc3HAHis5 |
| GFP encoding sequence inserted in Ttc3HAHis5, Amp | This study | Ttc3GFP-His5 |
| N-terminal or internal 6xHA tagging, Amp, KanR | Euroscarf | pOM10 |
| Transient, conditional expression of Cre recombinase under GAL1 promoter, Amp, URA3 | Euroscarf | pSH47 |
| Gene deletion, Amp, KILEU2 | Euroscarf | pUG73 |
| Integratable plasmid, Amp, TRP1 | Rout Lab | p404GALL-GFP |
| C-terminal AID tagging, KanMX, Amp | Yeast NIG | BYP6740 |
| osTIR expression, integrative URA3, Amp | Yeast NIG | BYP6744 |
| Sequence-Based Reagents | ||
| GAAATGAACTATATATCCTATAATCCCTTTGGCGGGACTTGGACTTTCAAAGTCAATCCTTGCAGGTCGACAACCCTTAAT | This study | ZH127 |
| ACTCAAATCGTCTTCATCTATTTCCGCATCTTCTTCATTGACTAACCCCCAAATGCTAGAGCTAGAAGCGTAATCTGGAAC | This study | ZH128 |
| CCGCATCAGACAAATCAG | This study | ZH131 |
| AAACGAGCTCTCGAGAAC | This study | ZH132 |
| ATGACTAACGAAAAGGTCTGGATAGAGAAGTTGGATAATCCAACTCTTTCAGTGTTACCAGATCCGCAGGCTAACCGGAAC | This study | ZH150 |
| TTAAGCTGCTGCGGAGCTTCCACGAGCACCAGCACCTGAAGCAACTAGACTTATTTGCGCAGCTCGCTGTGAAGATCCCAG | This study | ZH151 |
| TATGAAGGATCCGGCCTGGAAGTTCTGTTCCA | This study | ZH90 |
| GCACCGAAGCTTTTTGTACAATTCATCCATAC | This study | ZH94 |
| Software and Algorithms | ||
| MaxQuant (version 1.2.2.5) | Cox et al., 2008 | http://www.coxdocs.org/doku.php?id=maxquant:start |
| UCSF Chimera, version 1.13 | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| Other | ||
| Mass Spectrometer | Thermo Fisher Scientific | Orbitrap Fusion |
| Mass Spectrometer | Thermo Fisher Scientific | Q Exactive Plus |
| Liquid Chromatograph | Thermo Fisher Scientific | Easy-nLC 1000 |
| Easy-Spray column | Thermo Fisher Scientific | ES800 |
| NuPage 4–12% Bis-Tris Gel 1.0mm × 10 well | Thermo Fisher Scientific | Cat.# NP0321Box |
| POROS R2 Reversed - Phase Resin | Thermo Fisher Scientific | Cat.# 1112906 |
| Omix Tips | Agilent | Cat.# A57003100 |
METHOD DETAILS
Yeast strains construction and culture
Standard procedures for culturing, maintenance, transformation, mating, sporulation, and selection were carried out according to the methods described in Yeast Protocols (Xiao, 2006). Unless otherwise specified, the cells were grown in synthetic complete (SC) medium, supplemented with 2% dextrose and 50mg/L of isotopically light or heavy lysine, and at 30°C with 220 rpm agitation.
A transformation cassette, carrying the TDH3 promoter, 3 tetracycline binding aptamers in the 5′UTR, and N-terminal GFP was amplified from Ttc3GFP-His5 construct and inserted upstream of either the NUP84 or the NUP157 coding sequence via homologous recombination. Positive transformants were selected on SD-His medium. The correct insertion as well as the retention of one wild-type copy of either the NUP84 or NUP157 gene in the diploid strain was verified by colony PCR. Thus a wild-type copy of Nup84 or Nup157 is always expressed and present, even upon inhibition of the GFP-tagged copy. The fact we used both tagged Nups and they gave very comparable results also controls for such Nup-specific effects. Lastly, proper nuclear rim and NPC localization of the GFP tagged Nup84 and Nup157 were confirmed by fluorescence microscopy and biochemical purification of the whole NPC through the GFP tag. Strain design is illustrated also in Figure S1A.
To construct the Nup145NC-fusion strain, a diploid DF5 strain was first transformed with a PCR cassette (amplified from pOM10, oligos ZH127, ZH128) integrating two LoxP sites flanking kanMX4 gene followed by 6xHA tag sequence at the site where Nup145 precursor protein auto-cleaves: F605-S606 (Teixeira et al., 1997, 1999). Positive transformants were selected on YPD+G418 medium. Targeted insertion was verified via colony PCR (ZH131, ZH132). Next, pSH47 plasmid, conditionally expressing Cre recombinase in galactose was transformed and the cells switched to a permissive condition. After the successful recombination between the two LoxP sites the cells were expected to lose the kanMX4 gene and start expressing the full-length Nup145N-C fusion protein with a 6xHA tag inserted at the cleavage site (inhibiting the autocleavage). To screen for recombinants the cells were plated on YPD and replica plated on YPD+G418 medium. Colonies that were viable on YPD but absent from YPD+G418 plates were analyzed by anti-HA western blot (WB) to verify the expression of Nup145NC fusion protein. Next, the strain was sporulated and 6 tetrads were dissected. Four haploid colonies resulting from each tetrad dissection were grown and subjected to immunoblotting to test for Nup145NC fusion protein expression. The mating type of positive clones was determined with mating type locus PCR (Huxley et al., 1990). Finally, the haploid strains of opposite mating type containing the Nup145N-C allele were mated and the successful mating verified by PCR (Huxley et al., 1990).
To generate diploid nup120 null strain the two haploid nup120 null strains from Aitchison et al. (1995a) were mated and single colonies were screened for the presence of both mating type loci by colony PCR.
SWY2870 was a generous gift from Prof. Susan Wente (Strawn et al., 2004a). To make the strain lysine auxotrophic, a single wild-type LYS2 gene was knocked out with a disruption cassette generated with PCR amplification from pUG73 (oligos ZH150, ZH151) and SD-Leu selection.
For C-terminal tagging of Nup170 or Nup116 with an auxin inducible degron (AID), a recombination cassette was amplified from BYP6740, transformed into Tc3-GFP-Nup84 and selected on YPD+G418; A BYP6744 construct expressing osTIR (auxin binding receptor) was then digested with StuI and integrated into the URA3 locus (Heider et al., 2015).
Plasmid construction
To swap the S. pombe His5 gene from pUG27 into pTDH3-tc3–3xHA instead of the KanR marker, both plasmids were digested with NcoI and SacI. DNA fragments were purified after agarose gel electrophoresis, and the insert from the pUG27 digest was ligated to the backbone from pTDH3-tc3–3xHA digest to obtain Ttc3HAHis5. To introduce the GFP sequence into the previous construct, GFP was PCR-amplified from p404GALL-GFP (oligos ZH90, ZH94); the PCR product and the Ttc3HAHis5 plasmid were subjected to a double digest with BamHI and HindIII. The backbone was treated with CIP. The backbone and insert were ligated to obtain Ttc3GFP-His5. Finally, the correct sequence of the GFP integration site was verified by sequencing.
Conditional gene expression
To repress the translation of GFP-Nup84 and GFP-Nup157, mRNA 0.2mg/mL of Cltc (chlortetracycline) HCl was added to the medium following the recommendation in the reference (Kotter et al., 2009). Cltc HCl was used instead of Tetracycline for its superior stability (Okerman et al., 2007). The expected degree of reduction of GFP-Nup84 or GFP-Nup157 amount over time was verified by anti-GFP immunoblotting. The rapid repression of GFP-Nup84 expression after ClTc HCl addition was further confirmed by concurrent labeling with 13C6 K (lysine) and monitoring the intensity of 13C6 K over isotopically light K in GFP-Nup84 peptides by ESI-MS/MS (electrospray ionization tandem mass spectrometry).
Metabolic labeling for turnover and exchange experiments
In order to label cells with 13C6 K, a single colony was inoculated into a 50mL pre-culture of synthetic complete dextrose dropout medium lacking histidine and lysine (SCD-HK), supplemented with 50mg/L of 13C6 K and grown overnight. The resulting culture was diluted at least 20-fold in the same medium and grown until OD 1. To exchange the metabolic label to light K, the cells were spun down at 5000 rpm for 5 min, the supernatant discarded and the pellet re-suspended in SCD-HK medium to OD 0.25, supplemented with 50mg/L of 12C6 K, with or without 0.2 mg/mL ClTc. The cells were grown for an additional 5 h, where for each hour a sample of cells was harvested and flash frozen in liquid nitrogen.
Nup170-AID degron activation experiments
Nup170-AID was inoculated into overnight pre-culture of heavy lysine supplemented synthetic defined medium. The following morning the stationary phase culture was inoculated into 2L of synthetic defined medium supplemented with heavy lysine and grown until OD 1.5. The culture was spun down, the cells resuspended in 6L of synthetic defined medium supplemented with light lysine, and the culture split into 4 flasks. 0.2mg/mL ClTc was added to the 1st and 3rd flasks, 0.7mM auxin was added to the 1st and 2nd flasks (the same volume of the auxin solvent ethanol was added to the 3rd and 4th flasks as a mock treatment). The cells were grown for 2h, harvested and processed for affinity capture and mass spectrometry. Samples from the 1st and 3rd flasks (the ones that received ClTc) were used to test the replacement/exchange of nups after Nup170-AID degradation. The samples from the 2nd and 4th flasks were used to evaluate turnover in samples with Nup170-AID induction.
For relative quantification of nucleoporin amounts in auxin degraded and undegraded cells, we performed metabolic labeling without a label switch as follows. Nup170-AID was inoculated into two overnight pre-cultures of synthetic defined medium with 50mg/L of isotopically light lysine in one and 50mg/L of heavy lysine in the other. 600uL of each saturated culture were inoculated into 1L of the same medium (light in light, heavy in heavy lysine) and grown until OD 0.5. 0.2mg/mL of ClTc was added to both cultures to stop the translation of GFP-Nup84. 0.7mM auxin was then added to the heavy lysine containing culture to induce Nup170-AID degradation. After 2 h (approximately one cell division) of growth both cultures were harvested, snap frozen in liquid nitrogen and cryogenically ground. The two powders were mixed at an equal ratio, and affinity capture and mass spectrometric analysis conducted on the combined cell material. The heavy to light ratios were quantified with MaxQuant and a custom Python script as for all quantification experiments in this study.
Affinity capture
The NPC affinity capture was carried out as before (Hakhverdyan et al., 2015) with minor modifications. Harvested cell mass was flash frozen in liquid nitrogen and ground to fine powder at cryogenic temperature (4 times 2 min, 200 rpm in a PM 100 Retsch planetary ball mill). 0.5g of the resulting disrupted cell material was resuspended in 2mL of the room temperature extraction buffer:
20 mM HEPES, pH 7.4, 150 mM NaCI, 250 mM sodium citrate, 1% v/v Triton X-100, 1× protease inhibitor cocktail (PIC) and 10 mM glutaraldehyde (GA).
A mild fixation with GA was implemented to minimize the protein exchange in the lysate, following published procedure (Subbotin and Chait, 2014). The reconstituted lysate was incubated on ice for 5 min, after which the GA cross linker was quenched by the addition of 100 mM Tris, pH 8. The lysate was clarified by centrifugation at 15k g for 10min. The supernatant was incubated with 25 μL of anti-GFP antibody-conjugated magnetic slurry for 30 min with agitation at 4°C. Bound beads were washed 3 times with 1mL of cold extraction buffer without PIC and GA. After the removal of the final wash the beads were re-suspended in 30 μL of 1× LDS (lithium dodecyl sulfate) loading buffer (Thermo Fisher) and incubated for 10 min at 70°C to elute the bound protein complexes. For the isolated NPC exchange experiments in Figure 1G, an I-DIRT protocol (Tackett et al., 2005) was adapted in which frozen powder from and equal amount of heavy labeled tagged cells was mixed with light labeled untagged cells and affinity captured as above, except in 1.5M Ammonium Acetate, pH 7, 1% Triton X-100 without GA stabilization, and the incubation with 25 μL of anti-GFP antibody-conjugated magnetic slurry being for for 60 min with agitation at 4°C.
In-gel digestion and peptide extraction
The samples were reduced for 10 min at 70°C with 20 mM DTT and alkylated with 100 mM iodoacetamide (IAM) for 30 min in the dark before running on the gel. The samples were run on a 4%–16% Bis-Tris gel (Thermo Fisher) at 200 V either for 55 min to fully resolve proteins or 3 min to gel-purify the sample from contaminants interfering with downstream MS, resulting in a “gel plug” (Kim et al., 2018). The gel was fixed for 5 min in 16% methanol, 10% acetic acid, washed thrice for 5 min in ddH2O, stained for 1 h with Coomassie blue R-250 stain and destained in water overnight. The image of the gel was recorded and either the protein bands or gel plugs (containing the entire affinity purified sample) were excised out of the gel, placed in Eppendorf tubes and chopped into 1mm3 pieces. The gel pieces were destained with 500 μL 50mM ammonium bicarbonate (AmBic), 50% acetonitrile (ACN) at 30°C, periodically replacing the destaining solution with a fresh aliquot until the Coomassie was completely removed. Following this, the gel pieces were dehydrated in 100% ACN and subsequent air drying, and then rehydrated in digestion solution (10 ng/μL of sequencing grade Trypsin in 50 mM AmBic) and incubated at 37°C overnight. To extract the digested peptides from the gel, 50uL of POROS R2 (reverse phase) beads slurry (Thermo Fisher) was added to the sample and incubated overnight at 4°C. The peptides bound to POROS beads were desalted on 10 μL C18 tips according to manufacturer’s instructions (OMIX tips, Agilent Technologies). To avoid particulates in the final sample, the POROS bead mixture was loaded on top of C18 resin and spun through at 1000 rpm for 1 min instead of pipetting up and down. Next, the peptides were eluted 2× each with 40% ACN, 0.1% trifluoroacetic acid (TFA) and 80% ACN, 0.1% TFA. Finally, the peptides were vacuum dried in SpeedVac.
Mass spectrometry
Peptides were resuspended in 5% (v/v) methanol, 0.2% (v/v) formic acid and loaded onto an EASY-Spray column (Thermo Fisher Scientific, ES800, 15cm × 75μm ID, PepMap C18, 3μm) via an EASY-nLC 1000 (Thermo Fisher Scientific). The column temperature was set to 35°C. Using a flow rate of 300 nl/min, peptides were eluted with a gradient of 4%–30% solvent B in 35 min, followed by 30%–80% B in 5 min, where solvent B was 0.1% (v/v) formic acid in acetonitrile and solvent A was 0.1% (v/v) formic acid in water. Peptides were ionized by electrospray as they eluted, with voltages of 1.7 – 2.5 kV applied to obtain stable spray. Either a Q Exactive Plus or Orbitrap Fusion (both Thermo Fisher Scientific) were used to perform online mass spectrometric analyses. For analyses performed on the Q Exactive Plus, the top 10 most intense ions in each full scan were fragmented by higher-energy collisional dissociation. Detailed parameters of the method are as follows: The parameters for full scans were: Microscans: 1; Resolution: 70,000; AGC target: 3e6; Maximum injection time: 500 msec; m/z scan range: 350–1500; Spectrum data type: profile. Parameters relevant to the selection of precursors for MS2: Exclude charge 1 and unassigned; Peptide match: preferred; Exclude isotopes: true; Dynamic exclusion: Exclude after 1 MS2 scan for 15 s, with mass tolerance 10 ppmThe parameters for MS2 scans were: Microscans: 1; Resolution: 17,500; AGC target: 1e5; Maximum injection time: 100 msec; Spectrum data type: centroid; Normalized collision energy: 30%; Isolation window: 2 m/z. For analyses performed on the Orbitrap Fusion, full scans were performed every 5 s. As time between full scans allowed, ions were fragmented by collision-induced dissociation in descending intensity order. Precursors were detected in the Orbitrap and fragments were detected in the ion trap. Detailed parameters of the method are as follows: The parameters for full scans were: Microscans: 1; Resolution: 60,000; AGC target: 2e5; Maximum injection time: 100 msec; m/z scan range: 300–1500; Use quadrupole isolation: true; Spectrum data type: profile. Parameters relevant to the selection of precursors for MS2: Monoisotopic precursor selection: true; Intensity threshold: 1e3; Include charge 2–6; Dynamic exclusion: Exclude after 1 MS2 scan for 15 s, with mass tolerance 10 ppm. The parameters for MS2 scans were: Microscans: 1; Isolation mode: quadrupole; AGC target: 7e3; Maximum injection time: 100 msec; Spectrum data type: centroid; Isolation window: 2 m/z; Collision energy: 35%; Activation Q: 0.25; Ion trap scan rate: rapid.
ODELAY phenotypic analysis
ODELAY experiments were performed as described previously (Herricks et al., 2017a, 2017b, 2020). Yeast cultures were grown in synthetic complete media with 2% glucose for 2–3 h until they reached logarithmic phase and then diluted to 0.05 OD600. These cultures were then spotted down onto 1% agarose pads formulated with synthetic complete media and 2% glucose. For benzyl alcohol experiments the media was supplemented with 0.2% vol/vol benzyl alcohol. Cells were imaged at either 30°C or 37°C for 48 h. The resulting images were processed with ODELAY-ODELAM software (Herricks et al., 2020). Doubling time boxplots were generated using the python package Matplotlib.
Chimera rendering of NPC dynamics maps
The UCSF Chimera package (https://www.cgl.ucsf.edu/chimera/) was used to render and color the whole NPC and individual complexes (Pettersen et al., 2004) using the map from Kim et al. (2018), and using LUT scales that quantitatively correspond to the values being mapped. For mutant / wild-type comparisons, any Nup with an absolute Z-score < 3 is considered unaltered (white).
QUANTIFICATION AND STATISTICAL ANALYSIS
Exchange and turnover rate calculation
The RAW mass spectrometric files were searched with the MaxQuant software (Cox and Mann, 2008) both to identify peptides and measure the heavy/light ratio of lysine containing peptides. For MaxQuant analysis, mostly default parameters were used with the following exceptions: the multiplicity was set to 2, for the light sample all amino acids were set to light, for the heavy sample “lys6” was selected; the yeast translated ORF sequences – https://www.yeastgenome.org:443/ – reversed sequences and contaminants database were searched; “Re-quantify” and “Match between runs” were enabled with the default parameters; and the “I=L” box was checked. The data were extracted from the master table outputted by MaxQuant called “Evidence.txt,” containing all the peptide identification and intensity measurement information. The analysis consisted of the following steps performed by a custom Python script:
The peptides mapping to the contaminants, decoy reversed sequence or those containing no lysine were filtered out.
- For the remaining peptides the heavy label fraction (HLF) was calculated by using heavy/light intensity ratio measurements produced by MaxQuant with the following conversion:
To calculate the HLF of a protein, the HLFs of its constituent peptides were averaged. For proteins with 4 or more peptides contributing to the HLF calculation, the outlier measurement was filtered out using the following criteria: if the HLF measurement of a peptide < Q1 (first quartile) − 1.5 × IQR (interquartile range) or > Q3 (third quartile) + (1.5 × IQR), that peptide measurement was excluded. No more than one peptide measurement was removed per protein.
We assumed the exchange of nucleoporins to be a first order reaction and approximated it with an exponential decay model. The average HLF values were log-transformed and linear regression performed on the time course data on the > = 3 data points available. An r correlation coefficient cutoff was set to 0.9 and the p value cut off to 0.05. Note, for proteins with low exchange rates the calculated slope was shallow and the p value (which is the test for the two-tailed probability for the null hypothesis that the slope = 0) was not a good indicator of goodness of fit. Hence, when the slope was small in absolute value (> = −0.05), a moderately low correlation value of 0.7 was accepted, without regards to a p value. Two experiments were conducted for each strain, and the slopes averaged.
The plots were started at 1 h, to exclude any potential initial lag due to a soluble pool of protein. Subcellular fractionations indicate that such pools are small (Rout et al., 2000; Rout and Blobel, 1993; Strambio-de-Castillia et al., 1995).
Finally, SGD annotations for all the protein hits that produced a successful model fit, were searched for keywords corresponding nuclear pore complex components and nucleocytoplasmic transport factors to group the data into 3 categories: nucleoporin, transport factor, and other (neither nucleoporin nor transport factor). The latter category usually corresponds to abundant cytosolic proteins that commonly co-isolate as contaminants in affinity capture experiments (Mellacheruvu et al., 2013).
Comparison of mutant and wild-type exchange rates
In order to determine if exchange rates were significantly altered in mutant experiments as compared to wild-type, Z-scores were calculated as follows. We took the ratio of slopes (exchange rates) of all the proteins in the “Other” group. Since we do not expect the proteins in this group to have altered exchange rates, we assumed all changes in this group to be random. A normal distribution was constructed using the average and standard deviation of these ratios. The ratios of nucleoporin exchange rates were compared to this distribution. We accepted a Z-score of > 3 (3 standard deviations away from the mean) to be potentially significant.
Quantification of relative nucleoporin amount change after degron induction
HLF was calculated as previously described. Next, an average mixing ratio (of heavy-labeled and light-labeled cell material) and standard deviation were calculated from the average HLF of proteins in the “Other” group. Again, we assumed that proteins in the “Other” group should not change in concentration based on auxin treatment (in support of which, the standard deviation is < 9%). Thus, we accepted measures of > 1 standard deviation away from the mean to be potentially significant.
Quantification of relative new nucleoporin association in degron-induced versus uninduced NPCs
For this experiment we calculated LLF the (light labeled fraction), which is 1-HLF. In this case the light labeled protein signifies new protein becoming associated with the NPC after degron induction. To obtain the relative difference in new protein between degron-induced and uninduced cells, we divided the LLF of auxin-treated (degron induced cells) by LLF of untreated cells. We performed this operation on all the proteins in the dataset. A normal distribution was constructed with the average and standard deviation of “Other” protein ratios. Next we compared the nucleoporin ratios to the distribution. We accepted a Z-score of > 3 (3 standard deviations away from the mean) to be potentially significant, as with analogous experiments (above).
Supplementary Material
Highlights.
General proteomics method to add dynamic dimension to macromolecular assemblies
Despite low turnover, nucleoporins show significant and wide degree of exchange
Nucleoporin exchange correlates with their structural role, not peripheral position
Nucleoporin exchange ensures maintenance and damage resilience in NPCs
ACKNOWLEDGMENTS
We are very grateful to Prof. Frederick Cross for critical feedback throughout this study. We acknowledge Ignacia Echeverria, Seung Joong Kim, and Andrej Sali for help with NPC visualization and structural calculations. This work was supported by NIH grants U54 GM103511 (to B.T.C., J.D.A., and M.P.R.), R01 AI141953 (to J.D.A.), R01 GM068803 (to M.M.), R01 GM112108 (to J.D.A. and M.P.R.), P41 GM109824 (to M.P.R., J.D.A., and B.T.C.), and P41 GM103314 (to B.T.C.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.11.032.
REFERENCES
- Aitchison JD, and Rout MP (2015). The interactome challenge. J. Cell Biol. 211, 729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitchison JD, Blobel G, and Rout MP (1995a). Nup120p: a yeast nucleoporin required for NPC distribution and mRNA transport. J. Cell Biol. 131, 1659–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitchison JD, Rout MP, Marelli M, Blobel G, and Wozniak RW (1995b). Two novel related yeast nucleoporins Nup170p and Nup157p: complementation with the vertebrate homologue Nup155p and functional interactions with the yeast nuclear pore–membrane protein Pom152p. J. Cell Biol. 131, 1133–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baade I, and Kehlenbach RH (2019). The cargo spectrum of nuclear transport receptors. Curr. Opin. Cell Biol. 58, 1–7. [DOI] [PubMed] [Google Scholar]
- Beck M, and Hurt E (2017). The nuclear pore complex: understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 18, 73–89. [DOI] [PubMed] [Google Scholar]
- Boisvert FM, Ahmad Y, Gierlinski M, Charriere F, Lamont D, Scott M, Barton G, and Lamond AI (2012). A quantitative spatial proteomics analysis of proteome turnover in human cells. Mol. Cell Proteomics 11, M111.011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambridge SB, Gnad F, Nguyen C, Bermejo JL, Krüger M, and Mann M (2011). Systems–wide Proteomic Analysis in Mammalian Cells Reveals Conserved, Functional Protein Turnover. J. Proteome Res. 10, 5275–5284. [DOI] [PubMed] [Google Scholar]
- Chial HJ, Rout MP, Giddings TH, and Winey M (1998). Saccharomyces cerevisiae Ndc1p is a shared component of nuclear pore complexes and spindle pole bodies. J. Cell Biol. 143, 1789–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiano R, Nagaraj N, Frühlich F, and Walther TC (2014). Global Proteome Turnover Analyses of the Yeasts S. cerevisiae and S. pombe. Cell Rep. 9, 1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- D’Angelo MA, Raices M, Panowski SH, and Hetzer MW (2009). Age-Dependent Deterioration of Nuclear Pore Complexes Causes a Loss of Nuclear Integrity in Postmitotic Cells. Cell 136, 284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle N (2001). Nuclear pore complexes form immobile networks and have a very low turnover in live mammalian cells. J. Cell Biol. 154, 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza CPC, Osmani AH, Hashmi SB, and Osmani SA (2004). Partial nuclear pore complex disassembly during closed mitosis in Aspergillus nidulans. Curr. Biol. 14, 1973–1984. [DOI] [PubMed] [Google Scholar]
- Deng J, Erdjument–Bromage H, and Neubert TA (2019). Quantitative Comparison of Proteomes Using SILAC. Curr. Protoc. Protein Sci. 95, e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilworth DJ, Suprapto A, Padovan JC, Chait BT, Wozniak RW, Rout MP, and Aitchison JD (2001). Nup2p dynamically associates with the distal regions of the yeast nuclear pore complex. J. Cell Biol. 153, 1465–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doye V, and Hurt EC (1995). Genetic approaches to nuclear pore structure and function. Trends Genet. 11, 235–241. [DOI] [PubMed] [Google Scholar]
- Emtage JL, Bucci M, Watkins JL, and Wente SR (1997). Defining the essential functional regions of the nucleoporin Nup145p. J. Cell Sci. 110, 911–925. [DOI] [PubMed] [Google Scholar]
- Fabre E, and Hurt E (1997). Yeast genetics to dissect the nuclear pore complex and nucleocytoplasmic trafficking. Annu. Rev. Genet. 31, 277–313. [DOI] [PubMed] [Google Scholar]
- Fernandez-Martinez J, Phillips J, Sekedat MD, Diaz-Avalos R, Velázquez–Muriel J, Franke JD, Williams R, Stokes DL, Chait BT, Sali A, et al. (2012). Structure–function mapping of a heptameric module in the nuclear pore complex. J. Cell Biol. 196, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Martinez J, Kim SJ, Shi Y, Upla P, Pellarin R, Gagnon M, Chemmama IE, Wang J, Nudelman I, Zhang W, et al. (2016). Structure and Function of the Nuclear Pore Complex Cytoplasmic mRNA Export Platform. Cell 167, 1215–1228.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J, Teimer R, Amlacher S, Kunze R, and Hurt E (2015). Linker Nups connect the nuclear pore complex inner ring with the outer ring and transport channel. Nat. Struct. Mol. Biol. 22, 774–781. [DOI] [PubMed] [Google Scholar]
- Folkmann AW, Noble KN, Cole CN, and Wente SR (2011). Dbp5, Gle1-IP6 and Nup159: a working model for mRNP export. Nucleus 2, 540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaik M, Flemming D, von Appen A, Kastritis P, Mücke N, Fischer J, Stelter P, Ori A, Bui KH, Baßler J, et al. (2015). Structural basis for assembly and function of the Nup82 complex in the nuclear pore scaffold. J. Cell Biol. 208, 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlick PJ, and Millward DJ (1972). An appraisal of techniques for the determination of protein turnover in vivo. Biochem. J. 129, 1P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffis ER, Altan N, and Lippincott–Schwartz J (2002). Nup98 is a mobile nucleoporin with transcription-dependent dynamics. Mol. Biol. Cell 13, 1282–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffis ER, Craige B, Dimaano C, Ullman KS, and Powers MA (2004). Distinct functional domains within nucleoporins Nup153 and Nup98 mediate transcription-dependent mobility. Mol. Biol. Cell 15, 1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünwald D, and Singer RH (2010). In vivo imaging of labelled endogenous β-actin mRNA during nucleocytoplasmic transport. Nature 467, 604–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakhverdyan Z, Domanski M, Hough LE, Oroskar AA, Oroskar AR, Keegan S, Dilworth DJ, Molloy KR, Sherman V, Aitchison JD, et al. (2015). Rapid, optimized interactomic screening. Nat. Methods 12, 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson S, Berthelot K, Fink B, McCarthy JEG, and Suess B (2003). Tetracycline–aptamer–mediated translational regulation in yeast. Mol. Microbiol. 49, 1627–1637. [DOI] [PubMed] [Google Scholar]
- Heath CV, Copeland CS, Amberg DC, Del Priore V, Snyder M, and Cole CN (1995). Nuclear pore complex clustering and nuclear accumulation of poly(A)+ RNA associated with mutation of the Saccharomyces cerevisiae RAT2/NUP120 gene. J. Cell Biol. 131, 1677–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heider MR, Gu M, Duffy CM, Mirza AM, Marcotte LL, Walls AC, Farrall N, Hakhverdyan Z, Field MC, Rout MP, et al. (2015). Subunit connectivity, assembly determinants and architecture of the yeast exocyst complex. Nat. Struct. Mol. Biol. 23, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herricks T, Dilworth DJ, Mast FD, Li S, Smith JJ, Ratushny AV, and Aitchison JD (2017a). One-Cell Doubling Evaluation by Living Arrays of Yeast, ODELAY! G3 (Bethesda) 7, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herricks T, Mast FD, Li S, and Aitchison JD (2017b). ODELAY: A Large-scale Method for Multi-parameter Quantification of Yeast Growth. J. Vis. Exp. (125), 55879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herricks T, Donczew M, Mast FD, Rustad T, Morrison R, Sterling TR, Sherman DR, and Aitchison JD (2020). ODELAM, rapid sequence-independent detection of drug resistance in isolates of Mycobacterium tuberculosis. eLife 9, e56613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer G, and Antonin W (2018). Nuclear Pore Complexes: Global Conservation and Local Variation. Curr. Biol. 28, R674–R677. [DOI] [PubMed] [Google Scholar]
- Huxley C, Green ED, and Dunham I (1990). Rapid assessment of S. cerevisiae mating type by PCR. Trends Genet. 6, 236. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Fernandez-Martinez J, Nudelman I, Shi Y, Zhang W, Raveh B, Herricks T, Slaughter BD, Hogan JA, Upla P, et al. (2018). Integrative structure and functional anatomy of a nuclear pore complex. Nature 555, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knockenhauer KE, and Schwartz TU (2016). The Nuclear Pore Complex as a Flexible and Dynamic Gate. Cell 164, 1162–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinski J, Mosalaganti S, von Appen A, Teimer R, DiGuilio AL, Wan W, Bui KH, Hagen WJH, Briggs JAG, Glavy JS, et al. (2016). Molecular architecture of the inner ring scaffold of the human nuclear pore complex. Science 352, 363–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter P, Weigand JE, Meyer B, Entian K–D, and Suess B (2009). A fast and efficient translational control system for conditional expression of yeast genes. Nucleic Acids Res. 37, e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DH, Stuwe T, Schilbach S, Rundlet EJ, Perriches T, Mobbs G, Fan Y, Thierbach K, Huber FM, Collins LN, et al. (2016). Architecture of the symmetric core of the nuclear pore. Science 352, aaf1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman SK, and Gerace L (2001). Nuclear pore complexes: dynamics in unexpected places. J. Cell Biol. 154, 17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson T, Franken H, Kosinski J, Kurzawa N, Zinn N, Sweetman G, Poeckel D, Ratnu VS, Schramm M, Becher I, et al. (2018). Systematic analysis of protein turnover in primary cells. Nat. Commun. 9, 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert J-P, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. (2013). The CRAPome: a contaminant repository for affinity purification–mass spectrometry data. Nat. Methods 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mészáros N, Cibulka J, Mendiburo MJ, Romanauska A, Schneider M, and Köhler A (2015). Nuclear Pore Basket Proteins Are Tethered to the Nuclear Envelope and Can Regulate Membrane Curvature. Dev. Cell 33, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakielny S, Shaikh S, Burke B, and Dreyfuss G (1999). Nup153 is an M9-containing mobile nucleoporin with a novel Ran-binding domain. EMBO J. 18, 1982–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niepel M, Molloy KR, Williams R, Farr JC, Meinema AC, Vecchietti N, Cristea IM, Chait BT, Rout MP, and Strambio-de-Castillia C (2013). The nuclear basket proteins Mlp1p and Mlp2p are part of a dynamic interactome including Esc1p and the proteasome. Mol. Biol. Cell 24, 3920–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niño CA, Guet D, Gay A, Brutus S, Jourquin F, Mendiratta S, Salamero J, Géli V, and Dargemont C (2016). Posttranslational marks control architectural and functional plasticity of the nuclear pore complex basket. J. Cell Biol. 212, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, and Kanemaki MT (2014). Rapid Depletion of Budding Yeast Proteins via the Fusion of an Auxin-Inducible Degron (AID). Curr. Protoc. Cell Biol. 64, 20.29.21–16. [DOI] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, and Kanemaki M (2009). An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922. [DOI] [PubMed] [Google Scholar]
- Oda Y, Huang K, Cross FR, Cowburn D, and Chait BT (1999). Accurate quantitation of protein expression and site–specific phosphorylation. Proc. Natl. Acad. Sci. USA 96, 6591–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okerman L, Van Hende J, and De Zutter L (2007). Stability of frozen stock solutions of beta–lactam antibiotics, cephalosporins, tetracyclines and quinolones used in antibiotic residue screening and antibiotic susceptibility testing. Anal. Chim. Acta 586, 284–288. [DOI] [PubMed] [Google Scholar]
- Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, and Mann M (2002a). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics 1, 376–386. [DOI] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, and Mann M (2002b). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386. [DOI] [PubMed] [Google Scholar]
- Onischenko E, Tang JH, Andersen KR, Knockenhauer KE, Vallotton P, Derrer CP, Kralt A, Mugler CF, Chan LY, Schwartz TU, et al. (2017). Natively Unfolded FG Repeats Stabilize the Structure of the Nuclear Pore Complex. Cell 171, 904–917.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pratt JM, Robertson DHL, Gaskell SJ, Riba–Garcia I, Hubbard SJ, Sidhu K, Oliver SG, Butler P, Hayes A, Petty J, et al. (2002). Stable isotope labelling in vivo as an aid to protein identification in peptide mass fingerprinting. Proteomics 2, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak C, Aitchison JD, and Wozniak RW (2014). The multifunctional nuclear pore complex: a platform for controlling gene expression. Curr. Opin. Cell Biol. 28, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabut G, Doye V, and Ellenberg J (2004). Mapping the dynamic organization of the nuclear pore complex inside single living cells. Nat. Cell Biol. 6, 1114–1121. [DOI] [PubMed] [Google Scholar]
- Rajoo S, Vallotton P, Onischenko E, and Weis K (2018). Stoichiometry and compositional plasticity of the yeast nuclear pore complex revealed by quantitative fluorescence microscopy. Proc. Natl. Acad. Sci. USA 115, E3969–E3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner GA, Hodel AE, and Powers MA (2007). Molecular determinants of binding between Gly-Leu-Phe-Gly nucleoporins and the nuclear pore complex. J. Biol. Chem. 282, 33968–33976. [DOI] [PubMed] [Google Scholar]
- Ribbeck K, and Görlich D (2001). Kinetic analysis of translocation through nuclear pore complexes. EMBO J. 20, 1320–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, and Blobel G (1993). Isolation of the yeast nuclear pore complex. J. Cell Biol. 123, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, Aitchison JD, Suprapto A, Hjertaas K, Zhao Y, and Chait BT (2000). The yeast nuclear pore complex: composition, architecture, and transport mechanism. J. Cell Biol. 148, 635–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüthnick D, Neuner A, Dietrich F, Kirrmaier D, Engel U, Knop M, and Schiebel E (2017). Characterization of spindle pole body duplication reveals a regulatory role for nuclear pore complexes. J. Cell Biol. 216, 2425–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas JN, Toyama BH, Xu T, Yates JR, and Hetzer MW (2012). Extremely Long-Lived Nuclear Pore Proteins in the Rat Brain. Science 335, 942, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siniossoglou S, Lutzmann M, Santos–Rosa H, Leonard K, Mueller S, Aebi U, and Hurt E (2000). Structure and assembly of the Nup84p complex. J. Cell Biol. 149, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AE, Slepchenko BM, Schaff JC, Loew LM, and Macara IG (2002). Systems analysis of Ran transport. Science 295, 488–491. [DOI] [PubMed] [Google Scholar]
- Strambio-de-Castillia C, Blobel G, and Rout MP (1995). Isolation and characterization of nuclear envelopes from the yeast Saccharomyces. J. Cell Biol. 131, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strawn LA, Shen T, Shulga N, Goldfarb DS, and Wente SR (2004). Minimal nuclear pore complexes define FG repeat domains essential for transport. Nat. Cell Biol. 6, 197–206. [DOI] [PubMed] [Google Scholar]
- Subbotin RI, and Chait BT (2014). A Pipeline for Determining Protein-Protein Interactions and Proximities in the Cellular Milieu. Mol. Cell Proteomics 13, 2824–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackett AJ, Degrasse JA, Sekedat MD, Oeffinger M, Rout MP, and Chait BT (2005). I-DIRT, a general method for distinguishing between specific and nonspecific protein interactions. J. Proteome Res. 4, 1752–1756. [DOI] [PubMed] [Google Scholar]
- Teimer R, Kosinski J, von Appen A, Beck M, and Hurt E (2017). A short linear motif in scaffold Nup145C connects Y-complex with pre-assembled outer ring Nup82 complex. Nat. Commun. 8, 1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira MT, Siniossoglou S, Podtelejnikov S, Bénichou JC, Mann M, Dujon B, Hurt E, and Fabre E (1997). Two functionally distinct domains generated by in vivo cleavage of Nup145p: a novel biogenesis pathway for nucleoporins. EMBO J. 16, 5086–5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira MT, Fabre E, and Dujon B (1999). Self-catalyzed cleavage of the yeast nucleoporin Nup145p precursor. J. Biol. Chem. 274, 32439–32444. [DOI] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR III, and Hetzer MW (2013). Identification of Long-Lived Proteins Reveals Exceptional Stability of Essential Cellular Structures. Cell 154, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Arrojo EDR, Lev-Ram V, Ramachandra R, Deerinck TJ, Lechene C, Ellisman MH, and Hetzer MW (2018). Visualization of long-lived proteins reveals age mosaicism within nuclei of postmitotic cells. J. Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson JR (2008). Cooperativity in macromolecular assembly. Nat. Chem. Biol. 4, 458–465. [DOI] [PubMed] [Google Scholar]
- Wolf DH, and Menssen R (2018). Mechanisms of cell regulation - proteolysis, the big surprise. FEBS Lett. 592, 2515–2524. [DOI] [PubMed] [Google Scholar]
- Xiao W (2006). Yeast Protocols , Second Edition (Humana Press; ). [Google Scholar]
- Yang W, Gelles J, and Musser SM (2004). Imaging of single-molecule translocation through nuclear pore complexes. Proc. Natl. Acad. Sci. USA 101, 12887–12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Seo H-S, Debler EW, Blobel G, and Hoelz A (2011). Structural and functional analysis of an essential nucleoporin heterotrimer on the cytoplasmic face of the nuclear pore complex. Proc. Natl. Acad. Sci. USA 108, 16571–16576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Files containing the input data and custom Python scripts are available upon request. Mass spectrometry raw datasets used in this study are publicly available in Zenodo (https://zenodo.org/) under DOI #: 10.5281/zenodo.4062150.