

Abstract
The nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 (NLRP3) inflammasome has been implicated in podocyte injury and glomerular sclerosis in response to hyperhomocysteinemia (hHcy). However, it remains unknown how the products of NLRP3 inflammasome in cytoplasm are secreted out of podocytes. In the present study, we tested whether exosome release serves as a critical mechanism to mediate the action of NLRP3 inflammasome activation in hHcy-induced glomerular injury. By various approaches, we found that hHcy induced NLRP3 inflammasome activation and neutrophil infiltration in glomeruli of WT/WT mice. Lysosome-MVB interaction in glomeruli remarkably decreased in WT/WT mice fed with FF diet, leading to elevation of urinary exosome excretion of these mice. Podocyte-derived exosomes containing pro-inflammatory cytokines increased in urine of WT/WT mice in response to hHcy. The release of inflammatory exosomes from podocytes was prevented by Smpd1 gene deletion but enhanced by podocyte-specific Smpd1 gene overexpression (Smpd1 encodes Asm in mice). Pathologically, hHcy-induced podocyte injury and glomerular sclerosis were blocked by Smpd1 gene knockout but amplified by podocyte-specific Smpd1 gene overexpression. Taken together, our results suggest that Asm-ceramide signaling pathway contributes to NLRP3 inflammasome activation and robust release of inflammatory exosomes in podocytes during hHcy, which together trigger local glomerular inflammation and sclerosis.
Keywords: exosome, acid sphingomyelinase, NLRP3 inflammasome, podocyte, glomerular inflammation, hyperhomocysteinemia
1. Introduction
The nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 (NLRP3) inflammasome activation in podocytes has been demonstrated to be an important pathogenic mechanism mediating hyperhomocysteinemia (hHcy)-induced glomerular injury and ultimate end stage renal disease (ESRD) [1]. However, NLRP3 inflammasomes are activated mainly in the cytosol and their products therefore appear not to be secreted out of podocytes via a classical, Golgi apparatus-mediated delivery pathway. It is imperative to understand how the NLRP3 inflammasome activation-derived products such as IL-1β, IL-18, and high mobility group protein B1 (HMGB1) are released out of podocytes to trigger the inflammatory response in glomeruli, leading to glomerular sclerosis. One possibility is that these pro-inflammatory products of NLRP3 inflammasome activation are secreted from podocytes in the form of exosomes, extracellular vesicles (EVs) released into the extracellular space [2, 3]. Exosomes are considered to be an important mediator of cell-to-cell communication, regulating both physiologic and pathophysiologic processes through their delivery of mRNAs, miRNAs, proteins, and other constituents to acceptor cells [4, 5]. In regard to glomerular diseases, increased exosome release has been recognized as a biomarker of glomerular diseases and is implicated as a pathogenic factor in kidney disease [6–11]. The idea that exosomes may also facilitate the secretion of pro-inflammatory products is supported by studies reporting that IL-1β can be a constituent of exosomes and may be released from cells in this form [12]. These studies raise the possibility that exosomes may deliver NLRP3 inflammasome products out of podocytes, leading to glomerular inflammation and sclerosis. However, the precise mechanism mediating inflammatory exosome release and thereby activating the local inflammatory response in glomeruli has not been determined.
With respect to the molecular mechanism of exosome release, it is considered to occur when multivesicular bodies (MVBs) fuse with the plasma membrane, releasing their contained exosomes into the extracellular space. A potential regulatory control point involves lysosome-mediated degradation of the MVBs, thereby controlling the fate of MVBs prior to secretion of exosomes [13–18]. Ceramide, the product of acid sphingomyelinase (Asm), and its metabolite, sphingosine-1-phosphate (S1P) may regulate exosome release at any of several potential control points, including exosome biogenesis, sorting of intraluminal vesicles (ILVs) into MVBs, MVB fusion and exosome budding [19–21]. In particular, ceramide and associated sphingolipids as a key regulator of lysosome trafficking and fusion with other vesicles are shown in different cell types [8, 22–27]. In previous studies, it has been demonstrated that ceramide contributes to the activation of NLRP3 inflammasomes in podocytes, glomerular inflammation, and consequent glomerular sclerosis during hHcy [28–30]. We considered that the Asm-ceramide signaling pathway may also be involved in inflammatory exosome release under this pathological condition. The present study tested the hypothesis that lysosomal Asm-ceramide signaling pathway may not only mediate NLRP3 inflammasome activation but also promote robust release of inflammatory exosomes to deliver inflammasome products from podocytes during hHcy, together triggering local glomerular inflammation and sclerosis.
2. Materials and Methods
2.1. Animals
Podocyte-specific Cre recombinase (PodoCre) mice were obtained from the Jackson Laboratory [Bar Harbor, ME; B6.Cg-Tg(NPHS2-Cre)295Lbh/J; stock number 008205]. Smpd1trg mice are with the floxed STOP cassette inserted between the beta-actin fusion promoter and mouse cDNA, which were obtained from Erich Gulbins (University of Duisburg-Essen, Essen, Germany). Smpd1−/− breeding pairs were obtained from Dr. Phillip Hylemon’s laboratory at the VCU [31]. The Smpd1trg/PodoCre mice and their littermates were on a C57/Bl6 background. Eight-week-old male WT/WT, Smpd1−/−, and Smpd1trg/PodoCre mice were used in the present study. To speed up the damaging effects of hHcy on glomeruli, all mice were uninephrectomized, as we described previously [32]. This model has been demonstrated to induce glomerular damage unrelated to the uninephrectomy and arterial blood pressure, but specific to hHcy. After allowing 1 week for surgery recovery, mice were fed either normal chow or folate-free (FF) diet (Dyets Inc., Bethlahem, PA) for 8 weeks. All protocols were approved by the Institutional Animal Care and Use Committee of the Virginia Commonwealth University.
2.2. Mouse genotyping
Each mouse used in the in vivo studies was genotyped for the Smpd1trg gene and Cre recombinase gene to confirm podocyte-specific gene overexpression of acid sphingomylinase before use in experiments. Briefly, genomic DNA extracted from the tail was subjected to PCR amplification using taq DNA polymerase (Invitrogen, Inc., Grand Island, NY). Using a Bio-Rad iCycler, PCR was performed using a validated protocol provided by Jackson Laboratories: denaturing the DNA at 94°C for 3 minutes, followed by a first round of 12 cycles: 94°C for 20 seconds, 64°C for 30 seconds (−0.5°C per cycle), 72°C for 35 seconds, and then a second round of 25 cycles: 94°C for 20 seconds, 58°C for 30 seconds, 72°C for 35 seconds, and a final extension step at 72°C for 2 minutes. Smpd1 gene deletion was confirmed by PCR using primers, 5′-GGCTACCCGTGATATTGC-3′ (forward) and 5′-AGCCGTGTCCTCTTCCTTAC-3′ (reverse) [33]. The Smpd1wt and Smpd1trg genes were detected using primers of 5′-TGAGGCAGGAGAATCGCTTGAACC-3′ (forward) and 5′-GCCTCCCAGATGCTAATAGGTTGTGG-3′ (reverse). The internal control gene was detected using primers of 5′-CTAGGCCACAGAATTGAAAGATCT-3′ (forward) and 5′-GTAGGTGGAAATTCTAGCATCATCC-3′ (reverse). The Cre recombinase gene was detected using primers of 5′-GCGGTCTGGCAGTAAAAACTATC-3′ (forward) and 5′-GTGAAACAGCATTGCTGTCACTT-3′ (reverse). The PCR products were separated by gel electrophoresis on a 3% agarose gel, visualized by ethidium bromide fluorescence, and compared with a 100-bp DNA ladder (New England Biosystems, Ipswich, MA).
2.3. Immunofluorescent staining
Frozen slides with mouse kidney tissue were fixed in acetone, blocked, then incubated with primary antibodies, including anti-podocin antibody (1:100; Sigma-Aldrich, St. Louis, MO), anti-desmin antibody (1:100; Invitrogen, Carlsbad, CA), anti-Asm antibody (1:100; LSBio, Seattle, WA), anti-Cre antibody (1:100; Millipore, Temecula, CA), anti-ceramide antibody (1:100; ALEXIS, Farmingdale, NY), anti-NLRP3 antibody (1:50; Abcam, Cambridge, MA), anti-ASC antibody (1:50; Santa Cruz Biotechnology, Dallas, TX), anti-Ly6G antibody (1:50; Abcam, Cambridge, MA), anti-VPS16 antibody (1:50; proteintech, Rosemont, IL), and anti-Lamp-1 antibody (1:50; Abcam, Cambridge, MA), overnight at 4°C. Immunofluorescent staining was accomplished by incubating slides with Alexa-488 or Alexa-594-labeled secondary antibodies (Invitrogen, Carlsbad, CA) for 1 hour at room temperature [34]. Slides were washed, mounted, and observed by a confocal laser scanning microscope (FluoView FV1000, Olympus, Tokyo, Japan). Image Pro Plus 6.0 (Media Cybernetics, Bethesda, MD) was used to analyze colocalization which was expressed as Pearson correlation coefficient (PCC).
2.4. Immunohistochemistry
Kidneys were embedded with paraffin, 5 μm sections were cut and mounted onto microscope slides. After heat-induced antigen retrieval, washing with 3% hydrogen peroxide, and 30 min blocking with fetal bovine serum, slides were incubated with anti-IL-1β antibody (Abcam, Cambridge, MA), and then diluted in PBS with 4% fetal bovine serum overnight. The sections were washed with PBS and incubated with biotinylated IgG (1:200) for 1 h at room temperature, then with streptavidin-HRP for 30 min. Each kidney section was then stained with DAB for 1 min followed by counterstaining with hemotoxyline for 5 min. The slides were mounted and observed under a microscope [34].
2.5. Nanoparticle tracking analysis
Nanoparticle tracking analysis (NTA) measurements were performed with a NanoSight NTA3.2 Dev Build 3.2.16 (Malvern Instruments Ltd., UK), equipped with a sample chamber with a 638-nm laser and a Viton fluoroelastomer O-ring. The samples were injected in the sample chamber with sterile syringes (BD, New Jersey, USA) until the liquid reached the tip of the nozzle. All measurements were performed at room temperature. The screen gain and camera level were 10 and 13, respectively. Each sample was measured at standard measurement, 30 s with manual shutter and gain adjustments. Three measurements of each sample were performed. 3D figures were exported from the software. Particles sized between 50 and 140 nm were calculated [35]. At the end of the 8-week treatment, mice were placed in metabolic cages for 24 hours to collect urine samples. After NTA, we compared urinary exosome excretion of different groups of mice in 24 hours ((urinary exosome concentration × urine volume)/mouse body weight) vs. WT/WT-ND.
2.6. Purification and concentration of urinary exosomes
At the end of the 8-week treatment, mice were placed in metabolic cages for 24 hours to collect urine samples. Exosome purification and concentration system (ExoJuice, Columbus, OH, USA) was used to purify and concentrate urine samples for analysis. After normalization of the volume of urine samples, the centrifugation at 12,000 g for 30 min was performed to remove cell debris from urine samples. The supernatant was put into an ultracentrifuge tube and 200 μL of Exojuice reagent was added to the bottom of the centrifuge tube. After using the centrifuge at 100,000 g for 70 min, the bottom 500 μL of liquid was collected. When using the SW28 rotor for this step, about 230 mL of the culture supernatant was concentrated to 3 mL in one run. 3 ml of crudely extracted exosomes was mixed to 2.5 mL of sterile PBS (pH 7.2, filtered through a 0.2 μm filter) and added to a centrifuge tube. Then, 500 μL of Exojuice was added to the bottom of this tube. After using the centrifuge at 100,000 g for another 70 min, the centrifuge tube was took out and 300 μL of liquid (1) was discarded. 200 μL of liquid (2) was collected for a higher purity exosomes. Dialysis was performed to remove Exojuice reagent from the purified exosomes with dialysis membrane with 1KD cutoff. 200 μL of exosome extract was added to the dialysis bag against 100 mL of 1 × PBS buffer for 2 hr. Then, purified urinary exosomes were collected in the dialysis bag for analysis.
2.7. Western blot analysis
Western blot analysis was performed as described previously [36]. After boiling for 5 min at 95°C in a 4× loading buffer, purified urinary exosomes were subjected to SDS-PAGE, transferred onto a PVDF membrane and blocked by solution with dry milk. Then, the membrane was probed with anti-CD63 antibody (1:1000; Abcam, Cambridge, MA) overnight at 4 °C followed by incubation with horseradish peroxidase-labeled IgG (1:5000). The immunoreactive bands were detected by chemiluminescence methods and visualized on Kodak Omat X-ray films. Densitometric analysis of the images obtained from X-ray films was performed using the Image J software (NIH, Bethesda, MD, USA).
2.8. Transmission electron microscopy
For transmission electron microscopy (TEM) analysis of ultrastructural changes in podocytes, mouse kidneys were perfused with a fixative containing 3% glutaraldehyde and 4% paraformaldehyde in 0.1 mol/L phosphate buffer. After fixation and dehydration with ethanol, the samples were embedded in Durcupan resin for ultrathin sectioning by the Virginia Commonwealth University microscopy core facility [34]. One glomerulus was randomly chosen from each sample for analysis. The proportion of the podocyte slits, where the linear image of the slit diaphragm was detectable, was counted [37].
2.9. Glomerular morphological examination
Fixed kidney tissues were paraffin-embedded, sectioned, and stained with periodic acid–Schiff (PAS). Fifty glomeruli per slide were counted under a light microscope and scored as 0–4 (0: no lesion, 1: sclerosis <25%, 2: sclerosis of 25% to 50%, 3: sclerosis of 50% to 75%, 4: sclerosis >75%) by an observer who was blind to treatment groups. Glomerular sclerosis was expressed as glomerular damage index (GDI), which was calculated by the formula ((N1 ×1) + (N2 ×2) + (N3 ×3) + (N4 ×4))/n. N1, N2, N3, and N4 represent the numbers of glomerular damage grades 1, 2, 3, and 4, respectively, and n represents the total scored number of glomeruli [38].
2.10. Urinary protein and albumin measurements
Total urinary protein was determined spectrophotometrically by the Bradford assay (Sigma-Aldrich). Urinary albumin concentration was measured by a commercially available mouse albumin ELISA kit (Bethyl Laboratories, Montgomery, TX) [34].
2.11. Statistical analysis
SigmaPlot 14.0 was used for statistical analysis of data. All values are expressed as mean ± SEM. Significant differences among multiple groups were examined using ANOVA followed by a Student-Newman-Keuls test. P<0.05 was considered statistically significant.
3. Results
3.1. Characterization of podocyte-specific overexpression of Smpd1 gene in Smpd1trg/Podocre mice
To examine the pathological relevance of Asm-ceramide signaling pathway in podocytes for glomerular sclerosis during hHcy, a podocyte-specific Smpd1 gene overexpression (Smpd1trg/Podocre) mouse strain was generated. These mice and their littermates were characterized using several genetic, molecular, and biochemical approaches. As shown in Fig. 1A, PCR analysis of DNA from Smpd1trg/Podocre mice demonstrated the presence of all three tested targets: the Smpd1trg gene (360 bp), the internal control gene (324 bp), and the Cre recombinase gene (100 bp). In contrast, Smpd1trg/Podowt mice showed only the Smpd1trg gene and the internal control gene, whereas WT/WT mice tested positive for only the internal control gene. Confocal microscopy showed enhanced colocalization of podocin, a marker for podocytes, and Asm in glomeruli of Smpd1trg/Podocre mice, compared with WT/WT and Smpd1trg/Podowt mice (Fig. 1B). Correspondingly, the colocalization of podocin and Asm product, ceramide, was remarkably increased by podocyte-specific Smpd1 gene overexpression (Fig. 1C). To further confirm the tissue-specific Smpd1 gene overexpression in podocytes, Smpd1trg/Podocre mice were mated with ROSA mice to produce Smpd1trg/Podocre/ROSA mice. Although the green fluorescence emitted by enhanced green fluorescence protein (EGFP) was undetectable in either Smpd1trg/Podocre or ROSA mice, the colocalization of EGFP and podocin in glomeruli of the Smpd1trg/Podocre/ROSA mice was remarkable (Fig. 1D). This is explained by selective activation of EGFP in the cross-bred strain. Altogether, these results demonstrate podocyte-specific overexpression of Smpd1 gene in Smpd1trg/Podocre mice.
Figure 1.
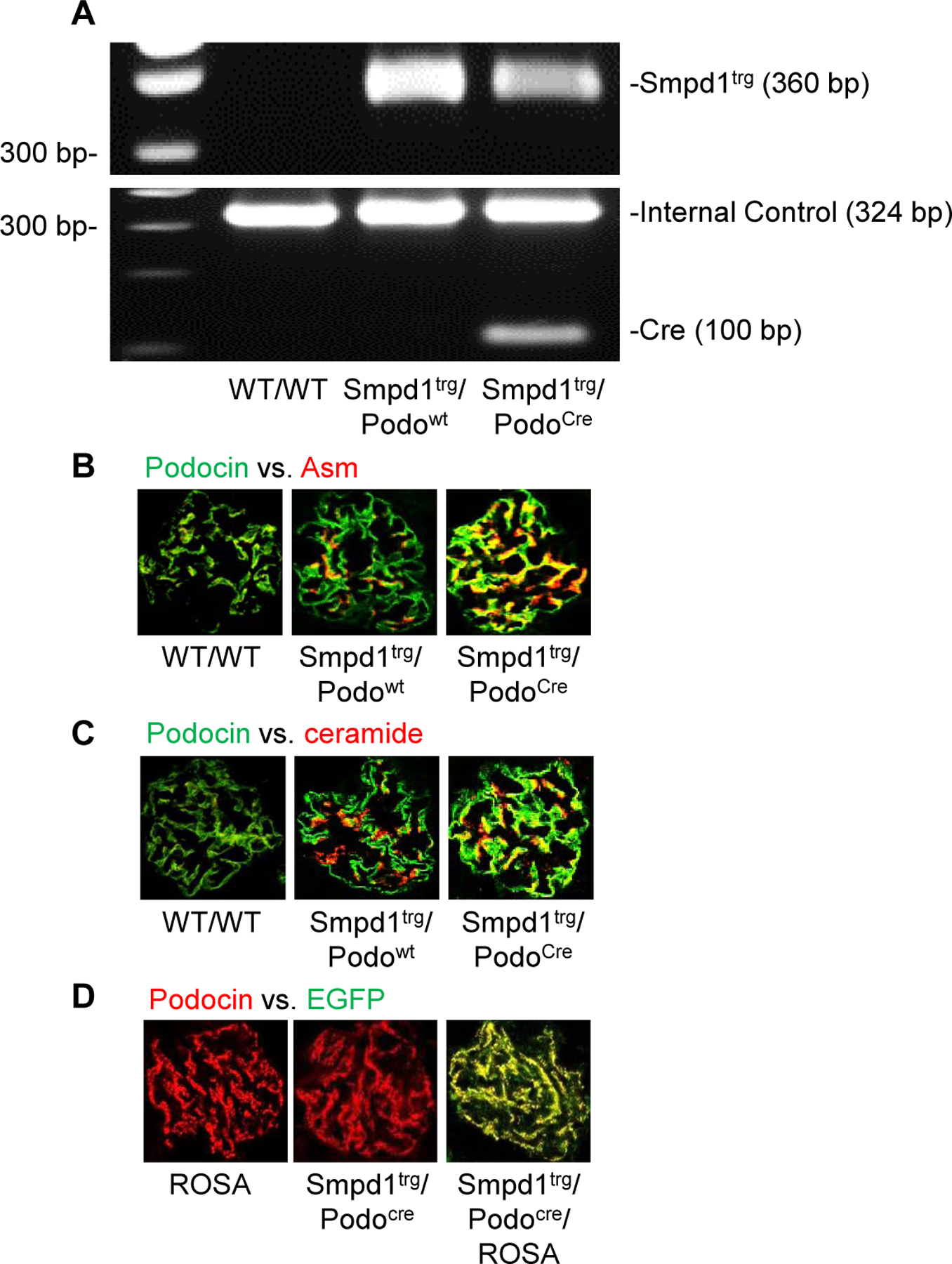
Characterization of Smpd1trg/Podocre mice. A. Representative gel showing detection of Smpd1 transgene and Cre recombinase gene by PCR Genotyping. B. Representative images showing the colocalization of podocin (green fluorescence) and Asm (red fluorescence) in glomeruli of different groups of mice (n=6). C. Representative images showing the colocalization of podocin (green fluorescence) and ceramide (red fluorescence) in glomeruli of different groups of mice (n=6). D. Representative images showing the colocalization of EGFP (green fluorescence) and podocin (red fluorescence) in glomeruli of different groups of mice (n=4).
3.2. Smpd1 gene deletion abolished hHcy-induced inflammasome activation and consequent inflammatory response in glomeruli
To determine whether Asm activity in podocytes plays an important role in NLRP3 inflammasome activation and consequent inflammatory response in glomeruli of mice during hHcy, we fed WT/WT, Smpd1−/−, and Smpd1trg/Podocre mice with normal diet (ND) or folate free (FF) diet for 8 weeks. As shown in Fig. 2A, the glomeruli of WT/WT mice on the FF diet had remarkably elevated colocalization of NLRP3 with ASC compared with ND-fed WT/WT mice. The formation of NLRP3 inflammasome in glomeruli was prevented by Smpd1 gene deletion. On the contrary, podocyte-specific Smpd1 gene overexpression significantly enhanced the assembly of NLRP3 inflammasome in glomeruli of Smpd1trg/Podocre mice compared with WT/WT mice on both diets. Consistently, IL-1β production was significantly increased in glomeruli of WT/WT mice on the FF diet compared with the ND, but this change was blocked by Smpd1 gene knockout in Smpd1−/− mice. However, podocyte-specific Smpd1 gene overexpression markedly aggravated glomerular IL-1β production in Smpd1trg/Podocre mice compared with WT/WT mice on both diets. Immunofluorescent staining of Ly6G, a neutrophil marker, revealed more infiltration of neutrophil in glomeruli of WT/WT mice on FF diet compared with WT/WT mice on ND. The infiltration of neutrophil in glomeruli was significantly enhanced by podocyte-specific Smdp1 gene overexpression in Smpd1trg/Podocre mice on either diet. Nevertheless, elevation of Ly6G staining in glomeruli was not observed in Smpd1−/− mice during hHcy (Fig. 2C). These findings confirm that Asm activity in podocytes contributes to NLRP3 inflammasome formation and activation, leading to infiltration of neutrophil in glomeruli of mice during hHcy.
Figure 2.
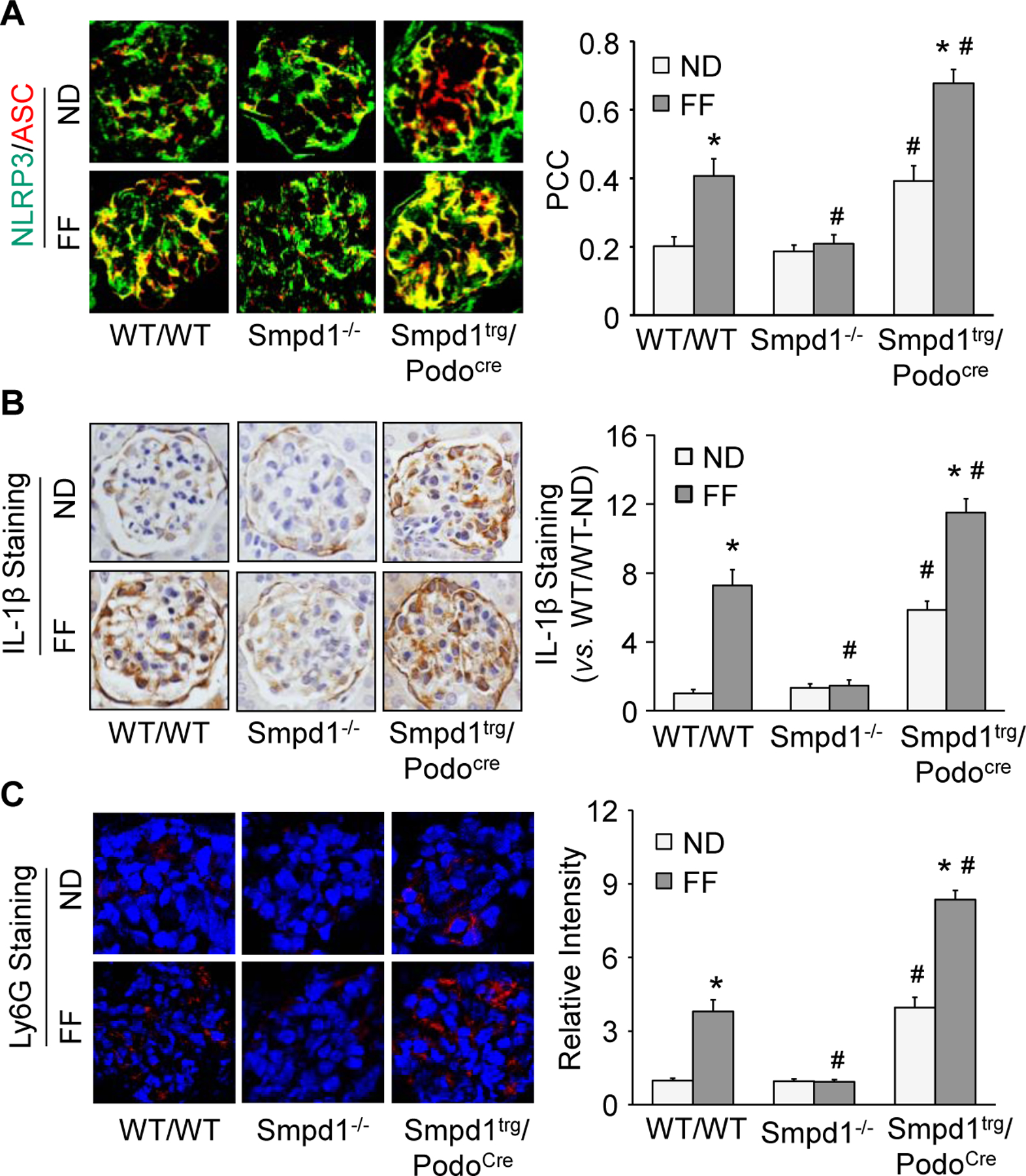
Podocyte-specific Smpd1 gene overexpression enhanced glomerular NLRP3 inflammasome activation and inflammation in mice during hHcy. A. Representative images and summarized data showing the colocalization of NLRP3 (green fluorescence) and ASC (red fluorescence) in glomeruli of different groups of mice (n=3–8). B. Representative images and summarized data showing the immunohistochemical staining of IL-1β in glomeruli of different groups of mice (n=5–6). C. Representative images and summarized data showing the immunofluorescent staining of Ly6G in glomeruli of different groups of mice (n=4). * P<0.05 vs. ND group. # P<0.05 vs. WT/WT group.
3.3. Decreased glomerular lysosome-MVB interaction during hHcy
To explore the mechanism by which inflammasome products are released out of podocytes to induce inflammatory response in glomeruli during hHcy, we tested whether exosomes mediate the secretion of inflammatory cytokines produced by activated NLRP3 inflammasomes in podocytes. As shown in Fig. 3A, VPS16, an MVB marker, and Lamp-1, a lysosome marker, showed colocalization in glomeruli of WT/WT mice, indicating an interaction of MVBs and lysosomes under normal conditions. Interestingly, induction of hHcy by the FF diet resulted in a decrease in this interaction compared to WT/WT mice fed the ND. The decrease in lysosome-MVB interaction in glomeruli during hHcy was prevented by Smpd1 gene knockout in Smpd1−/− mice. Podocyte-specific Smpd1 gene overexpression, however, significantly reduced the colocalization of VPS16 and Lamp-1 in Smpd1trg/PodoCre mice on both diets. Using nanoparticle tracking analysis (NTA), we found that hHcy largely increased urinary exosomes in WT/WT mice, but not in Smpd1−/− mice. Compared with WT/WT mice, podocyte-specific Smpd1 gene overexpression significantly enhanced urinary exosome excretion in Smpd1trg/Podocre mice on both diets (Fig. 3B). To confirm the origin of exosomes detected in mouse urine by NTA, we detected CD63, an exosome marker, and podocin, a podocyte marker, in urine of mice. Western blot analysis was performed to detect the levels of CD63 in purified urinary exosomes. Mouse podocin ELISA kit (Biomatik, Cambridge, Canada) was used to detect the levels of podocin in purified urinary exosomes. It was found that WT/WT mice fed with FF diet had remarkably higher levels of CD63 and podocin in their urine compared with WT/WT mice on ND, which was blocked by Smpd1 gene knockout in Smpd1−/− mice. However, Smpd1 gene overexpression in podocytes significantly enhanced the elevation of CD63 and podocin in urine of Smpd1trg/Podocre mice on both diets compared with WT/WT mice (Fig. 3C and 3D). These results demonstrate that exosome release from podocytes is enhanced during hHcy, which is attributed to Asm activity in these cells.
Figure 3.
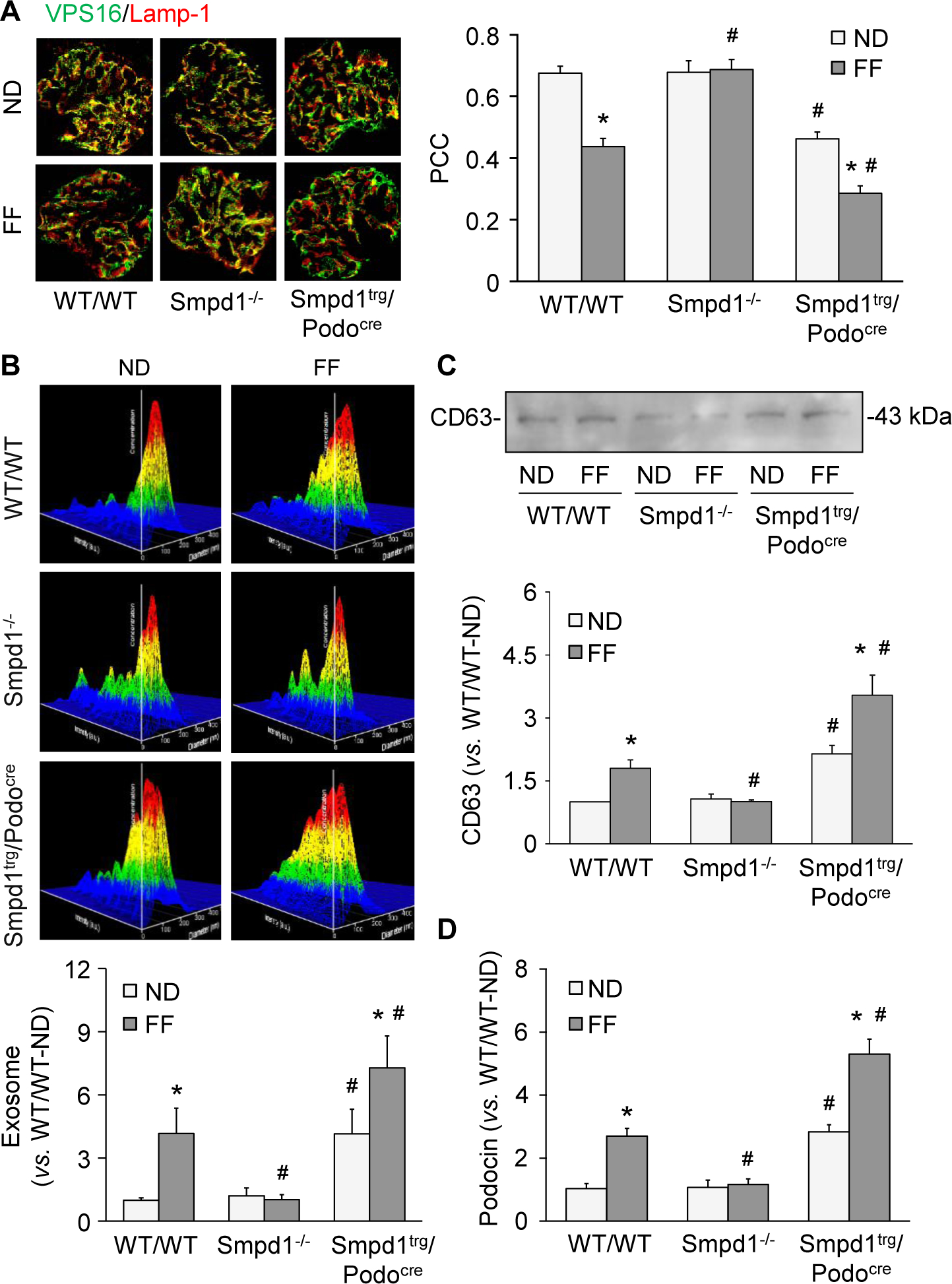
Decreased glomerular lysosome-MVB interaction during hHcy. A. Representative images and summarized data showing the colocalization of VPS16 (green fluorescence) and Lamp-1 (red fluorescence) in glomeruli of different groups of mice (n=5–8). B. Representative images and summarized data showing the urinary exosome excretion in different groups of mice (n=7–13). C. Representative Western blot images and summarized data showing the urinary levels of CD63 in different groups of mice (n=5–6). D. Urinary levels of podocin in different groups of mice (n=6). * P<0.05 vs. ND group. # P<0.05 vs. WT/WT group.
3.4. Enhanced release of inflammatory exosomes during hHcy
Using confocal microscopy, we next examined colocalization of IL-1β and a marker for MVBs, VPS16, to assess the potential for NLRP3 inflammasome products to be secreted from podocytes in the form of exosomes during hHcy. In the WT/WT mice fed the ND diet, there was evidence of basal colocalization of IL-1β and VPS16, which was significantly enhanced in the mice with hHcy (Fig. 4A). Compared with WT/WT mice, the Smpd1trg/Podocre mice exhibited higher degrees of colocalization of IL-1β and VPS16 in glomeruli after both the ND and FF diet. However, the effect of FF diet on the colocalization of IL-1β and VPS16 was blocked by Smpd1 gene knockout in Smpd1−/− mice. Using an alternative approach, the NLRP3 inflammasome-derived cytokines, IL-1β and IL-18, were measured in exosomes isolated from urines of different groups of mice (Figs. 4B and 4C for IL-1β and IL-18, respectively). Consistent with the colocalization results in Fig. 4A, both IL-1β and IL-18 were increased significantly by the FF diet in both WT/WT and Smpd1trg/Podocre mice and overall, the levels of both cytokines in urinary exosomes were enhanced significantly regardless of the diet in the Smpd1trg/Podocre mice compared to their respective WT/WT groups. In contrast, this enhancing effect of hHcy on the levels of IL-1β and IL-18 in the urinary exosomes was absent in Smpd1−/− mice.
Figure 4.
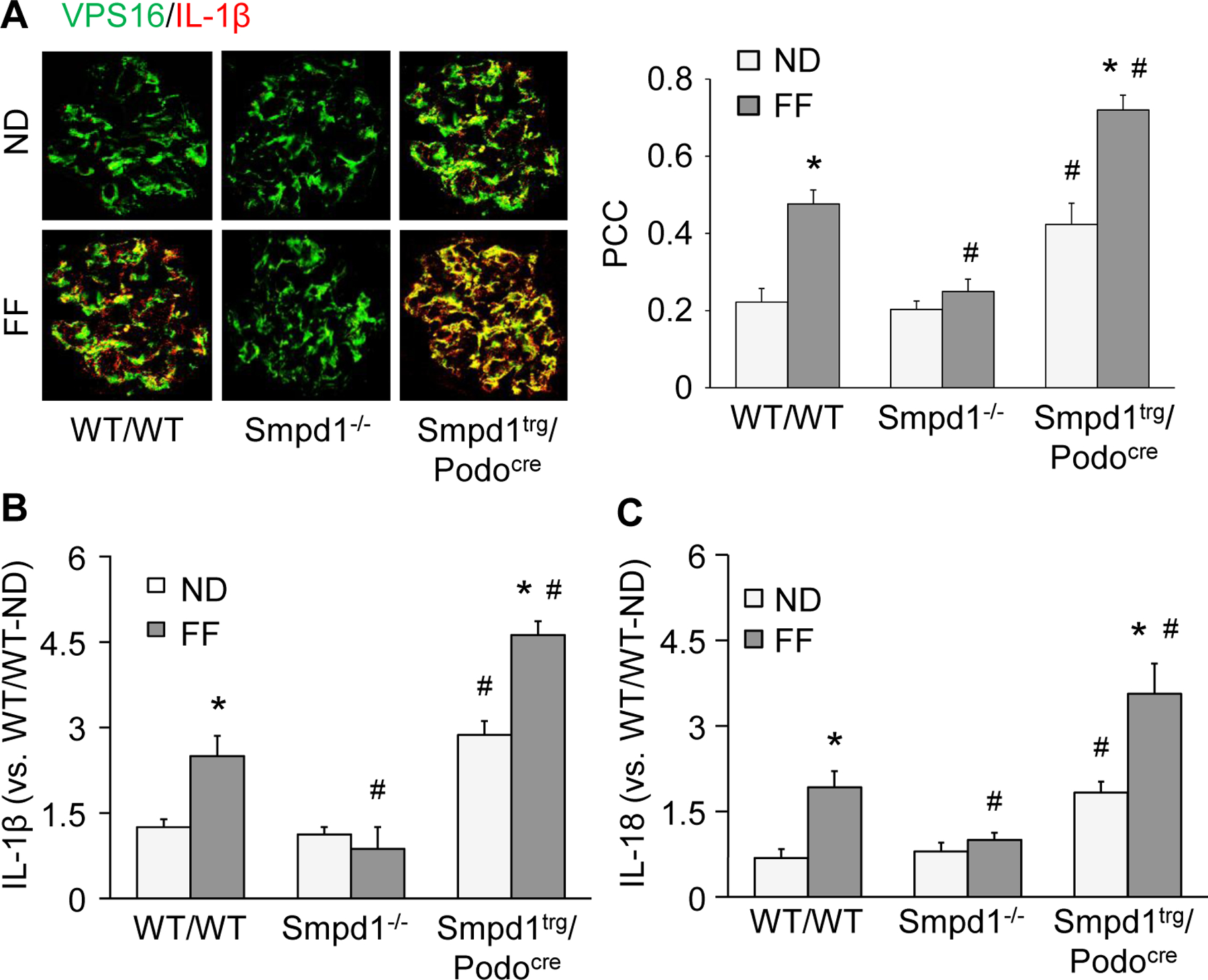
Enhanced release of inflammatory exosomes during hHcy. A. Representative images and summarized data showing the colocalization of VPS16 (green fluorescence) and IL-1β (red fluorescence) in glomeruli of different groups of mice (n=8). B. Urinary levels of IL-1β in different groups of mice (n=4). C. Urinary levels of IL-18 in different groups of mice (n=5–8). * P<0.05 vs. ND group. # P<0.05 vs. WT/WT group.
3.5. Role of Asm in podocyte injury and foot process effacement during hHcy
As shown in Fig. 5A and 5B, the WT/WT mice with hHcy showed remarkable decrease in podocin and obvious increase in desmin compared with the WT/WT mice fed with ND. These responses were also evident in the Smpd1trg/Podocre mice. The overexpression of Smpd1 in podocytes decreased podocin expression and increased desmin expression even in Smpd1trg/Podocre mice on the ND diet and these effects were further enhanced by the FF diet. However, hHcy-induced changes in podocin and desmin were not observed in Smpd1−/− mice. TEM analysis was performed to investigate the ultrastructural changes in podocytes in different groups of mice. Whereas podocytes of WT/WT mice on the ND showed intact foot processes of podocytes, marked foot process effacement was observed in the FF diet-treated WT/WT mice. Podocytes of Smpd1−/− mice on the FF diet had relatively normal ultrastructure. Foot process effacement of podocytes was observed in the Smpd1trg/Podocre mice on ND. Podocyte-specific Smpd1 gene overexpression amplified podocyte foot process effacement induced by hHcy (Fig. 5C).
Figure 5.
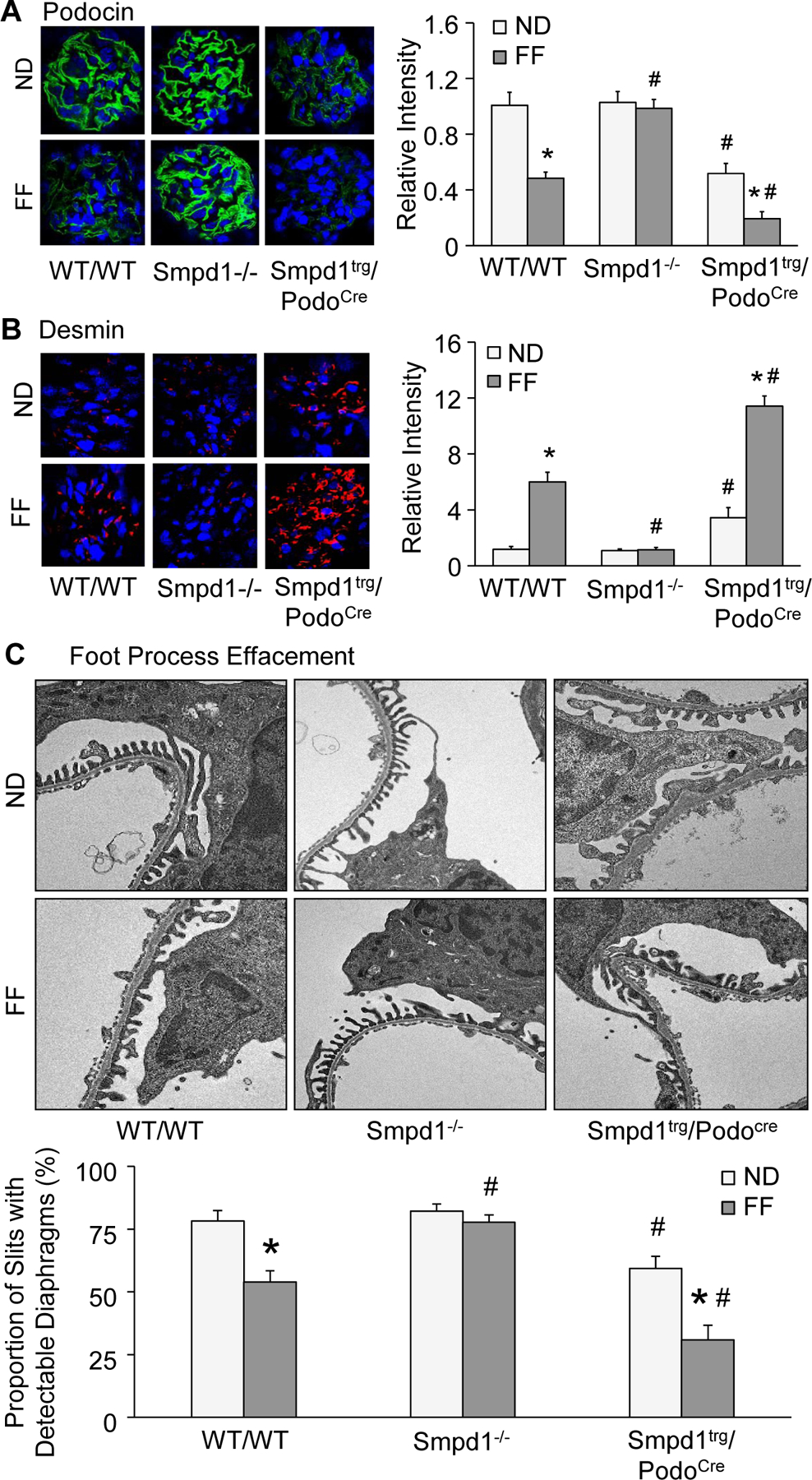
Podocyte-specific Smpd1 gene overexpression enhanced podocyte injury and glomerular sclerosis in mice during hHcy. A. Representative images and summarized data showing the immunofluorescent staining of podocin in glomeruli of different groups of mice (n=6). B. Representative images and summarized data showing the immunofluorescent staining of desmin in glomeruli of different groups of mice (n=6). C. Representative electron microscopic images and summarized data showing foot processes in podocytes of different groups of mice (n=4). * P<0.05 vs. ND group. # P<0.05 vs. WT/WT group.
3.6. Smpd1 gene knockout prevented proteinuria and glomerular sclerosis during hHcy
As shown in Fig. 6A, WT/WT mice on FF diet exhibited severe proteinuria compared to WT/WT mice on ND. Proteinuria in response to the FF diet was prevented by Smpd1 gene knockout in Smpd1−/− mice. Podocyte-specific Smpd1 gene overexpression, however, significantly aggravated proteinuria in mice, both after the ND and FF diets. By PAS staining, morphological examinations showed sclerotic changes in glomeruli of WT/WT mice on the FF diet (Fig. 6B). Correspondingly, the glomerular damage index increased significantly in mice fed the FF diet. Smpd1 gene knockout totally blocked the glomerular damage induced by the FF diet. On the contrary, overexpression of Smpd1 gene in podocytes significantly enhanced glomerular damage in mice on both diets.
Figure 6.
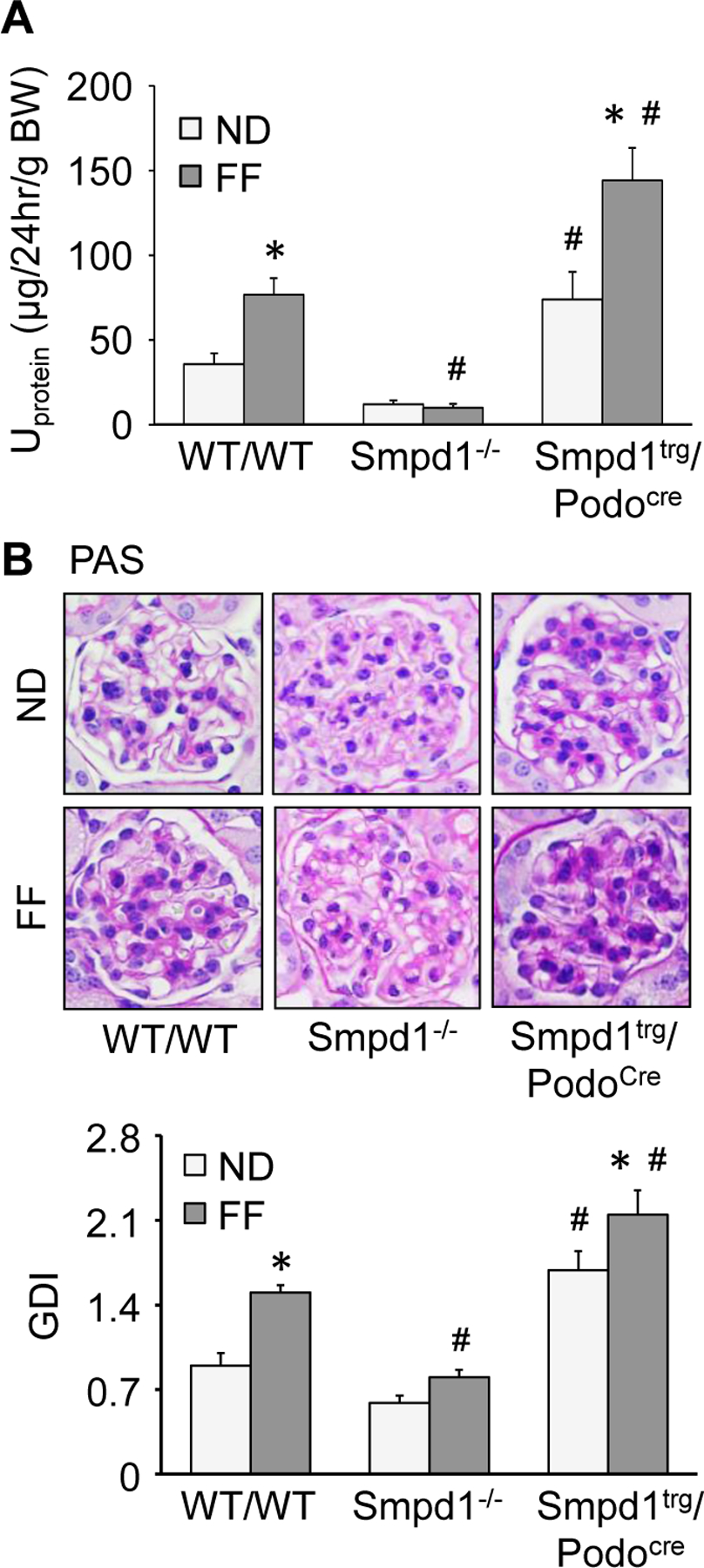
Smpd1 gene knockout prevented proteinuria and glomerular sclerosis during hHcy. A. Urinary protein excretion of different groups of mice (n=8–13). B. Representative images and summarized data showing the glomerular morphological changes (periodic acid-Schiff staining) of different groups of mice (n=6). * P<0.05 vs. ND group. # P<0.05 vs. WT/WT group.
4. Discussion
The major goal of the present study was to determine whether Asm-ceramide signaling pathway contributes to NLRP3 inflammasome activation and inflammatory exosome release in podocytes and thereby lead to podocyte injury and subsequent glomerular sclerosis during hHcy. Our results demonstrated that hHcy induced inflammatory exosome release from podocytes, which was attributed to NLRP3 inflammasome activation and inhibited lysosome-MVB interaction in these cells. The inflammatory exosome release was accompanied by neutrophil infiltration in glomeruli. Also, podocyte damage, proteinuria, and glomerular sclerosis were found in mice with hHcy. These pathological changes were enhanced by podocyte-specific Smpd1 gene overexpression but blocked by Smpd1 gene knockout. Our findings indicate that increased exosome release due to reduced lysosome-MVB interaction may be a mechanism mediating the secretion of NLRP3 inflammasome products in podocytes, leading to glomerular inflammation and sclerosis during hHcy. Asm-ceramide signaling pathway may control NLRP3 inflammasome activation and lysosome-MVB interaction and thereby determine the release of inflammatory exosomes in podocytes during hHcy.
It has been shown that chronic injury in glomeruli during hHcy is attributed to the activation of NLRP3 inflammasome, which is composed of three major proteins, including a NOD-like receptor NLRP3, an adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and caspase-1 [1]. NLRP3 acts as the sensor to recognize both endogenous and exogenous danger signals [39–41], which leads to recruitment and aggregation of ASC and caspase-1 to form a protein complex, where caspase-1 is activated. The active caspase-1 not only proteolytically cleaves IL-1β and IL-18 into their biologically active form, but also produces other danger molecules like damage-associated molecular patterns (DAMPs). Both will act to turn on inflammatory response and induce cell dysfunction or death via pyroptosis [42–45]. NLRP3 inflammasome activation has been shown to be a crucial mechanism mediating hHcy-induced glomerular injury and ultimate ESRD, which are associated with podocyte injury, glomerular inflammation, and ultimate sclerosis [46–48]. As mentioned above, however, NLRP3 inflammasomes are activated mainly in the cytosol and their products may not be secreted out of podocytes via a classical and Golgi apparatus-mediated delivery pathway, it is imperative to study how NLRP3 inflammasome activation-derived products such as IL-1β, IL-18, and high mobility group protein B1 (HMGB1) are released out of podocytes to trigger the inflammatory response and ultimate glomerular injury. Given the important role of Asm and its product ceramide in glomerular injury induced by hHcy [28–30] and in lysosome trafficking and fusion with other vesicles [8, 22–27], the present study tested whether lysosomal Asm-ceramide contributes to NLRP3 inflammasome activation and robust release of inflammatory exosomes in podocytes during hHcy, which trigger glomerular inflammation and sclerosis.
By various approaches, enhancement of NLRP3 inflammasome activation and subsequent neutrophil infiltration was found in glomeruli of WT/WT mice fed with FF diet. The enhanced NLRP3 inflammasome activation and inflammatory response were even seen in glomeruli of Smpd1trg/Podocre mice on ND and exaggerated in these mice on FF diet. However, in Smpd1−/− mice these pathological changes were blocked. These results clearly suggest that Asm-ceramide signaling pathway contributes to hHcy-induced NLRP3 inflammasome activation in podocytes and glomerular inflammation. In this regard, there are some reports that Asm and its product ceramide are involved in NLRP3 inflammasome activation and glomerular injury during hHcy [28–30]. The role of ceramide in glomerular injury induced by hHcy was found to be associated with the production of O2.− and activation of NLRP3 inflammasomes in podocytes and consequent glomerular inflammation [30, 49, 50]. However, it remains unclear which types of glomerular cells determine the pathological role of Asm-ceramide signaling pathway in glomerular injury induced by hHcy. To our knowledge, the results from the present study provide the first experimental evidence that hHcy-induced NLRP3 inflammasome activation in glomeruli is attributed to Asm-ceramide signaling pathway in podocytes, which may be an important pathogenic mechanism responsible for glomerular injury during hHcy.
Another interesting finding of the present study is that hHcy inhibited lysosome-MVB interaction in glomeruli and enhanced urinary exosome excretion in mice. The MVBs in glomeruli and exosomes in urine were found to contain increased inflammatory cytokines in mice fed with FF diet. These pathological changes were blocked in Smpd1−/− mice but exaggerated in Smpd1trg/Podocre mice. These results imply that increased release of exosomes may be one of the mechanisms mediating the secretion of inflammatory cytokines as the products of NLRP3 inflammasome in podocytes during hHcy. In addition to enhancement of NLRP3 inflammasome activation, Asm-ceramide signaling pathway contributes to hHcy-induced glomerular inflammation and sclerosis through regulation of lysosome-MVB interaction and inflammatory exosome release in podocytes. Consistent with our findings, previous studies have demonstrated that ceramide and its metabolites, Sph-1-phosphate (S1P) participate in exosome biogenesis, sorting ILVs into MVBs, budding of exosome or MVB fusion to membrane for release of exosomes [19–21]. In particular, ceramide and associated sphingolipids as a key regulator of lysosome trafficking and fusion with other vesicles are shown in different cell types [8, 22–27]. Although a recent study has shown that exosome may mediate the secretion of inflammatory cytokines in glomeruli during D-ribose stimulation [51], the results from the present study provide the first experimental evidence that hHcy damages lysosome-MVB interaction in glomeruli and amplifies urinary excretion of exosomes containing inflammatory cytokines, which are regulated by Asm-ceramide signaling pathway in podocytes. In previous studies, it has been shown that translocation of lysosomal Asm to plasma membrane and consequent lipid raft clustering contribute to NADPH oxidase aggregation and activation, leading to elevated reactive oxygen species (ROS) production in response to different pathological stimuli, such as Hcy, visfatin, and Fas ligand [52–55]. Also, we have reported that NADPH oxidase-derived ROS triggers NLRP3 inflammasome activation in podocytes during hHcy [47]. It is possible that Hcy-induced Asm activation enhances ROS production by NADPH oxidase and thereby inhibits lysosome-MVB interaction, leading to elevated inflammatory exosome secretion form podocytes. Our further experiments confirmed that inflammatory exosome release was associated with podocyte injury, glomerular sclerosis, and proteinuria during hHcy. It was found that podocyte-specific Smpd1 gene overexpression enhanced these pathological changes. On the contrary, Smpd1 gene deletion protected podocytes and glomeruli from hHcys-induced injury. These findings provide evidence that targeting the inflammatory exosome release may be a novel strategy to prevent the development of podocyte injury and glomerular sclerosis during hHcy. To further confirm the involvement of Asm-ceramide signaling pathway in hHcy-induced glomerular inflammation and sclerosis, we treated four-week-old male WT/WT, Smpd1−/−, and Smpd1trg/Podocre mice with ND or FF diet for 2 weeks. Then, mouse kidneys were harvested for sectioning. Since uninephrectomy was not performed on these mice and the treatment of FF diet was short, podocyte injury was not detected in these mice. Our results showed that expression of Asm in podocytes was significantly elevated by hHcy (Supplementary Fig. 1). Moreover, glomerular ceramide accumulation was found in mice with hHcy (Supplementary Fig. 2). These results suggest that hHcy-induced upregulation of Asm expression in podocytes leads to elevation of glomerular ceramide in mice. Glomerular ceramide accumulation may contribute to NLRP3 inflammasome activation and robust release of inflammatory exosomes in podocytes during hHcy, which together trigger local glomerular inflammation and sclerosis.
Previous studies have shown that active caspase-1 cleaves GSDMD to release GSDMD-N, leading to the formation of GSDMD pores on the plasma membrane. The GSDMD pores mediate the release of inflammatory cytokines [56]. A recent study has demonstrated that caspase-11/4 and GSDMD-mediated pyroptosis is activated and involved in podocyte loss under hyperglycemia condition and the development of diabetic nephropathy [57]. However, it remains unknown whether hHcy induces the formation of GSDMD pores on the plasma membrane of podocytes. Moreover, inhibition of microRNA-15a has been found to increase the levels of Asm and GSDMD in endothelial cells [58]. Nevertheless, these is no evidence of any interaction between Asm and GSDMD. In our future study, it would be interesting to test whether Asm-ceramide signaling pathway affects GSDMD and thereby determines the secretion of inflammatory cytokines from podocytes during hHcy. Another remaining question is whether ASM-ceramide signaling pathway altered exosome release through regulation of NLRP3 inflammasome activation. In this regard, canonical activators of NLRP3 inflammasome, such as ATP, monosodium urate, β-glucan, and nigericin, have been found to induce secretion of EVs [59]. It has been demonstrated that calcium influx can induce both NLRP3 inflammasome and EV secretion [59]. As a well-established inducer of NLRP3 inflammasome, cathepsin has been shown to be involved in the secretion of EVs [60]. It has been reported that NLRP3 and ASC are required for exosome release in response to stimulation of P2X7 receptor, although the results are arguable [61]. Our findings in this study may not determine whether exosome release was affected by NLRP3 inflammasome in podocytes during hHcy. In our future study, it would be interesting to test whether the inhibition of NLRP3 inflammasome activation can block the enhancement of exosome release from podocytes during hHcy.
In summary, the present study demonstrated that hHcy stimulated release of inflammatory exosomes containing the products of NLRP3 inflammasome from podocytes, which initiated glomerular inflammation, leading to more serious glomerular injury and ultimate sclerosis. Asm-ceramide signaling pathway contributed to glomerular pathology induced by hHcy through enhancement of NLRP3 inflammasome activation and inhibition of lysosome-MVB interaction. These results may establish a new concept that exosomes mediate the secretion of NLRP3 inflammasome products and thereby recruit immune cells in glomeruli, leading to podocyte injury and glomerular sclerosis during hHcy.
Supplementary Material
Highlights.
Exosomes mediate the secretion of NLRP3 inflammasome products from podocyte.
Smpd1 gene overexpression enhances inflammatory exosome release from podocytes.
Smpd1 gene knockout inhibits inflammatory exosome release from podocytes.
Asm contributes to exosome release via inhibition of lysosome-MVB interaction.
Acknowledgement
This study was supported by grants DK054927 and DK120491 from National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest statement
None of the authors have conflict of interest.
References
- 1.Martinon F; Mayor A; Tschopp J, The inflammasomes: guardians of the body. Annu Rev Immunol 2009, 27, 229–65. [DOI] [PubMed] [Google Scholar]
- 2.Colombo M; Raposo G; Thery C, Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 2014, 30, 255–89. [DOI] [PubMed] [Google Scholar]
- 3.Schorey JS; Harding CV, Extracellular vesicles and infectious diseases: new complexity to an old story. J Clin Invest 2016, 126, (4), 1181–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eitan E; Suire C; Zhang S; Mattson MP, Impact of lysosome status on extracellular vesicle content and release. Ageing research reviews 2016, 32, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Balkom BW; Pisitkun T; Verhaar MC; Knepper MA, Exosomes and the kidney: prospects for diagnosis and therapy of renal diseases. Kidney Int 2011, 80, (11), 1138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erdbrugger U; Le TH, Extracellular Vesicles in Renal Diseases: More than Novel Biomarkers? J Am Soc Nephrol 2016, 27, (1), 12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hara M; Yanagihara T; Kihara I; Higashi K; Fujimoto K; Kajita T, Apical cell membranes are shed into urine from injured podocytes: a novel phenomenon of podocyte injury. Journal of the American Society of Nephrology : JASN 2005, 16, (2), 408–16. [DOI] [PubMed] [Google Scholar]
- 8.Lee H; Han KH; Lee SE; Kim SH; Kang HG; Cheong HI, Urinary exosomal WT1 in childhood nephrotic syndrome. Pediatr Nephrol 2012, 27, (2), 317–20. [DOI] [PubMed] [Google Scholar]
- 9.Lytvyn Y; Xiao F; Kennedy CR; Perkins BA; Reich HN; Scholey JW; Cherney DZ; Burger D, Assessment of urinary microparticles in normotensive patients with type 1 diabetes. Diabetologia 2017, 60, (3), 581–584. [DOI] [PubMed] [Google Scholar]
- 10.Stahl AL; Johansson K; Mossberg M; Kahn R; Karpman D, Exosomes and microvesicles in normal physiology, pathophysiology, and renal diseases. Pediatric nephrology 2019, 34, (1), 11–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tkaczyk M; Baj Z, Surface markers of platelet function in idiopathic nephrotic syndrome in children. Pediatric nephrology 2002, 17, (8), 673–7. [DOI] [PubMed] [Google Scholar]
- 12.Rubartelli A; Cozzolino F; Talio M; Sitia R, A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J 1990, 9, (5), 1503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boulanger CM; Loyer X; Rautou PE; Amabile N, Extracellular vesicles in coronary artery disease. Nat Rev Cardiol 2017, 14, (5), 259–272. [DOI] [PubMed] [Google Scholar]
- 14.Chistiakov DA; Orekhov AN; Bobryshev YV, Extracellular vesicles and atherosclerotic disease. Cellular and molecular life sciences : CMLS 2015, 72, (14), 2697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hessvik NP; Llorente A, Current knowledge on exosome biogenesis and release. Cell Mol Life Sci 2018, 75, (2), 193–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baixauli F; Lopez-Otin C; Mittelbrunn M, Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Frontiers in immunology 2014, 5, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fader CM; Sanchez D; Furlan M; Colombo MI, Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 2008, 9, (2), 230–50. [DOI] [PubMed] [Google Scholar]
- 18.Murrow L; Debnath J, ATG12-ATG3 connects basal autophagy and late endosome function. Autophagy 2015, 11, (6), 961–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kajimoto T; Okada T; Miya S; Zhang L; Nakamura S, Ongoing activation of sphingosine 1-phosphate receptors mediates maturation of exosomal multivesicular endosomes. Nature communications 2013, 4, 2712. [DOI] [PubMed] [Google Scholar]
- 20.Trajkovic K; Hsu C; Chiantia S; Rajendran L; Wenzel D; Wieland F; Schwille P; Brugger B; Simons M, Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, (5867), 1244–7. [DOI] [PubMed] [Google Scholar]
- 21.Yuyama K; Sun H; Mitsutake S; Igarashi Y, Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. The Journal of biological chemistry 2012, 287, (14), 10977–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alvarez-Erviti L; Seow Y; Schapira AH; Gardiner C; Sargent IL; Wood MJ; Cooper JM, Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiology of disease 2011, 42, (3), 360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui Y; Luan J; Li H; Zhou X; Han J, Exosomes derived from mineralizing osteoblasts promote ST2 cell osteogenic differentiation by alteration of microRNA expression. FEBS Lett 2016, 590, (1), 185–92. [DOI] [PubMed] [Google Scholar]
- 24.Li CM; Hong SB; Kopal G; He X; Linke T; Hou WS; Koch J; Gatt S; Sandhoff K; Schuchman EH, Cloning and characterization of the full-length cDNA and genomic sequences encoding murine acid ceramidase. Genomics 1998, 50, (2), 267–74. [DOI] [PubMed] [Google Scholar]
- 25.Li PL; Zhang Y; Abais JM; Ritter JK; Zhang F, Cyclic ADP-Ribose and NAADP in Vascular Regulation and Diseases. Messenger 2013, 2, (2), 63–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liebau MC; Braun F; Hopker K; Weitbrecht C; Bartels V; Muller RU; Brodesser S; Saleem MA; Benzing T; Schermer B; Cybulla M; Kurschat CE, Dysregulated autophagy contributes to podocyte damage in Fabry’s disease. PLoS One 2013, 8, (5), e63506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorber D, Importance of cardiovascular disease risk management in patients with type 2 diabetes mellitus. Diabetes Metab Syndr Obes 2014, 7, 169–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yi F; Chen QZ; Jin S; Li PL, Mechanism of homocysteine-induced Rac1/NADPH oxidase activation in mesangial cells: role of guanine nucleotide exchange factor Vav2. Cell Physiol Biochem 2007, 20, (6), 909–18. [DOI] [PubMed] [Google Scholar]
- 29.Yi F; Jin S; Zhang F; Xia M; Bao JX; Hu J; Poklis JL; Li PL, Formation of lipid raft redox signalling platforms in glomerular endothelial cells: an early event of homocysteine-induced glomerular injury. J Cell Mol Med 2009, 13, (9B), 3303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yi F; Zhang AY; Janscha JL; Li PL; Zou AP, Homocysteine activates NADH/NADPH oxidase through ceramide-stimulated Rac GTPase activity in rat mesangial cells. Kidney Int 2004, 66, (5), 1977–87. [DOI] [PubMed] [Google Scholar]
- 31.Gupta S; Natarajan R; Payne SG; Studer EJ; Spiegel S; Dent P; Hylemon PB, Deoxycholic acid activates the c-Jun N-terminal kinase pathway via FAS receptor activation in primary hepatocytes. Role of acidic sphingomyelinase-mediated ceramide generation in FAS receptor activation. The Journal of biological chemistry 2004, 279, (7), 5821–8. [DOI] [PubMed] [Google Scholar]
- 32.Li G; Chen Z; Bhat OM; Zhang Q; Abais-Battad JM; Conley SM; Ritter JK; Li PL, NLRP3 inflammasome as a novel target for docosahexaenoic acid metabolites to abrogate glomerular injury. Journal of lipid research 2017, 58, (6), 1080–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horinouchi K; Erlich S; Perl DP; Ferlinz K; Bisgaier CL; Sandhoff K; Desnick RJ; Stewart CL; Schuchman EH, Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nature genetics 1995, 10, (3), 288–93. [DOI] [PubMed] [Google Scholar]
- 34.Li G; Kidd J; Kaspar C; Dempsey S; Bhat OM; Camus S; Ritter JK; Gehr TWB; Gulbins E; Li PL, Podocytopathy and Nephrotic Syndrome in Mice with Podocyte-Specific Deletion of the Asah1 Gene: Role of Ceramide Accumulation in Glomeruli. The American journal of pathology 2020, 190, (6), 1211–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li G; Huang D; Hong J; Bhat OM; Yuan X; Li PL, Control of lysosomal TRPML1 channel activity and exosome release by acid ceramidase in mouse podocytes. American journal of physiology. Cell physiology 2019, 317, (3), C481–C491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li G; Li CX; Xia M; Ritter JK; Gehr TW; Boini K; Li PL, Enhanced epithelial-to-mesenchymal transition associated with lysosome dysfunction in podocytes: role of p62/Sequestosome 1 as a signaling hub. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 2015, 35, (5), 1773–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patrakka J; Lahdenkari AT; Koskimies O; Holmberg C; Wartiovaara J; Jalanko H, The number of podocyte slit diaphragms is decreased in minimal change nephrotic syndrome. Pediatric research 2002, 52, (3), 349–55. [DOI] [PubMed] [Google Scholar]
- 38.Abais JM; Xia M; Li G; Gehr TW; Boini KM; Li PL, Contribution of endogenously produced reactive oxygen species to the activation of podocyte NLRP3 inflammasomes in hyperhomocysteinemia. Free radical biology & medicine 2014, 67, 211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cruz CM; Rinna A; Forman HJ; Ventura AL; Persechini PM; Ojcius DM, ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem 2007, 282, (5), 2871–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halle A; Hornung V; Petzold GC; Stewart CR; Monks BG; Reinheckel T; Fitzgerald KA; Latz E; Moore KJ; Golenbock DT, The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 2008, 9, (8), 857–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nour AM; Yeung YG; Santambrogio L; Boyden ED; Stanley ER; Brojatsch J, Anthrax lethal toxin triggers the formation of a membrane-associated inflammasome complex in murine macrophages. Infect Immun 2009, 77, (3), 1262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen GY; Nunez G, Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010, 10, (12), 826–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamkanfi M, Emerging inflammasome effector mechanisms. Nat Rev Immunol 2011, 11, (3), 213–20. [DOI] [PubMed] [Google Scholar]
- 44.Martinon F; Burns K; Tschopp J, The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002, 10, (2), 417–26. [DOI] [PubMed] [Google Scholar]
- 45.Srinivasula SM; Poyet JL; Razmara M; Datta P; Zhang Z; Alnemri ES, The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem 2002, 277, (24), 21119–22. [DOI] [PubMed] [Google Scholar]
- 46.Zhang C; Boini KM; Xia M; Abais JM; Li X; Liu Q; Li PL, Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension 2012, 60, (1), 154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abais JM; Zhang C; Xia M; Liu Q; Gehr TW; Boini KM; Li PL, NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxidants & redox signaling 2013, 18, (13), 1537–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abais JM; Xia M; Li G; Chen Y; Conley SM; Gehr TW; Boini KM; Li PL, Nod-like receptor protein 3 (NLRP3) inflammasome activation and podocyte injury via thioredoxin-interacting protein (TXNIP) during hyperhomocysteinemia. The Journal of biological chemistry 2014, 289, (39), 27159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conley SM; Abais-Battad JM; Yuan X; Zhang Q; Boini KM; Li PL, Contribution of guanine nucleotide exchange factor Vav2 to NLRP3 inflammasome activation in mouse podocytes during hyperhomocysteinemia. Free Radic Biol Med 2017, 106, 236–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Conley SM; Abais JM; Boini KM; Li PL, Inflammasome Activation in Chronic Glomerular Diseases. Curr Drug Targets 2017, 18, (9), 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong J; Bhat OM; Li G; Dempsey SK; Zhang Q; Ritter JK; Li W; Li PL, Lysosomal regulation of extracellular vesicle excretion during d-ribose-induced NLRP3 inflammasome activation in podocytes. Biochimica et biophysica acta. Molecular cell research 2019, 1866, (5), 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bao JX; Jin S; Zhang F; Wang ZC; Li N; Li PL, Activation of membrane NADPH oxidase associated with lysosome-targeted acid sphingomyelinase in coronary endothelial cells. Antioxidants & redox signaling 2010, 12, (6), 703–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bao JX; Xia M; Poklis JL; Han WQ; Brimson C; Li PL, Triggering role of acid sphingomyelinase in endothelial lysosome-membrane fusion and dysfunction in coronary arteries. American journal of physiology. Heart and circulatory physiology 2010, 298, (3), H992–H1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boini KM; Zhang C; Xia M; Han WQ; Brimson C; Poklis JL; Li PL, Visfatin-induced lipid raft redox signaling platforms and dysfunction in glomerular endothelial cells. Biochimica et biophysica acta 2010, 1801, (12), 1294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boini KM; Xia M; Li C; Zhang C; Payne LP; Abais JM; Poklis JL; Hylemon PB; Li PL, Acid sphingomyelinase gene deficiency ameliorates the hyperhomocysteinemia-induced glomerular injury in mice. The American journal of pathology 2011, 179, (5), 2210–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chauhan D; Vande Walle L; Lamkanfi M, Therapeutic modulation of inflammasome pathways. Immunological reviews 2020, 297, (1), 123–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu CZ; Li FY; Lv XF; Ma MM; Li XY; Lin CX; Wang GL; Guan YY, Endophilin A2 regulates calcium-activated chloride channel activity via selective autophagy-mediated TMEM16A degradation. Acta pharmacologica Sinica 2020, 41, (2), 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rana S; Espinosa-Diez C; Ruhl R; Chatterjee N; Hudson C; Fraile-Bethencourt E; Agarwal A; Khou S; Thomas CR Jr.; Anand S, Differential regulation of microRNA-15a by radiation affects angiogenesis and tumor growth via modulation of acid sphingomyelinase. Scientific reports 2020, 10, (1), 5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cypryk W; Nyman TA; Matikainen S, From Inflammasome to Exosome-Does Extracellular Vesicle Secretion Constitute an Inflammasome-Dependent Immune Response? Frontiers in immunology 2018, 9, 2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rintahaka J; Lietzen N; Ohman T; Nyman TA; Matikainen S, Recognition of cytoplasmic RNA results in cathepsin-dependent inflammasome activation and apoptosis in human macrophages. Journal of immunology 2011, 186, (5), 3085–92. [DOI] [PubMed] [Google Scholar]
- 61.Qu Y; Ramachandra L; Mohr S; Franchi L; Harding CV; Nunez G; Dubyak GR, P2X7 receptor-stimulated secretion of MHC class II-containing exosomes requires the ASC/NLRP3 inflammasome but is independent of caspase-1. Journal of immunology 2009, 182, (8), 5052–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.