

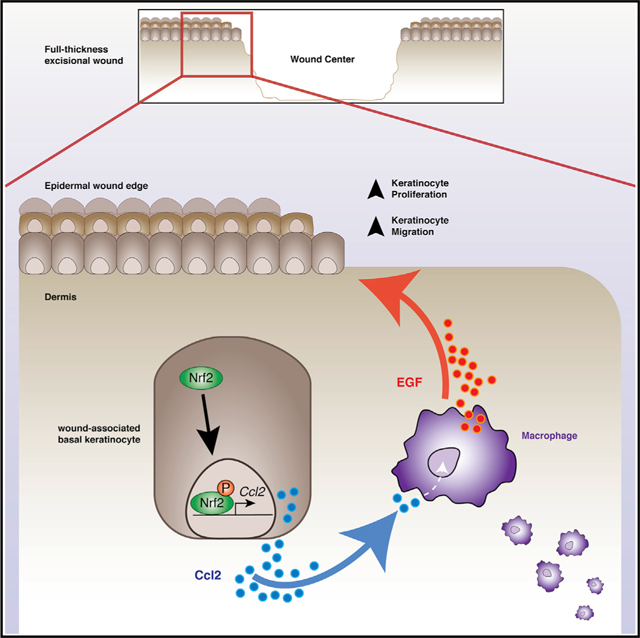
SUMMARY
Unveiling the molecular mechanisms underlying tissue regeneration provides new opportunities to develop treatments for diabetic ulcers and other chronic skin lesions. Here, we show that Ccl2 secretion by epidermal keratinocytes is directly orchestrated by Nrf2, a prominent transcriptional regulator of tissue regeneration that is activated early after cutaneous injury. Through a unique feedback mechanism, we find that Ccl2 from epidermal keratinocytes not only drives chemotaxis of macrophages into the wound but also triggers macrophage expression of EGF, which in turn activates basal epidermal keratinocyte proliferation. Notably, we find dysfunctional activation of Nrf2 in epidermal keratinocytes of diabetic mice after wounding, which partly explains regenerative impairments associated with diabetes. These findings provide mechanistic insight into the critical relationship between keratinocyte and macrophage signaling during tissue repair, providing the basis for continued investigation of the therapeutic value of Nrf2.
In Brief
Villarreal-Ponce et al. show that diabetic wounds fail to activate Nrf2 in keratinocytes, which functions in promoting an early regenerative response in basal keratinocytes by mediating macrophage trafficking through release of Ccl2. Keratinocyte-derived Ccl2 promotes macrophage production of EGF to induce keratinocyte proliferation to promote wound repair.
Graphical Abstract
INTRODUCTION
Nonhealing diabetic wounds impose a major global socioeconomic burden, with an estimated cost of $40.5 billion annually (Sen, 2019). During tissue repair, adult epithelial stem cells (EpSCs) of the skin initiate molecular programs of migration, proliferation, and terminal differentiation to reestablish the impermeable skin barrier (Fuchs, 2007). Although EpSCs in the basal epidermis have a prominent role in wound repair, their regenerative capacity is influenced by regulatory networks mediated by local immune responses, which are often altered by disease (Chen et al., 2015; Deng et al., 2012; Godwin et al., 2013; Naik et al., 2018). Particularly, macrophages play a critical role in the inflammatory, proliferative, and remodeling phases of repair by signaling to various resident stem and progenitor cells of the skin (Hsu et al., 2014; Wynn and Vannella, 2016). These macrophage-dependent regenerative cues are necessary to activate the wound microenvironment, because macrophage ablation studies demonstrate their contribution to regenerative processes such as angiogenesis and reepithelialization (Godwin et al., 2013). In addition to the growing list of cytokines released by macrophages during repair, macrophages are essential sources of chemokines, matrix metalloproteases, and mediators of growth (Krzyszczyk et al., 2018; Wynn and Vannella, 2016). However, the precise communication circuitry by which macrophages become activated to facilitate a regenerative response is not yet clearly understood.
Oxidative damage caused by chronic hyperglycemia contributes to the pathogenesis and progression of many diabetic complications, including impaired wound healing (Brownlee, 2005; Giacco and Brownlee, 2010). As a result, Nrf2 (NF-E2-related factor 2), a master regulator of oxidative stress response, has been implicated in a range of diabetic complications, including diabetic nephropathy and pancreatic βcell dysfunction (Giacco and Brownlee, 2010). Under basal conditions, Nrf2 is constitutively marked for ubiquitination by its inhibitor Keap1, an adaptor component of the Cullin 3-based ubiquitin E3 ligase complex, and degraded (Tonelli et al., 2018). In conditions of elevated stress, Nrf2 undergoes nuclear translocation, in which it transcriptionally regulates expression of target genes by binding to specific gene regulatory motifs identified as antioxidant response elements (AREs) (Tonelli et al., 2018).
In the context of diabetic healing, our work has shown that dysregulated Nrf2 activity largely stems from Keap1 dysfunction, which impairs its nuclear translocation and regenerative activity (Soares et al., 2016). Moroever, forced global activation of Nrf2 is shown to improve the healing capacity of diabetic wounds (Rabbani et al., 2017, 2018; Soares et al., 2016). However, the spatiotemporal dynamics of Nrf2 and the molecular mechanisms it regulates to prompt a regenerative response remain elusive. Studies using both global Nrf2 knockout mice and transgenic mice constitutively expressing dominant-negative mutants show no major healing abnormalities (auf dem Keller et al., 2006; Braun et al., 2002). Although these models have described important features of Nrf2 function, germline Nrf2 deletion may permit activation of compensatory redundant mechanisms that may obscure its total function. This fact is revealed through studies showing that Nrf2 suppression delays corneal and diabetic wound healing (Hayashi et al., 2013; Li et al., 2019; Long et al., 2016).
Here, incorporating transgenic mouse models to genetically ablate epidermal Nrf2 acutely, we describe Nrf2 in EpSCs as a crucial mediator of wound repair that is responsible for regulating onset of an immunogenic signature that directs and sustains the early regenerative response after injury. Importantly, we uncover a signaling circuit by which epidermal Nrf2 regulates chemokine Ccl2 expression to mediate macrophage trafficking and direct macrophage production of epidermal growth factor (EGF), which in turn signals for EpSC activation. Inactivation of epidermal Nrf2 impairs wound closure partly from reduced reepithelialization, highlighting the significance of this regulatory circuit in tissue repair. Our results reveal an essential regulatory network in wound repair by which Nrf2 and Ccl2 in epidermal keratinocytes regulate macrophage trafficking and tissue regeneration.
RESULTS
Dysfunctional Nrf2 Activation in Diabetic Wounds Occurs Primarily in the Basal Epithelium
Leprdb/db (db/db) diabetic mice show impaired wound-healing capacity in full-thickness excisional wound models (Michaels et al., 2007; Rabbani et al., 2018; Soares et al., 2016). Given that oxidative stress must be tightly regulated for physiological wound repair, we created stented, 10 mm full-thickness excisional wounds on the dorsum of 6- to 8-week-old nondiabetic wild-type (WT) and db/db mice to analyze Nrf2 expression in whole wounds over time. Using qPCR, we found an increase in Nrf2 expression in both WT and db/db wounds over time (Figure S1A). Next, we used antibodies that detect the active form of Nrf2 (Nrf2 S40-P) to address whether reduced Nrf2 activity accounts for impaired redox balance in diabetic mice. Specifically, phosphorylation of Ser-40 by protein kinase C (PKC) dissociates Nrf2 from its cytoplasmic inhibitor Keap1 to promote its translocation into the nucleus (Huang et al., 2002). Through western blot, we found that compared with WT wounds, db/db wounds showed a marked reduction in Nrf2 S40-P (Figures 1A and S1B). Dysfunctional Nrf2 activity in db/db wounds was confirmed by mRNA and protein expression of classical Nrf2 gene targets Nqo1 and Sod2 (Figures 1B, 1C, and S1C).
Figure 1. Nrf2 Activation Is Dysfunctional in Epidermal Keratinocytes of Diabetic Wounds.

(A) Western blot (WB) for active Nrf2 on whole-wound lysates from WT and db/db mice (10 DPW). Erk2 is a loading control. n = 5 and 4, respectively.
(B and C) qPCR of (B) Sod2 and (C) Nqo1 in whole wounds from WT and db/db mice (10 DPW). Data were normalized to 18S. n = 3.
(D) Immunofluorescence (IF) staining of active Nrf2 at the epithelial edge in WT and db/db wounds (10 DPW). Active Nrf2 (green), K14 (red), and DAPI (blue). The inset area depicts active Nrf2 in dermis. n = 4. Scale, 100 μm.
(E) Quantification of active Nrf2 IF labeling in K14+ basal keratinocytes (10 DPW). n = 4.
(F) Quantification of active Nrf2 IF labeling in dermal cells (10 DPW). n = 4.
(G) IF of 8-OHdG in intact and wounded skin from WT and db/db mice. 8-OHdG (red), K14 (green), and DAPI (blue). n = 3. Scale, 100 μm.
Data in (E) and (F) were quantified from >3 independent high-powered fields (20×). Data are represented as mean ± standard deviation (SD). Unpaired t tests were used to analyze statistical significance in (B), (C), (E), and (F). ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Recent imaging and lineage tracing studies defined distinct zones of epithelial cellular activity, namely, regions of migration and proliferation, that reside adjacent to the wound margin (Aragona et al., 2017). We used the Nrf2 S40-P antibody to immune-fluorescently label cellular compartments within the diabetic wound microenvironment that showed impaired Nrf2 activity. Compared with nondiabetic WT wounds, db/db wounds showed an ~75% reduction in Nrf2 S40-P staining within the nuclei of the K14+ EpSC-containing basal epidermal wound edge (Figures 1D and 1E), here defined as the epithelial region between the wound edge and the most proximal hair follicle. Comparatively, the difference in dermal Nrf2 S40-P expression in db/db wounds was mild but significantly reduced compared with expression nondiabetic wounds (Figures 1D, inset, and 1F). Reduced Nrf2 activity in the basal epithelium of db/db mice correlated with increased detection of 8-OHdG, a marker of ROS-dependent DNA damage, in the epidermis of both intact and wounded db/db skin (Figure 1G). These results show that Nrf2 dysfunction occurs primarily in the basal epidermis of diabetic mice, which exhibits elevated oxidative damage in both homeostatic and regenerative events.
Acute Deletion of Nrf2 in the Basal Epithelium Impairs Wound Repair
To investigate the functional significance of epidermal Nrf2 in wound repair, we generated transgenic mice to conditionally inactivate Nrf2 in the adult K14+ EpSC-containing basal epidermis upon administration of tamoxifen (TAM) (Figure 2A). The mutant Nrf2 gene lacks exon 5, which encodes the DNA binding domain and results in a nonfunctional form of the transcription factor (Kong et al., 2011; Reddy et al., 2011). TAM-induced Nrf2 deletion in heterozygous and homozygous mutants (Nrf2Δ/+Ker and Nrf2Δ/ΔKer, respectively) resulted in reduced mRNA/protein expression of Nrf2 and classical Nrf2 gene targets (Figures 2B, 2C, S2A, and S2B; data not shown). However, we found no morphological differences in epithelial thickness or dermal collagen maturity in TAM-induced Nrf2Δ/+Ker or Nrf2Δ/ΔKer intact skin when compared with WT control littermates (K14-Cre-ERneg, hereafter abbreviated as Nrf2+/+Ker) (Figures S2C and S2D). In contrast, when subjected to full-thickness excisional wounding, both Nrf2Δ/+Ker and Nrf2Δ/ΔKer mice displayed a marked healing impairment that showed the impact of even partial allelic deletion of epidermal Nrf2 during repair (Figures 2D and 2E). Wound closure rates in Nrf2Δ/+Ker and Nrf2Δ/ΔKer mice were noticeably delayed by 7 days post-wounding (DPW), and approximated the pathological healing rate in diabetic mice (Figure 2E). Specifically, delayed repair in Nrf2Δ/ΔKer mice, measured histologically and by the timing of scab detachment, persisted until complete reepithelialization (Figure 2F). Using area-under-the-curve analysis to describe the regenerative potential of the wound (wound burden), we found Nrf2Δ/ΔKer wounds to maintain a prolonged healing defect (Figure 2G).
Figure 2. Nrf2 Deletion from Interfollicular Basal Epidermal Keratinocytes Impairs Wound Repair.

(A) Strategy used to generate Nrf2Δ/+Ker and Nrf2Δ/ΔKer mice and dissect wound-healing dynamics.
(B) WB for MnSOD and Nqo1 on whole-wound lysates from Nrf2Δ/+Ker and Nrf2Δ/ΔKer mice (10 DPW). Erk2 is a loading control. n = 4.
(C) Quantification of (B).
(D) Representative images of healing dynamics in Nrf2+/+Ker, Nrf2Δ/+Ker, Nrf2Δ/ΔKer, and db/db mice. n = 10, 10, 10, and 7, respectively.
(E–G) Wound area over time (E), wound closure (F), and wound burden analyses (G) in Nrf2+/+Ker, Nrf2Δ/+Ker, Nrf2Δ/ΔKer, and db/db mice. n = 10, 10, 10, and 7, respectively. Wound burden is described by the area under curve in (E).
(H) Epithelial gap measurements in Nrf2+/+Ker and Nrf2Δ/ΔKer mice (10 DPW). n = 13 and 15.
(I) Quantification of Ki-67+ keratinocytes at the epithelial wound edge in Nrf2+/+Ker and Nrf2Δ/ΔKer mice (10 DPW). Quantified from independent high-powered fields (20×). n = 4.
(J) Granulation tissue measurements in Nrf2+/+Ker and Nrf2Δ/ΔKer mice over time. n = 11 and 8, respectively.
(K) Strategy used to generate Nrf2Δ/+HFSC and Nrf2Δ/ΔHFSC mice and dissect healing dynamics.
(L) Representative photographs of wound-healing dynamics in Nrf2+/+HFSC and Nrf2Δ/ΔHFSC mice. n = 4.
(M–O) Wound area over time (M), wound closure (N), and wound burden analyses (O) in Nrf2+/+HFSC and Nrf2Δ/ΔHFSC mice. n = 4.
Data are represented as mean ± SD. For (C), (H)–(J), (N), and (O), unpaired t tests were used to analyze statistical significance between the two experimental groups. For (E)–(G) and (M), two-way ANOVA with multiple comparisons was used to analyze statistical significance of open wounds between groups on respective days after wounding. ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Having observed an intermediate healing delay in Nrf2Δ/+Ker mice, we carried out subsequent analyses between Nrf2+/+Ker and Nrf2Δ/ΔKer mice to maximize our understanding of the regenerative function of epidermal Nrf2. Delayed wound repair is linked to reduced activation of wound-edge keratinocytes, which fail to propagate and migrate to reepithelialize the wound (Keyes et al., 2016). To identify whether epidermal Nrf2 loss affects keratinocyte-autologous wound responses, we measured the epithelial gap at early and intermediate stages of repair to analyze reepithelialization dynamics. At both stages, the epithelial gap underneath the eschar extended farther in Nrf2Δ/ΔKer wounds than in TAM-induced controls (Figures 2H, S2E, and S2L), suggesting that reduced migration and/or proliferation at least partially contributes to the healing delay. To determine whether delayed reepithelialization resulted from reduced keratinocyte migration, Nrf2+/+Ker and Nrf2Δ/ΔKer transgenic mice were crossed with RO-SA26mTmG reporter mice (Muzumdar et al., 2007) to trace the migratory capacity of GFP+ keratinocytes from TAM-induced R26mTmG;Nrf2+/+Ker and R26mTmG;Nrf2Δ/ΔKer skin explants after mitomycin C treatment (Figure S2F). Area measurements, representative of the distance traveled by GFP+ keratinocytes, revealed R26mTmG;Nrf2Δ/ΔKer keratinocytes show reduced ability to migrate (Figure S2G). We followed this analysis with an assessment of basal EpSC proliferation. Using immunofluorescence (IF) staining, we found reduced Ki-67+ proliferating basal epithelial cells at the Nrf2Δ/ΔKer wound edge (Figures 2I and S2H). Reduced proliferation did not occur at the expense of increased apoptosis, because we did not detect a noticeable difference in the number of cleaved caspase-3+ apoptotic basal keratinocytes (Figure S2I). The findings demonstrate that delayed wound closure partly corresponds to decreased migration and proliferation of the epithelial wound edge.
We also characterized keratinocyte-extrinsic impairments affected by loss of epidermal Nrf2. Granulation tissue formation describes an essential component of wound repair that encompasses the production of new connective tissue and blood vessels within the wound bed (Ellis et al., 2018). Histological evaluation of the granulation tissue area within the wound bed showed that compared with Nrf2+/+Ker wounds, epidermal Nrf2 loss led to reduced granulation tissue formation (Figures 2J and S2J). As depicted by trichrome staining we also observed reduced deposition and organization of collagens in Nrf2Δ/ΔKer wounds(Figure S2K). Cutaneous lesions in Nrf2Δ/ΔKer mice shared other histological features with those from diabetic mice, most prominently reduced neovascularization, which was quantified via immunostaining of CD31+ vascular tissue in the wound bed (Figure S2L). Altogether, these results provide compelling evidence that wound-healing impairments in epidermal Nrf2 null wounds are rooted in both keratinocyte-autologous and nonkeratinocyte-intrinsic effects.
Deletion of Nrf2 in HFSCs Is Dispensable for Wound Repair
Hair follicle stem cells (HFSCs) residing in the skin epithelial bulge are regarded as key contributors to wound repair (Ito et al., 2005; Joost et al., 2018). Recent studies reveal that constitutive genetic activation of Nrf2 in K5+ keratinocytes accelerates wound repair kinetics from expanded proliferation and migratory capacity of the pilosebaceous unit (Muzumdar et al., 2019). Having detected a prominent healing delay in K14+ epidermal Nrf2 mutants, we wondered whether our observed delay stemmed from impaired Nrf2 activity in HFSCs. Immunofluorescent labeling showed strong nuclear staining of Nrf2 in keratinocytes labeled positive for K15, a marker of HFSCs, confirming that this EpSC subset expresses Nrf2 in wounded skin (Figure S3A). We then created full-thickness excisional wounds on transgenic mice in which functional deletion of Nrf2 can be conditionally induced in HFSCs (K15-CrePR1;Nrf2fl/fl) upon RU486 treatment (Figures 2K and S3B) (Morris et al., 2004). After RU486 treatment during late first telogen/early second anagen (Alonso and Fuchs, 2006; Müller-Röver et al., 2001), we found that acute Nrf2 deletion in HFSCs (Nrf2Δ/ΔHFSC) resulted in similar wound closure kinetics to those of WT control littermates (Nrf2+/+HFSC), represented by comparable reepithelialization rate and time to wound closure (Figures 2L–2N). Although gross wound closure was unaffected, Nrf2Δ/ΔHFSC wounds displayed a mild and temporary, early repair delay that increased the wound burden (Figure 2O). Interestingly, we did not observe delays in hair cycle progression after depilation (data not shown). Collectively, the data from these experiments indicate that acute deletion of Nrf2 from the basal, EpSC-containing interfollicular epidermis, but not in HFSCs, markedly impairs wound healing and produces clinical features observed in diabetic healing.
Epidermal Nrf2 Controls Keratinocyte-Mediated Paracrine Signaling Networks
To gain insight into critical wound response mechanisms regulated by epidermal Nrf2, we microdissected an ~2.5 mm concentric ring around the 10 mm wound and used flow cytometry to isolate wound-associated keratinocytes from Nrf2+/+Ker and Nrf2Δ/ΔKer mice for RNA sequencing (RNA-seq) (Figure 3A). We isolated this population using α6-integrin (Linneg; CD49fpos) for its strong expression in epithelial cells and reported ability to discern basal keratinocytes (Figure S4A) (Sonnenberg et al., 1991). Differential expression analysis was carried out at 5 DPW, by reasoning that early Nrf2 upregulation likely corresponds with the impaired wound-healing kinetics observed in Nrf2Δ/ΔKer mice (Figures 2E and 2F). Our analysis revealed 278 differentially expressed genes (DEGs) (±2-fold; q < 0.05; n ≥ 2), 229 of which were downregulated in Nrf2Δ/ΔKer keratinocytes (Figure 3B). Using chromatin immunoprecipitation (ChIP) enrichment analysis (ChEA) and ChIP-X enrichment analysis (Lachmann et al., 2010) to computationally identify likely candidates responsible for the transcriptome-wide expression changes, we identified Nrf2 as the top putative transcriptional regulator of our dataset (Figures S4B and S4C), suggesting our identified DEGs are primary and directly dependent on Nrf2 activity.
Figure 3. Nrf2 Deletion from Epidermal Keratinocytes Affects Paracrine Responses Critical for Tissue Regeneration.
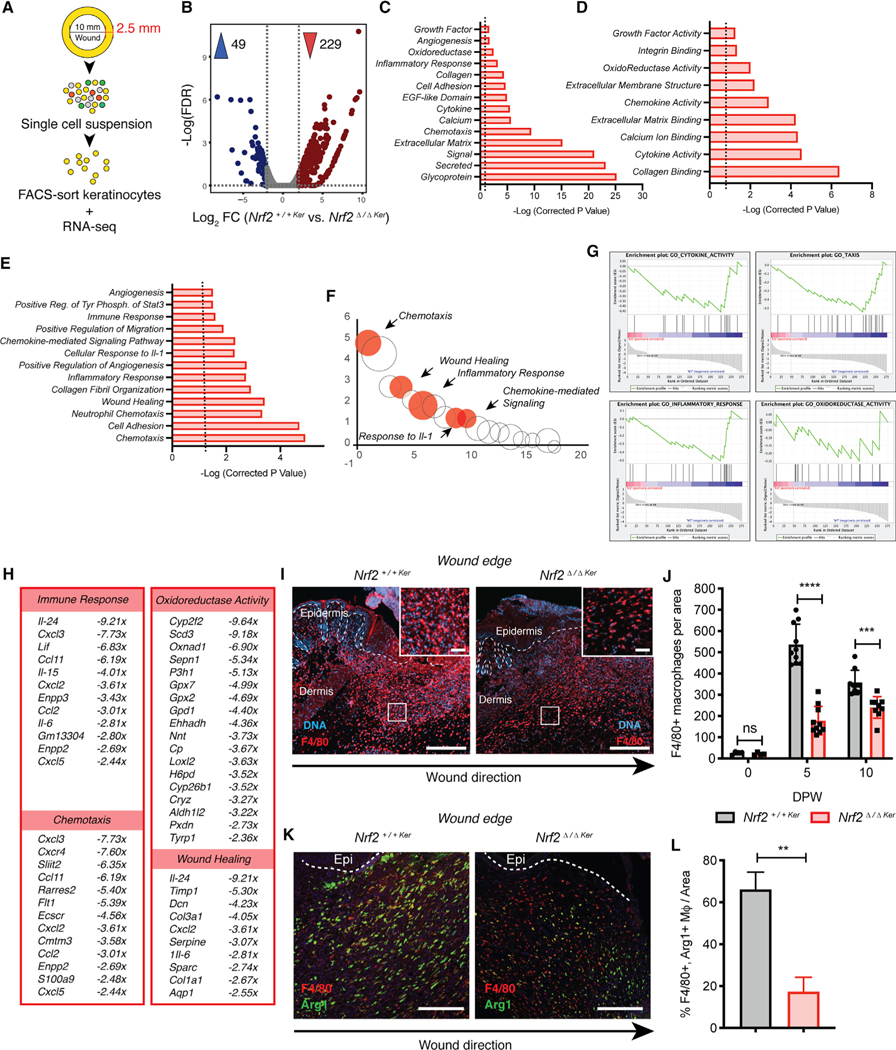
(A) Strategy used to isolate keratinocytes for bulk RNA-seq. RNA sorted from each experimental group was pooled for analysis.
(B) Volcano plot depicting mean DEG expression between Nrf2+/+Ker and Nrf2Δ/ΔKer (±2-fold; q < 0.05; n ≥ 2).
(C–E) DAVID Gene Ontology (GO) analysis of downregulated DEGs from Nrf2Δ/ΔKer wound keratinocytes depicting (C) cell compartments, (D) molecular functions, and (E) biological processes (±2-fold; p < 0.05).
(F) Bubble chart comparing overlap of gene classifications shown in (E). The bubble size depicts the gene number in each classification (±2-fold; p < 0.05). Select classifications are marked.
(G) GSEA of downregulated DEGs from Nrf2Δ/ΔKer wound keratinocytes for select gene sets: (1) cytokine activity, (2) taxis, (3) inflammatory response, and (4) oxidoreductase activity (p < 0.05).
(H) Display of ontologies and fold expression change of downregulated DEGs from Nrf2Δ/ΔKer wound keratinocytes (±2-fold; p < 0.05).
(I) IF labeling of macrophages trafficked into Nrf2+/+Ker and Nrf2Δ/ΔKer wounds (5 DPW). F4/80 (red) and DAPI (blue). n = 10. Scale, 100 μm; inset scale, 30 μm.
(J) Quantification of F4/80+ IF labeling in Nrf2+/+Ker and Nrf2Δ/ΔKer wounds at various times. n = 4 (0 DPW), n = 10 (5 DPW), and n = 9 (10 DPW).
(K) IF of M2 macrophages trafficked into wounds of Nrf2+/+Ker and Nrf2Δ/ΔKer mice (10 DPW). F4/80 (red), Arg1 (green), and DAPI (blue). n = 10. Scale, 100 μm.
(L) Quantification of (K).
IF in (J) and (L) was quantified from independent high-powered fields (20×) in specified independent biological replicates. Data are represented as mean ± SD. Unpaired t tests were used to analyze statistical significance in (J) and (L). ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Next, we evaluated core reparative processes affected by grouping DEGs by ontological terms (p < 0.05): cell compartment (CC), molecular function (MF), and biological process (BP). As expected, we found reduced expression of genes encoding proteins with classical oxidoreductase function, including Oxnad1, Gpx2, and Gpx7 (Figures 3C and 3D). Inspection through leading edge analysis revealed that many downregulated DEGs in Nrf2Δ/ΔKer wound keratinocytes influenced keratinocyte-mediated paracrine signaling functions, including inflammation, immune cell guidance, extracellular structure, and angiogenesis (p < 0.05) (Figures 3C–3E). We found minimal overlap of chemotaxis-related gene signatures (Figure 3F), suggesting that epidermal Nrf2 uniquely regulates these paracrine functions. We also employed gene set enrichment analysis (GSEA) to produce unbiased predictions of gene profile enrichment in Nrf2Δ/ΔKer mutants. GSEA corroborated our findings, showing Nrf2 loss leads to a reduction in taxis, cytokine activity, inflammatory response, and oxidoreductase activity (p < 0.05) (Figure 3G). Among representative genes implicated in governing immune cell taxis and other chemotaxis-related ontological terms were Ccl2 (Mcp1), Ccl1 (Eotaxin), Cxcl2, Cxcl3, Cxcl5, Cxcr4, and Il-6 (Figure 3H).
From the observed impact epidermal Nrf2 loss has on signaling for immune cell trafficking, we applied an unbiased immunolabeling approach using sagittal sections of wounded skin to analyze the overall immune response, focusing on immune cell surface marker expression, morphology, and localization at early and intermediate stages of wound repair. First, variance in Ly6g+ neutrophil influx by 2 DPW could not specifically explain the wound repair anomaly between Nrf2+/+Ker and Nrf2Δ/ΔKer mice (Figure S4D). Next, because of the known role of macrophages in inflammation and tissue remodeling within the wound microenvironment (Krzyszczyk et al., 2018), we compared the relative numbers of F4/80+ macrophages recruited from the peripheral blood to the site of injury. No differences in macrophage abundance were noted in intact skin from Nrf2+/+Ker and Nrf2Δ/ΔKer mice (Figure 3J). In contrast, high-powered IF field analysis showed that although both Nrf2+/+Ker and Nrf2Δ/ΔKer wounds showed an influx of F4/80+ macrophages by 5 DPW (n = 10) (Figures 3I and 3J), there was a global ~80% reduction in the number and density of F4/80+ macrophages in Nrf2Δ/ΔKer wounds, primarily near the epithelial wound edge (Figures 2J, 3I, and S4E). A 30% reduction in macrophage density persisted in Nrf2Δ/ΔKer wounds by 10 DPW (Figure 3J), suggesting that epidermal Nrf2 deletion results in delayed macrophage trafficking that impairs healing.
Although macrophages display greater complexity in vivo, they can be functionally categorized into pro-inflammatory (M1) and anti-inflammatory/tissue remodeling (M2) phenotypes through differential expression of surface markers (Italiani and Boraschi, 2014; Krzyszczyk et al., 2018). To examine whether epidermal Nrf2 deletion also affects M1-to-M2 polarization, we immunolabeled sagittal sections of wounded skin to quantitate the percentage of M2 (F4/80+; Arg1+) macrophages in Nrf2+/+Ker and Nrf2Δ/ΔKer wounds. We found reduced abundance of M2 macrophages in Nrf2Δ/ΔKer wounds compared with Nrf2+/+Ker controls (Figures 3K and 3L), suggesting that in addition to macrophage taxis, Nrf2 may regulate keratinocyte signaling networks that influence M1-to-M2 polarization. The finding indicates a possible undocumented role of Nrf2 in M1-to-M2 polarization. Altogether, these data indicate that epidermal keratinocyte-mediated signaling of inflammation is a critical component of wound repair that is regulated by Nrf2 activity.
Direct Regulation of Ccl2 by Epidermal Nrf2 Reverses Wound-Healing Defects in Epidermal Nrf2 Null and Diabetic Mice
Macrophage homeostasis and recruitment depends on the stimulatory effects of Ccl2 (Chen et al., 2015; Dewald et al., 2005; Gschwandtner et al., 2019). In acute inflammatory conditions, trafficking of macrophages from the bone marrow is strongly governed by the Ccl2/Ccr2 signaling axis. However, an understanding of how Ccl2 is regulated in wound repair is incomplete. Having identified epidermal Nrf2 as an early regulator of Ccl2 expression through our RNA-seq data, we used a gain-of-function approach to define whether Nrf2 is key for chemokine Ccl2 production in keratinocytes. To test this hypothesis, we treated primary WT keratinocyte cultures with pharmacological Nrf2 activator CDDO-IM (Aleksunes et al., 2010; Reisman et al., 2009) before measuring expression of Nrf2 S40-P and Ccl2 through immunocytochemistry and qPCR. WT keratinocytes treated with CDDO-IM (100 nM for 1.5 h) markedly increased Nrf2 S40-P and Ccl2 mRNA/protein levels compared with controls after 24 h (Figures 4A–4C and S5A). Next, we investigated the dynamic control of Nrf2 on the Ccl2 promoter in wound-associated keratinocytes in vivo. Using ARE as the recognized Nrf2 binding motif (Chorley et al., 2012; Malhotra et al., 2010), we found a putative Nrf2 binding site 1.8 kb upstream of the Ccl2 transcriptional start sites (TSS) (Figure 4D). Using ChIP-PCR, we found Nrf2 binds to this site in wound keratinocytes by 5 DPW but does not in intact keratinocytes (Figure 4D). We confirmed the fidelity of our analysis by showing Nrf2 binding to a promoter proximal ARE-motif-containing site on known Nrf2 gene target Nqo1, but not on a nonspecific site (Figure 4D). These results indicate that Nrf2 functions dynamically in wound-associated keratinocytes to regulate expression of chemokine Ccl2.
Figure 4. Epidermal Nrf2 Directly Regulates Ccl2 to Recruit Macrophages in Response to Wounding and Rescues Wound Repair Debilitations in Nrf2Δ/ΔKer and db/db Mice.

(A) IF labeling of active Nrf2 and Ccl2 in primary WT keratinocytes cultures with or without CDDO-IM. Active Nrf2 (green), Ccl2 (yellow), Keratin 14 (purple), and DAPI (blue). n = 4. Scale, 100 μm.
(B) Quantification of active Nrf2 in (A). Nrf2 was quantified from 2 independent high-powered fields. n = 4.
(C) Quantification of Ccl2 in (A). Ccl2 was quantified as the mean fluorescence intensity from 2 independent high-powered fields. n = 4.
(D) ARE motif distribution at the Nqo1 and Ccl2 promoter. Nrf2 binding in keratinocytes isolated from intact and wounded skin (5 DPW). Binding is represented as fold enrichment to the immunoglobulin G (IgG) control. n = 3.
(E) Strategy used to assess Ccl2 rescue of Nrf2Δ/ΔKer healing impairments.
(F) Representative photographs of healing dynamics in Nrf2Δ/ΔKer +Veh. and Nrf2Δ/ΔKer +Ccl2 wounds (n = 7 and 10). A comparison is also made with untreated Nrf2+/+Ker wounds (n = 10).
(G–I) Wound area over time (G), wound closure (H), and wound burden analysis (I) in Nrf2Δ/ΔKer +Veh., Nrf2Δ/ΔKer +Ccl2, and Nrf2+/+Ker mice. n = 7, 10, and 10, respectively.
(J) IF labeling of macrophages in Nrf2Δ/ΔKer wounds treated with or without Ccl2 at 5 DPW. F4/80 (red) and DAPI (blue). n = 4. Scale, 100 μm.
(K) Quantification of (J). Labeling was quantified from independent high-powered fields (20×) in independent biological replicates.
(L) Quantification of F4/80+ IF in WT and db/db wounds at different time points. n = 4 and 3 (0 DPW), n = 9 and 10 (5 DPW), and n = 7 (10 DPW). Quantified from independent high-powered fields (20×) in independent biological replicates.
(M) qPCR of Ccl2 in WT and db/db wound keratinocytes at 5 DPW. Data were normalized to 18S. n = 3.
(N)Representative photographs of healing dynamics in db/db + Veh. and db/db + Ccl2 wounds (n = 7 and 10). Repair was also compared with Nrf2+/+Ker wounds (n = 10).
(O–Q) Wound area over time (O), wound closure (P), and wound burden analysis (Q) in db/db + Veh., db/db + Ccl2, and Nrf2+/+Ker mice. n = 7, 10, and 10, respectively.
Data are represented as mean ± SD. Unpaired t tests were used to analyze statistical significance in (B)–(D) and (K)–(M). For (G)–(I) and (N)–(Q), two-way ANOVA with multiple comparisons was used to analyze statistical significance of open wounds between groups on respective days after wounding. ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Next, to probe into the functional relationship between dysregulated Nrf2 activation of Ccl2 and impaired wound healing, we asked whether exogenous Ccl2 could rescue the healing impairments in Nrf2Δ/ΔKer mice (Figure 4E). Exogenous treatment of recombinant Ccl2 (50 ng/application; every other day until 5 DPW) fell within the reported physiological Ccl2 concentration range in normal skin wounds (Dipietro et al., 2001). When Nrf2Δ/ΔKer wounds were subjected to a vehicle-only treatment (Nrf2Δ/ΔKer + Veh.), we found healing kinetics to closely mimic the delays associated with epidermal Nrf2 deletion (Figure 4F). However, treatment of Nrf2Δ/ΔKer wounds with exogenous Ccl2 (Nrf2Δ/ΔKer + Ccl2) significantly rescued reparative morbidities to nearly those of Nrf2+/+Ker counterparts (Figures 4F–4I). Compared with controls, Nrf2Δ/ΔKer + Ccl2 wounds showed a substantial wound repair rate increase by 7 DPW (Figure 4G) and accelerated wound time to closure by ~56% (Figure 4H). In addition, wound burden analyses showed exogenous Ccl2 corrected the regenerative potential of Nrf2Δ/ΔKer wounds by ~30% (Figure 4I), indicating that Ccl2 dysregulation is at least partially responsible for the wound-healing impairments caused by loss of epidermal Nrf2. Next, we immunolabeled sagittal sections of Nrf2Δ/ΔKer + Veh. and Nrf2Δ/ΔKer + Ccl2 wounds at early stages of repair to see whether exogenous Ccl2 restored F4/80+ macrophage infiltration. Contrasting macrophage trafficking in Nrf2Δ/ΔKer + Veh. wounds, high-powered IF field analysis showed considerable restoration in F4/80+ macrophage infiltration in Ccl2-treated wounds (Figures 4J and 4K), suggesting that the wound-healing phenotype in Nrf2Δ/ΔKer partly results from deficient macrophage trafficking.
Relevantly, studies show that diabetes impairs the responsiveness of inflammatory macrophages to exogenous stimuli (Hesketh et al., 2017; Khanna et al., 2010; Maruyama et al., 2007; Moore et al., 1997). To test whether macrophages fail to infiltrate diabetic wounds, we analyzed macrophage density in nondiabetic WT and db/db wounds during early and intermediary repair stages by generating a ratio of F4/80+ macrophages present at each stage to those in intact skin. We found that although abundance of F4/80+ macrophages was elevated in intact db/db skin, trafficking of these macrophages to the site of injury was reduced in db/db mice at 5 and 10 DPW (Figures 4L and S5B). Compared with WT controls, we also found reduced Ccl2 expression in flow-sorted wound-associated keratinocytes isolated from db/db wounds at 5 DPW (Figure 4M), indicating the likelihood that dysfunctional regulation of Ccl2 by Nrf2 affects macrophage trafficking in diabetic wounds. Next, we sought to establish whether topical application of Ccl2 could be used as a broad-spectrum diabetic wound repair therapy that functions by boosting macrophage trafficking. Interestingly, recent studies find Ccl2 improves wound-healing dynamics in streptozotocin (STZ)-induced mouse models of type 1 diabetes by promoting neovascularization and collagen accumulation (Ishida et al., 2019). Similar to our findings in Nrf2Δ/ΔKer mice, topical Ccl2 treatment improved wound-healing kinetics of db/db mice. Ccl2 treatment significantly reduced the time to heal by enhancing the rate of repair (Figures 4N–4Q). The enhanced rate of repair in db/db mice treated with Ccl2 did not reach the rescued repair rate observed in Nrf2Δ/ΔKer + Ccl2 mice, suggesting that additional factors contribute to delayed repair in this multi-factorial metabolic disease. Collectively, the data from these experiments indicate that Nrf2 orchestrates a prominent regenerative response through regulation of Ccl2 in epidermal keratinocytes.
Ccl2 Facilitates EGF Production in Macrophages Promoting Keratinocyte Activity
After showing that reduced EpSC activity contributes to healing complications in Nrf2Δ/ΔKer mice (Figure 2I), we sought to investigate macrophage-dependent mechanisms that affect the regenerative response. Macrophage secretion of EGF is important in facilitating macrophage-keratinocyte crosstalk and stimulating reepithelialization (Hancock et al., 1988). To begin testing whether reduced Nrf2Δ/ΔKer EpSC regenerative activity results from dampened macrophage-keratinocyte paracrine signaling, we analyzed global EGF expression in Nrf2+/+Ker and Nrf2Δ/ΔKer wounds at 10 DPW, a time point at which we found reduced EpSC proliferation. Compared with controls, we found reduced EGF expression in Nrf2Δ/ΔKer wounds (Figure 5A). Previous studies describe a Ccl2/EGF positive feedback loop in the neoplastic setting, in which Ccl2 stimulates EGF production in macrophages to stimulate epidermal growth (Gao et al., 2016). To test whether Ccl2 regulates EGF production in normal macrophages, we cultured primary WT undifferentiated bone marrow cells with macrophage colony stimulating factor (M-CSF) to generate bone-marrow-derived macrophages (BMDMs) that were subsequently treated with exogenous Ccl2. qPCR analysis showed BMDMs treated with Ccl2 had an ~4-fold increase in EGF expression over control BMDMs (Figure 5B). Next, we used an isolated system to identify the significance of EGF production in Ccl2-stimulated macrophages on keratinocyte proliferation. Given that physical interaction is not required for EGF signaling, we cultured BMDMs with/without exogenous Ccl2 and subsequently transferred the conditioned media (CM) to WT keratinocyte cultures to assess the effect on proliferation. Compared with controls, keratinocytes cultured in CM from Ccl2-treated macrophages exhibited greater Ki-67 staining (Figure 5C). We did not find that exogenous Ccl2 treatment alone affected keratinocyte proliferation (Figure 5D) nor after knocking down Ccl2 expression in keratinocyte cultures with Ccl2-targeting small interfering RNAs (siRNAs) (Figures 5E and 5F). EGF expression was not induced in keratinocyte cultures with Ccl2 treatment alone or with Nrf2 activator CDDO-IM (Figures S5C and S5D).
Figure 5. Ccl2 Induces Production of EGF in Macrophages that Signals for Keratinocyte Proliferation.

(A) qPCR of EGF in WT and db/db wounds at 10 DPW. Data were normalized to 18S. n = 3.
(B) qPCR of EGF in Ccl2-treated primary BMDMs cultured ex vivo. Data were normalized to 18S. n = 3.
(C) IF/quantification of Ki-67 in WT keratinocytes cultured with conditioned media (CM) from macrophage cultures treated with or without recombinant Ccl2. Ki-67 (green) and DAPI (blue). n = 3. Scale, 100 μm.
(D) IF/quantification of Ki-67 in WT keratinocytes cultured with or without recombinant Ccl2. Ki-67 (green) and DAPI (blue). n = 3. Scale, 100 μm.
(E) qPCR of Ccl2 in WT keratinocytes cultures with shCcl2 or negative control. Data were normalized to 18S. n = 3.
(F) IF/quantification of Ki-67 in WT keratinocytes cultured with shCcl2 or negative control. Ki-67 (green) and DAPI (blue). n = 3. Scale, 100 μm.
(G) IF/quantification of Ki-67 in PD153035-treated WT keratinocyte cultures before CM from macrophage cultures treated with Ccl2. Ki-67 (green) and DAPI (blue). n = 3. Scale, 100 μm.
(H) Migration of K14Cre-ER;mTmG skin explant cultures pretreated with or without PD153035 before addition of CM from macrophage cultures treated with Ccl2. Ki-67 (green) and DAPI (blue). n = 3. Scale, 100 μm.
(I) qPCR of EGF in Nrf2Δ/ΔKer + Veh. and Nrf2Δ/ΔKer + Ccl2 wounds at 10 DPW. Data were normalized to 18S. n = 3.
(J) IF/quantification of Ki-67+ keratinocytes at the epithelial wound edge of Nrf2Δ/ΔKer + Veh. and Nrf2Δ/ΔKer + Ccl2 at 10 DPW. Ki-67 (green) and DAPI (blue). n = 3. Scale, 100 μm.
Data are represented as mean ± SD. Unpaired t tests were used to analyze statistical significance in (A)–(G). ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To confirm EGF as a major product of Ccl2-treated macrophage CM that is responsible for driving keratinocyte activity, we analyzed proliferation in keratinocyte cultures pretreated with PD153035 (250 nM for 24 h), a known potent and specific inhibitor of EGF receptor (EGFR) signaling, before the addition of CM from Ccl2-treated BMDMs. Compared with controls, PD153035 treatment attenuated keratinocyte proliferation, suggesting that EGF is a key macrophage-secreted factor that regulates keratinocyte behavior (Figure 5G). Next, because EGF is also known to facilitate repair by stimulating keratinocyte migration (Haase et al., 2003; Seeger and Paller, 2015), we analyzed the effects of CM from Ccl2-treated macrophages on keratinocyte migration using skin explants from mitomycin C-treated, TAM-induced K14Cre-ER;R26mTmG mice that were treated with or without PD153035. We found that PD153035 significantly reduced keratinocyte migration in explant cultures treated with CM from Ccl2-treated macrophages (Figure 5H).
Next, having identified the capacity of Ccl2 to rescue Nrf2Δ/ΔKer wound repair defects (Figures 4F–4I), we asked whether EGF expression was restored within the wound microenvironment upon Ccl2 treatment. We found that compared with controls, Ccl2 treatment elevated EGF levels in Nrf2Δ/ΔKer wounds (Figure 5I). We then quantified Ki-67+ proliferating keratinocytes at the epidermal wound edge in Ccl2 + Veh. and Nrf2Δ/ΔKer + Veh. wounds. Compared with controls, Ccl2 treatment resulted in a significant increase in the number of Ki-67+ keratinocytes in Nrf2Δ/ΔKer wounds (Figure 5J). Thus, Ccl2 appears to be an epidermal Nrf2-regulated factor that guides both macrophage trafficking and EGF production to support keratinocyte regenerative activity.
DISCUSSION
Coordinated behavior of the cellular constituents within the wound microenvironment is critical to promoting effective tissue repair. Disruption of these mechanisms, as in nonhealing diabetic wounds, results in an impaired regenerative response. Previous studies describe the Nrf2 signaling pathway as a promising therapeutic axis to target chronic wound debilitations (Rabbani et al., 2017, 2018; Soares et al., 2016). However, detailed spatiotemporal and mechanistic evidence on how Nrf2 promotes repair remains elusive. In our study, we describe dysfunctional activity of Nrf2 in keratinocytes of diabetic wounds and examine its function during wound repair. Here, we uncover epidermal Nrf2 as an activator of the Nrf2-Ccl2-EGF signaling axis by which keratinocytes produce a regenerative response that promotes macrophage trafficking and signaling for reepithelialization.
Although the role of Ccl2 as a macrophage chemoattractant and facilitator of tissue repair is becoming evident (Ishida et al., 2019; Low et al., 2001), there remains a significant knowledge gap in how Ccl2 is regulated and the mechanisms it governs during repair. Global Ccl2 deletion results in wound repair delays that reiterate key features of our observed Nrf2Δ/ΔKer phenotype, including reduced macrophage trafficking, reepithelialization, and neovascularization (Low et al., 2001). Interestingly, Ccl2 normalizes collagen abundance and angiogenesis (Ishida et al., 2019), which not only fortifies the relevance of the Nrf2-Ccl2 regulatory circuit but also gives insight into additional functions Ccl2 may regulate (Gschwandtner et al., 2019). Molecular studies show Ccl2 signals through the JAK2/STAT3 (Mellado et al., 1998), mitogen-activated protein kinase (MAPK) (Cambien et al., 2001), and phosphatidylinositol 3-kinase (PI3K) signaling pathways (Turner et al., 1998). EGF regulation by Ccl2 was demonstrated in the neoplastic setting (Gao et al., 2016), but its physiological relevance, particularly in repair, had not been verified. We provide evidence for the positive regulation of EGF by Ccl2 in macrophages, which contributes to wound reepithelialization. Because EGFR/MAPK signaling is shown to activate Nrf2, the Nrf2-Ccl2-EGF signaling axis underscores a possible positive regenerative feedback loop that is supported by this axis.
Wound repair transpires through overlapping phases that depend on the activity and communication of various cell types (Gurtner et al., 2008; Khanna et al., 2010). Our work uncovers epidermal Nrf2 as a mediator of tissue regeneration with a role in managing keratinocyte-macrophage crosstalk and classifies the epidermis as a prominent conductor of repair. Although we analyzed several acute Cre drivers to specify the epithelial subsets at which Nrf2 exerts its greatest regenerative effects, our transcriptome-wide analysis will require deeper analysis to assess specific K14+ sub-compartments that engender these paracrine signals. This is notable, because other cellular compartments of the wound microenvironment show varied reliance on Nrf2 activity. Recent studies showed dermal Nrf2 has a dispensable role in wound repair (Hiebert et al., 2018). In germ-line dermal Nrf2 knockout mice, macrophage, T cell, and B cell frequencies are unaffected after wounding, despite dermal regulation of an extravasation signature (Hiebert et al., 2018). However, constitutive activation of dermal Nrf2 accelerates wound closure, suggesting pleiotropic Nrf2 functions that may contribute to repair.
Although the consequences of disrupting Nrf2 in keratinocytes in vitro have been explored (Braun et al., 2002; Long et al., 2016; Pedersen et al., 2003), as has its role in promoting epithelial proliferation and migration during corneal repair (Hayashi et al., 2013), our observations differ from those of a previous study using transgenic mice expressing a germline dominant-negative Nrf2 mutant in the epidermis, which showed its dispensable role in wound repair (auf dem Keller et al., 2006). Consistent with these findings, we observe that epidermal Nrf2 loss impairs the inflammatory response. In contrast, however, we find acute Nrf2 deletion from the K14+ epidermis delays wound healing, demonstrating its significance in repair. The disparate findings may result from the concurrent expression of the dominant-negative mutant and endogenous Nrf2. We, among others (Braun et al., 2002), show that Nrf2 is upregulated in response to wounding. Upregulation of WT Nrf2, if substantial, is prone to competing with the dominant-negative mutant and may eliminate deleterious effects. In addition, our use of K14-CreER mice to acutely ablate Nrf2 expression in a specific spatiotemporal manner allows us to reduce the activation of potential compensatory mechanisms that may mask true gene function. In our study, acute deletion of the floxed Nfe2l2 locus (Reddy et al., 2011) was carried out using Cre-ER under control of the human K14 promoter, which is highly active in dividing cells of the epidermis and allowed efficient recombination.
What is the significance of keratinocyte-mediated macrophage trafficking in the hyperinflamed diabetic setting? It is known that components of the immune system are altered in diabetes, because it is characterized by hyperinflammation and elevated macrophage abundance in various tissues (Blakytny and Jude, 2006; Hesketh et al., 2017; Krzyszczyk et al., 2018; Shaw and Martin, 2009; Wetzler et al., 2000). We show that diabetic wounds fail to traffic macrophages from peripheral tissues, including those exhibiting increased macrophage density. This prompts questions on whether trafficking signals or macrophage recognition of these signals is dysfunctional in diabetes. There is evidence of the contribution of macrophage dysfunction in diabetic complications, as shown by their failure to activate and traffic to the peritoneal cavity after thioglycolate-induced inflammation (Maruyama et al., 2007) and their reduced expression of activation-associated markers after exposure to inflammatory conditions (Moore et al., 1997). However, although features of macrophage dysfunction contribute to regenerative impairments in diabetes, our work reveals keratinocyte signaling for macrophage trafficking to be dysregulated. Our study suggests Ccr2 activation is not compromised in db/db mice, because Ccl2 treatment rescues macrophage trafficking in Nrf2Δ/ΔKer mice. In addition, we provide insight into the potential role of the epidermal Nrf2 in regulating M1-to-M2 polarization. A defective M1-to-M2 transition is characteristic of the prolonged inflammatory state observed in diabetic wounds, because M2 macrophages promote an anti-inflammatory environment that is necessary for healing. However, the cues responsible for this differentiation are likely tightly regulated spatiotemporally, because M2 application delays wound repair in db/db mice. Thus, an understanding of how epidermal Nrf2 governs the cues underlying M1-to-M2 polarization is critical to better characterizing M1/M2-specific functions in repair and M1-to-M2 deficiencies in diabetes.
Overall, by combining analyses of both transgenic and diabetic mouse models, we uncover a role of epidermal Nrf2 in regulating a critical regenerative response in wound repair that is impaired in the diabetic setting. Our data suggest that targeting Ccl2, a downstream target of epidermal Nrf2, may be an effective treatment to improve diabetic wound repair complications.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel J. Ceradini (Daniel.Ceradini@nyulangone.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The bulk RNA-seq dataset generated during this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE159361.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal procedures followed guidelines that were approved by the New York University (NYU) Institutional Animal Care and Use Committee (IACUC) under protocol number IA16–000204. Both male and female mice (6–8-weeks-of-age) were used for full-thick-ness excisional wounding experiments, and were singly housed post-surgery within the NYU School of Medicine animal facility.
Diabetic (Lepr db/db) (Stock No. 000642) and nondiabetic C57BL/6J (wild-type) (Stock No. 000664) mice were purchased from Jackson Laboratories (Bar Harbor, ME). The hyperglycemic status of each diabetic mouse was measured using a glucometer and only those with blood glucose levels > 400mg/dl before wounding and at the time of analysis were used in the study. K14-CreER (Stock No. 005107) (Vasioukhin et al., 1999), K15-CrePR1 (Stock No. 005249) (Ito et al., 2005), and Rosa26 mTmG (Stock No. 007676) transgenic mice were all obtained from Jackson Laboratories. Transgenic mice carrying the floxed Nrf2 allele were previously described in Reddy et al. (2011) and were generously provided by Dr. Shyam Biswal (Johns Hopkins Bloomberg School of Public Health).
K14-specific and K15-specific, homozygous Nrf2 double-transgenic mice (K14 CreER; Nrf2 fl/fl and K15 CrePR1; Nrf2 fl/fl, respectably) were generated from crossing heterozygous mice produced from K14-CreER X Nrf2 fl/fl or K15-CrePR1 X Nrf2 fl/fl genetic crosses. Mouse genotype was determined through PCR-based assays using tail DNA as template and primers described in Table S1. To induce Nrf2 deletion in K14+ basal epidermis, mice were treated with Tamoxifen (Sigma Aldrich, T5648–5G) (dissolved in corn oil at 20mg/mL and injected intraperitoneally at a dosage of 0.1 mg/g body weight) for 10 consecutive days upon weaning at 3 weeks of age. RU486 (Mifepristone) (Sigma Aldrich, M8046–500mg) (1% solution in ethanol was applied to depilated dorsal skin) was used to induce Nrf2 deletion in K15+ HFSCs for 10 consecutive days. Hair depilation was performed during telogen. Both sexes were used in the study.
METHOD DETAILS
Wounding Procedures
Methodology for full-thickness excisional wound has been previously described in Gurtner et al. (2008) and was carried out with minor modifications. Briefly, mice were anaesthetized with isofluorane and a full-thickness excisional wound was created on the back-skin of 6–8-week-old mice using a 10 mm sterile punch biopsy that was splinted with circular silicone stents (15 mm external diameter, 10 mm internal diameter) (Grace Bio-Labs, Bend, 665581) to prevent contraction. Wounds were photographed and collected at indicated time-points for histology or assays described. For rescue experiments, recombinant mouse Ccl2 (R&D Systems, 479-JE-019/CF) was dissolved in DMEM:F12 (1:1) growth medium (Thermo Fisher, 11330032) and applied exogenously (50ng/application) every other day for the first five days post-wounding. Control wounds were treated with growth medium only. Wounds were photographed and collected at indicated time-points for histology.
Hair Plucking Procedures
Hairs were plucked from 5×5mm areas on the dorsal skin during telogen with forceps. Hair regeneration was analyzed over time through histology.
Tissue Processing for Flow Cytometry
Microdissection of a ~2.5mm concentric ring around the 10mm wound was carried out using a 15mm-diameter punch biopsy. Wounded skin was harvested at indicated time points and digested in 10u/mL Dispase (Thermo Fisher, 17105041) and 5ug/mL Accutase (Bio-Legend, 42301) before passing the suspension through a 70um pore cell trainer. Lineage-positive cells and dead cells/debris were excluded from the cell suspension utilizing the MACS Lineage Depletion Kit (Miltenyi, 130–090-858) and MACS Dead Cell Removal Kit (Miltenyi, 130–090-101) following the manufacturer’s instructions. Cell number and viability was assessed using trypan blue and a hemocytometer. Dissociated single cells were suspended in 5% FBS in 1x PBS and incubated with various antibody combinations for multi-parameter flow acquisition and analysis. Live keratinocytes were sorted using antibodies directed against the following surface makers: Lin- (CD5-, CD45R-, CD11b-, Cr-1-, 7–4-, Ter-119-) (Miltenyi, 130–090-858), CD49f+ (Bio-Legend, 313610) using a SY3200 4 laser/10 color cell sorter. HFSCs were sorted using antibodies directed against Lin-, CD49f+, and CD34+ (BD PharMingen, 560238).
In Vitro Primary Keratinocyte Cultures
Primary mouse keratinocytes were isolated for in vitro experiments as reported in Lichti et al. (2008) with minor modifications. Briefly, mouse back skin was harvested and digested in 10u/mL Dispase (GIBCO, Life Technologies). Keratinocytes were then isolated using 5ug/mL Accutase (Bio-Legend, 42301). After, 5×105 freshly isolated primary keratinocytes were cultured on glass bottom microwell culture dishes (MatTek; P35G-1.5–14-C) without fibroblast feeders, using low-Ca2+ keratinocyte media (Lonza, 192060) at 37C. For Nrf2 induction assays, 100nM CDDO-IM (Torcis, 473710) was added after 24h to primary keratinocyte cultures for 1.5h, before replacing with new media. For in vitro Ccl2 assays, 50ng/mL recombinant mouse Ccl2 (R&D Biosystems, 479-JE) was added to primary keratinocyte cultures for 24h, before fixing with 4% PFA and permeabilizing with 0.1% Triton-X for downstream analysis. Protein expression analysis was carried out following immunofluorescence staining protocols detailed below. In vitro primary keratinocyte cultures were imaged using BioTek Cytation 7 Cell Imaging Multi-Mode Reader (Agilent).
For conditioned media studies on keratinocyte activity, macrophage-conditioned media was harvested after 24h and applied to primary mouse keratinocyte cultures for 24h before fixing with 4% PFA and permeabilizing cells with 0.1% Triton-X for downstream analysis. For studies determining the significance of EGF in macrophage conditioned media, primary mouse keratinocytes were pretreated with 250nM PD 153035 (Abcam, Ab141839) for 24h prior to receiving macrophage conditioned media. For Ccl2 silencing studies, primary keratinocytes were treated with siRNAs specific for mouse Ccl2 (Thermo Fisher, AM16708, ID151586) and siRNA controls with the of Lipofectamine 3000 (Invitrogen, L3000015) following the manufacturer’s directions.
For ex vivo explant migration assays, 2mm circular punch biopsies were taken from Nrf2 +/+ Ker; mTmG and Nrf2 Δ/Δ Ker; mTmG following procedures delineated in Keyes et al. (2016). Briefly, subcutaneous fat was gently removed from skin biopsies with a scalpel then secured to the bottom of culture dishes with ~2uL of Growth Factor Reduced Matrigel (BD Biosciences, 354230), and submerged in low-Ca2+ keratinocyte media (Lonza, 192060) at 37C. After 7d, explant outgrowths were imaged and analyzed using ImageJ/FIJI software (Schneider et al., 2012).
Immunohistochemistry and Immunofluorescence Staining
Intact skin and cutaneous wound tissue samples were fixed in 4% PFA overnight at 4C. Paraffin sections (5um thickness) were prepared and stained with hematoxylin and eosin (H&E stain), Masson’s trichrome stain (Trichrome collagen three-color staining protocol), Elastin stain, or with the appropriate antibodies (immunofluorescence staining). For immunostaining, tissues were deparaffinized, rehydrated, and processed for antigen retrieval using Citri-solve Hybrid (Fisher, 04355121), Ethanol (Sigma, E7023), and DAKO Target Retrieval Solution (DAKO, S1700) following manufacturers directions. Tissue sections were subsequently blocked in 5% normal goat serum (Jackson, 0005–000-121) or normal donkey serum (Jackson, 017–000-121) for 1h at room temperature. The primary and secondary antibodies used are listed in Key Resources Table. For immunohistochemistry, sections were washed in TBST before incubation with HRP-conjugated secondary antibody (Abcam, Ab6721) for 1hr at room temperature and followed treatment with IMMPACT DAB (Vector Laboratories, SK-4105). Tissues stained for Immunofluorescence labeling were counterstained with nuclear dye Hoechst 33342 (Thermo Fisher, 62249). Labeling was quantified from independent high-powered fields (20x) in independent biological replicates. Images were taken with a Nikon fluorescence upright microscope (Nikon) or a Zeiss Axio Observer (Zeiss). Confocal images were taken with a Zeiss 710 confocal microscope (Zeiss).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 647 anti-CD49f | Bio-Legend | 313610; RRID:AB_493637 |
| Armenian Hamster anti-Ccl2 | Bio X Cell | BE0185; RRID:AB_10950302 |
| Chicken anti-K14 | Bio-Legend | Poly9060; RRID:AB_2565055 |
| Chicken anti-K15 | Bio-Legend | 833904; RRID:AB_2616894 |
| Donkey Anti-Chicken Cy5 | Jackson | 703-175-155; RRID:AB_2340365 |
| Donkey anti-Rabbit AF 488 | Jackson | 711-546-152; RRID:AB_2340619 |
| Donkey anti-Rabbit AF594 | Jackson | 711-585-152; RRID:AB_2340621 |
| Donkey anti-Rat Cy2 | Jackson | 703-175-155; RRID:AB_2340365 |
| FITC anti-CD34 | BD Biosciences | 560238; RRID:AB_1645242 |
| Goat anti-8-OHdG | Novus | NB600-1508SS; RRID:AB_787860 |
| Goat anti-Chicken AF594 | Jackson | 103-585-155; RRID:AB_2337391 |
| Goat anti-Rabbit HRP | Abcam | Ab6721; RRID:AB_955447 |
| Goat anti-Rat AF594 | Jackson | 112-585-155 |
| Mouse anti-Erk2 | Santa Cruz Biotechnology | SC-1647; RRID:AB_627547 |
| Rabbit anti-Arginase 1 | Protein Tech | 16001-1-AP; RRID:AB_2289842 |
| Rabbit anti-beta Actin | Abcam | Ab8227; RRID:AB_2305186 |
| Rabbit anti-Cleaved Casp3 | Abcam | Ab2302; RRID:AB_302962 |
| Rabbit anti-Keratin 10 | Bio-Legend | Poly 19054; RRID:AB_2616955 |
| Rabbit anti-Nqo1 | Abcam | Ab34173; RRID:AB_2251526 |
| Rabbit anti-Nrf2 phospho. S40 | Abcam | Ab76026; RRID:AB_1524049 |
| Rabbit anti-SOD2 | Abcam | Ab13533; RRID:AB_300434 |
| Rabbit IgG | Abcam | Ab172569 |
| Rat anti-Ccl2 | R&D Biosystems | Mab479-100; RRID:AB_2259770 |
| Rat anti-CD31 | BD Biosciences | 553370; RRID:AB_394816 |
| Rat anti-F4/80 | Abcam | Ab6640; RRID:AB_1140040 |
| Rat anti-Ki-67 | Bio-Legend | 652401; RRID:AB_11203533 |
| Rat anti-Ly-6G | BD Biosciences | 551459; RRID:AB_394206 |
| Chemicals, Peptides and Recombinant Proteins | ||
| CDDO-IM | Torcis | 473710 |
| Hoechst 33342 | Thermo Fisher | 62249 |
| M-CSF | Millipore | M9170 |
| Mifepristone | Sigma-Aldrich | M8046-500mg |
| PD 153035 | Abcam | Ab141839 |
| Recombinant Mouse Ccl2 | R&D Biosystems | 479-JE |
| Tamoxifen | Sigma-Aldrich | T5648-5G |
| Critical Commercial Assays | ||
| High Capacity cDNA Kit | Thermo Fisher | 4368814 |
| Lineage Cell Depletion Kit, mouse | Miltenyi | 130-090-858 |
| Lipofectamine 3000 | Invitrogen | L3000015 |
| MACS Dead Cell Removal Kit | Miltenyi | 130-090-101 |
| Nugen Trio RNA-seq Kit | Nugen | 0507 |
| QIA-Quick PCR Purification Kit | QIAGEN | 28104 |
| RN-Easy Plus Micro Kit | QIAGEN | 74034 |
| siRNA Silencer (mouse Ccl2) | Thermo Fisher | AM16708; ID151586 |
| siRNA Silencer (negative control) | Thermo Fisher | AM4611 |
| SSO Adv. Univ. SYBR Green Super Mix | Bio-Rad | 172-5271 |
| Deposited Data | ||
| Bulk RNA-seq data | This paper | GEO Series accession: GSE159361 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | Jackson Laboratory | 000664 |
| BKS.Cg-Dock7m =/ = Lepr db/J | Jackson Laboratory | 000642 |
| Tg(KRT14-cre/ERT)20Efu/J | Jackson Laboratory | 005107 |
| B6;SJL-Tg(Krt15-cre/PGR)22Cot | Jackson Laboratory | 005249 |
| Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J | Jackson Laboratory | 007676 |
| Nrf2flox/flox | Gift from Dr. Shyam Biswal | N/A |
| Oligonucleotides | ||
| Nqo1 ChIP ARE site F: 5′-CACTCAGCCGTGGGAAGT-3′ | Life Technologies | N/A |
| Nqo1 ChIP ARE site R: 5′-AGCAGAACGCAGCACGAAT-3′ | Life Technologies | N/A |
| Nqo1 ChIP NS site F: 5′-TCTGGGGACTTGGGTATCTG-3′ | Life Technologies | N/A |
| Nqo1 ChIP NS site R: 5′-TACGCTGTAGTGGTGGTGGA-3′ | Life Technologies | N/A |
| Ccl2 ChIP ARE site F: 5′-ACTACTGCCTCCAA TCCTGAAATAA-3′ | Life Technologies | N/A |
| Ccl2 ChIP ARE site R: 5′-GGACCCTCCTTTTGTTACCT-3′ | Life Technologies | N/A |
| 18 s F: 5′-CGGCTACCAGATCCAAGGAA-3′ | Life Technologies | N/A |
| 18 s R: 5′-GCTGGAATTACCGCGGCTG-3′ | Life Technologies | N/A |
| Ccl2 F: 5′-GCAGCAGGTGTCCCAAAGAA-3′ | Life Technologies | N/A |
| Ccl2 R: 5′-TTGGTTCCGATCCAGGTTTTT-3′ | Life Technologies | N/A |
| Please refer to Table S1 for complete listing of oligonucleotides used in the study. | N/A | |
| Software and Algorithms | ||
| Cluster 3.0 | (de Hoon et al., 2004) | http://bonsai.hgc.jp/~mdehoon/software/cluster/ |
| Enrichr | (Kuleshov et al., 2016) | https://maayanlab.cloud/Enrichr/ |
| Gene Set Enrichment Analysis | (Subramanian et al., 2005) | https://www.gsea-msigdb.org/gsea/index.jsp |
| Graph pad Prism 8 | Graph pad | N/A |
| ImageJ /FIJI | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Java Tree-View | (Saldanha, 2004) | http://jtreeview.sourceforge.net/ |
| RSEM | (Li and Dewey, 2011) | https://deweylab.github.io/RSEM/ |
| STAR Aligner | (Dobin et al., 2013) | https://code.google.com/archive/p/rna-star/ |
| Trimmomatic | (Bolger et al., 2014) | http://www.usadellab.org/cms/?page=trimmomatic |
| Other | ||
| Accutase | Bio-Legend | 42301 |
| Dispase | Thermo Fisher | 17105041 |
| DMEM: F12 (1:1) | Thermo Fisher | 11330032 |
| Citri-Solve | Fisher | 04355121 |
| DAKO Target Retrieval Solution | DAKO | S1700 |
| Dynabeads Protein A | Thermo Fisher | 10001D |
| Ethanol | Sigma | E7023 |
| Glass-bottom Micro-well dishes | Mat-Tek | P35G-1.5-14-C |
| IMMPACT DAB | Vector Laboratories | SK-4105 |
| KAPA Hot-Start Ready Mix with Dye | KAPA Biosystems | KK5609 |
| KBM-Gold Basal Growth Medium | Lonza | 192060 |
| Matrigel Membrane Matrix | BD Biosciences | 354230 |
| Protein Assay Reagent | Bio-Rad | 5000006 |
| Proteinase K | QIAGEN | 1017738 |
| PVDF Immobilon-FL membranes | Millipore | IPF00010 |
| Silicone Isolator Sheets | Grace Bio-Labs | 665581 |
| Trizol | Life Technologies | 15596018 |
| Normal Donkey Serum | Jackson | 017-000-121 |
| Normal Goat Serum | Jackson | 0005-000-121 |
| SSO-Adv. Universal SYBR Green Super mix | Bio-Rad | 172-5271 |
Protein Lysates and Western Blot
For protein expression analysis, total protein was extracted from wound tissue with radio-immunoprecipitation assay (RIPA) buffer {150mM NaCl, 50mM Tris HCl (pH 8.0), 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS) and homogenized using a bead mill homogenizer (Omni International). Protein concentrations were quantified using the Bio-Rad Protein Assay Reagent (Bio-Rad, 5000006). Equal amounts were run on 10% poly-acrylamide gels and transferred onto PVDF Immobilon-FL membranes (Millipore, IPF00010) prior to probing with antibodies. The antibodies used are listed in Key Resources Table.
Keratinocyte Chromatin Immunoprecipitation (ChIP)
ChIP was performed according to a previously described protocol (Gu et al., 2013) with some modifications. 20mm-diameter punch biopsies encompassing the circular 10mm full-excisional cutaneous wound were harvested and digested in 10u/mL Dispase (Thermo Fisher, 17105041), before gently separating epithelial layer from the underlying dermis and digesting with Accutase (Bio-Legend, 42301) to create a single cell suspension. Primary keratinocytes were then cross-linked with 1% formaldehyde for 10 minutes at 37C and quenched with 125mM Glycine for 5 minutes at room temperature. After washing, chromatin was sheared into ~100–500-bp fragments using a Bioruptor Sonicator (Diagenode Inc.) at “high” setting and cycling 30 s ON and 30 s OFF for 30 minutes. A small aliquot of the recovered sheared chromatin was reverse cross-linked, purified and used to measure chromatin DNA concentration (Input sample). Immunoprecipitation was performed by incubating 15ug of chromatin overnight, at 4C with control IgG (Abcam, Ab172569) or anti-phospho Nrf2 (S40) antibody (Abcam, Ab76026). Immuno-complexes were then purified with Dynabeads protein A (Thermo Fisher, 10001D), washed, and underwent reverse cross-linking by treatment with 200mM NaCl, containing proteinase K (QIAGEN, 1017738) overnight at 65C. ChIP DNA was recovered using the QIA-Quick PCR purification kit (QIAGEN, 28104). ChIP-CR primers were designed based on the location of putative core ARE motifs. Primer sequences are listed in Key Resources Table.
Bulk RNaseq Transcript Alignment, Quantification, and Filtering
For RNA-seq, RNA was isolated from flow cytometry-sorted cells using RN-Easy Plus Micro Kit (QIAGEN, 74034). cDNA library preparation used optimal quality RNA (RIN > 9). Full length cDNA library amplification was performed as previously described. Briefly, total RNA was reverse-transcribed and the resulting cDNA was pre-amplified using the Nugen Trio RNaseq Kit (Nugen Inc., 0507). Paired-end 50 (PE50) sequencing was performed on the Illumina HiSeq 4000 (Illumina). Paired-end reads were trimmed using Trimmomatic (version 0.35) (Bolger et al., 2014) with the following parameters HEADCROP:5, LEADING:20 TRAILING:20, SLIDINGWINDOW:4:17 MINLEN:25. Trimmed reads were aligned to the mouse genome (mm10/gencode.vM13) with STAR aligner (version 2.6.0c) (Dobin et al., 2013) and quantified using the RNA-seq by Expectation-Maximization algorithm (RSEM) (version 1.2.31) (Li and Dewey, 2011) with the following standard parameters: rsem-calculate-expression, –paired-end. Samples displaying > 78% alignment were considered for downstream analyses. Samples were filtered using Python (Version 3.7.1) and only genes expressed at minimum 1 TPM in at least one sample in all biological replicates were kept and considered for downstream analyses. Only protein coding-, lncRNA, and miRNA genes were considered for downstream analyses. A pseudo-count of 0.1 was added to TPM values. Samples were clustered with Cluster3.0 (de Hoon et al., 2004) and visualized with JavaTreeView (Saldanha, 2004). Downstream analysis of RNA-seq data was carried out using Enrichr (Kuleshov et al., 2016) and Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005).
Gene Expression Analysis
For RNA isolation, Trizol (Life; 15596018) was used per manufacturer’s instructions. cDNA was synthesized using 500ng of RNA was using High Capacity cDNA Kit (Thermo Fisher; 4368814) per manufacturer’s instructions. qPCR was performed using a QuantStudio7 Flex System (Applied Biosciences) using SSO Advanced Universal SYBR Green Super Mix (Bio-Rad; 172–5271). Genotyping PCR analysis was carried out using KAPA Hot-Start Ready Mix with Dye (Kapa Biosystems, KK5609). qPCR and genotyping primers are listed in Key Resources Table and Table S1.
Production of Bone Marrow Derived Macrophages
Bone marrow derived macrophages (BMDM) were generated as described in Weischenfeldt and Porse (2008) with minor modifications. Briefly, bone marrow cells were isolated from mouse femur and tibia by flushing with a 25-guage needle using DMEM: F12 (1:1) (Thermo Fisher, 4368814) and subsequently passed through a 70um pore filter. To induce BMDMs, bone marrow cells were cultured in DMEM: F12 (1:1) supplemented with 10% FBS, 1% Pen/Strep, with 20 units M-CSF (Millipore, M9170). After 1-week, recombinant mouse 50ng/mL Ccl2 (R&D Biosystems, 479-JE) was added BMDM cultures and harvested after 24h for RNA isolation. After 24h, macrophage-conditioned media was harvested and applied to primary mouse keratinocyte cultures for 24h before fixing with 4% PFA and permeabilizing cells with 0.1% Triton-X for downstream analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments were carried out using Graph Pad Prism (Graph-pad) and can be found in the figure legends and results sections. A minimum of three biological replicates were performed for each experimental condition both in vitro and ex vivo experiments, and are represented as mean ± standard deviation (SD). For in vivo experiments, at least three mice were used per group and experimental sample sizes were determined on the basis of prior power calculations. Additional experiments were performed when greater variation was observed, and data were pooled for analysis. Unpaired t tests were used to analyze studies between two experimental groups. Analysis of variance (ANOVA) was used to analyze studies with three or more experimental groups. Results with a p value < 0.05 were considered statistically significant.
Supplementary Material
Highlights.
Epidermal Nrf2 initiates a regenerative response through Ccl2 regulation
Ccl2 release by basal stem/progenitor keratinocytes prompts macrophage trafficking
Ccl2 regulates EGF production in macrophages trafficked to the injury site
EGF from macrophages stimulates basal keratinocyte proliferation
ACKNOWLEDGMENTS
The authors acknowledge the generosity and support of Dr. Shyam Biswal (Johns Hopkins Bloomberg School of Public Health) for providing the Nrf2fl/fl mice described in our study. We thank the NYU Pathology, Cytometry & Cell Sorting, and High Throughput cores for their support. We acknowledge grants from the ADA (1-16-ACE-08) for funding this research. Q.N. is also supported by NIH grants U01-AR073159 and R01-GM123731, NSF grants DMS1161621 and DMS1562176, the Jayne Koskinas Ted Giovanis Foundation for Health and Policy, and the NSF-Simons Foundation. C.F.G.-J. is supported by the UCI Chancellor’s ADVANCE Fellowship Program, NSF-Simons Fellowship, NSF grant DMS1763272, and the Simons Foundation (594598 to Q.N.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108417.
REFERENCES
- Aleksunes LM, Goedken MJ, Rockwell CE, Thomale J, Manautou JE, and Klaassen CD (2010). Transcriptional regulation of renal cytoprotective genes by Nrf2 and its potential use as a therapeutic target to mitigate cisplatin-induced nephrotoxicity. J. Pharmacol. Exp. Ther 335, 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso L, and Fuchs E. (2006). The hair cycle. J. Cell Sci 119, 391–393. [DOI] [PubMed] [Google Scholar]
- Aragona M, Dekoninck S, Rulands S, Lenglez S, Mascré G, Simons BD, and Blanpain C. (2017). Defining stem cell dynamics and migration during wound healing in mouse skin epidermis. Nat. Commun 8, 14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- auf dem Keller U, Huber M, Beyer TA, Kümin A, Siemes C, Braun S, Bugnon P, Mitropoulos V, Johnson DA, Johnson JA, et al. (2006). Nrf transcription factors in keratinocytes are essential for skin tumor prevention but not for wound healing. Mol. Cell. Biol 26, 3773–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakytny R, and Jude E. (2006). The molecular biology of chronic wounds and delayed healing in diabetes. Diabet. Med 23, 594–608. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S, Hanselmann C, Gassmann MG, auf dem Keller U, Born-Berclaz C, Chan K, Kan YW, and Werner S. (2002). Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol. Cell. Biol 22, 5492–5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee M. (2005). The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54, 1615–1625. [DOI] [PubMed] [Google Scholar]
- Cambien B, Pomeranz M, Millet MA, Rossi B, and Schmid-Alliana A. (2001). Signal transduction involved in MCP-1-mediated monocytic transendothelial migration. Blood 97, 359–366. [DOI] [PubMed] [Google Scholar]
- Chen CC, Wang L, Plikus MV, Jiang TX, Murray PJ, Ramos R, Guerrero-Juarez CF, Hughes MW, Lee OK, Shi S, et al. (2015). Organ-level quorum sensing directs regeneration in hair stem cell populations. Cell 161, 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, Xue P, Pi J, Kleeberger SR, and Bell DA (2012). Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 40, 7416–7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJL, Imoto S, Nolan J, and Miyano S. (2004). Open source clustering software. Bioinformatics 20, 1453–1454. [DOI] [PubMed] [Google Scholar]
- Deng B, Wehling-Henricks M, Villalta SA, Wang Y, and Tidball JG (2012). IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol 189, 3669–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, and Frangogiannis NG (2005). CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res 96, 881–889. [DOI] [PubMed] [Google Scholar]
- Dipietro LA, Reintjes MG, Low QEH, Levi B, and Gamelli RL (2001). Modulation of macrophage recruitment into wounds by monocyte chemoattractant protein-1. Wound Repair Regen. 9, 28–33. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis S, Lin EJ, and Tartar D. (2018). Immunology of Wound Healing. Curr. Dermatol. Rep 7, 350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E. (2007). Scratching the surface of skin development. Nature 445, 834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Wang FQ, Li HM, Yang JG, Ren J-G, He KF, Liu B, Zhang W, and Zhao YF (2016). CCL2/EGF positive feedback loop between cancer cells and macrophages promotes cell migration and invasion in head and neck squamous cell carcinoma. Oncotarget 7, 87037–87051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacco F, and Brownlee M. (2010). Oxidative stress and diabetic complications. Circ. Res 107, 1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godwin JW, Pinto AR, and Rosenthal NA (2013). Macrophages are required for adult salamander limb regeneration. Proc. Natl. Acad. Sci. USA 110, 9415–9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwandtner M, Derler R, and Midwood KS (2019). More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol 10, 2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B, Watanabe K, Sun P, Fallahi M, and Dai X. (2013). Chromatin effector Pygo2 mediates Wnt-notch crosstalk to suppress luminal/alveolar potential of mammary stem and basal cells. Cell Stem Cell 13, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, and Longaker MT (2008). Wound repair and regeneration. Nature 453, 314–321. [DOI] [PubMed] [Google Scholar]
- Haase I, Evans R, Pofahl R, and Watt FM (2003). Regulation of keratinocyte shape, migration and wound epithelialization by IGF-1- and EGF-dependent signalling pathways. J. Cell Sci 116, 3227–3238. [DOI] [PubMed] [Google Scholar]
- Hancock GE, Kaplan G, and Cohn ZA (1988). Keratinocyte growth regulation by the products of immune cells. J. Exp. Med 168, 1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi R, Himori N, Taguchi K, Ishikawa Y, Uesugi K, Ito M, Duncan T, Tsujikawa M, Nakazawa T, Yamamoto M, and Nishida K. (2013). The role of the Nrf2-mediated defense system in corneal epithelial wound healing. Free Radic. Biol. Med 61, 333–342. [DOI] [PubMed] [Google Scholar]
- Hesketh M, Sahin KB, West ZE, and Murray RZ (2017). Macrophage phenotypes regulate scar formation and chronic wound healing. Int. J. Mol. Sci 18, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiebert P, Wietecha MS, Cangkrama M, Haertel E, Mavrogonatou E, Stumpe M, Steenbock H, Grossi S, Beer HD, Angel P, et al. (2018). Nrf2-Mediated Fibroblast Reprogramming Drives Cellular Senescence by Targeting the Matrisome. Dev. Cell 46, 145–161.e10. [DOI] [PubMed] [Google Scholar]
- Hsu YC, Li L, and Fuchs E. (2014). Emerging interactions between skin stem cells and their niches. Nat. Med 20, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, and Pickett CB (2002). Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem 277, 42769–42774. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Kuninaka Y, Nosaka M, Furuta M, Kimura A, Taruya A, Yamamoto H, Shimada E, Akiyama M, Mukaida N, and Kondo T. (2019). CCL2-Mediated Reversal of Impaired Skin Wound Healing in Diabetic Mice by Normalization of Neovascularization and Collagen Accumulation. J. Invest. Dermatol 139, 2517–2527.e5. [DOI] [PubMed] [Google Scholar]
- Italiani P, and Boraschi D. (2014). From monocytes to M1/M2 macrophages: Phenotypical vs. functional differentiation. Front. Immunol 5, 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Liu Y, Yang Z, Nguyen J, Liang F, Morris RJ, and Cotsarelis G. (2005). Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med 11, 1351–1354. [DOI] [PubMed] [Google Scholar]
- Joost S, Jacob T, Sun X, Annusver K, La Manno G, Sur I, and Kasper M. (2018). Single-Cell Transcriptomics of Traced Epidermal and Hair Follicle Stem Cells Reveals Rapid Adaptations during Wound Healing. Cell Rep. 25, 585–597.e7. [DOI] [PubMed] [Google Scholar]
- Keyes BE, Liu S, Asare A, Naik S, Levorse J, Polak L, Lu CP, Nikolova M, Pasolli HA, and Fuchs E. (2016). Impaired Epidermal to Dendritic T Cell Signaling Slows Wound Repair in Aged Skin. Cell 167, 1323–1338.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, Bhasker V, Gordillo GM, Sen CK, and Roy S. (2010). Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS ONE 5, e9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Thimmulappa R, Craciun F, Harvey C, Singh A, Kombairaju P, Reddy SP, Remick D, and Biswal S. (2011). Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am. J. Respir. Crit. Care Med 184, 928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyszczyk P, Schloss R, Palmer A, and Berthiaume F. (2018). The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Front. Physiol 9, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44 (W1), W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, and Ma’ayan A. (2010). ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Yu H, Pan H, Zhou X, Ruan Q, Kong D, Chu Z, Li H, Huang J, Huang X, et al. (2019). Nrf2 suppression delays diabetic wound healing through sustained oxidative stress and inflammation. Front. Pharmacol. 10, 1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichti U, Anders J, and Yuspa SH (2008). Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat. Protoc 3, 799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M, Rojo de la Vega M, Wen Q, Bharara M, Jiang T, Zhang R, Zhou S, Wong PK, Wondrak GT, Zheng H, et al. (2016). An Essential Role of NRF2 in Diabetic Wound Healing. Diabetes 65, 780–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low QEH, Drugea IA, Duffner LA, Quinn DG, Cook DN, Rollins BJ, Kovacs EJ, and DiPietro LA (2001). Wound healing in MIP-1a(—/—) and MCP-1(—/—) mice. Am. J. Pathol 159, 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, and Biswal S. (2010). Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama K, Asai J, Ii M, Thorne T, Losordo DW, and D’Amore PA (2007). Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am. J. Pathol 170, 1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellado M, Rodríguez-Frade JM, Aragay A, del Real G, Martín AM, Vila-Coro AJ, Serrano A, Mayor F Jr., and Martínez-A C. (1998). The chemokine monocyte chemotactic protein 1 triggers Janus kinase 2 activation and tyrosine phosphorylation of the CCR2B receptor. J. Immunol 161, 805–813. [PubMed] [Google Scholar]
- Michaels J 5th, Churgin SS, Blechman KM, Greives MR, Aarabi S, Galiano RD, and Gurtner GC (2007). db/db mice exhibit severe wound-healing impairments compared with other murine diabetic strains in a silicone-splinted excisional wound model. Wound Repair Regen. 15, 665–670. [DOI] [PubMed] [Google Scholar]
- Moore K, Ruge F, and Harding KG (1997). T lymphocytes and the lack of activated macrophages in wound margin biopsies from chronic leg ulcers. Br. J. Dermatol 137, 188–194. [DOI] [PubMed] [Google Scholar]
- Morris RJ, Liu Y, Marles L, Yang Z, Trempus C, Li S, Lin JS, Sawicki JA, and Cotsarelis G. (2004). Capturing and profiling adult hair follicle stem cells. Nat. Biotechnol 22, 411–417. [DOI] [PubMed] [Google Scholar]
- Müller-Röver S, Handjiski B, van der Veen C, Eichmüller S, Foitzik K, McKay IA, Stenn KS, and Paus R. (2001). A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J. Invest. Dermatol 117, 3–15. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, and Luo L. (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. [DOI] [PubMed] [Google Scholar]
- Muzumdar S, Hiebert H, Haertel E, Ben-Yehuda Greenwald M, Bloch W, Werner S, and Schäfer M. (2019). Nrf2-Mediated Expansion of Pilosebaceous Cells Accelerates Cutaneous Wound Healing. Am. J. Pathol 189, 568–579. [DOI] [PubMed] [Google Scholar]
- Naik S, Larsen SB, Cowley CJ, and Fuchs E. (2018). Two to Tango: Dialog between Immunity and Stem Cells in Health and Disease. Cell 175, 908–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen TX, Leethanakul C, Patel V, Mitola D, Lund LR, Danø K, Johnsen M, Gutkind JS, and Bugge TH (2003). Laser capture microdissection-based in vivo genomic profiling of wound keratinocytes identifies similarities and differences to squamous cell carcinoma. Oncogene 22, 3964–3976. [DOI] [PubMed] [Google Scholar]
- Rabbani PS, Zhou A, Borab ZM, Frezzo JA, Srivastava N, More HT, Rifkin WJ, David JA, Berens SJ, Chen R, et al. (2017). Novel lipoproteoplex delivers Keap1 siRNA based gene therapy to accelerate diabetic wound healing. Biomaterials 132, 1–15. [DOI] [PubMed] [Google Scholar]
- Rabbani PS, Ellison T, Waqas B, Sultan D, Abdou S, David JA, Cohen JM, Gomez-Viso A, Lam G, Kim C, et al. (2018). Targeted Nrf2 activation therapy with RTA 408 enhances regenerative capacity of diabetic wounds. Diabetes Res. Clin. Pract 139, 11–23. [DOI] [PubMed] [Google Scholar]
- Reddy NM, Potteti HR, Mariani TJ, Biswal S, and Reddy SP (2011). Conditional deletion of Nrf2 in airway epithelium exacerbates acute lung injury and impairs the resolution of inflammation. Am. J. Respir. Cell Mol. Biol 45, 1161–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisman SA, Buckley DB, Tanaka Y, and Klaassen CD (2009). CDDO-Im protects from acetaminophen hepatotoxicity through induction of Nrf2-dependent genes. Toxicol. Appl. Pharmacol 236, 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ (2004). Java Treeview—extensible visualization of microarray data. Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger MA, and Paller AS (2015). The Roles of Growth Factors in Keratinocyte Migration. Adv. Wound Care (New Rochelle) 4, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen CK (2019). Human Wounds and Its Burden: An Updated Compendium of Estimates. Adv. Wound Care (New Rochelle) 8, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw TJ, and Martin P. (2009). Wound repair at a glance. J. Cell Sci 122, 3209–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MA, Cohen OD, Low YC, Sartor RA, Ellison T, Anil U, Anzai L, Chang JB, Saadeh PB, Rabbani PS, and Ceradini DJ (2016). Restoration of Nrf2 signaling normalizes the regenerative niche. Diabetes 65, 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg A, Calafat J, Janssen H, Daams H, van der Raaij-Helmer LMH, Falcioni R, Kennel SJ, Aplin JD, Baker J, Loizidou M, et al. (1991). Integrin α 6/β 4 complex is located in hemidesmosomes, suggesting a major role in epidermal cell-basement membrane adhesion. J. Cell Biol 113, 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli C, Chio IIC, and Tuveson DA (2018). Transcriptional Regulation by Nrf2. Antioxid. Redox Signal 29, 1727–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner SJ, Domin J, Waterfield MD, Ward SG, and Westwick J. (1998). The CC chemokine monocyte chemotactic peptide-1 activates both the class I p85/p110 phosphatidylinositol 3-kinase and the class II PI3K-C2α. J. Biol. Chem 273, 25987–25995. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Degenstein L, Wise B, and Fuchs E. (1999). The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc. Natl. Acad. Sci. USA 96, 8551–8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, and Porse B. (2008). Bone marrow-derived macrophages (BMM): Isolation and applications. CSH Protoc 2008, t5080. [DOI] [PubMed] [Google Scholar]
- Wetzler C, Kämpfer H, Stallmeyer B, Pfeilschifter J, and Frank S. (2000). Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: prolonged persistence of neutrophils and macrophages during the late phase of repair. J. Invest. Dermatol 115, 245–253. [DOI] [PubMed] [Google Scholar]
- Wynn TA, and Vannella KM (2016). Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44, 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The bulk RNA-seq dataset generated during this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE159361.