

Abstract
Background
Massively parallel sequencing (MPS) is a promising supplementary method for forensic casework in short tandem repeats (STRs) genotyping, owing to several advantageous features in comparison to traditional capillary electrophoresis (CE). However, the application of MPS in casework requires accessible datasets from the worldwide population to enrich the allele frequencies of sequence‐based STR genotypes.
Methods
In this study, we report the characterization of sequence‐based allele frequencies of 58 STRs from a Tibetan population comprising 120 unrelated individuals using the ForenSeq™ DNA Signature Prep Kit. A concordance study evaluating MPS and CE allele data was performed to ensure that MPS is compatible with current CE‐based forensic databases. The diversity of observed alleles, allele frequencies, and forensic parameters per locus by length (LB), sequence without flanking region (RSB), and sequence with flanking region (FSB) were analyzed and compared.
Results
The concordance study demonstrated a concordance rate exceeding 99%. The combined random match probability (RMP) for the 26 A‐STRs was 2.04 × 10–29, 1.93 × 10–31, and 9.56 × 10–33 for LB, RSB, and FSB, respectively. Similar trends were observed in other forensic parameters resulting from the increase in the number of unique alleles available. A total of 111 and 113 unique haplotypes in the Y‐STR loci were observed when using length‐based and sequence‐based alleles, respectively. In addition, we identified 35 novel alleles at 25 loci and 25 polymorphisms in the flanking regions at 17 STRs.
Conclusions
Our data suggest that MPS‐ and CE‐derived alleles are compatible. MPS‐based analysis of the STR data substantially increased the allele diversity and improved the forensic parameters, which clearly demonstrated the advantages of MPS in comparison to CE. With more pooled data and larger‐scale validation, MPS could play a valuable role in forensic genetics and might be an additional tool for routine casework.
Keywords: flanking region, forensic genetics, MPS, population genetics, STR
We characterized MPS‐STR data in a Tibetan population to enrich the allele frequencies of sequence‐based STR genotypes for the application of MPS in forensic casework. CE‐STR data were also analyzed and compared to MPS‐STR data, which clearly demonstrated the advantages and compatibility of MPS.
![]()
1. INTRODUCTION
Short tandem repeats (STRs) are known to be ubiquitous across the human genome, and present with sufficient variability to allow for the identification of individuals, making them ideal in forensic genetic applications (Butler, 2005; Jobling & Gill, 2004). STRs are routinely analyzed using capillary electrophoresis (CE), which is considered the gold standard for forensic genetics and has been widely recognized in criminal investigations and prosecutions for over two decades (Butler, 2015; Butler et al., 2004; Thompson et al., 2012). However, the CE method only detects amplicon length while overlooking potentially informative sequence variation. The high polymorphism rates of STRs are underutilized.
Massively parallel sequencing (MPS), also known as next‐generation sequencing (NGS), has been shown to have potential in forensic genetics over the past few years (Alvarez‐Cubero et al., 2017; Aly & Sabri, 2015; Børsting & Morling, 2015; Bruijns et al., 2018). The potentially informative sequence variations in STRs (both in the repeat and in the flanking regions) can be evaluated by MPS, broadening STR diversity and increasing the discrimination powers of analytical tests (Barrio et al., 2019; Churchill et al., 2017; Delest et al., 2020; Gettings et al., 2015, 2016; Hussing et al., 2019; Jäger et al., 2017; Khubrani et al., 2019; Kim et al., 2016, 2018; Novroski et al., 2016; Peng et al., 2020; Phillips, Devesse, et al., 2018; Phillips, Gettings, et al., 2018; Wang et al., 2020; Wendt et al., 2017). In addition, MPS allows for the simultaneous analysis of significantly more loci than CE as it is not limited by restrictions in size‐based separation or fluorescence dye detection (Li et al., 2017). The smaller amplicon sizes in MPS may also improve the analysis of challenging or degraded DNA samples (Elwick et al., 2019; Fattorini et al., 2017; Kuffel et al., 2020). Given these advantages MPS is a promising supplementary method for forensic casework.
The application of MPS in casework requires accessible datasets from the global population designed to enrich the allele frequencies of sequence‐based STR genotypes, as recommended by the International Society for Forensic Genetics (ISFG) (Parson et al., 2016; Phillips, Devesse, et al., 2018; Phillips, Gettings, et al., 2018). Several MPS‐STR population datasets have been reported in recent years (Churchill et al., 2017; Delest et al., 2020; Hussing et al., 2019; Khubrani et al., 2019; Kim et al., 2016, 2018; Novroski et al., 2016; Peng et al., 2020; Phillips, Devesse, et al., 2018; Phillips, Gettings, et al., 2018; Wang et al., 2020; Wendt et al., 2017). Tibetans are one of China's 56 ethnic groups and are indigenous to the Qinghai‐Tibet Plateau. In China, Tibetans are primarily distributed across the Tibet Autonomous Region in the Qinghai, western Sichuan Province, Diqing in Yunnan, and Gannan in Gansu. Additionally, some Tibetans live in India, Bhutan, the United States, Canada, Europe, Australia, and other parts of the world. In this study, we report the characterization of sequence‐based allele frequencies from 58 STRs in a Tibetan population comprising 120 unrelated individuals using the ForenSeq DNA Signature Prep Kit (Verogen, San Diego, CA, USA), an MPS panel validated by several researchers and laboratories worldwide (Guo et al., 2017; Köcher et al., 2018; Wu et al., 2019). Notably, the genotypes obtained using MPS must be consistent with those obtained by CE to ensure that these data are compatible with current forensic databases (Devesse et al., 2018, 2020). Therefore, prior to the characterization of the sequence variation and the allele frequencies, a concordance study between the two methods was performed in our study.
2. MATERIALS AND METHODS
2.1. DNA sampling, extraction, and quantification
Peripheral blood samples were collected using FTA cards from 120 unrelated male individuals who claimed to be indigenous Tibetans residing in Lhasa, the capital of Tibet, who could track their heritage by at least three generations. All individuals provided written informed consent and genomic DNA was extracted using the BioRobotEZ1 Advanced XL and EZ1 DNA Investigator kits (Qiagen) according to the manufacturer's instructions. DNA was then quantified using a Qubit 2.0 Fluorometer and a Qubit dsDNA HS Assay Kit (Thermo Fisher). This study was approved by the Fudan University ethics committee (2020016).
2.2. Library preparation and sequencing
Libraries were prepared using the ForenSeqTM DNA Signature Prep Kit according to the manufacturer’ s instructions (https://verogen.com/documentation/). Briefly, library preparation included an initial two‐step PCR, using 1 ng of template DNA, to amplify the target loci and facilitate indexed adapter enrichment which help during the purification, and normalization of these libraries in the next step. Primer Mix B was used to amplify 58 STRs and 172 SNPs (not reported in this study). The prepared libraries were then pooled and denatured, and sequencing was performed using the Miseq FGx instrument (Verogen). Pooled libraries were placed in a Miseq FGx reagent cartridge and a flow cell facilitated the release of the incorporation buffers and sequencing reagents in accordance with the standard protocol. Five sequencing runs were performed and a negative and positive amplification control (2800 M, Verogen) was added to each run.
2.3. Sequence data analysis
Sequence data were analyzed using ForenSeqTM Universal Analysis Software (UAS) version 1.3 with Verogen's default settings. The analytical threshold (AT) and interpretation threshold (IT) were set at 1.5% and 4.5%, respectively. The STR intra‐locus balance threshold was set at 60% and the stutter filter was adjusted to reflect the specific needs of each locus. The minimum AT and IT were set at 10 and 30 reads, respectively, and the UAS used 650 reads as the minimum threshold before applying the AT and IT values. All sequence data were exported to Microsoft Office Excel and reviewed manually. All alleles identified in the sequencing analysis were then integrated into the Excel documents for further analysis.
2.4. Concordance study
Four commercial CE‐based STR kits were used to evaluate the overlap between STR genotypes generated using MPS and CE methods. Autosomal STRs (A‐STRs) were typed using a PowerPlex 21 System (Promega) and AGCU 21 + 1 Multiplex PCR Amplification Kits (AGCU), whereas Y chromosome STRs (Y‐STRs) and X chromosome STRs (X‐STRs) were typed using the Yfiler™ Platinum PCR Amplification (Thermo Fisher) and AGCU X19 Multiplex PCR Amplification Kits (AGCU), respectively. All 58 STRs (27 A‐STRs, 22 Y‐STRs, and 7 X‐STRs) were covered apart from DYS505 and DYS612. PCR products were separated and evaluated using an ABI 3500XL Genetic Analyzer (Thermo Fisher) according to the manufacturer's instructions. The electrophoretic results were analyzed using GeneMapper® ID‐X software v1.4 (Thermo Fisher). Any discordance within the CE‐based typing was evaluated using the binary sequence alignment (BAM) file and the Integrative Genomics Viewer (IGV) (Thorvaldsdóttir et al., 2013).
2.5. Identification of sequence variants
Sequence variation and allele frequencies were calculated and integrated into Excel according to the ISFG recommendations (Parson et al., 2016; Phillips, Devesse, et al., 2018; Phillips, Gettings, et al., 2018), to allow the comparison of these data with the records of the STR Sequencing Project (Gettings et al., 2017) and various other previous studies (Barrio et al., 2019; Churchill et al., 2017; Delest et al., 2020; Devesse et al., 2018, 2020; Gettings et al., 2015, 2016; Hussing et al., 2019; Jäger et al., 2017; Khubrani et al., 2019; Kim et al., 2016, 2018; Novroski et al., 2016; Peng et al., 2020; Phillips, Devesse, et al., 2018; Phillips, Gettings, et al., 2018; Wang et al., 2020; Wendt et al., 2017). Sanger sequencing was used to verify any novel alleles using a BigDye1 Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher).
2.6. Forensic parameters and Y‐haplotype frequencies
The forensic parameters for all the A‐STRs were calculated using STRAF (Gouy & Zieger, 2017) and included by length (LB), sequence without flanking region (RSB), and sequence with flanking region (FSB). The forensic parameters included genotype count (N), allele count based on sequence (Nall), expected heterozygosity (Hexp) or genetic diversity (GD), polymorphism information content (PIC), random match probability (RMP), power of discrimination (PD), observed heterozygosity (Hobs), power of exclusion (PE), and typical paternity index (TPI). Allele frequencies (LB, RSB, and FSB,) were calculated in Excel (allele count/total). STRAF was also used to test for Hardy–Weinberg equilibrium (HWE), applying a Bonferroni correction for multiple comparisons.
The Y‐haplotype frequencies were calculated using the direct counting method incorporating LB, RSB, and FSB alleles. RMP was calculated using the equation P = ∑χ2, whereas discrimination power (DP) was calculated using DP = 1−∑χ2 and haplotype diversity (HD) was calculated using HD = n(1‐∑χ2)/n−1, where χ is the frequency of each Y‐STR haplotype.
3. RESULTS AND DISCUSSION
3.1. Sequencing results
This study produced 17,510,320 reads over five sequencing runs. No allele above the interpretation threshold was detected in any of the negative amplification controls. Sequencing metrics for each run are shown in Table S1. Data analyzed by UAS were exported to Microsoft Office Excel sheets for manual review.
Here, we identified a heterozygous imbalance in D22S1045 which may cause some allele dropout resulting in some heterozygotes appearing to be homozygotes. For this reason, no further analysis (concordance study, allele frequency, and forensic parameters) was performed for this locus. In addition, we identified a high rate of allelic dropout at DYS392 (5%, 6/120). The poor performance at these two loci is due to the data quality issues/interpretation challenges, regardless of total sample read counts according to previous reports (Just et al., 2017; Novroski et al., 2016; Peng et al., 2020), which was also noted by the manufacturer's protocol (Verogen, 2018). Besides D22S1045 and DYS392, the instances of allelic dropout were also observed at another three loci: PentaE (2/240), DYS448 (1/120), and DXS7132 (1/120). A complete list of all A‐STRs and Y‐STRs alleles identified in 120 individuals is detailed in Tables S3 and S5.
3.2. Concordance study
Concordance refers to the likelihood of obtaining the same allele calls using any one of multiple methods, in this case MPS and CE. Before being implemented in routine casework, MPS data should be shown to be compatible with current CE‐based forensic databases (Devesse et al., 2018, 2020). It is crucial to evaluate the consistency between these two methods. In our study, all loci in the ForenSeq DNA Signature Prep Kit were compared except for D22S1045, as explained above, and DYS505 and DYS612, which were not included in all of the CE kits used in this study. The CE‐based profiles of all A‐STRs and Y‐STRs are detailed in Tables S2 and S4.
Our data suggest that there is over 99% concordance between each of the data points evaluated in this experiment. In the instances of allelic dropout mentioned above, alleles were obtained by CE. We assumed that the allelic dropout at DYS392 was due to the defects of the ForenSeq DNA Signature Prep Kit. The allelic drop out at the other loci was likely associated with SNPs or other mutations within the MPS primer binding sites.
One discordance, at DYS439, was observed. With the exception of DYS385a‐b and DYS387S1, Y‐STRs are expected to present with only one allele. However, our sample data suggested that there was a duplicated allele at DYS439. This duplication was only detected by MPS, whereas the CE‐based genotyping appeared normal. This discordance could have been caused by differences in the primer sequences used for the MPS and CE assays, as described by Kwon et al. (2016).
There was an additional discordance at the PentD allele where the CE‐based genotyping called a 9.2, 11 pattern and the MPS‐based genotyping called a 10, 11 ([AAAGA]10, [AAAGA]11). The allele with the [AAAGA]10 repeat was subsequently confirmed after the BAM file was checked using the IGV program. No sequence variation in the flanking regions was detected using UAS. The sample was sequenced using primers designed to extend the detection in the flanking region. A rare indel (rs1176142838) in the downstream flanking region was observed, which was determined to cause the discordance. The explanation is that the position of the three bases (TAA) deletion is outside of the bioinformatic recognition sites but within the CE amplified region. The original sequence data showed no discordance in the length of the amplicons between MPS‐based and CE‐based genotype. Thus, the discordance was due to bioinformatics configurations, as described by Gettings et al. (2016). Overall, the concordance rate between the two methods is extremely high and we can assume that the accidental discordances can be further reduced by improving bioinformatics analysis methods and expanding the available pool of MPS‐STR population data.
3.3. Sequence variation and diversity of observed alleles
Without considering D22S1045, the instances of allelic dropout, duplicated Y‐STR alleles, and the discordance described above, we were able to identify 10187 alleles from 120 individuals (A‐STRs: 6236 alleles, Y‐STRs: 3112 alleles, and X‐STRs: 839 alleles). MPS analysis of these STRs substantially increased the allele diversity, as demonstrated in previous studies (Barrio et al., 2019; Churchill et al., 2017; Delest et al., 2020; Gettings et al., 2015, 2016; Hussing et al., 2019; Jäger et al., 2017; Khubrani et al., 2019; Kim et al., 2016, 2018; Novroski et al., 2016; Peng et al., 2020; Phillips, Devesse, et al., 2018; Phillips, Gettings, et al., 2018; Wang et al., 2020; Wendt et al., 2017). The increase in the number of unique alleles varied from locus to locus.
For the A‐STR loci, 235 unique LB alleles, 325 unique RSB alleles, and 363 unique FSB alleles were observed. With sequence variation in the repeat and flanking regions identified in over 69% of the loci (18/26) demonstrating an increase in the number of unique alleles identified using MPS versus CE. D21S11 exhibited the highest diversity, with 33 unique alleles and TPOX displaying the lowest diversity, with only five unique alleles. The number of unique alleles per locus by LB, RSB, and FSB was compared to each other, as shown in Table 1a and Figure 1. Eight loci showed an increase in allele diversity due to variation within the repeat region sequence alone (D1S1656, D2S1338, D3S1358, D4S2408, D8S1179, D9S1122, D12S391, and FGA), whereas three loci exhibited increased allele diversity due to variations in only the flanking region (D5S818, D10S1248, and D16S539) and seven loci displayed an increase in allele diversity due to variations in the repeat and flanking region sequences (D2S441, D6S1043, D7S820, D13S317, D20S482, D21S11, and vWA). The eight remaining loci demonstrated no increase in allele diversity (D17S1301, D18S51, D19S433, CSF1PO, PentaD, PentaE, TH01, and TPOX). These results are similar but slightly different from those of previous studies (Barrio et al., 2019; Churchill et al., 2017; Delest et al., 2020; Gettings et al., 2015, 2016; Hussing et al., 2019; Jäger et al., 2017; Khubrani et al., 2019; Kim et al., 2016, 2018; Novroski et al., 2016; Peng et al., 2020; Phillips, Devesse, et al., 2018; Phillips, Gettings, et al., 2018; Wang et al., 2020; Wendt et al., 2017). Gettings et al. (2016) reported an increase in allele diversity at D19S433 and PentaE, whereas no sequence variation was observed at D7S820, D13S317, and D16S539 in their study (three populations, N = 183). Sequence variation was observed at D19S433 and PentaD in the study by Delest et al. (2020) (French population, N = 169), whereas in the study by Khubrani et al. (2019), sequence variation was observed at TH01 but not at D6S1043 (Arab population, N = 89). Novroski et al. (2016) reported sequence variations in all the A‐STRs except TPOX (four populations, N = 777). This suggests that sequence variations differ slightly between different populations, and these slight differences are likely due to the differences in genetic admix and the number of samples investigated.
TABLE 1A.
The number of unique alleles observed at 26 A‐STRs by LB, by RSB, and by FSB
| Locus | LB | RSB | FSB | Increase | %Increase |
|---|---|---|---|---|---|
| D12S391 | 11 | 30 | 30 | 19 | 172.73 |
| D21S11 | 13 | 32 | 33 | 20 | 153.85 |
| D13S317 | 8 | 9 | 20 | 12 | 150.00 |
| D2S1338 | 11 | 25 | 25 | 14 | 127.27 |
| D7S820 | 8 | 10 | 18 | 10 | 125.00 |
| D3S1358 | 6 | 13 | 13 | 7 | 116.67 |
| D20S482 | 6 | 8 | 13 | 7 | 116.67 |
| D8S1179 | 9 | 17 | 17 | 8 | 88.89 |
| D16S539 | 7 | 7 | 13 | 6 | 85.71 |
| D2S441 | 7 | 10 | 12 | 5 | 71.42 |
| D1S1656 | 9 | 14 | 14 | 5 | 55.56 |
| D9S1122 | 8 | 12 | 12 | 4 | 50.00 |
| D5S818 | 7 | 7 | 10 | 3 | 42.86 |
| D10S1248 | 6 | 6 | 8 | 2 | 33.33 |
| vWA | 8 | 10 | 10 | 2 | 25.00 |
| D4S2408 | 5 | 6 | 6 | 1 | 20.00 |
| D6S1043 | 15 | 17 | 17 | 2 | 13.33 |
| FGA | 15 | 16 | 16 | 1 | 6.67 |
| D17S1301 | 6 | 6 | 6 | 0 | 0.00 |
| D18S51 | 13 | 13 | 13 | 0 | 0.00 |
| D19S433 | 11 | 11 | 11 | 0 | 0.00 |
| CSF1PO | 8 | 8 | 8 | 0 | 0.00 |
| PentaD | 9 | 9 | 9 | 0 | 0.00 |
| PentaE | 18 | 18 | 18 | 0 | 0.00 |
| TH01 | 6 | 6 | 6 | 0 | 0.00 |
| TPOX | 5 | 5 | 5 | 0 | 0.00 |
| Total | 235 | 325 | 363 | 128 | 54.47 |
Abbreviations: A‐STRs, Autosomal STRs; FSB, sequence‐based alleles with flanking region; LB, length‐based alleles; RSB, sequence‐based alleles without flanking region.
FIGURE 1.
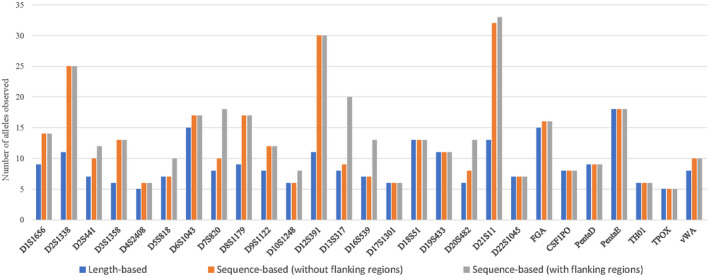
Allele diversity of A‐STRs based on length or sequence (with or without flanking regions)
There were 145 unique LB alleles, 203 unique RSB alleles, and 212 unique FSB alleles identified in our Y‐STR data. While there were 61 unique LB alleles, 78 unique RSB alleles, and 78 unique FSB alleles within the X‐STR samples. The increases in diversity for both the Y‐ and X‐STRs were smaller than that of the A‐STR loci. Approximately 53.8% of Y‐STRs (14/26) presented with increased allelic diversity, whereas only 42.9% (3/7) of the X‐STR loci exhibited any increases in diversity. Among these loci, nine of the Y‐STR loci gained in allele diversity due to variation in the repeat region sequence alone (DYF387S1, DYS389I, DYS389II, DYS439, DYS448, DYS481, DYS612, and DYS635). Three exhibited increased diversity caused by changes in the flanking regions alone (DYS19, DYS460, and Y‐GATA‐H4) and two Y‐STR loci displayed an increase in allele diversity due to variations in both the repeat and flanking region sequences (DYS390 and DYS437). All X‐STR loci with increased allele diversity displayed an increase in variation within the repeat region sequence (DXS10135, DXS7132, and DXS10103) but no other changes were evident. DYF387S1 presented with the highest diversity among the Y‐STR loci, with 26 unique alleles and DXS10135 was the most variable X‐STR locus, with 34 unique alleles. The lowest Y‐STR and X‐STR diversity was observed at DYS391, with three unique alleles, and DXS7423, with five unique alleles, respectively. The number of alleles per locus for LB, RSB, and FSB is summarized in Tables 1b,c, and Figure 2.
TABLE 1B.
The number of unique alleles observed at 24 Y‐STRs and 7 X‐STRs by LB, by RSB, and by FSB.
| Locus | LB | RSB | FSB | Increase | %Increase |
|---|---|---|---|---|---|
| DYS389II | 7 | 21 | 21 | 14 | 200.00 |
| DYF387S1 | 9 | 26 | 26 | 17 | 188.89 |
| DYS390 | 7 | 14 | 16 | 9 | 128.57 |
| DYS437 | 4 | 7 | 8 | 4 | 100.00 |
| DYS448 | 6 | 12 | 12 | 6 | 100.00 |
| Y‐GATA‐H4 | 4 | 4 | 8 | 4 | 100.00 |
| DYS635 | 6 | 10 | 10 | 4 | 66.67 |
| DYS612 | 9 | 12 | 12 | 3 | 33.33 |
| DYS460 | 4 | 4 | 5 | 1 | 25.00 |
| DYS19 | 5 | 5 | 6 | 1 | 20.00 |
| DYS389I | 5 | 6 | 6 | 1 | 20.00 |
| DYS439 | 5 | 6 | 6 | 1 | 20.00 |
| DYS481 | 13 | 15 | 15 | 2 | 15.38 |
| DYS385a‐b | 12 | 12 | 12 | 0 | 0.00 |
| DYS391 | 3 | 3 | 3 | 0 | 0.00 |
| DYS392 | 6 | 6 | 6 | 0 | 0.00 |
| DYS438 | 4 | 4 | 4 | 0 | 0.00 |
| DYS505 | 4 | 4 | 4 | 0 | 0.00 |
| DYS522 | 5 | 5 | 5 | 0 | 0.00 |
| DYS533 | 5 | 5 | 5 | 0 | 0.00 |
| DYS549 | 4 | 4 | 4 | 0 | 0.00 |
| DYS570 | 7 | 7 | 7 | 0 | 0.00 |
| DYS576 | 5 | 5 | 5 | 0 | 0.00 |
| DYS643 | 6 | 6 | 6 | 0 | 0.00 |
| Total | 145 | 203 | 212 | 67 | 46.215 |
Abbreviations: FSB, sequence‐based alleles with flanking region; LB, length‐based alleles; RSB, sequence‐based alleles without flanking region; Y‐STRs, Y chromosome STRs.
TABLE 1C.
The number of unique alleles observed at 7 X‐STRs by LB, by RSB, and by FSB.
| Locus | LB | RSB | FSB | Increase | %Increase |
|---|---|---|---|---|---|
| DXS10135 | 20 | 34 | 34 | 14 | 70.00 |
| DXS7132 | 8 | 10 | 10 | 2 | 25.00 |
| DXS10103 | 6 | 7 | 7 | 1 | 16.67 |
| DXS8378 | 6 | 6 | 6 | 0 | 0.00 |
| DXS10074 | 9 | 9 | 9 | 0 | 0.00 |
| DXS7423 | 5 | 5 | 5 | 0 | 0.00 |
| HPRTB | 7 | 7 | 7 | 0 | 0.00 |
| Total | 61 | 78 | 78 | 17 | 27.87 |
Abbreviations: FSB, sequence‐based alleles with flanking region; LB, length‐based alleles; RSB, sequence‐based alleles without flanking region; X‐STRs, X chromosome STRs.
FIGURE 2.
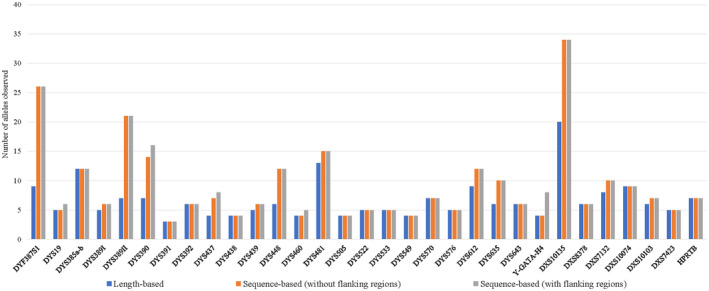
Allele diversity of Y‐ and X‐STRs based on length or sequence (with or without flanking regions)
3.4. Allele frequencies and forensic parameters
The observed LB, RSB, and FSB allele frequencies for all the STR loci (except D22S1045) from 120 Tibetan individuals were calculated using the counting method, as summarized in Tables S6–S8 (with those instances of discordance described above not included). All A‐STR loci allele data met HWE expectations after Bonferroni correction (α=0.05/26), as listed in Table S9, which means that the population data from this study can be considered representative.
The forensic parameters (described in Material and Methods) for each A‐STR locus obtained from the LB, RSB, and FSB data were evaluated using STRAF and are summarized in Table S9. The average GD/Hexp for all of the A‐STRs was 0.7785 when analyzed by length, 0.7979 when evaluated using the repeat region sequence, and 0.8106 when sequence variation in the flanking region was considered. The combined RMP for the 26 A‐STRs was 2.04 × 10–29, 1.93 × 10–31, and 9.56 × 10–33 for LB, RSB, and FSB, respectively. When we used the sequence variation in the repeat regions alone, the combined RMP was more than 105 times lower than that of the length‐based alleles. The addition of sequence variation in the repeat and flanking regions, allowed MPS analysis to reduce the combined RMP by over 2100 times compared to the CE method. Similar trends were observed in other forensic parameters resulting from the increase in the number of unique alleles available. A total of 111 and 113 unique haplotypes in the Y‐STR loci were observed when using length‐based and sequence‐based alleles, respectively. Sequence variations in the flanking region did not increase the number of unique haplotypes. While among the 111 unique length‐based allele haplotypes, 106, 5, and 2 were observed once, twice, and three times, respectively (Table S4). The RMP was 0.01, the DP was 0.99, and the HD was 0.9983. When sequence variation was considered, 108, 3, and 2 haplotypes were observed once, twice, and three times, respectively (Table S5) whereas RMP was reduced to 0.0096, DP was increased to 0.9904, and HD increased to 0.9987. Overall, the advantages of the MPS are reflected in the forensic parameters.
3.5. Novel alleles and SNPs in flanking regions
Among the 653 unique sequence‐based alleles (including the flanking region) observed in this study, 35 identified in 25 loci (12 A‐STRs loci, 11 Y‐STR loci, and 2 X‐STRs loci, respectively) were not recorded in the STR Sequencing Project (Gettings et al., 2017) and have never been reported in any of the previous studies (Barrio et al., 2019; Churchill et al., 2017; Delest et al., 2020; Devesse et al., 2018, 2020; Gettings et al., 2015, 2016; Hussing et al., 2019; Jäger et al., 2017; Khubrani et al., 2019; Kim et al., 2016, 2018; Novroski et al., 2016; Peng et al., 2020; Phillips, Devesse, et al., 2018; Phillips, Gettings, et al., 2018; Wang et al., 2020; Wendt et al., 2017). Sanger sequencing was performed to verify these novel sequence‐based alleles (data not provided) and all 35 are listed in Table 2.
TABLE 2.
Thirty‐five novel alleles of STR loci observed in this study.
| Locus | MPS allele (following the nomenclature recommended by IFSG) |
|---|---|
| D5S818 | D5S818 [CE6]‐GRCh38‐Chr5‐123775543‐123775606 [ATCT]6 |
| D6S1043 | D6S1043 [CE20.3]‐GRCh38‐Chr6‐91740160‐91740292 [ATCT]6[ATGT][ATCT]2[ATC][ATCT]11 91740273‐A |
| D6S1043 | D6S1043 [CE21.3]‐GRCh38‐Chr6‐91740160‐91740292 [ATCT]6[ATGT][ATCT]2[ATC][ATCT]12 91740273‐A |
| D7S820 | D7S820 [CE11]‐GRCh38‐Chr7‐84160191‐84160297 [TATC]9[TGTC][TATC] 84160204‐A |
| D9S1122 | D9S1122 [CE17]‐GRCh38‐Chr9‐77073809‐77073880 [TAGA][TCGA][TAGA]15 |
| D10S1248 | D10S1248 [CE14]‐GRCh38‐Chr10 129294226‐129294318 [GGAA]14 129294238‐A |
| D10S1248 | D10S1248 [CE15]‐GRCh38‐Chr10 129294226‐129294318 [GGAA]15 129294243‐A |
| D12S391 | D12S391 [CE21]‐GRCh38‐Chr12‐12296981‐12297189 [AGAT]4[AGGT][AGAT]9[AGAC]6[AGAT] |
| D13S317 | D13S317 [CE14]‐GRCh38‐Chr13‐82147986‐82148107 [TATC]14 82148069‐T 82148073‐T |
| D13S317 | D13S317 [CE15]‐GRCh38‐Chr13‐82147986‐82148107 [TATC]15 82148069‐T |
| D16S539 | D16S539 [CE14]‐GRCh38‐Chr16‐86352664‐86352781 [GATA]14 86352761‐C |
| D18S51 | D18S51 [CE7]‐GRCh38‐Chr18‐63281662‐63281796 [AGAA]7 |
| D20S482 | D20S482 [CE14]‐GRCh38‐Chr20‐4525674‐4525771 [AGAT]4[ATAT][AGAT]9 |
| D20S482 | D20S482 [CE15]‐GRCh38‐Chr20‐4525674‐4525771 [AGAT]14[AGAC] |
| D21S11 | D21S11 [CE29]‐GRCh38‐Chr21‐19181939‐19182111 [TCTA]6[TCTG]5[TCTA]3 TA [TCTA]3 TCA[TCTA]2 TCCATA [TCTA]10 19182101‐T |
| D21S11 | D21S11 [CE30.2]‐GRCh38‐Chr21‐19181939‐19182111 [TCTA]5[TCTG]7[TCTA]3 TA [TCTA]2 TCA[TCTA]2 TCCATA [TCTA]10 TA[TCTA] |
| D21S11 | D21S11 [CE31.2]‐GRCh38‐Chr21‐19181939‐19182111 [TCTA]5[TCTG]7[TCTA]2 TA [TCTA]3 TCA[TCTA]2 TCCATA [TCTA]11 TA[TCTA] |
| D21S11 | D21S11 [CE33.2]‐GRCh38‐Chr21‐19181939‐19182111 [TCTA]5[TCTG]6[TCTA]3 TA [TCTA]4 TCA[TCTA]2 TCCATA [TCTA]12 TA[TCTA] |
| FGA | FGA [CE29]‐GRCh38‐Chr4‐154587713‐154587840 [GGAA]2[GGAG][AAAG]21[AGAA][AAAA][GAAA]3 |
| DYS19 | DYS19 [CE14]‐GRCh38‐ChrY‐9684267‐9684443 [TCTA]11 CCTA [TCTA]3 9684269‐T |
| DYS390 | DYS390 [CE19]‐ChrY‐GRCh38 15162096‐15163170 [TAGA]11[CAGA]8 |
| DYS390 | DYS390 [CE23]‐ChrY‐GRCh38 15162096‐15163170 [TAGA]3[CAGA][TAGA]10[CAGA]9 |
| DYS389I | DYS389I [CE13]‐ChrY‐GRCh38 12500387‐12500513 [TAGA]9[CAGA]4 |
| DYS389II | DYS389II [CE29]‐ChrY‐GRCh38 12500448‐12500633 [TAGA]9[CAGA]4N48 [TAGA]11[CAGA]5 |
| DYS439 | DYS439 [CE11]‐ChrY‐GRCh38 12403461‐12403587 [GATA]11 12403513‐G 12403514‐A 12403515‐T |
| DYS460 | DYS460 [CE9]‐ChrY‐GRCh38 18888810‐18889046 [TATC]9 1888811‐G |
| DYS481 | DYS481 [CE17]‐ChrY‐GRCh38 8558313‐8558408 [CTT]17 |
| DYS635 | DYS635 [CE23]‐ChrY‐GRCh38 12258755‐12258975 [TAGA]12 [TACA]3 [TAGA]2 [TACA]2 [TAGA]4 |
| DYF387S1 | DYF387S1 [CE40]‐GRCh38‐ChrY‐23785347‐23785521 [AAAG]3[GTAG][GAAG]4[AAAG]2[GAAG][AAAG]2[GAAG]8[AAAG]19 |
| DYF387S1 | DYF387S1 [CE42]‐GRCh38‐ChrY‐23785347‐23785521 [AAAG]3[GTAG][GAAG]4[AAAG]2[GAAG][AAAG]2[GAAG]10[AAAG]19 |
| Y‐GATA‐H4 | Y‐GATA‐H4 [CE10]‐ChrY‐GRCh38 16631624‐16631759 [TCTA]10 16631721‐T |
| DXS10135 | DXS10135 [CE14]‐GRCh38‐ChrX‐9338302‐9338520 [AAGA]3 N7 [AAGA]10[AAAG] |
| DXS10135 | DXS10135 [CE26]‐GRCh38‐ChrX‐9338302‐9338520 [AAGA]3 N7 [AAGA]15[AAGG][AAGA]4[AAGG][AAGA][AAAG] |
| DXS10135 | DXS10135 [CE30]‐GRCh38‐ChrX‐9338302‐9338520 [AAGA]3 N7 [AAGA]19[AAGG]2[AAGA]3[AAGG][AAGA][AAAG] |
| DXS7132 | DXS7132 [CE16]‐GRCh38‐ChrX‐65435623‐65435778 [TAGA]15[CAGA] |
Abbreviations: CE, capillary electrophoresis; MPS, massively parallel sequencing.
In total, 24 SNPs and one InDel were observed in the flanking region of 17 STRs (Table 3) 6 of which were not recorded in the Single‐Nucleotide Polymorphism database (dbSNP, https://www.ncbi.nlm.nih.gov/snp/). Among the 25 sequence variations in the flanking region, 15 SNPs were found within 10 A‐STRs, whereas variations in the flanking regions of the Y‐STRs and X‐STRs were not as frequent. Nine SNPs and one deletion were observed in the flanking regions of six Y‐STR loci and one X‐STR locus, respectively.
TABLE 3.
SNPs and InDels observed in flanking regions at 58 STRs using UAS.
| Locus | Variation | Position (GRCh38/hg38) | dbSNP ID | Wild | Mutant | Count |
|---|---|---|---|---|---|---|
| D2S441 | SNP | Chr2: 68011922 | rs74640515 | G | A | 25 |
| D5S818 | SNP | Chr5: 123775552 | rs73801920 | C | A | 54 |
| D6S1043 | SNP | Chr6: 91740273 | rs529713981 | G | A | 4 |
| D7S820 | SNP | Chr7: 84160204 | rs7789995 | T | A | 222 |
| D7S820 | SNP | Chr7: 84160286 | rs16887642 | G | A | 42 |
| D10S1248 | SNP | Chr10: 129294238 | rs1279061683 | G | A | 1 |
| D10S1248 | SNP | Chr10: 129294243 | rs563636310 | T | A | 1 |
| D13S317 | SNP | Chr13: 82148069 | rs9546005 | A | T | 120 |
| D13S317 | SNP | Chr13: 82148073 | rs202043589 | A | T | 23 |
| D16S539 | SNP | Chr16: 86352692 | rs563997442 | C | G | 2 |
| D16S539 | SNP | Chr16: 86352761 | rs11642858 | A | C | 77 |
| D20S482 | SNP | Chr20: 4525681 | rs561985213 | G | A | 2 |
| D20S482 | SNP | Chr20: 4525680 | rs77560248 | C | T | 29 |
| D21S11 | SNP | Chr21: 19182101 | rs1051967683 | C | T | 1 |
| vWA | SNP | Chr12: 5983970 | rs75219269 | A | G | 1 |
| DYS19 | SNP | ChrY: 9684269 | Null | G | T | 1 |
| DYS390 | SNP | ChrY: 15163163 | rs758940870 | T | C | 2 |
| DYS437 | SNP | ChrY: 12346421 | Null | G | A | 9 |
| DYS439 | SNP | ChrY: 12403513 | rs1042036966 | A | G | 1 |
| DYS439 | SNP | ChrY: 12403514 | Null | G | A | 1 |
| DYS439 | SNP | ChrY: 12403515 | Null | A | T | 1 |
| DYS460 | SNP | ChrY: 1888811 | Null | T | G | 1 |
| Y‐GATA‐H4 | SNP | ChrY: 16631721 | rs765275581 | C | T | 1 |
| Y‐GATA‐H4 | SNP | ChrY: 16631756 | Null | A | G | 59 |
| DXS10135 | Deletion | ChrX: 9338410–9338416 | rs201630737 | AAGAAGA | AGA | 1 |
Abbreviations: dbSNP, Single‐Nucleotide Polymorphism database; Null, No record in dbSNP.
Most sequence variations in the flanking regions were found to be present in low proportions. However, SNPs in the flanking regions of some of the A‐STR loci were so frequent that they could be observed in a large proportion of sequence‐based alleles. D7S820 was shown to exhibit the highest proportion of sequence‐based alleles with flanking SNPs (rs7789995: 222/240 and rs16887642: 42/240) with D13S317 following a similar pattern (rs9546005: 120/240 and rs202043589: 23/240). The variations at Y‐GATA‐H4 (ChrY: 16631756) were observed in almost half of the samples (59/120), although this SNP was not recorded in the dbSNP. DYS439 also had an interesting SNP allele, DYS439 [CE11]‐ChrY‐GRCh38 12403461‐12403587 [GATA]11 12403513‐G 12403514‐A 12403515‐T) which included three coterminous SNPs within its flanking region. This allele can also be referred to as DYS439 [CE11]‐ChrY‐GRCh38 12403461‐12403587 [GATA]12 12403513‐12403516 Del, which suggests that there was an additional repeat [GATA] within the repeat region and the sequence variation in the flanking region was a four base (AGAA) deletion. Here, we use the first name to allow overlap with the CE databases. These novel alleles and polymorphisms in the flanking regions can further enrich the MPS‐STR population data.
4. CONCLUSION
Here, we report the MPS‐STR profile data for the Tibetan population. The diversity of the observed alleles, allele frequencies, and forensic parameters per locus by length, sequence without flanking region, and sequence with flanking region were analyzed and compared and clearly demonstrate the advantages of MPS in comparison to CE. To ensure compatibility between the MPS data and the current CE‐based forensic databases, we completed a concordance study that demonstrated a concordance rate of more than 99% with some exceptions which were subjected to further analysis. Our data suggest that MPS‐ and CE‐derived alleles are compatible and we were able to identify 35 novel alleles at 25 loci using the MPS method.
In conclusion, with more pooled data and larger‐scale validation, MPS could play a valuable role in forensic genetics and might be an additional tool for routine casework.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
AUTHORS’ CONTRIBUTIONS
J.X. and X.Z. involved in the study design. C.Z. involved in sample collection. K.M., Y.C., and Q.Y. involved in experimental work. H.L. and G.S. involved in data analysis. H.L. involved in the writing of the initial manuscript. J.X. and X.Z. involved in revision. All authors reviewed the manuscript.
Supporting information
Tables S1‐S9
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (Grant No. 81801880), the Shanghai Sailing Program (Grant No. 18YF1421400), the Shanghai Municipal Natural Science Foundation (Grant No. 18ZR1435000), and the Special Project for Basic Science and Technology Research of the Ministry of Public Security (2018GABJC32).
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Alvarez‐Cubero, M. J. , Saiz, M. , Martínez‐García, B. , Sayalero, S. M. , Entrala, C. , Lorente, J. A. , & Martinez‐Gonzalez, L. J. (2017). Next generation sequencing: An application in forensic sciences? Annals of Human Biology, 44(7), 581–592. 10.1080/03014460.2017.1375155 [DOI] [PubMed] [Google Scholar]
- Aly, S. M. , & Sabri, D. M. (2015). Next generation sequencing (NGS): a golden tool in forensic toolkit. Archives of Forensic Medicine and Criminology, 65(4), 260–271. 10.5114/amsik.2015.61029 [DOI] [PubMed] [Google Scholar]
- Barrio, P. A. , Martín, P. , Alonso, A. , Müller, P. , Bodner, M. , Berger, B. , Parson, W. , & Budowle, B. (2019). Massively parallel sequence data of 31 autosomal STR loci from 496 Spanish individuals revealed concordance with CE‐STR technology and enhanced discrimination power. Forensic Science International: Genetics, 42, 49–55. 10.1016/j.fsigen.2019.06.009 [DOI] [PubMed] [Google Scholar]
- Børsting, C. , & Morling, N. (2015). Next generation sequencing and its applications in forensic genetics. Forensic Science International: Genetics, 18, 78–89. 10.1016/j.fsigen.2015.02.002 [DOI] [PubMed] [Google Scholar]
- Bruijns, B. , Tiggelaar, R. , & Gardeniers, H. (2018). Massively parallel sequencing techniques for forensics: A review. Electrophoresis, 39(21), 2642–2654. 10.1002/elps.201800082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler, J. M. (2005). Forensic DNA typing: biology, technology, and genetics of STR markers, 2nd ed. Elsevier Academic Press. [Google Scholar]
- Butler, J. M. (2015). The future of forensic DNA analysis. Philosophical Transactions of the Royal Society B: Biological Sciences, 370(1674), 20140252– 10.1098/rstb.2014.0252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler, J. M. , Buel, E. , Crivellente, F. , & McCord, B. R. (2004). Forensic DNA typing by capillary electrophoresis using the ABI Prism 310 and 3100 genetic analyzers for STR analysis. Electrophoresis, 25(10–11), 1397–1412. 10.1002/elps.200305822 [DOI] [PubMed] [Google Scholar]
- Churchill, J. D. , Novroski, N. M. M. , King, J. L. , Seah, L. H. , & Budowle, B. (2017). Population and performance analyses of four major populations with Illumina's FGx Forensic Genomics System. Forensic Science International: Genetics, 30, 81–92. 10.1016/j.fsigen.2017.06.004 [DOI] [PubMed] [Google Scholar]
- Delest, A. , Godfrin, D. , Chantrel, Y. , Ulus, A. , Vannier, J. , Faivre, M. , Hollard, C. , & Laurent, F. X. (2020). Sequenced‐based French population data from 169 unrelated individuals with Verogen's ForenSeq DNA signature prep kit. Forensic Science International: Genetics, 47, 102304. 10.1016/j.fsigen.2020.102304 [DOI] [PubMed] [Google Scholar]
- Devesse, L. , Ballard, D. , Davenport, L. , Riethorst, I. , Mason‐Buck, G. , & Syndercombe, C. D. (2018). Concordance of the ForenSeq™ system and characterisation of sequence‐specific autosomal STR alleles across two major population groups. Forensic Science International: Genetics, 34, 57–61. [DOI] [PubMed] [Google Scholar]
- Devesse, L. , Davenport, L. , Borsuk, L. , Gettings, K. , Mason‐Buck, G. , Vallone, P. M. , Syndercombe Court, D. , & Ballard, D. (2020). Classification of STR allelic variation using massively parallel sequencing and assessment of flanking region power. Forensic Science International: Genetics, 48, 102356. 10.1016/j.fsigen.2020.102356 [DOI] [PubMed] [Google Scholar]
- Elwick, K. , Bus, M. M. , King, J. L. , Chang, J. , Hughes‐Stamm, S. , & Budowle, B. (2019). Utility of the Ion S5™ and MiSeq FGx™ sequencing platforms to characterize challenging human remains. Legal Medicine (Tokyo), 41, 101623. 10.1016/j.legalmed.2019.08.001 [DOI] [PubMed] [Google Scholar]
- Fattorini, P. , Previderé, C. , Carboni, I. , Marrubini, G. , Sorçaburu‐Cigliero, S. , Grignani, P. , Bertoglio, B. , Vatta, P. , & Ricci, U. (2017). Performance of the ForenSeqTM DNA Signature Prep kit on highly degraded samples. Electrophoresis, 38(8), 1163–1174. 10.1002/elps.201600290 [DOI] [PubMed] [Google Scholar]
- Gettings, K. B. , Aponte, R. A. , Vallone, P. M. , & Butler, J. M. (2015). STR allele sequence variation: Current knowledge and future issues. Forensic Science International: Genetics, 18, 118–130. 10.1016/j.fsigen.2015.06.005 [DOI] [PubMed] [Google Scholar]
- Gettings, K. B. , Borsuk, L. A. , Ballard, D. , Bodner, M. , Budowle, B. , Devesse, L. , King, J. , Parson, W. , Phillips, C. , & Vallone, P. M. (2017). STRSeq: A catalog of sequence diversity at human identification Short Tandem Repeat loci. Forensic Science International: Genetics, 31, 111–117. 10.1016/j.fsigen.2017.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gettings, K. B. , Kiesler, K. M. , Faith, S. A. , Montano, E. , Baker, C. H. , Young, B. A. , Guerrieri, R. A. , & Vallone, P. M. (2016). Sequence variation of 22 autosomal STR loci detected by next generation sequencing. Forensic Science International: Genetics, 21, 15–21. 10.1016/j.fsigen.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy, A. , & Zieger, M. (2017). STRAF‐A convenient online tool for STR data evaluation in forensic genetics. Forensic Science International: Genetics, 30, 148–151. 10.1016/j.fsigen.2017.07.007 [DOI] [PubMed] [Google Scholar]
- Guo, F. , Yu, J. , Zhang, L. , & Li, J. (2017). Massively parallel sequencing of forensic STRs and SNPs using the Illumina® ForenSeq™ DNA Signature Prep Kit on the MiSeq FGx™ Forensic Genomics System. Forensic Science International: Genetics, 31, 135–148. 10.1016/j.fsigen.2017.09.003 [DOI] [PubMed] [Google Scholar]
- Hussing, C. , Bytyci, R. , Huber, C. , Morling, N. , & Børsting, C. (2019). The Danish STR sequence database: duplicate typing of 363 Danes with the ForenSeq™ DNA Signature Prep Kit. International Journal of Legal Medicine, 133(2), 325–334. 10.1007/s00414-018-1854-0 [DOI] [PubMed] [Google Scholar]
- Jäger, A. C. , Alvarez, M. L. , Davis, C. P. , Guzmán, E. , Han, Y. , Way, L. , Walichiewicz, P. , Silva, D. , Pham, N. , Caves, G. , Bruand, J. , Schlesinger, F. , Pond, S. J. K. , Varlaro, J. , Stephens, K. M. , & Holt, C. L. (2017). Developmental validation of the MiSeq FGx Forensic Genomics System for Targeted Next Generation Sequencing in Forensic DNA Casework and Database Laboratories. Forensic Science International: Genetics, 28, 52–70. 10.1016/j.fsigen.2017.01.011 [DOI] [PubMed] [Google Scholar]
- Jobling, M. A. , & Gill, P. (2004). Encoded evidence: DNA in forensic analysis. Nature Reviews Genetics, 5(10), 739–751. 10.1038/nrg1455 [DOI] [PubMed] [Google Scholar]
- Just, R. S. , Moreno, L. I. , Smerick, J. B. , & Irwin, J. A. (2017). Performance and concordance of the ForenSeq™ system for autosomal and Y chromosome short tandem repeat sequencing of reference‐type specimens. Forensic Science International: Genetics, 28, 1–9. 10.1016/j.fsigen.2017.01.001 [DOI] [PubMed] [Google Scholar]
- Khubrani, Y. M. , Hallast, P. , Jobling, M. A. , & Wetton, J. H. (2019). Massively parallel sequencing of autosomal STRs and identity‐informative SNPs highlights consanguinity in Saudi Arabia. Forensic Sci Int Genet, 43, 102164. 10.1016/j.fsigen.2019.102164 [DOI] [PubMed] [Google Scholar]
- Kim, E. H. , Lee, H. Y. , Yang, I. S. , Jung, S. E. , Yang, W. I. , & Shin, K. J. (2016). Massively parallel sequencing of 17 commonly used forensic autosomal STRs and amelogenin with small amplicons. Forensic Science International: Genetics, 22, 1–7. 10.1016/j.fsigen.2016.01.001 [DOI] [PubMed] [Google Scholar]
- Kim, S. Y. , Lee, H. C. , Chung, U. , Ham, S. K. , Lee, H. Y. , Park, S. J. , Roh, Y. J. , & Lee, S. H. (2018). Massive parallel sequencing of short tandem repeats in the Korean population. Electrophoresis, 39(21), 2702–2707. 10.1002/elps.201800090 [DOI] [PubMed] [Google Scholar]
- Köcher, S. , Müller, P. , Berger, B. , Bodner, M. , Parson, W. , Roewer, L. , & Willuweit, S. (2018). Inter‐laboratory validation study of the ForenSeq™ DNA Signature Prep Kit. Forensic Science International: Genetics, 36, 77–85. 10.1016/j.fsigen.2018.05.007 [DOI] [PubMed] [Google Scholar]
- Kuffel, A. , Gray, A. , & Nic, D. N. (2020). Human Leukocyte Antigen alleles as an aid to STR in complex forensic DNA samples. Science & Justice, 60(1), 1–8. 10.1016/j.scijus.2019.09.003 [DOI] [PubMed] [Google Scholar]
- Kwon, S. Y. , Lee, H. Y. , Kim, E. H. , Lee, E. Y. , & Shin, K. J. (2016). Investigation into the sequence structure of 23 Y chromosomal STR loci using massively parallel sequencing. Forensic Sci Int Genet, 25, 132–141. 10.1016/j.fsigen.2016.08.010 [DOI] [PubMed] [Google Scholar]
- Li, H. , Zhao, X. , Ma, K. , Cao, Y. , Zhou, H. , Ping, Y. , Shao, C. , Xie, J. , & Liu, W. (2017). Applying massively parallel sequencing to paternity testing on the Ion Torrent Personal Genome Machine. Forensic Science International: Genetics, 31, 155–159. 10.1016/j.fsigen.2017.09.007 [DOI] [PubMed] [Google Scholar]
- Novroski, N. M. M. , King, J. L. , Churchill, J. D. , Seah, L. H. , & Budowle, B. (2016). Characterization of genetic sequence variation of 58 STR loci in four major population groups. Forensic Science International: Genetics, 25, 214–226. 10.1016/j.fsigen.2016.09.007 [DOI] [PubMed] [Google Scholar]
- Parson, W. , Ballard, D. , Budowle, B. , Butler, J. M. , Gettings, K. B. , Gill, P. , Gusmão, L. , Hares, D. R. , Irwin, J. A. , King, J. L. , Knijff, P. , Morling, N. , Prinz, M. , Schneider, P. M. , Neste, C. V. , Willuweit, S. , & Phillips, C. (2016). Massively parallel sequencing of forensic STRs: Considerations of the DNA commission of the International Society for Forensic Genetics (ISFG) on minimal nomenclature requirements. Forensic Science International: Genetics, 22, 54–63. 10.1016/j.fsigen.2016.01.009 [DOI] [PubMed] [Google Scholar]
- Peng, D. , Zhang, Y. , Ren, H. , Li, H. , Li, R. , Shen, X. , Wang, N. , Huang, E. , Wu, R. , & Sun, H. (2020). Identification of sequence polymorphisms at 58 STRs and 94 iiSNPs in a Tibetan population using massively parallel sequencing. Scientific Reports, 10(1), 12225. 10.1038/s41598-020-69137-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, C. , Devesse, L. , Ballard, D. , van Weert, L. , de la Puente, M. , Melis, S. , Álvarez Iglesias, V. , Freire‐Aradas, A. , Oldroyd, N. , Holt, C. , Syndercombe Court, D. , Carracedo, Á. , & Lareu, M. V. (2018). Global patterns of STR sequence variation: Sequencing the CEPH human genome diversity panel for 58 forensic STRs using the Illumina ForenSeq DNA Signature Prep Kit. Electrophoresis, 39(21), 2708–2724. 10.1002/elps.201800117 [DOI] [PubMed] [Google Scholar]
- Phillips, C. , Gettings, K. B. , King, J. L. , Ballard, D. , Bodner, M. , Borsuk, L. , & Parson, W. (2018). “The devil’s in the detail”: Release of an expanded, enhanced and dynamically revised forensic STR Sequence Guide. Forensic Science International: Genetics, 34, 162–169. 10.1016/j.fsigen.2018.02.017 [DOI] [PubMed] [Google Scholar]
- Thompson, R. , Zoppis, S. , & McCord, B. (2012). An overview of DNA typing methods for human identification: past, present, and future. Methods in Molecular Biology, 830, 3–16. 10.1007/978-1-61779-461-2_1 [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir, H. , Robinson, J. T. , & Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Briefings in Bioinformatics, 14(2), 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verogen , (2018). MiseqTM DNA signature prep reference Guide, Document#VD2018005 Rev. A.
- Wang, Z. , Wang, L. , Liu, J. , Ye, J. , & Hou, Y. (2020). Characterization of sequence variation at 30 autosomal STRs in Chinese Han and Tibetan populations. Electrophoresis, 41(3–4), 194–201. 10.1002/elps.201900278 [DOI] [PubMed] [Google Scholar]
- Wendt, F. R. , King, J. L. , Novroski, N. M. M. , Churchill, J. D. , Ng, J. , Oldt, R. F. , McCulloh, K. L. , Weise, J. A. , Smith, D. G. , Kanthaswamy, S. , & Budowle, B. (2017). Flanking region variation of ForenSeq™ DNA Signature Prep Kit STR and SNP loci in Yavapai Native Americans. Forensic Science International: Genetics, 28, 146–154. 10.1016/j.fsigen.2017.02.014 [DOI] [PubMed] [Google Scholar]
- Wu, J. , Li, J. L. , Wang, M. L. , Li, J. P. , Zhao, Z. C. , Wang, Q. , Yang, S. D. , Xiong, X. , Yang, J. L. , & Deng, Y. J. (2019). Evaluation of the MiSeq FGx system for use in forensic casework. International Journal of Legal Medicine, 133(3), 689–697. 10.1007/s00414-018-01987-x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1‐S9
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.