

Abstract
Background
Targeted next‐generation sequencing is an efficient tool to identify pathogenic mutations of hereditary deafness. The molecular pathology of deaf patients in southwestern China is not fully understood.
Methods
In this study, targeted next‐generation sequencing of 127 deafness genes was performed on 84 deaf patients. They were not caused by common mutations of GJB2 gene, including c.35delG, c.109 G>A, c.167delT, c.176_191del16, c.235delC and c.299_300delAT.
Results
In the cohorts of 84 deaf patients, we did not find any candidate pathogenic variants in 14 deaf patients (16.7%, 14/84). In other 70 deaf patients (83.3%, 70/84), candidate pathogenic variants were identified in 34 genes. Of these 70 deaf patients, the percentage of “Solved” and “Unsolved” patients was 51.43% (36/70) and 48.57% (34/70), respectively. The most common causative genes were SLC26A4 (12.9%, 9/70), MT‐RNR1 (11.4%, 8/70), and MYO7A (2.9%, 2/70) in deaf patients. In “Unsolved” patients, possible pathogenic variants were most found in SLC26A4 (8.9%, 3/34), MYO7A (5.9%, 2/34), OTOF (5.9%, 2/34), and PDZD7 (5.9%, 2/34) genes. Interesting, several novel recessive pathogenic variants were identified, like SLC26A4 c.290T>G, SLC26A4 c.599A>G, PDZD7c.490 C>T, etc.
Conclusion
In addition to common deafness genes, like GJB2, SLC26A4, and MT‐RNR1 genes, other deafness genes (MYO7A, OTOF, PDZD7, etc.) were identified in deaf patients from southwestern China. Therefore, the spectrum of deafness genes in this area should be further studied.
Keywords: hereditary deafness, mutations, targeted next‐generation sequencing
In addition to common deafness genes, like GJB2, SLC26A4, and MT‐RNR1 genes, other deafness genes (MYO7A, OTOF, PDZD7, etc.) were identified in deaf patients from southwestern China.
1. INTRODUCTION
Deafness (or hearing loss) is a disease characterized by partial or total inability to hear. It can be inherited (Duman & Tekin, 2012; Meena & Ayub, 2017; da Silva & Duarte, 1995). Most of the heritable deafness is inherited by an autosomal recessive pattern. Autosomal dominant, X‐linked, mitochondrial, and digenic inheritance were also observed in heritable deafness. More than 100 genes have been identified in heritable deafness. Targeted next‐generation sequencing enabled us to efficiently screen mutations in several deafness genes compared to PCR‐based Sanger sequencing (Lin et al., 2012; Yang et al., 2013). It evolutionarily improves the clinical diagnosis rate of heritable deafness (Shearer & Smith, 2012; Vona et al., 2014). Several studies showed that it is an efficient tool to identify causative genes in deaf patients (Miyagawa et al., 2013, 2015). Different cultures, ethnicities, and living environments lead to various genetic backgrounds in the world (Donovan, 1984). Yunnan province, a region in southwestern China, has a higher attitude compared to other plain areas of China. The populations in this region might have their unique features, and the molecular epidemiology of deaf patients here is still unclear now.
In the present study, a panel containing 127 deafness genes was generated. These genes were associated with nonsyndromic, syndromic, and mitochondrial deafness. Eighty‐four deaf patients were subjected to 127 deafness genes sequencing. We aimed to preliminarily analyze the molecular epidemiology of deaf patients from southwestern China.
2. MATERIALS AND METHODS
2.1. Statement of ethics, patients, and DNA preparation
Peripheral blood samples of deaf patients were collected in First People's Hospital of Yunnan Province from 2016 to 2019. The study was approved by the Ethics Committee of First People's Hospital of Yunnan Province (Affiliated Hospital of Kunming University of Science and Technology). Each patient in this study signed informed consent. By E.Z.N.A.® Blood DNA Kit (cat. no. D3392‐02; Omega Bio‐tek, Inc.), genomic DNA was extracted from the peripheral blood leukocytes.
2.2. Targeted sequence capture and high‐throughput sequencing
One hundred and twenty‐seven deafness genes (Table S1) were sequenced and captured in this study. The experimental procedures were referred to our previously published article (Li et al., 2016; Li et al., 2019). Briefly, probe capture panels (Roche NimbleGen Inc.) were designed for 127 deafness genes, and total size for targeted regions (exons, splicing sites, and immediate flanking intron sequences of 100 bp) was 619167 bp. DNA was fragmented, and the fragmented DNA was end‐repaired and ligated to the adapter oligonucleotides. After that, PCR was used to make a library preenrichment amplification. The qualified libraries were used for the capture of fragments of 127 genes. The capture of 127 genes was performed according to the NimbleGen capture protocol. The captured fragments were further sequenced by HiSeq 2500 Analyzers (Illumina).
2.3. Data filtering, read mapping, variant detection, and analysis pipeline
Data filtering, read mapping, and variant detection were based on our previous descriptions (Li et al., 2016). Briefly, following the Illumina Pipeline (version 1.3.4), primary data were achieved after image analyses, error estimation, and base calling. Data were further filtered, and clean reads were kept. Clean reads were aligned to the UCSC hg19 reference genome by the BWA (Burrows‐Wheeler Aligner) Multi‐Vision software package. SOAPsnp software (Li et al., 2009) and GATK Indel Genotyper (http://www.broadinstitute.org/gsa/wiki/index.php/, The Genome Analysis Toolkit) were used to detect SNPs (single‐nucleotide polymorphisms) and indels (insertion‐deletions).
Missense, indel, nonsense and splice‐site variants in each patient were selected out and screened for quality. The frequency of these variants was further referred to 1000 Genomes public variant databases, dbSNP, and ExAC databases. The known disease‐causing mutations were determined by the HGMD database (Stenson et al., 2014).
2.4. Molecular modeling
Amino acids sequences of candidate causative genes were referred to the GenBank database. 3D protein structure of candidate pathogenic genes was modeled by RaptorX web server (Wang et al., 2016). PyMOL was used to view 3D structure of proteins which was constructed by RaptorX. Structure of mutated and wild‐type protein was compared.
3. RESULTS
Total of 84 deaf patients participated in this study, including 70 simplex probands and 14 multiplex probands. They are clinically diagnosed with deafness. Environmental causes were excluded in these patients. Most of them were congenital profound deafness (>95 dB). Of these patients, two patients were diagnosed with Charge syndrome and Usher syndrome 1B, respectively.
Patients participated in this study were previously screened by common mutations in GJB2 gene, including c.35delG, c.109 G>A, c.167delT, c.176_191del16, c.235delC and c.299_300delAT. Only the negative patients of GJB2 common mutations were subjected to 127 deafness genes sequencing. The lists of genes are shown in Table S1. The targeted region was 619167 bp. Sequencing data showed high quality. The average sequencing depth was approximately 200 X. Several variants were detected in each patient, including SNPs and indels. In further data analysis, variants with minor allele frequency (MAF)>0.01 were filtered out. Subsequently, nonsynonymous and splicing site variants were selected out for the next data analysis. Splicing site variants located in 100 bp upstream and 100 bp downstream of the exon–intron or intron–exon boundary. At last, candidate pathogenic variants were identified by combining analysis of genotype and clinical phenotype of each deaf patient. According to the ACMG guideline, candidate variants were defined as “pathogenic,” “likely pathogenic,” “variants of unknown significance (VUS),” “likely benign,” and “benign.”
In total 84 deaf patients, we did not find any pathogenic variants in 14 patients (16%). In other 74 patients (84% of total patients), we found candidate pathogenic variants which were directly or possibly associated with phenotypes of patients, as shown in Figure 1a. Both monogenic and digenic deafness were considered during analysis. Candidate pathogenic variants could be classified as three inheritance modes: autosomal dominant, autosomal recessive, maternally inherited (mitochondrial). Digenic inheritance of deafness was not observed in our deafness patients. Subsequently, based on the descriptions by Yan et al., (2016), we classified deaf patients harboring candidate variants as “Solved” and “Unsolved” groups, respectively. “Solved” patients were caused by “pathogenic” or “likely pathogenic” variants, while “Unsolved” patients harbored at least one “VUS” variant. The percentage of “Solved” and “Unsolved” deaf patients was 51.43% (36/70) and 48.57% (34/70), respectively. In multiplex families, the solved rate in the patients’ group of “Solved” and “Unsolved” were 36.11% (13/36) and 20.59% (7/34), respectively. “Unsolved” deaf patients were divided into two groups: “Unsolved 1” and “Unsolved 2.” Deaf patients in “Unsovled1” group harbored candidate pathogenic variants that could possibly explain their pathology. Candidate pathogenic variants identified in “Unsolved2” patients could not possibly explain their pathology. Some specific candidate pathogenic variants in “Unsolved” patients of multiplex families are highlighted in Table 2. Compared with simplex families, these variants in multiplex families were more likely to be associated with deafness.
FIGURE 1.
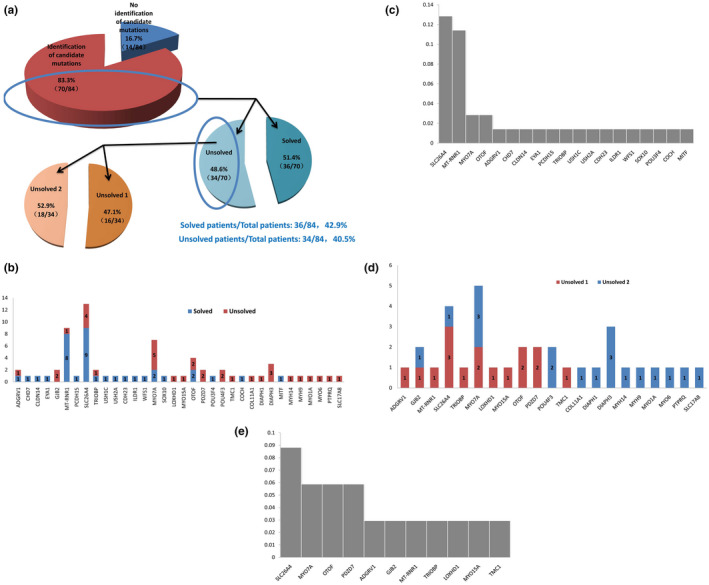
(a) Percentage of patients in “Solved” and “Unsolved” groups. (b and c). Candidate causative genes in “Solved” and “Unsolved” groups. The number of patients was annotated in each gene group. (d and e) Candidate causative genes in “Unsolved1” and “Unsolved2” groups. The number of patients was annotated in each gene group.
TABLE 2.
Variants in the “Unsolved” (“Unsolved 1” and “Unsolved 2”) patients
| Gene | Unsolved | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Transcript | Genotype | Chromosome | SIFT | Polyphen2 | ExAC database | Multiplex/simplex family | Zygosity | Unsolved 1/unsolved 2 | ACMG | |
| COL11A1 | 1 | NM_080630 | EX1_EX65 DUP | — | — | — | — | Simplex | Het | Unsolved 2 | VUS |
| DIAPH1 | 1 | NM_001079812 | c.2983C>T | chr5:140908277 | Damaging | — | — | Simplex | Het | Unsolved 2 | VUS |
| DIAPH3 | 1 | NM_001042517 | c.2608‐3T>C | chr13:60435673 | — | — | 0.00005802 | Simplex | Het | Unsolved 2 | VUS |
| 1 | NM_001042517 | c.3124C>T | chr13:60384961 | Damaging | Probably damaging | — | Multiplex | Het | Unsolved 2 | VUS | |
| 1 | NM_001042517 | c.1100G>A | chr13:60566632 | Damaging | Probably damaging | 0.00006994 | Simplex | Het | Unsolved 2 | VUS | |
| PDZD7 | 2 | NM_024895 | c.490C>T | chr10:102783245 | Damaging | Probably damaging | 0.00004959 | Simplex | Hom | Unsolved 1 | VUS |
| GJB2 | 1 | NM_004004 | c.109G>A | chr13:20763612 | — | Probably damaging | 0.006587 | Multiplex | Het | Unsolved 1 | Pathogenic |
| c.471G>A | chr13:20763250 | Tolerated | Benign | 0.000008261 | Het | VUS | |||||
| 1 | NM_004004 | c.235delC | chr13:20763486 | — | — | 0.0003625 | Simplex | Het | Unsolved 2 | Pathogenic | |
| ADGRV1 | 1 | NM_032119 | c.2241‐10A>T | chr5:89938443 | — | — | — | Simplex | Het | Unsolved 1 | Likely pathogenic |
| c.13637T>A | chr5:90079858 | Damaging | Possibly damaging | — | Het | VUS | |||||
| LOXHD1 | 1 | NM_144612 | c.1468C>T | chr18:44172511 | — | — | — | Multiplex | Het | Unsolved 1 | Pathogenic |
| c.977A>G | chr18:44181337 | Tolerated | — | 0.0003579 | Het | likely benign | |||||
| MT‐RNR1 | 1 | NC_012920 | m.681T>C | Simplex | Hom | Unsolved 1 | VUS | ||||
| MYH14 | 1 | NM_001077186 | c.475G>A | chr19:50720941 | Damaging | Probably damaging | 0.00001657 | Simplex | Het | Unsolved 2 | VUS |
| MYH9 | 1 | NM_002473 | c.1897C>T | chr22:36702600 | Tolerated | Benign | — | Multiplex | Het | Unsolved 2 | VUS |
| MYO15A | 1 | NM_016239 | c.9243_9251delinsAAGGGGGG | chr17:18062933..18062941 | — | — | — | Simplex | Het | Unsolved 1 | Pathogenic |
| c.3952G>A | chr17:18030399 | Damaging | Probably damaging | 0.000008324 | Het | VUS | |||||
| MYO1A | 1 | NM_005379 | c.235G>T | chr12:57441501 | Tolerated | Probably damaging | 0.002871 | Simplex | Het | Unsolved 2 | Likely benign |
| MYO6 | 1 | NM_004999 | c.2672C>T | chr6:76599787 | Damaging | Possibly damaging | 0.0001422 | Simplex | Het | Unsolved 2 | VUS |
| MYO7A | 1 | NM_000260 | c.1622C>T | chr11:76873966 | Damaging | Probably damaging | — | Simplex | Het | Unsolved 2 | VUS |
| 1 | NM_000260 | c.2558G>A | chr11:76890971 | Damaging | Probably damaging | — | Multiplex | Het | Unsolved 1 | VUS | |
| 2 | NM_000260 | c.1142C>T | chr11:76871270 | Tolerated | Possibly damaging | 0.000235 | Simplex | Het | Unsolved 2 | VUS | |
| 1 | NM_000260 | c.541C>T | chr11:76867776 | — | — | — | Simplex | Het | Unsolved 1 | Likely pathogenic | |
| NM_000260 | c.617G>A | chr11:76867932 | Damaging | Probably damaging | 0.00014 | Het | VUS | ||||
| OTOF | 1 | NM_194248 | c.1790C>T | chr2:26703667 | Tolerated | Possibly damaging | 0.000008474 | Simplex | Het | Unsolved 1 | VUS |
| c.829G>T | chr2:26717878 | Tolerated | Probably damaging | — | Het | VUS | |||||
| 1 | NM_194248 | c.1273C>T | chr2:26706449 | Damaging | Probably damaging | 0.00002 | Simplex | Het | Unsolved 1 | Pathogenic | |
| NM_194248 | c.3983G>C | chr2:26693501 | Damaging | Probably damaging | — | Het | VUS | ||||
| POU4F3 | 1 | NM_002700 | c.593G>A | chr5:145719583 | Damaging | Probably damaging | — | Simplex | Het | Unsolved 2 | VUS |
| 1 | NM_002700 | c.909G>T | chr5:145719899 | Simplex | Het | Unsolved 2 | VUS | ||||
| PTPRQ | 1 | NM_001145026 | c.1786A>G | chr12:80889054 | Damaging | Benign | — | Simplex | Het | Unsolved 2 | VUS |
| SLC17A8 | 1 | NM_139319 | c.760T>G | chr12:100795638 | Damaging | Probably damaging | — | Simplex | Het | Unsolved 2 | VUS |
| SLC26A4 | 1 | NM_000441 | c.919‐2A>G | chr7:107323898 | — | — | 0.0003058 | Multiplex | Het | Unsolved 1 | Pathogenic |
| c.290T>G | chr7:107303866 | Damaging | Probably damaging | — | Het | VUS | |||||
| 1 | c.1746 delG | chr7:107341584 | — | — | — | Simplex | Het | Unsolved 1 | Pathogenic | ||
| c.2110G>A | chr7:107350519 | Tolerated | Benign | — | Het | VUS | |||||
| 1 | c.2027T>A | chr7:107342495 | Damaging | Probably damaging | — | Simplex | Het | Unsolved 1 | Pathogenic | ||
| c.599A>G | chr7:107314792 | Damaging | Probably damaging | — | Het | VUS | |||||
| 1 | c.919‐2A>G | chr7:107323898 | — | — | 0.000306 | Multiplex | Het | Unsolved 2 | Pathogenic | ||
| TMC1 | 1 | NM_138691 | c.373A>C | chr9:75355045 | Tolerated | Benign | 0.00001654 | Simplex | Het | Unsolved 1 | VUS |
| c.1449A>C | chr9:75407151 | Tolerated | Possibly damaging | — | Het | VUS | |||||
| TRIOBP | 1 | NM_001039141 | c.1283C>G | chr22:38119846 | Damaging | Possibly damaging | 0.0001739 | Simplex | Het | Unsolved 1 | Likely benign |
| c.4442C>T | chr22:38130785 | Tolerated | Benign | 0.0001354 | Het | VUS | |||||
Specific candidate pathogenic variants in “Unsolved” patients of multiplex families (in bold).
“Exac Database,” Exome Aggregation Consortium (ExAC) database.
Abbreviations: ACMG, a technical standard of the American College of Medical Genetics and Genomics; unsolved1, deafness patients in unsovled1 group. In this group, deafness patients harbored candidate pathogenic variants that could possibly explain their pathology; unsolved2, deafness patients in unsovled2 group. In this group, deafness patients harbored the candidate pathogenic variants that could not explain their pathology; VUS, variant of unknown significance; likely benign, variant of likely benign.
In the “Solved” group, most of the causative genes were SLC26A4 and MT‐RNR1 (Table 1 and Figure 1b,c). Other causative genes were also identified, like MYO7A, SOX10, CHD7, etc. Nine patients were caused by mutations in the SLC26A4 gene. SLC264 c.919‐2A>G was a frequent mutation. Eight patients were caused by the MT‐RNR1 gene, all of them were caused by homogeneous m.1555A>G mutation. Two patients were caused by compound heterozygous mutations in the MYO7A gene. Mutations were c.1679A>C, c.6115G>C, c.2183T>C and c.2187 +2_+8 delTGAGCAC. Two patients harbored mutations in OTOF gene. Other patients were caused by each one different gene. There were two patients with syndromic deafness in “Solved” patients. They were Charge syndrome and Usher syndrome 1B, respectively. A patient with Charge syndrome harbored CHD7 c.1714C>T mutation. Compound mutations of MYO7A c.1679 A>C and c.6115 G>C were identified in a patient with Usher syndrome 1B.
TABLE 1.
Variants in the “Solved” patients
| Gene | Solved | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Transcript | Genotype | Chromosome | SIFT | Polyphen2 | ExAC database | Multiplex/simplex family | Zygosity | Solved | ACMG | |
| ADGRV1 | 1 | NM_032119 | c.8551G>T | chr5:90001381 | — | — | — | Simplex | Het | Solved | Pathogenic |
| c.12631C>T | chr5:90073825 | — | — | — | Het | Pathogenic | |||||
| CHD7 | 1 | NM_017780 | c.1714C>T | chr8:61693607 | — | — | — | Simplex | Het | Solved | Pathogenic |
| CLDN14 | 1 | NM_001146077 | c.202C>T | chr21:37833792 | — | — | — | Simplex | Hom | Solved | Pathogenic |
| POU3F4 | 1 | NM_000307 | EX1 DEL | — | — | — | — | Multiplex | Hemi | Solved | Pathogenic |
| MT‐RNR1 | 1 | NC_012920 | m.1555A>G | Simplex | Hom | Solved | Pathogenic | ||||
| 7 | NC_012920 | m.1555A>G | Multiplex | Hom | Solved | Pathogenic | |||||
| MYO7A | 1 | NM_000260 | c.1679A>C | chr11:76874023 | Damaging | Probably damaging | — | Simplex | Het | Solved | Likely pathogenic |
| c.6115G>C | chr11:76922260 | Damaging | Probably damaging | — | Het | Likely pathogenic | |||||
| 1 | NM_000260 | c.2183T>C | chr11:7688650 | Damaging | Probably damaging | — | Simplex | Het | Solved | Likely pathogenic | |
| c.2187 +2_+8 delTGAGCAC |
chr11:7688651 2..768865186 |
— | — | — | Het | Pathogenic | |||||
| PCDH15 | 1 | NM_033056 | c.1864_1865 insTA | chr10:55892687..55892688 | — | — | — | Simplex | Het | Solved | Pathogenic |
| c.2220+1G>A | chr10:55826516 | — | — | — | Het | Pathogenic | |||||
| SLC26A4 | 2 | NM_000441 | c.919‐2A>G | chr7:107323898 | — | — | 0.000306 | Simplex | Hom | Solved | Pathogenic |
| 1 | c.919‐2A>G | chr7:107323898 | — | — | 0.0003058 | Multiplex | Het | Solved | Pathogenic | ||
| c.915_916 insG | chr7:1073237966..107323797 | — | — | 0.00001650 | Het | Pathogenic | |||||
| 1 | c.919‐2A>G | chr7:107323898 | — | — | 0.000306 | Multiplex | Hom | Solved | Pathogenic | ||
| 1 | c.2027T>A | chr7:107342495 | Damaging | Damaging | — | Simplex | Het | Solved | Pathogenic | ||
| c.2168A>G | chr7:107350577 | Probably damaging | Probably damaging | 0.0001238 | Het | Pathogenic | |||||
| 1 | c.919‐2A>G | chr7:107323898 | — | — | 0.0003058 | Multiplex | Het | Solved | Pathogenic | ||
| EX5_6 DEL | — | — | — | — | Het | Pathogenic | |||||
| 1 | c.1991C>T | chr7:107342459 | Tolerated | Damaging | — | Multiplex | Het | Solved | Pathogenic | ||
| c.2168A>G | chr7:107350577 | Probably damaging | Probably damaging | 0.0001238 | Het | Pathogenic | |||||
| 1 | c.1174A>T | chr7:107330593 | Damaging | Probably damaging | 0.000008242 | Simplex | Hom | Solved | Pathogenic | ||
| 1 | c.2168A>G | chr7:107350577 | Probably damaging | Probably damaging | 0.0001238 | Simplex | Het | Solved | Pathogenic | ||
| EX1_3 DEL | — | — | — | Het | Pathogenic | ||||||
| TRIOBP | 1 | NM_001039141 | c.1342C>T | chr22:38119905 | — | — | — | Simplex | Het | Solved | Pathogenic |
| c.2074C>T | chr22:38120637 | — | — | — | Het | Pathogenic | |||||
| USH1C | 1 | NM_153676 | c.388‐1G>A | chr11:17548879 | — | — | — | Simplex | Hom | Solved | Pathogenic |
| USH2A | 1 | NM_206933 | c.99_100insT | chr1:216595579..216595580 | — | — | — | Simplex | Het | Solved | Pathogenic |
| c.11660G>A | chr1:215914768 | — | — | — | Het | Pathogenic | |||||
| OTOF | 1 | NM_194248 | c.2236C>T | chr2:26700596 | Tolerated | Benign | 0.00001 | Simplex | Hom | Solved | Likely pathogenic |
| 1 | NM_194248 | c.4747C>T | chr2:26688592 | Damaging | Probably damaging | 0.00002471 | Multiplex | Hom | Solved | Likely pathogenic | |
| ILDR1 | 1 | NM_175924 | c.206C>A | chr3:121725861 | Tolerated | Probably damaging | 0.00004 | Simplex | Hom | Solved | Pathogenic |
| CDH23 | 1 | NM_022124 | c.6442G>A | chr10:73553127 | Tolerated | Probably damaging | — | Simplex | Hom | Solved | Likely pathogenic |
| EYA1 | 1 | NM_000503 | EX1_18 DEL | — | — | — | — | Simplex | Het | Solved | Pathogenic |
| SOX10 | 1 | NM_006941 | c.378C>A | chr22:38378414 | — | — | — | Simplex | Het | Solved | Likely pathogenic |
| WFS1 | 1 | NM_006005 | c.1356_1371delGCCCTACACGCGCAGG | chr4:6302878..6302893 | — | — | — | Simplex | Het | Solved | Likely pathogenic |
| COCH | 1 | NM_004086 | c.847G>A | chr14:31354713 | Tolerated | Probably damaging | — | Simplex | Het | Solved | Likely pathogenic |
| MITF | 1 | NM_198159 | c.1021C>G | chr3:70008431 | Damaging | Probably damaging | — | Simplex | Het | Solved | Likely pathogenic |
Abbreviations: ACMG, a technical standard of the American College of Medical Genetics and Genomics; Exac Database, Exome Aggregation Consortium (ExAC) database.
In the “Unsolved” group, we found 21 possibly causative genes (Table 2 and Figure 1D,E), including MYO7A, DIAPH3, PDZD7, OTOF, SLC26A4, GPR98, etc. In the dominant inheritance model, we found candidate pathogenic variants in COCH, DIAPH1, DIAPH3, MYH14, MYH9, MYO1A, MYO6, MYO7A, POU4F3, PTPRQ, and SLC17A8 genes. In the recessive inheritance model, candidate pathogenic variants were identified in GJB2, ADGRV1, LOXHD1, MYO15A, OTOF, PDZD7, SLC26A4, TMC1, and TRIOBP genes. In the mitochondrial inheritance model, m.681T>C was identified in the MT‐RNR1 gene. Nonsynonymous variants were predicted by bioinformatics tools: SIFT and Polyphen‐2. Candidate variants that were predicted to be both tolerated and benign were GJB2 c.471G>A, LOXHD1 c.977A>G, MYH9 c.1897C>T, SLC26A4 c.2110G>A, TMC1 c.373A>C, and TRIOBP c.4442C>T. Four patients were associated with the MYO7A gene. PDZD7 c.490C>T, a recurrent pathogenic variant, was identified in two deaf patients. SLC26A4 c.290T>G and c.599A>G were novel likely pathogenic variants.
To further analyze candidate pathogenic variants classified as “VUS,” the 3D protein structure of each mutant were constructed by Raptor X web server, as shown in Figure S1. ADGRV1 p.Leu4546His, LOXHD1 p.Asn326Ser, MYO15A p.Gly1318Ser, MYO7A p.Thr381Met, TMC1 p.Lys125Gln might result in alteration (increase or disappearance) of hydrogen bonds. PDZD7 p.Arg164Trp, TMC1 p.Glu483Asp, MYO7A p.Arg853His might lead to the adaptation of α‐Helix or β‐sheet. GJB2 p.Met157Ile, MYO7A p.Pro541Leu, OTOF p.Thr597Met, OTOF p.Arg1583Cys might not have an influence on hydrogen bonds, α‐Helix, and β‐sheet. We could not analyze some candidate pathogenic variants, like TRIOBP p.Ser1481Phe, since we could not generate the 3D protein structure of its corresponding wild‐type protein. Representative 3D protein structure of candidate pathogenic variants in SLC26A4, MYO7A, and PDZD7 genes is shown in Figure 2.
FIGURE 2.
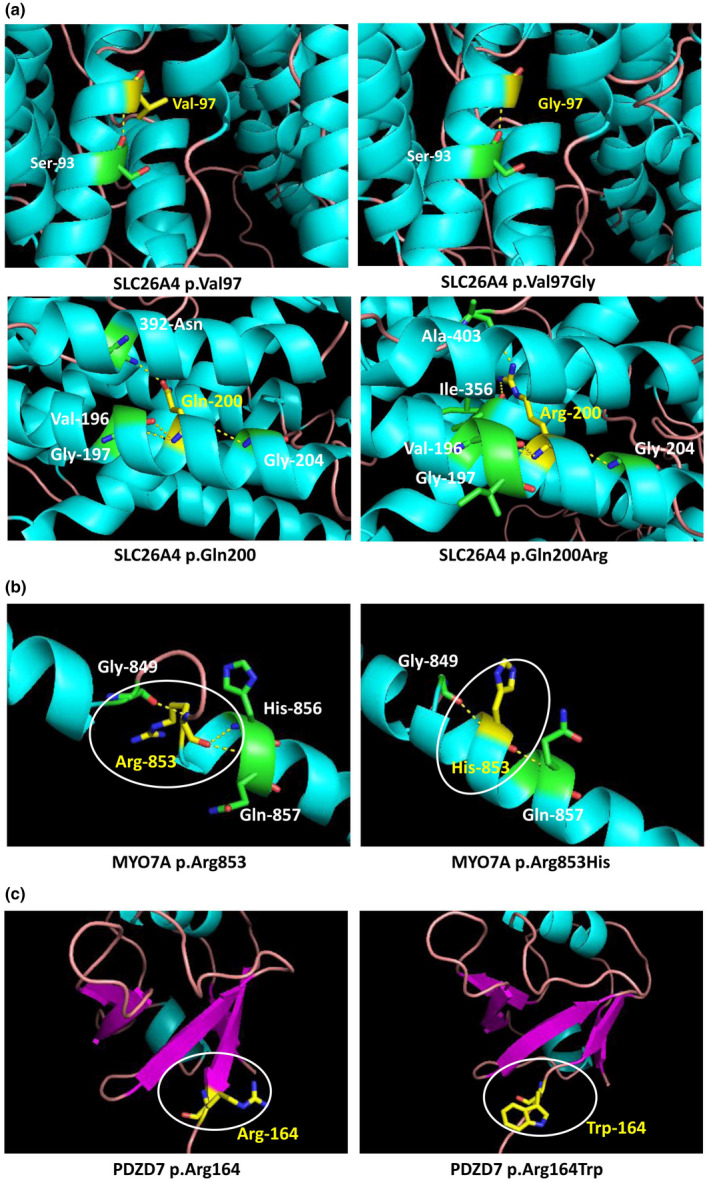
Three‐dimensional (3D) protein structure prediction and analysis of mutants in genes, like SLC26A4 (a), MYO7A (b), and PDZD7 (c) genes. These genes were shown to contain variants of unknown significance (VUS) in the present study. Hydrogen bonds were predicted by PyMOL software and shown in yellow dotted lines. α‐Helix and β‐sheet were colored as blue and red, respectively.
Generally, autosomal dominant, autosomal recessive, and maternally (mitochondrial) inheritance were observed in our cohorts of deaf patients. Candidate pathogenic variants were identified in 34 deafness genes, like SLC26A4, MT‐RNR1, MYO7A, OTOF, and PDZD7 genes.
4. DISCUSSION
In the present study, by high throughput sequencing of 127 deafness genes, we aimed to preliminarily analyze causative genes of Southwestern China in a cohort of 84 deaf patients from Southwestern China. The deafness genes in this area were rarely studied. In addition to GJB2, SLC26A4, and MT‐RNR1 genes (common deafness genes), we found candidate pathogenic variants in 34 deafness genes, like MYO7A, PDZD7, and OTOF genes.
In our cohorts of deaf patients, we found 34 genes related to deafness (Tables 1,2). The number of “Solved” patients was 34, while that of “Unsolved” patients was 36. The percentage of “Unsolved” patients in total deaf patients was higher than that of “Solved” patients. “Unsolved” patients harbored “VUS” and “likely benign” variants. Some of these variants had no allele frequency data in 1000 Genome (Clarke et al., 2012) and ExAC databases. It might be caused by genetic background differences. Altitude is closely related to oxygen density. In the present study, patients were all from Yunnan province, China. The average altitude of Yunnan province is higher than the coastal areas of China. It might lead to genetic background differences between people living in coastal areas and Yunnan province. This might be an explanation for the identification of several “VUS” variants in our cohorts of deaf patients. Several studies have shown that people in different regions have different genetic backgrounds, especially for ethnicities (Liu et al., 2002; Manzoli et al., 2016; Miyagawa et al., 2013; Yan et al., 2016). This suggested that allele frequency database of deafness genes in local people should be established. It might be very helpful. In addition, functional studies should be performed on some recurrent candidate pathogenic variants, like PDZD7 c.490 C>T.
GJB2, SLC26A4, and MT‐RNR1 genes have been identified in Chinese deaf patients of different regions (Chan & Chang, 2014; Lu et al., 2010; Shen et al., 2011; Tsukada et al., 2015). They are thought to be common deafness genes in Chinese. The distributions of uncommon deafness genes are still unclear in different populations and regions. In the study of uncommon deafness genes by Yan et al., (2016), MYO15A, USH2A, MYO7A, MYO6, and TRIOBP were their most identified genes. The results of the present study might have some difference from that of Yan et al., (2016). Although we also identified the MYO7A gene in our cohorts of deaf patients, the frequency of other candidate causative genes in our deaf patients was different from that of Yan et al., (2016), like OTOF, PDZD7, and DIAPH3 genes. The result suggested that the distribution of deafness genes might have its features in deaf patients from Southwestern China. The conclusion needs to be further confirmed, since the sample size was limited in the present study. In addition, candidate pathogenic variants in multiplex families were not tested in affected family members, since we were not able to collect their samples. It was another limitation of the present study.
Heritable deafness is a complicated disease. More than 200 deafness genes have been reported (Angeli et al., 2012; Liu et al., 2006; Petersen, 2002). Here, 127 deafness genes screening was applied to 84 deaf patients from southwestern China. Only half of the deaf patients (“Solved” patients) could be clearly diagnosed. Therefore, 127 deafness genes screening had its limitation in clinical application.
By 127 deafness genes screening, no candidate pathogenic mutations/variants were detected in 14 deaf patients. Whether these patients harbored other pathogenic mutations was still unknown. Whole‐exome/genome sequencing has been widely used to identify uncommon deafness genes (Berg et al., 2011; Choi et al., 2009; Ormond et al., 2010; Yang, Muzny, et al., 2013). To further explore molecular pathology of these patients, whole‐exome/genome sequencing might be very effective. Our results also indicated the possibility that some deaf patients from southwestern China might be caused by other deafness genes. It should be further explored.
In summary, the present study provided a preliminary overview of the spectrum of mutations in deaf patients from Southwestern China. Thirty‐four genes were found to have candidate pathogenic variants in our cohorts of patients. The findings will be helpful in the prevention and molecular diagnosis of deafness.
CONFLICT OF INTEREST
The authors declared no conflicts of interest.
AUTHORS’ CONTRIBUTIONS
Study concepts: Yunlong Li and Baosheng Zhu. Literature research: Yunlong Li. Clinical information collection: Jie Su, Jingman Zhang, Jiahong Pei, Dongmei Li, and Yinhong Zhang. Data acquisition: Jingyu Li and Menglang Chen. Data analysis/interpretation: Yunlong Li Jingyu Li, Menglang Chen. Manuscript preparation: Yunlong Li. Manuscript revision/review: Yunlong Li, Jie Su, Jingman Zhang, Jiahong Pei, Dongmei Li, Yinhong Zhang, and Baosheng Zhu. Manuscript final version approval: Yunlong Li and Baosheng Zhu.
Supporting information
Table S1
Fig S1
Funding information
The present study was supported by the National Natural Science Foundation of China (81860190), the Joint Special Research Funds of Kunming Medical University (2017FE468 [‐010]), the Foundation of Medical Discipline Leaders Program of Health and Family Planning Commission of Yunnan Province (D‐201668), the Personnel Training Project of Yunnan Province (2017HB043) and the funds from Institute of Clinical Genetics in Yunnan Province (2018NS0251).
DATA AVAILABILITY STATEMENT
Data available on request from the authors.
REFERENCES
- Angeli, S. , Lin, X. , & Liu, X. Z. (2012). Genetics of hearing and deafness. Anatomical Record (Hoboken), 295(11), 1812–1829. 10.1002/ar.22579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, J. S. , Khoury, M. J. , & Evans, J. P. (2011). Deploying whole genome sequencing in clinical practice and public health: meeting the challenge one bin at a time. Genetics in Medicine, 13(6), 499–504. 10.1097/GIM.0b013e318220aaba [DOI] [PubMed] [Google Scholar]
- Chan, D. K. , & Chang, K. W. (2014). GJB2‐associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope, 124(2), E34–E53. 10.1002/lary.24332 [DOI] [PubMed] [Google Scholar]
- Choi, M. , Scholl, U. I. , Ji, W. , Liu, T. , Tikhonova, I. R. , Zumbo, P. , Nayir, A. , Bakkaloğlu, A. , Özen, S. , Sanjad, S. , Nelson‐Williams, C. , Farhi, A. , Mane, S. , & Lifton, R. P. (2009). Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proceedings of the National Academy of Sciences of the United States of America, 106(45), 19096–19101. 10.1073/pnas.0910672106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, L. , Zheng‐Bradley, X. , Smith, R. , Kulesha, E. , Xiao, C. , Toneva, I. , Vaughan, B. , Preuss, D. , Leinonen, R. , Shumway, M. , Sherry, S. , & Flicek, P. (2012). The 1000 Genomes Project: data management and community access. Nature Methods, 9(5), 459–462. 10.1038/nmeth.1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva, E. O. , & Duarte, A. R. (1995). Genetic deafness: a brief review. Jornal de Pediatria, 71(6), 297–302. 10.2223/jped.796 [DOI] [PubMed] [Google Scholar]
- Donovan, J. L. (1984). Ethnicity and health: a research review. Social Science and Medicine, 19(7), 663–670. 10.1016/0277-9536(84)90237-5 [DOI] [PubMed] [Google Scholar]
- Duman, D. , & Tekin, M. (2012). Autosomal recessive nonsyndromic deafness genes: a review. Frontiers in Bioscience, 17, 2213–2236. 10.2741/4046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R. , Li, Y. , Fang, X. , Yang, H. , Wang, J. , Kristiansen, K. , & Wang, J. (2009). SNP detection for massively parallel whole‐genome resequencing. Genome Research, 19(6), 1124–1132. 10.1101/gr.088013.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Su, J. , Ding, C. , Yu, F. , & Zhu, B. (2019). Identification of four novel mutations in MYO7A gene and their association with nonsyndromic deafness and Usher Syndrome 1B. International Journal of Pediatric Otorhinolaryngology, 120, 166–172. 10.1016/j.ijporl.2019.02.021 [DOI] [PubMed] [Google Scholar]
- Li, Y. , Zhu, B. , Su, J. , Yin, Y. , & Yu, F. (2016). Identification of SLC26A4 mutations p. L582LfsX4, p.I188T and p.E704K in a Chinese family with large vestibular aqueduct syndrome (LVAS). International Journal of Pediatric Otorhinolaryngology, 91, 1–5. 10.1016/j.ijporl.2016.08.026 [DOI] [PubMed] [Google Scholar]
- Lin, X. , Tang, W. , Ahmad, S. , Lu, J. , Colby, C. C. , Zhu, J. , & Yu, Q. (2012). Applications of targeted gene capture and next‐generation sequencing technologies in studies of human deafness and other genetic disabilities. Hearing Research, 288(1–2), 67–76. 10.1016/j.heares.2012.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Ouyang, X. , & Yan, D. (2006). The Genetic Deafness in Chinese Population. Journal of Otology, 1(1), 1–10. [Google Scholar]
- Liu, X. , Xia, X. , Ke, X. , Ouyang, X. , Du, L. I. , Liu, Y. U. , Angeli, S. , Telischi, F. , Nance, W. , Balkany, T. , & Xu, L. I. (2002). The prevalence of connexin 26 (GJB2) mutations in the Chinese population. Human Genetics, 111(4–5), 394–397. 10.1007/s00439-002-0811-6 [DOI] [PubMed] [Google Scholar]
- Lu, J. , Li, Z. , Zhu, Y. I. , Yang, A. , Li, R. , Zheng, J. , Cai, Q. , Peng, G. , Zheng, W. , Tang, X. , Chen, B. , Chen, J. , Liao, Z. , Yang, L. I. , Li, Y. , You, J. , Ding, Y. U. , Yu, H. , Wang, J. , … Guan, M.‐X. (2010). Mitochondrial 12S rRNA variants in 1642 Han Chinese pediatric subjects with aminoglycoside‐induced and nonsyndromic hearing loss. Mitochondrion, 10(4), 380–390. 10.1016/j.mito.2010.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoli, G. N. , Bademci, G. , Acosta, A. X. , Felix, T. M. , Cengiz, F. B. , Foster, J. 2nd , & Tekin, M. (2016). Targeted resequencing of deafness genes reveals a founder MYO15A variant in Northeastern Brazil. Annals of Human Genetics, 80(6), 327–331. 10.1111/ahg.12177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meena, R. , & Ayub, M. (2017). Genetics of human hereditary hearing impairment. Journal of Ayub Medical College, Abbottabad, 29(4), 671–676. [PubMed] [Google Scholar]
- Miyagawa, M. , Naito, T. , Nishio, S. Y. , Kamatani, N. , & Usami, S. (2013). Targeted exon sequencing successfully discovers rare causative genes and clarifies the molecular epidemiology of Japanese deafness patients. PLoS One, 8(8), e71381. 10.1371/journal.pone.0071381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagawa, M. , Nishio, S. Y. , Ikeda, T. , Fukushima, K. , & Usami, S. (2013). Massively parallel DNA sequencing successfully identifies new causative mutations in deafness genes in patients with cochlear implantation and EAS. PLoS One, 8(10), e75793. 10.1371/journal.pone.0075793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagawa, M. , Nishio, S. Y. , Kumakawa, K. , & Usami, S. (2015). Massively parallel DNA sequencing successfully identified seven families with deafness‐associated MYO6 mutations: the mutational spectrum and clinical characteristics. The Annals of Otology, Rhinology, and Laryngology, 124(Suppl 1), 148S–157S. 10.1177/0003489415575055 [DOI] [PubMed] [Google Scholar]
- Ormond, K. E. , Wheeler, M. T. , Hudgins, L. , Klein, T. E. , Butte, A. J. , Altman, R. B. , Ashley, E. A. , & Greely, H. T. (2010). Challenges in the clinical application of whole‐genome sequencing. Lancet, 375(9727), 1749–1751. 10.1016/S0140-6736(10)60599-5 [DOI] [PubMed] [Google Scholar]
- Petersen, M. B. (2002). Non‐syndromic autosomal‐dominant deafness. Clinical Genetics, 62(1), 1–13. 10.1034/j.1399-0004.2002.620101.x [DOI] [PubMed] [Google Scholar]
- Shearer, A. E. , & Smith, R. J. (2012). Genetics: advances in genetic testing for deafness. Current Opinion in Pediatrics, 24(6), 679–686. 10.1097/MOP.0b013e3283588f5e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Z. , Zheng, J. , Chen, B. , Peng, G. , Zhang, T. , Gong, S. , Zhu, Y. I. , Zhang, C. , Li, R. , Yang, L. I. , Zhou, J. , Cai, T. , Jin, L. , Lu, J. , & Guan, M.‐X. (2011). Frequency and spectrum of mitochondrial 12S rRNA variants in 440 Han Chinese hearing impaired pediatric subjects from two otology clinics. Journal of Translational Medicine, 9, 4. 10.1186/1479-5876-9-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson, P. D. , Mort, M. , Ball, E. V. , Shaw, K. , Phillips, A. , & Cooper, D. N. (2014). The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Human Genetics, 133(1), 1–9. 10.1007/s00439-013-1358-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada, K. , Nishio, S. Y. , Hattori, M. , & Usami, S. (2015). Ethnic‐specific spectrum of GJB2 and SLC26A4 mutations: their origin and a literature review. The Annals of Otology, Rhinology, and Laryngology, 124(Suppl 1), 61S–76S. 10.1177/0003489415575060 [DOI] [PubMed] [Google Scholar]
- Vona, B. , Müller, T. , Nanda, I. , Neuner, C. , Hofrichter, M. A. H. , Schröder, J. , Bartsch, O. , Läßig, A. , Keilmann, A. , Schraven, S. , Kraus, F. , Shehata‐Dieler, W. , & Haaf, T. (2014). Targeted next‐generation sequencing of deafness genes in hearing‐impaired individuals uncovers informative mutations. Genetics in Medicine, 16(12), 945–953. 10.1038/gim.2014.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Li, W. , Liu, S. , & Xu, J. (2016). RaptorX‐Property: a web server for protein structure property prediction. Nucleic Acids Research, 44(W1), W430–W435. 10.1093/nar/gkw306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, D. , Tekin, D. , Bademci, G. , Foster, J. , Cengiz, F. B. , Kannan‐Sundhari, A. , Guo, S. , Mittal, R. , Zou, B. , Grati, M. , Kabahuma, R. I. , Kameswaran, M. , Lasisi, T. J. , Adedeji, W. A. , Lasisi, A. O. , Menendez, I. , Herrera, M. , Carranza, C. , Maroofian, R. , … Tekin, M. (2016). Spectrum of DNA variants for non‐syndromic deafness in a large cohort from multiple continents. Human Genetics, 135(8), 953–961. 10.1007/s00439-016-1697-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, T. , Wei, X. , Chai, Y. , Li, L. , & Wu, H. (2013). Genetic etiology study of the non‐syndromic deafness in Chinese Hans by targeted next‐generation sequencing. Orphanet Journal of Rare Diseases, 8, 85. 10.1186/1750-1172-8-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Muzny, D. M. , Reid, J. G. , Bainbridge, M. N. , Willis, A. , Ward, P. A. , Braxton, A. , Beuten, J. , Xia, F. , Niu, Z. , Hardison, M. , Person, R. , Bekheirnia, M. R. , Leduc, M. S. , Kirby, A. , Pham, P. , Scull, J. , Wang, M. , Ding, Y. , … Eng, C. M. (2013). Clinical whole‐exome sequencing for the diagnosis of mendelian disorders. New England Journal of Medicine, 369(16), 1502–1511. 10.1056/NEJMoa1306555 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Fig S1
Data Availability Statement
Data available on request from the authors.