

Abstract
Tumour progression locus 2 (TPL‐2) kinase mediates Toll‐like receptor (TLR) activation of ERK1/2 and p38α MAP kinases in myeloid cells to modulate expression of key cytokines in innate immunity. This study identified a novel MAP kinase‐independent regulatory function for TPL‐2 in phagosome maturation, an essential process for killing of phagocytosed microbes. TPL‐2 catalytic activity was demonstrated to induce phagosome acidification and proteolysis in primary mouse and human macrophages following uptake of latex beads. Quantitative proteomics revealed that blocking TPL‐2 catalytic activity significantly altered the protein composition of phagosomes, particularly reducing the abundance of V‐ATPase proton pump subunits. Furthermore, TPL‐2 stimulated the phosphorylation of DMXL1, a regulator of V‐ATPases, to induce V‐ATPase assembly and phagosome acidification. Consistent with these results, TPL‐2 catalytic activity was required for phagosome acidification and the efficient killing of Staphylococcus aureus and Citrobacter rodentium following phagocytic uptake by macrophages. TPL‐2 therefore controls innate immune responses of macrophages to bacteria via V‐ATPase induction of phagosome maturation.
Keywords: DMXL1, macrophage phagosome, Staphylococcus aureus, TPL‐2 kinase, V‐ATPase
Subject Categories: Immunology, Signal Transduction
In addition to the well‐established role of TPL‐2 in regulating cytokine expression in response to TLR signaling, TPL‐2 also regulates phagosome maturation via the V‐ATPase to control a bacterial infection.
Introduction
A key component of the innate immune response involves the killing of phagocytosed microbes, such as bacteria, by macrophages (Pauwels et al, 2017). Phagocytosis internalises bacteria into membrane‐bound vacuoles in the cytoplasm called phagosomes (Flannagan et al, 2012). The nascent phagosome is innocuous, and to kill internalised bacteria, the phagosome must mature via an ordered series of membrane fusion and fission events with endosomes and ultimately with lysosomes to form the phagolysosome, a potent microbicidal organelle (Flannagan et al, 2009). During the maturation process, phagosomes become increasingly acidic, highly oxidative and enriched with proteases and hydrolases that can degrade the internalised bacteria (Flannagan et al, 2009). Phagosome acidification is mediated by V‐ATPases, ATP‐dependent proton pumps, which are recruited to the phagosome during maturation (Lukacs et al, 1990; Sun‐Wada et al, 2009). Acidification inhibits bacterial growth (Downey et al, 1999; Ip et al, 2010), activates cathepsin proteases that degrade internalised bacteria (Yates et al, 2005) and promotes NOX2 production of reactive oxygen species (ROS) that damage bacterial proteins, lipids and nucleic acids (Savina et al, 2006).
Bacterial infection also engages macrophage Toll‐like receptors (TLRs) on the plasma and endosomal membranes, triggering intracellular signalling pathways that control activation of NF‐κB transcription factors and each of the major mitogen‐activated protein (MAP) kinases (extracellular signal‐regulated kinases 1 and 2 [ERK1/2], Jun amino‐terminal kinases 1 and 2 [JNK1/2] and p38α) (Arthur & Ley, 2013). Together, these stimulate the expression of multiple genes, the products of which can directly target the invading bacterium (e.g. antimicrobial peptides and NOS2) or induce recruitment of additional immune cells (e.g. cytokines and chemokines) (Smale, 2010; Kawasaki & Kawai, 2014). This inflammatory response is essential for killing of bacteria by macrophages and also the subsequent induction of the adaptive immune response (Nau et al, 2002).
TLR activation of ERK1/2 and p38α in macrophages is mediated by TPL‐2 (tumour progression locus 2, also known as MAP3K8), a MAP 3‐kinase that directly phosphorylates and activates the MAP 2‐kinases MKK1, 2, 3 and 6 (Gantke et al, 2012; Pattison et al, 2016). TPL‐2 is critical for inflammatory immune responses to invading bacteria, fungi and viruses. TPL‐2 forms a stoichiometric complex with NF‐κB1 p105 and A20 binding inhibitor of NF‐κB (ABIN)‐2 in unstimulated macrophages (Lang et al, 2004). NF‐κB1 p105 functions as an inhibitor of TPL‐2 and TLR activation of ERK1/2, and p38α requires the liberation of TPL‐2 from p105‐mediated inhibition (Beinke et al, 2003, 2004). This results from p105 proteolysis by the proteasome triggered by IκB kinase (IKK)‐induced p105 phosphorylation (Belich et al, 1999). ABIN‐2 is also released from p105 after TLR stimulation but is not required for TPL‐2 activation, and its function in innate immune responses remains unclear.
TPL‐2 expression is required for efficient immune responses to Citrobacter rodentium, decreasing the bacterial burden and dissemination to the liver and spleen (Acuff et al, 2017). TPL‐2 deficiency reduces neutrophil recruitment to the colon at peak infection. In vitro experiments have shown that TPL‐2 expression in neutrophils is required for LPS activation of ERK1/2 and induction of tumour necrosis factor (TNF) secretion, similar to that in macrophages (Acuff et al, 2017). TPL‐2 is also required for efficient killing of phagocytosed C. rodentium, although phagocytic uptake of bacteria is TPL‐2‐independent (Acuff et al, 2017). However, the mechanism by which TPL‐2 regulates bacterial killing has not been established. A recent study by one of our groups using quantitative mass spectrometry demonstrated that TPL‐2 catalytic activity potentially regulates intracellular trafficking. Tpl2 D270A kinase‐dead mutation reduced the phosphorylation of several proteins linked to endocytosis, vesicle trafficking and GTPase signalling in LPS‐stimulated macrophages (Pattison et al, 2016). Together, these results raised the interesting possibility that TPL‐2 promotes the killing of internalised bacteria by controlling phagosome maturation in myeloid cells.
In the present study, the potential regulation of phagosome maturation by TPL‐2 in macrophages was investigated. TPL‐2 catalytic activity was found to induce macrophage phagosome proteolytic activity and phagosome acidification, both independently of its ability to activate MAP kinase signalling. These effects were mediated by direct regulation of V‐ATPase assembly and function via controlling the phosphorylation of the V‐ATPase‐associated regulatory protein DMXL1. Consistent with these findings, TPL‐2 catalytic activity was necessary for efficient macrophage killing of phagocytosed Staphylococcus aureus and Citrobacter rodentium. These results indicate that TPL‐2 has two key functions in the innate immune response of macrophages to bacterial infection. TPL‐2 mediates TLR activation of MAP kinase signalling to control inflammation by modulation of gene expression and directly promotes bacterial killing by MAP kinase‐independent induction of phagosome acidification.
Results
TPL‐2 catalytic activity promotes macrophage phagosome maturation
To investigate the role of TPL‐2 signalling in phagosome maturation, bone marrow‐derived macrophages (BMDMs) were generated from Tpl2 D270A/D270A knock‐in mice, which express catalytically inactive TPL‐2 (Sriskantharajah et al, 2014). Consistent with earlier experiments using Tpl2 −/− neutrophils (Acuff et al, 2017), phagocytic uptake of fluorescently labelled beads by Tpl2 D270A/D270A BMDMs was similar to wild‐type (WT) controls (Fig 1A). To investigate whether TPL‐2 regulated phagosome maturation, bulk intra‐phagosomal proteolysis was monitored using latex beads coupled to the fluorescent substrate DQ green BSA (Yates & Russell, 2008; Russell et al, 2009). Alexa Fluor 594 labelling of beads was used to normalise phagocytic uptake. Real‐time measurements showed that phagosome proteolysis was significantly reduced in Tpl2 D270A/D270A BMDMs compared to WT controls (Fig 1B).
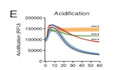
Figure 1. Tpl2[D270A] mutation impairs phagosome maturation.
- Phagocytic uptake of fluorescently labelled latex beads by WT and Tpl2[D270A] (Tpl2 D270/D270A) BMDMs. Intracellular fluorescence was monitored following uptake of AF488 latex beads by BMDMs (n = 4 wells).
- Intra‐phagosomal proteolysis in WT and Tpl2[D270A] BMDMs was assayed following uptake of DQ Green BSA / AF594 latex beads. As positive assay control, BMDMs were separately pre‐treated with 100 μg/ml leupeptin for 1 h to inhibit serine‐cysteine proteases (n = 4 wells).
- Cathepsin activity assay in WT and Tpl2[D270A] BMDMs 0.5 h after uptake of latex beads. BMDMs were stained with the Magic Red cathepsin L substrate (red). Average fluorescence intensity of the cathepsin probe per cell was quantified (n = 40–51 cells).
- Intra‐phagosomal acidification in WT and Tpl2[D270A] BMDMs was monitored following uptake of BCECF‐coupled latex beads. BMDMs were pre‐treated with 1 μM bafilomycin A1 for 15 min to inhibit V‐ATPases (n = 4 wells).
- pH assay in WT and Tpl2[D270A] BMDMs upon 0.5 h after incubation with latex beads. BMDMs were stained with the LysoTracker Red DND‐99 dye (red). Average fluorescence intensity of the LysoTracker Red probe per cell was quantified (n = 95–126 cells).
- Intra‐phagosomal oxidative burst in WT and Tpl2[D270A] BMDMs was assayed following uptake of OxyBURST Green BSA / AF594 latex beads. BMDMs were pre‐treated with 1 μM bafilomycin A1 for 15 min to inhibit ROS production (n = 4 wells).
- Reactive oxygen species assay in WT and Tpl2[D270A] BMDMs upon 0.5 h after incubation with latex beads. BMDMs were stained with the ROS Deep Red dye. Average fluorescence intensity of the ROS Deep Red probe per cell was quantified (n = 80–110 cells).
Data information: One representative experiment out of three shown. Error bars and shaded areas represent SEM. ****P < 0.0001. Panels (B, D, F) Paired Mann–Whitney t‐test; All differences relative to WT are ****. Panels (C, E, G) Student’s unpaired t‐test.
Next, a confocal microscopy assay was used to monitor cathepsin protease activation following phagocytic uptake of latex beads by Tpl2 D270A/D270A BMDMs compared to WT control cells, using a fluorescently labelled cathepsin L target peptide. Thirty mins after bead uptake, phagosome cathepsin activity was significantly reduced by Tpl2 D270A mutation (Fig 1C). These results showed that TPL‐2 catalytic activity is required to increase protease activity inside phagosomes of primary macrophages.
During maturation, the lumens of phagosomes become increasingly acidic due to the action of V‐ATPases that are recruited by fusion of phagosomes initially with endocytic vesicles and ultimately lysosomes (Kinchen & Ravichandran, 2008). Intra‐phagosomal acidification was assayed using latex beads coupled to the pH indicator BCECF (Russell et al, 1995). Real‐time measurements showed that phagosomal acidification was significantly decreased in Tpl2 D270A/D270A BMDMs compared to WT BMDMs (Fig 1D). Tpl2 D270A mutation reduced phagosome acidification to a similar degree to the pre‐treatment of WT BMDMs with the V‐ATPase inhibitor bafilomycin A1 (Fig 1D). Consistent with these results, analysis of phagosomal pH by staining with LysoTracker Red, a dye that labels acidic compartments, and confocal microscopy demonstrated that Tpl2 D270A mutation reduced LysoTracker Red staining following latex bead uptake (Fig 1E).
The production of reactive oxygen species (ROS) in phagosomes by NAPDH oxidase is essential for killing internalised bacteria and is dependent on an intra‐phagosomal proton gradient by V‐ATPases (Mantegazza et al, 2008). Consistent with its inhibitory effects on phagosome acidification, Tpl2 D270A mutation significantly reduced ROS generation in BMDMs after uptake of latex beads coupled to the fluorescence reporter substrate OxyBURST Green BSA (Fig 1F) (Vanderven et al, 2009). Tpl2 D270A mutation also significantly reduced ROS generation detected microscopically using ROS Deep Red dye following latex bead uptake (Fig 1G).
Together, these results show that TPL‐2 catalytic activity promotes the maturation of phagosomes in macrophages.
TPL‐2 induces phagosome maturation independently of MAP kinase activation
TPL‐2’s established functions in innate immunity are mediated by activation of MAP kinase pathways (Gantke et al, 2011). However, uptake of latex beads by BMDMs did not induce ERK1/2 and p38α activation, as detected by phospho‐antibody immunoblotting, suggesting that TPL‐2 modulated phagosome maturation independently of its ability to activate MAP kinases. Genetic and pharmacological experiments were used to investigate whether MAP kinase activation was required for TPL‐2 catalytic activity to promote phagosome maturation.
IKK triggers proteolysis of NF‐κB1 p105 by phosphorylating Ser930 and Ser935 in the p105 PEST region (Lang et al, 2003). Nfkb1 SSAA mutation, which changes both serines to alanine, blocks IKK‐induced p105 proteolysis, thereby preventing release of TPL‐2 and ABIN‐2 from p105 and blocking TPL‐2 activation of ERK1/2 and p38α MAP 2 kinases (Yang et al, 2012; Pattison et al, 2016). Phagosome proteolysis and acidification, however, were not affected by Nfkb1 SSAA mutation (Fig 2A and B). Importantly, however, both processes were reduced by Nfkb1 SSAA Tpl2 D270A compound mutation confirming their dependence on TPL‐2 catalytic activity (Fig 2A and B). Inhibition of ERK1/2 phosphorylation in Nfkb1 SSAA and Nfkb1 SSAA Tpl2 D270A BMDMs following LPS stimulation was confirmed by immunoblotting (Fig 2C). In line with these genetic data, simultaneous pharmacological inhibition of ERK1/2 (PD0325901) and p38α (VX‐745) activity did not alter phagosome proteolysis or acidification (Fig 2D and E). In contrast, TPL‐2 inhibition with the small molecule inhibitor C34 (Wu et al, 2009) significantly reduced both processes to a similar degree as Tpl2 D270A mutation (Fig 2D and E). Substantial reductions of MAP kinase activation by pharmacological inhibitors were confirmed by immunoblotting of LPS‐stimulated BMDM lysates (Fig 2F).
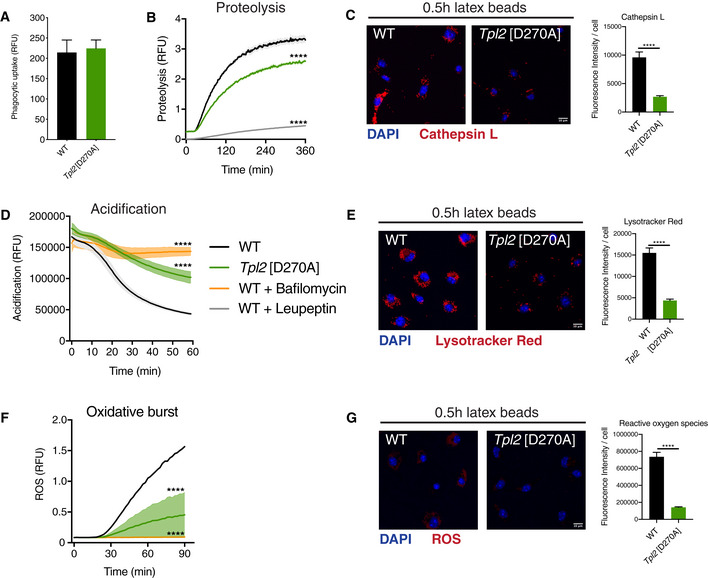
Figure 2. Tpl2[D270A] mutation promotes phagosome maturation independently of MAP kinase activation.
-
A–FExperiments were performed using murine BMDMs. (A) Intra‐phagosomal proteolysis in WT, Nfkb1[SSAA] (Nfkb1 SSAA/SSAA), and Nfkb1[SSAA]/Tpl2[D270A] BMDMs were monitored as in Fig 1B (n = 4 wells). (B) Intra‐phagosomal acidification in WT, Nfkb1[SSAA], and Nfkb1[SSAA]/Tpl2[D270A] BMDMs was monitored as in Fig 1C (n = 4 wells). (C) Cell extracts from LPS‐stimulated Nfkb1[SSAA] and Nfkb1[SSAA]/Tpl2[D270A] BMDMs were immunoblotted for the indicated antigens. (D) Intra‐phagosomal proteolysis in WT and inhibitor‐treated BMDMs (n = 4 wells) was monitored as in Fig 1B. BMDMs were pre‐treated with 0.1 μM PD0325901 (10 min) to inhibit MEK1, pre‐treated with 1 μM VX‐745 (1 h) to inhibit p38α, and pre‐treated with 10 μM C34 (1 h) to inhibit TPL‐2. (E). Intra‐phagosomal acidification in WT and inhibitor‐treated BMDMs (see (D) for conditions) was assayed as in Fig 1C (n = 4 wells). (F) Cell extracts from LPS‐stimulated, inhibitor‐treated WT, and Tpl2[D270A] BMDMs were immunoblotted for the indicated antigens. 1 μM bafilomycin A1 was added to inhibit V‐ATPases.
-
G–KExperiments were performed using human primary monocyte‐derived macrophages. (G) Intra‐phagosomal proteolysis in human macrophages was monitored following uptake of DQ Green BSA / AF594 latex beads. TPL‐2 catalytic kinase activity was blocked by pre‐treatment with 10 μM C34 for 1 h (n = 4 wells). (H) Intra‐phagosomal acidification in human macrophages was monitored following uptake of BCECF‐coupled latex beads (n = 4 wells). (I) Intra‐phagosomal proteolysis in human macrophages was assayed as in Fig 2G. MAP kinase activity was inhibited by combinatorial pre‐treatment with 0.1 μM PD0325901 (MEK1 inhibition) for 10 min and 1 μM VX‐745 (p38 inhibition) for 1 h (n = 4 wells). (J) Intra‐phagosomal acidification in human macrophages was monitored as in Fig 2H (n = 4 wells). (K) Cell extracts from LPS‐stimulated, inhibitor‐treated primary human macrophages were immunoblotted for p‐ERK1/2, ERK1/2, p‐p38, p38, and HSP90.
Data information: One representative experiment out of three shown. Error bars and shaded areas represent SEM. ****P < 0.0001. Panels (A, B, D, E, G, H, I, J) Paired Mann–Whitney t‐test; all differences relative to WT are ****.
TPL‐2 catalytic activity therefore stimulated phagosome maturation in macrophages independently of MAP kinase activation (via unknown substrates). The inhibitory effects of C34 on phagosome proteolysis and acidification in WT BMDMs also demonstrated that the effects of TPL‐2 catalytic activity on phagosome maturation were mediated acutely following latex bead uptake.
To investigate the role of TPL‐2 in phagosome maturation in human primary macrophages, macrophages were generated from monocytes isolated from peripheral blood by culturing in GM‐CSF and phagosome maturation was assayed as for BMDMs. C34 inhibition of TPL‐2 catalytic activity reduced intra‐phagosomal proteolysis (Fig 2G) and acidification (Fig 2H) in monocyte‐derived human macrophages compared to cells pre‐treated with vehicle control. Pharmacological inhibition of MEK1/2 (PD0325901) (Ciuffreda et al, 2009) and p38α (VX‐745) (Duffy et al, 2011) did not alter either phagosome proteolysis (Fig 2I) or acidification (Fig 2J), although these inhibitors potently inhibited phosphorylation of ERK1/2 and p38α following LPS stimulation (Fig 2K). These experiments showed that MAP kinase‐independent stimulation of phagosome maturation by TPL‐2 catalytic activity was conserved between mouse and human primary macrophages.
TPL‐2 catalytic activity regulates the protein composition of phagosomes
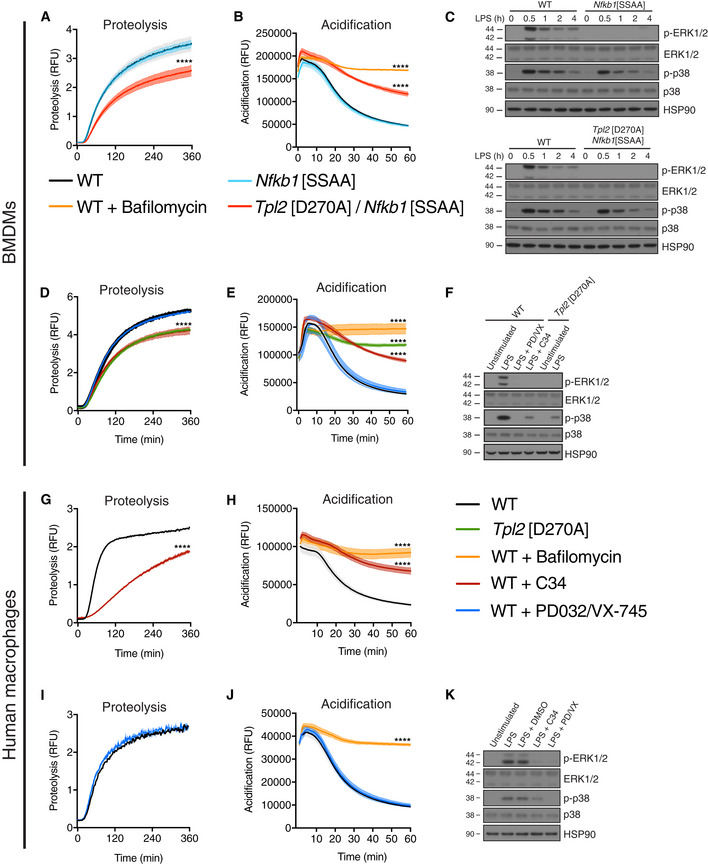
To further study the molecular mechanisms by which TPL‐2 kinase activity promotes phagosome maturation, the composition of phagosomes isolated from Tpl2 D270A/D270A and WT BMDMs following uptake of latex beads was analysed by mass spectrometry. Tpl2 D270A mutation significantly altered the composition of the phagosome proteome (Fig 3A and B, Dataset EV1). Phagosome abundance of numerous Rab GTPases was decreased by Tpl2 D270A mutation (Fig 3A). These included RAB5 and RAB7, which have key roles in regulating the maturation of phagosomes (Vieira et al, 2003; Huynh et al, 2007). The abundance of LAMP1, which is essential for late phagosome/lysosome fusion, was also decreased in Tpl2 D270A/D270A phagosomes compared with WT cells (Fig 3A and D). The levels of seven V‐ATPase subunits, including V‐ATPase subunits D, D1 and E1, were also significantly reduced (Fig 3C). This finding was particularly interesting given the inhibitory effect of Tpl2 D270A mutation on phagosome acidification. Phagosome abundance of four members of the LAMTOR protein complex, a key regulator of lysosomal trafficking (Colaço & Jäättelä, 2017), was also reduced in Tpl2 D270A/D270A BMDMs compared to controls. Immunoblotting confirmed that Tpl2 D270A mutation reduced levels of RAB5 GTPase and LAMP‐1 associated with purified phagosomes (Fig 3D). The effect of TPL‐2 catalytic activity on the abundance of phagosomal proteins was selective since abundance of numerous proteins on phagosomes, including vimentin, SNARE, vacuolar protein sorting (VPS) and ESCRT proteins, was similar between WT and Tpl2 D270A/D270A BMDMs (Figs 3D and EV1).
Figure 3. Tpl2[D270A] mutation alters the protein composition of phagosomes.
- Heatmap of selected proteins that were significantly downregulated in BMDMs from Tpl2[D270A] mice relative to WT (P < 0.05). Selected hits were grouped into clusters according to their molecular functions.
- Gene set enrichment analysis (GSEA) of significantly downregulated biological processes in phagosomal fractions. Changes of Tpl2[D270A] phagosomes relative to WT. Dot colour; enrichment score. Dot size; statistical significance.
- Protein intensities of V‐ATPase subunits from phagosomes purified from WT and Tpl2[D270A] BMDMs, (n = 3 biological replicates).
- Protein intensities of RAB5 and LAMP‐1 from phagosome proteome analysis of WT and Tpl2[D270A] BMDMs (n = 3 biological replicates) (left). Immunoblot of isolated phagosomes from WT and Tpl2[D270A] BMDMs probed for RAB5, LAMP‐1, and vimentin. Phagosomal fractions of two biological replicates were pooled. One representative experiment out of two shown (right) (n = 2).
Data information: Data were analysed by Student’s t‐test. Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure EV1. Tpl2[D270A] mutation does not alter abundance of several key phagosomal proteins on isolated phagosomes.
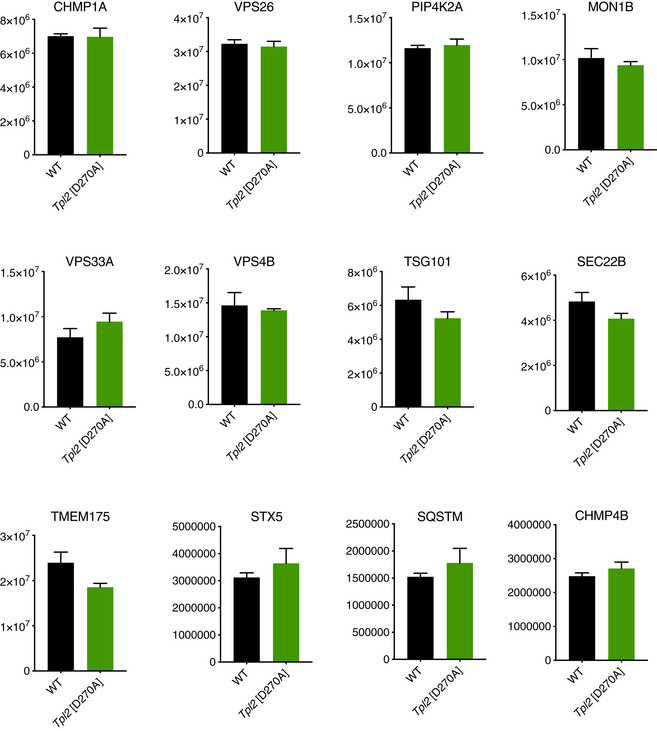
Protein intensities of selected proteins from phagosomes purified from WT and Tpl2[D270A] BMDMs (n = 3 biological replicates). Error bars represent SEM.
Consistent with these changes in phagosome composition, gene set enrichment analyses to identify significantly downregulated biological processes revealed that Tpl2 D270A mutation decreased the abundance of proteins involved in vesicle‐mediated transport, receptor‐mediated transport, ion transport, vesicle proteolysis and regulation of pH (Fig 3B). These processes are all important in the generation of mature phagolysosomes (Flannagan et al, 2009; Pauwels et al, 2017).
Together, these experiments indicated that blocking TPL‐2 catalytic activity substantially changed the composition of phagosomes in BMDMs, reducing the abundance of several proteins with key roles in regulating different stages of phagosome maturation. Importantly, this analysis revealed that TPL‐2 catalytic activity promoted the recruitment of V‐ATPase subunits to phagosomes during their maturation suggesting a possible mechanism for TPL‐2 control of phagosome acidification.
TPL‐2 induces phosphorylation of DMXL1, a component of the V‐ATPase complex
To further investigate the mechanism by which TPL‐2 kinase activity regulated phagosome maturation, the phosphoproteome in Tpl2 D270A/D270A and WT BMDMs was characterised 30 min after phagocytic uptake of latex beads. TMT (tandem mass tag) labelling and mass spectrometry were used to quantify differences in phosphopeptide abundance (Dataset EV2).
The phospho‐site most highly downregulated by Tpl2 D270A mutation was Ser141 on TPL‐2 itself (Fig 4A), a known autophosphorylation site (Stafford et al, 2006; Xu et al, 2018). Two of the next most downregulated phospho‐sites were Ser1903 and Ser1904 of DMXL1, which were both reduced over threefold. Phosphorylation of DMXL1 on Ser434, Ser915, Thr1373 and Ser1900 was not altered by Tpl2 D270A mutation indicating that TPL‐2 catalytic activity did not change DMXL1 protein abundance (Fig 4A).
Figure 4. TPL‐2 induces phosphorylation of DMXL1, a V‐ATPase regulatory protein.
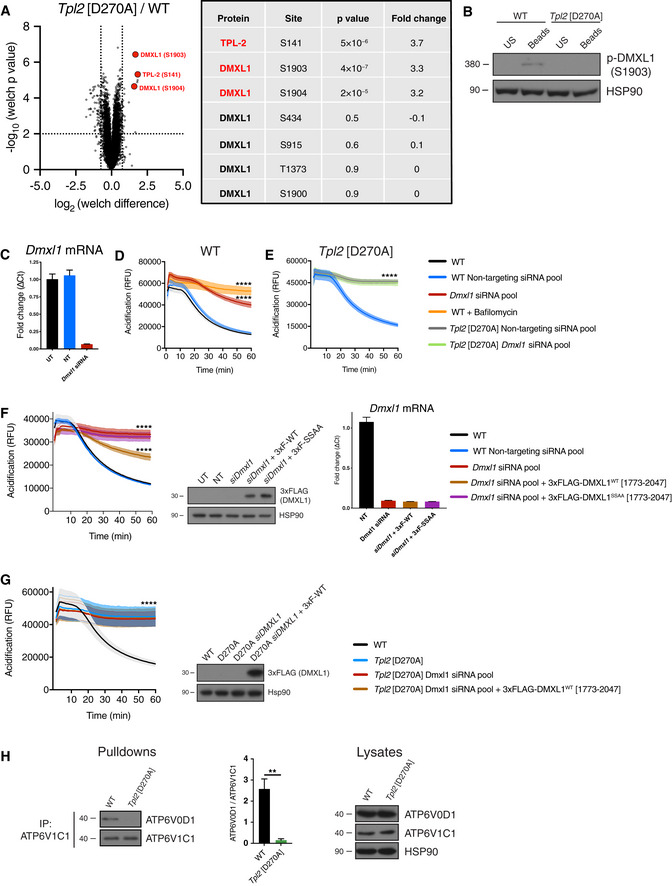
-
ATPL‐2‐dependent phosphoproteome following phagocytosis of latex beads (0.5 h) was determined by TMT mass spectrometry. Volcano plot representing the significance (‐log10 P‐values after Welch’s t‐test) versus phosphorylation fold change (Welch difference ratios) between WT and Tpl2[D270A] BMDMs. Five biological replicates were analysed per genotype (n = 5). Three of the most highly and significantly downregulated phospho‐sites in Tpl2[D270A] BMDMs relative to WT, as well as unaltered DMXL1 phospho‐sites, are shown.
-
BTotal cell lysates from WT and Tpl2[D270A] BMDMs 0.5 h after incubation with latex beads were immunoblotted for phospho‐DMXL1 (S1903) and HSP90 (loading control).
-
C–FDmxl1 was knocked down in WT iBMDMs or Tpl2[D270A] iBMDMs by RNA interference using a SMARTpool ON‐TARGETplus siRNA for 48 h. ON‐TARGETplus non‐targeting pool functioned as siRNA control. (C) qRT–PCR analysis of RNA extracted from iBMDMs was used to check the efficiency of Dmxl1 knockdown (D, E). Dmxl1 mRNA levels were normalised to Hprt mRNA levels and fold changes calculated (ΔCt values) (n = 4). (D) Intra‐phagosomal acidification was assayed following uptake of BCECF‐coupled latex beads by WT iBMDMs. As a control, BMDMs were pre‐treated with 1 μM bafilomycin A1 for 15 min to directly block V‐ATPase function (n = 4 wells). (E) Intra‐phagosomal acidification was assayed following uptake of BCECF‐coupled latex beads by Tpl2[D270A] iBMDMs (n = 4 wells). (F) Simultaneous with Dmxl1 siRNA knockdown, WT iBMDMs were co‐transfected with plasmids expressing either 3xFLAG‐DMXL1 (1,773–2,047) or 3xFLAG‐DMXL1 S1903A/S1904A (1,773–2,047). Intra‐phagosomal acidification was monitored as in Fig 4E (n = 4 wells). Immunoblot analysis of total cell lysates for FLAG demonstrated similar expression levels of the two DMXL1 polypeptides in iBMDMs. qRT–PCR analysis of RNA extracted from iBMDMs confirmed that Dmxl1 mRNA was efficiently knocked down (n = 4) (right) in cells expressing either 3xFLAG‐DMXL1 (1,773–2,047) or 3xFLAG‐DMXL1 S1903A/S1904A (1,773–2,047).
-
GSimultaneous with Dmxl1 siRNA knockdown, Tpl2[D270A] iBMDMs were co‐transfected with a plasmid expressing 3xFLAG‐DMXL1 (1,773–2,047). Intra‐phagosomal acidification was monitored (n = 4 wells) (left). Immunoblot analysis of total cell lysates for FLAG demonstrated strong of 3xFLAG‐DMXL1 (1,773–2,047) in iBMDMs (right).
-
HWT and Tpl2[D270A] BMDMs were treated with latex beads (1:50) for 30 min, and ATP6V1C1 was immunoprecipitated from cell extracts. Immunoblot analysis of eluates for ATP6V1C1 and co‐immunoprecipitated ATP6V0D1 (left). ATP6V1C1 binding to ATP6V0D1 was quantified from three independent experiments (centre, n = 3). Immunoblot analysis of total cell lysates for both V‐ATPase subunits and HSP90 (right).
Data Information: (B–H) One representative experiment out of three shown. Error bars and shaded areas represent SEM. ****P < 0.0001. Panels (D–G) Paired Mann–Whitney t‐test. All differences relative to WT are ****. US, unstimulated; UT, untransfected; NT, non‐targeting siRNA pool. Panel (H) Student’s unpaired t‐test. **P < 0.01.
A phospho‐Ser1903 DMXL1‐peptide antibody was generated and immunoblotting was used to validate the MS results. This analysis demonstrated that phagocytic uptake of latex beads by WT BMDMs induced the phosphorylation of DMXL1 on Ser1903, and this was blocked in Tpl2 D270A/D270A BMDMs (Fig 4B). A recent study demonstrated that DMXL1, a WD40 repeat protein, interacts with the V‐ATPase complex and is required for acidification of endocytic vesicles (Merkulova et al, 2015). Together, these results raised the interesting possibility that TPL‐2 might promote phagosome acidification by inducing the phosphorylation of DMXL1.
The role of DMXL1 in intracellular vesicle acidification was previously studied in a kidney cell line by siRNA knockdown (Merkulova et al, 2015). Immortalised BMDMs (iBMDMs) from WT C57BL/6 and Tpl2 D270A/D270A mice were used to investigate whether DMXL1 was required for phagosome maturation in macrophages. Initial experiments determined whether TPL‐2 catalytic activity was required for phagosome maturation in iBMDMs similar to primary BMDMs. This was monitored following uptake of fluorescently labelled latex beads, initially by testing the effect of blocking TPL‐2 catalytic activity in WT iBMDMs pharmacologically. C34 TPL‐2 inhibitor significantly reduced phagosome proteolysis (Fig EV2A) and acidification (Fig EV2B). Importantly, phagosome proteolysis (Fig EV2A) and acidification (Fig EV2B) were also significantly impaired in Tpl2 D270A/D270A iBMDMs relative to WT iBMDMs. These results showed that TPL‐2 catalytic activity promoted phagosome maturation in immortalised macrophages, similar to primary BMDMs.
Figure EV2. TPL‐2 kinase activity promotes phagosomal proteolysis and acidification in iBMDMs.
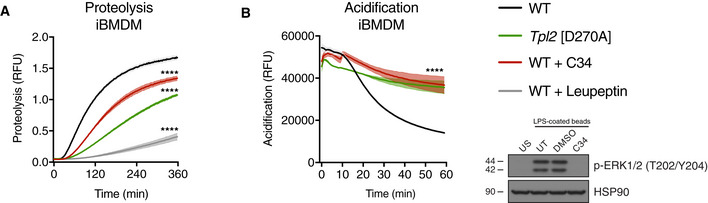
- Intra‐phagosomal proteolysis of DQ Green BSA / AF594 latex beads in iBMDMs pre‐treated with 10 μM C34 TPL‐2 inhibitor for 1 h (n = 4 wells). WT cells were pre‐treated with leupeptin to block proteolysis.
- Intra‐phagosomal acidification of BCECF‐coupled latex beads in iBMDMs pre‐treated with 10 μM C34 TPL‐2 inhibitor for 1 h (n = 4 wells).
Data information: One representative experiment out of three shown. Shaded areas represent SEM. ****P < 0.0001. Paired Mann–Whitney t‐test. All differences relative to WT are ****.
To investigate whether DMXL1 was required for phagosome acidification, Dmxl1 mRNA in WT iBMDMs was depleted by over 90% using Dmxl1 siRNA oligonucleotides (Fig 4C). Following uptake of BCECF latex beads, phagosome acidification was significantly reduced compared to WT iBMDMs transfected with non‐targeting siRNAs (Fig 4D). DMXL1 depletion also decreased phagosome proteolysis monitored using DQ green BSA latex beads. (Fig EV3A). These results demonstrated that DMXL1 expression was required for phagosome acidification in macrophages. The observation that DMXL1 expression was also required for phagosome proteolysis was consistent with the known role of V‐ATPase‐mediated acidification for the induction of proteolytic activity in phagosomes (Lennon‐Duménil et al, 2002; Kinchen & Ravichandran, 2008). Depletion of DMXL2, a paralog of DMXL1, did not alter phagosome acidification or proteolysis in macrophages (Fig EV3B–D).
Figure EV3. TPL‐2 regulates phagosome proteolysis via DMXLI.

-
ADmxl1 was knocked down in WT iBMDMs by RNA interference using a SMARTpool ON‐TARGETplus siRNA for 48 h. ON‐TARGETplus non‐targeting pool functioned as siRNA control. Intra‐phagosomal proteolysis was assayed following uptake of DQ Green BSA / AF594 latex beads (n = 4 wells).
-
B–DDmxl2 was knocked down in WT iBMDMs by RNA interference using a SMARTpool ON‐TARGETplus siRNA for 48 h. ON‐TARGETplus non‐targeting pool functioned as siRNA control. (B) Intra‐phagosomal proteolysis was assayed following uptake of DQ Green BSA / AF594 latex beads (n = 4 wells). (C) Intra‐phagosomal acidification was assayed following uptake of BCECF‐coupled latex beads (n = 4 wells). (D) qRT–PCR analysis of RNA extracted from iBMDMs was used to check the efficiency of Dmxl2 knockdown (A and B). Dmxl2 mRNA levels were normalised to Hprt mRNA levels and fold changes calculated (ΔCt values) (n = 4).
-
EDmxl1 was knocked down in Tpl2[D270A] iBMDMs by RNA interference using a SMARTpool ON‐TARGETplus siRNA for 48 h. ON‐TARGETplus non‐targeting pool functioned as siRNA control. Intra‐phagosomal proteolysis was assayed following uptake of DQ Green BSA / AF594 latex beads (n = 4 wells).
-
FSimultaneous with Dmxl1 siRNA knockdown, WT iBMDMs were co‐transfected with plasmids expressing either 3xFLAG‐DMXL1 (1,773–2,047) or 3xFLAG‐DMXL1 S1903A/S1904A (1,773–2,047). Intra‐phagosomal proteolysis was assayed following uptake of DQ Green BSA / AF594 latex beads (n = 4 wells).
-
GSimultaneous with Dmxl1 siRNA knockdown, Tpl2[D270A] iBMDMs were co‐transfected with a plasmid expressing 3xFLAG‐DMXL1 (1,773–2,047). Intra‐phagosomal proteolysis was monitored (n = 4 wells).
Data information: (A‐G) One representative experiment out of three shown. Error bars and shaded areas represent SEM. ****P < 0.0001. Paired Mann–Whitney t‐test. All differences relative to WT are ****. UT, untransfected; NT, non‐targeting siRNA pool.
To study whether TPL‐2 and DMXL1 function in the same pathway, the effect of DMXL1 depletion on phagosome maturation was monitored in Tpl2 D270A/D270A iBMDMs. DMXL1 siRNA knockdown did not alter phagosome acidification (Fig 4E) relative to transfection with a non‐targeting siRNA pool. Similarly, DMXL1 depletion in Tpl2 D270A/D270A iBMDMs did not alter phagosomal proteolysis compared to Tpl2 D270A/D270A iBMDMs transfected with a non‐targeting siRNA pool (Fig EV3E). These results were consistent with TPL‐2 catalytic activity promoting phagosome acidification and proteolysis via DMXL1.
To investigate the role of TPL‐2‐regulated DMXL1 phosphorylation in DMXL1 function, DMXL1‐depleted iBMDMs were transfected with an expression plasmid encoding a 274 amino acid DMXL1 fragment containing the two TPL‐2‐regulated phosphorylation sites. Expression of 3xFLAG‐DMXL1WT[1,773–2,047] partially rescued the defect in phagosome acidification induced by DMXL1 knockdown (Fig 4F). Expression of 3xFLAG‐DMXL1WT[1,773–2,047] also partially rescued impaired phagosome proteolysis resulting from DMXL1 depletion (Fig EV3F). In contrast, expression of 3xFLAG‐DMXL1S1903A/S1904A[1,773–2,047], lacking the TPL‐2 regulated phosphorylation sites, had no effect on phagosome acidification (Fig 4F) or phagosome proteolysis (Fig EV3F). Furthermore, expression of 3xFLAG‐DMXL1WT[1,773–2,047] did not rescue phagosome acidification (Fig 4G) or phagosome proteolysis (Fig EV3G) in DMXL1‐depleted Tpl2 D270A/D270A iBMDMs. These findings indicate that TPL‐2‐mediated phosphorylation of DMXL1, and not DMXL1 expression alone, was required to drive phagosome maturation.
V‐ATPase function is controlled by the reversible association of the integral membrane V0 subcomplex with peripheral membrane V1 subcomplex to form active V0/V1 holoenzyme (Kane, 2012). V0/V1 assembly is controlled by the RAVE (regulator of H+‐ATPase of vacuolar and endosomal membranes) complex, which includes DMXL2 (Sethi et al, 2010). TPL‐2 regulation of phagosome acidification via DMXL1, a paralog of DMXL2 with 54% amino acid identity, raised the possibility that TPL‐2 may phagosome acidification by regulating V‐ATPase assembly. To investigate this, the ATP6V1C1 component of the V1 subcomplex was immunoprecipitated from lysates of Tpl2 D270A/D270A and WT BMDMs following uptake of latex beads. Immunoblotting demonstrated that co‐immunoprecipitation of ATP6V0D1, a component of the V0 subcomplex, was significantly decreased by Tpl2 D270A mutation (Fig 4H). These results indicate that TPL‐2 catalytic activity promotes V‐ATPase assembly in macrophages.
Together, the results in this section suggest that TPL‐2 stimulates phagosome acidification in macrophages by regulating the phosphorylation of DMXL1 to control the assembly of active V‐ATPase complexes.
TPL‐2 catalytic activity induces phagosome maturation following phagocytic uptake of Staphylococcus aureus
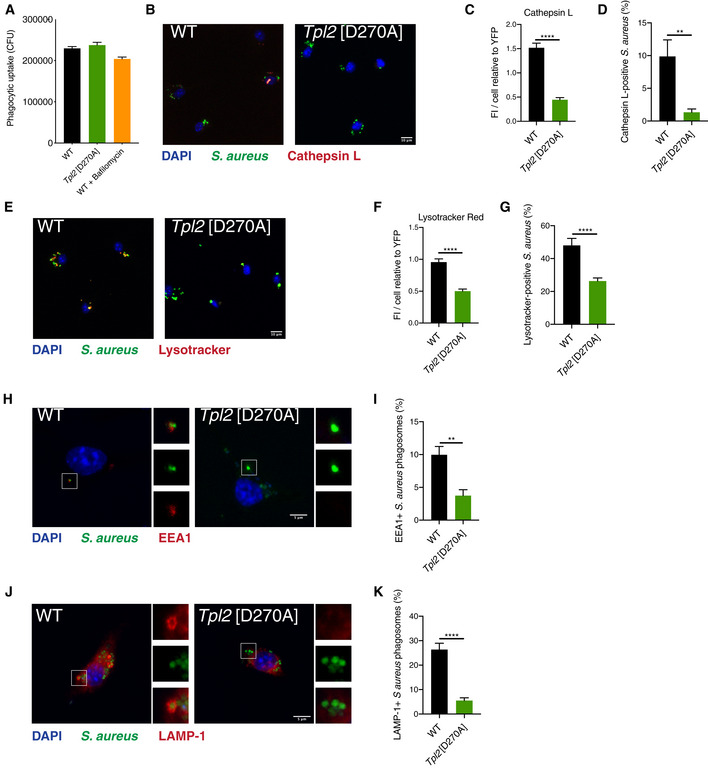
Phagocytosis is a critical component of the innate immune response to bacterial infection. To investigate whether TPL‐2 regulates phagosome maturation following phagocytic uptake of bacteria, BMDMs were infected with S. aureus. Tpl2 D270A mutation did not alter the phagocytic uptake of YFP‐labelled S. aureus (Fig 5A), consistent with results using latex beads. However, the total levels of active cathepsin per YFP+ cell were significantly reduced, as detected using a labelled cathepsin L peptide substrate and confocal microscopy (Fig 5B and C). Furthermore, the percentages of cathepsin L‐positive S. aureus phagosomes in Tpl2 D270A/D270A BMDMs were decreased approximately five‐fold compared to WT BMDMs (Fig 5D).
Figure 5. Tpl2[D270A] mutation impairs maturation of Staphylococcus aureus phagosomes.
-
A–KBMDMs of the indicated genotypes were infected with YFP‐S. aureus (MOI 10) for 1 h. (A) Phagocytic uptake (CFU) of YFP‐labelled S. aureus into BMDMs of indicated genotypes 1 h post‐infection (n = 12; 2 biological and 6 technical replicates per condition per experiment). (B) Representative images of YFP+ BMDMs labelled with Magic Red cathepsin L substrate (red) and DAPI (nuclear stain) 1 h post‐infection. (C) Quantification of Magic Red cathepsin L staining from panel (A). Average fluorescence intensity of Magic Red cathepsin L substrate per cell relative to fluorescence intensity of YFP‐S. aureus (n = 26–29 cells). (D) Quantification of Magic Red cathepsin L staining co‐localised with YFP‐S. aureus from panel (A) (n = 26–29 cells). (E) Representative images of YFP+ BMDMs labelled with LysoTracker Red DND‐99 dye (red) and DAPI, 1 h post‐infection with YFP‐S. aureus (green). (F) Quantification of LysoTracker Red signal from panel (D). Average fluorescence intensity of LysoTracker Red per cell relative to fluorescence intensity of YFP‐S. aureus (n = 66–98 cells). (G) Quantification of LysoTracker Red co‐localisation with YFP‐S. aureus from panel (D) (n = 66–98 cells). (H) Representative images of BMDMs stained with an anti‐EEA1 antibody (red) and DAPI, 1 h post‐infection with YFP‐S. aureus (green). (I) Quantification of EEA1+ S. aureus‐containing phagosomes from panel (G) (n = 27 cells). (J) Representative images of BMDMs stained with an anti‐LAMP‐1 antibody (red) and DAPI, 1 h post‐infection with YFP‐S. aureus (green). (K) Quantification of LAMP‐1+ S. aureus phagosomes from panel (I) (n = 25 cells).
Data information: One representative experiment out of three shown. Error bars represent SEM. P < 0.01, ****P < 0.0001. Student’s unpaired t‐test.
Phagosome acidification was also analysed following S. aureus uptake by LysoTracker Red staining and confocal microscopy. Tpl2 D270A mutation decreased the fluorescence intensity for LysoTracker Red per YFP+ cell (Fig 5E and F). The percentage of LysoTracker Red+ phagosomes containing S. aureus was also significantly reduced (Fig 5G). The effect of TPL‐2 catalytic activity on phagosome maturation was also investigated using specific markers phagosome maturation. Co‐localisation of YFP+ S. aureus with the early endosomal antigen 1 (EEA1), a marker of early phagosomes, was reduced in Tpl2 D270A/D270A BMDMs compared to WT cells (Fig 5H and I). Recruitment of LAMP‐1, a marker for late phagosomes and phagolysosomes, to YFP+ S. aureus phagosomes was also reduced by Tpl2 D270A mutation (Fig 5J and K).
Together, these results indicated that TPL‐2 catalytic activity in macrophages promotes maturation of phagosomes containing internalised S. aureus, stimulating phagosome acidification and cathepsin activation. These results confirmed our earlier findings monitoring the effects of TPL‐2 signalling on phagosome maturation in macrophages following uptake of latex beads.
Tpl2 D270A mutation impairs killing of phagocytosed bacteria
Following engulfment, bacterial killing by macrophages is dependent on the maturation of phagosomes to generate phagolysosomes that have microbicidal activity. To investigate whether Tpl2 D270A mutation impaired the killing of phagocytosed bacteria, BMDMs were infected with S. aureus and the bacterial load was measured 8h after infection by enumerating CFUs.
Bacterial killing was significantly reduced in Tpl2 D270A/D270A BMDMs compared to WT cells (Fig 6A). Consistent with this, the fluorescence intensity of YFP+ S. aureus was higher in mutant cells compared to WT controls (Fig 6B). Importantly, the degree by which Tpl2 D270A mutation impaired bacterial killing was comparable to that detected by treatment of WT BMDMs with the V‐ATPase inhibitor bafilomycin A1 (Fig 6A), which has pronounced inhibitory effects on phagosome maturation (Yoshimori et al, 1991; Bidani et al, 2000).
Figure 6. Tpl2[D270A] mutation impairs macrophage killing of S. aureus .
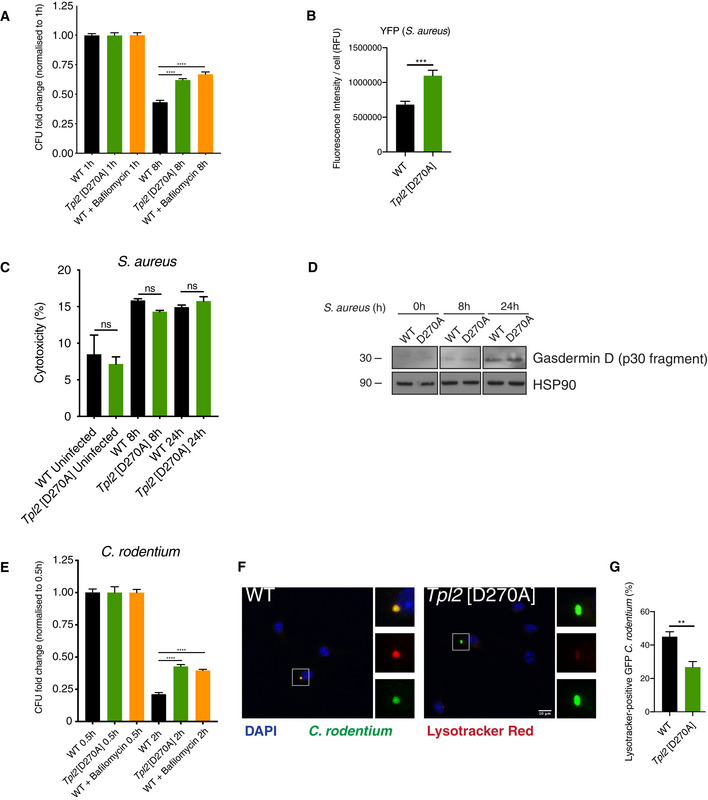
- BMDMs of the indicated genotypes were infected with YFP‐S. aureus (MOI 10) for 0.5 h. S. aureus CFU were assessed at 8 h after infection and normalised to CFU at 1 h (n = 12; 2 biological and 6 technical replicates per condition per experiment). As a control, WT cells were treated with bafilomycin A to block V‐ATPase acidification of phagosomes and phagosome proteolysis.
- Quantification of YFP‐S. aureus average fluorescence intensity per cell from Fig 5 panel (A) (n = 66–85 cells).
- Cell death following was assayed by measuring LDH release from cytosol of BMDMs 8 and 24 h post‐infection with S. aureus (MOI 10; n = 4 wells).
- Whole cell extracts from WT and Tpl2[D270A] BMDMs following S. aureus infection (MOI 10) were immunoblotted for the gasdermin D p30 cleavage product. HSP90 was used as a loading control.
- BMDMs of the indicated genotypes were infected with GFP‐C. rodentium (MOI 2) for 0.5 h. C. rodentium CFU were assessed at 2 h after infection and normalised to CFU at 0.5 h (n = 12; 2 biological and 6 technical replicates per condition per experiment). As a control, WT cells were treated with bafilomycin A.
- Representative images of GFP+ BMDMs labelled with LysoTracker Red DND‐99 dye (red) and DAPI, 1 h post‐infection with GFP‐C. rodentium (green).
- Quantification of LysoTracker Red co‐localisation with GFP‐C. rodentium from panel (F) (n = 54–63 cells).
Data information: One representative experiment out of three shown. Error bars represent SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001, not significant (ns). Panels (A, B, C, E, G) one‐way ANOVA.
The partial inhibitory effect of bafilomycin A1 on bacterial killing implied that BMDMs also employed a phagosome‐independent mechanism to kill S. aureus. This was likely due to the escape of bacteria from phagosomes and/or injection of pore‐forming toxins into the cytosol, which result in activation of inflammasomes and induction of pyroptosis (Seilie & Wardenburg, 2017). Consistent with this, S. aureus infection decreased the viability of BMDMs (Fig 6C) but this was not altered by Tpl2 D270A mutation, as indicated by comparable levels of released lactate dehydrogenase (Fig 6C). Furthermore, gasdermin D p30 immunoblotting demonstrated that S. aureus infection induced similar levels of pyroptosis in Tpl2 D270A/D270A and WT cells (Fig 6D). These data show that decreased bacterial killing in Tpl2 D270A/D270A BMDMs compared to controls is not due to changes in host cell death and that pyroptosis of BMDMs induced by S. aureus infection is independent of TPL‐2 catalytic activity.
To determine whether TPL‐2 catalytic activity promoted the killing of Gram‐negative bacteria, BMDMs were infected with Citrobacter rodentium. Phagocytic uptake of bacteria was similar between Tpl2 D270A/D270A and WT BMDMs (Fig 6E). Measurement of the bacterial load at 2 h showed that Tpl2 D270A mutation impaired the killing of C. rodentium, to a similar extent as bafilomycin A1 treatment (Fig 6E). Acidification of phagosomes containing GFP‐C. rodentium was monitored by confocal microscopy and LysoTracker Red staining. Tpl2 D270A mutation significantly decreased the percentage of LysoTracker Red+ phagosomes containing C. rodentium (Fig 6F and G).
TPL‐2 catalytic activity is therefore required for the acidification of phagosomes containing Gram‐positive or Gram‐negative bacteria following infection of macrophages and for efficient killing of these bacteria.
Discussion
Macrophage killing of phagocytosed bacteria is critical for controlling bacterial infections in the initial phase of an innate immune response. The recruitment of V‐ATPases to phagosomes during their maturation is required for phagosome acidification, which is essential for further maturation to generate microbicidal phagolysosomes containing active cathepsin proteases (Kinchen & Ravichandran, 2008). In this study, genetic and pharmacological experiments demonstrated that TPL‐2 is a positive regulator of phagosome acidification and proteolytic activity in mouse and human primary macrophages after uptake of latex beads. Consistent with this, following infection of macrophages by S. aureus, TPL‐2 kinase activity stimulates acidification and activation of cathepsins in phagosomes containing internalised bacteria, promoting their efficient killing.
The stimulation of MAP kinase signalling in myeloid cells by TPL‐2 during innate immune responses is well established (Gantke et al, 2012). TPL‐2 is essential for activation of ERK1/2 MAP kinases by multiple TLRs in macrophages (Banerjee et al, 2006) and also contributes to TLR activation of p38α MAP kinase (Pattison et al, 2016). However, pharmacological and genetic experiments herein showed that TPL‐2 stimulated phagosome acidification and proteolysis independently of MAP kinase activation. Phosphoproteomic analyses identified that TPL‐2 catalytic activity is required for Ser1903 and Ser1904 phosphorylation of DMXL1 in macrophages following phagocytic uptake of latex beads. DMXL1 is a recently identified V‐ATPase interacting protein that regulates V‐ATPase‐mediated acidification of endocytic vesicles in a kidney cell line (Merkulova et al, 2015). Knockdown experiments demonstrated that DMXL1 was also required for phagosome acidification (and proteolysis) in macrophages after latex bead internalisation, while DMXL2 was dispensable. Expression of a DMXL1 fragment containing the Ser1903/Ser1904 phosphorylation sites rescued the acidification defect in DMXL1‐deficient WT macrophages but not DMXL1‐deficient Tpl2 D270A/D270A macrophages. Together, these results indicate that TPL‐2 induced phosphorylation of DMXL1 Ser1903 and Ser1904 promotes V‐ATPase‐mediated acidification of phagosomes.
V‐ATPases consist of a peripheral membrane subcomplex called V1, which contains the sites of ATP hydrolysis, associated with an integral membrane subcomplex called V0, which encompasses the proton pore. Initial studies in yeast showed that V‐ATPases are regulated by the reversible interaction of V0 and V1 subcomplexes; release of V1 into the cytosol inhibits both ATPase and proton transport activities (Parra & Kane, 1998). DMXL1 is homologous to DMXL2 (also known as Rabconnectin‐3) that binds to the cytosolic V1 subcomplex of V‐ATPases and promotes its association with membrane‐bound V0 V‐ATPase subcomplex to form an active V0/V1 holoenzyme following Notch stimulation (Nagano et al, 2002; Sethi et al, 2010). Our results show that TPL‐2 catalytic activity promotes V0/V1 association following uptake of latex beads by macrophages, suggesting that TPL‐2 stimulates phagosome acidification by inducing the assembly of active V‐ATPase complexes via control of DMXL1 phosphorylation.
A phospho‐Ser1903 DMXL1 antibody showed that the phosphorylation of DMXL1 is induced by TPL‐2 following macrophage uptake of latex beads. This implies that scavenger receptors involved in phagocytosis of latex beads (Sulahian et al, 2008) trigger a TPL‐2‐dependent signalling pathway that leads to Ser1903 phosphorylation of DMXL1. In future studies, it will be important to determine whether DMXL1 Ser1903 is phosphorylated directly by TPL‐2 itself or a TPL‐2‐regulated kinase. The failure of Nfkb1 SSAA mutation to block TPL‐2 induction of phagosome maturation raises the possibility that this pathway induces DMXL1 phosphorylation while TPL‐2 still complexed with NF‐κB1 p105 and ABIN‐2 (Yang et al, 2012; Pattison et al, 2016). However, it cannot be ruled out that phosphorylation of p105 on sites other than those targeted by the IKK complex induces p105 proteolysis following phagocytic uptake of latex beads, releasing TPL‐2 from p105 to control DMXL1 phosphorylation.
Killing of S. aureus, measured by CFU assay, showed that Tpl2 D270A mutation reduced the efficiency of BMDM killing of internalised bacteria. This correlated with reduced acidification and cathepsin activation in phagosomes containing fluorescently labelled bacteria. In addition, a decreased fraction of phagosomes containing fluorescently labelled bacteria stained positive for the early and late markers for phagosome maturation, EEA1 and LAMP‐1, respectively. Together, these results showed that Tpl2 D270A mutation impaired the maturation of phagosomes containing internalised S. aureus bacteria, which explained the reduced efficiency of bacterial killing in Tpl2 D270A/D270A BMDMs compared to WT controls. TPL‐2 catalytic activity was also required for acidification of C. rodentium phagosomes and efficient killing of C. rodentium suggesting that TPL‐2‐dependent induction of phagosome maturation is important in innate immune responses to Gram‐positive and Gram‐negative bacterial species.
The inhibitory effect of Tpl2 D270A mutation on S. aureus killing was of a similar magnitude to that observed with WT cell treatment with bafilomycin A1, which directly inhibited V‐ATPase function. Since bafilomycin A1 has pronounced inhibitory effects on phagosome maturation and endosomal trafficking (Yamamoto et al, 1998), these experiments showed that in vitro killing of S. aureus by BMDMs was only partially mediated by phagolysosomes. In both WT and Tpl2 D270A/D270A BMDMs, the phagosome‐independent killing of internalised bacteria was likely mediated by inflammasome activation and pyroptosis induced by escape of bacteria into the cytosol or bacterial toxins (Man et al, 2017). Assay of cell death (LDH release) and gasdermin D cleavage demonstrated that Tpl2 D270A mutation did not alter pyroptosis induced by S. aureus infection of macrophages (Aglietti et al, 2016). Cleaved gasdermin D has been shown to directly kill both intracellular and extracellular bacteria (Liu et al, 2016). Staphylococcus aureus released from permeabilised cells was also exposed to bactericidal gentamicin in the culture medium. Together, our experiments show that decreased phagosome‐mediated killing does not prevent S. aureus‐induced pyroptosis and that host cell death cannot explain the effects of Tpl2 D270A mutation on killing of S. aureus.
TPL‐2, therefore, has two key roles in the innate immune response of macrophages to bacterial infection. TPL‐2 activation of MAP kinase pathways, triggered by TLRs at the cell surface, and, independently of MAP kinase activation, TPL‐2 stimulates V‐ATPase‐mediated acidification of phagosomes to promote more efficient bacterial killing in phagolysosomes. These processes function together to limit bacterial replication in innate immune responses.
Materials and Methods
Mice
Mouse strains were bred in a specific‐pathogen‐free environment at the Francis Crick Institute (London, United Kingdom), and all experiments carried out in accordance with regulations of the UK Home Office. Map3k8 D270A/D270A, Nfkb1 SSAA/SSAA, and Nfkb1 SSAA/SSAA Map3k8 D270A/D270A have been described previously (Sriskantharajah et al, 2009, 2014). All mouse strains were fully backcrossed on to a C57BL/6Jax background (The Jackson Laboratory).
Reagents
PD0325901 (0.1 μM; pre‐treatment for 10 min) was produced by the Division of Signal Transduction Therapy (DSTT), University of Dundee. VX‐745 (1 μM; pre‐treatment for 1 h) was purchased from Selleckchem. C34 (10 μM; pre‐treatment for 1 h) was from MedChemExpress. Leupeptin (100 μg/ml; pre‐treatment for 1 h) and bafilomycin A1 (1 μM; pre‐treatment for 15 min) were purchased from Sigma‐Aldrich. Lipopolysaccharide (LPS) from Salmonella minnesota R595 was from Enzo Life Sciences.
Plasmids
3xFLAG‐DMXL1 (1,773–2,047) and 3xFLAG‐DMXL1 S1903A/S1904A (1,773–2,047) were generated by the Division of Signal Transduction Therapy (DSTT), University of Dundee.
Antibodies
Gasdermin D antibody (#G7422) was purchased from Sigma‐Aldrich. Antibodies against p‐ERK1/2 T202/Y204 (#9101), ERK1/2 (#9102), p‐p38 T180/Y182 (clone D3F9, #4511), p38 (#9212) and RAB5 (clone C8B1, #3547) were obtained from Cell Signaling Technology. HSP90 (clone H‐114, #sc‐7947), ATP6V0D1 (clone D4, #sc‐393322) and ATP6V1C1 (clone H5, #sc‐166848) antibodies were from Santa Cruz Biotechnology. EEA1 antibody (clone 14, #610457) was from BD Biosciences. LAMP‐1 (clone 1D4B, #AB_528127) and vimentin (clone H5, #AB_528506) antibodies were from Developmental Studies Hybridoma Bank, University of Iowa. Custom‐made phospho‐antibody against p‐DMXL‐1 S1903 (Bleed 2, #DA048) was generated by the Division of Signal Transduction Therapy (DSTT), University of Dundee. All primary antibodies for Western blotting were used at 1:1,000 dilution, while primary antibodies for immunofluorescence were used at 1:200 dilution. Secondary IgG(H + L) antibodies for Western blotting were purchased from Southern Biotech and used at 1:5,000. Secondary Alexa Fluor antibodies for immunofluorescence were purchased from Thermo Fisher Scientific and used at 1:1,000.
Mouse macrophages
Bone marrow‐derived macrophages (BMDMs) were grown from femurs of 6‐ to 12‐week‐old WT and knock‐in C57BL/6 mice (Kaiser et al, 2009). Bone marrow cells were extracted and cultured in complete medium comprising RPMI‐1640 (Sigma‐Aldrich) plus 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l‐glutamine, 50 μM β‐mercaptoethanol and 20% L929 cell supernatant. Cells were differentiated in bacteriological Sterilin dishes (Thermo Fisher Scientific). BMDMs were fed with complete medium on day 4 and harvested on day 7. Cells were seeded in complete RPMI‐1640 medium minus L929 cell supernatant. BMDMs were seeded in tissue culture‐treated Nunc dishes (Thermo Fisher Scientific). Immortalised BMDMs (iBMDMs) were generated by infection of BMDMs with J2 virus containing both v‐raf and v‐myc oncogenes following a published protocol (Gandino & Varesio, 1990). iBMDMs were cultured in complete RPMI‐1640 containing 10% L929 cell supernatant.
Human monocyte‐derived macrophages
Blood from healthy donors was used in this study, which was approved by the NHS Health Research Authority and complied with the Human Tissue Act 2004. Human peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats provided by NHS Blood and Transplant Colindale. PBMCs were isolated from peripheral blood by density gradient centrifugation using Ficoll‐Paque Plus (GE Healthcare). PBMCs were labelled with CD14 MicroBeads (MACS Miltenyi Biotec). CD14+ PBMCs were applied to, washed and eluted from LS columns (MACS Miltenyi Biotec) according to manufacturer instructions. CD14+ PBMCs were seeded at 2 × 107 cells per 140 mm Nunc dish (Sigma‐Aldrich) in RPMI‐1640 medium containing 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l‐glutamine and 50 ng/ml human GM‐CSF (STEMCELL Technologies). Cells were fed on day 4 and resulting macrophages harvested on day 7.
Phagocytic uptake assay
Carboxylated silica beads were coupled to Alexa Fluor 488 succinimidyl ester (Thermo Fisher Scientific) (Yates & Russell, 2008). BMDMs were incubated with coupled beads at 1:300 in 5% FBS in Dulbecco’s phosphate‐buffered saline (DPBS) for 30 min at 37°C. External beads were removed by washing and extracellular fluorescence quenched using trypan blue. Cell‐associated fluorescence intensities were measured in a CLARIOstar (BMG Labtech) microplate reader at excitation/emission wavelengths 490/520 nm.
Phagosome proteolysis assay
Bead coupling procedures and proteolysis assays were adapted from protocols developed by the Valenzeno and Russell laboratories (Russell et al, 1995; Yates et al, 2005; Yates & Russell, 2008). DQ Green BSA (Thermo Fisher Scientific) and Alexa Fluor 594 succinimidyl ester (Thermo Fisher Scientific) were coupled to carboxylated 3 µm silica beads (Kisker Biotech), as described (Yates et al, 2005; Yates & Russell, 2008). BMDMs were seeded on 96‐well plates (Greiner CELLSTAR®) at 1 × 105 cells/well and cultured overnight. DQ Green BSA/Alexa Fluor 594‐coupled silica beads were diluted 1:300 in 5% FBS in DPBS and incubated with cells for 3 min at RT. External beads were removed by washing with pre‐warmed 5% FBS in DPBS, and real‐time fluorescence intensities of cells were measured at ex/em 490/520 nm and at ex/em 590/620 nm at 37°C using a CLARIOstar (BMG Labtech) reader at 2min intervals over 6 h. The calibration fluorophore Alexa Fluor 594 was used to normalise against phagocytic bead uptake. Plots were generated from DQ Green BSA to Alexa Fluor 594 ratios.
Phagosome acidification assay
Silica beads were coupled to BCECF acid (2′,7′‐Bis‐(2‐Carboxyethyl)‐5‐(and‐6)‐Carboxyfluorescein) (Thermo Fisher Scientific) (Russell et al, 1995). BCECF‐coupled beads were diluted 1:200 in DPBS + 5% FBS and incubated with cells for 3 min at RT. After washing to remove external beads, real‐time fluorescence intensities were measured at ex/em 485/520 nm at 37°C using a PHERAstar Plus (BMG Labtech) reader at 1 min intervals over 1 h. BCECF is a pH indicator with optimal fluorescence at pH 7. As phagosomal pH rapidly decreases, fluorescence intensity of the bead‐coupled pH indicator BCECF decreases at 485nm/520nm.
Oxidative burst assay
OxyBURST Green H2HFF BSA (Thermo Fisher Scientific) and Alexa Fluor 594 succinimidyl ester (Thermo Fisher Scientific) were coupled to carboxylated 3 µm silica beads (Kisker Biotech), as described (Vanderven et al, 2009). OxyBURST Green BSA/Alexa Fluor 594‐coupled silica beads were diluted 1:200 in 5% FBS in DPBS and incubated with BMDMs for 3 min at RT. External beads were removed by washing and replaced with pre‐warmed 5% FBS in DPBS. Real‐time fluorescence intensities of cells were measured at ex/em 480/520 nm and at ex/em 590/620 nm at 37°C using a CLARIOstar (BMG Labtech) reader at 2 min intervals over 90 min. Oxidation of H2HFF‐OxyBURST generates a fluorescein‐based product, which emits a signal at 520 nm upon excitation at 480 nm (Vanderven et al, 2009).
Phagosome isolation
Phagosomes were isolated from BMDMs as previously described (Trost et al, 2009; Dill et al, 2015) Briefly, 0.93 µM blue‐dyed carboxylated polystyrene beads (Estapor Merck Chimie SAS) were diluted 1:50 in complete RPMI‐1640 medium (without L929 cell supernatant) and induced for 30 min with 1 × 107 BMDMs per 10 cm Nunc dish. BMDMs were washed and lysed in hypotonic buffer by Dounce homogenisation. Nuclei and cell debris were removed by centrifugation 1,000 ×g for 5 min at 4°C. Phagosomes were then isolated using a discontinuous sucrose gradient. Phagosome fractions were separated in a SW41Ti swinging bucket rotor in an Optima™ L‐90 K ultracentrifuge (Beckman Coulter) at 72,300 ×g for 1 h at 4°C. Blue‐coloured latex bead‐containing phagosomes were collected from the interface of the 10 and 25% sucrose fractions.
Proteomic sample preparation of phagosomes
Isolated phagosomes were lysed in 5% SDS, 50 mM triethylammonium bicarbonate (TEAB) pH 7.5 (S‐Trap), sonicated and protein concentration determined using the BCA Protein Assay Kit (Pierce Protein). Samples were reduced in 10 mM tris(2‐carboxyethyl)phosphine for 30 min at RT followed by alkylation in 10 mM iodoacetamide for 30 min. Samples were acidified and digested on an S‐trap spin column by addition of porcine trypsin at 1:10 (Pierce) for 2 h at 47°C. Peptides were eluted in 50 mM TEAB pH 8.0, 0.2% formic acid and 0.2% formic acid in 50% acetonitrile, respectively.
Label‐free MS acquisition of phagosome proteomes
Peptide samples were separated on an Ultimate 3000 RSLC system (Thermo Scientific) with a C18 PepMap, serving as a trapping column (2 cm × 100 µm ID, PepMap C18, 5 µm particles, 100 Å pore size) followed by a 50 cm EASY‐Spray column (50 cm × 75 µm ID, PepMap C18, 2 µm particles, 100 Å pore size) (Thermo Scientific). Buffer A contained 0.1% formic acid and Buffer B 80% acetonitrile, 0.1% formic acid. Peptides were separated with a linear gradient of 1–35% (Buffer B) over 120 min followed by a step from 35 to 90% acetonitrile, 0.1% formic acid in 0.5 min at 300 nl/min and held at 90% for 4 min. The gradient was then decreased to 1% Buffer B in 0.5 min at 300 nl/min for 10 min. Mass spectrometric identification was performed on an Orbitrap QE HF mass spectrometer (Thermo Scientific) operated “TopN” data‐dependent mode in positive ion mode. FullScan spectra were acquired in a range from 400 m/z to 1,500 m/z, at a resolution of 120,000 (at 200 m/z), with an automated gain control (AGC) of 1 × 106 and a maximum injection time of 50 ms. Charge state screening is enabled to exclude precursors with a charge state of 1. For MS/MS fragmentation, the minimum AGC was set to 5,000 and the most intense precursor ions were isolated with a quadrupole mass filter width of 1.6 m/z and 0.5 m/z offset. Precursors were subjected to higher‐energy collisional dissociation (HCD) fragmentation that was performed in one‐step collision energy of 25%. MS/MS fragments ions were analysed in the Orbitrap mass analyser with a 15,000 resolution at 200 m/z.
Sample preparation for Tandem Mass Tag (TMT) phosphoproteome analysis of BMDMs
BMDMs from WT or Tpl2 D270A/D270A mice were incubated with carboxylated polystyrene beads as for phagosome isolations. Protein content was estimated by protein assay, and a 200 μg aliquot of each sample was taken for further processing. Samples were reduced with 10 mM dithiothreitol for 25 min at 56°C and then alkylated with 20 mM iodoacetamide for 30 min at RT. The alkylation reaction was quenched with an additional 10 mM dithiothreitol, and then, each sample was diluted with 50 mM HEPES to reduce the urea concentration to < 2 M prior to digestion. Proteolytic digestion was carried out by the addition of 4 μg LysC (FUJIFILM Wako Pure Chemical Corporation) and incubated at 37°C for 2.5 h followed by the addition of 10 μg trypsin (Pierce) and overnight incubation at 37°C. After acidification, C18 MacroSpin columns (Nest Group) were used to clean up the digested peptide solutions and the eluted peptides dried by vacuum centrifugation. Samples were resuspended in 50 mM HEPES and labelled using the 0.8 mg Tandem Mass Tag 10plex isobaric reagent kit (Thermo Scientific) resuspended in acetonitrile. Labelling reactions were quenched with hydroxylamine, and a pool was made containing all of the samples. Acetonitrile content was removed from the pooled TMT solution by vacuum centrifugation and then acidified before using a Sep‐Pak C18 (Waters) to clean up the labelled peptide pool prior to phosphopeptide enrichment. The eluted TMT‐labelled peptides were dried by vacuum centrifugation, and phosphopeptide enrichment was subsequently carried out using the sequential metal oxide affinity chromatography (SMOAC) strategy with High Select TiO2 and Fe‐NTA enrichment kits (Thermo Scientific). Eluates were immediately dried by vacuum centrifugation after enrichment and then combined prior to fractionation with the Pierce High pH Reversed‐Phase Peptide Fractionation Kit (Thermo Scientific). All fractions were dried by vacuum centrifugation and stored at −80°C until the day of MS analysis.
MS data acquisition and processing for TMT phosphoproteome
Dried TMT‐labelled phosphopeptide fractions were resuspended in 0.1% trifluoroacetic acid and then analysed by online nanoflow LC‐MS/MS using an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific) coupled to an Ultimate 3000 RSLCnano (Thermo Scientific). 15 µl of sample was loaded via autosampler into a 20 µl sample loop and pre‐concentrated onto an Acclaim PepMap 100 75 µm × 2 cm nanoviper trap column with loading buffer, 2% v/v acetonitrile, 0.05% v/v trifluoroacetic acid, 97.95% water (Optima grade, Fisher Scientific) at a flow rate of 7 µl/min for 6 min in the column oven held at 40°C. Peptides were gradient eluted onto a C18 75 µm × 50 cm, 2 µm particle size, 100 Å pore size, reversed‐phase EASY‐Spray analytical column (Thermo Scientific) at a flow rate of 275 nl/min and with the column temperature held at 40°C, and a spray voltage of 2,100 v using the EASY‐Spray Source (Thermo Scientific). Gradient elution buffers were A 0.1% v/v formic acid, 5% v/v DMSO, 94.9% v/v water and B 0.1% v/v formic acid, 5% v/v DMSO, 20% v/v water, 74.9% v/v acetonitrile (all Optima grade, Fisher Scientific aside from DMSO, Honeywell Research Chemicals). The gradient elution profile used was 2% B to 25% B over 120 min, increasing to 40% B over the following 25 min; then, the column was washed 90% B and re‐equilibrated to 2% B to complete the 180 min run. Two replicate injections were made for each fraction with different fragmentation methods based on the MS2 HCD and MSA SPS MS3 strategies described (Jiang et al, 2017). The acquired raw mass spectrometric data were processed in MaxQuant (Cox & Mann, 2008) (version 1.6.2.10) for peptide and protein identification; the database search was performed using the Andromeda search engine against the Mus Musculus reference proteome canonical sequences from UniProtKB (UP000000589 download August 2017). Fixed modifications were set as Carbamidomethyl (C) and variable modifications set as Oxidation (M) and Phospho (STY). The estimated false discovery rate was set to 1% at the peptide, protein and site levels. A maximum of two missed cleavages were allowed. Reporter ion MS2 or Reporter ion MS3 was appropriately selected for each raw file. Other parameters were used as preset in the software. The MaxQuant output file PhosphoSTY Sites.txt, an FDR‐controlled site‐based table compiled by MaxQuant from the relevant information about the identified peptides, was imported into Perseus (v1.4.0.2) for data evaluation.
Immunofluorescence
BMDMs were seeded on 13 × 1.5 mm round coverslips (VWR International) at 1 × 105 cells in a 24‐well plate and rested overnight. Cells were treated as described in figure legends. After treatments, BMDMs were washed in PBS and fixed in eBioscience™ IC fixation buffer (Thermo Fisher Scientific) for 10 min at RT. BMDMs were permeabilised in 0.1% Triton X‐100 in PBS for 4 min, washed and then blocked in 0.4% gelatin from cold water fish skin (Sigma‐Aldrich) in PBS for 10 min. Following incubation in 0.1% sodium borohydride pH 8.0 (Sigma‐Aldrich) for 10 min, cells were stained with primary antibody solution (1:200) for 1 h, followed by three washes. BMDMs were incubated in secondary antibody solution (1:1,000) for 30 min. Nuclei were stained by incubation with 4′,6‐diamidino‐2‐phenylindole (DAPI) (1:1,000) for 15 min. Washed coverslips were mounted on Corning microscope slides (Merck) using Fluoromount mounting solution (Sigma‐Aldrich). Slides were visualised under a Zeiss Inverted LSM 880 Axio Observer confocal laser scanning microscope (ZEISS) using a ZEISS 40× Plan‐Apochromat (numerical aperture = 1.3) DIC M27 oil immersion objective. Where indicated, a ZEISS 63x Plan‐Apochromat (numerical aperture = 1.4) DIC UV‐VIS‐IR M27 oil immersion objective was used.
Cathepsin activity assay
Following treatment, BMDMs cultured on coverslips (as described above) were washed twice with DPBS and then incubated with the Magic Red cathepsin L substrate (ICT942, Bio‐Rad Antibodies) in 5% FBS in DPBS at a final dilution of 1:250 for 1 h at 37°C. Following washing, BMDMs were fixed, stained with DAPI and imaged as detailed above. Fluorescence of the Magic Red cathepsin L substrate was monitored at ex/em 592/628 nm.
LysoTracker red
Following treatment, BMDMs cultured on coverslips were washed twice with DPBS and then incubated with LysoTracker Red DND‐99 (100nM final concentration; L7528, Thermo Fisher Scientific) in DPBS + 5% FBS for 1 h at 37°C. The immunostaining protocol was performed as described above, and fluorescence of LysoTracker Red was monitored at ex/em 577/590 nm. LysoTracker Red is an acidotropic red‐fluorescent‐dye, which stains acidic cellular compartments.
ROS assay
Following treatment, BMDMs cultured on coverslips were washed twice with DPBS and then incubated with ROS Deep Red dye (100 nM final concentration; ab186029, Abcam) in DPBS + 5% FBS for 1 h at 37°C. The immunostaining protocol was performed as described above, and fluorescence of ROS Deep Red dye was monitored at ex/em 650/675 nm. ROS Deep Red is a cell permeable Deep Red‐fluorescent dye, which reacts with superoxides and hydroxyl radicals.
Image analysis
Images were analysed using ImageJ (Fiji) version 2.0.0 (National Institutes of Health, USA). Fluorescence intensities per BMDM were quantified by creating masks of cell outlines and determining the integrated density (IntDen). Ratios of integrated densities for two different markers in the same masked area were used to determine relative fluorescence intensity. Coloc 2, a Fiji plugin, was used to quantify co‐localisation. To calculate Pearson’s correlation coefficients and Manders’ coefficients, Coloc 2 parameters were set to bisectional threshold regression with a point spread function of 3.0 and Costes randomisations of 100. Coefficients above the Coloc 2 autothreshold were selected for data analysis. For LAMP‐1 staining, S. aureus bacteria were automatically counted by normalising the YFP fluorescence threshold across all fields of view and counting particles > 0.2 µm. Percentages of YFP‐S. aureus associated with LAMP‐1 rings were determined by analysing > 100 BMDMs including > 600 S. aureus bacteria per genotype from at least 40 random fields of view in three independent experiments.
siRNA‐mediated knockdown of Dmxl1
iBMDMs were seeded at 5 × 104 cells/well in a tissue culture‐treated Nunc 24‐well plate and at 1 × 104 cells/well in a 96‐well plate (Greiner CELLSTAR®) in complete RPMI‐1640 containing 10% L929 cell supernatant and cultured overnight at 37°C. All siRNA stocks were reconstituted in siRNA buffer (Horizon Discovery) to 20 μM. SMARTpool ON‐TARGETplus siRNA was used for Dmxl1 (L‐055471‐01‐0005, Horizon Discover) alongside an ON‐TARGETplus non‐targeting pool as siRNA control (D‐001810‐10‐20, Horizon Discovery). Cells were transfected with siRNAs using Viromer Green (Lipocalyx) according to manufacturer instructions. Macrophages were transfected with siRNA at a final concentration of 50 nM. After 48 h culture, 96‐well plates were used for bead assays, while 24‐well plates were used for qRT–PCR analysis to confirm Dmxl1 mRNA knockdown. In Dmxl1 rescue experiments, Dmxl1 siRNA was co‐transfected with plasmids expressing either 3xFLAG‐DMXL1 (1773‐2047) or 3xFLAG‐DMXL1 S1903A/S1904A (1,773–2,047). Each well in a 24‐well and 96‐well plate was co‐transfected with 125 ng and 50 ng cDNA, respectively.
Quantitative real‐time PCR (qRT–PCR)
Following siRNA knockdown, iBMDMs were lysed in 350 μl Qiagen RLT lysis buffer (Thermo Fisher Scientific) containing β‐mercaptoethanol at 1:100 (v/v). RNA was isolated using the Qiagen RNeasy Mini Kit (Thermo Fisher Scientific) and Qiagen RNase‐free DNase set (Thermo Fisher Scientific), following manufacturer instructions. Isolated RNA was reverse transcribed to cDNA using the SuperScript VILO cDNA Synthesis Kit (Invitrogen). qRT–PCR was prepared in MicroAmp Optical 384‐well plates (Applied Biosystems) with a total reaction volume of 7 μl, which contained 2 μl cDNA, 3.5 μl TaqMan Universal PCR Master Mix (Applied Biosystems), 0.35 μl TaqMan FAM‐coupled probe and 1.15 μl nuclease‐free water. TaqMan FAM‐MGB probe Mm01261785_m1 was used for amplification of Dmxl1 (Thermo Fisher Scientific). qRT–PCR was performed on a QuantStudio 3 and 5 (Applied Biosystems) set to comparative CT mode, and Hprt (Mm01545399_m1) was used as a housekeeping gene for expression level normalisation.
SDS–PAGE and immunoblotting
Macrophages were lysed in 50 mM Tris–HCl pH 7.5, 150 mM sodium chloride, 1% Triton X‐100, 10 mM sodium fluoride, 1 mM sodium pyrophosphate, 10 mM β‐glycerophosphate, 2 mM EDTA, 100 μM sodium orthovanadate, 10% glycerol, supplemented with one EDTA‐free protease inhibitor cocktail tablet (Roche). Protein concentration in cleared cell extracts was quantified using the Coomassie Protein Assay Kit (Pierce). Lysates were mixed with 5X sample buffer (250 mM Tris–HCl pH 6.8, 32.5% glycerol, 5% SDS, 5% β‐mercaptoethanol) and heated for 10 min at 90°C. Samples were subjected to SDS–PAGE on a 10% acrylamide gel.
After electrophoresis, proteins were transferred onto PVDF membranes (Bio‐Rad Laboratories) using the Blot Turbo Transfer System (Bio‐Rad Laboratories). Membranes were blocked with 5% (w/v) milk powder in PBS plus 0.05% Tween‐20 for 1 h. Membranes were incubated with primary antibodies overnight at 4°C, washed four times in PBS plus 0.05% Tween‐20 and then incubated with horseradish‐peroxidase (HRP)‐conjugated secondary antibodies. After four PBS plus 0.05% Tween‐20 washes, membranes were incubated with Amersham ECL Western Blotting Detection Reagent (GE Healthcare Life Sciences) or Immobilon Western Chemiluminescent HRP Substrate (Millipore) to visualise protein levels following exposure of X‐ray film (Scientific Laboratory Supplies).
V‐ATPase co‐immunoprecipitation
IgG Sepharose resin (Sigma‐Aldrich, 15 μg packed beads) was incubated with 2.5 mg cell lysate and 2 μg/ml ATP6V1C1 antibody for 16 h at 4°C. Beads were washed four times with lysis buffer and once with sterile water to remove any buffering capacity. Beads were dried with a flat gel‐loading tip. Immunoprecipitated proteins were released by glycine elution. Briefly, beads were incubated with glycine elution buffer at 1,400 rpm for 3 min, centrifuged at 14,000 g for 1 min at RT, and the eluate was transferred to a new Eppendorf tube. Elution was repeated three times. To neutralise the acidic pH, 1 M Tris base pH 8 was added at a final concentration of 20% (v/v). Eluates were mixed with 5× sample buffer and heated for 10 min at 90°C. Samples were subjected to SDS–PAGE.
Bacterial colony‐forming unit (CFU) assay
BMDMs were seeded in antibiotic‐free RPMI‐1640 medium (Sigma‐Aldrich) containing 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate and 50 μM β‐mercaptoethanol. YFP‐Staphylococcus aureus (S. aureus strain RN6390) was cultured in Luria‐Bertani medium supplemented with 10 μg/ml chloramphenicol (Sigma‐Aldrich) for 16 h at 37°C with constant shaking at 200 rpm. GFP‐Citrobacter rodentium (C. rodentium strain ICC683) was cultured in Luria‐Bertani medium supplemented with 50 μg/ml nalidixic acid (Sigma‐Aldrich) and 100 μg/ml chloramphenicol (Sigma‐Aldrich). Staphylococcus aureus culture was diluted in antibiotic‐free RPMI‐1640 medium and added to BMDMs at an MOI of 10:1, while C. rodentium culture was added at an MOI of 2:1. Plates were centrifuged at 100× g for 5 min at RT to facilitate phagocytic uptake.
Following bacterial infection, BMDMs were incubated for 25 min at 37°C to allow phagocytosis. BMDMs were washed in pre‐warmed PBS and incubated with RPMI‐1640 medium containing 50 μg/ml gentamicin (Sigma‐Aldrich) up to 1 h at 37°C. After 1 h, medium was replaced with RPMI‐1640 medium containing 10 μg/ml gentamicin to block division of extracellular bacteria. After further culture for 7 h, BMDMs were washed in ice‐cold PBS and lysed in 1 ml 0.1% Triton X‐100 in PBS for 5 min. For C. rodentium, phagocytic uptake was normalised to a 0.5 h time point and bacterial killing was monitored at 2 h. Lysates were diluted in PBS, plated on antibiotic‐free agar plates (The Francis Crick Institute) and incubated at 37°C overnight. Colony‐forming units were automatically counted using the Synbiosis aCOLyte (7510/SYN).
Cytotoxicity assay
BMDMs of indicated genotypes were seeded in 24‐well Nunc plates and infected with S. aureus (MOI of 10:1) for the indicated times in antibiotic‐free RPMI‐1640 medium containing 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate and 50 μM β‐mercaptoethanol. Supernatants were collected, centrifuged at 10,000 g for 2 min at RT to remove cell debris. As MAX control, an uninfected 24‐well plate was transferred to −80°C for complete cell lysis. The Cytotoxicity Detection Kit (11 644 793 001, Roche) was used according to manufacturer instructions. Briefly, 100 μl supernatant was combined with 100 μl substrate for 10 min at RT protected from light. 100 μl 0.16 M sulphuric acid was added to terminate the assay. LDH (lactate dehydrogenase) activities in supernatants were measured at 490 nm, with a reference wavelength set to 650 nm.
Statistical analysis
All data analyses were performed using GraphPad Prism software (version 8.2.1). Unless otherwise stated in figure legends, data were compared using a two‐way ANOVA. Error bars represent standard errors of the mean (SEM). Statistical significance of data is represented by asterisks (*) corresponding to P value thresholds: not significant (ns) P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Author contributions
FB performed the majority of experiments. AH helped establish phagosome bead assays. TT provided advice about S. aureus infection of macrophages and CFU measurement. AH and TH performed phagosome isolations together with FB. HRF assisted with sample preparation for phosphoproteome mass spectrometry. HRF and APS co‐designed and performed phosphoproteome mass spectrometry experiments. PC and JP performed data analysis for phagosome proteomics. JJ managed and maintained mouse colonies. MT provided advice for the project and edited the manuscript. FB and SCL designed the experiments and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Review Process File
Acknowledgements
We are grateful to The Francis Crick Institute Biological Research, Advanced Light Microscopy and Media Preparation Science Technology Platforms for their help over the course of this work. We also thank Professor David W. Holden (Imperial College London) and Dr. Max Gutierrez (The Francis Crick Institute) for advice on bacterial infections, Professor Tracy Palmer (Newcastle University) for providing the YFP‐S. aureus strain, Professor Gad Frankel (Imperial College London) for providing the GFP‐C. rodentium strain, Dr José Luis Marin Rubio (Newcastle University) for assistance with phagosome isolations, Dr Antonio Tedeschi and Dr Jeremy Carlton (The Francis Crick Institute) for advice about vesicle trafficking and Louise Blair as well as other members of the Ley Laboratory for technical advice and discussions throughout these studies. This work was supported by The Francis Crick Institute, which is funded by the Medical Research Council, Cancer Research UK and the Wellcome Trust. Felix Breyer was funded on a Boehringer Ingelheim Fonds PhD Fellowship. Matthias Trost was funded through a Wellcome Trust Investigator Award (215542/Z/19/Z).
The EMBO Journal (2021) 40: e106188.
Data availability
The mass spectrometry phagosome proteomics data have been deposited to the ProteomeXchange (Deutsch et al, 2017) Consortium via the PRIDE (www.ebi.ac.uk/pride) (Perez‐Riverol et al, 2019) partner repository, with the dataset identifier PXD020401. The phosphoproteomics dataset identifier is PXD020247.
References
- Acuff NV, Li X, Elmore J, Rada B, Watford WT (2017) Tpl2 promotes neutrophil trafficking, oxidative burst, and bacterial killing. J Leukoc Biol 101: 1325–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC (2016) GsdmD p30 elicited by caspase‐11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA 113: 7858–7863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JSC, Ley SC (2013) Mitogen‐activated protein kinases in innate immunity. Nat Rev Immunol 13: 679–692 [DOI] [PubMed] [Google Scholar]
- Banerjee A, Gugasyan R, McMahon M, Gerondakis S (2006) Diverse Toll‐like receptors utilize Tpl2 to activate extracellular signal‐regulated kinase (ERK) in hemopoietic cells. Proc Natl Acad Sci USA 103: 3274–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinke S, Deka J, Lang V, Belich MP, Walker PA, Howell S, Smerdon SJ, Gamblin SJ, Ley SC (2003) NF‐κB1 p105 negatively regulates TPL‐2 MEK kinase activity. Mol Cell Biol 23: 4739–4752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinke S, Robinson MJ, Hugunin M, Ley SC (2004) Lipopolysaccharide activation of the TPL‐2/MEK/extracellular signal‐regulated kinase mitogen‐activated protein kinase cascade is regulated by IκB kinase‐induced proteolysis of NF‐κB1 p105. Mol Cell Biol 24: 9658–9667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belich MP, Salmerón A, Johnston LH, Ley SC (1999) TPL‐2 kinase regulates the proteolysis of the NF‐kappaB‐inhibitory protein NF‐kappaB1 p105. Nature 397: 363–368 [DOI] [PubMed] [Google Scholar]
- Bidani A, Reisner BS, Haque AK, Wen J, Helmer RE, Tuazon DM, Heming TA (2000) Bactericidal activity of alveolar macrophages is suppressed by V‐ATPase inhibition. Lung 178: 91–104 [DOI] [PubMed] [Google Scholar]
- Ciuffreda L, Del Bufalo D, Desideri M, Di Sanza C, Stoppacciaro A, Ricciardi MR, Chiaretti S, Tavolaro S, Benassi B, Bellacosa A et al (2009) Growth‐inhibitory and antiangiogenic activity of the MEK inhibitor PD0325901 in malignant melanoma with or without BRAF mutations. Neoplasia 11: 720–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaço A, Jäättelä M (2017) Ragulator‐a multifaceted regulator of lysosomal signaling and trafficking. J Cell Biol 216: 3895–3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez‐Riverol Y, Ternent T, Campbell DS, Bernal‐Llinares M, Okuda S, Kawano S et al (2017) The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res 45: D1100–D1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill BD, Gierlinski M, Härtlova A, Arandilla AG, Guo M, Clarke RG, Trost M (2015) Quantitative proteome analysis of temporally resolved phagosomes following uptake via key phagocytic receptors. Mol Cell Proteomics 14: 1334–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey GP, Botelho RJ, Butler JR, Moltyaner Y, Chien P, Schreiber AD, Grinstein S (1999) Phagosomal maturation, acidification, and inhibition of bacterial growth in nonphagocytic cells transfected with FcγRIIA receptors. J Biol Chem 274: 28436–28444 [DOI] [PubMed] [Google Scholar]
- Duffy JP, Harrington EM, Salituro FG, Cochran JE, Green J, Gao H, Bemis GW, Evindar G, Galullo VP, Ford PJ et al (2011) The discovery of VX‐745: a novel and selective p38α kinase inhibitor. ACS Med Chem Lett 2: 758–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan RS, Cosío G, Grinstein S (2009) Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol 7: 355–366 [DOI] [PubMed] [Google Scholar]
- Flannagan RS, Jaumouillé V, Grinstein S (2012) The cell biology of phagocytosis. Annu Rev Pathol Mech Dis 7: 61–98 [DOI] [PubMed] [Google Scholar]
- Gandino L, Varesio L (1990) Immortalization of macrophages from mouse bone marrow and fetal liver. Exp Cell Res 188: 192–198 [DOI] [PubMed] [Google Scholar]
- Gantke T, Sriskantharajah S, Ley SC (2011) Regulation and function of TPL‐2, an IB kinase‐regulated MAP kinase kinase kinase. Cell Res 21: 131–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantke T, Sriskantharajah S, Sadowski M, Ley SC (2012) IκB kinase regulation of the TPL‐2/ERK MAPK pathway. Immunol Rev 246: 168–182 [DOI] [PubMed] [Google Scholar]
- Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S (2007) LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J 26: 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip WKE, Sokolovska A, Charriere GM, Boyer L, Dejardin S, Cappillino MP, Yantosca LM, Takahashi K, Moore KJ, Lacy‐Hulbert A et al (2010) Phagocytosis and phagosome acidification are required for pathogen processing and MyD88‐dependent responses to Staphylococcus aureus . J Immunol 184: 7071–7081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Bomgarden R, Brown J, Drew DL, Robitaille AM, Viner R, Huhmer AR (2017) Sensitive and accurate quantitation of phosphopeptides using TMT isobaric labeling technique. J Proteome Res 16: 4244–4252 [DOI] [PubMed] [Google Scholar]
- Kaiser F, Cook D, Papoutsopoulou S, Rajsbaum R, Wu X, Yang H, Grant S, Ricciardi‐castagnoli P, Tsichlis PN, Ley SC et al (2009) TPL‐2 negatively regulates interferon‐beta production in macrophages and myeloid dendritic cells. J Exp Med 206: 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane P (2012) Targeting reversible disassembly as a mechanism of controlling V‐ATPase activity. Curr Protein Pept Sci 13: 117–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T, Kawai T (2014) Toll‐like receptor signaling pathways. Front Immunol 5: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchen JM, Ravichandran KS (2008) Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol 9: 781–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang V, Janzen J, Fischer GZ, Soneji Y, Beinke S, Salmerón A, Hay RT, Ben‐Neriah Y, Ley SC (2003) BTrCP‐mediated proteolysis of NF‐kB1 p105 requires phosphorylation of p105 serines 927 and 932. Mol Cell Biol 23: 402–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang V, Symons A, Watton SJ, Janzen J, Soneji Y, Howell S, Ley SC, Beinke S (2004) ABIN‐2 forms a ternary complex with TPL‐2 and NF‐ κ B1 p105 and is essential for TPL‐2 protein stability. Mol Cell Biol 24: 5235–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon‐Duménil AM, Bakker AH, Maehr R, Fiebiger E, Overkleeft HS, Rosemblatt M, Ploegh HL, Lagaudrière‐Gesbert C (2002) Analysis of protease activity in live antigen‐presenting cells shows regulation of the phagosomal proteolytic contents during dendritic cell activation. J Exp Med 196: 529–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J (2016) Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs GL, Rotstein OD, Grinstein S (1990) Phagosomal acidification is mediated by a vacuolar‐type H+‐ATPase in murine macrophages. J Biol Chem 265: 21099–21107 [PubMed] [Google Scholar]
- Man SM, Karki R, Kanneganti TD (2017) Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 277: 61–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza AR, Savina A, Vermeulen M, Pérez L, Geffner J, Hermine O, Rosenzweig SD, Faure F, Amigorena S (2008) NADPH oxidase controls phagosomal pH and antigen cross‐presentation in human dendritic cells. Blood 112: 4712–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkulova M, Păunescu TG, Azroyan A, Marshansky V, Breton S, Brown D (2015) Mapping the H + ( V ) ‐ATPase interactome : identification of proteins involved in trafficking, folding, assembly and phosphorylation. Sci Rep 5: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano F, Kawabe H, Nakanishi H, Shinohara M, Deguchi‐Tawarada M, Takeuchi M, Sasaki T, Takai Y (2002) Rabconnectin‐3, a novel protein that binds both GDP/GTP exchange protein and GTPase‐activating protein for Rab3 small G protein family. J Biol Chem 277: 9629–9632 [DOI] [PubMed] [Google Scholar]
- Nau GJ, Richmond JFL, Schlesinger A, Jennings EG, Lander ES, Young RA (2002) Human macrophage activation programs induced by bacterial pathogens. Proc Natl Acad Sci USA 99: 1503–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra KJ, Kane PM (1998) Reversible association between the V1 and V0 domains of yeast vacuolar H+‐ATPase is an unconventional glucose‐induced effect. Mol Cell Biol 18: 7064–7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison MJ, Mitchell O, Flynn HR, Chen C‐S, Yang H‐T, Ben‐Addi H, Boeing S, Snijders AP, Ley SC (2016) TLR and TNF‐R1 activation of the MKK3/MKK6‐p38 axis in macrophages is mediated by TPL‐2 kinase. Biochem J 473: 2845–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels AM, Trost M, Beyaert R, Hoffmann E (2017) Patterns, receptors, and signals: regulation of phagosome maturation. Trends Immunol 38: 407–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DA, Pottier RH, Valenzeno DP (1995) In vivo spectroscopic properties of the fluorescent pH indicator biscarboxyethyl carboxyfluorescein. J Photochem Photobiol B Biol 29: 17–22 [DOI] [PubMed] [Google Scholar]
- Russell DG, Vanderven BC, Glennie S, Mwandumba H, Heyderman RS (2009) The macrophage marches on its phagosome: dynamic assays of phagosome function. Nat Rev Immunol 9: 594–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon‐Duménil AM, Seabra MC, Raposo G, Amigorena S (2006) NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126: 205–218 [DOI] [PubMed] [Google Scholar]
- Seilie ES, Wardenburg JB (2017) Staphylococcus aureus pore‐forming toxins: the interface of pathogen and host complexity. Semin Cell Dev Biol 72: 101–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi N, Yan Y, Quek D, Schupbach T, Kang Y (2010) Rabconnectin‐3 is a functional regulator of mammalian notch signaling. J Biol Chem 285: 34757–34764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale ST (2010) Selective transcription in response to an inflammatory stimulus. Cell 140: 833–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriskantharajah S, Belich MP, Papoutsopoulou S, Janzen J, Tybulewicz V, Seddon B, Ley SC (2009) Proteolysis of NF‐κB1 p105 is essential for T cell antigen receptor‐induced proliferation. Nat Immunol 10: 38–47 [DOI] [PubMed] [Google Scholar]
- Sriskantharajah S, Gückel E, Tsakiri N, Kierdorf K, Brender C, Ben‐Addi A, Veldhoen M, Tsichlis PN, Stockinger B, O’Garra A et al (2014) Regulation of experimental autoimmune encephalomyelitis by TPL‐2 kinase. J Immunol 192: 3518–3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford MJ, Morrice NA, Peggie MW, Cohen P (2006) Interleukin‐1 stimulated activation of the COT catalytic subunit through the phosphorylation of Thr290 and Ser62. FEBS Lett 580: 4010–4014 [DOI] [PubMed] [Google Scholar]
- Sulahian TH, Imrich A, DeLoid G, Winkler AR, Kobzik L (2008) Signaling pathways required for macrophage scavenger receptor‐mediated phagocytosis: analysis by scanning cytometry. Respir Res 9: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun‐Wada GH, Tabata H, Kawamura N, Aoyama M, Wada Y (2009) Direct recruitment of H+‐ATPase from lysosomes for phagosomal acidification. J Cell Sci 122: 2504–2513 [DOI] [PubMed] [Google Scholar]
- Trost M, English L, Lemieux S, Courcelles M, Desjardins M, Thibault P (2009) The phagosomal proteome in interferon‐γ‐activated macrophages. Immunity 30: 143–154 [DOI] [PubMed] [Google Scholar]
- Vanderven BC, Yates RM, Russell DG (2009) Intraphagosomal measurement of the magnitude and duration of the oxidative burst. Traffic 10: 372–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira OV, Bucci C, Harrison RE, Trimble WS, Lanzetti L, Gruenberg J, Schreiber AD, Stahl PD, Grinstein S (2003) Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3‐kinase. Mol Cell Biol 23: 2501–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Green N, Hotchandani R, Hu Y, Condon J, Huang A, Kaila N, Li HQ, Guler S, Li W et al (2009) Selective inhibitors of tumor progression loci‐2 (Tpl2) kinase with potent inhibition of TNF‐α production in human whole blood. Bioorganic Med Chem Lett 19: 3485–3488 [DOI] [PubMed] [Google Scholar]
- Xu D, Matsumoto ML, McKenzie BS, Zarrin AA (2018) TPL2 kinase action and control of inflammation. Pharmacol Res 129: 188–193 [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y (1998) Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomesand lysosomesin rat hepatomacell line. Cell Struct Funct 23: 33–42 [DOI] [PubMed] [Google Scholar]
- Yang H‐T, Papoutsopoulou S, Belich M, Brender C, Janzen J, Gantke T, Handley M, Ley SC (2012) Coordinate regulation of TPL‐2 and NF‐kB signaling in macrophages by NF‐kB1 p105. Mol Cell Biol 32: 3438–3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates RM, Hermetter A, Russell DG (2005) The kinetics of phagosome maturation as a function of phagosome/lysosome fusion and acquisition of hydrolytic activity. Traffic 6: 413–420 [DOI] [PubMed] [Google Scholar]
- Yates RM, Russell DG (2008) Real‐time spectrofluorometric assays for the lumenal environment of the maturing phagosome. Methods Mol Biol 445: 311–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashir Y (1991) Bafilomycin A1, a specific inhibitor of vacuolar‐type H+‐ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 266: 17707–17712 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Review Process File
Data Availability Statement
The mass spectrometry phagosome proteomics data have been deposited to the ProteomeXchange (Deutsch et al, 2017) Consortium via the PRIDE (www.ebi.ac.uk/pride) (Perez‐Riverol et al, 2019) partner repository, with the dataset identifier PXD020401. The phosphoproteomics dataset identifier is PXD020247.