

Abstract
BNIP3 is a mitophagy receptor with context‐dependent roles in cancer, but whether and how it modulates melanoma growth in vivo remains unknown. Here, we found that elevated BNIP3 levels correlated with poorer melanoma patient’s survival and depletion of BNIP3 in B16‐F10 melanoma cells compromised tumor growth in vivo. BNIP3 depletion halted mitophagy and enforced a PHD2‐mediated downregulation of HIF‐1α and its glycolytic program both in vitro and in vivo. Mechanistically, we found that BNIP3‐deprived melanoma cells displayed increased intracellular iron levels caused by heightened NCOA4‐mediated ferritinophagy, which fostered PHD2‐mediated HIF‐1α destabilization. These effects were not phenocopied by ATG5 or NIX silencing. Restoring HIF‐1α levels in BNIP3‐depleted melanoma cells rescued their metabolic phenotype and tumor growth in vivo, but did not affect NCOA4 turnover, underscoring that these BNIP3 effects are not secondary to HIF‐1α. These results unravel an unexpected role of BNIP3 as upstream regulator of the pro‐tumorigenic HIF‐1α glycolytic program in melanoma cells.
Keywords: BNIP3, ferritinophagy, HIF‐1α, melanoma, metabolism
Subject Categories: Autophagy & Cell Death, Cancer, Metabolism
Mitophagy receptor BNIP3 facilitates skin cancer by supporting HIF1‐1α protein stability.

Introduction
Metabolic plasticity is a common trait of established tumors including melanoma, an aggressive skin cancer relying on heightened autophagy for stress adaptation and progression (Kimmelman & White, 2017; Rinaldi et al, 2018; Di Leo et al, 2019). Emerging evidence indicates that oncogenic drivers co‐opt autophagy to endow cancer cells with the metabolic versatility required for their bioenergetic and anabolic needs and to adjust to the fluctuating microenvironment (Galluzzi et al, 2015; Kimmelman & White, 2017). During tumorigenesis, lysosomal cargo degradation and recycling of nutrients through autophagy operate as fundamental cancer cell‐autonomous mechanisms to mitigate metabolic stress, by supplying fuel sources feeding into multiple metabolic and biosynthetic pathways (Kimmelman & White, 2017). Furthermore, the selective targeting and degradation of mitochondria, through the process of mitophagy, help cancer cells to preserve the cellular pool of healthy and respiring mitochondria (Vara‐Perez et al, 2019a). However, how mitophagy and its regulators functionally interface with oncogene‐driven pathways orchestrating metabolic networks in cancer cells remains ill‐defined.
The hypoxia‐inducible factor 1 alpha (HIF‐1α) target BCL‐2 19 kDa interacting protein 3 (BNIP3) is an outer mitochondria‐associated atypical BH3‐only member of the B‐cell lymphoma 2 (BCL2) family that operates as a mitophagy receptor. BNIP3 harbors an LC3B‐interacting region (LIR) motif, which facilitates autophagosomal targeting of the mitochondria for lysosomal degradation (Vara‐Perez et al, 2019a). Under hypoxia, HIF‐1α‐driven BNIP3 expression contributes to reducing the overall cellular oxygen consumption by eliciting mitophagy, thus favoring metabolic adaptation to low oxygen concentrations (Lee et al, 2020).
High BNIP3 and HIF‐1α levels often correlate with poor patient prognosis in a number of cancer types, suggesting that BNIP3 contributes to the tumor‐promoting role of HIF‐1α in these cancers. However, the precise mechanism of action of BNIP3 is largely unknown and appears to be mainly tumor type‐specific (Vara‐Perez et al, 2019a). In an MMTV‐PyMT breast cancer model, loss of BNIP3 resulted in reactive oxygen species (ROS)‐mediated stabilization of HIF‐1α, due to the inability of cancer cells to perform mitophagy, which then fostered a HIF‐1α‐driven glycolytic shift and a more invasive breast cancer phenotype (Chourasia et al, 2015). This study therefore revealed a tumor suppressor role for BNIP3 by restricting HIF‐1α signaling through mitophagy. Instead, BNIP3 levels positively correlated with poor prognosis in uveal melanoma (Jiang et al, 2018) and BNIP3 supported melanoma cell’s migratory abilities (Maes et al, 2014a) and their glutamine dependency (Vara‐Perez et al, 2019b) in vitro. Together with the fact that constitutive HIF‐1α levels have also been associated with poor prognosis in melanoma (Martínez‐García et al, 2017), these studies suggest that BNIP3 and HIF‐1α jointly coordinate key metabolic and signaling nodes driving the aggressive phenotype of melanoma cells, yet whether and how the loss of BNIP3 affects HIF‐1α signaling and melanoma growth in vivo have not been addressed. To shed light on these questions, we studied the effects of BNIP3 deletion on melanoma cell’s mitochondria quality control, metabolic profile, and growth in vivo.
Results
BNIP3 expression in melanoma is associated with poor prognosis
To assess the impact of BNIP3 levels on the prognosis of melanoma patients, we interrogated the skin cutaneous melanoma (SKCM) cohort from The Cancer Genome Atlas (TCGA) database comprising 469 melanoma samples. We compared SKCM patients harboring “high” BNIP3 levels versus “low” BNIP3 levels using different gene expression cutoffs based on the distribution of BNIP3 expression levels throughout the whole cohort (Fig 1A, Appendix Fig S1A and B). This analysis showed that SKCM patients with higher BNIP3 expression exhibited significantly reduced overall survival (OS). Only a subset of patients (with BNIP3 expression in median ± 10% log2 range, equivalent to 91 out of 455 patients) did not conform to this trend, possibly due to a lack of sufficiently different BNIP3 expression relative to median cutoff (Appendix Fig S1A and B). BNIP3 expression did not differ between primary (103 patients) and metastatic specimens (366 patients) (Fig 1B). In both patient groups, there was a consistent trend (albeit non‐significant) associating higher BNIP3 levels with lower OS (Appendix Fig S1C–H).
Figure 1. BNIP3 levels in melanoma patients inversely correlate with prognosis.
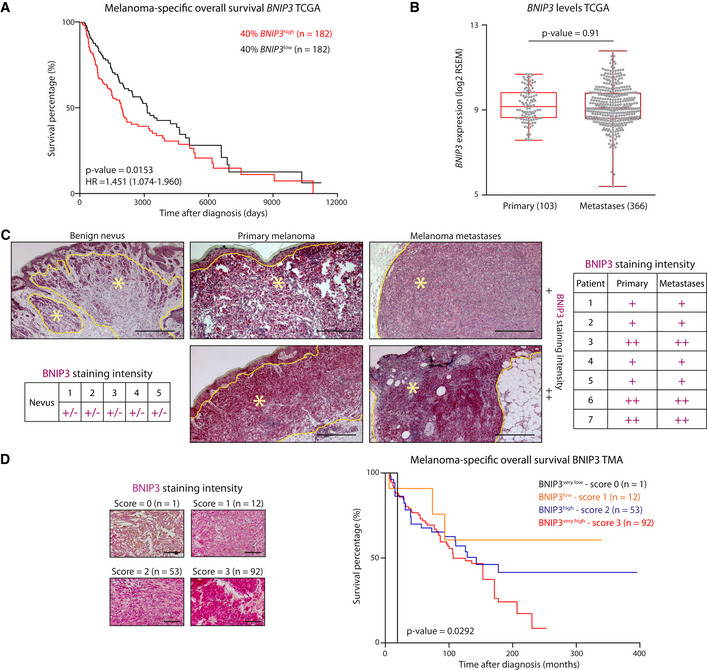
- Kaplan–Meier plots representing the survival difference between SKCM patients stratified according to the 40% highest or lowest BNIP3 expression.
- BNIP3 levels in primary melanoma and metastases within the SKCM TCGA cohort, using Mann–Whitney’s non‐parametric test. The box represents the median and the 25th–75th percentile with the whiskers representing the minimum and maximum values.
- Representative images of BNIP3 immunostaining in a benign nevus (n = 5), primary melanoma and their paired metastasis (n = 7). In the representative pictures, nevi or tumor tissue is circled in yellow and highlighted with a yellow asterisk (*). Histopathological scoring of BNIP3 protein levels in nevi and melanoma patients with paired primary and metastases harboring weak and in gradient (+/−), weak and homogeneous (+) or strong, and homogeneous (++) BNIP3 protein levels based on histopathological staining intensity and distribution scoring. Scale bars represent 150 µm.
- Representative images of BNIP3 immunohistochemistry in TMAs containing (n = 158) melanoma metastases and related Kaplan–Meier plot of survival difference comparing melanoma patients with different BNIP3 histopathological scores. Scale bars represent 50 µm.
Data information: Both Kaplan–Meier curves (A, D) were analyzed using the log‐rank (Mantel–Cox) test.
To substantiate the TCGA analyses at the protein level, we immunohistochemically stained a subset of benign nevi (n = 5) and paired (n = 7) primary and metastatic skin cutaneous melanoma patient samples (Fig 1C; Appendix Table S1). The histopathological analysis revealed a gradient pattern of BNIP3 expression in benign nevi coherent with the maturation pattern of the nevus (Fig 1C). As the melanocytes in the nevus mature toward a more neural phenotype in the deeper part of the lesion, they presented a lower intensity of BNIP3 compared with the melanocytes toward the dermo‐epidermal junction (Fig 1C). In contrast, BNIP3 expression was uniformly distributed in all primary melanoma sections, in agreement with the absence of maturation that is typical of melanoma lesions. Melanomas were divided by BNIP3 expression in two groups: those with an intensity comparable to the junctional part of the benign nevi (+, Fig 1C) and those with the strongest positivity (++, Fig 1C). We observed a strong concordance between the primary and their corresponding metastases: a stronger BNIP3 expression in the primary melanoma was found also in the associated metastatic lesion (Fig 1C). Together, these data suggest that the prognostic impact of BNIP3 in these SKCM patient cohorts may be driven by the primary tumor samples.
Furthermore, analysis of tissue microarrays (TMAs) consisting of 158 cases of metastatic melanomas (Appendix Table S1) stratified according to increasing BNIP3 staining intensity (Fig 1D) highlighted an inverse relationship between patients’ OS and BNIP3 staining intensity, whereby higher BNIP3 expression was associated to lower OS (Fig 1D). It should be noted that nearly 92% (145/158) of the metastatic melanoma samples showed a BNIP3 medium (score 2)‐to‐high (score 3) staining intensity, further underscoring a strong association between high BNIP3 levels and disease progression. This analysis supports an inverse relationship between BNIP3 expression and SKCM patients’ OS, as previously found in uveal melanoma (Jiang et al, 2018).
Together, these data show that increased BNIP3 expression is an early event during melanoma progression to metastasis.
Loss of BNIP3 delays melanoma growth in vivo
Melanoma cells lacking essential autophagy genes, such as ATG5, have been shown to display reduced growth (Maes et al, 2014b; Mgrditchian et al, 2017), but how BNIP3 deficiency affects melanoma growth in vivo remains ill‐defined. BNIP3 functions as a mitochondrial receptor for the recruitment of the autophagosomal machinery to the mitochondria (Vara‐Perez et al, 2019a). Since autophagosome formation requires ATG5 (Galluzzi et al, 2015), we asked to what extent defects caused by BNIP3 silencing in melanoma cells were epistatic to defects in general autophagy.
Hence, we subcutaneously transplanted BNIP3‐ or ATG5‐competent (shCntl) or deficient (shBNIP3 or shATG5, respectively) B16‐F10 cells (Fig EV1A) in immunocompetent (syngeneic) mice and collected tumors (named CntlKD, BNIP3KD, and ATG5KD from here onwards to distinguish them from the cell lines) when they reached 1,000 mm3 (Fig 2A and B). All further analysis was performed in size‐matched tumors. Not only B16‐F10‐BNIP3KD tumors showed a significant and pronounced growth delay compared with B16‐F10‐CntlKD but also to the B16‐F10‐ATG5KD counterparts (Fig 2B). Consistent with previous studies (Maes et al, 2014b), B16‐F10‐ATG5KD tumors displayed a trend toward increased cell death (Fig 2C and D) rather than a decrease in proliferation (Fig 2E and F). In contrast, B16‐F10‐BNIP3KD tumors showed a smaller proportion of actively proliferating cells (Fig 2E and F), in line with the defective proliferation observed in vitro (Fig EV1B). As expected, B16‐F10‐ATG5KD tumors displayed a more diffuse LC3B immunostaining, indicating impaired autophagosome formation (Fig 2G and Fig EV1C). In contrast, loss of BNIP3 caused an increase in the tumoral LC3B punctuated pattern (Fig 2G) suggestive of an altered LC3B turnover or enhanced autophagic flux in vivo. In vitro analysis showed that in contrast to the shATG5 cells, in the shBNIP3 cells basal autophagic flux was indeed functional, as indicated by (i) the accumulation of LC3B‐II by Western blot when lysosomal acidification was altered by Bafilomycin A1 (BafA; Fig EV1C) and (ii) upon transfection with the autophagy flux reporter RFP‐GFP‐LC3, with the presence of red fluorescent puncta (identifying autophagolysosomes which have lost GFP‐fluorescence upon acidification) turning into yellow puncta (identifying undegraded autophagolysosomes) upon BafA treatment (Fig EV1D and E). Additionally, we found that BafA treatment increased cell death predominantly in BNIP3‐silenced cells (Fig EV1F), suggesting that silencing BNIP3 levels renders melanoma cells particularly reliant on autophagy/lysosomal degradation for their survival, as also shown in previous studies using a breast cancer model (Chourasia et al, 2015). Interestingly, Map1lc3b and Sqstm1 (coding for the autophagic mediators LC3B and p62, respectively) were significantly increased both in vitro (Fig EV1G) and in vivo (Fig 2H and I). This suggests that BNIP3 downregulation in melanoma cells might stimulate a compensatory transcriptional mechanism sustaining elevated expression of pro‐autophagic genes. Consistent with this, along with Map1lc3b and Sqstm1, which in murine cells have been shown to belong to TFEB’s transcriptional program (Wang et al, 2019), shBNIP3 cells displayed elevated protein levels of TFEB (Fig EV1H).
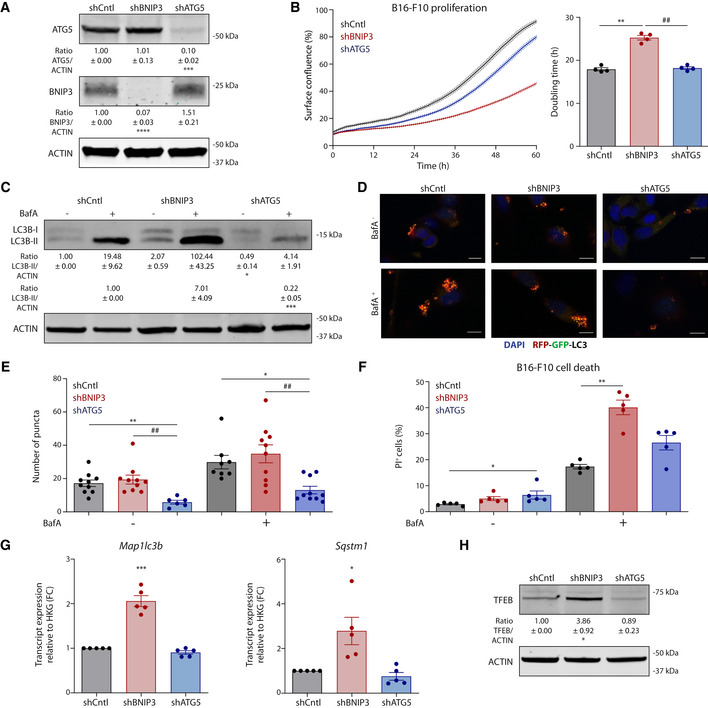
Figure EV1. BNIP3 removal from melanoma cells relents their growth in vitro (related to Fig 2).
-
AImmunoblot detection of ATG5, BNIP3 and ACTIN levels from normoxic lysates (n = 4) of B16‐F10 cells collected 48 h after plating.
-
BProliferation curves and corresponding doubling time of B16‐F10 cells (n = 4) calculated using the Incucyte technology.
-
CImmunoblot detection of LC3B (I–II) and ACTIN levels from lysates of B16‐F10 cells cultured alone or in the presence of the autophagic blocker BafA (5 nM) for 24 h.
-
D, EConfocal microscopy imaging (D) and corresponding quantification (E) of the B16‐F10 cells transiently transfected with the autophagy flux reporter RFP‐GFP‐LC3 and cultured alone or in the presence of the autophagic blocker BafA (5 nM) for 24 h. Scale bars represent 10 µm. N = 10 for shCntl Baf‐, shBNIP3 Baf−, shBNIP3 Baf+ and shATG5 Baf+. N = 6 for shATG5 Baf−. N = 8 for shCntl Baf+ condition. Both BafA‐ and BafA+ conditions were analyzed using a Kruskal–Wallis with Dunn’s multiple comparisons test.
-
FQuantification of dead (PI+) B16‐F10 cells cultured alone or in the presence of BafA (10 nM) for 24 h (n = 5). BafA‐ conditions were analyzed using Friedman’s test with Dunn’s multiple comparisons test whereas BafA+ conditions were analyzed using a RM one‐way ANOVA (Geisser–Greenhouse correction) with Holm–Sidak’s multiple comparisons test.
-
GMap1lc3b and Sqstm1 transcript levels from lysates of normoxic B16‐F10 cells (n = 5). Transcript expression is represented as the fold change relative to their corresponding shCntl value, and they were analyzed using a one‐sample t‐test against shCntl.
-
HImmunoblot detection of TFEB and ACTIN levels from normoxic lysates of B16‐F10 cells collected 48 h after plating (n = 5).
Data information: All quantitative data are mean ± SEM. Densitometric quantifications of all protein levels relative to ACTIN are shown below each corresponding band and analyzed using a one‐sample t‐test against shCntl (A, C, G, H) or shCntl BafA+ (C). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 when compared against shCntl. ##P < 0.01 when comparing shBNIP3 against shATG5. Unless otherwise specified, each graph or blot represents n = 4 biologically independent experiments and they were analyzed using an RM one‐way ANOVA (Geisser–Greenhouse correction) with Holm–Sidak’s multiple comparisons test.
Figure 2. BNIP3 removal from melanoma cells relents tumor growth in vivo .
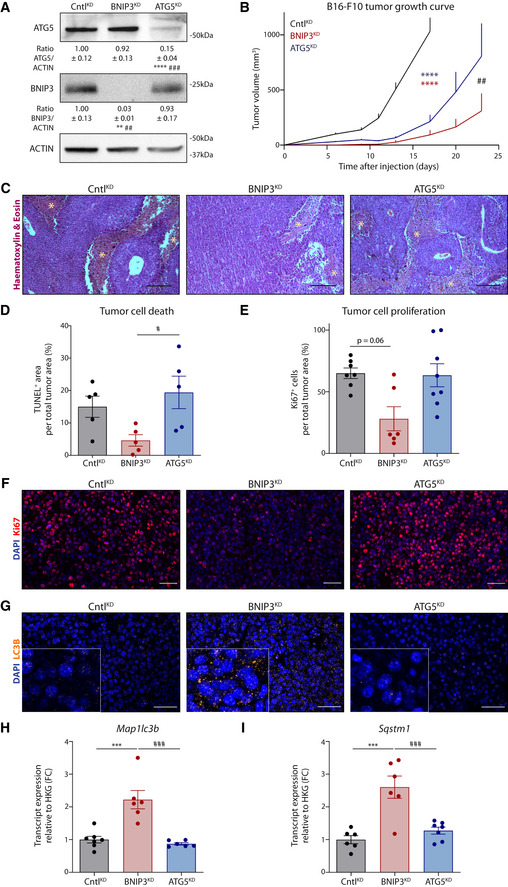
- Immunoblot detection of ATG5 and BNIP3 from lysates of CntlKD (n = 5), BNIP3KD (n = 5), and ATG5KD (n = 7) B16‐F10 tumors. Densitometric quantifications relative to ACTIN levels are shown below each corresponding band.
- Growth curves of B16‐F10 tumors generated with cells transduced with either empty vector (CntlKD), shRNA against BNIP3 (BNIP3KD), or ATG5 (ATG5KD) (n = 7 per condition) and analyzed with a two‐way ANOVA with Tukey’s multiple comparisons test.
- Hematoxylin and eosin staining of sections representative of CntlKD, BNIP3KD, or ATG5KD B16‐F10 tumors. Necrotic areas are highlighted with an asterisk (*).
- Quantification of TUNEL+ tumor area (representing the fraction of dying/dead cell population) of CntlKD, BNIP3KD, or ATG5KD B16‐F10 tumors (n = 5 per condition).
- Quantification of the Ki67+ cells within the tumor area (representing the fraction of proliferative cell populations in non‐necrotic areas) of CntlKD (n = 7), BNIP3KD (n = 6), and ATG5KD (n = 8) B16‐F10 tumors.
- Immunofluorescent staining for Ki67 (red) representative of CntlKD, BNIP3KD, or ATG5KD B16‐F10 tumor sections.
- Immunofluorescent staining for LC3B (orange granularity) representative of CntlKD, BNIP3KD, or ATG5KD B16‐F10 tumor sections. A zoomed‐in area is shown in the lower‐left corner.
- Map1lc3b transcript levels from lysates of B16‐F10 tumors [CntlKD (n = 7), BNIP3KD (n = 6), and ATG5KD (n = 6)].
- Sqstm1 transcript levels from lysates of B16‐F10 tumors [CntlKD (n = 6), BNIP3KD (n = 6), and ATG5KD (n = 7)].
Data information: Scale bars represent 200 µm (C) and 50 µm (F, G). All quantitative data are mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001 when compared against CntlKD. #P < 0.05, ##P < 0.01, ###P < 0.001 when comparing BNIP3KD against ATG5KD. Unless otherwise specified, groups were analyzed using a one‐way ANOVA with Tukey’s multiple comparisons test (A, D, H, I) or with Kruskal–Wallis with Dunn’s multiple comparisons test (E).
Together, these data highlight the reliance of melanoma cells on BNIP3 for tumor growth in vivo.
BNIP3 loss impairs mitochondria clearance and rewires the glycolytic metabolism of melanoma cells
Both the in vitro data and the histological analysis of the tumors described above suggest that loss of BNIP3 may affect melanoma growth by altering specific cancer cell‐autonomous processes.
Previous studies in melanoma (Maes et al, 2014a) and breast (Chourasia et al, 2015) cancer cells indicated the relevance of BNIP3 for mitochondria clearance and homeostasis. Consistent with these studies, under baseline conditions, BNIP3 silencing significantly impaired mitophagy, as shown by the reduced co‐localization of GFP‐LC3 with the mitochondrial marker TOMM20 (Fig 3A). BNIP3‐deprived cells displayed a fragmented mitochondrial network, which was associated with loss of mitochondrial membrane potential and an overall accumulation of intracellular ROS (Fig EV2A–C), as reported previously (Maes et al, 2014a). BNIP3 silencing resulted in the accumulation of the outer mitochondrial membrane mitophagy receptor NIX, a functional BNIP3 homolog (Vara‐Perez et al, 2019a; Lee et al, 2020), in the absence of BafA, whereas BNIP3‐proficient cells displayed increased amounts of both mitophagy receptors upon blockade of lysosomal degradation (Fig EV2D). In contrast, while ATG5 silencing increased the presence of both mitochondrial receptors, NIX knockdown did not alter BNIP3 levels above control (Fig EV2E), suggesting that these mitochondrial proteins undergo degradation in association with BNIP3‐regulated mitophagy. Together, while these results do not exclude the contribution of alternative clearance pathways (Saita et al, 2013; Vincow et al, 2019), they indicate that the absence of BNIP3 disturbs mitochondrial homeostasis leading to the accumulation of dysfunctional mitochondria, under baseline/replete conditions.
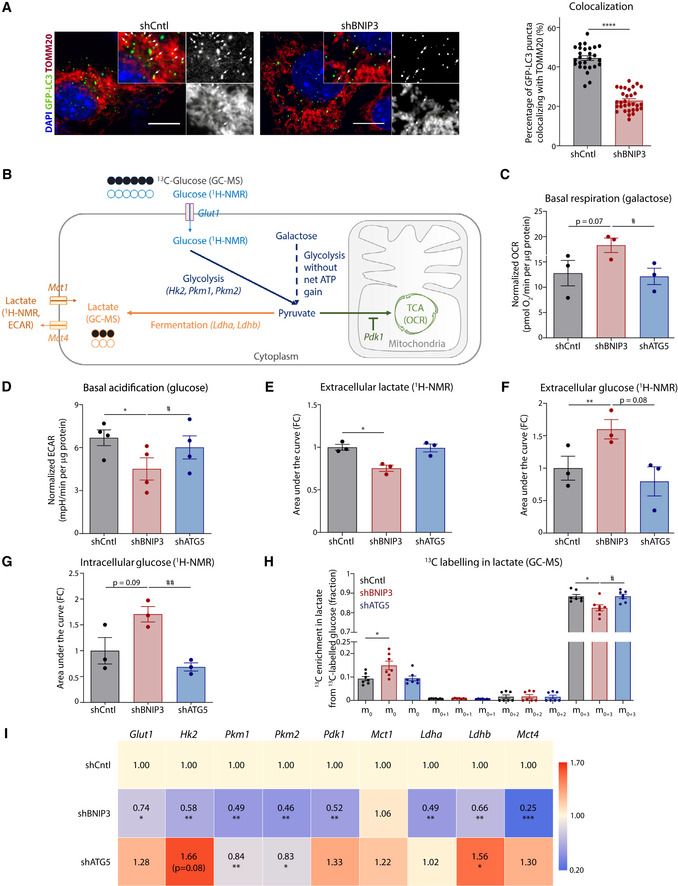
Figure 3. BNIP3 depletion impairs mitochondrial clearance and rewires the glycolytic metabolism of melanoma cells.
-
AConfocal microscopy images of co‐localization analysis of the BNIP3‐proficient (shCntl, n = 27 cells) or deficient (shBNIP3, n = 31 cells) B16‐F10 cells transiently transfected with GFP‐LC3 and immunostained for TOMM20 (mitochondrial marker). Co‐localization events of GFP‐LC3 puncta (green) with TOMM20 (red) are highlighted with white arrows. The percentage of GFP‐LC3 puncta colocalizing with TOMM20 was calculated in n cells from 3 biologically independent experiments and analyzed with Mann–Whitney’s non‐parametric test. Scale bars represent 10 µm.
-
BSchematic representation of the metabolic approaches used to characterize B16‐F10 cells 1H‐NMR stands for proton‐based nuclear magnetic resonance, GC‐MS for gas chromatography coupled to mass spectrometry, OCR for oxygen consumption rate and ECAR for extracellular acidification rate.
-
CBasal OCR per µg of protein of B16‐F10 cells in the presence of galactose (25 mM galactose, 2 mM glutamine) medium using the Seahorse technology (n = 3) analyzed using an RM one‐way ANOVA (Geisser–Greenhouse correction) with Holm–Sidak’s multiple comparisons test.
-
DBasal ECAR per µg of protein of B16‐F10 cells assayed in glucose (25 mM glucose, 2 mM glutamine) medium using the Seahorse technology.
-
E–GExtracellular lactate (E), extracellular (F) and intracellular (G) glucose levels from B16‐F10 cells detected using 1H‐NMR (n = 3) analyzed using a RM one‐way ANOVA (Geisser–Greenhouse correction) with Holm–Sidak’s multiple comparisons test.
-
H13C incorporation in intracellular lactate from B16‐F10 cells fed with 13C‐labeled glucose for 24 h and detected using GC‐MS (n = 7). The graph shows the percentage of lactate detected without any carbon isotopically labeled (m0), with one (m0+1), two (m0+2), or three (m0+3) carbons labeled. m0 and m0+3 conditions were analyzed using Friedman’s test with Dunn’s multiple comparisons test.
-
IGlut1, Hk2, Pkm1, Pkm2, Pdk1, Mct1, Ldha, Ldhb, and Mct4 transcript levels measured by qPCR from the corresponding B16‐F10 cell lysates. Transcript expression is represented as the fold change relative to their corresponding shCntl value. Heatmap shows the average values in each condition (n = 4), and they were analyzed with a one‐sample t‐test only against shCntl, except for Glut1 shATG5 that was analyzed with a Wilcoxon rank test.
Data information: All quantitative data are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 when compared against shCntl. #P < 0.05, ##P < 0.01, when comparing shBNIP3 against shATG5. Unless otherwise specified, each graph represents n = 4 biologically independent experiments and it was analyzed using an RM one‐way ANOVA (Geisser–Greenhouse correction) with Holm–Sidak’s multiple comparisons test.
Figure EV2. BNIP3 loss affects mitochondrial homeostasis (related to Fig 3).
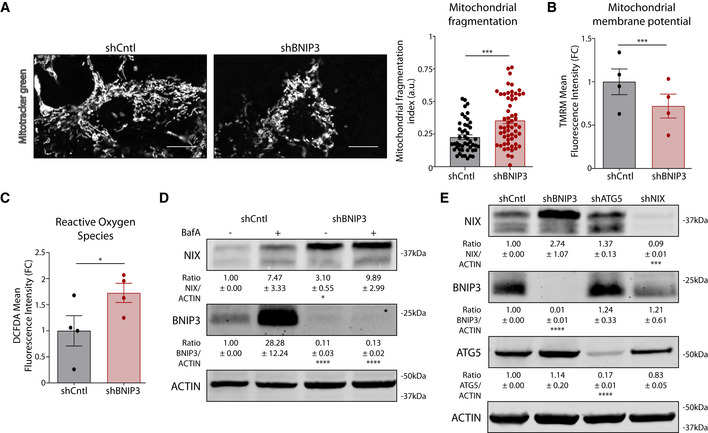
- Representative confocal microscopy analysis of mitochondrial morphology in shCntl (n = 47 cells) or shBNIP3 (n = 58 cells) B16‐F10 cells incubated 30 min with MitoTracker green coming from 3 biologically independent experiments and analyzed with Mann–Whitney’s non‐parametric test. Scale bars represent 10 µm.
- Intracellular TMRM mean fluorescence intensity in B16‐F10 cells 24 h after plating.
- Intracellular DCFDA mean fluorescence intensity in B16‐F10 cells 24 h after plating.
- Immunoblotting for NIX, BNIP3, and ACTIN protein levels in lysates from B16‐F10 cells cultured alone or in presence of the autophagic blocker BafA (5 nM) for 24 h (n = 4).
- Immunoblotting for NIX, BNIP3, ATG5, and ACTIN protein levels in normoxic lysates from B16‐F10 cells collected 48 h after plating (n = 3).
Data information: All quantitative data are mean ± SEM. Densitometric quantifications of all protein levels relative to ACTIN levels are shown below the corresponding band are were analyzed using a one‐sample t‐test. *P < 0.05, ***P < 0.001, ****P < 0.0001 when compared against shCntl. Unless otherwise specified, each graph represents n = 4 biologically independent experiments and were analyzed using a paired t‐test.
Defects in mitochondrial clearance or general autophagy not only impact mitochondrial functionalities but could alter the metabolic performance of melanoma cells. Considering that in BNIP3‐silenced cells mitochondrial clearance and homeostasis were impaired but autophagic flux was still operational, we then evaluated the effects of BNIP3 or ATG5 depletion on the metabolic profiles of melanoma cells by combining different metabolomic approaches (Fig 3B). ShBNIP3 cells respired equally well as their shCntl counterparts in the presence of full medium (25 mM glucose, 2 mM glutamine) whereas shATG5 cells showed an overall decrease in basal oxygen consumption rate (OCR; Appendix Fig S2A), as reported in previous studies (Kimmelman & White, 2017; Jiao et al, 2018). However, under galactose, which forces the cells to rely further on mitochondrial oxidative phosphorylation (OXPHOS) (Aguer et al, 2011), shBNIP3 melanoma cells exhibited an increased basal OCR compared with their control and shATG5 counterparts (Fig 3C, Appendix Fig S2B). Interestingly, we observed a significant reduction in extracellular acidification rate (ECAR, which is often used as a proxy for lactate release) in BNIP3‐deprived cells (Fig 3D), suggestive of impaired glycolytic activity.
To further validate these findings and discriminate the impact of defective BNIP3 or ATG5 expression on the metabolic profile of melanoma cells, we next analyzed melanoma cells’ steady‐state metabolism using complementary approaches: by proton nuclear magnetic resonance (1H‐NMR; steady‐state) and by gas chromatography coupled to mass spectrometry (GC‐MS; steady‐state 13C glucose labeling). ShBNIP3 cells secreted (Fig 3E, Appendix Fig S2C) significantly less lactate, whereas they accumulated glucose both extracellularly and intracellularly (Fig 3F and G) as assessed by 1H‐NMR. Furthermore, 13C‐labeled lactate (m0+3 isotopologue) was significantly reduced in shBNIP3 cells when compared to both shCntl and shATG5 conditions (Fig 3H), indicating that less glucose is contributing to lactate production, as expected from cells with defective glycolysis. Altogether, these results indicate that—opposite to the metabolic effects observed upon blockade of canonical autophagy—silencing BNIP3 in melanoma cells results in a decreased glycolytic and fermentation ability.
We then evaluated whether this specific BNIP3 effect could be the result of a transcriptional downregulation of key components of the glycolytic pathway. Consistent with this, we found that the expression level of several glycolytic enzymes (Hk2, Pdk1, Pkm1, Pkm2, Ldha, and Ldhb), as well as of the glucose and lactate transporters Glut1 and Mct4, respectively, was significantly diminished in BNIP3‐deficient cells (Fig 3I). Consistent with previous reports (Jiao et al, 2018), ATG5 removal induced a trend toward an upregulation—rather than downregulation—of some components of this pathway (Fig 3I), in line with the higher glycolytic metabolism of these autophagy‐compromised melanoma cells.
Collectively, these data support a specific role for BNIP3 in promoting the glycolytic metabolism of these melanoma cells.
BNIP3 maintains HIF‐1α levels in melanoma in vivo
Our findings that BNIP3 depletion in melanoma cells reduced glycolytic fermentation and forced cells to rely on OXPHOS seemed at first inconsistent with the accumulation of their abnormal mitochondria and with the glycolytic shift usually observed in cells with compromised mitophagy (Vara‐Perez et al, 2019a). Since the metabolic effects of BNIP3 were orchestrated at the transcriptional level, we then postulated that depletion of BNIP3 could cause the downregulation of a master transcription factor regulating glycolysis in melanoma cells. We focused on HIF‐1α, rather than on other transcription factors, for several reasons including: (i) Glut1, Hk2, Pdk1, Pkm2, and Ldha are well‐established HIF‐1α targets and HIF‐1α is a master regulator of the Warburg effect in melanoma (Soni & Padwad, 2017; Lee et al, 2020); (ii) melanoma cells harbor constitutively high levels of HIF‐1α (Kuphal et al, 2010; Martínez‐García et al, 2017) suggesting regulatory mechanisms of HIF‐1α stability operating under normoxia, and last but not least (iii) BNIP3 is a well‐established HIF‐1α target (Lee et al, 2020).
BNIP3 silencing, but not the knockdown of ATG5, caused a significant downregulation of HIF‐1α protein levels, which was accompanied by a reduction in the LDHA protein expression (Fig 4A). Lentiviral transduction of a myc‐tagged BNIP3 full‐length construct in the shBNIP3 cells led to a partial recovery of BNIP3 protein levels (Appendix Fig S3A), likely due to fast protein turnover of the BNIP3 full length‐tagged mutant under baseline conditions (Park et al, 2013). In spite of this, we observed a proportional rescue of HIF‐1α protein levels and its target LDHA (Appendix Fig S3B), thus supporting the specific effects of BNIP3 on HIF‐1α levels.
Figure 4. BNIP3 supports normoxic HIF‐1α in melanoma.
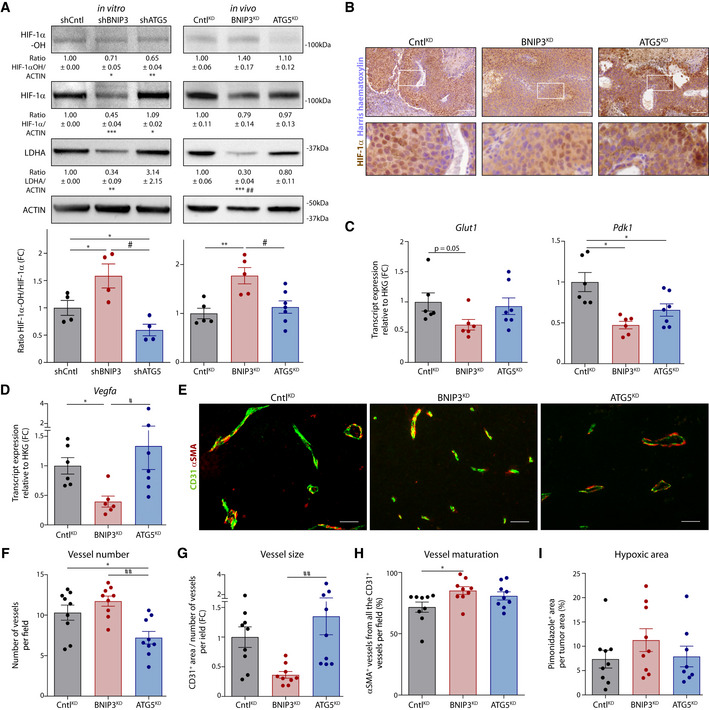
-
AImmunoblot detection of Hydroxylated HIF‐1α (HIF‐1α‐OH), total HIF‐1α, LDHA, and ACTIN protein levels from lysates of B16‐F10 cells (n = 4) or B16‐F10 tumors [CntlKD (n = 5), BNIP3KD (n = 5), and ATG5KD (n = 7)]. The HIF‐1α‐OH /HIF‐1α ratio is shown in a bar graph below. Densitometric quantifications relative to ACTIN levels are shown below each corresponding band. Analysis was performed as indicated in the Data Information section, unless for HIF‐1α/Actin ratio (in vivo), which was analyzed with Kruskal–Wallis non‐parametric test with Dunn’s multiple comparisons test, and LDHA/Actin ratio for shATG5 condition (in vitro), which was analyzed with Wilcoxon’s rank test.
-
BImmunohistochemical staining for HIF‐1α (brown) representative from CntlKD, BNIP3KD and ATG5KD, B16‐F10 tumor tissues. Higher magnification of the highlighted (with a white square) stained sections is shown below.
-
C, DGlut1 (C), Pdk1 (C), and Vegfa (D) transcript levels from lysates of B16‐F10 tumors [CntlKD (n = 6), BNIP3KD (n = 6), and ATG5KD (n = 7)]. Glut1 and Vegfa were analyzed with Kruskal–Wallis non‐parametric test with Dunn’s multiple comparisons test.
-
E–HDouble immunostaining images for CD31 and αSMA (E) and quantification of corresponding tumor vessel number (F), size (G), and maturation (H) in B16‐F10 tumors (n = 9 per cohort). Vessel maturation (H) was analyzed with Kruskal–Wallis non‐parametric test with Dunn’s multiple comparisons test.
-
IQuantification of hypoxic (pimonidazole positive) area in B16‐F10 tumor tissues [CntlKD (n = 9), BNIP3KD (n = 9), and ATG5KD (n = 8)].
Data information: All quantitative data are mean ± SEM. Scale bars (B, E) represent 50 µm. *P < 0.05, **P < 0.01, ***P < 0.001 when compared against shCntl/ CntlKD. #P < 0.05, ##P < 0.01 when comparing shBNIP3/ BNIP3KD against shATG5/ATG5KD. In vitro WB were analyzed using a one‐sample t‐test against shCntl whereas the hydroxylation rate was analyzed using an RM one‐way ANOVA (Geisser–Greenhouse correction) with Holm–Sidak’s multiple comparisons test unless otherwise stated. In vivo experiments were analyzed using a one‐way ANOVA with Tukey’s multiple comparisons test unless otherwise stated.
We then assessed the effects of BNIP3 silencing on HIF‐1α levels in vivo as detected by Western blotting of tumor lysates (comprising normoxic and hypoxic areas) (Fig 4A). HIF‐1α staining showed a fainter pattern in BNIP3KD tumors than in CntlKD or ATG5KD (Fig 4B). The effects of BNIP3 silencing on HIF‐1α were mitigated but not completely abrogated under hypoxia (Appendix Fig S3C), suggesting that BNIP3‐regulated mechanisms become overshadowed by the powerful HIF‐1α stabilization under low oxygen levels (1% O2). Yet, qPCR analysis from tumor tissues showed a concomitant reduction in the levels of various key HIF‐1α target genes including Glut1, Pdk1, and Vegfa (Fig 4C and D). Along with the reduced HIF‐1α and LDHA protein levels detected by immunohistochemistry and Western blot in BNIP3‐silenced tumors, these data show that the BNIP3‐HIF‐1α axis is still operational in vivo. In contrast, shATG5 cells or B16‐F10‐ATG5KD tumors remained HIF‐1α proficient both in vitro and in vivo (Fig 4A). These BNIP3 effects were not caused by alterations of Hif1a mRNA levels (Appendix Fig S3D), suggesting that the activity of the oxygen‐sensing prolyl hydroxylases (PHDs) might be differently regulated.
Prolyl hydroxylases hydroxylate the alpha subunit of HIF‐1 in two proline residues, tagging HIF‐1α for proteasomal degradation (Lee et al, 2020). We then looked at the ratio hydroxylated/total HIF‐1α (by Western blotting) both in vitro and in vivo. Consistent with this, HIF‐1α was more hydroxylated (on P564) upon BNIP3 knockdown in vitro and in vivo (Fig 4A, Appendix Fig S3B). Moreover, proteasomal blockade via MG132 treatment rescued HIF‐1α, but not LDHA, protein levels (Appendix Fig S3E), as hydroxylated HIF‐1α cannot enter the nucleus to activate transcription (Semenza, 2007). Increased HIF‐1α hydroxylation and downregulation of its main target genes were also observed in two human early‐stage melanoma cell lines, the BRAFWT 530 and BRAFV600E mutant WM35, upon BNIP3 silencing (Fig EV3A–D), although these BNIP3‐mediated effects were of a lower magnitude as compared to the murine B16‐F10 cell line. Interestingly, analysis of TCGA data from the BNIP3 high and BNIP3 low melanoma patient clusters (Fig 1B) showed that, irrespective of HIF1A transcript levels that were unchanged in both cohorts (Fig EV3E), there was a significant association between low expression levels of BNIP3, reduced HIF‐1α transcriptional signature, and higher OS (Fig EV3E). This trend is in stark contrast to what was reported in breast cancer patients (Chourasia et al, 2015), where low BNIP3 levels and high HIF1A predicted poor patient survival better than high HIF1A alone. These observations suggest the existence of a cancer type‐specific functional outcome of the BNIP3‐HIF‐1α axis.
Figure EV3. BNIP3 stabilizes normoxic HIF‐1α on a post‐translational level in human melanoma cells (related to Fig 4).
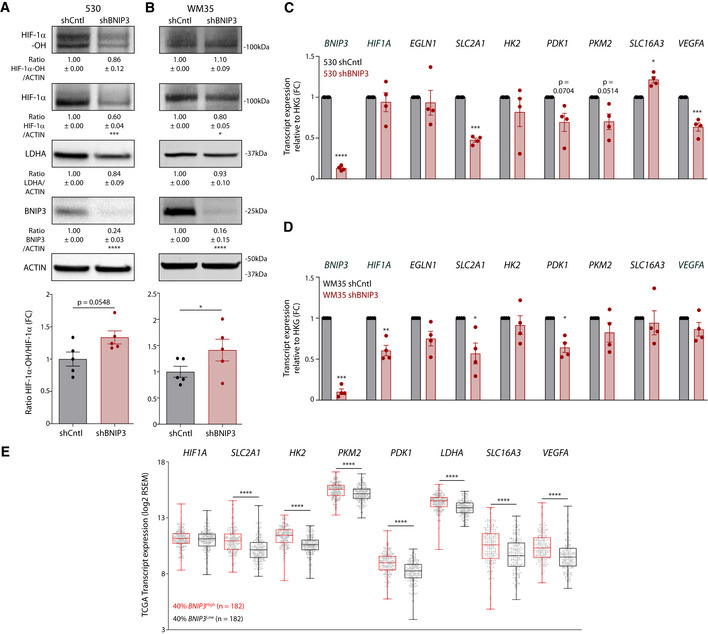
-
A, BImmunoblot detection of Hydroxylated HIF‐1α (HIF‐1α‐OH), total HIF‐1α, LDHA, BNIP3, and ACTIN levels from normoxic lysates of the early‐stage melanoma cell lines 530 (A) or WM35 (B) transduced with either empty vector (shCntl) or shRNA against BNIP3 (shBNIP3) (n = 5). Hydroxylation levels were analyzed using a paired t‐test.
-
C, DBNIP3, HIF1Α, EGLN1(PHD2), SLC2A1 (GLUT1), HK2, PDK1, PKM2, SLC16A3 (MCT4), and VEGFA transcript levels from normoxic lysates (n = 4) of the early‐stage melanoma cell lines 530 (C) or WM35 (D) transduced with either empty vector (shCntl) or shRNA against BNIP3 (shBNIP3). Transcript expression is represented as the fold change relative to their corresponding shCntl value and they were analyzed using a one‐sample t‐test against shCntl.
-
EHIF1Α, SLC2A1 (GLUT1), HK2, PKM2, PDK1, LDHA, SLC16A3 (MCT4), and VEGFA transcript level comparison between BNIP3 high (40%, n = 182) and BNIP3 low (40%, n = 182) in all melanoma patients within the SKCM TCGA cohort, using the unpaired Mann–Whitney’s non‐parametric test. The box represents the median and the 25th–75th percentile with the whiskers representing the minimum and maximum values.
Data information: All quantitative data are mean ± SEM. Densitometric quantifications of all protein levels relative to ACTIN levels are shown below the corresponding band, and they were analyzed using a one‐sample t‐test against shCntl. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 when compared against shCntl.
Given that the downregulation of HIF‐1α signaling included the expression of Vegfa, the main effector of HIF‐1α driven angiogenesis (Lee et al, 2020), both in vitro and in vivo (Fig 4D, Appendix Fig S3F), we questioned whether loss of BNIP3 was associated with defects in tumor angiogenesis. We then analyzed tumoral vessel morphology and maturation by (co)staining tumor sections with the endothelial cell marker CD31 and the mural cell marker αSMA, identifying vessel coverage by pericytes (Fig 4E). In B16‐F10‐BNIP3KD tumors, while the number of vessels per field slightly increased, the vessel size was significantly diminished compared with control tumors and even more compared with B16‐F10‐ATG5KD (Fig 4F and G). Double staining for CD31 and αSMA did not reveal gross, albeit significant, differences in vessel maturation across conditions (Fig 4H) and tumors displayed similar levels of hypoxia (Fig 4I).
In aggregate, these results corroborate the view that the specific depletion of BNIP3 restrains HIF‐1α signaling both in vitro and in vivo.
BNIP3 silencing exacerbates PHD2‐mediated HIF‐1α downregulation through modulation of intracellular iron levels
Key effects of the depletion of BNIP3 in melanoma cells were associated both in vitro and in vivo with changes in HIF‐1α hydroxylation. We then analyzed the role of the PHDs and the factors that could elevate their HIF‐1α hydroxylating activity in response to BNIP3 deprivation. We focused on PHD2 because, among the three PHD isoforms, PHD2 showed a specific trend toward an upregulation upon loss of BNIP3 on RNA (Fig EV4A) and protein level both in vitro and in vivo (Fig 5A). Moreover, previous studies on the differential selectivity among PHDs indicated PHD2 as the main HIF‐1α hydroxylating enzyme (Di Conza et al, 2017; Lee et al, 2020).
Figure EV4. BNIP3 reintroduction, but not metabolites, rescue normoxic HIF‐1α levels in BNIP3‐deficient melanoma cells (related to Figs 5 and 6).
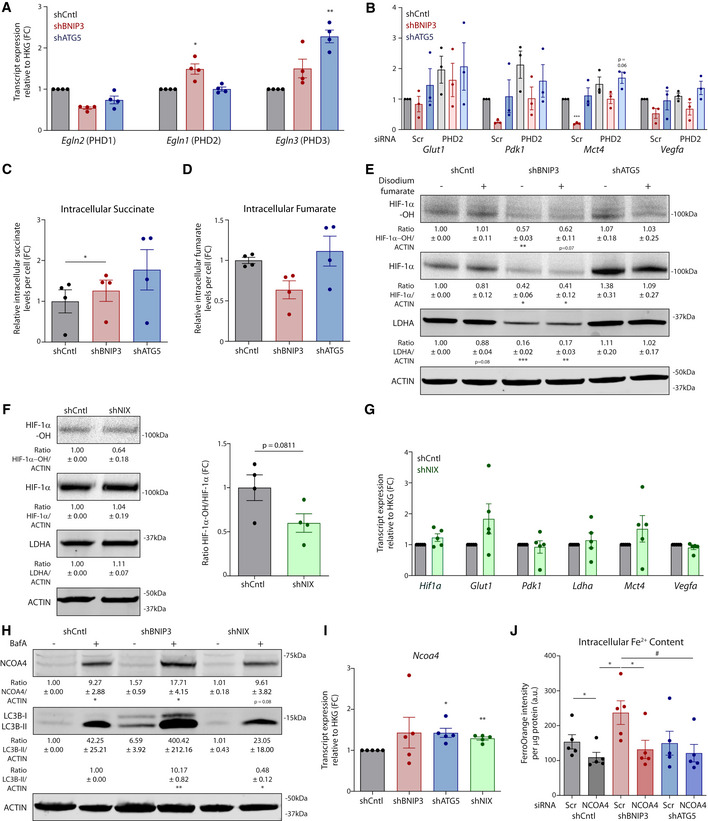
-
AEgln2, Egln1, and Egln3 transcript levels from lysates of normoxic B16‐F10 cells, and they were analyzed using a one‐sample t‐test against shCntl (n = 4) except Phd1 for shBNIP3, which was analyzed using Wilcoxon rank test.
-
BGlut1, Pdk, Mct4, and Vegfa transcript levels from lysates of B16‐F10 cells cultured in presence of non‐targeting (Scr) or siRNA against PHD2 (n = 3).
-
C, DRelative intracellular succinate (C) and fumarate (D) levels from B16‐F10 cells detected using GC‐MS (n = 4). Data were analyzed using an RM one‐way ANOVA (Geisser–Greenhouse correction) with Holm–Sidak’s multiple comparisons test.
-
EImmunoblot detection of Hydroxylated HIF‐1α (HIF‐1α‐OH), total HIF‐1α, LDHA, and ACTIN protein levels from normoxic lysates of B16‐F10 cells cultured in presence or absence of di‐sodium fumarate (4 mM) for 24 h (n = 3).
-
FImmunoblot detection of Hydroxylated HIF‐1α (HIF‐1α‐OH), total HIF‐1α, LDHA, and ACTIN levels from normoxic lysates of B16‐F10 cells transduced with either empty vector (shCntl) or shRNA against NIX (shNIX) (n = 4). Hydroxylation levels were analyzed using a paired t‐test.
-
GHif1a, Glut1, Pdk1, Ldha, Mct4, and Vegfa transcript levels from normoxic lysates of B16‐F10 cells transduced with either empty vector (shCntl) or shRNA against NIX (shNIX) (n = 5).
-
HImmunoblot detection of NCOA4, LC3B, and ACTIN levels from normoxic lysates of B16‐F10 cells transduced with either empty vector (shCntl) or shRNA against BNIP3 (shBNIP3) or NIX (shNIX) and cultured in the presence or absence of BafA 5 µM (n = 4).
-
INcoa4 transcript levels from normoxic lysates of B16‐F10 cells transduced with either empty vector (shCntl) or shRNA against BNIP3 (shBNIP3), ATG5 (shATG5), or NIX (shNIX) (n = 5).
-
JRelative intracellular Fe2+ levels measured with FerroOrange in normoxic B16‐F10 cells transfected in presence of non‐targeting siRNA sequences (Scr) or siRNA against NCOA4 (n = 5). Data were analyzed using a RM one‐way ANOVA (Geisser–Greenhouse correction) with Holm–Sidak’s multiple comparisons test.
Data information: All quantitative data are mean ± SEM. Densitometric quantifications of protein levels relative to ACTIN are shown below each corresponding band. Transcript expression is represented as the fold change relative to their corresponding shCntl value. Densitometric quantifications and qPCR data were analyzed using a one‐sample t‐test against shCntl. *P < 0.05, **P < 0.01, ***P < 0.001 when compared against shCntl. #P < 0.05 when comparing shBNIP3 against shATG5.
Figure 5. BNIP3 stabilizes normoxic HIF‐1α on a post‐translational level in melanoma.
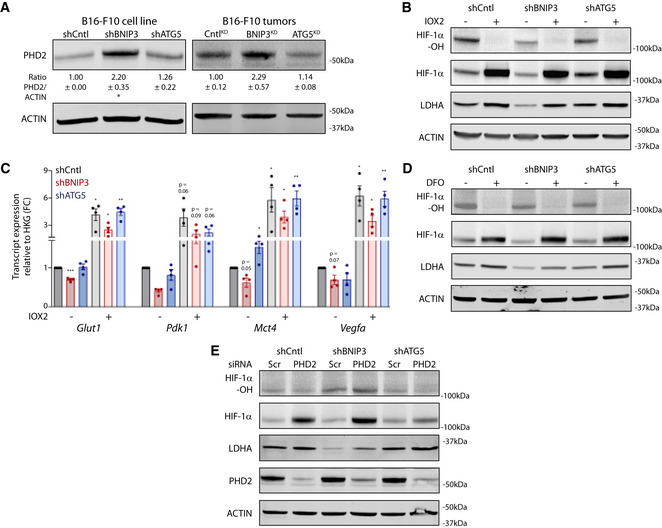
- Immunoblot detection of PHD2 and ACTIN protein levels from lysates of normoxic cultures (n = 4) or B16‐F10 tumors [CntlKD (n = 5), BNIP3KD (n = 7), and ATG5KD (n = 6)]. Densitometric quantifications relative to ACTIN levels are shown below each corresponding band. In vitro data were analyzed using a one‐sample t‐test against shCntl whereas the in vivo data were analyzed using a one‐way ANOVA with Tukey’s multiple comparisons test.
- Immunoblot detection of Hydroxylated HIF‐1α (HIF‐1α‐OH), total HIF‐1α, LDHA, and ACTIN protein levels from normoxic lysates of B16‐F10 cells cultured alone or in the presence of the PHD2 inhibitor IOX2 (100 µM) for 24 h.
- Glut1, Pdk1, Mct4, and Vegfa transcript levels from B16‐F10 cells cultured alone or in the presence of the PHD2 inhibitor IOX2 (100 µM) for 24 h (n = 4). Data were analyzed using a one‐sample t‐test against shCntl, except for Pdk1 shBNIP3 IOX2‐ which was analyzed using the non‐parametric Wilcoxon test.
- Immunoblot detection of Hydroxylated HIF‐1α (HIF‐1α‐OH), total HIF‐1α, LDHA, and ACTIN protein levels from normoxic lysates of B16‐F10 cells cultured alone or in the presence of the iron chelator DFO (200 µM) for 24 h.
- Immunoblot detection of Hydroxylated HIF‐1α (HIF‐1α‐OH), total HIF‐1α, LDHA, PHD2, and ACTIN protein levels from normoxic lysates of B16‐F10 cells transfected in presence of non‐targeting siRNA sequences (Scr) or siRNA against PHD2.
Data information: All quantitative data are mean ± SEM. Densitometric quantifications of protein levels relative to ACTIN for (B, D, E) are shown in Appendix Table S2. *P < 0.05, **P < 0.01, ***P < 0.001 when compared against shCntl. (B, D, E) provide a representative blot from n = 3 biologically independent experiments.
The reaction catalyzed by PHD2 requires alpha‐ketoglutarate (αKG) as donor substrate, iron (Fe2+), and oxygen as co‐factors and yields succinate and CO2 together with the hydroxylated proteins (Lee et al, 2020). We first validated the functional role of PHD2 in our settings by chemical and genetic interference.
The PHD2‐chemical inhibitor IOX2 (Chowdhury et al, 2013) and the iron chelator deferoxamine (DFO) significantly decreased the ratio HIF‐1α‐OH/HIF‐1α levels in all the cell lines (Fig 5B–D, Appendix Table S2) and recovered HIF‐1α signaling in BNIP3‐silenced cells to a similar extent as observed in shCntl and ATG5‐depleted melanoma cells (Fig 5B–D, Appendix Table S2). The siRNA‐based PHD2 knockdown (Fig 5E and Fig EV4B, Appendix Table S2) exerted similar rescuing effects though their magnitude was milder as compared to the PHD2 pharmacological inhibition and depended on the PHD2 knockdown efficiency. These results together indicate PHD2 as the predominant HIF‐1α hydroxylating enzyme in these melanoma cells.
Beyond increased expression, we also examined possible mechanisms underlying the exacerbated PHD2 hydroxylating activity upon loss of BNIP3. The TCA metabolites succinate and fumarate have been described as PHD inhibitors (Koivunen et al, 2007; Lee et al, 2020). Considering the metabolic alterations observed under the condition of BNIP3 silencing, we first evaluated whether changes in the intracellular concentration of these metabolites could explain the BNIP3 effects on HIF‐1α. Succinate levels were significantly higher in shBNIP3 cells, which would however suggest PHD2 inhibition and higher HIF‐1α levels (Koivunen et al, 2007; Lee et al, 2020). Instead, intracellular fumarate levels were reduced, although not significantly, by BNIP3 knockdown (Fig EV4C and D). However, the addition of exogenous disodium‐fumarate (Fig EV4E) failed to rescue the levels of HIF‐1α in the BNIP3‐silenced cells. Together, these data confute (but do not rule out) that changes in the concentration of TCA intermediates operate as main mechanisms promoting the increased downregulation of HIF‐1α in BNIP3‐depleted melanoma cells.
We then reasoned that the specific changes in mitochondrial homeostasis, oxygen consumption, and clearance induced by the loss of BNIP3 could underlie the effector mechanism promoting PHD2‐mediated HIF‐1α downregulation. The emerging crosstalk between mitochondria clearance, autophagy, iron homeostasis, and HIF‐1α signaling suggested that BNIP3 could control PHD2 activity through changes in the intracellular iron content. In line with this, only BNIP3‐depleted cells—but not autophagy‐compromised cells—displayed a significantly higher content of intracellular Fe2+ as imaged and quantitated by FerroOrange, a probe reacting specifically with intracellular Fe2+ ions (Fig 6A). Furthermore, to validate this finding we performed highly sensitive capillary electrophoresis combined with inductively coupled plasma mass spectrometry(CE‐ICP‐MS), from shCntl, shBNIP3, and shATG5 melanoma cells, which allowed for quantitative Fe2+/Fe3+ speciation analysis (Michalke et al, 2019). This analysis showed that while the total iron content across these cell lines remained constant (Fig 6B; Appendix Table S3), shBNIP3 cells harbored a significantly higher proportion of Fe2+ when compared to Fe3+ (Fig 6B, Appendix Table S3).
Figure 6. BNIP3 modulates intracellular iron content to support HIF‐1α levels in melanoma cells.
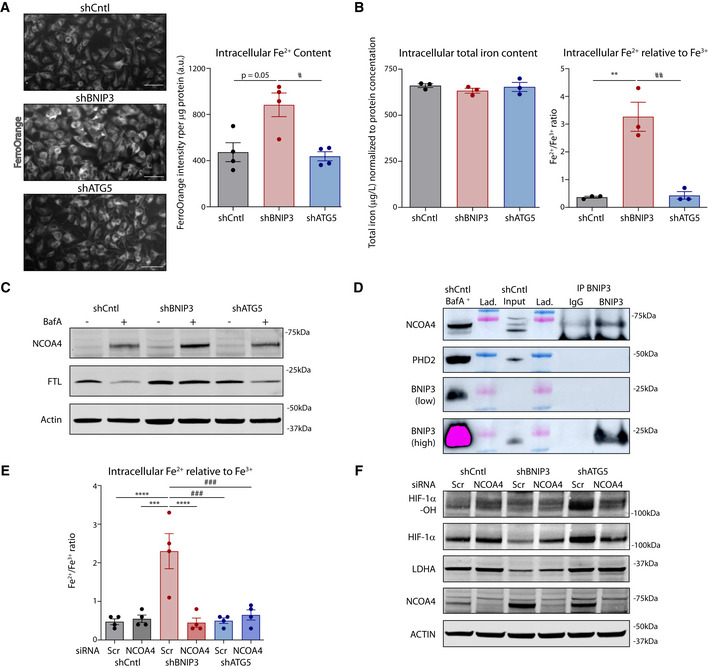
- Relative intracellular Fe2+ levels in normoxic B16‐F10 cells (n = 4). Representative pictures of FerroOrange intensity are shown on the left and analyzed using a RM one‐way ANOVA (Geisser–Greenhouse correction) with Holm–Sidak’s multiple comparisons test. Scale bars represent 50 µm.
- Intracellular total iron content (left) and Fe2+ to Fe3+ ratio (right) measured by CE‐ICP‐MS in normoxic B16‐F10 cells (n = 3). Data were analyzed using a one‐way ANOVA with Tukey’s multiple comparisons test.
- Immunoblot detection of NCOA4, FTL, and ACTIN levels from normoxic lysates of B16‐F10 cells cultured in the presence or absence of BafA 5 nM for 24 h (n = 3).
- Immunoblot detection of NCOA4, PHD2, and BNIP3 levels after immunoprecipitation of endogenous BNIP3 protein from normoxic lysates of B16‐F10 shCntl cells (representative of n = 3 immunoprecipitations). A lysate from B16‐F10 shCntl cells cultured in the presence of BafA was used for band reference. Lad. stands for the reference ladder.
- Intracellular Fe2+ to Fe3+ ratio measured by CE‐ICP‐MS in normoxic B16‐F10 cells transfected in presence of non‐targeting siRNA sequences (Scr) or siRNA against NCOA4 (n = 4). Data were analyzed using a one‐way ANOVA with Tukey’s multiple comparisons test.
- Immunoblot detection of Hydroxylated HIF‐1α (HIF‐1α‐OH), total HIF‐1α, LDHA, NCOA4, and ACTIN levels from normoxic lysates of B16‐F10 cells transfected in presence of non‐targeting siRNA sequences (Scr) or siRNA against NCOA4 (n = 4).
Data information: All quantitative data are mean ± SEM. Densitometric quantifications of protein levels relative to ACTIN for (C, F) are shown in Appendix Table S2. **P < 0.01, ***P < 0.001, ****P < 0.0001 when compared against shCntl. #P < 0.05, ##P < 0.01, ###P < 0.001 when comparing shBNIP3 against shATG5.
The cytoplasmic pool of labile iron is regulated by ferritinophagy, which utilizes the nuclear receptor coactivator‐4 (NCOA4) to target ferritin to the lysosomes through the autophagosome, thereby leading to the export of Fe2+ in the cytoplasm (Mancias et al, 2014). NCOA4, as well as other receptors for specific autophagic cargo, has been shown to be among the most rapidly degraded proteins (Mejlvang et al, 2018), although a comprehensive understanding of the degradation pathways contributing to the overall control of NCOA4 turnover is still missing.
We then compared NCOA4 and ferritin (typified by its light chain, FTL) protein expression in the absence or presence of BafA to unravel its lysosomal turnover. In line with this, baseline levels of NCOA4 in the melanoma cells were increased by altering lysosomal acidification and degradation upon treatment with BafA (Fig 6C, Appendix Table S2), while this was not the case when ATG5 was silenced. NCOA4 can be degraded also by alternative ATG8 (Goodwin et al, 2017) and ATG5/ATG7 (Mejlvang et al, 2018)‐independent pathways, suggesting that a similar mechanism operates under basal conditions in these melanoma cells. In the presence of BafA, we observed reduced levels of FTL as compared to non‐BafA‐treated cells, which may highlight the contribution of alternative degradative (proteasomal) pathways as observed previously (De Domenico et al, 2006; Du et al, 2019; Gammella et al, 2020). However, and irrespective of this, both NCOA4 and FTL accumulation were higher in shBNIP3 cells compared with shCntl or ATG5‐depleted cells upon alkalinization of the lysosomes (Fig 6C, Appendix Table S2), thus suggesting that loss of BNIP3 enhances ferritinophagy.
We then questioned whether the knockdown of the close BNIP3 homolog and mitophagy receptor NIX (Vara‐Perez et al, 2019a; Lee et al, 2020) would phenocopy the effects of the BNIP3 silencing on HIF‐1α signaling and intracellular iron content. However, the absence of NIX did not affect either HIF‐1α stabilization (Fig EV4F), HIF‐1α‐driven transcription (Fig EV4G) or NCOA4 turnover (Fig EV4H), thus demonstrating that the observed effects are BNIP3 specific.
We then investigated whether BNIP3 regulated Ncoa4 levels. However, BNIP3 knockdown did not affect Ncoa4 mRNA levels (Fig EV4I) while it increased PHD2 (Egln1) expression (Fig EV4A). Further, we explored the possibility that BNIP3 could directly interact with NCOA4 or PHD2, thereby regulating their signaling functions. To this end, we immunoprecipitated endogenous BNIP3 in B16‐F10 shCntl cells and probed for the presence of NCOA4 or PHD2 by Western blotting. Because baseline levels of NCOA4 in the input were nearly undetectable, shCntl cells treated with BafA to block NCOA4 turnover, thereby increasing its detection, were loaded on the same gel as reference. Notably, the immunoprecipitation of endogenous BNIP3 pulled down NCOA4 but not PHD2 (Fig 6D). This suggests that BNIP3 regulates the availability of the cytosolic pool of NCOA4 that can be engaged in ferritinophagy under normoxic conditions, by its interaction with NCOA4.
We then questioned whether the heightened ferritinophagy in the BNIP3‐depleted condition could contribute to enhancing the PHD2‐mediated degradation of HIF‐1α. To this end, we downregulated NCOA4 by siRNA‐mediated knockdown and analyzed iron content and HIF‐1α levels in these melanoma cells by using both FerroOrange (Fig EV4J) and CE‐ICP‐MS‐based Fe2+/Fe3+ speciation analysis (Fig 6E). In line with this, NCOA4 silencing diminished intracellular Fe2+ levels (Fig 6E and Fig EV4J and Appendix Table S4) and completely abolished the striking increase in the Fe2+/Fe3+ ratio observed in the BNIP3‐depleted cells. NCOA4 silencing also partially rescued the levels of HIF‐1α and its downstream target LDHA in the BNIP3‐depleted melanoma cells, whereas did not exert major effects in the shCntl and shATG5 (Fig 6F, Appendix Table S2).
Although we cannot exclude that other mechanisms controlling overall iron metabolism are affected, these data indicate that BNIP3 depletion in melanoma cells is associated with an exacerbated ferritinophagy flux, which increases the bioavailability of free iron operating as the main co‐factor for the PHD2‐mediated HIF‐1α hydroxylation.
Rescuing HIF‐1α levels compromises the anti‐melanoma effects of BNIP3 deficit
We next wished to elucidate the extent to which increased HIF‐1α degradation in BNIP3‐silenced melanoma cells was responsible for metabolic alterations and impaired tumor growth in vivo.
To this end, we transduced the melanoma cell lines with constructs overexpressing either a decoy protein (luciferase, Luc) or a human HIF‐1α mutant which is no longer susceptible to PHD‐mediated degradation, since both proline residues targeted for hydroxylation have been mutated into alanine residues (HIF‐1α‐AA), thereby leading to a constitutive HIF‐1α transcriptional activation (Di Conza et al, 2017).
Successful expression of the human HIF‐1α‐AA mutant in the murine B16‐F10 cell lines (Fig EV5A and B) elevated overall HIF‐1α levels, but not its hydroxylation, in shCntl and corrected HIF‐1α protein levels in BNIP3‐silenced cells, without overtly causing an overexpression (Fig 7A). Most importantly, in the BNIP3‐silenced melanoma cells (Fig 7A), HIF‐1α‐AA expression significantly rescued (i) HIF‐1α glycolytic and pro‐angiogenic (Vegfa) target genes/proteins (Fig 7A and B) without affecting their basal PHD2 levels (Fig EV5C; which also suggests that the increase in PHD2 in BNIP3 silenced cells is possibly an adaptive downstream response to changes in iron homeostasis, rather than downstream of HIF‐1α) and (ii) induced a drop in OCR concomitant with a rise in ECAR, thus restoring glycolysis‐driven metabolism (Fig 7C and D, and Fig EV5D).
Figure EV5. Recovery of HIF‐1α levels in BNIP3‐silenced cells restores melanoma glycolytic phenotype and growth in vitro (related to Fig 7).
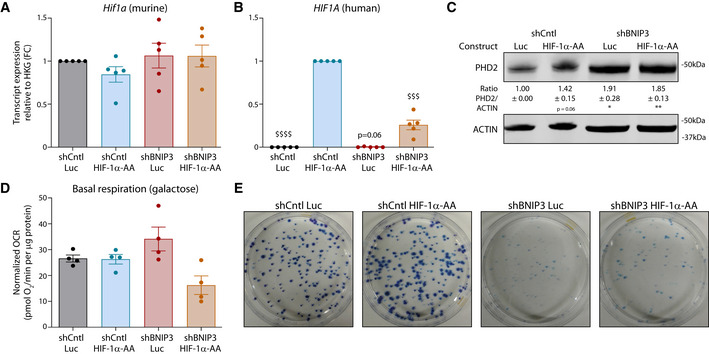
-
A, BMurine (A) and human (B) HIF1Α transcript levels measured by qPCR in B16‐F10 cell lysates collected 48 h after plating (n = 5). They were analyzed against shCntl Luc (A) or shCntl HIF‐1α‐AA (B) using a one‐sample t‐test except human HIF1A in shBNIP3 Luc that was analyzed with Wilcoxon rank test.
-
CImmunoblot detection of PHD2 and ACTIN levels from B16‐F10 cell lysates collected 48 h after plating (n = 4). Densitometric quantifications are shown below each corresponding band they were analyzed using a one‐sample t‐test against shCntl Luc.
-
DBasal respiration rates of B16‐F10 cells assayed in galactose (25 mM galactose, 2 mM glutamine) medium using the Seahorse technology (n = 4).
-
EClonogenic growth of B16‐F10 cells 10 days after seeding.
Data information: All quantitative data are mean ± SEM. Transcript expression is represented as the fold change relative to their corresponding shCntl value. *P < 0.05 **P < 0.01 when compared against shCntl Luc and $$$ P < 0.001, $$$$P < 0.0001 when compared against shCntl HIF‐1α‐AA.
Figure 7. Recovery of HIF‐1α levels in BNIP3‐silenced cells restores melanoma glycolytic phenotype and growth in vivo .
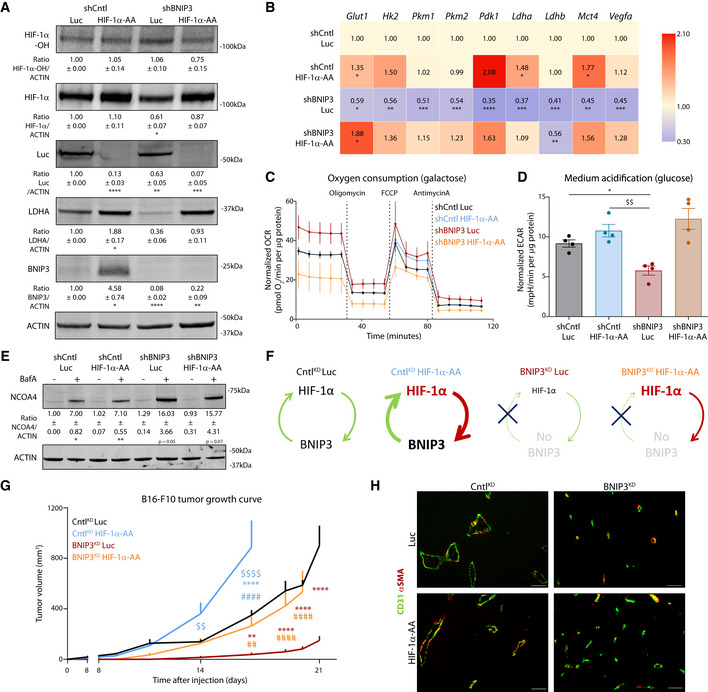
-
AImmunoblot detection of Hydroxylated HIF‐1α (HIF‐1α‐OH), total HIF‐1α, Luc, LDHA, BNIP3, and ACTIN levels from normoxic lysates of shCntl and shBNIP3 cells expressing either Luc or HIF‐1α‐AA and collected 48 h after plating (n = 4). Densitometric quantifications relative to ACTIN levels are shown below each corresponding band and they were analyzed with a one‐sample t‐test against shCntlLuc except for LDHA in shBNIP3 Luc, which was analyzed with Wilcoxon rank test.
-
BGlut1, Hk2, Pkm1, Pkm2, Pdk1, Ldha, Ldhb, Mct4, and Vegfa transcript levels measured by qPCR in B16‐F10 cell lysates collected 48 h after plating (n = 5). Transcript expression is represented as the fold change relative to their corresponding shCntl value. Heatmap shows the average per condition, and values were analyzed with a one‐sample t‐test against shCntl Luc except for Pdk1 in shCntl HIF‐1a‐AA that was analyzed with Wilcoxon rank test.
-
C, DOCR (C) and ECAR (D) per µg of protein of B16‐F10 cells assayed in medium containing either 25 mM galactose (C) or 25 mM glucose (D) medium using the Seahorse technology (n = 4).
-
EImmunoblot detection of NCOA4 and ACTIN levels from normoxic lysates of shCntl and shBNIP3 cells expressing either Luc or HIF‐1α‐AA, collected and treated for 24 h in the absence or presence of 5 nM BafA (n = 3). Densitometric quantifications relative to ACTIN levels are shown below each corresponding band, and they were analyzed with a one‐sample t‐test against shCntl Luc.
-
FScheme explaining the feedforward loop between HIF‐1α and BNIP3 in the different tumor conditions (G).
-
GGrowth curves of B16‐F10 tumors generated by shCntl or shBNIP3 cells expressing Luciferase (Luc) or an undegradable HIF‐1α (HIF‐1α‐AA) construct (n = 7 for CntlKD Luc/BNIP3KD HIF‐1α‐AA, n = 8 CntlKD HIF‐1α‐AA and n = 5 for BNIP3KD Luc) and analyzed with a two‐way ANOVA with Tukey’s multiple comparisons test.
-
HRepresentative double immunostaining for CD31 and αSMA in B16‐F10 tumors (n = 5 per condition). Scale bars represent 50 µm.
Data information: All quantitative data are mean ± SEM. *P < 0.05, **/$$/##P < 0.01, ***P < 0.001, ****/$$$$/####P < 0.0001 where (*) represents comparisons against shCntl Luc/ CntlKD Luc, ($) against shCntl HIF‐1α‐AA/ CntlKD HIF‐1α‐AA and (#) against shBNIP3 Luc/ BNIP3KD Luc. Unless otherwise specified, graphs were analyzed using a RM one‐way ANOVA (Geisser–Greenhouse correction) with Holm–Sidak’s multiple comparisons test.
In contrast, BNIP3‐mediated effects on the lysosomal NCOA4 turnover (ferritinophagy flux) were not affected by the expression of HIF‐1α‐AA (Fig 7E). Taken together, these data show that in the BNIP3‐depleted cells the glycolytic defect is secondary to HIF‐1α downregulation and suggest that the effects on ferritinophagy are BNIP3‐coordinated upstream events, ultimately stimulating PHD2 expression and function.
The expression of HIF‐1α‐AA in control cells fostered their clonogenic growth in comparison with the shCntl Luc cells but did not exert major effects on the colony formation defects of the BNIP3‐deficient cells (Fig EV5E). This is likely explained by the reported inability of BNIP3‐depleted melanoma cells to form clones in vitro, due to the major effects of BNIP3 on the cytoskeleton (Maes et al, 2014a). We next evaluated the tumor growth ability and impact on the tumor microenvironment of the HIF‐1α‐AA expressing shCntl (CntlKD HIF‐1α‐AA) and of the shBNIP3 melanoma cells (BNIP3KD HIF‐1α‐AA) and compared them with their respective empty‐vector expressing shCntl (CntlKD Luc) and shBNIP3 melanoma cells (BNIP3KD Luc) (schematically shown in Fig 7F). Remarkably, compared with the BNIP3KD Luc which showed the observed severe tumor growth impairment, the BNIP3KD HIF‐1α‐AA cells recovered their tumor‐forming ability to a similar degree as the CntlKD Luc (Fig 7G). The growth of the CntlKD HIF‐1α‐AA melanoma cells with fully operational HIF‐1α signaling including the enforced expression of BNIP3 (Fig 7A), possibly mimicking a pseudo‐hypoxic status, was significantly exacerbated in these in vivo settings (Fig 7G), further suggesting that a feedforward BNIP3‐HIF‐1α axis reinforces its pro‐tumorigenic action (Fig 7F).
In line with the finding that expression of HIF‐1α‐AA in shCntl and BNIP3‐silenced melanoma cells promoted or rescued prevalently their glycolytic phenotype (Fig 7A–D), we did not observe major morphological or functional changes in the vasculature of the HIF‐1α‐AA expressing tumors that could explain their heightened growth rate (Fig 7H). Instead, the accelerated growth rate of the CntlKD HIF‐1α‐AA tumors further suggests that beyond the tumor‐promoting role of glucose metabolism in melanoma, a feedforward BNIP3‐HIF‐1α axis supports tumor growth through additional mechanisms.
Together these results show that, secondary to the increase in NCOA4‐mediated ferritinophagy induced by the depletion of BNIP3, PHD2‐mediated downregulation of HIF‐1α ablates tumor glycolysis and growth in BNIP3‐silenced melanoma cells.
Discussion
In this study, we show that BNIP3 supports HIF‐1α stabilization and its pro‐tumorigenic program in melanoma. We propose a model in which BNIP3, due to its ability to regulate the intracellular availability of iron by directly controlling NCOA4‐mediated ferritinophagy, maintains HIF‐1α‐driven glycolytic program and establishes a feedforward BNIP3‐HIF‐1α axis that fosters melanoma growth. This bidirectional loop between BNIP3 and HIF‐1α is an unexpected finding of this study, given that BNIP3 is a hypoxia‐responsive gene thought to operate as a downstream target of HIF‐1α rather than being a positive regulator of this transcription factor.
BNIP3 supports HIF‐1α glycolytic program and melanoma growth
Mechanistic studies comprehensively examining the role of BNIP3 in cancer are scarce. In this study, we show that depletion of BNIP3 decreases mitochondria clearance causing the accumulation of dysfunctional mitochondria. Despite this, BNIP3‐silenced melanoma cells still retain their OXPHOS ability in combination with dysregulated glycolysis and compromised fermentation. This is in contrast to the effects mediated by the reduction in general autophagy in the ATG5‐silenced melanoma cells, which result in the stimulation of aerobic glycolysis, as observed in other studies (Kimmelman & White, 2017; Jiao et al, 2018). We show that the reduced aerobic glycolysis of the BNIP3‐silenced cells is secondary to their inability to support normoxic HIF‐1α levels and its overall transcriptional glycolytic program. Furthermore, rescuing the basal expression of HIF‐1α (by expressing an undegradable HIF‐1α mutant) in BNIP3‐silenced cells restores their glycolytic capability and corrects their delayed tumor growth in vivo.
Thus, compromising mitochondrial clearance while concomitantly preventing aerobic glycolysis in melanoma cells through the removal of BNIP3 has oncosuppressive ability. Consistent with this, studies in multiple tumor mouse models have shown that defects in mitophagy can cause tumor regression (Porporato et al, 2018; Vara‐Perez et al, 2019a). In our melanoma model, BNIP3‐silenced cells are still able to sustain their heightened autophagic flux, which ultimately supports their survival, at least in vitro. Future studies using autophagy blockers in vivo will be important to understand whether inhibiting autophagic flux contributes to accelerate tumor cell death and completely block the growth (or relapse) of the BNIP3‐depleted tumors.
Finally, the metabolic rewiring observed in the BNIP3‐silenced cells could impact the tumor microenvironment. We found that BNIP3‐depleted tumors have smaller tumor vessels, which together with lower LDHA levels (reduced acidosis), could render the tumor microenvironment less immunosuppressive (Cascone et al, 2018). Considering that melanoma glycolysis is associated with immune resistance to adoptive T‐cell therapy (Cascone et al, 2018), the potential contribution of BNIP3 on immune resistance mechanisms (and T cell effector functions, in particular) should be explored in future studies.
Notably, in an MMTV‐PyMT breast cancer model, loss of BNIP3 was shown to mitigate mitophagy, elevate ROS, and maintain autophagic flux under normoxic conditions (Chourasia et al, 2015), as observed in this study. Yet, in the context of breast cancer, BNIP3 had a pro‐tumorigenic role. This in vivo dichotomous BNIP3 role is related to HIF‐1α. In the MMTV‐PyMT breast cancer model, mitochondrial dysfunction promotes—rather than suppresses—HIF‐1α stabilization and aerobic glycolysis (Chourasia et al, 2015). The stabilization of HIF‐1α eventually feeds breast cancer growth, angiogenesis, and dissemination (Chourasia et al, 2015), supporting the view that BNIP3 restricts breast cancer progression. A clear understanding of these discrepancies is still lacking and may reflect additional roles of BNIP3 that are cancer cell type‐specific or related to alterations of other catabolic pathways, such as ferritinophagy (see below) or to cancer cell‐specific mitochondrial circuitries.
Irrespective of this unknown, and supporting our patient and mechanistic data, high BNIP3 levels have been shown to promote cancer cell survival and metastatic dissemination in uveal melanomas (Jiang et al, 2018). Our TCGA and TMA data show that, in early‐stage melanomas and further in metastasis, BNIP3 levels are elevated, correlate with the HIF‐1α transcriptional signature and associate with shorter patient survival. A limitation of this analysis is the inability to discriminate normoxic and hypoxic HIF‐1α stabilization in vivo since it is very difficult to assess the origin of HIF‐1α levels in whole tissue proteomic or transcriptomic analysis (such as the TCGA or our own tumor lysates). Despite this, BNIP3 could be considered as a novel prognostic biomarker for at least a subset of melanoma patients. Moreover, since HIF‐1α levels contribute to therapeutic failure in melanoma (Zbytek et al, 2013), a better insight into the BNIP3‐mediated pathways regulating the HIF‐1α glycolytic program could be important to design new therapeutic avenues.
Lack of BNIP3 enhances PHD2‐mediated HIF‐1α degradation by stimulating ferritinophagy
Using pharmacological and genetic tools, we show that BNIP3 depletion increases the hydroxylation of HIF‐1α both in vitro and in vivo by elevating the expression and activity of PHD2. This BNIP3‐regulated mechanism operates prevalently under conditions of oxygen availability and is mitigated when HIF‐1α is firmly stabilized by hypoxia. HIF‐1α is classically associated with the transcriptional response to oxygen deprivation (Lee et al, 2020); however, in the past years, several studies have highlighted mechanisms of normoxic or pseudo‐hypoxic HIF‐1α stabilization in cancer (Semenza, 2010). High normoxic HIF‐1α levels in glioblastoma cells were supported by the accumulation of glycolytic metabolites, such as pyruvate and lactate (De Saedeleer et al, 2012). Together with pyruvate, other metabolites such as succinate or fumarate (Hewitson et al, 2007; Lee et al, 2020) have been proposed as indirect HIF‐1α stabilizers, although the precise mechanism remains controversial (Hewitson et al, 2007). Similar mechanisms were described in response to the loss of PDK1 in head and neck squamous cell carcinoma (McFate et al, 2008) or PDK2 in lung and breast cancer cells (Sutendra et al, 2013). Although we cannot rule out that, upon loss of BNIP3, subtle changes in the concentrations of cellular metabolites affecting PHD2 activity may further accentuate HIF‐1α downregulation, our data suggest that deregulated iron metabolism is a crucial BNIP3‐associated effector mechanism.
Ferritinophagy is activated largely in response to changes in the cytoplasmic concentration of Fe2+ and is controlled by the cargo receptor NCOA4 (Santana‐Codina & Mancias, 2018). We found that BNIP3 silencing in melanoma cells exacerbated the lysosomal turnover of NCOA4, resulting in an increase in the cytoplasmic Fe2+/Fe3+ ratio. Moreover, we also found that BNIP3 and NCOA4 physically interact and that the removal of NCOA4 rescued iron levels in BNIP3‐silenced cells. Hence, the elevated iron level of the BNIP3‐depleted cells is likely resulting from increased availability of cytosolic NCOA4 engaged in homeostatic ferritinophagy, which is caused by disrupting BNIP3‐NCOA4 interaction. Intracellularly, iron is mobilized by ferritinophagy and by both mitophagy and non‐canonical (e.g. endosomal microautophagy) lysosomal pathways, but the molecular mediators of these pathways are still elusive (Goodwin et al, 2017; Mejlvang et al, 2018; Santana‐Codina & Mancias, 2018). It is tempting to propose that, as a consequence of BNIP3 depletion and the disrupted interaction between BNIP3 and NCOA4, the elevated turnover of NCOA4‐mediated ferritinophagy compensates for the reduced mitophagy in order to supply levels of Fe2+ compatible with the maintenance of vital mitochondria OXPHOS functions (Vara‐Perez et al, 2019a). This is an intriguing possibility that requires further investigation.
Boosting the pool of labile iron then favors PHD2 activity and HIF‐1α degradation, as shown by the complete rescue of HIF‐1α levels and downstream targets in the BNIP3‐silenced cells treated with the iron chelator DFO. Although our data support a role for PHD2 as an important effector of the BNIP3‐HIF‐1α axis, we cannot exclude that elevation of intracellular iron in the BNIP3‐depleted cells favors the concerted activity of other prolyl hydroxylases and may affect other iron‐dependent enzymes and pathways.
Lastly, the elevation of the labile iron pool observed upon loss of BNIP3 suggests that melanoma cells harboring high levels of BNIP3 may be less vulnerable to ferroptosis, an emerging iron‐mediated and lipid peroxidation‐driven necrotic cell death with possible therapeutic implications in melanoma (Tsoi et al, 2018). These are interesting conjectures that need further molecular investigations in order to identify the iron‐sensing mechanisms and effectors regulated by BNIP3.
In summary, this study unravels an unexpected loop whereby BNIP3 and HIF‐1α mutually reinforce each‐other pro‐tumorigenic function in melanoma, which may provide the basis for new therapeutic options.
Materials and Methods
Experimental design
The objective of this study was to determine the role of BNIP3 in melanoma progression. We investigated mRNA and protein expression of BNIP3 in melanoma patient‐derived material by analysis of public RNAseq datasets together with IHC‐TMA and paired primary‐metastases samples. Subcutaneous implantation of a murine melanoma cell line with different BNIP3 or ATG5 levels in syngeneic mice aimed to establish the pro‐tumorigenic function of BNIP3 in vivo and the potential difference between BNIP3 and a canonical autophagy gene. In vitro/in vivo studies using immuno(cyto/histo)chemistry, flow cytometry, metabolic flux readouts (Seahorse, 1H‐NMR, GC‐MS), protein and transcript levels, and subsequent mechanistic validation studies evaluated the role of BNIP3 in melanoma progression, with particular focus on the regulatory feedforward loop of HIF‐1α.
Chemicals and reagents
Cell culture reagents (unless otherwise stated), antimycin A, β‐mercaptoethanol, bovine serum albumin (BSA), 3‐[(3‐Cholamidopropyl)dimethylammonio]‐1‐propanesulfonate hydrate (CHAPS), carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP), deferoxamine mesylate (DFO), D‐glucose, dimethyl sulfoxide (DMSO), disodium‐fumarate, galactose, gelatin from bovine skin, glycerol, glycine, gelatin, 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES), hydrogen peroxide solution, IOX2, methylene blue, oligomycin, polybrene, sodium docecyl sulfate (SDS), Tris, Triton X, and Tween 20 were obtained from Sigma‐Aldrich (Bornem, Belgium). Sodium deoxycholate, NP‐40, phenylmethane sulfonyl fluoride or phenylmethylsulfonyl fluoride (PMSF), glutamine and orthovanadate were purchased from Sigma‐Aldrich (Taufkirchen, Germany). FeCl3‐ 6H2O standard was from Sigma‐Aldrich Chemie (Steinheim, Germany). Sodium chloride (NaCl), potassium chloride (KCl), magnesium chloride (MgCl2), and calcium chloride (CaCl2) were purchased from ACROS organics (Fisher Scientific, USA). Chloroform, ethanol, methanol, and xylene were obtained from VWR International (Oud‐Heverlee, Belgium). Bafilomycin A (BafA) was purchased from Sanbio (Uden, The Netherlands). Puromycin dihydrochloride was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Hygromycin B was obtained from Invivogen (San Diego, CA, USA). Tetramethylammoniumhydroxide (TMAH) and HCl suprapure were purchased from Merck (Darmstadt, Germany). Argonliquid was purchased from Air Liquide (Düsseldorf, Germany). FeCl2‐4H2O was purchased from AppliChem GmbH (Darmstadt, Germany). The cOmplete™ Protease Inhibitor Cocktail was from Roche, Mannheim, Germany. Whenever concentrations are included throughout the methods, they refer to the final concentration.
Cell culture
Melanoma cell lines B16‐F10 (murine) and WM35 (human) were obtained from ATCC. WM35 was validated by STR profiling. Murine cell lines cannot be reliably validated by STR profiling at this moment. Patient‐derived early‐stage human melanoma cell line 530 was a kind gift from Dr. G. N. P. van Muijen (Radboud University, The Netherlands). Murine melanoma cells (B16‐F10) were maintained in RPMI‐1640 medium containing 10% (v/v) Fetal Bovine Serum (FBS; Hyclone, Thermo Fisher Scientific) and penicillin (100 units/ml)/streptomycin (0.1 mg/ml). Human melanoma cells 530 and WM35 were maintained in Dulbecco’s Modified Eagles Medium (DMEM) supplemented with 10% (v/v) FBS, 2 mM glutamine and penicillin (100 units/ml)/streptomycin (0.1 mg/ml). All cells were maintained routinely in 5% CO2 and 95% air at 37°C. Cells were routinely checked for mycoplasma contaminations using the Plasmotest kit (Invivogen) according to the manufacturer’s instructions.
For protein and RNA extraction, cells were seeded at a density of 150,000 (B16‐F10 shCntl, B16‐F10 shATG5), 200,000 (all human melanoma cell lines), or 300,000 (B16‐F10 shBNIP3) cells/ well in a 6‐well plate (Greiner Bio‐One, Vilvoorde, Belgium) and cultured for 48 h (normoxic condition). For hypoxic conditions (1.5% O2, 5% CO2, 93.5% N2), the cells were kept in an Invivo2400 hypoxia workstation (Ruskinn, Heusden‐Zolder, Belgium) for 24 h after 24 h of plating. In case of a 24‐h treatment, cell medium was refreshed after 24 h of plating including the chemical of interest [IOX2 (in DMSO, 100 µM), BafA (in DMSO, 5 or 10 nM), or DFO (in milliQ, 200 µM)] for another 24 h until cell collection for protein or RNA extraction.
Generation of shRNA stable clones of melanoma cells
Lentiviral shRNA vectors against human BNIP3 or murine BNIP3, ATG5, and NIX were purchased from the MISSION® shRNA library (Sigma‐Aldrich). An empty pLKO.1‐puro control vector was used as a control (shCntl). To generate lentiviral particles, HEK 293T cells were seeded in 10 cm2 dishes at 1.5 × 106 cells per 6 ml and transfected the following day by the calcium phosphate method with 4 µg of pLKO.1‐puro carrying the respective shRNAs or with empty pLKO.1‐puro. Each transfection also included 1.2 µg of a plasmid encoding VSV‐G (pMD2‐VSV‐G, Tronolab) and 2.6 µg of a plasmid encoding packaging proteins (pCMVdR8.9, Tronolab). VSV‐G pseudotyped virus was collected 48 h after transfection, passed through 0.45 µm filters (Millipore, Burlington, MA, USA), and then added to the exponentially growing melanoma cell cultures in the presence of 8 mg/ml of polybrene. The cells were expanded and selected by puromycin treatment (10 µg/ml). Target downregulation was confirmed by immunoblotting and qPCR.
Generation of undegradable HIF‐1α and BNIP3 stable clones of B16‐F10 melanoma cells
Human HIF‐1α mutant cDNA was PCR‐amplified from the plasmid pLenti/V5‐HIF‐1α‐mPPN and cloned into in‐house pCHMWS‐eGFP‐ires‐hygro after removal of eGFP. Murine Myc‐BNIP3 sequence was obtained from Addgene plasmid #100796. The cDNA was ordered as a gBlocks Gene Fragment at IDT (Leuven, Belgium). These fragments were cloned into our in‐house pCHMWS‐GFP‐ires‐hygro transfer plasmid via BamHI and NheI restriction sites after removal of eGFP. In both cases, Firefly luciferase was used as decoy protein in the control cell lines. Lentiviral vectors were produced by the Leuven Viral Vector Core as described in (Ibrahimi et al, 2009). 100,000 cells/well were plated in a 24‐well plate, and viral transduction was performed during 48 h. Cells were selected with hygromycin (200 µg/ml) until all non‐transduced cells died. Target overexpression was confirmed by immunoblotting and qPCR.
Cell transfection
Cells were transiently transfected with different immunofluorescently labeled LC3B constructs: RFP‐EGFP‐LC3 tandem (Addgene plasmid #21074) and LC3B‐GFP (Addgene plasmid #22405) using Trans‐IT X2 (Mirus Bio LLC, Madison, WI, USA) transfection reagent in serum‐free DMEM medium according to the manufacturer’s indications. 24 h after transfection, the transfection medium was removed and was replaced by full medium to let the cells recover. After 24 h, surviving cells were seeded in glass coverslips (Thermo Fisher Scientific) coated with 1% (w/v) gelatin in PBS and allowed to grow for an additional 24 h before fixation. When necessary, BafA was added 4 h before fixation.
Cells were transiently transfected with 40 µM siRNA from the SMARTpool: ONtarget plus library from Dharmacon (Lafayette, CO, USA) either non‐targeting (siScr, D‐001810‐10‐05), targeting murine PHD2 (siPHD2, L‐040757‐01‐0005) or murine NCOA4 (siNCOA4, L‐049515‐01‐0005) using Dharmafect1 (Dharmacon) transfection reagent in serum‐free medium according to the manufacturer’s indications. 24 h after transfection, a second transfection was performed to ensure enough protein degradation. 24 h after the second transfection, medium was removed and was replaced by full medium to let the cells recover. 36 h after the change of medium, surviving cells were collected for protein and RNA extraction. 12 h after the change of medium, cells were plated for the FerroOrange assay in a dark 96‐well plate (see below).
Mice experiments
Animal procedures were approved by the Institutional Animal Care and Research Advisory Committee of the KU Leuven (ECD P237/2015) and were performed following the institutional and national guidelines and regulations. Female C57BL/6 mice of 6 weeks of age were provided by the KU Leuven mouse facility or were purchased from Janvier (France). For all experiments, mice were group‐housed in standard cages under a 12 h light/dark cycle with ad libitum access to water and food. Only females were used in the experiments. To assess subcutaneous tumor growth, 150,000 B16‐F10 murine melanoma cells proficient or deficient for ATG5 and BNIP3 were injected subcutaneously into the right flanks of immunocompetent syngeneic (C57BL/6) mice. Tumor volumes were measured at least three times per week with a caliper using the formula V = π * [D*d*t]/6, where D is the major tumor axis, d is the minor tumor axis and t is the thickness of the tumor. The mice were sacrificed when tumors reached a maximum size of 1,000 mm3 depending on the experimental setup. Tumors were collected for histological analysis, gene expression, or protein analysis. To detect tumor hypoxia, tumor‐bearing mice were injected 60 mg/kg pimonidazole hydrochloride (Hypoxyprobe kit, #HP3‐200Kit, Chemicon‐Millipore, Billerica, MA, USA) 1 h before tumor collection.
Immunoblotting
A modified Laemli sample buffer (125 mM Tris–HCl, pH 6.8 buffer containing 2% SDS and 20% glycerol) containing 10% (v/v) protease and phosphatase inhibitors (Pierce Protease Inhibitor/Phosphatase Inhibitor Tablets, Thermo Fisher Scientific) was used to lysate the cells. Frozen tumors were smashed in Laemli buffer with a metal mortar (Usbeck Metal Products, Radevormwald, Germany), centrifuged 15 min, at 20.000 g at 10°C, and the supernatant was collected for protein assays. Protein concentration in cell and tumor lysates was determined using the BCA protein assay reagent (Pierce, Thermo Fisher Scientific). Precision Plus Protein™ ladder (#1610374, Bio‐Rad Laboratories, Hercules, CA, USA) was used for protein size reference. Samples were separated by SDS–PAGE on the Criterion system (Bio‐Rad Laboratories) on a 4%–12% Bis‐TRIS gel and electrophoretically transferred to Amersham Protran 2 μm‐pored nitrocellulose paper (GE Healthcare, Chicago, IL, USA). The blots were blocked for 60 min at RT in TBS‐T buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.1% Tween‐20) containing 5% non‐fat dry milk (w/v) and then incubated with selected primary and secondary antibody solutions prepared in blocking buffer unless otherwise stated.
Primary antibodies for ATG5 (1:1000, #12994), murine BNIP3 (1:1000, #3769), HIF‐1α‐OH P564 (1:500, #3434), LC3B (1:1,000, #3868), LDHA (1:1,000, #2012), NIX (1:1,000, #12396), PHD2 (1:1,000, #4835), and TFEB (1:500, #4240) were purchased from Cell Signaling Technologies (Danvers, MA, USA) and prepared in 5% (w/v) BSA in TBS‐T buffer. HIF‐1α antibody (1:1,000, #10006421) was purchased from Cayman Chemicals (Ann Arbor, MI, USA). FTL antibody (1:1,000, #ab109373) was purchased from Abcam (Cambridge, UK). Human BNIP3 (1:1,000, #HPA003015) is from the Sigma‐Aldrich’s Protein Atlas antibody repertoire. Luciferase antibody (1:1,000, NB600‐307) is from Novus Biologicals (Centenials, CO, USA). (Beta‐)Actin (1:2,000, #A5441) is from Sigma‐Aldrich. NCOA4 (1:1,000, A302‐272A) was purchased from Bethyl Laboratories (Montgomery, TX, USA).
Appropriate secondary antibodies for chemical (HRP‐based) detection were from Cell Signalling Technologies and for infrared detection were from Thermo Fisher Scientific. Signal detection was performed using the Typhoon infrared‐imaging system (GE Healthcare) or ChemidocTM MP system (Bio‐Rad Laboratories) using the ECL solution from Pierce (Thermo Fisher Scientific). Whenever necessary, membranes were stripped for 20 min in ReBlot Plus Mild Antibody Stripping Solution (Millipore) 1X in dH2O and reblocked in 5% milk in TBS‐T before the addition of the new primary antibody. Quantifications by densitometry of the bands were calculated using the software Image Studio (Li‐Cor Biosciences, Lincoln, NE, USA).
Immunoprecipitation of endogenous BNIP3
48 h after plating, B16‐F10 shCntl cells were collected through scraping and lysed in lysis buffer (1% (w/v) CHAPS, 50 mM Tris–HCl, 100 mM KCl, 150 mM NaCl, 10% (v/v) protease inhibitor) for 30 min at 4°C. Cells were centrifuged to remove debris for 5 min (max. speed, 4°C), and the supernatant was collected for protein quantification with the BCA protein assay reagent. 1,250 μg of proteins were precleared for 1 h at room temperature (RT) using the Protein A/G‐agarose beads (#sc2003, Santa Cruz Technologies). From the precleared solution, 500 μg of proteins were combined with 4 μl from either an isotype immunoglobulin G (IgG) control (#sc2025, Santa Cruz Technologies) or anti‐BNIP3 (#ab10433, Abcam) primary antibody overnight (ON) at 4°C.
Protein–antibody complexes were captured by the addition of Protein AG Magnetic Beads (#88802, Pierce, Thermo Scientific) for 1.5 h at RT. Protein AG Magnetic Beads with captured protein–antibody complexes were washed three times with lysis buffer. Proteins were eluted in loading buffer, boiled at 100°C for 5 min, and loaded on a gel for Western blot analysis. 50 μg of precleared protein solution were used as input, and 50 μg of a B16‐F10 shCntl treated with BafA lysate were used as band reference. For the immunoprecipitation blots, HRP‐based detection using as secondary antibody the Veriblot antibody (#ab131366, Abcam; 1:1,000) and the Clarity and Clarity Max solutions (Bio‐Rad Laboratories) to develop the membranes in an Amersham Imager 600 (GE Healthcare).
RNA extraction, cDNA generation, and qPCR
Cells destined for RNA extraction were collected in RLTplus buffer (Qiagen, Hilden, Germany) supplemented 1/100 (v/v) with β‐mercaptoethanol. RNA extraction was performed using the RNeasy Mini Kit (Qiagen) following the manufacturer’s indications. Tumor RNA was extracted first by homogenizing the tissue with a Precellys 24 tissue homogenizer (Bertin Instruments, Montigny‐le‐Bretonneux, France) in the presence of TRIzol™ (Life Technologies, Thermo Fisher Scientific) and precipitating it with chloroform. After centrifugation, RNA was further purified using the PureLink™ RNA Mini Kit (Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instructions.
RNA concentration was measured using a Nanodrop 3000 (Thermo Fisher Scientific) and cDNA was synthesized using 0.5 µg RNA per sample as template and the Quantitect Kit (Qiagen) following the manufacturer’s instructions. After diluting the resulting cDNA 1:5 in sterile water, expression analysis was performed using home‐designed or literature‐based primers (Integrated DNA Technologies, Haasrode, Belgium) and ORA Sybr Green Mastermix (Highqu, Kraichtal, Germany) in an ABI7500 (Applied Biosystems, Thermo Fisher Scientific) machine. Primer sequences are available in Appendix Table S5. CT values were calculated using the software of the machine and the geometric mean of two housekeeping genes (Hprt and Ppib for mouse, HPRT and PPIB for human) was used for normalization. Transcript expression was calculated using the formula: Expression Value = , where GOI stands for “Gene Of Interest” and HKG for “Housekeeping Genes”. Afterward, the fold change relative to their respective control was calculated.
Determination of proliferation, cell death, and colony formation
Proliferation curves were generated using an IncuCyte ZOOM system (Essen BioScience), starting from cells seeded at a density of 3,000 cells per well in a 96‐well plate (Greiner, Vilvoorde, Belgium), based on phase‐contrast images taken at 2‐h intervals for the duration of the experiments. The interval comprising 10–80% of confluence was used to calculate the doubling time of the cells per well using the exponential fitting provided by Prism software (GraphPad Software, La Jolla, CA, USA).
Cell death was determined by the propidium iodide (PI) exclusion method. Briefly, cells were trypsinized at the indicated time points and incubated with the vital dye PI (Sigma‐Aldrich) for 15 min at RT. PI‐positive (dead) cells were quantified via flow cytometry (Attune Cytometer, Invitrogen, Life Technologies, Paris, France) and analyzed using FlowJo (BD Biosciences).
For the colony formation assay, cells were plated as single‐cell suspension at very low density (500 cells in 15 ml medium on a 10 cm dish). After growth for 10 days, cells were fixed in methanol containing 1% (w/v) methylene blue.
Determination of mitochondrial parameters
Mitochondrial depolarization and ROS levels were detected separately after incubation of the cells with either 20 nM TMRM (Molecular probes, Invitrogen) or 7.5 mM CM‐H2DCFDA (Molecular probes, Invitrogen), respectively, 30 min at 37°C. Fluorescence was measured using an Attune flow cytometer and analyzed using FlowJo software by gating first alive cells in the FSC/SSC display and singlets in the FSC‐A/FSC‐H display. Mean fluorescence intensity (MFI) was calculated from the final population and plotted as fold change of the corresponding shCntl MFI value.
Mitochondrial morphology was detected after incubation of the cells with 100 nM MitoTracker Green (Molecular Probes, Invitrogen) 30 min at 37°C. After one wash in PBS, live cells were imaged in Krebs solution (containing 150 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 11.6 mM HEPES (pH 7.3), 11.5 mM glucose, and 1.5 mM CaCl2), using an LSM880 confocal microscope in a temperature, CO2‐controlled incubator (Carl Zeiss, Munich, Germany).
Immunostaining and Immunohistochemistry
Cells were grown and treated on gelatin‐coated glass coverslips (as described before). Cells were fixed with 4% PFA, permeabilized with 0.1% Triton X‐100 (v/v) in PBS, blocked with 5% (v/v) FBS, 1% (w/v) BSA, 2.25% (w/v) glycine in PBS and incubated with selected primary (antiTOMM20, 1:100, #ab186735, Abcam) and secondary antibodies (1:200, #A27040, Life Technologies) in 5% (v/v) FBS, 1% (w/v) BSA in PBS. Finally, nuclei were stained with DAPI (10 μg/ml, Life Technologies) and cells were mounted using Prolong Gold antifade reagent (Life Technologies).
Mouse tissue samples were fixed in 3–4% PFA v/v in PBS (VWR) overnight at 4°C, dehydrated, and embedded in paraffin using an Excelsior AS Tissue Processor and HistoStar Embedding Workstation (Thermo Scientific). Sections of 7 μm of thickness obtained from the paraffin‐embedded tissues with a Microm HM340E microtome (Thermo Scientific) were mounted on Superfrost Adhesion slides (Thermo Fisher Scientific) and routinely stained with hematoxylin and eosin (H&E; Diapath, Martinengo, Italy) for histopathological examination.
Briefly, slides were deparaffinized in xylene and then rehydrated in ethanol series (100%, 96%, and 70%) and distilled water. Epitope retrieval was performed in 1× citrate buffer (Dako, Heverlee, Belgium) using the 2100 Retriever (Aptum Biologics, Southampton, UK). Inhibition of endogenous peroxidase was achieved by incubating the slides in 0.3% hydrogen peroxide in methanol for 20 min at −20°C. Sections were blocked in TNB blocking buffer (TSA kit, Perkin Elmer, Life Sciences, Zaventem, Belgium) supplemented with 20% (v/v) goat serum (Sigma‐Aldrich) for 1 h at RT and then incubated overnight at 4°C with the primary antibody in TNB buffer: anti‐Ki67 (1:100, #MA5‐14520, Thermo Fisher Scientific), anti‐HIF‐1α (1:300, #10006421, Cayman Chemicals), anti‐LC3B (1:100, #3868, Cell Signaling Technologies), anti‐CD31 (1:100, #550274, BD Biosciences), anti‐α‐smooth muscle actin‐Cy3 (1:250, #C6198, Sigma‐Aldrich), or anti‐hydroxyprobe (1:100, HP3‐200Kit, Chemicon‐Millipore).
For the immunoperoxidase technique, the Vectastain Elite ABC kit (Vector Labs, Peterborough, UK) combined with the chromogen ImmPACT DAB Peroxidase substrate kit (Vector Labs) were used according to the manufacturer’s protocol. Slides were counterstained in Harris Haematoxylin (Diapath), dehydrated in ethanol series, cleared in xylene, and permanently mounted with a resinous mounting medium (DPX, Diapath). For immunofluorescence, sections were then incubated with the appropriate peroxidase‐labeled IgGs (Dako), followed by amplification with Cy3 or Cy5‐ tyramide signal amplification (TSA) kit (Perkin Elmer). DAPI was used as nuclear counterstain and the slides were mounted with Prolong Gold antifade reagent. TNT buffer was used as washing buffer in between steps.
Cell death was detected and quantitated via the transferase‐mediated dUTP nick end‐labeling (TUNEL) technique using the “In situ Cell Death Detection” Kit (Roche, USA) according to the manufacturer’s instructions.
Imaging and analysis
For cell death and hypoxia measurements, 10 × mosaic pictures were taken with an Olympus IX73 (Olympus, Berchem, Belgium) and the positive area was quantified using Olympus’ CellSense imaging software and expressed as a percentage of the total area of the analyzed tumor. For proliferation measurements, 20 × mosaic pictures were taken with an AxioScan.ZI (Carl Zeiss) and the positive cells were quantified using QuPath software (Bankhead et al, 2017) and expressed as a percentage of total cell number within the area of the analyzed tumor. For the LC3B immunohistological staining, the pictures were acquired with the LSM780 confocal microscope, 25 × objective (Carl Zeiss, Munich, Germany).
For the co‐localization and the mitochondrial morphology analysis, the pictures were acquired with the LSM880 confocal microscope, 60 × objective (Carl Zeiss).
For the GFP‐LC3‐TOMM20 co‐localization analysis, images were quantified by manual counting using ImageJ/Fiji software. GFP‐LC3 puncta were defined as round structures found only in the green channel with no corresponding structure in the other channels. Co‐localization analysis was performed by counting all GFP‐LC3 puncta per cell overlapping with a mitochondrial structure, defined by TOMM20 staining and calculating the percentage of colocalizing puncta over the total number/cell. The mitochondrial morphology was analyzed with the macro “Mito‐morphology” from ImageJ/Fiji Software. Since the confocal pictures contained a variable number of cells, these values were considered non‐parametric for statistical purposes and outlier values were removed using the Rout method before statistical analysis.
Metabolic measurements
Seahorse
Cells were plated at a density of 30,000 (shCntl, shATG5) and 50,000 (shBNIP3) cells per well of an XFp Extracellular Flux Analyzer culture plate in culture medium overnight. Before the assay, cells were switched to the Mitostress assay medium (Seahorse Biosciences, North Billerica, MA, USA) containing either glucose or galactose (25 mM) and supplemented with 2 mM glutamine and 1 mM sodium pyruvate (Sigma‐Aldrich), following the manufacturer instructions. Real‐time extracellular acidification (ECAR) and oxygen consumption (OCR) during the Mitostress assay were measured with the XFp Extracellular Flux Analyzer (Seahorse Biosciences) following the manufacturer instructions. Following the Mitostress assay, 8 µM oligomycin, 4.5 µM FCCP and 5 µM antimycin A diluted in assay medium were added whenever appropriate. Once the run was finished, all the medium was removed from the well and all the cells were collected in 10 µl of Laemli buffer for BCA‐based protein quantification (as explained before).
1H‐NMR‐based metabolic measurements
Cells were plated and grown to confluency in T175 flasks. 24 h before collecting the samples, medium was replaced by fresh new ones. Both cellular extracts and culture media were considered for the 1H‐NMR measurements. First, the 24 h culture media were removed from each flask and stored (−80°C). The remaining adherent cells were washed twice using 14 ml cold D‐PBS (Sigma‐Aldrich), quenched using 6 ml of cold methanol, and collected using a scraper. Cells were de novo quenched by immersing the pellet into liquid nitrogen for a few seconds before storage (−80°C). Next, a methanol: water: Chloroform 1:0.9:1 (6 ml) extraction was carried out to separate polar metabolites from macromolecules. The polar phase was dried using a SpeedVacuum and then resuspended in 700 μl phosphate buffer (0.2 M Na2HPO4/0.04 M NaH2PO4, pH 7.4) prepared in a mixture of H2O/D2O (80/20; v/v). Samples were centrifuged at 13,000 g for 10 min. 50 μl of a 7 mM solution of 3‐trimethylsilyl propionic‐2,2,3,3‐d4 acid (TSP) reference prepared in 100% deuterium oxide was added to 650 μl of each supernatant. Finally, 700 μl of this final mixture was transferred into 5 mm NMR tubes prior to analysis. For the culture media preparation, 500 μl of each sample was mixed with 250 μl of phosphate buffer. The samples were prepared as described above, with addition of 14 mM TSP solution. Acquisition of the 1H‐NMR spectra was processed on a Bruker 500‐MHz Advance spectrometer for 1H, using the NOESYPRESAT‐1D pulse sequence and 256 scans. For the spectra processing, both phases and baseline of each 1H‐NMR spectra were corrected using the MestreLab Research 10.0.2 software (Mestrelab Research, S.L, Santiago de Compostela, Spain). The water peak region ranging from 4.20 to 5.50 ppm was excluded, and spectra were then binned in small subregions of 0.04 ppm width called descriptors, giving rise to 220 descriptors for each spectrum. For each descriptor, the area under the curve (AUC) was calculated to obtain numerical data in all spectra. Each descriptor value was then divided by the total area of the spectrum for normalization. Data were processed for multivariate data analysis using the software SIMCA‐P + 12.0 (Umetrics, Umeå, Sweden). Principal component analysis (PCA) followed by a partial least square discriminant analysis (PLS‐DA) were carried out on the dataset. Only the descriptors with a Variable Importance in the Projection (VIP) above 1 were considered in this study. Their corresponding metabolites were identified using the Chenomx NMR suite software (version 8.1.1).
GC‐MS‐based metabolic measurements
Metabolite extraction and derivatization method
Metabolite quenching and extraction were performed as described before (Elia et al, 2017). Briefly, samples were collected in 800 µl of 60% ice‐cold methanol containing 90 ng/ml of glutaric acid as an internal standard. Subsequently, a volume of 500 µl of precooled chloroform was added to each sample. During the collection procedure, the samples were always refrigerated in a mixture of dry and wet ice. The samples were then vortexed for 10 min at 4°C and centrifuged for 10 min (max. speed, 4°C) to achieve phase separation. After centrifugation, the metabolites were separated into two phases divided by a protein layer: polar metabolites in the methanol/water (upper) phase and the lipid fraction in the chloroform (lower) phase. The upper and lower phases were collected in separate tubes and dried at 4°C overnight using a vacuum concentrator.
The samples were derivatized and measured as described before (Elia et al, 2017; Lorendeau et al, 2017). Briefly, polar metabolites were resuspended and derivatized in 20 µl of 20 mg/ml methoxyamine in pyridine per sample for 90 min at 37°C. Subsequently, 15 µl of N‐(tert‐butyldimethylsilyl)‐N‐methyl‐trifluoroacetamide, with 1% tert‐butyldimethylchlorosilane were added to 7.5 µl of each derivative and incubated for 60 min at 60°C.
The metabolites were analyzed by gas chromatography (7890A GC system) coupled to mass spectrometry (5975C Inert MS system) from Agilent Technologies. Metabolites were separated with a DB35MS column (30 m, 0.25 mm, 0.25 µm) using a carrier gas flow of helium fixed at 1 ml/min for the analysis of polar metabolites. A volume of 1 µl of sample was injected with a split ratio 1–3 with an inlet temperature set at 270°C. For the detection of polar metabolites, the gradient started at 100°C for 1 min ramped to 105°C at 2.5°C/min, then to 240°C at 3.5°C/min and finally to 320°C at 22°C/min. For the measurement of metabolites by mass spectrometry, the temperatures of the quadrupole and the source were set at 150°C and 230°C, respectively. An electron impact ionization fixed at 70 eV was applied and either a scan or a selected‐ion monitoring (SIM) mode was used for the measurement of polar metabolites.
After the acquisition by GC‐MS, an in‐house MATLAB M‐file was used to extract mass distribution vectors and integrated raw ion chromatograms. The natural isotope distribution was also corrected using the method developed by Fernandez et al, as described before (Elia et al, 2017; Lorendeau et al, 2017). The peak area was subsequently normalized to the internal standard glutaric acid and to cell number to account for sample loss during the metabolite extraction procedure and differences in cell density in the samples, respectively.
Intracellular Fe2+ measurements (FerroOrange)
Intracellular iron content was measured using the dye FerroOrange (F374‐10, Dojindo EU GmbH, Munich, Germany) according to the manufacturer’s instructions. Briefly, B16‐F10 cells were seeded in 100 µl of medium (20,000 cells per well for shCntl/shATG5 and 30,000 shBNIP3) and incubated overnight. After washing with HBSS with 10 mM HEPES (HBSS‐HEPES buffer) three times, cells were incubated with FerroOrange 1 µM in HBSS‐HEPES buffer for 30 min. After the incubation, the plate was immediately read using a Synergy H1 M Multi‐Mode reader (BioTek, Winooski, VT, USA), with the excitation set at 543 nm and emission at 580 nm. Afterward, if microscopy images were needed, cells were imaged in the wells using an Olympus IX73 microscope with a 20 × objective. Both wells containing cells without dye and dye in empty wells were used for background correction of the plate reader intensity. For protein normalization, background wells seeded in parallel that were not incubated with the dye were used. All the cells were collected in 10 µl of Laemli buffer for BCA‐based protein quantification (as explained before).
Intracellular total iron and iron redox species (Fe2+, Fe3+) measurements (CE‐ICP‐MS)
Cells were lysed in modified RIPA lysis buffer containing PBS pH 7.4, 0.5% sodium deoxycholate, 1% NP‐40 containing 1 mM PMSF, 1 mM orthovanadate, and 1× cOmplete™ Protease Inhibitor on ice for 45 min with gentle agitation. Cell lysates were centrifuged at 10,000 g for 10 min, and supernatants were sent on dry ice to Helmholtz Center Munich. Speciation and quantification of Fe2+, Fe3+, and total iron were performed by capillary electrophoresis inductively coupled plasma mass spectrometry (CE‐ICP‐MS) as described previously (Michalke et al, 2019). Briefly, samples were analyzed on a "PrinCe 706” CE system equipped with an uncoated capillary (85 cm × 50 µm ID) and a laboratory‐constructed CE‐ICP‐MS interface for element selective iron quantification of separated iron redox species at ICP‐DRC‐MS. Fe2+/Fe3+ separation and quantification were performed in 20 mM HCl‐electrolyte at + 25 kV separation voltage and 56Fe isotope detection at ICP‐DRC‐MS. DRC technology with NH3 as reaction gas was employed for interference‐free detection of the 56Fe isotope. Data were presented as Fe2+/Fe3+ ratio, and absolute values are presented in Appendix Tables S3 and S4.
Bioinformatic analysis (TCGA)
RNAseq level3 data and associated patient information from the SKCM cohort were downloaded from the TCGA website on 04/Nov/2016 and it contained 469 SKCM cases. 15 samples were discarded because of lack of available follow‐up data, and one was discarded due to data disparity. The remaining 453 cases were analyzed using the R environment. Briefly, samples were separated in primary or metastatic according to the TCGA information (if necessary) and ranked according to their log2 transformed BNIP3 levels using the function ntile. Once the expression groups were formed, Kaplan–Meier analysis or associated gene analysis (log2 transformed) was performed using GraphPad Prism.
Patient sample acquisition and processing
Biopsies from benign nevi, primary, and metastatic melanoma were collected at the University Hospital of Leuven with patients’ informed consent, in accordance with the Declaration of Helsinki and with the approval of the Medical Ethics Committee of the University Hospital. The use of samples for the present study was approved by the Medical Ethics Committee of the University Hospital (project number S58320). The TMA consisting of 181 cases of melanoma metastases was constructed previously at the UZ Leuven (van Kempen et al 2016). From the original 181 cases, 16 did not have tissue available and the other 7 did not have complete survival data. These 23 cases were excluded from the analysis, resulting in 158 cases. Appendix Table S1 compiles the clinical information associated with both the paired melanoma slides and the TMA samples. Immunohistochemistry was performed on the Leica BOND‐MAX automatic immunostainer (Leica Microsystems). Antigen retrieval was performed on board using the Bond Epitope Retrieval Solution 2 (pH 9.0; Leica) according to the manufacturer’s instructions followed by 5 min of peroxidase blocking, 30 min of anti‐BNIP3 (1:100 dilution; HPA003015, Protein Atlas, Sigma‐Aldrich) primary antibody incubation, 30 min of secondary antibody (Dako, ready to use), and 30 min of AEC (Dako, ready to use) or Bond Polymer Refine Red Detection (Leica) as substrate, both resulting in pink/red immunoreactivity. Melanoma‐specific overall survival was defined as the interval from diagnosis of the primary tumor to death from melanoma or date of the last follow‐up. Sections were analyzed by conventional light microscopy, and the results were analyzed in a semiquantitative manner both for paired slides (+/−, weak and gradient staining; +, weak and homogeneous staining; ++, intense and homogeneous staining) and TMA (0, absence of staining; 1, weak staining; 2, moderate staining; 3, intense staining).
Statistics
Data represent mean ± SEM of pooled biologically independent experiments, and each experiment was repeated at least three times. n values represent the number of independent experiments performed (in vitro data), the number of animals per condition (in vivo), or the number of patients per condition (patient‐derived data). Normality was checked using both Shapiro–Wilk and Kolmogorov–Smirnov tests. To account for the inter‐experimental technical variation among biological replicates from the in vitro experiments, the values coming from the same experiment were considered as matched values and, consequently, analyzed with the corresponding paired test, as specified in the figure legend. Statistical significance was calculated using the statistical software Prism software (GraphPad Software). A P < 0.05 was considered significant.
Author contributions
Experiment design, analysis, and manuscript writing: MV‐P and PA; Experiments: MV‐P, HM, ER, and KR; Data analysis: MV‐P, HM, and ER; Performing and analyzing GC‐MS steady‐state stable isotope metabolic experiments: MR and S‐MF; Performing and analyzing confocal imaging: MV‐P and MLS; Generation of the Luc, BNIP3‐FL, and HIF1α‐AA overexpressing plasmids: CVH; Performing and analyzing the CE‐ICP‐MS experiments: VV and BM; Performing and analyzing the 1H‐NMR metabolic experiments: CS and JMC; Staining, scoring, and analyzing the human patent material: MV‐P, JW, FMB, and JJO; Assistance in TCGA data analysis: ADG; Assistance in tumor vasculature staining and analysis: AIO and MM; Helpful comments and discussion: WA, PV, JVS, S‐MF, and MM; Project and funding: PA; Manuscript revision and comments: All the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgments
We would like to thank Sofie Van Eygen, Diede Houbaert, Jelle Verhoeven, Maarten Ganne, and Jens Serneels for excellent technical assistance and Dr. Marco Schaaf, Dr. Blanca Felipe‐Abrio, Dr. Marco Spinazzi, Dr. Stefan Vinckier, and Dr. David Nittner for expert advice. We would like to thank Prof. Dr. L. Kirshenbaum (University of Manitoba, Canada) for his excellent discussion and comments. We would also like to thank Prof. Dr. T. Acker (Justus Liebig University, Germany) for kindly providing the plasmid pLenti/V5‐HIF‐1α‐mPPN and Dr. G. N. P. van Muijen (Radboud University, The Netherlands) for the 530 early‐stage melanoma cell line. M.V.P. is the recipient of an FWO Doctoral Fellowship from the Flemish Research Foundation (FWO‐Vlaanderen, 1186019N), Belgium. P.A. is supported by grants from the Flemish Research Foundation (FWO‐Vlaanderen; G076617N, G049817N, G070115N), the EOS consortium (30837538, with A.D.G.) and Stichting tegen Kanker (FAF‐F/2018/1252). P.A., J.V.S, P.V., and W. A. are jointly supported by KU Leuven (C16/15/073). E.R received a fellowship of the Kom Op tegen Kanker (Stand up to Cancer), the Flemish cancer society. M.R. has received consecutive postdoc fellowships from FWO and Stichting tegen Kanker. S.M.F. acknowledges KU Leuven Methusalem Co‐funding. J.W. received a postdoctoral research fellowship from Kom op tegen Kanker (Stand up to Cancer), the Flemish Cancer Society, and is currently funded by a postdoctoral fellowship from Stichting tegen Kanker (Foundation against Cancer), the Belgian Cancer Society. The work of B.M. and V.V. was financially supported by the Deutsche Forschungsgemeinschaft (DFG) through the Priority Program “Ferroptosis: from Molecular Basics to Clinical Applications” (SPP 2306). Images were recorded on a Zeiss LSM880—Airyscan or on a Zeiss LSM 780—SP Mai Tai HP DS (both from the Cell and Tissue Imaging Cluster, CIC), supported by Hercules AKUL/15/37_GOH1816N, Hercules AKUL/11/37, and FWO G.0929.15 to Pieter Vanden Berghe, University of Leuven.
The EMBO Journal (2021) 40: e106214.
Data availability
This study includes no data deposited in external repositories.
References
- Aguer C, Gambarotta D, Mailloux RJ, Moffat C, Dent R, McPherson R, Harper M‐E (2011) Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS One 6: e28536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG et al (2017) QuPath: Open source software for digital pathology image analysis. Sci Rep 7: 16878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A et al (2018) Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab 27: 977–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS et al (2015) Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep 16: 1145–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R, Candela‐Lena JI, Chan MC, Greenald DJ, Yeoh KK, Tian Y‐M, McDonough MA, Tumber A, Rose NR, Conejo‐Garcia A et al (2013) Selective small molecule probes for the hypoxia inducible factor (HIF) prolyl hydroxylases. ACS Chem Biol 8: 1488–1496 [DOI] [PubMed] [Google Scholar]
- Di Conza G, Trusso Cafarello S, Loroch S, Mennerich D, Deschoemaeker S, Di Matteo M, Ehling M, Gevaert K, Prenen H, Zahedi RP et al (2017) The mTOR and PP2A pathways regulate PHD2 phosphorylation to fine‐tune HIF1α levels and colorectal cancer cell survival under Hypoxia. Cell Rep 18: 1699–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Domenico I, Vaughn MB, Li L, Bagley D, Musci G, Ward DM, Kaplan J (2006) Ferroportin‐mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J 25: 5396–5404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, Ren X, An Y, Wu Y, Sun W et al (2019) DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med 131: 356–369 [DOI] [PubMed] [Google Scholar]
- Elia I, Broekaert D, Christen S, Boon R, Radaelli E, Orth MF, Verfaillie C, Grünewald TGP, Fendt S‐M (2017) Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat Commun 8: 15267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Bravo‐San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V et al (2015) Autophagy in malignant transformation and cancer progression. EMBO J 34: 856–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammella E, Lomoriello IS, Conte A, Freddi S, Alberghini A, Poli M, Sigismund S, Cairo G, Recalcati S (2020) Unconventional endocytosis and trafficking of Transferrin Receptor induced by iron. Mol Biol Cell mbcE20020129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin JM, Dowdle WE, DeJesus R, Wang Z, Bergman P, Kobylarz M, Lindeman A, Xavier RJ, McAllister G, Nyfeler B et al (2017) Autophagy‐independent lysosomal targeting regulated by ULK1/2‐FIP200 and ATG9. Cell Rep 20: 2341–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitson KS, Liénard BMR, McDonough MA, Clifton IJ, Butler D, Soares AS, Oldham NJ, McNeill LA, Schofield CJ (2007) Structural and mechanistic studies on the inhibition of the hypoxia‐inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J Biol Chem 282: 3293–3301 [DOI] [PubMed] [Google Scholar]
- Ibrahimi A, Vande Velde G, Reumers V, Toelen J, Thiry I, Vandeputte C, Vets S, Deroose C, Bormans G, Baekelandt V et al (2009) Highly efficient multicistronic lentiviral vectors with peptide 2A sequences. Hum Gene Ther 20: 845–860 [DOI] [PubMed] [Google Scholar]
- Jiang Z, Yu F, Li M (2018) Upregulation of BCL2 19 kD protein‐interacting protein 3 (BNIP3) is predictive of unfavorable prognosis in uveal melanoma. Med Sci Monit 24: 4711–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao L, Zhang H‐L, Li D‐D, Yang K‐L, Tang J, Li X, Ji J, Yu Y, Wu R‐Y, Ravichandran S et al (2018) Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy 14: 671–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kempen LCL, Redpath M, Elchebly M, Klein KO, Papadakis AI, Wilmott JS, Scolyer RA, Edqvist P‐H, Ponten F, Schadendorf D et al (2016) The protein phosphatase 2A regulatory subunit PR70 is a gonosomal melanoma tumor suppressor gene. Sci Transl Med 8: 369ra177 [DOI] [PubMed] [Google Scholar]
- Kimmelman AC, White E (2017) Autophagy and tumor metabolism. Cell Metab 25: 1037–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J (2007) Inhibition of hypoxia‐inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem 282: 4524–4532 [DOI] [PubMed] [Google Scholar]
- Kuphal S, Winklmeier A, Warnecke C, Bosserhoff A‐K (2010) Constitutive HIF‐1 activity in malignant melanoma. Eur J Cancer 46: 1159–1169 [DOI] [PubMed] [Google Scholar]
- Lee P, Chandel NS, Simon MC (2020) Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol 21: 268–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leo L, Bodemeyer V, De Zio D (2019) The complex role of autophagy in melanoma evolution: new perspectives from mouse models. Front Oncol 9: 1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorendeau D, Rinaldi G, Boon R, Spincemaille P, Metzger K, Jäger C, Christen S, Dong X, Kuenen S, Voordeckers K et al (2017) Dual loss of succinate dehydrogenase (SDH) and complex I activity is necessary to recapitulate the metabolic phenotype of SDH mutant tumors. Metab Eng 43: 187–197 [DOI] [PubMed] [Google Scholar]
- Maes H, Van Eygen S, Krysko DV, Vandenabeele P, Nys K, Rillaerts K, Garg AD, Verfaillie T, Agostinis P (2014a) BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis 5: e1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J et al (2014b) Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 26: 190–206 [DOI] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC (2014) Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509: 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐García MÁ, Riveiro‐Falkenbach E, Rodríguez‐Peralto JL, Nagore E, Martorell‐Calatayud A, Campos‐Rodríguez F, Farré R, Hernández Blasco L, Bañuls Roca J, Chiner Vives E et al (2017) A prospective multicenter cohort study of cutaneous melanoma: clinical staging and potential associations with HIF‐1α and VEGF expressions. Melanoma Res 27: 558–564 [DOI] [PubMed] [Google Scholar]
- McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O et al (2008) Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem 283: 22700–22708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejlvang J, Olsvik H, Svenning S, Bruun J‐A, Abudu YP, Larsen KB, Brech A, Hansen TE, Brenne H, Hansen T et al (2018) Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J Cell Biol 217: 3640–3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, Van Moer K, Kreis S, Guerin C, Buart S et al (2017) Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5‐dependent manner. Proc Natl Acad Sci USA 114: E9271–E9279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalke B, Willkommen D, Venkataramani V (2019) Iron redox speciation analysis using capillary electrophoresis coupled to inductively coupled plasma mass spectrometry (CE‐ICP‐MS). Front Chem 7: 136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CW, Hong SM, Kim E‐S, Kwon JH, Kim K‐T, Nam HG, Choi KY (2013) BNIP3 is degraded by ULK1‐dependent autophagy via MTORC1 and AMPK. Autophagy 9: 345–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato PE, Filigheddu N, Pedro JMB‐S, Kroemer G, Galluzzi L (2018) Mitochondrial metabolism and cancer. Cell Res 28: 265–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi G, Rossi M, Fendt S‐M (2018) Metabolic interactions in cancer: cellular metabolism at the interface between the microenvironment, the cancer cell phenotype and the epigenetic landscape. Wiley Interdiscip Rev Syst Biol Med 10: e1397 [DOI] [PubMed] [Google Scholar]
- De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O, Sonveaux P (2012) Lactate activates HIF‐1 in oxidative but not in Warburg‐phenotype human tumor cells. PLoS One 7: e46571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saita S, Shirane M, Nakayama KI (2013) Selective escape of proteins from the mitochondria during mitophagy. Nat. Commun. 4: 1410 [DOI] [PubMed] [Google Scholar]
- Santana‐Codina N, Mancias JD (2018) The role of NCOA4‐mediated ferritinophagy in health and disease. Pharmaceuticals 11: 114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2007) Hypoxia‐Inducible factor 1 (HIF‐1) pathway. Sci STKE 2007: cm8 [DOI] [PubMed] [Google Scholar]
- Semenza GL (2010) Defining the role of hypoxia‐inducible factor 1 in cancer biology and therapeutics. Oncogene 29: 625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni S, Padwad YS (2017) HIF‐1 in cancer therapy: two decade long story of a transcription factor. Acta Oncol 56: 503–515 [DOI] [PubMed] [Google Scholar]
- Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JMR, McMurtry MS, Michelakis ED (2013) Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 32: 1638–1650 [DOI] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X et al (2018) Multi‐stage differentiation defines melanoma subtypes with differential vulnerability to drug‐induced iron‐dependent oxidative stress. Cancer Cell 33: 890–904.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vara‐Perez M, Felipe‐Abrio B, Agostinis P (2019a) Mitophagy in cancer: a tale of adaptation. Cells 8: 493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vara‐Perez M, Maes H, Van Dingenen S, Agostinis P (2019b) BNIP3 contributes to the glutamine‐driven aggressive behavior of melanoma cells. Biol Chem 400: 187–193 [DOI] [PubMed] [Google Scholar]
- Vincow ES, Thomas RE, Merrihew GE, Shulman NJ, Bammler TK, MacDonald JW, MacCoss MJ, Pallanck LJ (2019) Autophagy accounts for approximately one‐third of mitochondrial protein turnover and is protein selective. Autophagy 15: 1592–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y‐T, Li X, Chen J, McConnell BK, Chen L, Li P‐L, Chen Y, Zhang Y (2019) Activation of TFEB ameliorates dedifferentiation of arterial smooth muscle cells and neointima formation in mice with high‐fat diet. Cell Death Dis 10: 676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zbytek B, Peacock DL, Seagroves TN, Slominski A (2013) Putative role of HIF transcriptional activity in melanocytes and melanoma biology. Dermatoendocrinol 5: 239–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
This study includes no data deposited in external repositories.