

Abstract
DNA synthesis during homologous recombination is highly mutagenic and prone to template switches. Two‐ended DNA double‐strand breaks (DSBs) are usually repaired by gene conversion with a short patch of DNA synthesis, thus limiting the mutation load to the vicinity of the DSB. Single‐ended DSBs are repaired by break‐induced replication (BIR), which involves extensive and mutagenic DNA synthesis spanning up to hundreds of kilobases. It remains unknown how mutagenic BIR is suppressed at two‐ended DSBs. Here, we demonstrate that BIR is suppressed at two‐ended DSBs by proteins coordinating the usage of two ends of a DSB: (i) ssDNA annealing proteins Rad52 and Rad59 that promote second end capture, (ii) D‐loop unwinding helicase Mph1, and (iii) Mre11‐Rad50‐Xrs2 complex that promotes synchronous resection of two ends of a DSB. Finally, BIR is also suppressed when Sir2 silences a normally heterochromatic repair template. All of these proteins are particularly important for limiting BIR when recombination occurs between short repetitive sequences, emphasizing the significance of these mechanisms for species carrying many repetitive elements such as humans.
Keywords: break‐induced replication, double‐strand break, DSB end resection, homologous recombination, ssDNA annealing
Subject Categories: DNA Replication, Repair & Recombination
Coordinated usage of both DNA break ends via annealing proteins Rad52/59, D‐loop‐unwinding Mph1 helicase and the MRX complex ensures their repair by gene conversion, rather than by BIR operating at single‐ended breaks.
Introduction
Two‐ended double‐strand breaks (DSBs) are repaired by either non‐homologous end‐joining (NHEJ) or homologous recombination (HR). In the most basic HR pathway by gene conversion, one of the DSB ends invades the homologous template, forming a displacement loop (D‐loop) that primes short‐patch new DNA synthesis. Repair is completed when the newly synthesized strand is unwound and anneals to the second end of the DSB leading to conservative inheritance of newly synthesized strands (Ira et al, 2006) (Fig 1A). This mechanism, called synthesis‐dependent strand annealing (SDSA), is used in mitotically growing cells. Alternatively, the second end of a DSB anneals to the extended D‐loop leading to the formation of a double Holliday junction (dHJ), an intermediate common in meiotic recombination and essential for crossover outcomes (Symington et al, 2014). In repair of one‐ended DSBs or when homology within the genome is present only for one end of the DSB, the single end invades the template and initiates extensive repair‐specific DNA synthesis that can reach even the end of the chromosome. This pathway, called break‐induced replication (BIR), proceeds via D‐loop migration and leads to conservative inheritance of the newly synthesized strands (Fig 1A) (Donnianni & Symington, 2013; Saini et al, 2013). Both SDSA and BIR lead to a significant increase of point mutations along the entire length of new DNA synthesis (McGill et al, 1989; Ponder et al, 2005; Hicks et al, 2010; Deem et al, 2011; Shee et al, 2012; Saini et al, 2013; Sakofsky et al, 2014); however, the mutation load is far greater in BIR because of the increased length of repair‐specific and low‐fidelity DNA synthesis. In addition, frequent template switches are common in BIR within the first 10 kb of the strand invasion site (Smith et al, 2007; Anand et al, 2014; Stafa et al, 2014). The increase of point mutations during DSB repair is attributed to the exposure of long single‐strand DNA (ssDNA). Exposed ssDNA is prone to mutagenesis, and unwinding of the newly synthesized strand from its template decreases mismatch repair efficiency (Saini et al, 2013; Sakofsky et al, 2014). The frequent template switches in BIR are likely due to the intrinsic instability of the D‐loop intermediate (Smith et al, 2007; Piazza et al, 2019). In diploids, the extensive use of BIR poses a threat to genome integrity by creating new mutations, template switches, and the loss of heterozygosity. In both diploids and haploids, BIR also generates nonreciprocal translocations by template switching between dispersed homologous repeats (Anand et al, 2014). In humans, BIR is less characterized; but similar to yeast, extensive repair‐specific DNA synthesis depends on POLD3 (yeast Pol32) and PIF1 (yeast Pif1), and conservative inheritance of newly synthesized DNA was observed during telomere recombination and during mitotic DNA synthesis (MiDAS) that likely proceeded by BIR (Lydeard et al, 2007; Costantino et al, 2013; Wilson et al, 2013; Bhowmick et al, 2016; Roumelioti et al, 2016; Macheret et al, 2020; Li et al, 2021). Moreover, BIR and the related microhomology‐mediated BIR (MMBIR) is likely the mechanisms underlying many forms of genome instability in mammalian cells (Lee et al, 2007; Hastings et al, 2009; Carvalho et al, 2013; Sakofsky et al, 2015; Li et al, 2020). At least some of the nonrecurrent structural variations that involve large copy number gains are accompanied by hypermutations, implicating BIR as the mechanism (Beck et al, 2019).
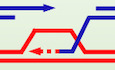
Figure 1. Role of Rad59 and DNA binding domain of Rad52 in suppressing BIR at two‐ended DSBs.
- Models of DSB repair by SDSA and BIR.
- Schematic of the H‐150 assay. Blue boxes depict homologous sequence on one end of the DSB and green boxes depict homology on the other end. A DSB is induced at modified MATa locus (Chr. III). Strand invasion occurs within the “Z” sequence (blue box) and after copying Yα‐inc (dashed line) and X sequence (150 bp), the X sequence is used to capture the second end during SDSA. When copying continues to the end of chromosome via BIR (dashed line), an acentric chromosome forms and an unrepaired chromosome segment remains, leading to cell death.
- Representative Southern blots showing DSB repair products in WT, rad59Δ, rad52‐R70A, and rad59Δ rad52‐R70A cells. DNA was digested with Bsp1286I and probed with a MAT‐distal sequence (yellow box in panel B). A detailed map of restriction cut sites and expected size for different repair products are illustrated in Appendix Fig S1.
- Percentage of BIR products among repair outcomes by 6 h are shown. (Mean ± SD; n = 3).
- Viability of indicated strains is shown (mean ± SD; n = 3).
- Graph shows repair efficiency among all cells by 6 h (left) and represents the sum of DSB repair by BIR and SDSA compared to the uncut parental band at 0 h (mean ± SD; n = 3). Graph shows repair by BIR and SDSA at 6 h after break induction in mutant cells compared to repair products in WT, which are set to 100% (right).
- Schematic of H‐150 no‐gap assay, where 150 bp of Yα homology on the second end of the break is immediately adjacent to the break site.
- Representative Southern blots showing DSB repair products in indicated mutants in H‐150 no‐gap assay.
- Percentage of BIR product among all repair products by 6 h are shown. (Mean ± SD; n = 3).
- Viability of indicated strains (mean ± SD; n = 3).
- Graphs showing repair efficiency compared to uncut parental band (left) (mean ± SD; n ≥ 3) or compared to the repair products in WT (right) at 6 h after DSB induction. See Fig 1F legend for details.
Data information: Welch’s unpaired t‐test was used to determine the P‐value in all panels.
Source data are available online for this figure.
How cells limit the use of BIR in repair of two‐ended breaks is not well understood. When homology on one of two ends of a DSB is short (46–150 bp), BIR contributes to or even outcompetes SDSA (Malkova et al, 2005; Mehta et al, 2017). Also, elimination of the D‐loop destabilizing enzyme Mph1, (Sun et al, 2008; Prakash et al, 2009; Stafa et al, 2014; Piazza et al, 2019), likely stabilizes D‐loops, resulting in increased BIR outcomes, at least when homology is short (Mehta et al, 2017). Consistently, BIR is suppressed when Mph1 is overexpressed (Luke‐Glaser & Luke, 2012; Stivison et al, 2020).
Here, we hypothesized that enzymes ensuring coordination of the usage of two DSB ends in repair may prevent BIR. Proteins important for annealing, D‐loop unwinding, initial resection, and maintaining two DSB ends together were examined in this study. Annealing of ssDNA in yeast is mediated by Rad52 and Rad59 (Mortensen et al, 1996; Sugiyama et al, 1998). Both purified enzymes show annealing activity, but only Rad52 can anneal complementary ssDNA in the presence of RPA (Petukhova et al, 1999; Wu et al, 2006). Rad59 interacts with Rad52 and stimulates the annealing activity of Rad52 in suboptimal conditions, likely by mitigating the negative impact of Rad51 binding to Rad52 on ssDNA annealing (Davis & Symington, 2001; Wu et al, 2008; Gallagher et al, 2020). In cells, Rad52 is required for single‐strand annealing, whereas Rad59 seems to be particularly important for annealing of shorter and not completely homologous sequences (Sugawara et al, 2000). Purified yeast or human Rad52 mediates second end capture to an extended joint molecule (McPherson et al, 2004; McIlwraith et al, 2005; Sugiyama et al, 2006; McIlwraith & West, 2008; Nimonkar et al, 2009). Analysis of recombination intermediates in meiosis also supports the later role of Rad52‐mediated annealing in both crossover and noncrossover pathways (Lao et al, 2008).
Here, we tested whether the elimination of the annealing activity, that is required for the second end capture to form double Holliday junctions or to complete SDSA, can unleash BIR during repair of two‐ended breaks. Besides the role of “annealases” in controlling BIR during repair of two‐ended DSBs, we also tested the possible function of Mph1 which disrupts the extended D‐loop in the SDSA pathway (Sun et al, 2008; Prakash et al, 2009), and the Mre11‐Rad50‐Xrs2 complex (MRX) responsible for initial end resection (reviewed in (Symington, 2016)) and maintaining two ends of a DSB together (Kaye et al, 2004; Lobachev et al, 2004). Finally, we examined the effect of the chromatin state of the donor template on the competition between BIR and SDSA. Multiple intra‐ and interchromosomal DNA recombination assays with a broad range of homology sizes (150 bp to over 100 kb) were used to test the function of these enzymes in the competition between SDSA and BIR.
Results
Rad59‐ and Rad52‐mediated ssDNA annealing suppresses BIR during repair of DSB
Initial steps of both SDSA and BIR involve strand invasion of one DSB end that results in formation of a displacement loop (D‐loop). Gene conversion via SDSA is completed when the D‐loop is extended, and the newly synthesized strand is displaced from its template and anneals to the second DSB end (Fig 1A). In BIR, the D‐loop migrates even to the end of the chromosome, creating initially a long ssDNA strand that is then converted into a double‐stranded product (Saini et al, 2013). Previous work showed that 46, 50 bp, or even 150 bp homology on the second end is too short for efficient second end capture and for completion of SDSA, resulting in high level of BIR (Deem et al, 2008; Mehta et al, 2017). In these ectopic recombination assays, an additional potential impediment for second end capture was the presence of ~700 bp non‐homologous gap between homologous sequences (Mehta et al, 2017). Here, we have used a number of new assays with much longer homology ranging from 150 bp to over 100 kb, and assays with or without the gap to study the role of proteins mediating annealing and other enzymes in the competition between SDSA and BIR for DSB repair. We hypothesized that decreased annealing activity would prevent the completion of gene conversion and allow the D‐loop to extend further to finish the repair by BIR.
To test the role of ssDNA annealing in the choice of repair pathway between BIR and SDSA at two‐ended DSBs, a separation‐of‐function mutant rad52‐R70A was introduced. This mutant is severely defective in DNA binding and ssDNA annealing, but capable of loading Rad51 properly to initiate strand exchange (Shi et al, 2009). Additionally, rad59Δ and rad59Δ rad52‐R70A double mutants were constructed. All these mutants were previously shown to reduce the SSA repair pathway which relies entirely on ssDNA annealing (Ivanov et al, 1996; Mortensen et al, 1996; Sugiyama et al, 1998; Petukhova et al, 1999; Davis & Symington, 2001; Wu et al, 2006; Gallagher et al, 2020).
We initially used a previously developed assay, a modified intrachromosomal mating‐type switching to study competition between SDSA and BIR (Mehta et al, 2017). A DSB is induced by HO endonuclease at the MAT a locus and repaired by recombination with an HMLα‐inc template, the resulting MATα‐inc product is not cleaved by HO endonuclease. The second template for mating‐type switching, HMR a, was deleted. The difference between native MAT switching and this recombination assay is that the homology size of the second DSB end is shortened from ~1,400 bp to 150 bp (H‐150 assay) (Fig 1B). The successful capture of the short second end leads to gene conversion of MAT a to MATα‐inc and produces viable colonies. In the event that the second end is not captured, DNA synthesis via migrating D‐loop (BIR) continues to the end of the chromosome (12 kb distance), including an apparent crossover: HML/MATα‐inc. The BIR product is inviable, as BIR forms an acentric chromosome fragment and leaves unrepaired the chromosome fragment carrying the centromere. SDSA and BIR products can be distinguished by different restriction fragment sizes on a Southern blot (Fig 1C). A detailed map of restriction cut sites and expected sizes for different repair products (BIR, SDSA, and crossovers) are illustrated in Appendix Fig S1. We note that crossovers accompanying gene conversion do not contribute to repair in this assay, because we do not observe the reciprocal crossover product. As previously shown in this H‐150 assay in wild‐type cells, SDSA dominated with ~82% contribution to DSB repair, with the remaining 18% being BIR products. Eliminating Rad59 reduced the contribution of the SDSA pathway to ~13%, while in rad52‐R70A or rad59Δ rad52R70A mutants, SDSA was further reduced to ~1% (Fig 1C and D). We note that while an important role of Rad59 and Rad52R70 in SDSA was shown previously, the contribution of BIR had not been tested (Bai et al, 1999). A switch to BIR is consistent with a marked decrease in the mutants’ viability, as BIR is a lethal event in this assay (Fig 1E). However, not all SDSA events were channeled to BIR in the annealing mutants, as overall product formation (BIR + SDSA) 6 h after break induction was decreased when compared to wild‐type cells (Fig 1F). This finding suggests that either BIR is decreased or delayed in annealing mutants, or that short 150 bp homology on the second end interferes with BIR. Both of these possibilities are tested below.
Besides the well‐established role of Rad52 in loading Rad51 and ssDNA annealing, Rad52 and its DNA‐binding domain also negatively regulate extensive resection, particularly in fission yeast (Yan et al, 2019). It is unlikely that the role of Rad52 in resection suppresses BIR, because the rad52‐R70A mutant shows only a minor increase in the rate of extensive resection, much less than that observed in the equivalent fission yeast DNA‐binding mutant rad52‐R45A (Appendix Fig S2A and Yan et al, 2019). Second, the deletion of DOT1 that causes a ~2‐fold increase in extensive resection rate (Chen et al, 2012) does not alter SDSA/BIR contribution to DSB repair (Appendix Fig S2B). Finally, we note that in rad52Δ or rad51Δ strains, no repair by BIR or SDSA was observed (Appendix Fig S2C).
In the mating‐type switching assay and its derivative H‐150 (Fig 1B), the Ya sequence is replaced by Yα‐inc. Because the Ya sequence is non‐homologous with the template, the invading strand must synthesize across about 700‐bp Yα‐inc gap to reach 150 bp homology with the second end. It is possible that this gap and/or the non‐homologous tail on the resected DSB end interferes with engaging the second end in recombination. To eliminate these constraints, we tested recombination between an HMLα‐inc and truncated MATα, where only 150 bp of Yα remains to the left of the DSB. In this assay, homology is present immediately at both DSB ends (H‐150 no‐gap assay, Fig 1G, Appendix Fig S1). In wild‐type cells, SDSA dominated with > 95% of total DSB repair, while in annealing‐defective rad59Δ and rad52‐R70A mutants, SDSA dropped to ~55 and 1%, respectively, while BIR increased (Fig 1H–K). The double mutant rad59Δ rad52‐R70A showed a similar phenotype as the rad52‐R70A single mutant. Similar to the H‐150 assay, the overall repair efficiency decreased in annealing mutants (Fig 1K). Together, these results confirm that annealing activity suppresses BIR at DSBs with short homology, either with or without a non‐homologous gap between homologous sequences. By comparing results obtained in H‐150 assays with or without the gap, we also conclude that the gap and/or the presence of a non‐homologous tail likely interfere with the second end capture in SDSA and therefore increase the usage of BIR, at least when homology on the second end is short (Appendix Fig S3).
BIR mildly decreases in annealing‐defective mutants
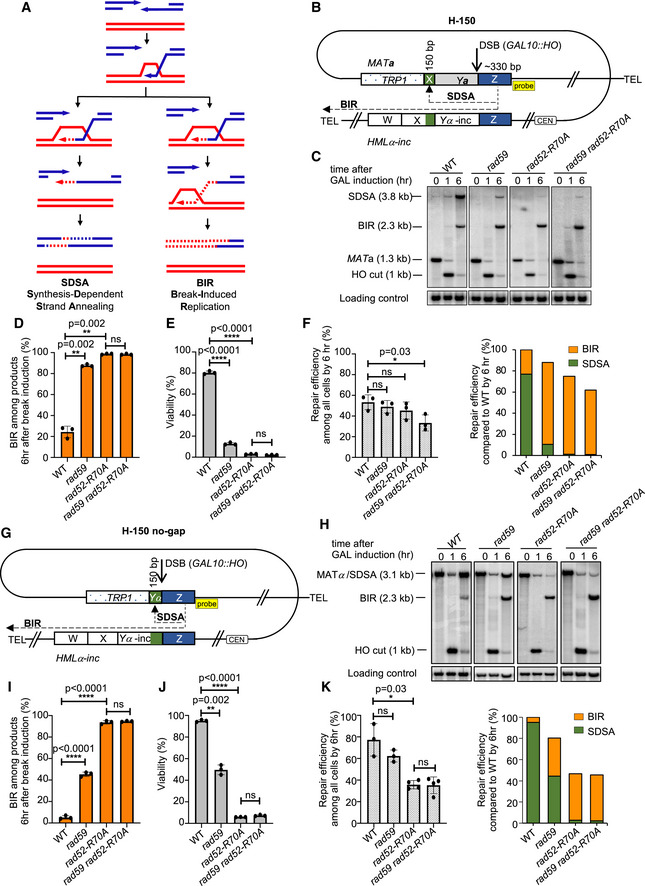
To test whether BIR itself is affected by any of the annealing mutations, we constructed a new system, called H‐0, where all homology to HMLα‐inc on the second end of the break was eliminated (Fig EV1A). As confirmed by Southern blot, BIR is the sole pathway of DSB repair and cells do not survive, as BIR is lethal in this system (Fig EV1B and C). In annealing‐defective rad52‐R70A mutant strain, we observed a decrease of BIR product formation 6 h after DSB induction to ~60% of the wild‐type levels (Fig EV1D). Again, the double mutant rad59Δ rad52‐R70A showed a similar phenotype as rad52‐R70A single mutant. These results suggest that BIR tested here is mildly deficient in annealing‐defective mutants.
Figure EV1. BIR efficiency is mildly decreased in annealing mutants.
- Schematic of the H‐0 BIR assay. DSB is induced at a modified MATa locus (Chr. III). Strand invasion occurs within the “Z” sequence (blue box) and DNA synthesis continues to the end of the chromosome.
- Representative Southern blots showing DSB repair products in WT, rad59Δ, rad52‐R70A, and rad59Δ rad52‐R70A cells. DNA was digested with Bsp1286I and probed with a MAT‐distal sequence (yellow box in panel A).
- Viability of indicated strains is shown (mean ± SD; n = 3).
- Repair efficiency compared to parental MATa at time 0 h (left) and repair efficiency of indicated mutants compared to WT by 6 h (right) (mean ± SD; n ≥ 3).
- Representative Southern blots showing BIR repair product in H‐0 system in WT and indicated mutant cells.
- Repair efficiency compared to parental MATa at time 0 h (left) and repair efficiency of indicated mutants compared to WT by 6 h (right) (mean ± SD; n ≥ 3).
- Model presenting the possible function of DNA binding/annealing domain of Rad52 in stabilizing the D‐loop. Rad52 stabilizes/extends D‐loop by three‐strand exchange (left) or Rad52 anneals a 3′ invading strand unwound from its template back to disrupted, but still RPA‐bound, D‐loop (right).
Data information: Welch’s unpaired t‐test was used to determine the P‐value in all panels. *P‐value 0.01 to 0.05, significant; **P‐value 0.001 to 0.01, very significant; ***P‐value 0.0001 to 0.001, extremely significant; ****P < 0.0001, extremely significant; P ≥ 0.05, not significant (ns).
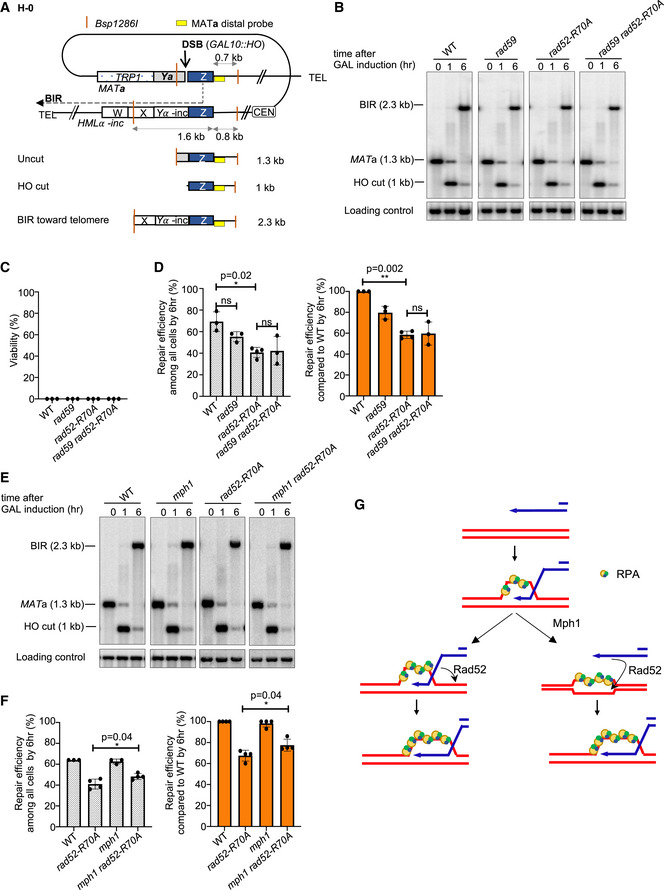
In the H‐0 assay, while there is no homology on the second end of the break, the homology on the invading end is also limited to ~300 bp. We therefore tested the role of annealing in the BIR assay where the invading end has no homology constraint. We employed an established BIR assay that involves recombination between a truncated and a complete chromosome III, where one DSB end has an extensive homology (~200 kb) with the template but the centromere‐distal end shares only 46 bp homology (Fig EV2A) (Malkova et al, 2005). In this disomic system, there was no loss of viability after HO induction in the mutant strains. Approximately 80% of the wild‐type cells use BIR for repair, during which the break end with extensive homology invades into the homologous chromosome and copies over 100 kb to the end of the chromosome. The remaining cells use the short homologous sequence at the second DSB end to complete the repair by SDSA. BIR and SDSA products can be followed by the presence of ADE1 markers and resistance to G418 (KANMX marker). Consistent with previous assays, rad52‐R70A or rad59Δ rad52‐R70A derivatives nearly eliminated SDSA among the products (< 1%) while BIR increased to about 90% (Fig EV2B). Notably, overall DSB repair by BIR is more efficient in this assay when compared to the H‐0 assay, as only 6–8% of the Rad52‐R70A mutant cells lacking strand annealing activity failed to repair the break, resulting in chromosome loss. Thus, with much longer homology available to the invading strand, the Rad52 DNA binding domain is nearly dispensable for the completion of BIR.
Figure EV2. Analysis of BIR efficiency in a disomic BIR system in annealing mutants.
- Schematic of the disomic BIR assay. DSB is induced at a modified MATa locus on a truncated chromosome III, with only 46 bp homology to the right of the DSB. Strand invasion occurs within MAT sequences within the full‐length chromosome III and replication continues until the end of the template chromosome. Chromosome ends are marked with ADE1, ADE3, KANMX, and HPHMX to distinguish different repair products or chromosome loss.
- Percentage of different repair products and chromosome loss. At least 500 colonies from YEP‐Galactose plates were scored per mutant.
One possible function of the DNA binding domain of Rad52 in BIR when homology is short could be related to the stability of the initial D‐loop. Annealases related to Rad52, such as RecT or λ beta, were shown to promote three‐strand exchange (Hall & Kolodner, 1994; Li et al, 1998), an activity that could extend the heteroduplex DNA. Alternatively, Rad52 could anneal a 3’ invading strand dissociated from its template back to the disrupted ‐ but still RPA‐bound ‐ D‐loop (Fig EV1G). Interestingly, deletion of Mph1, a D‐loop disrupting helicase, slightly increased BIR efficiency in the rad52‐R70A strain in the H‐0 assay, although not to WT levels (Fig EV1E and F). Thus, annealing activity may counteract Mph1 by stabilizing the D‐loop, at least when strand invasion is within short homologous sequences. A model presenting possible functions of Rad52’s DNA binding domain in BIR is summarized in Fig EV1G.
Rad59‐ and Rad52‐mediated annealing suppresses BIR at DSBs in regular MAT switching
The above BIR/SDSA competition assays had only 46–150 bp homology on the second DSB end. To test the role of annealing in a system with longer homology, we investigated regular MAT switching, between MAT a and HMLα‐inc. Here, the homology on the second end is ~1,400 bp (strain H‐1400) (Fig 2A). Southern blot analysis demonstrates that wild‐type cells are nearly 100% efficient in repair, and neither BIR nor crossover contributes to repair. However, annealing‐deficient mutants rad59Δ and particularly rad52‐R70A increased BIR to ~30% and 60–70%, respectively (Fig 2B and C). As expected, viability is significantly reduced in annealing mutants due to a switch to lethal repair by BIR and inability to complete SDSA (Fig 2D). These results also demonstrate the importance of the optimal annealing mediated by Rad59 for the very fundamental event in yeast’s life cycle, mating‐type switching.
Figure 2. Rad52 and Rad59 suppress BIR during MAT switching.
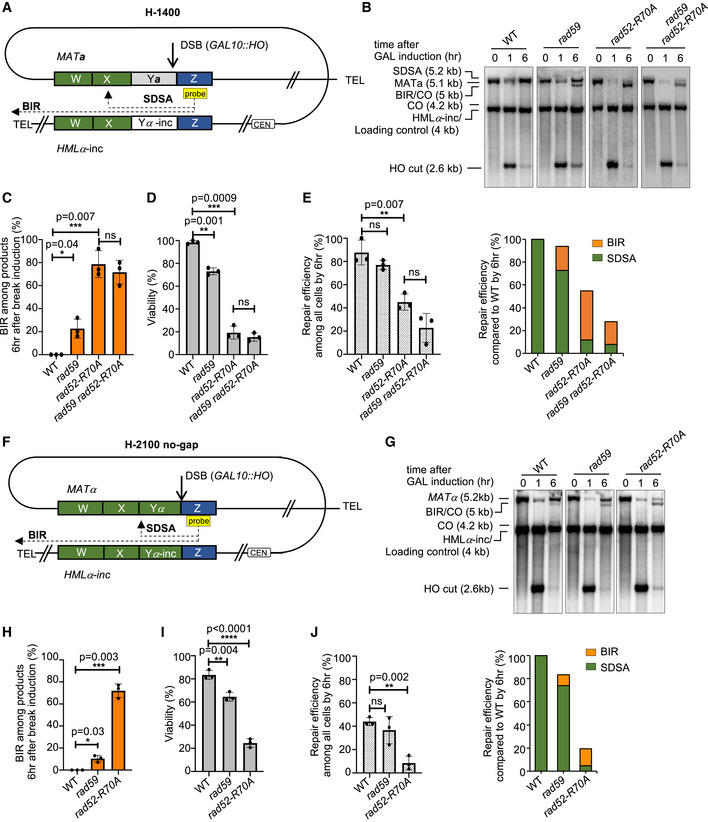
- Schematic of the H‐1400 assay. A DSB is induced at MATa and repaired by recombination with HMLα‐inc. There is ~ 1,400 bp homology between MATa and HMLα‐inc sequences on the second end (green box).
- Representative Southern blots showing DSB repair products in WT, rad59Δ, rad52‐R70A, and rad59Δ rad52‐R70A cells. DNA was digested with XhoI and EcoRI and probed with a Z sequence (yellow box). A detailed map of restriction cut sites and expected size for different repair products are illustrated in Appendix Fig S1.
- Percentage of BIR among all repair products by 6 h (mean ± SD; n = 3).
- Viability of indicated strains (mean ± SD; n = 3).
- Graphs show analysis of repair efficiency compared to uncut parental band (left) (mean ± SD; n = 3) and repair efficiency compared to WT (right) at 6 h after DSB induction (right). See Fig 1F for details.
- Schematic of the H‐2100 no‐gap assay. A DSB is induced at MATα and repaired by recombination with HMLα‐inc. There is ~ 2,100 bp homology between MATα and HMLα‐inc sequences on the second end (green box).
- Representative Southern blots showing DSB repair products in WT, rad59Δ, and rad52‐R70A. DNA was digested with XhoI and EcoRI and probed with a Z sequence (yellow box). A detailed map of restriction cut site and expected size for different repair products are illustrated in Appendix Fig S1.
- Percentage of BIR product among repair products by 6 h are shown (mean ± SD; n = 3).
- Viability of indicated strains (mean ± SD; n = 3).
- Graphs show analysis of repair efficiency compared to uncut parental band (left) (mean ± SD; n = 3) and repair efficiency compared to WT (right) at 6 h after DSB induction (right). See Fig 1F for details.
Data information: Welch’s unpaired t‐test was used to determine the P‐value in all panels. *P‐value 0.01 to 0.05, significant; **P‐value 0.001 to 0.01, very significant; ***P‐value 0.0001 to 0.001, extremely significant; ****P < 0.0001, extremely significant; P ≥ 0.05, not significant (ns).
To eliminate the possible constraint of the gap and the non‐homologous Ya tail, we tested recombination between MATα and HMLα‐inc where there is no Y sequence heterology (Fig 2F). In this case, the homology on the second end is further increased to 2,100 bp (H‐2100 no‐gap) and is immediately next to the break. As these are MATα cells expressing Matα2, the cis‐acting recombination enhancer (RE) that facilitates repair in MAT a cells is inactive (Wu & Haber, 1996); therefore, the repair efficiency by 6 h is reduced compared to the H‐1400 assay. We note that in all other assays tested here with HMLα‐inc being the recombination template, RE is active. Interestingly, weak bands corresponding to both crossover products (< 5%) in wild‐type cells were evident; such crossover products were not observed in recombination between MAT a and HMLα‐inc. Thus, the non‐homologous Y sequence during MAT switching prevents lethal intrachromosomal crossovers in ~5% of the cells. Importantly in this assay, the BIR contribution increases in both rad59Δ and rad52‐R70A, and viability decreases accordingly (Fig 2G–I). We noted above that BIR product has the same size as the larger of two crossover products. Considering that both crossover products form at the same time and the intensity of the shorter crossover product band is very weak or absent in annealing mutants, we conclude that BIR and not crossover is responsible for the increased intensity of this band in rad59Δ and rad52‐R70A cells. In this assay, extended homology is present immediately on both sides of the break; thus, both ends could invade template early and prime BIR, one toward the telomere and one toward the centromere. However, BIR toward the centromere was not observed in the annealing mutants in this or any of the assays tested here with HMLα‐inc serving as a template (Figs 2B and G, and EV3A). In all these assays, the Z‐end invades and primes DNA synthesis toward the telomere. To test whether the centromere itself imposes a long‐range negative impact on BIR, we constructed an additional HR‐0 recombination assay (Fig EV3B). In this assay, a DSB is induced at the MATα locus. The Z‐end of MATα shares 230 bp homology with the HMR a‐inc template, while the homology on the other end was eliminated entirely and the HML donor was deleted. Upon strand invasion, the Z‐end primes DNA synthesis toward the centromere. Interestingly, only up to 3% of cells that induced the break synthesized a few kb of dsDNA in the direction of the centromere as measured 6 or even 10 h after DSB induction. Thus, BIR‐specific DNA synthesis is more efficient in the direction toward the telomere than the centromere. However, nearly complete lack of DNA synthesis in the direction of centromere is not related to the centromere itself, because the break at the MAT locus separates the chromosome fragment containing the invading Z‐end and the template from the one carrying the centromere (Fig EV3B). Thus, some yet to be characterized chromatin or topological features may inhibit DNA synthesis at specific loci and orientation or, alternatively, proximity of telomeres have positive effect on BIR‐specific DNA synthesis.
Figure EV3. Analysis of BIR toward the centromere.
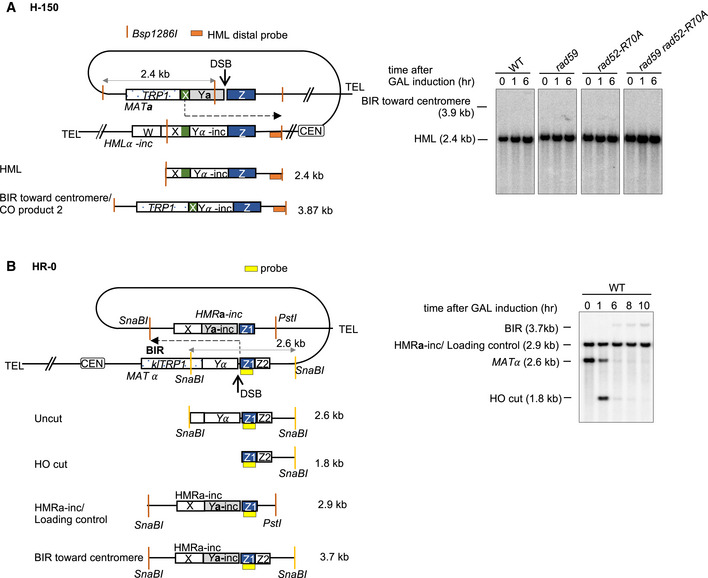
- Schematic of H‐150 assay with position of HML distal probe (left) and representative Southern blot (right) are shown. No BIR product was detected in the direction of the centromere.
- Schematic of HR‐0 assay with sites of restriction enzymes, position of probe, and size of different bands (left) and representative Southern blot (right) are shown. Weak BIR product is visible.
We also note that in the H‐2100 or H‐1400 assays, only 15–30% of rad52‐R70A mutant cells repair the break by BIR, respectively (Fig 2E and J). This level of repair by BIR is far below the one observed in cells that lack any homology on the second end of a DSB (H‐0, 60%). Thus, longer homology on the second end, particularly when it is located right next to the DSB end (H‐2100), decreases BIR efficiency in annealing mutants. It is possible that with increased homology both DSB ends might simultaneously invade the template or Y‐end invades first, which preclude BIR.
Increased homology partially alleviates the need for Rad52‐ and Rad59‐mediated ssDNA annealing in recombination and in suppressing BIR
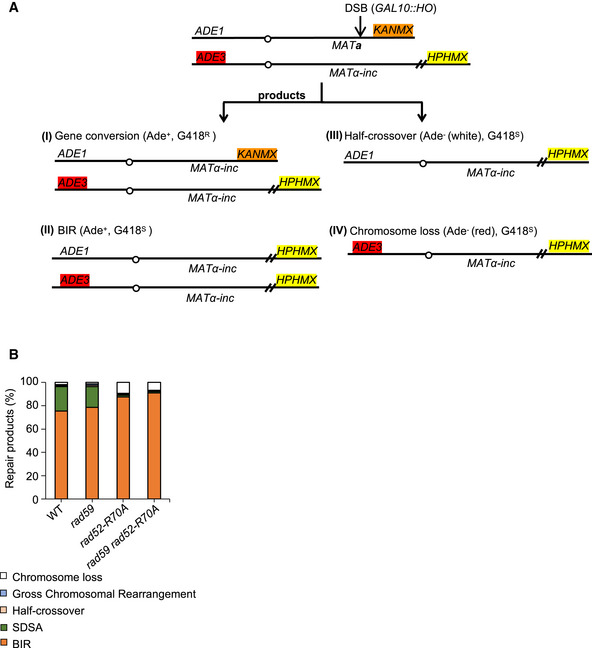
Using intrachromosomal MAT switching assays, we found that second end capture through ssDNA annealing stimulated by Rad59 and Rad52 is important to suppress the BIR pathway during the repair of two‐ended DSBs. All of these assays have homology ranging from 150 bp to ~2 kb. Next, we tested whether Rad52 annealing activity restricts BIR in allelic recombination between two chromosomes III, where homology on each end is over 100 kb. To follow different products of DSB repair or chromosome loss in the allelic system, chromosome ends were marked with TRP1, LEU2, and KANMX (Fig 3A). LEU2 and KANMX markers were inserted at subtelomeric regions of the right ends of the two chromosomes III. BIR products are viable and can be genetically distinguished from gene conversion with or without crossover and from chromosome loss (Fig 3A). In wild‐type cells, fewer than 1% of cells repair the DSB by BIR. However, BIR increases ~6‐ to ~18‐fold in rad59Δ, rad52‐R70A, and rad59Δ rad52‐R70A mutant cells (Fig 3B, Appendix Table S1) while viability remains the same as in wild type (100%).
Figure 3. Longer homology mitigates the loss of Rad52 DNA binding domain and Rad59.
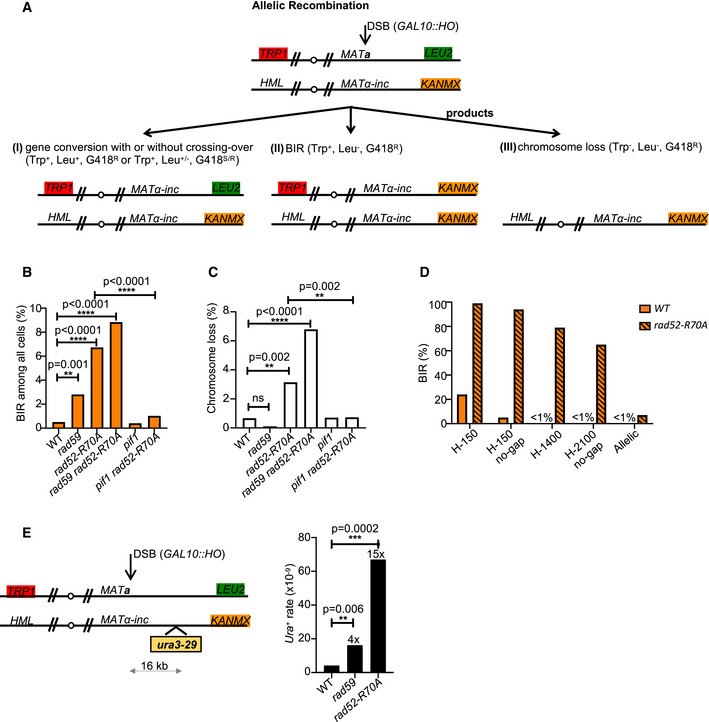
- Schematic of allelic recombination between two chromosomes III. A DSB is induced at MATa and repaired by recombination with the homologous chromosome carrying MATα‐inc. Chromosome ends are marked with TRP1, LEU2, and KANMX to distinguish different repair products or chromosome loss. Trp +, Leu −, G418R colonies are scored as BIR products. Reciprocal sectored colonies (Trp +, Leu −/+ G418R/S) represent gene conversion with crossover.
- Percentage of BIR product among all cells are shown.
- Percentage of chromosome loss of indicated mutants.
- Schematic of mutation analysis during allelic recombination (left). Mutation reporter cassette is inserted within the repair template 16 kb away from strand invasion site. Mutation rate (right) of WT, rad59Δ, rad52‐R70A was calculated as previously described (Saini et al, 2013).
Data information: Chi‐square test is used to determine the P‐value in Fig 3B and C, number of colonies tested per mutant is indicated in Appendix Table S1. Mutation rates in Fig 3E are reported as the median value, and statistical comparisons between median mutation rates were performed using the Mann–Whitney U test. *P‐value 0.01 to 0.05, significant; **P‐value 0.001 to 0.01, very significant; ***P‐value 0.0001 to 0.001, extremely significant; ****P < 0.0001, extremely significant; P ≥ 0.05, not significant (ns).
The helicase Pif1 is important for BIR as it facilitates D‐loop migration (Wilson et al, 2013). As expected, elimination of Pif1 significantly reduced BIR in rad52‐R70A mutant cells during allelic recombination (Fig 3B). Compared to MAT switching assays, we found that repair in the allelic system was far more efficient in all annealing mutants with only up to 7% chromosome loss (Fig 3C). With all assays taken together, it is clear that increased homology reduces the need for Rad52‐mediated annealing for basic gene conversion (Fig 3D). It is possible that with longer homologous sequences, spontaneous Rad52‐independent annealing is sufficient to complete DSB repair (Ozenberger & Roeder, 1991).
The increased BIR in annealing mutants is accompanied by an increase of mutations
BIR is an extremely mutagenic process owing to its specific mechanism limiting mismatch repair (Deem et al, 2011; Saini et al, 2013). To test whether increased BIR contribution to DSB repair between fully homologous chromosomes in annealing mutants is mutagenic, we inserted the ura3‐29 reporter gene 16 kb away from the break on the template chromosome and tested the level of mutations. We observed a 4‐ and 15‐fold increase of mutation rate in rad59Δ and rad52‐R70A mutants, respectively, when compared to WT cells (Fig 3E). This result is consistent with the fact that the proportion of BIR among repair products increases in annealing mutants.
Role of Mph1 helicase in suppressing BIR at two‐ended breaks
Mph1 is capable of unwinding D‐loops formed during recombination and thus stimulates the SDSA pathway in budding and fission yeast (Sun et al, 2008; Prakash et al, 2009). Mph1 is also involved in D‐loop unwinding during template switches in BIR (Stafa et al, 2014). Here, we constructed mutants defective in both D‐loop unwinding and annealing, and tested BIR and SDSA contributions to the repair of two‐ended breaks.
Previously it was demonstrated that elimination of Mph1 increases BIR in the repair of two‐ended breaks when homology on the second end is short (Luke‐Glaser & Luke, 2012; Mehta et al, 2017); however, this increase was much smaller than in annealing mutants. Here, we used recombination assays with longer homology to test the role of Mph1 in competition between SDSA and BIR. In regular mating‐type switching where the second end carries 1.4 kb homology (H‐1400 assay), crossovers are not visible among the products of MAT switching in wild‐type cells (Fig 2B), while in mph1∆, two weak crossover bands are observed. This result further supports the role of Mph1 in suppressing crossover outcomes (Fig 4A) (Prakash et al, 2009). The longer crossover band that also corresponds to the BIR product has a slightly stronger intensity than the shorter crossover band, suggesting that there is a very mild stimulation of BIR in mph1Δ mutant cells. Accordingly, the viability of the mph1∆ cells is comparable to the wild‐type cells (Fig 4B and C). However, the elimination of Mph1 in annealing mutants, rad59Δ or rad52‐R70A further increased the BIR contribution to the repair, or nearly entirely switched repair to BIR (Fig 4A–D). In allelic recombination, the elimination of Mph1 by itself did not increase BIR events at all. However, loss of Mph1 in annealing mutants increased the BIR level to ~12% in triple mph1∆ rad59∆ rad52‐R70A mutant cells (Fig 4E, Appendix Table S1). We conclude that with extensive homology, Mph1 by itself does not change BIR frequency; however, it suppresses BIR when ssDNA annealing activity is compromised.
Figure 4. Role of Mph1 in suppressing BIR.
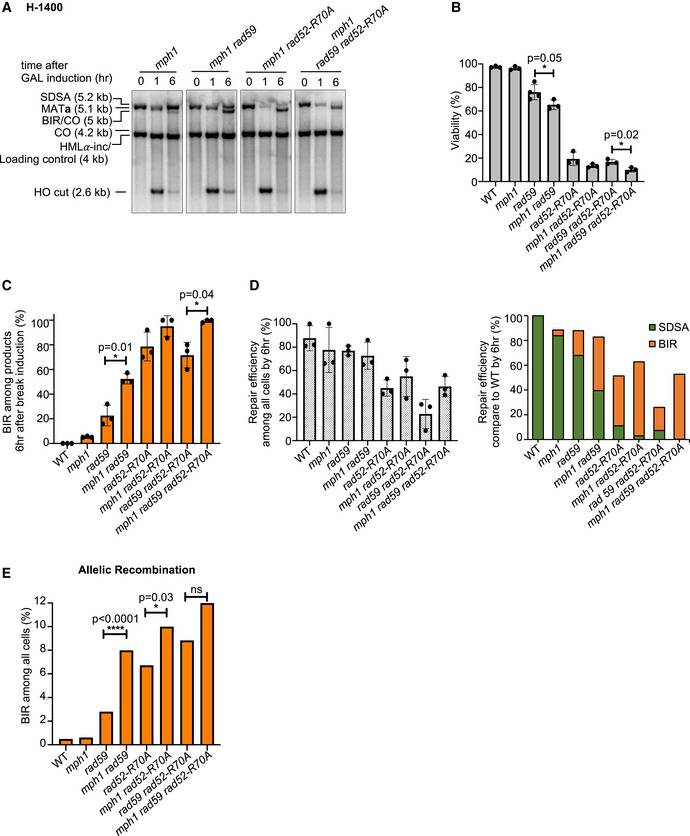
- Representative Southern blots showing DSB repair products of indicated mutants in regular mating‐type switching (H‐1400 assay).
- Viability of indicated mutants is shown. (Mean ± SD; n = 3).
- Percentage of BIR product among all repair products by 6 h are shown. (Mean ± SD; n = 3).
- Graphs show analysis of repair efficiency compared to uncut parental band (left) (mean ± SD; n = 3) and repair efficiency compared to WT (right) at 6 h after DSB induction (right). See Fig 1F for details.
- Percentage of BIR among all repair products in allelic recombination.
Data information: Welch’s unpaired t‐test was used to determine the P‐value in (B, C, D). Chi‐square test is used to determine the P‐value in Fig 4E. Number of colonies tested per mutant is indicated in Appendix Table S1. *P‐value 0.01 to 0.05, significant; **P‐value 0.001 to 0.01, very significant; ***P‐value 0.0001 to 0.001, extremely significant; ****P < 0.0001, extremely significant; P ≥ 0.05, not significant (ns).
Sir2 suppresses BIR with a heterochromatic donor when homology is short
The HML locus serves as a template for recombination in MAT switching in most of the assays described here (H‐150, H‐1400, H‐2100). HML is heterochromatic and silenced by the Sir2 histone deacetylase (Weiss & Simpson, 1998). Highly positioned nucleosomes could potentially affect the D‐loop migration or D‐loop unwinding and therefore impact BIR and SDSA choice. To determine whether the heterochromatic state of the recombination template affects the competition between BIR and SDSA, we constructed sir2∆ derivatives of the H‐150, H‐150 no‐gap, and H‐1400 assays, in which the HML template is unsilenced. The presence of the HMLα‐inc allele prevents the cleavage of HML when it is unsilenced. In the H‐150 assay, the contribution of BIR increased from ~20% up to ~55%, and the viability of sir2Δ decreased accordingly, owing to the lethality of BIR events (Fig 5A). Similarly, BIR increased by twofold in the H‐150 no‐gap system (Fig 5B). We note that sir2∆ cells in H‐150 assay carrying HMLα‐inc and MAT a express both mating‐type genes. Previous studies have shown that cells expressing both MAT a1 and HMLα2 genes exhibit some mating‐type specific effects on DNA repair (Valencia‐Burton et al, 2006). However, the effect of deleting SIR2 on the competition between SDSA and BIR is not related to mating‐type per se, as the increase of BIR was also observed in sir2Δ mutant cells in the system that expresses only α genes (H‐150 no‐gap, Fig 5A and B). In regular mating‐type switching (H‐1400 system), we also observe a minimal BIR increase in the sir2Δ mutant (~2%) and no change in viability (Fig 5C). We conclude that closed chromatin structure within the template has a role in suppressing BIR at DSBs, but mostly when homology at the second end is short.
Figure 5. Role of Sir2 in suppressing BIR during repair of two‐ended breaks.
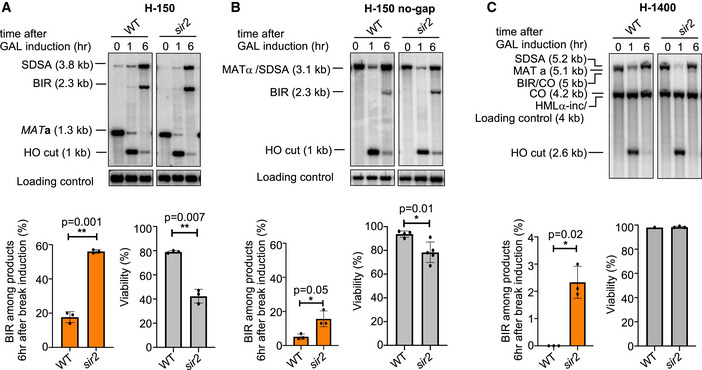
-
A–CRepresentative Southern blots showing DSB repair products (top), percentage of BIR among repair products (bottom left), and viability (bottom right) of WT and sir2∆ in the H‐150 assay (A), H‐150 no‐gap assay (B), or native mating‐type switching H‐1400 assay (C). (Mean ± SD; n = 3). Welch’s unpaired t‐test was used to determine the P‐value in panels A‐C.
Data information: *P‐value 0.01 to 0.05, significant; **P‐value 0.001 to 0.01, very significant; ***P‐value 0.0001 to 0.001, extremely significant; ****P < 0.0001, extremely significant; P ≥ 0.05, not significant (ns).
In these three assays, the HML template is within silent chromatin and SIR2 deletion leads to unsilencing of the HML template. Next, we used an established assay in which the template is within the unsilenced region of the genome and the deletion of SIR2 should have no impact on the chromatin state of the template. An HO‐induced DSB at the URA3 gene on chromosome V is repaired with a partial URA3 region inserted on the opposite arm of the chromosome (Anand et al, 2014). The homology on each side of the break (“R” and “A” end) is 300 bp (Fig EV4A). In wild‐type cells, only 0.8% of the cells repair the break by BIR (Anand et al, 2014). While we observed an over 30‐fold increase of BIR in rad52‐R70A annealing mutant cells, we did not see any change in BIR frequency in sir2Δ cells in this system, suggesting that the role of Sir2 in BIR regulation is related to the chromatin state of the template (Fig EV4B). A closed chromatin structure within the template could promote SDSA by slowing down the D‐loop migration and/or making the D‐loop more prone to unwinding. We favor the second possibility as the kinetics of BIR in sir2Δ cells, tested in H‐0 system, are only modestly increased when compared to wild‐type cells (Fig EV4C).
Figure EV4. Analysis of Sir2 role in repair pathway choice when the template is within unsilenced region.
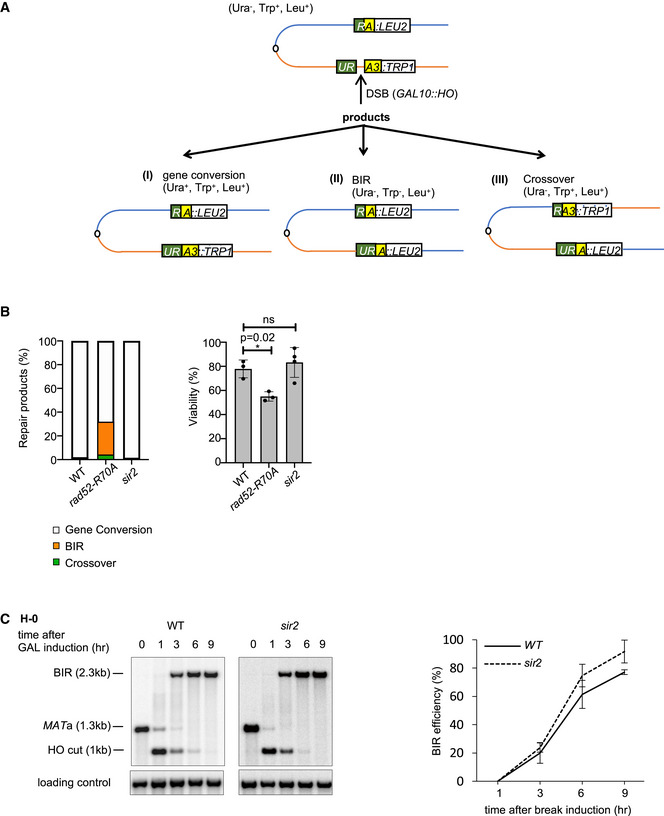
- Schematic of recombination assay on chromosome V, where a break is induced at URA3 and is repaired with a partial URA3 region (“RA”) fragment inserted on the other arm of the chromosome V.
- Percentage of different repair products (left) and viability (right) of WT, rad52‐R70A, and sir2Δ cells (mean ± SD; n = 3).
- Representative of Southern blots (left) and graph showing kinetics of BIR in H‐0 assay (right) of WT and sir2Δ cells (mean ± SD; n = 3).
Data information: Welch’s unpaired t‐test was used to determine the P‐value in all panels.
The MRX complex suppresses BIR at two‐ended DSBs
The MRX complex plays an important role in the initial DSB end resection (reviewed in (Symington, 2016)) and in DSB ends tethering (Kaye et al, 2004; Lobachev et al, 2004; Jain et al, 2016a). To test the role of the MRX complex in regulation of BIR during the repair of two‐ended DSBs, we used the H‐150 and H‐150 no‐gap assays. In mre11Δ mutant cells, we observed about 2‐ to 3‐fold increase of BIR in both assays (Fig 6A and B). Even when homology on the second end of the break is extensive in the allelic recombination assay, BIR still increased modestly in mre11Δ/mre11Δ (Fig EV5A). Colonies that showed a marker pattern typical for BIR products (Leu−, Trp+, and G418R) were further evaluated by CHEF and Southern blot to make sure that these are products of expected BIR size. 19 of 21 colonies tested by CHEF carried the expected BIR product size (Fig EV5B) while the remaining two colonies corresponded to chromosomal rearrangements. We conclude that MRX suppresses BIR events during repair of two‐ended DSBs.
Figure 6. Role of the MRX complex in suppressing BIR during repair of two‐ended breaks.
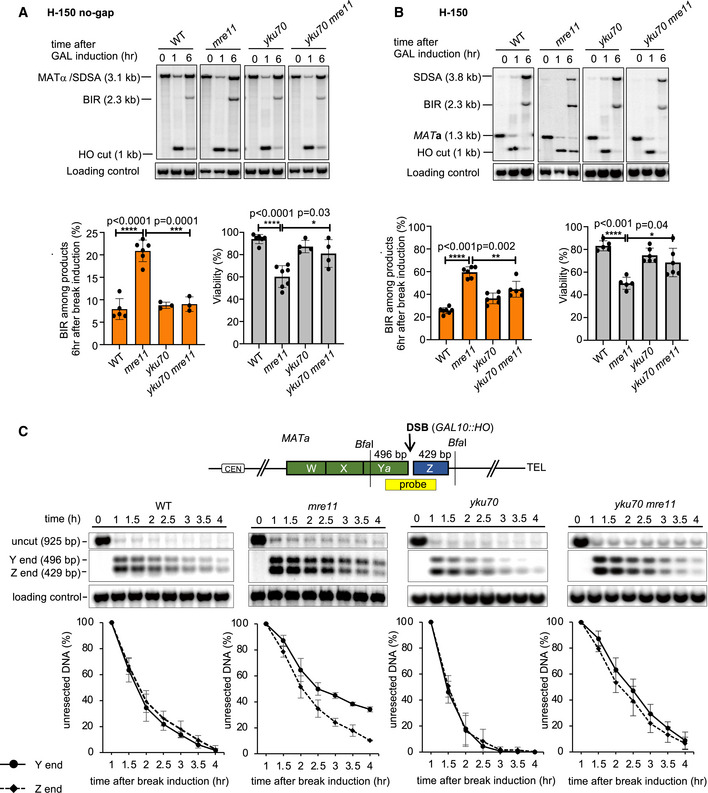
-
A, BRepresentative Southern blots showing DSB repair products (top), percentage of BIR among repair products (bottom left), and viability (bottom right) of indicated mutants in the H‐150 no‐gap assay (A) or H‐150 assay (B). (Mean ± SD; n ≥ 3). Welch’s unpaired t‐test was used to determine the P‐value in panels A and B.
-
CSchematic of resection assay with sites of restriction enzyme BfaI cleavage and location of probe (top), representative of Southern blots, and graph showing kinetics of resection two break end (bottom) (mean ± SD; n = 3) in WT, mre11Δ, yku70Δ, and yku70Δ mre11Δ.
Data information: *P‐value 0.01 to 0.05, significant; **P‐value 0.001 to 0.01, very significant; ***P‐value 0.0001 to 0.001, extremely significant; ****P < 0.0001, extremely significant; P ≥ 0.05, not significant (ns).
Source data are available online for this figure.
Figure EV5. MRX complex suppresses BIR in allelic recombination.
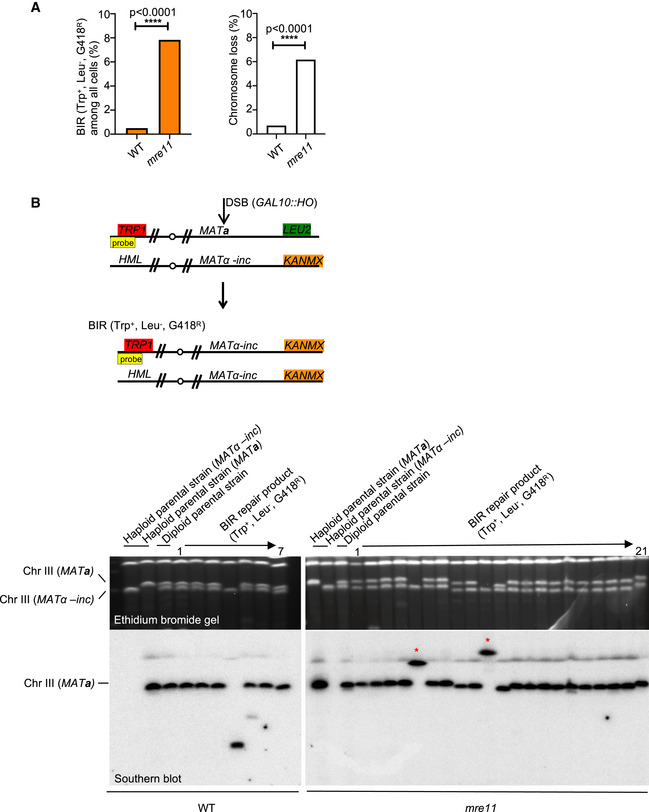
- Percentage of BIR and chromosome loss in WT and mre11∆ cells in allelic recombination. Chi‐square test is used to determine the P‐value, number of colonies tested per mutant is indicated in Appendix Table S1.
- Schematic of allelic assay with a TRP1 probe (yellow box) used for Southern blot analysis. Ethidium bromide gel (top) and Southern blot with TRP1 probe (bottom) showing chromosome III of haploid parental strains, diploid, and BIR products of WT (left) and mre11∆ cells (right). “*” depicts gross chromosomal arrangement event.
Data information: *P‐value 0.01 to 0.05, significant; **P‐value 0.001 to 0.01, very significant; ***P‐value 0.0001 to 0.001, extremely significant; ****P < 0.0001, extremely significant; P ≥ 0.05, not significant (ns).
Mre11 promotes synchronous resection of two DSB ends
The MRX complex could coordinate both DSB ends in repair and suppress BIR by tethering two DSB ends or by promoting the synchronous resection of two DSB ends. It was previously suggested that MRX plays a role in synchronous resection of DSB ends (Westmoreland et al, 2009; Westmoreland & Resnick, 2013). Here, we tested whether both HO‐induced DSB ends at the MAT locus were synchronously resected in wild‐type and in mre11Δ mutant cells. The initial resection was monitored every 30 min up to 4 h after DSB induction using Southern blots and a probe specific for the MAT locus (Fig 6C). We note that in a population of cells, the asynchrony of resection can be observed only if the average resection kinetics of the centromere proximal DSB end (Y‐end) and telomere proximal DSB end (Z‐end) are different. Since resection cannot be monitored in individual cells using this method, stochastic asynchrony at two break ends could not be detected. As shown in Fig 6C, both ends of the break are resected with equal kinetics in wild‐type cells, while in mre11Δ cells the resection of the Y‐end is slower. In ~15–25% of mre11Δ cells, where the Z‐end remained unresected by 3–4 h, we cannot test whether the Y‐end was resected. Therefore, while Z‐end is resected faster overall, we could not exclude the possibility that the Y‐end is resected faster in some cells. Asynchrony of resection observed in mre11Δ cells may explain the increase of BIR observed at two‐ended DSBs.
The resection defect of MRX mutants can be partially suppressed by the elimination of the Ku complex (Lee et al, 1998; Mimitou & Symington, 2010; Shim et al, 2010). Consistent with previous publications, the deletion of the YKU70 increased resection kinetics, and this increase was observed on both ends of the break (Fig 6C). Also, yku70Δ partially suppressed the defect of resection in mre11Δ cells on the Y‐end, but surprisingly it had nearly no effect on the Z‐end where resection remained as slow as in mre11Δ single mutant cells (Fig 6C). This result suggests that the Ku complex by itself or in combination with other proteins forms a stronger barrier for MRX‐independent resection on the Y‐end. In mre11Δ cells in both the H‐150 and H‐150 no‐gap assays, the deletion of YKU70 lead to a significant decrease of BIR. This result suggests that the synchronous resection of two DSB ends is important to reduce BIR events. Altogether, the role of MRX in suppressing BIR at two‐ended breaks is related to synchronous resection rather than tethering of two ends of the break. However, it is possible that these two functions may be related to each other. We note that expression of MRE11‐NLS in xrs2Δ mutant strain restores ends resection and DSB repair by SDSA but not ends tethering. This result provides further support for the role of resection rather than end tethering in proper DSB repair (Oh et al, 2018).
Discussion
DNA breaks can be repaired by many different pathways of recombination, some of which are mutagenic and normally suppressed. BIR is one of the pathways that drives genome instability as it results in mutations, nonreciprocal translocations, and loss of heterozygosity (reviewed by (Sakofsky & Malkova, 2017)). Here, we aimed to understand the mechanism that limits the use of BIR in the repair of two‐ended breaks. We identified several independent mechanisms that impair BIR, as shown in Fig 7. Two‐ended breaks are usually repaired by the SDSA mechanism in growing cells, resulting in a short‐patch DNA synthesis by polymerase δ (Ira et al, 2006; Maloisel et al, 2008; Guo et al, 2017). An essential step in the repair is to engage both ends of a DSB, thus eliminating the possibility that the invading end primes excessive DNA synthesis up to the end of the chromosome via BIR. We hypothesized that by eliminating activities that coordinate the usage of two ends of a DSB in SDSA, the contribution of BIR would increase. We found that the loss of ssDNA annealing activity in rad52‐R70A or rad59Δ increases BIR at two‐ended breaks. We propose that compromised ssDNA annealing prevents the second end from annealing to the extended and displaced 3′ strand in the SDSA mechanism, or from capturing the extended D‐loop in a double Holliday junction mechanism. In these scenarios, BIR synthesis via a migrating D‐loop can reach the end of the chromosome. However, decreased annealing has a milder effect on overall repair by gene conversion and BIR when homology at the second end is extensive. This result leads to an intriguing question: How are two‐ended breaks repaired in mutants severely deficient in ssDNA annealing? It is possible that spontaneous annealing is sufficient for gene conversion when homology is long, as proposed previously in SSA (Ozenberger & Roeder, 1991). Alternatively, with extended homology on both sides of the DSB, both ends could invade the template and be resolved as a gene conversion, thus reducing the chance that the D‐loop migrates to the end of the chromosome. Finally, we cannot exclude the possibility that there is an additional protein capable of weak ssDNA annealing during recombination, such as Mcm10 (Mayle et al, 2019). Notably in fission yeast, Rad52 annealing activity seems to be mostly dispensable for DSB repair, as rad52∆ cells are resistant to DNA damage so long as cells are capable of loading Rad51 in a Rad52‐independent manner (Osman et al, 2005).
Figure 7. Model presenting multiple mechanisms restraining Break‐Induced Replication at two‐ended DNA double‐strand breaks.
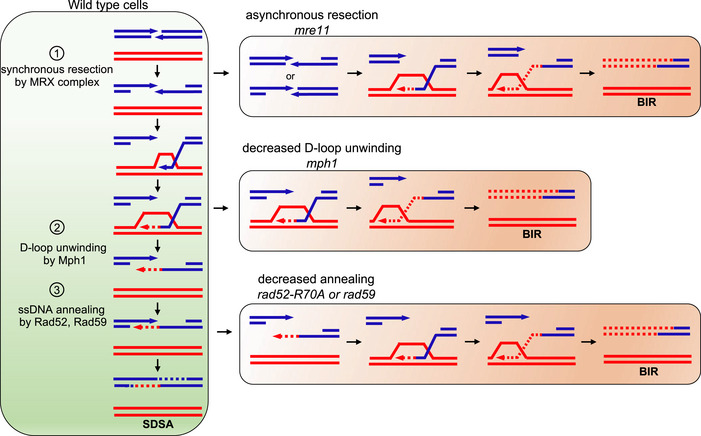
Several proteins coordinating the usage of two ends of a DSB suppress BIR: (1) Mre11‐Rad50‐Xrs2 complex (MRX) that promotes synchronous resection of two ends of a DSB, (2) D‐loop unwinding helicase Mph1 that stimulates SDSA, and (3) ssDNA annealing proteins Rad52 and Rad59 that promote second end capture. All of these proteins are particularly important to reduce BIR when recombination occurs between short repetitive sequences.
DNA helicases also coordinate the use of two ends of a DSB. Mph1 unwinds the extended 3’ strand that subsequently anneals with the second DSB end during SDSA. A more stable D‐loop in the mph1Δ mutant cells likely results in increased D‐loop migration and BIR as previously proposed (Luke‐Glaser & Luke, 2012; Mehta et al, 2017). Furthermore, mph1∆ cells increase BIR when gene conversion is not in competition (Jain et al, 2016b), presumably by preventing unwinding of the initial strand invasion intermediate. BIR suppression by Mph1 in competition with gene conversion is particularly strong with shorter homologous sequences at the second DSB end. It is likely that besides Mph1, other DNA helicases also coordinate the use of two DSB ends. Yeast Srs2 is capable of unwinding D‐loops, particularly when they are longer (Liu et al, 2017). Consequently, the elimination of Srs2 leads to an increase of crossover products (Ira et al, 2003; Robert et al, 2006; Mitchel et al, 2013). Thus, besides Mph1, Srs2 may also suppress BIR. However, we have not tested Srs2 here because Srs2 itself is very important for BIR (Elango et al, 2017).
The effect of mutating enzymes regulating BIR frequency is partially (e.g., annealing mutants) or entirely (e.g., mph1Δ) mitigated by increased homology. This observation suggests that the control of BIR at two‐ended breaks by enzymes coordinating the engagement of two DSB ends is essential in recombination between short repeated sequences and, thus, in genomes carrying many repetitive elements, such as human genome. Longer homology on the second end of the break likely increases the chance of the initiation of recombination by second end of the DSB. Recently, we have shown that head‐on collisions by transcription prevent BIR and it is likely that assembling the repair apparatus on the opposite end may also inhibit BIR (Liu et al, 2021).
Another protein complex that may coordinate the use of two ends of a DSB is the MRX complex. Mutants deficient in MRX show frequent (10–20%) separation of two ends of a DSB (Kaye et al, 2004; Lobachev et al, 2004; Jain et al, 2016a). This result suggests that MRX is important for holding two DSB ends together. The MRX complex also coordinates the resection at ionizing radiation‐induced DSB ends (Westmoreland et al, 2009; Westmoreland & Resnick, 2013), and the synchronous resection of two ends is likely important to engage them in HR at the same time. Indeed, we found that in the absence of Mre11, repair of two‐ended DSBs by BIR increases. Careful analysis of resection showed that two ends of the break are not processed synchronously in mre11Δ cells. In a population of cells, the telomere proximal end of the break is processed faster. Deletion of YKU70 in mre11Δ cells restores synchronous resection of the two ends and decreases the BIR level among the products. Thus, we conclude that the Ku complex by itself or in combination with other proteins or specific DNA sequences forms a barrier for MRX‐independent resection more efficiently on one of two DSB ends. This barrier results in asynchronous formation of ssDNA and increase of BIR. We note that we cannot exclude the possibility that tethering of DSB ends by MRX also contributes to regulation of SDSA/BIR as synchronous resection of two ends may require ends tethering.
Mating‐type switching normally occurs between the HO‐cleaved euchromatic MAT locus and the heterochromatic donor HML. We found that Sir2’s maintenance of this heterochromatic state is important for suppressing BIR, particularly when homology is short. It is possible that histone deacetylation and the highly ordered nucleosomes within HML’s chromatin structure interfere with D‐loop migration, thus resulting in easier D‐loop disruption. Polδ, which is responsible for lagging strand synthesis during replication, usually stops DNA synthesis at the point where the interaction between DNA and histones is strongest (Smith & Whitehouse, 2012). Thus it is possible that Polδ‐mediated DNA synthesis in BIR is less effective within dense chromatin. We note also that BIR is a pathway used to extend telomeres in telomerase deficient cells (review in (Lee et al, 2020)), and telomeres reside within heterochromatin. However, telomere silencing is weaker when compared to HML silencing (review in Haber, 2012), so BIR through telomeric regions might be easier than through the HML locus. Alternatively, the chromatin state at subtelomeric regions could change during telomere length crisis.
Together this work determines the function of many recombination enzymes in suppressing one of the most mutagenic DSB repair pathways, BIR. Rad52 is highly conserved, but in many eukaryotes it plays a less prominent role in HR. In these organisms, additional annealing enzymes evolved with a role in HR such as FANCA in humans, the annealing helicase SMARCAL1 in flies, or Bhr2, the BRCA2 homolog in U. maydis (Mazloum & Holloman, 2009; Holsclaw & Sekelsky, 2017; Benitez et al, 2018). The MRX complex and D‐loop unwinding enzymes are also well conserved between yeast and humans. These enzymes likely prevent mutagenic BIR in higher eukaryotes in a similar way as observed in yeast, particularly when recombination occurs between short homologous repeats.
This work also shows that BIR is not efficient everywhere in the genome. Even in the same HML locus, BIR is efficient in the direction of the telomere and inefficient or absent in the direction of the centromere, but is apparently not dependent on the centromere itself. Favoring BIR toward telomere over centromere has implications for genome stability. Since telomeric distal regions are devoid of essential genes, the outcome of mutagenic BIR toward telomere is more tolerable. BIR toward the telomere could also be beneficial evolutionarily, as the duplication and exchange of genetic material in subtelomeric regions could increase the diversity of subtelomeric gene families (Brown et al, 2010). Chromosomal features determining BIR efficiency at specific loci or direction remain unknown. It remains to be determined whether some local topology, cohesion, or other chromosome features interfere with extensive D‐loop migration required for BIR.
Materials and Methods
Strains and plasmids
All stains used in this study are presented in Appendix Table S2.
To study competition between SDSA and BIR using intrachromosomal recombination, we used YAM033 and its derivatives listed in Appendix Table S2 (ho ade3::GAL10::HO HMLα‐inc MAT a hmr::ADE1 bar1::ADE3 nej1::KANMX ade1 leu2,3‐112 trp1::hisG ura3‐52 thr4 lys5) (Mehta et al, 2017). To construct strains with different homology size on one end of the DSB at MAT, complete/partial deletions of W, X, Yα were made by integrating Candida glabrata TRP1, amplified out from YAM033, (primers listed in Appendix Table S3). To make the H‐150 assay, complete deletion of W, and partial deletion of X within MAT a, leaving 150 bp, was constructed as described (Mehta et al, 2017). To make the “H‐150 no‐gap” assay, we created a complete deletion of W, X, and most of the Yα sequence within MATα, leaving just 150 bp of Yα. For the H‐0 assay, all W and X sequences were completely deleted in MAT a. The H‐1400 assay represents regular MAT switching between MAT a and HMLα‐inc, while the H‐2100 assay represents recombination between MATα and HMLα‐inc.
Another recombination on chromosome V system was further used to test the competition between SDSA and BIR. The strain yRA97 was gifted from Haber lab (hoΔ mat::hisG hmlΔ::hisG HMRa‐stk ura3Δ851 trp1Δ63 leu2Δ::KAN hmrΔ::ADE3 ade3::GAL10::HO can1Δ::UR::HOcs::A3::TRP1 RA::LEU2) (Anand et al, 2014).
To study the outcomes of the allelic recombination, we used derivatives of JKM139 (ho hml::ADE1 MAT a hmr::ADE1 ade1 leu2‐3,112 lys5 trp1::hisG ura3‐52 ade3::GAL10::HO) and NP633 (MATα‐inc HMLα‐inc hmr::ADE1 bar1::ADE3 ade1 leu2,3‐112 trp1::hisG ura3‐52 thr4 lys5 ade3::GAL10::HO). To mark the ends of chromosome III in JKM139, a kiTRP1 marker was inserted at location 17130, and a LEU2 marker was inserted at position 296670 to create strain NP676, and in NP633 the KANMX marker was inserted at position 296670 to generate strain NP643. DNA repair genes listed in Appendix Table S2 were deleted in NP676 and NP643, and resulting haploid mutant strains were crossed to obtain diploids for allelic recombination analysis.
To estimate the mutation rate ~16 kb away from the DSB end during allelic recombination, we first eliminated ura3‐52 from strains NP676 and NP643 using CRISPR/Cas9 methods to construct NP729 and NP728 respectively. Plasmid NP633 was made by cloning gRNA targeting URA3 (sequence listed in Appendix Table S3) into plasmid bRA89 after digestion with BplI as published (Anand et al, 2017). The deletion of ura3‐52 was obtained by transforming cells with plasmid NP633 and URA3 deletion template DNA. The deletion was confirmed by PCR and Sanger sequencing. The ura3‐29‐HPH fragment was generated by PCR amplification using primers listed in Appendix Table S3 and inserted ~16 kb distal to MATα‐inc in the donor parent strain NP728, resulting in strain NP754. The ura3‐29 mutation reporter (Shcherbakova & Pavlov, 1996) can revert to Ura+ via three different base substitutions at one C‐G pair. Haploid strains NP729 and NP754 were crossed to obtain diploid NP757, and mutation rates were estimated as previously described (Saini et al, 2013). Diploid strains lacking DNA repair enzymes were constructed for mutation rate analysis as described above and are presented in Appendix Table S2.
Strains carrying BIR assay between two chromosomes III are derivatives of strain AM1003 (MAT a ‐LEU2‐tel/MAT a ‐inc ade1 met13 ura3 leu2‐3,112/leu2 thr4 lys5 hml::ADE1/hml::ADE3 hmr::HYG ade3::GAL‐HO FS2::NAT/FS2) (Deem et al, 2008).
Strain to study DNA synthesis during BIR toward the centromere (HR‐0) was constructed by deleting all W and X sequences at MATα. A DSB is induced at MAT locus, invasion at Z sequence of HMR a ‐inc template leads to repair by BIR.
Mutagenesis of arginine 70 to alanine (R70A) in RAD52 to make DNA binding‐defective rad52‐R70A mutant was done using CRISPR‐Cas9 methods. We note that RAD52 contains multiple putative start codons (Mortensen et al, 2002). In this work, RAD52 sequence starts from the first start codon and the annealing mutants correspond to R70A. In modified and shorter by 33 residues RAD52 gene in SGD database, it begins at the third ATG and annealing mutant corresponds to R37A. gRNA targeting RAD52 was cloned into plasmid bRA90 to construct plasmid NP511. Cells were then transformed with NP511 and rad52‐R70A template DNA to construct the rad52‐R70A mutant. Mutation was then confirmed by Sanger sequencing.
Viability assays
To determine the viability, cells were grown overnight in YEP‐raffinose medium (1% yeast extract, 2% peptone, 2% raffinose), and ~100 cells were plated onto YEPD and YEP‐galactose plates. Colonies were counted 3–5 days after plating. The proportion of viable cells was estimated by dividing the number of colony‐forming units on YEP‐galactose plates by that on YEPD plates. Statistical comparison between mutants was performed using Welch’s unpaired t‐test and Prism 8 software.
Growth and induction of DSB in yeast
Yeast cells were grown overnight in YEPD (1% yeast extract, 2% peptone, 2% dextrose) and transferred to YEP‐raffinose medium (1% yeast extract, 2% peptone, 2% raffinose) for ~16 h. HO was induced when the cell density was ~1 × 107 cells/ml by adding galactose to a final concentration of 2%. Samples were collected for DNA isolation just before and at different time points following the addition of galactose.
Analysis of BIR and SDSA by Southern blot
DNA was isolated by glass bead disruption using a standard phenol extraction protocol. DNA was digested with either Bsp1286I (H‐0 and H‐150, H‐150 no‐gap system) or EcoRI and XhoI (H‐1400 and H‐2100 system) and separated on 0.8% agarose gels. Southern blotting and hybridization with radiolabeled DNA probes were carried out as described previously (Church & Gilbert, 1984). Southern blots were probed with a 32P‐labeled MAT a‐distal or Z fragment. All primers used to amplify the probes are listed in Appendix Table S3. All quantitative densitometric analysis was done using ImageQuant TL 5.2 software (GE Healthcare Life Sciences). To control for differences in the amount of DNA loaded into each lane, each fragment of interest was normalized to a loading control fragment: ACT1 fragment (H‐0, H‐150, and H‐150 no‐gap) or Z fragment of HMLα‐inc H‐1400 and H‐2100 system). Percentage of BIR among the products measured at 6 h after DSB induction was calculated as the percentage of the pixel intensity of the band corresponding to BIR to the total pixel intensity of the bands corresponding to both BIR and SDSA products. In rare cases, a very weak crossover band was observed. In these cases, the intensity of the shorter crossover band was subtracted from the band that corresponds to both the longer crossover band and BIR. Repair efficiency among all cells of each mutant was calculated as the percentage of normalized pixel intensity of bands corresponding to both BIR and SDSA products at 6 h to the normalized pixel intensity of the parental band at 0 h. Repair efficiency compared to WT was measured as the percentage of normalized pixel intensity of bands corresponding to either BIR or SDSA at 6 h of a mutant to the normalized pixel intensity of bands corresponding to BIR and SDSA at 6 h of WT cells. Statistical comparison of the percentage of BIR and repair efficiency between different mutants was performed using Welch’s unpaired t‐test (Prism 8 software).
Analysis of allelic recombination assay
Allelic recombination between chromosomes III was analyzed using diploid strains where each parental haploid strain carried chromosome III with different markers to follow different repair products, as described in Fig 3. The assay was performed by growing cells overnight in YEP‐ raffinose medium, and ~50–70 cells were plated onto YEPD and YEP‐galactose plates. After 3–5 days, replica plating on tryptophan dropout, leucine dropout, or YEPD plates supplemented with G418 to score different repair outcomes. BIR is scored as Trp +, Leu −, G418R, chromosome loss is scored as Trp −, Leu −, G418R. Gene conversion without crossing‐over is scored as Trp +, Leu +, G418R. Gene conversion with crossover will generate Trp+, Leu−, G418R/Trp +, Leu +, G418S sectored colonies, and Trp +, Leu +, G418R colonies. Since gene conversion without crossover is also scored as Trp +, Leu +, G418 R colonies, the counted number of Trp +, Leu −, G418 R/Trp +, Leu +, G418 S sectored colonies is then doubled to reflex the actual number of gene conversion with crossover and the number of gene conversion without crossover will be then subtracted accordingly. χ 2 analysis was used to perform a statistical comparison between mutants for individual classes of events using Prism 8 software.
Mutation analysis
To determine mutation frequency associated with BIR, cells were grown overnight in YEPD, followed by growth in YEP‐raffinose to the concentration of ~1 × 107 cells/ml. Breaks were induced by adding galactose to a final concentration of 2%, and cells were incubated for 6 h. ~3–5 × 108 cells were plated on uracil dropout plate(s) before (0 h) and 6 h after galactose addition. The number of colonies was counted after 3–5 days. The mutant frequency was calculated as f = m/N (m is the total number of mutants in a culture, and N is the total number of cells in a culture. Since the experimental strains did not divide or underwent a single division between 0 h and 6 h, the rate of mutations after galactose treatment was determined using a simplified version of the Drake equation: µ6 = (ϕ6–f0), where f6 and f0 are the Ura+ mutation frequencies at times 6 h and 0 h, respectively. Rates are reported as the median value, and statistical comparisons between median mutation rates were performed using the Mann–Whitney U test as previously described (Saini et al, 2013).
CHEF analysis of allelic recombination products
To analyze BIR repair products scored by plating assay, chromosomal DNA plugs were prepared and separated on 1% agarose gel at 6 V/cm for 48 h (initial time = 20 s, final time = 30 s) by using the CHEF DRII apparatus (Bio‐Rad), followed by Southern blotting and hybridization with a TRP1 sequence as a probe.
Analysis of allelic BIR
BIR assay using a disomic strain with an extra, truncated copy of chromosome III was previously described (Saini et al, 2013). The BIR assay was performed by growing cells overnight in 2% raffinose medium, and ~100 cells were plated on YEP‐galactose plates to induce the break and on YEPD as control. Different repair outcomes were score after 3–5 days by replica plating on adenine dropout, YEPD plates supplemented with G418, or clonNAT to score different repair outcomes.
Analysis of recombination assay on chromosome V
A same number of cells were plated on YEPD to get the total cell count and on YEP‐galactose plates for HO induction. Cells that grew on YEP‐galactose were counted and replica‐plated to replica plating on uracil, tryptophan, or leucine dropout plates to screen for different repair outcomes.
Resection analysis
Resection analysis was done as previously described (Zhu et al, 2008). Cells were cultured overnight in raffinose to the concentration of 1 × 107/ml. Cells were collected at 0 h before galactose was added to the culture to the final concentration of 2% to induce the break. Cells were then collected every 30 min (Fig 6) or every 2 h (Appendix Fig S2). DNA isolated by glass bead disruption using a standard phenol extraction protocol was digested with BfaI or EcoRI. Southern blot and hybridization with radiolabeled DNA MAT and loading control BPL1 were used to monitor 5′ strand resection beyond the BfaI or EcoRI site. All quantitative densitometric analysis was done using ImageQuant TL 5.2 software (GE Healthcare Life Sciences).
Author contributions
NP constructed strains, designed, conducted, and analyzed data. ZY helped with construction of Rad52 point mutant, YY helped with resection analysis, and MFA helped with analysis of repair events in yRA97 strain. NP, AM, and JEH and GI designed experiments, analyzed the data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 6
Acknowledgements
We thank members of the Ira laboratory for critical reading of the manuscript. This work was funded by grants from the National Institute of Health (GM080600 and GM125650 to G.I., R35 GM127029 to J.E.H., R35GM127006 to A.M.).
The EMBO Journal (2021) 40: e104847.
Data availability
This study includes no data deposited in external repositories.
References
- Anand R, Beach A, Li K, Haber J (2017) Rad51‐mediated double‐strand break repair and mismatch correction of divergent substrates. Nature 544: 377–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand RP, Tsaponina O, Greenwell PW, Lee CS, Du W, Petes TD, Haber JE (2014) Chromosome rearrangements via template switching between diverged repeated sequences. Genes Dev 28: 2394–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Davis AP, Symington LS (1999) A novel allele of RAD52 that causes severe DNA repair and recombination deficiencies only in the absence of RAD51 or RAD59. Genetics 153: 1117–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Carvalho CMB, Akdemir ZC, Sedlazeck FJ, Song X, Meng Q, Hu J, Doddapaneni H, Chong Z, Chen ES et al (2019) Megabase length hypermutation accompanies human structural variation at 17p11.2. Cell 176: 1310–1324 e1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitez A, Liu W, Palovcak A, Wang G, Moon J, An K, Kim A, Zheng K, Zhang Y, Bai F et al (2018) FANCA promotes DNA double‐strand break repair by catalyzing single‐strand annealing and strand exchange. Mol Cell 71: 621–628.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick R, Minocherhomji S, Hickson ID (2016) RAD52 facilitates mitotic DNA synthesis following replication stress. Mol Cell 64: 1117–1126 [DOI] [PubMed] [Google Scholar]
- Brown CA, Murray AW, Verstrepen KJ (2010) Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr Biol 20: 895–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Pehlivan D, Ramocki MB, Fang P, Alleva B, Franco LM, Belmont JW, Hastings PJ, Lupski JR (2013) Replicative mechanisms for CNV formation are error prone. Nat Genet 45: 1319–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Cui D, Papusha A, Zhang X, Chu CD, Tang J, Chen K, Pan X, Ira G (2012) The Fun30 nucleosome remodeller promotes resection of DNA double‐strand break ends. Nature 489: 576–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church GM, Gilbert W (1984) Genomic sequencing. Proc Natl Acad Sci USA 81: 1991–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD (2013) Break‐induced replication repair of damaged forks induces genomic duplications in human cells. Science 343: 88–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AP, Symington LS (2001) The yeast recombinational repair protein Rad59 interacts with Rad52 and stimulates single‐strand annealing. Genetics 159: 515–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem A, Barker K, Vanhulle K, Downing B, Vayl A, Malkova A (2008) Defective break‐induced replication leads to half‐crossovers in Saccharomyces cerevisiae. Genetics 179: 1845–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A (2011) Break‐induced replication is highly inaccurate. PLoS Biol 9: e1000594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnianni RA, Symington LS (2013) Break‐induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci USA 110: 13475–13480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elango R, Sheng Z, Jackson J, DeCata J, Ibrahim Y, Pham NT, Liang DH, Sakofsky CJ, Vindigni A, Lobachev KS et al (2017) Break‐induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat Commun 8: 1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher DN, Pham N, Tsai AM, Janto AN, Choi J, Ira G, Haber JE (2020) A Rad51‐independent pathway promotes single‐strand template repair in gene editing. PLoS Genet 16: e1008689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Hum YF, Lehner K, Jinks‐Robertson S (2017) Regulation of hetDNA Length during mitotic double‐strand break repair in yeast. Mol Cell 67: 539–549.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber JE (2012) Mating‐type genes and MAT switching in Saccharomyces cerevisiae. Genetics 191: 33–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall SD, Kolodner RD (1994) Homologous pairing and strand exchange promoted by the Escherichia coli RecT protein. Proc Natl Acad Sci USA 91: 3205–3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR (2009) A microhomology‐mediated break‐induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks WM, Kim M, Haber JE (2010) Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 329: 82–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsclaw JK, Sekelsky J (2017) Annealing of complementary DNA sequences during double‐strand break repair in drosophila is mediated by the ortholog of SMARCAL1. Genetics 206: 467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, Haber JE (2003) Srs2 and Sgs1‐Top3 suppress crossovers during double‐strand break repair in yeast. Cell 115: 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Satory D, Haber JE (2006) Conservative inheritance of newly synthesized DNA in double‐strand break‐induced gene conversion. Mol Cell Biol 26: 9424–9429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Sugawara N, Fishman‐Lobell J, Haber JE (1996) Genetic requirements for the single‐strand annealing pathway of double‐strand break repair in Saccharomyces cerevisiae . Genetics 142: 693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Sugawara N, Haber JE (2016a) Role of double‐strand break end‐tethering during gene conversion in Saccharomyces cerevisiae . PLoS Genet 12: e1005976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Sugawara N, Mehta A, Ryu T, Haber JE (2016b) Sgs1 and Mph1 helicases enforce the recombination execution checkpoint during DNA double‐strand break repair in Saccharomyces cerevisiae . Genetics 203: 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye JA, Melo JA, Cheung SK, Vaze MB, Haber JE, Toczyski DP (2004) DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr Biol 14: 2096–2106 [DOI] [PubMed] [Google Scholar]
- Lao JP, Oh SD, Shinohara M, Shinohara A, Hunter N (2008) Rad52 promotes postinvasion steps of meiotic double‐strand‐break repair. Mol Cell 29: 517–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR (2007) A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247 [DOI] [PubMed] [Google Scholar]
- Lee JJ, Lee J, Lee H (2020) Alternative paths to telomere elongation. Semin Cell Dev Biol 10.1016/j.semcdb.2020.11.003 [DOI] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE (1998) Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94: 399–409 [DOI] [PubMed] [Google Scholar]
- Li S, Wang H, Jehi S, Li J, Liu S, Wang Z, Truong L, Chiba T, Wang Z, Wu X (2021) PIF1 helicase promotes break‐induced replication in mammalian cells. EMBO J 40: e104509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, Khurana E, Waszak S, Korbel JO, Haber JE et al (2020) Patterns of somatic structural variation in human cancer genomes. Nature 578: 112–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Karakousis G, Chiu SK, Reddy G, Radding CM (1998) The beta protein of phage lambda promotes strand exchange. J Mol Biol 276: 733–744 [DOI] [PubMed] [Google Scholar]
- Liu J, Ede C, Wright WD, Gore SK, Jenkins SS, Freudenthal BD, Todd Washington M, Veaute X, Heyer WD (2017) Srs2 promotes synthesis‐dependent strand annealing by disrupting DNA polymerase delta‐extending D‐loops. Elife 6: 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Yan Z, Osia BA, Twarowski J, Sun L, Kramara J, Lee SL, Kumar S, Elango R, Li H et al (2021) Tracking break‐induced replication shows that it stalls at roadblocks. Nature 590: 655–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobachev K, Vitriol E, Stemple J, Resnick MA, Bloom K (2004) Chromosome fragmentation after induction of a double‐strand break is an active process prevented by the RMX repair complex. Curr Biol 14: 2107–2112 [DOI] [PubMed] [Google Scholar]
- Luke‐Glaser S, Luke B (2012) The Mph1 helicase can promote telomere uncapping and premature senescence in budding yeast. PLoS One 7: e42028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard JR, Jain S, Yamaguchi M, Haber JE (2007) Break‐induced replication and telomerase‐independent telomere maintenance require Pol32. Nature 448: 820–823 [DOI] [PubMed] [Google Scholar]
- Macheret M, Bhowmick R, Sobkowiak K, Padayachy L, Mailler J, Hickson ID, Halazonetis TD (2020) High‐resolution mapping of mitotic DNA synthesis regions and common fragile sites in the human genome through direct sequencing. Cell Res 30: 997–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, Naylor ML, Yamaguchi M, Ira G, Haber JE (2005) RAD51‐dependent break‐induced replication differs in kinetics and checkpoint responses from RAD51‐mediated gene conversion. Mol Cell Biol 25: 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloisel L, Fabre F, Gangloff S (2008) DNA polymerase delta is preferentially recruited during homologous recombination to promote heteroduplex DNA extension. Mol Cell Biol 28: 1373–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle R, Langston L, Molloy KR, Zhang D, Chait BT, O'Donnell ME (2019) Mcm10 has potent strand‐annealing activity and limits translocase‐mediated fork regression. Proc Natl Acad Sci USA 116: 798–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazloum N, Holloman WK (2009) Second‐end capture in DNA double‐strand break repair promoted by Brh2 protein of Ustilago maydis. Mol Cell 33: 160–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill C, Shafer B, Strathern J (1989) Coconversion of flanking sequences with homothallic switching. Cell 57: 459–467 [DOI] [PubMed] [Google Scholar]
- McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, West SC (2005) Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell 20: 783–792 [DOI] [PubMed] [Google Scholar]
- McIlwraith MJ, West SC (2008) DNA repair synthesis facilitates RAD52‐mediated second‐end capture during DSB repair. Mol Cell 29: 510–516 [DOI] [PubMed] [Google Scholar]
- McPherson JP, Lemmers B, Chahwan R, Pamidi A, Migon E, Matysiak‐Zablocki E, Moynahan ME, Essers J, Hanada K, Poonepalli A et al (2004) Involvement of mammalian Mus81 in genome integrity and tumor suppression. Science 304: 1822–1826 [DOI] [PubMed] [Google Scholar]
- Mehta A, Beach A, Haber JE (2017) Homology requirements and competition between gene conversion and break‐induced replication during double‐strand break repair. Mol Cell 65: 515–526: e513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS (2010) Ku prevents Exo1 and Sgs1‐dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J 29: 3358–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchel K, Lehner K, Jinks‐Robertson S (2013) Heteroduplex DNA position defines the roles of the Sgs1, Srs2, and Mph1 helicases in promoting distinct recombination outcomes. PLoS Genet 9: e1003340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen UH, Bendixen C, Sunjevaric I, Rothstein R (1996) DNA strand annealing is promoted by the yeast Rad52 protein. Proc Natl Acad Sci USA 93: 10729–10734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen UH, Erdeniz N, Feng Q, Rothstein R (2002) A molecular genetic dissection of the evolutionarily conserved N terminus of yeast Rad52. Genetics 161: 549–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Sica RA, Kowalczykowski SC (2009) Rad52 promotes second‐end DNA capture in double‐stranded break repair to form complement‐stabilized joint molecules. Proc Natl Acad Sci USA 106: 3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Lee SJ, Rothstein R, Symington LS (2018) Xrs2 and Tel1 independently contribute to MR‐mediated DNA tethering and replisome stability. Cell Rep 25: 1681–1692.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman F, Dixon J, Barr AR, Whitby MC (2005) The F‐Box DNA helicase Fbh1 prevents Rhp51‐dependent recombination without mediator proteins. Mol Cell Biol 25: 8084–8096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozenberger BA, Roeder GS (1991) A unique pathway of double‐strand break repair operates in tandemly repeated genes. Mol Cell Biol 11: 1222–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukhova G, Stratton SA, Sung P (1999) Single strand DNA binding and annealing activities in the yeast recombination factor Rad59. J Biol Chem 274: 33839–33842 [DOI] [PubMed] [Google Scholar]
- Piazza A, Shah SS, Wright WD, Gore SK, Koszul R, Heyer WD (2019) Dynamic processing of displacement loops during recombinational DNA repair. Mol Cell 73: 1255–1266.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponder RG, Fonville NC, Rosenberg SM (2005) A switch from high‐fidelity to error‐prone DNA double‐strand break repair underlies stress‐induced mutation. Mol Cell 19: 791–804 [DOI] [PubMed] [Google Scholar]
- Prakash R, Satory D, Dray E, Papusha A, Scheller J, Kramer W, Krejci L, Klein H, Haber JE, Sung P et al (2009) Yeast Mph1 helicase dissociates Rad51‐made D‐loops: implications for crossover control in mitotic recombination. Genes Dev 23: 67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert T, Dervins D, Fabre F, Gangloff S (2006) Mrc1 and Srs2 are major actors in the regulation of spontaneous crossover. EMBO J 25: 2837–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumelioti FM, Sotiriou SK, Katsini V, Chiourea M, Halazonetis TD, Gagos S (2016) Alternative lengthening of human telomeres is a conservative DNA replication process with features of break‐induced replication. EMBO Rep 17: 1731–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, Malkova A (2013) Migrating bubble during break‐induced replication drives conservative DNA synthesis. Nature 502: 389–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Ayyar S, Deem AK, Chung WH, Ira G, Malkova A (2015) Translesion polymerases drive microhomology‐mediated break‐induced replication leading to complex chromosomal rearrangements. Mol Cell 60: 860–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Malkova A (2017) Break induced replication in eukaryotes: mechanisms, functions, and consequences. Crit Rev Biochem Mol Biol 52: 395–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Roberts SA, Malc E, Mieczkowski PA, Resnick MA, Gordenin DA, Malkova A (2014) Break‐induced replication is a source of mutation clusters underlying kataegis. Cell Rep 7: 1640–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova PV, Pavlov YI (1996) 3'–>5' exonucleases of DNA polymerases epsilon and delta correct base analog induced DNA replication errors on opposite DNA strands in Saccharomyces cerevisiae . Genetics 142: 717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shee C, Gibson JL, Rosenberg SM (2012) Two mechanisms produce mutation hotspots at DNA breaks in Escherichia coli . Cell Rep 2: 714–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi I, Hallwyl SC, Seong C, Mortensen U, Rothstein R, Sung P (2009) Role of the Rad52 amino‐terminal DNA binding activity in DNA strand capture in homologous recombination. J Biol Chem 284: 33275–33284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim EY, Chung WH, Nicolette ML, Zhang Y, Davis M, Zhu Z, Paull TT, Ira G, Lee SE (2010) Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J 29: 3370–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE, Llorente B, Symington LS (2007) Template switching during break‐induced replication. Nature 447: 102–105 [DOI] [PubMed] [Google Scholar]
- Smith DJ, Whitehouse I (2012) Intrinsic coupling of lagging‐strand synthesis to chromatin assembly. Nature 483: 434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafa A, Donnianni RA, Timashev LA, Lam AF, Symington LS (2014) Template switching during break‐induced replication is promoted by the Mph1 helicase in Saccharomyces cerevisiae. Genetics 196: 1017–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stivison EA, Young KJ, Symington LS (2020) Interstitial telomere sequences disrupt break‐induced replication and drive formation of ectopic telomeres. Nucleic Acids Res 48: 12697–12710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Ira G, Haber JE (2000) DNA length dependence of the single‐strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double‐strand break repair. Mol Cell Biol 20: 5300–5309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Kantake N, Wu Y, Kowalczykowski SC (2006) Rad52‐mediated DNA annealing after Rad51‐mediated DNA strand exchange promotes second ssDNA capture. EMBO J 25: 5539–5548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, New JH, Kowalczykowski SC (1998) DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single‐stranded DNA. Proc Natl Acad Sci USA 95: 6049–6054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Nandi S, Osman F, Ahn JS, Jakovleska J, Lorenz A, Whitby MC (2008) The FANCM ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during DNA double‐strand break repair. Mol Cell 32: 118–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS (2016) Mechanism and regulation of DNA end resection in eukaryotes. Crit Rev Biochem Mol Biol 51: 195–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, Lisby M (2014) Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae . Genetics 198: 795–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia‐Burton M, Oki M, Johnson J, Seier TA, Kamakaka R, Haber JE (2006) Different mating‐type‐regulated genes affect the DNA repair defects of Saccharomyces RAD51, RAD52 and RAD55 mutants. Genetics 174: 41–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss K, Simpson RT (1998) High‐resolution structural analysis of chromatin at specific loci: Saccharomyces cerevisiae silent mating type locus HMLalpha. Mol Cell Biol 18: 5392–5403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmoreland J, Ma W, Yan Y, Van Hulle K, Malkova A, Resnick MA (2009) RAD50 is required for efficient initiation of resection and recombinational repair at random, gamma‐induced double‐strand break ends. PLoS Genet 5: e1000656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmoreland JW, Resnick MA (2013) Coincident resection at both ends of random, gamma‐induced double‐strand breaks requires MRX (MRN), Sae2 (Ctp1), and Mre11‐nuclease. PLoS Genet 9: e1003420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Kwon Y, Xu Y, Chung WH, Chi P, Niu H, Mayle R, Chen X, Malkova A, Sung P et al (2013) Pif1 helicase and Poldelta promote recombination‐coupled DNA synthesis via bubble migration. Nature 502: 393–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Haber JE (1996) A 700 bp cis‐acting region controls mating‐type dependent recombination along the entire left arm of yeast chromosome III. Cell 87: 277–285 [DOI] [PubMed] [Google Scholar]
- Wu Y, Kantake N, Sugiyama T, Kowalczykowski SC (2008) Rad51 protein controls Rad52‐mediated DNA annealing. J Biol Chem 283: 14883–14892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Sugiyama T, Kowalczykowski SC (2006) DNA annealing mediated by Rad52 and Rad59 proteins. J Biol Chem 281: 15441–15449 [DOI] [PubMed] [Google Scholar]
- Yan Z, Xue C, Kumar S, Crickard JB, Yu Y, Wang W, Pham N, Li Y, Niu H, Sung P et al (2019) Rad52 restrains resection at DNA double strand break ends in yeast. Mol Cell 76: 699–711.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G (2008) Sgs1 helicase and two nucleases dna2 and exo1 resect DNA double‐strand break ends. Cell 134: 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 6
Data Availability Statement
This study includes no data deposited in external repositories.