

Abstract
HIV‐1 latency is a major obstacle to achieving a functional cure for AIDS. Reactivation of HIV‐1‐infected cells followed by their elimination via immune surveillance is one proposed strategy for eradicating the viral reservoir. However, current latency‐reversing agents (LRAs) show high toxicity and low efficiency, and new targets are needed to develop more promising LRAs. Here, we found that the histone chaperone CAF‐1 (chromatin assembly factor 1) is enriched on the HIV‐1 long terminal repeat (LTR) and forms nuclear bodies with liquid–liquid phase separation (LLPS) properties. CAF‐1 recruits epigenetic modifiers and histone chaperones to the nuclear bodies to establish and maintain HIV‐1 latency in different latency models and primary CD4+ T cells. Three disordered regions of the CHAF1A subunit are important for phase‐separated CAF‐1 nuclear body formation and play a key role in maintaining HIV‐1 latency. Disruption of phase‐separated CAF‐1 bodies could be a potential strategy to reactivate latent HIV‐1.
Keywords: CAF‐1, epigenetic regulation, HIV‐1 latency, nuclear body, phase separation
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Microbiology, Virology & Host Pathogen Interaction
Enrichment of histone chaperone CAF‐1 on long terminal repeats of integrated HIV‐1 retrovirus recruits epigenetic and chromatin modifiers involved in viral latency via liquid‐liquid phase separation.
Introduction
HIV‐1/AIDS is incurable because of HIV‐1 latency (Chun et al, 1997; Finzi et al, 1997; Wong et al, 1997). HIV‐1 proviruses are temporarily silenced within infected resting CD4+ T cells and reactivated along with CD4+ T‐cell activation. Although combined antiretroviral therapy (cART) suppresses HIV‐1 effectively, the treatment has to be lifelong, as HIV‐1 viremia quickly rebounds upon the interruption of cART. Thus, complete eradication of HIV‐1‐infected cells seems to be the best strategy to cure patients. To this end, several functional cure strategies have been proposed to achieve long‐term suppression of HIV‐1 replication and remission of HIV‐1 viremia (Deeks, 2012; Liu et al, 2015; Elsheikh et al, 2019). Nevertheless, no matter what kinds of functional cure strategies will be adopted, comprehensively elucidating the mechanisms of HIV‐1 latency is the prerequisite to develop new or improve existed therapeutic interventions.
The mechanisms of HIV‐1 latency refer to multiple aspects including the specificities of integration sites, epigenetic regulations, transcriptional control, and post‐transcriptional regulations (Mbonye & Karn, 2017; Khoury et al, 2018). Most of the proviruses tend to integrate into the intron of actively transcribed genes. Some latently infected cells with integration hotspots undergo clonal expansion (Schröder et al, 2002; Maldarelli et al, 2014; Wagner et al, 2014; Cohn et al, 2015). To enter latent status for HIV‐1 proviruses, the expression of both viral Tat and cellular transcription factors including NF‐κB, Sp1, AP‐1, NFAT1, and TFIIH should be decreased, and transcription suppressors such as LSF, YY1, CTIP2, and TRIM28 could be recruited to HIV‐1 promoters (Nabel & Baltimore, 1987; Perkins et al, 1993; Kinoshita et al, 1998; Yang et al, 1999; Ping & Rana, 2001; He & Margolis, 2002; Kim et al, 2006; Marban et al, 2007; Razooky et al, 2015; Ma et al, 2019). Along with transcriptional inhibition, the HIV‐1 promoter undergoes multiple suppressive epigenetic modifications. Chromatin “eraser” modifiers, such as histone deacetylases HDAC1 and HDAC2, remove the active marks acetyls from histone lysine residues (Marban et al, 2007). Deacetylated histone H3 Lysine 9 (H3K9) is further methylated by chromatin “writers”, such as G9a, SUV39H1, and GLP (Chéné et al, 2007; Imai et al, 2010; Ding et al, 2013). H3K27 is methylated by EZH2 (Friedman et al, 2011). H4K20 is methylated by SMYD2 (Boehm et al, 2017). Both H3K9me3 and H3K27me3 as well as H4K20me1 contribute to HIV‐1 latency (Ruelas & Greene, 2013). These suppressive epigenetic marks are ultimately maintained by chromatin “readers”, which include three heterochromatin proteins (HP1α, HP1β, HP1γ) for H3K9me3, five CBX paralogs (CBX2, CBX4, CBX6, CBX7, CBX8) for H3K27me3, L3MBTL1 for H4K20me1 (Chéné et al, 2007; Boehm et al, 2017; Khan et al, 2018). Apart from histone modifications, DNA methylation, which is modified by DNMT1 and maintained by MBD2, is also found to contribute to HIV‐1 latency in several latency cell lines and HIV‐1‐infected patients (Blazkova et al, 2009; Kauder et al, 2009; Trejbalová et al, 2016).
Cellular processes, as well as HIV‐1 latency, have long been thought to be regulated unidimensionally by different cellular components. However, with the rapid development of super‐resolution imaging and chromosome conformation capture technologies, a plethora of membrane‐less condensates have been found to regulate gene expression in the spatiotemporally multi‐dimensional manner (Dekker et al, 2013; Banani et al, 2017). Physicochemical studies of these non‐membrane‐enclosed compartments have revealed that membrane‐less condensates have liquid‐like properties and are coalesced via phase separation (Hyman et al, 2014). The driving forces of liquid–liquid phase separation (LLPS) are the weak multivalent interactions which are mediated by low‐complexity intrinsically disordered regions (IDRs) within corresponding compartment components (Brangwynne et al, 2015). BRD4, MED1, OCT4, and GCN4, all of which harbor IDRs, form LLPS droplets to link super‐enhancers (SEs) and gene activation (Boija et al, 2018; Sabari et al, 2018). HP1, SUV39H1, and TRIM28 form LLPS droplets to link heterochromatic H3K9me3 and gene suppression (Larson et al, 2017; Strom et al, 2017; Sanulli et al, 2019; Wang et al, 2019). More other components with LLPS properties include paraspeckle component NEAT1, Polycomb body component CBX2, PML body component PML (Banani et al, 2016; Yamazaki et al, 2018; Plys et al, 2019). The fate of these LLPS condensates is highly influenced by different modifications. The hyperphosphorylation of RNA polymerase II (RNAP II) C‐terminal domain switches RNAP II from transcriptional condensates to splicing condensates (Lu et al, 2018; Guo et al, 2019). The acetylation of DDX3X, the methionine oxidation of Pbp1, and the phosphorylation of FUS inhibit the formation of corresponding phase‐separated condensates (Monahan et al, 2017; Kato et al, 2019; Saito et al, 2019). The SUMOylation of PML promotes the formation of phase‐separated condensates (Banani et al, 2016). Apart from the LLPS of cellular components, measles virus (MeV) and vesicular stomatitis virus (VSV) also form inclusion bodies with LLPS properties (Heinrich et al, 2018; Zhou et al, 2019). Although multitudes of cellular or viral proteins or RNAs have been found to phase separate into membrane‐less organelles, the precise function of LLPS on different cellular and viral processes is still enigmatic (Alberti et al, 2019).
Epigenetic modifiers and maintainers, some of which also have LLPS properties as we mentioned above, have been studied intensively these years (Larson et al, 2017; Strom et al, 2017; Plys et al, 2019; Sanulli et al, 2019; Wang et al, 2019). However, how and when distinct epigenetic proteins are recruited to target genes and HIV‐1 promoter are still less defined. Histone chaperones, which contribute to epigenetic memory and genome stability, can act as landing pads for multiple epigenetic proteins to alter the global epigenetic landscape (De Koning et al, 2007; Groth et al, 2007). DNA clamp PCNA recruits DNMT1 and HDAC1 to methylate DNA CpGs and deacetylate histone lysines, respectively (Chuang et al, 1997; Milutinovic et al, 2002). Chromatin assembly factor 1 (CAF‐1), which deposits newly synthesized H3‐H4 onto replicating DNA, has been found to recruit SETDB1, SUV39H1, HDAC1/2, KDM1A, MBD1, and HP1α to the long terminal repeats (LTRs) of endogenous retrotransposons or endogenous retroviruses to establish and maintain the suppressive epigenetic modifications in mouse pluripotent stem cells (Murzina et al, 1999; Reese et al, 2003; Loyola et al, 2009; Cheloufi et al, 2015; Hatanaka et al, 2015; Yang et al, 2015). Some histone chaperones also participate in HIV‐1 latency. The chromatin reassembly factor (CRF) FACT contributes to HIV‐1 latency by decoying Tat and subsequently interfering with the association of Tat with P‐TEFb complex (Huang et al, 2015). Another CRF named Spt6 forms a complex with LEDGF/p75 and Iws1 and aggregates on the latent HIV‐1 LTR to maintain suppressive chromatin marks (Gérard et al, 2015). Due to the clustered epigenetic protein‐bound nature of histone chaperones, we hypothesize that a pivotal histone chaperone may exist to orchestrate most of the suppressive epigenetic proteins together to synergistically mediate HIV‐1 latency.
In this study, we revealed that histone chaperone CAF‐1 is enriched on HIV‐1 LTR and maintains HIV‐1 latency in several in vitro latency models and in resting CD4+ T cells from individuals on cART. The depletion of CHAF1A subunit of CAF‐1 results in the loss of multiple suppressive epigenetic marks including H3K9me3, H4K20me3, and methyl‐CpGs, as well as the accumulation of several active epigenetic marks including H3K4me3, H3K36me2, and acetyl‐lysines. Further proteomics, biophysical, and biochemical assays characterized that multitudes of suppressive epigenetic proteins and histone chaperones are recruited by CAF‐1 and form nuclear condensates with LLPS properties. We also identified a few of key amino acids within CHAF1A IDRs which mediate the LLPS of CAF‐1 body. Most importantly, the mutation of key amino acids not only dissolves CAF‐1 body but also eliminates CAF‐1 body‐mediated HIV‐1 latency. We speculate that CAF‐1 body could be a core factor which organizes the suppressive elements and maintains HIV‐1 latency.
Results
CAF‐1 promotes HIV‐1 latency
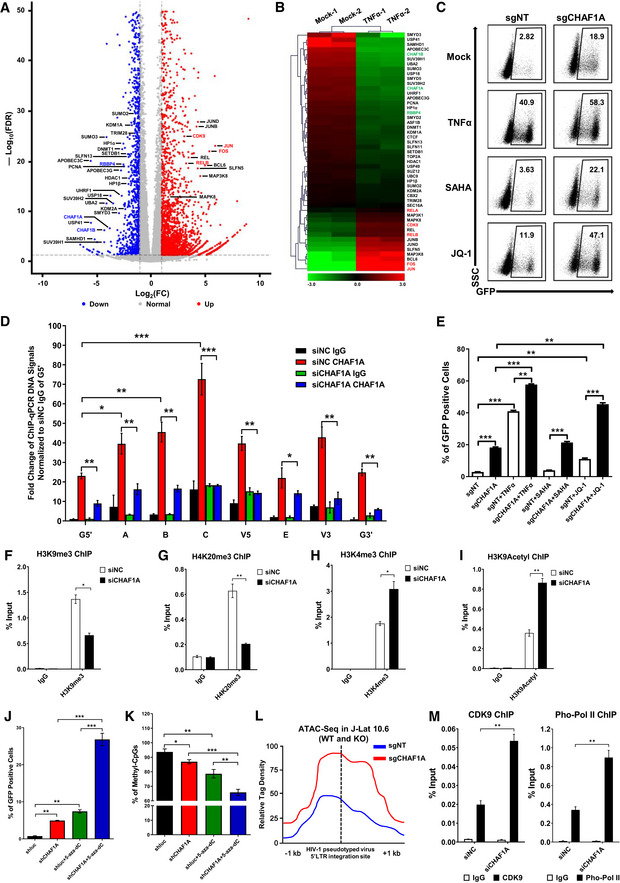
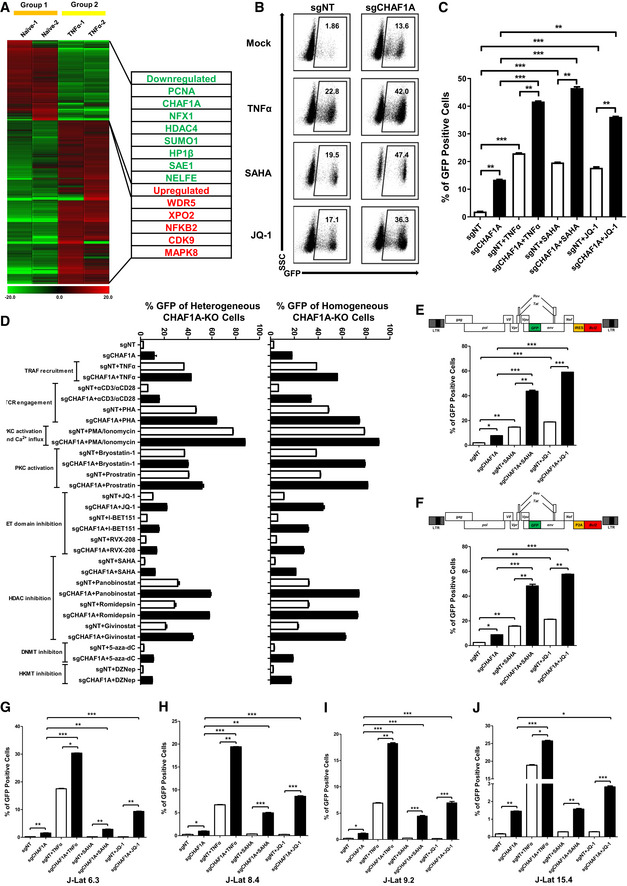
To find potential targets which might contribute to HIV‐1 latency, we compared gene expression in unstimulated and TNFα‐stimulated HIV‐1 latency cell line J‐Lat 10.6 which harbors an integrated full‐length HIV‐1 pseudotyped provirus (Jordan et al, 2003). The expression of GFP, which is inserted into the HIV‐1 nef open reading frame, is significantly upregulated upon TNFα stimulation. Utilizing RNA‐Seq and mass spectrometry (MS) analyses, we found that several transcriptional factors, including JUN, were significantly upregulated in the presence of TNFα signaling (Figs 1A and B, and EV1A). The expression of SEC16A, the third intron of which harbors HIV‐1 pseudotyped provirus integration site, was unchanged. Conversely, several suppressive epigenetic proteins were significantly downregulated. Especially, CHAF1A, CHAF1B, and RBBP4, which are subunits of CAF‐1 complex, were simultaneously downregulated (Fig 1A). Given that CAF‐1 shows suppressive effect on gene expression, we speculated that CAF‐1 might contribute to HIV‐1 latency (Murzina et al, 1999; Reese et al, 2003; Loyola et al, 2009; Cheloufi et al, 2015; Hatanaka et al, 2015; Yang et al, 2015). As CHAF1A is the major subunit of CAF‐1, hereafter we conducted further experiments on CHAF1A. We knocked out CHAF1A in J‐Lat 10.6 and found that the depletion of CHAF1A upregulated HIV‐1 expression in the heterogeneous knockout cell line and the homogeneous knockout cell line (Figs 1C and E, and EV1B and C, Appendix Fig S1A and B). HIV‐1 reactivation was enhanced much higher when supplemented with histone deacetylase (HDAC) inhibitor SAHA, bromodomain and extra‐terminal (BET) domain inhibitor JQ‐1, and other analogous LRAs (Fig EV1D). These results were well‐repeated in other monoclonal latency model cell lines including J‐Lat 6.3, 8.4, 9.2, 15.4, and several heterogeneous latency model cell lines (Fig EV1, EV2, EV3, EV4, EV5, Appendix Fig S1C–E).
Figure 1. CAF‐1 promotes HIV‐1 latency.
-
ARNA‐Seq of naïve and TNFα‐stimulated J‐Lat 10.6 cells. Differentially expressed genes were filtered with log2FC of 1 and P value FDR cutoff of 0.05. Upregulated and downregulated genes were labeled as red and blue dots, respectively. Representative genes were labeled aside corresponding dots.
-
BRNA‐Seq result as in (A). Significantly changed genes were sorted out and plotted as heatmap. SEC16A indicated unchanged gene.
-
CThe GFP‐positive percentages of monoclonal sgCHAF1A and sgNT J‐Lat 10.6 cell lines were shown in the top right corner of each flow cytometry figure. TNFα, SAHA, and JQ‐1 were used as supplements.
-
DChIP assay with antibody against CHAF1A was performed in J‐Lat 10.6 cells. All the ChIP‐qPCR DNA signals were normalized to siNC IgG of G5′. G5′ represented cellular DNA and viral 5′LTR junction; A: Nucleosome 0 assembly site; B: Nucleosome free region; C: Nucleosome 1 assembly site; V5: Viral 5′LTR and gag leader sequence junction; E represented envelope; V3: Viral poly purine tract and 3′LTR junction; G3′ represented viral 3′LTR and cellular DNA junction.
-
EThe statistical graph of the result in (C).
-
F–IChIP assays with antibodies against H3K9me3, H4K20me3, H3K4me3, and H3K9Acetyl were performed as in (D). Only “C” position signals were showed and normalized to Input.
-
J, KJ‐Lat 8.4 cells were treated with shCHAF1A lentiviruses and 5‐aza‐dC. The GFP‐positive cells and HIV‐1 LTR methyl‐CpGs percentages of different groups were plotted in (J) and (K), respectively.
-
LATAC‐Seq was performed in WT and CHAF1A‐KO J‐Lat 10.6 cells. The relative tag densities of the pseudotyped HIV‐1 5′LTR integration site in each group were calculated. The highest tag density was set as 100. Figure showed 2 kb range centered the 5′LTR integration site. Dashed line represented HIV‐1 pseudotyped virus 5′LTR integration site.
-
MChIP assays with antibodies against CDK9 and Pho‐Pol II were performed in siNC and siCHAF1A J‐Lat 10.6 cells. ChIP signals in each group were normalized to input.
Data information: Data represented mean ± SEM in triplicate. P‐values were calculated by Student’s t‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure EV1. CAF‐1 promotes HIV‐1 latency.
-
AMass spectrometry (MS) result of naïve and TNFα‐stimulated J‐Lat 10.6 cells. Significantly changed genes were plotted as heatmap. Representative genes were shown in table aside the heatmap. The heatmap scale represented fold change of gene expression (Min: −20 fold; Max: 20 fold).
-
B, CThe GFP‐positive percentages of heterogeneous sgCHAF1A and sgNT J‐Lat 10.6 cell lines were shown as flow cytometry figure and statistical histogram. TNFα, SAHA and JQ‐1 were used as supplements.
-
DFifteen LRAs targeting eight signaling pathways were used in heterogeneous and homogeneous CHAF1A‐KO J‐Lat 10.6 cell lines.
-
E, FTwo pseudotyped HIV‐1 latency cell lines (J‐Lat‐NIB and J‐Lat‐NPB) were treated with sgCHAF1A lentiviruses, SAHA and JQ‐1. The GFP‐positive percentages of each group were measured. The backbones of each pseudotyped HIV‐1 were also shown.
-
G–JJ‐Lat 6.3, 8.4, 9.2 and 15.4 were treated as in Fig 1C. The reactivation efficiencies of each group were indicated by the percentages of GFP‐positive cells.
Data information: Data represented mean ± SEM in triplicate. P‐values were calculated by Student’s t‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
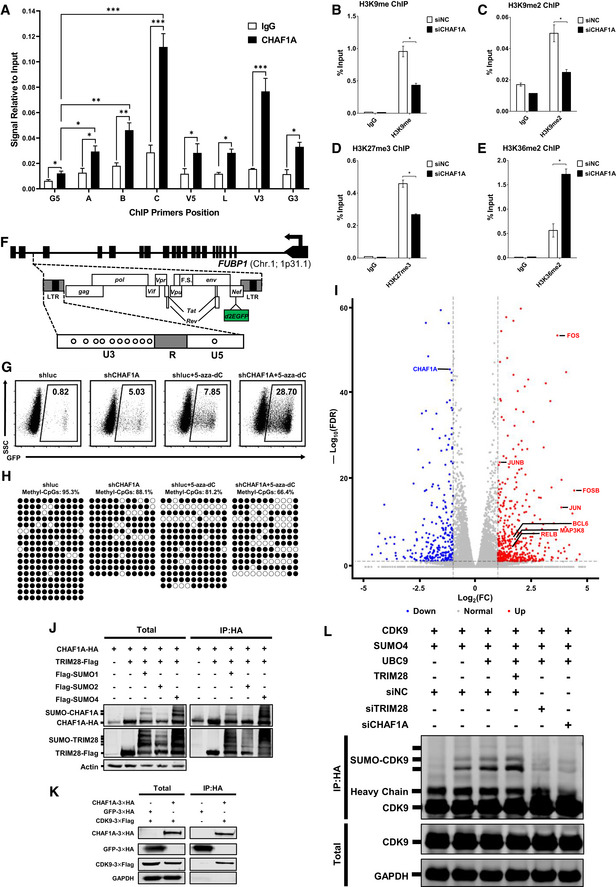
Figure EV2. CAF‐1 alters the epigenetic status of HIV‐1 promoter and enriches SUMOylation system.
- A
-
B–EChIP assays with antibodies against H3K9me, H3K9me2, H3K27me3 and H3K36me2 were performed as in Fig 1F.
-
FThe schematic of CpGs on HIV‐1 LTR of J‐Lat 8.4. Eleven CpGs were shown in circles.
-
G, HThe flow cytometry figures and Methyl‐CpGs graphs of J‐Lat 8.4 which were treated with shCHAF1A and 5‐aza‐dC, corresponding to statistical histograms in Fig 1J and K.
-
IRNA‐Seq result of wild‐type and CHAF1A‐KO J‐Lat 10.6. Differentially expressed genes were filtered with log2FC of 1 and PvalueFDR cutoff of 0.05. Upregulated and downregulated genes were labeled as red and blue dots, respectively. Representative genes were labeled aside corresponding dots.
-
JFlag‐tagged TRIM28, SUMO1, SUMO2 and SUMO4 were co‐overexpressed with HA‐tagged CHAF1A. HA‐tagged CHAF1A was IP with anti‐HA beads. Both total and IP samples were IB with anti‐HA, anti‐Flag and anti‐Actin antibodies.
-
KHA‐tagged CHAF1A and HA‐tagged GFP were co‐overexpressed with Flag‐tagged CDK9. HA‐tagged proteins were immunoprecipitated (IP) with anti‐HA beads. Both total and IP samples were immunoblotted (IB) with anti‐HA, anti‐Flag and anti‐GAPDH antibodies.
-
LHA‐tagged CDK9 was co‐overexpressed with Flag‐tagged SUMO4, UBC9, and TRIM28. siRNAs targeting TRIM28 and CHAF1A treated CDK9‐overexpressed HeLa cells, respectively. CDK9 was IP with anti‐HA beads. Both total and IP samples were IB with anti‐HA, anti‐Flag and anti‐GAPDH antibodies.
Data information: Data represented mean ± SEM in triplicate. P‐values were calculated by Student’s t‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
Source data are available online for this figure.
Figure EV3. CAF‐1 forms nuclear bodies.
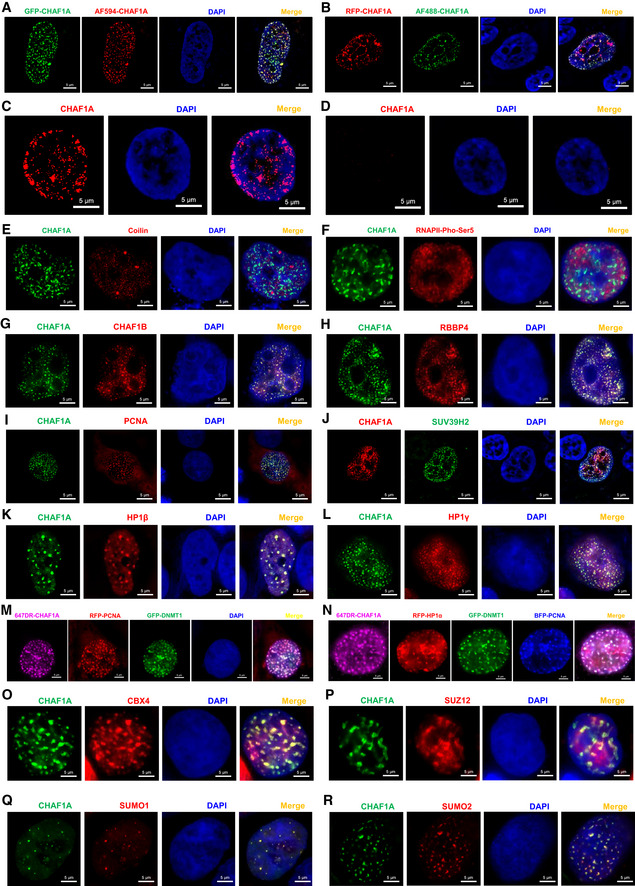
-
AGFP‐tagged CHAF1A was imaged with AF594‐tagged antibody against CHAF1A.
-
BRFP‐tagged CHAF1A was imaged with AF488‐tagged antibody against CHAF1A.
-
C, DThe endogenous CHAF1A was imaged before (C) and after (D) CHAF1A knockdown in J‐Lat 10.6 cells.
-
E, FCoilin and Pho‐Ser5‐RNAP II were immunostained with corresponding antibodies and imaged with CHAF1A, respectively.
-
G–LCHAF1B, RBBP4, PCNA, SUV39H2, HP1β, and HP1γ was co‐overexpressed with CHAF1A, respectively. SIM images were captured for each combination.
-
MRFP‐tagged PCNA and GFP‐tagged DNMT1 were imaged with AF647‐tagged antibody against CHAF1A simultaneously. DAPI was used to dye DNA.
-
NRFP‐tagged HP1α, GFP‐tagged DNMT1, and BFP‐tagged PCNA were imaged with AF647‐tagged antibody against CHAF1A simultaneously.
-
O–RCBX4, SUZ12, SUMO1, and SUMO2 were imaged with CHAF1A, respectively.
Data information: Scale bar represented 5 μm. At least three images were captured for each sample.
Figure EV4. The influence of different CHAF1A mutants on CAF‐1 body and the epigenetic status of HIV‐1 promoter.
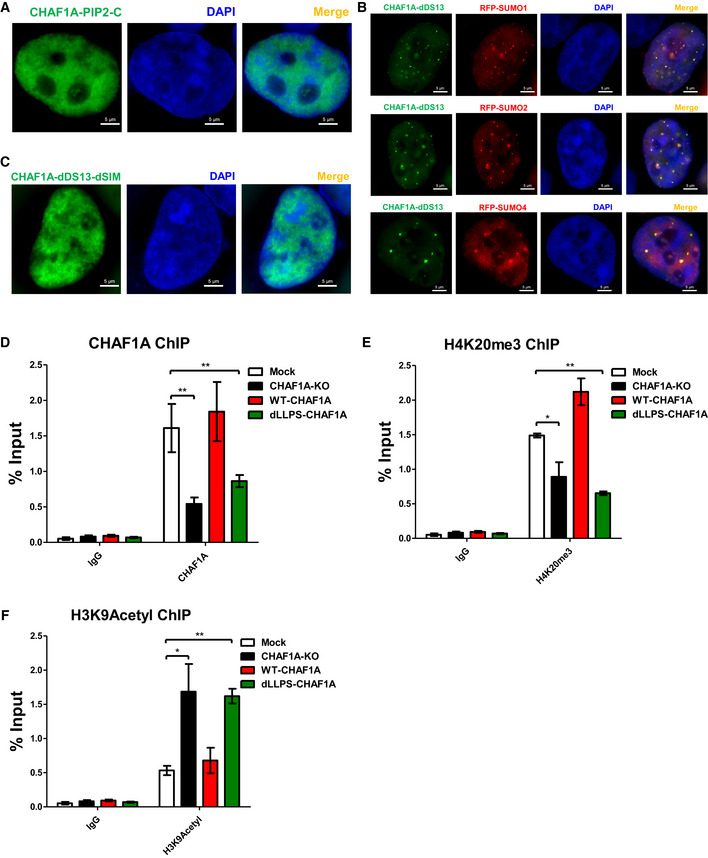
-
AThe distribution of CHAF1A‐PIP2‐C. PIP2: the second PCNA‐interacting protein motif which located after KER. C: CHAF1A C‐terminal.
-
BThe co‐localization of RFP‐tagged SUMO1, SUMO2 and SUMO4 with GFP‐tagged CHAF1A‐dDS13.
-
CThe distribution of CHAF1A‐dDS13‐dSIM.
-
D–FThe enrichment of CHAF1A, H4K20me3 and H3K9Acetyl on HIV‐1 LTR in different CHAF1A status.
Data information: The scale bar in (A–C) represented 5 μm. Data in (D–F) represented mean ± SEM in triplicate. P‐values were calculated by Student’s t‐test. *P < 0.05, **P < 0.01.
Figure EV5. CHAF1A depletion reactivates latent HIV‐1 in primary CD4+ T cells.
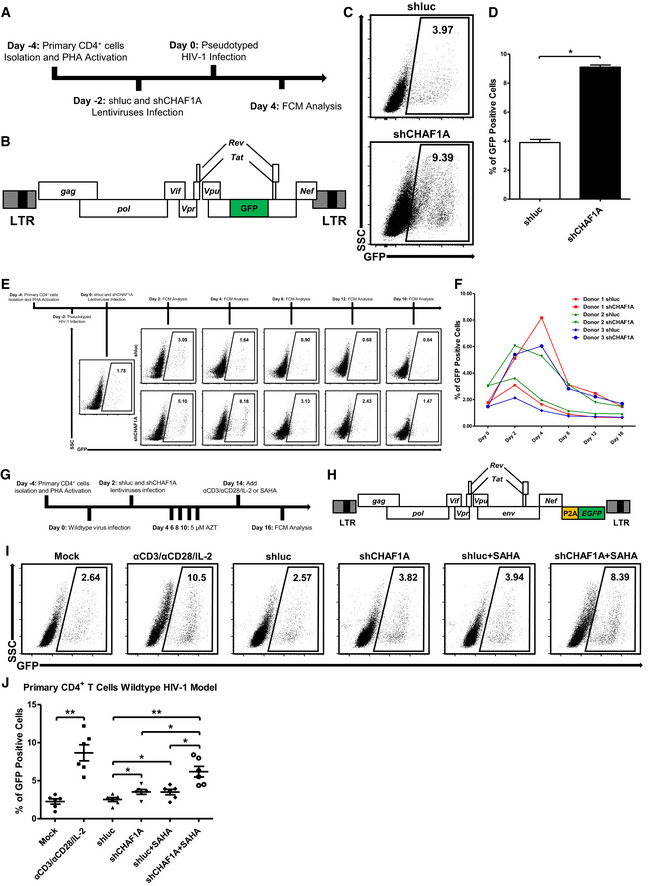
-
AThe procedure of CHAF1A‐mediated HIV‐1 integration restriction. On Day −4, primary CD4+ T cells were isolated from PBMCs of healthy donors, followed by the activation of PHA. On Day −2, endogenous CHAF1A was knocked down by shCHAF1A lentiviruses. Two days later, shluc‐ and shCHAF1A‐treated cells were proceeded to the infection of pseudotyped HIV‐1. Another 4 days later, the percentages of GFP‐positive cells, which indicated the reactivation efficiency, were measured by FCM.
-
BThe schematic of pseudotyped HIV‐1 backbone used in (A).
-
C, DThe FCM figures and statistical histogram of shluc‐ and shCHAF1A‐treated groups.
-
EThe procedure of CHAF1A‐mediated delay of HIV‐1 entering into latency. On Day −4, primary CD4+ T cells were isolated and activated by PHA. On Day −2, activated CD4+ T cells were washed and infected with pseudotyped HIV‐1. Another 2 days later, cells were infected with shluc or shCHAF1A lentiviruses. In the following 2 weeks, the percentages of GFP‐positive cells were measured by FCM every 4 days.
-
FThe statistical histogram of the results in (E). Data showed results from three different healthy donors.
-
GThe procedure of CHAF1A‐mediated HIV‐1 latency in wild‐type HIV‐1 latency model. The procedure was similar as in Fig 7A, except that the virus used in (G) was wild‐type HIV‐1 and the infected cells were treated with 5 μM AZT during the 2 weeks of resting status.
-
HThe schematic of GFP‐conjugated wild‐type HIV‐1 backbone used in (G).
-
I, JThe representative FCM figures and statistical scatter plot of the results in (G).
Data information: Data in (D) represented mean ± SEM in triplicate. P‐values were calculated by Student’s t‐test. Data in (J) represented mean ± SEM in sextuplicate. P‐values were calculated by Mann–Whitney U‐test. *P < 0.05, **P < 0.01.
CAF‐1 depletion alters the epigenetic status of HIV‐1 promoter
CAF‐1 has been found to recruit several suppressive epigenetic proteins to the LTRs of endogenous retroviruses to maintain suppressive epigenetic marks (Murzina et al, 1999; Reese et al, 2003; Loyola et al, 2009; Cheloufi et al, 2015; Hatanaka et al, 2015; Yang et al, 2015). To identify whether it influenced the epigenetic status of HIV‐1 5′‐LTR, we inspected the global epigenetic marks on HIV‐1 LTR upon CHAF1A knockdown. Based on chromatin immunoprecipitation (ChIP) assay, we observed that CHAF1A was enriched on HIV‐1 LTR, especially the nucleosome one (Nuc‐1) assembly site (Dataset EV1). The enrichment of CHAF1A on HIV‐1 LTR was lost upon CHAF1A knockdown (Figs 1D and EV2A, and Appendix Figs S1F, G and S2A). We found that H3K9me, H3K9me2, and H3K9me3 were significantly decreased in the absence of CHAF1A (Figs 1F, and EV2B and C, and Appendix Fig S2B). CHAF1A depletion also induced significant H4K20me3 downregulation and a slight decrease of H3K27me3 (Figs 1G and EV2D, and Appendix Fig S2C). Conversely, active marks including H3K4me3, H3K9Acetyl, and H3K36me2 were significantly increased when knocking down CHAF1A (Figs 1H and I, and EV2E, and Appendix Fig S2D–G). Besides, we also inspected the DNA methylation level of HIV‐1 LTR upon CHAF1A knockdown in J‐Lat 8.4 (Fig EV2F, and Appendix Fig S1H). The methylation of LTR in J‐Lat 8.4 is much higher than that in other monoclonal latency model cell lines. We found that the depletion of CHAF1A reactivated HIV‐1 expression in J‐Lat 8.4 (Figs 1J and EV2G). Besides, the CpG methylation of LTR was downregulated when knocking down CHAF1A (Figs 1K and EV2H). The downregulation was more significant when combined with the use of 5‐aza‐2‐deoxycytidine (5‐aza‐dC) which was widely used to inhibit DNMT1 methyltransferase activity.
As a complemental experiment to investigate other potential mechanisms which CAF‐1 might adopt to promote HIV‐1 latency, we also inspected the HIV‐1 promoter transcriptional activity by utilizing ATAC‐Seq to probe the chromatin accessibility of HIV‐1 promoter upon CHAF1A knockout. We found that the transposable tag density, which indicated the accessible region, was increased on HIV‐1 promoter when knocking out CHAF1A (Fig 1L). P‐TEFb subunit CDK9, as well as super‐phosphorylated RNAP II, were also significantly enriched on HIV‐1 LTR when depleting CHAF1A (Fig 1M, and Appendix Fig S2H and I). We also investigated which transcription factors were influenced by CHAF1A. We compared global gene expression between wild‐type J‐Lat 10.6 and monoclonal CHAF1A‐depletion J‐Lat 10.6 by RNA‐Seq. The expression levels of FOS and JUN, both of which enhanced HIV‐1 transcription, were significantly upregulated upon CHAF1A depletion (Fig EV2I). Taken together, we hypothesized that CAF‐1 might contribute to HIV‐1 latency by manipulating suppressive epigenetic modifications and transcriptional control.
CAF‐1 enriches many suppressive epigenetic proteins
To elucidate the mechanisms of CAF‐1‐mediated epigenetic and transcriptional suppression of HIV‐1, we utilized target‐specific MS to systematically identify CAF‐1‐bound proteins. To avoid the exposure of degradation signals presented on both CHAF1A and CHAF1B subunits, we overexpressed both subunits in HeLa cells to form heterodimer which stabilized each subunit (Kaufman et al, 1995). Transfected cells were harvested and followed by the enrichment of CHAF1A and CHAF1B as well as corresponding co‐immunoprecipitated proteins. We used SDS–PAGE to separate the enriched proteins and excised distinct bands followed by in‐gel digestion by trypsin (Fig 2A). The digested peptides were characterized by nanoscale LC‐MS/MS and annotated by PEAKS Studio. Nearly 1,664 proteins were identified as candidates enriched by CAF‐1 at a significant threshold below 10−5 (Fig 2B). Most of the enriched proteins were nucleic acid‐binding proteins (Fig 2C). We used STRING network analysis to qualify their interconnectivity with the interaction confidence of 0.7 (Fig 2D). The highly interconnected proteins were further proceeded to k‐means clustering. We found that most of these proteins belonged to four subclusters: chromatin binding, epigenetic modification, chromatin remodeling, and ubiquitylation and SUMOylation (Fig 2E–H). To investigate whether these proteins contributed to HIV‐1 suppression, we knocked down each gene in TZM‐bl cell lines which harbored an integrated luciferase under the control of HIV‐1 promoter. We found that over 70% of genes significantly restricted HIV‐1 promoter‐driven luciferase expression (Fig 2I, Dataset EV2). All of the three subunits of CAF‐1 complex suppressed HIV‐1 expression. Several proteins including SETDB1, SUV39H1, SUV39H2, G9a, GLP, HP1α, HP1β, and HP1γ involved in the establishment and maintenance of H3K9 methylation. Another group of proteins including HDAC1, HDAC2, CHD3, CHD4, MTA2, MBD2 were parts of nucleosome remodeling deacetylase (NuRD) complex which participated in the deacetylation of histone lysines. DNMT1 and MBD1 accomplished and maintained DNA methylation, respectively. Interestingly, H3K27me3 “writer” EZH2, H3K27me3 “reader” CBX4, H3K4me3 “eraser” KDM1A, and H3K36me2 “eraser” KDM2A also inhibited HIV‐1 promoter activity (Fig 2I). Collectively, the above MS analysis and HIV‐1 promoter activity assays were highly consistent with the epigenetic modification variation upon CAF‐1 depletion. We speculated that CAF‐1 recruited multiple suppressive epigenetic modifiers and maintainers to the HIV‐1 LTR and established and maintained the suppressive epigenetic status of HIV‐1 promoter.
Figure 2. CAF‐1 enriches many suppressive epigenetic proteins.
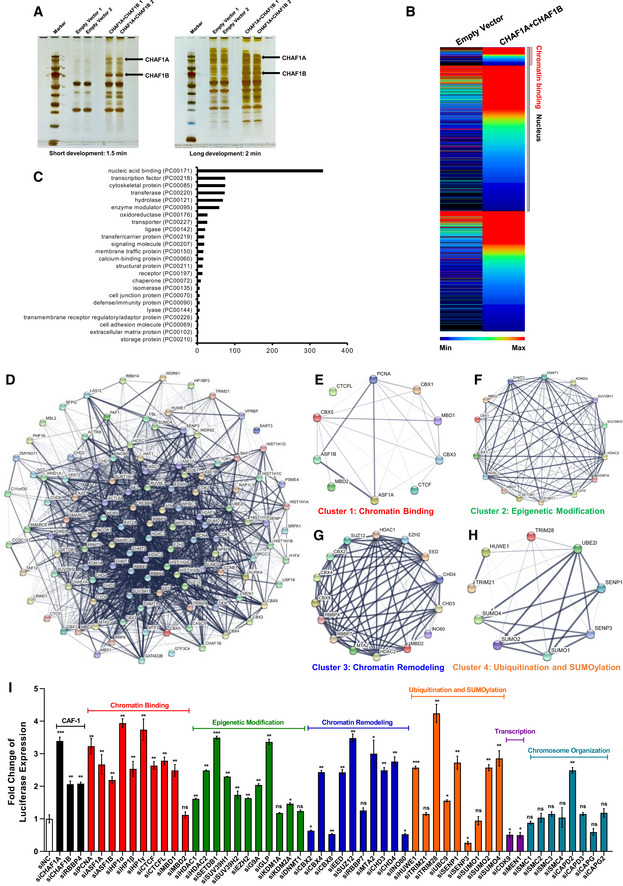
-
AHA‐tagged CHAF1A and CHAF1B were co‐overexpressed in HEK293T cells. Forty‐eight hours post‐transfection, cells were lysed and immunoprecipitated (IP) with anti‐HA beads. The control groups were transfected with empty vector. The IP samples were proceeded to SDS–PAGE and developed with silver staining. Data showed both short and long development results. The whole lane of each group was cut into several gel slices and proceeded to in‐gel digestion and LC‐MS/MS.
-
BThe heatmap of CHAF1A‐ and CHAF1B‐enriched proteins. Gray bar‐covered genes represented nucleus proteins. Pink bar‐covered genes represented chromatin binding proteins.
-
CGene Ontology (GO) analysis of significantly enriched proteins utilizing PANTHER classification system.
-
DSTRING network analysis of enriched proteins with the interaction confidence of 0.7.
-
E–Hk‐means clustering of significantly enriched proteins. Four major clusters were classified: chromatin binding, epigenetic modification, chromatin remodeling, ubiquitination, and SUMOylation.
-
IsiRNAs targeting each CAF‐1 subunits and corresponding enriched genes were transfected into TZM‐bl cell lines, respectively. The fold change of luciferase expression of each groups was normalized to siNC.
Data information: Data represented mean ± SEM in triplicate. P‐values were calculated by Student’s t‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
In addition, CAF‐1 has been found to coexist with SUMO E3 ligase TRIM28 on the LTRs of endogenous retroviruses (Yang et al, 2015). In our MS data, we found that CAF‐1 enriched TRIM28. The Co‐IP assay showed that CAF‐1 bound not only wild‐type TRIM28 but also the SUMOylated TRIM28 (Fig EV2J). We also noticed that transcription factor CDK9 was also among CAF‐1‐enriched candidates. The enrichment was well‐validated by Co‐IP experiment (Fig EV2K). Our group previously found that TRIM28 is recruited to the HIV‐1 LTR to inhibit P‐TEFb‐mediated transcriptional activation through SUMOylating CDK9 (Ma et al, 2019). Therefore, we investigated whether CAF‐1 influenced CDK9 SUMOylation. We found that the depletion of CHAF1A significantly decreased TRIM28‐mediated CDK9 SUMOylation (Fig EV2L). In our preceding data, we had demonstrated that CAF‐1 depletion resulted in higher chromatin accessibility as well as higher CDK9 and super‐phosphorylated RNAP II enrichment on HIV‐1 LTR. Taken together, our results indicated that CAF‐1 also restricted HIV‐1 transcription by recruiting TRIM28 to SUMOylate CDK9.
CAF‐1 forms nuclear bodies
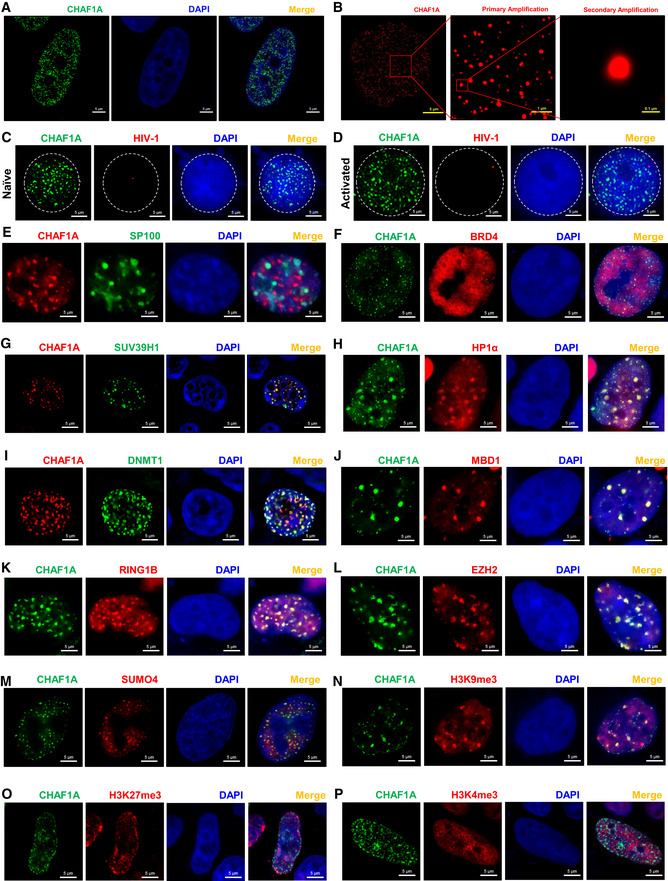
Numerous nuclear bodies have been found to dynamically modulate nuclear organization and function (Mao et al, 2011). To our surprise, CAF‐1 also formed hundreds of nuclear condensates with size ranging from 0.1 to 1.5 μm (Fig 3A, Movie EV1, Appendix Fig S3A and B). CAF‐1 bodies were well‐confirmed in multiple cell types utilizing different fluorescent fusion proteins‐tagged exogenous CHAF1A and different antibodies‐immunofluorescent endogenous CHAF1A (Fig EV3A and B). CAF‐1 bodies were clearly visualized in multiple imaging platforms including confocal microscopy, super‐resolution Structured Illumination Microscopy (SIM), and continuous Stochastic Optical Reconstruction Microscopy (cSTORM) (Fig 3A and B, Movies EV2 and EV3, Appendix Fig S4A and B). The depletion of CHAF1A eliminated CAF‐1 body (Fig EV3C and D). We next conducted immunolabeling‐based fluorescence in situ hybridization (ImmunoFISH) assay to investigate the relationship between CAF‐1 bodies and HIV‐1 provirus (Lusic et al, 2013; Marini et al, 2015). We found that the HIV‐1 genomic DNA was co‐localized with the CAF‐1 body in latent status (Fig 3C, Appendix Figs S3C and S4C). Upon activation, the HIV‐1 genomic DNA was far away from CAF‐1 bodies (Fig 3D, Appendix Figs S3D and S4D). To investigate whether CAF‐1 bodies were novel nuclear foci, we imaged CAF‐1 body with other canonical nuclear bodies simultaneously. We found that CAF‐1 body was distinct from traditional suppressive nuclear bodies including PML body and Cajal body, and classical transcription modulation nuclear bodies including coactivator condensates and RNA Pol II condensates (Figs 3E and F, and EV3, EV4, EV5E and F, Appendix Fig S3E and F). In our preceding MS and Co‐IP data, we have identified many CAF‐1‐enriched proteins which referred to different functional layers. Apart from CHAF1B and RBBP4, which were the other two subunits of CAF‐1, many heterochromatin‐related proteins co‐localized with CHAF1A included H3K9me3 modifiers and maintainers SUV39H1, SUV39H2, HP1α, HP1β, and HP1γ (Figs 3G and H, and EV3, EV4, EV5, Appendix Fig S3G and H), as described previously (Quivy et al, 2004; Houlard et al, 2006). Unsurprisingly, DNA clamp PCNA, DNA methylation modifier DNMT1, and maintainer MBD1 also co‐localized with CHAF1A (Figs 3I and J, and EV3, EV4, EV5M and N, Appendix Fig S3I and J). Interestingly, we also found that some polycomb repressive complex (PRC) proteins including PRC1 components RING1B and CBX4, and PRC2 components EZH2 and SUZ12, were also co‐localized with CHAF1A (Figs 3K and L, and EV3, EV4, EV5O and P, Appendix Fig S3K and L). SUMO paralogs SUMO1, SUMO2, and SUMO4 were also within CAF‐1 bodies (Figs 3M, and EV3, EV4, EV5Q and R, Appendix Fig S3M). Besides, we also inspected the correlation of CAF‐1 body with chromatin compartments. Interestingly, although CAF‐1 body showed significant co‐localization with H3K9me3 other than H3K4me3, CAF‐1 body also showed moderate co‐localization with H3K27me3 (Fig 3N–P and Appendix Fig S3N–P). The quantitative co‐localization analysis results were well consistent with the above imaging data (Appendix Fig S3Q–S). These results indicated that CAF‐1 mediated HIV‐1 latency by forming versatile suppressive nuclear bodies with multi‐layer components.
Figure 3. CAF‐1 forms nuclear bodies.
-
ASuper‐resolution SIM image of CHAF1A in HEK293T cells. DAPI was used to dye DNA which was colored into blue. FITC‐tagged antibody was used to label endogenous CHAF1A which was colored into green.
-
BSuper‐resolution cSTORM image of CHAF1A in HEK293T cells. Two degrees of amplification were performed to show CAF‐1 bodies.
-
C, DImmunoFISH images of CAF‐1 bodies and HIV‐1 genomic DNA within J‐Lat 10.6. Naïve cells were treated with DMSO. Activated cells were acquired by treating with TNFα.
-
ERFP‐tagged CHAF1A and GFP‐tagged PML body component SP100 were co‐overexpressed in HEK293T cells and imaged by SIM.
-
FGFP‐tagged CHAF1A and RFP‐tagged BRD4 were co‐overexpressed in HEK293T cells and imaged by SIM.
-
G–MSUV39H1, HP1α, DNMT1, MBD1, RING1B, EZH2, and SUMO4 were co‐overexpressed with CHAF1A in HEK293T cells, respectively. SIM imaging was performed for each combination.
-
N–PGFP‐tagged CHAF1A was overexpressed in HEK293T cells. CF568‐tagged antibodies against H3K9me3, H3K27me3, and H3K4me3 were used to treat IF samples, respectively. CHAF1A and each histone modification were imaged by SIM.
Data information: The scale bar of each SIM image represented 5 μm. The scale bars of amplified STORM images in (B) represented 1 μm (primary amplification) and 0.1 μm (secondary amplification). All the samples were imaged to obtain at least three images.
CAF‐1 bodies are phase‐separated nuclear condensates
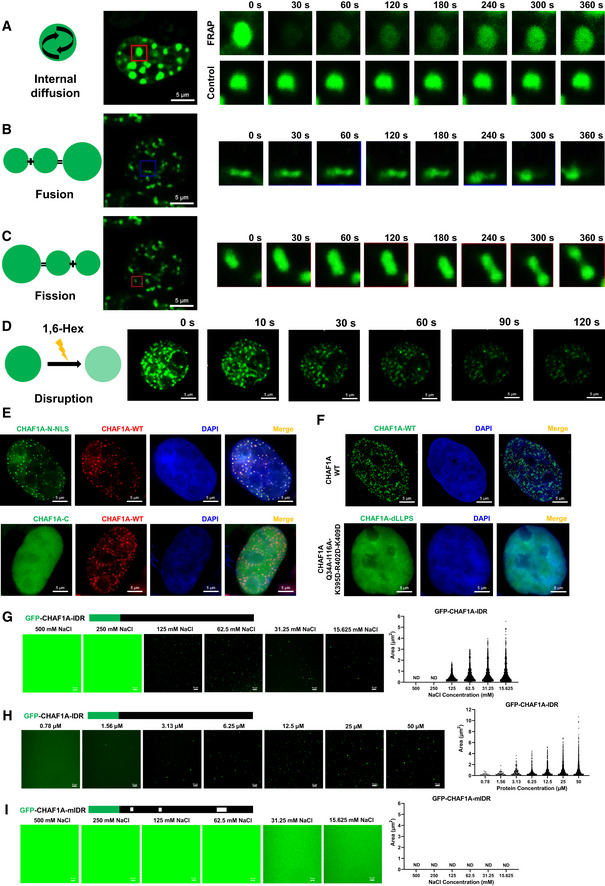
Until now, there is no crystal structure of CHAF1A, although CAF‐1 has been discovered for 30 years (Smith & Stillman, 1989). It has been proposed that the large intrinsically disordered regions (IDRs) within protein often result in the failure of crystallization (Dunker et al, 2001). We found that 72.47% of CHAF1A protein sequence was disordered, which belonged to six IDRs (Appendix Fig S5A). The weak multivalent interactions mediated by IDRs are the driving force of liquid–liquid phase separation (LLPS) (Brangwynne et al, 2015). Therefore, we speculated that CAF‐1 nuclear bodies might be coalesced through LLPS. We found that CAF‐1 bodies were spherical (Appendix Fig S4E). We performed fluorescence recovery after photobleaching (FRAP) experiment on GFP‐tagged CHAF1A and found that the fluorescence recovered quickly, which indicated that CAF‐1 bodies were internally diffused (Fig 4A and Appendix Fig S4F). Besides, we observed that two CAF‐1 bodies were able to fuse together (Fig 4B). Conversely, one CAF‐1 body was able to split into two smaller ones (Fig 4C). Internal diffusion, fusion, and fission were typical features of liquid‐like condensates (Alberti et al, 2019). To investigate whether CAF‐1 bodies were sensitive to 1,6‐hexanediol which was a common compound used to disrupt phase‐separated condensates, we treated GFP‐CHAF1A‐expressing cells with 1,6‐hexanediol and found that CAF‐1 bodies were dissolved quickly upon treatment (Fig 4D) (Sabari et al, 2018). We also evaluated CAF‐1 bodies phase separation upon the treatment with the crowding agent PEG, sucrose, and sorbitol, all of which were known to destabilize weak electrostatic interactions involved in proteins phase separation (Cai et al, 2019; Lu et al, 2020). We found that CAF‐1 bodies within cells existed in physiological condition and were unchanged upon the hyperosmotic stresses of PEG, sucrose, or sorbitol treatment (Appendix Fig S4G–J). All of the above results indicated that CAF‐1 body was liquid–liquid phase‐separated condensates.
Figure 4. CAF‐1 bodies are phase‐separated nuclear condensates.
-
AFRAP images of CAF‐1 bodies to indicate the internal diffusion property. GFP‐tagged CHAF1A was overexpressed in HEK293T cells. Twenty‐four hours later, live cells were proceeded to time series imaging. One of the CAF‐1 bodies was bleached with intense laser pulse. Images of live cell were captured every 4 s. One of the unbleached CAF‐1 bodies was used as control. Data represented eight time points. At least three cells of each sample were treated with intense laser pulse to obtain bleached CAF‐1 bodies.
-
BRepresentative images of two CAF‐1 bodies fusing into one.
-
CRepresentative images of one CAF‐1 body splitting into two.
-
DLive cells were imaged every 2 s in time series. 1,6‐hexanediol was added into the cell incubation well carefully at time point of 5th second. Images were captured for 3 min.
-
EGFP‐tagged CHAF1A‐N‐NLS and CHAF1A‐C were imaged with RFP‐tagged CHAF1A, respectively. N: CHAF1A N‐terminal. NLS: nuclear localization signal of CHAF1A, which was presented within C‐terminal. C: CHAF1A C‐terminal.
-
FUpper panel showed WT‐CHAF1A bodies. Lower panel showed dLLPS‐CHAF1A which was CHAF1A‐Q34A‐I116A‐K395D‐R402D‐K409D.
-
G–IGFP‐tagged CHAF1A‐IDR and CHAF1A‐mIDR were purified in vitro. Ten micromole of each protein was incubated with gradient droplet formation buffer. The droplets of CHAF1A‐IDR appeared in NaCl concentration of 125 mM or lower (G). No CHAF1A‐mIDR droplet was detected within any NaCl concentration (I). The volumes of CHAF1A‐IDR droplets increased with the increase of protein concentration (H). Left subpanels within each panel represented images of protein droplets in different buffers. Right subpanels within each panel represented statistical analysis results of droplets areas and numbers.
Data information: The scale bar of images in (A–F) represented 5 μm. The scale bar in (G–I) represented 10 μm.
To identify the key protein segments or amino acids which mediated the LLPS of CAF‐1 body, we constructed over 90 CHAF1A mutants and captured over 150 corresponding super‐resolution SIM images (Dataset EV3, Appendix Fig S5). The bimolecular fluorescence complementation (BiFC) results showed that CHAF1A biomacromolecules were brought into close proximity, which indicated that the coalescence of CHAF1A molecules was not indirect (Appendix Fig S6A). Both the N‐ and C‐terminals of CHAF1A were able to co‐immunoprecipitated with full‐length CHAF1A (Appendix Fig S6B). However, only the N‐terminal of CHAF1A formed similar nuclear puncta and co‐localized with CAF‐1 body (Fig 4E). The N‐terminal of CHAF1A harbored three IDRs which we named disordered segment 1 (DS1), DS2, and DS3. We found that neither the sole deletion of each DS nor the simultaneous deletion of two DSs dissolved CAF‐1 body. Thus, we inspected the LLPS ability of each nuclear localization signal (NLS)‐conjugated DS. We found that all of the three DSs within N‐terminal were able to form nuclear puncta independently, although the size and number of these bodies have changed slightly (Dataset EV3). Subsequently, we inspected the key amino acids which mediated LLPS within each DS in the absence of the other two DSs. We found that the PCNA‐interacting protein motif 1 (PIP1) within DS1, the SUMO‐interacting motif (SIM) within DS2, and the KER domain within DS3 contributed to the LLPS of each DSs, respectively. Q34A (the thirty‐fourth Glutamine residue substituted with Alanine residue) or F39A mutation within PIP1 dissolved DS1‐mediated nuclear puncta formation. Any of the SIM residue mutations which were I116A, I117A, D118A, L119A, and T120A was able to dissolve DS2‐mediated droplets formation. Based on the prediction, KER within DS3, which is interspaced with many lysine, glutamic acid, and arginine residues, tended to form a coiled‐coil structure (Appendix Fig S6C) (Lupas et al, 1991; Wolf et al, 1997). The coiled‐coil has been found to promote the LLPS of PCA nuclear body in Arabidopsis (Fang et al, 2019). We speculated that kinking the coiled‐coil might be able to dissolve nuclear bodies. Therefore, we substituted residues within the coiled‐coil interface with negatively charged aspartic acid (D) residues, which broke the coiled‐coil structure (Straussman et al, 2007). We found that the simultaneous mutation of K395D, R402D, and K409D within KER was able to dissolve DS3‐mediated nuclear body formation. Eventually, we combined the mutations within each DS and found that five mutations which were Q34A, I116A, K395D, R402D, and K409D were able to dissolve CAF‐1 bodies (Fig 4F).
To identify whether CHAF1A IDRs formed phase‐separated droplets in vitro, we purified the N‐terminal of CHAF1A (CHAF1A‐IDR) and each DS within N‐terminal (CHAF1A‐DS1, CHAF1A‐DS2, and CHAF1A‐DS3) (Appendix Fig S4K). We found that CHAF1A‐IDR, CHAF1A‐DS1, and CHAF1A‐DS3 were able to form droplets along with the decrease of NaCl concentration (Fig 4G, Appendix Fig S6D, E and H). CHAF1A‐IDR formed bigger droplets in higher protein concentrations (Fig 4H). The disability of CHAF1A‐DS2 droplet formation was not surprising as the SIM‐mediated droplet formation relied on stoichiometrically sufficient SUMO proteins (Appendix Fig S6G) (Banani et al, 2016). We also purified the corresponding segment mutants (CHAF1A‐mIDR, CHAF1A‐mDS1, and CHAF1A‐mDS3) (Appendix Fig S4K). These mutants harbored mutations which dissolved CAF‐1 bodies in vivo. We found that these mutated CHAF1A segments were unable to form droplets in vitro, either (Fig 4I, Appendix Fig S6F and I). The above results further confirmed that CAF‐1 bodies were coalesced directly by multivalent interactions of key residues within CHAF1A molecules.
CHAF1A is the leading factor of CAF‐1 body
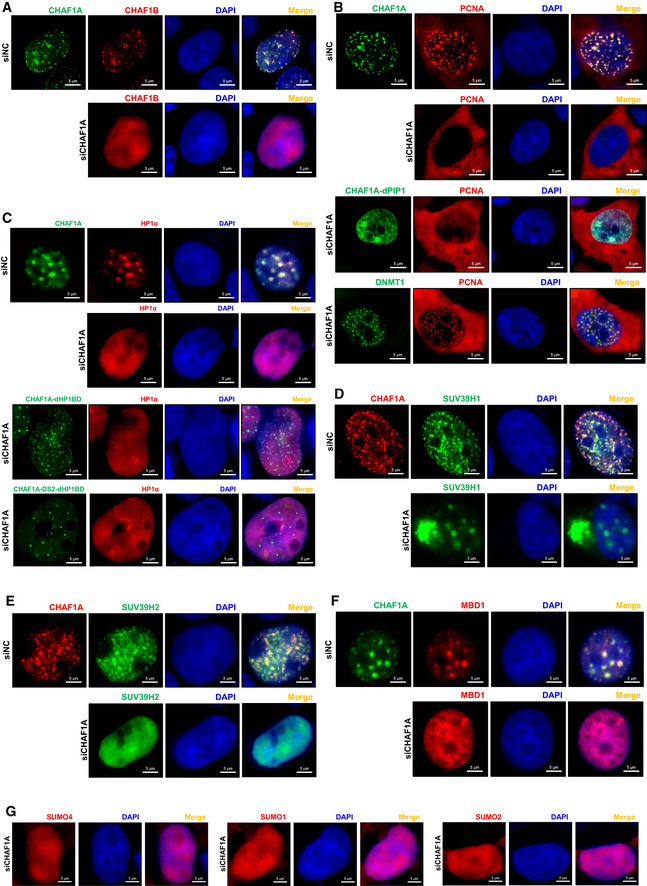
To investigate whether other CAF‐1 body components would influence CHAF1A‐mediated LLPS of CAF‐1 body, we knocked down each CHAF1A‐enriched protein. We found that CAF‐1 bodies were not dissolved in the absence of each enriched protein (Dataset EV4). CAF‐1 bodies were not influenced by other kinds of nuclear bodies, either. On the contrary, the depletion of CHAF1A significantly influenced the body formation of other CAF‐1 body components. We found that neither CHAF1B nor PCNA was able to form nuclear puncta in the absence of CHAF1A (Fig 5A and B, Appendix Fig S7A and B). In our preceding results, we have found that the PCNA‐interacting motif PIP1 within CHAF1A partially contributed to the LLPS of CAF‐1 body. Here, we found that PIP1 deletion did not abort CAF‐1 bodies because of the contribution of the other functional domains including SIM and KER within CHAF1A (Fig 5B, Appendix Fig S7C). However, PCNA‐formed nuclear bodies disappeared in the absence of PIP1. The PIP2 which was located after KER domain did not contribute to the body formation of CHAF1A (Fig EV4A). Interestingly, PCNA bodies were rescued by overexpressed DNMT1 in the absence of CHAF1A (Fig 5B, Appendix Fig S7D). We also found that HP1α molecules were diffused within nuclei in the absence of CHAF1A, although HP1α has been found to undergo LLPS previously (Fig 5C, Appendix Fig S7E; Larson et al, 2017; Strom et al, 2017). The similar phenomenon of CHAF1A depletion‐mediated HP1α bodies alteration was also found in mouse embryonic stem cells (Houlard et al, 2006). The mutation of the HP1‐binding domain (HP1BD) within CHAF1A did not dissolve CHAF1A‐mediated body formation but eliminated HP1α bodies (Fig 5C, Appendix Fig S7F, G and N). The size and number of SUV39H1 bodies were dramatically changed when knocking down CHAF1A, resulting in much less and bigger SUV39H1 nuclear bodies (Fig 5D, Appendix Fig S7H). Large amounts of SUV39H1 molecules were even hijacked within cytoplasm and formed big speckles (Appendix Fig S7O and P). Without CHAF1A, both SUV39H2 and MBD1 bodies were also disappeared (Fig 5E and F, Appendix Fig S7I, J and Q). The quantitative co‐localization analysis results were well consistent with the above imaging data (Appendix Fig S7K–Q). Based on our above results, CAF‐1 bodies might coalesce SUV39H1, SUV39H2, and HP1 paralogs to accomplish the maintenance of heterochromatin. Previously, we have found that CHAF1A co‐localized with SUMO molecules. SIM within DS2 partially contributed to the CAF‐1 body formation. Here, we found that without DS1 and DS3, DS2‐formed nuclear bodies co‐localized with three SUMO analogs (Fig EV4B). The deletion of SIM dissolved DS2‐mediated body formation (Fig EV4C). Upon CHAF1A depletion, none of the SUMO paralogs was able to form nuclear puncta (Fig 5G). These results confirmed that DS2‐mediated body formation was SUMO‐dependent, and CAF‐1 might manipulate nuclear SUMOylation apparatus by forming SUMOylation‐dependent bodies. Taken together, these results indicated that the core factor of CAF‐1 body was CHAF1A, the absence or mutation of which influenced the body formation of several enriched proteins.
Figure 5. CHAF1A is the leading factor of CAF‐1 body.
-
AThe distribution of CHAF1B in the presence of CHAF1A (upper panel) and in the absence of endogenous CHAF1A (lower panel).
-
BFirst panel: the co‐localization of PCNA and CHAF1A. Second panel: the distribution of PCNA in the absence of endogenous CHAF1A. Third panel: the distribution of PCNA in the presence of CHAF1A‐dPIP1 mutant and the absence of endogenous CHAF1A. Fourth panel: the co‐localization of PCNA and overexpressed DNMT1 in the absence of endogenous CHAF1A.
-
CUpper two panels: the distribution of HP1α in the presence or absence of CHAF1A. Lower two panels: the distribution of HP1α in the presence of CHAF1A‐dHP1BD or CHAF1A‐DS2‐dHP1BD. Both images of lower two panels were in the absence of endogenous CHAF1A.
-
D–FThe distribution of SUV39H1 (D), SUV39H2 (E) and MBD1 (F) in the presence or absence of CHAF1A.
-
GThe distribution of SUMO paralogs in the absence of CHAF1A.
Data information: The scale bar within each image represented 5 μm.
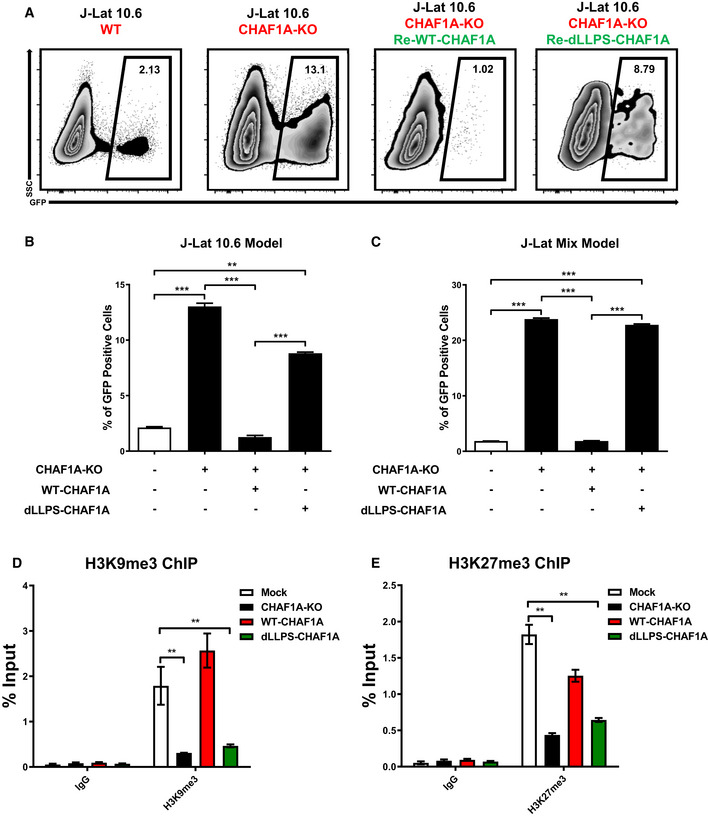
As the CAF‐1 body coalesced multiple suppressive proteins to epigenetically and transcriptionally promote HIV‐1 latency, we wondered whether the deficiency of LLPS property of CAF‐1 body would influence its function on HIV‐1. We performed rescue experiments in a monoclonal CHAF1A knockout cell line named P1D9. The reintroduce of wild‐type CHAF1A rescued HIV‐1 suppression to the basal level (Fig 6A and B). However, the LLPS‐deficient CHAF1A, which harbored five mutations dissolved CAF‐1 body, was unable to suppress HIV‐1. These results were well‐repeated in heterogeneous Jurkat latency model (Fig 6C). To identify whether LLPS‐deficient CHAF1A influenced histone modifications, we inspected the epigenetic status of HIV‐1 LTR upon different CHAF1A‐expressing situations. CHAF1A‐KO‐mediated H3K9me3, H3K27me3, and H4K20me3 downregulation was rescued upon the reintroduce of wild‐type CHAF1A (Figs 6D and E, and EV4D and E). However, the reintroduction of LLPS‐deficient CHAF1A was unable to rescue the downregulation in CHAF1A‐KO cells. Besides, we also found that wild‐type CHAF1A, instead of LLPS‐deficient CHAF1A, was able to rescue CHAF1A‐KO‐mediated H3K9Acetyl upregulation (Fig EV4F). The LLPS deficiency of CAF‐1 body depended on CHAF1A mutation which also disabled the body formation of other CAF‐1 body components as we have mentioned above. Thus, the dysfunction of the LLPS‐deficient CAF‐1 body on HIV‐1 latency was a comprehensive perturbation which was triggered by the LLPS‐deficient CHAF1A mutants.
Figure 6. LLPS‐deficient CAF‐1 body is unable to mediate HIV‐1 latency.
-
AFlow cytometry figures of CAF‐1 body rescue experiment. First figure: Wild‐type J‐Lat 10.6. Second figure: CHAF1A knockout J‐Lat 10.6. Third figure: CHAF1A‐KO J‐Lat 10.6 was re‐introduced with wild‐type CHAF1A. Fourth figure: CHAF1A‐KO J‐Lat 10.6 was re‐introduced with dLLPS‐CHAF1A which harbored five mutations. The percentages of GFP‐positive cells were labeled in the top right corner of each figure.
-
BThe statistical histogram of the results in (A).
-
CRescue experiment in heterogeneous Jurkat latency model which were performed as in (A).
-
D, EThe enrichment of H3K9me3 and H3K27me3 on HIV‐1 LTR in different CHAF1A status. The reactivation efficiency in each group was indicated by the percentage of GFP‐positive cells.
Data information: Data represented mean ± SEM in triplicate. P‐values were calculated by Student’s t‐test. **P < 0.01, ***P < 0.001.
CHAF1A depletion reactivates latent HIV‐1 from HIV‐1‐infected individuals
To investigate whether CAF‐1 body promoted the establishment and maintenance of HIV‐1 latency in latently infected primary CD4+ T cells, we knocked down CHAF1A in several primary CD4+ T‐cell latency models and resting CD4+ T cells isolated from HIV‐1‐infected individuals. Firstly, we evaluated whether CAF‐1 bodies contributed to the establishment of HIV‐1 latency in CD4+ T cells by knocking down CHAF1A followed by pseudotyped HIV‐1 infection. In our preceding ATAC‐Seq experiment, we have found that the depletion of CHAF1A resulted in higher chromatin accessibility, which might facilitate HIV‐1 integration. Here, we found that the percentage of pseudotyped virus‐infected CD4+ T cells was much higher in CHAF1A depletion group than that in control group (Fig EV5A–D, Appendix Fig S8A and B). The absence of CHAF1A also significantly delayed pseudotyped HIV‐1 entering into latency (Fig EV5E and F, Appendix Fig S8C). These results indicated that the vanguard of CAF‐1 body could be a prerequisite to effectively establish HIV‐1 latency. Subsequently, we evaluated whether CAF‐1 body contributed to the maintenance of HIV‐1 latency by firstly infecting HIV‐1 followed by depleting CHAF1A. We found that the absence of CHAF1A significantly upregulated the expression of latently infected pseudotyped HIV‐1 (Fig 7A–C, Appendix Fig S8D). The upregulation of HIV‐1 expression was enhanced much higher when supplemented with LRA SAHA. We also constructed a wild‐type HIV‐1 latency model by infecting activated CD4+ T cells with HIV‐1, followed by the treatment of AZT to prevent persistent infection. Both envelope and Nef of the virus used here were intact. The expression of GFP, which was in frame with Nef and spaced by a P2A sequence, indicated the intensity of HIV‐1 latency reactivation. We found that CHAF1A depletion also significantly reactivated wild‐type latent HIV‐1 (Fig EV5G–J, Appendix Fig S8E). The combination of CHAF1A knockdown and SAHA stimulation were able to activate more viruses. To confirm the contribution of CAF‐1 body on HIV‐1 latency in resting CD4+ T cells isolated from HIV‐1‐infected individuals who underwent suppressive cART for over 6 months, we knocked down CHAF1A in latently infected CD4+ T cells. The amount of intracellular HIV‐1 RNAs significantly increased upon αCD3/αCD28 stimulation, which indicated active HIV‐1 expression (Fig 7D, Appendix Fig S8F). CHAF1A depletion resulted in the reactivation of considerable amount of HIV‐1 RNAs. The combination of CHAF1A knockdown and SAHA stimulation was able to reactivate more HIV‐1 RNAs compared with individual intervention. To prove that the RNAs of reactivated HIV‐1 were not transcribed from a few of integrated HIV‐1, we quantified the genetic diversities of reactivated HIV‐1 of different groups. The genetic diversity potentially reflected the diversity of HIV‐1 integration sites. We found that the depletion of CHAF1A was able to reactivate more genetically diversified HIV‐1 than SAHA (Fig 7E). The genetic diversity indexes of combined usage of CHAF1A depletion and SAHA stimulation were much higher than separate treatment only. Taken together, these results indicated that CAF‐1 body promoted the establishment and maintenance of HIV‐1 latency in latently infected CD4+ T cells.
Figure 7. CHAF1A depletion reactivates latent HIV‐1 from HIV‐1‐infected individuals.
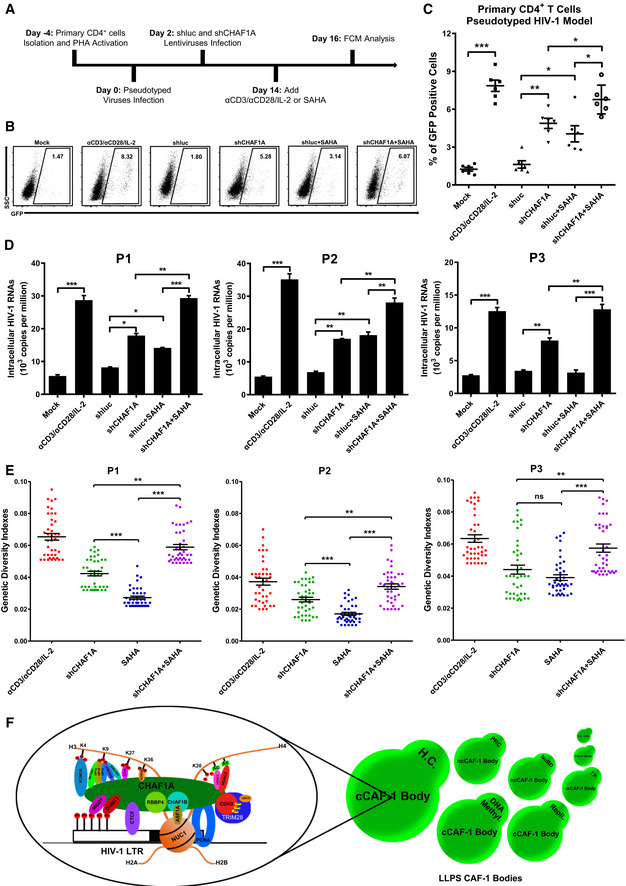
- The procedure of CHAF1A depletion‐mediated HIV‐1 reactivation in primary CD4+ T cells. On Day −4, primary CD4+ T cells were isolated from PBMCs of healthy donors, and activated by PHA. On Day 0, activated CD4+ T cells were washed and infected with pseudotyped HIV‐1. Another 2 days later, cells were treated with shluc and shCHAF1A lentiviruses, respectively. Two weeks later, one part of naïve CD4+ T cells was treated with αCD3/αCD28/IL‐2. One part of shluc‐treated cells and one part of shCHAF1A‐treated cells were further treated with 500 nM SAHA. On day 16, all the cells were proceeded to FCM analysis.
- The flow cytometry figures of each group. The reactivation efficiency was indicated by the GFP‐positive cells percentages which were labeled on the top right corner.
- The statistical scatter plot of the results in (B).
- The amounts of intracellular HIV‐1 RNAs of latently infected primary CD4+ T cells which were isolated from PBMCs of HIV‐1‐infected individuals. αCD3/αCD28/IL‐2 treatment was performed as positive control. shCHAF1A lentiviruses were packaged to knock down endogenous CHAF1A. shluc lentiviruses treatment was performed as negative control. SAHA was added as LRA supplement.
- Envelope V1–V3 region of HIV‐1 RNAs in (D) was reverse‐transcribed and PCR‐amplified. The PCR products were proceeded to TA‐cloning. At least 60 single clones were picked from each group and sequenced. These sequences and the standard HIV‐1 sequence HXB2 were aligned. Genetic diversity index of each sequence was calculated for each group.
- The schematic of CAF‐1 body‐mediated HIV‐1 latency. Detailed information for the schematic was indicated in discussion.
Data information: Data in (C) represented mean ± SEM in sextuplicate. P‐values were calculated by Mann–Whitney U‐test. Data in (D) represented mean ± SEM in triplicate. P‐values were calculated by Student’s t‐test. Data in (E) represented mean ± SEM (n = 40 for each group within each patient sample). P‐values were calculated by Mann–Whitney U‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
CAF‐1 was originally identified as a factor affecting chromatin assembly by depositing newly synthesized H3‐H4 onto replicating DNA. It was eventually re‐defined as a chromatin silencing complex to orchestrate heterochromatin maintenance (Smith & Stillman, 1989; Murzina et al, 1999; Quivy et al, 2004; Houlard et al, 2006; Loyola et al, 2009; Wang et al, 2018). Utilizing multitudes of modern biochemical and high‐through sequencing technologies, a previous study has unveiled its delicate roles in safeguarding cell identity (Cheloufi & Hochedlinger, 2017). The manipulation of CAF‐1 dramatically facilitates cell reprogramming and lineage conversion (Tursun et al, 2011; Cheloufi et al, 2015). Besides, the depletion of CAF‐1 results in the expression of endogenous retrotransposons and endogenous retroviruses in mouse pluripotent stem cells (Hatanaka et al, 2015; Yang et al, 2015). In this study, we identified that CAF‐1 also suppresses the invaded exogenous HIV‐1 and ultimately promotes HIV‐1 latency by forming versatile suppressive nuclear bodies with LLPS properties.
Multi‐component CAF‐1 bodies with multi‐functions
Many independent studies which reported previously have unveiled the enigmatic function of CAF‐1 on cellular processes within different cell types. Our systematic investigation of CAF‐1‐mediated HIV‐1 latency has significantly extended CAF‐1 function on exogenous viruses in differentiated cells. Our microscopic imaging and physiochemical studies showed that CAF‐1 forms nuclear bodies which contain multiple suppressive proteins besides conventional CAF‐1 subunits (CHAF1A, CHAF1B, and RBBP4). Although CHAF1A is the largest subunit of CAF‐1 body and is not an enzyme, it acts as a landing pad to recruit many enzymes which refer to multiple functional layers. Specific genes and HIV‐1 LTR can be recognized by CAF‐1 body components which coalesce other body components. Based on our investigation, we propose a model of CAF‐1 body‐mediated HIV‐1 latency (Fig 7F). CAF‐1 is enriched on HIV‐1 LTR and forms liquid‐like nuclear condensates by three distinct IDRs of CHAF1A subunit. Multiple suppressive epigenetic modifiers and maintainers, which refer to different epigenetic modulation layers, are recruited to HIV‐1 LTR region lead by CAF‐1 body with LLPS properties. Consequently, HDAC1 deacetylates acetyls from newly assembled or pre‐existing histone lysine residues. SETDB1, SUV39H1, or SUV39H2 modify deacetylated histone lysine residues with H3K9me, H3K9me2, and H3K9me3. EZH2 modifies deacetylated lysine residues with H3K27me3. CAF‐1 body recruits KDM1A and KDM2A to demethylate active marks H3K4me3 and H3K36me2, respectively. Suppressive epigenetic mark H4K20me3 is also maintained by CAF‐1 body on HIV‐1 LTR. Apart from histone modifications, CAF‐1 body also significantly co‐localized with DNMT1 which methylates DNA CpGs on HIV‐1 LTR. The above established suppressive modifications are further maintained by corresponding “readers”. H3K9me3 is maintained by HP1α, HP1β, and HP1γ, while CpGs methylation is maintained by MBD1.
Interestingly, we also noticed that CAF‐1 body recruits H3K27me3 modifier EZH2 and maintainer CBX4, although CAF‐1 body shows only moderate co‐localization with H3K27me3 foci. We propose that CAF‐1 bodies may recruit EZH2 to establish moderate H3K27me3 without forming H3K27me3‐mediated heterochromatin. Another possibility is that CAF‐1 bodies may utilize EZH2 to establish H3K27me3 foci in adjacent chromatin, as we have found that H3K9me3 foci and H3K27me3 foci are usually adjacent to each other. We also found that CAF‐1 body facilitates H4K20me3 modification, although CAF‐1 body does not co‐localize with H4K20me3, the modifier of which should be investigated in the future.
Protein modifications also contribute to the heterogeneity of multi‐component CAF‐1 bodies. We found that the SIM of CHAF1A contributes to the DS2‐mediated LLPS, the recruitment of TRIM28 on HIV‐1 LTR, and the co‐localization with SUMO paralogs. CHAF1A itself can also be SUMOylated, which may facilitate SUMOylation‐dependent proteins recruitment (Xiao et al, 2015). Many CAF‐1 body components also can be SUMOylated based on our previous MS data (Ma et al, 2019). The SUMO puncta are dissolved upon CHAF1A depletion. Besides, we have found that TRIM28 and SUMO molecules mediate the transcriptional‐pausing of RNAP II on HIV‐1 LTR (Ma et al, 2019). The wild‐type and SUMOylated TRIM28 are recruited by CAF‐1 body to hijack P‐TEFb, resulting in transcriptional suppression of HIV‐1.
CAF‐1 bodies and chromatin biological processes
Recent work on the LLPS‐mediated gene regulation has shown that coactivators including MED1 and BRD4, and transcription factors including OCT4 and GCN4, form phase‐separated transcriptional condensates to efficiently facilitate gene expression. Besides, P‐TEFb recruits RNAP II to a phase‐separated compartment and enable efficient Pol II hyperphosphorylation and Pol II‐mediated elongation. Several chromosome conformation capture (3C) work indicates that different distal genes are recruited together by transcriptional machinery and simultaneously regulated by shared distal DNA elements within same TADs and shared DNA loops. All the work indicates that phase‐separated proteins form big nuclear condensates and coalesce distal genes and DNA elements to efficiently manipulate the expression of multiple genes. Previous work has shown that CAF‐1 promotes heterochromatin formation (Quivy et al, 2004; Houlard et al, 2006; Loyola et al, 2009; Wang et al, 2018). Our physicochemical work here indicates that CAF‐1 bodies recruit several H3K9me3 modifiers and maintainers and show significant co‐localization with these epigenetic proteins and corresponding heterochromatin marks H3K9me3. In addition, we found that CAF‐1 bodies also recruit and co‐localize with PRC components EZH2 and CBX4, which “writes” and “reads” heterochromatin marks H3K27me3, respectively. Besides, CAF‐1 bodies also show moderate co‐localization with H3K27me3. H3K9me3 and H3K27me3 are typical marks of constitutive heterochromatin and facultative heterochromatin, respectively (Trojer & Reinberg, 2007; Saksouk et al, 2015). The prerequisite of both heterochromatins is hypoacetylation (Casas‐Delucchi et al, 2012; Watts et al, 2018). As CAF‐1 also maintains histone hypoacetylation by recruiting corresponding modifiers HDAC1 and HDAC2, we believe that CAF‐1 bodies participate in the formation of both constitutive and facultative heterochromatins. Taken together, CAF‐1 bodies form numerous liquid‐like nuclear condensates and recruit different epigenetic proteins and could subsequently lead to chromatin compaction and formation of heterochromatin. Thousands of genes are fitted within the small nucleus and positioned in different nuclear compartments. It will be extremely inefficient to regulate each gene individually. Based on our work, we suspect that phase‐separated nuclear condensates guided by CAF‐1 could facilitate the formation of 3D genomic architecture by condensing the chromatin and promoting the compartmentalization of chromatin and synergistically regulate multiple genes, which greatly increases the efficiency of gene regulation.
Canonical and non‐canonical CAF‐1 bodies
None of proteins exists in isolation in the intracellular environment. They form a large internal interaction network and regulate cellular processes synergistically. If each kind of nuclear proteins forms thousands of isolated nuclear puncta, the nucleus would be extremely crowd and various biological processes will be restricted. Thus, there may be some core factors which regulate the liquid‐like phases of the others. All these nuclear bodies could exist semi‐discontinuously and shuttle between different condensates. We have found that the size and distribution of CAF‐1 bodies are variable, and the major component of CAF‐1 body is CHAF1A which possesses LLPS properties and influences the body formation of many recruited proteins. However, some components such as HP1α, SUV39H1, and TRIM28 also have been reported to possess LLPS properties and form distinct liquid puncta in vitro. Bioinformatics prediction has shown that many of the CAF‐1 body components harbor large IDRs and have the potential of phase separation. The multi‐phasic property of CAF‐1 hetero‐condensates indicates that CAF‐1 bodies may be semi‐discontinuous. Combined with the visualization data reported previously and newly found imaging results which we reported here, we propose that CAF‐1 bodies are kinds of multi‐layer complexes which include both canonical and non‐canonical CAF‐1 bodies (Fig 7F). We found that CHAF1A shows significant co‐localization with HP1α, HP1β, HP1γ, SUV39H1, and SUV39H2, as well as corresponding H3K9me3. The depletion of CHAF1A disables the body formation of these proteins. Although SUV39H1 body still exists upon CHAF1A depletion, its number and shape have significantly changed. We cannot exclude the possibility that SUV39H1 enters into another liquid‐like hetero‐condensates. Thus, these H3K9me3‐related proteins and CHAF1A form the canonical CAF‐1 bodies which could mediate heterochromatin formation, more specifically, H3K9me3‐containing constitutive heterochromatin formation. Another canonical CAF‐1 body refers to DNA methylation. The depletion of CHAF1A dissolves PCNA bodies and Methyl‐DNA reader MBD1 bodies. Conversely, the depletion of DNMT1 does not dissolve MBD1 bodies and MBD1 bodies do not co‐localize with DNMT1, either. Interestingly, we found that the DNMT1 body is unchanged upon CHAF1A depletion, although they show significant co‐localization. Besides, the CHAF1A depletion‐mediated the elimination of PCNA body can be rescued by overexpressed DNMT1. These results indicate that DNMT1 may also have the LLPS property and substitute CHAF1A to function on PCNA, resulting in the entry of PCNA into DNMT1‐guided bodies. CHAF1B and PCNA also show significant co‐localization with CHAF1A and are well‐characterized CHAF1A partners which mediate DNA replication and histone deposition. Obviously, they represent another canonical CAF‐1 bodies which mediate replication.
CHAF1A also guides the formation of many non‐canonical CAF‐1 bodies. We found that CHAF1A co‐immunoprecipitates and co‐localizes with several PRC components including RING1B, CBX4, EZH2, and SUZ12. CHAF1A also shows moderate co‐localization with H3K27me3. The depletion of CHAF1A also induces the demethylation of H3K27 on HIV‐1 LTR, which is unable to be rescued by LLPS‐deficient CHAF1A. These results indicate that CAF‐1 also participates in the maintenance of H3K27me3. Recently, CAF‐1 also has been reported to participate in the establishment of H3K27me3 during cell fate determination (Cheng et al, 2019). However, the PRC components and corresponding H3K27me3‐containing heterochromatin still form nuclear puncta upon CHAF1A depletion, although we cannot exclude the possibility that the depletion of CHAF1A may induce the re‐distribution of H3K27me3. As PRC components also form many non‐classical complexes, we propose that CAF‐1 guides the formation of non‐canonical CAF‐1 bodies, which crosstalk with PRCs and maintain H3K27me3‐containing facultative heterochromatin. Besides, we found that many NuRD complex components including RBBP4, HDAC1, HDAC2, and MBD2 also co‐immunoprecipitate or co‐localize with CHAF1A. Thus, CAF‐1 guides the formation of another non‐canonical CAF‐1 bodies which crosstalk with NuRD, which facilitate the deacetylation of histone lysines.
CAF‐1‐guided body is a core factor contributing to HIV‐1 latency
The concept of “shock and kill” was proposed several years ago (Deeks, 2012). However, we are still far behind to find an efficient and low‐toxic shock agent which is also called latency‐reversing agent (LRA) (Spivak & Planelles, 2018). Almost all of the tested LRAs target only one of the latency contributors. Several lines of evidence have shown that targeting only one contributor will only reactivate few particular HIV‐1. Thus, combined “shock” strategy which utilizes two or more LRAs targeting different latency contributors may greatly reactivate more HIV‐1. However, more LRAs treatment also results in more side effects. Developing LRAs targeting the core factor of HIV‐1 latency should be more valuable.
In our study, we have provided evidence that the phase‐separated CAF‐1 body orchestrates majority, if not all, suppressive proteins to epigenetically and transcriptionally promote HIV‐1 latency. Targeting its core subunit CHAF1A will be able to overcome the restriction of several suppressive epigenetic modifications and TRIM28‐mediated transcriptional‐pausing. Development of LRAs targeting CAF‐1 body will not only remove multiple suppressive epigenetic modifications, but also activate transcriptional initiation and elongation. In addition, the chromatin and genomic architecture around HIV‐1 become more accessible, which facilitates the entry of transcription machinery. If so, latent HIV‐1 could be activated and exposed to immunosurveillance, leading to the full eradication of viral reservoir.
Materials and Methods
Study participants
Blood samples of chronically HIV‐1‐infected participants were obtained from Department of Infectious Diseases in Guangzhou 8th People’s Hospital, Guangzhou. Both the Ethics Review Board of Sun Yat‐sen University and the Ethics Review Board of Guangzhou 8th People’s Hospital approved this study. All the participants were given written informed consent with approval of the Ethics Committees. The enrollment of HIV‐1‐infected participants was based on the criteria of long‐term suppression of HIV‐1 viremia by cART, which means undetectable plasma HIV‐1 RNAs (< 50 copies/ml) and high CD4+ T‐cell number (more than 350 cells/mm3) for at least 6 months.
Blood samples of healthy participants were obtained from Guangzhou Blood Center. We did not have any interaction with the healthy participants or protected information. Therefore, no informed consent was required.
Cell lines and primary cultures
HEK293T, HeLa, and Jurkat cells were obtained from ATCC. TZM‐bl cells were obtained from NIH AIDS Reagent Program. J‐Lat 6.3, 8.4, 9.2, 10.6, and 15.4 cells, which were originally constructed from Dr. Eric Verdin (The Buck Institute for Research on Aging, Novato, CA, USA) Laboratory, were obtained from Dr. Robert F. Siliciano ((Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, USA) Laboratory. Adherent cells including HEK293T, HeLa, and TZM‐bl were cultured in DMEM supplemented with 1% penicillin‐streptomycin (Thermo Fisher) and 10% FBS (Thermo Fisher). Suspension cells including Jurkat, J‐Lat 6.3, 8.4, 9.2, 10.6, and 15.4 were cultured in RPMI 1640 supplemented with 1% penicillin‐streptomycin and 10% FBS.
Peripheral blood mononuclear cells (PBMCs) and primary CD4+ T cells were isolated and purified from study participants and cultured in RPMI 1640 supplemented with 1% penicillin‐streptomycin and 10% FBS. Primary CD4+ T cells were supplied with 1/1,000 ~ 1/10,000 recombinant human interleukin 2 (IL‐2) (R&D) to maintain proliferation viability.
All cells were cultured in sterile incubator at 37°C and 5% CO2. All cells have been tested for mycoplasma utilizing PCR assay and confirmed to be mycoplasma‐free.
Microbe strains
Escherichia coli DH5α (Takara), HB101 (Takara), BL21 (Takara), and Stbl3 (Thermo Fisher) were grown in LB medium with indicated antibiotic at 37°C. DH5α was used to propagate most of the protein‐coding constructs. HB101 and Stbl3 were used to propagate lentivirus‐based constructs. BL21 was used to express and purify prokaryon‐expressed proteins.
HIV‐1 infectious molecular clone pNL4‐3 was obtained from NIH AIDS Reagent Program. All the related engineered clones including pNL4‐3‐P2A‐EGFP (NPG), pNL4‐3‐ΔEnv/EGFP, pNL4‐3‐ΔEnv/ΔNef‐d2EGFP, pNL4‐3‐ΔEnv/d2EGFP‐Nef‐IRES‐Bcl2 (NIB), and pNL4‐3‐ΔEnv/d2EGFP‐Nef‐P2A‐Bcl2 (NPB) were generated from pNL4‐3. All the HIV‐1 clones were amplified and purified from HB101 and used to transfect HEK293T cells. The viral package and infection of these HIV‐1 clones were conducted in Biosafety Level 3 (BSL‐3) Laboratory.
CRISPR‐CAS9‐sgRNA‐mediated knockout
The endogenous CHAF1A in Jurkat cells and corresponding latency cell lines was knocked out by CRISPR‐CAS9 system. sgRNA targeting dummy guide (sgNT: 5′‐ACGGAGGCTAAGCGTCGCAA‐3′) was used as negative control (Sanjana et al, 2014). sgRNAs targeting CHAF1A were screened from six sequences. The most efficient one (sgCHAF1A: 5′‐CTCGGGCCACTCGTCAGCTC‐3′) was chosen and proceeded to conduct all the knockout experiments (Sanjana et al, 2014). Target sequences were cloned into lentiCRISPRv2.0 (Addgene plasmid # 52961). Pseudotyped viruses were packaged in HEK293T cells by co‐transfecting 3 μg of VSV‐G glycoprotein‐expressing vector, 6 μg of lentiviral packaging construct pCMVΔ8.2, and 6 μg of CAS9/sgRNA‐expressing construct lentiCRISPRv2.0 using Lipofectamine 2000 (Thermo Fisher) according to the manufacture’s instruction. VSV‐G glycoprotein‐expression vector was obtained from Addgene (Addgene plasmid # 12259) (Zufferey et al, 1997). pCMVΔR8.2 was a kindly gift from Dr. Didier Trono (School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland) (Zufferey et al, 1997). Viruses were concentrated by PEG 6000. Target cells were spin‐infected with sgNT lentiviruses and sgCHAF1A lentiviruses, respectively. The infected cells were treated with 1 μg/ml puromycin (Sigma‐Aldrich) to eliminate uninfected cells. Three days post‐treatment, cells were transferred into fresh RPMI 1640. The reactivation efficiency was indicated by the percentage of GFP‐positive cells. The knockout efficiency was confirmed by Western blot and genomic DNA PCR. The homogeneous sgCHAF1A cell line was selected by limiting dilution assay. The monoclonal cells were identified by genomic DNA PCR followed by Sanger sequencing. The synergistic reactivation of LRAs and sgCHAF1A to HIV‐1‐infected cells was conducted by treating knockout cell lines with indicated LRAs.
siRNA and shRNA‐mediated knockdown
For the knockdown of CHAF1A, negative control siRNA (siNC) was purchased from RiboBio (Guangzhou, China). siCHAF1A‐1 (5′‐CCGACTCAATTCCTGTGTAAA‐3′), siCHAF1A‐2 (5′‐CCATAAGGTCCGCCAGAAA‐3′), and siCHAF1A‐3 (5′‐GCAGGACAGTTGGAGTGAA‐3′) were used as mixture and have been validated by company to ensure that at least one siRNA was able to knock down CHAF1A up to 75%. siRNAs which were used for knocking down CHAF1A in rescue experiments targeted the 3′UTR of CHAF1A mRNA. These siRNAs included siCHAF1A‐4 (5′‐GCTGGATTAGTTCCTTTGA‐3′), siCHAF1A‐5 (5′‐ATATGAATCTGCCCTTTAA‐3′) and siCHAF1A‐6 (5′‐TAAATGAATTGAAGCGTCA‐3′). siRNA library targeting CAF‐1 body components was purchased from RiboBio (Guangzhou, China). Each gene was knocked down by three different siRNAs. For adherent cells including HEK293T, HeLa, and TZM‐bl, siRNAs were transfected utilizing Lipofectamine RNAiMAX (Thermo Fisher) according to the manufacturer’s instruction. For suspension cells including Jurkat and corresponding latency cell lines, siRNAs were nucleofected utilizing nucleofector solution (Lonza) according to the manufacturer’s instruction. The knockdown efficiency was confirmed by qPCR and Western blot. The percentages of viable cells were quantitated by measuring the percentages of amine‐reactive fluorescent dye non‐permeant cells.
For shRNA‐mediated CHAF1A knockdown, shRNA targeting luciferase (shluc: 5′‐ACCGCCTGAAGTCTCTGATTAA‐3′) was used as negative control (Rousseaux et al, 2018). The shRNA target sequence against CHAF1A mRNA was: 5′‐CCGACTCAATTCCTGTGTAAA‐3′. Target sequences were cloned to pLKO.3G‐RFP which was derived from pLKO.3G (Addgene plasmid # 14748). shRNA lentiviruses were packaged and concentrated as CAS9/sgRNA lentiviruses. Targeted cells were spin‐infected with shRNA lentiviruses. Specific LRAs were added 48 h later or 2 weeks later. The infection efficiency was indicated by the percentage of RFP‐positive cells. The knockdown efficiency was confirmed by qPCR and Western blot. The reactivation efficiency for GFP‐tagged HIV‐1 was validated by the percentage of GFP‐positive cells. The reactivation efficiency for clinical samples was validated by the amount of intracellular HIV‐1 RNAs.
ChIP‐qPCR
Chromatin immunoprecipitation (ChIP) was performed according to the manufacturer’s instruction (CST). Briefly, four million cells were harvested for each immunoprecipitation (IP). Cells were crosslinked with 1% formaldehyde (Sigma‐Aldrich) for 10 min followed by quenching with 125 mM glycine solution for 5 min. Cell pellets which were deposited by centrifuging were lysed in ice‐cold Buffer A (CST) supplemented with DTT and Protease inhibitor cocktail (PIC) for 10 min. The nuclei were pelleted by centrifuging and resuspended in ice‐cold Buffer B (CST) supplemented with DTT. Nuclei pellets were centrifuged down and resuspended in 100 μl of Buffer B supplemented with DTT and 0.5 μl of micrococcal nuclease (CST) per IP preparation. The digestion was performed at 37°C for 20 min followed by stopping with 50 mM EDTA. Pelleted nuclei were resuspended in 100 μl of ChIP Buffer (CST) supplemented with PIC and incubated on ice for 10 min. The nuclei were lysed by sonication with three sets of 20‐s pulses at 40% amplitude. Pellets were cooled on ice for 30 s between pulses. The lysates were clarified by centrifuging for 10 min. One‐tenth of the supernatant which contained digested chromatin was proceeded to DNA purification. The size distribution and concentration of chromatin DNA were determined by electrophoresis and OD260 measurement, respectively.
For each IP reaction, approximately 10 μg of chromatin was diluted into 500 μl of ChIP Buffer supplemented with PIC. Ten microliters of diluted chromatin were used as input sample. Immunoprecipitation antibodies against normal rabbit IgG (CST, 2729), CHAF1A (Proteintech, 17037‐1‐AP), H3K9me (Abcam, ab9045), H3K9me2 (Abcam, ab1220), H3K9me3 (Abcam, ab8898), H4K20me3 (Active Motif, 39671), H3K4me3 (Abcam, ab8580), H3K9Acetyl (Abcam, ab4441), CDK9, Pho‐RNAP II (Abcam, ab5095), H3K27me3 (Abcam, ab6002), H3K36me2 (Abcam, ab9049), Histone H3 (CST, 4620), Histone H4 (Abcam, ab177840), and RNAP II (Abcam, ab26721) were added into each diluted chromatin in indicated concentrations. IP reaction was conducted on rotator at 4°C for at least 6 h. ChIP‐Grade Protein G Magnetic Beads (CST) were added to each IP reaction and incubated for another 2 h at 4°C while rotating. The protein G magnetic beads were pelleted and washed on a magnetic separation rack. Chromatin enriched by protein G magnetic beads was eluted by ChIP Elution Buffer (CST). All the chromatin from each group including input samples was proceeded to DNA purification by DNA purification column.
Target DNA fragments in each group were quantitated by real‐time quantitative PCR. For ChIP‐qPCR in Jurkat and related cells, HIV‐1 provirus fragments were quantitated on the following regions. G5′: Cellular DNA and viral 5′LTR junction; A: Nucleosome 0 assembly site; B: Nucleosome free region; C: Nucleosome 1 assembly site; V5: Viral 5’LTR and gag leader sequence junction; E: envelope region; V3: Viral poly purine tract and 3’LTR junction; G3′: Viral 3’LTR and cellular DNA junction. For ChIP‐qPCR in TZM‐bl cells, A, B, C, V5, and V3 were as in Jurkat cells. G5: Cellular DNA and viral 5′LTR junction; G3: Viral 3′LTR and cellular DNA junction; L: Luciferase region. The primer pairs used to amplify these regions were showed as previously (Ma et al, 2019). All the ChIP‐qPCR DNA signals were normalized to siNC IgG of G5′ or G5.
DNA methylation assay
J‐Lat 8.4 cells were treated with shCHAF1A or 5‐aza‐dC. One week post‐infection or drug treatment, cells were harvested. Genomic DNA of each group was extracted utilizing tissue DNA kit (OMEGA) according to the manufacture’s instruction. One microgram of genomic DNA was proceeded to sodium bisulfite conversion of unmethylated cytosines utilizing EpiTect Bisulfite Kit (QIAGEN) according to the manufacture’s instruction. Bisulfite‐converted DNA was purified by spin‐column and eluted with 20 μl of ddH2O. HIV‐1 LTR of bisulfite‐treated DNA was PCR‐amplified by two rounds of nested PCR with the following primer pairs. 1st round primer pair: 5′‐TGGTAGAATTATATATTAGGGTTAGGGATT‐3’ and 5′‐CACCCATCTCTCTCCTTCTAACCTC‐3′. 2nd round primer pair: 5′‐AGAGTAAGTAGAAGAGGTTAATGAAGGAGA‐3′ and 5′‐AAATCTAAAAAATCTCTAATTACCAAAATC‐3′. The PCR products were TA‐cloned into pMD‐18T vector. At least 30 single clones were picked for each sample. Sequencing results were aligned with standard unmethylated and converted DNA and quantified using the Quantification Tool for Methylation Analysis (QUMA) software (Kumaki et al, 2008).
SUMOylation assay
To test the recruitment of TRIM28 and corresponding SUMOylated TRIM28 by CHAF1A, Flag‐tagged SUMO1, SUMO2, and SUMO4 were co‐overexpressed with Flag‐tagged TRIM28 and HA‐tagged CHAF1A, respectively. Cell lysates were IP with anti‐HA beads and WB with anti‐HA and anti‐Flag antibodies. The sizes of wild‐type CHAF1A and TRIM28 were similar as corresponding predicted sizes. The SUMOylated targets were about 20 kD, 40 kD, or 60 kD larger than predicted size. To test the influence of CHAF1A on TRIM28‐mediated CDK9 SUMOylation, CDK9 was co‐overexpressed with SUMO4, UBC9, and TRIM28. siRNAs targeting TRIM28 and CHAF1A were transfected to knock down each target. SUMOylation status of CDK9 was indicated by the amount of SUMO‐CDK9.
Co‐immunoprecipitation
Specific combinations of proteins co‐overexpression were conducted in HeLa cells. HA‐tagged and Flag‐tagged proteins constructs were transfected into HeLa cells. Forty‐eight hours later, cells were harvested and lysed in NP‐40 lysis buffer (10 mM Tris–HCl buffered at pH 7.5, 150 mM NaCl, 0.5% NP‐40, 1% Triton X‐100, 10% Glycerol, 2 mM EDTA, 1 mM NaF, 1 mM Na3VO4) supplemented with 1/100 PIC (Sigma‐Aldrich), 2 M N‐ethylmaleimide (NEM) (Selleckchem) and 1/1,000 DNase I (Thermo Fisher). Cell lysates were IP with anti‐HA or anti‐Flag beads. At least 6 h post‐incubation with beads, proteins which were enriched by indicated beads were washed five times with ice‐cold STN IP wash buffer (10 mM Tri‐HCl buffered at pH 7.5, 150 mM NaCl, 0.5% NP‐40, 0.5% Triton X‐100). IP samples were eluted by boiling with 5 × protein SDS–PAGE loading buffer at 100°C for 10 min. Western blots were conducted with antibodies against HA (MBL, PM020) and Flag (MBL, M180‐3). GAPDH (Proteintech, 10494‐1‐AP) or Actin (CST, 4967) was IB with corresponding antibodies and used as internal reference. Both 680RD goat anti‐mouse IgG antibody (LI‐COR Biosciences, 926‐68070) and 800CW goat anti‐rabbit IgG antibody (LI‐COR Biosciences, 926‐32211) were chosen as secondary antibodies. The antibody‐incubated membranes were developed with Odyssey CLX Imager (LI‐COR Biosciences) and analyzed by Image Studio Lite Ver 4.0 (LI‐COR Biosciences).
SIM imaging
To prepare samples which were used for super‐resolution Structured Illumination Microscopy (SIM) imaging, HEK293T cells, HeLa cells, and Jurkat cells were plated into Lab‐Tek II chambered cover glass (Thermo Fisher) which was treated with poly‐lysine (Sigma‐Aldrich). Twenty‐four hours later, cells were fixed with 3% paraformaldehyde (Electron Microscopy Sciences)/0.1% glutaraldehyde (Electron Microscopy Sciences) followed by permeabilized with 0.2% Triton X‐100 (Sigma‐Aldrich). Cells were further blocked in 10% normal donkey serum (NDS) (Jackson ImmunoResearch)/0.05% Triton X‐100 for 90 min at room temperature (RT). Blocked samples were proceeded to specific primary antibodies incubation and fluorescently labeled secondary antibodies incubation. After incubation, cells were washed with 1% NDS/0.05% Triton X‐100 for five times. To indicate chromatin DNA, cells were treated with 4′, 6‐diamidino‐2‐phenylindole, dihydrochloride (DAPI) (Thermo Fisher) solution. Samples were washed for five times after DAPI incubation and stored at 4°C or imaged with SIM. For cells which were transfected with GFP‐, RFP‐, or BFP‐tagged proteins, samples were directly fixed and permeabilized without blocking and antibodies incubation. DAPI was added to dye DNA. If cells were grown on cover glass (Matsunami), samples were prepared in 6‐well plate. After dying DNA with DAPI, the cover glass with cells was mounted on the glass slide with the Antifade Mountant (Thermo Fisher).
Prepared samples were imaged on an Eclipse Ti inverted microscope equipped with a CFI Apo TIRF objective (1.49 NA, oil immersion), a NIS‐Elements AR software, an sCMOS camera (Hamamatsu Flash 4.0, 6.5 μm × 6.5 μm pixel size), and four lasers including SIM 405, SIM 488, SIM 561, and SIM 647. The original images were acquired with 512 × 512 resolution and reconstructed to form the SIM image with 1,024 × 1,024 resolution. The lateral resolution of the SIM image is 115 nm and the axial resolution is 300 nm. For 3D‐Stack‐SIM, Z‐step size was set to 0.20 μm per stack. For each focal plane, 15 images (five phases, three angles, 3D‐SIM mode) were captured with the NIS‐Elements. SIM images were reconstructed and analyzed with the N‐SIM module of the NIS‐Elements AR software (Nikon).
STORM imaging
To acquire higher resolution, samples were prepared for super‐resolution continuous STochastic Optical Reconstruction Microscopy (cSTORM) imaging. Samples were prepared as SIM samples preparation, except the following differences. After fixation, cells were reduced with 0.1% NaBH4 (Sigma‐Aldrich) for 7 min at room temperature while shaking, followed by washing with PBS for three times at room temperature, 5 min per wash. Specific secondary antibodies designed for STORM were used. These antibodies included Donkey Anti‐Mouse IgG H&L (Alexa Fluor 647) Antibody (Abcam, ab150107), Donkey Anti‐Rabbit IgG H&L (CF 568) Antibody (Biotium, 20803‐500 μl), Donkey Anti‐Rabbit IgG H&L (Alexa Fluor 647) Antibody (Abcam, ab150075), and Donkey Anti‐Mouse IgG H&L (CF 568) Antibody (Biotium, 20802‐500 μl). After secondary antibodies incubation, cells were post‐fixed for 10 min with 3% paraformaldehyde/0.1% glutaraldehyde. DAPI and Hoechst were not allowed to dye DNA according to cSTORM protocol. cSTORM imaging buffer was freshly prepared. Each well of Lab‐Tek II chambered cover glass was added with 700 μl of imaging buffer which was able to be used for imaging for over 2 h. For photoswitchable fluorescent protein (PsFP)‐tagged proteins, such as mEos2‐CHAF1A and mEos2‐DNMT1, transfected samples were simply prepared as in SIM and imaged without cSTORM imaging buffer. pmEos2‐C1 was a kindly gift from Dr. Jie Yao (School of Medicine, Sun Yat‐sen University, Guangzhou, China) (Yao et al, 2011).
STORM Samples were imaged utilizing a Nikon super‐resolution N‐STORM microscope equipped with a high‐numerical‐aperture (high‐NA) 100 × oil‐immersion objective (Nikon CFI SR Apochromat TIRF 100 × oil, 1.49 NA), a high‐sensitivity and high‐resolution sCMOS camera (Hamamatsu Flash 4.0, 6.5 μm × 6.5 μm pixel size, and an 0.4 × relay lens to match the pixel size under STORM mode), and four lasers with excitation wavelengths of 405, 488, 561, and 647 nm. Laser of 405 nm was used as activation laser. Lasers including 488, 561, and 647 nm were used as reporter lasers. The lateral resolution of the cSTORM image is 20 nm, and the axial resolution is 50 nm. Nikon perfect focus system (PFS) was used to maintain the z position during the acquisition. 20,000 to 25,000 frames were taken for each channel. Single‐molecule localization was obtained by Gaussian fitting utilizing the STORM plug‐in of NIS‐Elements AR software considering both drift and chromatic aberrations.
ImmunoFISH
Immunolabeling‐based fluorescence in situ hybridization (ImmunoFISH) assays were conducted to investigate the relationship between CAF‐1 bodies and HIV‐1 provirus. Suspended naïve and activated J‐Lat 10.6 cells were grown on coverslips pretreated with poly‐lysine for at least 4 h, followed by the quick treatment with 0.3 × PBS. Immobile cells were fixed with 4% paraformaldehyde/PBS for 10 min, permeabilized with 0.5% Triton X‐100/PBS for 10 min while shaking, and blocked with 4% BSA/PBS for 1 h while shaking. Blocked cells were proceeded to the incubation with rabbit anti‐CHAF1A primary antibodies for 1 h and goat anti‐rabbit FITC secondary antibodies for 1 h. Subsequently, cells were washed with PBS/T, post‐fixed with 4% paraformaldehyde/PBS at room temperature (RT) for 10 min and permeabilized again with 0.5% Triton X‐100/PBS at RT for 10 min. After washing with PBS/T, cells were soaked in 20% glycerol/PBS for at least 1 h. Cells were proceeded to six rounds of freezing and thawing in liquid nitrogen and glycerol/PBS, followed by incubating with freshly prepared 0.1 N HCl for 10 min. Cells were permeabilized again with 0.5% Triton X‐100/PBS for 10 min and stabilized in 2 × SSC (Sigma‐Aldrich) for 5 min. After stabilizing, cells were treated with freshly prepared RNase A/2 × SSC (100 μg/ml) for 30 min at 37°C. Cells were stabilized in 2 × SSC again, followed by soaking in hybridization solution (50% formamide/2 × SSC) at 4°C overnight.
The probes of HIV‐1 DNA were obtained by nick translation utilizing the FISH Tag DNA kit (Thermo Fisher) according to the manufacturer’s recommendations. Briefly, 5 µg of pNL4‐3 plasmids were used for nick translation reaction to enzymatically incorporate amine‐modified nucleotides into the probe templates. The amine‐modified DNA was purified by DNA‐binding column and precipitated by ethanol. Subsequently, the purified DNA was labeled with Alexa Fluor 594 dye. Fluorescent dye‐labeled DNA was column purified and ethanol precipitated in the presence of 10 μl Cot‐1 DNA (Thermo Fisher) and 10 μl herring sperm DNA (Sigma‐Aldrich). The DNA pellet was dissolved in 10 μl 100% formamide and incubated for 10 min at 37°C while shaking. The DNA solution was added with 10 μl 4 × SSC/20% Dextran and mixed thoroughly. Total probes solution was denatured at 95°C for 5 min, followed by quickly inserting into ice to incubate for 3 min. The prepared dye‐labeled DNA was ready for hybridization.
Two microliter DNA probe which was prepared as above was diluted with 4 μl hybridization buffer (50% formamide/10% Dextran/2 × SSC). After vortex and centrifugation, the diluted probes were denatured at 95°C for 5 min, followed by quickly inserting into ice to incubate for 3 min. DNA probes were loaded onto the glass slide and covered with coverslips which were adhered with FISH samples prepared as above. The edges were sealed with rubber cement. After drying, the slide was denatured at 77.5°C water bath for 6 min and cooled to 37°C gradually. The hybridization was carried out in a box floating at 37°C water bath for 48 h, followed by incubating at 42°C for another 24 h. The rubber cement was carefully removed after hybridization FISH samples with probes. The coverslip was washed in 2 × SSC at 37°C for four times, followed by washing with 2 × SSC at RT once. Subsequently, the coverslip was washed twice with 0.5 × SSC at 56°C for 5 min. After last washing, cells were equilibrated in PBS and stained with DAPI. Samples were briefly washed with PBS for 5 times and mounted on glass slide with Antifade Mountant (Thermo Fisher). After incubating mounted samples at RT for 24 h, samples were imaged with SIM as described above.
FRAP
Fluorescence recovery after photobleaching (FRAP) experiment was conducted to identify the internal diffusion property of CAF‐1 body. GFP‐tagged CHAF1A was transfected into HEK293T cells. Twenty‐four hours later, cells were imaged under Nikon Confocal A1 with laser 488 nm. Two CAF‐1 bodies which were indicated by GFP‐tagged CHAF1A were circled as regions of interest (ROI). One ROI was set as negative control (unbleached region), and the other was bleached with 100% laser power. Both pre‐bleached and post‐bleached images were captured every 4 s. The images were captured for 5 min. Fluorescence intensities in each time point were recorded and analyzed with NIS‐Elements AR software. The background intensity was subtracted. The fluorescence intensities in each time point were normalized to pre‐bleaching time point.
Protein purification
GFP, GFP‐CHAF1A‐IDR, GFP‐CHAF1A‐DS1, GFP‐CHAF1A‐DS2, GFP‐CHAF1A‐DS3, GFP‐CHAF1A‐mIDR, GFP‐CHAF1A‐DS1‐Q34A, and GFP‐CHAF1A‐DS3‐KERDB were cloned to pET28a vector which contained His tag. Plasmid sequences for CHAF1A mutants will be made available upon request. These constructs were transformed into BL21 (Takara). Single clone was amplified in LB with kanamycin at 37°C while shaking. When the OD600 of bacterial solution reached 0.4–0.6, the solution was transferred to 16°C and added with isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) (Takara) to induce the expression of target proteins. Another 18 h later, protein‐expressing bacteria were harvested by centrifuging and washed once with PBS. 500 ml of bacterial solution was concentrated in 25 ml of Buffer A (50 mM Tris–HCl, pH 7.5, 500 mM NaCl) supplemented with PIC. Resuspended bacteria were lysed by sonication for 20 min in ice. The sonication was conducted by 5‐s pulse and 5‐s pause in 70% of maximum power. After sonication, lysates were centrifuged and filtered to remove residual bacteria and insoluble components. Ni‐NTA agarose (QIAGEN) was added into the protein solution and enriched His‐tagged proteins. After 1 h of incubation at 4°C while rotating, the protein solution was passed through the filter, leaving only Ni‐NTA agarose and corresponding enriched proteins. The agarose was washed with 30 ml of Buffer B (50 mM Tris–HCl, pH 7.5, 500 mM NaCl, 10 mM Imidazole), 30 ml of Buffer C (50 mM Tris–HCl, pH 7.5, 500 mM NaCl, 20 mM Imidazole), and 30 ml of Buffer D (50 mM Tris–HCl, pH 7.5, 500 mM NaCl, 50 mM Imidazole). Then, proteins on agarose were eluted with 2 ml of Buffer E (50 mM Tris–HCl, pH 7.5, 500 mM NaCl, 100 mM Imidazole), 2 ml of Buffer F (50 mM Tris–HCl, pH 7.5, 500 mM NaCl, 200 mM Imidazole), and 2 ml of Buffer G (50 mM Tris–HCl, pH 7.5, 500 mM NaCl, 500 mM Imidazole). The eluate was combined and concentrated utilizing 10 kD or 30 kD ultrafiltration devices. The residual imidazole was removed by buffer replacement with Buffer H (50 mM Tris–HCl, pH 7.5, 500 mM NaCl, 10% Glycerol, 1 mM DTT). The replacement was conducted by centrifuging. The concentrated and buffer‐replaced protein solution was stored at −80°C.
In vitro droplet formation assay
In vitro purified proteins were proceeded to in vitro droplet formation experiment. Proteins were added into different NaCl gradient of droplet formation buffers. Buffer I (50 mM Tris–HCl, pH 7.5, 10% Glycerol, 1 mM DTT) was used to dilute NaCl. Droplet formation buffers included Buffer J (50 mM Tris–HCl, pH 7.5, 250 mM NaCl, 10% Glycerol, 1 mM DTT), Buffer K (50 mM Tris–HCl, pH 7.5, 125 mM NaCl, 10% Glycerol, 1 mM DTT), Buffer L (50 mM Tris–HCl, pH 7.5, 62.5 mM NaCl, 10% Glycerol, 1 mM DTT), Buffer M (50 mM Tris–HCl, pH 7.5, 31.25 mM NaCl, 10% Glycerol, 1 mM DTT), and Buffer N (50 mM Tris–HCl, pH 7.5, 15.625 mM NaCl, 10% Glycerol, 1 mM DTT). These protein solutions with various NaCl concentration and indicated protein concentration were quickly imaged under Nikon Confocal A1. The scan size was 1,024 × 1,024 utilizing CFI Apo TIRF objective (1.49 NA, oil immersion). Laser 488 nm was used to capture droplets.
HIV‐1 latency model construction
Different HIV‐1 latency models were built in Jurkat cells, HEK293T cells, and primary CD4+ T cells. In Jurkat cells, pseudotyped HIV‐1 constructs including pNL4‐3‐ΔEnv/d2EGFP‐Nef‐IRES‐Bcl2 (NIB) and pNL4‐3‐ΔEnv/d2EGFP‐Nef‐P2A‐Bcl2 (NPB)were used to build latency models. Three micrograms of VSV‐G constructs and 9 μg of pseudotyped virus constructs were co‐transfected into HEK293T cells to package pseudotyped viruses followed by the concentration of these viruses by PEG 6000. About 1 × 106 Jurkat cells were infected with pseudotyped viruses to ensure that no more than 20% of Jurkat cells were infected, which were indicated by the percentage of GFP‐positive cells. Forty‐eight hours post‐infection, GFP‐negative cells were sorted by fluorescence‐activated cell sorting (FACS) and cultured for another week to recovery. One week later, sorted GFP‐negative cells were activated by TNFα followed by the sorting of GFP‐positive cells. These activated GFP‐positive cells were cultured for 2 weeks to recovery and proliferate. Theoretically, these ever GFP‐positive cells would be GFP‐negative without TNFα stimulation. If GFP‐positive cells were persistent after 2 weeks of culturing, the GFP‐negative cells were sorted again. The persistent GFP‐negative cells were activated by TNFα again followed by the sorting of activated GFP‐positive cells. After several rounds of cell sorting, over 50% of infected cells would be TNFα‐specific, which meant almost 95% of infected cells were GFP‐negative without TNFα stimulation; over 50% of infected cells would be GFP‐positive after TNFα treatment. We named this cell population by the pseudotyped HIV‐1 latency model which were sensitive to TNFα and most LRAs including SAHA and JQ‐1.
HEK293T HIV‐1 latency model was built similarly. Pseudotyped HIV‐1 construct NL4‐3‐ΔEnv/EGFP was packaged and used to infect HEK293T cells. Unlike J‐Lat models, GFP‐positive HEK293T cells were sorted instead of GFP‐negative cells. About 99% of these GFP‐positive cells would be negative in HEK293T cells after 4 weeks of culturing. These acquired latent cells showed modest sensitivity to common LRAs. Genomic DNA PCR indicated that almost all the cells had been infected, which meant that most of these cells were in deep latency. Latently infected cells were treated with siRNAs targeting CHAF1A and constructs expressing wild‐type or dLLPS CHAF1A.
Primary CD4+ T‐cell‐based latency models had been partially described in corresponding figure legends. Generally, isolated primary CD4+ T cells were activated by PHA or αCD3/αCD28. Packaged viruses such as NL4‐3‐P2A‐EGFP (NPG) and NL4‐3‐ΔEnv/ΔNef‐d2EGFP were used to infect activated CD4+ T cells. Theoretically, 5–15% of CD4+ T cells would be GFP‐positive 2 days post‐infection. After 2 weeks of resting (for NPG model, AZT was added to prevent persistent HIV‐1 infection), most of the infected cells were GFP‐negative, which meant these infected cells had entered into latency.
HIV‐1 RNAs quantitation
Intracellular RNAs from clinical samples were extracted with TRIzol reagent (Thermo Fisher) according to the manufacture’s instruction. Residual DNA was removed by DNase (Promega). A specific reverse primer was used to reversely transcribe HIV‐1 RNA: 5′‐GCTTCAGCAAGCCGAGTCCTGCGTC‐3′ (Liu et al, 2016). qPCR was performed for specific reverse‐transcribed HIV‐1 cDNA with primer pairs: HIVTotRNA Forward Primer: 5′‐CTGGCTAACTAGGGAACCCACTGCT‐3′ and HIVTotRNA Reverse Primer: 5′‐GCTTCAGCAAGCCGAGTCCTGCGTC‐3′. An in vitro transcribed HIV‐1 RNA was used as the external control for measuring cell‐associated viral RNAs. The C t value of each sample was converted to mass and further converted to HIV‐1 RNA copies. The final expression of intracellular HIV‐1 RNAs was represented as 103 copies of viral RNA per million CD4+ T cells for each sample.
Genetic diversity analysis
To quantitate the genetic diversity of reactivated HIV‐1, the V1–V3 regions of HIV‐1 envelope were specifically evaluated. HIV‐1 RNAs of different groups were reversely transcribed by primer ES8B: 5′‐CACTTCTCCAATTGTCCCTCA‐3′ (Geng et al, 2016). A nested PCR was conducted to amplify V1–V3 region with the following primer pairs: 1st round Nest PCR Forward Primer (E00): 5′‐TAGAAAGAGCAGAAGACAGTGGCAATGA‐3′, 1st round Nest PCR Reverse Primer (ES8B): 5′‐CACTTCTCCAATTGTCCCTCA‐3′; 2nd round Nest PCR Forward Primer (E20): 5′‐GGGCCACACATGCCTGTGTACCCACAG‐3′, 2nd round Nest PCR Reverse Primer (E115): 5′‐AGAAAAATTCCCCTCCACAATTAA‐3′. Single genome amplification method was performed by obtaining 30 independent PCR products from each sample. The PCR products were TA‐ligated into pMD‐18T vector. At least 60 single clones were picked from each group and proceeded to Sanger sequencing. Sequences were aligned using MUSCLE. The sequences with ambiguous positions were excluded. The average genetic distance between one given clone and the relevant entire population was calculated by MEGA 7 and represented as genetic diversity index. The Mann–Whitney U‐test was performed to compare the genetic diversity indexes between different groups using Prism 5.
RNA‐Seq
J‐Lat 10.6 cells were stimulated by TNFα. Twenty‐four hours later, both naïve J‐Lat 10.6 cells and TNFα‐stimulated cells were lysed and proceeded to RNA extraction. RNAs from wild‐type J‐Lat 10.6 cells and CHAF1A‐KO J‐Lat 10.6 cells were extracted similarly. The qualities of these RNAs were evaluated by NanoDrop (Thermo Fisher) and BioAnalyzer 2100 (Aglient). The RNA‐Seq libraries were performed with TruSeq Stranded mRNA Library Prep Kit (Illumina) and sequenced with HiSeq X Ten (Illumina) under PE150 protocol. RNA‐Seq reads were trimmed, filtered, and quality‐controlled by FastQC (Babraham Institute). The reads were aligned to the human reference genome NCBI build 38 (GRCh38) by Hisat2, followed by calculating the reads per kilobase per million mapped reads (RPKM) (Kim et al, 2015). Differentially expressed genes were filtered by DEGseq (Bioconductor) with log2FC of 1 and PvalueFDR cutoff of 0.05. Filtered genes were plotted as volcanoplots by gplots (R Foundation).
Mass spectrometry
To identify differentially expressed proteins upon TNFα stimulation, naïve and TNFα‐stimulated J‐Lat 10.6 cells were proceeded to mass spectrometry (MS). Briefly, cells were lysed with NP‐40 lysis buffer (10 mM Tris–HCl buffered at pH 7.5, 150 mM NaCl, 0.5% NP‐40, 1% Triton X‐100, 10% Glycerol, 2 mM EDTA, 1 mM NaF, 1 mM Na3VO4) supplemented with PIC, DNase, and RNase for 30 min on ice. Lysates were boiled with 4 × protein SDS–PAGE loading buffer (Takara) at 100°C for 15 min. Each sample was separated with 4–12% gradient protein gel (Thermo Fisher). The gel was dyed with silver stain kit (Sigma‐Aldrich). Eight gel slices of each sample were cut out and proceeded to in‐gel digestion. Each gel slice was destained utilizing silver stain kit and treated with 10 mM DTT followed by the treatment of 55 mM iodoacetamide. The gels were further washed with 25 mM NH4HCO3 and 25 mM NH4HCO3 in 50% ACN to remove residual DTT or iodoacetamide, followed by desiccation with vacuum. One hundred nanogram trypsin (Thermo Fisher) was added to each dry gel and incubated overnight at 37°C to digest proteins to small peptides. Digested peptides were extracted with the following extraction solutions orderly: 50% ACN/5%TFA, 75% ACN/0.1% TFA, and 100% ACN. All the extracts were subjected to vacuum to remove the solvent. The peptides were desalted and enriched by C18 ZipTip (Millipore), and re‐dissolved in 50% ACN containing 0.1% TFA, followed by vacuum to remove the solvent. Twelve microliters of 0.01% formic acid were used to resolve the peptides and proceeded to nanoscale LC‐MS/MS. Peptide was separated by 20‐cm‐long analytical columns (ID 75 μm, Polymicro Avantes) packed in house with Luna 3.0 u C18 (2) 100A (Phenomenex) with a 90‐min gradient from 3 to 90% acetonitrile in 0.1% formic acid and a flow rate of 300 nl/min. Data‐dependent acquisition mode with a top‐ten method was used to operate the mass spectrometer. Full‐scan MS spectra were obtained with a target value of 3E6, a resolution of 70,000, with a scan range from 300 to 1,800 m/z. HCD tandem MS/MS spectra were obtained with a target value of 1E6, a resolution of 17,500, and a normalized collision energy of 25%. Unknown charges or charges lower than two and higher than eight were excluded.
To identify CAF‐1 body components, HA‐tagged CHAF1A and CHAF1B were co‐overexpressed in HeLa cells. CHAF1A and CHAF1B were able to form heterodimer which stabilized each subunit. Forty‐eight hours post‐transfection, cells were harvested and IP with anti‐HA beads. Enriched proteins were washed and eluted by boiling with 4 × protein SDS–PAGE loading buffer. Proteins of each samples were separated by 4–12% gradient SDS–PAGE gel. Eight slices were cut out for each sample. The in‐gel digestion and LC‐MS/MS analysis were the same as TNFα‐stimulated samples. The generated raw data were annotated and analyzed utilizing PEAKS Studio. Significantly changed proteins were presented as heatmap and classified by PANTHER classification system. STRING network and k‐means clustering were used to specifically present highly interconnected proteins.
ATAC‐Seq
Approximately 30,000 cells of sgNT and sgCHAF1A J‐Lat 10.6 were harvested for building ATAC‐Seq library. The ATAC‐Seq library was built with TruePrep DNA Library Prep Kit V2 (Vazyme) according to the manufacture’s instruction. Briefly, cells of each group were lysed with 50 μl of ice‐cold lysis buffer (10 mM Tris–HCl buffered at pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Igepal CA‐630) for 10 min on ice, followed by centrifuging to remove supernatant. Transposition reaction mix was used to resuspend nuclei pellet and incubated at 37°C for 30 min. The transposed DNA was purified by VAHTS DNA Clean Beads (Vazyme) and PCR‐amplified. The amplified ATAC‐Seq library was purified with VAHTS DNA Clean Beads and eluted with 30 μl ddH2O. The library quality was evaluated by Qubit 3.0 Fluorometer (Thermo Fisher) and BioAnalyzer 2100 (Aglient), and sequenced with HiSeq X Ten (Illumina) under the PE150 protocol. ATAC‐Seq reads were trimmed, filtered, and quality‐controlled by FastQC. Both the HIV‐1 genome and human genome were analyzed for accessibility. The reads were aligned to HIV‐1 reference genome K03455, M38432 (Version K03455.1) by Bowtie2, followed by rearranging with Samtools (Li et al, 2009; Langmead & Salzberg, 2012). The human genome was aligned to human reference genome GRCh38. The integration sites of HIV‐1 pseudotyped viruses in J‐Lat 10.6 have been identified by genome walking strategy. Cellular DNA and viral 5′LTR junction was different from viral 3′LTR and cellular DNA junction, although 5′LTR and 3′LTR showed significant similarity. Only the reads containing both HIV‐1 5′LTR and human integration junction fragments were sorted. Thus, the read density centered HIV‐1 5′LTR could be calculated and discriminated from 3′LTR. The tag peaks were visualized by Igvtools (Broad Institute). Tag density of different groups was calculated by normalizing to the total mapped reads. The highest tag density was set as 100. Relative tag densities of two kilobases range centered HIV‐1 5′LTR integration sites were calculated and compared with sgNT.
Data and statistical analysis
All the statistical details of specific experiments, which included the statistical tests used, number of samples, mean values, standard errors of the mean (SEM), and P‐values derived from indicated tests, had been described in the figure legends and showed in the figures. Statistical analyses were conducted utilizing GraphPad Prism 5 or Microsoft Excel. The network analysis and clustering analysis were conducted with STRING. Triplicate, sextuplicate, and other replicate data were presented as mean ± SEM. A value of P < 0.05 was considered to be statistically significant and represented as asterisk (*). Value of P < 0.01 was considered to be more statistically significant and represented as double asterisks (**). Value of P < 0.001 was considered to be the most statistically significant and represented as triple asterisks (***). For comparison between two treatments, a Student’s t‐test was used. For the comparison of highly heterogeneous data including HIV‐1 reactivation of clinical samples and genetic diversity index experiment, a Mann–Whitney U‐test was used.
Author contributions
XM and HZ conceived and designed the study; XM, JL, and ZT optimized the methods and analyzed high‐throughput sequencing data; XM, TC, ZP, ZW, TY, LW, GL, MZ, MT, YG, XZ, YL, TP, and HZ performed the experiments; JL, GL, YG, LL, XT, and ZT provided analysis software and clinical samples; XM, YG, ZT, and HZ proofread the whole manuscript; XM, and HZ wrote, reviewed and edited the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Movie EV1
Movie EV2
Movie EV3
Source Data for Expanded View and Appendix
Review Process File
Acknowledgements
This work was supported by the National Special Research Program of China for Important Infectious Diseases (2018ZX10302103 and 2017ZX10202102), the Important Key Program of Natural Science Foundation of China (81730060) and the Joint‐innovation Program in Healthcare for Special Scientific Research Projects of Guangzhou (201803040002) to H.Z. This work was also supported by the National Postdoctoral Program for Innovative Talents and the General Program of China Postdoctoral Science Foundation (BX20190398 and 2019M663215) to X.M. This work was also supported by the National Natural Science Foundation of China (81702856) to G.L. This work was also supported by the Pearl River S&T Nova Program of Guangzhou (201806010118) to T.P.
The EMBO Journal (2021) 40: e106632.
Data availability
The RNA‐Seq data from this publication have been deposited to the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166337) and assigned the identifier (GSE166337). The MS proteomics data from this publication have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD024172) via the iProX partner repository (https://www.iprox.org/page/project.html?id=IPX0002805000) and assigned the identifier (PXD024172). Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author, Dr. Hui Zhang (zhangh92@mail.sysu.edu.cn). Plasmid sequences for CHAF1A mutants will be made available upon request. Purified proteins for in vitro experiments can be provided upon execution of a material transfer agreement (MTA) with inquiries directed to Dr. Hui Zhang.
References
- Alberti S, Gladfelter A, Mittag T (2019) Considerations and challenges in studying liquid‐liquid phase separation and biomolecular condensates. Cell 176: 419–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani SF, Rice AM, Peeples WB, Lin Y, Jain S, Parker R, Rosen MK (2016) Compositional control of phase‐separated cellular bodies. Cell 166: 651–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, Rosen MK (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18: 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazkova J, Trejbalova K, Gondois‐Rey F, Halfon P, Philibert P, Guiguen A, Verdin E, Olive D, Van Lint C, Hejnar J et al (2009) CpG methylation controls reactivation of HIV from latency. PLoS Pathog 5: e1000554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm D, Jeng M, Camus G, Gramatica A, Schwarzer R, Johnson JR, Hull PA, Montano M, Sakane N, Pagans S et al (2017) SMYD2‐mediated histone methylation contributes to HIV‐1 latency. Cell Host Microbe 21: 569–579.e566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM et al (2018) Transcription factors activate genes through the phase‐separation capacity of their activation domains. Cell 175: 1842–1855.e1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne Clifford P, Tompa P, Pappu Rohit V (2015) Polymer physics of intracellular phase transitions. Nat Phys 11: 899–904 [Google Scholar]
- Cai D, Feliciano D, Dong P, Flores E, Gruebele M, Porat‐Shliom N, Sukenik S, Liu Z, Lippincott‐Schwartz J (2019) Phase separation of YAP reorganizes genome topology for long‐term YAP target gene expression. Nat Cell Biol 21: 1578–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas‐Delucchi CS, van Bemmel JG, Haase S, Herce HD, Nowak D, Meilinger D, Stear JH, Leonhardt H, Cardoso MC (2012) Histone hypoacetylation is required to maintain late replication timing of constitutive heterochromatin. Nucleic Acids Res 40: 159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S, Elling U, Hopfgartner B, Jung YL, Murn J, Ninova M, Hubmann M, Badeaux AI, Euong Ang C, Tenen D et al (2015) The histone chaperone CAF‐1 safeguards somatic cell identity. Nature 528: 218–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S, Hochedlinger K (2017) Emerging roles of the histone chaperone CAF‐1 in cellular plasticity. Curr Opin Genet Dev 46: 83–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chéné Id, Basyuk E, Lin Y‐L, Triboulet R, Knezevich A, Chable‐Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P et al (2007) Suv39H1 and HP1γ are responsible for chromatin‐mediated HIV‐1 transcriptional silencing and post‐integration latency. EMBO J 26: 424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Zhang X, Wang Y, Gan H, Xu X, Lv X, Hua X, Que J, Ordog T, Zhang Z (2019) Chromatin Assembly Factor 1 (CAF‐1) facilitates the establishment of facultative heterochromatin during pluripotency exit. Nucleic Acids Res 47: 11114–11131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang LS‐H, Ian H‐I, Koh T‐W, Ng H‐H, Xu G, Li BFL (1997) Human DNA‐(Cytosine‐5) methyltransferase‐PCNA complex as a target for p21WAF1 . Science 277: 1996–2000 [DOI] [PubMed] [Google Scholar]
- Chun T‐W, Stuyver L, Mizell SB, Ehler LA, Mican JAM, Baseler M, Lloyd AL, Nowak MA, Fauci AS (1997) Presence of an inducible HIV‐1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci USA 94: 13193–13197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn Lillian B, Silva Israel T, Oliveira Thiago Y, Rosales Rafael A, Parrish Erica H, Learn Gerald H, Hahn Beatrice H, Czartoski Julie L, McElrath MJ, Lehmann C et al (2015) HIV‐1 integration landscape during latent and active infection. Cell 160: 420–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Koning L, Corpet A, Haber JE, Almouzni G (2007) Histone chaperones: an escort network regulating histone traffic. Nat Struct Mol Biol 14: 997–1007 [DOI] [PubMed] [Google Scholar]
- Deeks SG (2012) Shock and kill. Nature 487: 439–440 [DOI] [PubMed] [Google Scholar]
- Dekker J, Marti‐Renom MA, Mirny LA (2013) Exploring the three‐dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet 14: 390–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding D, Qu X, Li L, Zhou X, Liu S, Lin S, Wang P, Liu S, Kong C, Wang X et al (2013) Involvement of histone methyltransferase GLP in HIV‐1 latency through catalysis of H3K9 dimethylation. Virology 440: 182–189 [DOI] [PubMed] [Google Scholar]
- Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW et al (2001) Intrinsically disordered protein. J Mol Graph Model 19: 26–59 [DOI] [PubMed] [Google Scholar]
- Elsheikh MM, Tang Y, Li D, Jiang G (2019) Deep latency: a new insight into a functional HIV cure. EBioMedicine 45: 624–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Wang L, Ishikawa R, Li Y, Fiedler M, Liu F, Calder G, Rowan B, Weigel D, Li P et al (2019) Arabidopsis FLL2 promotes liquid–liquid phase separation of polyadenylation complexes. Nature 569: 265–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R et al (1997) Identification of a reservoir for HIV‐1 in patients on highly active antiretroviral therapy. Science 278: 1295–1300 [DOI] [PubMed] [Google Scholar]
- Friedman J, Cho W‐K, Chu CK, Keedy KS, Archin NM, Margolis DM, Karn J (2011) Epigenetic silencing of HIV‐1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J Virol 85: 9078–9089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng G, Liu B, Chen C, Wu K, Liu J, Zhang Y, Pan T, Li J, Yin Y, Zhang J et al (2016) Development of an attenuated tat protein as a highly‐effective agent to specifically activate HIV‐1 latency. Mol Ther 24: 1528–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gérard A, Ségéral E, Naughtin M, Abdouni A, Charmeteau B, Cheynier R, Rain J‐C, Emiliani S (2015) The Integrase cofactor LEDGF/p75 associates with Iws1 and Spt6 for postintegration silencing of HIV‐1 gene expression in latently infected cells. Cell Host Microbe 17: 107–117 [DOI] [PubMed] [Google Scholar]
- Groth A, Rocha W, Verreault A, Almouzni G (2007) Chromatin challenges during DNA replication and repair. Cell 128: 721–733 [DOI] [PubMed] [Google Scholar]
- Guo YE, Manteiga JC, Henninger JE, Sabari BR, Dall’Agnese A, Hannett NM, Spille J‐H, Afeyan LK, Zamudio AV, Shrinivas K et al (2019) Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572: 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatanaka Y, Inoue K, Oikawa M, Kamimura S, Ogonuki N, Kodama EN, Ohkawa Y, Tsukada Y‐i, Ogura A (2015) Histone chaperone CAF‐1 mediates repressive histone modifications to protect preimplantation mouse embryos from endogenous retrotransposons. Proc Natl Acad Sci USA 112: 14641–14646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Margolis DM (2002) Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV‐1) by the HIV‐1 repressor YY1 and HIV‐1 activator tat. Mol Cell Biol 22: 2965–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich BS, Maliga Z, Stein DA, Hyman AA, Whelan SPJ (2018) Phase transitions drive the formation of vesicular stomatitis virus replication compartments. MBio 9: e02290‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlard M, Berlivet S, Probst AV, Quivy J‐P, Héry P, Almouzni G, Gérard M (2006) CAF‐1 Is essential for heterochromatin organization in pluripotent embryonic cells. PLoS Genet 2: e181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Santoso N, Power D, Simpson S, Dieringer M, Miao H, Gurova K, Giam C‐Z, Elledge SJ, Zhu J (2015) FACT proteins, SUPT16H and SSRP1, are transcriptional suppressors of HIV‐1 and HTLV‐1 that facilitate viral latency. J Biol Chem 290: 27297–27310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, Jülicher F (2014) Liquid‐liquid phase separation in biology. Annu Rev Cell Dev Biol 30: 39–58 [DOI] [PubMed] [Google Scholar]
- Imai K, Togami H, Okamoto T (2010) Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV‐1 latency and its reactivation by BIX01294. J Biol Chem 285: 16538–16545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Bisgrove D, Verdin E (2003) HIV reproducibly establishes a latent infection after acute infection of T cells in vitro . EMBO J 22: 1868–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Yang Y‐S, Sutter BM, Wang Y, McKnight SL, Tu BP (2019) Redox state controls phase separation of the yeast ataxin‐2 protein via reversible oxidation of its methionine‐rich low‐complexity domain. Cell 177: 711–721.e718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E (2009) Epigenetic regulation of HIV‐1 latency by cytosine methylation. PLoS Pathog 5: e1000495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman PD, Kobayashi R, Kessler N, Stillman B (1995) The p150 and p60 subunits of chromatin assemblyfactor I: A molecular link between newly synthesized histories and DNA replication. Cell 81: 1105–1114 [DOI] [PubMed] [Google Scholar]
- Khan S, Iqbal M, Tariq M, Baig SM, Abbas W (2018) Epigenetic regulation of HIV‐1 latency: focus on polycomb group (PcG) proteins. Clin Epigenet 10: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G, Darcis G, Lee MY, Bouchat S, Van Driessche B, Purcell DFJ, Van Lint C (2018) The molecular biology of HIV latency, In HIV vaccines and cure: the path towards finding an effective cure and vaccine, Zhang L, Lewin SR (eds), pp 187–212. Singapore: Springer Singapore; [DOI] [PubMed] [Google Scholar]
- Kim YK, Bourgeois CF, Pearson R, Tyagi M, West MJ, Wong J, Wu S‐Y, Chiang C‐M, Karn J (2006) Recruitment of TFIIH to the HIV LTR is a rate‐limiting step in the emergence of HIV from latency. EMBO J 25: 3596–3604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita S, Chen BK, Kaneshima H, Nolan GP (1998) Host control of HIV‐1 parasitism in T cells by the nuclear Factor of activated T cells. Cell 95: 595–604 [DOI] [PubMed] [Google Scholar]
- Kumaki Y, Oda M, Okano M (2008) QUMA: quantification tool for methylation analysis. Nucleic Acids Res 36: W170–W175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, Narlikar GJ (2017) Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547: 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP (2009) The sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Ma X, Liu B, Chen C, Zhang H (2015) HIV‐1 functional cure: will the dream come true? BMC Med 13: 284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Zou F, Lu L, Chen C, He D, Zhang X, Tang X, Liu C, Li L, Zhang H (2016) Chimeric antigen receptor T cells guided by the single‐chain Fv of a broadly neutralizing antibody specifically and effectively eradicate virus reactivated from latency in CD4+ T lymphocytes isolated from HIV‐1‐infected individuals receiving suppressive combined antiretroviral therapy. J Virol 90: 9712–9724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyola A, Tagami H, Bonaldi T, Roche D, Quivy JP, Imhof A, Nakatani Y, Dent SYR, Almouzni G (2009) The HP1α–CAF1–SetDB1‐containing complex provides H3K9me1 for Suv39‐mediated K9me3 in pericentric heterochromatin. EMBO Rep 10: 769–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Yu D, Hansen AS, Ganguly S, Liu R, Heckert A, Darzacq X, Zhou Q (2018) Phase‐separation mechanism for C‐terminal hyperphosphorylation of RNA polymerase II. Nature 558: 318–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Wu T, Gutman O, Lu H, Zhou Q, Henis YI, Luo K (2020) Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat Cell Biol 22: 453–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J (1991) Predicting coiled coils from protein sequences. Science 252: 1162–1164 [DOI] [PubMed] [Google Scholar]
- Lusic M, Marini B, Ali H, Lucic B, Luzzati R, Giacca M (2013) Proximity to PML nuclear bodies regulates HIV‐1 latency in CD4+ T cells. Cell Host Microbe 13: 665–677 [DOI] [PubMed] [Google Scholar]
- Ma X, Yang T, Luo Y, Wu L, Jiang Y, Song Z, Pan T, Liu B, Liu G, Liu J et al (2019) TRIM28 promotes HIV‐1 latency by SUMOylating CDK9 and inhibiting P‐TEFb. Elife 8: e42426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, Spindler J, Ferris AL, Mellors JW, Kearney MF et al (2014) Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345: 179–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao YS, Zhang B, Spector DL (2011) Biogenesis and function of nuclear bodies. Trends Genet 27: 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, Aunis D, Rohr O (2007) Recruitment of chromatin‐modifying enzymes by CTIP2 promotes HIV‐1 transcriptional silencing. EMBO J 26: 412–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini B, Kertesz‐Farkas A, Ali H, Lucic B, Lisek K, Manganaro L, Pongor S, Luzzati R, Recchia A, Mavilio F et al (2015) Nuclear architecture dictates HIV‐1 integration site selection. Nature 521: 227–231 [DOI] [PubMed] [Google Scholar]
- Mbonye U, Karn J (2017) The molecular basis for human immunodeficiency virus latency. Annu Rev Virol 4: 261–285 [DOI] [PubMed] [Google Scholar]
- Milutinovic S, Zhuang Q, Szyf M (2002) Proliferating cell nuclear antigen associates with histone deacetylase activity, integrating DNA replication and chromatin modification. J Biol Chem 277: 20974–20978 [DOI] [PubMed] [Google Scholar]
- Monahan Z, Ryan VH, Janke AM, Burke KA, Rhoads SN, Zerze GH, O'Meally R, Dignon GL, Conicella AE, Zheng W et al (2017) Phosphorylation of the FUS low‐complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J 36: 2951–2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzina N, Verreault A, Laue E, Stillman B (1999) Heterochromatin dynamics in mouse cells: interaction between chromatin assembly factor 1 and HP1 proteins. Mol Cell 4: 529–540 [DOI] [PubMed] [Google Scholar]
- Nabel G, Baltimore D (1987) An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326: 711–713 [DOI] [PubMed] [Google Scholar]
- Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ (1993) A cooperative interaction between NF‐kappa B and Sp1 is required for HIV‐1 enhancer activation. EMBO J 12: 3551–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping Y‐H, Rana TM (2001) DSIF and NELF interact with RNA polymerase II elongation complex and HIV‐1 tat stimulates P‐TEFb‐mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation. J Biol Chem 276: 12951–12958 [DOI] [PubMed] [Google Scholar]
- Plys AJ, Davis CP, Kim J, Rizki G, Keenen MM, Marr SK, Kingston RE (2019) Phase separation of Polycomb‐repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev 33: 799–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivy J‐P, Roche D, Kirschner D, Tagami H, Nakatani Y, Almouzni G (2004) A CAF‐1 dependent pool of HP1 during heterochromatin duplication. EMBO J 23: 3516–3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razooky Brandon S, Pai A, Aull K, Rouzine Igor M, Weinberger Leor S (2015) A hardwired HIV latency program. Cell 160: 990–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese BE, Bachman KE, Baylin SB, Rountree MR (2003) The methyl‐CpG binding protein MBD1 interacts with the p150 subunit of chromatin assembly factor 1. Mol Cell Biol 23: 3226–3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseaux MWC, Revelli J‐P, Vázquez‐Vélez GE, Kim J‐Y, Craigen E, Gonzales K, Beckinghausen J, Zoghbi HY (2018) Depleting Trim28 in adult mice is well tolerated and reduces levels of α‐synuclein and tau. ELife 7: e36768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruelas Debbie S, Greene Warner C (2013) An integrated overview of HIV‐1 latency. Cell 155: 519–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC et al (2018) Coactivator condensation at super‐enhancers links phase separation and gene control. Science 361: eaar3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Hess D, Eglinger J, Fritsch AW, Kreysing M, Weinert BT, Choudhary C, Matthias P (2019) Acetylation of intrinsically disordered regions regulates phase separation. Nat Chem Biol 15: 51–61 [DOI] [PubMed] [Google Scholar]
- Saksouk N, Simboeck E, Déjardin J (2015) Constitutive heterochromatin formation and transcription in mammals. Epigenet Chrom 8: 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanulli S, Trnka MJ, Dharmarajan V, Tibble RW, Pascal BD, Burlingame AL, Griffin PR, Gross JD, Narlikar GJ (2019) HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 575: 390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder ARW, Shinn P, Chen H, Berry C, Ecker JR, Bushman F (2002) HIV‐1 integration in the human genome favors active genes and local hotspots. Cell 110: 521–529 [DOI] [PubMed] [Google Scholar]
- Smith S, Stillman B (1989) Purification and characterization of CAF‐I, a human cell factor required for chromatin assembly during DNA replication in vitro . Cell 58: 15–25 [DOI] [PubMed] [Google Scholar]
- Spivak AM, Planelles V (2018) Novel latency reversal agents for HIV‐1 cure. Annu Rev Med 69: 421–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straussman R, Ben‐Ya’acov A, Woolfson DN, Ravid S (2007) Kinking the coiled coil – negatively charged residues at the coiled‐coil interface. J Mol Biol 366: 1232–1242 [DOI] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH (2017) Phase separation drives heterochromatin domain formation. Nature 547: 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejbalová K, Kovářová D, Blažková J, Machala L, Jilich D, Weber J, Kučerová D, Vencálek O, Hirsch I, Hejnar J (2016) Development of 5’ LTR DNA methylation of latent HIV‐1 provirus in cell line models and in long‐term‐infected individuals. Clin Epigenet 8: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Reinberg D (2007) Facultative heterochromatin: is there a distinctive molecular signature? Mol Cell 28: 1–13 [DOI] [PubMed] [Google Scholar]
- Tursun B, Patel T, Kratsios P, Hobert O (2011) Direct conversion of C. elegans germ cells into specific neuron types. Science 331: 304–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner TA, McLaughlin S, Garg K, Cheung CYK, Larsen BB, Styrchak S, Huang HC, Edlefsen PT, Mullins JI, Frenkel LM (2014) Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 345: 570–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Liu X, Gao Y, Yang L, Li C, Liu W, Chen C, Kou X, Zhao Y, Chen J et al (2018) Reprogramming of H3K9me3‐dependent heterochromatin during mammalian embryo development. Nat Cell Biol 20: 620–631 [DOI] [PubMed] [Google Scholar]
- Wang L, Gao Y, Zheng X, Liu C, Dong S, Li R, Zhang G, Wei Y, Qu H, Li Y et al (2019) Histone modifications regulate chromatin compartmentalization by contributing to a phase separation mechanism. Mol Cell 76: 646–659.e6 [DOI] [PubMed] [Google Scholar]
- Watts BR, Wittmann S, Wery M, Gautier C, Kus K, Birot A, Heo D‐H, Kilchert C, Morillon A, Vasiljeva L (2018) Histone deacetylation promotes transcriptional silencing at facultative heterochromatin. Nucleic Acids Res 46: 5426–5440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E, Kim PS, Berger B (1997) MultiCoil: A program for predicting two‐and three‐stranded coiled coils. Protein Sci 6: 1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JK, Hezareh M, Günthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD (1997) Recovery of replication‐competent HIV despite prolonged suppression of plasma viremia. Science 278: 1291–1295 [DOI] [PubMed] [Google Scholar]
- Xiao Z, Chang J‐G, Hendriks IA, Sigurðsson JO, Olsen JV, Vertegaal ACO (2015) System‐wide analysis of SUMOylation dynamics in response to replication stress reveals novel small ubiquitin‐like modified target proteins and acceptor lysines relevant for genome stability. Mol Cell Proteomics 14: 1419–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Souquere S, Chujo T, Kobelke S, Chong YS, Fox AH, Bond CS, Nakagawa S, Pierron G, Hirose T (2018) Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol Cell 70: 1038–1053.e1037 [DOI] [PubMed] [Google Scholar]
- Yang X, Chen Y, Gabuzda D (1999) ERK MAP kinase links cytokine signals to activation of latent HIV‐1 infection by stimulating a cooperative interaction of AP‐1 and NF‐κB. J Biol Chem 274: 27981–27988 [DOI] [PubMed] [Google Scholar]
- Yang Bin X, El Farran CA, Guo Hong C, Yu T, Fang Hai T, Wang Hao F, Schlesinger S, Fen SY, Seah YFS, Goh GYL et al (2015) Systematic identification of factors for provirus silencing in embryonic stem cells. Cell 163: 230–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Fetter RD, Hu P, Betzig E, Tjian R (2011) Subnuclear segregation of genes and core promoter factors in myogenesis. Genes Dev 25: 569–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Su JM, Samuel CE, Ma D (2019) Measles virus forms inclusion bodies with properties of liquid organelles. J Virol 93: e00948‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D (1997) Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo . Nat Biotechnol 15: 871–875 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Movie EV1
Movie EV2
Movie EV3
Source Data for Expanded View and Appendix
Review Process File
Data Availability Statement
The RNA‐Seq data from this publication have been deposited to the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166337) and assigned the identifier (GSE166337). The MS proteomics data from this publication have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD024172) via the iProX partner repository (https://www.iprox.org/page/project.html?id=IPX0002805000) and assigned the identifier (PXD024172). Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author, Dr. Hui Zhang (zhangh92@mail.sysu.edu.cn). Plasmid sequences for CHAF1A mutants will be made available upon request. Purified proteins for in vitro experiments can be provided upon execution of a material transfer agreement (MTA) with inquiries directed to Dr. Hui Zhang.