

Abstract
Drug resistance is prevalent across many diseases, rendering therapies ineffective with severe financial and health consequences. Rather than accepting resistance after the fact, proactive strategies need be incorporated into the drug design and development process to minimize the impact of drug resistance. These strategies can be derived from our experience with viral disease targets where multiple generations of drugs had to be developed to combat resistance and avoid antiviral failure. Significant efforts including experimental and computational structural biology, medicinal chemistry and machine learning have focused on understanding the mechanisms and structural basis of resistance against direct-acting antiviral (DAA) drugs. Integrated methods show promise for being predictive of resistance and potency. In this review, we give an overview of this research for HIV-1, HCV and IAV, and the lessons learned from resistance mechanisms of DAAs. These lessons translate into rational strategies to avoid resistance in drug design, which can be generalized and applied beyond viral targets. While resistance may not be completely avoidable, rational drug design can and should incorporate strategies at the outset of drug development to decrease the prevalence of drug resistance.
Graphical Abstract
1. Introduction: drug resistance and viral infections
The emergence and spread of drug resistance is a global threat to public health and requires immediate action.1–2 Drug resistance is prevalent in antimicrobials, which include antibiotics, antivirals, antifungals and antiprotozoals. This prevalence of resistance leads to higher medical cost, longer treatment duration, increased patient mortality, and severe financial consequences. In fact, drug resistance is estimated to cost society greater than 55 billion dollars annually.3 Moreover, 700,000 people die globally per year as a result of antimicrobial resistance, which may increase to over 10 million by 2050, if no concerted effort is made to combat this threat.4–5
Each year, viral infections cause millions of deaths and engender efforts to develop novel antivirals. With rapid spread of outbreaks and pandemics on the rise such as Ebola or COVID-19, there is a growing need and interest to design effective therapeutic agents against viral diseases. Over 200 viral species are known to infect humans. Yet, there are FDA-approved direct-acting antiviral (DAA) drugs that target only 9 of these viruses: Human Immunodeficiency virus (HIV), Hepatitis B virus (HBV), Hepatitis C virus (HCV), Human Cytomegalovirus (HCMV), Herpes Simplex virus (HSV), Human Papilloma virus (HPV), Respiratory Syncytial virus (RSV), Varicella-Zoster virus (VZV) and Influenza virus (IAV).6 Currently the quest for DAAs to target SARS-CoV-2 is ongoing, with over 1 million associated deaths worldwide. However, to date antivirals only exist in the regular treatment of HIV, HCV and IAV. Thus, these viruses serve as ideal systems to understanding drug resistance mechanisms and developing strategies to combat antiviral failure.
Drug development efforts for antivirals often have not included strategies to pre-emptively avoid resistance, as small molecule inhibitors are typically rationally designed using natural substrates of the target protein as starting points. This ligand-based drug design approach has successfully been used to discover potent lead compounds and direct-acting antiviral (DAA) drugs against HIV-1 protease, reverse transcriptase and integrase, HCV NS3/4A protease and NS5B polymerase, and influenza neuraminidase. High-throughput screening (HTS)7–9 is often the first step in drug discovery and this process can be streamlined if a focused or knowledge-based library can be constructed with compounds that had success against that specific target class.7, 10 HTS is combined subsequently with medicinal chemistry efforts for hit-to-lead optimization with analogs for structure-activity relationship (SAR) studies where chemical structure of a compound is correlated with biological activity. Iterative rounds of optimization are often necessary to improve binding affinity, cellular potency, selectivity, toxicity, and pharmacokinetic properties.11–12 While the combination of HTS and extensive SAR studies has been very successful in drug discovery in the absence of structural studies (including the discovery of HIV-1 protease inhibitor tipranavir, HIV-1 integrase inhibitor raltegravir, and the HCV NS5A inhibitor daclatasvir), incorporating structure-based drug design (SBDD)13–14 can accelerate drug discovery. SBDD in combination with HTS and medicinal chemistry optimization has yielded many antivirals.15–17 However incorporating strategies to avoid resistance in antiviral discovery is not common.
SBDD traditionally focuses solely on increasing ligand interactions with the target to disrupt the activity without considering potential heterogeneity of the target or resistance. Viruses have a wide genetic diversity from single polymorphisms to large divergences leading to different subtypes (i.e. genotypes in HCV, clades in HIV, and strains in influenza virus and even variations between flaviviruses and coronaviruses).18–20 Polymorphisms among genotypes of HCV underlie the differential efficacy of protease inhibitors.21 Even when an antiviral has high potency against a certain wild-type strain, resistance can evolve. The single digit nanomolar or even picomolar22–24, potency against the wildtype target enzyme has led to FDA approval of multiple chemically similar inhibitors against the same target.6 However, this has resulted in cross-resistance 25–28 where a single mutation selected against one inhibitor can confer resistance to others in the same class. Often combination therapy, which involves simultaneously inhibiting more than one target, works effectively to suppress viral replication. Nevertheless, if for any reason the drug combination is not effective and the virus is allowed to replicate in the presence of these drugs then the selective pressure on the target still permits the emergence of a resistant viral population. Thus, disrupting an antiviral target’s activity is necessary but not sufficient for developing a robust drug with a low probability of resistance. The high rates of antiviral drug resistance suggest that our current paradigm for drug development needs improvement (Figure 1). Pre-emptively restricting the evolutionary pathways to resistance requires further understanding of the underlying molecular mechanisms, including the structural and dynamic changes due to resistance mutations in the protein–inhibitor system. Improved drug design strategies should proactively consider viral evolution to reduce rates of resistance.
Figure 1.
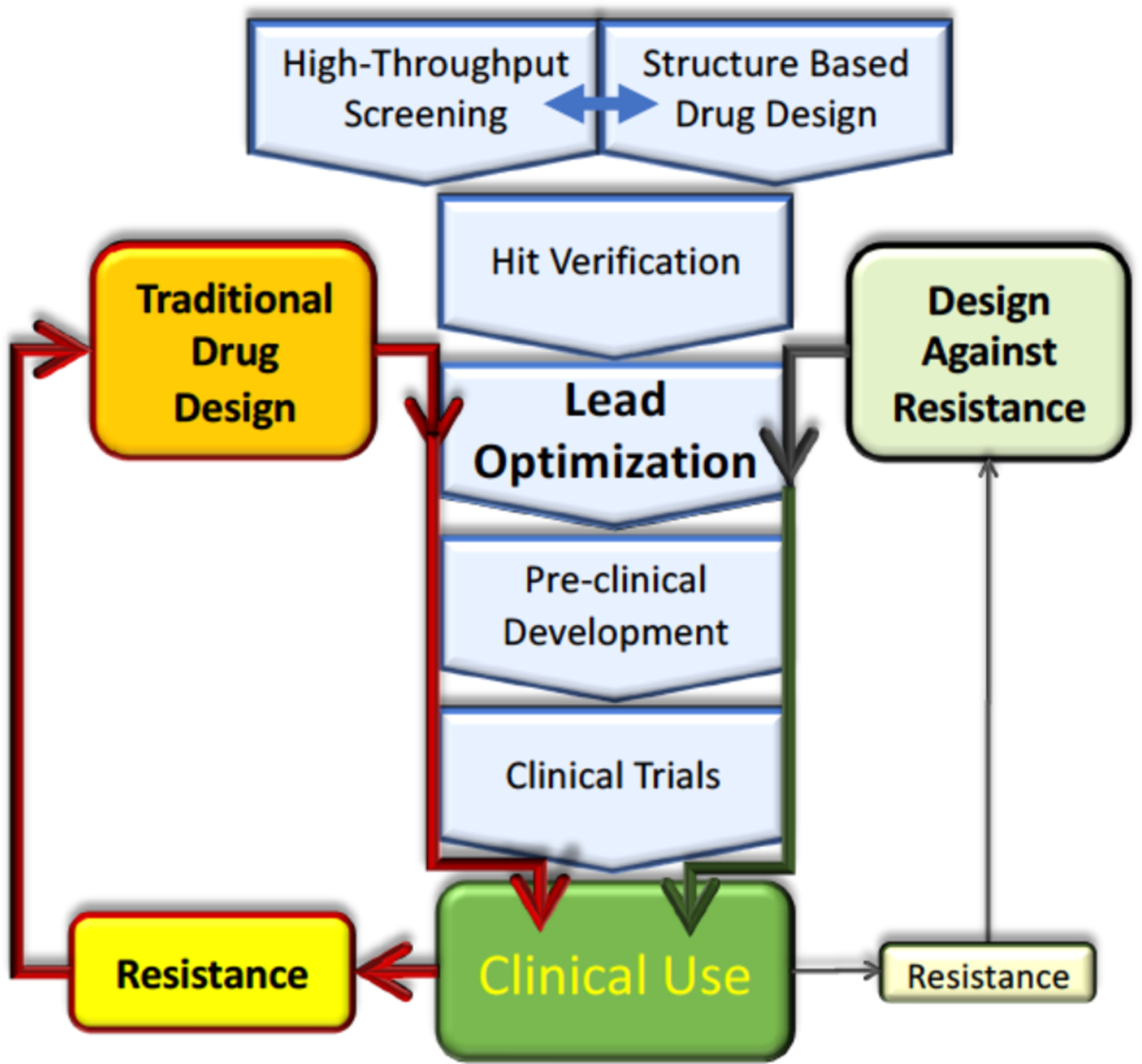
Traditional drug design and development process can result in clinical resistance, which renders therapies obsolete. Incorporating strategies to avoid resistance in the lead optimization of drug design can minimize the probability of resistance.
In this review, we focus on HIV, HCV and IAV as clinical resistance exists to DAAs against each of these viruses. We begin with a brief review of current antivirals against these three viruses. Next, the structural and dynamic mechanisms by which mutations in the antiviral target confer resistance is discussed where we focus primarily on the extensive data on the viral proteases of HIV and HCV, with brief references to other systems. We then describe how elucidation of these mechanisms has been incorporated in both SBDD and medicinal chemistry strategies to avoid resistance. Finally, we describe how integrative computational methods combine experimental data with structural information and conformational dynamics into a comprehensive drug design strategy to avoid drug resistance. These methods and strategies developed to avoid resistance in viral proteases should generally be applicable to other quickly evolving enzymatic drug targets where susceptibility to drug resistance is a necessary consideration to ensure the effectiveness of the therapeutic.
2. Viruses targeted with antivirals
2.1. Human immunodeficiency virus (HIV)
Currently ~38 million people million globally are living with Human Immunodeficiency Virus type 1 (HIV-1) with approximately 40,000 new infections in the United States yearly.29 DAAs against HIV-1 inhibit critical proteins in the viral life cycle including the reverse transcriptase (RT), integrase, and protease. Two classes of RT inhibitors have been approved by the FDA: nucleoside RT inhibitors (NRTIs) and non-nucleoside RT inhibitors (NNRTIs). NRTIs are competitive inhibitors that bind at the active site while NNRTIs bind allosterically and inhibit the enzyme with a noncompetitive or uncompetitive mechanism.30 The first FDA-approved NRTI, zidovudine/AZT, was developed using traditional drug discovery approaches.31 Since then nine additional NRTIs have been FDA approved, the latest of which was emtricitabine in 2003.
Another antiviral target is HIV-1 integrase, which catalyzes the integration of viral DNA into the host genome. A combination of HTS and SBDD approaches led to development of raltegravir, a competitive integrase stand-transfer inhibitor approved by the FDA in 2007.32–33 Medicinal chemistry efforts led to the discovery and FDA approval of dolutegravir in 2013, and recently another integrase inhibitor bictegravir (Figure 2). As with most viral small molecule inhibitors, resistance to integrase inhibitors have emerged, with mutations in integrase that decrease inhibitor potency.34
Figure 2.
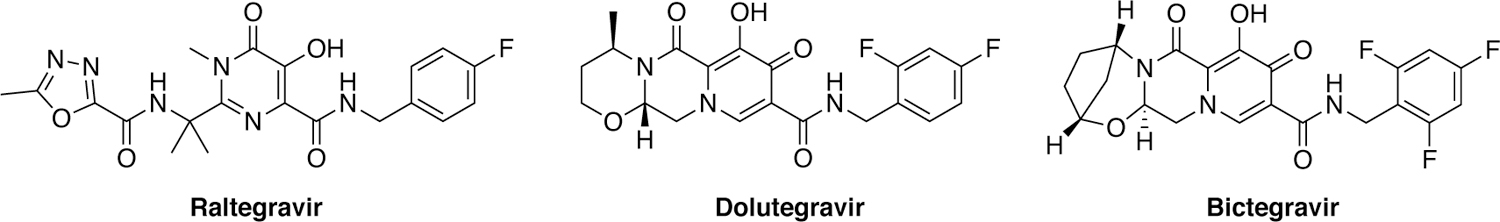
Structures of HIV-1 integrase inhibitors.
After DNA integration, replication and virion assembly occur leading to immature virions budding off from the host cell. Viral maturation requires processing of the Gag and Gag-Pol polyproteins by HIV-1 protease. Inhibiting HIV-1 protease prevents the formation of mature and thus infectious virus. Leveraging modern drug discovery techniques, the first protease inhibitor (PI), saquinavir, was designed as a peptidomimetic transition-state analogue. Since the FDA approval of saquinavir in 1995, a combination of SBDD and medicinal chemistry approaches led to the development of nine FDA-approved PIs, all of which are peptidomimetics with the exception of tipranavir (Figure 3).35 The latest and most potent of the FDA approved PIs is darunavir (DRV), which retains potency against many single and double resistance mutations.36 DRV has a high barrier to resistance and > 7 mutations are necessary in vitro for the drug to become ineffective.37 In fact, there is no evidence of appreciable resistance to single or double mutant variants. Clinical resistance to DRV does occur albeit rare and is often in the setting of cross-resistance with treatment-experienced patients.38–39
Figure 3.
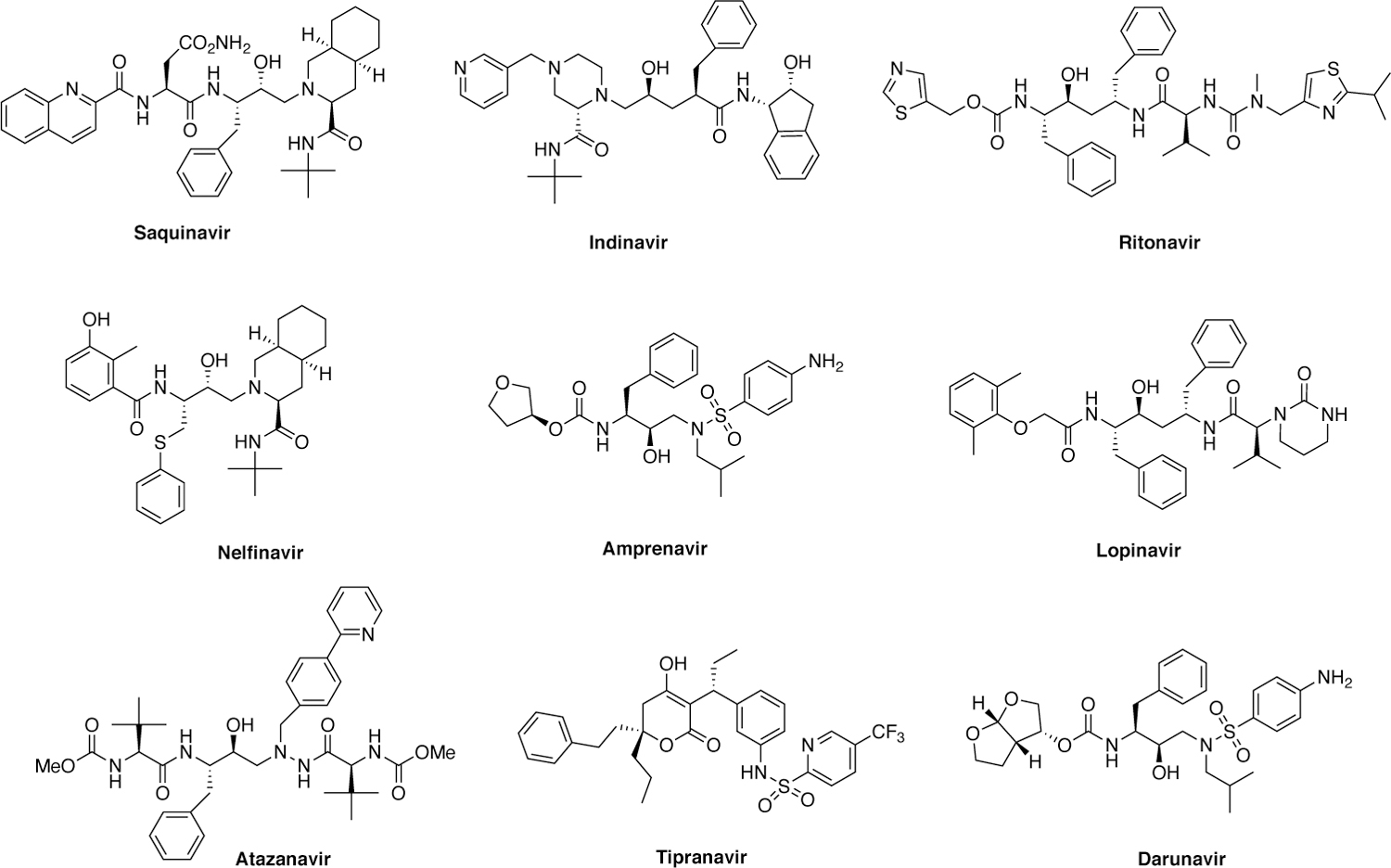
Structures of HIV-1 protease inhibitors.
HIV-1 infected individuals need to be treated with combination therapies as individual DAAs are highly susceptible to resistance. DAAs target viral proteins which can readily mutate due to error-prone activity of the RT, lack of proof-reading functionality, high replication rate and host factors (APOBEC3s) thereby providing the opportunity for resistance to be selected. Resistance is exacerbated by poor patient compliance leading to suboptimal DAA concentrations that cannot suppress viral replication and thus evolution of resistant variants. Six years post infection, a single HIV-1 infected patient can possess the viral genetic diversity of the entire influenza strain of 1996.40 The immense genetic diversity can result in rapid selection of resistance to individual DAAs, requiring combination therapies to effectively suppress the viral population and prevent resistance. The combination of drugs with distinct mechanism of action effectively reduces HIV viral load in patients, in some to undetectable levels.41
2.2.2. Hepatitis C virus (HCV)
Similar to HIV-1, HCV is a global health problem, with over 71 million people being chronically infected worldwide. HCV is the leading cause of chronic liver disease, liver cirrhosis and hepatocellular carcinoma. HCV was historically known as a “silent killer” as many people are unaware of their infection and eventually 80% of patients progress to chronic liver disease.42 HCV has seven known genotypes (GT 1–7), each of which is further divided into subtypes.18 This diversity has presented a challenge in developing effective pan-genotypic therapies. Over the last several years, effective DAAs have been developed against essential proteins in the viral life cycle including the NS3/4A protease, NS5A assembly protein and NS5B polymerase. Lessons from decades of work on HIV-1 DAAs have been applied to HCV, most notably using multiple drugs as a combination therapy to prevent or at least minimize the emergence of resistance. Unlike with HIV-1 which causes incurable life-long chronic infection, HCV infected patients can be cured with combination therapy. The most recent combinations achieve 95% or higher cure rates in certain patient populations, but genotypic differences and resistance mutations can still cause treatment failure.
The first FDA-approved antiviral drugs against HCV were inhibitors of the NS3/4A protease.43–44 This protease is responsible for cleaving the viral polyprotein into structural and nonstructural proteins that are critical in viral replication and maturation. A major turning point in HCV protease inhibitor development occurred in 1998, when the N-terminal cleavage product of a designed (consensus) substrate peptide DDIVPC-OH was identified as a weak competitive inhibitor.45–46 Pharmaceutical companies exploited this hexapeptide scaffold by extensive SAR exploration and SBDD, which led to the development of the first-in-class NS3/4A protease inhibitor ciluprevir, a macrocyclic inhibitor that exhibited nanomolar antiviral activity.47–49 Unfortunately, ciluprevir was discontinued due to cardiotoxicity.50 Further inhibitor development using SBDD has led to the FDA approval of 7 protease inhibitors with more in clinical development (Figure 4).6, 51 First generation protease inhibitors had activity against mainly only GT1, the most prevalent genotype in the West. Most recent inhibitors grazoprevir, glecaprevir, and voxilaprevir are extremely potent and exhibit pan-genotypic activity.52–53
Figure 4.
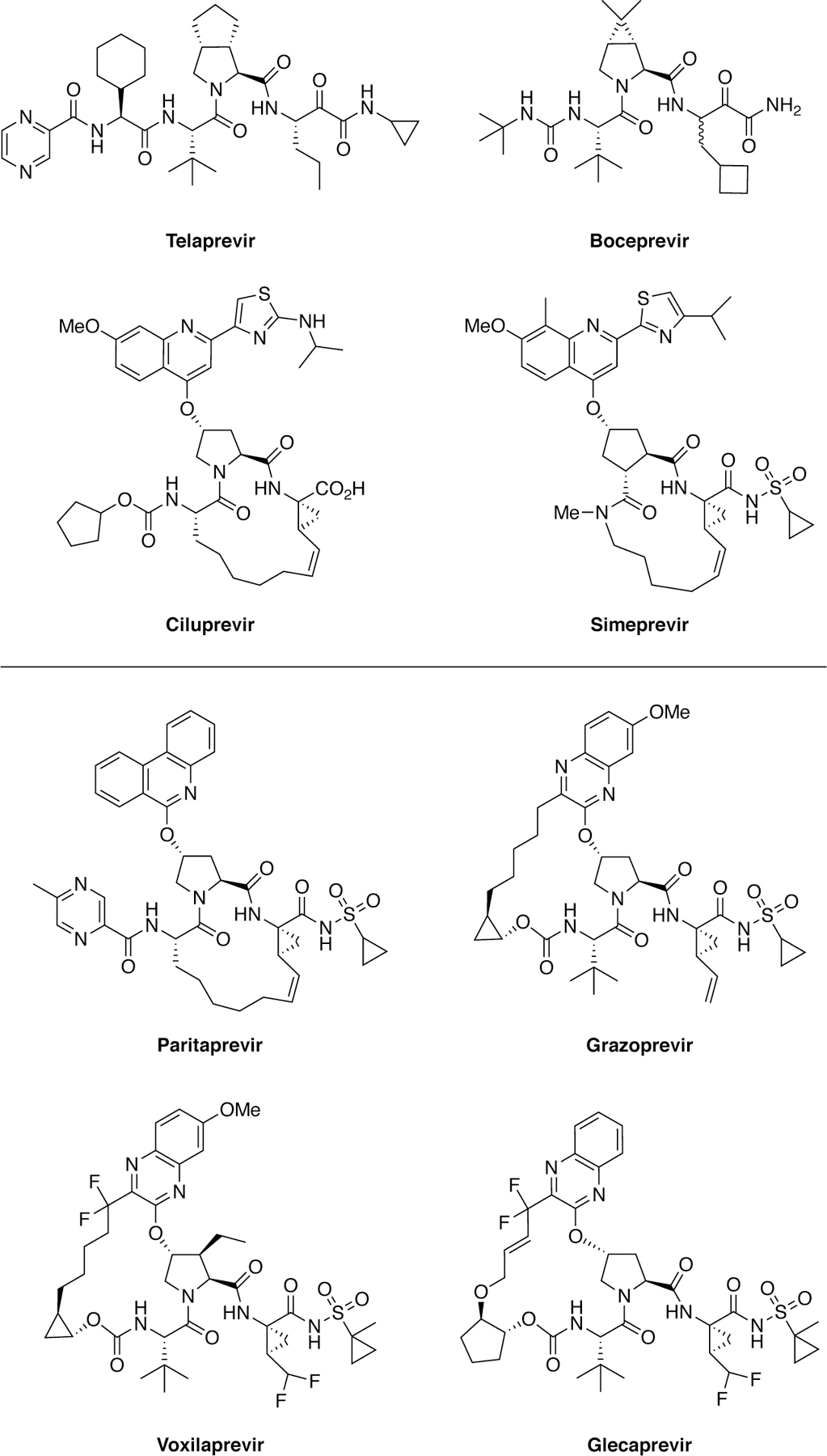
Chemical structures of first-generation (top) and latest generation (bottom) HCV NS3/4A protease inhibitors.
The NS5B is the RNA-dependent RNA polymerase which is responsible for viral replication. The first combination therapy that achieved a cure for HCV infection was Harvoni® which contained NS5B inhibitor sofosbuvir.54 There are two FDA-approved NS5B inhibitors, sofosbuvir and dasabuvir (Figure 5), with others in clinical development.6 Sofosbuvir is a nucleoside analogue that gets incorporated into the nascent RNA chain, which induces a chain termination event to stop active transcription.55 Sofosbuvir was discovered through SAR studies of nucleoside analogues and replicon–based screening assays where it exhibited nanomolar activity against the wildtype enzyme.56 Similarly, NS5B inhibitor dasabuvir was discovered through a HTS campaign of libraries of compounds that inhibit the activity of recombinant polymerase in vitro.57 Unlike sofosbuvir, dasabuvir is a non-nucleotide small molecule that allosterically binds to the polymerase preventing conformational changes that are necessary for viral RNA replication.58 Dasabuvir has a low barrier to resistance given its allosteric binding site that can mutate without affecting substrate nucleotide binding.59 In contrast, the nucleoside NS5B inhibitors exhibit pan-genotypic activity and a higher barrier to resistance, and clinical failure is rare with sofosbuvir containing combination therapies.
Figure 5.
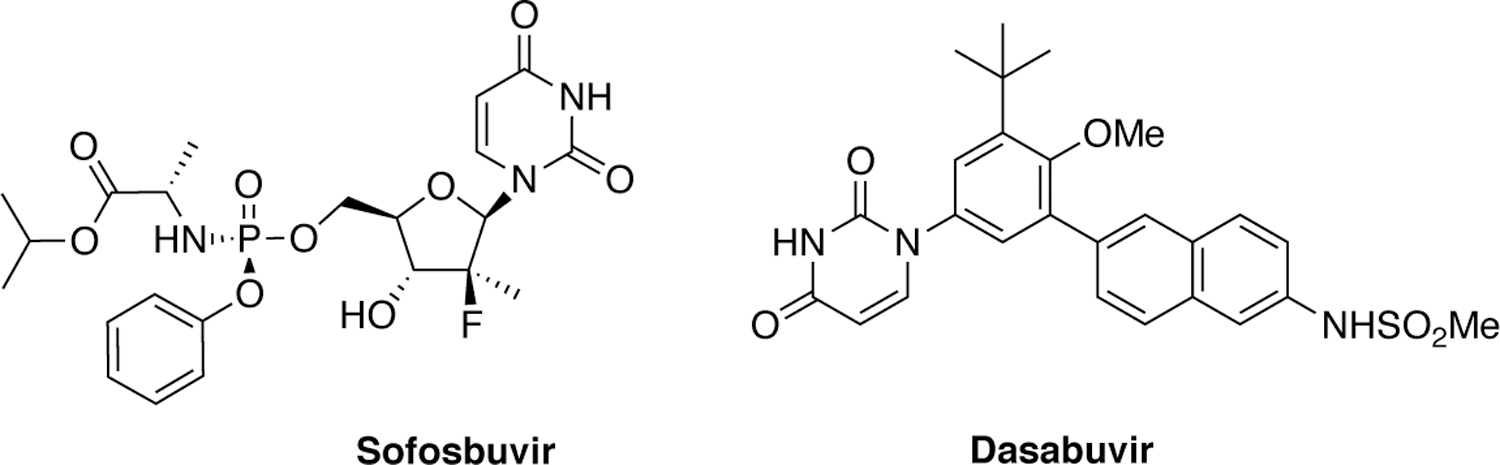
Chemical structures of HCV NS5B polymerase inhibitors.
The last class of HCV DAAs is the NS5A inhibitors, which block RNA replication and virion assembly/release.60 This class includes FDA-approved inhibitors daclatasvir, ledipasvir, ombitasvir, elbasvir, velpatasvir, and pibrentasvir (Figure 6). The initial lead molecule was identified by a phenotypic HTS and optimized by extensive SAR studies, leading to the discovery of first-in-class HCV NS5A inhibitor daclatasvir.
Figure 6.
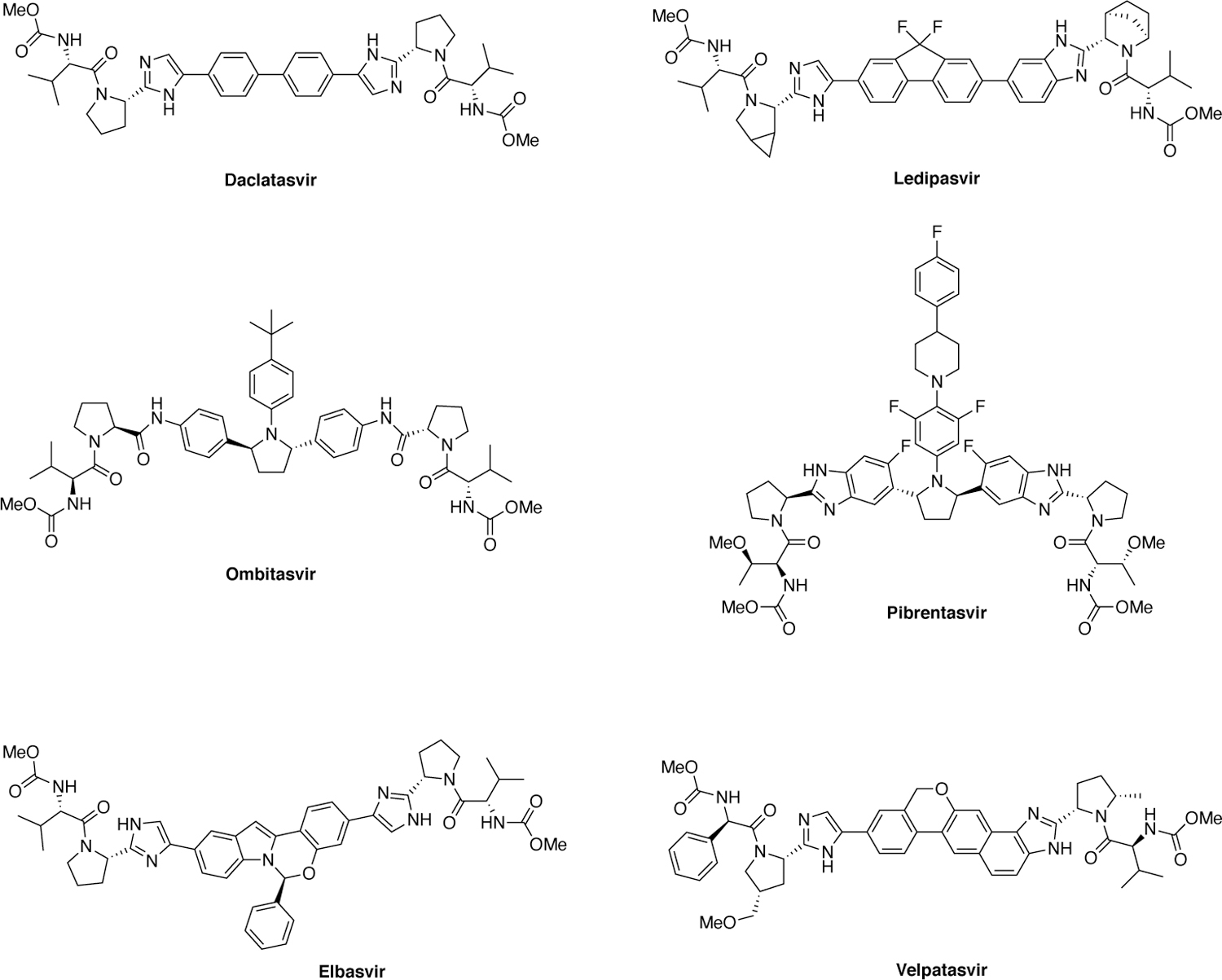
Chemical structures of HCV NS5A inhibitors.
Despite improved treatment options and outcomes61–63 with combination therapies,64–68 drug resistance remains a problem. Even the most recent DAA combinations fail to cure some patients.27, 63, 69 Especially for DAA-experienced patients, baseline polymorphisms among diverse genotypes and preexisting resistance-associated substitutions negatively impact treatment outcomes.62–63, 70 Thus, strategies to thwart resistance need to be applied in the design and development of novel antiviral therapeutics.
2.2.3. Influenza virus (IAV)
The CDC estimates that influenza infects between 39–56 million people annually in the United States, resulting in over 410,000 hospitalizations and 12,000–60,000 deaths.71 The annual influenza vaccine provides preventative measure against seasonal influenza. However, vaccine effectiveness can be low due to antigenic drift, high mutation rate, and mismatch between vaccine and circulating strains.72–73 As a result, DAA options are needed for combating influenza infection especially in vulnerable populations such as those with underlying conditions.
There are currently three FDA-approved DAAs against IAV, which target neuraminidase (NA) on the surface of the viral particles (Figure 7). NA is a sialidase that cleaves the terminal sialic acid from glycoproteins to release the budding virus from the surface of infected host cells.74–76 In 1999, the FDA approved two competitive active site NA inhibitors, oral oseltamivir, inhaled zanamivir, and in 2014 peramivir for intravenous administration.77–80
Figure 7.
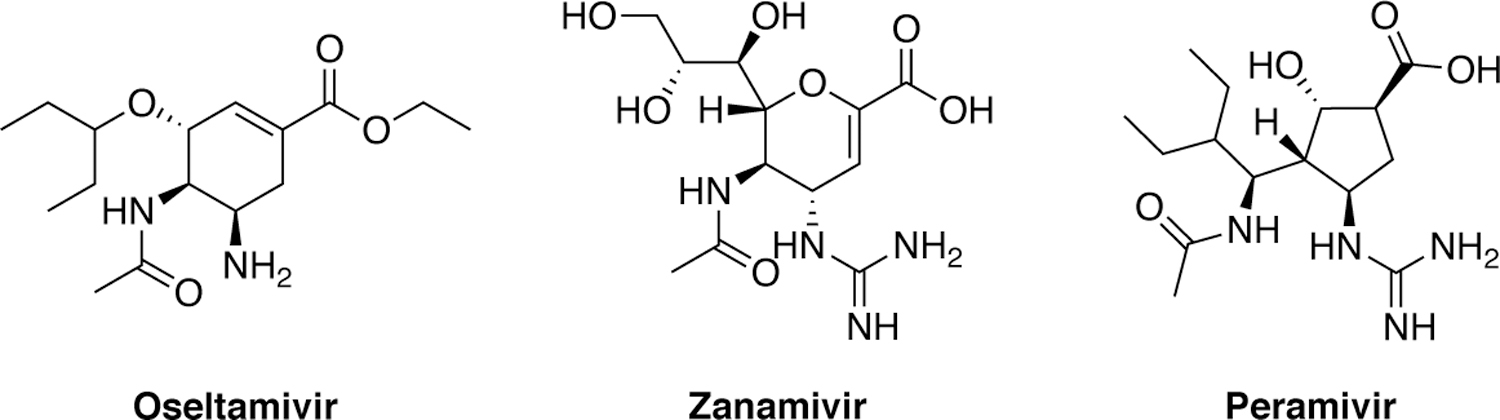
Chemical structures of influenza neuraminidase inhibitors.
Influenza NA inhibitors were rationally designed based on the molecular features of substrates and to optimize binding interactions in the active site; however, resistant variants that avoid inhibition but still cleave the substrate sialic acid have emerged.78–81 Although sialic acid binding sites between subtypes are highly homologous, subtype specific patterns of drug resistance have emerged against NA inhibitors.82–85 Type A influenza is divided into subtypes based on two surface proteins, hemagglutinin (HA) and NA. Type A is the most prevalent and is subdivided into two subtypes that predominate in human infection, N1 and N2. Differential patterns of drug resistance mutations have been observed clinically in both N1 and N2, and identified experimentally through in vitro and in vivo experiments.81
Recently in 2018, baloxavir targeting the cap-dependent endonuclease of the PA subunit of IAV has been approved by the FDA. Although effective against strains resistant to the other three drugs that target NA, baloxavir has a low barrier to resistance. A single substitution such as the common I38T can confer baloxavir resistance.86 Antivirals that target other essential IAV proteins, ideally in combination with those with orthogonal resistance profiles, are likely needed for an effective influenza therapy that is less susceptible to resistance, if pre-existing resistance prevails within a pandemic strain.
3. Mechanisms of drug resistance
On the molecular level, multiple mechanisms can lead to drug resistance and decrease the potency of antiviral therapies. Many viruses are masterfully erroneous and thus acquire random mutations during replication. Patients infected with these viruses develop a heterogeneous population of viral species known as quasi-species. Often, a patient when first diagnosed will already be infected with a heterogeneous viral population.87–89 When therapy begins mutations are selected that prevent inhibitor binding while maintaining viral replication.90–91 The emergence of these resistant variants eventually renders once-effective drugs obsolete.92 This is especially problematic as traditional inhibitor design paradigms focus on the wild-type target only and do not incorporate strategies to evade resistance. Thus, without pre-emptive methods rapidly evolving disease targets, the acquisition of resistance is almost inevitable given enough time.
3.1. Sites of drug resistance mutations
Drug resistance mutations in a therapeutic target often occur initially around the drug binding site, where primary resistance is conferred through a physical change in the direct contacts with the inhibitor. For a competitive active site inhibitor, mutations causing this primary resistance is usually located at or adjacent the active site (Figure 8). For high levels of resistance to occur against potent inhibitors, these primary active site mutations are often supplemented with mutations that are distal to the active site. These distal mutations, often referred to as secondary mutations, were thought to primarily be compensatory and preserve enzymatic function lost due to primary mutations; however, recent data have shown these distal mutations can contribute significantly to the loss of inhibitor potency.93–97
Figure 8.
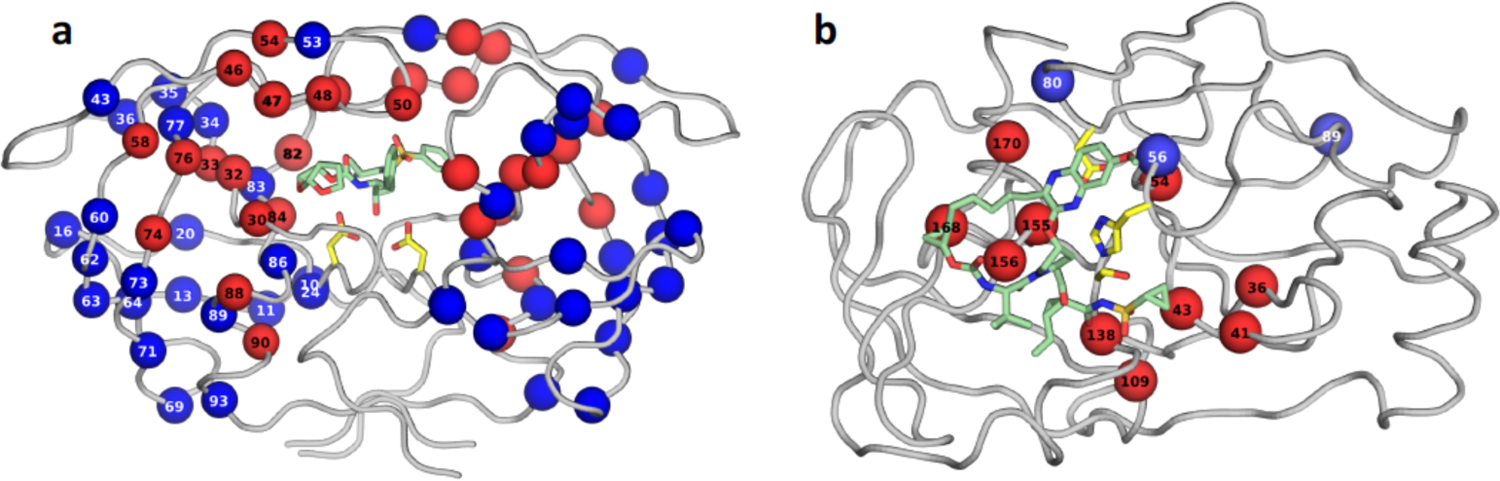
Residues that are sites of primary (red spheres) and secondary (blue spheres) resistance mutations displayed on the structures of (a) HIV-1 protease, and (b) HCV NS3/4A protease. The protease backbone is represented as gray tubes, side chains of catalytic residues are shown as yellow sticks, and inhibitor bound at the active site is displayed as green sticks. Primary resistance mutations are mostly at the active site where the inhibitor binds but many resistance mutations can occur distal from the active site.
3.2. Substrate recognition versus inhibitor binding
Resistance mutations in the drug target are selected to tip the balance in favor of substrate recognition over inhibitor binding. Substrate recognition has been extensively studied in HIV-1 and HCV NS3/4A proteases (Figure 9).98–101 HIV-1 protease cleaves at least 10 sites along the Gag and the Gag-Pro-Pol polyproteins to release proteins that are necessary in the viral life cycle.102 Similarly, HCV protease cleaves four sites and two innate immune adapters. These viral substrates share little amino acid sequence homology and thus are highly diverse.100 If the substrates have little sequence homology, how is a viral protease able to recognize and cleave the appropriate site along the polyprotein with specificity? Moreover, how are proteases with multiple mutations able to process the substrate sequences necessary for viral maturation and evade inhibition by small molecules? The answer lies in the shape the substrates adopt when bound to protease, and not the exact amino acid sequence.
Figure 9.
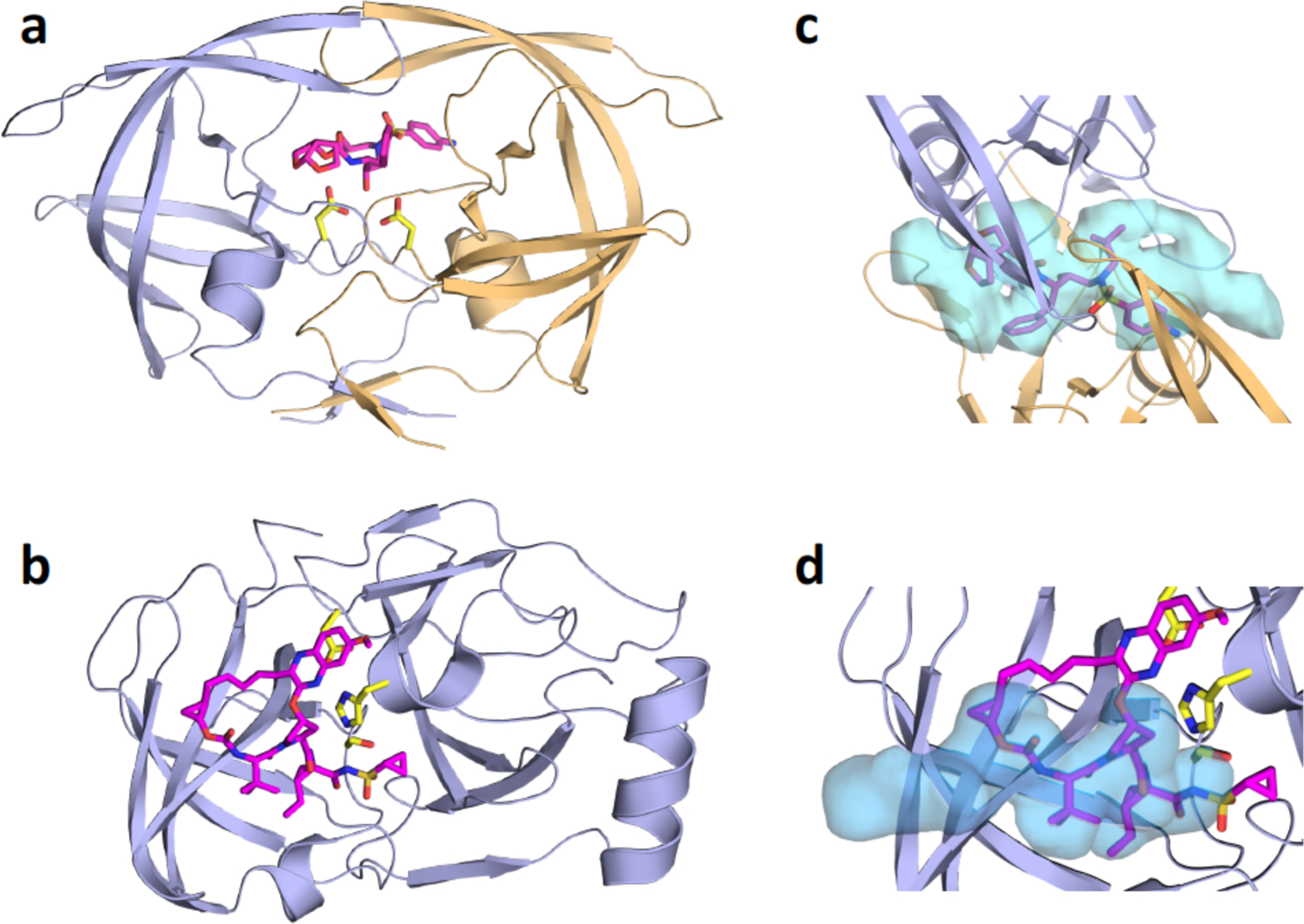
Crystal structures of inhibitor-bound viral proteases, and the substrate envelopes determined through substrate-bound cocrystal structures. a) HIV-1 protease bound to darunavir (DRV; magenta sticks). The two protein chains that comprise the homodimeric protease are in light violet and gold cartoon representation, with DRV bound at the active site. b) HCV NS3/4A protease (light violet cartoon) with grazoprevir (GZR; magenta sticks) bound at the active site. c) HIV-1 protease substrate envelope (blue volume) and fit of DRV within the envelope. d) HCV NS3/4A protease substrate envelope (blue volume) and fit of GZR within the envelope.
Crystal structures of substrate-protease complexes showed that HIV-1 viral substrates occupy a consensus volume or shape in the active site, now termed the substrate envelope (Figure 9c).100 The substrate envelope for any enzyme can be determined by solving cocrystal structures with endogenous substrates that the enzyme has to process for biological function, and provides key molecular insights into substrate recognition and resistance.103 Similarly, superposition of substrate-protease complexes of HCV protease showed that substrates adopt a conserved volume in the protease active site (Figure 9d).99–100 Therefore, this consensus volume or substrate envelope is the basis for molecular recognition of substrates for HIV-1 and HCV viral substrates.
3.3. Substrate envelope and primary active site resistance mutations
Comparison of the substrate envelope with inhibitor binding elucidated molecular mechanisms of resistance due to active site mutations. Active site residues that make critical interactions with substrates such as catalytic residues, are essential and would prohibit substrate processing if mutated. Instead, as structures of inhibitor-protease complexes revealed, resistance mutations occur where inhibitors protrude beyond the substrate envelope and make contact with residues that are not essential for substrate recognition.98–99, 104 These mutations differentially impact inhibitor binding while maintaining substrate processing. As each inhibitor may protrude at different sites from the envelope, selected resistance mutations are inhibitor specific. This is evident in HCV NS3/4A protease inhibitors where resistance mutation patterns are largely determined by the P2 extended moiety.98
In HCV, resistance to protease inhibitors are common due to mutations in the S2 and S4 subsites where the inhibitors protrude beyond the substrate envelope and make van der Waals (vdW) contacts with primarily three active site residues: Arg155, Ala156 and Asp168 (Figure 10). These three residues contact the large P2 heterocyclic moiety, an extension to the PIs that significantly improves inhibitor potency, and the P4 capping group.105–106 Arg155 and Asp168 form a critical salt bridge that provides additional hydrophobic surface necessary for inhibitor binding. This salt bridge stabilizes Arg155 in a conformation that allows potent inhibitor binding, often through aromatic stacking with the inhibitor (Figure 10a–c). Disruption of this electrostatic network as a result of substitutions R155K or D168A, or loss of stacking on Arg155 underlies the mechanism of resistance for most first and second generation HCV PIs.100 The third generation inhibitors managed to largely avoid susceptibility to mutations at these two sites, as the P2 moiety packs on the catalytic triad and avoids packing on Arg155, as in grazoprevir (Figure 10d). The related third generation inhibitors glecaprevir52, 68, 107 and voxilaprevir108 (Figure 4) maintain excellent potency across viral genotypes coordinating water structure and sharing the same binding mode with grazoprevir.109 However, the rigid macrocycle in all these PIs still causes susceptibility to changes at 168, and more significantly to mutation of Ala156 to a larger Val or Thr which causes a steric clash with the macrocycle. Thus, significant primary resistance occurs for all the HCV PIs where these inhibitors protrude beyond the substrate envelope and contact residues that can tolerate mutations as they are not essential for substrate recognition and turnover.
Figure 10.
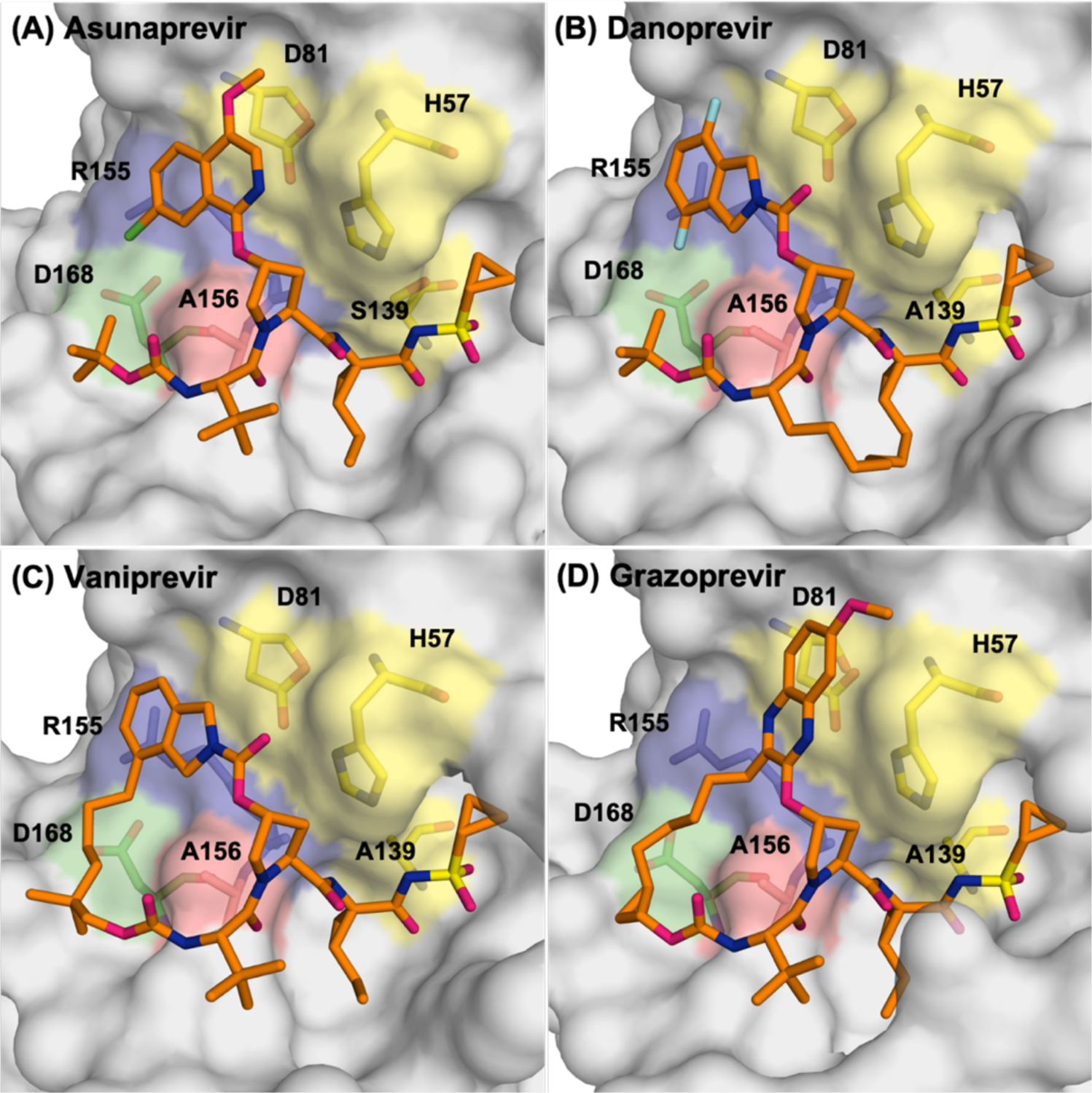
Comparison of the binding conformations of second-generation HCV NS3/4A protease inhibitors asunaprevir (A), danoprevir (B) and vaniprevir (C) with third generation inhibitor grazoprevir (D).
Similarly, in HIV-1 protease, individual inhibitors contact specific active site residues beyond the substrate envelope, which are primary sites of drug resistance mutations selected both in the laboratory and in clinic. For instance, D30N is a signature mutation to nelfinavir, I47A to lopinavir, I50L/V for amprenavir/darunavir/atazanavir, G48V to saquinavir, and V82A/I/F to saquinavir/ritonavir.110 These mutations directly impact inhibitor binding by altering and reducing intermolecular contacts necessary for inhibiting the enzyme (Figure 9), but still allow continuing to recognize and process substrates. DRV is the FDA approved inhibitor that fits best within the substrate envelope and is the least susceptible to resistance. DRV retains picomolar inhibition against primary resistance mutations such as I84V.111 However, DRV is still susceptible to the combined accumulation of mutations proximal and distal to the active site. In a recent study, these distal mutations were shown to perturb the dynamic conformational ensemble of the protease, propagating changes to the active site to severely impact DRV binding.95 Thus, the substrate envelope not only explains substrate specificity but also provides a framework for understanding the mechanism of resistance due to active site mutations.
As with protease inhibitors, resistance to HIV-1 RT inhibitors has been observed. Single mutations around the active site, or the hydrophobic allosteric site of RT can confer significant resistance to NRTIs and NNRTIs, respectively.112 For instance, the M184V/I mutations, close to the active site, decrease susceptibility to the NRTIs lamivudine (3TC) and emtricitabine (FTC) by more than 100-fold. 113–114 In addition, the K65R mutation is highly clinically relevant and has been shown to reduce susceptibility to lamivudine and emtricitabine by 5- to 10-fold115–116 and to tenofovir (TDF) ~2-fold.117 Conserved allosteric sites can also be viable drug targets but are highly susceptible to resistance mutations as is the case for NNRTIs for which single site mutations can compromise affinity. For example, the K103N decreases susceptibility to nevirapine and efavirenz,118–121 Y181C decreases susceptibility to nevirapine,122–123 efavirenz,124 etravirine,125 and rilpivirine126 and G190A decreases susceptibility to nevirapine and efavirenz.118–120 Resistance in HIV-1 RT will be reviewed in a companion article within this issue and thus is not expanded here.
Much like the protease and RT, certain mutations in the integrase are clinically relevant and cause significant levels of resistance to commonly used integrase inhibitors. Mutations N155H and Q148K/R/H decrease susceptibility to raltegravir and elvitegravir. More recent cryo-EM structures of second generation integrase inhibitors, dolutegravir and bictegravir, show how these inhibitors are less susceptable to the resistance that impacted first generation inhibitors, in part by their high affinity, stabilization of the optimal binding geometry and ligand extension to better fill the active site.127–128 G140S is a compensatory mutation34, 129–130 that rescues a replication defect caused by Q148H mutation (Figure 11).
Figure 11.
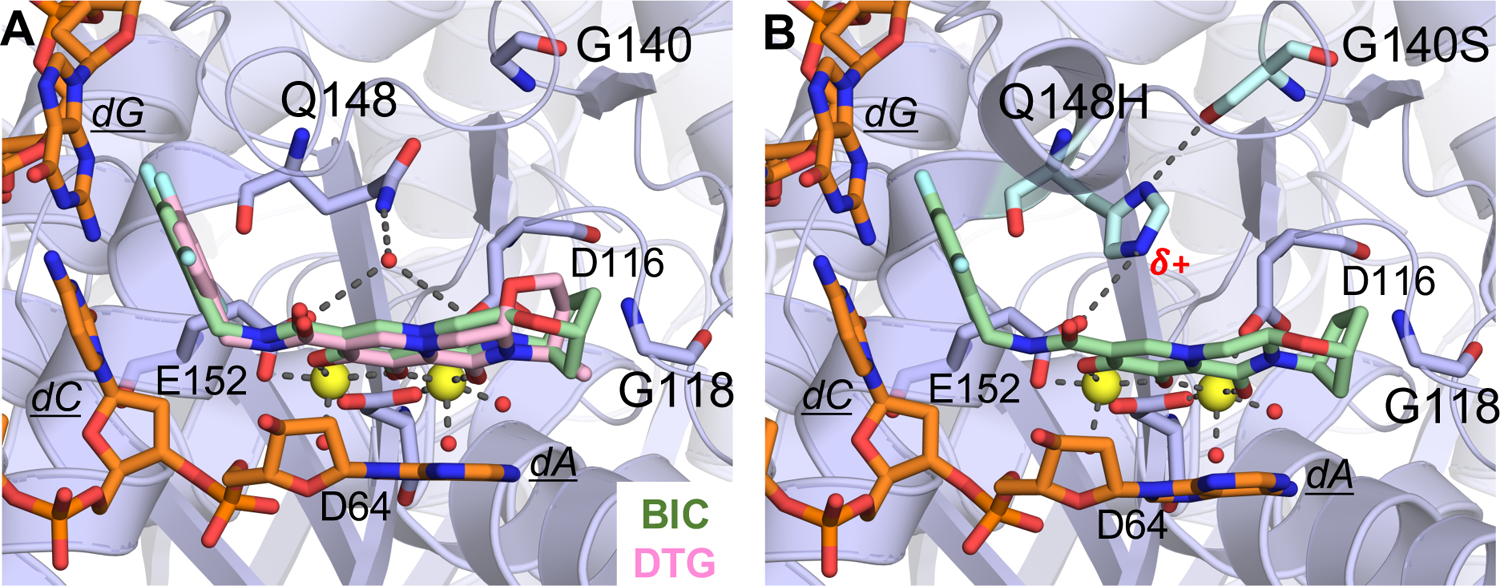
Active site of the Simian immunodeficiency virus (from red-capped mangabeys) intasome in complex with bictegravir (BIC) and dolutegravir (DTG); protein, DNA, and drug are shown as sticks. Yellow spheres represent Mg2+ ions, and water molecules are shown as small red spheres. (A) Superposition of BIC (green) (PDB-ID: 6RWM) and DTG (pink) (PDB-ID: 6RWN) bound structures with protein and DNA shown in orange. (B) Q148H/G140S variant bound to BIC (PDB-ID: 6RWO).127
3.4. Resistance via altered dynamics – applying parallel molecular dynamics (pMD)
Many mutations in a drug target occur remote from the active site of the enzyme and their role in resistance has remained controversial. One mechanism by which distal mutations contribute to resistance is via altering the conformational dynamics of the enzyme-inhibitor system. In the past decade, the strategy of pMD was developed to collectively analyze a series of MD simulations of similar yet distinct molecular complexes to decipher conformational and dynamic differences responsible for changes in molecular recognition due to resistance. pMD simulations are performed on complexes with varied protein sequence and/or inhibitor identity to unravel structural and dynamic properties that underlie coupled changes in molecular recognition, binding affinity and resistance. With this powerful strategy drug resistance mutations were observed to cause alterations in everything from water structure to physical interactions and correlated fluctuations.93–94, 96–97, 131–140 This method was applied to a wide variety of enzymes including the viral proteases of HIV,94, 131, 134–135, 139, 141–144 HCV,93, 145–146 Dengue virus,137–138 and influenza neuraminidase,136 successfully providing insights into the mechanisms of resistance.
Traditional drug design paradigms do not consider the role of dynamics in molecular recognition. Instead, the modus operandi is identifying a lead compound and optimizing by either brute force trial and error or with some insights from the 3D structure if available, with the goal of obtaining desirable activity against the target. However, as pMD studies have indicated, the conformational ensemble and dynamics of the inhibitor–protein complex may be key to potency and resistance. Better understanding and computational tools are needed to incorporate these considerations into drug design to improve potency and avoid drug resistance.
3.5. Dynamic substrate envelope in molecular recognition and resistance
Substrate recognition and processing by enzymes involve a series of dynamic events where the protein will adopt different conformational states. This is not surprising as most proteins are inherently flexible and sample a conformational dynamic ensemble in their native states. These conformational dynamics occur on various time scales and are essential for function. Their substrates are often also naturally flexible, especially for peptide substrates. In the bound complex, the conformational freedom of the substrate and enzyme can be coupled and the dynamics of the overall system is critical for molecular recognition.
The substrate envelope model was initially developed using crystallographic structures and provided crucial insights into drug resistance. Protein crystallography has been invaluable to visualizing, understanding and targeting with small molecules of many proteins. Crystallography has even been able to capture some dynamic movements of the HIV-1 protease at high resolution.37, 147 Still, a great deal of information is missed by crystallography as the structure is only a snapshot of the protein or complex within the confines of the crystal lattice. While crystal structures give some insight into short-range protein dynamics, complementary methods are needed to fully elucidate the details of conformational flexibility.148
The substrate envelope was redefined to include the role of protein dynamics by analyzing substrate-protease complexes of HIV-1 using pMD simulations.132, 148 HIV-1 substrate-protease complexes were simulated and conformational dynamic ensemble analyzed.101, 148–150 Most of the HIV-1 substrate-protease molecular interactions observed in the crystal structures were conserved across the dynamic trajectories. Accordingly, the dynamic substrate envelope, calculated over thousands of substrate conformers from the MD simulations, reproduced main characteristics of the static envelope. In addition, the dynamic substrate envelope gives a probabilistic volume that accounts for substrate flexibility101, 151 and a more realistic representation of the consensus volume that the substrates occupy in the active site.
The dynamic substrate envelope as a predictive tool of resistance has been applied to other viral proteins including HCV protease and neuraminidase. For HCV protease, the dynamic substrate envelope analysis was consistent with that of the static substrate envelope revealing the mechanism of resistance to common resistance-associated substitutions.151 In influenza neuraminidase, the dynamic substrate envelope explained differential patterns of resistance for N1 and N2 despite highly homologous active site (Figure 12).136 Mutations often occur at residues I222, S246 and H274 in N1 and E119 in N2 to confer resistance to NA inhibitors. Intermolecular interactions, especially van der Waals contacts, with these residues are crucial for inhibitor but not substrate binding.82, 136 In these viral proteins, further analysis of the dynamic substrate envelope revealed that inhibitors do not optimally occupy the remaining space in the substrate envelope, which for rapidly evolving disease targets presents opportunities to design inhibitors with improved potency and substrate mimicry.
Figure 12.
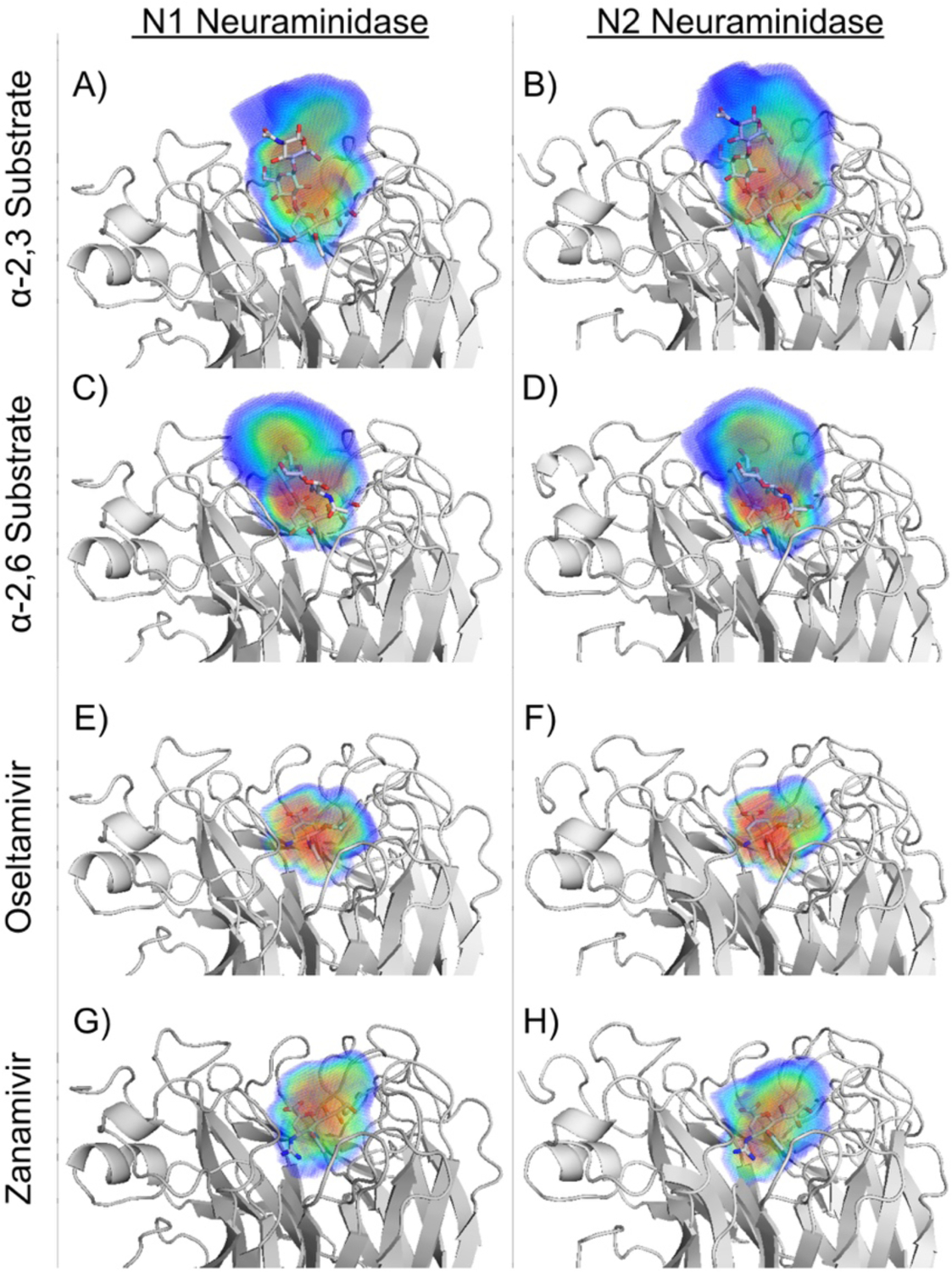
Dynamic substrate and inhibitor envelopes for N1 and N2 influenza NA. The ligands are in gray sticks, and the probabilistic volume distribution for the envelopes is represented using a rainbow color spectrum from red to blue to indicate more to less occupied regions. The left and right columns are for subtypes N1 and N2 NA, respectively. Dynamic substrate envelope of (A, B) α−2,3 and (C, D) α−2,6 substrates and the inhibitor envelopes of (E, F) oseltamivir and (G, H) zanamivir.
Reproduced with permission from Ref 136 Copyright © 2016, American Chemical Society.
3.6. Protein dynamics and role of distal mutations in conferring resistance
Protein dynamics have been shown to play a key role in drug resistance.93–97, 131–133, 135–136, 140, 152–153 Proteins are dynamic and conformational flexibility is required for biological activity and substrate molecular recognition. Mutations not only at the active site but throughout the enzyme can lead to changes in protein dynamics to impact substrate recognition and inhibition. The flexibility and conformation of loops and other structural elements in the enzyme active site can affect catalytic activity and susceptibility to resistance. For example in HIV-1 protease, the flaps need to open and then close down on the substrate, which is coupled with an extensive rearrangement of hydrophobic residues in the core of the enzyme, known as hydrophobic sliding 133, 152. Alterations in the core that affect hydrophobic sliding can thus impact flexibility of the flaps, which control access of substrates and inhibitors to the active site.133, 152
Similarly in influenza neuraminidase, the dynamic 150s-loop controls the size of the active site and flexibility of this loop varies across subtypes.154–155 This loop contains a catalytic residue, Asp151, and therefore, the flexibility and conformation is directly related to substrate processing. Thus, understanding the dynamics of the target of interest and key interactions with substrate is important in inhibitor design.
Importance of conformational dynamics in viral resistance has also been reported for the HIV-1 gp41/gp120 envelope fusion protein. The gp120 variants that have faster fusion kinetics are more resistant to the fusion inhibitor enfuvirtide.156–157 The mechanism of this specific enfuvirtide resistance was shown to accelerate the first step of entry, by mutations altering the conformational dynamics of the CD4-bound Env, limiting the enfuvirtide susceptible conformation that is available for binding.158
3.6.1. Distal mutations and dynamics in HIV-1 protease
Mutations that are distal to the active site in the target enzyme are often observed in resistant variants. As these mutations occur at amino acids that do not physically interact with the inhibitor, the assumption has been that distal mutations merely compensate for the enzyme activity lost due to primary drug resistance mutations. However, recent evidence shows that distal mutations also contribute to resistance.94–97, 159 Although distal changes cannot directly alter intermolecular interactions with the substrate or inhibitor, they exert their effect indirectly by altering the protein’s dynamics. Mutations far from the active site in HIV-1 protease change the dynamic ensemble of the protease and are particularly relevant in evolution of resistance to highly potent inhibitors such as DRV. While DRV is highly potent against wild-type enzyme and fits well within the substrate envelope, high-level resistance to DRV has been observed in patient isolates and in vitro selection variants.94, 96, 119, 160–161 These variants achieve resistance to DRV through a combination of mutations proximal and distal to the active site.
In a study evaluating five highly mutated patient variants with 19–24 mutations, from the HIV Drug Resistance Database160–161 distal mutations were shown to cause alterations in the protein’s internal hydrogen-bonding network.94 Another study investigated a clinical variant using pMD to elucidate the role of distal mutations in resistance where one distal mutation weakened inhibitor potency and another restored substrate processivity in conferring resistance.162 pMD analysis of a series of protease variants each with a single distal mutation bound to DRV revealed that a distal mutation can result in significant rearrangement of the protein and hydrogen bond network that propagates to critical active site residues that interact with the inhibitor. This idea that the effect of distal mutations propagates to pivotal active site residues has been termed as the “network hypothesis.” 94, 134, 162 More recent studies with highly mutated and highly DRV resistant variants confirmed the role of distal mutations in altering the dynamics of the enzyme-inhibitor complex.94–97
Under inhibitor selective pressure, in vitro viral selection experiments159 have generated a highly DRV resistant variant with 11 mutations.95 Much like clinical variants, in vitro selection variants need to accumulate a combination of active site and distal mutations for high-level resistance. With 3 active site and 8 distal mutations, this selection variant reduced DRV’s low picomolar inhibition to near micromolar inhibition, a 152,000-fold decrease in potency. Protease variants engineered to harbor subsets of these 11 mutations revealed that the distal mutations were critical in achieving high-level resistance. Crystal structures of these complexes did not reveal a potential molecular mechanism to account for this loss of affinity. In contrast pMD simulations reflected altered the molecular mechanism through altered dynamics of the enzyme-inhibitor complex that correlated with the experimental loss of affinity, this loss was observed in a variety of physical characteristics including the increase in root mean square fluctuation of DRV and a decrease in the van der Waals contact DRV makes with the enzyme (Figure 13) 95.
Figure 13.
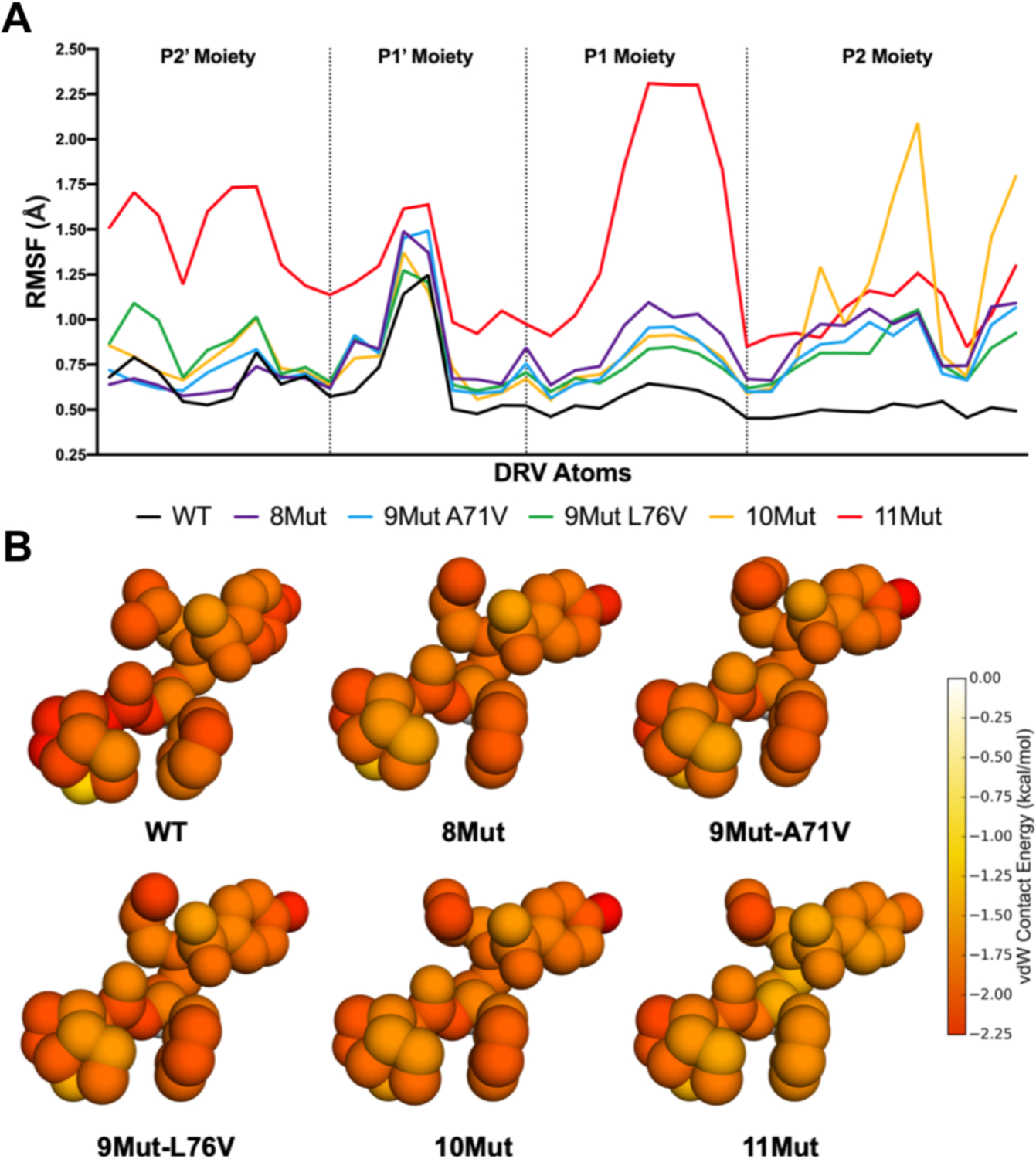
A) Root-mean-square fluctuation (RMSF) of DRV atoms grouped by moiety monitored during MD simulations bound to WT and resistant HIV-1 protease variants. B) Packing around DRV in complex with WT protease and resistant variants. Total per atom protease–DRV vdW contact energies mapped onto the respective DRV crystal structure, with red indicating more contacts.
Reproduced with permission from Ref 95 Copyright © 2019, American Chemical Society.
3.6.2. Dynamics, resistance and genotypic differences in HCV protease
In HCV NS3/4A protease, substitutions distal from the inhibitor binding site either due to resistance mutations or genotypic differences can decrease inhibitor potency. Variants with double or triple substitutions including a distal mutation have been observed in clinic. These distal substitutions, such as V36M or Y156H, propagate their effects to the active site and impact the conformational dynamics of the enzyme-inhibitor complex in conferring resistance.93, 145, 163 This was most dramatically shown in the elucidation of the differential potency for the HCV protease inhibitors between genotypes 1a and 3a, where inhibitors lost up to three orders of magnitude in potency. This loss of potency against genotype 3a was recapitulated by introducing three site mutations near the substrate binding site, where the two genotypes varied, into the genotype 1a enzyme. Co-crystal structures showed virtually no difference in the binding conformation, and only in the comparison pMD simulations were variations observed where a decrease in correlated dynamic fluctuations were observed between the inhibitor and the genotype-3 enzyme (Figure 14).93
Figure 14.
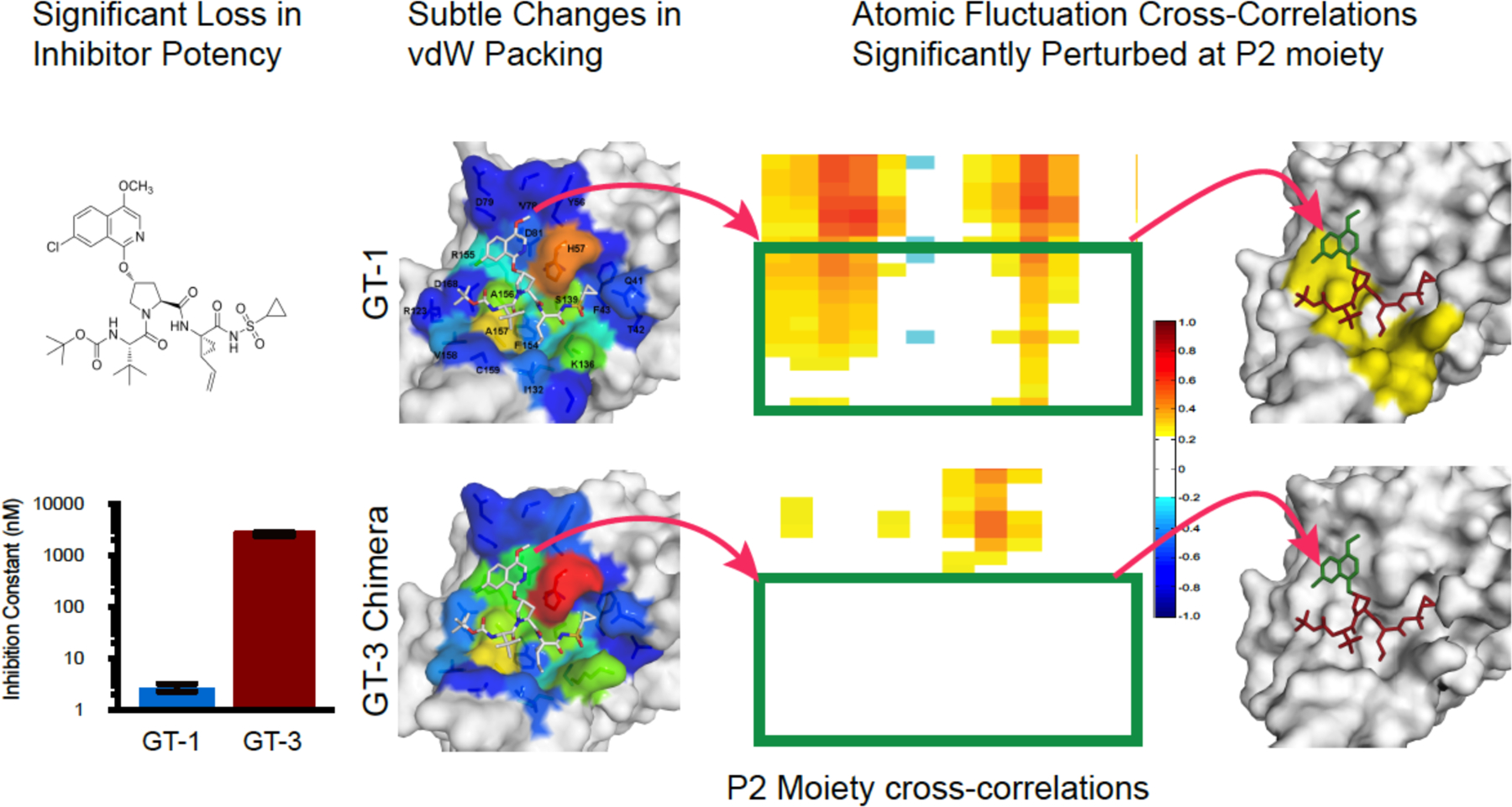
HCV NS3/4A inhibitors can lose significant potency against genotype 3 (GT-3). Although the crystal structures show only subtle changes in inhibitor binding and van der Waals (vdW) packing, pMD simulations revealed significant perturbations in inhibitor–protease dynamics, evident in loss of cross-correlations between atomic fluctuations of the inhibitor and active site residues.
Reproduced with permission from Ref 93 Copyright © 2016, American Chemical Society.
Overall, distal mutations can change protein dynamics and in turn protein-ligand interactions. However, teasing out the molecular mechanisms underlying resistance requires consideration of dynamic effects. More comprehensive integrated methods with a large number of variants and/or inhibitors leveraging machine learning and pMD96 are needed to better understand the role of distal mutations in resistance. Such analysis and tools may lead to better incorporation of protein conformational dynamics into the drug design process.
3.6.3. The role of hydration in drug resistance
Water structure also likely plays a direct role in conferring resistance. When the hydration structure was investigated using extended pMD for HIV-1 protease,140 water was found to occupy certain specific sites in hydrating the protein-inhibitor complex, and the water sites surrounding the bound inhibitor DRV were asymmetric on the concave surface of the active site. Comparison with wild-type HIV-1 protease revealed that key interactions between water molecules and the protease were altered in a drug resistant variant (Figure 15), indicating that modulation of solvent-solute interactions likely plays a key role in conveying drug resistance.140
Figure 15.
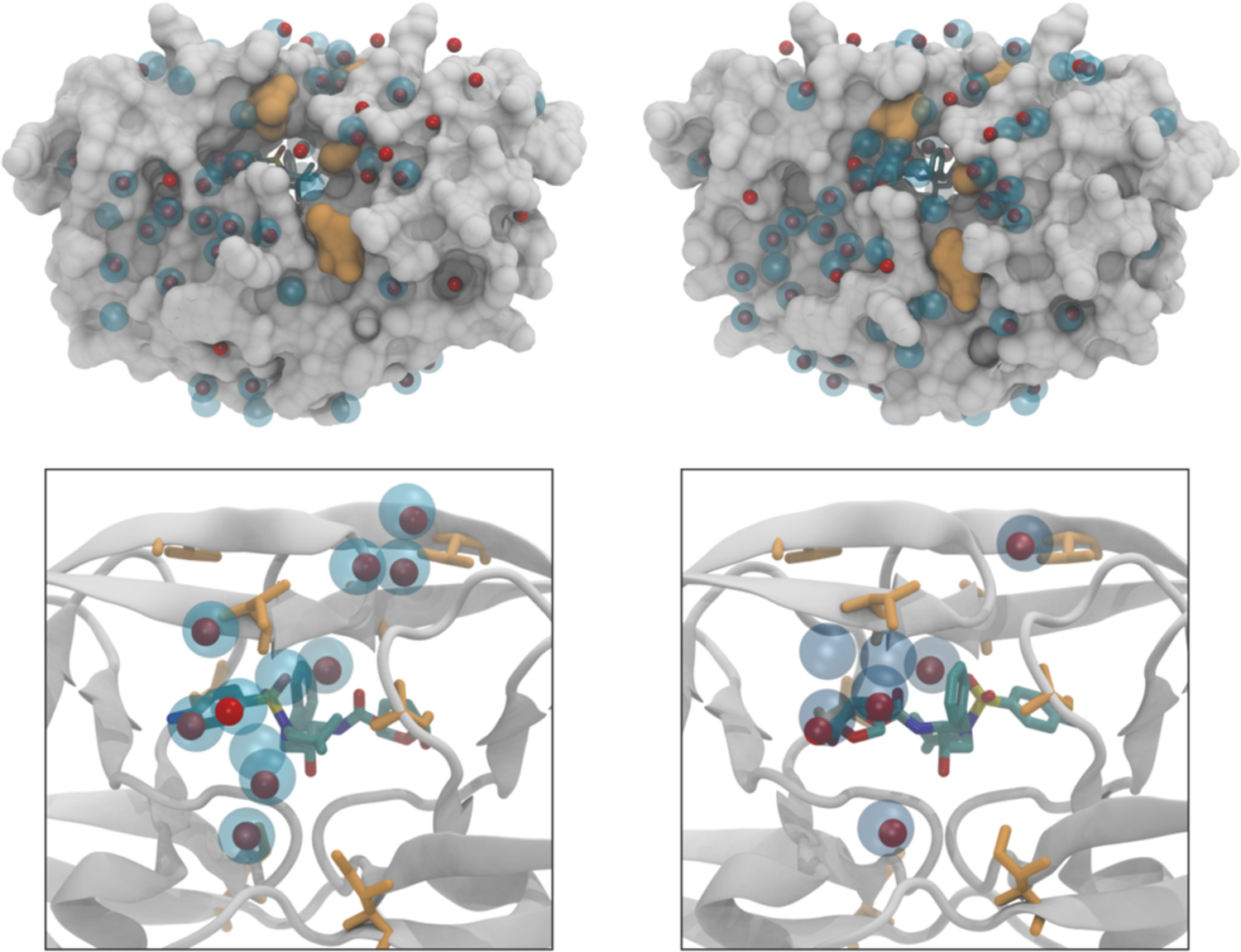
Water structure around the inhibitor-protein complex can be impacted by drug resistance mutations. Water sites for wild-type HIV-1 protease (blue spheres) compared to those in a resistant variant (red spheres). Location of mutations are indicated in orange.
Reproduced with permission from Ref 140 Copyright © 2018, American Chemical Society.
4. Strategies to avoid resistance in structure-based drug design
In development of new inhibitors, having high affinity against the wildtype protein is essential but not enough especially in the case of a rapidly evolving disease where lead compounds need to have high potency not only against the wildtype protein but also against various genotypes and resistant variants. Compounds that mimic a transition state, establish evolutionarily conserved interactions with active site residues, form a covalent bond, and fit within the substrate envelope along with an optimized inhibitor scaffold will likely be less susceptible to drug resistance.164–165 While resistance may not be completely avoidable, rational drug design can incorporate strategies at the outset of drug development to evade resistance (Figures 1 and 16).
Figure 16.
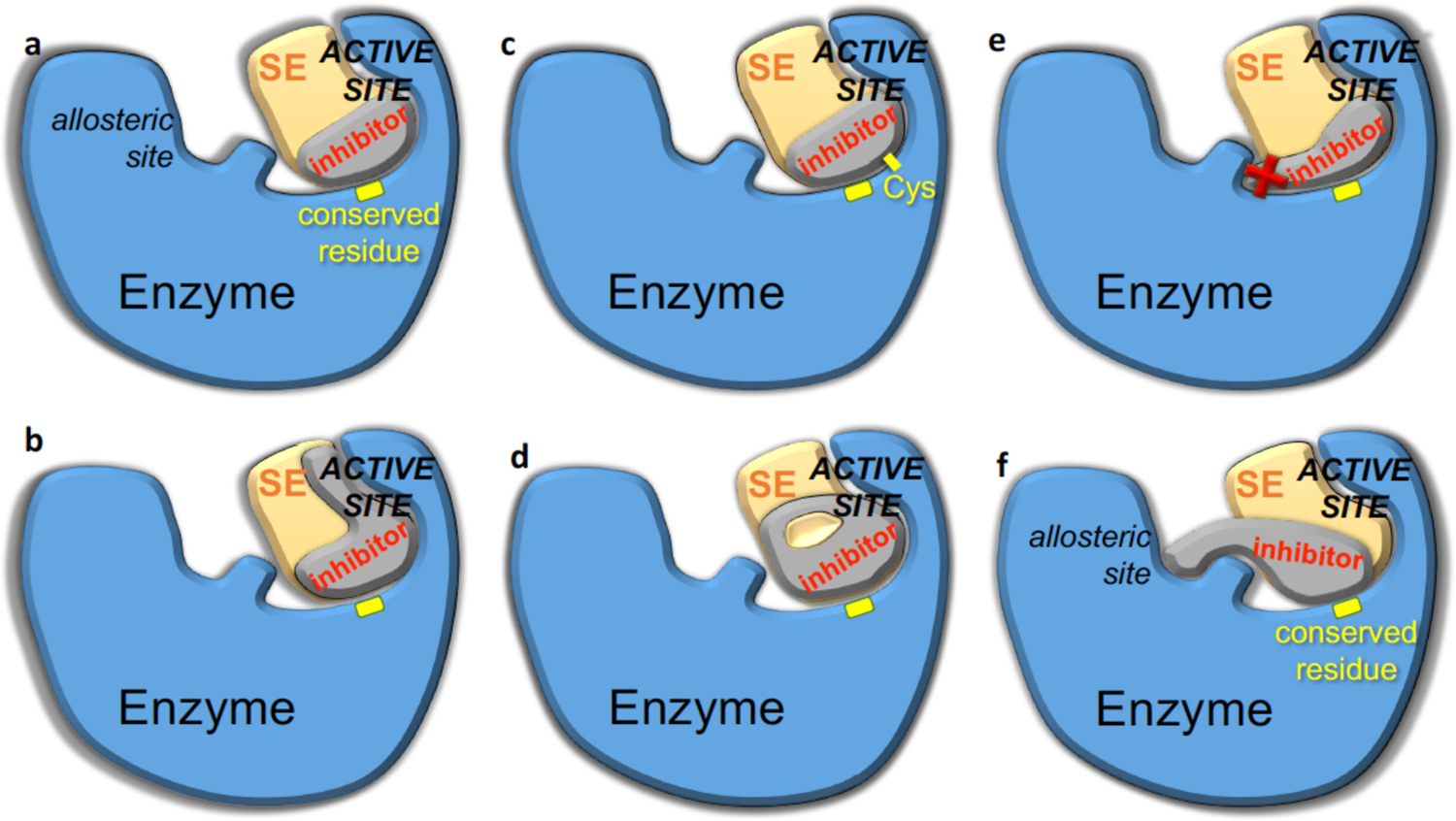
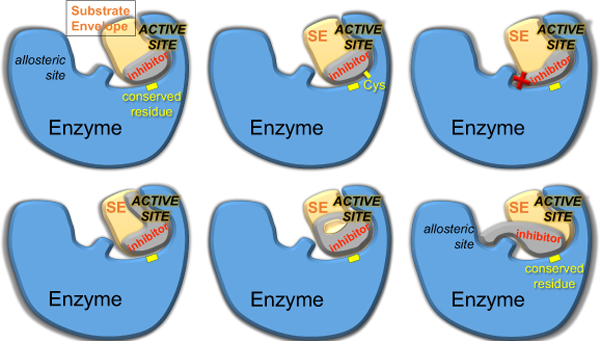
Strategies to minimize the probability of resistance in rational structure-based drug design. The structure of enzyme-inhibitor complex and the substrate envelope (SE) can guide the design to enhance potency while avoiding resistance. The inhibitor should/can be designed to a) contact evolutionarily conserved residues of the enzyme; b) extend within the SE to exploit unleveraged space and interactions; c) covalently attach to the enzyme, such as through cysteine side chains (Cys) at or near the active site; d) incorporate a macrocycle while staying within the SE; e) avoid protruding beyond the SE to contact residues that can mutate; f) target conserved allosteric and active site residues.
4.1. Substrate envelope guided inhibitor design to avoid resistance
A rational structure-based strategy for designing inhibitors that are less susceptible to resistance is to fit within the substrate envelope (Figure 16a).164–165 The substrate envelope is the basis for molecular recognition of substrates by viral enzymes, as explained above, and adequately explains selection of active site resistance mutations. Inhibitors that protrude beyond the substrate envelope and contact vulnerable positions, more critical for inhibitor binding than substrate turnover, select for mutations at these active site residues. To avoid this vulnerability, a potent inhibitor that fits within the substrate envelope is highly advantageous. Such an inhibitor would retain potency as mutations that disrupt inhibitor binding would also have a negative effect on enzymatic activity and thus viral fitness.
Proof of concept of this strategy is exemplified with DRV, which is the most potent FDA-approved HIV-1 protease inhibitors with picomolar inhibition of wild type enzyme.36 DRV fits very well within the HIV-1 substrate envelope, although this constraint was not used in its design and development.164 DRV mimics certain aspects of the three-dimensional shape conserved among protease substrates, and makes key interactions with backbone atoms of protease active site residues. While limiting an inhibitor to a confined volume may raise concern of the ability to achieve high potency, DRV is a glaring example that single digit picomolar activity is possible while fitting in the substrate envelope.164
The substrate envelope can guide the design of inhibitor not only against the wild type but also mutant variants of the target enzyme. Under evolutionary pressure, highly resistant variants of HIV-1 protease are observed to co-evolve with mutations at the cleavage sites, including the p1-p6 and CA-p2.166–168 Structural and dynamic analysis of these co-evolved substrates demonstrated that substrate co-evolution reinforces and maintains the substrate structure and dynamics rather than altering them.132, 135, 149, 151 This further validates the essential role of substrate recognition in viral evolution.
The substrate envelope has been used as a constraint during inhibitor development yielding inhibitors with reduced susceptibility to resistance across multiple targets.169–170 Analogous HIV-1 protease inhibitors were designed to either respect (fit within) or violate the envelope by protruding beyond the consensus volume. Inhibitors that fit within the substrate envelope had better activity against multi-drug resistant variants whereas their analogous counterparts exhibited high potency only against the wildtype enzyme.169 The substrate envelope was also used to design HIV-1 integrase inhibitors to fill the same consensus volume as DNA substrates leading to inhibitors with better efficacy against a panel of known integrase resistant variants.171
In addition to fitting within the substrate envelope, filling must be optimal. Analysis of the dynamic substrate envelope of HCV protease revealed that the P1–P5 substrate positions are quite conserved with the P6 position being the most dynamic.101 The scaffold of all current HCV protease inhibitors only spans the P4–P1’ positions with small P4 capping groups. However, there is adequate volume remaining in the substrate envelope for further inhibitor optimization. Rational design of two series of HCV protease inhibitors that extend in the P4–P5 direction, yielded robust inhibitors with improved activity against drug resistant variants.172 The P4 amino acid position is not conserved but is often occupied by a hydrophobic residue.173 Thus, the inhibitors in this study that had enhanced hydrophobic packing in the S4 pocket and avoided an energetically frustrated pocket performed the best (Figure 17) .172 Properly filling of nonpolar pockets is well known to be beneficial to inhibitor binding and selectivity for the target.174–175 This strategy complements substrate mimicry of conserved binding interactions, and may help designing inhibitors with improved potency.
Figure 17. Filling the S4 subsite of the HCV NS3/4A protease active site.
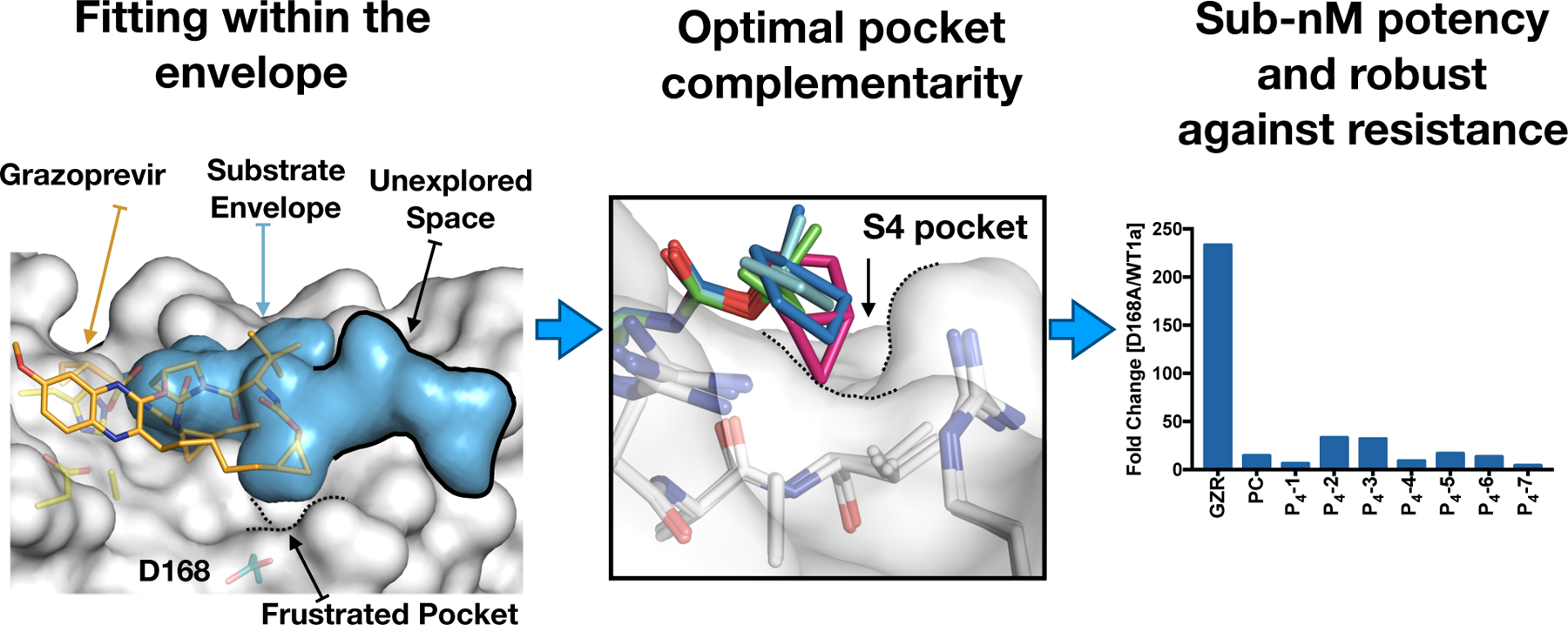
Comparison of grazoprevir binding at the active site reveals unexplored space in the substrate envelope (blue volume) where the inhibitors can be extended. Mutation at residue D168 confers significant resistance when the S4 pocket is not optimally filled by the inhibitor. Modification of the moiety to optimally fill the pocket resulted in potent inhibitors that retained potency against D168A mutation, with significantly smaller fold-changes compared to grazoprevir. Reproduced with permission from Ref 172 Copyright 2020 The American Society for Microbiology under Creative Commons Attribution 4.0 International license (https://creativecommons.org/licenses/by/4.0/)
Does the target have to rapidly evolve for the substrate envelope to be useful in drug design? Does the target need to recognize multiple substrates? In the case of HIV and HCV protease which inherently have to recognize diverse substrates with no amino acid sequence motif, they do so by binding to a conserved consensus volume. While other targets likely do the same, this is not a prerequisite for using the substrate envelope to guide inhibitor design. More generally, the substrate envelope has been applied to a diverse set of rapidly evolving disease targets including abl kinase, chitinase, thymidylate synthase, dihydrofolate reductase and neuraminidase.148, 170 This demonstrates the broad utility of the substrate envelope. If only one natural substrate is known for a target, a substrate envelope can and should be calculated. The envelope can be used to understand the molecular interactions of the substrate-target complex. Thus, the premise of this strategy is to gain a detailed understanding of substrate-target interactions that are essential in the enzymatic or biological activity and conserve those interactions in inhibitor design while avoiding unessential contacts that may cause vulnerability to resistance, thereby decreasing the probability of resistance.
4.2. Leveraging evolutionarily constrained regions of target active site
The second strategy in inhibitor design is to target evolutionarily constrained or conserved regions in the active site of the target. The substrate envelope is a constraint that can be applied at the outset of inhibitor design, and adhering to the substrate envelope has demonstrated success in numerous studies.170, 176 However, is it possible to violate the substrate envelope but still avoid resistance? The answer lies in exploiting a target’s sequence conservation and how critical a conserved region is in biological function. Most enzymes have critical residues that are indispensable to carry out their biological function such as the catalytic residues. With or without selective pressure, these residues are invariant as mutation is not tolerated without disrupting activity (Figure 16). Designing inhibitors that pack on these invariant residues is one strategy that can be used to avoid resistance.
These invariant and/or critical residues can be determined by mutational analysis coupled with activity assays to map the fitness landscape. Mutational analysis of HCV polymerase demonstrated that four amino acid sequence motifs are crucial for polymerase activity.177 Specifically, mutations at residue Gly283 disrupted activity of the polymerase.178 Sofosbuvir was the first FDA-approved HCV polymerase inhibitor.56 Interestingly, sofosbuvir forms a hydrogen bond with residue Ser282 which positions Gly283 to form a hydrogen bond with the residue of the template that base pairs with the nucleotide substrate. This hydrogen bond network is conserved with the natural substrates but sofosbuvir becomes incorporated into the growing RNA, inducing chain termination. While sofosbuvir is near-resistance proof, the S282T resistance mutation has been observed in clinical failure.179 However, this mutation disrupts the extensive hydrogen bond network and results in stalling of the enzyme – a severe fitness cost leading to rapid reversion to wildtype in absence of selective pressure.178 Thus, in drug design, exploitation of conserved critical residues is a great strategy as mutations that disrupt such residues will not only affect inhibitor binding but biological activity as well.
Another good example of this strategy is grazoprevir, an HCV NS3/4A protease inhibitor, and the two related inhibitors glecaprevir52, 68, 107 and voxilaprevir 108 (Figure 4). Grazoprevir’s P2 moiety extends well beyond the substrate envelope, but stacks on the invariant catalytic triad residues (Figures 10). Modification of the P2 extended moiety of grazoprevir yields inhibitors with improved resistance profiles.180 Even though grazoprevir protrudes beyond the substrate envelope, leveraging interactions with the conserved catalytic residues reduces susceptibility to resistance.
4.3. Targeting protein backbone interactions to avoid resistance
Designing inhibitors that make backbone interactions with the target residues is another strategy to minimize the probability of drug resistance. This concept exploits the fact that amino acid substitutions alter the side chains of mutated residues, but not the backbone atoms. Inhibitors designed to have strong hydrogen bonds with backbone atoms of the protein would likely conserve interactions in the mutant variant – thus retaining potency. This has been demonstrated in both HIV-1 reverse transcriptase and HIV-1 protease inhibitors where compound scaffolds were modified to have optimized moieties for hydrogen bonding interactions with backbone atoms. In both cases, the resulting inhibitors had high potency against wildtype and resistant variants.181–184 Loss of a hydrogen bond to the protein backbone is less likely to occur than to the side chain when a protein is mutated. Still, there are caveats to this approach. Steric clashes due to protein shifts or rearrangement from distal mutations can result in changes in the inhibitor binding mode, disrupting the backbone interactions which are highly constrained and dependent on distance and angles. This strategy may be most useful when combined with the aforementioned strategies where backbone interactions are optimized to mimic natural substrate binding while fitting in the substrate envelope.
4.5. Allosteric inhibition
Another strategy against resistance that can be considered is the design of allosteric inhibitors (Figure 16f). These are drugs that bind to sites outside of the natural substrate binding site. NNRTI and some HCV polymerase inhibitors are allosteric and have demonstrated success as therapeutics.58, 185–186 However, these inhibitors often have low barriers to resistance mutations at the allosteric site as these sites are less conserved and not necessary for natural substrate recognition.70, 187 One consideration in using this strategy is that drugs should have an “anchor” moiety that interacts with invariant or essential residues. This will help sustain potency when the inhibitor needs to adapt against a mutated variant. Additionally, dual targeting of the allosteric and active site simultaneously or use of covalent inhibition of the allosteric site are alternative strategies that can be used to raise the barriers to resistance.188 A detailed understanding of the interdependence of the allosteric site and substrate processing is fundamental in utilizing this design strategy.
5. Chemical optimization of inhibitors to avoid resistance
5.1. Designing inhibitors beyond DRV to target HIV-1 protease
Since the FDA approval of DRV as a potent antiviral in 2006, significant efforts have been made both by pharmaceutical companies and academic groups to develop HIV-1 protease inhibitors with improved potency and resistance profiles.22 These efforts have been either to further optimize the DRV scaffold or discover novel scaffolds to enhance the bioavailability so as to avoid ritonavir boosting or potentially to develop long-acting HIV-1 protease inhibitors. Many of these efforts have focused on exploring analogues of DRV with modifications at various inhibitor moieties including the bis-THF.189 The motivations for these modifications have varied from introducing additional enzyme–inhibitor binding interactions,190 providing what was thought to be a “solvent anchor”,191 optimally filling the substrate envelope,192 to optimizing and maximizing backbone hydrogen bonding (Figure 18).182, 193 Many of these modifications resulted in significant improvement in potency compared to DRV, even to highly resistant variants, however several still have susceptibility to active site mutations.22
Figure 18.
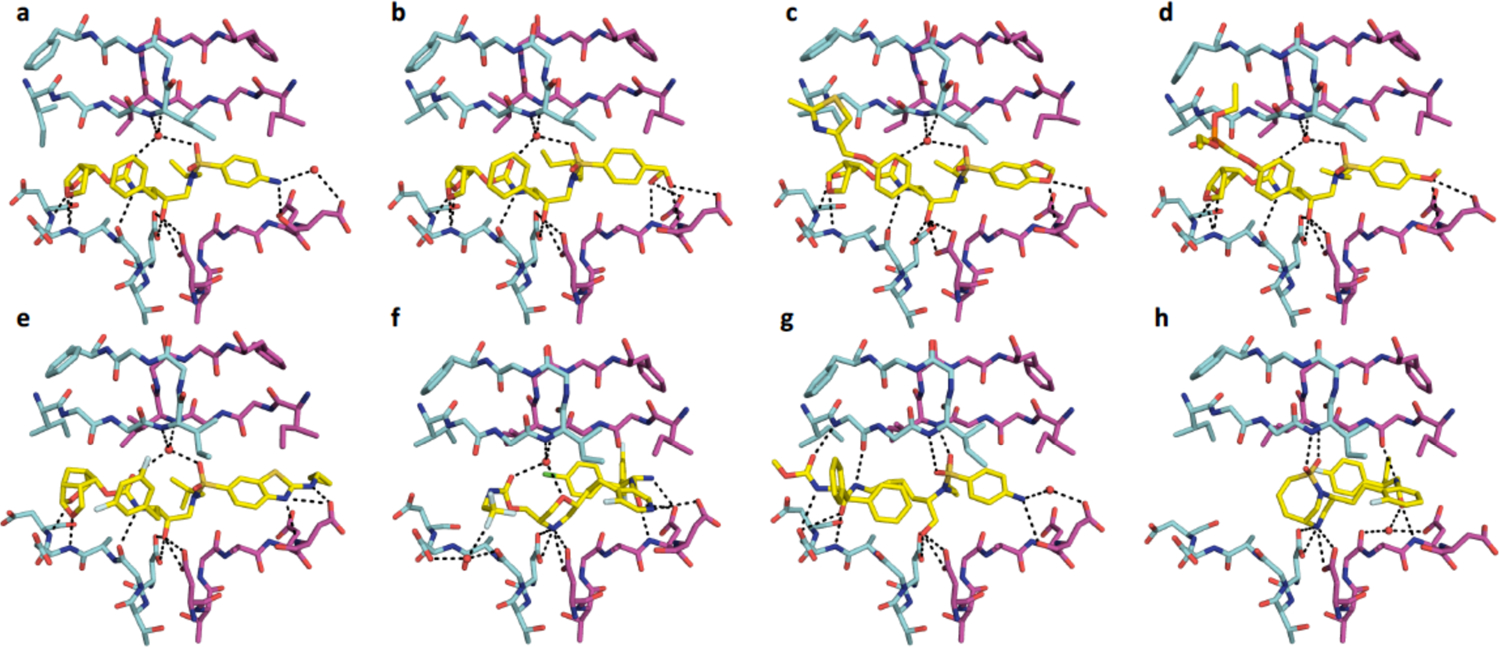
Comparison of recently designed “4th” generation inhibitors and their intermolecular hydrogen bonding interactions in the active site of HIV-1 protease crystal structures (A) DRV (PDB-ID 1T3R), (B) U8-dH (PDB-ID 60Y1), (C) brecanavir (PDB-ID 3FDE), (D) GS-8374 (PDB-ID 2I4W), (E) GRL-142 (PDB-ID 5TYS), (F) MK-8718 (PDB-ID 5IVT), (G) PL-100 (PDB-ID 2QMP), and (H) MK-8718 analogue (PDB-ID 6B3H).
DRV with the bis-THF moiety was originally pioneered by Ghosh and coworkers194–195 who continued to improve potency by introducing modifications at various inhibitor positions including diverse substitutions at the bis-THF moiety.22, 193 Their more recent efforts have focused on developing novel bi- and tri-cyclic ether moieties to further improve binding in the S2 subsite of HIV-1 protease compared to bis-THF. This work has resulted in the discovery of several novel P2 ligands (Figure 19) including the 6–5–5 ring-fused crown-like tetrahydropyranofuran (Crn-THF)196–197 and octahydrocyclopentylpyranofuran, an umbrella-like tetrahydropyranofuran (Umb-THF),198 and cyclohexane fused-tetrahydrofuranofuran (Chf-THF) moieties.199 These and other novel P2 moieties have been explored in combination with modifications at the P1 and P2’ positions in the DRV scaffold providing HIV-1 protease inhibitors with improved potency against multidrug-resistant HIV-1 variants. In most cases, the novel P2 moieties resulted in enhanced hydrophobic interactions in the S2 subsite. The increased hydrophobicity allowed these moieties to be combined with more polar ligands, such as the cyclopentyl-aminobenzothizole (Cp-Abt) moiety, to be incorporated at the P2’ position, improving polar and vdW interactions in the S2’ subsite. The resulting HIV-1 protease inhibitors exhibited excellent antiviral potency and resistance profiles. In particular, GRL-142, which contains the Crn-THF and the Cp-Abt moieties at the P2 and P2’ positions, respectively in combination with a 3,5-difluorophenyl group at the P1 position, showed sub-nanomolar antiviral potency against highly PI-resistant HIV-1 variants including DRV-resistant strains.200 GRL-142 also showed extremely high genetic barrier to the emergence of resistance and enhanced central nervous system penetration.
Figure 19.
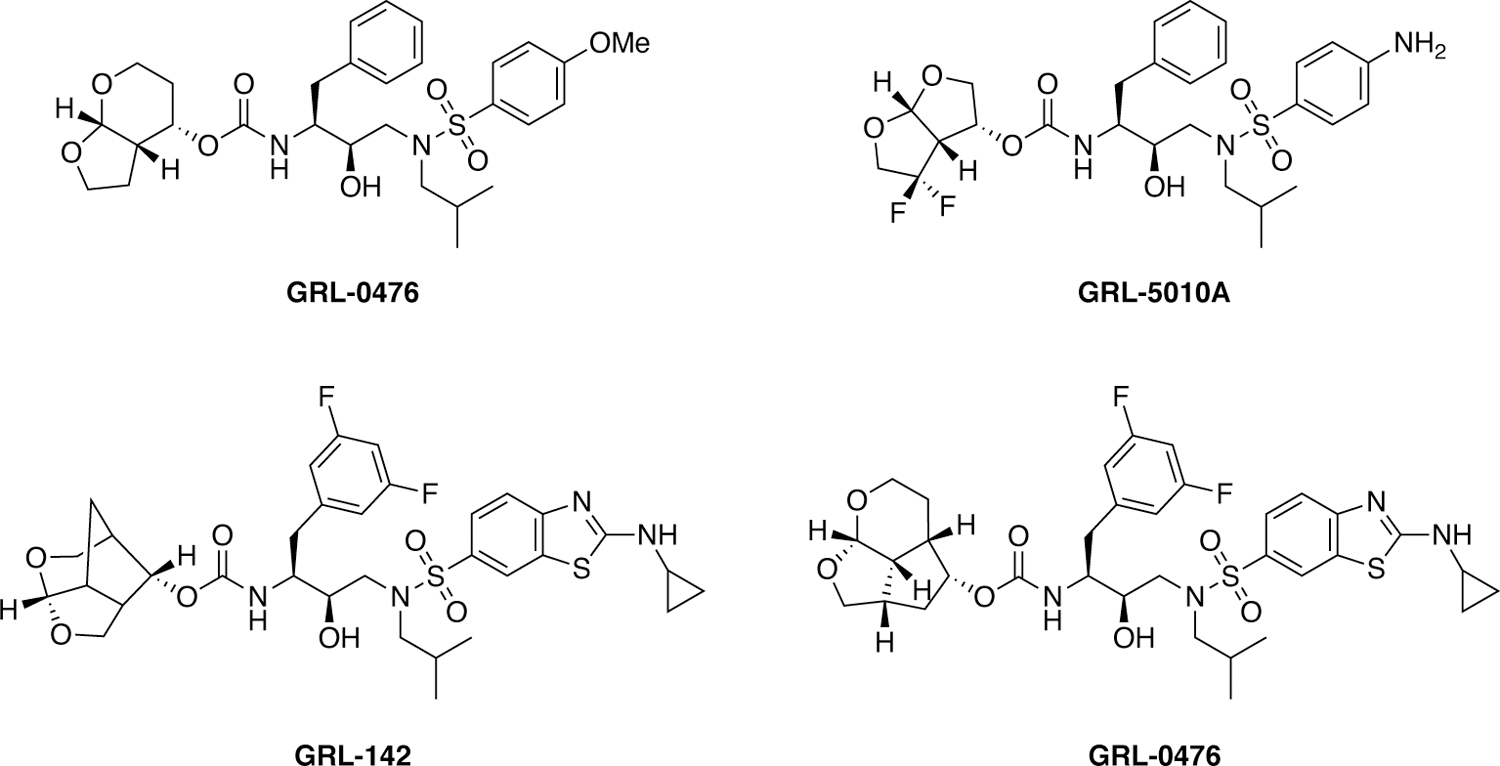
Chemical structures of HIV-1 protease inhibitors containing novel bi- and tri-cyclic ether moieties as P2 ligands.
The substrate envelope was used to design a series of highly potent HIV-1 protease inhibitors with improved resistance profiles.192 The designed inhibitors shared a common chemical scaffold with DRV but used various P1′ and P2′ chemical moieties that optimally fill the substrate envelope (Figure 20). These inhibitors retained robust binding to multi-drug resistant protease variants and displayed exceptional antiviral potencies against a panel of 12 drug-resistant HIV-1 strains.192 Additionally, viral selection demonstrated their ability to retain potency with greater than 10 mutations required for high levels of resistance.159 The substrate envelope-guided design strategy was used to further improve the resistance profile of HIV-1 protease inhibitors by optimizing hydrogen bonding and van der Waals interactions with the protease.201 Stereoisomers of 4-(1-hydroxyethyl)benzene and 4-(1,2-dihydroxyethyl)benzene moieties were explored as novel P2′ ligands to enhance hydrogen bonding interactions in the S2′ subsite of HIV-1 protease, as represented by U8-mH and U8-dH. Co-crystal structures revealed unique polar interactions, including a network of direct and water-mediated hydrogen bonding with the backbone and side chain atoms of D29′ and D30′. Notably, and as aimed, the (R)-4-(1,2-dihydroxyethyl)benzene moiety makes hydrogen bonding interactions in the S2′ subsite, closely mimicking the polar interactions of the substrates.201 These inhibitors fit within the substrate envelope and maintain excellent potency against highly drug-resistant HIV-1 strains representing the spectrum of clinically relevant multidrug-resistant viruses. The retained potency and flatter resistance profiles compared to DRV indicate that inhibitor flexibility along with the substrate envelope permits the inhibitors to adapt to drug-resistant variants of HIV-1 protease.
Figure 20.
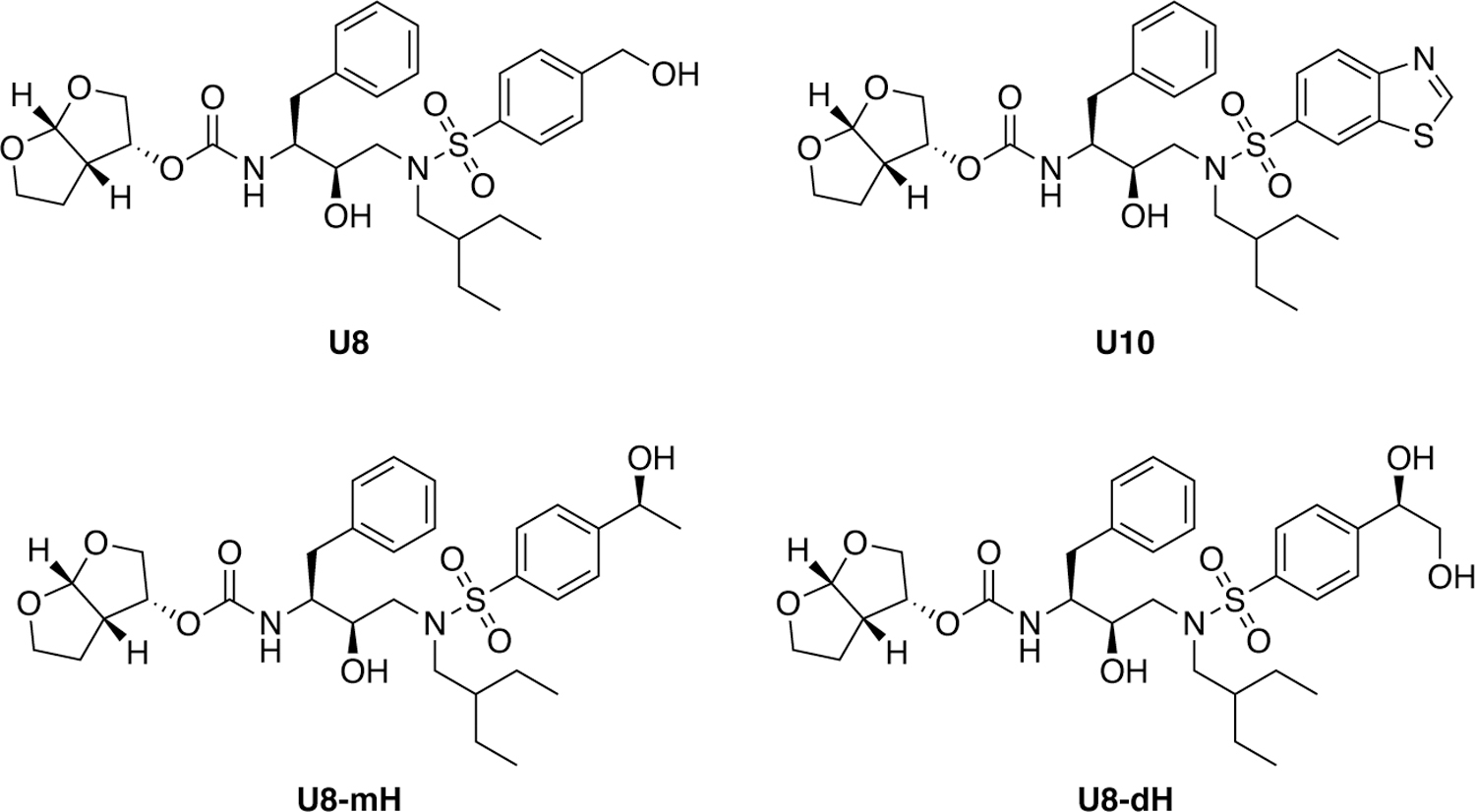
Chemical structures of substrate envelope-designed HIV-1 protease inhibitors.
Displacement of critical water molecules can also be a strategy for avoiding resistance. HIV-1 protease inhibitors containing a novel lysine sulfonamide scaffold were discovered through high throughput screening and exhibited this property.202 Originally, lysine derivatives were identified as potent hits against HIV-1 protease, which after modifications provided compounds with nanomolar potency.203 Further iterative rounds of optimization led to the discovery of PL-100, a novel HIV-1 protease inhibitor that exhibited potent antiviral activity (Figure 21) against a large selection of patient-derived HIV-1 isolates resistant to other protease inhibitors.204 This class of compounds was further optimized with modifications at the epsilon position of the lysine core to provide PL-100 analogues with improved potency.205 The distinct potency profile of PL-100 compared to other peptidomimetic HIV-1 protease inhibitors likely arises from the novel binding mode of lysine sulfonamide compounds where the sulfonamide moiety displaces the conserved flap water molecule (PDB 2QMP).
Figure 21.
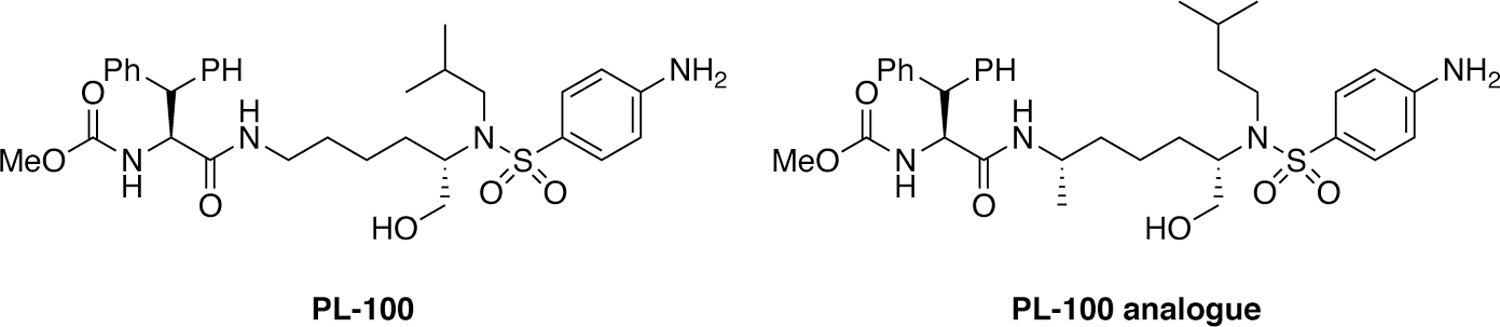
Chemical structures of lysine sulfonamide-based HIV-1 protease inhibitors.
A team at GlaxoSimthKine explored modifications at the P1 and P1′ position in the DRV scaffold to improve potency against drug resistant HIV-1 strains through the introduction of additional enzyme-inhibitor binding interactions.206–207 Iterative SAR studies and lead optimization identified a series of exceptionally potent HIV-1 protease inhibitors containing ether-linked aryl and heteroaryl moieties attached to the P1 phenyl group, culminating in the discovery of clinical candidate brecanavir (Figure 22).190, 208 Brecanavir exhibited femtomolar enzyme inhibition and maintained low nanomolar antiviral potency against a panel of 10 multidrug-resistant HIV-1 strains. Co-crystal structure of brecanavir with wild-type HIV-1 protease (PDB 2FDE) revealed that the 2-methylthiazole moiety at the P1 position binds in a solvent-exposed cleft between Pro81 and Phe53 making extensive vdW interaction with these and other residues. The clinical development of brecanavir was halted due to formulation issues.
Figure 22.
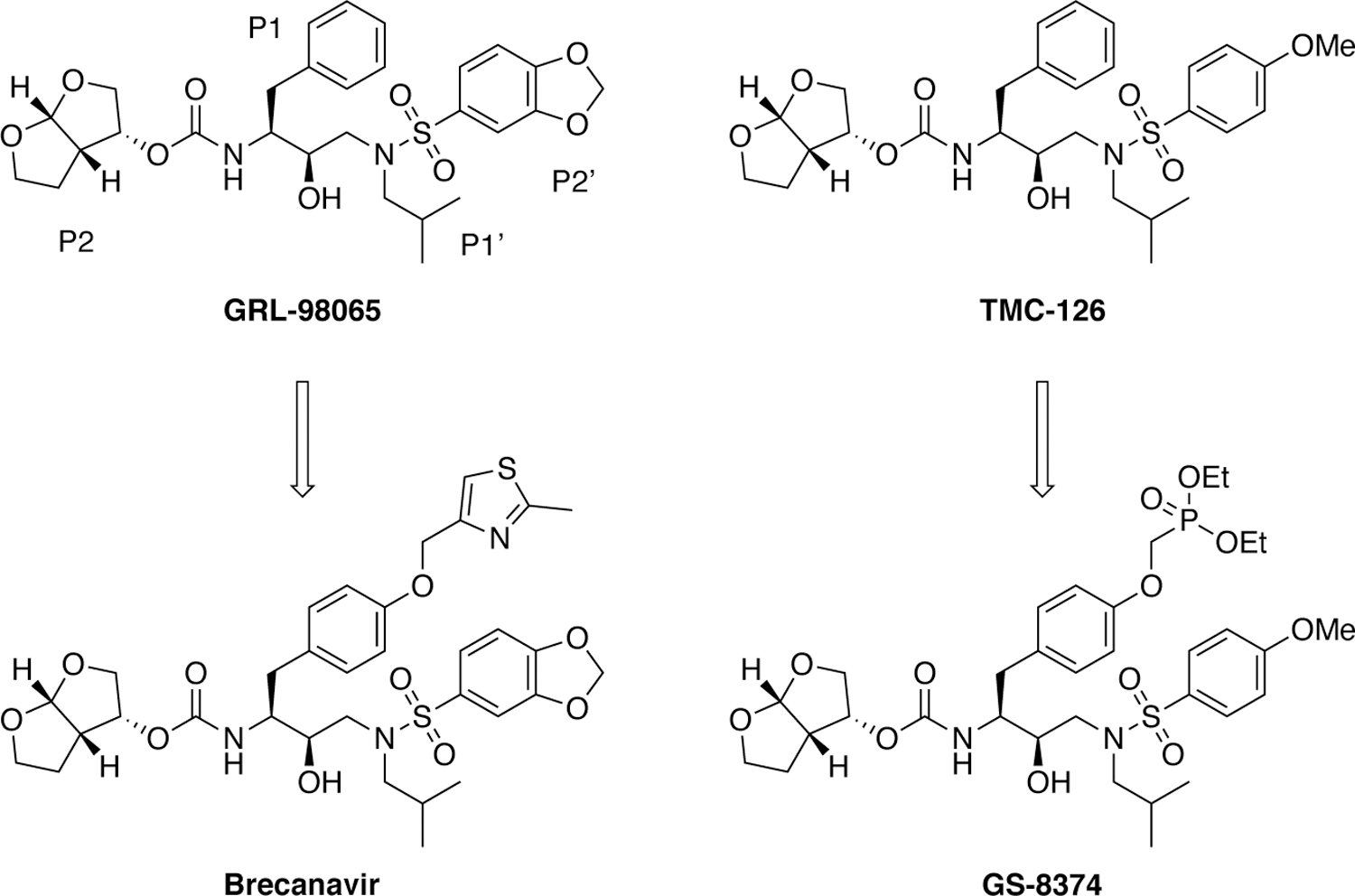
Chemical structures of P1 modified DRV analogues as HIV-1 protease inhibitors.
Incorporation of a polar phosphonate group at the P1 moiety of TMC-126 scaffold was found to improve the resistance profile of resulting analogues (Figure 22).191 Optimization of the phosphonate moiety and linker provided an orally bioavailable HIV-1 protease inhibitor GS-8374 that maintained excellent antiviral potency against a panel of clinically important PI-resistant HIV-1 strains.209–210 The phosphonate moiety was designed to be solvent exposed with no interactions with the protease resides. This “solvent anchoring” was proposed to provide entropic advantage to binding of the inhibitor, allowing it to adapt to changes in the protease active site caused by mutations.191 However, a close examination of the co-crystal structures of GS-8374 (PDB 2I4W) revealed that the phosphonate moiety bound to the protease similar to the thiazole moiety in brecanavir, with one of the ethyl groups making van der Waals interactions with largely invariant glycines in the flap. Although solvent exposed, the additional van der Waals contacts of the phosphonate moiety to invariant residues within the flap residues may contribute to the superior resistance profile of GS-8374.
An alternate approach to combat drug resistance in inhibitor design is to improve pharmacokinetic properties and metabolic stability. These can be achieved either by incorporating novel structural features or using novel inhibitor scaffolds to provide protease inhibitors that potentially do not require ritonavir boosting. Recently researchers at Merck used a structure-based design approach to develop novel HIV-1 protease inhibitors containing a morpholine core as the aspartyl binding group (Figure 23).211 Structure-guided optimization led to the discovery of MK-8718 that exhibited potent antiviral activity and a favorable pharmacokinetic profile. Further efforts to improve antiviral potency identified a bicyclic piperazine sulfonamide core designed to displace the conserved flap water. The resulting HIV-1 protease inhibitor showed picomolar binding affinity to wild-type HIV-1 protease and low nanomolar antiviral potency.212
Figure 23.
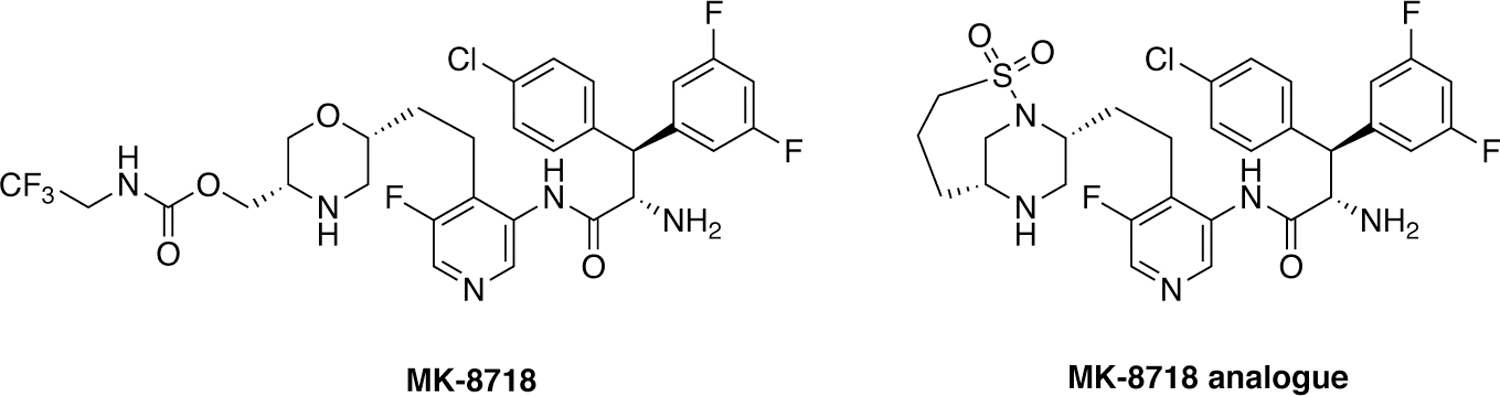
HIV-1 protease inhibitors with a morpholine and a bicyclic piperazine sulfonamide cores as aspartyl binding groups.
In addition pharmaceutical companies have made significant efforts to improve the ADME properties of approved HIV-1 protease inhibitors by prodrug and formulation approaches.213–215 Efforts have been made to develop new protease inhibitors suitable for long-acting formulations that may be both potent and less susceptible to resistance as there would be much less likelihood of treatment troughs during a patient’s therapy.216
5.2. Macrocyclization of HCV NS3/4A inhibitors
Another strategy to improve potency, and hopefully suppress resistance, is preorganization of a ligand in its bioactive conformation either by intramolecular interactions or macrocyclization of the inhibitor scaffold (Figure 16d). Although the Lipinski’s “rule of five” is often used in traditional “small” molecule drug design, these criteria are not universal to all targets.217 Many targets are “difficult-to-drug” with “rule of five” compliant ligands. Using macrocyclic compounds allows targeting of binding sites that are large, lipophilic, or highly polar, flexible, flat or even featureless as seen in HCV protease with its shallow binding site.173, 218 Macrocycles are able to stabilize inhibitors in a bioactive conformation which could be leveraged to take advantage of interactions with essential residues in the target protein. Moreover, macrocyclic compounds can have improved cellular permeability and target selectivity, key properties needed for a lead compound.219 Macrocycles add rigidity to a compound and reduce the entropic penalty of binding by conformationally constraining the ligand, which can increase binding affinity.220 However, conformational flexibility is also important especially to be able to adapt to changes due to mutations. Thus, when using this strategy, there must be a balance between flexibility and rigidity as compounds still need to be able to adapt to changes by having some degree of flexibility to target the binding surface and avoid resistance.
In HCV protease, the addition of the macrocycle to connect the P1–P3 or P2–P4 positions on the scaffold greatly enhances inhibitor potency.104, 221–222 The first HCV NS3/4A protease inhibitor that was in clinical trials was the P1–P3 macrocyclic BILN-2061, which was dropped due to toxicity. This was then followed by the first FDA-approved inhibitors that were linear ketoamides (telaprevir and boceprevir) but side effects, lack of potency and susceptibility to resistance limited their clinical use. All subsequent FDA-approved inhibitors contained macrocycles. In addition to the extended P2 moiety, the particular macrocycle is a major structural feature impacting protease inhibitor resistance profiles.98, 101, 104 The P1–P3 macrocycle, as in simeprevir and paritaprevir, is largely within the substrate envelope, but many of the extended P2 moieties in the first and second generation protease inhibitors packed on the ionic network of R155 and D168 and were susceptible to mutations (Figure 11).98 In contrast, the P2 quinoxaline moiety in the P2–P4 macrocyclic inhibitor grazoprevir successfully avoided interactions with these residues largely through packing on the catalytic triad. Grazoprevir was the first FDA-approved inhibitor with a P2–P4 macrocycle. Yet the rigidity induced by the macrocycle is a double-edged sword, as A156T mutation causes a steric clash and D168A causes a loss of packing, leading to high levels of resistance. Following grazoprevir, glecaprevir and voxilaprevir107–108, 221, 223 utilized very similar scaffolds, which renders all of the latest generation HCV protease inhibitors susceptible to significant cross-resistance.
Changing the macrocycle location to the P1–P3 circumvents the resistance susceptibility patterns caused by the P2–P4 macrocycle. Analysis of inhibitor dynamics with either macrocyclic connection demonstrated that the P1–P3 compounds have more conformational flexibility and can adapt to the A156T mutation.104 Thus, in inhibitor design rigidity and flexibility need to optimally balance tight binding interactions that contribute to high potency while retaining conformational flexibility to adapt to perturbations at the binding site.
Efforts to improve potency of P1–P3 macrocyclic HCV NS3/4A protease inhibitors against drug resistant variants initially focused on modifications of the P2 quinoline moiety and the P4 capping group.224–225 Reducing the size of the P2 moiety and incorporating conformational flexibility together with modifications at the P4 position resulted in significant improvement in potency against key resistant variants R155K and D168V (Figure 25).225 Although the optimized P2 moiety still bound to the wild-type protease in a conformation similar to that of the second generation inhibitors, the conformational flexibility allowed the inhibitor to adapt to perturbation caused by D168V mutation by shifting the position of the quinoline moiety toward catalytic residues.226
Figure 25.
Chemical structures of HCV NS3/4A protease inhibitors with P1-P3 macrocycles and modifications at the P2 and P4 moieties.
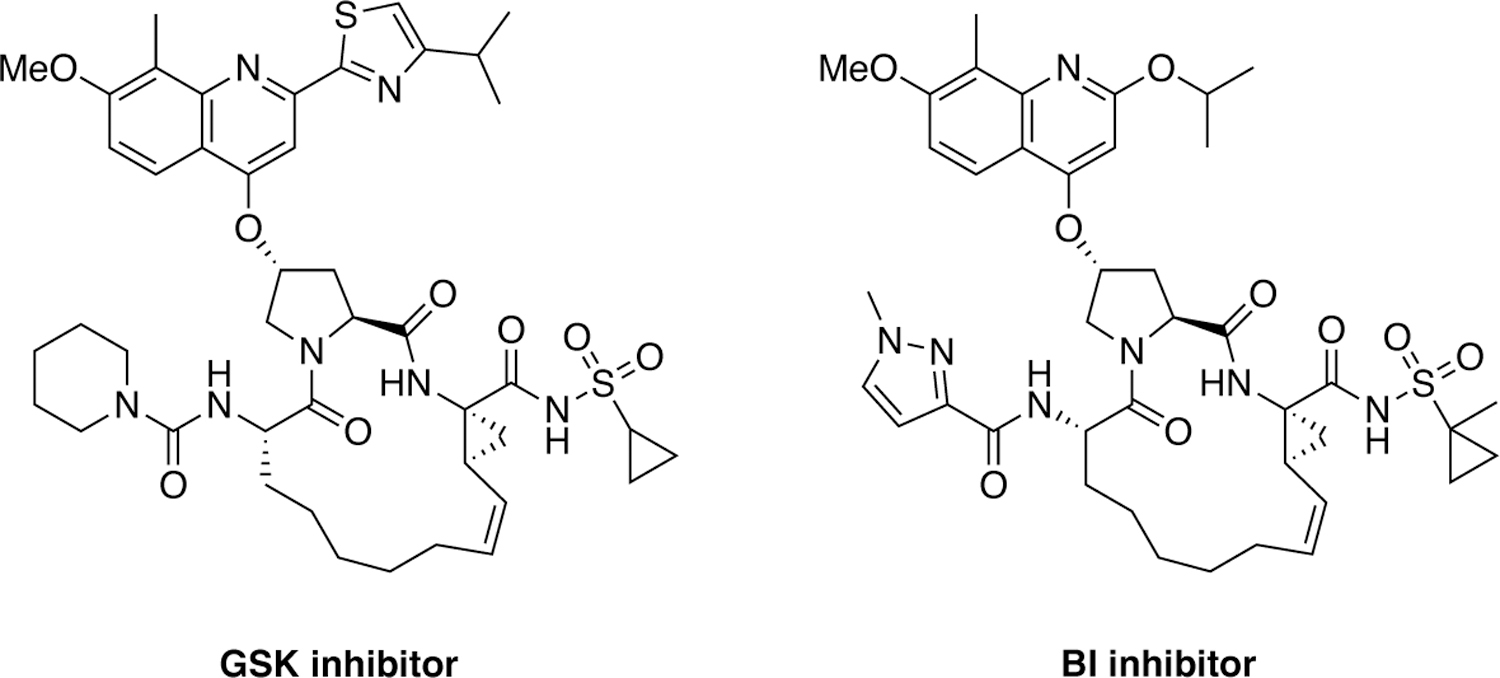
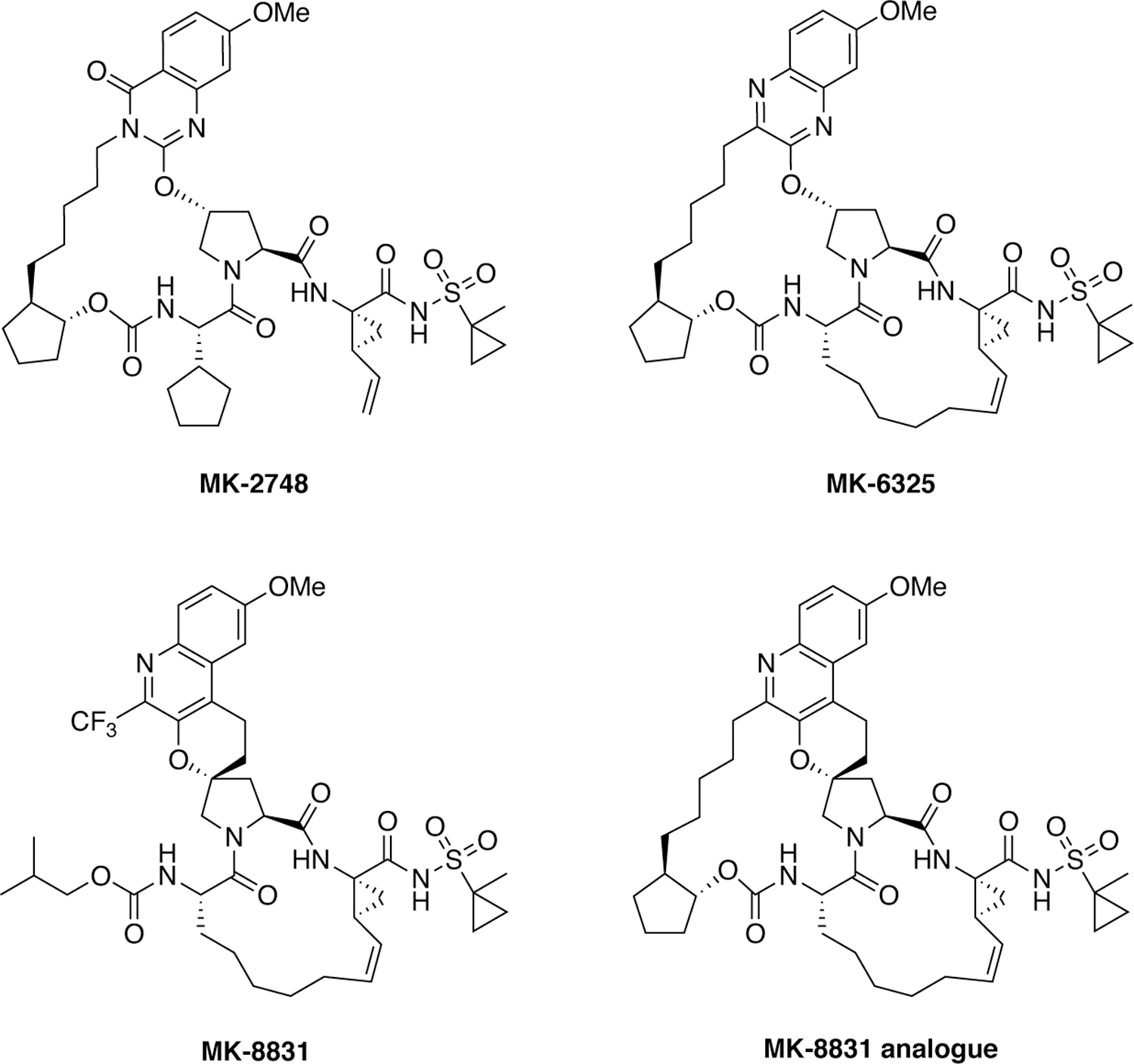
Since the discovery of grazoprevir several next-generation NS3/4A protease inhibitors have been discovered by Merck that exhibit pan genotypic activity (Figure 26). The strategy of conformationally constraining the P2 moiety was pursued and additional inhibitor features were introduced to further improve the potency and resistance profiles leading to the discovery of a quinazolinone-based P2–P4 macrocyclic inhibitor MK-2748 and a quinoxaline-based bis-macrocyclic inhibitor MK-6325 containing both P1–P3 and P2–P4 macrocycles.227–228 Compared to grazoprevir, MK-6325 showed improved pan-genotypic potency profile including against genotype 3, and maintained low nanomolar potency against major drug resistant HCV variants including A156T. Subsequent efforts to develop structurally distinct molecules with an alternate core led to the discovery of a novel class of HCV NS3/4A protease inhibitors containing a unique spirocyclic-proline structural motif.229 The P1–P3 macrocyclic compound MK-8831 and the bis-macrocyclic analogue exhibited excellent pan-genotypic activity and maintained potency against key drug resistant HCV variants.230
Figure 26.
HCV NS3/4A protease inhibitors with conformationally constrained P2 moieties.
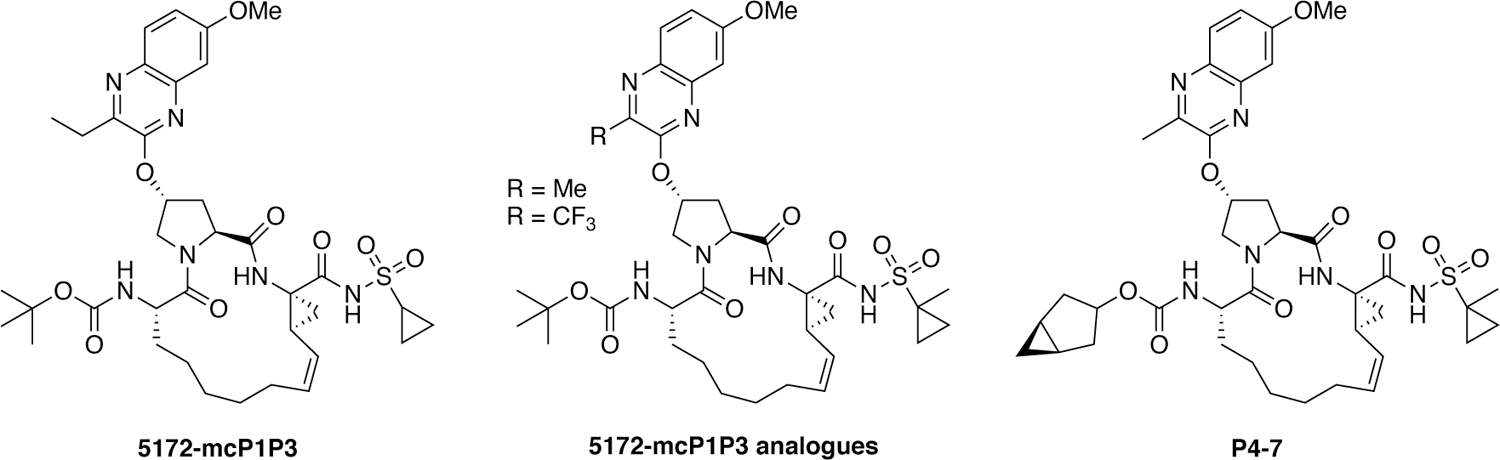
A substrate envelope-guided design strategy has been reported for improving the resistance profile of HCV NS3/4A protease inhibitors.180 This structure-guided design strategy incorporates the substrate envelope constraint and understanding of the mechanisms of drug resistance in inhibitor design which led to the discovery of inhibitors with significantly improved potency and resistance profiles. P1–P3 macrocyclic analogues of grazoprevir were designed by incorporating diverse quinoxalines at the P2 position that predominantly interact with the invariant catalytic triad of the protease (Figure 27). Exploration of SAR showed that inhibitors with small hydrophobic substituents at the 3-position of P2 quinoxaline maintain better potency against drug resistant variants, likely due to reduced interactions with residues in the S2 subsite. In contrast, inhibitors with larger groups at this position were highly susceptible to mutations at R155, A156 and D168. These findings support that inhibitors designed to interact with evolutionarily constrained regions of the protease, while avoiding interactions with residues not essential for substrate recognition, are less likely to be susceptible to drug resistance and can maintain potency.231
Figure 27.
HCV NS3/4A protease inhibitors with flexible P2 quinoxaline moieties.
5.3. Covalent inhibitors
Covalent inhibitors constitute another strategy to improve potency against drug resistant variants by including a “warhead” that permits covalent binding to the target (Figure 2c). Covalent inhibitors have made a resurgence recently in drug development. Many drugs used to treat human diseases rely on non-covalent intermolecular interactions including hydrophobics, electrostatics, van der Waals, and hydrogen bonds to bind to the target.232 In covalent inhibition, the small molecule binds to the target via a covalent linkage, which can be either reversible or irreversible. Irreversible covalent inhibitors can be an effective strategy in drug resistance as covalent bond formation may be possible even if the target mutates to decrease binding affinity. This is the case unless the residue being targeted by the covalent warhead mutates or a neighboring residue changes causing a steric hindrance. Covalent inhibitors have been successfully used in multiple viral targets including HIV-1 reverse transcriptase, influenza neuraminidase, and HCV protease with effect against some drug resistant variants.233–235 In fact, the first FDA-approved HCV protease inhibitors were the reversible covalent inhibitors boceprevir and telaprevir.44, 236 These inhibitors offer many advantages including high efficiency requiring lower drug doses and specificity. However, given their long duration of action, they are often highly susceptible to off-target effects and toxicity especially if desired specificity is not achieved.237 Therefore, while adding covalency to inhibitors is a viable approach to combat drug resistance, high target specificity, affinity and interactions with invariant residues should be integrated with the design of covalent inhibitors.
6. Integrative computational methods to evaluate inhibitor potency and resistance
6.1. Correlation of structural changes from mutations with variation of inhibitors
Decreasing the probability of drug resistance in drug design, as discussed above, involves many different aspects and various characteristics of the inhibitor and specific interactions with the enzyme target. Traditional structure-based drug design strategies typically leverage information from protein crystal structures to optimize a small molecule lead. However, this analysis can be extended to only a few variants and relies heavily on experience and subjective evaluation. To optimally consider resistance in drug design, integrative computational methods that take into account the physical interaction of the drug with the target are essential. In the case of HIV-1 protease, even for single primary active site mutations involving hydrophobic changes (at residues including I50, V82, and I84 that line the S1/S1' pocket) a collective computational analysis provided key insights (Figure 28). Through enzyme inhibition assays and a series of 12 crystal structures, the susceptibility of DRV and two potent analogues to primary S1/S1' mutations was analyzed111. The DRV analogues had modifications at the hydrophobic P1' moiety to better occupy the unexploited space in the S1' pocket where the primary mutations were located. Collective analysis of protease-inhibitor interactions in the crystal structures using principle component analysis was able to distinguish inhibitor identity and relative potency solely based on van der Waals contacts, indicating that such approaches can be a useful tool for distinguishing resistance and inhibitor potency.
Figure 28.
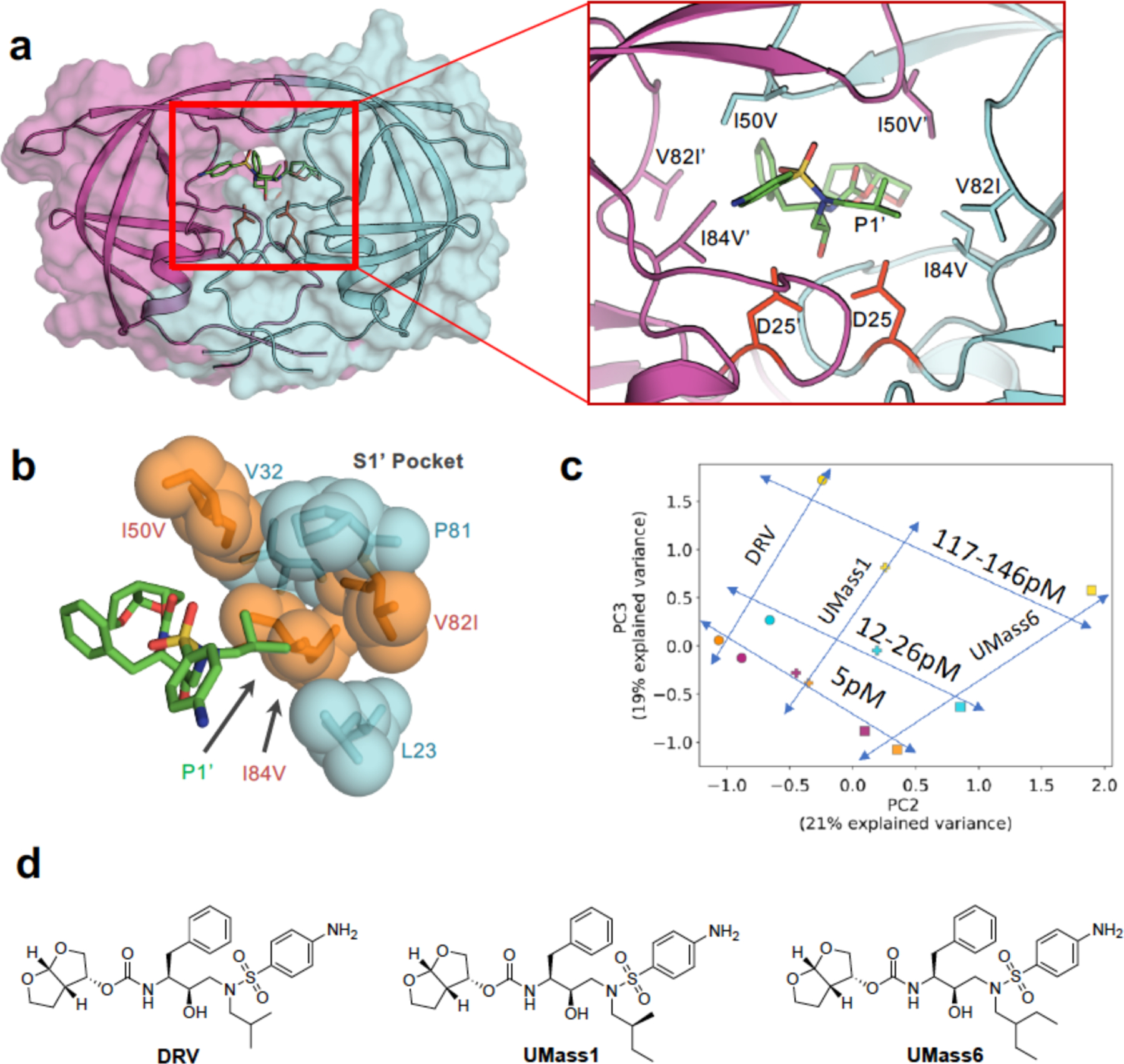
a) Co-crystal structure of DRV (green sticks) bound to WT HIV-1 protease. The two chains (cyan and magenta) are shown as a cartoon with a transparent surface. D25/D25’ catalytic aspartates (red) are displayed as sticks. A-Insert) Residues that contribute to primary drug resistance, shown as sticks. b) Spherical representation of residues that make up the S1’ subsite / P1’ pocket. Variable residues are shown in orange. c) Principle component analysis of van der Waals (vdW) interactions in 12 protease-inhibitor complexes with varying mutations and potency. The protease–inhibitor pairs plotted according to second and third principle components, PC2 versus PC3. The lines are shown only to guide the eye. d) Structures of inhibitors with modifications at the P1’ position that display varying susceptibility to S1’ mutations.
Reproduced with permission from Ref 111 Copyright © 2019, American Chemical Society.
6.2. pMD differentiates interdependence of functional group modifications in inhibitors.
Molecular recognition is a highly interdependent process. Subsite couplings within the active site of proteases are most often revealed through conditional amino acid preferences in substrate recognition. However, the potential effect of these couplings on inhibition and thus inhibitor design is largely unexplored. pMD of HIV-1 protease complexes of DRV and 11 analogs192 that were designed to fit within the substrate envelope through modifications at the P1’ and P2’ sites, discovered the interdependence of how modifications at P1’ altered packing at P2’ but the reverse was not the case139. These dynamic relationships intricately link the HIV-1 protease subsites and are critical to understanding the coupled recognition of inhibitor binding. More broadly, the interdependency of subsite recognition within an active site requires consideration in the selection of chemical moieties in drug design rather than independent optimization of chemical moieties of an inhibitor.
6.3. Structural inhibitor fingerprints and potency
Protein-inhibitors interaction fingerprints are high-level presentations of these complexes, where the binding mode of a small molecule inhibitor is encoded in a series of physical interactions. The exact composition of such a fingerprint can vary significantly from simple binary representations that inform about the presence or absence of a particular interaction, to quantitative measurement of the strength of interactions between protein and ligand atoms. 238–239 Protein-ligand interaction fingerprints have been used extensively to characterize virtual screening results to identify small molecules that share a common mode of interaction with known binders.240 Fingerprinting has been used to characterize conserved binding modes across families of potential drug targets such as human kinases and G protein-coupled receptors.241–242 One major advantage of fingerprinting is the ability to simultaneously evaluate a large number of protein-ligand complexes, which can identify signature interactions to guide structure-based drug design. Comparison of susceptible and resistant protein variants through their ligand interaction fingerprints can also reveal which interactions to avoid.239
Using these techniques coupled with recent advances in high-resolution structure determination, computational power, and machine learning methodology, it is becoming more tractable to elucidate the structural basis of drug potency. The applicability of machine learning models to drug design is limited by the interpretability of the resulting models in terms of feature importance. A good test to evaluate inhibitor diversity and machine learning models to predict ligand affinity is HIV-1 protease with the many co-crystal structures associated potencies. This was performed243 after hierarchical clustering (Figure 29) of HIV-1 protease inhibitors by distinct chemical core structures. Explicit features including protein-inhibitor interactions were extracted as three-dimensional fingerprints. A gradient boosting machine learning model with this explicit feature attribution was able to predict binding affinity with high accuracy based on specific van der Waals interactions of key protein residues which are pivotal for the predicted potency. Protein-specific and interpretable prediction models could guide the optimization of many small molecule drugs for improved potency.
Figure 29.
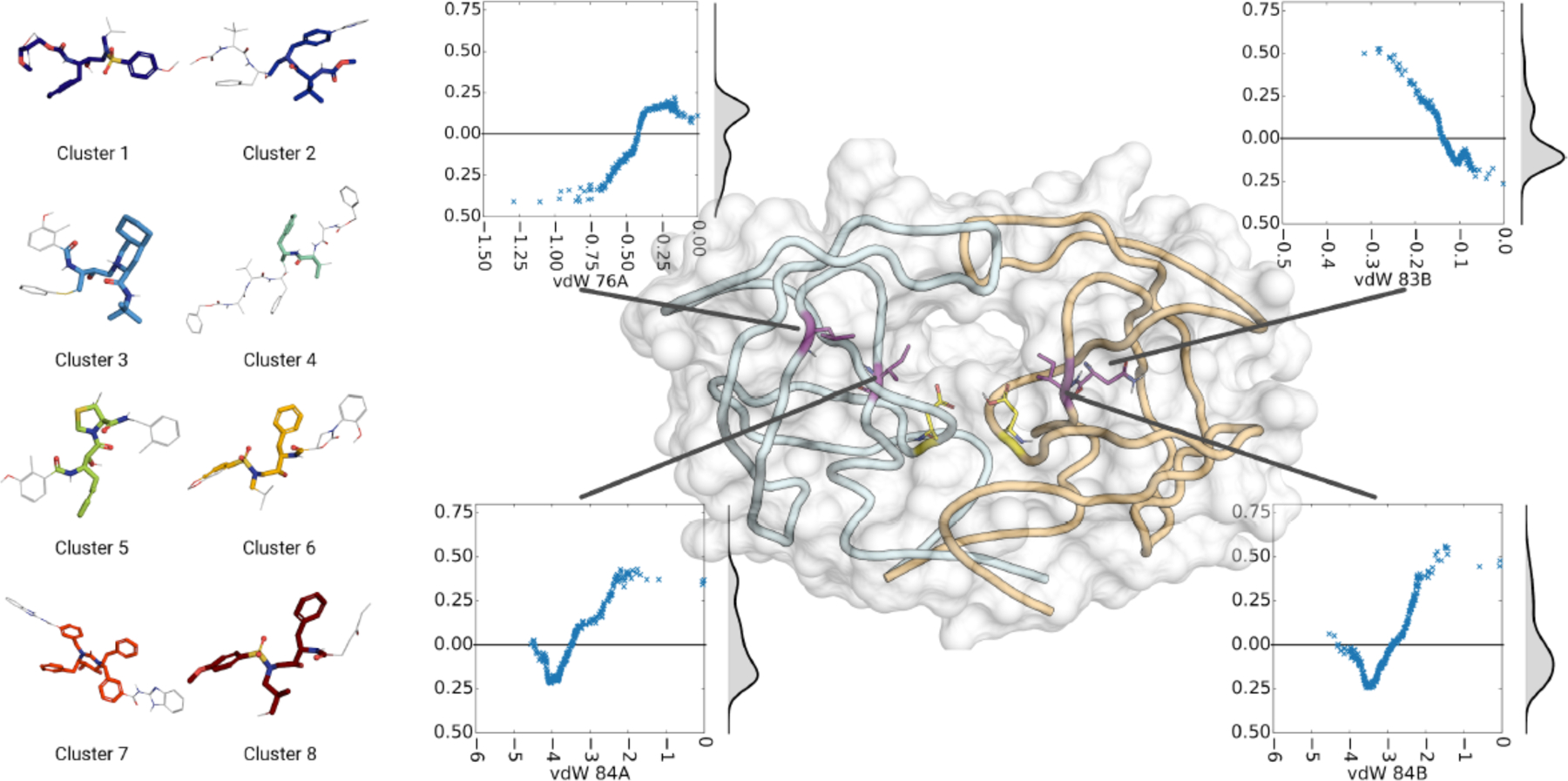
Structural fingerprints of HIV-1 protease inhibitors. (left) Representative structures for each cluster of inhibitors. The minimum common substructure is colored to highlight. (right) Key residues with effect of vdW contacts on predictions. In the center panel: Structure of HIV-1 protease where catalytic aspartic acids in the center of the active site are shown in yellow. Key residues are highlighted in purple. Inserts show the effect of changes in van der Waals contacts of those residues on the predicted binding free energy. Density distribution, computed by gaussian kernel density estimate, is displayed next to scatter plot.
Reproduced with permission from Ref 243 Copyright © 2019, American Chemical Society.
6.4. Integration of potency and pMD with machine learning for evaluating resistance
Protein-ligand interaction fingerprints are highly complementary to pMD analysis. Molecular dynamics simulations allow considering the flexibility and dynamics of the protein-ligand complex and the trajectories can be used for quantitative description of interactions. The fingerprints derived from pMD can include mean values or the distribution of values observed over the simulation to account for the dynamic properties of the protein-ligand complex. The combination of molecular interaction fingerprinting, pMD, and machine learning has been used successfully to predict the free energy of solvation, partition coefficients, and protein-ligand binding affinity.96, 244–245 For HIV-1 protease variants, the combination of pMD and interaction fingerprinting successfully identified signature interactions that drive resistance against the third-generation protease inhibitor DRV.96 A series of 28 susceptible and resistant protease variants were characterized by pMD, followed by computation of inter- and intra-molecular interaction fingerprints. Regression analysis was performed to identify key features that are highly correlated with the loss of inhibitor potency. Strikingly, only four distinct molecular interactions were required and sufficient to predict the loss of binding free energy in resistant variants to within 1 kcal/mol. These features included both intermolecular interactions and interactions within the enzyme and serve as bellwethers for loss of potency. The ability of interaction fingerprinting to not only predict the loss of binding affinity but also identify conserved mechanisms of drug resistance can be exploited in drug design strategies to avoid resistance.
7. Conclusions
Drug resistance is a major public health problem that needs serious attention. With pandemics like CoVID-19 and the rapid spread of new viral strains, the rational design of therapeutics with the goals of thwarting resistance should be considered at the outset of design. To avoid resistance, a detailed analysis of the biological function and natural substrate recognition of the target enzyme is critical. Drugs that leverage evolutionary constrained regions of the target and fit within the substrate envelope will have higher barriers to resistance. The goal is to not only consider the wildtype protein but also likely variants that may evolve, any sites of vulnerability in the target that may mutate to weaken inhibitor binding while maintaining substrate recognition. Rational drug design also needs to incorporate protein and inhibitor dynamics, water structure, and predictive models that can leverage machine learning to accelerate the design and development of robust drugs that last. The strategies discussed in this review are broadly applicable beyond antivirals, and these lessons are particularly relevant for rapidly evolving disease targets. If we stay ahead of “unavoidable” evolution with rational, inventive and intelligent drug design, we can greatly reduce the probability of drug resistance and design inhibitors that will last against our most dangerously evolving viruses and diseases.
Figure 24.
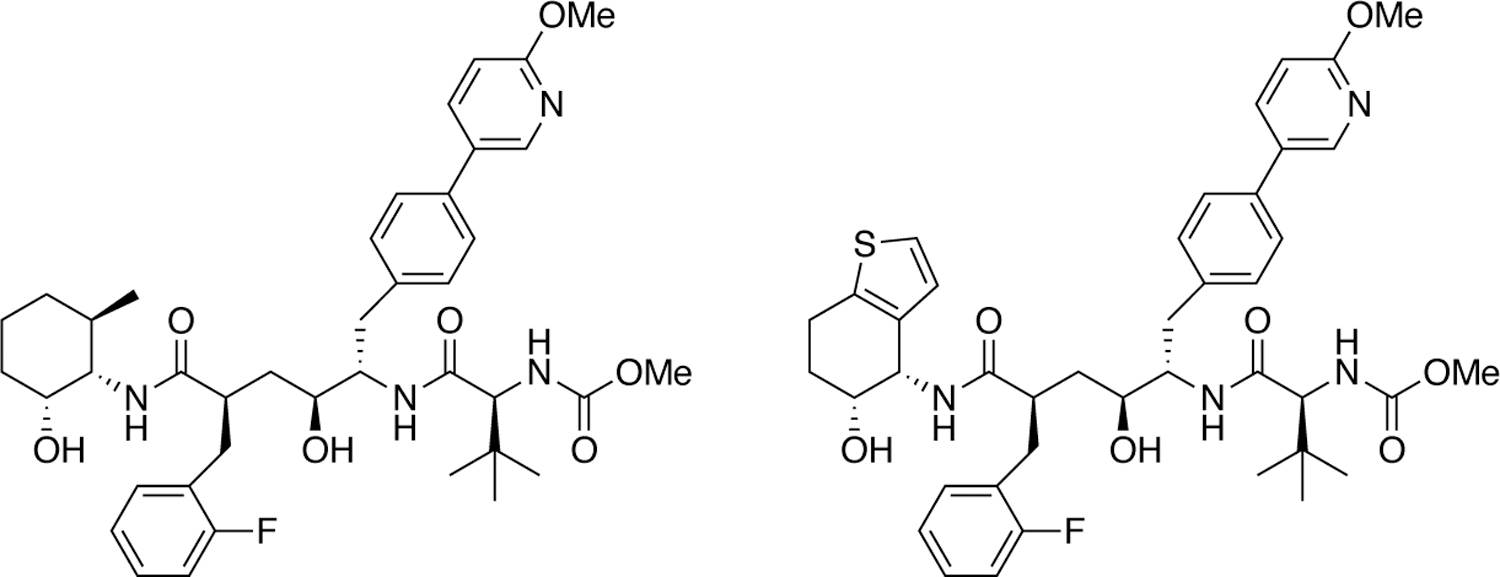
HIV-1 protease inhibitors designed for long-acting injectable drug applications.
Acknowledgements
We thank all past and present members of the Schiffer Lab and our collaborators who contributed to our understanding of drug resistance. Our research has been supported by NIH grants P01 GM109767, R01 AI085051, F31 GM131635, F31 GM119345, F31 GM111101, F31 GM103259, R01 GM135919, R21 AI149716
Biography
Ashley N. Matthew graduated from Xavier University of Louisiana with a BS degree in Chemistry and joined the MD/PhD program at UMass Medical School in 2012. Her PhD thesis research on drug resistance in HCV NS3/4A protease inhibitors was supported by an NIH F31 fellowship. Dr. Matthew was the first to receive the Chancellor’s Award in both Research and Medicine at her graduation in Spring 2020, after which she started the urology residency program at Virginia Commonwealth University.
Florian Leidner received his BS degree in Biology from Friedrich-Alexander-University Erlangen-Nürnberg in Germany. While pursuing his MS and PhD degrees from the same university with Dr. Yves Muller, he worked at UMass Medical School under the mentorship of Dr. Celia Schiffer. He has recently completed his PhD thesis research on drug resistance in HIV-1 protease, with a focus on computational and structural analysis.
Gordon J Lockbaum has a BA degree in Chemistry from Amherst College. After working as a chemistry teacher at Worcester Academy (2012–2015), he joined UMass Medical School for his graduate studies. He has recently received his PhD degree in Biomedical Sciences and in his thesis research determined dozens of crystal structures of protein variants, toward understanding the molecular determinants of resistance to guide the design of better drugs.
Mina Henes started research in the Schiffer lab in his senior year at the Worcester Polytechnic Institute as part of his graduation project, and continued after his graduation (2018) with a BS degree in Biochemistry. He contributed to various research projects both experimentally and computationally before moving to Emory University School of Medicine to join the MD/PhD program in Fall 2020.
Jacqueto Zephyr is a PhD student at UMass Medical School where his research focuses on inhibiting the proteases of HCV and flaviviruses that infect humans. Before starting his PhD studies in 2015, he worked as a research assistant at University of Chicago and as an intern at Stony Brook University in New York where he also obtained a BS degree in Chemistry (2014). His current thesis research, where he uses various techniques from organic synthesis to crystallography, is supported by an NIH F31 fellowship.
Shurong Hou completed her undergraduate studies at the Fudan University in China. After obtaining her Bachelor’s degree in Biomedical Sciences (2014), she joined the graduate program at UMass Medical School. While her thesis research mainly focused on APOBEC3 enzymes, she contributed to multiple projects in the Schiffer lab with computational and structural analysis providing key insights. She has recently obtained her PhD degree.
Nages Rao Desaboini completed his MS in Organic Chemistry (2009) from Osmania University Hyderabad, India and obtained PhD in Synthetic Organic Chemistry (2017) under the supervision of Dr. Parthasarathi Das from Indian Institute of Integrative Medicine (IIIM-CSIR)-Jammu, India (awarded by AcSIR, New-Delhi). In Jan 2018, he joined the Schiffer Lab as a postdoctoral researcher at UMass Medical School, where his research work was focused on the synthesis of inhibitors to target resistant variants under the guidance of Drs. Akbar Ali and Celia Schiffer. He is currently a postdoctoral researcher with Dr. Stephen Miller at the same institute.
Jennifer Timm is a structural biologist who crystallized multiple genotypes of HCV NS3/4A protease during her postdoctoral work at the Schiffer lab (2018–2019). After graduating in 2010 from University of Konstanz in Germany with a BS in Biological Sciences and spending her senior year as a visiting student at the California Institute of Technology, she completed her PhD work at the University of York in England. Dr. Timm is currently a postdoctoral researcher at Rutgers University in New Brunswick working on the ENIGMA project investigating the origins of catalysis and evolution of proteins as nanomachines.
Linah N. Rusere received her PhD degree in Organic Chemistry (2014) at Purdue University in Indiana under the mentorship of Dr. Arun Ghosh. She majored in Biochemistry at Union College in New York and worked as a teaching/research assistant during both her BS and PhD studies. After joining UMass Medical School in 2014, she has worked on multi-step synthesis and biological evaluation of numerous compounds as inhibitors to target viral proteases of HIV-1, HCV, and Dengue virus proteases. Dr. Rusere has been working as a research chemist at Raybow Pharmeceutical USA since 2019.
Debra A. Ragland worked on drug resistance in HIV-1 protease during her research with Dr. Celia Schiffer at UMass Medical School in 2012–2017. She received an NIH F31 fellowship that supported her PhD research. Dr. Ragland has a BS degree in Chemistry (2011) from North Carolina Agricultural & Technical State University. After obtaining her PhD degree at UMass Medical School, she taught chemistry as a lecturer at Clemson University in South Carolina. Dr. Ragland is currently at the University of North Carolina as the Assistant Director of Diversity Affairs and the NIH funded IMSD program, which aims to diversify the biomedical research workforce.
Janet L. Paulsen has a double major in Biochemistry and Chemistry from the Washington State University and a PhD degree in Pharmaceutical Science from the University of Connecticut under the mentorship of late Dr. Amy Anderson. After a year as a research fellow at the Harvard Medical School, she joined the Schiffer lab as a postdoctoral associate in 2014. Her postdoctoral work focused on HIV-1 protease using structural biology and computational techniques to understand drug resistance. Dr. Paulsen currently works as a scientist at Schrodinger in the San Francisco Bay Area.
Kristina Prachanronarong majored in Chemical Engineering at Brown University and joined the MD/PhD program at UMass Medical School in 2009. During her PhD thesis work with Dr. Schiffer, she worked on influenza virus with a focus on drug resistance mutations and inhibitors of the surface glycoprotein neuraminidase. After graduating with MD/PhD degrees, she moved to New York City for residency at Icahn School of Medicine at Mount Sinai where she currently works as a resident physician in diagnostic radiology.
Djade I. Soumana obtained his BS degree in Biochemistry and Molecular Biology from UMass Amherst after which he joined the PhD program at UMass Medical School. During her thesis research with Dr. Celia Schiffer, he used crystallography and enzymology to investigate drug resistance mechanisms in HCV NS3/4A protease. He was an NIH Ruth Kirschtein pre-doctoral fellow until he graduated in 2015, and an NIH-IRTA fellow at NIAID until 2016. While working at GE Healthcare, he pursued his MBA degree from Boston University Questrom School of Business. Dr. Soumana currently works at Cytiva in Cambridge, MA.
Ellen A. Nalivaika is a senior research scientist who has worked with Dr. Celia Schiffer since the Schiffer lab was founded at UMass Medical School. After graduating from Assumption College, she started working as a junior Research Associate at UMass Medical School and joined the Schiffer lab in 1997. In addition to managing the lab, she is an expert in molecular biology and biochemistry including protein purification, crystallization and characterization by a multitude of experimental techniques. She has contributed to various projects in the Schiffer lab related to drug resistance.
Nese Kurt Yilmaz is an Associate Professor of Biochemistry and Molecular Pharmacology at UMass Medical School, and has been working closely with Dr. Celia Schiffer since she joined the faculty at 2011. She has completed her undergraduate and graduate studies in Chemical Engineering at Bogazici University in Istanbul and was a visiting fellow at UMass Medical School with Dr. Schiffer during her PhD studies. After obtaining her PhD degree, she worked as a postdoctoral scholar and then a research scientist at University of Wisconsin-Madison with Dr. Silvia Cavagnero investigating protein folding using biophysical spectroscopy. Her current research focuses on protein conformational dynamics, biomolecular structure, and molecular basis of drug resistance. Dr. Kurt Yilmaz received the UMass GSBS faculty award for research mentoring in 2018.
Akbar Ali is an Associate Professor of Biochemistry and Molecular Pharmacology and has been at UMass Medical School since 2002. He received his Ph.D. in Organic Chemistry under a DAAD (German Academic Exchange Service) split Ph.D. program, working with Prof. Viqar Uddin Ahmed at University of Karachi and Prof. Jürgen Liebscher at Humboldt-Universität in Germany. He did post-doctoral training with Prof. A. Paul Krapcho at University of Vermont and Prof. Tariq M. Rana at UMass Medical School. Dr. Ali uses medicinal chemistry approaches to target drug-resistant pathogens such as HIV and HCV, flaviviruses, and cancers. Working with Prof. Celia Schiffer, he focuses on optimizing the resistance profile of HIV and HCV protease inhibitors.
Celia Schiffer has been on the faculty at UMass Medical School since 1997, and is a Professor of Biochemistry and Molecular Pharmacology and Director of the Institute for Drug Resistance which she founded in 2009. Dr. Schiffer is a structural biologist and biophysicist. She has a BA in Physics from University of Chicago (1986) and received her PhD in Biophysics from University of California San Francisco (1992). Her postdoctoral training was at the ETH Zurich (1992–94) and Genentech, Inc. (1994–1997) before joining the faculty at UMMS as an Assistant Professor. In 2019 she became the Gladys Smith Martin Chair in Oncology.
Dr. Schiffer’s scientific contributions are in defining the field of drug resistance and developing framework to avoid drug resistance from the very initial inhibitor design phase. She provides thought leadership bridging interdisciplinary fields and discovering the parallels between how resistance occurs and potentially could be averted for all evolving diseases.
Bibliography
- 1.World Health Organization. Fact Sheet: Antimicrobial Resistance 2018.
- 2.Editors PM, Antimicrobial Resistance: Is the World UNprepared? PLoS Med 2016, 13, e1002130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dadgostar P, Antimicrobial Resistance: Implications and Costs. Infect Drug Resist 2019, 12, 3903–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collier P; O’Neill LJ, Two years on: an update on achievement towards the recommendations of the antimicrobial resistance report. J Appl Microbiol 2018, 125, 308–312. [DOI] [PubMed] [Google Scholar]
- 5.O’neill J, Antimicrobial resistance: tackling a crisis for the health and wealth of nations. Rev. Antimicrob. Resist 2014, 20, 1–16. [Google Scholar]
- 6.De Clercq E; Li G, Approved antiviral drugs over the past 50 years. Clinical microbiology reviews 2016, 29, 695–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes JP; Rees S; Kalindjian SB; Philpott KL, Principles of early drug discovery. British journal of pharmacology 2011, 162, 1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fox S; Farr-Jones S; Sopchak L; Boggs A; Nicely HW; Khoury R; Biros M, High-throughput screening: update on practices and success. Journal of biomolecular screening 2006, 11, 864–869. [DOI] [PubMed] [Google Scholar]
- 9.Dandapani S; Rosse G; Southall N; Salvino JM; Thomas CJ, Selecting, acquiring, and using small molecule libraries for high‐throughput screening. Current protocols in chemical biology 2012, 4, 177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lage OM; Ramos MC; Calisto R; Almeida E; Vasconcelos V; Vicente F, Current screening methodologies in drug discovery for selected human diseases. Marine drugs 2018, 16, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dahlin JL; Walters MA, The essential roles of chemistry in high-throughput screening triage. Future medicinal chemistry 2014, 6, 1265–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lombardino JG; Lowe JA, The role of the medicinal chemist in drug discovery— then and now. Nature Reviews Drug Discovery 2004, 3, 853–862. [DOI] [PubMed] [Google Scholar]
- 13.Anderson AC, The process of structure-based drug design. Chemistry & biology 2003, 10, 787–797. [DOI] [PubMed] [Google Scholar]
- 14.Van Montfort RL; Workman P, Structure-based drug design: aiming for a perfect fit. Essays in biochemistry 2017, 61, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volarath P; Harrison RW; Weber IT, Structure based drug design for HIV protease: from molecular modeling to cheminformatics. Current topics in medicinal chemistry 2007, 7, 1030–1038. [DOI] [PubMed] [Google Scholar]
- 16.Kaur M; Rawal RK; Rath G; Goyal AK, Structure Based Drug Design: Clinically Relevant HIV-1 Integrase Inhibitors. Current topics in medicinal chemistry 2018, 18, 2664–2680. [DOI] [PubMed] [Google Scholar]
- 17.Babu YS; Chand P; Bantia S; Kotian P; Dehghani A; El-Kattan Y; Lin T-H; Hutchison TL; Elliott AJ; Parker CD, BCX-1812 (RWJ-270201): discovery of a novel, highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drug design. Journal of medicinal chemistry 2000, 43, 3482–3486. [DOI] [PubMed] [Google Scholar]
- 18.Gower E; Estes C; Blach S; Razavi-Shearer K; Razavi H, Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol 2014, 61, S45–S57. [DOI] [PubMed] [Google Scholar]
- 19.Spira S; Wainberg MA; Loemba H; Turner D; Brenner BG, Impact of clade diversity on HIV-1 virulence, antiretroviral drug sensitivity and drug resistance. Journal of Antimicrobial Chemotherapy 2003, 51, 229–240. [DOI] [PubMed] [Google Scholar]
- 20.Rejmanek D; Hosseini PR; Mazet JA; Daszak P; Goldstein T, Evolutionary dynamics and global diversity of influenza A virus. Journal of virology 2015, 89, 10993–11001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asselah T; Hassanein T; Waked I; Mansouri A; Dusheiko G; Gane E, Eliminating hepatitis C within low-income countries - The need to cure genotypes 4, 5, 6. J Hepatol 2018, 68, 814–826. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh AK; Osswald HL; Prato G, Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. Journal of medicinal chemistry 2016, 59, 5172–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee W-G; Gallardo-Macias R; Frey KM; Spasov KA; Bollini M; Anderson KS; Jorgensen WL, Picomolar inhibitors of HIV reverse transcriptase featuring bicyclic replacement of a cyanovinylphenyl group. Journal of the American Chemical Society 2013, 135, 16705–16713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu L; Bi Y; Wu Y; Zhang S; Qi J; Li Y; Lu X; Zhang Z; Lv X; Yan J, Structure-based tetravalent zanamivir with potent inhibitory activity against drug-resistant influenza viruses. Journal of medicinal chemistry 2016, 59, 6303–6312. [DOI] [PubMed] [Google Scholar]
- 25.Miller V; de Béthune M-P; Kober A; Stürmer M; Hertogs K; Pauwels R; Stoffels P; Staszewski S, Patterns of resistance and cross-resistance to human immunodeficiency virus type 1 reverse transcriptase inhibitors in patients treated with the nonnucleoside reverse transcriptase inhibitor loviride. Antimicrobial agents and chemotherapy 1998, 42, 3123–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riemenschneider M; Senge R; Neumann U; Hüllermeier E; Heider D, Exploiting HIV-1 protease and reverse transcriptase cross-resistance information for improved drug resistance prediction by means of multi-label classification. BioData mining 2016, 9, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wyles DL; Luetkemeyer AF, Understanding hepatitis C virus drug resistance: clinical implications for current and future regimens. Topics in antiviral medicine 2017, 25, 103. [PMC free article] [PubMed] [Google Scholar]
- 28.Serre SB; Jensen SB; Ghanem L; Humes DG; Ramirez S; Li Y-P; Krarup H; Bukh J; Gottwein JM, Hepatitis C virus genotype 1 to 6 protease inhibitor escape variants: in vitro selection, fitness, and resistance patterns in the context of the infectious viral life cycle. Antimicrobial agents and chemotherapy 2016, 60, 3563–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.www.hiv.gov HIV Basics overview: data & trends https://www.hiv.gov/hiv-basics/overview/data-and-trends/statistics.
- 30.Sluis-Cremer N; Tachedjian G, Mechanisms of inhibition of HIV replication by non-nucleoside reverse transcriptase inhibitors. Virus research 2008, 134, 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitsuya H; Weinhold KJ; Furman PA; St Clair MH; Lehrman SN; Gallo RC; Bolognesi D; Barry DW; Broder S, 3’-Azido-3’-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proceedings of the National Academy of Sciences 1985, 82, 7096–7100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hazuda D; Blau CU; Felock P; Hastings J; Pramanik B; Wolfe A; Bushman F; Farnet C; Goetz M; Williams M, Isolation and characterization of novel human immunodeficiency virus integrase inhibitors from fungal metabolites. Antiviral Chemistry and Chemotherapy 1999, 10, 63–70. [DOI] [PubMed] [Google Scholar]
- 33.Temesgen Z; Siraj DS, Raltegravir: first in class HIV integrase inhibitor. Therapeutics and clinical risk management 2008, 4, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mouscadet J-F; Tchertanov L, Raltegravir: molecular basis of its mechanism of action. European journal of medical research 2009, 14, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mehandru S; Markowitz M, Tipranavir: a novel non-peptidic protease inhibitor for the treatment of HIV infection. Expert opinion on investigational drugs 2003, 12, 1821–1828. [DOI] [PubMed] [Google Scholar]
- 36.Surleraux DL; Tahri A; Verschueren WG; Pille GM; de Kock HA; Jonckers TH; Peeters A; De Meyer S; Azijn H; Pauwels R; de Bethune MP; King NM; Prabu-Jeyabalan M; Schiffer CA; Wigerinck PB, Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J Med Chem 2005, 48, 1813–22. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y; Liu Z; Brunzelle JS; Kovari IA; Dewdney TG; Reiter SJ; Kovari LC, The higher barrier of darunavir and tipranavir resistance for HIV-1 protease. Biochemical and biophysical research communications 2011, 412, 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El Bouzidi K; White E; Mbisa JL; Sabin CA; Phillips AN; Mackie N; Pozniak AL; Tostevin A; Pillay D; Dunn DT, HIV-1 drug resistance mutations emerging on darunavir therapy in PI-naive and-experienced patients in the UK. Journal of Antimicrobial Chemotherapy 2016, 71, 3487–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Meyer S; Azijn H; Surleraux D; Jochmans D; Tahri A; Pauwels R; Wigerinck P; de Béthune M-P, TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrobial agents and chemotherapy 2005, 49, 2314–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ndung’u T; Weiss RA, On HIV diversity. Aids 2012, 26, 1255–1260. [DOI] [PubMed] [Google Scholar]
- 41.Zdanowicz MM, The pharmacology of HIV drug resistance. American journal of pharmaceutical education 2006, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hajarizadeh B; Grebely J; Dore GJ, Epidemiology and natural history of HCV infection. Nat. Rev. Gastroenterol. Hepatol 2013, 10, 553–562. [DOI] [PubMed] [Google Scholar]
- 43.Grillot A-L; Farmer LJ; Rao BG; Taylor WP; Weisberg IS; Jacobson IM; Perni RB; Kwong AD, Discovery and Development of Telaprevir. In Antiviral Drugs, John Wiley & Sons, Inc.: 2011; pp 207–224. [Google Scholar]
- 44.Lin C; Kwong AD; Perni RB, Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect Disord Drug Targets 2006, 6, 3–16. [DOI] [PubMed] [Google Scholar]
- 45.Llinas-Brunet M; Bailey M; Fazal G; Goulet S; Halmos T; Laplante S; Maurice R; Poirier M; Poupart MA; Thibeault D; Wernic D; Lamarre D, Peptide-based inhibitors of the hepatitis C virus serine protease. Bioorg Med Chem Lett 1998, 8, 1713–8. [DOI] [PubMed] [Google Scholar]
- 46.Steinkuhler C; Biasiol G; Brunetti M; Urbani A; Koch U; Cortese R; Pessi A; De Francesco R, Product inhibition of the hepatitis C virus NS3 protease. Biochemistry 1998, 37, 8899–905. [DOI] [PubMed] [Google Scholar]
- 47.Ingallinella P; Altamura S; Bianchi E; Taliani M; Ingenito R; Cortese R; De Francesco R; Steinkuhler C; Pessi A, Potent peptide inhibitors of human hepatitis C virus NS3 protease are obtained by optimizing the cleavage products. Biochemistry 1998, 37, 8906–14. [DOI] [PubMed] [Google Scholar]
- 48.Ingallinella P; Bianchi E; Ingenito R; Koch U; Steinkuhler C; Altamura S; Pessi A, Optimization of the P’-Region of Peptide Inhibitors of Hepatitis C Virus NS3/4A Protease. Biochemistry 2000, 39, 12898–12906. [DOI] [PubMed] [Google Scholar]
- 49.Llinàs-Brunet M; Bailey M; Fazal G; Ghiro E; Gorys V; Goulet S; Halmos T; Maurice R; Poirier M; Poupart M-A, Highly potent and selective peptide-based inhibitors of the hepatitis C virus serine protease: towards smaller inhibitors. Bioorganic & medicinal chemistry letters 2000, 10, 2267–2270. [DOI] [PubMed] [Google Scholar]
- 50.Lamarre D; Anderson PC; Bailey M; Beaulieu P; Bolger G; Bonneau P; Bos M; Cameron DR; Cartier M; Cordingley MG; Faucher AM; Goudreau N; Kawai SH; Kukolj G; Lagace L; LaPlante SR; Narjes H; Poupart MA; Rancourt J; Sentjens RE; St George R; Simoneau B; Steinmann G; Thibeault D; Tsantrizos YS; Weldon SM; Yong CL; Llinas-Brunet M, An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 2003, 426, 186–189. [DOI] [PubMed] [Google Scholar]
- 51.McCauley JA; McIntyre CJ; Rudd MT; Nguyen KT; Romano JJ; Butcher JW; Gilbert KF; Bush KJ; Holloway MK; Swestock J; Wan BL; Carroll SS; DiMuzio JM; Graham DJ; Ludmerer SW; Mao SS; Stahlhut MW; Fandozzi CM; Trainor N; Olsen DB; Vacca JP; Liverton NJ, Discovery of vaniprevir (MK-7009), a macrocyclic hepatitis C virus NS3/4a protease inhibitor. J. Med. Chem 2010, 53, 2443–2463. [DOI] [PubMed] [Google Scholar]
- 52.Pearlman BL; Hinds AE, novel antivirals for hepatitis C—sofosbuvir/velpatasvir/voxilaprevir, glecaprevir/pibrentasvir. Alimentary pharmacology & therapeutics 2018, 48, 914–923. [DOI] [PubMed] [Google Scholar]
- 53.Lawitz E; Yang JC; Stamm LM; Taylor JG; Cheng G; Brainard DM; Miller MD; Mo H; Dvory-Sobol H, Characterization of HCV resistance from a 3-day monotherapy study of voxilaprevir, a novel pangenotypic NS3/4A protease inhibitor. Antivir. Ther 2017. [DOI] [PubMed]
- 54.Afdhal N; Reddy KR; Nelson DR; Lawitz E; Gordon SC; Schiff E; Nahass R; Ghalib R; Gitlin N; Herring R; Lalezari J; Younes ZH; Pockros PJ; Di Bisceglie AM; Arora S; Subramanian GM; Zhu Y; Dvory-Sobol H; Yang JC; Pang PS; Symonds WT; McHutchison JG; Muir AJ; Sulkowski M; Kwo P, Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N. Engl. J. Med 2014, 370, 1483–1493. [DOI] [PubMed] [Google Scholar]
- 55.Bhatia HK; Singh H; Grewal N; Natt NK, Sofosbuvir: A novel treatment option for chronic hepatitis C infection. Journal of pharmacology & pharmacotherapeutics 2014, 5, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sofia MJ; Bao D; Chang W; Du J; Nagarathnam D; Rachakonda S; Reddy PG; Ross BS; Wang P; Zhang H-R, Discovery of a β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. Journal of medicinal chemistry 2010, 53, 7202–7218. [DOI] [PubMed] [Google Scholar]
- 57.Liu Y; Lim BH; Jiang WW; Flentge CA; Hutchinson DK; Madigan DL; Randolph JT; Wagner R; Maring CJ; Kati WM, Identification of aryl dihydrouracil derivatives as palm initiation site inhibitors of HCV NS5B polymerase. Bioorganic & medicinal chemistry letters 2012, 22, 3747–3750. [DOI] [PubMed] [Google Scholar]
- 58.Tomei L; Altamura S; Bartholomew L; Bisbocci M; Bailey C; Bosserman M; Cellucci A; Forte E; Incitti I; Orsatti L, Characterization of the inhibition of hepatitis C virus RNA replication by nonnucleosides. Journal of virology 2004, 78, 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Di Maio V; Cento V; Mirabelli C; Artese A; Costa G; Alcaro S; Perno C; Ceccherini-Silberstein F, Hepatitis C virus genetic variability and the presence of NS5B resistance-associated mutations as natural polymorphisms in selected genotypes could affect the response to NS5B inhibitors. Antimicrobial agents and chemotherapy 2014, 58, 2781–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guedj J; Dahari H; Rong L; Sansone ND; Nettles RE; Cotler SJ; Layden TJ; Uprichard SL; Perelson AS, Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Proceedings of the National Academy of Sciences 2013, 110, 3991–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Falade-Nwulia O; Suarez-Cuervo C; Nelson DR; Fried MW; Segal JB; Sulkowski MS, Oral direct-acting agent therapy for hepatitis c virus infection: a systematic review. Ann. Intern. Med 2017, 166, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baumert TF; Berg T; Lim JK; Nelson DR, Status of direct-acting antiviral therapy for hepatitis C virus infection and remaining challenges. Gastroenterology 2019, 156, 431–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ghany M; Morgan T, Hepatitis C Guidance 2019 Update: American Association for the Study of Liver Diseases-Infectious Diseases Society of America Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection. Hepatology (Baltimore, Md.) 2020, 71, 686–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Afdhal N; Zeuzem S; Kwo P; Chojkier M; Gitlin N; Puoti M; Romero-Gomez M; Zarski J-P; Agarwal K; Buggisch P; Foster GR; Bräu N; Buti M; Jacobson IM; Subramanian GM; Ding X; Mo H; Yang JC; Pang PS; Symonds WT; McHutchison JG; Muir AJ; Mangia A; Marcellin P, Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N. Engl. J. Med 2014, 370, 1889–1898. [DOI] [PubMed] [Google Scholar]
- 65.Ferenci P; Bernstein D; Lalezari J; Cohen D; Luo Y; Cooper C; Tam E; Marinho RT; Tsai N; Nyberg A; Box TD; Younes Z; Enayati P; Green S; Baruch Y; Bhandari BR; Caruntu FA; Sepe T; Chulanov V; Janczewska E; Rizzardini G; Gervain J; Planas R; Moreno C; Hassanein T; Xie W; King M; Podsadecki T; Reddy KR, ABT-450/r–ombitasvir and dasabuvir with or without ribavirin for HCV. N. Engl. J. Med 2014, 370, 1983–1992. [DOI] [PubMed] [Google Scholar]
- 66.Feld JJ; Jacobson IM; Hézode C; Asselah T; Ruane PJ; Gruener N; Abergel A; Mangia A; Lai C-L; Chan HL, Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. New England Journal of Medicine 2015, 373, 2599–2607. [DOI] [PubMed] [Google Scholar]
- 67.Zeuzem S; Ghalib R; Reddy KR; Pockros PJ; Ari ZB; Zhao Y; Brown DD; Wan S; DiNubile MJ; Nguyen B-Y, Grazoprevir–elbasvir combination therapy for treatment-naive cirrhotic and noncirrhotic patients with chronic hepatitis C virus genotype 1, 4, or 6 infection: a randomized trial. Annals of internal medicine 2015, 163, 1–13. [DOI] [PubMed] [Google Scholar]
- 68.Kwo PY; Poordad F; Asatryan A; Wang S; Wyles DL; Hassanein T; Felizarta F; Sulkowski MS; Gane E; Maliakkal B; Overcash JS; Gordon SC; Muir AJ; Aguilar H; Agarwal K; Dore GJ; Lin CW; Liu R; Lovell SS; Ng TI; Kort J; Mensa FJ, Glecaprevir and pibrentasvir yield high response rates in patients with HCV genotype 1–6 without cirrhosis. J. Hepatol 2017, 67, 263–271. [DOI] [PubMed] [Google Scholar]
- 69.Wyles D; Weiland O; Yao B; Weilert F; Dufour J-F; Gordon SC; Stoehr A; Brown A; Mauss S; Zhang Z, Retreatment of patients who failed glecaprevir/pibrentasvir treatment for hepatitis C virus infection. Journal of hepatology 2019, 70, 1019–1023. [DOI] [PubMed] [Google Scholar]
- 70.Pawlotsky J-M, Hepatitis C virus resistance to direct-acting antiviral drugs in interferon-free regimens. Gastroenterology 2016, 151, 70–86. [DOI] [PubMed] [Google Scholar]
- 71.CDC Disease Burden of Influenza.
- 72.Boivin S; Cusack S; Ruigrok RW; Hart DJ, Influenza A virus polymerase: structural insights into replication and host adaptation mechanisms. Journal of Biological Chemistry 2010, 285, 28411–28417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carrat F; Flahault A, Influenza vaccine: the challenge of antigenic drift. Vaccine 2007, 25, 6852–6862. [DOI] [PubMed] [Google Scholar]
- 74.Noll H; Aoyagi T; Orlando J, The structural relationship of sialidase to the influenza virus surface. Virology 1962, 18, 154–157. [DOI] [PubMed] [Google Scholar]
- 75.Palese P; Tobita K; Ueda M; Compans RW, Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 1974, 61, 397–410. [DOI] [PubMed] [Google Scholar]
- 76.Air GM, Influenza neuraminidase. Influenza and other respiratory viruses 2012, 6, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moscona A, Neuraminidase inhibitors for influenza. New England Journal of Medicine 2005, 353, 1363–1373. [DOI] [PubMed] [Google Scholar]
- 78.Lew W; Chen X; Kim CU, Discovery and development of GS 4104 (oseltamivir) an orally active influenza neuraminidase inhibitor. Current medicinal chemistry 2000, 7, 663–672. [DOI] [PubMed] [Google Scholar]
- 79.Smee DF; Sidwell RW, Peramivir (BCX-1812, RWJ-270201): potential new therapy for influenza. Expert opinion on investigational drugs 2002, 11, 859–869. [DOI] [PubMed] [Google Scholar]
- 80.Woods J; Bethell R; Coates J; Healy N; Hiscox S; Pearson B; Ryan D; Ticehurst J; Tilling J; Walcott S, 4-Guanidino-2, 4-dideoxy-2, 3-dehydro-N-acetylneuraminic acid is a highly effective inhibitor both of the sialidase (neuraminidase) and of growth of a wide range of influenza A and B viruses in vitro. Antimicrobial Agents and Chemotherapy 1993, 37, 1473–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nguyen HT; Fry AM; Gubareva LV, Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antiviral therapy 2012, 17, 159. [DOI] [PubMed] [Google Scholar]
- 82.McKimm‐Breschkin JL, Influenza neuraminidase inhibitors: antiviral action and mechanisms of resistance. Influenza and other respiratory viruses 2013, 7, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Meijer A; Rebelo-de-Andrade H; Correia V; Besselaar T; Drager-Dayal R; Fry A; Gregory V; Gubareva L; Kageyama T; Lackenby A, Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2012–2013. Antiviral research 2014, 110, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Abed Y; Baz M; Boivin G, Impact of neuraminidase mutations conferring influenza resistance to neuraminidase inhibitors in the N1 and N2 genetic backgrounds. Antiviral therapy 2006, 11, 971. [PubMed] [Google Scholar]
- 85.Orozovic G; Orozovic K; Lennerstrand J; Olsen B, Detection of resistance mutations to antivirals oseltamivir and zanamivir in avian influenza A viruses isolated from wild birds. PLoS One 2011, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Takashita E; Kawakami C; Ogawa R; Morita H; Fujisaki S; Shirakura M; Miura H; Nakamura K; Kishida N; Kuwahara T; Ota A; Togashi H; Saito A; Mitamura K; Abe T; Ichikawa M; Yamazaki M; Watanabe S; Odagiri T, Influenza A(H3N2) virus exhibiting reduced susceptibility to baloxavir due to a polymerase acidic subunit I38T substitution detected from a hospitalised child without prior baloxavir treatment, Japan, January 2019. Euro Surveill 2019, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Domingo E; Baranowski E; Ruiz-Jarabo CM; Martín-Hernández AM; Sáiz JC; Escarmís C, Quasispecies structure and persistence of RNA viruses. Emerging infectious diseases 1998, 4, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martell M; Esteban JI; Quer J; Genesca J; Weiner A; Esteban R; Guardia J; Gomez J, Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J. Virol 1992, 66, 3225–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu Y; Jia L; Su B; Li H; Li Z; Han J; Zhang Y; Zhang T; Li T; Wu H, The genetic diversity of HIV-1 quasispecies within primary infected individuals. AIDS research and human retroviruses 2020. [DOI] [PubMed]
- 90.Domingo E; Holland J, RNA virus mutations and fitness for survival. Annual review of microbiology 1997, 51, 151–178. [DOI] [PubMed] [Google Scholar]
- 91.Domingo E; de Ávila AI; Gallego I; Sheldon J; Perales C, Viral fitness: history and relevance for viral pathogenesis and antiviral interventions. Pathogens and disease 2019, 77, ftz021. [DOI] [PubMed] [Google Scholar]
- 92.Paolucci S; Baldanti F; Campanini G; Zavattoni M; Cattaneo E; Dossena L; Gerna G, Analysis of HIV drug-resistant quasispecies in plasma, peripheral blood mononuclear cells and viral isolates from treatment-naive and HAART patients. J. Med. Virol 2001, 65, 207–217. [DOI] [PubMed] [Google Scholar]
- 93.Soumana DI; Kurt Yilmaz N; Ali A; Prachanronarong KL; Schiffer CA, Molecular and dynamic mechanism underlying drug resistance in genotype 3 hepatitis C NS3/4A protease. J. Am. Chem. Soc 2016, 138, 11850–11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ragland DA; Whitfield TW; Lee SK; Swanstrom R; Zeldovich KB; KurtYilmaz N; Schiffer CA, Elucidating the Interdependence of Drug Resistance from Combinations of Mutations. J. Chem. Theory Comput 2017, 13, 5671–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Henes M; Lockbaum GJ; Kosovrasti K; Leidner F; Nachum GS; Nalivaika EA; Lee SK; Spielvogel E; Zhou S; Swanstrom R; Bolon DNA; Kurt Yilmaz N; Schiffer CA, Picomolar to Micromolar: Elucidating the Role of Distal Mutations in HIV-1 Protease in Conferring Drug Resistance. ACS Chem. Biol 2019, 14, 2441–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Leidner F; Kurt Yilmaz N; Schiffer CA, Deciphering complex mechanisms of resistance and loss of potency through coupled molecular dynamics and machine learning. bioRxiv 2020, 2020.06.08.139105. [DOI] [PMC free article] [PubMed]
- 97.Whitfield TW; Ragland DA; Zeldovich KB; Schiffer CA, Characterizing Protein-Ligand Binding Using Atomistic Simulation and Machine Learning: Application to Drug Resistance in HIV-1 Protease. J Chem Theory Comput 2020, 16, 1284–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Romano KP; Ali A; Aydin C; Soumana D; Özen A; Deveau LM; Silver C; Cao H; Newton A; Petropoulos CJ; Huang W; Schiffer CA, The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLoS Pathog 2012, 8, e1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Romano KP; Ali A; Royer WE; Schiffer CA, Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 20986–20991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Romano KP; Laine JM; Deveau LM; Cao H; Massi F; Schiffer CA, Molecular mechanisms of viral and host cell substrate recognition by hepatitis C virus NS3/4A protease. J. Virol 2011, 85, 6106–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Özen A; Sherman W; Schiffer CA, Improving the resistance profile of hepatitis C NS3/4A inhibitors: dynamic substrate envelope guided design. J. Chem. Theory Comput 2013, 9, 5693–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Coffin J; Swanstrom R, HIV pathogenesis: dynamics and genetics of viral populations and infected cells. Cold Spring Harbor perspectives in medicine 2013, 3, a012526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Prabu-Jeyabalan M; Nalivaika E; Schiffer CA, Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [DOI] [PubMed] [Google Scholar]
- 104.Ali A; Aydin C; Gildemeister R; Romano KP; Cao H; Özen A; Soumana D; Newton A; Petropoulos CJ; Huang W; Schiffer CA, Evaluating the role of macrocycles in the susceptibility of hepatitis C virus NS3/4A protease inhibitors to drug resistance. ACS Chem. Biol 2013, 8, 1469–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsantrizos YS; Bolger G; Bonneau P; Cameron DR; Goudreau N; Kukolj G; LaPlante SR; Llinas-Brunet M; Nar H; Lamarre D, Macrocyclic inhibitors of the NS3 protease as potential therapeutic agents of hepatitis C virus infection. Angew. Chem. Int. Ed. Engl 2003, 42, 1356–1360. [DOI] [PubMed] [Google Scholar]
- 106.LaPlante SR; Nar H; Lemke CT; Jakalian A; Aubry N; Kawai SH, Ligand bioactive conformation plays a critical role in the design of drugs that target the hepatitis C virus NS3 protease. J. Med. Chem 2013, 57, 1777–1789. [DOI] [PubMed] [Google Scholar]
- 107.Ng TI; Tripathi R; Reisch T; Lu L; Middleton T; Hopkins TA; Pithawalla R; Irvin M; Dekhtyar T; Krishnan P; Schnell G; Beyer J; McDaniel KF; Ma J; Wang G; Jiang LJ; Or YS; Kempf D; Pilot-Matias T; Collins C, In vitro antiviral activity and resistance profile of the next-generation hepatitis c virus NS3/4A protease inhibitor glecaprevir. Antimicrob. Agents Chemother 2017. [DOI] [PMC free article] [PubMed]
- 108.Rodriguez-Torres M; Glass S; Hill J; Freilich B; Hassman D; Di Bisceglie AM; Taylor JG; Kirby BJ; Dvory-Sobol H; Yang JC; An D; Stamm LM; Brainard DM; Kim S; Krefetz D; Smith W; Marbury T; Lawitz E, GS-9857 in patients with chronic hepatitis C virus genotype 1–4 infection: a randomized, double-blind, dose-ranging phase 1 study. J Viral Hepat 2016, 23, 614–22. [DOI] [PubMed] [Google Scholar]
- 109.Timm J; Kosovrasti K; Henes M; Leidner F; Hou S; Ali A; Kurt Yilmaz N; Schiffer CA, Molecular and Structural Mechanism of Pan-Genotypic HCV NS3/4A Protease Inhibition by Glecaprevir. ACS Chem Biol 2020, 15, 342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Šašková KG; Kožíšek M; Lepšík M; Brynda J; Řezáčová P; Václavíková J; Kagan RM; Machala L; Konvalinka J, Enzymatic and structural analysis of the I47A mutation contributing to the reduced susceptibility to HIV protease inhibitor lopinavir. Protein Science 2008, 17, 1555–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lockbaum GJ; Leidner F; Rusere LN; Henes M; Kosovrasti K; Nachum GS; Nalivaika EA; Ali A; Yilmaz NK; Schiffer CA, Structural Adaptation of Darunavir Analogues against Primary Mutations in HIV-1 Protease. ACS Infect Dis 2019, 5, 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shafer RW; Dupnik K; Winters MA; Eshleman SH, A Guide to HIV-1 Reverse Transcriptase and Protease Sequencing for Drug Resistance Studies. HIV Seq Compend 2001, 2001, 1–51. [PMC free article] [PubMed] [Google Scholar]
- 113.Miller V; Ait-Khaled M; Stone C; Griffin P; Mesogiti D; Cutrell A; Harrigan R; Staszewski S; Katlama C; Pearce G; Tisdale M, HIV-1 reverse transcriptase (RT) genotype and susceptibility to RT inhibitors during abacavir monotherapy and combination therapy. AIDS 2000, 14, 163–71. [DOI] [PubMed] [Google Scholar]
- 114.Harrigan PR; Stone C; Griffin P; Najera I; Bloor S; Kemp S; Tisdale M; Larder B, Resistance profile of the human immunodeficiency virus type 1 reverse transcriptase inhibitor abacavir (1592U89) after monotherapy and combination therapy. CNA2001 Investigative Group. J Infect Dis 2000, 181, 912–20. [DOI] [PubMed] [Google Scholar]
- 115.Whitcomb JM; Parkin NT; Chappey C; Hellmann NS; Petropoulos CJ, Broad nucleoside reverse-transcriptase inhibitor cross-resistance in human immunodeficiency virus type 1 clinical isolates. J Infect Dis 2003, 188, 992–1000. [DOI] [PubMed] [Google Scholar]
- 116.Borroto-Esoda K; Parkin N; Miller MD, A comparison of the phenotypic susceptibility profiles of emtricitabine and lamivudine. Antivir Chem Chemother 2007, 18, 297–300. [DOI] [PubMed] [Google Scholar]
- 117.Margot NA; Johnson A; Miller MD; Callebaut C, Characterization of HIV-1 Resistance to Tenofovir Alafenamide In Vitro. Antimicrob Agents Chemother 2015, 59, 5917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bacheler LT; Anton ED; Kudish P; Baker D; Bunville J; Krakowski K; Bolling L; Aujay M; Wang XV; Ellis D; Becker MF; Lasut AL; George HJ; Spalding DR; Hollis G; Abremski K, Human immunodeficiency virus type 1 mutations selected in patients failing efavirenz combination therapy. Antimicrob Agents Chemother 2000, 44, 2475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Reuman EC; Rhee SY; Holmes SP; Shafer RW, Constrained patterns of covariation and clustering of HIV-1 non-nucleoside reverse transcriptase inhibitor resistance mutations. J Antimicrob Chemother 2010, 65, 1477–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gulick RM; Ribaudo HJ; Shikuma CM; Lustgarten S; Squires KE; Meyer WA 3rd; Acosta EP; Schackman BR; Pilcher CD; Murphy RL; Maher WE; Witt MD; Reichman RC; Snyder S; Klingman KL; Kuritzkes DR; Team ACTGSA, Triple-nucleoside regimens versus efavirenz-containing regimens for the initial treatment of HIV-1 infection. N Engl J Med 2004, 350, 1850–61. [DOI] [PubMed] [Google Scholar]
- 121.Margot NA; Lu B; Cheng A; Miller MD; Study T, Resistance development over 144 weeks in treatment-naive patients receiving tenofovir disoproxil fumarate or stavudine with lamivudine and efavirenz in Study 903. HIV Med 2006, 7, 442–50. [DOI] [PubMed] [Google Scholar]
- 122.Richman D; Shih CK; Lowy I; Rose J; Prodanovich P; Goff S; Griffin J, Human immunodeficiency virus type 1 mutants resistant to nonnucleoside inhibitors of reverse transcriptase arise in tissue culture. Proc Natl Acad Sci U S A 1991, 88, 11241–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wu H; Zhang HJ; Zhang XM; Xu HF; Wang M; Huang JD; Zheng BJ, Identification of drug resistant mutations in HIV-1 CRF07_BC variants selected by nevirapine in vitro. PLoS One 2012, 7, e44333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Winslow DL; Garber S; Reid C; Scarnati H; Baker D; Rayner MM; Anton ED, Selection conditions affect the evolution of specific mutations in the reverse transcriptase gene associated with resistance to DMP 266. AIDS 1996, 10, 1205–9. [DOI] [PubMed] [Google Scholar]
- 125.Vingerhoets J; Azijn H; Fransen E; De Baere I; Smeulders L; Jochmans D; Andries K; Pauwels R; de Bethune MP, TMC125 displays a high genetic barrier to the development of resistance: evidence from in vitro selection experiments. J Virol 2005, 79, 12773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Azijn H; Tirry I; Vingerhoets J; de Bethune MP; Kraus G; Boven K; Jochmans D; Van Craenenbroeck E; Picchio G; Rimsky LT, TMC278, a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild-type and NNRTIresistant HIV-1. Antimicrob Agents Chemother 2010, 54, 718–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cook NJ; Li W; Berta D; Badaoui M; Ballandras-Colas A; Nans A; Kotecha A; Rosta E; Engelman AN; Cherepanov P, Structural basis of second-generation HIV integrase inhibitor action and viral resistance. Science 2020, 367, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Passos DO; Li M; Jozwik IK; Zhao XZ; Santos-Martins D; Yang R; Smith SJ; Jeon Y; Forli S; Hughes SH; Burke TR Jr.; Craigie R; Lyumkis D, Structural basis for strand-transfer inhibitor binding to HIV intasomes. Science 2020, 367, 810–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Steigbigel RT; Cooper DA; Kumar PN; Eron JE; Schechter M; Markowitz M; Loutfy MR; Lennox JL; Gatell JM; Rockstroh JK; Katlama C; Yeni P; Lazzarin A; Clotet B; Zhao J; Chen J; Ryan DM; Rhodes RR; Killar JA; Gilde LR; Strohmaier KM; Meibohm AR; Miller MD; Hazuda DJ; Nessly ML; DiNubile MJ; Isaacs RD; Nguyen BY; Teppler H; Teams BS, Raltegravir with optimized background therapy for resistant HIV-1 infection. N Engl J Med 2008, 359, 339–54. [DOI] [PubMed] [Google Scholar]
- 130.Hatano H; Lampiris H; Fransen S; Gupta S; Huang W; Hoh R; Martin JN; Lalezari J; Bangsberg D; Petropoulos C; Deeks SG, Evolution of integrase resistance during failure of integrase inhibitor-based antiretroviral therapy. J Acquir Immune Defic Syndr 2010, 54, 389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cai Y; Myint W; Paulsen JL; Schiffer CA; Ishima R; Kurt Yilmaz N, Drug Resistance Mutations Alter Dynamics of Inhibitor-Bound HIV-1 Protease. J. Chem. Theory. Comput 2014, 10, 3438–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Özen A; Haliloğlu T; Schiffer CA, HIV-1 protease and substrate coevolution validates the substrate envelope as the substrate recognition pattern. J. Chem. Theory Comput 2012, 8, 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mittal S; Cai Y; Nalam MN; Bolon DN; Schiffer CA, Hydrophobic core flexibility modulates enzyme activity in HIV-1 protease. J. Am. Chem. Soc 2012, 134, 4163–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ragland DA; Nalivaika EA; Nalam MN; Prachanronarong KL; Cao H; Bandaranayake RM; Cai Y; Kurt-Yilmaz N; Schiffer CA, Drug resistance conferred by mutations outside the active site through alterations in the dynamic and structural ensemble of HIV-1 protease. J. Am. Chem. Soc 2014, 136, 11956–11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ozen A; Lin KH; Kurt Yilmaz N; Schiffer CA, Structural basis and distal effects of Gag substrate coevolution in drug resistance to HIV-1 protease. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 15993–15998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Prachanronarong KL; Ozen A; Thayer KM; Yilmaz LS; Zeldovich KB; Bolon DN; Kowalik TF; Jensen JD; Finberg RW; Wang JP; Kurt-Yilmaz N; Schiffer CA, Molecular Basis for Differential Patterns of Drug Resistance in Influenza N1 and N2 Neuraminidase. J. Chem. Theory Comput 2016, 12, 6098–6108. [DOI] [PubMed] [Google Scholar]
- 137.Lin KH; Nalivaika EA; Prachanronarong KL; Yilmaz NK; Schiffer CA, Dengue Protease Substrate Recognition: Binding of the Prime Side. ACS Infect. Dis 2016, 2, 734–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lin KH; Ali A; Rusere L; Soumana DI; Kurt Yilmaz N; Schiffer CA, Dengue Virus NS2B/NS3 Protease Inhibitors Exploiting the Prime Side. J. Virol 2017, 91, e00045–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Paulsen JL; Leidner F; Ragland DA; Kurt Yilmaz N; Schiffer CA, Interdependence of inhibitor recognition in HIV-1 protease. J. Chem. Theory. Comput 2017, 13, 2300–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Leidner F; Kurt Yilmaz N; Paulsen J; Muller YA; Schiffer CA, Hydration Structure and Dynamics of Inhibitor-Bound HIV-1 Protease. J. Chem. Theory. Comput 2018, 14, 2784–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cai Y; Schiffer CA, Decomposing the energetic impact of drug resistant mutations in HIV-1 protease on binding DRV. J. Chem. Theory Comput 2010, 6, 1358–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cai Y; Yilmaz NK; Myint W; Ishima R; Schiffer CA, Differential Flap Dynamics in Wild-type and a Drug Resistant Variant of HIV-1 Protease Revealed by Molecular Dynamics and NMR Relaxation. J. Chem. Theory Comput 2012, 8, 3452–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Henes M; Lockbaum GJ; Kosovrasti K; Leidner F; Nachum GS; Nalivaika EA; Lee SK; Spielvogel E; Zhou S; Swanstrom R; Bolon DNA; Kurt Yilmaz N; Schiffer CA, Picomolar to Micromolar: Elucidating the Role of Distal Mutations in HIV-1 Protease in Conferring Drug Resistance. ACS Chem. Biol 2019, doi: 10.1021/acschembio.9b00370. [DOI] [PMC free article] [PubMed]
- 144.Ishima R; Kurt Yilmaz N; Schiffer CA, NMR and MD studies combined to elucidate inhibitor and water interactions of HIV-1 protease and their modulations with resistance mutations. J. Biomol. NMR 2019, 73, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Matthew AN; Leidner F; Newton A; Petropoulos CJ; Huang W; Ali A; KurtYilmaz N; Schiffer CA, Molecular Mechanism of Resistance in a Clinically Significant Double-Mutant Variant of HCV NS3/4A Protease. Structure 2018, 26, 1360–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Soumana DI; Kurt Yilmaz N; Prachanronarong KL; Aydin C; Ali A; Schiffer CA, Structural and thermodynamic effects of macrocyclization in HCV NS3/4A inhibitor MK-5172. ACS Chem. Biol 2016, 11, 900–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Agniswamy J; Shen CH; Aniana A; Sayer JM; Louis JM; Weber IT, HIV-1 protease with 20 mutations exhibits extreme resistance to clinical inhibitors through coordinated structural rearrangements. Biochemistry 2012, 51, 2819–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Özen A; Schiffer CA, Integrating Evolution of Drug Resistance into Drug Discovery: Lessons from the Viral Proteases of HIV‐1 and HCV. Structural Biology in Drug Discovery: Methods, Techniques, and Practices 2020, 521–543.
- 149.Özen A; Lin K-H; Kurt Yilmaz N; Schiffer CA, Structural basis and distal effects of Gag substrate coevolution in drug resistance to HIV-1 protease. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 15993–15998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Özen A; Schiffer CA, Substrate‐envelope‐guided design of drugs with a high barrier to the evolution of resistance. Handbook of Antimicrobial Resistance 2014.
- 151.Ozen A; Haliloglu T; Schiffer CA, Dynamics of preferential substrate recognition in HIV-1 protease: redefining the substrate envelope. Journal of molecular biology 2011, 410, 726–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Foulkes-Murzycki JE; Scott WRP; Schiffer CA, Hydrophobic sliding: a possible mechanism for drug resistance in human immunodeficiency virus type 1 protease. Structure 2007, 15, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Galiano L; Ding F; Veloro AM; Blackburn ME; Simmerling C; Fanucci GE, Drug pressure selected mutations in HIV-1 protease alter flap conformations. Journal of the American Chemical Society 2009, 131, 430–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Amaro RE; Swift RV; Votapka L; Li WW; Walker RC; Bush RM, Mechanism of 150-cavity formation in influenza neuraminidase. Nature communications 2011, 2, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Russell RJ; Haire LF; Stevens DJ; Collins PJ; Lin YP; Blackburn GM; Hay AJ; Gamblin SJ; Skehel JJ, The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 2006, 443, 45–49. [DOI] [PubMed] [Google Scholar]
- 156.Reeves JD; Lee F-H; Miamidian JL; Jabara CB; Juntilla MM; Doms RW, Enfuvirtide resistance mutations: impact on human immunodeficiency virus envelope function, entry inhibitor sensitivity, and virus neutralization. Journal of virology 2005, 79, 4991–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Miller MD; Hazuda DJ, HIV resistance to the fusion inhibitor enfuvirtide: mechanisms and clinical implications. Drug resistance updates 2004, 7, 89–95. [DOI] [PubMed] [Google Scholar]
- 158.Dingens AS; Arenz D; Overbaugh J; Bloom JD, Massively Parallel Profiling of HIV-1 Resistance to the Fusion Inhibitor Enfuvirtide. Viruses 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Spielvogel E; Lee S-K; Zhou S; Lockbaum GJ; Henes M; Sondgeroth A; Kosovrasti K; Ali A; Yilmaz NK; Schiffer CA; Swanstrom R, Selection of HIV-1 for Resistance to Fourth Generation Protease Inhibitors Reveals Two Independent Pathways to High-Level Resistance. bioRxiv 2019, 837120. [DOI] [PMC free article] [PubMed]
- 160.Rhee SY; Gonzales MJ; Kantor R; Betts BJ; Ravela J; Shafer RW, Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res 2003, 31, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shafer RW, Rationale and uses of a public HIV drug-resistance database. J Infect Dis 2006, 194 Suppl 1, S51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Henes M; Kosovrasti K; Lockbaum GJ; Leidner F; Nachum GS; Nalivaika EA; Bolon DNA; Kurt Yilmaz N; Schiffer CA; Whitfield TW, Molecular Determinants of Epistasis in HIV-1 Protease: Elucidating the Interdependence of L89V and L90M Mutations in Resistance. Biochemistry 2019, 58, 3711–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Özen A; Lin K-H; Romano KP; Tavella D; Newton A; Petropoulos CJ; Huang W; Aydin C; Schiffer CA, Resistance from Afar: Distal Mutation V36M Allosterically Modulates the Active Site to Accentuate Drug Resistance in HCV NS3/4A Protease. bioRxiv 2018, 452284.
- 164.King NM; Prabu-Jeyabalan M; Nalivaika EA; Schiffer CA, Combating susceptibility to drug resistance: lessons from HIV-1 protease. Chem Biol 2004, 11, 1333–8. [DOI] [PubMed] [Google Scholar]
- 165.Kurt Yilmaz N; Swanstrom R; Schiffer CA, Improving viral protease inhibitors to counter drug resistance. Trends Microbiol 2016, 24, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kolli M; Ozen A; Kurt-Yilmaz N; Schiffer CA, HIV-1 protease-substrate coevolution in nelfinavir resistance. J Virol 2014, 88, 7145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kolli M; Stawiski E; Chappey C; Schiffer CA, Human immunodeficiency virus type 1 protease-correlated cleavage site mutations enhance inhibitor resistance. J Virol 2009, 83, 11027–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kolli M; Lastere S; Schiffer CA, Co-evolution of nelfinavir-resistant HIV-1 protease and the p1-p6 substrate. Virology 2006, 347, 405–9. [DOI] [PubMed] [Google Scholar]
- 169.Shen Y; Altman MD; Ali A; Nalam MNL; Cao H; Rana TM; Schiffer CA; Tidor B, Testing the substrate-envelope hypothesis with designed pairs of compounds. ACS Chem. Biol 2013, 8, 2433–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kairys V; Gilson MK; Lather V; Schiffer CA; Fernandes MX, Toward the design of mutation‐resistant enzyme inhibitors: further evaluation of the substrate envelope hypothesis. Chemical biology & drug design 2009, 74, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhao XZ; Smith SJ; Maskell DP; Métifiot M; Pye VE; Fesen K; Marchand C; Pommier Y; Cherepanov P; Hughes SH, Structure-guided optimization of HIV integrase strand transfer inhibitors. Journal of medicinal chemistry 2017, 60, 7315–7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Matthew AN; Zephyr J; Rao DN; Henes M; Kamran W; Kosovrasti K; Hedger AK; Lockbaum GJ; Timm J; Ali A, Avoiding Drug Resistance by Substrate Envelope-Guided Design: Toward Potent and Robust HCV NS3/4A Protease Inhibitors. Mbio 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lin C, HCV NS3–4A serine protease. Hepatitis C viruses: genomes and molecular biology 2006, 52, 55. [Google Scholar]
- 174.Zürcher M; Diederich F, Structure-based drug design: exploring the proper filling of apolar pockets at enzyme active sites. The Journal of organic chemistry 2008, 73, 4345–4361. [DOI] [PubMed] [Google Scholar]
- 175.Hongmao S, A practical guide to rational drug design Woodhead Publishing: 2015. [Google Scholar]
- 176.Tuske S; Sarafianos SG; Clark AD; Ding J; Naeger LK; White KL; Miller MD; Gibbs CS; Boyer PL; Clark P, Structures of HIV-1 RT–DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nature structural & molecular biology 2004, 11, 469–474. [DOI] [PubMed] [Google Scholar]
- 177.Lohmann V; Körner F; Herian U; Bartenschlager R, Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. Journal of virology 1997, 71, 8416–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kulkarni AS; Damha MJ; Schinazi RF; Mo H; Doehle B; Sagan SM; Götte M, A complex network of interactions between S282 and G283 of hepatitis c virus nonstructural protein 5B and the template strand affects susceptibility to sofosbuvir and ribavirin. Antimicrobial agents and chemotherapy 2016, 60, 2018–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Walker A; Filke S; Lübke N; Obermeier M; Kaiser R; Häussinger D; Timm J; Bock HH, Detection of a genetic footprint of the sofosbuvir resistance-associated substitution S282T after HCV treatment failure. Virology journal 2017, 14, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Matthew AN; Zephyr J; Hill CJ; Jahangir M; Newton A; Petropoulos CJ; Huang W; Kurt-Yilmaz N; Schiffer CA; Ali A, Hepatitis c virus NS3/4A protease inhibitors incorporating flexible P2 quinoxalines target drug resistant viral variants. J. Med. Chem 2017, 60, 5699–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wang X; Zhang J; Huang Y; Wang R; Zhang L; Qiao K; Li L; Liu C; Ouyang Y; Xu W, Design, synthesis, and biological evaluation of 1-[(2-benzyloxyl/alkoxyl) methyl]-5-halo-6-aryluracils as potent HIV-1 non-nucleoside reverse transcriptase inhibitors with an improved drug resistance profile. Journal of medicinal chemistry 2012, 55, 2242–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ghosh AK; Anderson DD; Weber IT; Mitsuya H, Enhancing Protein Backbone Binding—A Fruitful Concept for Combating Drug‐Resistant HIV. Angewandte Chemie International Edition 2012, 51, 1778–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hao G-F; Yang G-F; Zhan C-G, Structure-based methods for predicting target mutation-induced drug resistance and rational drug design to overcome the problem. Drug discovery today 2012, 17, 1121–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ghosh A; Chapsla B; Weber I; Mitsuya H, Design of protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Acc. Chem. Res 2008, 41, 78–86. [DOI] [PubMed] [Google Scholar]
- 185.Schauer GD; Huber KD; Leuba SH; Sluis-Cremer N, Mechanism of allosteric inhibition of HIV-1 reverse transcriptase revealed by single-molecule and ensemble fluorescence. Nucleic acids research 2014, 42, 11687–11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tomei L; Altamura S; Bartholomew L; Biroccio A; Ceccacci A; Pacini L; Narjes F; Gennari N; Bisbocci M; Incitti I, Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. Journal of Virology 2003, 77, 13225–13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Asahchop EL; Wainberg MA; Sloan RD; Tremblay CL, Antiviral drug resistance and the need for development of new HIV-1 reverse transcriptase inhibitors. Antimicrobial agents and chemotherapy 2012, 56, 5000–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Nussinov R; Tsai C-J, The design of covalent allosteric drugs. Annual review of pharmacology and toxicology 2015, 55, 249–267. [DOI] [PubMed] [Google Scholar]
- 189.Gulnik SV; Eissenstat M, Approaches to the design of HIV protease inhibitors with improved resistance profiles. Curr. Opin. HIV AIDS 2008, 3, 633–641. [DOI] [PubMed] [Google Scholar]
- 190.Miller JF; Andrews CW; Brieger M; Furfine ES; Hale MR; Hanlon MH; Hazen RJ; Kaldor I; McLean EW; Reynolds D; Sammond DM; Spaltenstein A; Tung R; Turner EM; Xu RX; Sherrill RG, Ultra-potent P1 modified arylsulfonamide HIV protease inhibitors: the discovery of GW0385. Bioorg. Med. Chem. Lett 2006, 16, 1788–1794. [DOI] [PubMed] [Google Scholar]
- 191.Cihlar T; He GX; Liu X; Chen JM; Hatada M; Swaminathan S; McDermott MJ; Yang ZY; Mulato AS; Chen X; Leavitt SA; Stray KM; Lee WA, Suppression of HIV-1 protease inhibitor resistance by phosphonate-mediated solvent anchoring. J. Mol. Biol 2006, 363, 635–647. [DOI] [PubMed] [Google Scholar]
- 192.Nalam MN; Ali A; Reddy GS; Cao H; Anjum SG; Altman MD; Yilmaz NK; Tidor B; Rana TM; Schiffer CA, Substrate envelope-designed potent HIV-1 protease inhibitors to avoid drug resistance. Chem. Biol 2013, 20, 1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ghosh AK; Chapsal BD; Weber IT; Mitsuya H, Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Acc. Chem. Res 2008, 41, 78–86. [DOI] [PubMed] [Google Scholar]
- 194.Ghosh AK; Kincaid JF; Cho W; Walters DE; Krishnan K; Hussain KA; Koo Y; Cho H; Rudall C; Holland L; Buthod J, Potent HIV protease inhibitors incorporating high-affinity P2-ligands and (R)-(hydroxyethylamino)sulfonamide isostere. Bioorg Med Chem Lett 1998, 8, 687–90. [DOI] [PubMed] [Google Scholar]
- 195.Ghosh AK; Ramu Sridhar P; Kumaragurubaran N; Koh Y; Weber IT; Mitsuya H, Bis-tetrahydrofuran: a privileged ligand for darunavir and a new generation of HIV protease inhibitors that combat drug resistance. ChemMedChem 2006, 1, 939–950. [DOI] [PubMed] [Google Scholar]
- 196.Ghosh AK; Rao KV; Nyalapatla PR; Osswald HL; Martyr CD; Aoki M; Hayashi H; Agniswamy J; Wang YF; Bulut H; Das D; Weber IT; Mitsuya H, Design and development of highly potent HIV-1 protease inhibitors with a crown-like oxotricyclic core as the P2-ligand to combat multidrug-resistant HIV variants. J. Med. Chem 2017, 60, 4267–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ghosh AK; Rao KV; Nyalapatla PR; Kovela S; Brindisi M; Osswald HL; Sekhara Reddy B; Agniswamy J; Wang YF; Aoki M; Hattori SI; Weber IT; Mitsuya H, Design of highly potent, dual-acting and central-nervous-system-penetrating HIV-1 protease inhibitors with excellent potency against multidrug-resistant HIV-1 variants. ChemMedChem 2018, 13, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ghosh AK; P, R. N.; Kovela S; Rao KV; Brindisi M; Osswald HL; Amano M; Aoki M; Agniswamy J; Wang YF; Weber IT; Mitsuya H, Design and synthesis of highly potent HIV-1 protease inhibitors containing tricyclic fused ring systems as novel P2 ligands: structure-activity studies, biological and X-ray structural analysis. J. Med. Chem 2018, 61, 4561–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ghosh AK; Kovela S; Osswald HL; Amano M; Aoki M; Agniswamy J; Wang YF; Weber IT; Mitsuya H, Structure-Based Design of Highly Potent HIV-1 Protease Inhibitors Containing New Tricyclic Ring P2-Ligands: Design, Synthesis, Biological, and X-ray Structural Studies. J Med Chem 2020, 63, 4867–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Aoki M; Hayashi H; Rao KV; Das D; Higashi-Kuwata N; Bulut H; Aoki-Ogata H; Takamatsu Y; Yedidi RS; Davis DA; Hattori SI; Nishida N; Hasegawa K; Takamune N; Nyalapatla PR; Osswald HL; Jono H; Saito H; Yarchoan R; Misumi S; Ghosh AK; Mitsuya H, A novel central nervous system-penetrating protease inhibitor overcomes human immunodeficiency virus 1 resistance with unprecedented aM to pM potency. Elife 2017, 6, e28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rusere LN; Lockbaum GJ; Lee SK; Henes M; Kosovrasti K; Spielvogel E; Nalivaika EA; Swanstrom R; Yilmaz NK; Schiffer CA; Ali A, HIV-1 protease inhibitors incorporating stereochemically defined P2’ ligands to optimize hydrogen bonding in the substrate envelope. J. Med. Chem 2019, 62, 8062–8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Stranix BR; Lavallee JF; Sevigny G; Yelle J; Perron V; LeBerre N; Herbart D; Wu JJ, Lysine sulfonamides as novel HIV-protease inhibitors: Nepsilon-acyl aromatic alpha-amino acids. Bioorg Med Chem Lett 2006, 16, 3459–62. [DOI] [PubMed] [Google Scholar]
- 203.Bouzide A; Sauve G; Yelle J, Lysine derivatives as potent HIV protease inhibitors. Discovery, synthesis and structure-activity relationship studies. Bioorg Med Chem Lett 2005, 15, 1509–13. [DOI] [PubMed] [Google Scholar]
- 204.Dandache S; Sevigny G; Yelle J; Stranix BR; Parkin N; Schapiro JM; Wainberg MA; Wu JJ, In vitro antiviral activity and cross-resistance profile of PL-100, a novel protease inhibitor of human immunodeficiency virus type 1. Antimicrob. Agents Chemother 2007, 51, 4036–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jones KL; Holloway MK; Su HP; Carroll SS; Burlein C; Touch S; DiStefano DJ; Sanchez RI; Williams TM; Vacca JP; Coburn CA, Epsilon substituted lysinol derivatives as HIV-1 protease inhibitors. Bioorg Med Chem Lett 2010, 20, 4065–8. [DOI] [PubMed] [Google Scholar]
- 206.Miller JF; Brieger M; Furfine ES; Hazen RJ; Kaldor I; Reynolds D; Sherrill RG; Spaltenstein A, Novel P1 chain-extended HIV protease inhibitors possessing potent anti-HIV activity and remarkable inverse antiviral resistance profiles. Bioorg Med Chem Lett 2005, 15, 3496–500. [DOI] [PubMed] [Google Scholar]
- 207.Miller JF; Furfine ES; Hanlon MH; Hazen RJ; Ray JA; Robinson L; Samano V; Spaltenstein A, Novel arylsulfonamides possessing sub-picomolar HIV protease activities and potent anti-HIV activity against wild-type and drug-resistant viral strains. Bioorg Med Chem Lett 2004, 14, 959–63. [DOI] [PubMed] [Google Scholar]
- 208.Hazen R; Harvey R; Ferris R; Craig C; Yates P; Griffin P; Miller J; Kaldor I; Ray J; Samano V; Furfine E; Spaltenstein A; Hale M; Tung R; St Clair M; Hanlon M; Boone L, In vitro antiviral activity of the novel, tyrosyl-based human immunodeficiency virus (HIV) type 1 protease inhibitor brecanavir (GW640385) in combination with other antiretrovirals and against a panel of protease inhibitor-resistant HIV. Antimicrob. Agents Chemother 2007, 51, 3147–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Callebaut C; Stray K; Tsai L; Williams M; Yang ZY; Cannizzaro C; Leavitt SA; Liu X; Wang K; Murray BP; Mulato A; Hatada M; Priskich T; Parkin N; Swaminathan S; Lee W; He GX; Xu L; Cihlar T, In vitro characterization of GS-8374, a novel phosphonate-containing inhibitor of HIV-1 protease with a favorable resistance profile. Antimicrob Agents Chemother 2011, 55, 1366–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.He G-X; Yang Z-Y; Williams M; Callebaut C; Cihlar T; Murray BP; Yang C; Mitchell ML; Liu H; Wang J; Arimilli M; Eisenberg E; Stray KM; Tsai LK; Hatada M; Chen X; Chen JM; Wang Y; Lee MS; Strickley RG; Iwata Q; Zheng X; Kim CU; Swaminathan S; Desai MC; Lee WA; Xu L, Discovery of GS-8374, a potent human immunodeficiency virus type 1 protease inhibitor with a superior resistance profile. MedChemComm 2011, 2, 1093–1098. [Google Scholar]
- 211.Bungard CJ; Williams PD; Ballard JE; Bennett DJ; Beaulieu C; Bahnck-Teets C; Carroll SS; Chang RK; Dubost DC; Fay JF; Diamond TL; Greshock TJ; Hao L; Holloway MK; Felock PJ; Gesell JJ; Su HP; Manikowski JJ; McKay DJ; Miller M; Min X; Molinaro C; Moradei OM; Nantermet PG; Nadeau C; Sanchez RI; Satyanarayana T; Shipe WD; Singh SK; Truong VL; Vijayasaradhi S; Wiscount CM; Vacca JP; Crane SN; McCauley JA, Discovery of MK-8718, an HIV Protease Inhibitor Containing a Novel Morpholine Aspartate Binding Group. ACS Med Chem Lett 2016, 7, 702–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Bungard CJ; Williams PD; Schulz J; Wiscount CM; Holloway MK; Loughran HM; Manikowski JJ; Su HP; Bennett DJ; Chang L; Chu XJ; Crespo A; Dwyer MP; Keertikar K; Morriello GJ; Stamford AW; Waddell ST; Zhong B; Hu B; Ji T; Diamond TL; Bahnck-Teets C; Carroll SS; Fay JF; Min X; Morris W; Ballard JE; Miller MD; McCauley JA, Design and synthesis of piperazine sulfonamide cores leading to highly potent HIV-1 protease inhibitors. ACS Med. Chem. Lett 2017, 8, 1292–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.M, A. M. S.; Mandlekar S; Desikan S; Ramar T; Subramani L; Annadurai M; Desai SD; Sinha S; Jenkins SM; Krystal MR; Subramanian M; Sridhar S; Padmanabhan S; Bhutani P; Arla R; Singh S; Sinha J; Thakur M; Kadow JF; Meanwell NA, Design, Synthesis, and Pharmacokinetic Evaluation of Phosphate and Amino Acid Ester Prodrugs for Improving the Oral Bioavailability of the HIV-1 Protease Inhibitor Atazanavir. J. Med. Chem 2019, 62, 3553–3574. [DOI] [PubMed] [Google Scholar]
- 214.DeGoey DA; Grampovnik DJ; Flosi WJ; Marsh KC; Wang XC; Klein LL; McDaniel KF; Liu Y; Long MA; Kati WM; Molla A; Kempf DJ, Water-soluble prodrugs of the human immunodeficiency virus protease inhibitors lopinavir and ritonavir. J. Med. Chem 2009, 52, 2964–2970. [DOI] [PubMed] [Google Scholar]
- 215.Furfine ES; Baker CT; Hale MR; Reynolds DJ; Salisbury JA; Searle AD; Studenberg SD; Todd D; Tung RD; Spaltenstein A, Preclinical pharmacology and pharmacokinetics of GW433908, a water-soluble prodrug of the human immunodeficiency virus protease inhibitor amprenavir. Antimicrob. Agents Chemother 2004, 48, 791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kesteleyn B; Amssoms K; Schepens W; Hache G; Verschueren W; Van De Vreken W; Rombauts K; Meurs G; Sterkens P; Stoops B; Baert L; Austin N; Wegner J; Masungi C; Dierynck I; Lundgren S; Jonsson D; Parkes K; Kalayanov G; Wallberg H; Rosenquist A; Samuelsson B; Van Emelen K; Thuring JW, Design and synthesis of HIV-1 protease inhibitors for a long-acting injectable drug application. Bioorg. Med. Chem. Lett 2013, 23, 310–317. [DOI] [PubMed] [Google Scholar]
- 217.Lipinski CA, Lead-and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technologies 2004, 1, 337–341. [DOI] [PubMed] [Google Scholar]
- 218.Doak BC; Zheng J; Dobritzsch D; Kihlberg J, How beyond rule of 5 drugs and clinical candidates bind to their targets. Journal of medicinal chemistry 2016, 59, 2312–2327. [DOI] [PubMed] [Google Scholar]
- 219.Doak BC; Kihlberg J, Drug discovery beyond the rule of 5-Opportunities and challenges Taylor & Francis: 2017. [DOI] [PubMed] [Google Scholar]
- 220.Mallinson J; Collins I, Macrocycles in new drug discovery. Future medicinal chemistry 2012, 4, 1409–1438. [DOI] [PubMed] [Google Scholar]
- 221.Harper S; McCauley JA; Rudd MT; Ferrara M; DiFilippo M; Crescenzi B; Koch U; Petrocchi A; Holloway MK; Butcher JW; Romano JJ; Bush KJ; Gilbert KF; McIntyre CJ; Nguyen KT; Nizi E; Carroll SS; Ludmerer SW; Burlein C; DiMuzio JM; Graham DJ; McHale CM; Stahlhut MW; Olsen DB; Monteagudo E; Cianetti S; Giuliano C; Pucci V; Trainor N; Fandozzi CM; Rowley M; Coleman PJ; Vacca JP; Summa V; Liverton NJ, Discovery of MK-5172, a macrocyclic hepatitis C virus NS3/4a protease inhibitor. ACS Med. Chem. Lett 2012, 3, 332–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Vendeville S; Nilsson M; de Kock H; Lin TI; Antonov D; Classon B; Ayesa S; Ivanov V; Johansson PO; Kahnberg P; Eneroth A; Wikstrom K; Vrang L; Edlund M; Lindstrom S; Van de Vreken W; McGowan D; Tahri A; Hu L; Lenz O; Delouvroy F; Van Dooren M; Kindermans N; Surleraux D; Wigerinck P; Rosenquist A; Samuelsson B; Simmen K; Raboisson P, Discovery of novel, potent and bioavailable proline-urea based macrocyclic HCV NS3/4A protease inhibitors. Bioorg. Med. Chem. Lett 2008, 18, 6189–6193. [DOI] [PubMed] [Google Scholar]
- 223.Childs-Kean LM; Brumwell NA; Lodl EF, Profile of sofosbuvir/velpatasvir/voxilaprevir in the treatment of hepatitis C. Infection and Drug Resistance 2019, 12, 2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kazmierski WM; Hamatake R; Duan M; Wright LL; Smith GK; Jarvest RL; Ji JJ; Cooper JP; Tallant MD; Crosby RM; Creech K; Wang A; Li X; Zhang S; Zhang YK; Liu Y; Ding CZ; Zhou Y; Plattner JJ; Baker SJ; Bu W; Liu L, Discovery of novel urea-based hepatitis C protease inhibitors with high potency against protease-inhibitor-resistant mutants. J Med Chem 2012, 55, 3021–6. [DOI] [PubMed] [Google Scholar]
- 225.Moreau B; O’Meara JA; Bordeleau J; Garneau M; Godbout C; Gorys V; Leblanc M; Villemure E; White PW; Llinàs-Brunet M, Discovery of hepatitis C virus NS3–4A protease inhibitors with improved barrier to resistance and favorable liver distribution. J. Med. Chem 2014, 57, 1770–1776. [DOI] [PubMed] [Google Scholar]
- 226.O’Meara JA; Lemke CT; Godbout C; Kukolj G; Lagacé L; Moreau B; Thibeault D; White PW; Llinàs-Brunet M, Molecular mechanism by which a potent hepatitis C virus NS3-NS4A protease inhibitor overcomes emergence of resistance. J. Biol. Chem 2013, 288, 5673–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Shah U; Jayne C; Chackalamannil S; Velázquez F; Guo Z; Buevich A; Howe JA; Chase R; Soriano A; Agrawal S; Rudd MT; McCauley JA; Liverton NJ; Romano J; Bush K; Coleman PJ; Grisé-Bard C; Brochu M-C; Charron S; Aulakh V; Bachand B; Beaulieu P; Zaghdane H; Bhat S; Han Y; Vacca JP; Davies IW; Weber AE; Venkatraman S, Novel quinoline-based P2–P4 macrocyclic derivatives as pan-genotypic HCV NS3/4a protease inhibitors. ACS Med. Chem. Lett 2014, 5, 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Rudd MT; Butcher JW; Nguyen KT; McIntyre CJ; Romano JJ; Gilbert KF; Bush KJ; Liverton NJ; Holloway MK; Harper S; Ferrara M; DiFilippo M; Summa V; Swestock J; Fritzen J; Carroll SS; Burlein C; DiMuzio JM; Gates A; Graham DJ; Huang Q; McClain S; McHale C; Stahlhut MW; Black S; Chase R; Soriano A; Fandozzi CM; Taylor A; Trainor N; Olsen DB; Coleman PJ; Ludmerer SW; McCauley JA, P2-Quinazolinones and bis-macrocycles as new templates for next-generation hepatitis C virus NS3/4A protease inhibitors: discovery of MK-2748 and MK-6325. ChemMedChem 2015, 10, 727–735. [DOI] [PubMed] [Google Scholar]
- 229.Neelamkavil SF; Agrawal S; Bara T; Bennett C; Bhat S; Biswas D; Brockunier L; Buist N; Burnette D; Cartwright M; Chackalamannil S; Chase R; Chelliah M; Chen A; Clasby M; Colandrea VJ; Davies IW; Eagen K; Guo Z; Han Y; Howe J; Jayne C; Josien H; Kargman S; Marcantonio K; Miao S; Miller R; Nolting A; Pinto P; Rajagopalan M; Ruck RT; Shah U; Soriano A; Sperbeck D; Velazquez F; Wu J; Xia Y; Venkatraman S, Discovery of MK-8831, A Novel Spiro-Proline Macrocycle as a Pan-Genotypic HCV-NS3/4a Protease Inhibitor. ACS Med Chem Lett 2016, 7, 111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Velazquez F; Chelliah M; Clasby M; Guo Z; Howe J; Miller R; Neelamkavil S; Shah U; Soriano A; Xia Y; Venkatraman S; Chackalamannil S; Davies IW, Design and Synthesis of P2-P4 Macrocycles Containing a Unique Spirocyclic Proline: A New Class of HCV NS3/4A Inhibitors. ACS Med Chem Lett 2016, 7, 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Matthew AN; Zephyr J; Nageswara Rao D; Henes M; Kamran W; Kosovrasti K; Hedger AK; Lockbaum GJ; Timm J; Ali A; Kurt Yilmaz N; Schiffer CA, Avoiding Drug Resistance by Substrate Envelope-Guided Design: Toward Potent and Robust HCV NS3/4A Protease Inhibitors. mBio 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Moshafi MH; Ghasemshirazi S; Abiri A, The Art of Suicidal Molecular Seduction for Targeting Drug Resistance. Medical Hypotheses 2020, 109676. [DOI] [PubMed]
- 233.Chan AH; Lee W-G; Spasov KA; Cisneros JA; Kudalkar SN; Petrova ZO; Buckingham AB; Anderson KS; Jorgensen WL, Covalent inhibitors for eradication of drug-resistant HIV-1 reverse transcriptase: From design to protein crystallography. Proceedings of the National Academy of Sciences 2017, 114, 9725–9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kim J-H; Resende R; Wennekes T; Chen H-M; Bance N; Buchini S; Watts AG; Pilling P; Streltsov VA; Petric M, Mechanism-based covalent neuraminidase inhibitors with broad-spectrum influenza antiviral activity. Science 2013, 340, 71–75. [DOI] [PubMed] [Google Scholar]
- 235.Pan Q; Peppelenbosch MP; Janssen HL; de Knegt RJ, Telaprevir/boceprevir era: from bench to bed and back. World Journal of Gastroenterology: WJG 2012, 18, 6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Malcolm BA; Liu R; Lahser F; Agrawal S; Belanger B; Butkiewicz N; Chase R; Gheyas F; Hart A; Hesk D; Ingravallo P; Jiang C; Kong R; Lu J; Pichardo J; Prongay A; Skelton A; Tong X; Venkatraman S; Xia E; Girijavallabhan V; Njoroge FG, SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob. Agents Chemother 2006, 50, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Bauer RA, Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug discovery today 2015, 20, 1061–1073. [DOI] [PubMed] [Google Scholar]
- 238.Deng Z; Chuaqui C; Singh J, Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein− ligand binding interactions. Journal of medicinal chemistry 2004, 47, 337–344. [DOI] [PubMed] [Google Scholar]
- 239.Leidner F; Kurt Yilmaz N; Schiffer CA, Target-Specific Prediction of Ligand Affinity with Structure-Based Interaction Fingerprints. Journal of chemical information and modeling 2019, 59, 3679–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Da C; Stashko M; Jayakody C; Wang X; Janzen W; Frye S; Kireev D, Discovery of Mer kinase inhibitors by virtual screening using Structural Protein–Ligand Interaction Fingerprints. Bioorganic & medicinal chemistry 2015, 23, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Van Linden OP; Kooistra AJ; Leurs R; De Esch IJ; De Graaf C, KLIFS: a knowledge-based structural database to navigate kinase–ligand interaction space. Journal of medicinal chemistry 2014, 57, 249–277. [DOI] [PubMed] [Google Scholar]
- 242.Vass M; Kooistra AJ; Ritschel T; Leurs R; de Esch IJ; de Graaf C, Molecular interaction fingerprint approaches for GPCR drug discovery. Current opinion in pharmacology 2016, 30, 59–68. [DOI] [PubMed] [Google Scholar]
- 243.Leidner F; Kurt Yilmaz N; Schiffer CA, Target-Specific Prediction of Ligand Affinity with Structure-Based Interaction Fingerprints. J Chem Inf Model 2019, 59, 3679–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Riniker S, Molecular dynamics fingerprints (MDFP): machine learning from MD data to predict free-energy differences. Journal of chemical information and modeling 2017, 57, 726–741. [DOI] [PubMed] [Google Scholar]
- 245.Wang S; Riniker S, Use of molecular dynamics fingerprints (MDFPs) in SAMPL6 octanol–water log P blind challenge. Journal of computer-aided molecular design 2019, 1–11. [DOI] [PubMed]